 Rachida Ammari1,2,3
Rachida Ammari1,2,3


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Syst. Neurosci. , 13 June 2011
volume 5 - 2011 | https://doi.org/10.3389/fnsys.2011.00043
This article is part of the Research Topic Basal Ganglia X - Proceedings of the 10th Triennial Meeting of the International Basal Ganglia Society View all 47 articles
Excessive burst firing in the dopamine-depleted basal ganglia correlates with severe motor symptoms of Parkinson’s disease that are attenuated by high frequency electrical stimulation of the subthalamic nucleus (STN). Here we test the hypothesis that pathological bursts in dopamine-deprived basal ganglia are generated within the STN and transmitted to globus pallidus neurons. To answer this question we recorded excitatory synaptic currents and potentials from subthalamic and pallidal neurons in the basal ganglia slice (BGS) from dopamine-depleted mice while continuously blocking GABAA receptors. In control mice, a single electrical stimulus delivered to the internal capsule or the rostral pole of the STN evoked a short duration, small amplitude, monosynaptic EPSC in subthalamic neurons. In contrast, in the dopamine-depleted BGS, this monosynaptic EPSC was amplified and followed by a burst of polysynaptic EPSCs that eventually reverberated three to seven times, providing a long lasting response that gave rise to bursts of EPSCs and spikes in GP neurons. Repetitive (10–120 Hz) stimulation delivered to the STN in the dopamine-depleted BGS attenuated STN-evoked bursts of EPSCs in pallidal neurons after several minutes of stimulation but only high frequency (90–120 Hz) stimulation replaced them with small amplitude EPSCs at 20 Hz. We propose that the polysynaptic pathway within the STN amplifies subthalamic responses to incoming excitation in the dopamine-depleted basal ganglia, thereby transforming the STN into a burst generator and entraining pallidal neurons in pathogenic bursting activities. High frequency stimulation of the STN prevents the transmission of this pathological activity to globus pallidus and imposes a new glutamatergic synaptic noise on pallidal neurons.
Patients suffering from Parkinson’s disease, as well as animal models of Parkinson’s, exhibit abnormally synchronized neuronal oscillatory activity at multiple levels of the basal ganglia–cortical loop, particularly within the subthalamic nucleus (STN; Bergman et al., 1994); for review (Hammond et al., 2007). In vitro, these pathological oscillations have been observed in organotypic co-cultures of globus pallidus (GP, the rodent equivalent of the external segment of the globus pallidus in primates) and STN with frontomedial cortex and dorsolateral striatum (Plenz and Kitaï, 1999), but have so far been absent from ex vivo preparations (Wilson et al., 2006). In this co-culture preparation which lacked dopaminergic neurons of the substantia nigra (SN), STN, and GP neurons spontaneously produced synchronized oscillating bursts. Disconnection of the STN from the cortex and the striatum did not abolish synchronized oscillatory bursting, whereas interruption of the STN from GP suppressed bursting in both STN and GP. This led to the conclusion that GP bursts, although weaker than STN bursts, were required for synchronized oscillatory burst generation (Plenz and Kitaï, 1999), probably via STN rebound excitation (Baufreton et al., 2005; Bevan et al., 2006, 2007). Bursting activity was attributed to a central pacemaker system consisting of the interconnected GABAergic GP and glutamatergic STN neurons. Interestingly, 1 h after disconnection of the STN from the GP, long lasting and uncorrelated burst activity built up in the STN, although spontaneous activity was almost absent in the isolated GP (Plenz and Kitaï, 1999).
These observations can be explained by an alternative hypothesis, namely that abnormal bursts are generated by STN neurons alone (Ryan and Sanders, 1993; Murer et al., 1997). We and others have suggested that a polysynaptic glutamatergic network inside the STN amplifies the STN excitatory drive onto GP (Ammari et al., 2010), entopeduncular nucleus (Ammari et al., 2010) and substantia nigra reticulata (SNr; Shen and Johnson, 2006; Ammari et al., 2010) neurons under certain conditions. In the present paper we have blocked GABAA synaptic transmission to decrease the impact of the reciprocal GP-STN pathway, while recording synaptic currents from patch-clamped STN and GP neurons in the basal ganglia slice (BGS; Beurrier et al., 2006) from control or reserpine-treated mice.
We now report that dopamine deprivation causes the STN to generate large bursts of EPSCs (compound EPSCs) in response to a single stimulation of its afferents in the internal capsule (IC). These bursts consist of a series of polysynaptic EPSCs, and may reverberate several times without further stimulation. Both the primary and reverberating bursts propagate to GP neurons. High frequency stimulation of the STN (STN-HFS) dramatically weakens transmission of compound EPSCs to GP neurons and replaces them with small amplitude EPSCs at 20 Hz. We propose that the polysynaptic glutamatergic network within the STN would normally be partly under the inhibitory control of dopaminergic signaling (Shen and Johnson, 2000) and therefore relatively difficult to activate (Shen and Johnson, 2006; Ammari et al., 2010). In contrast, when dopamine-deprived, the polysynaptic STN network becomes the central generator of bursts in the basal ganglia, in particular when activated by afferent excitations. We show that the transmission of bursts from STN to GP neurons and probably to the other target neurons of the STN as well, is dramatically weakened by continuous stimulation of STN neurons at approximately 100 Hz, causing depression of glutamatergic synaptic transmission.
Experiments were performed in slices obtained from naive or reserpine-treated 13- to 28-day-old C57Bl6 mice. Experiments were carried out in accordance with European Communities Council Directive of 24 November 1986 (86/609/EEC) for care of laboratory animals. Reserpine (5 mg/kg, i.p.) and α-methyl-p-tyrosine (a tyrosine hydroxylase inhibitor; 250 mg/kg, i.p.) were injected at 20 and 4 h, respectively, before preparation of slices to deplete dopaminergic terminals and prevent new synthesis of dopamine (Moody and Spear, 1992). In their cages, the injected mice showed a complete lack of spontaneous activity and when placed on the grid they were unable to grasp it.
Mice were killed by decapitation under isofluorane anesthesia and 400-μm-thick oblique parasagittal slices were cut with an angle of 10 ± 2° to obtain BGS as previously described (Beurrier et al., 2006). For the slicing procedure, the solution contained (in mM) 110 choline, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3, 7 glucose. After a recovery period, the slices were recorded in a submersion-type chamber at room temperature with standard ACSF saturated with 95% O2/5% CO2 and containing (in mM) 126 NaCl, 3.5 KCl, 1.2 NaH2PO4, 1.3 MgCl2, 2.4 CaCl2, 25 NaHCO3, 12 glucose.
Cells were visualized with infrared-differential interference optics (BMX Olympus). We performed patch-clamp recordings in the whole-cell or cell-attached configuration using the Multiclamp 700A amplifier, the Digidata 1344A interface and PClamp9 software (Axon Instruments, Foster City, CA, USA). For voltage and current clamp recordings, patch electrodes (6–10 MΩ) contained (in mM): 120 K-gluconate, 5 KCl, 1 CaCl2, 10 HEPES, 10 EGTA, 2.5 MgATP, 0.5 NaGTP and 2 QX-314, pH 7.2–7.4 (275–285 mOsm). Biocytin (5 mg/ml) was added to the pipette solution and osmolarity corrected when necessary so that the recorded neurons were filled with biocytin (0.4%) for post hoc morphological analysis. We identified STN neurons by their localization, GP neurons by their localization, I–V curves, and morphology (Ammari et al., 2010).
Synaptic currents were evoked in the STN and GP neurons using a bipolar stimulating electrode (33 kΩ impedance in response to 1 V at 1000 Hz, separation of the two electrodes: 100 μm, Phymep NEX). Rectangular pulses (100 μs duration) of constant current (50–800 μA) were delivered every 30 s unless otherwise stated. For 10–120 Hz STN stimulation we used a stimulator made in the laboratory (UMR CNRS, 5293) to deliver constant-current pulses, as previously described (Garcia et al., 2003). Stimulation was applied between the two poles of the electrode. It consisted of negative current pulses of fixed duration (100 μs), fixed frequency (10–120 Hz), and intensity adjusted between 100 and 300 μA (0.5–1.5 V). Stimulation was applied over periods of several minutes. To remove artifacts, a sample and hold electronic device was built in the laboratory as previously described (Garcia et al., 2003). Synchronized with the stimulator, the artifact suppressor allowed the recorded signal to be held at the level before the current pulse and to restore the signal a few microseconds later. Comparison of recording traces before and after artifact suppression allowed checking that the sample-and-hold procedure did not consistently affect EPSCs recordings.
Drugs were prepared as concentrated stock solutions and diluted in ACSF for bath application immediately prior to use. Gabazine, a selective GABAA receptor antagonist; (±)-2-amino-5-phosphonopentanoic acid (AP5, a selective NMDA receptor antagonist), 6-cyano-7nitroquinoxaline 2, 3-dione (CNQX, an AMPA-KA receptors antagonist), were all purchased from Sigma Aldrich (France).
Evoked and spontaneous EPSCs were quantified using pClamp 9 software (Axon Instruments, Foster City, CA, USA). We quantified the total charge (in nA.ms), latency (ms), duration (ms), peak amplitude (pA), and time to peak (ms) of EPSCs. Spontaneous and HFS-evoked EPSCs were analyzed off-line with Mini Analysis program (Synaptosoft 6.0), to determine their frequency, amplitude, and pattern. All detected currents were then visually inspected to reject artifactual events.
Average values are presented as means ± SEM and we performed statistical comparisons with Mann–Whitney rank sum test (SigmaStat 3.1). We set the level of significance as P < 0.05 (*); (**) for P < 0.01; (***) for P < 0.001.
All recordings were performed in the continuous presence of Gabazine (10 μM) in order to block GABAA synaptic transmission in the BGS.
In control BGS, IC or rostral STN stimulation (100–500 μA, 100 μs) evoked in STN neurons small amplitude (peak amplitude: −105 ± 5 pA, time to peak 10.3 ± 0.7 ms) and short duration (35 ± 2 ms) EPSCs with a mean latency of 2.9 ± 0.1 ms (total charge: 0.80 ± 0.05 nA.ms) in whole-cell recorded STN neurons (VH = −70 mV, n = 7). The corresponding EPSP recorded in current clamp mode, was of longer duration (116 ± 6 ms at Vm = −70 mV, range 58–225 ms, n = 7), due to the activation of depolarizing voltage-gated currents in the STN neuronal membrane. It gave rise to only one or two spikes (Vm = −60 mV; Figure 1A, left).
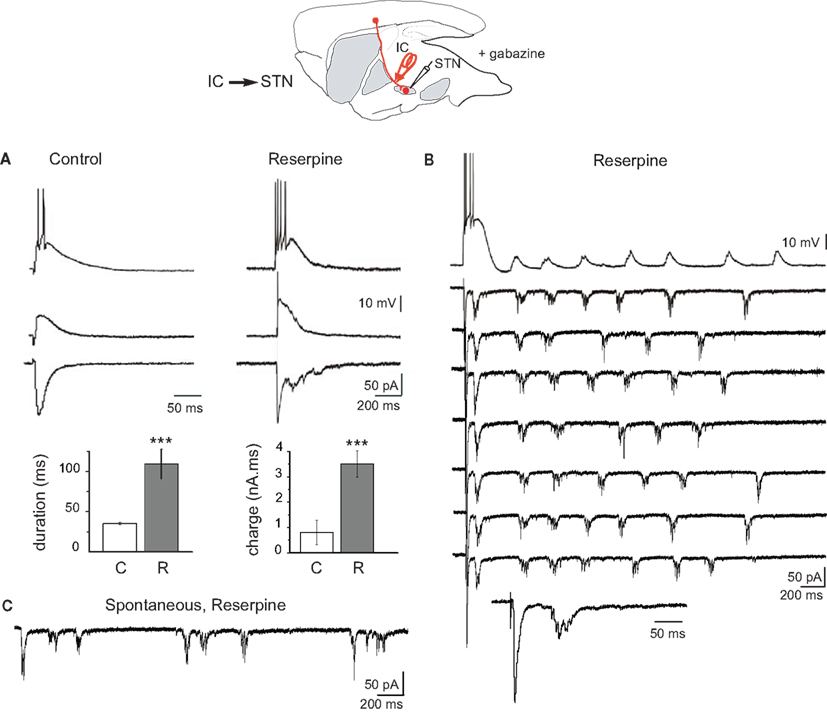
Figure 1. Response of STN neurons to internal capsule stimulation in control and dopamine-depleted BGS in the continuous presence of gabazine (10 μM). Top: schematic illustration of the experimental design with the stimulation (red) and recording sites. (A) Examples of evoked glutamatergic EPSPs (current clamp traces at Vm = −60 mV (top) and Vm = −70 mV (middle) and EPSCs (voltage clamp traces at VH = −70 mV, bottom) recorded from the same STN neuron in response to a single shock (100 μs, 300 μA) delivered to the internal capsule (IC) in the BGS from control (left) and reserpine-treated (right) mice. Note the difference in time calibration between left and right traces. Histograms of the duration and total charge of the IC-evoked STN EPSC recorded in BGS from control (C, white) and reserpine-treated (R, gray) mice (n = 7 and 11, respectively). (B) Example trace of reverberant glutamatergic EPSPs (top trace, Vm = −70 mV) and EPSCs (seven traces, VH = −70 mV) recorded from the same STN neuron in response to a single shock (100 μs, 85 μA) delivered to the internal capsule at 0.03 Hz (arrowhead). The 8th trace is the first part of the voltage clamp response at an expanded time scale to show the early EPSC. (C) Spontaneous bursts of glutamatergic EPSCs (VH = −70 mV) recorded from a STN neuron in a BGS from a reserpine-treated mouse.
After reserpine treatment, the IC or STN-evoked EPSC in STN neurons had a lower threshold (60–100 μA instead of 200–300 μA), was intensity-dependent and strikingly amplified (Figure 1A, right). It had a higher amplitude (−166 ± 11 pA) and a longer duration (109 ± 18 ms) than the EPSC recorded in control STN (n = 11; P < 0.001 for both). Overall, DA depletion increased the total charge approximately fourfold (3.5 ± 0.5 nA.ms; Figure 1A, bottom histograms). In corresponding current clamp recordings, the IC or STN-evoked EPSP was also largely increased in duration (543 ± 14 ms at Vm = −70 mV, range 360–730 ms) and gave rise to trains of spikes (4–14 spikes) at 35 ± 1 Hz (n = 7). Sometimes, the evoked EPSCs reverberated in STN neurons up to seven times after a single, brief IC stimulation (Figure 1B). The total mean duration of the reverberations after a single shock delivered to the IC was 1117 ± 103 ms and their mean frequency was 4.5 ± 0.5 Hz (total charge: 20 ± 1 nA.ms, n = 5). Interestingly, spontaneous, repetitive bursts of EPSCs were also recorded from STN neurons in cell-attached (not shown) or whole-cell (Figure 1C) configuration in the absence of any stimulation. Evoked and spontaneous bursts of EPSCs were totally abolished by the application of glutamatergic antagonists (APV 50 μM + CNQX 10 μM; data not shown). We next tested whether STN bursts propagate to GP neurons.
Previous studies showed that brief high intensity STN stimulation evoked long lasting compound EPSCs in target neurons of the STN (GP, EP, and SNr neurons) in sagittal or horizontal slices from control mice (Shen and Johnson, 2006; Ammari et al., 2010). These responses consisted of an early monosynaptic EPSC followed by a barrage of polysynaptic EPSCs which dramatically prolonged the response up to several hundreds of milliseconds. The late polysynaptic glutamatergic component depended on the presence of the STN cell bodies in the slice. When the same experiment was performed in DA-depleted BGS we observed that the threshold STN stimulation for the generation of the late barrage of polysynaptic EPSCs in GP neurons was lowered by a factor of two to three (from 200 to 60–100 μA, single pulse, 100 μs duration). At intensities 1.5 times the threshold of the late component, the duration, and total charge of the late component in reserpine slices were increased compared to control slices (non-significant change from 510 ± 141 ms to 739 ± 150 ms for the duration and significant increase from 5.3 ± 1.0 to 19.4 ± 1.2 nA.ms for the charge, P < 0.001; n = 12 GP control and n = 5 GP reserpine; Figures 2A,C). In cell-attached and whole-cell current clamp recordings, compound EPSCs gave rise to long lasting bursts of spikes in GP neurons (Figure 2B).
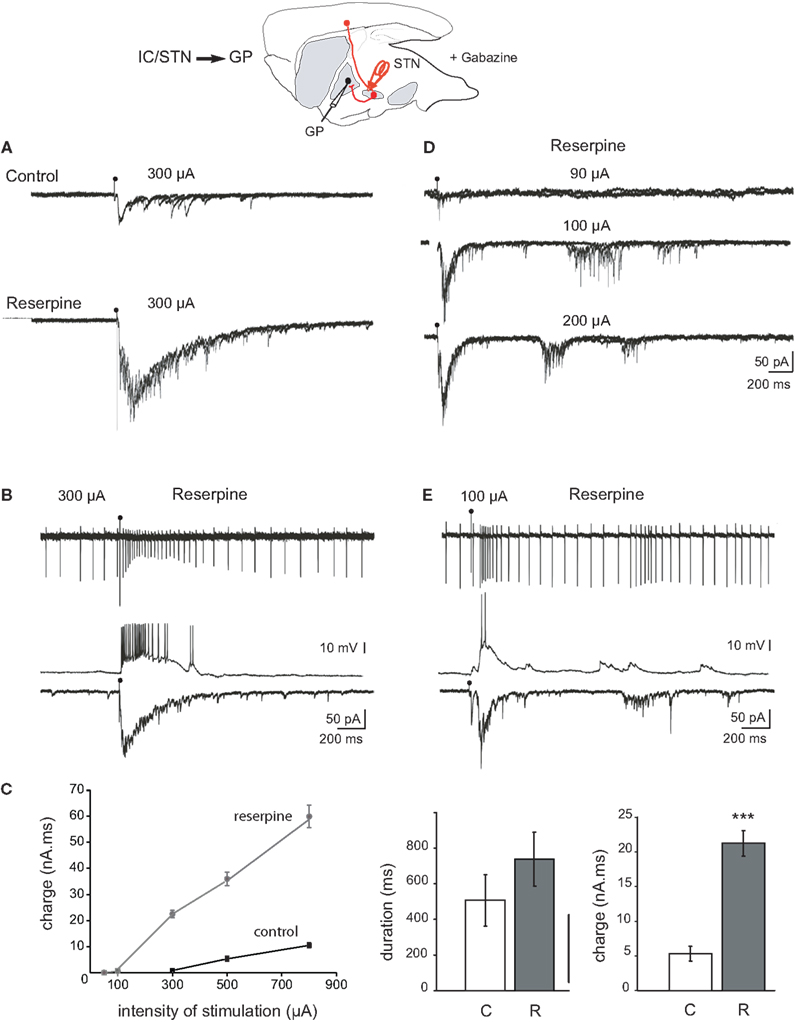
Figure 2. Response of GP neurons to stimulation of the internal capsule or rostral STN in control and dopamine-depleted BGS. Top: Schematic illustration of the experimental design with the stimulation (red) and recording sites. (A) Example traces from GP cells recorded from control BGS (top) and BGS from a reserpinized mouse (bottom). A single shock (100 μs, 300 μA) was delivered with the same stimulating electrode to the rostral pole of the STN at the time of the artifact. It evoked compound glutamatergic EPSCs (VH = −70 mV, three superimposed traces). (B) Recordings from the same GP neuron in cell-attached current clamp mode (top), whole-cell current clamp mode (middle, Vm = −60 mV) and whole-cell voltage clamp mode (bottom, VH = −70 mV) in a BGS from a reserpinized mouse. A single shock delivered to the rostral pole of the STN (100 μs, 300 μA) evoked a long lasting train of spikes, an EPSP giving rise to a long lasting train of spikes and a compound EPSC. (C) Total charge (nA.ms) of the compound EPSCs recorded in (A) as a function of the intensity of stimulation (μA; black circles: control, gray circles: reserpine). Histograms of the duration (left), and charge (right) of the compound EPSCs evoked in control (white, C) and reserpine (gray, R) conditions with the same stimulating electrode and at the same intensity of stimulation (300 μA). (D) Reverberating bursts of EPSCs recorded from the same GP neuron in a BGS from a reserpinized mouse. A single shock (100 μs, arrow head) was delivered to the rostral pole of the STN at the indicated intensities. (E) Example trace of a reverberant glutamatergic train of spikes (cell-attached, top), EPSPs (whole-cell, Vm = −60 mV, middle) and bursts of EPSCs (whole-cell, VH = −70 mV, bottom) recorded from the same GP neuron in a BGS from a reserpinized mouse, in response to a single shock (100 μs, 85 μA) delivered to the rostral pole of the STN.
The late polysynaptic component of the compound EPSC could reverberate three to five times at a mean frequency of 4.8 ± 0.6 Hz in response to a single and brief (100 μs) STN stimulation (total duration of the reverberations: 1480 ± 194 ms, total charge 31 ± 2.8 nA.ms, n = 5; Figure 2D). These reverberating bursts of EPSCs gave rise to reverberant bursts of spikes in cell-attached recordings (Figure 2E). Finally, spontaneous, APV/CNQX-sensitive bursts of EPSCs were sometimes recorded in the absence of any stimulation in DA-depleted GP neurons (not shown). We next tested the role of high frequency STN stimulation (STN-HFS) on the activity of the STN-GP glutamatergic synapses in the DA-depleted BGS.
We recorded the glutamatergic activity of GP neurons during stimulation of the STN at 8–10 or 90–120 Hz. We used the voltage clamp mode (VH = −70 mV) to analyze the STN-GP synaptic glutamatergic transmission without participation of depolarization-gated membrane currents. The same parameters of STN stimulation (100 μs, 300 μA) that at 0.03 Hz reliably evoked long lasting compound EPSCs in GP neurons in the DA-depleted BGS (mean amplitude −176.7 ± 8.7 pA, n = 5), only evoked short duration EPSCs after 3–5 min of 8–10 Hz stimulation (steady state; Figure 3A). In the 100-ms interval between the 8–10 Hz stimuli, we observed several small amplitude EPSCs (Figures 3A bottom,C). They had a mean amplitude of 20.1 ± 0.5 pA) and a mean frequency of 42 ± 2 Hz (n = 1087 events). STN-HFS at 90–120 Hz for 3–5 min evoked significantly smaller EPSCs (12.9 ± 0.2 pA) at a significantly lower mean frequency (19.6 ± 3.0 Hz, n = 1010 events; Figures 3B–D). The dramatic weakening of the STN-GP synaptic transmission by STN-HFS is also supported by the dramatic reduction of mean charge of the evoked EPSCs: from 3500 ± 500 pA.ms (reserpine, see above) to 78 ± 2 pA.ms (10 Hz) and 49 ± 1 pA.ms (100 Hz).
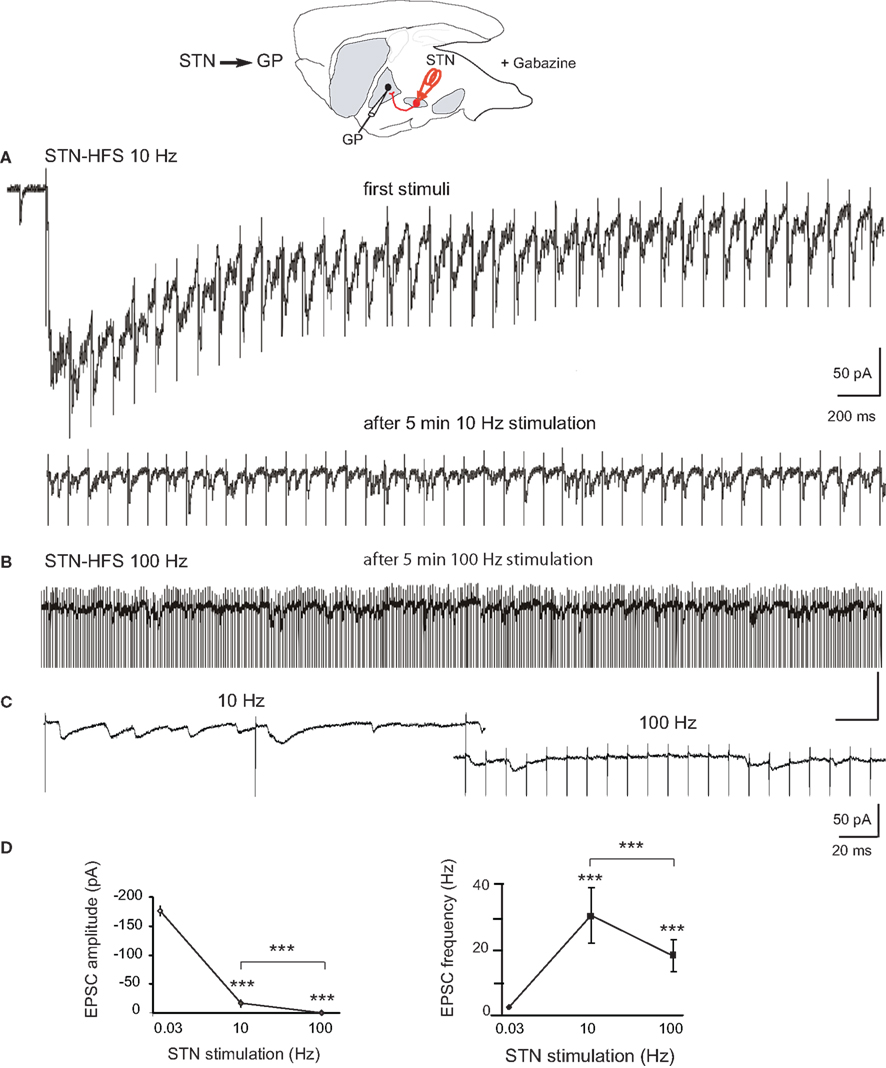
Figure 3. Effects of STN-HFS on the EPSCs evoked in GP neurons in dopamine-depleted BGS. Top: Schematic illustration of the experimental design with the stimulation (red) and recording sites. (A) STN-HFS at 10 Hz evoked EPSCs at 10 Hz. Note that HFS causes an initial shift in holding current that is caused by time-dependent summation of the late components of compound EPSCs. After 5 min of 10 Hz stimulation, the EPSCs evoked in the same GP neuron had a lower amplitude. (B) STN-evoked glutamatergic EPSCs recorded in the same GP neuron as in (A) during STN-HFS at 100 Hz. (C): Close up of traces under 10 and 100 Hz STN-HFS. (D) Averaged data showing the effects of 10 and 100 Hz STN stimulation compared to 0.03 Hz (control) on the amplitude and frequency of the EPSCs evoked in DA-depleted GP neurons. Each data point is the mean ± SEM. For the response at 0.03 Hz we considered the complex response as a whole and plotted its peak amplitude and frequency as if it were a single EPSC.
We also compared these EPSCs evoked by STN-HFS (HFSEPSCs) to spontaneous EPSCs (sEPSCs) recorded before the stimulation from the same GP neurons (Figures 4A,B). Spontaneous EPSCs from DA-depleted GP neurons had a mean amplitude of 17.9 ± 1.5 pA (range: 2.8–48.2 pA) with a mean frequency of 5.5 ± 1.2 Hz (range inter sEPSC intervals: 1.5–1653 ms, n = 1085 events; Figures 4A,C). STN-HFS at 100 Hz evoked HFSEPSCs of significantly smaller amplitude than that of sEPSCs (0 Hz) and of HFSEPSCs recorded during 10 Hz (notice that the mean amplitudes of EPSCs were not significantly different between 0 and 10 Hz, P = 0.1; Figure 4C). Finally, 100 Hz STN-HFS narrowed the distribution of HFSEPSCs amplitude since it suppressed the occurrence of HFSEPSCs over 40 pA (range 2.2–38 pA) compared to 10 Hz STN-HFS (2.1–92 pA; Figure 4D). But both 10 and 100 Hz STN-HFS strikingly narrowed the distribution of HFSEPSCs frequency compared to that of sEPSCs (inter HFSEPSC intervals: 1.2–181 ms for 10 Hz and 2.7–273 ms for 100 Hz; Figure 4E). We could not analyze the pattern (bursty or not) of the HFSEPSCs due to the small amplitude of many of these events.
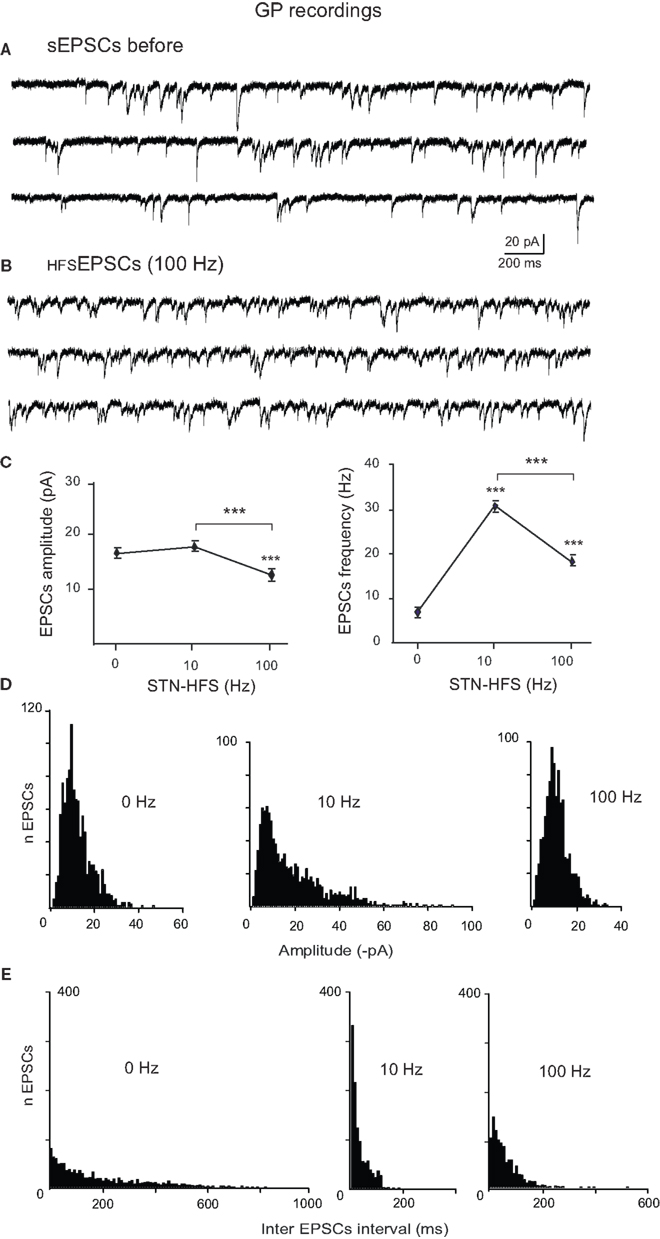
Figure 4. Comparison of spontaneous glutamatergic EPSCs recorded before and during STN-HFS. (A) Three example traces showing the spontaneous EPSCs (sEPSCs) recorded in voltage clamp mode (VH = −70 mV) from a GP neuron in a BGS from a reserpinized mouse. (B) Three example traces from the same GP neuron as in (A) showing the EPSCs recorded during STN-HFS at 100 Hz (HFSEPSCs, filtered traces without stimulation artifacts). (C) Average data showing the effects of 10 and 100 Hz STN stimulation compared to before HFS (0 Hz) on the amplitude and frequency of the EPSCs recorded in DA-depleted GP neurons. Each data point is the mean ± SEM. Histograms of the amplitudes [bin 1 pA, (D)] and inter EPSCs intervals [bin 10 ms, (E)] for the spontaneous EPSCs (0 Hz) and the HFSEPSCs recorded during 10 or 100 Hz STN stimulation in the same GP neuron, as indicated.
We show that DA-depleted STN neurons without GABAA synaptic feedback from GP and EP neurons generate bursts of EPSCs lasting several hundreds of milliseconds in response to a single and brief activation of their glutamatergic afferents in the IC. These EPSCs, together with depolarizing voltage-gated currents, give rise to bursts of propagating spikes and activate GP neurons that in turn generate bursts of spikes. Moreover, a single stimulus produced repetitive bursts along the STN-GP pathway due to the reverberation of IC-evoked bursts in STN neurons, suggesting that the STN alone becomes responsible for the well-known bursting pattern recorded in vivo in the parkinsonian state. Feedback IPSCs from GP neurons may synchronize and amplify cortically evoked bursts in STN neurons (Baufreton and Bevan, 2008) but are not instrumental for repetitive bursting.
We propose that the dopaminergic synapses afferent to STN neurons control the afferent glutamatergic synapses to the STN and the glutamatergic polysynaptic network within the STN. As previously shown in control condition (Ammari et al., 2010), this dopaminergic control can be counterbalanced by a strong excitatory input to the STN, allowing the STN polysynaptic network generating a long lasting excitation of its target neurons. Likewise, STN neurons can provide, when needed, a widespread and time-locked reset of target neurons activity (Frank, 2006). In the chronic absence of dopaminergic control, this reset would become repetitive and out of control. This would explain why HFS at the level of the STN remains an efficient way to ameliorate parkinsonian symptoms (Benabid et al., 2009).
There are two sequential amplifications of incoming excitation in the STN: one occurs at the level of synaptic connections to and/or between STN neurons, and the other is intrinsic to the STN neuronal membrane. The STN is innervated by dopamine-containing pathways that arise from the substantia nigra compacta (SNc) and the ventral tegmental area (Brown et al., 1979; Meibach and Katzman, 1979; Hassani et al., 1997). Tyrosine hydroxylase positive axons form asymmetric synapses and release DA in a Ca2+-dependent manner in the STN (Cragg et al., 2004). Dopamine D1- and D2-like receptor subtypes are both expressed in the STN (Mansour et al., 1990; Flores et al., 1999). Dopamine reduces the amplitude of evoked glutamate EPSCs of control STN neurons by over 30% in vitro (Shen and Johnson, 2000). Although the precise site of action of dopamine is unknown, removal of this inhibition could explain the increased STN response to incoming excitation in DA-depleted state.
The second amplification is achieved by the depolarizing voltage-dependent currents of the STN membrane activated by EPSPs. These include the NMDA (Standaert et al., 1994), Ca2+ (Beurrier et al., 1999; Song et al., 2000), persistent Na+ (Bevan and Wilson, 1999; Beurrier et al., 2000) and Ca2+-activated depolarizing cationic (Beurrier et al., 1999; Zhu et al., 2004) currents which all prolong the STN firing period in response to the stimulation of glutamatergic afferents (Otsuka et al., 2001; Kass and Mintz, 2006). Kass and Mintz (2006) showed that STN neurons produce various patterns of activity when synaptically activated: silence, classical tonic firing, plateau potentials, burst responses, and rhythmic bursts. This allows STN neurons to adapt their pattern of activity to the environmental context. How these intrinsic membrane currents are affected by the chronic reduction of dopaminergic synaptic transmission is largely unknown (Baufreton and Bevan, 2008). Overall, the resultant effect of dopamine depletion is an increased impact of cortical afferents on neuronal activity in the STN (Magill et al., 2001).
In vivo studies also suggest that the cortico-STN pathway malfunctions in the chronic absence of dopamine. The coherent oscillations between cortical regions and STN neurons recorded from anaesthetized, chronically 6-OHDA-lesioned rats (Sharott et al., 2005; Mallet et al., 2008) and un-medicated parkinsonian patients (Eusebio et al., 2009) are predominantly led by activity in the cortex (Magill et al., 2001; Litvak et al., 2011). The glutamatergic hyperdirect pathway is the likely candidate, but in vivo trans-striatal influences via the external globus pallidus (Bevan et al., 2002) cannot be excluded. Malfunction of the cortico-STN pathway, possibly reinforced by the feedback STN-cortex (Degos et al., 2008) and local STN-STN (Shen and Johnson, 2006; Ammari et al., 2010) glutamatergic pathways, in the absence of dopamine, is also supported by the beneficial effect of high frequency stimulation of motor cortical areas on parkinsonian motor symptoms (Drouot et al., 2004; Fregni et al., 2006; Gradinaru et al., 2009; Lefaucheur, 2009).
In their dynamic model of STN neurons, Gillies and Willshaw (2004) predicted that when recurrent glutamatergic synapses between STN neurons are rare (3% of connectivity among STN neurons; Kita et al., 1983) and/or of low synaptic weight, direct cortical drive can push large interconnected regions of the STN into a uniform high state as observed in vivo (Magill et al., 2004). As a consequence, glutamatergic STN neurons may therefore not only provide a drive to target neurons in their resting low state (Robledo and Feger, 1990), but also a widespread reset when stimulated into the high activity state. This situation has been experimentally described in vitro in all target neurons of the STN (SNr, GP, and EP neurons) in response to high intensity STN stimulation (Ammari et al., 2010). When the synaptic weight of recurrent collaterals is increased, the model predicts that oscillatory behavior can emerge. If we hypothesize that dopamine controls the weight of recurrent collateral synapses in the STN, this model explains the present results in the dopamine-depleted state.
The mechanisms of action of STN-HFS on the activity of STN target neurons have been rarely analyzed from in vivo or ex vivo preparations from animal models of Parkinson’s disease. In MPTP-treated monkeys, STN-HFS drives the activity of GP (Hashimoto et al., 2003) and SNr (Galati et al., 2006) neurons, whereas it inhibits SNr neurons in 6-hydroxydopamine lesioned rats (Benazzouz et al., 2000). In neuroleptic-treated rats, STN-HFS, at parameters that reverse catalepsy in freely moving animals, either excited or inhibited the firing of SNr neurons. However, the net resultant effect was a reshaping of their firing into a tonic and regular mode (Degos et al., 2005; Deniau et al., 2010). We have previously shown that the polysynaptic late component of the STN-evoked compound EPSC in GP neurons is suppressed by 10 Hz stimulation (Ammari et al., 2010) as in SNr neurons (Shen and Johnson, 2008) in control slices. The present results show that 10 Hz stimulation depresses the late component of the compound EPSC in DA-depleted GP neurons, but leaves some late EPSCs during the interstimulus interval. STN-HFS at 100 Hz is more efficient at weakening the transmission along the STN-GP pathway because it reduces the amplitude of all EPSCs. This form of depression could result from desensitization (Jones and Westbrook, 1996), internalization (Carroll et al., 1999) and surface trafficking (Choquet, 2010) of AMPA receptors.
In previous studies we showed that STN-HFS suppresses endogenous firing of STN somas and drives their activity probably via antidromic activation of their efferent axons (Garcia et al., 2003, 2005). The present study shows that orthodromically, STN-HFS not only suppresses the transmission of cortically evoked, pathological oscillations from STN to GP neurons but also introduces small amplitude HFS-evoked, postsynaptic EPSCs, that could be considered as a “stimulated resting” glutamatergic noise. This new regular synaptic noise under HFS could serve to regularize GP neuronal firing and re-establish synaptic integrative properties of GP neurons.
We propose that dopamine depletion transforms the STN into a central generator of bursts in which glutamatergic, rather than GABAergic, inputs to the STN play a central role. Transmission of these pathological oscillations to target neurons of the STN is readily interrupted by STN-HFS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by Institut National de la Santé et de la Recherche Médicale (Inserm), Centre National de la Recherche Scientifique (CNRS), and grant from ERA-Net Neuron (PhysDBS). Rachida Ammari was supported by a PhD grant from the Ministère de la Recherche et de la technologie (MRT) and Fondation pour la Recherche Médicale (FRM).
Ammari, R., Lopez, C., Bioulac, B., Garcia, L., and Hammond, C. (2010). Subthalamic nucleus evokes similar long lasting glutamatergic excitations in pallidal, entopeduncular and nigral neurons in the basal ganglia slice. Neuroscience 166, 808–818.
Baufreton, J., Atherton, J. F., Surmeier, D. J., and Bevan, M. D. (2005). Enhancement of excitatory synaptic integration by GABAergic inhibition in the subthalamic nucleus. J. Neurosci. 25, 8505–8517.
Baufreton, J., and Bevan, M. D. (2008). D-2-like dopamine receptor-mediated modulation of activity-dependent plasticity at GABAergic synapses in the subthalamic nucleus. J. Physiol. (Lond.) 586, 2121–2142.
Benabid, A. L., Chabardes, S., Torres, N., Piallat, B., Krack, P., Fraix, V., and Pollak, P. (2009). Functional neurosurgery for movement disorders: a historical perspective. Prog. Brain Res. 175, 379–391.
Benazzouz, A., Gao, D. M., Ni, Z. G., Piallat, B., Bouali-Benazzouz, R., and Benabid, A. L. (2000). Effect of high-frequency stimulation of the subthalamic nucleus on the neuronal activities of the substantia nigra pars reticulata and ventrolateral nucleus of the thalamus in the rat. Neuroscience 99, 289–295.
Bergman, H., Wichmann, T., Karmon, B., and DeLong, M. R. (1994). The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J. Neurophysiol. 72, 507–520.
Beurrier, C., Ben-Ari, Y., and Hammond, C. (2006). Preservation of the direct and indirect pathways in an in vitro preparation of the mouse basal ganglia. Neuroscience. 140, 77–86.
Beurrier, C., Bioulac, B., and Hammond, C. (2000). Slowly inactivating sodium current (INap) underlies single-spike activity in rat subthalamic neurons. J. Neurophysiol. 83, 1951–1957.
Beurrier, C., Congar, P., Bioulac, B., and Hammond, C. (1999). Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J. Neurosci. 19, 599–609.
Bevan, M. D., Atherton, J. F., and Baufreton, J. (2006). Cellular principles underlying normal and pathological activity in the subthalamic nucleus. Curr. Opin. Neurobiol. 16, 621–628.
Bevan, M. D., Hallworth, N. E., and Baufreton, J. (2007). GABAergic control of the subthalamic nucleus. Prog. Brain Res. 160, 173–188.
Bevan, M. D., Magill, P. J., Terman, D., Bolam, J. P., and Wilson, C. J. (2002). Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 25, 525–531.
Bevan, M. D., and Wilson, C. J. (1999). Mechanisms underlying spontaneous oscillation and rhythmic firing in rat subthalamic neurons. J. Neurosci. 19, 7617–7628.
Brown, L. L., Markman, M. H., Wolfson, L. I., Dvorkin, B., Warner, C., and Katzman, R. (1979). A direct role of dopamine in the rat subthalamic nucleus and an adjacent intrapeduncular area. Science 206, 1416–1418.
Carroll, R. C., Lissin, D. V., von, Z. M., Nicoll, R. A., and Malenka, R. C. (1999). Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat. Neurosci. 2, 454–460.
Choquet, D. (2010). Fast AMPAR trafficking for a high-frequency synaptic transmission. Eur. J. Neurosci. 32, 250–260.
Cragg, S. J., Baufreton, J., Xue, Y., Bolam, J. P., and Bevan, M. D. (2004). Synaptic release of dopamine in the subthalamic nucleus. Eur. J. Neurosci. 20, 1788–1802.
Degos, B., Deniau, J. M., Le, C. J., Mailly, P., and Maurice, N. (2008). Evidence for a direct subthalamo-cortical loop circuit in the rat. Eur. J. Neurosci. 27, 2599–2610.
Degos, B., Deniau, J. M., Thierry, A. M., Glowinski, J., Pezard, L., and Maurice, N. (2005). Neuroleptic-induced catalepsy: electrophysiological mechanisms of functional recovery induced by high-frequency stimulation of the subthalamic nucleus. J. Neurosci. 25, 7687–7696.
Deniau, J. M., Degos, B., Bosch, C., and Maurice, N. (2010). Deep brain stimulation mechanisms: beyond the concept of local functional inhibition. Eur. J. Neurosci. 32, 1080–1091.
Drouot, X., Oshino, S., Jarraya, B., Besret, L., Kishima, H., Remy, P., Dauguet, J., Lefaucheur, J. P., Dolle, F., Conde, F., Bottlaender, M., Peschanski, M., Keravel, Y., Hantraye, P., and Palfi, S. (2004). Functional recovery in a primate model of Parkinson’s disease following motor cortex stimulation. Neuron 44, 769–778.
Eusebio, A., Pogosyan, A., Wang, S., Averbeck, B., Gaynor, L. D., Cantiniaux, S., Witjas, T., Limousin, P., Azulay, J. P., and Brown, P. (2009). Resonance in subthalamo-cortical circuits in Parkinson’s disease. Brain 132, 2139–2150.
Flores, G., Liang, J. J., Sierra, A., Martinez-Fong, D., Quirion, R., Aceves, J., and Srivastava, L. K. (1999). Expression of dopamine receptors in the subthalamic nucleus of the rat: characterization using reverse transcriptase-polymerase chain reaction and autoradiography. Neuroscience 91, 549–556.
Frank, M. J. (2006). Hold your horses: a dynamic computational role for the subthalamic nucleus in decision making. Neural. Netw. 19, 1120–1136.
Fregni, F., Boggio, P. S., Santos, M. C., Lima, M., Vieira, A. L., Rigonatti, S. P., Silva, M. T., Barbosa, E. R., Nitsche, M. A., and Pascual-Leone, A. (2006). Noninvasive cortical stimulation with transcranial direct current stimulation in Parkinson’s disease. Mov. Disord. 21, 1693–1702.
Galati, S., Mazzone, P., Fedele, E., Pisani, A., Peppe, A., Pierantozzi, M., Brusa, L., Tropepi, D., Moschella, V., Raiteri, M., Stanzione, P., Bernardi, G., and Stefani, A. (2006). Biochemical and electrophysiological changes of substantia nigra pars reticulata driven by subthalamic stimulation in patients with Parkinson’s disease. Eur. J. Neurosci. 23, 2923–2928.
Garcia, L., Audin, J., D’Alessandro, G., Bioulac, B., and Hammond, C. (2003). Dual effect of high-frequency stimulation on subthalamic neuron activity. J. Neurosci. 23, 8743–8751.
Garcia, L., D’Alessandro, G., Fernagut, P. O., Bioulac, B., and Hammond, C. (2005). The impact of high frequency stimulation parameters on the pattern of discharge of subthalamic neurons. J. Neurophysiol. 94, 3362–3369.
Gillies, A., and Willshaw, D. (2004). Models of the subthalamic nucleus. The importance of intranuclear connectivity. Med. Eng. Phys. 26, 723–732.
Gradinaru, V., Mogri, M., Thompson, K. R., Henderson, J. M., and Deisseroth, K. (2009). Optical deconstruction of parkinsonian neural circuitry. Science 324, 354–359.
Hammond, C., Bergman, H., and Brown, P. (2007). Pathological synchronization in Parkinson’s disease: networks, models and treatments. Trends Neurosci. 30, 357–364.
Hashimoto, T., Elder, C. M., Okun, M. S., Patrick, S. K., and Vitek, J. L. (2003). Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J. Neurosci. 23, 1916–1923.
Hassani, O. K., Francois, C., Yelnik, J., and Feger, J. (1997). Evidence for a dopaminergic innervation of the subthalamic nucleus in the rat. Brain Res. 749, 88–94.
Jones, M. V., and Westbrook, G. L. (1996). The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 19, 96–101.
Kass, J. I., and Mintz, I. M. (2006). Silent plateau potentials, rhythmic bursts, and pacemaker firing: three patterns of activity that coexist in quadristable subthalamic neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 183–188.
Kita, H., Chang, H. T., and Kitai, S. T. (1983). The morphology of intracellularly labeled rat subthalamic neurons: a light microscopic analysis. J. Comp. Neurol. 215, 245–257.
Lefaucheur, J. P. (2009). Treatment of Parkinson’s disease by cortical stimulation. Expert Rev. Neurother. 9, 1755–1771.
Litvak, V., Jha, A., Eusebio, A., Oostenveld, R., Foltynie, T., Limousin, P., Zrinzo, L., Hariz, M. I., Friston, K., and Brown, P. (2011). Resting oscillatory cortico-subthalamic connectivity in patients with Parkinson’s disease. Brain 134, 359–374.
Magill, P. J., Bolam, J. P., and Bevan, M. D. (2001). Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus-globus pallidus network. Neuroscience 106, 313–330.
Magill, P. J., Sharott, A., Bevan, M. D., Brown, P., and Bolam, J. P. (2004). Synchronous unit activity and local field potentials evoked in the subthalamic nucleus by cortical stimulation. J. Neurophysiol. 92, 700–714.
Mallet, N., Pogosyan, A., Sharott, A., Csicsvari, J., Bolam, J. P., Brown, P., and Magill, P. J. (2008). Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J. Neurosci. 28, 4795–4806.
Mansour, A., Meador-Woodruff, J. H., Bunzow, J. R., Civelli, O., Akil, H., and Watson, S. J. (1990). Localization of dopamine D2 receptor mRNA and D1 and D2 receptor binding in the rat brain and pituitary: an in situ hybridization-receptor autoradiographic analysis. J. Neurosci. 10, 2587–2600.
Meibach, R. C., and Katzman, R. (1979). Catecholaminergic innervation of the subthalamic nucleus: evidence for a rostral continuation of the A9 (substantia nigra) dopaminergic cell group. Brain Res. 173, 364–368.
Moody, C. A., and Spear, L. P. (1992). Effects of acute dopamine depletion on responsiveness to D1 and D2 receptor agonists in infant and weanling rat pups. Psychopharmacology (Berl.) 107, 39–49.
Murer, M. G., Riquelme, L. A., Tseng, K. Y., and Pazo, J. H. (1997). Substantia nigra pars reticulata single unit activity in normal and 60HDA-lesioned rats: effects of intrastriatal apomorphine and subthalamic lesions. Synapse 27, 278–293.
Otsuka, T., Murakami, F., and Song, W. J. (2001). Excitatory postsynaptic potentials trigger a plateau potential in rat subthalamic neurons at hyperpolarized states. J. Neurophysiol. 86, 1816–1825.
Plenz, D., and Kitaï, S. T. (1999). A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature 400, 677–682.
Robledo, P., and Feger, J. (1990). Excitatory influence of rat subthalamic nucleus to substantia nigra pars reticulata and the pallidal complex: electrophysiological data. Brain Res. 518, 47–54.
Ryan, L. J., and Sanders, D. J. (1993). Subthalamic nucleus lesion regularizes firing patterns in globus pallidus and substantia nigra pars reticulata neurons in rats. Brain Res. 626, 327–331.
Sharott, A., Magill, P. J., Harnack, D., Kupsch, A., Meissner, W., and Brown, P. (2005). Dopamine depletion increases the power and coherence of beta-oscillations in the cerebral cortex and subthalamic nucleus of the awake rat. Eur. J. Neurosci. 21, 1413–1422.
Shen, K. Z., and Johnson, S. W. (2000). Presynaptic dopamine D2 and muscarine M3 receptors inhibit excitatory and inhibitory transmission to rat subthalamic neurones in vitro. J. Physiol. 525(Pt 2), 331–341.
Shen, K. Z., and Johnson, S. W. (2006). Subthalamic stimulation evokes complex EPSCs in the rat substantia nigra pars reticulata in vitro. J. Physiol. 573, 697–709.
Shen, K. Z., and Johnson, S. W. (2008). Complex EPSCs evoked in substantia nigra reticulata neurons are disrupted by repetitive stimulation of the subthalamic nucleus. Synapse 62, 237–242.
Song, W. J., Baba, Y., Otsuka, T., and Murakami, F. (2000). Characterization of Ca(2+) channels in rat subthalamic nucleus neurons. J. Neurophysiol. 84, 2630–2637.
Standaert, D. G., Testa, C. M., Young, A. B., and Penney, J. B. Jr. (1994). Organization of N-methyl-D-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J. Comp. Neurol. 343, 1–16.
Wilson, C. L., Cash, D., Galley, K., Chapman, H., Lacey, M. G., and Stanford, I. M. (2006). Subthalamic nucleus neurones in slices from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mice show irregular, dopamine-reversible firing pattern changes, but without synchronous activity. Neuroscience 143, 565–572.
Keywords: basal ganglia, subthalamic nucleus, Parkinson, high frequency stimulation, burst firing, basal ganglia slice
Citation: Ammari R, Bioulac B, Garcia L and Hammond C (2011) The subthalamic nucleus becomes a generator of bursts in the dopamine-depleted state. Its high frequency stimulation dramatically weakens transmission to the globus pallidus. Front. Syst. Neurosci. 5:43. doi: 10.3389/fnsys.2011.00043
Received: 29 March 2011; Accepted: 01 June 2011;
Published online: 13 June 2011.
Edited by:
James M. Tepper, Rutgers, The State University of New Jersey, USAReviewed by:
Steven W. Johnson, Oregon Health & Science University, USACopyright: © 2011 Ammari, Bioulac, Garcia and Hammond. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Constance Hammond, INMED UMR 901 Inserm, 163 route de Luminy, BP 13, 13273 Marseille Cédex 09, France. e-mail:aGFtbW9uZEBpbm1lZC51bml2LW1ycy5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.