 Alan Talevi
Alan Talevi Carolina L. Bellera
Carolina L. Bellera
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Nat. Prod. , 15 March 2024
Sec. Informatics and Computational Methods
Volume 3 - 2024 | https://doi.org/10.3389/fntpr.2024.1367537
The clustering of small molecules is of considerable importance for computer-aided drug discovery and virtual screening applications. The structure of chemical data in appropriate subspaces of the chemical space is relevant to sample datasets in a representative manner, to generate small libraries with wide or narrow chemical coverage (depending on the specific goals), and to guide the selection of subsets of in silico hits that are submitted for experimental confirmation. In the field of natural products, identifying regions of the chemical space where bioactive compounds congregate and understanding the relationship between biosynthetic gene clusters and the molecular structure of secondary metabolites may have a direct impact on natural product discovery and engineering. Here, we briefly discuss general approximations and available resources for the clustering of small molecules, and how the clustering of small molecules can be boosted by the application of novel clustering approximations, namely subspace clustering and multi-view clustering, which represent opposite philosophies of the clustering paradigm. We present some specific applications of small molecule clustering in the field of natural products, and analyze how a chemogenomic perspective may be particularly embodied in the field of natural products.
Clustering (grouping a set of objects by different criteria of similarity and/or distance within a spatial representation) is an inherent capacity of the human brain and a pre-condition for complex thinking (Seger and Miller, 2010). The tendency of the human mind to group things in classes or categories is clearly reflected in the structure of language itself: common nouns refer to people, animals, or things of the same class or species, whereas proper nouns distinguish specific objects from any other of the same class or species. Perceptual grouping is performed according to principles of proximity, similarity (by shape, size, color, texture, odor, or taste), continuity (when objects are arranged in continuous lines or curves) and common fate (objects that move at the same speed or direction are perceived as part of a continuous), among others (Brooks and Wagemans, 2014).
For example, if we present a series of chemical structures to a person, even if he or she has not received any training in chemistry, it is possible that he or she will be able to identify structural patterns and differentiate compounds from different chemical families. Although perceptual grouping is performed at a remarkable speed, it is evident that it has limitations when dealing with large-scale data, for example, big datasets, and that it is often convenient to realize the clustering of small molecules in an automated manner with the assistance of algorithms and information technologies.
Therefore, clustering of small molecules is of great importance in the field of computer-aided drug discovery and to guide virtual and wet screening applications. The structure of chemical data in appropriate subspaces of the chemical space is highly relevant to sample datasets in a representative or biased manner, depending on the pursued goals. This is useful for several tasks. First, stratified splitting of datasets into representative training and validation sets is routinely performed when building QSAR/cheminformatics machine learning models. This practice may obey different reasons: it ensures maximal coverage of the chemical space by training data, which broadens the generalizability and applicability domain of the models (Leonard and Roy, 2006; Hadipour et al., 2022), and it can also be used to avoid compound biases and overconfident validation results by allocating extremely similar compounds in both the training and validation sets (Mayr et al., 2018; Lopez-Del Rio et al., 2019). Second, structuring data in the chemical space can be useful either to explore unexplored regions for novel active compounds (Virshup et al., 2013; Domingo-Fernández et al., 2023) or, conversely, to focus on specific already-explored regions to generate preferred screening collections or focused libraries (Böcker et al., 2005; Harris et al., 2011). Third, small-molecule clustering can be used to guide the selection of subsets of in silico hits submitted to experimental confirmation (Prada Gori et al., 2022a). Because of budget limitations, the number of in silico hits that emerge from a virtual screening campaign frequently exceeds the number that can be synthesized, isolated, or acquired and assayed experimentally. Clustering and subsequent representative sampling can ensure the coverage of as much chemical diversity as possible with those relatively scarce predicted hits sent to wet assays. Furthermore, the same principle may be used to expedite structure-based virtual screening, without exhaustive docking of every molecule in a large or ultra-large chemical library (Yang et al., 2021).
Broadly speaking, clustering approaches (including those used to group small molecules) can be classified into hierarchical and non-hierarchical algorithms.
In hierarchical clustering, the data are partitioned or grouped serially (Everitt et al., 2011a): agglomerative (or bottom-up) approximations proceed by a series of successive fusions of the N objects into groups (eventually merging them all together in the last step); divisive (or top-down) methods, in contrast, separate the N objects successively into smaller groups (eventually resulting in isolated elements in the last step). Hierarchical clustering is most often represented as a hierarchical tree or dendrogram (various leveled representations of clusters). A significant limitation of these approaches is that once individuals have been merged or separated, this cannot be revised or reverted in the subsequent steps of the algorithms. Classic examples of hierarchical clustering methods include single linkage, complete linkage, and Ward linkage, with Ward linkage being commonly used in the context of chemical library clustering (Murtagh and Contreras, 2017). Another example is the Maximal Common Substructure (MCS) clustering approach, which agglomerates compounds based on their common subgraph of the greatest cardinality, and has been implemented at Chemaxon’s Jchem (https://chemaxon.com/).
Non-hierarchical approaches, on the other hand, do not have a tree-like, progressive structure and regularly (but not always) require (pre)specification of the number of clusters to be obtained; in optimization approaches, the clusters are refined in successive steps by either minimizing or maximizing some numerical criterion, as in the popular K-means approach (MacQueen et al., 1967). A common incognita when using these approaches is how to decide on the number of clusters that will be considered and what compounds will be used as initial seeds to start the procedure. The first choice is often made in a rather systematic way, by plotting the value of a “goodness of clustering” criterion against the number of k groups: large changes of levels in the plot, such as in the elbow method, are usually taken as an indication of a particular number of groups (Everitt et al., 2011b), as seen in recent examples in the field (Prada Gori et al., 2022a; Hadipour et al., 2022). Butina clustering, a sphere-exclusion method based on similarity coefficients, is another example of a frequently used classic non-hierarchical approach (Butina, 1999).
Notably, small molecules are often represented by high-dimensional feature representations (i.e., a large pool of global and/or local molecular features), which, for the sake of visual representation and/or reduction of computational cost, are often pre-processed using dimensionality reduction techniques, such as Principal Component Analysis (PCA) (Prada Gori et al., 2022a) or the Uniform Manifold Approximation and Projection (UMAP) approaches (Prada Gori et al., 2022a; Hernández-Hernández and Ballester, 2023), which provide a low dimensional projection of the data. Hadipour et al., 2022 recently reported an open-source deep clustering approach, in which different dimensionality-reduction techniques were concatenated. First, they resort to PCA on sets of global and local molecular features, resulting in a representation comprising 243 learned features, which was later subjected to further dimensionality reduction using autoencoders.
It is noteworthy that the assessment of the quality of the clustering is often omitted or performed informally, for example, by simple visual inspection of the results, as in the elbow method. It is essential to emphasize the importance of resorting to quantitative metrics to assess the goodness of the clustering output. The silhouette coefficient (Rousseeuw, 1987) and the Calinski–Harabasz score (Caliński and Harabasz, 1974) are good examples of such metrics; they approach optimal values when the clustering procedure results in compact clusters (low within-cluster distances) that are well separated from each other (high between-cluster distances).
Fortunately, there is currently a wide range of freely available resources that offer hierarchical and/or non-hierarchical small molecule clustering tools, either via web servers or code. Among them, we may mention ChemBioServer Karatzas et al., 2021, ChemMine Tools Backman et al., 2011, DeepClustering (Hadipour et al., 2022), ChemmineR Cao et al., 2008, RDKit (Hernández-Hernández and Ballester, 2023), IRaPCA (Prada Gori et al. 2022b), SOMoC (Prada Gori et al. 2022b), and ChiCA (Prada Gori et al. 2022a) (see Table 1 for additional details).
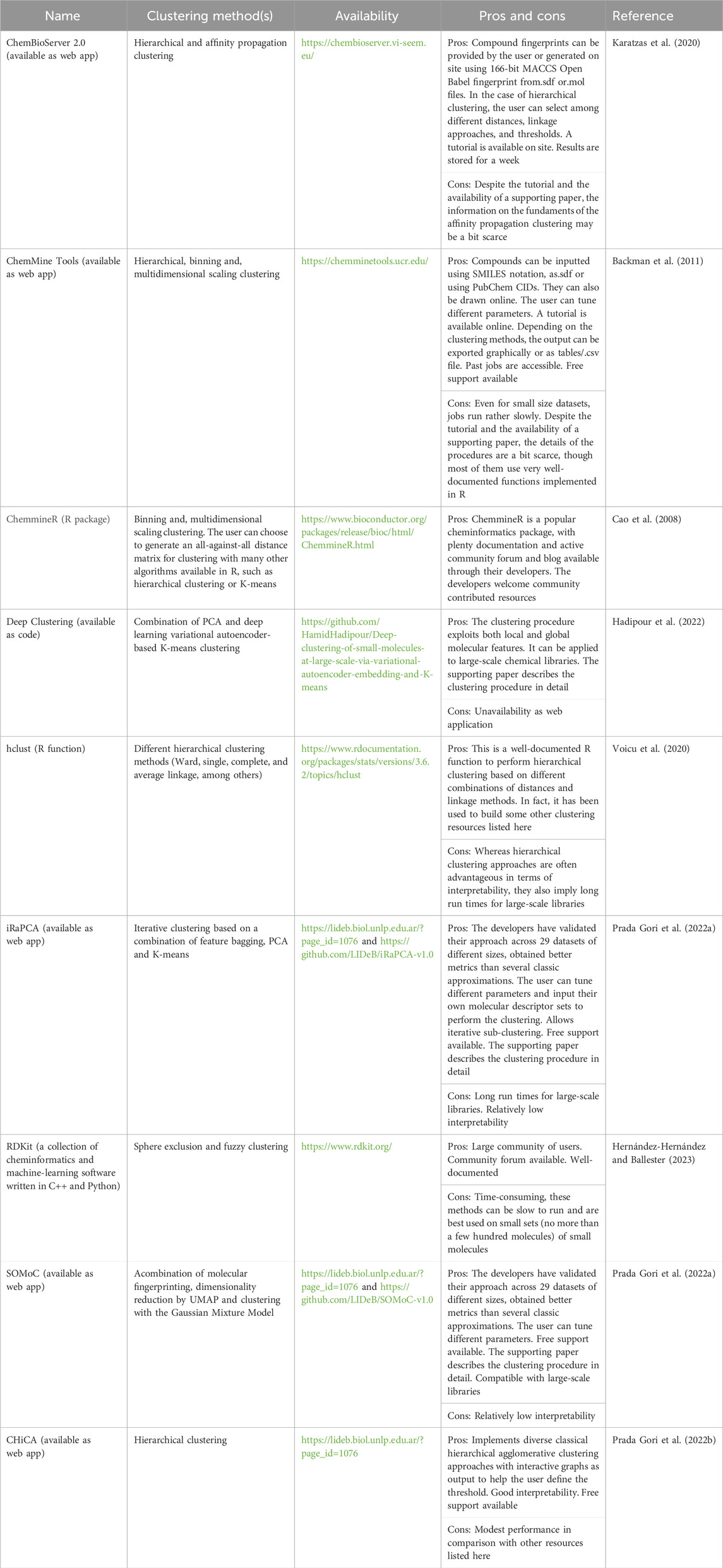
TABLE 1. A selection of freely available resources to perform small molecule clustering. A subjective appraisal of their advantages and disadvantages of each tool is included.
Let us say we have been given the (simple?) task of clustering the following three objects: a human, a dolphin, and a shark. Some people may argue that the dolphin and the shark should be grouped together, based on criteria of shape, color, living environment, external organs, etc. Other people may propose that dolphins are more like humans based, for instance, on the similarities between their internal organs. Both these arguments are valid. The example illustrates that how things are clustered depends on the features that are used for the clustering exercise: if we consider different sets of features, we will obtain different results from the application of the clustering algorithms. On the other hand, it is also evident that, whereas multiple classifications of the data are possible depending on the features used to represent such data, some classification criteria are more useful than others. For instance, a classification of books based on subject matter would be of much wider use than a classification based on the color of the book’s binding (Everitt et al., 2011c).
The former discussion brings about a fundamental question related to clustering: does it exist a ground truth, that is to say, an actual, natural, or absolute structure of the data? We believe it does not (particularly if we intend to use clustering as an unsupervised approximation, in which case no labelling of the data is considered a priori). However, not all possible representations of the data are equally useful, and their usefulness depends on the value judgment of the user.
The precedent concepts are taken into consideration by clustering approaches that incorporate feature-selection steps, and by subspace clustering approximations, although these approaches have been rarely applied in the field of small molecule clustering (Rivera-Borroto et al., 2011). Subspace clustering represents an extension of feature selection that attempts to find clusters in different subspaces of the same dataset (as in the case of feature selection, subspace clustering requires evaluation criteria to choose relevant subspaces, which in the case of unsupervised clustering may well be the previously mentioned silhouette coefficient and Calinski–Harabasz score). For example, starting from descriptor sets provided by the user, the iRaPCA approach (Prada Gori et al., 2022a) randomly explores possible subspaces where adequate clustering can be found, as judged by the silhouette coefficient or other metric. Moreover, each resulting cluster may be further explored iteratively to identify proper sub-clusters. Interestingly, iRaPCA exhibited consistent and almost optimal behavior in benchmarking exercises across 29 datasets of variable sizes, even without iterations (Prada Gori et al., 2022a; Prada Gori et al., 2022b).
However, mainly in line with the idea of an underlying ideal structure of the data (in which case any view of the data would originate from an underlying latent space), multi-view clustering has also attracted much attention recently (although it has been mostly overlooked in the field of cheminformatics) (Zhang et al., 2020; Guo et al., 2022; Cao and Xie, 2024). The idea here is that different views or representations of data may be integrated or combined because of their complementarity (which may be of particular interest in the case of supervised clustering) or based on their consensus.
It is well known that natural products display greater chemical diversity and complexity (including grater stereochemical content) than drugs from completely synthetic origins (Stratton et al., 2015). This makes total synthesis of drugs of natural origin particularly challenging and difficult to scale. Furthermore, fractionation (e.g., to perform bioassay-guided identification of promising compounds), isolation and characterization of natural drug candidates is also time- and resource-intensive (especially in the case of scarce natural products with limited sample amount) (Kumarasamy, 2012; Kuranaga et al., 2020) and crude extracts are comparatively unfriendly to undertake high-throughput screening campaigns due to assay interference and issues associated with dereplication, reconstitution and liquid handling (Schmid et al., 1999; Henrich and Beutler, 2013). Since only a fraction of natural products can be practically explored in the short to midterm, drug discovery efforts need to be prioritized towards natural products with higher discovery potentials (Tao et al., 2015). This may be accomplished by sampling a variety of taxonomy or chemical diversity, by focusing on unexploited chemical regions or, on the contrary, by submitting to experimental assays samples that are congregated in regions with high bioactivity potential (Henrich and Beutler, 2013). In any case, the design of focused or privileged libraries and thereby the use of small molecule clustering approaches is of special interest in the field of natural products.
For instance, Tao et al., 2015 resorted to hierarchical clustering (deriving molecular scaffold tress and molecular fingerprinting trees based on the complete linkage approach) and found that natural product leads corresponding to either approved drugs of natural origin or natural drug candidates in clinical trials emerge from pre-existing drug productive clusters, suggesting that focusing on already known drug productive clusters could enhance drug discovery potential. Hagan and Kell, in contrast, used a sphere exclusion approach on about 196K compounds emerging from the union of the UNPD database and the Dictionary of Natural Products database (O'Hagan and Kell, 2018). After disregarding unusual structures, they resorted to hierarchical K-Means clustering to analyze what sizes should a subset library of the initial collection have to provide representative coverage of the entire database.
Chemogenomics has been defined as the investigation of classes of compounds or focused libraries against families of functionally related proteins (Kubinyi, 2006). The three basic principles underlying chemogenomic analysis are as follows: a) similar small molecules are likely to bind to the same target; b) similar targets are likely to share ligands; and c) ligands with a similar interaction signature are likely to elicit similar phenotypic responses. In other words, known and/or predicted associations between ligands (e.g., through small molecule clustering) and targets (e.g., through sequence-, structure- or binding site-matching tools) are used to reveal hidden associations. Chemogenomics may have specific applications in the field of drug discovery, from the search for bioactive analogs within a target family to target deconvolution, from on-target drug repurposing to the investigation of subtype selectivity. For instance, the last release of TDR Targets integrated genomic data from diverse microorganisms (with a focus on those that cause tropical infections) with information on bioactive compounds (Urán Landaburu et al., 2020). Based on a drug-target network, the database currently includes network-driven target prioritizations and novel visualizations of network subgraphs that display chemical- and target-similarity neighborhoods with target-compound bioactivity links, which may be used to propose novel druggable targets and explore new repurposing opportunities in the field of neglected diseases. The Computational Analysis of Novel Drug Opportunities (CANDO) is another good example of chemogenomic analysis. It departs from a traditional reverse docking approach but incorporates a systems pharmacology perspective by considering both ligand similarity and the similarity between the interaction signatures of two ligands against a large panel of targets as a possible indication of similar phenotypic effects (Minie et al., 2014). All this, of course, may find straightforward applications in the field of natural product drug discovery.
However, there are possible exclusive implementations of the chemogenomics approach in the field of natural products. The key here is the increasing knowledge of biosynthetic gene clusters and sub-clusters (the latter being responsible for the biosynthesis of a specific chemical moiety in a natural product) in plants and microorganisms (Polturak and Osbourn, 2021; Louwen et al., 2023). In the same manner that inter-species associations between biosynthetic genes can be used to guide the discovery of unnoticed bioactive natural products (Bauman et al., 2021), clustering of secondary metabolites, for instance, may provide clues on secondary metabolic pathways (based on a “guilt by association” principle, similar secondary metabolites are likely to emerge from similar biosynthetic pathways), which are often transcriptionally silent under typical laboratory growth conditions (Kwon et al., 2021). This can have immediate applications in bioengineering oriented to obtention of natural products in artificial settings.
Based on the results in other fields of knowledge, it is possible that cheminformatics clustering tools will benefit, in the new few years, from the implementation of feature selection, subspace clustering, and multi-view clustering tools, which have been so far scarcely applied (though with promising results) in the field of small molecules.
Beyond their general applications in the field of cheminformatics, small-molecule clustering tools hold promise for the discovery of bioactive natural products, which often exhibit intrinsic difficulties to achieve scalable synthesis or purification that allow their characterization. The laborious obtention of the isolated amounts of natural products required for bioassays and the challenges presented by crude and fractioned extracts in relation to screening technologies make particularly relevant the obtention of small focused or privileged libraries with high discovery potential. This task can be efficiently addressed by use of small molecule clustering approaches. Depending on the particular goal of the investigation, research may focus on unexplored regions of the natural product chemical space (if the focus is novelty) or in already known productive regions where bioactive products tend to converge.
The increasing knowledge on biosynthetic gene clusters and sub-clusters and the fact that biosynthetic pathways may remain silent in artificial/laboratory conditions suggest that small molecule clustering may find specific applications in chemogenomics and bioengineering, as similar molecules are often synthesized by similar biosynthetic routes.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
AT: Funding acquisition, Investigation, Writing–original draft, Writing–review and editing. CB: Funding acquisition, Investigation, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors thank Agencia I+D+I (PICT 2019-1075, PICT 2019-00984, PICT 2021-0404) and CONICET PIP-2022-11220210100030C) for funding their research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AT declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Backman, T. W., Cao, Y., and Girke, T. (2011). ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res. 39, W486–W491. doi:10.1093/nar/gkr320
Bauman, K. D., Butler, K. S., Moore, B. S., and Chekan, J. R. (2021). Genome mining methods to discover bioactive natural products. Nat. Prod. Rep. 38, 2100–2129. doi:10.1039/d1np00032b
Böcker, A., Derksen, S., Schmidt, E., Teckentrup, A., and Schneider, G. (2005). A hierarchical clustering approach for large compound libraries. J. Chem. Inf. Model 45, 807–815. doi:10.1021/ci0500029
Brooks, J. L. “Traditional and new principles of perceptual grouping”. In: J. Wagemans, edi-tor. The oxford handbook of perceptual organization. Oxford: Oxford University Press (2014). p. 57–87.
Butina, D. (1999). Unsupervised data base clustering based on daylight’s fingerprint and tanimoto similarity: a fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 39, 747–750. doi:10.1021/ci9803381
Caliński, T., and Harabasz, J. (1974). A dendrite method for cluster analysis. Commun. Stat-Theory Methods 3, 1–27. doi:10.1080/03610927408827101
Cao, Y., Charisi, A., Cheng, L. C., Jiang, T., and Girke, T. (2008). ChemmineR: a compound mining framework for R. Bioinformatics 24, 1733–1734. doi:10.1093/bioinformatics/btn307
Cao, Z., and Xie, X. (2024). Structure learning with consensus label information for multi-view unsupervised feature selection. Expert Syst. Appl. 238, 121893. doi:10.1016/j.eswa.2023.121893
Domingo-Fernández, D., Gadiya, Y., Mubeen, S., Healey, D., Norman, B. H., and Colluru, V. (2023). Exploring the known chemical space of the plant kingdom: insights into taxonomic patterns, knowledge gaps, and bioactive regions. J. Cheminform 15, 107. doi:10.1186/s13321-023-00778-w
Everitt, B. S., Landau, S., Leese, M., and Stahl, D. (2011a). Cluster analysis. 5th Edition, 1. West Sussex: John Wiley and Sons, 71.
Everitt, B. S., Landau, S., Leese, M., and Stahl, D. (2011b). Cluster analysis. 5th Edition, 1. West Sussex: John Wiley and Sons, 126.
Everitt, B. S., Landau, S., Leese, M., and Stahl, D. (2011c). Cluster analysis. 5th Edition, 1. West Sussex: John Wiley and Sons, 7.
Guo, J., Sun, Y., Gao, J., Hu, Y., and Yin, N. (2022). Rank consistency induced multiview subspace clustering via low-rank matrix factorization. EEE Trans. Neural Netw. Learn Syst. 33, 3157–3170. doi:10.1109/tnnls.2021.3071797
Hadipour, H., Liu, C., Davis, R., Cardona, S. T., and Hu, P. (2022). Deep clustering of small molecules at large-scale via variational autoencoder embedding and K-means. BMC Bioinforma. 23 (Suppl. 4), 132. doi:10.1186/s12859-022-04667-1
Harris, C. J., Hill, R. D., Sheppard, D. W., Slater, M. J., and Stouten, P. F. (2011). The design and application of target-focused compound libraries. Comb. Chem. High. Throughput Screen 14, 521–531. doi:10.2174/138620711795767802
Henrich, C. J., and Beutler, J. A. (2013). Matching the power of high throughput screening to the chemical diversity of natural products. Nat. Prod. Rep. 30, 1284–1298. doi:10.1039/c3np70052f
Hernández-Hernández, S., and Ballester, P. J. (2023). On the best way to cluster NCI-60 molecules. Biomolecules 13, 498. doi:10.3390/biom13030498
Karatzas, E., Zamora, J. E., Athanasiadis, E., Dellis, D., Cournia, Z., and Spyrou, H. M. (2020). ChemBioServer 2.0: an advanced web server for filtering, clustering and networking of chemical compounds facilitating both drug discovery and repurposing. Bioinformatics 36, 2602–2604. doi:10.1093/bioinformatics/btz976
Kubinyi, H. (2006). “Chemogenomics in drug discovery,” in Chemical Genomics. Editors S. Jaroch, and H. Weinmann (Springer, Berlin, Heidelberg: Ernst Schering Research Foundation Workshop), Vol. 58.
Kumarasamy, Y. (2012). Scaling-up of natural products isolation. Methods Mol. Biol. 864, 465–472. doi:10.1007/978-1-61779-624-1_18
Kuranaga, T., Minote, M., Morimoto, R., Pan, C., Ogawa, H., and Kakeya, H. (2020). Highly sensitive labeling reagents for scarce natural products. ACS Chem. Biol. 15, 2499–2506. doi:10.1021/acschembio.0c00517
Kwon, M. J., Steiniger, C., Cairns, T. C., Wisecaver, J. H., Lind, A. L., Pohl, C., et al. (2021). Beyond the biosynthetic gene cluster paradigm: genome-wide coexpression networks connect clustered and unclustered transcription factors to secondary metabolic pathways. Microbiol. Spectr. 9, e0089821. doi:10.1128/spectrum.00898-21
Leonard, J. T., and Roy, K. (2006). On selection of training and test sets for the development of predictive QSAR models. QSAR Comb. Sci. 25, 235–251. doi:10.1002/qsar.200510161
Lopez-Del Rio, A., Nonell-Canals, A., Vidal, D., and Perera-Lluna, A. (2019). Evaluation of cross-validation strategies in sequence-based binding prediction using deep learning. J. Chem. Inf. Model 59, 1645–1657. doi:10.1021/acs.jcim.8b00663
Louwen, J. J. R., Kautsar, S. A., van der Burg, S., Medema, M. H., and van der Hooft Jjj, (2023). iPRESTO: automated discovery of biosynthetic sub-clusters linked to specific natural product substructures. PLoS Comput. Biol. 19, e1010462. doi:10.1371/journal.pcbi.1010462
MacQueen, J. (1967). “Some methods for classification and analysis of multivariate observations,” in Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability Volume 1. Nerkeley. Editors L. M. Le Cam, and J. Neyman (University of California Press), 281–297.
Mayr, A., Klambauer, G., Unterthiner, T., Steijaert, M., Wegner, J. K., Ceulemans, H., et al. (2018). Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. ChEML. Chem. Sci. 9, 5441–5451. doi:10.1039/c8sc00148k
Minie, M., Chopra, G., Sethi, G., Horst, J., White, G., Roy, A., et al. (2014). CANDO and the infinite drug discovery frontier. Drug Discov. Today 19, 1353–1363. doi:10.1016/j.drudis.2014.06.018
Murtagh, F., and Contreras, P. (2017). Algorithms for hierarchical clustering: an overview, II, 7. Wiley Interdiscip Rev Data Min Knowl Discov, e1219.
O'Hagan, S., and Kell, D. B. (2018). Analysing and navigating natural products space for generating small, diverse, but representative chemical libraries. Biotechnol. J. 13, 201700503. doi:10.1002/biot.201700503
Polturak, G., and Osbourn, A. (2021). The emerging role of biosynthetic gene clusters in plant defense and plant interactions. PLoS Pathog. 17, e1009698. doi:10.1371/journal.ppat.1009698
Prada Gori, D. N., Alberca, L. N., Rodriguez, S., Llanos, M. A., Bellera, C. L., Talevi, A., et al. (2022b). LIDeB tools: a Latin American resource of freely available, open-source cheminformatics apps. Artif. Intell. Life Sci. 2, 100049. doi:10.1016/j.ailsci.2022.100049
Prada Gori, D. N., Llanos, M. A., Bellera, C. L., Talevi, A., and Alberca, L. N. (2022a). iRaPCA and SOMoC: development and validation of web applications for new approaches for the clustering of small molecules. J. Chem. Inf. Model 62, 2987–2998. doi:10.1021/acs.jcim.2c00265
Rivera-Borroto, O. M., Marrero-Ponce, Y., García-de la Vega, J. M., and Grau-Ábalo Rdel, C. (2011). Comparison of combinatorial clustering methods on pharmacological data sets represented by machine learning-selected real molecular descriptors. J. Chem. Inf. Model 51, 3036–3049. doi:10.1021/ci2000083
Rousseeuw, P. J. (1987). Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65. doi:10.1016/0377-0427(87)90125-7
Schmid, I. I., Sattler, I. I., Grabley, S., and Thiericke, R. (1999). Natural products in high throughput screening: automated high-quality sample preparation. J. Biomol. Screen 4, 15–25. doi:10.1177/108705719900400104
Seger, C. A., and Miller, E. K. (2010). Category learning in the brain. Annu. Rev. Neurosci. 33, 203–219. doi:10.1146/annurev.neuro.051508.135546
Stratton, C. F., Newman, D. J., and Tan, D. S. (2015). Cheminformatic comparison of approved drugs from natural product versus synthetic origins. Bioorg Med. Chem. Lett. 25, 4802–4807. doi:10.1016/j.bmcl.2015.07.014
Tao, L., Zhu, F., Qin, C., Zhang, C., Chen, S., Zhang, P., et al. (2015). Clustered distribution of natural product leads of drugs in the chemical space as influenced by the privileged target-sites. Sci. Rep. 5, 9325. doi:10.1038/srep09325
Urán Landaburu, L., Berenstein, A. J., Videla, S., Maru, P., Shanmugam, D., Chernomoretz, A., et al. (2020). TDR targets 6: driving drug discovery for human pathogens through intensive chemogenomic data integration. Nucleic Acids Res. 48, D992–D1005. doi:10.1093/nar/gkz999
Virshup, A. M., Contreras-García, J., Wipf, P., Yang, W., and Beratan, D. N. (2013). Stochastic voyages into uncharted chemical space produce a representative library of all possible drug-like compounds. J. Am. Chem. Soc. 135, 7296–7303. doi:10.1021/ja401184g
Voicu, A., Duteanu, N., Voicu, M., Vlad, D., and Dumitrascu, V. (2020). The rcdk and cluster R packages applied to drug candidate selection. J. Cheminform 12, 3. doi:10.1186/s13321-019-0405-0
Yang, Y., Yao, K., Repasky, M. P., Leswing, K., Abel, R., Shoichet, B. K., et al. (2021). Efficient exploration of chemical space with docking and deep learning. J. Chem. Theory Comput. 17, 7106–7119. doi:10.1021/acs.jctc.1c00810
Keywords: biosynthetic gene cluster (BGC), secondary metabolites, clustering, small molecules, virtual screening, subspace clustering, multi-view clustering, chemogenomics
Citation: Talevi A and Bellera CL (2024) Clustering of small molecules: new perspectives and their impact on natural product lead discovery. Front. Nat. Produc. 3:1367537. doi: 10.3389/fntpr.2024.1367537
Received: 08 January 2024; Accepted: 05 March 2024;
Published: 15 March 2024.
Edited by:
Marcus Scotti, Federal University of Paraíba, BrazilReviewed by:
Chun-Wei Tung, National Health Research Institutes, TaiwanCopyright © 2024 Talevi and Bellera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alan Talevi, YWxhbnRhbGV2aUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.