 Claire N. J. Meunier1
Claire N. J. Meunier1 Pascal Chameau
Pascal Chameau Philippe M. Fossier
Philippe M. Fossier
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Synaptic Neurosci. , 01 February 2017
Volume 9 - 2017 | https://doi.org/10.3389/fnsyn.2017.00002
This article is part of the Research Topic Neuromodulatory state-dependent plasticity View all 9 articles
The prefrontal cortex (PFC) is involved in cognitive tasks such as working memory, decision making, risk assessment and regulation of attention. These functions performed by the PFC are supposed to rely on rhythmic electrical activity generated by neuronal network oscillations determined by a precise balance between excitation and inhibition balance (E/I balance) resulting from the coordinated activities of recurrent excitation and feedback and feedforward inhibition. Functional alterations in PFC functions have been associated with cognitive deficits in several pathologies such as major depression, anxiety and schizophrenia. These pathological situations are correlated with alterations of different neurotransmitter systems (i.e., serotonin (5-HT), dopamine (DA), acetylcholine…) that result in alterations of the E/I balance. The aim of this review article is to cover the basic aspects of the regulation of the E/I balance as well as to highlight the importance of the complementarity role of several neurotransmitters in the modulation of the plasticity of excitatory and inhibitory synapses. We illustrate our purpose by recent findings that demonstrate that 5-HT and DA cooperate to regulate the plasticity of excitatory and inhibitory synapses targeting layer 5 pyramidal neurons (L5PyNs) of the PFC and to fine tune the E/I balance. Using a method based on the decomposition of the synaptic conductance into its excitatory and inhibitory components, we show that concomitant activation of D1-like receptors (D1Rs) and 5-HT1ARs, through a modulation of NMDA receptors, favors long term potentiation (LTP) of both excitation and inhibition and consequently does not modify the E/I balance. We also demonstrate that activation of D2-receptors requires functional 5-HT1ARs to shift the E-I balance towards more inhibition and to favor long term depression (LTD) of excitatory synapses through the activation of glycogen synthase kinase 3β (GSK3β). This cooperation between different neurotransmitters is particularly relevant in view of pathological situations in which alterations of one neurotransmitter system will also have consequences on the regulation of synaptic efficacy by other neurotransmitters. This opens up new perspectives in the development of therapeutic strategies for the pharmacological treatment of neuronal disorders.
The prefrontal cortex (PFC) plays an important role in the processing of cognitive functions such attention, memory, decision making and more specifically in working memory (Fuster, 2001; Miller and Cohen, 2001). Its ability to integrate and process sensory information is ensured by a neuronal network receiving numerous afferent connections from sensory areas and making efferent connections to motor area and subcortical area (Miller and Cohen, 2001; Uylings et al., 2003). The higher-order brain functions performed by the PFC are supposed to rely on rhythmic electrical activity generated by neuronal network oscillations. Indeed, cortical networks are permanently active and their activity patterns, depending on the wakefulness or sleep states, are characterized by slow oscillations of the neuronal membrane potential (so-called up and down states) during slow wave sleep and by fluctuations around a depolarized level (persistent up state) during REM sleep and wakefulness (Steriade et al., 2001). Membrane potential fluctuations, the above described phase transitions between up and down state, can be spontaneously generated by local (surrounding) cortical synaptic connections. The up state is initiated by the recruitment of neurons through recurrent excitation and is regulated by local feedback inhibition, pyramidal and non-pyramidal neurons receiving strong barrages of synaptic input that cause membrane depolarization and action potential firing (up state). The down state is characterized by neuronal hyperpolarization, a reduction in synaptic bombardment and a reduction of action potential activity (Sanchez-Vives and McCormick, 2000; Timofeev et al., 2000; Compte et al., 2003; Shu et al., 2003a). This transition to the down state may result from synaptic depression or the buildup of intracellular calcium and sodium concentrations that activate potassium channels in neurons of the local network (Contreras et al., 1996; Sanchez-Vives and McCormick, 2000; Timofeev et al., 2000). These cortical network rhythms are determined by a precise balance between excitation and inhibition that results from the coordinated activities of recurrent excitation and feedback and feedforward inhibition (Shu et al., 2003b; Haider et al., 2006). The initiation of the up state is characterized by an increase in excitatory conductance followed by an increase in inhibitory conductance. Following onset and during the up state, excitatory and inhibitory conductances remain proportionally increased and dynamically balanced (Haider et al., 2006). This balanced activity keeps neurons at a noisy and elevated level of depolarization near firing threshold and prevents aberrant network activity. In addition to being spontaneously generated (Sanchez-Vives and McCormick, 2000; Timofeev et al., 2000; Shu et al., 2003b), both transitions from up to down and from down to up can be triggered by sensory input and activation of afferent inputs (Anderson et al., 2000; Petersen et al., 2003; Shu et al., 2003a; MacLean et al., 2005; Rigas and Castro-Alamancos, 2007).
Within cortical networks, interneurons are in charge of the dynamic adjustment of the level of excitation, and their role in the maintenance of a dynamic balance between excitation and inhibition balance (E-I balance) is essential in cortical function (Shu et al., 2003b). The E-I balance is a value that reflects the activity of neuronal network at one time (Haider and McCormick, 2009). A proper E-I balance is essential for physiological processes such as sensory perception, short term memory, long term memory and development (Saghatelyan et al., 2001; Egorov et al., 2002; Monier et al., 2003; Zhang et al., 2011). Alterations in maintaining the E-I balance have been observed in several pathologies such as epilepsy and autism. For instance, an increase in GABAergic transmission leading to a change in excitation has been observed in epileptic tissue (Cossart et al., 2001). In the case of autism, the E-I balance appears to vary in the direction of greater excitation (Rubenstein and Merzenich, 2003; Powell, 2004). These pathologies, currently considered as polygenic and multifactorial disorders, are characterized by a disruption of normal cortical connections with aberrant synaptic function and disorganized neurotransmitter interactions (Stephan et al., 2009). For instance, dysfunctions of dopaminergic, glutamatergic and serotoninergic systems have been associated with the pathophysiology of schizophrenia and major depression (Marek, 2007; Kantrowitz and Javitt, 2012). Although the pathophysiology of schizophrenia has been associated with alterations of the dopaminergic system (Creese et al., 1976; Meisenzahl et al., 2008), others suggest that the use of antipsychotic drugs acting at both the serotonergic and dopamine (DA) systems must be considered (Meltzer et al., 2004; Newman-Tancredi et al., 2007; Jones and McCreary, 2008; Newman-Tancredi, 2010; Meltzer and Massey, 2011; Newman-Tancredi and Kleven, 2011). Several studies have suggested that an imbalance between DA and serotonin (5-HT) systems could be the cause of adverse side effects observed during treatment of some pathologies (Borah and Mohanakumar, 2007; Carta et al., 2007; Navailles et al., 2010). These observations offer the possibility of a new therapeutic approach which consists in combining drugs that target several receptors to neuromodulators in the treatment of schizophrenia and major depression in order to reassess a proper E-I balance required for harmonious brain functions.
Synaptic plasticity is a fundamental property of the brain which allows the nervous system to adapt its response to experience. In this review article, we first briefly address the general mechanisms of synaptic plasticity leading to long term potentiation (LTP) or Depression of excitatory and inhibitory synapses and we introduce the notion of E-I balance. We also illustrate how the method we developed to determine the E-I balance offers the possibility to study simultaneously the plasticity of excitatory and inhibitory inputs on a layer 5 pyramidal neuron (L5PyN). The second part of this review article highlights our recent findings regarding the cooperation between serotoninergic and dopaminergic systems within the PFC in the orientation of synaptic plasticity towards potentiation or depression.
Synaptic plasticity is a fundamental property of neuronal circuits which endows the nervous system the ability to adapt its responses to a changing environment (sensory information). It confers the brain the capacity to adapt its behavior and to anticipate and control executive functions (Feldman, 2009). First postulated by Hebb (1949), the concept of synaptic plasticity was substantiated by Bliss and Lomo (1973) who demonstrated that, in hippocampus, a high frequency stimulation (HFS) of excitatory synapses induced a persistent enhancement of excitatory neurotransmission, the so called LTP. This extensively studied synaptic property is supposed to be the basic mechanism of learning and memory. It is initiated within a short period of time (a few seconds), it is stable and persistent after induction (for several weeks in vivo), it is initiated by frequencies of stimulation (theta burst) similar to the neuronal rhythmic activity observed in cortical networks engaged in various information processing tasks. Long term depression (LTD), the opposite of LTP, corresponds to a long-lasting decrease of synaptic efficacy (several hours) in response to low frequency stimulations (Dudek and Bear, 1992; Bear and Malenka, 1994).
These two forms of long-lasting synaptic plasticity do not oppose each other but rather are considered to be complementary in memory formation. For instance, it has been shown, in hippocampus, that LTP and LTD have respective roles in encoding spatial information (Kemp and Manahan-Vaughan, 2004). LTD is necessary to acquire a new object location in a new environment while LTP is associated with spatial exploration. Other studies have suggested that LTD could contribute to the acquisition of new information by weakening previously strengthened synapses and by preventing interferences with this new incoming information (Etkin et al., 2006; Nicholls et al., 2008; Malleret et al., 2010).
Most of the studies conducted over the past decades relate to the plasticity of excitatory synapses and it is rather recently that attention has been focused on the plasticity of inhibitory synapses. In the following chapters, we will summarize the knowledge on these two aspects of long-term synaptic plasticity.
First discovered in hippocampus in the mid-eighties, LTP of excitatory synapses is generally considered as being a process requiring the activation of post (or pre) synaptic NMDA receptors and an increase in intracellular calcium that leads to an increase in synaptic efficacy after HFS of presynaptic terminals (Malenka and Nicoll, 1999). The most generally described mechanism to induce LTP is the activation of post-synaptic AMPA and NMDA receptors following a massive release of glutamate at the synaptic cleft induced by HFS of the presynaptic afferents. The activation of post-synaptic AMPA receptors results in a strong depolarization of the post-synaptic element and removes the magnesium blockade of the NMDA receptors (Kleckner and Dingledine, 1988; Burnashev et al., 1992; Calabresi et al., 1992) allowing cations to flow through the activated NMDA receptors. The high calcium permeability of NMDA receptors results in a calcium influx into the post-synaptic compartment that leads to changes in synaptic strength through a well described mechanism involving the activation of the calmodulin-dependent protein kinase II (CaMKII) and the subsequent phophorylation of AMPA receptors (increased open probability) and/or the trafficking of AMPA receptors to the post-synaptic membrane. Other kinases have been reported to be involved in the trafficking of AMPA receptors. Recent studies have shown that LTP can be induced at the presynaptic level either through a modulation of pre-synaptic NMDA receptors or through neuromodulators (NO, BDNF) released from the post-synaptic site acting at the presynaptic level. Following a theta-burst stimulation in hippocampus, McGuinness et al. (2010) have shown that action potentials at the synapse evoke a calcium influx through voltage-dependent calcium channel (VDCC) that induces glutamate release which can activate presynaptic NMDA receptors. This together with the removal of the Mg2+ block of NMDA receptors by depolarization results in an increase in intracellular calcium concentration and an enhanced glutamate release. Other mechanisms have been proposed to be responsible for the induction of LTP at excitatory synapses. The best characterized is the regulation of synaptic strength by retrograde messengers such as nitric oxide (NO). As shown in hippocampus, calcium ions entering the post-synaptic element through NMDA receptors activate NO synthase to produce NO from L-arginine. NO diffuses back to the presynaptic element and activates guanylate cyclase (GC) leading to the synthesis of cGMP and the activation of PKG and an increase in glutamate release (Costa et al., 2011). More recent studies reported that post-synaptic protein synthesis can also play a role in the regulation of the pre-synaptic mechanisms involved in persistent forms of LTP (Johnstone and Raymond, 2013). It has also been suggested that postsynaptic activation of the translation promotor mTOR complex 1 (mTORC1) results in the synthesis of the retrograde messenger BDNF and an enhancement of neurotransmitter release (Henry et al., 2012). Although NMDA receptors have a key role in the induction of LTP, it has been demonstrated in the visual cortex and in hippocampus (Johnston et al., 1992) that other players can trigger the calcium signal responsible for LTP induction. Grover and Teyler (1994) have shown that, in hippocampus, pairing presynaptic stimulation with post synaptic depolarization or with action potential firing results in a post synaptic calcium influx through L-type VDCC responsible for the induction of LTP. Other forms of NMDA-independent LTP have also been reported in rodent cortex that requires the activation of metabotropic glutamate receptors (Wilsch et al., 1998; Huemmeke et al., 2002).
The induction of LTD of excitatory synapses is mainly determined by the magnitude of the post-synaptic increase in intracellular calcium concentration (Mulkey and Malenka, 1992). A smaller increase in intracellular calcium concentration evoked by the activation of NMDA receptors will lead to the activation of a signaling cascade involving phosphatases such as calcineurin and PP1 (Lisman and Zhabotinsky, 2001) which results in the de-phosphorylation of synaptic AMPA receptors and their internalization causing a reduction of synaptic efficacy. Other forms of LTD involve the activation of metabotropic glutamate receptors followed by an increase in intracellular calcium concentration. Depending on the receptor subtype different signaling pathways are engaged. In pyramidal neurons from the neocortex (Czarnecki et al., 2007), mGluR1 receptors activation causes an increase in intracellular calcium that leads to the activation of protein kinase C (PKC) and AMPA receptors internalization. It has also been suggested that activation of mGluR5 or mGluR1 receptors, respectively in striatum and in nucleus accumbens, results in an increase in post-synaptic calcium concentration and in the synthesis of endocanabinoïds (eCBs) which activate presynaptic CB1R to decrease glutamate release (Chevaleyre et al., 2006). Finally another form of LTD has been described in visual cortex where LTD is induced by coincident activation of presynaptic NMDA auto-receptors and of CB1R, activated by retrograde signaling of eCBs (Sjöström et al., 2003).
Post synaptic changes of GABAergic neurotransmission can be explained by: (i) a modulation of GABA receptors activity by protein kinases such as PKC, CaMKII or by phosphatases such as calcineurine (Kittler and Moss, 2003; Houston et al., 2009); (ii) trafficking of GABAA receptors at the synapse (Saliba et al., 2007; Arancibia-Cárcamo and Kittler, 2009; Castillo et al., 2011). As for excitatory synapses, modulations of postsynaptic calcium concentration are supposed to play a role in the induction of inhibitory synaptic plasticity. It has been demonstrated in hippocampus that LTP of inhibitory synaptic transmission is induced by an increase in post-synaptic calcium concentration resulting from the activation of GABAB receptors and the subsequent synthesis of IP3 leading to the release of calcium from the IP3-sensitive calcium store (Gaiarsa and Ben-Ari, 2006). It has also been demonstrated that, in the case of heterosynaptic plasticity, different sources of calcium (VGCC, ionotropic NMDA receptors, metabotropic mGluR1 glutamate receptors) contribute to the increases in post-synaptic calcium concentration responsible for changes in synaptic efficacy (Goldberg et al., 2003; Nyíri et al., 2003; Topolnik et al., 2005). This variety of post-synaptic calcium sources allows a precise control of calcium signaling and a fine tuning of synaptic plasticity (Camiré and Topolnik, 2012).
The best characterized form of presynaptic plasticity of GABAergic synapse involves the activation of presynaptic receptors by retrograde messengers such as: eCBs, BDNF and NO. eCB-mediated synaptic plasticity has been described in several brain regions such as hippocampus (Chevaleyre and Castillo, 2003), amygdala (Marsicano et al., 2002; Azad et al., 2004), striatum (Adermark and Lovinger, 2009), visual cortex (Jiang et al., 2010) and cerebellum (Pitler and Alger, 1992; Llano et al., 1994). The induction of eCB-mediated plasticity is initiated by glutamate release from presynaptic terminals which induces the post-synaptic synthesis of eCBs. eCBs retrogradely activate CB1R located on presynaptic afferents from GABAergic interneurons (heterosynaptic iLTD) to decrease GABA release through a signaling cascade which inhibits the cAMP/protein kinase A (PKA) pathway (Heifets and Castillo, 2009). Whereas eCBs induce i-LTD, other retrograde messengers such as BDNF and NO can induce i-LTP. It has been shown in visual cortex and hippocampus (Lu, 2003; Gubellini et al., 2005; Sivakumaran et al., 2009) that the activation of NMDA receptors and/or VGCC and the subsequent increase in intracellular calcium concentration in the post synaptic element can result in the synthesis of BDNF or NO which increases GABA release (Nugent et al., 2007).
Neurons in the cerebral cortex consist of a majority of excitatory (glutamatergic) pyramidal neurons (75%–80%) making synaptic contacts both locally (local network) and over long distances (across distinct cortical areas) and of inhibitory (GABAergic) interneurons (25%–20%) making extensive local connections. Within a given local cortical network, neurons receive inputs from neighboring neurons which form recurrent excitatory and feedforward and feedback inhibitory circuits (Peters and Kara, 1985; White, 1989; Abeles, 1991; Markram et al., 2004). The great diversity of interneurons, in terms of their connectivity and their functional properties, allows them to control cortical excitation leading to a dynamic equilibrium between excitation and inhibition (Shu et al., 2003b). This dynamic control of excitation that regulates cortical network activity is performed through feedback and feedforward inhibition (Isaacson and Scanziani, 2011). Feedforward inhibition occurs when excitatory afferents activate first interneurons, prior to activating principal neurons, and results in a reduced excitation of the target neuron. This well described mechanism (Isaacson and Scanziani, 2011) contributes to the weakening of excitatory inputs. Feedback inhibition takes place when active principal neurons excite inhibitory interneurons which, in a feedback loop, inhibit principal neurons themselves (for instance, see Silberberg and Markram, 2007; Berger et al., 2010) to ensure the stability to the network.
It is remarkable to note that despite the diversity of neuronal networks, the relative contribution of excitatory and inhibitory input conductance to a given neuron is dynamically maintained at comparable values across different cortical layers and different cortical areas to guarantee a proper balance between excitation and inhibition (E-I balance). The E-I balance, determined as the ratio between excitatory and inhibitory input conductance onto a neuron evoked by an electrical stimulation of afferents (in vitro) or by sensory stimuli (in vivo), is maintained at approximately 20% excitation and 80% inhibition (Le Roux et al., 2006; Monier et al., 2008; Lucas-Meunier et al., 2009; Zhang et al., 2011; Xue et al., 2014; den Boon et al., 2015). This dynamic E-I balance is thought to result from the coordinated activities of direct and recurrent excitation and (feed-forward and feedback) inhibition and reflects the activity of neuronal networks at a given moment (Haider and McCormick, 2009; Isaacson and Scanziani, 2011). A proper balance between excitatory and inhibitory inputs onto cortical neurons is essential to maintain the stability of cortical networks in order to perform cognitive functions such as memory and sensory information processing. For instance, it determines proper cortical network rhythms responsible for higher order cognitive functions (Shu et al., 2003b; Haider et al., 2006). It is also essential for physiological processes such as short term memory and long term memory (Saghatelyan et al., 2001; Egorov et al., 2002). Cortical responses elaborated by L5PyNs depend on the balance between excitatory and inhibitory inputs perceived (Borg-Graham et al., 1998; Wehr and Zador, 2003). Balanced excitatory and inhibitory neurotransmission also appears to be fundamental in the fine tuning of neuronal responses to specific sensory features.
It is conceivable to think that any alterations of excitation or inhibition will generate aberrant information processing in the cortex (Haider and McCormick, 2009) and will lead to pathological situations. Indeed, disturbances in the E-I balance are associated with a broad spectrum of neuropsychiatric and neurological diseases, such as autism, schizophrenia and epilepsy (Cobos et al., 2005; Lewis et al., 2005; Rubenstein, 2010). It has been observed that, in epilepsy, an altered maturation of chandelier cells (interneurons) interferes with the activity of pyramidal neurons (Marco et al., 1996), leading to an unbalanced E-I ratio (Cossart et al., 2005). It is also hypothesized that, in schizophrenia, a dysfunction of NMDA receptors (Coyle, 2006) changes the level of excitation and consequently the E-I balance (Kehrer et al., 2008). Finally, a dysregulation of the E-I balance has also been observed in autism (Rubenstein and Merzenich, 2003; Powell, 2004; Rippon et al., 2007), Rett syndrome (Dani et al., 2005) and Tourette syndrome (Singer and Minzer, 2003).
Given that neuronal networks require a proper E-I balance to perform cognitive functions, how does network stability deal with synaptic plasticity? In other words, how to learn within dynamically balanced networks? The learning process might not only be considered not only as a gain or a loss of efficacy at a given synapse but also as dynamic changes of both excitatory and inhibitory synaptic strength. This homeostatic maintenance of excitation and inhibition balance may be a determining factor in the regulation of neuronal input-output function and information processing (learning) within a given neuronal network (Daoudal and Debanne, 2003; Staff and Spruston, 2003; Marder and Buonomano, 2004). Although the effects of concerted regulation of excitation and inhibition are still poorly understood, it has been suggested that balanced or imbalanced changes in synaptic strength of excitatory and inhibitory inputs have a different impact on the neuronal input-output function and consequently on neuronal firing. Indeed the two parameters, threshold and gain (rate of change or sensitivity of the input-output function), that characterize the probability of a neuron to fire action potentials as a function of stimulus intensity can be modulated by the ratio between excitation and inhibition. By combining a computational model with experimental data obtained in hippocampus, Carvalho and Buonomano (2009) have shown that imbalanced plasticity of excitatory synaptic inputs affects the threshold while the E-I balance affect the gain allowing neurons to optimize their information processing.
The method we use to determine the E-I balance is based on the continuous measurement of evoked postsynaptic currents and the decomposition of the conductance dynamics into its excitatory and inhibitory components (Monier et al., 2003, 2008). This continuous and simultaneous determination of both excitatory and inhibitory inputs also offers the possibility to determine the changes in excitatory and inhibitory synaptic strength occurring after applying a high frequency stimulus evoking long-term synaptic plasticity. We have shown that, in rat visual cortex and in mouse PFC, HFSs of layer 2–3 (theta-burst stimulation), not only induced LTP of excitatory inputs measured from L5PyNs but also LTP of inhibitory inputs resulting in an E-I balance that remains equal to the control situation (Le Roux et al., 2006; Meunier et al., 2013). These results are in accordance with the notion of homeostatic regulation which mainly involves a dynamic adjustment of excitatory and inhibitory circuits (Turrigiano and Nelson, 2004). Because alterations in several neurotransmitter systems have been correlated with cognitive deficits resulting in impairments of learning and memory, we will focus our attention on the importance of the cooperation between different neuromodulators in the modulation of the plasticity of excitatory and inhibitory synapses. Based on our recent findings, we will highlight the importance of the coordinated action of 5-HT and DA in regulating the interaction between excitation and inhibition in the PFC, a cortical area involved in higher order cognitive functions.
5-HT is the most widely distributed neuromodulator in the brain (Dahlström and Fuxe, 1964; Steinbusch, 1981). 5-HT is implicated in the regulation of many physiological functions such as mood, sleep, vigilance, cognitive functions, learning and memory. In the brain, serotoninergic axons originating from the raphe nucleus make synapses “en passant” and release 5-HT from varicosities (Oleskevich and Descarries, 1990) to activate a great diversity of 5-HT receptors (up to 16 types of receptors have been identified; Bockaert et al., 2006). We will focus here our attention on the role of the metabotropic 5-HT1A receptor (5-HT1AR) which is predominant in the PFC (Santana et al., 2004).
5-HT1ARs have been identified both on serotoninergic neurons (autoreceptors) where they regulate the release of 5-HT through a negative feedback and on neuronal targets of serotoninergic neurons where they function as heteroreceptors. In the cortex, the majority of postsynaptic 5-HT1ARs (50%–60%) are expressed in glutamatergic neurons. In L5PyNs, 5-HT1ARs are located in the soma, in the initial axonal part (Czyrak et al., 2003; Cruz et al., 2004; Santana et al., 2004) and in dendrites (Kia et al., 1996; Riad et al., 2000). We have shown for instance that in the cerebral cortex this specific distribution of 5-HTRs on pyramidal neurons is important to control output signals from the cortex (Moreau et al., 2010). In the PFC, a weaker proportion (25%) of 5-HT1ARs is also expressed in GABAergic interneurons projecting onto the dendrites of pyramidal cells (Santana et al., 2004). From this distribution of 5-HT1ARs in the PFC, it appears that 5-HT can regulate the excitability of both glutamatergic and GABAergic neurons (Andrade, 2011; Puig and Gulledge, 2011) but the modulatory effects of 5-HT1ARs activation are not clearly understood.
Several pathologies such as mood disorders, anxiety, psychosis and fear are associated with serotonergic disorders and dysfunction of 5-HT1ARs. For instance, it has been shown that the number of postsynaptic 5-HT1ARs is decreased in depressive or anxious human brain (Shively et al., 2006; Lanzenberger et al., 2007; Akimova et al., 2009). One of the most commonly used pharmacological treatments of anxiety disorders is the use of selective serotonin reuptake inhibitors (SSRIs; Kasper et al., 2005). However, failures in SSRI treatment seems to be associated with the polymorphism of the gene encoding 5-HT1AR (Lemonde et al., 2003; Czesak et al., 2012) which might differently affect the nature (or type) of pre- vs. post-synaptic 5-HT1AR (Bortolozzi et al., 2012). A better understanding of the role of post-synaptic 5-HT1AR in the PFC is a prerequisite to the design of more selective psychoactive drugs.
Dopaminergic fibers, mainly originating from the Substantia nigra, are widely distributed in all areas of the PFC and target layer 2 and layer 5 (Emson and Koob, 1978; Callier et al., 2003; Van De Werd et al., 2010). The effects of DA are mediated by the D1-class receptors (D1 and D5 receptors) and by the D2-class receptors (D2, D3 and D4 receptors; Beaulieu and Gainetdinov, 2011). Activation of the D1 class enhances the activity of adenylate cyclase whereas activation of the D2 class inhibits it (Girault and Greengard, 2004). In rat PFC, D1-like receptors (D1Rs) and D2Rs are localized on dendritic spines of L5PyNs (Gaspar et al., 1995; Negyessy and Goldman-Rakic, 2005; Paspalas and Goldman-Rakic, 2005). D2Rs are also present on GABAergic interneurons (Santana et al., 2009) while D1Rs are mainly expressed in the parvalbulmin-positive subtype of GABAergic interneurons (Glausier et al., 2009). Such a distribution of D1 and D2 receptors is again in favor of a modulatory role of DA on neuronal networks in the PFC.
Many electrophysiological studies have shown that activation of DA D1Rs favors the induction of LTP at hippocampal-PFC synapses by increasing NMDAR-mediated responses in PFC (Gurden et al., 2000; Chen et al., 2004). This enhancement of NMDA current (Shih, 2004; Tseng and O’Donnell, 2004) is explained by either the externalization of NMDA receptors (Dunah and Standaert, 2001; Dunah et al., 2004) or by their phosphorylation. D1Rs and NMDAR are co-localized in pyramidal neurons of the PFC and a direct interaction between the C-terminal domain of D1R and NR1-NR2A subunits of the NMDAR has been proposed to explain the increase in NMDA current (Kruse et al., 2009). Activation of D2Rs results in a decrease in NMDA currents (Zheng et al., 1999; Wang et al., 2003) presumably through the inhibition of the CaMKII by the PKA and the subsequent internalization of NMDARs (Wang et al., 2003). Alternatively, another signaling cascade leading to the internalization of the NR2B subunit of the NMDAR and a decrease in NMDA current has been proposed. It involves, in the PFC, an increase in the activity of the glycogen-synthase kinase-3 (GSK3) after D2R activation (Beaulieu et al., 2008; Li et al., 2009; Skinbjerg et al., 2009; Beaulieu and Gainetdinov, 2011; Sutton and Rushlow, 2011).
It is of particular interest to note that in the treatment of schizophrenia, new therapeutic strategies consist in targeting both 5-HT1ARs and DA receptors by combining either 5-HT1ARs agonists with D2Rs antagonists (Newman-Tancredi, 2010) or 5-HT1A agonists with a D1R agonist and a D2R antagonist to prevent positive and negative symptoms of schizophrenia (Newman-Tancredi and Kleven, 2011). These clinical observations, highlight the importance of taking into account the interactions between serotoninergic and dopaminergic systems and their consequences on the physiology of the PFC, a brain area particularly associated with depressive disorders and schizophrenia.
5-HT, through the activation of postsynaptic 5-HT1AR, is known to regulate the excitability of glutamatergic and GABAergic neurons in the PFC (Andrade, 2011; Puig and Gulledge, 2011). 5-HT modulates the induction of plasticity, depending on the 5-HT receptor subtype and brain regions (Kemp and Manahan-Vaughan, 2004). In the rat PFC, consequences of HFS on synaptic plasticity are rather complicated given that tetanic stimulations in superficial layers can induce either LTP or LTD or no plasticity. Indeed, it has been reported that such a HFS protocol can induce LTD in about one half of the cells and LTP in about one third of the cells, the remaining cells did not display any change of synaptic properties (Hirsch and Crepel, 1990, 1991; Nowicky and Bindman, 1993; Matsuda et al., 2006).
To explore the role of 5-HT1ARs in the modulation of the plasticity of excitatory and inhibitory synaptic inputs to L5PyNs of the PFC, we compared the excitatory and inhibitory conductances evoked by electrical stimulations of layer 2/3 between wild-type (129/Sv WT) and 5-HT1AR-knock out (5-HT1AR-KO) mice. These transgenic mice are considered as a model for anxiety disorder (Ramboz et al., 1998) and have been designed to better understand human psychiatric conditions where a profound decrease of 5-HT1AR expression has been demonstrated (Sargent et al., 2000; Bhagwagar et al., 2004; Shively et al., 2006; Lanzenberger et al., 2007; Akimova et al., 2009).
Our experimental approach, based on the simultaneous determination of excitatory (E) and inhibitory (I) conductance in L5PyNs, also allows the determination of the E-I balance in the PFC. Electrical stimulations of layer 2/3 (low frequency stimulation, 0.05 Hz) evoked complex postsynaptic current in L5PyNs (Figure 1A). The evoked total synaptic conductance (gT) was extracted (see Le Roux et al., 2006) and decomposed into excitatory (gE) and inhibitory (gI) conductance (Figure 1A). This allowed further evaluation of the relative contribution of evoked excitatory and inhibitory inputs reaching the soma of the recorded pyramidal neuron (Le Roux et al., 2006; Moreau et al., 2010). Calculated integrals of excitatory (IntgE) and inhibitory (IntgI) conductance were expressed as percentages of the integral of the total conductance (IntgT; Figure 1B). Analysis of recorded neurons showed that the E-I balance was significantly different in 5-HT1AR-KO mutants (23%–77%) compared to 129/Sv WT mice (20%–80%; Figure 1B). In control 129/Sv WT mice and in the presence of WAY 100635 (a 5-HT1AR selective antagonist) the E-I balance was shifted towards more excitation (24%–76%), a value similar to what we observed in 5HT1AR-KO mice (Meunier et al., 2013). These results show that L5PyNs in 129/Sv mice express functional 5HT1ARs and that 5HT1ARs play a crucial role in tuning the E-I balance in L5PyNs of the PFC.
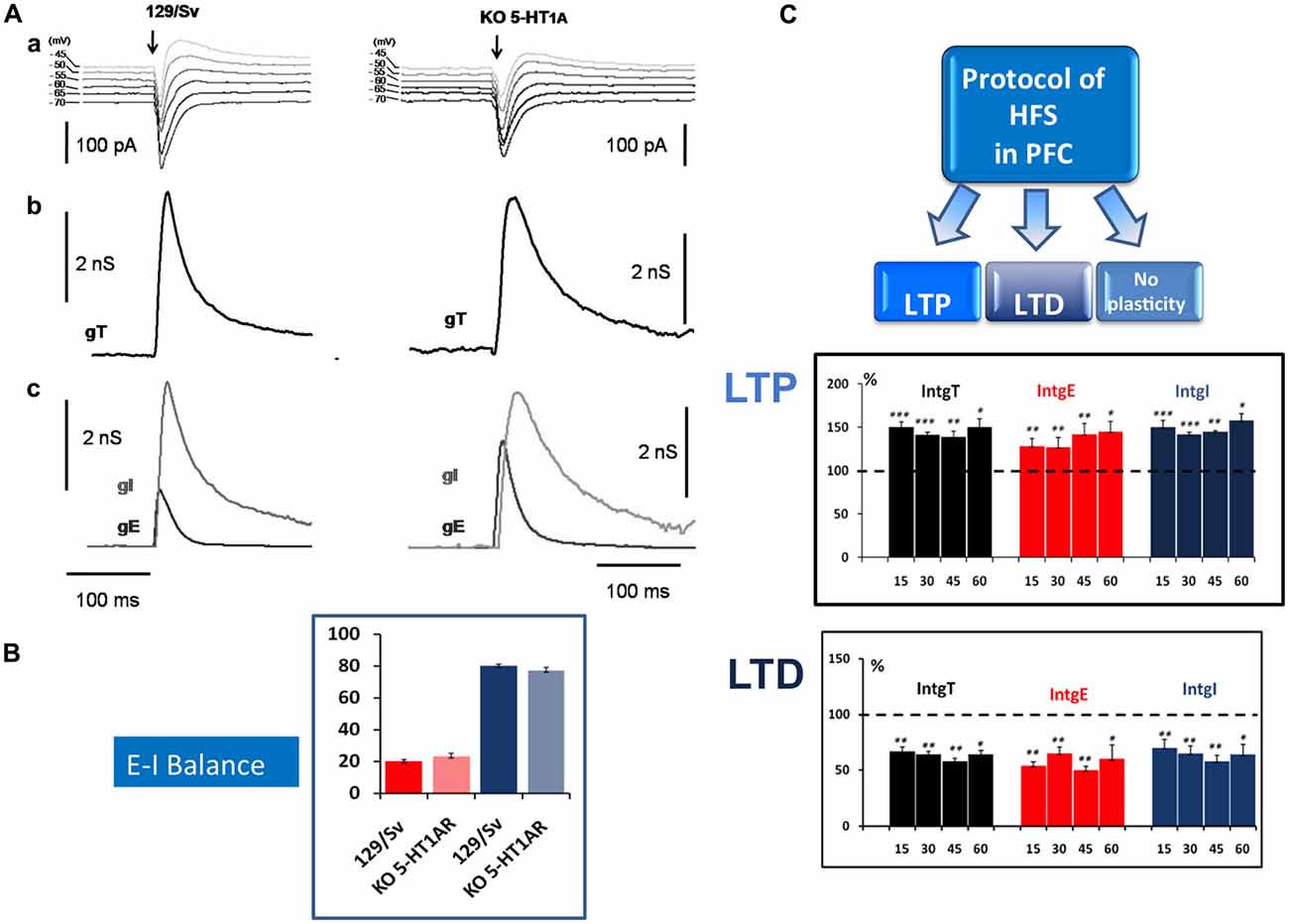
Figure 1. Determination of the Excitation-Inhibition balance (E-I balance) in the prefrontal cortex (PFC) and analysis of synaptic plasticity. (A) Brief description of the method to determine the E-I balance. (a) Representative current traces of synaptic responses (upper part) to layer 2–3 stimulation (arrow) recorded from a layer 5 pyramidal neuron (L5PyN) under voltage-clamp at various holding potentials in 129/Sv and KO 5-HT1A mice. (b) The second row represents the corresponding total conductance changes (gT) at the somatic level (for a complete description of the method, see Le Roux et al., 2006; Lucas-Meunier et al., 2009). (c) gT was then decomposed into its Excitatory (gE) and the Inhibitory (gI) components (third row). (B) The percentage of excitation and inhibition is expressed as the ratio of its integral value integrals of excitatory (IntgE) and inhibitory (IntgI) to the total conductance IntgT to determine the E-I balance. We observed a significant shift of the E-I balance towards more excitation between 129/Sv mice (20%–80%) and 5-HT1AR-KO mice (23%–77%; Meunier et al., 2013). (C) High frequency stimulation (HFS) protocol (Theta burst) in layer 2–3 of the PFC induced in L5PyNs, long term potentiation (LTP) or long term depression (LTD) or no plasticity of the responses. LTP (upper insert) was recorded 15 min, 30 min, 45 min and 60 min after the stimulation. Histograms represent relative changes (compared to the control before HFS: 100%) of IntgT, IntgE, IntgI. Synaptic conductances were determined under control conditions or after HFS protocol with a low frequency of stimulation (0.05 Hz). LTD (lower insert) was recorded in the same condition. *p < 0.05, **p < 0.01, ***p < 0.001.
We observed that, in both strains, HFS (theta burst) induced either LTP or LTD of both excitatory and inhibitory conductance or no plasticity (Figure 1C). These changes in synaptic efficacy were due to the activation of NMDARs since their blockade with D-L-AP5 prevented the induction of synaptic plasticity. When comparing 129/Sv WT and 5HT1AR-KO mice showing LTP of excitatory and inhibitory synaptic transmission, we observed that the absence of functional 5HT1AR resulted in a reduced potentiation of excitatory synaptic transmission while the potentiation of inhibition remained unaffected (Meunier et al., 2013). The use of a specific 5-HT1AR antagonist confirmed the role of 5-HT1ARs in the modulation of LTP of excitation in the PFC. We also observed that the proportion (calculated from the whole neuronal population) of L5PyNs displaying either LTP or LTD or no change were different between 129/Sv WT and 5-HT1AR-KO mice. In 5-HT1AR-KO mice, the percentage of neurons displaying LTP was significantly increased compared to 129/Sv WT mice (Figure 2) whereas the percentage of neurons showing LTD remained identical. Our results indicate that 5-HT1ARs play an important role in the orientation of the synaptic plasticity of L5PyNs in the PFC towards either LTP or LTD or no plasticity (Meunier et al., 2013).
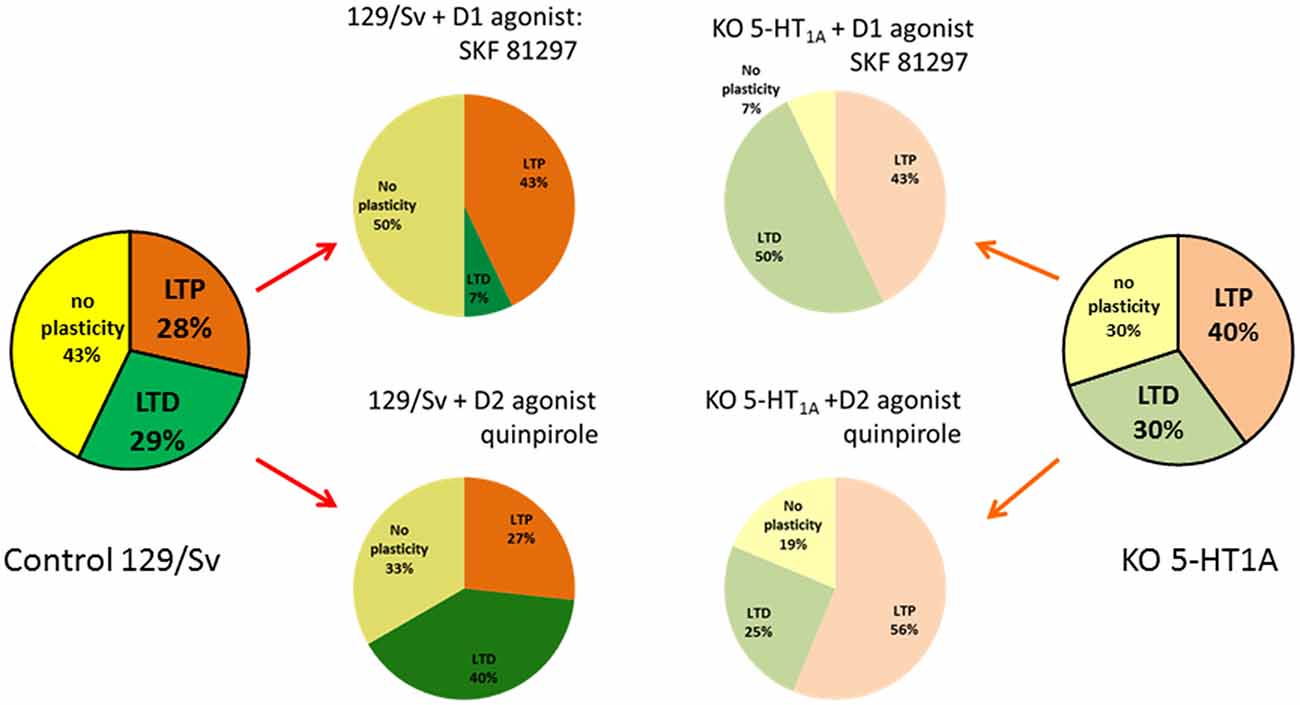
Figure 2. Schematic summary of the orientation of the plasticity in L5PyNs of the PFC. Ratio of L5PyNs displaying LTP, LTD or no plasticity calculated 45 min after HFS under different conditions. In each case, the analysis of the neuronal population results from 15 independent experiments. Left and right charts were obtained from control 129/Sv mice or from 5-HT1AR-knock out (5-HT1AR-KO) mice. Note that the ratio of L5PyNs displaying LTP was significantly increased in 5-HT1AR-KO mice (see Meunier et al., 2013). In 129/Sv mice, the D1-like receptors (D1Rs) agonist, SKF 81297, markedly reduced the proportion of L5PyNs displaying LTD while it enhanced the ratio of L5PyNs displaying LTP (Meunier et al., 2015). The D2Rs agonist, quinpirole, markedly increased the ratio of L5PyNs displaying LTD (from Meunier et al., 2017). In 5-HT1AR-KO mice, the D1Rs agonist, SKF 81297, markedly increased the proportion of L5PyNs displaying LTD while it reduced the ratio of L5PyNs displaying LTP. Activation of D2R with quinpirole had no significant effect on the ratio of L5PyNs displaying LTD.
It is established that different forms of synaptic plasticity such as LTP and LTD are induced by NMDAR activation (Stanton, 1996) that leads to the control of the trafficking of AMPA receptors. The externalization (Malenka and Nicoll, 1999; Malinow and Malenka, 2002) or the internalization (Beattie et al., 2000; Hanley and Henley, 2005; Fernández-Monreal et al., 2012) of AMPA receptors are commonly considered to be responsible for LTP and LTD induction respectively. The orientation of the synaptic plasticity towards LTP or LTD relying on AMPARs trafficking is known to be correlated with the magnitude of the dendritic calcium signal (Cormier et al., 2001). We observed that the activation of 5-HT1ARs enhanced evoked NMDA currents, indicating that 5-HT1ARs modulate NMDARs (Meunier et al., 2013). Therefore we propose that the increase in the percentage of neurons displaying LTP observed in the absence of functional 5-HT1ARs (5-HT1AR-KO mice) could result from a reduced calcium influx through NMDARs. So it appears that, in the PFC, 5-HT1ARs limit the induction of LTP and favor LTD by modulating the NMDA currents.
At hippocampal-PFC synapses, DA (acting at D1Rs) is known to favor LTP induction (Gurden et al., 2000; Chen et al., 2004). We have shown recently (Meunier et al., 2015) that a high frequency protocol of stimulation (HFS) in the presence of the D1R agonist SKF 81297 results in an increase of the population of L5PyNs displaying LTP in 129/Sv mice (Figure 2). By contrast, we observed an increase in the amount of neurons showing LTD when D1R were activated in the absence of functional 5-HT1ARs (KO). This cooperation between 5-HT1ARs and D1Rs to determine the direction of the synaptic plasticity was confirmed by a pharmacological approach (Meunier et al., 2015). We also observed in 129/Sv mice, an increase in the number of L5PyNs in the “no plasticity” class after D1Rs activation (Figure 2).
This orientation of the plasticity could be the result of a modulation of NMDA-mediated currents by D1Rs and 5-HT1ARs. A strong increase in intracellular calcium concentration within the dendritic spines of L5PyNs would facilitate LTP induction whereas a weaker calcium signal would facilitate LTD induction. D1Rs activation is known to modulate NMDARs and this modulation relies on the expression of different NMDARs subunits (NR2B vs. NR2A; Liu et al., 2006; Varela et al., 2009). The subunit composition of NMDARs determines the amplitude and the duration of the transient calcium signal that will lead direct synaptic plasticity towards either LTP or LTD (Mulkey and Malenka, 1992; Huang et al., 2010). We therefore make the assumption that the cooperation between 5-HT1ARs and D1Rs would set the magnitude of the calcium influx through NMDARs (Meunier et al., 2015). The resulting calcium signal at the dendrite would lead to the modulation of the CamKII activity (Chen et al., 2007; Ashpole et al., 2012; Coultrap et al., 2014) to control the trafficking of AMPARs at the synapse and consequently determine the direction of the synaptic plasticity. When D1Rs are activated in the absence of functional 5-HT1ARs (5-HT1AR-KO mice), the increased proportion of neurons showing LTD responses (Meunier et al., 2015) could then be the result of reduced NMDA currents due to changes in NMDARs subunits composition.
It cannot be excluded that other signaling pathways following 5-HT1ARs and D1Rs activation play a role in AMPARs trafficking and in the modulation of synaptic plasticity. Following 5-HT1AR activation (Polter and Li, 2010) as well as D1R activation (Gurden et al., 2000; Huang et al., 2004; Kruse et al., 2009), the cAMP/PKA signaling cascade could also be involved in regulating the incorporation of AMPARs at the synapse.
There is also growing evidence that D1/D5R stimulation leads to Gq-dependent activation of PLC, IP3-mediated Ca2+ release and CamKII activation (Chen et al., 2007). It is important to note that several studies have reported that metabotropic receptor activation coupled to PLC stimulation lead to LTD induction (Choi et al., 2005). In the 5-HT1ARs-KO mice, the D1Rs effects could mostly rely on the regulation of PLC activity.
DA is known to modulate NMDAR- and AMPAR-mediated currents, neuronal excitability and synaptic plasticity towards LTD through the activation of D2Rs. Both DA and 5-HT can regulate neuronal activity via a common signaling pathway involving the regulation of the activity of GSK3, a serine/threonine kinase playing an important role in the regulation of several receptors for neurotransmitters (Chen et al., 2009).
Two isoforms of GSK3 have been identified (GSK3α and GSK3β; Woodgett, 1990, 2001) which can be inactivated through their phosphorylation by Akt (Cross et al., 1995; Frame et al., 2001). It has been reported that, in the PFC and the striatum, the activation of D2Rs induces the activation of GSK3 (Beaulieu et al., 2004, 2008; Chen et al., 2009; Beaulieu and Gainetdinov, 2011). This activation of GSK3β has been shown to promote the internalization of the NR2B subunit of NMDARs leading in the rat hippocampus to the induction of LTD (Peineau et al., 2007) while LTP could be favored by higher levels of phosphorylation of GSK3β (Hooper et al., 2007; Peineau et al., 2007). It has also been observed that GSK3 can modulate NMDARs and AMPARs through either 5-HT1AR or 5-HT2AR activation (Li et al., 2004, 2007).
Importantly, like 5-HT1ARs and D2Rs, GSK3 is also a molecular target in the treatment of neuropsychiatric disorders such as major depression and anxiety (Meltzer et al., 2003; Meltzer and Massey, 2011). We therefore investigated the possible cooperation between 5-HT1AR and D2R in the modulation of synaptic plasticity within the PFC. We have shown that the activation of D2Rs and the downstream activation of GSK3 favor the induction of LTD of excitatory synaptic transmission (Meunier et al., 2017). In addition, we demonstrated that this control of synaptic plasticity via GSK3 requires the presence of functional 5-HT1ARs. We therefore made the hypothesis of cooperation between 5-HT1AR and D2R, via the regulation of GSK3, to modulate excitatory synaptic transmission. This hypothesis is supported by recent observations showing that D2R and 5-HT1AR can modulate the phosphorylation or the dephosphorylation of GSK3β (the dephosphorylated form being the active form).
The pathway downstream D2R activation leads, via the regulation of cyclic AMP production and β-arrestin activity, to the dephosphorylation (deactivation) of Akt and consequently to the dephosphorylation (activation) of GSK3 by the protein phosphatase 2A (PP2A Beaulieu et al., 2004, 2005, 2008, 2009; Beurel et al., 2015). There are apparent contradictory observations regarding the regulation of GSK3β by 5-HT receptors. It has been reported, in rat hippocampus, that the activation of 5-HT1ARs increases the phosphorylation of GSK3β by Akt, promoting the inactivated form (Polter et al., 2012). Others have suggested that 5-HT1ARs activation could activate GSK3 via the phosphorylation of Akt by PP2A (Hsiung et al., 2008). We have shown, in the PFC of 5-HT1AR-KO mice, an increase in the phosphorylated form of GSK3 (Meunier et al., 2017). This suggests that in the PFC, the activation of 5-HT1ARs promotes the active form of GSK3 and leads to the internalization of AMPARs and to the induction of LTD of excitatory synapses.
Our results highlight the importance of the cooperation between D2Rs and 5-HT1ARs in the regulation of the E-I balance and of the plasticity of excitatory synapses in the PFC. 5-HT2AR activation is also known to have a clear excitatory effect on L5PyNs and layer 2–3 GABAergic interneurons in the PFC (Andrade, 2011; Celada et al., 2013) to modulate NMDARs and AMPARs through GSK3 (Li et al., 2004, 2007). It it thus very likely that 5-HT2AR activation plays a role in the modulation of the E-I balance and in the modulation of long-term synaptic plasticity by DA. The functional dimerization between D2R and 5-HT2AR (Franklin and Carrasco, 2012) that can be modulated by cannabinoid receptors, introduces another level of complexity that has to be taken in account. Recent findings indicate a prominent co-localization of D2Rs and 5-HT1ARs in the neurons of the PFC compared to other brain regions (Łukasiewicz et al., 2016) suggesting that D2Rs-5-HT1ARs heteromers may be expressed in the PFC. When expressed in HEK cells (Łukasiewicz et al., 2016), it appears that the activation of such heteromers recruits metabolic pathways different from those downstream the activation of homomeric 5-HT1A or D2 receptors. This could open up new therapeutic strategies based on the selective activation of one or the other metabolic pathways to improve the treatment of pathologies such as schizophrenia.
Based on our own research, we focused here our attention on the role of 5-HT1ARs in the dopaminergic modulation of long-term synaptic plasticity. The modulatory effect of D1 or D2R on excitatory synaptic plasticity depends on the presence of functional 5-HT1ARs. It appears that 5-HT1AR cooperate with either D1R or D2R to direct the long term plasticity towards either LTP (D1Rs) or LTD (D2R). We assume that it is the cooperation of 5-HT1ARs with either D1Rs or D2Rs that will determine the magnitude of the Ca2+ influx through NMDARs evoked by high frequency synaptic stimulation and consequently favor either LTP or LTD. As illustrated in Figure 3, we consider the Ca2+ transient within dendritic spines as a key element of long-term synaptic plasticity. However it remains to determine how the recruitment of different signaling pathways following the activation of D1Rs, D2Rs and 5-HT1ARs leads to the regulation of AMPAR trafficking at the synapse which determines the synaptic strength. The cooperation between 5-HT and DA receptors in the modulation of the long-term plasticity of excitatory synaptic inputs may have consequences on neuronal output (spiking behavior) and information processing within the PFC. The absence of modulatory effects on inhibitory synaptic inputs would result in imbalanced changes in synaptic strength in favor of excitation affecting the threshold of neuronal input-output functions (Carvalho and Buonomano, 2009).
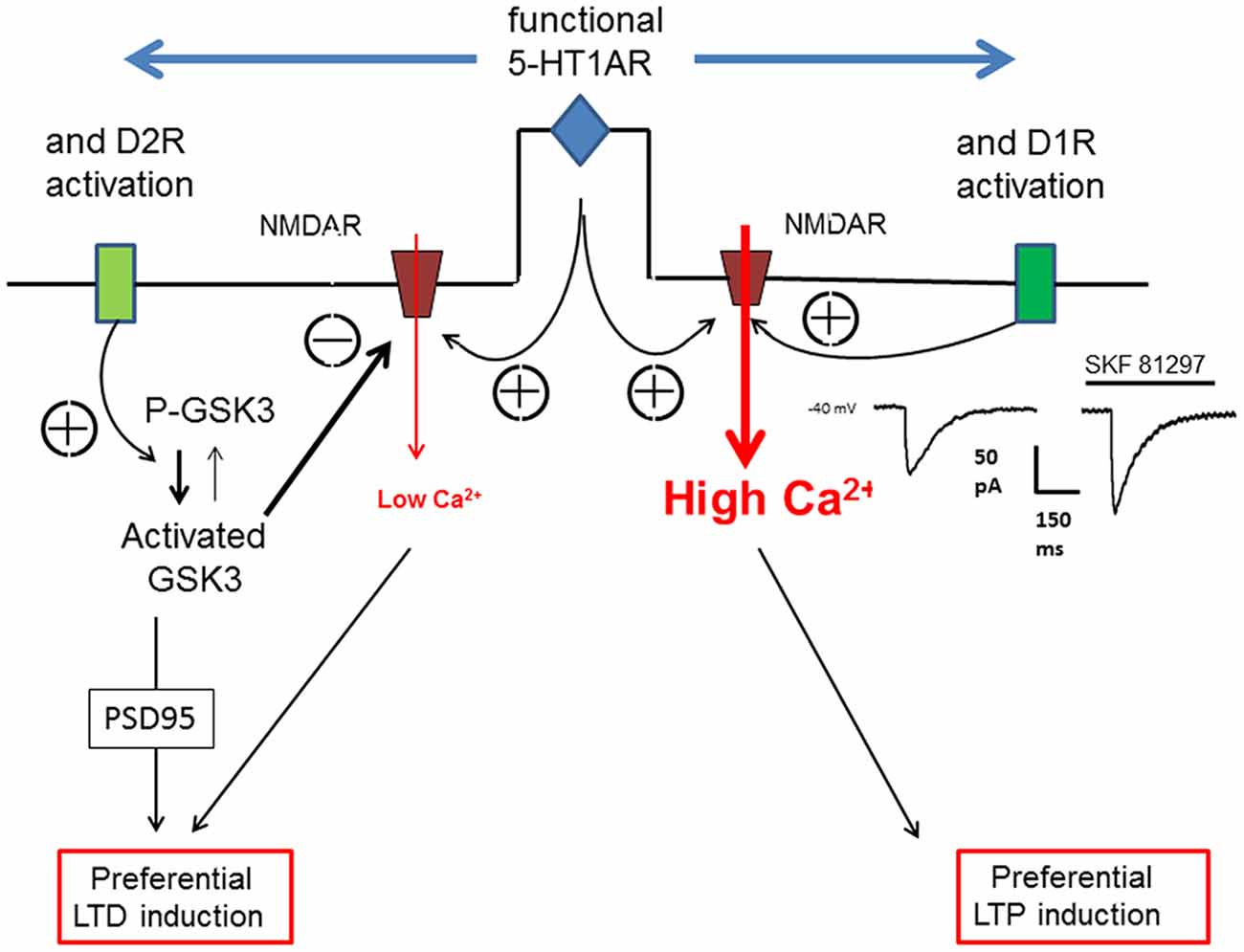
Figure 3. Schematic view of the action of 5-HT1AR on D1R or D2R to orientate the plasticity of an excitatory synapse in the PFC. The orientation of synaptic plasticity by dopamine (DA) acting at either D1Rs or D2Rs is dependent on the concomitant activation of 5-HT1ARs. We assume that the induction of either LTP or LTD of excitatory synapses depends on the magnitude of calcium transient (controlling AMPARs trafficking) within the dendritic spines which is determined by the calcium influx through NMDARs. Therefore, an increased calcium transient (High Ca2+) following 5-HT1AR and D1R activation (see the NMDA current on the right inset) would lead to LTP whereas a reduced calcium transient (low Ca2+) following 5-HT1AR and D2R activation would lead, via the activation of glycogen-synthase kinase-3 (GSK3), to LTD.
GSK3β is considered as a key player in synaptic plasticity (reviewed in Bradley et al., 2012). It is therefore important to understand the factors regulating GSK3β activity in the perspective of adapting therapy for several neurological and psychiatric disorders in which a dysregulation of GSK3β activity has been reported (Beurel et al., 2015). Mood disorders (major depressive disorder and bipolar disorder) are characterized by a disruption of the fine equilibrium between 5-HT and DA systems regulating the balance between the inactive and the active form of GSK3β at the synaptic level. Many therapeutic strategies using Li+ as a blocker of GSK3ß activity have been considered as an attempt to resettle such a balance (Jope, 2011). Our results suggest that in the case of pathologies such as major depression for which a reduction of the number of 5-HT1ARs is observed, new therapeutic strategies resetting the equilibrium between the active and the inactive form of GSK3ß could rather consist in acting at both 5-HT and DA receptors.
CNJM designed and conducted the experiments, wrote the article. PC wrote the article. PMF designed the experiments and wrote the article.
Centre National de la Recherche Scientifique (CNRS) is our institution which has provided basic funding.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abeles, M. (1991). Corticonomics: Neuronal Circuits of the Cerebral Cortex. Cambridge, MA: Cambridge University Press.
Adermark, L., and Lovinger, D. M. (2009). Frequency-dependent inversion of net striatal output by endocannabinoid-dependent plasticity at different synaptic inputs. J. Neurosci. 29, 1375–1380. doi: 10.1523/JNEUROSCI.3842-08.2009
Akimova, E., Lanzenberger, R., and Kasper, S. (2009). The serotonin-1A receptor in anxiety disorders. Biol. Psychiatry 66, 627–635. doi: 10.1016/j.biopsych.2009.03.012
Anderson, J., Lampl, I., Reichova, I., Carandini, M., and Ferster, D. (2000). Stimulus dependence of two-state fluctuations of membrane potential in cat visual cortex. Nat. Neurosci. 3, 617–621. doi: 10.1038/75797
Andrade, R. (2011). Serotonergic regulation of neuronal excitability in the prefrontal cortex. Neuropharmacology 61, 382–386. doi: 10.1016/j.neuropharm.2011.01.015
Arancibia-Cárcamo, I. L., and Kittler, J. T. (2009). Regulation of GABAA receptor membrane trafficking and synaptic localization. Pharmacol. Ther. 123, 17–31. doi: 10.1016/j.pharmthera.2009.03.012
Ashpole, N. M., Song, W., Brustovetsky, T., Engleman, E. A., Brustovetsky, N., Cummins, T. R., et al. (2012). Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J. Biol. Chem. 287, 8495–8506. doi: 10.1074/jbc.m111.323915
Azad, S. C., Monory, K., Marsicano, G., Cravatt, B. F., Lutz, B., Zieglgänsberger, W., et al. (2004). Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J. Neurosci. 24, 9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004
Bear, M. F., and Malenka, R. C. (1994). Synaptic plasticity: LTP and LTD. Curr. Opin. Neurobiol. 4, 389–399. doi: 10.1016/0959-4388(94)90101-5
Beattie, E. C., Carroll, R. C., Yu, X., Morishita, W., Yasuda, H., von Zastrow, M., et al. (2000). Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 3, 1291–1300. doi: 10.1038/81823
Beaulieu, J., and Gainetdinov, R. R. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217. doi: 10.1124/pr.110.002642
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2009). Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 49, 327–347. doi: 10.1146/annurev.pharmtox.011008.145634
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273. doi: 10.1016/j.cell.2005.05.012
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., et al. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U S A 101, 5099–5104. doi: 10.1073/pnas.0307921101
Beaulieu, J.-M., Zhang, X., Rodriguiz, R. M., Sotnikova, T. D., Cools, M. J., Wetsel, W. C., et al. (2008). Role of GSK3β in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U S A 105, 1333–1338. doi: 10.1073/pnas.0711496105
Berger, T. K., Silberberg, G., Perin, R., and Markram, H. (2010). Brief bursts self-inhibit and correlate the pyramidal network. PLoS Biol. 8:e1000473. doi: 10.1371/journal.pbio.1000473
Beurel, E., Grieco, S. F., and Jope, R. S. (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131. doi: 10.1016/j.pharmthera.2014.11.016
Bhagwagar, Z., Rabiner, E. A., Sargent, P. A., Grasby, P. M., and Cowen, P. J. (2004). Persistent reduction in brain serotonin1A receptor binding in recovered depressed men measured by positron emission tomography with [11C]WAY-100635. Mol. Psychiatry 9, 386–392. doi: 10.1038/sj.mp.4001401
Bliss, T. V., and Lomo, T. (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 232, 331–356. doi: 10.1113/jphysiol.1973.sp010273
Bockaert, J., Claeysen, S., Bécamel, C., Dumuis, A., and Marin, P. (2006). Neuronal 5-HT metabotropic receptors: fine-tuning of their structure, signaling, and roles in synaptic modulation. Cell Tissue Res. 326, 553–572. doi: 10.1007/s00441-006-0286-1
den Boon, F. S., Werkman, T. R., Schaafsma-Zhao, Q., Houthuijs, K., Vitalis, T., Kruse, C. G., et al. (2015). Activation of type-1 cannabinoid receptor shifts the balance between excitation and inhibition towards excitation in layer II/III pyramidal neurons of the rat prelimbic cortex. Pflugers Arch. 467, 1551–1564. doi: 10.1007/s00424-014-1586-z
Borah, A., and Mohanakumar, K. P. (2007). Long-term L-DOPA treatment causes indiscriminate increase in dopamine levels at the cost of serotonin synthesis in discrete brain regions of rats. Cell. Mol. Neurobiol. 27, 985–996. doi: 10.1007/s10571-007-9213-6
Borg-Graham, L. J., Monier, C., and Frégnac, Y. (1998). Visual input evokes transient and strong shunting inhibition in visual cortical neurons. Nature 393, 369–373. doi: 10.1038/30735
Bortolozzi, A., Castañé, A., Semakova, J., Santana, N., Alvarado, G., Cortés, R., et al. (2012). Selective siRNA-mediated suppression of 5-HT1A autoreceptors evokes strong anti-depressant-like effects. Mol. Psychiatry 17, 612–623. doi: 10.1038/mp.2011.92
Bradley, C. A., Peineau, S., Taghibiglou, C., Nicolas, C. S., Whitcomb, D. J., Bortolotto, Z. A., et al. (2012). A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 5:13. doi: 10.3389/fnmol.2012.00013
Burnashev, N., Schoepfer, R., Monyer, H., Ruppersberg, J. P., Günther, W., Seeburg, P. H., et al. (1992). Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science 257, 1415–1419. doi: 10.1126/science.1382314
Calabresi, P., Pisani, A., Mercuri, N. B., and Bernardi, G. (1992). Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur. J. Neurosci. 4, 929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x
Callier, S., Snapyan, M., Le Crom, S., Prou, D., Vincent, J.-D., and Vernier, P. (2003). Evolution and cell biology of dopamine receptors in vertebrates. Biol. Cell 95, 489–502. doi: 10.1016/s0248-4900(03)00089-3
Camiré, O., and Topolnik, L. (2012). Functional compartmentalisation and regulation of postsynaptic Ca2+ transients in inhibitory interneurons. Cell Calcium 52, 339–346. doi: 10.1016/j.ceca.2012.05.001
Carta, M., Carlsson, T., Kirik, D., and Björklund, A. (2007). Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 130, 1819–1833. doi: 10.1093/brain/awm082
Carvalho, T. P., and Buonomano, D. V. (2009). Differential effects of excitatory and inhibitory plasticity on synaptically driven neuronal input-output functions. Neuron 61, 774–785. doi: 10.1016/j.neuron.2009.01.013
Castillo, P. E., Chiu, C. Q., and Carroll, R. C. (2011). Long-term plasticity at inhibitory synapses. Curr. Opin. Neurobiol. 21, 328–338. doi: 10.1016/j.conb.2011.01.006
Celada, P., Puig, M. V., and Artigas, F. (2013). Serotonin modulation of cortical neurons and networks. Front. Integr. Neurosci. 7:25. doi: 10.3389/fnint.2013.00025
Chen, L., Bohanick, J. D., Nishihara, M., Seamans, J. K., and Yang, C. R. (2007). Dopamine D1/5 receptor-mediated long-term potentiation of intrinsic excitability in rat prefrontal cortical neurons: Ca2+-dependent intracellular signaling. J. Neurophysiol. 97, 2448–2464. doi: 10.1152/jn.00317.2006
Chen, G., Greengard, P., and Yan, Z. (2004). Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc. Natl. Acad. Sci. U S A 101, 2596–2600. doi: 10.1073/pnas.0308618100
Chen, L., Salinas, G. D., and Li, X. (2009). Regulation of serotonin 1B receptor by glycogen synthase kinase-3. Mol. Pharmacol. 76, 1150–1161. doi: 10.1124/mol.109.056994
Chevaleyre, V., and Castillo, P. E. (2003). Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38, 461–472. doi: 10.1016/S0896-6273(03)00235-6
Chevaleyre, V., Takahashi, K. A., and Castillo, P. E. (2006). Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 29, 37–76. doi: 10.1146/annurev.neuro.29.051605.112834
Choi, S. Y., Chang, J., Jiang, B., Seol, G. H., Min, S. S., Han, J. S., et al. (2005). Multiple receptors coupled to phospholipase C gate long-term depression in visual cortex. J. Neurosci. 25, 11433–11443. doi: 10.1523/JNEUROSCI.4084-05.2005
Cobos, I., Calcagnotto, M. E., Vilaythong, A. J., Thwin, M. T., Noebels, J. L., Baraban, S. C., et al. (2005). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059–1068. doi: 10.1038/nn1499
Compte, A., Sanchez-Vives, M. V., McCormick, D. A., and Wang, X. J. (2003). Cellular and network mechanisms of slow oscillatory activity (<1 Hz) and wave propagations in a cortical network model. J. Neurophysiol. 89, 2707–2725. doi: 10.1152/jn.00845.2002
Contreras, D., Timofeev, I., and Steriade, M. (1996). Mechanisms of long-lasting hyperpolarizations underlying slow sleep oscillations in cat corticothalamic networks. J. Physiol. 494, 251–264. doi: 10.1113/jphysiol.1996.sp021488
Cormier, R. J., Greenwood, A. C., and Connor, J. A. (2001). Bidirectional synaptic plasticity correlated with the magnitude of dendritic calcium transients above a threshold. J. Neurophysiol. 85, 399–406.
Cossart, R., Bernard, C., and Ben-Ari, Y. (2005). Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 28, 108–115. doi: 10.1016/j.tins.2004.11.011
Cossart, R., Dinocourt, C., Hirsch, J. C., Merchan-Perez, A., De Felipe, J., Ben-Ari, Y., et al. (2001). Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat. Neurosci. 4, 52–62. doi: 10.1038/82900
Costa, C., Tozzi, A., Siliquini, S., Galletti, F., Cardaioli, G., Tantucci, M., et al. (2011). A critical role of NO/cGMP/PKG dependent pathway in hippocampal post-ischemic LTP: modulation by zonisamide. Neurobiol. Dis. 44, 185–191. doi: 10.1016/j.nbd.2011.06.015
Coultrap, S. J., Freund, R. K., O’Leary, H., Sanderson, J. L., Roche, K. W., Dell’Acqua, M. L., et al. (2014). Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep. 6, 431–437. doi: 10.1016/j.celrep.2014.01.005
Coyle, J. T. (2006). Substance use disorders and Schizophrenia: a question of shared glutamatergic mechanisms. Neurotox. Res. 10, 221–233. doi: 10.1007/bf03033359
Creese, I., Burt, D. R., and Snyder, S. H. (1976). Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192, 481–483. doi: 10.1126/science.3854
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789. doi: 10.1038/378785a0
Cruz, D. A., Eggan, S. M., Azmitia, E. C., and Lewis, D. A. (2004). Serotonin1A receptors at the axon initial segment of prefrontal pyramidal neurons in schizophrenia. Am. J. Psychiatry 161, 739–742. doi: 10.1176/appi.ajp.161.4.739
Czarnecki, A., Birtoli, B., and Ulrich, D. (2007). Cellular mechanisms of burst firing-mediated long-term depression in rat neocortical pyramidal cells. J. Physiol. 578, 471–479. doi: 10.1113/jphysiol.2006.123588
Czesak, M., Le François, B., Millar, A. M., Deria, M., Daigle, M., Visvader, J. E., et al. (2012). Increased serotonin-1A (5-HT1A) autoreceptor expression and reduced raphe serotonin levels in deformed epidermal autoregulatory factor-1 (Deaf-1) gene knock-out mice. J. Biol. Chem. 287, 6615–6627. doi: 10.1074/jbc.m111.293027
Czyrak, A., Czepiel, K., Mackowiak, M., Chocyk, A., and Wedzony, K. (2003). Serotonin 5-HT1A receptors might control the output of cortical glutamatergic neurons in rat cingulate cortex. Brain Res. 989, 42–51. doi: 10.1016/s0006-8993(03)03352-3
Dahlström, A., and Fuxe, K. (1964). Localization of monoamines in the lower brain stem. Experientia 20, 398–399. doi: 10.1007/bf02147990
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U S A 102, 12560–12565. doi: 10.1073/pnas.0506071102
Daoudal, G., and Debanne, D. (2003). Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learn. Mem. 10, 456–465. doi: 10.1101/lm.64103
Dudek, S. M., and Bear, M. F. (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. U S A 89, 4363–4367. doi: 10.1073/pnas.89.10.4363
Dunah, A. W., Sirianni, A. C., Fienberg, A. A., Bastia, E., Schwarzschild, M. A., and Standaert, D. G. (2004). Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol. 65, 121–129. doi: 10.1124/mol.65.1.121
Dunah, A. W., and Standaert, D. G. (2001). Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J. Neurosci. 21, 5546–5558.
Egorov, A. V., Hamam, B. N., Fransén, E., Hasselmo, M. E., and Alonso, A. A. (2002). Graded persistent activity in entorhinal cortex neurons. Nature 420, 173–178. doi: 10.1038/nature01171
Emson, P. C., and Koob, G. F. (1978). The origin and distribution of dopamine-containing afferents to the rat frontal cortex. Brain Res. 142, 249–267. doi: 10.1016/0006-8993(78)90634-0
Etkin, A., Alarcón, J. M., Weisberg, S. P., Touzani, K., Huang, Y. Y., Nordheim, A., et al. (2006). A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron 50, 127–143. doi: 10.1016/j.neuron.2006.03.013
Feldman, D. E. (2009). Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55. doi: 10.1146/annurev.neuro.051508.135516
Fernández-Monreal, M., Brown, T. C., Royo, M., and Esteban, J. A. (2012). The balance between receptor recycling and trafficking toward lysosomes determines synaptic strength during long-term depression. J. Neurosci. 32, 13200–13205. doi: 10.1523/JNEUROSCI.0061-12.2012
Frame, S., Cohen, P., and Biondi, R. M. (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 7, 1321–1327. doi: 10.1016/s1097-2765(01)00253-2
Franklin, J. M., and Carrasco, G. A. (2012). Cannabinoid-induced enhanced interaction and protein levels of serotonin 5-HT2A and dopamine D2 receptors in rat prefrontal cortex. J. Psychopharmacol. 26, 1333–1347. doi: 10.1177/0269881112450786
Fuster, J. M. (2001). The prefrontal cortex—an update: time is of the essence. Neuron 30, 319–333. doi: 10.1016/S0896-6273(01)00285-9
Gaiarsa, J. L., and Ben-Ari, J. (2006). “Long-term plasticity at inhibitory synapses: a phenomenon that has been overlooked,” in The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology, eds J. T. Kittler and S. J. Moss (Boca Raton, FL: CRC Press), Chapter 2, 23–36.
Gaspar, P., Bloch, B., and Le Moine, C. (1995). D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur. J. Neurosci. 7, 1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x
Girault, J.-A., and Greengard, P. (2004). The neurobiology of dopamine signaling. Arch. Neurol. 61, 641–644. doi: 10.1001/archneur.61.5.641
Glausier, J. R., Khan, Z. U., and Muly, E. C. (2009). Dopamine D1 and D5 receptors are localized to discrete populations of interneurons in primate prefrontal cortex. Cereb. Cortex 19, 1820–1834. doi: 10.1093/cercor/bhn212
Goldberg, J. H., Yuste, R., and Tamas, G. (2003). Ca2+ imaging of mouse neocortical interneurone dendrites: contribution of Ca2+-permeable AMPA and NMDA receptors to subthreshold Ca2+dynamics. J. Physiol. 551, 67–78. doi: 10.1113/jphysiol.2003.042598
Grover, L. M., and Teyler, T. J. (1994). Activation of NMDA receptors in hippocampal area CA1 by low and high frequency orthodromic stimulation and their contribution to induction of long-term potentiation. Synapse 16, 66–75. doi: 10.1002/syn.890160108
Gubellini, P., Ben-Ari, Y., and Gaïarsa, J.-L. (2005). Endogenous neurotrophins are required for the induction of GABAergic long-term potentiation in the neonatal rat hippocampus. J. Neurosci. 25, 5796–5802. doi: 10.1523/JNEUROSCI.0824-05.2005
Gurden, H., Takita, M., and Jay, T. M. (2000). Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J. Neurosci. 20:RC106.
Haider, B., Duque, A., Hasenstaub, A. R., and McCormick, D. A. (2006). Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J. Neurosci. 26, 4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006
Haider, B., and McCormick, D. A. (2009). Rapid neocortical dynamics: cellular and network mechanisms. Neuron 62, 171–189. doi: 10.1016/j.neuron.2009.04.008
Hanley, J., and Henley, J. (2005). PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 24, 3266–3278. doi: 10.1038/sj.emboj.7600801
Hebb, D. O. (1949). Temperament in chimpanzees: method of analysis. J. Comp. Physiol. Psychol. 42, 192–206. doi: 10.1037/h0056842
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306. doi: 10.1146/annurev.physiol.010908.163149
Henry, F. E., McCartney, A. J., Neely, R., Perez, A. S., Carruthers, C. J. L., Stuenkel, E. L., et al. (2012). Retrograde changes in presynaptic function driven by dendritic mTORC1. J. Neurosci. 32, 17128–17142. doi: 10.1523/JNEUROSCI.2149-12.2012
Hirsch, J. C., and Crepel, F. (1990). Use-dependent changes in synaptic efficacy in rat prefrontal neurons in vitro. J. Physiol. 427, 31–49. doi: 10.1113/jphysiol.1990.sp018159
Hirsch, J. C., and Crepel, F. (1991). Blockade of NMDA receptors unmasks a long-term depression insynaptic efficacy in rat prefrontal neurons in vitro. Exp. Brain Res. 85, 621–624. doi: 10.1007/bf00231747
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., et al. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86. doi: 10.1111/j.1460-9568.2006.05245.x
Houston, C. M., He, Q., and Smart, T. G. (2009). CaMK II phosphorylation of the GABAA receptor: receptor subtype- and synapse-specific modulation. J. Physiol. 587, 2115–2125. doi: 10.1113/jphysiol.2009.171603
Hsiung, S., Tin, A., Tamir, H., Franke, T. F., and Liu, K. (2008). Inhibition of 5-HT 1A receptor-dependent cell survival by cAMP/protein kinase A: role of protein phosphatase 2A and Bax. J. Neurosci. Res. 86, 2326–2338. doi: 10.1002/jnr.21676
Huang, F. S., Abbas, A. K., Li, R., Afanasenkau, D., and Wigström, H. (2010). Bidirectional synaptic plasticity in response to single or paired pulse activation of NMDA receptors. Neurosci. Res. 67, 108–116. doi: 10.1016/j.neures.2010.02.005
Huang, Y. Y., Simpson, E., Kellendonk, C., and Kandel, E. R. (2004). Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc. Natl. Acad. Sci. U S A 101, 3236–3241. doi: 10.1073/pnas.0308280101
Huemmeke, M., Eysel, U. T., and Mittmann, T. (2002). Metabotropic glutamate receptors mediate expression of LTP in slices of rat visual cortex. Eur. J. Neurosci. 15, 1641–1645. doi: 10.1046/j.1460-9568.2002.02002.x
Isaacson, J. S., and Scanziani, M. (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. doi: 10.1016/j.neuron.2011.09.027
Jiang, B., Huang, S., de Pasquale, R., Millman, D., Song, L., Lee, H.-K., et al. (2010). The maturation of GABAergic transmission in visual cortex requires endocannabinoid-mediated LTD of inhibitory inputs during a critical period. Neuron 66, 248–259. doi: 10.1016/j.neuron.2010.03.021
Johnston, D., Williams, S., Jaffe, D., and Gray, R. (1992). NMDA-receptor-independent long-term potentiation. Annu. Rev. Physiol. 54, 489–505. doi: 10.1146/annurev.physiol.54.1.489
Johnstone, V. P. A., and Raymond, C. R. (2013). Postsynaptic protein synthesis is required for presynaptic enhancement in persistent forms of long-term potentiation. Front. Synaptic Neurosci. 5:1. doi: 10.3389/fnsyn.2013.00001
Jones, C. A., and McCreary, A. C. (2008). Serotonergic approaches in the development of novel antipsychotics. Neuropharmacology 55, 1056–1065. doi: 10.1016/j.neuropharm.2008.05.025
Jope, R. S. (2011). Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front. Mol. Neurosci. 4:16. doi: 10.3389/fnmol.2011.00016
Kantrowitz, J., and Javitt, D. C. (2012). Glutamatergic transmission in schizophrenia: from basic research to clinical practice. Curr. Opin. Psychiatry 25, 96–102. doi: 10.1097/YCO.0b013e32835035b2
Kasper, S., Stein, D. J., Loft, H., and Nil, R. (2005). Escitalopram in the treatment of social anxiety disorder: randomised, placebo-controlled, flexible-dosage study. Br. J. Psychiatry 186, 222–226. doi: 10.1192/bjp.186.3.222
Kehrer, C., Maziashvili, N., Dugladze, T., and Gloveli, T. (2008). Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 1:6. doi: 10.3389/neuro.02.006.2008
Kemp, A., and Manahan-Vaughan, D. (2004). Hippocampal long-term depression and long-term potentiation encode different aspects of novelty acquisition. Proc. Natl. Acad. Sci. U S A 101, 8192–8197. doi: 10.1073/pnas.0402650101
Kia, H. K., Brisorgueil, M. J., Hamon, M., Calas, A., and Vergé, D. (1996). Ultrastructural localization of 5-hydroxytryptamine1A receptors in the rat brain. J. Neurosci. Res. 46, 697–708. doi: 10.1002/(sici)1097-4547(19961215)46:6<697::aid-jnr7>3.0.co;2-a
Kittler, J. T., and Moss, S. J. (2003). Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol. 13, 341–347. doi: 10.1016/s0959-4388(03)00064-3
Kleckner, N. W., and Dingledine, R. (1988). Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241, 835–837. doi: 10.1126/science.2841759
Kruse, M. S., Prémont, J., Krebs, M.-O., and Jay, T. M. (2009). Interaction of dopamine D1 with NMDA NR1 receptors in rat prefrontal cortex. Eur. Neuropsychopharmacol. 19, 296–304. doi: 10.1016/j.euroneuro.2008.12.006
Lanzenberger, R. R., Mitterhauser, M., Spindelegger, C., Wadsak, W., Klein, N., Mien, L.-K., et al. (2007). Reduced serotonin-1A receptor binding in social anxiety disorder. Biol. Psychiatry 61, 1081–1089. doi: 10.1016/j.biopsych.2006.05.022
Lemonde, S., Turecki, G., Bakish, D., Du, L., Hrdina, P. D., Bown, C. D., et al. (2003). Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J. Neurosci. 23, 8788–8879.
Le Roux, N., Amar, M., Baux, G., and Fossier, P. (2006). Homeostatic control of the excitation-inhibition balance in cortical layer 5 pyramidal neurons. Eur. J. Neurosci. 24, 3507–3518. doi: 10.1111/j.1460-9568.2006.05203.x
Lewis, D. A., Hashimoto, T., and Volk, D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324. doi: 10.1038/nrn1648
Li, X., Rosborough, K. M., Friedman, A. B., Zhu, W., and Roth, K. A. (2007). Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int. J. Neuropsychopharmacol. 10, 7–19. doi: 10.1017/s1461145706006547
Li, Y.-C., Xi, D., Roman, J., Huang, Y.-Q., and Gao, W.-J. (2009). Activation of glycogen synthase kinase-3 β is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J. Neurosci. 29, 15551–15563. doi: 10.1523/JNEUROSCI.3336-09.2009
Li, X., Zhu, W., Roh, M. S., Friedman, A. B., Rosborough, K., and Jope, R. S. (2004). In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuropsychopharmacology 29, 1426–1431. doi: 10.1038/sj.npp.1300439
Lisman, J. E., and Zhabotinsky, A. M. (2001). A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31, 191–201. doi: 10.1016/S0896-6273(01)00364-6
Liu, X. Y., Chu, X. P., Mao, L. M., Wang, M., Lan, H. X., Li, M. H., et al. (2006). Modulation of D2R-NR2B interactions in response to cocaine. Neuron 52, 897–909. doi: 10.1016/j.neuron.2006.10.011
Llano, I., DiPolo, R., and Marty, A. (1994). Calcium-induced calcium release in cerebellar Purkinje cells. Neuron 12, 663–673. doi: 10.1016/0896-6273(94)90221-6
Lu, B. (2003). BDNF and activity-dependent synaptic modulation. Learn. Mem. 10, 86–98. doi: 10.1101/lm.54603
Lucas-Meunier, E., Monier, C., Amar, M., Baux, G., Frégnac, Y., and Fossier, P. (2009). Involvement of nicotinic and muscarinic receptors in the endogenous cholinergic modulation of the balance between excitation and inhibition in the young rat visual cortex. Cereb. Cortex 19, 2411–2427. doi: 10.1093/cercor/bhn258
Łukasiewicz, S., Błasiak, E., Szafran-Pilch, K., and Dziedzicka-Wasylewska, M. (2016). Dopamine D2 and serotonin 5-HT1A receptor interaction in the context of the effects of antipsychotics–in vitro studies. J. Neurochem. 137, 549–560. doi: 10.1111/jnc.13582
MacLean, J. N., Watson, B. O., Aaron, G. B., and Yuste, R. (2005). Internal dynamics determine the cortical response to thalamic stimulation. Neuron 48, 811–823. doi: 10.1016/j.neuron.2005.09.035
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation–a decade of progress? Science 285, 1870–1874. doi: 10.1126/science.285.5435.1870
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126. doi: 10.1146/annurev.neuro.25.112701.14275
Malleret, G., Alarcon, J. M., Martel, G., Takizawa, S., Vronskaya, S., Yin, D., et al. (2010). Bidirectional regulation of hippocampal long-term synaptic plasticity and its influence on opposing forms of memory. J. Neurosci. 30, 3813–3825. doi: 10.1523/JNEUROSCI.1330-09.2010
Marco, P., Sola, R. G., Pulido, P., Alijarde, M. T., Sánchez, A., Ramón y Cajal, S., et al. (1996). Inhibitory neurons in the human epileptogenic temporal neocortex. An immunocytochemical study. Brain 119, 1327–1347. doi: 10.1093/brain/119.4.1327
Marder, C. P., and Buonomano, D. V. (2004). Timing and bbalanceof inhibition enhance the effect of long-term potentiation on cell firing. J. Neurosci. 24, 8873–8884. doi: 10.1523/JNEUROSCI.2661-04.2004
Marek, G. J. (2007). Serotonin and dopamine interactions in rodents and primates: implications for psychosis and antipsychotic drug development. Int. Rev. Neurobiol. 78, 165–192. doi: 10.1016/s0074-7742(06)78006-0
Markram, H., Toledo-Rodriguez, M., Wang, Y., Gupta, A., Silberberg, G., and Wu, C. (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807. doi: 10.1038/nrn1519
Marsicano, G., Moosmann, B., Hermann, H., Lutz, B., and Behl, C. (2002). Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J. Neurochem. 80, 448–456. doi: 10.1046/j.0022-3042.2001.00716.x
Matsuda, Y., Marzo, A., and Otani, S. (2006). The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J. Neurosci. 26, 4803–4810. doi: 10.1523/JNEUROSCI.5312-05.2006
McGuinness, L., Taylor, C., Taylor, R. D. T., Yau, C., Langenhan, T., Hart, M. L., et al. (2010). Presynaptic NMDARs in the hippocampus facilitate transmitter release at theta frequency. Neuron 68, 1109–1127. doi: 10.1016/j.neuron.2010.11.023
Meisenzahl, E. M., Schmitt, G., Gründer, G., Dresel, S., Frodl, T., la Fougère, C., et al. (2008). Striatal D2/D3 receptor occupancy, clinical response and side effects with amisulpride: an iodine-123-iodobenzamide SPET study. Pharmacopsychiatry 41, 169–175. doi: 10.1055/s-2008-1076727
Meltzer, H. Y., Li, Z., Kaneda, Y., and Ichikawa, J. (2003). Serotonin receptors, their key role in drugs to treat schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 1159–1172. doi: 10.1016/j.pnpbp.2003.09.010
Meltzer, H. Y., and Massey, B. W. (2011). The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr. Opin. Pharmacol. 11, 59–67. doi: 10.1016/j.coph.2011.02.007
Meltzer, C. C., Price, J. C., Mathis, C. A., Butters, M. A., Ziolko, S. K., Moses-Kolko, E., et al. (2004). Serotonin 1A receptor binding and treatment response in late-life depression. Neuropsychopharmacology 29, 2258–2265. doi: 10.1038/sj.npp.1300556
Meunier, C. N. J., Amar, M., Lanfumey, L., Hamon, M., and Fossier, P. (2013). 5-HT1A receptors direct the orientation of plasticity in layer 5 pyramidal neurons of the mouse prefrontal cortex. Neuropharmacology 71, 37–45. doi: 10.1016/j.neuropharm.2013.03.003
Meunier, C. N., Callebert, J., Cancela, J-M., and Fossier, P. (2015). Effect of dopaminergic D1 receptors on plasticity is dependent of serotoninergic 5-HT1A receptors in L5-pyramidal neurons of the prefrontal cortex. PLoS One 10:e0120286. doi: 10.1371/journal.pone.0120286
Meunier, C. N., Cancela, J. M., and Fossier, P. (2017). Lack of GSK3β activation and modulation of synaptic plasticity by dopamine in 5-HT1A-receptor KO mice. Neuropharmacology 113, 124–136. doi: 10.1016/j.neuropharm.2016.09.025
Miller, E. K., and Cohen, J. D. (2001). An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 24, 167–202. doi: 10.1146/annurev.neuro.24.1.167
Monier, C., Chavane, F., Baudot, P., Graham, L. J., and Frégnac, Y. (2003). Orientation and direction selectivity of synaptic inputs in visual cortical neurons: a diversity of combinations produces spike tuning. Neuron 37, 663–680. doi: 10.1016/s0896-6273(03)00064-3
Monier, C., Fournier, J., and Frégnac, Y. (2008). In vitro and in vivo measures of evoked excitatory and inhibitory conductance dynamics in sensory cortices. J. Neurosci. Methods 169, 323–365. doi: 10.1016/j.jneumeth.2007.11.008
Moreau, A. W., Amar, M., Le Roux, N., Morel, N., and Fossier, P. (2010). Serotoninergic fine-tuning of the excitation-inhibition balance in rat visual cortical networks. Cereb. Cortex 20, 456–467. doi: 10.1093/cercor/bhp114
Mulkey, R. M., and Malenka, R. C. (1992). Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9, 967–975. doi: 10.1016/0896-6273(92)90248-c
Navailles, S., Benazzouz, A., Bioulac, B., Gross, C., and De Deurwaerdère, P. (2010). High-frequency stimulation of the subthalamic nucleus and L-3,4-dihydroxyphenylalanine inhibit in vivo serotonin release in the prefrontal cortex and hippocampus in a rat model of Parkinson’s disease. J. Neurosci. 30, 2356–2364. doi: 10.1523/JNEUROSCI.5031-09.2010
Negyessy, L., and Goldman-Rakic, P. S. (2005). Subcellular localization of the dopamine D2 receptor and coexistence with the calcium-binding protein neuronal calcium sensor-1 in the primate prefrontal cortex. J. Comp. Neurol. 488, 464–475. doi: 10.1002/cne.20601
Newman-Tancredi, A. (2010). The importance of 5-HT1A receptor agonism in antipsychotic drug action, rationale and perspectives. Curr. Opin. Investig. Drugs 11, 802–812.
Newman-Tancredi, A., Cussac, D., and Depoortere, R. (2007). Neuropharmacological profile of bifeprunox: merits and limitations in comparison with other third-generation antipsychotics. Curr. Opin. Investig. Drugs 8, 539–554.
Newman-Tancredi, A., and Kleven, M. S. (2011). Comparative pharmacology of antipsychotics possessing combined dopamine D2 and serotonin 5-HT1A receptor properties. Psychopharmacology (Berl) 216, 451–473. doi: 10.1007/s00213-011-2247-y
Nicholls, R. E., Alarcon, J. M., Malleret, G., Carroll, R. C., Grody, M., Vronskaya, S., et al. (2008). Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron 58, 104–117. doi: 10.1016/j.neuron.2008.01.039
Nowicky, A. V., and Bindman, L. J. (1993). The nitric oxide synthase inhibitor, N-monomethyl-L-arginine blocks induction of a long-term potentiation-like phenomenon in rat medial frontal cortical neurons in vitro. J. Neurophysiol. 70, 1255–1259.
Nugent, F. S., Penick, E. C., and Kauer, J. A. (2007). Opioids block long-term potentiation of inhibitory synapses. Nature 446, 1086–1090. doi: 10.1038/nature05726
Nyíri, G., Stephenson, F. A., Freund, T. F., and Somogyi, P. (2003). Large variability in synaptic N-methyl-D-aspartate receptor density on interneurons and a comparison with pyramidal-cell spines in the rat hippocampus. Neuroscience 119, 347–363. doi: 10.1016/s0306-4522(03)00157-x
Oleskevich, S., and Descarries, L. (1990). Quantified distribution of the serotonin innervation in adult rat hippocampus. Neuroscience 34, 19–33. doi: 10.1016/0306-4522(90)90301-j
Paspalas, C. D., and Goldman-Rakic, P. S. (2005). Presynaptic D1 dopamine receptors in primate prefrontal cortex: target-specific expression in the glutamatergic synapse. J. Neurosci. 25, 1260–1267. doi: 10.1523/JNEUROSCI.3436-04.2005
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., et al. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron 53, 703–717. doi: 10.1016/j.neuron.2007.01.029
Peters, A., and Kara, D. (1985). The neuronal composition of area 17 of rat visual cortex. I. The pyramidal cells. J. Comp. Neurol. 234, 218–241. doi: 10.1002/cne.902340208
Petersen, C. C., Hahn, T. T., Mehta, M., Grinvald, A., and Sakmann, B. (2003). Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex. Proc. Natl. Acad. Sci. U S A 100, 13638–13643. doi: 10.1073/pnas.2235811100
Pitler, T. A., and Alger, B. E. (1992). Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J. Neurosci. 12, 4122–4132.
Polter, A. M., and Li, X. (2010). 5-HT1A receptor-regulated signal transduction pathways in brain. Cell. Signal. 22, 1406–1412. doi: 10.1016/j.cellsig.2010.03.019
Polter, A. M., Yang, S., Jope, R. S., and Li, X. (2012). Functional significance of glycogen synthase kinase-3 regulation by serotonin. Cell. Signal. 24, 265–271. doi: 10.1016/j.cellsig.2011.09.009
Powell, K. (2004). Opening a window to the autistic brain. PLoS Biol. 2:E267. doi: 10.1371/journal.pbio.0020267
Puig, M. V., and Gulledge, A. T. (2011). Serotonin and prefrontal cortex function: neurons, networks, and circuits. Mol. Neurobiol. 44, 449–464. doi: 10.1007/s12035-011-8214-0
Ramboz, S., Oosting, R., Amara, D. A., Kung, H. F., Blier, P., Mendelsohn, M., et al. (1998). Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc. Natl. Acad. Sci. U S A 95, 14476–14481. doi: 10.1073/pnas.95.24.14476
Riad, M., Garcia, S., Watkins, K. C., Jodoin, N., Doucet, E., Langlois, X., et al. (2000). Somatodendritic localization of 5-HT1A and preterminal axonal localization of 5-HT1B serotonin receptors in adult rat brain. J. Comp. Neurol. 417, 181–194. doi: 10.1002/(sici)1096-9861(20000207)417:2<181::aid-cne4>3.3.co;2-1
Rigas, P., and Castro-Alamancos, M. A. (2007). Thalamocortical Up states: differential effects of intrinsic and extrinsic cortical inputs on persistent activity. J. Neurosci. 27, 4261–4272. doi: 10.1523/JNEUROSCI.0003-07.2007
Rippon, G., Brock, J., Brown, C., and Boucher, J. (2007). Disordered connectivity in the autistic brain: challenges for the “new psychophysiology”. Int. J. Psychophysiol. 63, 164–172. doi: 10.1016/j.ijpsycho.2006.03.012
Rubenstein, J. L. (2010). Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr. Opin. Neurol. 23, 118–123. doi: 10.1097/wco.0b013e328336eb13
Rubenstein, J. L. R., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183x.2003.00037.x
Saghatelyan, A. K., Dityatev, A., Schmidt, S., Schuster, T., Bartsch, U., and Schachner, M. (2001). Reduced perisomatic inhibition, increased excitatory transmission and impaired long-term potentiation in mice deficient for the extracellular matrix glycoprotein tenascin-R. Mol. Cell. Neurosci. 17, 226–240. doi: 10.1006/mcne.2000.0922
Saliba, R. S., Michels, G., Jacob, T. C., Pangalos, M. N., and Moss, S. J. (2007). Activity-dependent ubiquitination of GABAA receptors regulates their accumulation at synaptic sites. J. Neurosci. 27, 13341–13351. doi: 10.1523/JNEUROSCI.3277-07.2007
Sanchez-Vives, M. V., and McCormick, D. A. (2000). Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 3, 1027–1034. doi: 10.1038/79848
Santana, N., Bortolozzi, A., Serrats, J., Mengod, G., and Artigas, F. (2004). Expression of serotonin1A and serotonin2A receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb. Cortex 14, 1100–1109. doi: 10.1093/cercor/bhh070
Santana, N., Mengod, G., and Artigas, F. (2009). Quantitative analysis of the expression of dopamine D1 and D2 receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb. Cortex 19, 849–860. doi: 10.1093/cercor/bhn134
Sargent, P. A., Kjaer, K. H., Bench, C. J., Rabiner, E. A., Messa, C., Meyer, J., et al. (2000). Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: effects of depression and antidepressant treatment. Arch. Gen. Psychiatry 57, 174–180. doi: 10.1001/archpsyc.57.2.174
Shih, J. C. (2004). Cloning, after cloning, knock-out mice, and physiological functions of MAO A and B. Neurotoxicology 25, 21–30. doi: 10.1016/s0161-813x(03)00112-8
Shively, C. A., Friedman, D. P., Gage, H. D., Bounds, M. C., Brown-Proctor, C., Blair, J. B., et al. (2006). Behavioral depression and positron emission tomography-determined serotonin 1A receptor binding potential in cynomolgus monkeys. Arch. Gen. Psychiatry 63, 396–403. doi: 10.1001/archpsyc.63.4.396
Shu, Y., Hasenstaub, A., Badoual, M., Bal, T., and McCormick, D. A. (2003a). Barrages of synaptic activity control the gain and sensitivity of cortical neurons. J. Neurosci. 23, 10388–10401.
Shu, Y., Hasenstaub, A., and McCormick, D. A. (2003b). Turning on and off recurrent balanced cortical activity. Nature 423, 288–293. doi: 10.1038/nature01616
Silberberg, G., and Markram, H. (2007). Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53, 735–746. doi: 10.1016/j.neuron.2007.02.012
Singer, H. S., and Minzer, K. (2003). Neurobiology of Tourette’s syndrome: concepts of neuroanatomic localization and neurochemical abnormalities. Brain Dev. 25, S70–S84. doi: 10.1016/s0387-7604(03)90012-x
Sivakumaran, S., Mohajerani, M. H., and Cherubini, E. (2009). At immature mossy-fiber-CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J. Neurosci. 29, 2637–2647. doi: 10.1523/JNEUROSCI.5019-08.2009
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654. doi: 10.1016/s0896-6273(03)00476-8
Skinbjerg, M., Namkung, Y., Halldin, C., Innis, R. B., and Sibley, D. R. (2009). Pharmacological characterization of 2-methoxy-N-propylnorapomorphine’s interactions with D2 and D3 dopamine receptors. Synapse 63, 462–475. doi: 10.1002/syn.20626
Staff, N. P., and Spruston, N. (2003). Intracellular corelate of EPSP-spike potentiation in CA1 pyramidal neurons is controlled by GABAergic modulation. Hippocampus 13, 801–805. doi: 10.1002/hipo.10129
Stanton, P. K. (1996). LTD, LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus 6, 35–42. doi: 10.1002/(SICI)1098-1063(1996)6:1<35::AID-HIPO7>3.0.CO;2-6
Steinbusch, H. W. (1981). Distribution of serotonin-immunoreactivity in the central nervous system of the rat—cell bodies and terminals. Neuroscience 6, 557–618. doi: 10.1016/0306-4522(81)90146-9
Stephan, K. E., Friston, K. J., and Frith, C. D. (2009). Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr. Bull. 35, 509–527. doi: 10.1093/schbul/sbn176
Steriade, M., Timofeev, I., and Grenier, F. (2001). Natural waking and sleep states: a view from inside neocortical neurons. J. Neurophysiol. 85, 1969–1985.
Sutton, L. P., and Rushlow, W. J. (2011). The effects of neuropsychiatric drugs on glycogen synthase kinase-3 signaling. Neuroscience 199, 116–124. doi: 10.1016/j.neuroscience.2011.09.056
Timofeev, I., Grenier, F., Bazhenov, M., Sejnowski, T. J., and Steriade, M. (2000). Origin of slow cortical oscillations in deafferented cortical slabs. Cereb. Cortex 10, 1185–1199. doi: 10.1093/cercor/10.12.1185
Topolnik, L., Congar, P., and Lacaille, J.-C. (2005). Differential regulation of metabotropic glutamate receptor- and AMPA receptor-mediated dendritic Ca2+ signals by presynaptic and postsynaptic activity in hippocampal interneurons. J. Neurosci. 25, 990–1001. doi: 10.1523/JNEUROSCI.4388-04.2005
Tseng, K. Y., and O’Donnell, P. (2004). Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J. Neurosci. 24, 5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5, 97–107. doi: 10.1038/nrn1327
Uylings, H. B. M., Groenewegen, H. J., and Kolb, B. (2003). Do rats have a prefrontal cortex? Behav. Brain Res. 146, 3–17. doi: 10.1016/j.bbr.2003.09.028
Varela, J. A., Hirsch, S. J., Chapman, D., Leverich, L. S., and Greene, R. W. (2009). D1/D5 modulation of synaptic NMDA receptor currents. J. Neurosci. 29, 3109–3119. doi: 10.1523/JNEUROSCI.4746-08.2009
Wang, X., Zhong, P., Gu, Z., and Yan, Z. (2003). Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J. Neurosci. 23, 9852–9861.
Van De Werd, H. J. J. M., Rajkowska, G., Evers, P., and Uylings, H. B. M. (2010). Cytoarchitectonic and chemoarchitectonic characterization of the prefrontal cortical areas in the mouse. Brain Struct. Funct. 214, 339–353. doi: 10.1007/s00429-010-0247-z
Wehr, M., and Zador, A. M. (2003). Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature 426, 442–446. doi: 10.1038/nature02116
Wilsch, V. W., Behnisch, T., Jäger, T., Reymann, K. G., and Balschun, D. (1998). When are class I metabotropic glutamate receptors necessary for long-term potentiation? J. Neurosci. 18, 6071–6080.
Woodgett, J. R. (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9, 2431–2438.
Woodgett, J. R. (2001). Judging a protein by more than its name, GSK-3. Sci. STKE 2001:re12. doi: 10.1126/stke.2001.100.re12
Xue, M., Atallah, B. V., and Scanziani, M. (2014). Equalizing excitation-inhibition ratios across visual cortical neurons. Nature 511, 596–600. doi: 10.1038/nature13321
Zhang, Z., Jiao, Y. Y., and Sun, Q. Q. (2011). Developmental maturation of excitation and inhibition balance in principal neurons across four layers of somatosensory cortex. Neuroscience 174, 10–25. doi: 10.1016/j.neuroscience.2010.11.045
Keywords: prefrontal cortex, serotonin, dopamine, LTP and LTD, neuromodulation
Citation: Meunier CNJ, Chameau P and Fossier PM (2017) Modulation of Synaptic Plasticity in the Cortex Needs to Understand All the Players. Front. Synaptic Neurosci. 9:2. doi: 10.3389/fnsyn.2017.00002
Received: 25 October 2016; Accepted: 13 January 2017;
Published: 01 February 2017.
Edited by:
Jack Mellor, University of Bristol, UKReviewed by:
Ricardo C. Araneda, University of Maryland, College Park, USACopyright © 2017 Meunier, Chameau and Fossier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe M. Fossier, cGhpbGlwcGUuZm9zc2llckB1LXBzdWQuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.