 Jesse J. Langille
Jesse J. Langille Richard E. Brown
Richard E. Brown- Department of Psychology and Neuroscience, Dalhousie University, Halifax, NS, Canada
Trettenbrein (2016) has argued that the concept of the synapse as the locus of memory is outdated and has made six critiques of this concept. In this article, we examine these six critiques and suggest that the current theories of the neurobiology of memory and the empirical data indicate that synaptic activation is the first step in a chain of cellular and biochemical events that lead to memories formed in cell assemblies and neural networks that rely on synaptic modification for their formation. These neural networks and their modified synaptic connections can account for the cognitive basis of learning and memory and for memory deterioration in neurological disorders. We first discuss Hebb’s (1949) theory that synaptic change and the formation of cell assemblies and phase sequences can link neurophysiology to cognitive processes. We then examine each of Trettenbrein’s (2016) critiques of the synaptic theory in light of Hebb’s theories and recent empirical data. We examine the biochemical basis of memory formation and the necessity of synaptic modification to form the neural networks underlying learning and memory. We then examine the use of Hebb’s theories of synaptic change and cell assemblies for integrating neurophysiological and cognitive conceptions of learning and memory. We conclude with an examination of the applications of the Hebb synapse and cell assembly theories to the study of the neuroscience of learning and memory, the development of computational models of memory and the construction of “intelligent” robots. We conclude that the synaptic theory of memory has not met its demise, but is essential to our understanding of the neural basis of memory, which has two components: synaptic plasticity and intrinsic plasticity.
Introduction
When Pavlov first discovered classical conditioning, he was faced with the problem of how to interpret the behavior of his dogs. Some of his students and colleagues tried to interpret their behavior in terms of the subjective, introspective psychology of the day (Windholz, 1995). Pavlov, however, was determined to use an objective physiological interpretation and rejected subjective psychological terms. Once Pavlov’s lectures were translated into English (Pavlov, 1927, 1928a,b), Guthrie (1930) and Lashley (1930, 1931) published critiques which pointed out that Pavlov’s theories of brain and behavior ignored psychological concepts, such as attention and intelligence. Pavlov did not take these critiques lightly and wrote a lengthy reply (Pavlov, 1932). The Pavlov-Lashley debates regarding the neural basis of memory continued long after the death of Pavlov in 1936 (Lashley and Wade, 1946) and it was not until Hebb (1949) developed his neuropsychological theory that Pavlov’s physiological approach was integrated with the psychological concepts of perception and attention in the study of learning and memory.
Neurophysiological theories of synaptic plasticity, based on the experimental phenomena of long-term potentiation (LTP) and long-term depression (LTD), are now used to explain the neural basis of learning and memory (Nabavi et al., 2014; Yang et al., 2016). However, Trettenbrein (2016) has called into question the theory that synaptic change provides the neural basis for learning and memory, stating that: “Tentative evidence from a wide variety of work in neuroscience seems to provide support for the idea that the synapse is an ill fit when looking for the brain’s basic memory mechanism.” In this article, we examine Trettenbrein’s (2016) critiques of the synaptic theory of learning and memory and show that both theoretical and empirical research supports the concept that synaptic plasticity is an essential part of the complex cellular and molecular neurobiological changes which form the neural basis of learning and memory.
Trettenbrein’s Critiques of the Neurophysiologic Explanation of Learning and Memory
Trettenbrein’s critique is that the synapse-centered view of learning and memory is ill-focused and that the neurobiological basis of learning and memory is still far from understood. There are six critiques of the synaptic plasticity theory of memory in Trettenbrein’s article, which will be addressed here: (1) the synapse may not be the sole locus of learning and memory; (2) a synaptic locus of memory does not fit well with philosophical and cognitive theories of learning and memory; (3) memories survive despite synapse destruction and synaptic and (or) protein turn-over; (4) evidence from spatial training suggests that there is a need to separate learning from memory; (5) existing learning mechanisms cannot explain information that is encoded in a single trial (Gallistel and Balsam, 2014); and (6) memory may be sub-cellular in nature. Trettenbrein (2016) makes the argument that the concept of the synapse as the locus of memory is not tenable and that a paradigm shift is necessary. However, he does not provide a new paradigm, except to suggest that “the memory mechanism is (sub-) molecular in nature” (page 3). The purpose of our article is to argue that there is a considerable literature on the neurobiology of learning and memory that shows the importance of synaptic plasticity as the first step in the chain of cellular and biochemical events involved in memory formation, and that, once memories are formed, synaptic modification is essential for their expression. We couch our discussion in terms of Hebb’s (1949) neuropsychological theory.
Donald O. Hebb’s Theory of Learning and Memory
Hebb’s (1949) theory postulated that the neurophysiological changes underlying learning and memory occur in three stages: (1) synaptic changes; (2) formation of a “cell assembly”; and (3) formation of a “phase sequence,” which link the neurophysiological changes underlying learning and memory as studied by physiologists to the study of thought and “mind” as conceived by cognitive psychologists. As pointed out by Trettenbrein (2016), Hebb’s neurophysiological postulate (Hebb, 1949, page 62) states that:
“When an axon of cell A is near enough to excite a cell B and repeatedly or persistently takes part in firing it, some growth process or metabolic change takes place in one or both cells such that A’s efficiency, as one of the cells firing B, is increased.”
The “cell assembly” (Hebb, 1949, pages 69–74) is a set of neurons and the pathways connecting them, which act together, such that a stimulus activating pathway 1 will activate a reverberating circuit of N pathways (in Hebb’s example, n = 15). A cell assembly is a hypothetical reverberating system, proposed as a mediating process, an element of thought, capable of holding an excitation and thus of bridging a gap in time between stimulus and response (Hebb, 1972, pages 295 and 304). A series of cell assemblies, connected by neural activity over time is a “Phase Sequence,” which provides the neural basis for a “train of thought” from one cell assembly to another (Hebb, 1949, pages 79–106). The cell assembly thus “relates the individual nerve cell to psychological phenomenon” such that “a bridge has been thrown across the great gap between the details of neurophysiology and the molar conceptions of psychology” (Hebb, 1949, page 101). Hebb then elaborated on how this theory could account for learning and memory, how new learning could be associated with previous learning, and how “quick learning” (perhaps similar to the single trial learning of Gallistel and Balsam (2014)) might occur (Hebb, 1949, chapter 8). Hebb’s cell assembly theory thus showed how differences between psychologists and physiologists, who often use different definitions for the same phenomena, could be reconciled into a theory of the neurophysiological basis of learning and memory. It is important to note that Hebb’s postulate quoted above contains two concepts: synaptic plasticity and “some growth process or metabolic change” in the neuron, which has been termed “intrinsic plasticity” (Sehgal et al., 2013; Titley et al., 2017).
Resolution of Recent Critiques Using Modern Neurophysiological Research
We believe that the critique of synaptic plasticity theory proposed by Trettenbrein (2016) can be resolved using Hebb’s synaptic theory, research based on cell assemblies as components of neural networks, and current research on the cellular and molecular basis of memory formation to indicate the essential nature of synaptic plasticity in understanding the neurobiology of learning and memory. The six critiques proposed by Trettenbrein (2016) will be addressed sequentially.
The Synapse May Not be the Sole Locus of Learning and Memory
Trettenbrein (2016) describes the reservations held by some cognitive neuroscientists that the synapse is the “sole” locus of memory. While the synapse is an essential and highly studied component in the learning and memory process, it is not viewed by neurophysiologists as the sole locus of memory nor are its changes viewed as the sole basis of memory (Josselyn et al., 2015; Lisman et al., 2018). Memory is not represented by change at a single synapse, but by a series of processes involving molecular, biochemical, cellular and circuit level changes in widespread constellations of neurons throughout the brain. Specifically, when a strong stimulus occurs in the external environment, it drives high frequency stimulus trains (tetanic activity) in the neurons of a particular cell assembly, which, through their simultaneous synaptic activity, represent particular elements of the external stimulus (Buzsáki, 2010). Such a cell assembly or neural network has been identified for fear memory (Butler et al., 2015, 2018).
Propagation of strong excitatory currents through the synapses activate biochemical changes within neurons that lead to the strengthening of the synaptic connections within the circuit, or cell assembly. More precisely, N-methyl-D-aspartate receptor (NMDAR) induced calcium transients initiate intracellular signaling cascades leading to up-regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), synaptic growth and phosphorylation mediated increases in AMPAR conductance at the post-synaptic compartment of synapses between cells participating in a given network (Lee et al., 2003; Bailey and Kandel, 2008; Henley and Wilkinson, 2013). Both pre-synaptic and post-synaptic changes are important in the strengthening of synapses between neurons comprising cell assemblies (Costa et al., 2017). Subsequent activity in the neurons of this cell assembly, with the connections between cells now demonstrating an increased synaptic efficacy, represents the elementary building blocks of learning, memory and other cognitive processes (Choi et al., 2018). Many of these cell assemblies firing consecutively form a phase sequence which connects the individual elements to produce a more complete mental representation, or train of thought (Almeida-Filho et al., 2014). Once the synaptic connections in the cell assemblies forming a phase sequence are modified by experience, subsequent activation of this specific array of neurons results in the experiential recollection of the memory stored in the constellation of modified synaptic connections (Nabavi et al., 2014; Josselyn et al., 2015; Butler et al., 2018).
Synaptic change is thus the first step in a series of events which link molecular activity at the synapse and the subsequent intracellular biochemical cascades and cellular changes to the cognitive aspects of memory. The neurobiological basis of memory exists as a series of synapse-specific molecular and biochemical changes, including de novo protein synthesis, phosphorylation, up-regulation of synaptic receptors and synaptic growth within and between cell assemblies which results in long-term changes to synaptic efficacy (Lee et al., 2003; Bailey and Kandel, 2008; Henley and Wilkinson, 2013; Jarome and Helmstetter, 2014). The synapse is the location where these biochemical changes are initially manifested and is altered through the intracellular changes and, therefore, is an integral part of the neurobiology of memory (Mayford et al., 2012). Any memories stored in the neurons of the cell assembly or phase sequence are expressed through synaptic modifications (Caroni et al., 2014; Khalaf and Gräff, 2016; Knoblauch and Sommer, 2016). Thus, the synapse is not the sole locus of memory nor is it the neurobiological basis of memory, but it is an integral component of the memory process. The locus of memory is the particular set of neurons comprising a cell assembly or phase sequence connected by synapses which are activated and modified by experience. Memory formation is the result of both synaptic plasticity and intracellular (intrinsic) plasticity (Titley et al., 2017; Lisman et al., 2018).
A Synaptic Locus of Memory Does Not Fit Well With Philosophical and Cognitive Theories of Learning and Memory
Gallistel and Matzel (2014) and Trettenbrein (2016) stated that there was an incongruity between the theories of cognitive neuroscience and the idea of the synapse serving as the locus of memory. More precisely, he suggested that the synapse may be too complex to underlie a process as fundamental and essential as memory and that the elementary unit of memory is more likely a molecular or submolecular unit (Gallistel and King, 2009). As stated in “The Synapse May Not be the Sole Locus of Learning and Memory” section, memory formation has two components; synaptic changes within a cell assembly and intracellular biochemical changes. Memory involves many molecular and submolecular components, from the activation of second messenger systems, protein kinases and nuclear binding proteins to the transcription of DNA and translation of RNA, both locally at the synapses and in the nucleus and soma of the neurons (Fernandez-Moya et al., 2014; Jarome and Helmstetter, 2014). The resulting proteins and associated molecular changes serve as the neurobiological basis of memory, but the locus at which these sub-cellular changes are expressed is the synapse, itself a complex biochemical structure (Craig et al., 2006), indicating the important role the synapse and the network of synapses forming a cell assembly plays in both the formation and expression of memories in the neural tissue (Butler et al., 2015; Josselyn et al., 2015; Choi et al., 2018).
Memories Survive Despite Synapse Destruction and Synaptic/Protein Turn-Over
An important critique proposed by Trettenbrein (2016) is that memories often persist despite synapse destruction and for durations outlasting the turnover of the synapse-modifying proteins which, according to synaptic theory, store memories. Many memories survive extreme brain remodeling such as occurs in insect metamorphosis, planarian brain regeneration, and mammalian hibernation (Blackiston et al., 2015). These seemingly incompatible findings are not at odds with a synaptic theory of memory. Hebb (1949; page 129) suggested that once a memory phase sequence has been formed from a set of cell assemblies, it becomes independent of any particular sensory stimulation; thus, a memory formed from visual stimuli may be activated by tactile or auditory stimuli. Experimental evidence shows that the synaptic weights of a predictable set of synapses are altered during learning and these modifications in synaptic weight are part of the processes underlying learning (Butler et al., 2015, 2018). Yet, subsequent destruction of the modified synapse rarely eliminates learning (Takeuchi et al., 2014; Trettenbrein, 2016). This is because a memory is not represented in a single modified synapse, but in a network of simultaneously activated cell assemblies and phase sequences connected by synaptic activity (Buzsáki, 2010). Memories are thus represented by networks of many synapses which are distributed widely throughout the brain and memory expression exists as a pattern of neural activity within a particular constellation of neurons (Josselyn et al., 2015).
Individual neurons are, as a consequence of their inputs, endowed with mnemonic receptive fields representing “bits” of information. Temporally contiguous or contingent stimuli cause coincidently active neurons (a particular constellation of which possess the mnemonic fields configuring to represent the stimulus) to have their connections strengthened via mechanisms of Hebbian spike-timing-dependent synaptic plasticity. Although numerous intracellular processes occur upstream it is this downstream, experience-dependent synaptic re-weighting which is ultimately responsible for the redirecting of information flow linking neurons, and thus the bits of information represented by their mnemonic fields, together into concepts (Hebbian cell assemblies) and associations (phase sequences). Thus, memory expression is an emergent network property resulting from distributed neural activity and destruction of a single synapse may weaken a memory, but does not cause the loss of an entire memory. Learning can produce persistent, sometimes even trans-generational (Lim and Brunet, 2013; Dias et al., 2015), epigenetic modifications as part of the memory formation process and these epigenetic marks may provide the basis of the intrinsic plasticity which allows memories to persist, in some capacity, following synapse destruction (Rajasethupathy et al., 2012; Khalaf and Gräff, 2016; Poo et al., 2016).
Synapses regularly turn over as do the proteins that strengthen these synapses during learning. How then can memories persist for years or even a lifetime, in the face of regular synapse and protein turnover? There are two, non-mutually exclusive, explanations for this. First, as discussed above, memory does not exist as a single modified synapse, therefore even the recycling and regular removal of certain connections allows a great many of the synapses in the cell assembly representing a memory to remain, allowing the memory, even if weakened, to persist. Attardo et al. (2015) found that dendritic spines were turned over on time scales comparable with those of systems level memory consolidation. Thus, the transience of individual dendritic spines in the hippocampus may serve to ensure this rapid acquisition system remains labile to encode future experience, and that information is not redundantly represented in the brain, once information has been shuttled to the neocortex. Neocortical dendritic spines have a heightened permanency relative to those in the hippocampus (Yang et al., 2009), suggesting that spine turnover is not a hindrance to the synaptic theory of memory. The second explanation focuses on the persistence of memory in the face of rapid protein turnover. Strong stimulation of a synapse (for example, by late-phase LTP, L-LTP) leads to the synthesis of an atypical protein kinase C isoform known as protein kinase M Zeta (PKMZ; Neves et al., 2008; Sacktor, 2008). PKMZ has been called the “memory molecule” as it maintains the molecular and biochemical alterations at the synapse laid down during learning, allowing the memory trace to persist despite continued protein turnover (Glanzman, 2013), providing the basis of Hebb’s “reverberating circuit.” For example, AMPAR up-regulation, a common synaptic modification during memory formation which promotes increased synaptic efficacy, is short-lived and is readily susceptible to down-regulation by endocytic internalization. PKMZ both prevents AMPAR endocytosis (Yao et al., 2008; Hardt et al., 2014) and ensures that AMPAR up-regulation continues, maintaining the elevated synaptic efficacy produced by prior learning (Migues et al., 2010). This allows the memory which persists, in part, through sustained biochemical changes at the synapse, to be carried forward in time for durations exceeding the turnover of individual macromolecules.
But what happens when PKMZ, itself a protein susceptible to degradation, is broken down? The answer to this is what makes PKMZ a candidate for a “memory molecule.” PKMZ activity is self-sustaining since a continual positive feedback loop exists between the kinase activity and de novo PKMZ synthesis (Kwapis and Helmstetter, 2014). Once up-regulated, PKMZ molecules are autonomous and constitutively active owing to the activity of a catalytic domain and the lack of dependence on second messengers or auto-inhibition due to the absence of regulatory domains (Hernandez et al., 2003). In addition to its synaptic role PKMZ also acts in the nucleus to promote memory maintenance through epigenetic and transcriptional regulation (Ko et al., 2016). Mechanistically, the intracellular cascades activated during intense learning (LTP) drive the translocation of molecules, such as cyclic adenosine mono-phosphate (cAMP)-dependent protein kinase A and CaMKII, to the nucleus where they increase cAMP responsive element binding protein (CREB)-regulated CRE-driven transcription (Kandel, 2012) as shown in Figure 1, producing mRNA’s of memory-related proteins, including PKMZ (Lisman, 2017b). Although many of these PKMZ mRNAs bind RNA-binding proteins, such as staufen 1, and translocate to afferent neurites (Doyle and Kiebler, 2011), some are translated in free cytoplasmic ribosomes in the soma. These somatically translated PKMZ molecules then undergo importin-α mediated nuclear translocation where they phosphorylate CREB-binding protein (CBP). CBP regulates the accessibility to and transcription of proteins involved in synaptic potentiation and memory maintenance through its histone acetyltransferase and transcriptional co-activator activity, respectively (Ko et al., 2016). As long as PKMZ remains active and there is an absence of forces which terminate its activity (such as LTD), it will continue to sustain the biochemical changes at the synapse which serve as the neurobiological basis of memory, allowing the memory to persist for durations far exceeding the turnover of its component molecules. This mechanism may also underlie the stability of memories following brain remodeling (Blackiston et al., 2015). Additionally, memory persistence may be conferred, in part, by structural changes and growth at the synapse (Bailey et al., 2015), as structural changes last longer than many of the synaptic plasticity-related biochemical modifications (as discussed in “Synaptic Remodeling During Memory Formation” section). Thus, a synaptic theory of memory can account for the persistence of memories in spite of synaptic and protein turnover.
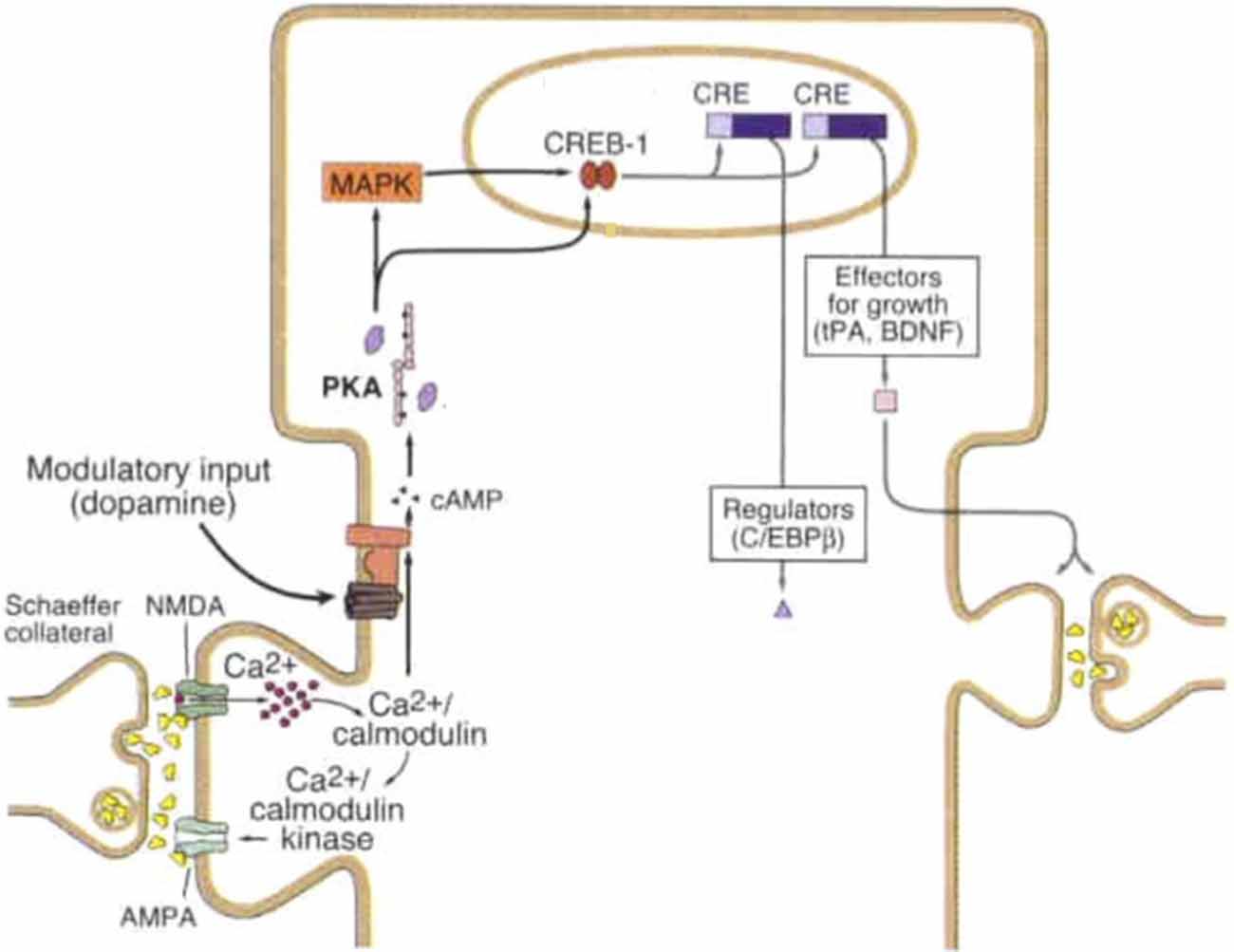
Figure 1. Late-long-term potentiation (L-LTP) mechanism at Schaeffer collateral synapse. Strong synaptic stimulation results in high magnitude calcium (Ca2+) influx, binding of calcium to calcium calmodulin which subsequently binds to calcium/calmodulin kinase and activates adenylyl cyclase (AC). AC generates the second messenger cyclic adenosine mono-phosphate (cAMP), cAMP activates PKA which translocates to the nucleus and activates mitogen activated protein kinase (MAPK) both of which turn on the transcriptional activator cAMP responsive element binding protein-1 (CREB-1). Upon CREB-1 binding to CRE transcription produces synapse modifying gene products including regulatory and growth proteins. Modulatory input, shown here as dopaminergic, can facilitate activation of AC and subsequent synaptic strengthening. Figure modified from Kandel (2001) License number 4363260804716.
Evidence From Spatial Training Suggests That There Is a Need to Separate Learning From Memory
Trettenbrein (2016) notes the need to separate learning from memory, citing literature showing that hippocampus-dependent spatial memory formation can occur even when NMDARs, which are thought to be necessary for LTP, are blocked (Saucier and Cain, 1995; Lüscher and Malenka, 2012). However, not all forms of hippocampus-dependent learning are NMDAR-dependent. For example, LTP in the hippocampal mossy-fiber pathway is NMDAR-independent (Johnston et al., 1992), yet LTP in this pathway is involved in spatial memory (Rekart et al., 2007). Thus, the involvement of the mossy fiber pathways in spatial memory offers an explanation for how learning can occur in the absence of NMDARs under certain conditions. NMDAR-independent synaptic potentiation is commonly categorized as a form of non-Hebbian synaptic plasticity. Kato et al. (2009) demonstrated that synaptic potentiation can occur in response to strong post-synaptic depolarization, without the requisite pre-synaptic activity seen in Hebbian models of plasticity. This form of synaptic plasticity is clearly NMDAR independent (as NMDARs require coincident pre- and post-synaptic activity) and instead utilizes post-synaptic voltage-dependent calcium channels. Additional forms of non-Hebbian learning are known to exist, including cerebellar learning which can result from a timing protocol opposite that of the spike-timing-dependent plasticity (STDP) originally postulated by Hebb (Hebb, 1949; Piochon et al., 2013). Even though cerebellar Purkinje cells can learn the interval timing involved in temporal memory traces, this does not rule out the synaptic theory of memory as the memory trace may be “coupled to a specific subset of synapses or dendritic compartments, but it is not exclusively reliant on a change in the strength of those synapses” (Jirenhed et al., 2017, page 6131). The problem with this argument is that even if “single Purkinje cells can learn response sequences” these cells rely on modified synaptic connections with other cells in order to express these memories. Therefore, non-Hebbian models of synaptic plasticity provide a mechanism by which learning can proceed, in certain cases, in the face of NMDAR blockade.
It is also difficult to separate learning from memory on theoretical grounds as learning and memory are inextricable parts of a continuum. Learning is the acquisition of new information or the modification of existing information, skills, etc., and that information must be represented in neural tissue, even if temporarily as occurs in sensory and working memory (Miller et al., 1960; Cowan, 2008; Bradley and Pearson, 2012). Synaptic plasticity provides a model for describing the continuity between learning and memory. The more incidental the learning (i.e., the less frequently the material being learned is rehearsed), the weaker and more transient the synaptic change by which that learning will be manifest, termed short-term potentiation (STP). On the other hand, more frequent or intense learning bouts will cause information to be stored in a series of persistent synaptic modifications, known as LTP. These stages (STP and LTP) are not discrete, binary states, but form a continuum (Volianskis and Jensen, 2003; Volianskis et al., 2013). Variability in learning intensity and frequency can cause activated synapses to become more or less potentiated, shifting memory strength and duration accordingly. This indicates that learning and memory are inextricable parts of a continuum, where the duration and strength of a memory is graded by the frequency and intensity of learning bouts and memory cannot occur without learning (Takeuchi et al., 2014), except in trans-generational epigenetic memories (Lim and Brunet, 2013; Dias et al., 2015). Therefore, it is not theoretically feasible to separate learning from memory as they are inextricable parts of a continuum, as discussed in “Existing Learning Mechanisms Cannot Explain Information That is Encoded in a Single Trial” and “Memory May Be Sub-Cellular in Nature” sections.
Existing Learning Mechanisms Cannot Explain Information That Is Encoded in a Single Trial
Traditional approaches to associative learning suggest that the number of trials and the CS-US interval are the primary determinants of learning (Hawkins et al., 2006), supporting Hebb (1949) principle that the repeated, simultaneous activation of a synapse leads to the strengthening of that synapse. Gallistel and Balsam (2014), cited in Trettenbrein (2016), suggest that it is physiologically difficult to explain how synaptic learning mechanisms can encode information after a single trial. However, one trial inhibitory avoidance conditioning induces LTP in the CA1 neurons of the hippocampus (Whitlock et al., 2006) and a single exposure to a novel environment can stimulate LTP and activate the immediate-early gene Arc in CA3 neurons of the hippocampus (Miyashita et al., 2009). Novel stimuli also stimulate the release of neuromodulatory transmitters such as dopamine and acetylcholine which contribute to the synaptic modifications underlying one trial learning (Takeuchi et al., 2016). Therefore, a theory which regards the synapse as integral to memory formation can address this criticism. Bissière et al. (2003) and Marowsky et al. (2005) have shown that dopamine, acting as a neuromodulator, facilitates Hebbian LTP in the lateral amygdala during aversive learning. Furthermore, Bush et al. (2010) describe the ability of beta-noradrenergic receptors (β-ARs) to modulate threat learning, perhaps through influencing STDP (Pawlak et al., 2010). Johansen et al. (2014) demonstrated that in learning following very few trials, Hebbian plasticity mechanisms alone were insufficient to produce plastic changes in the lateral amygdala. Learning successfully occurred, however, when these Hebbian plasticity mechanisms were paired with the co-activation of β-ARs, emphasizing the importance of neuromodulation in this form of learning. Theoretically, while a single small shock may produce STP (early E-LTP), a larger shock and the concomitant neuromodulation, will result in a more sustained potentiation (long L-LTP). In sum, neuromodulation and the activation of immediate early genes, working in concert with Hebbian plasticity, provide a mechanistic explanation for how instances of single trial learning are explained by a synaptic theory of memory.
Memory May Be Sub-cellular in Nature
Trettenbrein’s (2016), critique of synaptic theory and in support of a submolecular basis for memory, quotes from Gallistel and King (2009, page 282) that “our skepticism rests most strongly on the fact that the synapse is a circuit-level structure, a structure that it takes two different neurons and a great many molecules to realize. It seems to us likely for a variety of reasons that the elementary unit in the memory mechanism will prove to be a molecular or sub-molecular structural unit.” This statement contains two points, which need to be addressed independently. First, the fact that the synapse is a structure involving neuronal connections is not a burden to a synaptic theory of memory but an asset. Associative memories are formed through a rewiring of brain circuits (Fuster, 1997; Chau et al., 2014; Van Ooyen and Butz-Ostendorf, 2017) such that the collection of neurons (cell assemblies) representing stimulus “A” and those representing stimulus “B” are either connected by de novo synaptogenesis (Le Bé and Markram, 2006; Kwon and Sabatini, 2011); have existing synapses between these neurons potentiated; or a combination of both (Choi et al., 2018). Subsequent activity in “A” encoding neurons can then, through spreading activation, elicit activity in the neurons encoding “B” thus providing a neural link between the cell assemblies (a phase sequence) representing each of these two originally discrete stimuli (Anderson, 1983). As discussed in “Evidence From Spatial Training Suggests That There Is a Need to Separate Learning From Memory” section, learning and memory occur along a continuum, so the fact that synaptic encoding of a memory requires “a great many molecules to realize” is a benefit as it allows for the gradation of synaptic, and in turn memory, strength along a continuum in accordance with the salience of the information, or effortfulness of original learning (Liu et al., 2017). Furthermore, the biochemical and structural complexity of the glutamatergic synapse, actualized by these “great many molecules,” allows for several distinct forms of plasticity: (i) STP; (ii) early or late LTP (E-LTP or L-LTP); (iii) LTD; (iv) synaptic scaling; and (v) distance-dependent scaling (Volianskis et al., 2013; Lisman, 2017a). This diversity helps to explain how various types, durations and strengths of memory can be represented in the brain. So, the nature of the synapse as a circuit-level structure and the number of molecules involved in producing synaptic change are not hindrances to a synaptic theory of memory but strengths.
Second, considerable evidence exists for the intracellular chain of biochemical events, which lead to memory formation. As described by Kandel (Kandel, 2001; Kandel et al., 2014) and others (Guzman-Karlsson et al., 2014), synaptic activity triggers intracellular second messengers such as cyclic AMP, protein kinases, MEK-ERK pathways, CAMKII, CREB and binding to DNA for transcription of genetic information to mRNA and translation of this information for new protein synthesis; an overview of elements common to synaptic potentiation pathways is included as Figure 1. These intracellular pathways modulate synaptic activity by inducing changes in the quantity (Fleming and England, 2010) and conformational status of AMPARs (Lee et al., 2000); the surface area of the post-synaptic density, and in turn the size of the axon-spine interface (Desmond and Levy, 1986, 1988); the volume of the dendritic spine head (Matsuzaki et al., 2004; Bozdagi et al., 2010); and the growth of new synapses (Kandel, 2001). In addition, retrograde signals, such as nitric oxide (Padamsey and Emptage, 2013), are generated post-synaptically and travel to the presynaptic compartment where they elicit alterations as part of the potentiation process. These presynaptic alterations act to alter the probability and latency of presynaptic neurotransmitter release, modulate the magnitude of quantal release (Larkman et al., 1992; Sola et al., 2004; Enoki et al., 2009), the size of the pool of readily available synaptic vesicles, the quantity and responsiveness of presynaptic calcium channels and the set point of presynaptic calcium sensors (Kaeser and Regehr, 2014; Costa et al., 2017). These presynaptic modifications can act bi-directionally, to potentiate or depress synaptic efficacy (Fioravante and Regehr, 2011; Regehr, 2012; Yang and Calakos, 2013). Finally, CREB-mediated intrinsic plasticity can be described as being submolecular and has recently been implicated in processes of memory allocation, linking and integration (Josselyn and Frankland, 2018; Lisman et al., 2018; Sehgal et al., 2018). Thus, the suggestion put forth in Trettenbrein (2016) that memory is submolecular in nature is correct, but incomplete.
An additional piece of evidence supporting a synaptic plasticity model of memory is that both memory and synaptic plasticity consist of at least two discrete temporal stages. Short-term memory (using the neurophysiological definition) lasts for ~1–2 h. E-LTP, also, lasts for ~1–2 h and results from temporary modifications of pre-existing proteins which are sustained for as long as kinase activity (e.g., CaMKII) exerts dominance over phosphatase activity. Once dominance has shifted towards phosphatase activity, the biochemical substrates underlying the memory are rapidly removed and the memory is lost (Huang, 1998; Genoux et al., 2002; Munton et al., 2004). Long-term memory duration varies but is generally considered to persist for longer than 2 h and can last for days, weeks, years or even a lifetime. L-LTP, not coincidentally, lasts for durations comparable to those described for long-term memory (Costa-Mattioli et al., 2008). The protein synthesis underlying L-LTP is responsible not only for generating a greater number of synaptic potentiation promoting proteins and structural components but also for generating the persistent kinases capable of allowing these long-term memories to be maintained for such impressive durations (Jalil et al., 2015). Thus, this temporal overlap serves as evidence that the synaptic change occurring during E-LTP serves as the neurobiological correlate of short-term memory formation while those synaptic changes involved in L-LTP represent the neurobiological correlate of long-term memory formation.
A final component of Trettenbrein (2016) and Gallistel and King’s (2009) argument that memories might be submolecular is that, “neural computation is demonstrably incredibly fast, therefore making it much more likely that the memory mechanism is (sub-) molecular in nature so that computational machinery and memory can be located in close physical proximity in order to minimize the distance over which a signal has to be transmitted.” If it is assumed that by “computation” the authors are referring to the ability of the organism to take in, process and utilize information, then indeed memory is fast, but this rapidity can be explained while maintaining adherence to a synaptic theory of memory. The intracellular signaling cascades, protein synthesis and morphological changes that represent memory in a synaptic theory are indeed slow to occur, but one need not wait for these changes in order to utilize the memory. Memory is initially encoded in the mnemonic fields of a particular constellation of prefrontal cortical neurons as a reverberatory pattern of activity (Lorente De Nó, 1933; Hebb, 1949; Funahashi et al., 1989; Wang, 2005; Lara and Wallis, 2015). These electrical reverberations induce synaptic E-LTP which lays down information as a series of temporary, transcription independent, kinase activity-dependent, biochemical alterations (Huang, 1998) capable of maintaining information in the brain’s connectivity until the more permanent, transcription-dependent L-LTP-dependent processes can better establish this memory in the neural tissue (Vickers and Wyllie, 2007). Thus, a synaptic theory of memory ensures that the information is rapidly available for immediate computational usage, despite the temporal void that exists between the acquisition of this information and its later stabilization through the longer lasting de novo protein synthesis and morphological changes involved in consolidation. Once again, this is evidence that one need not stray from a synaptic theory in order to explain the complexities of memory.
Memory formation involves both genetic and epigenetic processes. The transcription of genetic information from DNA to mRNA involves histone acetylation, histone methylation and DNA methylation (Mazzio and Soliman, 2012; Zovkic and Sweatt, 2013). We believe these intracellular changes highlighted by Trettenbrein (2016) while implicated in memory formation and sustenance cannot provide a synapse independent explanation of memory. Such changes lack a means of modifying the flow of electrical activity and thus information in the brain necessary for the expression of experience-dependent memory. These changes instead regulate the expression of various genes the products of which re-weight synapses allowing a sustained redirecting of neural activity. The laying down of these epigenetic marks during learning and the influence that they then exert on both Hebbian and non-Hebbian synaptic plasticity has become a fruitful area of inquiry (Guzman-Karlsson et al., 2014; Kim and Kaang, 2017). These epigenetic processes allow modulation of memories by other chemical messengers, including neurotransmitters (dopamine, serotonin, etc.), hormones (corticosterone, androgens), neuropeptides (oxytocin, ACTH), and even cytokines (IL-1, IL-6, TNFα, etc.; Song et al., 2003; Hunter, 2012; Rajasethupathy et al., 2012; Trollope et al., 2012; del Rey et al., 2013; Tuesta and Zhang, 2014; Reizel et al., 2015; Haas et al., 2016; Madej et al., 2017; Prieto and Cotman, 2017), all of which can be activated by external and internal stimuli, including emotions, motivations, thoughts and memories (Davis et al., 1994; Berger et al., 2009).
In addition, the reactivation of stored memories causes them to become transiently deconsolidated and labile, providing an opportunity to amend preexisting memories with new information. These retrieved memories are then reconsolidated, leading to the confounding of original memories with new information and the storing of false memories (McKenzie and Eichenbaum, 2011; Ramirez et al., 2013; Almeida-Correa and Amaral, 2014). When a memory is explicitly retrieved, it is done so by driving reverberatory activity through the experientially modified synapses in the cell assemblies and phase sequences within which the memory is represented. This subjects it to the operations of working memory where it is Fragile and susceptible to alteration (Lee et al., 2017). The activated synapses produce intracellular biochemical changes and protein synthesis, as discussed in “Synaptic Remodeling During Memory Formation” section, which remodel the synapse in a manner consistent with the pattern of activity experienced at the synapse (i.e., according to the principles of STDP; Lee et al., 2011). The remodeling of the synapses in the cell assemblies and phase sequences underlying a memory can incorporate new information, or change the existing information, comprising the retrieved memory (Bonin and De Koninck, 2015; Hu and Schacher, 2015; Kastner et al., 2016). Thus, the neurophysiological basis of reconsolidation provides support for a synaptic theory of memory.
Synaptic Remodeling During Memory Formation
As stated by Hebb (1949), “some growth process or metabolic change” takes place to increase the efficacy of cell A on the firing of cell B. The activation of intracellular biochemical pathways, gene expression, epigenetic factors, and protein synthesis leads to the remodeling (strengthening or weakening) of synapses in cell assemblies. There are many different types of growth processes and/or metabolic changes that lead to changes in synaptic strength, including: (1) excitatory; (2) inhibitory; (3) size of the synapse; (4) ion channels; (5) dendrites, boutons, et cetera (De Roo et al., 2008; Caroni et al., 2014; Bailey et al., 2015; Yang et al., 2016; Choi et al., 2018). Distinct activity patterns drive specific intracellular biochemical signaling cascades which serve to adaptively remodel the synapse during activity-dependent synaptic plasticity (Colicos, 2009, pages 50–52). As an example, adaptive, biologically relevant information which needs to be remembered in order to optimally organize behavior and cognition at a later time point is likely to drive tetanic activity through a particular set of synapses, producing large post-synaptic calcium transients. Calcium then binds to calcium binding proteins, including calmodulin, which activates various effectors including nNOS, CaMKII, post-synaptic adenylyl cyclase (AC), etc. These effectors then activate downstream kinases, genes and other proteins capable of remodeling the pre- and post-synaptic elements in such a way to confer an increased synaptic efficacy (Wong et al., 1999; Kandel, 2012; Choi et al., 2018). These remodeling changes, including the growth of pre-existing and the formation of new dendritic spines are correlated in magnitude with the degree of training (Xu et al., 2009; Yang et al., 2009). Thus, intracellular metabolic changes function to modulate synaptic remodeling and growth processes during memory formation.
Reconciliation of the Differences Between Physiologists and Psychologists About the Role of Synaptic Plasticity in Learning and Memory
Our critique of Trettenbrein (2016) has focused on the term “demise” of the synaptic theory of memory. We believe that the synaptic theory of memory has not died but that there are two components of this theory: synaptic plasticity and intra-cellular biochemical changes. At issue is whether “memory” consists of the synaptic changes activated by intracellular biochemical changes OR whether memory consists of the intracellular biochemical changes expressed via synaptic plasticity. Our argument is that memory, as conceived by Hebb, consists inseparably of both synaptic plasticity and “intrinsic plasticity” of the neurons (Sehgal et al., 2013; Titley et al., 2017; Lisman et al., 2018).
Synaptic Change and the Formation of Cell Assemblies Are Fundamental for Theories of Memory
Hebb (1949, 1959) realized that his theory would need revision in the light of new discoveries, but the fact that his ideas on synaptic plasticity (Favero et al., 2014), cell assemblies (Lansner, 2009; Wallace and Kerr, 2010) and phase sequences (Almeida-Filho et al., 2014) continue to stimulate new research and discussion is a tribute to his prescience. For example, Hebb’s ideas continue to stimulate new research on the physiological mechanisms of learning and memory (Schrader et al., 2002; Lisman et al., 2011; Johansen et al., 2014), learning and development (Munakata and Pfaffly, 2004), memory span (Oberauer et al., 2015), decision making (Wang, 2012) and language learning (Wennekers et al., 2006). Posner and Rothbart (2004, 2007) went as far as to suggest that Hebb’s ideas should be used to integrate the disparate branches of Psychology and Neuroscience.
Cell Assemblies Have Been Verified by Neuroimaging
Hebb’s theories on the neurophysiological basis of learning and memory integrate synaptic neurophysiology with psychological concepts such as attention, perception, thought and mind—the concepts which Pavlov avoided in his objective approach to memory. Hebb’s theory effectively integrated Pavlov’s concepts of the physiology of learning with Lashley’s (1932) criticism that Pavlov ignored psychological concepts. Neuroimaging studies have shown the usefulness of Hebb’s ideas for understanding both the psychological and physiological mechanisms of memory. Memory processes have been shown by fMRI and other neuroimaging methods to be distributed across many cortical areas (Miyamoto et al., 2014). For example, Christophel et al. (2017) showed that different cortical neural networks are activated in different types of working memory, and O’Neil et al. (2012) found that different cortical regions were activated in recognition memory.
Hebb’s Synaptic Learning Rule and Cell Assembly Theory Is Used in Computational Neuroscience and Robotics
Hebb’s concept of cell assemblies and phase sequences have been used to develop theories of the cortical control of behavior (Palm et al., 2014), network theories of memory (Fuster, 1997) and computer models of memory processes (Lansner, 2009). According to Palm et al. (2014; page 560), “the further development of cell assembly theory was mainly driven by neurophysiological and biophysical findings concerning the basic neuronal mechanisms and the detailed temporal processes of neuronal activation and interaction on one hand and by computational arguments and requirements on the other.” Cell assembly theory has resulted in the development of anatomical features that underlie the location of memory storage in the cortex (see Palm et al., 2014). Computer models of the brain use Hebbian learning rules and cell assemblies (Wennekers, 2007) to build neural networks based on spike timing dependent plasticity (Markram et al., 2011). The role of Hebbian theory in neural network modeling will continue to grow in importance (Buzsáki, 2010; Fotouhi et al., 2015). Furthermore, Hebbian learning rules and cell assemblies are now used in robotics (Wang et al., 2009; Calderon et al., 2013) and it is now possible for Hebbian learning rules to control brain-robot interfaces in neurorehabilitation (Gomez-Rodriguez et al., 2011; Takeuchi and Izumi, 2015).
Abnormalities in Synaptic Plasticity Underlie Cognitive and Motor Dysfunctions Pain Mechanisms and Drug Addiction
The activation of the network of synaptic connections in a cell assembly requires changes in synaptic strength to establish the connectivity of the neurons in the cell assembly. Cell assemblies are essentially a collection of activated synapses and the sufficiently strong activation of these synapses causes biochemical changes in the neurons of the cell assembly. Thus, biochemical changes and gene activation within the neurons of a cell assembly are required to maintain memories (Li et al., 2016). These involve complex interactions between excitatory and inhibitory synapses (see Barron et al., 2017). The biochemical changes in the neurons of a cell assembly that are activated by transient changes in synaptic activity involve epigenetic mechanisms including chromatin remodeling which drives changes in the transcription and translation of information in the DNA, protein synthesis and cellular changes underlying learning and memory formation (Vogel-Ciernia and Wood, 2014). As stated by Hebb in 1949, the synaptic changes following repeated stimulation at a synapse lead to “some growth process or metabolic change… in one or both cells such that A’s efficiency, as one of the cells firing B, is increased.” Much of the neuroscientific research on the cellular and molecular basis of memory in the last 70 years has been oriented to finding these growth processes and metabolic changes that underlie memory (Josselyn et al., 2015; Poo et al., 2016). Synaptic change is not limited to learning and memory, but forms the basis of neural changes in perception (Dan and Poo, 2006; Yang et al., 2011), pain (Luo et al., 2014) and drug addiction (Jones and Bonci, 2005; Kauer and Malenka, 2007; Lüscher, 2013).
Neurological disorders which involve cognitive or motor dysfunction are the result of synaptic abnormalities (Amorim et al., 2015; Kouroupi et al., 2017). Synaptic dysfunction underlies neurodevelopmental disorders such as autism, Rett syndrome, Down syndrome and ADHD (Pettem et al., 2013; Moretto et al., 2018) and a wide range of neurological disorders of adulthood and aging, including Alzheimer disease, Parkinson’s disease, Huntington’s disease and multiple sclerosis (Henstridge et al., 2016; Rosales-Reynoso et al., 2016; Forner et al., 2017; Torres et al., 2017). For example, tau tangles and beta-amyloid peptide are elevated, intra- and extracellularly respectively, in the brains of Alzheimer’s disease patients and are known to impair synaptic function and cause synapse loss (Forner et al., 2017). Specifically, the accumulation of beta-amyloid oligomers in Alzheimer’s disease impairs hippocampal LTP and memory consolidation (Figueiredo et al., 2013; Bilousova et al., 2016; Yang et al., 2017) in addition to causing reductions in dendritic spine density, AMPAR numbers, synaptic strength (Rodrigues et al., 2016) and synapse loss (Sheng et al., 2012). Impaired hippocampal LTP and consolidation may explain the difficultly in forming new, lasting memories (Weintraub et al., 2012), termed anterograde amnesia, while decreases in synaptic strength (and thus removal of the physical substrates of memories) and synapse loss may explain the erasure of past memories in retrograde amnesia, both of which are characteristic of Alzheimer’s disease (Beatty et al., 1988). Thus, a synaptic plasticity theory of memory can explain the memory impairments in neuropathologic conditions such as Alzheimer’s disease.
Contrary Opinions
The theory that synaptic connectivity forms the basis of cognitive functions is not universally accepted. In addition to Trettenbrein’s (2016) critique, models of learning and memory based on non-synaptic plasticity have been proposed (Mozzachiodi and Byrne, 2010; Cacha et al., 2017), but these appear to involve neuromodulatory actions which alter the parameters of LTP and LTD, and thus the efficacy of synaptic plasticity. In addition, Arshavsky (2006, 2017), Chen et al. (2014) and Bèdècarrats et al. (2018) have advocated a genomic rather than a synaptic theory of memory and argue that changes in individual neurons rather than interconnected neural networks form the basis of memory. Bèdècarrats et al. (2018) demonstrated that the transfer of RNA from trained to naïve animals is sufficient to induce behavioral correlates of memory and that these changes are dependent on DNA methylation. However, the modified expression of these gene products likely serves to change synaptic weights, an idea supported by Bèdècarrats et al.’s (2018) demonstration of an increase in sensory neuron excitability following RNA injection. While these genetic and epigenetic views are minority opinions, they cannot be ruled out. However, if cognitive processes reside in individual neurons or through genomic and epigenomic modifications, the only way that these cells can communicate with other neurons is through synaptic activity. Thus, even if one was to concede that memory may reside in individual cells or in genomic and epigenomic modifications, these cells form a cell assembly and the expression of that memory requires the activation of the synapses linking the cells. In order to facilitate the formation of a cell assembly, synaptic activation occurs and as a memory is consolidated into a cell assembly, synaptic modifications occur. Without these synaptic modifications, memories could not be expressed.
Summary and Conclusions
Trettenbrein’s (2016) critiques of the synaptic theory of memory can be answered by the published evidence for the synaptic theory of memory, as argued in this article. The synaptic theory of memory remains the most empirically plausible explanation for the neurobiological basis of memory, even if it may need modification (Jirenhed et al., 2017). In this article we have addressed the six critiques raised by Trettenbrein (2016) and through the use of recent neurophysiologic literature, we have demonstrated that synaptic change is only the first step in formation of the cell assemblies and phase sequences postulated by Hebb (1949), which together constitute the Hebbian account of memory. We have shown how the concepts of synaptic change and the cell assembly are used to understand the neuroanatomical, cellular, molecular and genetic bases of memory and of neurological disorders. We have also shown how neuroimaging studies, computer modeling and robotics have used the Hebbian learning rules and cell assemblies to develop computer learning and “intelligent” robots. Our critique of Trettenbrein (2016) has focused on the term “demise” (death, downfall, disappearance or final fate) of the synaptic theory of memory. We believe that the synaptic theory of memory has not died, but has gone from strength to strength. But there are two components of this theory: synaptic plasticity and intra-cellular biochemical changes. At issue is whether “memory” consists of the synaptic changes activated by intracellular biochemical changes OR whether memory consists of the intracellular biochemical changes expressed via synaptic plasticity. Our argument is that memory, as conceived by Hebb, consists of both synaptic plasticity and “intrinsic plasticity” of the neurons (Sehgal et al., 2013; Titley et al., 2017; Lisman et al., 2018). You cannot separate one from the other.
Author Contributions
JL and RB wrote the article. RB edited and supervised the project.
Funding
This work was supported by grant number 7441 from the Natural Sciences and Engineering Research Council (NSERC) of Canada to RB.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Almeida-Correa, S., and Amaral, O. B. (2014). Memory labilization in reconsolidation and extinction—evidence for a common plasticity system? J. Physiol. Paris 108, 292–306. doi: 10.1016/j.jphysparis.2014.08.006
Almeida-Filho, D. G., Lopes-dos-Santos, V., Vasconcelos, N. A. P., Miranda, J. G. V., Tort, A. B. L., and Ribeiro, S. (2014). An investigation of Hebbian phase sequences as assembly graphs. Front. Neural Circuits 8:34. doi: 10.3389/fncir.2014.00034
Amorim, I. S., Mitchell, N. L., Palmer, D. N., Sawiak, S. J., Mason, R., Wishart, T. M., et al. (2015). Molecular neuropathology of the synapse in sheep with CLN5 batten disease. Brain Behav. 5:e00401. doi: 10.1002/brb3.401
Anderson, J. R. (1983). A spreading activation theory of memory. J. Verbal Learn. Verbal Behav. 22, 261–295. doi: 10.1016/s0022-5371(83)90201-3
Arshavsky, Y. I. (2006). “The seven sins” of the Hebbian synapse: can the hypothesis of synaptic plasticity explain long-term memory consolidation. Prog. Neurobiol. 80, 99–113. doi: 10.1016/j.pneurobio.2006.09.004
Arshavsky, Y. I. (2017). Neurons versus networks: the interplay between individual neurons and neural networks in cognitive functions. Neuroscientist 23, 341–355. doi: 10.1177/1073858416670124
Attardo, A., Fitzgerald, J. E., and Schnitzer, M. J. (2015). Impermanence of dendritic spines in live adult CA1 hippocampus. Nature 523, 592–596. doi: 10.1038/nature14467
Bailey, C. H., and Kandel, E. R. (2008). Synaptic remodeling, synaptic growth and the storage of long-term memory in Aplysia. Prog. Brain Res. 169, 179–198. doi: 10.1016/s0079-6123(07)00010-6
Bailey, C. H., Kandel, E. R., and Harris, K. M. (2015). Structural components of synaptic plasticity and memory consolidation. Cold Spring Harb. Perspect. Biol. 7:a021758. doi: 10.1101/cshperspect.a021758
Barron, H. C., Vogels, T. P., Behrens, T. E., and Ramaswami, M. (2017). Inhibitory engrams in perception and memory. Proc. Natl. Acad. Sci. U S A 114, 6666–6674. doi: 10.1101/117085
Beatty, W. W., Salmon, D. P., Butters, N., Heindel, W. C., and Granholm, E. L. (1988). Retrograde amnesia in patients with Alzheimer’s disease or Huntington’s disease. Neurobiol. Aging 9, 181–186. doi: 10.1016/s0197-4580(88)80048-4
Bèdècarrats, A., Chen, S., Pearce, K., Cai, D., and Glanzman, D. L. (2018). RNA from trained Aplysia can induce an epigenetic engram for long-term sensitization in untrained Aplysia. eNeuro 5:ENEURO.0038-18.2018. doi: 10.1523/ENEURO.0038-18.2018
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An operational definition of epigenetics. Genes Dev. 23, 781–783. doi: 10.1101/gad.1787609
Bilousova, T., Miller, C. A., Poon, W. W., Vinters, H. V., Corrada, M., Kawas, C., et al. (2016). Synaptic amyloid-β oligomers precede p-Tau and differentiate high pathology control cases. Am. J. Pathol. 186, 185–198. doi: 10.1016/j.ajpath.2015.09.018
Bissière, S., Humeau, Y., and Lüthi, A. (2003). Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat. Neurosci. 6, 587–592. doi: 10.1038/nn1058
Blackiston, D. J., Shomrat, T., and Levin, M. (2015). The stability of memories during brain remodeling: a perspective. Commun. Integr. Biol. 8:e1073424. doi: 10.1080/19420889.2015.1073424
Bonin, R. P., and De Koninck, Y. (2015). Reconsolidation and the regulation of plasticity: moving beyond memory. Trends Neurosci. 38, 336–344. doi: 10.1016/j.tins.2015.04.007
Bozdagi, O., Wang, X.-B., Nikitczuk, J. S., Anderson, T. R., Bloss, E. B., Radice, G. L., et al. (2010). Persistence of coordinated LTP and dendritic spine enlargement at mature hippocampal CA1 synapses requires N-cadherin. J. Neurosci. 30, 9984–9989. doi: 10.1523/jneurosci.1223-10.2010
Bradley, C., and Pearson, J. (2012). The sensory components of high-capacity iconic memory and visual working memory. Front. Psychol. 3:355. doi: 10.3389/fpsyg.2012.00355
Bush, D. E., Caparosa, E. M., Gekker, A., and Ledoux, J. (2010). β-adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front. Behav. Neurosci. 4:154. doi: 10.3389/fnbeh.2010.00154
Butler, C. W., Wilson, Y. M., Gunnersen, J. M., and Murphy, M. (2015). Tracking the fear memory engram: discrete populations of neurons within amygdala, hypothalamus and lateral septum are specifically activated by auditory fear conditioning. Learn. Mem. 22, 370–384. doi: 10.1101/lm.037663.114
Butler, C. W., Wilson, Y. M., Oyrer, J., Karle, T. J., Petrou, S., Gunnersen, J. M., et al. (2018). Neurons specifically activated by fear learning in lateral amygdala display increased synaptic strength. eNeuro 5:ENEURO.0114–18.2018. doi: 10.1523/eneuro.0114-18.2018
Buzsáki, G. (2010). Neural syntax: cell assemblies, synapsembles and readers. Neuron 63, 362–385. doi: 10.1016/j.neuron.2010.09.023
Cacha, L. A., Ali, J., Rizvi, Z. H., Yupapin, P. P., and Poznanski, R. R. (2017). Nonsynaptic plasticity model of long-term memory engrams. J. Integr. Neurosci. 16, 493–509. doi: 10.3233/jin-170038
Calderon, D., Baidyk, T., and Kussul, E. (2013). Hebbian ensemble neural network for robot movement control. Opt. Mem. Neural Netw. 22, 166–183. doi: 10.3103/s1060992x13030028
Caroni, P., Chowdhury, A., and Lahr, M. (2014). Synapse rearrangements upon learning: from divergent-sparse connectivity to dedicated sub-circuits. Trends Neurosci. 37, 604–614. doi: 10.1016/j.tins.2014.08.011
Chau, L. S., Prakapenka, A. V., Zendeli, L., Davis, A. S., and Galvez, R. (2014). Training-dependent associative learning induced neocortical structural plasticity: a trace eyeblink conditioning analysis. PLoS One 9:e95317. doi: 10.1371/journal.pone.0095317
Chen, S., Cai, D., Pearce, K., Sun, P. Y. -W., Roberts, A. C., and Glanzman, D. L. (2014). Reinstatement of long-term memory following erasure of its behavioral and synaptic expression in Aplysia. Elife 3:e03896. doi: 10.7554/eLife.03896
Choi, J.-H., Sim, S. E., Kim, J.-I., Choi, D., Oh, J., Ye, S., et al. (2018). Interregional synaptic maps among engram cells underlie memory formation. Science 360, 430–435. doi: 10.1126/science.aas9204
Christophel, T. B., Klink, P. C., Spitzer, B., Roelfsema, P. R., and Haynes, J. D. (2017). The distributed nature of working memory. Trends Cogn. Sci. 21, 111–124. doi: 10.1016/j.tics.2016.12.007
Colicos, M. A. (2009). “Activity-dependent synaptic plasticity,” in Encyclopedia of Neuroscience, (Berlin, Heidelberg: Springer), 50–52.
Costa, R. P., Mizusaki, B. E. P., Sjostrom, P. J., and van Rossum, M. C. W. (2017). Functional consequences of pre- and postsynaptic expression of synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160153. doi: 10.1098/rstb.2016.0153
Costa-Mattioli, M., Sossin, W. S., Klann, E., and Sonenberg, N. (2008). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26. doi: 10.1016/j.neuron.2008.10.055
Cowan, N. (2008). What are the differences between long-term, short-term and working memory? Prog. Brain Res. 169, 323–338. doi: 10.1016/s0079-6123(07)00020-9
Craig, A. M., Graf, E. R., and Linhoff, M. W. (2006). How to build a central synapse: clues from cell culture. Trends Neurosci. 29, 8–20. doi: 10.1016/j.tins.2005.11.002
Dan, Y., and Poo, M. M. (2006). Spike timing-dependent plasticity: from synapse to perception. Physiol. Rev. 86, 1033–1048. doi: 10.1152/physrev.00030.2005
Davis, M., Rainnie, D., and Cassell, M. (1994). Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 17, 208–214. doi: 10.1016/0166-2236(94)90106-6
del Rey, A., Balschun, D., Wetzel, W., Randolf, A., and Besedovsky, H. O. (2013). A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6 and TNFα operates during long-term potentiation and learning. Brain Behav. Immun. 33, 15–23. doi: 10.1016/j.bbi.2013.05.011
De Roo, M., Kaluser, P., Garcia, P. M., Poglia, L., and Muller, D. (2008). Spine dynamics and synapse remodeling during LTP and memory processes. Prog. Brain Res. 169, 199–207. doi: 10.1016/s0079-6123(07)00011-8
Desmond, N. L., and Levy, W. B. (1986). Changes in the postsynaptic density with long-term potentiation in the dentate gyrus. J. Comp. Neurol. 253, 476–482. doi: 10.1002/cne.902530405
Desmond, N. L., and Levy, W. B. (1988). Synaptic interface surface area increases with long-term potentiation in the hippocampal dentate gyrus. Brain Res. 453, 308–314. doi: 10.1016/0006-8993(88)90171-0
Dias, B. G., Maddox, S. A., Klengel, T., and Ressler, K. J. (2015). Epigenetic mechanisms underlying learning and the inheritance of learned behaviors. Trends Neurosci. 38, 96–107. doi: 10.1016/j.tins.2014.12.003
Doyle, M., and Kiebler, M. A. (2011). Mechanisms of dendritic mRNA transport and its role in synaptic tagging. EMBO J. 30, 3540–3552. doi: 10.1038/emboj.2011.278
Enoki, R., Hu, Y.-L., Hamilton, D., and Fine, A. (2009). Expression of long-term plasticity at individual synapses in hippocampus is graded, bidirectional and mainly presynaptic; optical quantal analysis. Neuron 62, 242–253. doi: 10.1016/j.neuron.2009.02.026
Favero, M., Cangiano, A., and Busetto, G. (2014). Hebb-based rules of neural plasticity: are they ubiquitously important for the refinement of synaptic connections in development? Neuroscientist 20, 8–14. doi: 10.1177/1073858413491148
Fernandez-Moya, S. M., Bauer, K. E., and Kiebler, M. A. (2014). Meet the players: local translation at the synapse. Front. Mol. Neurosci. 7:84. doi: 10.3389/fnmol.2014.00084
Figueiredo, C. P., Clarke, J. R., Ledo, J. H., Ribeiro, F. C., Costa, C. V., Melo, H. M., et al. (2013). Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 33, 9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013
Fioravante, D., and Regehr, W. G. (2011). Short-term forms of presynaptic plasticity. Curr. Opin. Neurobiol. 21, 269–274. doi: 10.1016/j.conb.2011.02.003
Fleming, J. J., and England, P. M. (2010). AMPA receptors and synaptic plasticity: a chemist’s perspective. Nat. Chem. Biol. 6, 89–97. doi: 10.1038/nchembio.298
Forner, S., Baglietto-Vargas, D., Martini, A. C., Trujillo-Estrada, L., and LaFerla, F. M. (2017). Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci. 40, 347–357. doi: 10.1016/j.tins.2017.04.002
Fotouhi, M., Heidari, M., and Sharifitabar, M. (2015). Continuous neural network with windowed Hebbian learning. Biol. Cybern. 109, 321–332. doi: 10.1007/s00422-015-0645-7
Funahashi, S., Bruce, C. J., and Goldman-Rakic, P. S. (1989). Mnemonic coding of visual space in the monkey’s dorsolateral prefrontal cortex. J. Neurophysiol. 61, 331–349. doi: 10.1152/jn.1989.61.2.331
Fuster, J. M. (1997). Network memory. Trends Neurosci. 20, 451–459. doi: 10.1016/S0166-2236(97)01128-4
Gallistel, C. R., and Balsam, P. D. (2014). Time to rethink the neural mechanisms of learning and memory. Neurobiol. Learn. Mem. 108, 136–144. doi: 10.1016/j.nlm.2013.11.019
Gallistel, C. R., and King, A. P. (2009). Memory and the Computational Brain: Why Cognitive Science Will Transform Neuroscience. Chichester, NJ: Wiley-Blackwell. doi: 10.1002/9781444310498
Gallistel, C. R., and Matzel, L. D. (2014). The neuroscience of learning: beyond the Hebbian synapse. Annu. Rev. Psychol. 64, 169–200. doi: 10.1146/annurev-psych-113011-143807
Genoux, D., Haditsch, U., Knobloch, M., Michalon, A., Storm, D., and Mansuy, I. M. (2002). Protein phosphatase 1 is a molecular constraint on learning and memory. Nature 418, 970–975. doi: 10.1038/nature00928
Gomez-Rodriguez, M., Grosse-Wentrup, M., Hill, J., Gharabaghi, A., Scholkopf, B., and Peters, J. (2011). Towards brain-robot interfaces in stroke rehabilitation. IEEE Int. Conf. Rehabil. Robot. 2011:5975385. doi: 10.1109/icorr.2011.5975385
Guthrie, E. R. (1930). Conditioning as a principle of learning. Psychol. Rev. 37, 412–428. doi: 10.1037/h0072172
Guzman-Karlsson, M. C., Meadows, J. P., Gavin, C. F., Hablitz, J. J., and Sweatt, J. D. (2014). Transcriptional and epigenetic regulation of Hebbian and non-Hebbian plasticity. Neuropharmacology 80, 3–17. doi: 10.1016/j.neuropharm.2014.01.001
Haas, B. W., Filkowski, M. M., Cochran, R. N., Denison, L., Ishak, A., Nishitani, S., et al. (2016). Epigenetic modification of OXT and human sociability. Proc. Natl. Acad. Sci. U S A 113, E3816–E3823. doi: 10.1073/pnas.1602809113
Hardt, O., Nader, K., and Wang, Y. T. (2014). GluA2-dependent AMPA receptor endocytosis and the decay of early and late long-term potentiation: possible mechanisms for forgetting of short- and long-term memories. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130141. doi: 10.1098/rstb.2013.0141
Hawkins, R. D., Kandel, E. R., and Bailey, C. H. (2006). Molecular mechanisms of memory storage. Biol. Bull. 210, 174–191. doi: 10.2307/4134556
Hebb, D. O. (1959). “A neuropsychological theory,” in Psychology: A Study of A Science, (Vol. 1) ed. S. Koch (New York,NY: McGraw Hill), 622–643.
Henley, J. M., and Wilkinson, K. A. (2013). AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 15, 11–27.
Henstridge, C. M., Pickett, E., and Spires-Jones, T. L. (2016). Synaptic pathology: a shared mechanism in neurological disease. Ageing Res. Rev. 28, 72–84. doi: 10.1016/j.arr.2016.04.005
Hernandez, A. I., Blace, N., Crary, J. F., Serrano, P. A., Leitges, M., Libien, J. M., et al. (2003). Protein kinase Mζ synthesis from a brain mRNA encoding an independent protein kinase Cζ catalytic domain. J. Biol. Chem. 278, 40305–40316. doi: 10.1074/jbc.M307065200
Hu, J., and Schacher, S. (2015). Persistent associative plasticity at an identified synapse underlying classical conditioning becomes labile with short-term homosynaptic activation. J. Neurosci. 35, 16159–16170. doi: 10.1523/jneurosci.2034-15.2015
Huang, E. P. (1998). Synaptic plasticity: going through phases with LTP. Curr. Biol. 8, R350–R352. doi: 10.1016/s0960-9822(98)70219-2
Hunter, R. G. (2012). Epigenetic effects of stress and corticosteroids in the brain. Front. Cell. Neurosci. 6:18. doi: 10.3389/fncel.2012.00018
Jalil, S. J., Sacktor, T. D., and Shouval, H. Z. (2015). Atypical PKCs in memory maintenance: the roles of feedback and redundancy. Learn. Mem. 22, 344–353. doi: 10.1101/lm.038844.115
Jarome, T. J., and Helmstetter, F. J. (2014). Protein degradation and protein synthesis in long-term memory formation. Front. Mol. Neurosci. 7:61. doi: 10.3389/fnmol.2014.00061
Jirenhed, D. A., Rasmussen, A., Johnasson, F., and Hesslow, G. (2017). Learned response sequences in cerebellar Purkinje cells. Proc. Natl. Acad. Sci. U S A 114, 6127–6132. doi: 10.1073/pnas.1621132114
Johansen, J. P., Diaz-Mataix, L., Hamanaka, H., Ozawa, T., Ycu, E., Koivumaa, J., et al. (2014). Hebbian and neuromodulatory mechanisms interact to trigger associative memory formation. Proc. Natl. Acad. Sci. U S A 111, E5584–E5592. doi: 10.1073/pnas.1421304111
Johnston, D., Williams, S., Jaffe, D., and Gray, R. (1992). NMDA-receptor-independent long-term potentiation. Annu. Rev. Neurosci. 54, 489–505. doi: 10.1146/annurev.physiol.54.1.489
Jones, S., and Bonci, A. (2005). Synaptic plasticity and drug addiction. Curr. Opin. Pharmacol. 5, 20–25. doi: 10.1016/j.coph.2004.08.011
Josselyn, S. A., and Frankland, P. W. (2018). Memory allocation: mechanisms and function. Annu. Rev. Neurosci. 41, 389–413. doi: 10.1146/annurev-neuro-080317-061956
Josselyn, S. A., Köhler, S., and Frankland, P. W. (2015). Finding the engram. Nat. Rev. Neurosci. 16, 521–534. doi: 10.1038/nrn4000
Kaeser, P. S., and Regehr, W. G. (2014). Molecular mechanisms for synchronous, asynchronous and spontaneous neurotransmitter release. Annu. Rev. Physiol. 76, 333–363. doi: 10.1146/annurev-physiol-021113-170338
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Kandel, E. R. (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2. and CPEB. Mol. Brain 5:14. doi: 10.1186/1756-6606-5-14
Kandel, E. R., Dudai, Y., and Mayford, M. R. (2014). The molecular and systems biology of memory. Cell 157, 163–186. doi: 10.1016/j.cell.2014.03.001
Kastner, D. B., Schwalger, T., Ziegler, L., and Gerstner, W. (2016). A model of synaptic reconsolidation. Front. Neurosci. 10:206. doi: 10.3389/fnins.2016.00206
Kato, H. K., Watabe, A. M., and Manabe, T. (2009). Non-Hebbian synaptic plasticity induced by repetitive postsynaptic action potentials. J. Neurosci. 29, 11153–11160. doi: 10.1523/jneurosci.5881-08.2009
Kauer, J. A., and Malenka, R. C. (2007). Synaptic plasticity and addiction. Nat. Rev. Neurosci. 8, 844–858. doi: 10.1038/nrn2234
Khalaf, O., and Gräff, J. (2016). Structural, synaptic and epigenetic dynamics of enduring memories. Neural Plast. 2016:3425908. doi: 10.1155/2016/3425908
Kim, S., and Kaang, B.-K. (2017). Epigenetic regulation and chromatin remodeling in learning and memory. Exp. Mol. Med. 49:e281. doi: 10.1038/emm.2016.140
Knoblauch, A., and Sommer, F. T. (2016). Structural plasticity, effectual connectivity and memory in cortex. Front. Neurosci. 10:63. doi: 10.3389/fnana.2016.00063
Ko, H. G., Kim, J. I., Sim, S. E., Kim, T., Yoo, J., Choi, S. L., et al. (2016). The role of nuclear PKMZ in memory maintenance. Neurobiol. Learn. Mem. 135, 50–56. doi: 10.1016/j.nlm.2016.06.010
Kouroupi, G., Taoufik, E., Vlachos, I. S., Tsioras, K., Antoniou, N., Papastefanaki, F., et al. (2017). Defective synaptic connectivity and axonal neuropathology in a human Ipsc-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 114, E3679–E3688. doi: 10.1073/pnas.1617259114
Kwapis, J. L., and Helmstetter, F. J. (2014). Does PKMZ maintain memory? Brain Res. Bull. 105, 36–45. doi: 10.1016/j.brainresbull.2013.09.005
Kwon, H. B., and Sabatini, B. L. (2011). Glutamate induces de novo growth of functional spines in developing cortex. Nature 474, 100–104. doi: 10.1038/nature09986
Lansner, A. (2009). Associative memory models: from the cell-assembly theory to biophysically detailed cortex simulations. Trends Neurosci. 32, 178–186. doi: 10.1016/j.tins.2008.12.002
Lara, A. H., and Wallis, J. D. (2015). The role of prefrontal cortex in working memory: a mini review. Front. Syst. Neurosci. 9:173. doi: 10.3389/fnsys.2015.00173
Larkman, A., Hannay, T., Stratford, K., and Jack, J. (1992). Presynaptic release probability influences the locus of long-term potentiation. Nature 360, 70–73. doi: 10.1038/360070a0
Lashley, K. S. (1930). Basic neural mechanisms in behavior. Psychol. Rev. 37, 1–24. doi: 10.1037/h0074134
Lashley, K. S. (1931). Mass action in cerebral function. Science 73, 245–254. doi: 10.1126/science.73.1888.245
Lashley, K. S. (1932). Studies of cerebral function in learning VIII. A reanalysis of data on mass action in the visual cortex. J. Comp. Neurol. 54, 74–84. doi: 10.1002/cne.900540106
Lashley, K. S., and Wade, M. (1946). The Pavlovian theory of generalization. Psychol. Rev. 53, 72–87. doi: 10.1037/h0059999
Le Bé, J. V., and Markram, H. (2006). Spontaneous and evoked synaptic rewiring in the neonatal neocortex. Proc. Natl. Acad. Sci. U S A 103, 13214–13219. doi: 10.1073/pnas.0604691103
Lee, H. K., Barbarosie, M., Kameyama, K., Bear, M. F., and Huganir, R. L. (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959. doi: 10.1038/35016089
Lee, S., Kim, J., and Choi, S. (2011). in vitro synaptic reconsolidation in amygdala slices prepared from rat brains. Biochem. Biophys. Res. Commun. 407, 339–342. doi: 10.1016/j.bbrc.2011.03.019
Lee, J. L. C., Nader, K., and Schiller, D. (2017). An update on memory reconsolidation updating. Trends Cogn. Sci. 21, 531–545. doi: 10.1016/j.tics.2017.04.006
Lee, H. K., Takamiya, K., Han, J. S., Man, H., Kim, C. H., Rumbaugh, G., et al. (2003). Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643. doi: 10.1016/s0092-8674(03)00122-3
Li, M., Liu, J., and Tsien, J. Z. (2016). Theory of connectivity: nature and nurture of cell assemblies and cognitive computation. Front. Neural Circuits 10:34. doi: 10.3389/fncir.2016.00034
Lim, J. P., and Brunet, A. (2013). Bridging the transgenerational gap with epigenetic memory. Trends Genet. 29, 176–186. doi: 10.1016/j.tig.2012.12.008
Lisman, J. (2017a). Glutamatergic synapses are structurally and biochemically complex because of multiple plasiticity processes: long-term potentiation, long-term depression, short-term potentiation and scaling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160260. doi: 10.1098/rstb.2016.0260
Lisman, J. (2017b). Criteria for identifying the molecular basis of the engram (CaMKII, PKMzeta). Mol. Brain 10:55. doi: 10.1186/s13041-017-0337-4
Lisman, J., Cooper, K., Sehgal, M., and Silva, A. J. (2018). Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat. Neurosci. 21, 309–314. doi: 10.1038/s41593-018-0076-6
Lisman, J., Grace, A. A., and Duzel, E. (2011). A neoHebbian framework for episodic memory; role of dopamine-dependent late LTP. Trends Neurosci. 34, 536–547. doi: 10.1016/j.tins.2011.07.006
Liu, K. K. L., Hagan, M. F., and Lisman, J. E. (2017). Gradation (approx. 10 size states) of synaptic strength by quantal addition of structural modules. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160328. doi: 10.1098/rstb.2016.0328
Lorente De Nó, R. (1933). Vestibulo-ocular reflex arc. Arch. Neurol. Psychiatry 30, 245–291. doi: 10.1001/archneurpsyc.1933.02240140009001
Luo, C., Kuner, T., and Kuner, R. (2014). Synaptic plasticity in pathological pain. Trends Neurosci. 37, 343–355. doi: 10.1016/j.tins.2014.04.002
Lüscher, C., and Malenka, R. C. (2012). NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 4:a005710. doi: 10.1101/cshperspect.a005710
Lüscher, C. (2013). Drug-evoked synaptic plasticity causing addictive behavior. J. Neurosci. 33, 17641–17646. doi: 10.1523/JNEUROSCI.3406-13.2013
Madej, M. P., Töpfer, E., Boraschi, D., and Italiani, P. (2017). Different regulation of interleukin-1 production and activity in monocytes and macrophages: innate memory as an endogenous mechanism of IL-1 inhibition. Front. Pharmacol. 8:335. doi: 10.3389/fphar.2017.00335
Markram, H., Gerstner, W., and Sjöström, P. J. (2011). A history of spike-timing-dependent plasticity. Front. Synaptic. Neurosci. 3:4. doi: 10.3389/fnsyn.2011.00004
Marowsky, A., Yanagawa, Y., Obata, K., and Vogt, K. E. (2005). A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron 48, 1025–1037. doi: 10.1016/j.neuron.2005.10.029
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C., and Kasai, H. (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. doi: 10.1038/nature02617
Mayford, M., Siegelbaum, S. A., and Kandel, E. R. (2012). Synapses and memory storage. Cold Spring Harb. Perspect. Biol. 4:a005751. doi: 10.1101/cshperspect.a005751
Mazzio, E. A., and Soliman, K. F. A. (2012). Basic concepts of epigenetics. Epigenetics 7, 119–130. doi: 10.4161/epi.7.2.18764
McKenzie, S., and Eichenbaum, H. (2011). Consolidation and reconsolidation: two lives of memories. Neuron 71, 224–233. doi: 10.1016/j.neuron.2011.06.037
Migues, P. V., Hardt, O., Wu, D. C., Gamache, K., Sacktor, T. C., Wang, Y. T., et al. (2010). PKMZ maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat. Neurosci. 13, 630–634. doi: 10.1038/nn.2531
Miller, G. A., Galanter, E., and Pribram, K. H. (1960). Plans and the Structure of Behavior. New York, NY: Holt, Rinehart and Winston, Inc.
Miyamoto, K., Osada, T., and Adachi, Y. (2014). Remapping of memory encoding and retrieval networks: insights from neuroimaging in primates. Behav. Brain Res. 275, 53–61. doi: 10.1016/j.bbr.2014.08.046
Miyashita, T., Kubik, S., Haghighi, N., Steward, O., and Guzowski, J. F. (2009). Rapid activation of plasticity-associated gene transcription in hippocampal neurons provides a mechanism for encoding of one-trial experience. J. Neurosci. 29, 898–906. doi: 10.1523/JNEUROSCI.4588-08.2009
Moretto, E., Murru, L., Martano, G., Sassone, J., and Passafaro, M. (2018). Glutamatergic synapses in neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 84, 328–342. doi: 10.1016/j.pnpbp.2017.09.014
Mozzachiodi, R., and Byrne, J. H. (2010). More than synaptic plasticity: role of nonsynaptic plasticity in learning and memory. Trends Neurosci. 33, 17–26. doi: 10.1016/j.tins.2009.10.001
Munakata, Y., and Pfaffly, J. (2004). Hebbian learning and development. Dev. Sci. 7, 141–148. doi: 10.1111/j.1467-7687.2004.00331.x
Munton, R. P., Vizi, S., Isabelle, M., and Mansuy, M. (2004). The role of protein phosphtase-1 in the modulation of synaptic and structural plasticity. FEBS Lett. 567, 121–128. doi: 10.1016/s0014-5793(04)00506-x
Nabavi, S., Fox, R., Proulx, C. D., Lin, J. Y., Tsien, R. Y., and Malinow, R. (2014). Engineering a memory with LTD and LTP. Nature 511, 348–352. doi: 10.1038/nature13294
Neves, G., Cooke, S. F., and Bliss, T. V. P. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75. doi: 10.1038/nrn2303
O’Neil, E. B., Protzner, A. B., McCormick, C., McLean, D. A., Poppenk, J., Cate, A. D., et al. (2012). Distinct patterns of functional and effective connectivity between perirhinal cortex and other cortical regions in recognition memory and perceptual discrimination. Cereb. Cortex 22, 74–85. doi: 10.1093/cercor/bhr075
Oberauer, K., Jones, T., and Lewandowsky, S. (2015). The Hebb repetition effect in simple and complex memory span. Mem. Cognit. 43, 852–865. doi: 10.3758/s13421-015-0512-8
Padamsey, Z., and Emptage, N. (2013). Two sides to long-term potentiation: a view towards reconciliation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130154. doi: 10.1098/rstb.2013.0154
Palm, G., Knoblauch, A., Hauser, F., and Schuz, A. (2014). Cell assemblies in the cerebral cortex. Biol. Cybern. 108, 559–572. doi: 10.1007/s00422-014-0596-4
Pavlov, I. P. (1927). Conditioned Reflexes: An Investigation of the Physiological Activity of the Cerebral Cortex. London: Oxford University Press.
Pavlov, I. P. (1928a). Croonian lecture: certain problems in the physiology of the cerebral hemispheres. Philos. Trans. R. Soc. Lond. B Biol. Sci. 103, 97–110. doi: 10.1098/rspb.1928.0027
Pavlov, I. P. (1928b). Lectures on Conditioned Reflexes: Twenty-Five Years of Objective Study of the Higher Nervous Activity (Behavior) of Animals. Vol.1. Edited and Translated by W. Horsley Gantt. New York, NY: International Publishers.
Pavlov, I. P. (1932). The reply of a physiologist to psychologists. Psychol. Rev. 39, 91–127. doi: 10.1037/h0069929
Pawlak, V., Wickens, J. R., Kirkwood, A., and Kerr, J. N. (2010). Timing is not everything: neuromodulation opens the STDP gate. Front. Synaptic. Neurosci. 2:146. doi: 10.3389/fnsyn.2010.00146
Pettem, K. L., Yokomaku, D., Takahashi, H., Ge, Y., and Craig, A. M. (2013). Interaction between autism-linked MDGAs and neuroligins suppresses inhibitory synapse development. J. Cell Biol. 200, 321–336. doi: 10.1083/jcb.201206028
Piochon, C., Kruskal, P., MacLean, J., and Hansel, C. (2013). Non-Hebbian spike-timing-dependent plasticity in cerebellar circuits. Front. Neural Circuits 6:124. doi: 10.3389/fncir.2012.00124
Poo, M. M., Pignatelli, M., Ryan, T. J., Tonegawa, S., Bonhoeffer, T., Martin, K. C., et al. (2016). What is memory? The present state of the engram. BMC Biol. 14:40. doi: 10.1186/s12915-016-0261-6
Posner, M. I., and Rothbart, M. K. (2004). Hebb’s neural networks support the integration of psychological science. Can. Psychol. 45, 265–278. doi: 10.1037/h0086997
Posner, M. I., and Rothbart, M. K. (2007). Research on attention networks as a model for the integration of psychological science. Annu. Rev. Psychol. 58, 1–23. doi: 10.1146/annurev.psych.58.110405.085516
Prieto, G. A., and Cotman, C. W. (2017). Cytokines and cytokine networks target neurons to modulate long-term potentiation. Cytokine Growth Factor Rev. 34, 27–33. doi: 10.1016/j.cytogfr.2017.03.005
Rajasethupathy, P., Antonov, I., Sheridan, R., Frey, S., Sander, C., Tuschl, T., et al. (2012). A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149, 693–707. doi: 10.1016/j.cell.2012.02.057
Ramirez, S., Liu, X., Lin, P. A., Suh, J., Pignatelli, M., Redondo, R. L., et al. (2013). Creating a false memory in the hippocampus. Science 341, 387–391. doi: 10.1126/science.1239073
Regehr, W. G. (2012). Short-term presynaptic plasticity. Cold Spring Harb. Perspect. Biol. 4:a005702. doi: 10.1101/cshperspect.a005702
Reizel, Y., Spiro, A., Sabag, O., Skversky, Y., Hecht, M., Keshet, I., et al. (2015). Gender-specific postnatal demethylation and establishment of epigenetic memory. Genes Dev. 29, 923–933. doi: 10.1101/gad.259309.115
Rekart, J. L., Sandoval, C. J., Bermudez-Rattoni, F., and Routtenberg, A. (2007). Remodeling of hippocampal mossy fibers is selectively induced seven days after the acquisition of a spatial but not a cued reference memory task. Learn. Mem. 14, 416–421. doi: 10.1101/lm.516507
Rodrigues, E. M., Scudder, S. L., Goo, M. S., and Patrick, G. N. (2016). Aβ-induced synaptic alterations require the E3 ubiquitin ligase Nedd4–1. J. Neurosci. 36, 1590–1595. doi: 10.1523/JNEUROSCI.2964-15.2016
Rosales-Reynoso, M. A., Ochoa-Hernández, A. B., Juárez-Vázquez, C. I., and Barros-Núñez, P. (2016). Epigenetic mechanisms in the development of memory and their involvement in certain neurological diseases. Neurologia 31, 628–638. doi: 10.1016/j.nrl.2014.02.004
Sacktor, T. C. (2008). PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog. Brain Res. 169, 27–40. doi: 10.1016/s0079-6123(07)00002-7
Saucier, D., and Cain, D. P. (1995). Spatial learning without NMDA receptor-dependent long-term potentiation. Nature 378, 186–189. doi: 10.1038/378186a0
Schrader, L. A., Anderson, A. E., Varga, A. W., Levy, M., and Sweatt, J. D. (2002). The other half of Hebb: K+ channels and the regulation of neuronal excitability in the hippocampus. Mol. Neurobiol. 25, 51–66. doi: 10.1385/MN:25:1:051
Sehgal, M., Song, C., Ehlers, V. L., and Moyer, J. R. Jr. (2013). Learning to learn - intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol. Learn. Mem. 105, 186–199. doi: 10.1016/j.nlm.2013.07.008
Sehgal, M., Zhou, M., Lavi, A., Huang, S., Zhou, Y., and Silva, A. J. (2018). Memory allocation mechanisms underlie memory linking across time. Neurobiol. Learn. Mem. 153, 21–25. doi: 10.1016/j.nlm.2018.02.021
Sheng, M., Sabatini, B. L., and Südhof, T. C. (2012). Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 4:a005777. doi: 10.1101/cshperspect.a005777
Sola, E., Prestori, F., Rossi, P., Taglietti, V., and D’Angelo, E. (2004). Increased neurotransmitter release during long-term potentiation at mossy fibre-granule cell synapses in rat cerebellum. J. Physiol. 557, 843–861. doi: 10.1113/jphysiol.2003.060285
Song, C., Phillips, A. G., and Leonard, B. (2003). Interleukin 1 β enhances conditioned fear memory in rats: possible involvement of glucocorticoids. Eur. J. Neurosci. 18, 1739–1743. doi: 10.1046/j.1460-9568.2003.02886.x
Takeuchi, T., Duszkiewicz, A. J., and Morris, R. G. M. (2014). The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130288. doi: 10.1098/rstb.2013.0288
Takeuchi, T., Duszkiewicz, A. J., Sonneborn, A., Spooner, P. A., Yamasaki, M., Watanabe, M., et al. (2016). Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 537, 357–362. doi: 10.1038/nature19325
Takeuchi, N., and Izumi, S. (2015). Combinations of stroke neurorehabilitation to facilitate motor recovery: perspectives on Hebbian plasticity and homeostatic metaplasticity. Front. Hum. Neurosci. 9:349. doi: 10.3389/fnhum.2015.00349
Titley, H. K., Brunel, N., and Hansel, C. (2017). Toward a neurocentric niew of learning. Neuron 95, 19–32. doi: 10.1016/j.neuron.2017.05.021
Torres, V. I., Vallejo, D., and Inestrosa, N. C. (2017). Emerging synaptic molecules as candidates in the etiology of neurological disorders. Neural Plast. 2017:8081758. doi: 10.1155/2017/8081758
Trettenbrein, P. C. (2016). The demise of the synapse as the locus of memory: a looming paradigm shift? Front. Syst. Neurosci. 10:88. doi: 10.3389/fnsys.2016.00088
Trollope, A. F., Gutièrrez-Mecinas, M., Mifsud, K. R., Collins, A., Saunderson, E. A., and Reul, J. M. H. M. (2012). Stress, epigenetic control of gene expression and memory formation. Exp. Neurol. 233, 3–11. doi: 10.1016/j.expneurol.2011.03.022
Tuesta, L. M., and Zhang, Y. (2014). Mechanisms of epigenetic memory and addiction. EMBO J. 33, 1091–1103. doi: 10.1002/embj.201488106
Van Ooyen, A., and Butz-Ostendorf, M. (2017). The Rewiring Brain: A Computational Approach to Structural Plasticity in the Adult Brain. New York, NY: Academic Press.
Vickers, C. A., and Wyllie, D. J. A. (2007). Late-phase, protein synthesis-dependent long-term potentiation in hippocampal CA1 pyramidal neurones with destabilized microtubule networks. Br. J. Pharmacol. 151, 1071–1077. doi: 10.1038/sj.bjp.0707314
Vogel-Ciernia, A., and Wood, M. A. (2014). Neuron-specific chromatin remodeling: a missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 80, 18–27. doi: 10.1016/j.neuropharm.2013.10.002
Volianskis, A., Collingridge, G. L., and Jensen, M. S. (2013). The roles of STP and LTP in synaptic encoding. PeerJ 1:e3. doi: 10.7717/peerj.3
Volianskis, A., and Jensen, M. S. (2003). Transient and sustained types of long-term potentiation in the CA1 area of the rat hippocampus. J. Physiol. 550, 459–492. doi: 10.1113/jphysiol.2003.044214
Wallace, D. J., and Kerr, J. N. (2010). Chasing the cell assembly. Curr. Opin. Neurobiol. 20, 296–305. doi: 10.1016/j.conb.2010.05.003
Wang, X. J. (2005). Discovering spatial working memory fields in prefrontal cortex. J. Neurophysiol. 93, 3027–3028. doi: 10.1152/classicessays.00028.2005
Wang, Y., Wu, T., Orchard, G., Dudek, P., Rucci, M., and Shi, B. E. (2009). “Hebbian learning of visually directed reaching by a robot arm,” in Biomedical Circuits and Systems Conference (Beijing: BioCAS IEEE), 28–29.
Wang, X. J. (2012). Neural dynamics and circuit mechanisms of decision-making. Curr. Opin. Neurobiol. 22, 1039–1046. doi: 10.1016/j.conb.2012.08.006
Weintraub, S., Wicklund, A. H., and Salmon, D. P. (2012). The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006171. doi: 10.1101/cshperspect.a006171
Wennekers, T. (2007). A cell assembly model for complex behaviour. Neurocomputing 70, 1988–1992. doi: 10.1016/j.neucom.2006.10.079
Wennekers, T., Garagnani, M., and Pulvermüller, F. (2006). Language models based on Hebbian cell assemblies. J. Physiol. Paris 100, 16–30. doi: 10.1016/j.jphysparis.2006.09.007
Whitlock, J. R., Heynen, A. J., Shuler, M. G., and Bear, M. F. (2006). Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097. doi: 10.1126/science.1128134
Windholz, G. (1995). Pavlov on the conditioned reflex method and its limitations. Am. J. Psychol. 108, 575–588. doi: 10.2307/1423074
Wong, S. T., Athos, J., Figueroa, X. A., Pineda, V. V., Schaefer, M. L., Chavkin, C. C., et al. (1999). Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23, 787–798. doi: 10.1016/s0896-6273(01)80036-2
Xu, T., Yu, X., Perlik, A. J., Tobin, W. F., Zweig, J. A., Tennant, K., et al. (2009). Rapid formation and selective stabilization of synapses for enduring memories. Nature 462, 915–919. doi: 10.1038/nature08389
Yang, Y., and Calakos, N. (2013). Presynaptic long-term plasticity. Front. Synaptic. Neurosci. 5:8. doi: 10.3389/fnsyn.2013.00008
Yang, Y., Liu, D. Q., Huang, W., Deng, J., Sun, Y., Zuo, Y., et al. (2016). Selective synaptic remodeling of amygdalocortical connections associated with fear memory. Nat. Neurosci. 19, 1348–1355. doi: 10.1038/nn.4370
Yang, T., Li, S., Xu, H., Walsh, D. M., and Selkoe, D. J. (2017). Large soluble oligomers of amyloid ß-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J. Neurosci. 37, 152–163. doi: 10.1523/JNEUROSCI.1698-16.2016
Yang, G., Pan, F., and Gan, W. B. (2009). Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920–924. doi: 10.1038/nature08577
Yang, S., Weiner, B. D., Zhang, L. S., Cho, S. J., and Bao, S. (2011). Homeostatic plasticity drives tinnitus perception in an animal model. Proc. Natl. Acad. Sci. U S A 108, 14974–14979. doi: 10.1073/pnas.1107998108
Yao, Y., Kelly, M. T., Sajikumar, S., Serrano, P., Tian, D., Bergold, P. J., et al. (2008). PKMζ maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J. Neurosci. 28, 7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008
Keywords: Hebb, memory, synaptic theory, molecular mechanisms, epigenetics, neurological disorders
Citation: Langille JJ and Brown RE (2018) The Synaptic Theory of Memory: A Historical Survey and Reconciliation of Recent Opposition. Front. Syst. Neurosci. 12:52. doi: 10.3389/fnsys.2018.00052
Received: 22 May 2018; Accepted: 28 September 2018;
Published: 26 October 2018.
Edited by:
Federico Bermudez-Rattoni, Universidad Nacional Autónoma de México, MexicoReviewed by:
Jorge Medina, Universidad de Buenos Aires, ArgentinaJames W. Grau, Texas A&M University, United States
Copyright © 2018 Langille and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard E. Brown, cmVicm93bkBkYWwuY2E=
† Present address: Jesse J. Langille, McGill University, Montreal, QC, Canada