


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Syst. Neurosci. , 08 May 2012
Volume 6 - 2012 | https://doi.org/10.3389/fnsys.2012.00034
This article is part of the Research Topic Ringing ears: the neuroscience of tinnitus View all 23 articles
The understanding of tinnitus has progressed considerably in the past decade, but the details of the mechanisms that give rise to this phantom perception of sound without a corresponding acoustic stimulus have not yet been pinpointed. It is now clear that tinnitus is generated in the brain, not in the ear, and that it is correlated with pathologically altered spontaneous activity of neurons in the central auditory system. Both increased spontaneous firing rates and increased neuronal synchrony have been identified as putative neuronal correlates of phantom sounds in animal models, and both phenomena can be triggered by damage to the cochlea. Various mechanisms could underlie the generation of such aberrant activity. At the cellular level, decreased synaptic inhibition and increased neuronal excitability, which may be related to homeostatic plasticity, could lead to an over-amplification of natural spontaneous activity. At the network level, lateral inhibition could amplify differences in spontaneous activity, and structural changes such as reorganization of tonotopic maps could lead to self-sustained activity in recurrently connected neurons. However, it is difficult to disentangle the contributions of different mechanisms in experiments, especially since not all changes observed in animal models of tinnitus are necessarily related to tinnitus. Computational modeling presents an opportunity of evaluating these mechanisms and their relation to tinnitus. Here we review the computational models for the generation of neurophysiological correlates of tinnitus that have been proposed so far, and evaluate predictions and compare them to available data. We also assess the limits of their explanatory power, thus demonstrating where an understanding is still lacking and where further research may be needed. Identifying appropriate models is important for finding therapies, and we therefore, also summarize the implications of the models for approaches to treat tinnitus.
Over the last two decades, our understanding of tinnitus has increased greatly through the results of animal models of tinnitus. As tinnitus can often be related to cochlear damage, animal models have used acoustic trauma or ototoxic drugs to induce hearing loss and to study changes in the central auditory system. After hearing loss, a variety of changes that could contribute to tinnitus have been observed; most notably, increased spontaneous neuronal activity throughout the central auditory system (see Kaltenbach, 2011, for a review), but not at the level of the auditory nerve (AN) (Liberman and Dodds, 1984; Heinz and Young, 2004). Importantly, increases in spontaneous firing rates were correlated to behavioral signs of tinnitus in animals (Brozoski et al., 2002; Kaltenbach et al., 2004; Middleton et al., 2011), and they have been linked to decreases in inhibition (Dong et al., 2009; Middleton et al., 2011). Also an increase in the synchrony of the neuronal discharge has been observed in the auditory cortex after noise trauma (Norena and Eggermont, 2003). Furthermore, a reorganization of the tonotopic map in the auditory cortex has also been found after hearing loss (Rajan and Irvine, 1998; Rauschecker, 1999; Irvine et al., 2000; Komiya and Eggermont, 2000).
Human neuroimaging studies on tinnitus have also shown changes in spontaneous neuronal activity (Weisz et al., 2005, 2007; Lorenz et al., 2009) where spontaneous rhythmic brain activity displayed a decrease in power in the alpha band and increases in power in the delta and gamma frequency bands. Moreover, an association between tinnitus and reorganization of the tonotopic map in the auditory cortex has been reported (Mühlnickel et al., 1998). These studies in humans and animals show that tinnitus is not generated in the ear, but in the brain itself (Eggermont and Roberts, 2004; Roberts et al., 2010). However, the exact mechanisms that lead to the development of this phantom sensation have still remained elusive.
Further progress in understanding tinnitus has been made using computational models, which are the main topic of this review. Such models, also called “theories” or “hypotheses,” provide a motivational and interpretational framework for possibly diverse sets of data, and, ideally explain how the data fit together to yield a more complete understanding of tinnitus. Because the data on which models are based are from different levels, i.e., from the microscopic molecular and single-neuron level to the macroscopic levels of large-scale brain signals and behavior, computational models must capture these different levels to be in line with salient features of the respective data. Therefore, modeling at different levels is justified, but one also needs models that bridge across levels. Such models could also link research across different fields, for example research on humans and animals or research in vivo and in vitro.
However, not all models are equally valuable. To outline some basic criteria for “good” models, let us summarize some general principles, which might help to justify our selection of models in this review. Regardless of the level of modeling, a model should always be as simple as possible and be based on as few reasonable assumptions as is feasible. This rule determines the predictive power of a model, i.e., its ability to generate testable predictions on the outcome of future experiments. One such example could be how a specific type of hearing loss determines the tinnitus pitch or the tinnitus spectrum. Good models are falsifiable, and progress in understanding is closely related to ruling out models. Therefore, models should be quantitative and tell us how large a new effect should be, for example, what the loudness of the tinnitus is. Because of this need for verifiability, we have excluded qualitative models from this review. Quantitative or “computational” models also might permit a mathematical analysis and facilitate numerical simulations on a computer. In the following, we will evaluate each reviewed model based on these criteria.
The appropriate level of detail for a model is, in general, a highly controversial issue. An oversimplified or abstract model may ignore many experimental details and could provide misleading results; on the other hand, an excessively complex model can reproduce many experimental results but may lack predictive power because of too many unconstrained or “free” parameters and too many ad-hoc assumptions. The adequate detail of modeling, therefore, strongly depends on available data, i.e., on the physiology of the auditory system in general, and on tinnitus in particular. Interestingly, the available computational models on tinnitus, which are all described below, are rather more abstract than detailed.
In summary, a computational model of tinnitus should (1) explain how neural correlates of tinnitus could arise, (2) outline which mechanisms might be involved, and (3) predict how the processes that give rise to tinnitus can be suppressed or reversed. Before we summarize the available computational models of tinnitus, let us briefly introduce the basic mechanisms employed by these models.
Three main mechanisms have been explored in computational models of tinnitus: lateral inhibition, homeostatic plasticity, and gain adaptation.
Lateral inhibition is inhibition between neighboring neurons in a neuronal structure (Figure 1A). Functionally, in the auditory system it also means inhibition between frequency channels, i.e., inhibition between neurons whose characteristic frequency (CF) is close, but not identical. Lateral inhibition is ubiquitous in the brain, and it is assumed to be a basic mechanism of information processing in neural circuits. Lateral inhibition also plays a role in keeping neural networks balanced, and it can enhance the activity difference between neurons with high and low levels of activity in a neuronal network (Figures 1B,C). One example of lateral inhibition in the auditory system is its involvement in the sharpening of receptive fields. Lateral inhibition has been found at all central processing stages from the cochlear nucleus (Roberts and Trussell, 2010) to the auditory cortex (de la Rocha et al., 2008).
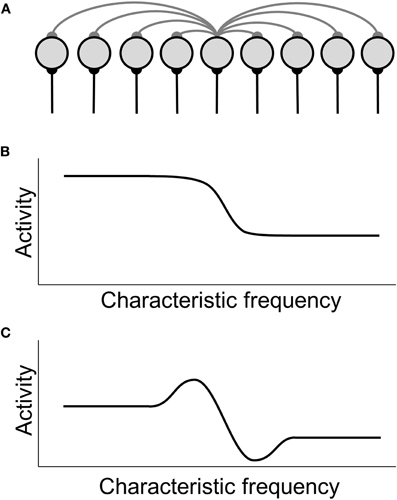
Figure 1. Schematic illustration of lateral-inhibition models. (A) Depiction of a layer of neurons with lateral inhibition. Neurons are represented by gray circles, lateral inhibitory connections by gray lines (only the inhibitory projections from the central neuron to its neighbors are shown), and excitatory afferents by black lines. (B) Hypothetical auditory activity pattern with a drop toward high frequencies, as it could for example occur in the spontaneous activity of the auditory nerve after noise-induced hearing loss. (C) Activity pattern in the lateral-inhibition network driven by the input shown in (B). An activity peak is generated at the edge of the input pattern but below the region of hearing loss.
Homeostatic plasticity is a plasticity mechanism that stabilizes the mean activity of neurons on time scales of hours to days (Turrigiano et al., 1998; Turrigiano, 1999). This mechanism sets the basic operating point of neurons and ensures that neurons are neither inactive nor too active when averaged over time windows of hours to days. In cell culture, homeostatic plasticity in response to activity deprivation has been shown to scale up the strength of excitatory synapses (Turrigiano et al., 1998) and increase intrinsic neuronal excitability (Desai et al., 1999), while the strength of inhibitory synapses was scaled down (Kilman et al., 2002). On the other hand, when activity was pathologically increased by blocking inhibition, excitatory synapses were scaled down, neuronal excitability was decreased, and inhibition was scaled up, which restored circuit activity back to a normal level (Turrigiano et al., 1998; Rannals and Kapur, 2011). Similar changes have also been observed in the auditory system in vivo after hearing loss, cochlear damage, or auditory deprivation (Suneja et al., 1998a,b; Oleskevich and Walmsley, 2002; Vale and Sanes, 2002; Muly et al., 2004; Vale et al., 2004; Caspary et al., 2005; Kotak et al., 2005; Whiting et al., 2009).
Gain adaptation adjusts the responses of single-neurons or neuronal circuits to their input, thus enabling neurons to cope with the wide dynamic range of natural signals. Gain adaptation occurs at various stages of the auditory pathway. Fast gain adaptation on the time scale of seconds has been observed for example in the AN (Wen et al., 2009) and the inferior colliculus (IC) (Dean et al., 2005, 2008). Such fast adaptation phenomena are usually caused by the activation of adaptation currents (Benda and Herz, 2003). Slower adaptation mechanisms on a time scale of minutes can involve modulation of ion channels (van Welie et al., 2004). On longer time scales of hours to days, gain adaptation can be seen as functionally equivalent to homeostatic plasticity.
In the following section, we present the main features of the computational models of neurophysiological correlates of tinnitus that have been proposed so far. As already mentioned, we have excluded qualitative models because they do not give rise to detailed predictions, which makes them hard to falsify. The remaining quantitative models are grouped by mechanisms and then presented in chronological order, to highlight the development of the different concepts.
The first computational model that addressed the question of how a neural correlate of tinnitus could arise in the central auditory system after noise-induced damage to structures of the inner ear was the auditory brainstem model proposed by Gerken (1996). In this model, lateral inhibition was the key mechanism responsible for generating a tinnitus-related pattern of neuronal activity. Another basic model assumption was that after noise-induced cochlear damage, the spontaneous activity of AN fibers is reduced in the high-frequency range, creating a drop in the profile of spontaneous activity along the tonotopic axis. The drop starts at CFs corresponding to the audiogram edge. When such a pattern of activity is processed by a neuronal structure with lateral inhibition, the neurons just below and at the edge receive less lateral inhibition than their counterparts at lower frequencies. In contrast, the neurons just above the edge receive more lateral inhibition than the other neurons with higher CFs. As a consequence, the edge in the activity profile is amplified, leading to a peak in the profile of spontaneous activity (Figure 1C). When this activity peak is interpreted by higher stages of the auditory system as sound-evoked activity, a tinnitus sensation is created. In the Gerken-model, lateral inhibition was assumed to occur at the level of the IC, and the model employed a feed-forward architecture. However, as the circuit of the IC was not explicitly modeled, the model can rather be seen as a generic demonstration of the effects of lateral inhibition. Moreover, Gerken did not assume plastic changes to take place in the auditory system after hearing loss. The model's achievement was to demonstrate that even though there is no direct indication of a neural correlate of tinnitus at the level of the AN, central processing of distorted AN output could give rise to tinnitus-related patterns of spontaneous activity. The basic prediction following from this and all other lateral inhibition models is that tinnitus will emerge almost instantaneously when the profile of spontaneous activity is changed by hearing loss, as no plastic changes are required. The resulting tinnitus pitch will be associated with the audiogram edge.
Also in 1996, lateral inhibition was proposed as a key factor to explain why most people start hearing phantom sounds after spending some time in a sound-proof booth (Kral and Majernik, 1996). Kral and Majernik used a neural network model with several layers, each with lateral inhibition, and they assumed stochastic spontaneous activity, such as the spontaneous activity of AN fibers, as an input to the network. Processing of this spontaneous activity by the feed-forward network with lateral inhibition resulted in several distinct activity peaks along the tonotopic axis, and the peaks occurred at random locations. Kral and Majernik proposed that these activity peaks could underlie the perception of tinnitus in absolute silence, and that in normal acoustic environments, the spontaneous activity is masked by ambient noise. Whether this mechanism could also account for the emergence of tinnitus after hearing loss was not investigated, but in principle the predictions of this model should match those of the Gerken-model.
Lateral inhibition was combined with plasticity in the central auditory system by Langner and Wallhäusser-Franke (1999). The model was set up as a multi-layer feed-forward network with lateral inhibition representing processing in the auditory brainstem and midbrain, with additional modulatory inputs representing feedback from the auditory cortex and amygdala. Specific details of the auditory brainstem and midbrain circuitry were omitted for simplicity. Their model was inspired by c-fos labeling data showing increased activity correlations between the auditory and the limbic system after salicylate administration. In the model, lateral inhibition in the first processing stages amplified unevenness in the tonotopic profile of spontaneous activity, which was caused by cochlear damage. As to be expected for a lateral inhibition model, the resulting activity peaks were located close to the edge of hearing loss, as it was there that the contrast between the output of the undamaged and the damaged parts of the cochlea produced the greatest unevenness in the spontaneous activity. The activity peaks were then further amplified by positive feedback at higher stages, which was attributed to the action of the auditory cortex and the amygdala. This feedback elevated the activity peaks substantially above the level of spontaneous activity, possibly generating a highly salient tinnitus percept.
The putative role of lateral inhibition in generating tinnitus-related patterns of neural activity was further explored by Bruce et al. (2003). They showed that also in a recurrent network of spiking model neurons, lateral inhibition could generate an activity peak at the edge of hearing loss. However, they also found that the generation of such a peak depended not only on the contrast between the levels of spontaneous activity in the healthy and the damaged region, but also on the overall level of input received by the network, and the time constants of excitation and inhibition. The time constants needed to be long enough, and the input rates high enough, so that an interaction between excitatory and inhibitory postsynaptic potentials could take place. For low input rates to the network, the enhancement of the edge was not significant.
The development of neural correlates of tinnitus at the level of the auditory cortex has been explored in a model by Dominguez et al. (2006). This model comprised a network of spiking model neurons with pyramidal cells and inhibitory interneurons. The thalamic stage was modeled through a network with lateral inhibition as employed by Bruce et al. (2003), and the network received random afferent input. Hearing loss was modeled by decreasing the firing rate of the inputs to the thalamus stage. After simultaneously increasing the strength of lateral excitatory connections and decreasing the strength of lateral inhibitory connections in the cortical network model in the region affected by the hearing loss, pyramidal neurons displayed increased spontaneous firing rates and increased synchrony. Additionally, the network displayed an activity peak in the region of hearing loss. Without this plasticity, the peak was located below the edge of hearing loss. Thus, the model by Dominguez et al. (2006) demonstrated that decreases in inhibition and increases in excitation, as observed in animals after the induction of hearing loss, can lead to the development of a neural correlate of tinnitus in a recurrent neuronal network. Moreover, if the peak in the profile of spontaneous activity was interpreted as the dominant tinnitus pitch, the model would predict a tinnitus pitch in the region of hearing loss.
How gain adaptation in the auditory system might give rise to the perception of phantom sounds was addressed by Parra and Pearlmutter (2007). They considered an abstract model organized in frequency channels. Gain adaptation was implemented by calculating a running average of input activity for each channel, which was then used as a normalization factor to scale the channel's output. If a channel did not receive input, for example due to hearing loss, its average input activity was close to the neuronal noise level (i.e., spontaneous activity), and the normalization factor was low. As a consequence, the output of this channel was scaled-up. Because also the spontaneous input activity was scaled by the low normalization factor, it was effectively amplified, leading to increased spontaneous activity in the output, which was interpreted as tinnitus. In addition to gain adaptation, Parra and Pearlmutter also analyzed the effects of lateral inhibition in their model. They showed that lateral inhibition combined with a steep audiogram slope could lead to a pronounced “tinnitus” peak in the profile of spontaneous activity. However, shallow audiogram slopes did not produce such peaks, which matched the experimental finding that for noise-induced hearing loss, tinnitus is associated with steep audiogram slopes (König et al., 2006). The pitch of the model tinnitus was then located in the region of hearing loss, at the “elbow” of the audiogram where hearing loss has reached a plateau.
Functional mechanisms to explain changes in excitation and inhibition after hearing loss and how these changes are connected to the development of tinnitus were studied by Schaette and Kempter (2006, 2008, 2009) in a model based on the physiology of the AN and the cochlear nucleus. This computational model showed that activity stabilization through homeostatic plasticity after hearing loss could account for changes in excitation and inhibition as well as for the development of increased spontaneous firing rates. The model assumed that hearing loss reduces AN activity with a concomitant reduction in excitatory drive to the central auditory system (Figure 2A). In order to stabilize mean neuronal activity, homeostatic plasticity then generated increased excitatory gain and reduced inhibitory gain in neurons downstream of the AN, restoring average neuronal activity to normal levels. However, as neurons became more excitable, spontaneous activity was amplified, leading to neuronal hyperactivity, which was interpreted as a tinnitus percept (Figure 2B). The model thus suggested that tinnitus could be an unwanted side-effect of a stabilization of neuronal activity levels in the central auditory system after hearing loss. Tinnitus pitch predicted from the audiograms of patients with noise-induced hearing loss was located in the region of hearing loss (Schaette and Kempter, 2009). Interestingly, in the model not all types and degrees of cochlear damage increased central spontaneous activity to comparable degrees. Loss of outer hair cells and moderate noise damage led to the greatest increases in spontaneous firing rates whereas inner hair cell loss and severe noise damage could even cause spontaneous firing rates to decrease (Schaette and Kempter, 2006, 2008). Moreover, different response types of model DCN projection neurons differed in their disposition for hyperactivity (Schaette and Kempter, 2008), indicating that not all central neurons might necessarily develop increased spontaneous firing rates after hearing loss.
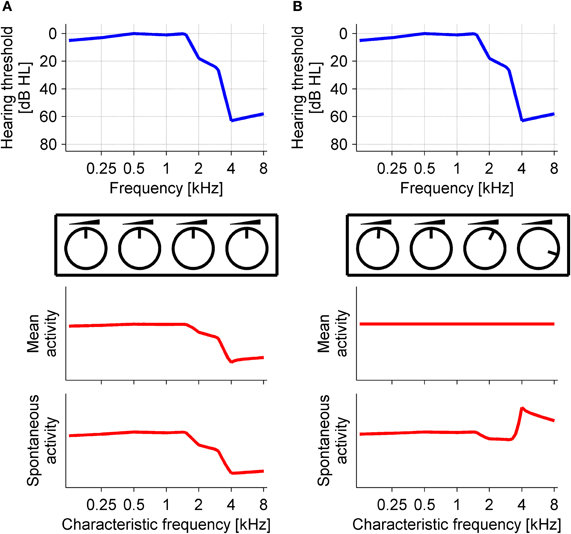
Figure 2. Schematic illustration of homeostatic plasticity models. The “knobs” represent the effective response gain of neurons in the central auditory system, determined by the strength of excitatory and inhibitory synapses as well as intrinsic neuronal excitability. (A) Before homeostatic plasticity: noise-induced hearing loss (example audiogram in the top panel) has reduced mean and spontaneous activity in the central auditory system (bottom panels). (B) After homeostatic plasticity: the response gain has been increased to restore the mean activity of central auditory neurons back to its target level. However, spontaneous activity is amplified through the increased gain, giving rise to increased spontaneous firing rates in the region of hearing loss.
The effects of homeostatic plasticity were also studied in the cortex-based model of Chrostowski et al. (2011), which built up on the model of Dominguez et al. (2006). As in the earlier model, they considered a network of spiking model neurons based on features of the auditory cortex, but only a simplified thalamic stage without lateral inhibition. The activity of the pyramidal cells of the cortical network was stabilized by homeostatic plasticity. When hearing loss was induced in the model by decreasing the activity of thalamic afferents, homeostatic plasticity increased the strength of excitatory projections onto the pyramidal neurons and decreased the strength of the inhibitory synapses. These changes lead to a combination of increased spontaneous firing rates and increased synchrony of the neuronal discharge in the pyramidal neurons of the model network. Interestingly, while the increase in spontaneous firing rates was rather uniform across the range of hearing loss, the greatest increase in synchrony occurred near the audiogram edge. This synchrony maximum was restricted to a relatively narrow range of CFs, and could thus be interpreted as giving rise to a tone-like tinnitus sensation, even though the hearing loss and the increases in spontaneous activity spanned a large frequency range. Moreover, the model also displayed traveling waves of excitation, which confirmed another study on homeostatic plasticity in cortical network models (Houweling et al., 2005).
The majority of the computational models of tinnitus employed firing-rate-based frameworks. Spiking neurons can be considered to represent a higher degree of biological realism, but it should be noted that in all studies the choice of model neurons corresponded to the type of neuronal data that was to be modeled: changes in the synchrony of the spontaneous neuronal activity, i.e., a measure where the timing of action potentials is important, have only been investigated in the auditory cortex, whereas the putative neuronal correlates of tinnitus in subcortical stages have only been reported in terms of average firing rates. Consequently, all models based on the auditory brainstem are firing-rate models (Gerken, 1996; Schaette and Kempter, 2006, 2008, 2009), and the cortex-based models employ spiking neurons (Dominguez et al., 2006; Chrostowski et al., 2011).
Models based on firing rates and on spikes provided similar results regarding the role of lateral inhibition, which basically amplifies edges. However, the spiking neuronal network by Bruce et al. (2003) highlighted an additional potential dependence on neuronal properties, i.e., the interplay between the effects of lateral inhibition and synaptic time constants, which was not apparent in the firing-rate models. For homeostatic plasticity, on the other hand, qualitatively similar results were obtained for feed-forward firing-rate and recurrent spiking models, demonstrating the robustness of the mechanism.
The models that refer to specific brain structures are phenomenological models that only contain simplified versions of the neuronal circuits they are representing (Dominguez et al., 2006; Schaette and Kempter, 2006, 2008, 2009; Chrostowski et al., 2011). The remaining more generic models are not based on a specific brain structure in the first place (Gerken, 1996; Kral and Majernik, 1996; Bruce et al., 2003; Parra and Pearlmutter, 2007). An evaluation of the effects of different kinds of cochlear damage beyond a mere threshold increase or simple activity reduction was only performed by Schaette and Kempter (2006, 2008, 2009). Thus, we can safely conclude that none of the models contains unnecessary detail. Moreover, all models are simple enough to be fully tractable, and they are also specific enough in their structure, assumptions, and predictions to be testable and falsifiable. In the following, we will discuss the implications of the models for putative mechanisms of tinnitus generation by comparing their predictions to experimental findings. We will assess the limitations of the current modeling approaches, and finally give an outlook for future directions.
The computational models that we have reviewed demonstrate that lateral inhibition, homeostatic plasticity, and gain adaptation could all in principle be involved in generating tinnitus-related neuronal activity patterns. Interestingly, all models focussed on how auditory input that was altered by hearing loss, induces changes in subsequent stages of the auditory system. This model feature indicates that not the process of cochlear damage as such, but rather the effects of the input signal to the auditory brain might generate tinnitus. This view could also explain how tinnitus is related to the kind and degree of hearing loss.
Computational models of tinnitus offer an explanation for the plastic changes that were observed in the central auditory system in animal models of tinnitus. Decreases in inhibition, for example, were found all along the auditory pathway, and this decrease can be explained through homeostatic plasticity or gain adaptation. In that respect, gain adaptation models (Parra and Pearlmutter, 2007) and homeostatic plasticity models (Schaette and Kempter, 2006, 2008, 2009; Chrostowski et al., 2011) suggest that tinnitus might not be the result of abnormal or aberrant plasticity, but rather that phantom sounds could arise as a side-effect of plasticity mechanisms that normally ensure proper function of the auditory brain. Plasticity triggered by hearing loss might simply produce unwanted effects when AN activity is pathologically altered, i.e., in a way that the plasticity mechanism is not designed to cope with.
Computational models of tinnitus must account for basic experimental findings. For example, in tonal tinnitus, a basic feature is its pitch, which is related to the shape of the audiogram. For noise-induced hearing loss, models based on lateral inhibition as the main mechanism (Gerken, 1996; Kral and Majernik, 1996) predict tinnitus pitch at the audiogram edge (Schaette and Kempter, 2009). Even though this relation between tinnitus pitch and the audiogram edge is supported by a recent study (Moore and Vinay, 2010), other studies report tinnitus pitch to be above the audiogram edge, i.e., within the region of hearing loss (Norena et al., 2002; König et al., 2006; Roberts et al., 2008; Pan et al., 2009; Sereda et al., 2011). Models based on homeostatic plasticity predict tinnitus pitch to be within the region of hearing loss (Schaette and Kempter, 2009) because activity stabilization through homeostatic plasticity leads to an elevation of central spontaneous activity in the frequency range that is affected by hearing loss (Figure 2B).
In general, lateral-inhibition models of tinnitus produce a “tinnitus” activity peak at a discontinuity or edge in the profile of spontaneous activity along the tonotopic axis (Figures 1B,C). However, not all kinds of cochlear damage produce such an edge. Pure loss of outer hair cells through ototoxic drugs like gentamycin or cisplatin, for example, increases the hearing thresholds but does not change the spontaneous firing rates of AN fibers (Dallos and Harris, 1978), yet tinnitus is a common side-effect of cisplatin chemotherapy (Sprauten et al., 2011). In that case, a model relying on lateral inhibition only, i.e., without additional plasticity, would not predict the occurrence of tinnitus. A homeostatic plasticity model or gain adaptation model, on the other hand, would predict the occurrence of a neural correlate of tinnitus also for pure loss of outer hair cells (Schaette and Kempter, 2006, 2008; Parra and Pearlmutter, 2007).
Evaluating model predictions in a new experimental setting is a particularly challenging test for any model. For example, plasticity models predict that tinnitus only occur when hearing is impaired, yet a significant fraction of tinnitus patients present with a normal audiogram (Barnea et al., 1990; Sanchez et al., 2005). This subgroup of tinnitus patients thus presents a considerable challenge for the hypotheses of tinnitus generation that have been formalized in the computational models summarized above. However, normal hearing thresholds do not necessarily indicate the absence of cochlear damage. In fact, it has been shown in mice that noise trauma that only leads to a temporary increase in the hearing thresholds still causes permanent damage to the synaptic contacts between inner hair cells and AN fibers (Kujawa and Liberman, 2009). When the AN stage of the homeostasis-hyperactivity model by Schaette and Kempter was adjusted to reflect this deafferentation of AN fibers, the model predicted the development of a neural correlate of tinnitus in response to the decrease in overall AN input (Schaette and McAlpine, 2011). This model result is supported by auditory brainstem response (ABR) data of tinnitus patients with normal audiograms, where a significant reduction of the amplitude of wave I of the ABR in conjunction with normal amplitudes of the centrally generated wave V was found, suggesting the presence of “hidden hearing loss” together with increased central gain (Schaette and McAlpine, 2011). Homeostasis models further predict that non-traumatic but prolonged reduction of auditory input, for example through an earplug, should lead to the occurrence of phantom sounds. This was tested experimentally in a study where participants with normal hearing and no tinnitus continuously wore an earplug for seven days. Eleven out of 18 participants perceived phantom sounds after seven days of wearing the earplug, and the phantom sounds disappeared after removing the earplug (Schaette et al., 2012).
We can conclude that a large body of evidence suggests that plasticity is a necessary ingredient of computational models of tinnitus, whereas models based on lateral inhibition only are not able to explain basic features of the data on tinnitus.
The computational models of tinnitus that have been proposed so far are almost exclusively focussed on the ascending auditory pathway. Feedback connections were omitted, and the extralemniscal pathway was not considered. Moreover, information processing and plasticity were mostly considered in a bottom-up fashion only. Top-down influences and modulation were addressed only by Langner and Wallhäusser-Franke (1999). The focus on bottom-up models can be explained by the fact that computational models need to be constrained by experimental data. The physiology of the classical ascending auditory pathway has been studied extensively whereas information about the function of feedback connections and also the extralemniscal pathways is still relatively scarce. Moreover, while a computational model needs to be as complex as necessary, ideally it should not be any more complex than required. If a phenomenon of interest can be accounted for by a simple model that captures the standard aspects of physiology, it is not necessary to include further structures and/or mechanisms that are not known well enough because such a model extension would introduce more and possibly unconstrained parameters.
In line with the idea of adequate simplicity, most models either focussed on a small part of the auditory pathway, such as the brainstem (Gerken, 1996; Schaette and Kempter, 2006, 2008, 2009) or the thalamus and cortex (Dominguez et al., 2006; Chrostowski et al., 2011), or models were not related to a particular brain region (Kral and Majernik, 1996; Parra and Pearlmutter, 2007). A unifying model that combines the aspects of these model classes could now be attempted. It would be especially interesting to see how increased spontaneous activity and activity stabilization in brainstem structures might interact with plasticity at the level of the auditory cortex. It is conceivable that less drastic changes in excitation and inhibition might be required to stabilize cortical activity when homeostatic mechanisms also increase activity in the sub-cortical processing stages. To study the interaction of subcortical and cortical levels, the respective time scales of changes are important. In any case, a combined model of brainstem, thalamus, and cortex could potentially also incorporate thalamic gating mechanisms, which have recently been implied to play a role in tinnitus (Rauschecker et al., 2010). Such unifying models might help to understand why hearing loss not always leads to tinnitus. This puzzling fact is especially important as animal studies have shown a direct relation between the degree of hearing loss and the development of putative neuronal correlates of tinnitus (Mulders et al., 2011).
At the cortical level, it might be an interesting future directive to quantitatively explore the reorganization of tonotopic maps and the relation of this phenomenon to tinnitus. Cortical reorganization can be induced by hearing loss (Irvine et al., 2000) and it was implicated as a contributor to tinnitus (Mühlnickel et al., 1998; Engineer et al., 2011), but it has not yet been explored in a computational model. Modeling studies not related to the auditory system showed that spike-timing-dependent plasticity could be the driving force for such reorganization (Song and Abbott, 2001; Young et al., 2007). An analysis of the interplay of reorganization and homeostatic plasticity could be especially interesting since recent experimental studies have reported different roles for reorganization, from promoting (Engineer et al., 2011) to reducing tinnitus (Yang et al., 2011).
So far, computational models of tinnitus have looked at neuronal activity at a microscopic level, as measured for example with microelectrodes. Another interesting aspect for future modeling studies would be to consider macroscopic signals like EEG and MEG and to include cortical rhythms. MEG studies in tinnitus patients showed that tinnitus is associated with a decrease in the power of the alpha rhythm and an increase in power in the delta frequency band (Weisz et al., 2005, 2007). Moreover, an increase in gamma power accompanied temporary tinnitus after noise exposure (Ortmann et al., 2011). Building up on models of the generation of cortical rhythms (Freyer et al., 2011) and on models that relate neuronal spiking activity to field potentials (Kuokkanen et al., 2010), future modeling studies on tinnitus could explore which parameter changes generate the observed changes in cortical rhythms, and then try to relate parameter changes to microscopic models of changes in spiking activity of neurons. Potentially, such an approach might help to bridge the gaps between animal models of tinnitus and human studies.
Before we provide an assessment of the predictions of the model for tinnitus treatments, we point out that all models we have discussed are basically bottom-up. Therefore, model predictions for tinnitus treatments also concern bottom-up approaches, and the models are not applicable to treatments employing top-down influences, like cognitive behavioral therapy.
The models based on homeostatic plasticity (Schaette and Kempter, 2006, 2008, 2009; Chrostowski et al., 2011) and gain control (Parra and Pearlmutter, 2007) make specific predictions for treatments employing acoustic or electric stimulation. These models predict that a stimulation strategy that succeeds in restoring normal AN activity should completely abolish tinnitus. For acoustic stimulation, this would correspond to the “perfect hearing aid,” and its effects would be similar to the disappearance of earplug-induced phantom sounds after removing the earplug (Schaette et al., 2012). However, the perfect hearing aid has not been invented yet. If AN activity cannot be restored, the concept of homeostatic plasticity suggests that a certain increase in AN activity should be sufficient to dampen the increased central gain and thus to reduce tinnitus. This dampening could be achieved by acoustic stimulation with noise that is spectrally matched to the hearing loss (Schaette and Kempter, 2006, 2008). Alternatively, for steeply sloping hearing loss, an amplification strategy could also try to smooth the transition from good to impaired hearing and thus to reduce the effective slope of the audiogram. This would lead to a spontaneous activity pattern with less pronounced peaks (Parra and Pearlmutter, 2007; Schaette and Kempter, 2009), corresponding to a reduction of the tinnitus salience.
In general, hearing aids and noise devices provide a certain degree of tinnitus relief (Trotter and Donaldson, 2008). However, on average the treatment success is quite limited. A prerequisite for all acoustic stimulation treatments is evoked activity in AN fibers and central auditory neurons. Further the stimulation device needs to be able to drive all frequency channels of the auditory system that are required for the treatment. However, both these assumptions might not be justified. One direct caveat for acoustic treatments is the limited frequency range of behind-the-ear devices that are commonly used to deliver the acoustic stimulation. Most behind-the-ear hearing aids and noise generators have an upper cut-off frequency in the range of 5–6 kHz, and tinnitus patients with a higher tinnitus pitch do in fact show less benefit from these devices, possibly because they do not receive adequate stimulation in their tinnitus frequency range (Schaette et al., 2010). In that case, the tinnitus models based on plasticity and gain control predict that treatment will not be effective. Furthermore, certain kinds of cochlear damage could also be major obstacles for acoustic stimulation strategies aimed at re-normalizing or at least increasing AN activity. A recent study found evidence for cochlear dead regions in 16 out of 20 participants with chronic tinnitus (Kiani et al., unpublished results). Tinnitus pitch was either at the dead region's edge frequency or inside the dead region. A cochlear dead region occurs when a stretch of the cochlea is devoid of functioning inner hair cells, and, as a consequence, the corresponding frequency channels of the auditory system cannot be stimulated acoustically. Moreover, “hidden hearing loss,” i.e., the deafferentation of AN fibers in tinnitus patients with a normal audiogram (Schaette and McAlpine, 2011), might also complicate a re-normalization of AN activity through acoustic stimulation.
In case of severe cochlear damage, electric stimulation of AN fibers, for example by means of a cochlear implant, could be another option. As long as a sufficient number of AN fibers can be stimulated, homeostatic plasticity and gain control models would also predict a reduction of tinnitus. This prediction is in line with the observation that cochlear implants can strongly reduce tinnitus (Punte et al., 2011), even generating long-lasting after-effects after the stimulation has been turned off (van de Heyning et al., 2008).
Computational models of tinnitus could be especially valuable as tools for understanding, evaluating and predicting the effects of drug treatments against tinnitus. Most of the drugs that have been recently tested for tinnitus increase inhibition in the brain. This treatment is motivated by animal studies that identified a correlation between reduced inhibition and increased spontaneous neuronal activity and tinnitus. At this point it is essential to determine whether decreased inhibition is truly the underlying cause for the development of tinnitus. The experimental data and predictions of computational models are consistent with the underlying cause being the average level of activity in the auditory system, which controls homeostatic plasticity, and thus also regulates inhibition. When inhibition is increased after hearing loss, for example by administering a drug like gabapentin, activity in the auditory system is reduced to an even greater degree than before. In that case, homeostatic plasticity would decrease the efficacy of inhibitory synapses further and also strengthen excitation, thus counterbalancing the effects of the drug. After the drug treatment has been ceased, there might even be an overshoot of activity if the drug is metabolized faster than the time constant of homeostatic plasticity. Such model-based considerations could help to explain why drugs like gabapentin are not more effective than placebo (Aazh et al., 2011).
Computational models of tinnitus opened up a functional view on plastic changes in the auditory system after hearing loss and their relation to tinnitus. Moreover, the quantitative approach used in computational modeling contributed to an assessment of different candidate mechanisms for the development of tinnitus, inspiring new experiments in order to test model predictions. In the future, a combination of brainstem and cortex models and an inclusion of feedback mechanisms could be important steps toward a more comprehensive model of tinnitus generation. Such theoretical approaches will complement and motivate further experimental studies, and a combined theoretical and experimental approach will contribute to the development of targeted tinnitus therapies in the future.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Warren Bakay for helpful comments on this manuscript. This work was supported by the British Tinnitus Association, by the Deutsche Forschungsgemeinschaft (DFG) through the SFB 618 “Theoretical Biology”, and the Bundesministerium für Bildung und Forschung (BMBF) through the Bernstein Center for Computational Neuroscience Berlin (01GQ1001A) and through the Bernstein Focus: Neuronal Basis of Learning (01GQ0972).
Aazh, H., El Refaie, A., and Humphriss, R. (2011). Gabapentin for tinnitus: a systematic review. Am. J. Audiol. 20, 151–158.
Barnea, G., Attias, J., Gold, S., and Shahar, A. (1990). Tinnitus with normal hearing sensitivity: extended high-frequency audiometry and auditory-nerve brain-stem-evoked responses. Audiology 29, 36–45.
Benda, J., and Herz, A. V. (2003). A universal model for spike-frequency adaptation. Neural Comput. 15, 2523–2564.
Brozoski, T. J., Bauer, C. A., and Caspary, D. M. (2002). Elevated fusiform cell activity in the dorsal cochlear nucleus of chinchillas with psychophysical evidence of tinnitus. J. Neurosci. 22, 2383–2390.
Bruce, I. C., Bajaj, H. S., and Ko, J. (2003). “Lateral-inhibitory-network models of tinnitus,” in 5th IFAC Symposium on Modelling and Control in Biomedical Systems. (Melbourne, Australia).
Caspary, D. M., Schatteman, T. A., and Hughes, L. F. (2005). Age-related changes in the inhibitory response properties of dorsal cochlear nucleus output neurons: role of inhibitory inputs. J. Neurosci. 25, 10952–10959.
Chrostowski, M., Yang, L., Wilson, H. R., Bruce, I. C., and Becker, S. (2011). Can homeostatic plasticity in deafferented primary auditory cortex lead to travelling waves of excitation? J. Comput. Neurosci. 30, 279–299.
Dallos, P., and Harris, D. (1978). Properties of auditory nerve responses in absence of outer hair cells. J. Neurophysiol. 41, 365–383.
de la Rocha, J., Marchetti, C., Schiff, M., and Reyes, A. D. (2008). Linking the response properties of cells in auditory cortex with network architecture: cotuning versus lateral inhibition. J. Neurosci. 28, 9151–9163.
Dean, I., Harper, N. S., and McAlpine, D. (2005). Neural population coding of sound level adapts to stimulus statistics. Nat. Neurosci. 8, 1684–1689.
Dean, I., Robinson, B. L., Harper, N. S., and McAlpine, D. (2008). Rapid neural adaptation to sound level statistics. J. Neurosci. 28, 6430–6438.
Desai, N. S., Rutherford, L. C., and Turrigiano, G. G. (1999). Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat. Neurosci. 2, 515–520.
Dominguez, M., Becker, S., Bruce, I., and Read, H. (2006). A spiking neuron model of cortical correlates of sensorineural hearing loss: spontaneous firing, synchrony, and tinnitus. Neural Comput. 18, 2942–2958.
Dong, S., Mulders, W. H., Rodger, J., and Robertson, D. (2009). Changes in neuronal activity and gene expression in guinea-pig auditory brainstem after unilateral partial hearing loss. Neuroscience 159, 1164–1174.
Eggermont, J. J., and Roberts, L. E. (2004). The neuroscience of tinnitus. Trends Neurosci. 27, 676–682.
Engineer, N. D., Riley, J. R., Seale, J. D., Vrana, W. A., Shetake, J. A., Sudanagunta, S. P., Borland, M. S., and Kilgard, M. P. (2011). Reversing pathological neural activity using targeted plasticity. Nature 470, 101–104.
Freyer, F., Roberts, J. A., Becker, R., Robinson, P. A., Ritter, P., and Breakspear, M. (2011). Biophysical mechanisms of multistability in resting-state cortical rhythms. J. Neurosci. 31, 6353–6361.
Gerken, G. M. (1996). Central tinnitus and lateral inhibition: an auditory brainstem model. Hear. Res. 97, 75–83.
Heinz, M. G., and Young, E. D. (2004). Response growth with sound level in auditory-nerve fibers after noise-induced hearing loss. J. Neurophysiol. 91, 784–795.
Houweling, A. R., Bazhenov, M., Timofeev, I., Steriade, M., and Sejnowski, T. J. (2005). Homeostatic synaptic plasticity can explain post-traumatic epileptogenesis in chronically isolated neocortex. Cereb. Cortex 15, 834–845.
Irvine, D. R., Rajan, R., and McDermott, H. J. (2000). Injury-induced reorganization in adult auditory cortex and its perceptual consequences. Hear. Res. 147, 188–199.
Kaltenbach, J. A., Zacharek, M. A., Zhang, J., and Frederick, S. (2004). Activity in the dorsal cochlear nucleus of hamsters previously tested for tinnitus following intense tone exposure. Neurosci. Lett. 355, 121–125.
Kilman, V., van Rossum, M. C., and Turrigiano, G. G. (2002). Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J. Neurosci. 22, 1328–1337.
Komiya, H., and Eggermont, J. J. (2000). Spontaneous firing activity of cortical neurons in adult cats with reorganized tonotopic map following pure-tone trauma. Acta Otolaryngol. 120, 750–756.
König, O., Schaette, R., Kempter, R., and Gross, M. (2006). Course of hearing loss and occurrence of tinnitus. Hear. Res. 221, 59–64.
Kotak, V. C., Fujisawa, S., Lee, F. A., Karthikeyan, O., Aoki, C., and Sanes, D. H. (2005). Hearing loss raises excitability in the auditory cortex. J. Neurosci. 25, 3908–3918.
Kral, A., and Majernik, V. (1996). On lateral inhibition in the auditory system. Gen. Physiol. Biophys. 15, 109–127.
Kujawa, S. G., and Liberman, M. C. (2009). Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 29, 14077–14085.
Kuokkanen, P. T., Wagner, H., Ashida, G., Carr, C. E., and Kempter, R. (2010). On the origin of the extracellular field potential in the nucleus laminaris of the barn owl (Tyto alba). J. Neurophysiol. 104, 2274–2290.
Langner, G., and Wallhäusser-Franke, E. (1999). “Computer simulation of a tinnitus model based on tinnitus activity in the auditory cortex,” in Sixth International Tinnitus Seminar, ed J. W. P. Hazell (Cambridge, UK: The Tinnitus and Hyperacusis Centre).
Liberman, M. C., and Dodds, L. W. (1984). Single-neuron labeling and chronic cochlear pathology. II. Stereocilia damage and alterations of spontaneous discharge rates. Hear. Res. 16, 43–53.
Lorenz, I., Muller, N., Schlee, W., Hartmann, T., and Weisz, N. (2009). Loss of alpha power is related to increased gamma synchronization-A marker of reduced inhibition in tinnitus? Neurosci. Lett. 453, 225–228.
Middleton, J. W., Kiritani, T., Pedersen, C., Turner, J. G., Shepherd, G. M., and Tzounopoulos, T. (2011). Mice with behavioral evidence of tinnitus exhibit dorsal cochlear nucleus hyperactivity because of decreased GABAergic inhibition. Proc. Natl. Acad. Sci. U.S.A. 108, 7601–7606.
Moore, B. C., and Vinay, S. (2010). The relationship between tinnitus pitch and the edge frequency of the audiogram in individuals with hearing impairment and tonal tinnitus. Hear. Res. 261, 51–56.
Mühlnickel, W., Elbert, T., Taub, E., and Flor, H. (1998). Reorganization of auditory cortex in tinnitus. Proc. Natl. Acad. Sci. U.S.A. 95, 10340–10343.
Mulders, W. H., Ding, D., Salvi, R., and Robertson, D. (2011). Relationship between auditory thresholds, central spontaneous activity, and hair cell loss after acoustic trauma. J. Comp. Neurol. 519, 2637–2647.
Muly, S. M., Gross, J. S., and Potashner, S. J. (2004). Noise trauma alters D-[3H]aspartate release and AMPA binding in chinchilla cochlear nucleus. J. Neurosci. Res. 75, 585–596.
Norena, A., Micheyl, C., Chery-Croze, S., and Collet, L. (2002). Psychoacoustic characterization of the tinnitus spectrum: implications for the underlying mechanisms of tinnitus. Audiol. Neurootol. 7, 358–369.
Norena, A. J., and Eggermont, J. J. (2003). Changes in spontaneous neural activity immediately after an acoustic trauma: implications for neural correlates of tinnitus. Hear. Res. 183, 137–153.
Oleskevich, S., and Walmsley, B. (2002). Synaptic transmission in the auditory brainstem of normal and congenitally deaf mice. J. Physiol. 540, 447–455.
Ortmann, M., Muller, N., Schlee, W., and Weisz, N. (2011). Rapid increases of gamma power in the auditory cortex following noise trauma in humans. Eur. J. Neurosci. 33, 568–575.
Pan, T., Tyler, R. S., Ji, H., Coelho, C., Gehringer, A. K., and Gogel, S. A. (2009). The relationship between tinnitus pitch and the audiogram. Int. J. Audiol. 48, 277–294.
Parra, L. C., and Pearlmutter, B. A. (2007). Illusory percepts from auditory adaptation. J. Acoust. Soc. Am. 121, 1632–1641.
Punte, A. K., Vermeire, K., Hofkens, A., De Bodt, M., De Ridder, D., and van de Heyning, P. (2011). Cochlear implantation as a durable tinnitus treatment in single-sided deafness. Cochlear Implants Int. 12(Suppl. 1), S26–S29.
Rajan, R., and Irvine, D. R. (1998). Neuronal responses across cortical field A1 in plasticity induced by peripheral auditory organ damage. Audiol. Neurootol. 3, 123–144.
Rannals, M. D., and Kapur, J. (2011). Homeostatic strengthening of inhibitory synapses is mediated by the accumulation of GABAA receptors. J. Neurosci. 31, 17701–17712.
Rauschecker, J. P. (1999). Auditory cortical plasticity: a comparison with other sensory systems. Trends Neurosci. 22, 74–80.
Rauschecker, J. P., Leaver, A. M., and Muhlau, M. (2010). Tuning out the noise: limbic-auditory interactions in tinnitus. Neuron 66, 819–826.
Roberts, L. E., Eggermont, J. J., Caspary, D. M., Shore, S. E., Melcher, J. R., and Kaltenbach, J. A. (2010). Ringing ears: the neuroscience of tinnitus. J. Neurosci. 30, 14972–14979.
Roberts, L. E., Moffat, G., Baumann, M., Ward, L. M., and Bosnyak, D. J. (2008). Residual inhibition functions overlap tinnitus spectra and the region of auditory threshold shift. J. Assoc. Res. Otolaryngol. 9, 417–435.
Roberts, M. T., and Trussell, L. O. (2010). Molecular layer inhibitory interneurons provide feedforward and lateral inhibition in the dorsal cochlear nucleus. J. Neurophysiol. 104, 2462–2473.
Sanchez, T. G., Medeiros, I. R., Levy, C. P., Ramalho Jda, R., and Bento, R. F. (2005). Tinnitus in normally hearing patients: clinical aspects and repercussions. Br. J. Otorhinolaryngol. 71, 427–431.
Schaette, R., and Kempter, R. (2006). Development of tinnitus-related neuronal hyperactivity through homeostatic plasticity after hearing loss: a computational model. Eur. J. Neurosci. 23, 3124–3138.
Schaette, R., and Kempter, R. (2008). Development of hyperactivity after hearing loss in a computational model of the dorsal cochlear nucleus depends on neuron response type. Hear. Res. 240, 57–72.
Schaette, R., and Kempter, R. (2009). Predicting tinnitus pitch from patients' audiograms with a computational model for the development of neuronal hyperactivity. J. Neurophysiol. 101, 3042–3052.
Schaette, R., König, O., Hornig, D., Gross, M., and Kempter, R. (2010). Acoustic stimulation treatments against tinnitus could be most effective when tinnitus pitch is within the stimulated frequency range. Hear. Res. 269, 95–101.
Schaette, R., and McAlpine, D. (2011). Tinnitus with a normal audiogram: physiological evidence for hidden hearing loss and computational model. J. Neurosci. 31, 13452–13457.
Schaette, R., Turtle, C., and Munro, K. J. (2012). Reversible induction of phantom auditory sensations through simulated unilateral hearing loss. PLoS One. doi: 10.1371/journal.pone.0035238
Sereda, M., Hall, D. A., Bosnyak, D. J., Edmondson-Jones, M., Roberts, L. E., Adjamian, P., and Palmer, A. R. (2011). Re-examining the relationship between audiometric profile and tinnitus pitch. Int. J. Audiol. 50, 303–312.
Song, S., and Abbott, L. F. (2001). Cortical development and remapping through spike timing-dependent plasticity. Neuron 32, 339–350.
Sprauten, M., Darrah, T. H., Peterson, D. R., Campbell, M. E., Hannigan, R. E., Cvancarova, M., Beard, C., Haugnes, H. S., Fossa, S. D., Oldenburg, J., and Travis, L. B. (2011). Impact of long-term serum platinum concentrations on neuro- and ototoxicity in cisplatin-treated survivors of testicular cancer. J. Clin. Oncol. 30, 300–307.
Suneja, S. K., Benson, C. G., and Potashner, S. J. (1998a). Glycine receptors in adult guinea pig brain stem auditory nuclei: regulation after unilateral cochlear ablation. Exp. Neurol. 154, 473–488.
Suneja, S. K., Potashner, S. J., and Benson, C. G. (1998b). Plastic changes in glycine and GABA release and uptake in adult brain stem auditory nuclei after unilateral middle ear ossicle removal and cochlear ablation. Exp. Neurol. 151, 273–288.
Trotter, M. I., and Donaldson, I. (2008). Hearing aids and tinnitus therapy: a 25-year experience. J. Laryngol. Otol. 122, 1052–1056.
Turrigiano, G. G. (1999). Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 22, 221–227.
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896.
Vale, C., and Sanes, D. H. (2002). The effect of bilateral deafness on excitatory and inhibitory synaptic strength in the inferior colliculus. Eur. J. Neurosci. 16, 2394–2404.
Vale, C., Juiz, J. M., Moore, D. R., and Sanes, D. H. (2004). Unilateral cochlear ablation produces greater loss of inhibition in the contralateral inferior colliculus. Eur. J. Neurosci. 20, 2133–2140.
van de Heyning, P., Vermeire, K., Diebl, M., Nopp, P., Anderson, I., and De Ridder, D. (2008). Incapacitating unilateral tinnitus in single-sided deafness treated by cochlear implantation. Ann. Otol. Rhinol. Laryngol. 117, 645–652.
van Welie, I., van Hooft, J. A., and Wadman, W. J. (2004). Homeostatic scaling of neuronal excitability by synaptic modulation of so-matic hyperpolarization-activated Ih channels. Proc. Natl. Acad. Sci. U.S.A. 101, 5123–5128.
Weisz, N., Moratti, S., Meinzer, M., Dohrmann, K., and Elbert, T. (2005). Tinnitus perception and distress is related to abnormal spontaneous brain activity as measured by magnetoencephalography. PLoS Med. 2:e153. doi: 10.1371/journal.pmed.0020153
Weisz, N., Muller, S., Schlee, W., Dohrmann, K., Hartmann, T., and Elbert, T. (2007). The neural code of auditory phantom perception. J. Neurosci. 27, 1479–1484.
Wen, B., Wang, G. I., Dean, I., and Delgutte, B. (2009). Dynamic range adaptation to sound level statistics in the auditory nerve. J. Neurosci. 29, 13797–13808.
Whiting, B., Moiseff, A., and Rubio, M. E. (2009). Cochlear nucleus neurons redistribute synaptic AMPA and glycine receptors in response to monaural conductive hearing loss. Neuroscience 163, 1264–1276.
Yang, S., Weiner, B. D., Zhang, L. S., Cho, S. J., and Bao, S. (2011). Homeostatic plasticity drives tinnitus perception in an animal model. Proc. Natl. Acad. Sci. U.S.A. 108, 14974–14979.
Keywords: tinnitus, computational model, hearing loss, homeostatic plasticity, lateral inhibition, gain adaptation
Citation: Schaette R and Kempter R (2012) Computational models of neurophysiological correlates of tinnitus. Front. Syst. Neurosci. 6:34. doi: 10.3389/fnsys.2012.00034
Received: 20 January 2012; Accepted: 17 April 2012;
Published online: 08 May 2012.
Edited by:
Jos J. Eggermont, University of Calgary, CanadaCopyright: © 2012 Schaette and Kempter. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Roland Schaette, Ear Institute, University College London, 332 Gray's Inn Road, London WC1X 8EE, UK. e-mail:ci5zY2hhZXR0ZUB1Y2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.