 Tsukasa Funane
Tsukasa Funane Heechul Jun
Heechul Jun Stephanie Sutoko
Stephanie Sutoko Takaomi C. Saido
Takaomi C. Saido Akihiko Kandori
Akihiko Kandori Kei M. Igarashi
Kei M. Igarashi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Syst. Neurosci., 25 August 2022
Volume 16 - 2022 | https://doi.org/10.3389/fnsys.2022.955178
This article is part of the Research TopicReconfiguration of Neuronal Ensembles Throughout LearningView all 5 articles
Clinical evidence suggests that the entorhinal cortex is a primary brain area triggering memory impairments in Alzheimer’s disease (AD), but the underlying brain circuit mechanisms remain largely unclear. In healthy brains, sharp-wave ripples (SWRs) in the hippocampus and entorhinal cortex play a critical role in memory consolidation. We tested SWRs in the MEC layers 2/3 of awake amyloid precursor protein knock-in (APP-KI) mice, recorded simultaneously with SWRs in the hippocampal CA1. We found that MEC→CA1 coordination of SWRs, found previously in healthy brains, was disrupted in APP-KI mice even at a young age before the emergence of spatial memory impairments. Intriguingly, long-duration SWRs critical for memory consolidation were mildly diminished in CA1, although SWR density and amplitude remained intact. Our results point to SWR incoordination in the entorhinal-hippocampal circuit as an early network symptom that precedes memory impairment in AD.
Spatial memory impairments, including wandering and loss of spatial senses, are one of the most prominent symptoms in Alzheimer’s disease (AD) (Hope et al., 1994). Although past studies led to significant understanding of molecular and cellular mechanisms of AD, it remains unclear what type of activity impairment causes spatial memory impairments. Histological and functional imaging studies in AD patients showed that the entorhinal cortex, rather than the hippocampus, is the earliest brain region to have activity loss and atrophy in early phases of AD (Van Hoesen et al., 1991; Khan et al., 2014). Using in vivo electrophysiological recording from the amyloid precursor protein knock-in (APP-KI) mouse model, we previously showed that grid cells in the medial entorhinal cortex (MEC), critical for spatial navigation and memory (Moser et al., 2015), are impaired even at a young age before the emergence of spatial memory impairments (Jun et al., 2020). These evidence strongly suggests that the entorhinal cortex is the primary driver of pathophysiology in AD, but it remains largely unclear how the functional degradation of MEC causes spatial memory impairments in AD.
In healthy brains, sharp-wave ripples (SWR) are network activity underlying memory consolidation (Wilson and McNaughton, 1994; Foster and Wilson, 2006; Girardeau et al., 2009; Jadhav et al., 2012; Buzsaki, 2015). Long-duration SWRs especially play a critical role in consolidation, supposedly due to their higher spatial information content from longer concurrent spike replays and larger number of neurons involved in each SWR event (Fernández-Ruiz et al., 2019). Although the hippocampal CA3 critically contributes to the generation of SWRs propagating to CA1 (Buzsaki, 1986; Sullivan et al., 2011), SWRs in the layers 2/3 of the MEC also contributes to the generation of long and multiplex SWRs in CA1 (Yamamoto and Tonegawa, 2017). In AD brains, recent in vivo electrophysiological recording studies started to show disrupted SWRs in the hippocampus. Lower density (or abundance) of SWRs (the number of SWR events per time) was found in CA1 of 5xFAD mice (Iaccarino et al., 2016), apolipoprotein E4 mice (Gillespie et al., 2016; Jones et al., 2019), 3xTg AD mice (Benthem et al., 2020), and rTg4510 tau mice (Ciupek et al., 2015; Witton et al., 2016). A recent recording study from 5xFAD mice at the older age with memory impairments observed shorter SWR duration in CA1 (Prince et al., 2021). However, no previous studies tested SWRs in the entorhinal cortex in AD. We thus simultaneously recorded SWRs in the MEC and CA1 of APP-KI mice. To understand the role of entorhinal cortex in the pathogenesis of SWRs, we focused on recording from preclinical stage, that is, young age before the emergence of spatial memory impairments (Dubois et al., 2016; Sasaguri et al., 2017).
Data are shown with ± standard error. The animal numbers and sampled neuron numbers (biological replicates) were designed to achieve a power of more than 0.8. Both sexes of animals, randomized and blinded analyses were used (see Table 1). We initially tested difference between sexes and found no statistical difference, and thus combined data from sexes. For statistical testing, data were first tested for normal distribution using the Kolmogorov-Smirnov test (p < 0.05 cut-off). As distributions were not normal, we used Wilcoxon rank-sum test. Kolmogorov-Smirnov test was also used for comparing distribution.

Table 1. Sample information.
Mice were maintained in standard housing conditions on a 12 h dark/light cycle with food and water provided ad libitum. As APP-KI mice have C57BL/6J background, we used C57BL/6J mice as control wild-type (WT) mice. Data were obtained from six WT mice and six APP knock-in mouse models (APPNL–G–F; Saito et al., 2014; Masuda et al., 2016) between 3 and 5 months of age. The electrical activity (local field potential, LFP) of neurons in the hippocampus and MEC were obtained. All procedures were conducted in accordance with the guidelines of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. Sixteen tetrodes with a total of 12 mice’s data, treated independently with records of effective neuronal activity, resulted in 125 independent data.
All mice received a custom drive of 64 channels targeting CA1 or MEC (Figure 1A). In one group of animals, eight limbs targeted hippocampal CA1, and another eight limbs targeted the MEC. The tetrodes were constructed from 17 μm polyimide-coated platinum-iridium wires (90–10%) (California Fine Wire) twisted in four. A total of 16 tetrodes (i.e., 64 electrodes) were implanted in each mouse. The electrode tips were plated with gold and the electrode impedance was reduced to 150–300 kΩ at 1 kHz. Animals were anesthetized with isoflurane (air flow: 0.8–1.0 l/min, 1% isoflurane, adjusted for physiological conditions). The subject received buprenorphine subcutaneously at the start of the surgical operation. The depth of anesthesia was determined by examining the tail and pinch reflexes as well as by breathing. During induction of anesthesia, the animals were secured to a Kopf stereotactic frame. A craniotomy was performed in the hippocampus (approximately 1 × 1 mm) at 2.5 mm AP 2.5 mm ML and in the entorhinal cortex (approximately 1 × 1 mm) 0.2 to 0.4 mm anterior to the midline and 3.5 mm ML. The dura mater was carefully removed, and electrodes were implanted. A stainless-steel screw fixed to the skull in the upper cerebellum served as the ground electrode. The recording drive was secured to the skull using dental cement.
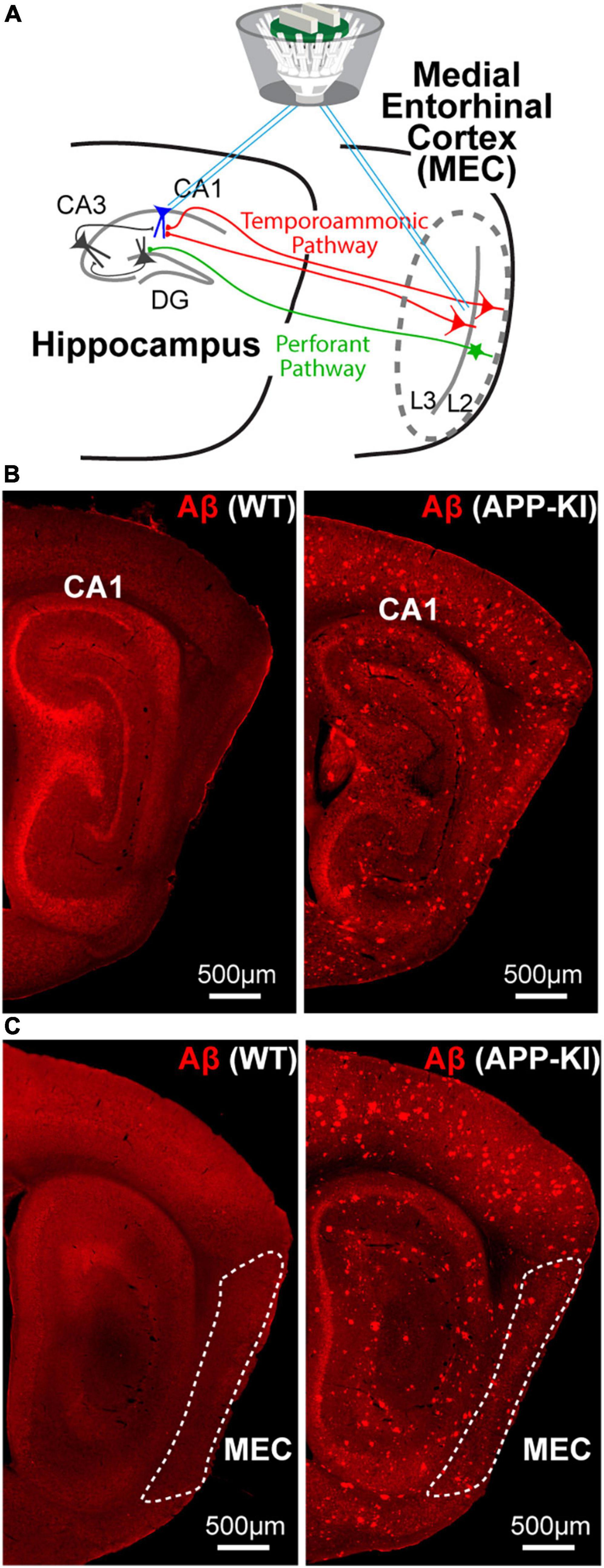
Figure 1. Simultaneous SWR recording from the MEC and hippocampus CA1 of young APP-KI mice. (A) A schematic of simultaneous SWR recording from medial entorhinal cortex (MEC) layer 2/3 and CA1 of the hippocampus. (B,C) Sagittal sections with anti-Aβ immunostaining for 3 mo WT mouse (left) and 3 mo APP-KI mouse (right).
Behavioral training was initiated 5 days after surgery, and data collection was performed on day 5 of training (Figure 2). The task required that the mice run on two 1-m-long linear tracks. Track A is a black acrylic enclosure decorated with a pattern of white boards across the track and black rubber floors on the inner wall. Track B is a white acrylic enclosure with clear black boards on the inside walls of the track and white sandpaper floors. During the remapping task, animals were trained to run in a sequential order of track A → track B → track B → track A. The two tracks were in the same room. The animals were given a 5-min rest between the session. The run was motivated by placing a cookie piece at the end of the linear track. Each session lasted 5–10 min. On the linear tracks, the mice ran 10 full laps (back and forth).
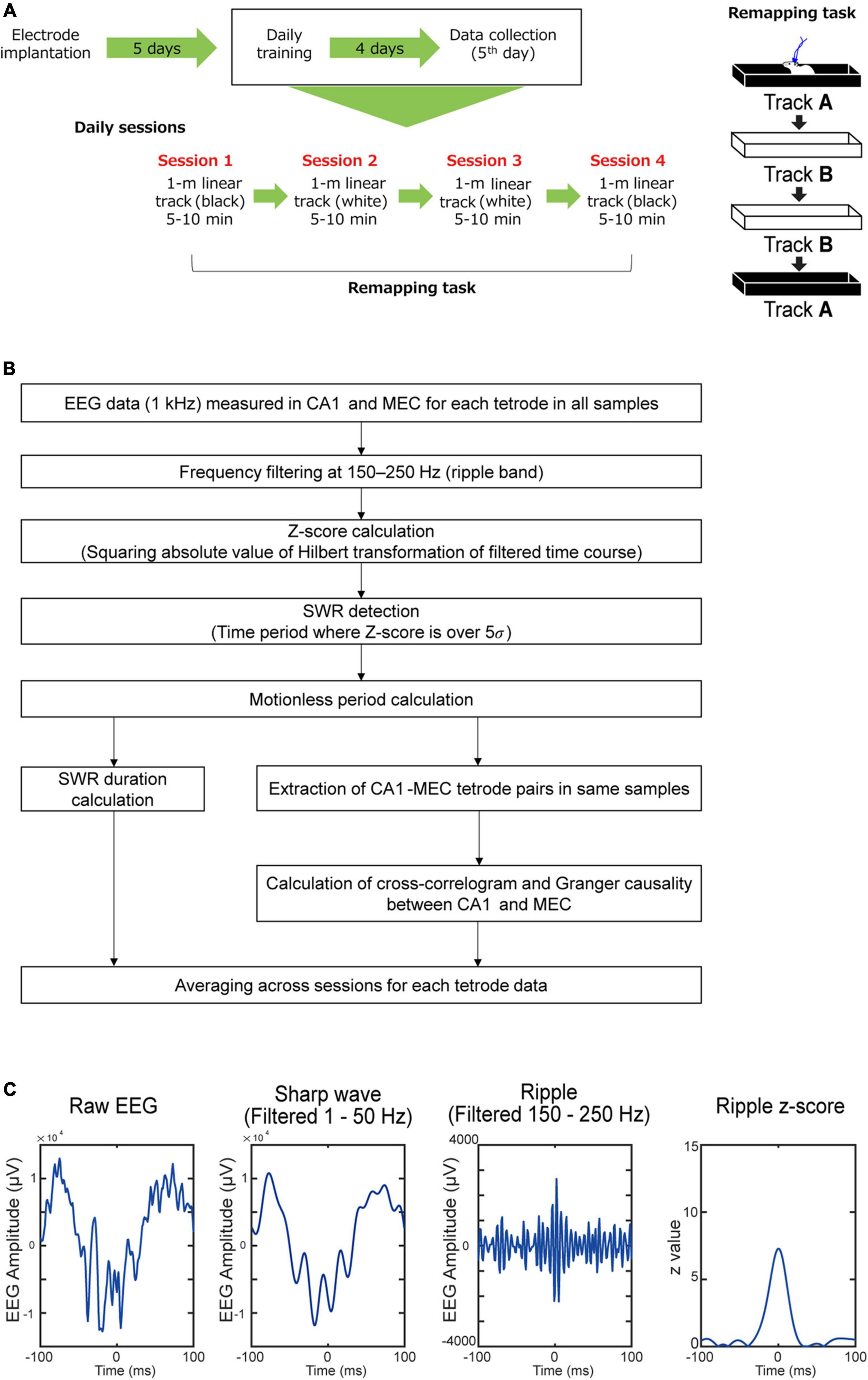
Figure 2. Experimental procedure. (A) In vivo electrophysiological recording were performed while APP-KI mice were running linear tracks. (B) Flow of the analysis used in the study. (C) An example SWR event defined by 150–250 Hz frequency band.
Electrode positions were confirmed by anesthetizing the drive-implanted mice using isoflurane and performing small electrolytic lesions through passing current (10 μA for 20 s) through the electrodes. Immediately after this, the mice received an overdose of isoflurane and were perfused intracardially with saline followed by 4% freshly depolymerized paraformaldehyde in phosphate buffer (PFA). The brains were extracted and stored in the same fixative overnight. After overnight cryoprotection in phosphate buffer saline with 30% sucrose at 4°C, tissue samples were embedded in O.C.T. mounting medium and sagittal sections (40 μm) were cut and stained with cresyl violet. All tetrodes were identified, and the tip of each electrode was found by comparison with adjacent sections. Only data from tetrodes in the hippocampal CA1 or the layer 2/3 of dorsal MEC was collected for analysis. The electrical lesions often made tip holes in the brain section larger so that they span across cell layers. To ensure that the LFP was recorded from cell layers (either in pyramidal cell layer in CA1 or layers 2/3 in the MEC) we used LFP data only from tetrodes that had spiking activity of principal neurons for LFP analyses. Some of the MEC sessions were recorded at positions 40 or 80 μm above the terminal point of electrodes, which were estimated to be within layers 2/3 in the MEC. If these sessions had spikes distinct from previous and following sessions, they were included as distinct data sets.
Local field potentials (LFPs) were recorded in a single-ended fashion using a skull ground in the upper cerebellum. The tetrodes were multi-channel, impedance matched, and connected to a single gain head stage (Neuralynx). The output of the headstage was transmitted to the data acquisition system (Neuralynx). Unit activity was amplified 3,000–5,000-fold, bandpass filtered at 600–6,000 Hz. The LFP signal was recorded at a sampling rate of 2,000 Hz per tetrode at a frequency range from 0 to 475 Hz. Notch filters were not applied. The animal’s position recording system tracked the positions of the two light emitting diodes (LEDs) on the head stage (sampling rate 50 Hz) with an overhead video camera.
Analyses were performed with custom MATLAB codes. The analysis flow used in the study is shown in detail in Figure 2. Each step of this flow is explained in the following sections. All data is shown as mean ± SE. All statistical testing assumed a non-parametric distribution and Wilcoxon rank sum test was used. We did not observe any statistical differences when data from males and females were compared, and thus combined them together.
The EEG data were converted to discrete-time analytic signals by Hilbert transformation after applying a frequency filter at 150–250 Hz (Gillespie et al., 2016) as a ripple frequency domain. The power absolute value of the discrete-time analytic signal was then acquired as a z-value. When the z-value exceeded 5σ (σ is standard deviation) (Cheng and Frank, 2008; Jones et al., 2019), it was detected as an SWR and used for later analysis. Using mouse movements tracked by LEDs, SWRs were further restricted to the period at speeds less than 2 cm/s (immobile state).
SWR duration was defined as the duration of EEG exceeding 5 times the standard deviation in z-values. SWR mean power (dB/Hz) was calculated by the mean of SWR power spectrum density (dB/Hz) among 150–250 Hz. The peak value of the SWR power spectrum was defined as the SWR frequency peak. SWR density (Hz) was calculated by the number of SWRs divided by total immobile-state duration. SWR density is the same as “SWR abundance” in some previous studies. This method defines SWR events using only ripple wave band width (150–250 Hz), and we further tested if sharp waves co-exist in these SWR events. Sharp wave events were independently defined as timepoints that exceeded 5σ in the EEG filtered at 1–50 Hz frequency band, and if a sharp wave event occurs within ±30 ms of a SWR event, it is considered as co-occurring (Maier et al., 2003). We found that 94.3 ± 1.6% of SWR events in the MEC and 96.9 ± 0.4% of SWR events in CA1 had co-occurring sharp waves in WT mice, and that 97.3 ± 0.6% of SWR events in the MEC and 96.1 ± 1.6% of SWR events in CA1 had co-occurring sharp waves in WT mice (Figure 2C).
The coordination of SWRs between MEC and CA1 was assessed using cross-correlation method (Nowak and Bullier, 2000). Briefly, the number of SWRs in the MEC was counted -100 to +100 ms from the time of SWRs detected in CA1. The SWR counts were then normalized by total immobile-state duration.
To further assess coordination between SWRs between MEC and CA1, the Granger causality analysis was used (Granger, 1969). In the z-value time series of SWR, all z-values were kept for time bins that surpass 5σ, whereas values were set to 0 for other bins. Presence or absence of Granger causality (GC) between the z-value time series from CA1 and MEC was tested. All z-scores above 5σ regardless of velocity were used in case the SWR time-course data become very sparse for the Granger causality analysis. Results from Granger causality test (α = 0.05, false discovery rate correction for multiple correction),1 for causal and 0 for no causal, were averaged for four linear track sessions. This result was further averaged among all tetrode combinations. The group data of wildtype and AD models were compared by Wilcoxon rank sum test. For the Granger causality analysis, we used the MATLAB multivariate Granger causality (MVGC) Toolbox (Seth, 2010; Barnett and Seth, 2014). Akaike’s information criterion (AIC) and Bayesian information criterion (BIC) were used for vector autoregression model order optimization. The model order was optimized among 1–50.
We performed in vivo electrophysiological recording simultaneously from layers 2/3 of the MEC and hippocampal CA1 in APP-KI mice at 3–5 months of age (mo; n = 6 mice) (Figure 1A). APP-KI mice have Amyloid-β (Aβ) deposition in the CA1 and MEC at 3 mo (Figures 1B,C), whereas spatial memory impairment starts after 7 mo (Saito et al., 2014; Sasaguri et al., 2017; Jun et al., 2020). Control recording was performed from C57BL/6J mice (referred to as WT mice; n = 6 mice). Electroencephalography (EEG) data were obtained while mice ran 1-m linear tracks. We used two black and white linear tracks, where we recorded spike activities during remapping of place cells and grid cells (Jun et al., 2020). In this study, we analyze EEG activity obtained from these recordings, and data from two linear track sessions were averaged for each electrode. EEG was filtered at 150–250 Hz band, and SWRs were detected at time points where z-scored amplitude exceeded 5 times standard deviation (Gillespie et al., 2016; Figure 2). Data from n = 29 and n = 25 independent electrodes were collected from the MEC and CA1 of APP-KI mice, respectively. In WT mice, n = 23 and n = 48 electrodes were respectively implanted in the MEC and CA1. Recorded layers in the MEC and CA1 were subsequently validated histologically (Supplementary Figures 1, 2). Figures 3A,B show representative SWR events recorded from CA1 of WT and APP-KI mice. Because long-duration SWRs in CA1 are critically involved in memory consolidation (Fernández-Ruiz et al., 2019), we first assessed the duration of SWRs. We found that duration of SWRs recorded in CA1 of APP-KI mice was significantly shorter than that in WT mice (Figure 3C, 26.2 ± 1.0 ms in APP-KI mice vs. 32.1 ± 1.6 ms in WT mice; p = 0.0042, Wilcoxon rank sum test). A fraction plot of SWRs as a function of SWR duration shows that a large proportion of SWRs in APP-KI mice exhibited shorter duration than SWRs in WT mice (Figures 3D,E; p = 0.019, Kolmogorov-Smirnov test). SWRs with their duration of more than 25 ms were fewer in APP-KI mice (Figure 3F; p = 0.011, Wilcoxon rank sum test). By contrast, other properties of CA1 SWRs remained intact. In our ∼30-min recording sessions, we detected comparable density of SWRs between WT and APP-KI mice (Figure 3G, 0.097 ± 0.006 and 0.114 ± 0.012 SWR events/s from WT and APP-KI mice, respectively; p = 0.43, Wilcoxon rank-sum test). SWR amplitude (p = 0.050), mean power (p = 0.17) and frequency (p = 0.14) were also comparable (Wilcoxon rank-sum test, Figures 3H–J). These results raise an intriguing hypothesis that long-duration SWRs are specifically impaired in CA1 of APP-KI mice at the preclinical stage.
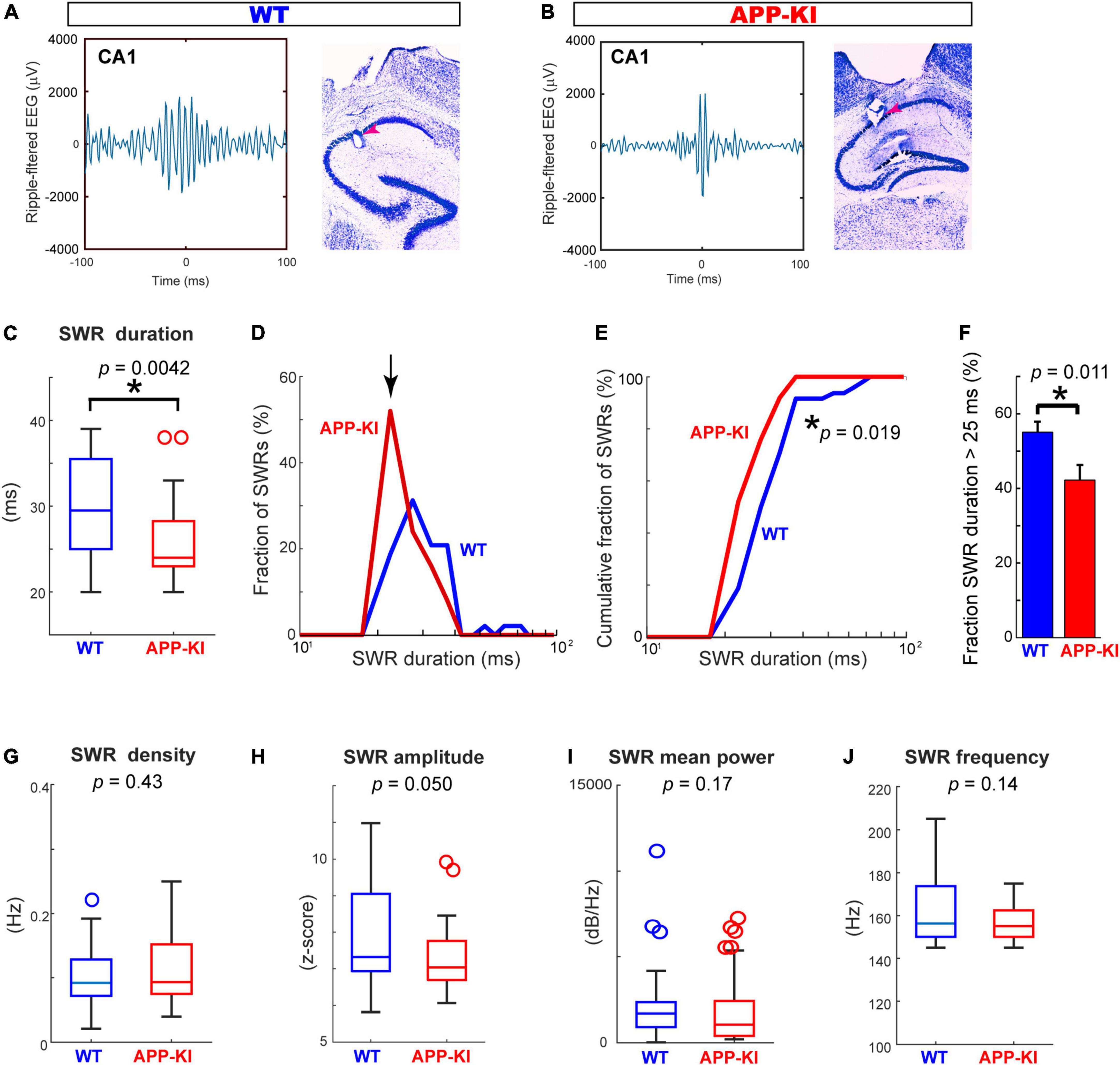
Figure 3. SWR duration was specifically diminished in CA1 of APP-KI mice. (A,B) Example SWR traces recorded from CA1 in WT (A) and APP-KI mice (B). (C) SWR duration in CA1. p = 0.0042, Wilcoxon rank-sum test. (D) Fraction of SWRs plotted as a function of SWR duration. APP-KI mice showed larger fraction of SWRs with short duration in CA1 (arrow). (E) Cumulative fraction of CA1 SWRs as a function of SWR duration; p = 0.019, Kolmogorov-Smirnov test. (F) Fraction of CA1 SWRs with duration of >25 ms; p = 0.011, Wilcoxon rank-sum test. (G–J) SWR properties in CA1. SWR density (G), SWR amplitude (H), SWR mean power (I) and SWR Frequency peak (J). P-values are from Wilcoxon rank-sum test. For all data in (C–J), n = 48 and n = 25 CA1 recordings in WT and APP-KI mice, respectively. *p < 0.05.
We next assessed properties of SWRs in the MEC (Figures 4A,B). In contrast to the diminished SWR duration in CA1, SWRs in the MEC showed comparable duration between APP-KI and WT mice (Figure 4C). While the distribution of SWR duration differs between APP-KI and WT mice (p = 0.0076, Kolmogorov-Smirnov test; Figures 4D,E), we also did not observe difference in the number of SWRs with duration >25 ms (Figure 4F). Furthermore, the density of SWR did not differ between WT and APP-KI mice (0.072 ± 0.019 and 0.053 ± 0.006 SWR events/s from WT and APP-KI mice, respectively; p = 0.62, Wilcoxon rank-sum test; Figure 4G). SWR amplitude (p = 0.88), mean power (p = 0.58) and frequency (p = 0.50) were all comparable between APP-KI and WT mice (Wilcoxon rank-sum test, Figures 4G–J). These data indicate that properties of individual SWR events remained unchanged in the MEC at preclinical stage, implying that generation mechanisms of SWR within the MEC are intact.
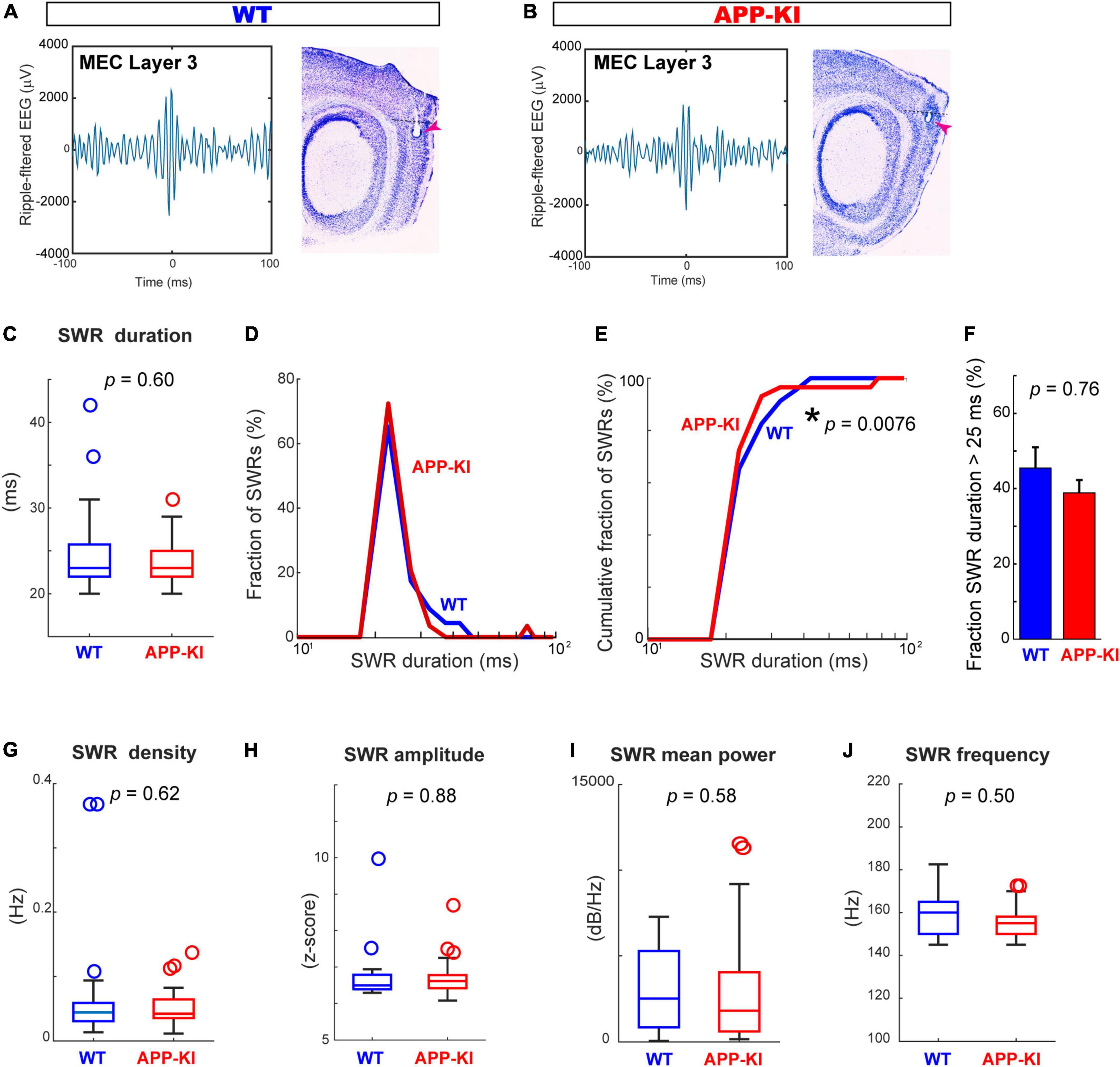
Figure 4. SWR characteristics in the MEC were comparable between APP-KI mice and WT mice. (A,B) Example SWR traces recorded from the MEC in WT (A) and APP-KI mice (B). (C) SWR duration in MEC. p = 0.60, Wilcoxon rank-sum test. (D) Fraction of MEC SWRs plotted as a function of SWR duration. (E) Cumulative fraction of MEC SWRs as a function of SWR duration; p = 0.0076, Kolmogorov-Smirnov test. (F) Fraction of MEC SWRs with duration of > 25 ms; p = 0.76, Wilcoxon rank-sum test. (G–J) SWR properties in CA1. SWR density (G), SWR amplitude (H), SWR mean power (I) and SWR Frequency peak (J). P-values are from Wilcoxon rank-sum test. For all data in (C–J), n = 23 and n = 29 CA1 recordings in WT and APP-KI mice, respectively. *p < 0.05.
The diminished duration of SWRs observed in CA1 raised an intriguing question as to whether the coordination of SWRs between the MEC and CA1, reported previously (Yamamoto and Tonegawa, 2017), were affected in APP-KI mice. We thus examined the coordination of SWR events between the MEC and CA1 using pairs of electrodes targeted in the two areas (n = 65 and n = 61 pairs from WT and APP-KI mice, respectively). Figures 5A,B show representative SWRs occurring simultaneously in the MEC and CA1. In healthy mice, SWRs in the MEC layers 2/3 are found to be preceding SWRs in the CA1 (Yamamoto and Tonegawa, 2017). However, the SWRs in the MEC of APP-KI mice appeared to be delayed from those in CA1 (Figure 5B). To assess the relative timing of SWRs between the MEC and CA1, we computed cross-correlograms of SWR counts recorded from the MEC around co-occurring SWRs in CA1 (Narayanan and Laubach, 2009; Figure 5C). WT mice showed SWRs in the MEC occurring 8.9 ± 5.6 ms prior to the SWRs in the CA1, consistent to the previous finding. By contrast, this advancement of SWRs in the MEC disappeared in APP-KI mice, with a peak of SWRs in MEC occurring 23.2 ± 4.6 ms after SWRs in the CA1 (p = 7.6 × 10–6, Wilcoxon rank-sum test; Figures 5C,D). To further test the idea that the MEC→CA1 SWR directionality is affected in APP-KI mice, we performed Granger causality analysis of SWR events for both MEC→CA1 and CA1→MEC directions (Granger, 1969). We found that MEC→CA1 Granger causality significantly decreased in APP-KI mice compared to that in WT (p = 2.1 × 10–4, Wilcoxon rank-sum test; Figure 5E), suggesting that the propagation of SWRs in the MEC→CA1 direction is impaired in APP-KI mice. By contrast, CA1cont Granger causality was comparable between APP-KI and WT mice (p = 0.080, Wilcoxon rank-sum test). The decreased MEC→CA1 Granger causality in APP-KI was also observed when BIC was used for the causality analysis (p = 0.0031, Wilcoxon rank-sum test), instead of Akaike information criterion. Together, these results indicated that MEC→CA1 coordination of SWRs was disrupted in APP-KI mice.
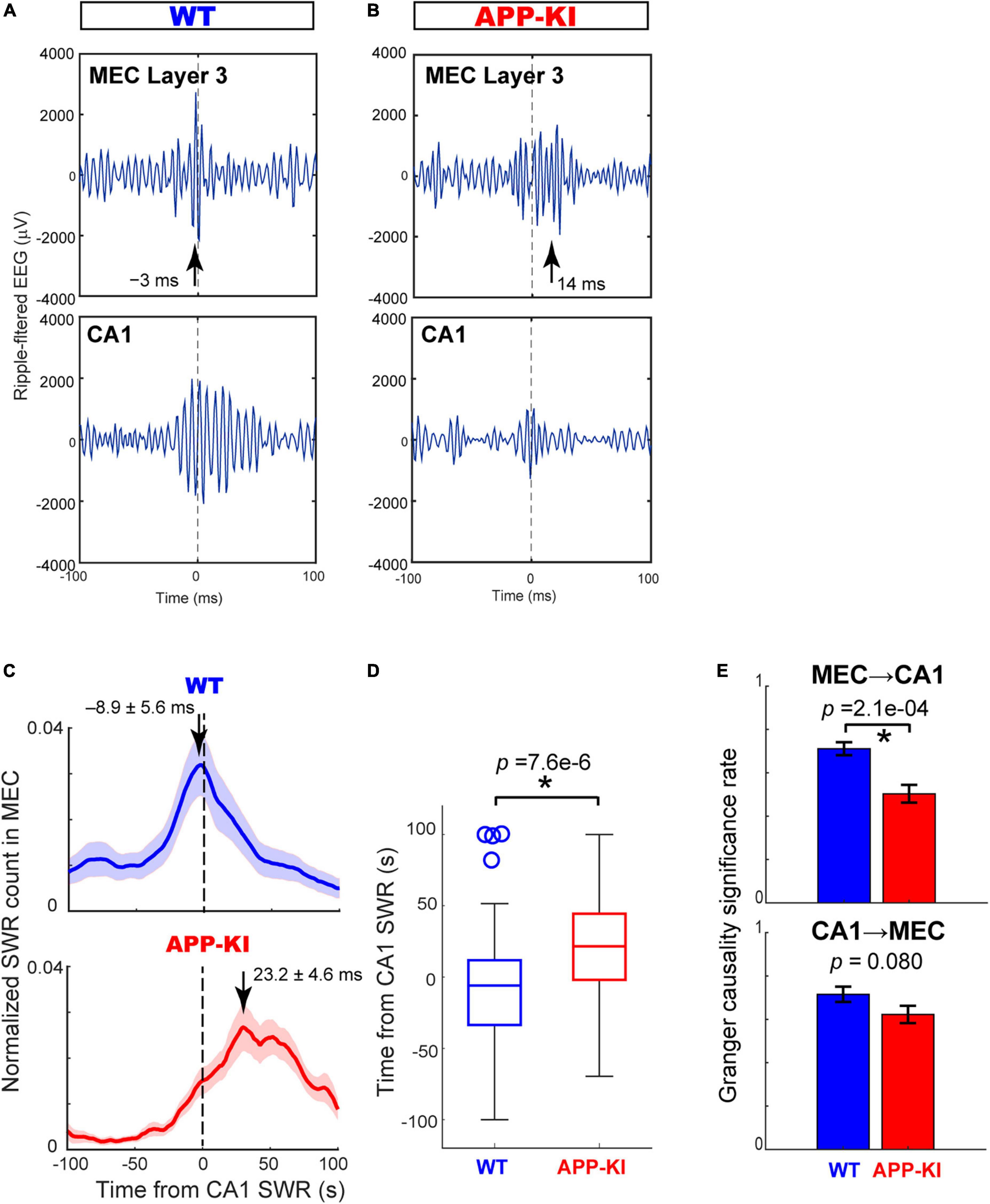
Figure 5. Cross-correlogram and Granger causality revealed impaired SWR coordination between MEC and CA1. (A,B) Example SWR traces occurring simultaneously in the MEC and CA1 in WT (A) and APP-KI mice (B). SWRs are aligned at the amplitude peaks of SWRs in CA1. Arrows depict peak times of SWRs in the MEC. (C) Normalized counts of SWRs in the MEC triggered by SWRs in CA1 for WT mice (n = 65 electrode pairs) and APP-KI mice (n = 61 electrode pairs). (D) Peak time of MEC SWRs relative to SWRs occurring in CA1. p = 7.6 × 106, Wilcoxon rank-sum test. (E) Significance rate obtained from Granger causality test on SWRs from MEC to CA1 (top, p = 2.1 × 104, Wilcoxon rank-sum test) and from CA1 to MEC (bottom, p = 0.080, Wilcoxon rank-sum test). *p < 0.05.
At the preclinical stage of APP-KI mice, we found (1) short-duration SWRs in the hippocampal CA1 and (2) impaired SWR coordination between the MEC and CA1. Fernandez-Ruiz and others previously showed in healthy brains that optogenetic prolongation of SWRs causally enhanced memory consolidation, indicating the critical role of long-duration SWRs in memory consolidation (Fernández-Ruiz et al., 2019). Long-duration SWRs would specifically provide higher spatial information from longer spike replays and larger number of neurons involved (Wilson and McNaughton, 1994; Foster and Wilson, 2006). Yamamoto and Tonegawa showed in healthy brains that long and multiplex SWRs in CA1 became disrupted when MEC input was optogenetically inhibited, demonstrating a critical role of SWRs in the MEC in supporting long-duration SWRs in the hippocampus (Yamamoto and Tonegawa, 2017). Although the mechanism of how MEC SWRs contribute to long-duration CA1 SWRs remains unknown, MEC inputs via the perforant pathway may modulate SWR generation in CA3 (Sullivan et al., 2011). Alternatively, MEC inputs by way of temporoammonic pathway may directly modulate SWRs in CA1. In either case, the disappearance of preceding SWRs in the MEC found in this study suggests that the MEC no longer contributes to the generation of long-duration hippocampal SWRs in APP-KI mice. The absence of long-duration SWRs may in turn cause memory consolidation abnormality at later stages of AD. Because SWR properties remained mostly intact in the MEC at the preclinical stage, it is likely that synaptic transmission from the MEC to the hippocampus (Dong et al., 2007; Palop et al., 2007), rather than SWR generation inside the MEC, becomes deteriorated and causes the SWR incoordination in APP-KI mice. Our finding of SWR incoordination at the preclinical stage points to a possible usage of entorhinal SWRs as an early biomarker for AD diagnosis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee at the University of California, Irvine.
TF, HJ, and KI contributed to conception and design of the study. TS developed the APP-KI mouse line. HJ and KI conducted animal experiments and data acquisition and preprocessed the data. TF, SS, HJ, TS, AK, and KI contributed to methodology and software. TF, HJ, SS, and KI performed the analyses. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by NIH R01 grants (R01MH121736, R01AG063864, and R01AG066806), PRESTO grant from Japan Science and Technology Agency (JPMJPR1681), Brain Research Foundation Fay-Frank Seed Grant (BRFSG-2017-04), Whitehall Foundation Research Grant (2017-08-01), BrightFocus Foundation Research grant (A2019380S), Alzheimer’s Association Research Grant (AARG-17-532932), and Donors Cure Foundation New Vision Award (CCAD201902) to KI. TS was supported by research grants from RIKEN Center for Brain Science, the Ministry of Education, Culture, Sports, Science and Technology, the Ministry of Health and Welfare, and AMED [JP18dm0207001, Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS)]. HJ was supported by the University of California, Irvine Medical Scientist Training Program (MSTP) (T32GM008620) and NIH F31 grant (1F31AG069500).
We thank Allen Bramian and Marjan Savadkohighodjanaki in the Igarashi lab for technical assistance.
TF, SS, and AK were employed by Hitachi, Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnsys.2022.955178/full#supplementary-material
Supplementary Figure 1 | List of MEC recording sessions in WT and APP-KI mice.
Supplementary Figure 2 | Histological validation of electrode positions in the MEC of WT and APP-KI mice.
Barnett, L., and Seth, A. K. (2014). The MVGC multivariate Granger causality toolbox: A new approach to Granger-causal inference. J. Neurosci. Methods 223, 50–68. doi: 10.1016/j.jneumeth.2013.10.018
Benthem, S. D., Skelin, I., Moseley, S. C., Stimmell, A. C., Dixon, J. R., Melilli, A. S., et al. (2020). Impaired Hippocampal-Cortical Interactions during Sleep in a Mouse Model of Alzheimer’s Disease. Curr. Biol. 30, 2588–2601.e5. doi: 10.1016/j.cub.2020.04.087
Buzsaki, G. (1986). Hippocampal sharp waves: Their origin and significance. Brain Res. 398, 242–252.
Buzsaki, G. (2015). Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 25, 1073–1188.
Cheng, S., and Frank, L. M. (2008). New Experiences Enhance Coordinated Neural Activity in the Hippocampus. Neuron 57, 303–313.
Ciupek, S. M., Cheng, J., Ali, Y. O., Lu, H. C., and Ji, D. (2015). Progressive functional impairments of hippocampal neurons in a tauopathy mouse model. J. Neurosci. 35, 8118–8131. doi: 10.1523/JNEUROSCI.3130-14.2015
Dong, H., Martin, M. V., Chambers, S., and Csernansky, J. G. (2007). Spatial relationship between synapse loss and beta-amyloid deposition in Tg2576 mice. J. Comp. Neurol. 500, 311–321. doi: 10.1002/cne.21176
Dubois, B., Hampel, H., Feldman, H. H., Scheltens, P., Aisen, P., Andrieu, S., et al. (2016). Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 12, 292–323. doi: 10.1016/j.jalz.2016.02.002
Fernández-Ruiz, A., Oliva, A., Oliveira, E. F. D., Rocha-Almeida, F., Tingley, D., and Buzsáki, G. (2019). Long-duration hippocampal sharp wave ripples improve memory. Science 364, 1082–1086. doi: 10.1126/science.aax0758
Foster, D. J., and Wilson, M. A. (2006). Reverse replay of behavioural sequences in hippocampal place cells during the awake state. Nature 440, 680–683.
Gillespie, A. K., Jones, E. A., Lin, Y. H., Karlsson, M. P., Kay, K., Yoon, S. Y., et al. (2016). Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron 90, 740–751. doi: 10.1016/j.neuron.2016.04.009
Girardeau, G., Benchenane, K., Wiener, S. I., Buzsaki, G., and Zugaro, M. B. (2009). Selective suppression of hippocampal ripples impairs spatial memory. Nat. Neurosci. 12, 1222–1223.
Granger, C. W. J. (1969). Investigating Causal Relations by Econometric Models and Cross-spectral Methods. Econometrica 37, 424–438.
Hope, T., Tilling, K. M., Gedling, K., Keene, J. M., Cooper, S. D., and Fairburn, C. G. (1994). The structure of wandering in dementia. Int. J. Geriatr. Psychiatry 9, 149–155.
Iaccarino, H. F., Singer, A. C., Martorell, A. J., Rudenko, A., Gao, F., Gillingham, T. Z., et al. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235.
Jadhav, S. P., Kemere, C., German, P. W., and Frank, L. M. (2012). Awake hippocampal sharp-wave ripples support spatial memory. Science 336, 1454–1458.
Jones, E. A., Gillespie, A. K., Yoon, S. Y., Frank, L. M., and Huang, Y. (2019). Early Hippocampal Sharp-Wave Ripple Deficits Predict Later Learning and Memory Impairments in an Alzheimer’s Disease Mouse Model. Cell Rep. 29, 2123–2133.e4. doi: 10.1016/j.celrep.2019.10.056
Jun, H., Bramian, A., Soma, S., Saito, T., Saido, T. C., and Igarashi, K. M. (2020). Disrupted Place Cell Remapping and Impaired Grid Cells in a Knockin Model of Alzheimer’s Disease. Neuron 107, 1095–1112.e6. doi: 10.1016/j.neuron.2020.06.023
Khan, U. A., Liu, L., Provenzano, F. A., Berman, D. E., Profaci, C. P., Sloan, R., et al. (2014). Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 17, 304–311. doi: 10.1038/nn.3606
Maier, N., Nimmrich, V., and Draguhn, A. (2003). Cellular and network mechanisms underlying spontaneous sharp wave-ripple complexes in mouse hippocampal slices. J. Physiol. 550, 873–887.
Masuda, A., Kobayashi, Y., Kogo, N., Saito, T., Saido, T. C., and Itohara, S. (2016). Cognitive deficit in single App knock-in mouse models. Neurobiol. Learn. Mem. 135, 73–82.
Moser, M. B., Rowland, D. C., and Moser, E. I. (2015). Place cells, grid cells, and memory. Cold Spring Harb. Perspect. Biol. 7:a021808.
Narayanan, N. S., and Laubach, M. (2009). Methods for studying functional interactions among neuronal populations. Methods Mol. Biol. 489, 135–165.
Nowak, L. G., and Bullier, J. (2000). “Cross correlograms for neuronal spike trains. Different types of temporal correlation in neocortex, their origin and significance,” in Time and the Brain, Conceptual advances in Brain Research, ed. R. Miller (Reading: Harwood Academic), 53–96.
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Prince, S. M., Paulson, A. L., Jeong, N., Zhang, L., Amigues, S., and Singer, A. C. (2021). Alzheimer’s pathology causes impaired inhibitory connections and reactivation of spatial codes during spatial navigation. Cell Rep. 35:109008. doi: 10.1016/j.celrep.2021.109008
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663.
Sasaguri, H., Nilsson, P., Hashimoto, S., Nagata, K., Saito, T., De Strooper, B., et al. (2017). APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 36, 2473–2487.
Seth, A. K. (2010). A MATLAB toolbox for Granger causal connectivity analysis. J. Neurosci. Methods 186, 262–273.
Sullivan, D., Csicsvari, J., Mizuseki, K., Montgomery, S., Diba, K., and Buzsaki, G. (2011). Relationships between hippocampal sharp waves, ripples, and fast gamma oscillation: Influence of dentate and entorhinal cortical activity. J. Neurosci. 31, 8605–8616. doi: 10.1523/JNEUROSCI.0294-11.2011
Van Hoesen, G. W., Hyman, B. T., and Damasio, A. R. (1991). Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus 1, 1–8.
Wilson, M. A., and McNaughton, B. L. (1994). Reactivation of hippocampal ensemble memories during sleep. Science 265, 676–679.
Witton, J., Staniaszek, L. E., Bartsch, U., Randall, A. D., Jones, M. W., and Brown, J. T. (2016). Disrupted hippocampal sharp-wave ripple-associated spike dynamics in a transgenic mouse model of dementia. J. Physiol. 594, 4615–4630. doi: 10.1113/jphysiol.2014.282889
Keywords: Alzheimer’s disease, hippocampus, entorhinal cortex, medial entorhinal cortex (MEC), sharp-wave ripples (SWRs)
Citation: Funane T, Jun H, Sutoko S, Saido TC, Kandori A and Igarashi KM (2022) Impaired sharp-wave ripple coordination between the medial entorhinal cortex and hippocampal CA1 of knock-in model of Alzheimer’s disease. Front. Syst. Neurosci. 16:955178. doi: 10.3389/fnsys.2022.955178
Received: 28 May 2022; Accepted: 09 August 2022;
Published: 25 August 2022.
Edited by:
Emilio Kropff, IIBBA-CONICET Leloir Institute Foundation, ArgentinaReviewed by:
Kenji Mizuseki, Osaka City University, JapanCopyright © 2022 Funane, Jun, Sutoko, Saido, Kandori and Igarashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tsukasa Funane, dHN1a2FzYS5mdW5hbmUuc2JAaGl0YWNoaS5jb20=; Kei M. Igarashi, a2VpLmlnYXJhc2hpQHVjaS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.