 Gregg W. Crabtree
Gregg W. Crabtree Joseph A. Gogos
Joseph A. Gogos- 1Department of Physiology and Cellular Biophysics, College of Physicians and Surgeons, Columbia University, New York, NY, USA
- 2Department of Neuroscience, College of Physicians and Surgeons, Columbia University, New York, NY, USA
Synaptic plasticity alters the strength of information flow between presynaptic and postsynaptic neurons and thus modifies the likelihood that action potentials in a presynaptic neuron will lead to an action potential in a postsynaptic neuron. As such, synaptic plasticity and pathological changes in synaptic plasticity impact the synaptic computation which controls the information flow through the neural microcircuits responsible for the complex information processing necessary to drive adaptive behaviors. As current theories of neuropsychiatric disease suggest that distinct dysfunctions in neural circuit performance may critically underlie the unique symptoms of these diseases, pathological alterations in synaptic plasticity mechanisms may be fundamental to the disease process. Here we consider mechanisms of both short-term and long-term plasticity of synaptic transmission and their possible roles in information processing by neural microcircuits in both health and disease. As paradigms of neuropsychiatric diseases with strongly implicated risk genes, we discuss the findings in schizophrenia and autism and consider the alterations in synaptic plasticity and network function observed in both human studies and genetic mouse models of these diseases. Together these studies have begun to point toward a likely dominant role of short-term synaptic plasticity alterations in schizophrenia while dysfunction in autism spectrum disorders (ASDs) may be due to a combination of both short-term and long-term synaptic plasticity alterations.
Introduction—Neuropsychiatric Disease Manifestations and Hypotheses of Neural Circuit Dysfunction
The recent advances over the past several decades in molecular biology, human genomics, and bioinformatics has helped to enable the identification of numerous candidate genes and distinct genetic mutations in a variety of human neuropsychiatric illnesses that may be the responsible agents for causing their symptomology. Based on the genetic mutations identified by human genomic analyses, many of these mutations have been introduced into the mouse genome to create genetic mouse models of these human diseases in the hope that the invasive and precisely refined experimentation permitted in mice will help to reveal the details of the molecular and neuronal dysfunction underlying these diseases. Despite the availability of these powerful experimental tools, the daunting molecular, anatomic, and functional complexity of the central nervous system demands that targeted and well-considered hypotheses be generated within this overwhelming complex system in order to direct experimental investigations to the systems and processes where dysfunction is most likely to be found in each unique disease.
When considering the underlying mechanistic causes of the dysfunction and symptomology of neuropsychiatric diseases, a thoughtful consideration of the most prominent features and most characteristic symptoms of each disease is a useful starting point for hypothesis generation. It has been proposed that the core commonality of dysfunction amongst neuropsychiatric diseases is a deficit in the information processing performed both by and between the various neural microcircuits located throughout the brain (Yizhar et al., 2011; Uhlhaas and Singer, 2012). Although multiple parameters work together to determine the computations performed by neural circuits, the synaptic plasticity that dynamically regulates synaptic function and hence the information flow between neurons at each of the millions of synapses throughout a neural microcircuit is thought to play a fundamental role in this microcircuit computation (Abbott and Regehr, 2004). While short-term synaptic plasticity leads to short-term changes in synaptic function (lasting from milliseconds to minutes) that regulate the moment-to-moment information flow through a neural circuit, long-term synaptic plasticity leads to persistent changes in synaptic function (lasting from hours to the lifetime of the synapse) that fundamentally and adaptively alters microcircuit function in response to activity indicating changing computational demands (Zucker and Regehr, 2002; Collingridge et al., 2010; Granger and Nicoll, 2013). Due to this critical role in controlling microcircuit computation, here we consider dysfunction in both long-term and short-term synaptic plasticity mechanisms as promising candidates underlying the neural microcircuit dysfunction that is likely responsible for neuropsychiatric disease symptomology (Table 1). While dysfunction in distinct neuropsychiatric diseases may result from alterations in either or both short-term and long-term synaptic plasticity, as a paradigm, studies of schizophrenia—where short-term synaptic plasticity alterations dominate—serve as an illustrative example of how targeted hypotheses of synaptic plasticity dysfunction can be formulated by beginning with the temporal nature of disease manifestations as a strategic starting point.
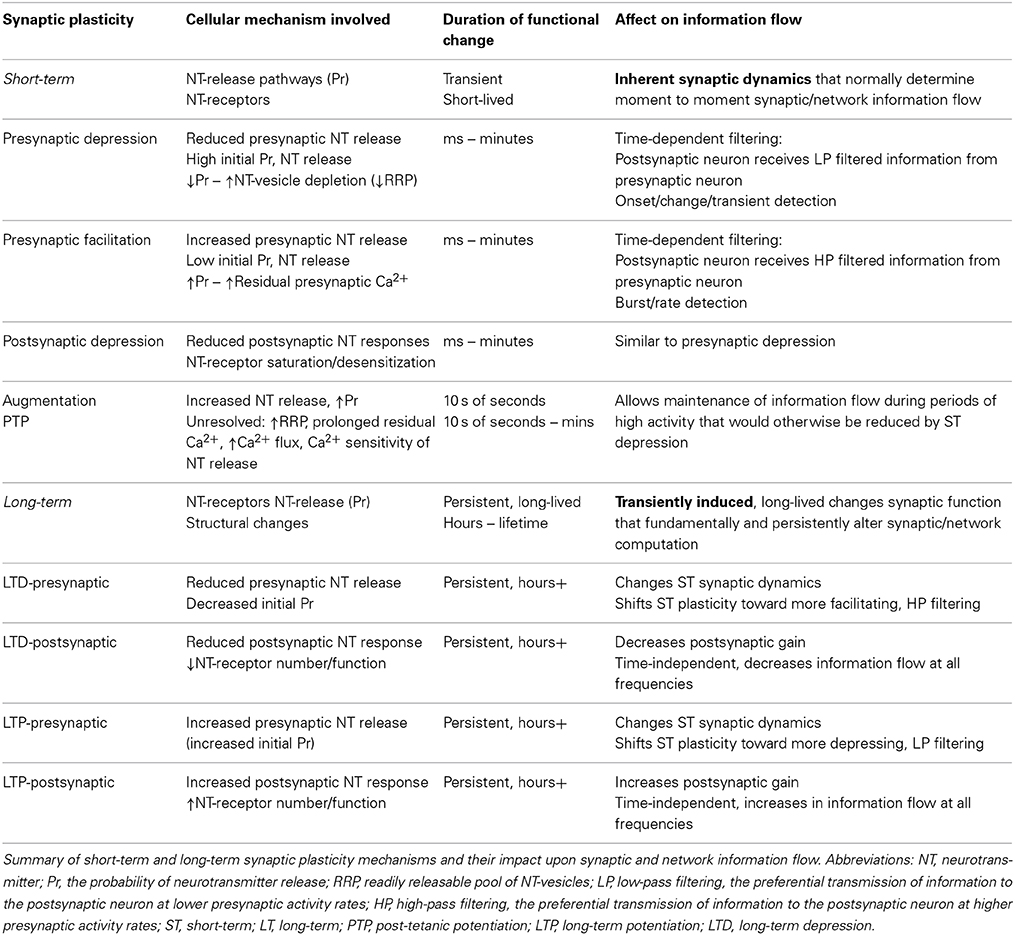
Table 1. Synaptic plasticity mechanisms and information flow.
Schizophrenia is a relatively common and severely debilitating psychiatric illness that is characterized by a somewhat variable yet core set of clinical symptomology. Although the most striking features of schizophrenics are their disorganized thinking and speech, delusions, and hallucinations (so-called positive symptoms of psychosis), the disease is also characterized by both “negative” symptoms (e.g., social withdrawal, blunted emotional range, lack of motivation, anhedonia) as well as severe cognitive deficits that may in fact contribute more substantially to functional disability and quality of life than the more prominent positive symptoms (Ranganath et al., 2000; Carter et al., 2008; American Psychiatric Association, 2013).
Current drug therapies, while substantially efficacious in treating positive symptoms, show little benefit in correcting the more disabling cognitive and negative symptoms and disease-preventative measures are currently non-existent (Buchanan et al., 2007; Brown and McGrath, 2011). Schizophrenia research targeted at understanding the precise molecular and neuronal mechanisms underlying disease dysfunction represents the most promising path toward more efficacious therapy and cure. To achieve these ends, genetic mouse models based upon robust candidate schizophrenia susceptibility genes identified by human genomic studies have recently been employed. These genetic mouse models are currently the best available analytical platform as they permit the invasive, high-resolution studies—impossible to conduct in human subjects—necessary to identify and elucidate the precise disease mechanisms present in neuropsychiatric diseases at the neuronal and molecular level (Arguello et al., 2010). At present, combining thoughtful consideration and analysis of human symptomology and the findings from mouse models based upon bone fide schizophrenia genetic risk variants has begun to suggest that, while long-term synaptic plasticity mechanisms may be relatively intact, there is an important and dominant role of short-term synaptic plasticity dysfunction in schizophrenia pathogenesis (Arguello and Gogos, 2012).
Although the clinical presentation and evolution of the schizophrenia can be highly variable, cognitive impairment frequently pre-dates diagnosis which often initially presents with the psychotic positive symptoms while negative symptoms usually become more prominent only at later times (O'Carroll, 2000). This evolution of symptomology strongly suggests that cognitive and psychotic symptomology may represent the primary disease process and be fundamental to understanding disease mechanisms. Significantly, both the specific cognitive deficits and psychotic features characteristic of schizophrenia suggest a fundamental problem with the moment-to-moment information processing performed by neural circuits within the brain (Compte et al., 2000; Mongillo et al., 2008). The cognitive deficits in schizophrenia are both pervasive and profound and range from impaired sensory processing to deficits in the cognitive control mechanisms necessary to manage and organize information (Barch, 2009; Dias et al., 2011; Lesh et al., 2011). Although schizophrenics show deficits in both working memory (a short-term memory process) and episodic memory (involving both short- and long-term processes), performance testing in human subjects suggests that both of these deficits likely involve an underlying deficit in short-term information management as episodic memory deficits can be overcome by employing certain encoding strategies indicating that long-term recall processes are largely intact (Holthausen et al., 2003; Ragland et al., 2005; Bonner-Jackson et al., 2008; Van Snellenberg, 2009).
The psychotic features of schizophrenia likewise point toward a deficit in the short-term processing of information. The nature of disorganized speech, hallucinations, and delusions all point to a moment-to-moment deficit in the instantaneous processing of on-going streams of information rather than a deficit involving long-term processes. Moreover, the drug-responsiveness of psychosis—as well as the ability of certain drugs and medications to induce psychosis in healthy individuals—further suggests a likely subtle defect in the dynamics of short-term information processing rather than a significant derangement or degeneration of microcircuit connectivity or architecture (Arguello and Gogos, 2012). Thus, taken together, both the psychosis and the nature of the cognitive deficits in schizophrenia strongly suggest pathologic alterations heavily weighted toward dysfunction in short-term information processing dynamics by neural microcircuitry. Given that the short-term, ongoing information processing performed by neural circuits is believed to be largely dictated by the short-term synaptic plasticity that controls the instantaneous dynamics of synaptic information flow strongly suggests that the neural circuit dysfunction in schizophrenia may to a large extent result from underlying alterations in short-term synaptic plasticity (Abbott and Regehr, 2004; Mongillo et al., 2008; Deng and Klyachko, 2011). Consistent with this hypothesis, studies in genetic mouse models of schizophrenia have found that the cognitive and synaptic plasticity alterations observed in these mice are dominated by deficits in working memory and short-term synaptic plasticity rather than in episodic memory and long-term synaptic plasticity (Koike et al., 2006; Kvajo et al., 2008; Stark et al., 2008; Sigurdsson et al., 2010; Drew et al., 2011b; Fénelon et al., 2011, 2013; Arguello and Gogos, 2012).
While at a biological top level it would seem necessary that disease dysfunction must follow gene dysfunction, the lack of full penetrance of schizophrenia within pedigrees—most notably the only 50% concordance between monozygotic twins—suggests that while gene dysfunction clearly plays a prominent role, other factors are also significantly contributing to disease genesis (Karayiorgou and Gogos, 1997). There is broad supporting data that environmental factors act both in concert with genetic background as well as epigenetically to enhance risk for schizophrenia (Prasad et al., 2010). Significant environmental risk factors predisposing to schizophrenia may include—but are not limited to—a variety of chronic, early-life social, emotional, and metabolic stressors, perinatal viral illness and hypoxic insults, and chronic, early-life use of some drugs of abuse, most significantly cannabis (Vilain et al., 2013; Schmitt et al., 2014). Despite the importance of identifying these risk factors, both risk genes and environmental factors still remain at a frustrating distance from a detailed, mechanistic understanding by which they lead to the deranged information processing at the level of the neural circuit dysfunction underlying disease (Lisman et al., 2008; Hu et al., 2010; Sigurdsson et al., 2010; Steullet et al., 2010; Arguello and Gogos, 2012; Filipović et al., 2013; Spellman and Gordon, 2014).
Although the molecular and neuronal dysfunction that leads to psychosis and schizophrenia remains poorly understood, theories of dysregulation of neurotransmitter systems including hyper-function of dopaminergic systems and hypo-function of glutamatergic systems (principally NMDA receptors) have been proposed as candidates underlying altered synaptic plasticity (Blum and Mann, 2002; Baldessarini and Tarazi, 2006). These theories, which were largely developed prior to the genomic era and were originally based upon clinical observations of responses to various drugs and medications that either precipitated or ameliorated psychosis, have been heavily investigated and at times supported by findings in a variety of models of schizophrenia (Mohn et al., 1999; Lahti et al., 2001; Dalmau et al., 2007; Manahan-Vaughan et al., 2008; Wiedholz et al., 2008; Ecker et al., 2009; Belforte et al., 2010; Carlén et al., 2012; Inta et al., 2014). But whether manipulations of these neurotransmitter systems simply phenotypically mimic the symptoms of schizophrenia or whether alterations in these NT systems are truly fundamental to the human disease process still remains unknown. Intriguingly, however, the genetic risk variants identified in most genomic studies have only rarely been found to be among those genes comprising the principal components of these neurotransmitter systems which suggests that if alterations of these NT systems are fundamentally involved in schizophrenia they must be so indirectly through the downstream actions of risk genes (Glatt et al., 2003). Thus, while it would likely be unwise not to explore the function of these NT systems in mouse models based upon human genetic risk variants, it may also be equally unwise to view these models without more unbiased eyes. We would suggest a broader and more measured investigative approach centered upon mouse models based upon bone fide genetic risk variants as a means to approach neural circuit dysfunction in disease though investigative analysis of the substrates most fundamentally involved in determining neural circuit function, principally synaptic plasticity and neural circuit architecture. By taking advantage of the unbiased nature of identifying genetic risk variants as a gateway to unravel disease-relevant neural circuit dysfunction, this approach may likely be the most fruitful and efficacious means to reveal the functional and molecular details of disease mechanisms required to create the most effective, targeted therapeutics.
This example of the hypothesis development and experimental approach for schizophrenia thus serves as a paradigm for the effective approach to the study of other neuropsychiatric diseases by showing how high-risk susceptibility genes found through human genomic studies can be used to develop genetic mouse models that not only recapitulate the behavioral deficits found in human disease but that further allow detailed analysis of the neuronal, synaptic, and neural circuit dysfunction underlying these behavioral alterations. As neural circuit dysfunction is a common underlying feature in neuropsychiatric disease, a research strategy focusing upon assessing the function of distinct neural circuits implicated in disease symptomology and the neuronal and synaptic mechanisms that underlie their function will likely provide the optimally productive approach for revealing the fundamental molecular mechanisms underlying neuropsychiatric disease (Lisman et al., 2008; Akil et al., 2010; Arguello and Gogos, 2012). While the unique symptomology of schizophrenia strongly suggests that neural circuit dysfunction may be predominantly due to alterations in short-term synaptic plasticity, the varied symptomology within the spectrum neuropsychiatric diseases can likewise be used to guide targeted experiments assessing the unique contributions of short-term and long-term synaptic plasticity mechanisms underlying neural circuit dysfunction in distinct neuropsychiatric diseases.
Potential Causes of Neural Circuit Dysfunction in Neuropsychiatric Disease
The commonality of dysfunction among the psychiatric diseases is a failure of the brain to process information appropriately in order to produce a normal and appropriate response to the demands of a given task or situation. While the specific details of how the brain accomplishes the processing of information remain understood at only the most rudimentary level, it seems apparent that the informational computation performed by neural circuits is critical to the interpretation of the world around us that ultimately leads to adaptive and advantageous behavioral responses (Uhlhaas and Singer, 2006, 2012; Yizhar et al., 2011). Although this review focuses exclusively upon the role of synaptic plasticity alterations in neuropsychiatric disease and their roles in neural microcircuit dysfunction, it is useful to first acknowledge and consider alternative paths that can also lead to the outcome of network dysfunction in neuropsychiatric disease. Neural microcircuit function is principally determined by three factors: (1) the network architecture that is defined by the synaptic connectivity between the neurons within a microcircuit, (2) the synaptic plasticity which dynamically regulates the strengths of these individual synapses and ultimately controls the information flow between the neurons in the network, and (3) the postsynaptic ensemble integration of these synaptic inputs that determines whether synaptic information received from presynaptic neurons successfully leads to an AP in the post-synaptic neuron. Thus, in considering potential neuronal causes of neural circuit dysfunction, it is possible to envision a comprehensive yet manageable set of pathologic scenarios that could lead to impaired neural circuit function in neuropsychiatric disease.
In perhaps the simplest scenario, the architecture and connectivity of the neural circuit could be normal and the intrinsic function of the neurons and synapses within the circuit could be normal while some outside factor could be causing neural circuit dysfunction. That both electrolyte imbalances as well as exogenous drugs and medications can alter neuronal function and acutely cause psychiatric symptomology suggests that similar mechanisms could play causal roles in neural circuit dysfunction in chronic neuropsychiatric disease (Webb and Gehi, 1981; Morton, 1999; Turjanski and Lloyd, 2005). Both the dopaminergic agonists and NMDA receptor antagonists that precipitate acute psychotic states that mimic the psychosis of schizophrenia appear to operate by this mechanism in the context of presumably otherwise normal basal neural circuit function (Lahti et al., 2001; Manahan-Vaughan et al., 2008; Ecker et al., 2009).
In another scenario in which neural circuit architecture is normal, alterations in either neuronal or synaptic function could lead to pathological alterations in neural circuit performance. Changes in either the excitability of the neurons within the circuit or the regulation by synaptic plasticity of the strengths of the synapses that define the functional connectivity of the circuit will alter the information flow through the circuit and thus lead to disruptions of neural circuit computation (Matveev and Wang, 2000; Zheng et al., 2004; Deng and Klyachko, 2011; Rotman et al., 2011; Yizhar et al., 2011; Ransdell et al., 2012). Current paradigms of schizophrenia and autism implicating the prominent alterations in synaptic plasticity observed in mouse models based on identified human genetic risk variants suggest that such functional changes alone could be largely responsible for neural circuit dysfunction (Markram and Markram, 2010; Arguello and Gogos, 2012).
In a complementary scenario in which both neuronal and synaptic function are normal, alterations in neural circuit architecture alone could similarly lead to alterations in the information processing performed by neural circuits. The neuron to neuron connectivity created by the synapses between neurons is a critical determinant in neural circuit function and alterations in this circuit architecture would have a significant impact upon information processing. Such alterations in connectivity in neural circuits likely play a central role in the neural circuit dysfunction in neurodegenerative diseases in which neuronal death and the inability to grow and maintain new synaptic connections would be expected to significantly alter neural circuit architecture (Hensch, 2005; Le Be and Markram, 2006; Pratt et al., 2008; Ricoy et al., 2011). Both the prominent progressive synaptic and neuronal degeneration observed in Alzheimer disease and the emerging role of deficient adult neurogenesis in major depression suggest that alterations in neural circuit architecture and the dynamic maintenance of that architecture may alone play substantial roles in neural circuit dysfunction (Selkoe et al., 2012; Miller and Hen, 2015).
In a final scenario, it is possible to have information processing abnormalities in the context of both normal neural circuit architecture and normal neuronal and synaptic function if the long-range connectivity between distinct neural circuits in different brain regions is altered. As complex behaviors and decisions frequently require the transmission of information processed in a neural circuit in one brain region to another circuit located in a distant brain region, the appropriate functional connectivity between these distinct neural circuits is critical to effective global information processing within the brain (Achard et al., 2006; Wang, 2010; Modha and Singh, 2010). Suggesting an important causal role in neuropsychiatric pathophysiology, prominent alterations in long-range connectivity between brain regions have long been implicated in major depression and have also more recently been implicated in schizophrenia, autism, and OCD (Lawrie et al., 2002; Nestler et al., 2002; Belmonte et al., 2004; Meyer-Lindenberg et al., 2005; Geschwind and Levitt, 2007; Murias et al., 2007; Welch et al., 2007; Krishnan and Nestler, 2010; Lynall et al., 2010; Markram and Markram, 2010; Sigurdsson et al., 2010; Ahmari et al., 2013; Brennan et al., 2013; Uhlhaas, 2013).
Furthermore, apart from the distinct scenarios presented above, it is possible—if not likely—that the information processing deficits present in neuropsychiatric illnesses could be caused by neural circuit dysfunction that is due to a combined dysfunction of the mechanisms presented in these individual scenarios. Despite this diversity of mechanisms potentially responsible for neural circuit dysfunction, here in this review we focus solely upon the possible role of alterations in synaptic plasticity as contributing to the dysfunction of information processing by neural circuits in neuropsychiatric diseases. Alterations in synaptic plasticity mechanisms represent particularly attractive candidates for information processing dysfunction in neuropsychiatric disease as their impact upon the dynamics of information flow through neural circuits appears particularly critical to the computation performed by these circuits (Abbott and Regehr, 2004; Mongillo et al., 2008; Deng and Klyachko, 2011). Although a detailed understanding of the information processing performed by neural circuits currently remains out of reach, convergent results from experimental and theoretical studies strongly suggest that synaptic plasticity—and in particular short-term synaptic plasticity—is fundamental in governing these computations (Markram and Tsodyks, 1996; Tsodyks and Markram, 1997; Milner et al., 1998; Dobrunz and Stevens, 1999; Martin et al., 2000; Abbott and Regehr, 2004; Whitlock et al., 2006; Mongillo et al., 2008; Deng and Klyachko, 2011).
In addition to synaptic plasticity mechanisms, however, there are many other forms of plasticity within neural systems that can also act upon neural circuits to modify their function. The growth of new synaptic connections, the birth of new neurons via neurogenesis that then become incorporated into existing circuitry, the modulatory impact of glial cells acting either alone or in concert with neurons, and the actions of neurotrophic factors released by either neurons or glia can all lead to significant changes in the function and architecture of neural circuits and have all been implicated in contributing to dysfunction in neuropsychiatric diseases (Gottschalk et al., 1998; Gu, 2002; Lamprecht and LeDoux, 2004; Ghashghaei et al., 2006; Le Be and Markram, 2006; Reif et al., 2006, 2007; Todd et al., 2006; Mao et al., 2009). As a comprehensive consideration of these diverse mechanisms is beyond the scope of this review, however, herein we limit our consideration exclusively to neuronal aspects of synaptic plasticity mechanisms that act acutely in a synaptic activity-dependent manner to modify the function of pre-existing synapses within neural circuits and the potential role of alterations in these mechanisms in the circuit dysfunction in neuropsychiatric disease.
Synaptic Plasticity as a Vantage Point for Psychiatric Disease
While there still remains a daunting knowledge gap between the symptomology of neuro-psychiatric disease and the causes of this symptomology at the synaptic, neuronal, and neural circuit level, considering how distinct forms of neuronal dysfunction might be reflected in disease symptomology can be cautiously applied toward hypothesis generation that can lead to targeted experiments in understanding the underlying dysfunction in these diseases. Broadly, neuropsychiatric diseases with both profound and persistent symptomology—such as the more severe neurodevelopmental and neurodegenerative disorders—suggest a more fundamental and pervasive underlying neuronal derangement while those diseases with more nuanced and episodic symptomology might suggest nearly normal neuronal, synaptic, and neural circuit function with dysregulation occurring at the level of the fine-tuning and modulation of neuronal, synaptic, and neural circuit function (Table 1).
In considering the cognitive deficits present in Alzheimer's disease (AD), for example, while there is some waxing and waning of symptomology, the memory deficits characteristic of AD dementia are largely persistent, pervasive, and progressive. While these memory deficits are possibly due in part to deficits of long-term plasticity, the major pathology underlying the dementia is likely to be predominantly the consequence of the neuronal death and degeneration that would fundamentally alter the connectivity of neural circuits involved in memory formation and storage (rather than due to alterations in the cellular mechanisms involved in LTP per se). The plausibility of this interpretation is supported by the broadly reported findings indicating that neuronal degeneration and death, pervasive alterations in neuronal and synaptic morphology, and deficits in synaptic maintenance are fundamental characteristics of AD pathology. Thus, while the memory deficits of AD do indeed imply neuronal and neural circuit dysfunction, these dysfunctions are likely a consequence of significant alterations in functional connectivity between neurons that affect neural circuit architecture rather than modulatory changes in neuronal function that serve to sculpt information processing in a largely intact and functional neural circuit. Taken together, both the symptomology and the profound degeneration and neuronal derangements present in AD dementia suggest that the underlying disease dysfunction in most cases is not likely primarily due to subtle changes in neuronal function such as alterations in synaptic plasticity mechanisms.
In contrast to the case of AD, the nature of the psychotic symptomology of both schizophrenia and bipolar disorder suggests the likelihood of a much more nuanced neuronal dysfunction that leads to information processing dysfunction. The severity of psychotic symptomology in these diseases tends to be much more episodic in nature suggesting periods of relatively normal neuronal and neural circuit function. This episodic nature coupled with the frequent responsiveness of psychosis to medication supports a much more subtle alteration in information processing, possibly in the context of both nearly normal neuronal and neural circuit function. Further supporting the “nearly normal” hypothesis of psychosis are the many drugs that can acutely lead to transient psychotic symptomology in healthy individuals that is clinically indistinguishable from the psychosis in psychiatric disease. As these drugs can induce psychosis in individuals with presumably normal baseline neuronal and neural circuit function, it is likely that psychosis in psychiatric disease can manifest in the context of relatively minor perturbations in neuronal function. The moment-to-moment information processing deficits that appear to be characteristic of psychosis suggest the possibility that short-term synaptic plasticity mechanisms—that control the computational rules within neural circuits—may represent the primary locus of dysfunction underlying this symptomology. In further support of likely short-term plasticity dysfunction in schizophrenia are the significant deficits in working memory in schizophrenia. While the exact neural substrates of working memory remain to be fully elucidated, both experimental as well as theoretical work suggest that working memory is supported largely by the short-term synaptic plasticity mechanisms at play within neural networks. Thus, both the psychotic features as well as the working memory deficits characteristic of schizophrenia strongly suggest that short-term synaptic plasticity mechanisms represent a promising target to explore for dysfunction, a notion supported by results from studies in mouse models based on valid schizophrenia risk genes which have most prominently displayed pronounced alterations in short-term synaptic plasticity (Arguello and Gogos, 2012).
With this framework in mind, we next examine the evidence for dysfunction in neuronal network performance and synaptic plasticity in neuropsychiatric diseases for which robust, corresponding genetic mouse models of disease have been created. Despite the recent explosive progress in the identification of an abundance of risk genes for various neuropsychiatric diseases, only a very small subset of these identified genetic variants have been found to be highly penetrant with respect to their ability to reliably lead to disease. As such, the creation of etiologically valid mouse models of disease based upon high-risk, highly penetrant genetic variants has only been possible to date in a small subset of neuropsychiatric diseases. Based upon these considerations, we limit our consideration here to two diseases with the strongest genetic mouse models: schizophrenia and autism.
Schizophrenia
Schizophrenia is a heterogeneous disorder with an approximate prevalence of 1% in most populations that most frequently presents in late adolescence to early adulthood and has a strong genetic component based upon the increased incidence observed in twin studies and within in familial pedigrees (McGuffin et al., 1995; Karayiorgou and Gogos, 1997; Sullivan et al., 2003; Lichtenstein et al., 2009a). Schizophrenia is characterized by positive symptoms (disordered thought and speech, delusions, and hallucinations—classic psychotic features), negative symptoms (blunted emotional range, anhedonia, social withdrawal, decreased motivation), and cognitive deficits (deficits in executive function, attention, and both episodic and working memory) that together define the illness (O'Carroll, 2000; Forbes et al., 2009; American Psychiatric Association, 2013). While the causes of most cases of schizophrenia remain unknown, a combination of genetic and environmental factors appears to collaborate in its pathogenesis. A wide array of schizophrenia candidate risk genes and genetic mutations have been identified—from both rare, syndromic and familial cases as well as through large genomic association studies—and have implicated genes with diverse function including those that are either known to or have since been shown to affect synaptic function (Frankle et al., 2003; Gogos and Gerber, 2006; Lisman et al., 2008; Xu et al., 2008, 2011, 2012; Stefansson et al., 2009; Drew et al., 2011a; Girard et al., 2011; Ripke et al., 2011, 2013; Rodriguez-Murillo et al., 2012). While a subset of genomic studies has suggested that genetic risk variants may be potentially biased toward postsynaptically expressed genes, mouse models based upon the most relevant and highly penetrant risk variants have most often shown alterations in short-term synaptic plasticity, which strongly implicates a presynaptic site of dysfunction (Kirov et al., 2012; Fromer et al., 2014; Hall et al., 2014; but see Frankle et al., 2003; Xu et al., 2012).
Abnormalities in the functional connectivity between microcircuits in different brain regions have been put forth as an important pathophysiological mechanisms underlying dysfunction in schizophrenia and functional imaging and EEG studies in schizophrenia as well as studies in mouse models support this possibility (Ford et al., 2002; Lawrie et al., 2002; Meyer-Lindenberg et al., 2005; Esslinger et al., 2009; Stephan et al., 2009; Sigurdsson et al., 2010; Brennan et al., 2013; Uhlhaas, 2013). Assessment of network function in schizophrenia with EEG has shown increased local microcircuit activity as evidenced by enhanced gamma-band oscillations under resting, unstimulated conditions (Kikuchi et al., 2011; Spencer, 2012). In contrast to the increased local microcircuit activity at rest, during a variety of cognitively demanding tasks local microcircuit activity is significantly reduced as indicated by a reduction in gamma oscillations during these tasks compared to healthy controls (Spencer et al., 2003; Ford et al., 2008; Hirano et al., 2008; Haenschel et al., 2009; Minzenberg et al., 2010). Furthermore, some of the observed network alterations have also been seen in both unaffected monozygotic twins and healthy first-degree relatives of schizophrenics suggesting that local circuit dysfunction may represent a significant intermediate functional endophenotype (Hong et al., 2008; Hall et al., 2011). Finally, in vivo plasticity studies conducted on schizophrenic sample groups have shown either reductions or complete absence of long-term plasticity evoked by transcranial stimulation indicating the possibility of significant impairments in long-term plasticity induction mechanisms (Fitzgerald et al., 2004; Daskalakis et al., 2008; Frantseva et al., 2008; Hasan et al., 2011, 2012a,b, 2013).
Despite the large number of schizophrenia risk genes and gene mutations implicated through various methodologies, only a subset of these targets appear to be able to confer by themselves a substantially increased risk of schizophrenia. Furthermore, amongst this subset the directionality and nature of the functional change (e.g., reduced expression or function vs. enhanced expression or function) caused by the genetic mutations that is required to create an accurate functional representation of human disease in mouse models is known for only a very small subset of these implicated genes. Here we discuss the findings in genetic mouse models of schizophrenia that meet these criteria. Intriguingly, studies among these mouse models have revealed that, while long-term synaptic plasticity is relatively intact, a striking preponderance of short-term plasticity alterations suggesting that the neural circuit dysfunction caused by these genetic risk variants may likely be due in large part to alterations in presynaptic function that fundamentally impact synaptic computation (Table 2).
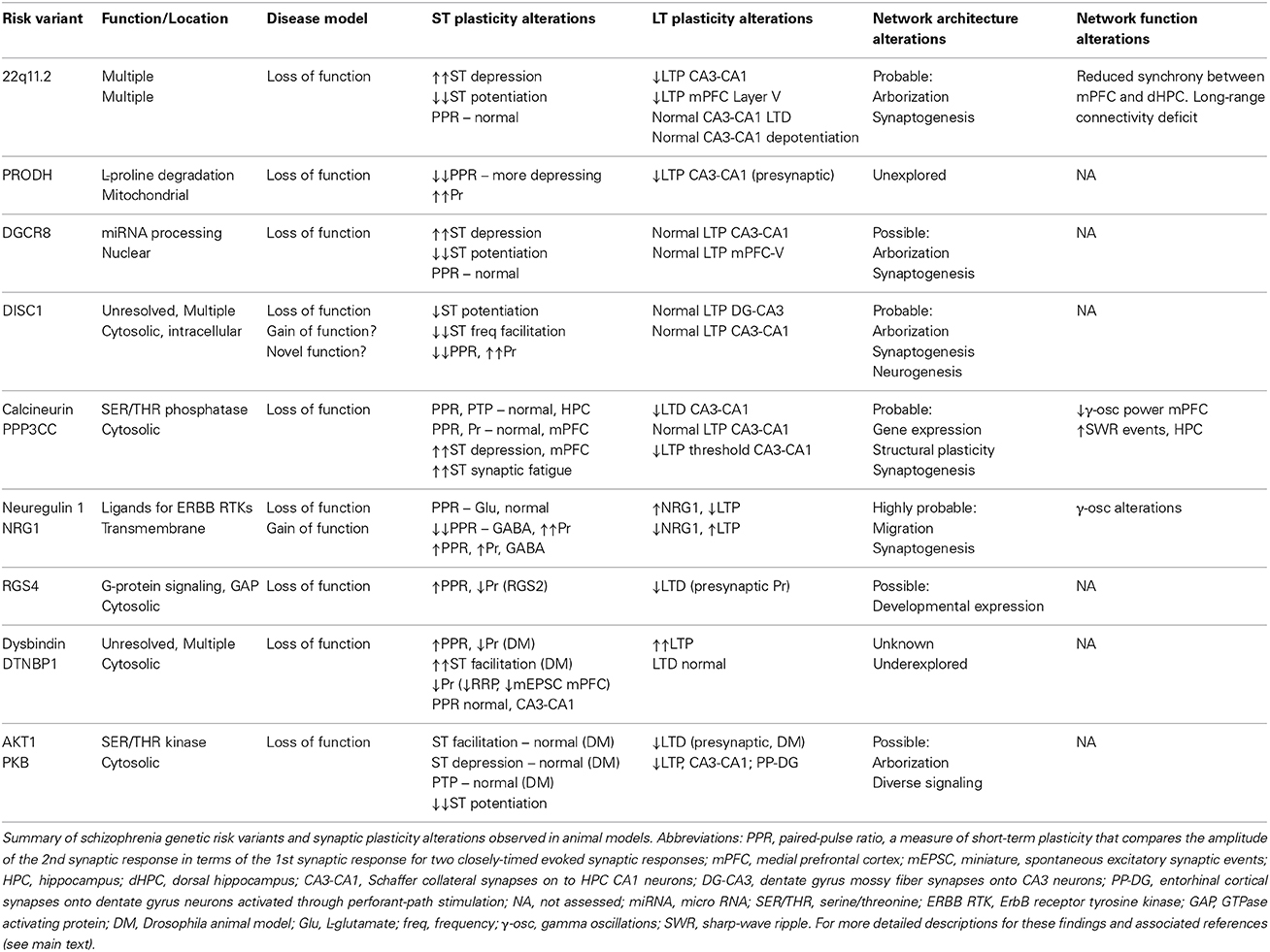
Table 2. Synaptic plasticity alterations in animal models of schizophrenia genetic risk variants.
The 22q11.2 Microdeletion and Related Genes
Microdeletions of the 22q11.2 locus are among the most common chromosomal abnormalities, occur predominantly de novo, and account for 1–2% of sporadic schizophrenia cases (Karayiorgou et al., 1995, 2010; McDonald-McGinn et al., 2001; Xu et al., 2008). Individuals with the 22q11.2 microdeletion are characterized by a high incidence of emotional problems and a spectrum of cognitive deficits, and approximately 30% eventually develop schizophrenia or schizoaffective disorder in adolescence or early adulthood (Pulver et al., 1994; Bearden et al., 2001; Woodin et al., 2001; Sobin et al., 2005; Chow et al., 2006).
A mouse model has been created to recapitulate the human deletion syndrome by creating an analogous deletion in the mouse genome that spans a segment syntenic to the 1.5-Mb human 22q11.2 microdeletion that results in the heterozygous deletion of 27 genes (Stark et al., 2008). Behavioral analysis of this mouse indicated both cognitive deficits, as manifested by impaired spatial working memory and contextual and cued fear learning, as well as deficits in sensorimotor gating—paralleling findings in schizophrenia—as demonstrated by reductions in pre-pulse inhibition (PPI) of acoustic startle (Swerdlow and Geyer, 1998; Stark et al., 2008). To further refine the interpretation of behavioral assays, results from novel object recognition and latent inhibition behavioral assays—that require hippocampal function but are largely independent of PFC function—indicated that purely hippocampal-dependent behaviors were intact suggesting that the primary functional deficit in these animals was largely restricted to dysfunction within the PFC (Fénelon et al., 2013).
Electrophysiological assessment of the 22q11.2-DS equivalent mouse showed that while nearly all forms of short-term and long-term plasticity tested in the hippocampus were unaltered (with the exception of modest reductions in 100 Hz induced LTP at CA3-CA1 synapses), synapses onto layer V neurons in the mPFC—although showing normal basic synaptic transmission and paired-pulse ratios—demonstrated enhanced synaptic depression during high-frequency stimulus trains and reductions in the initial phase of the synaptic potentiation (short-term potentiation) induced by the high frequency stimulus train for LTP induction (Drew et al., 2011b; Fénelon et al., 2013). Finally, in vivo recordings from mice performing a spatial working memory task have shown that the functional, synchronous coupling between the microcircuits in the hippocampus and the PFC is significantly reduced during working memory performance in deletion mice suggesting that the working memory deficits observed in these mice might be related to this disconnectivity (Sigurdsson et al., 2010). Overall these findings of cognitive deficits in the face of largely normal hippocampal electrophysiological function suggest that the primary dysfunction in the deletion syndrome mouse is likely due to a primary functional deficit in PFC microcircuit function and the connectivity between the PFC and the hippocampus. In addition to these findings in mice that employ the full 27 gene microdeletion, in order to more precisely dissect the underlying dysfunction, further evaluations have been performed in mice in which only single genes from within this region have been deleted individually.
DGCR8
DGCR8 is a gene within the 22q11.2 microdeletion whose gene product is involved with processing micro-RNAs (mi-RNAs) within the nucleus which are involved with repressing translation of their target mRNAs (Tomari and Zamore, 2005; Karayiorgou et al., 2010). While homozygous deletion of DGCR8 is lethal, heterozygous deletion such as occurs in the 22q11.2 deletion syndrome results in the reduction of numerous mi-RNAs leading to enhanced expression of their targets due to the loss of mi-RNA repression (Stark et al., 2008; Xu et al., 2013). Mice missing one copy of the DGCR8 gene (DGCR8± mice) demonstrate reduced PPI of acoustic startle, impaired performance in spatial working memory, but, in contrast to deletion-syndrome mice, both normal contextual and cued fear learning (Stark et al., 2008). Similar to electrophysiology findings in the deletion syndrome mouse, measures of both short-term and long-term plasticity were found to be normal at CA3-CA1 synapses in the hippocampus of DGCR8(±) mice, while synapses onto layer V neurons in the mPFC showed normal paired-pulse ratios (suggesting normal initial Pr) but enhanced synaptic depression during high-frequency stimulus trains and reductions in the initial phase of the synaptic potentiation induced by high frequency trains of stimuli (Fénelon et al., 2011). Thus, heterozygous deletion of DGCR8 alone seems to be able to account for most of the electrophysiological deficits found in the deletion syndrome mouse while the absence of fear-learning deficits suggests the involvement of other genes within the 27-gene deletion in these behaviors.
PRODH
PRODH is a gene within the 22q11.2 microdeletion whose gene product is involved with L-proline degradation within mitochondria and its homozygous or heterozygous deletion results in a gene-dose dependent elevation in L-proline in both the periphery and within the CNS (Phang et al., 2001; Karayiorgou et al., 2010). Human subjects with hypomorphic mutations in their PRODH gene have a disease called hyperprolinemia type I that is characterized by seizures and significantly increased incidence of schizophrenia and schizoaffective disorder (Phang et al., 2001; Jacquet et al., 2005; Raux et al., 2007; Clelland et al., 2011). Mice with homozygous functional deletion of the PRODH gene (PRODH knockdown mice) demonstrate reduced PPI of acoustic startle, normal performance in spatial working memory tasks, and reductions in both contextual and cued fear learning (Gogos et al., 1999; Paterlini et al., 2005). Electrophysiological assessment of CA3-CA1 hippocampal synapses with field recordings in PRODH-deficient mice showed increased evoked EPSPs and reduced paired-pulse ratios (depressing compared to facilitating in control animals at a 20 ms interval) both of which suggest increased initial probability in NT release (Pr) in PRODH-deficient mice (Paterlini et al., 2005). These mice also showed decreased CA3-CA1 LTP induction which was notable for a complete absence of its presynaptic component whereas control animals showed increased Pr after LTP induction (Zakharenko et al., 2003; Paterlini et al., 2005). Taken together these electrophysiological results suggest that PRODH deficiency leads to altered presynaptic function with increased initial Pr and an inability of LTP induction to lead to further increases in initial Pr. Interestingly, PRODH-deficient also mice showed evidence of dopaminergic dysregulation within the PFC as evidenced by enhanced hyperlocomotion and dopamine release within the PFC upon amphetamine challenge as well as a possibly compensatory enhanced expression of the dopamine degradative enzyme COMT (a gene located within the 22q11.2 microdeletion that would be expected to be compromised in its ability to undergo an equal compensatory upregulation in patients with the microdeletion). Overall, the findings in PRODH-deficient mice suggest primarily alterations in presynaptic function that—despite normal working memory performance in these mice—would be expected lead to significant alterations in synaptic filtering and network computation within the hippocampus (Tsodyks and Markram, 1997; Abbott and Regehr, 2004). Furthermore, although the underlying mechanism remains to be resolved, PRODH hypofunction also appears to lead to dopaminergic dysregulation and studies in schizophrenics that have revealed a synergy between PRODH and COMT hypofunction support this as a potentially important disease mechanism (Raux et al., 2007).
DISC1
Disrupted in schizophrenia 1 (DISC1) is a gene that was implicated in schizophrenia through its discovery as the gene disrupted by a chromosomal translocation that cosegregated with mental illness in a family pedigree prominently affected by schizophrenia, bipolar disorder, and major depression (St Clair et al., 1990; Millar et al., 2000; Blackwood et al., 2001). The precise cellular and biological role of the DISC1 gene product remains uncertain and resolution of this question has been hindered by the fact that its protein sequence appears to share no homology with other known proteins (Millar et al., 2000). To determine possible functions of Disc1, studies have identified numerous protein-binding partners of Disc1 the most promising of which suggest that Disc1 may play a significant role in modulating cellular function through alterations in cAMP levels and protein kinase function. Disc1 binds to and regulates the phosphodiesterase, PDE4b, and mice with a DISC1 mutation that models the consequences of the human chromosomal translocation demonstrate enhanced cAMP levels due to decrease PDE4 activity (Millar et al., 2005; Kvajo et al., 2011). Furthermore, Disc1 has also been shown to bind to and inhibit the function of the protein kinase GSK3β (Mao et al., 2009) leading to reduced phosphorylation of some of its protein targets. Although in early development Disc1 shows a broader expression pattern, in the adult brain prominent expression is restricted to the dentate gyrus and, to a lesser extent, the CA1 regions within the hippocampal microcircuitry (Austin et al., 2004). Based upon this expression pattern and given the potential role of adult neurogenesis in schizophrenia and other neuropsychiatric diseases, a number of studies have suggested a potentially important role for DISC1 in regulating microcircuit architecture through its regulation of neurogenesis in the dentate gyrus (Ghashghaei et al., 2006; Reif et al., 2006, 2007; Mao et al., 2009; Ming and Song, 2009; Inta et al., 2011; Kim et al., 2012). Within neurons, Disc1 has been shown to be present in multiple subcellular compartments including dendrites and dendritic spines as well as in growth cones and axonal terminals suggesting the possibility that Disc1 could regulate either postsynaptic or presynaptic function (Millar et al., 2005; Kirkpatrick et al., 2006; Taya et al., 2007; Bradshaw et al., 2008; Brandon and Sawa, 2011; Randall et al., 2014).
Using a genetic mouse model employing a DISC1 mutation that accurately recapitulates the consequences of the human chromosomal translocation, a comprehensive battery of behavioral analyses has revealed a pattern of cognitive deficits in these mice that appears to reflect cognitive deficits found in schizophrenia (Koike et al., 2006; Kvajo et al., 2008). Amongst this battery of cognitive behavioral assays, Disc1 mutant mice displayed an isolated deficit in spatial working memory as indicated by impaired performance on a delayed alternation t-maze task. Significantly, largely hippocampus-dependent behavioral tasks—contextual fear conditioning, novel object recognition, Morris water maze, and the eight-arm radial maze win-shift paradigm—all failed to reveal significant alterations in Disc1 mutant mice (Kvajo et al., 2008). Taken together, the results of these behavioral assays suggest an isolated deficit in the short-term maintenance and utilization of information—as opposed to information storage and retrieval—that likely results from information processing dysfunction in the PFC rather than in the hippocampus (Compte et al., 2000; Baddeley, 2012; Roux et al., 2012).
Electrophysiological assessment of granule cell mossy fiber synapses onto CA3 neurons—motivated by the high expression of Disc1 in the dentate gyrus—showed both normal basic synaptic transmission as well as normal LTP in response to 100 Hz stimulation (Austin et al., 2004; Kvajo et al., 2011). Measures of short-term plasticity at these synapses, however, revealed reduced paired-pulse ratios (suggesting increased Pr) and reductions in the short-term frequency facilitation characteristic of this synapse both pointing to selective deficits in short-term plasticity in Disc1 mutant mice (Nicoll and Schmitz, 2005). Electrophysiological assessment of CA3-CA1 hippocampal synapses, in contrast, showed both normal long-term and short-term plasticity with the exception of an isolated reduction in the initial, short-term synaptic potentiation induced by the 100 Hz stimulus train employed for LTP induction (Kvajo et al., 2008). Interestingly, this isolated deficit in short-term synaptic potentiation at CA3-CA1 synapses may potentially represent a significant pathophysiology for schizophrenia as a similar deficit in this short-term potentiation mechanism was observed at mPFC synapses in both the 22q11.2-DS equivalent mice and DGCR8 (+/−) mice (Fénelon et al., 2011, 2013). While assessment of the function of Disc1 at synapses within the PFC has not been reported in Disc1 mutant mice, a recent study employed in utero electroporation and optogenetic techniques to selectively examine the presynaptic role of Disc1 at synapses onto cortical layer II/III pyramidal neurons (Maher and LoTurco, 2012). This study provided evidence that Disc1 may positively regulate neurotransmitter release as Disc1 knockdown led to increased paired pulse ratios (consistent with reduced Pr) as well as disrupted kinetics of evoked synaptic responses potentially consistent with less synchronized NT release. Taken together, the results of these synaptic function studies suggest a limited role of Disc1 in long-term synaptic plasticity but a potentially significant role in regulating presynaptic short-term plasticity mechanisms that would be expected to significantly impact both synaptic and network computation (Abbott and Regehr, 2004).
Calcineurin
Calcineurin dysfunction has been implicated in the pathophysiology of schizophrenia (Gerber et al., 2003; Horiuchi et al., 2007; Liu et al., 2007) with reduced expression or function as the most likely responsible disease mechanism (Eastwood et al., 2005). Calcineurin is a cytosolic Ca2+-dependent protein phosphatase that regulates many cellular processes throughout the body including the CNS where is has been shown to regulate various neuronal processes including synaptic plasticity (Groth et al., 2003). Calcineurin-deficient mice display a number of behavioral abnormalities relevant to schizophrenia including severe reductions in working memory performance and deficits in the PPI of acoustic startle (Zeng et al., 2001; Miyakawa et al., 2003). Furthermore, these studies in mice with a forebrain specific calcineurin deficiency have described altered synaptic plasticity and neural network performance suggesting that hypofunction of calcineurin in schizophrenia could likewise alter synaptic plasticity and lead to disruptions of normal information processing (Cottrell et al., 2013; Suh et al., 2013).
It was originally reported that forebrain calcineurin deficiency led most notably to severe working memory deficits as well as to deficits in LTD at Schaffer collateral (CA3)—CA1 hippocampal synapses (Zeng et al., 2001). This study further showed that the frequency dependence of the stimulus required to induce long-term plasticity was shifted in calcineurin-deficient mice such that LTP (vs. LTD) induction was favored at lower than normal stimulus frequencies suggesting that deficiencies in LTD were permissive of LTP induction. Significantly, the LTD deficiency appeared to be due to an alteration of a post-synaptic mechanism since, in the genetic model employed, calcineurin was not deficient in the CA3 presynaptic neurons as these synapses. In further support of a post-synaptic mechanism, presynaptic function and short-term plasticity were unaltered as evidenced by normal PPR and post-tetanic potentiation (PTP). Although this study failed to identify the precise functional defect that led to LTD deficiency, the normal AMPA-R and NMDA-R mediated synaptic responses observed in calcineurin-deficient mice suggested possible dysfunction in downstream postsynaptic LTD induction mechanisms.
A follow-up study by this same group has recently described apparent network dysfunction in the hippocampus of freely behaving calcineurin-deficient mice (Suh et al., 2013). While in vivo recording from dorsal CA1 showed normal place fields and place cell activation during exploratory behavior, during rest periods place cells were overactive and temporally disorganized in their activity during sharp-wave ripple events (SWRs). The authors suggest that, as SWRs during rest are believed to orchestrate sequential “replay” of place cell activity in order to maintain spatial information, the absence of sequential replay of place cells during SWRs observed in calcineurin-deficient mice may underlie the severe spatial WM deficit previously described (Zeng et al., 2001). Finally, in an effort to link synaptic plasticity alterations to network dysfunction, the authors speculate that the LTD deficit present in CA1 might be responsible for the enhanced excitability within this neural circuitry that appeared to underlie the lack of organized “replay” of place cells during SWRs in calcineurin-deficient mice. Taken together the results of these two studies suggest calcineurin deficiency in schizophrenia would most likely have little impact upon short-term plasticity and synaptic filtering but would disrupt the ability to sculpt information processing by neural circuits by limiting the ability of LTD to down-regulate synaptic gain post-synaptically in order to appropriately control network excitability.
In contrast to the possible role of calcineurin in modulating postsynaptic plasticity in the hippocampus, another recent study in calcineurin-deficient mice has reported deficits in presynaptic function within the PFC (Cottrell et al., 2013). Although the cortical layer and synapse under study was not precisely clarified, these synapses in the PFC showed normal PPR and normal synaptic depression during 20 Hz stimulation; however, during 40 Hz stimulus trains calcineurin-deficient mice showed enhanced synaptic depression. Additional experiments in cultured neurons employing synapto-pHluorins revealed impaired synaptic vesicle exocytosis-endocytosis cycling suggesting that the enhanced synaptic depression was due to augmented synaptic fatigue that limited sustained NT release. Parallel experiments at hippocampal mossy fiber–CA3 synapses—where calcineurin remained expressed postsynaptically—showed similar deficits in sustained NT release indicating that presynaptic deficiency of calcineurin was responsible for the observed deficit. Further studies indicated that calcineurin-deficient mice also demonstrated neural circuit dysfunction as evidenced by reduced power of γ-oscillations in both PFC slices and in in vivo PFC recordings from mice exploring a novel environment. As γ-oscillations in the PFC are believed to be critical for working memory and other executive functions and as these high-frequency oscillations rely heavily upon sustained, high-frequency NT release, the observed deficits in network function suggest the possibility that the MW impairments in calcineurin-deficient mice may ultimately result from presynaptic NT release dysfunction. Thus, the combined results of this study provide a possible mechanism sequentially linking presynaptic NT release dysfunction, to neural circuit dysfunction, to cognitive and WM dysfunction that may be highly relevant to understanding how dysfunction in short-term synaptic plasticity could fundamentally underlie schizophrenia pathophysiology.
Taken together, the results from studies in calcineurin-deficient mice indicate prominent deficits in WM and synaptic plasticity while basic synaptic properties appear largely intact. While this series of studies suggests calcineurin may play different roles in regulating synaptic plasticity in different brain regions (postsynaptic responses in the hippocampus vs. presynaptic neurotransmitter release in the PFC), their findings consistently point toward network-dysfunction in the context of alterations in synaptic plasticity. Although none of these studies provides an unequivocal causal link between synaptic plasticity deficits and the observed network dysfunction, these results nonetheless compellingly suggest that calcineurin deficiency or hypofunction in schizophrenia may lead to similar synaptic plasticity alterations that would lead to significant changes in the information processing performed by neural circuits.
NRG1
Dysfunction in NRG1 signaling pathways has been implicated in the pathophysiology of a variety of neuropsychiatric diseases including schizophrenia (Stefansson et al., 2002; Bertram et al., 2007; Thomson et al., 2007; O'Donovan et al., 2008; Goes et al., 2009; Walker et al., 2010). The manifold NRG1 gene products are EGF-like ligands that signal through ERBB receptor tyrosine kinases—most relevantly through ERBB4—to regulate a large number of cellular processes including neuronal migration, synapse formation, myelination, synaptic transmission, and synaptic plasticity and both their normal signaling and altered signaling in neuropsychiatric diseases have been comprehensively reviewed recently (Mei and Nave, 2014). Although NRG1 has been implicated in an equivocal manner as a schizophrenia risk gene—primarily by human candidate gene studies—only a very small percentage of identified risk variants affect protein coding regions raising significant questions as to whether most identified non-coding variants lead to enhanced or decreased NRG1 signaling (Chen et al., 2006; Walss-Bass et al., 2006; Mei and Nave, 2014). As there is evidence supporting either enhanced or reduced NRG1 signaling in schizophrenia, no clear picture has emerged as to whether human NRG1 disease involves hypofunction or hyperfunction and this has led to the creation of distinct mouse models with opposing types of alterations in NRG1-ERBB signaling (Hashimoto et al., 2004; Petryshen et al., 2005; Hahn et al., 2006; Bertram et al., 2007; Parlapani et al., 2010; Shibuya et al., 2010). Despite the lack of a resolution of this very important issue that critically underlies the generation of valid genetic mouse models of disease, studies in both models of altered NRG1-ERBB signaling nonetheless reveal potential roles and sites of action for these molecules to alter synaptic plasticity and network function in schizophrenia.
Mouse models of both enhanced and reduced NRG1-ERBB signally display a variety of schizophrenia related behaviors. Mice with functional knockdown of NRG1 signaling show locomotor hyperactivity, impaired PPI of startle, impaired working memory, and abnormal social behaviors (Boucher et al., 2007; Chen et al., 2008; Ehrlichman et al., 2009; Shamir et al., 2012). Intriguingly, mice overexpressing components of NRG1-ERBB signaling display a similar pattern of behavioral alterations (Deakin et al., 2009, 2012; Yin et al., 2013; Luo et al., 2014). Importantly, at least some of these behaviors appear to result from semi-acute actions of NRG1-ERBB signaling—rather than complex developmental effects that could significantly impact neural circuit architecture—as perturbing or restoring expression acutely in adulthood can cause or partially reverse behavioral alterations, respectively (Yin et al., 2013; Agarwal et al., 2014; Luo et al., 2014). That many behavioral alterations appear due to acute actions of NRG1 suggests that underlying neural circuit dysfunction may be due to alterations in synaptic transmission and plasticity.
Until recently, the majority of studies exploring the impact of NRG1 signaling have focused upon the function of glutamatergic synapses where ERBB receptors co-localize with PSD95 suggesting a likely role in the regulation AMPA and NMDA receptors (Garcia et al., 2000; Huang et al., 2000; Hahn et al., 2006). Despite this fact, both pharmacological and genetic manipulation of NRG1 signaling does not appear to significantly affect basal glutamatergic synaptic transmission (Huang et al., 2000; Kwon et al., 2005; Bjarnadottir et al., 2007; Iyengar and Mott, 2008; Pitcher et al., 2008). Similarly—and in notable contrast to the findings from most other mouse models based upon schizophrenia risk genes—the preponderance of manipulations of NRG1 signaling have not revealed alterations in short-term glutamatergic synaptic plasticity suggesting NRG1 signaling may preferentially act postsynaptically at these synapses (Huang et al., 2000; Kwon et al., 2005; Iyengar and Mott, 2008; Pitcher et al., 2008; Chen et al., 2010; but see Bjarnadottir et al., 2007; Woo et al., 2007; Deakin et al., 2012). Conversely, NRG1 signaling has a significant effect upon both induction and expression of LTP with manipulations enhancing signaling suppressing LTP while reduced signaling facilitates LTP by removing an apparent tonic suppression (Huang et al., 2000; Kwon et al., 2005; Bjarnadottir et al., 2007; Pitcher et al., 2008; Chen et al., 2010; Agarwal et al., 2014, but see Deakin et al., 2012; Jiang et al., 2013). Although alterations in LTP at these synapses are usually interpreted as due to perturbations of glutamatergic synaptic physiology, complicating this interpretation of NRG1-ERBB signaling is accumulating evidence that NRG1 regulation of hippocampal LTP may be due to more powerful NRG1 facilitating actions at hippocampal GABA-ergic synapse (Chen et al., 2010; Shamir et al., 2012).
NRG1 signaling may particularly impact GABA-ergic transmission as its principal receptor, ErbB4, is selectively expressed in interneurons, particularly within the parvalbumin positive (PV) interneurons known to be fundamental to regulating synchronized network activity (Huang et al., 2000; Bartos et al., 2007; Fisahn et al., 2009; Vullhorst et al., 2009; Fazzari et al., 2010). Enhanced NRG1 signaling increases the amplitude of GABA-ergic synaptic events, most likely through a presynaptic mechanism that increases initial Pr to facilitate GABA release that further leads to altered short-term synaptic plasticity as manifested by enhanced short-term depression (Woo et al., 2007; Chen et al., 2010; Li et al., 2011). A potentially critical importance of the regulation of synaptic function and excitability specifically in PV interneurons by NRG1 is suggested by the alterations in network activity, behavior, and glutamatergic LTP that have been observed both in response to broad alterations in NRG1 signaling as well as when these manipulations are selectively confined to PV interneurons (Woo et al., 2007; Fisahn et al., 2009; Chen et al., 2010; Li et al., 2011; Andersson et al., 2012; Deakin et al., 2012; Shamir et al., 2012; Del Pino et al., 2013).
Overall, the findings from mouse models with a variety of manipulations of NRG1 signaling show prominent deficits in WM, synchronized neural circuit dysfunction likely due to PV interneuron dysfunction, and alterations in both long-term and short-term synaptic plasticity in the context of largely normal basal synaptic function. Furthermore, while on face the prevalence of LT vs. ST synaptic plasticity alterations observed seems at odds with results from mouse models based upon other schizophrenia risk genes, recent findings suggest that these LT plasticity alterations may in fact arise indirectly from altered presynaptic Pr in GABA-ergic neurons that display altered ST plasticity. Yet interpreting the causal impact of NRG1 signaling upon neural circuit function will require much further clarification because, in addition to its more acute effects upon synaptic transmission and synaptic plasticity, its developmental roles in neuronal migration, synapse formation, and myelination further suggests that altered NRG1 signaling in schizophrenia and other disease states may also lead to significant changes in neural circuit architecture.
RGS4
Reduced expression of the “regulator of g-protein signaling” protein RGS4 has been implicated in schizophrenia. Normally, RGS4 acts a negative regulator of GPCR signaling due to its GAP-activity (GTPase-activating protein) that serves to terminate cycles a G-protein signaling by converting active Gα subunits to their inactive state (De Vries et al., 2000). A number of studies have indicated that RGS4 deficiency leads to enhanced GPCR signaling and that this may ultimately lead to a decrease in the probability of NT release from presynaptic terminals (decreased presynaptic Pr). Such an alteration in presynaptic function would be expected to lead changes in short-term synaptic plasticity that would affect the computation of neural circuits suggesting that such functional changes could contribute to the dysfunction in schizophrenia.
The paper that best describes the possible synaptic mechanism and impact of RGS4 deficiency upon presynaptic function, however, is a report that examines the effects of RGS2 deficiency—rather than RGS4 deficiency—upon synaptic function in cultured hippocampal neurons (Han et al., 2006). This study shows that presynaptic RGS2 deficiency permits enhanced presynaptic mGluR signaling that leads to increased inhibition of presynaptic voltage-gated Ca2+ channels (most likely through direct Gβγ inhibition of N-type or P/Q-type channels). The reduced activity of these presynaptic Ca2+ channels in RGS2-deficient synapses results in reduced neurotransmitter release in response to APs. When RGS2 is present in presynaptic terminals these hippocampal synapses are either depressing or non-facilitating by paired-pulse paradigms, indicative of a high presynaptic Pr. RGS2-deficient synapses, however, show paired-pulse facilitation suggesting that in the absence of RGS2 the presynaptic Pr is decreased. In additional support of this action at voltage-gated Ca2+ channels, RGS2-deficient synapses required more extracellular Ca2+ for the same degree of NT release as WT synapses yet both synapses showed indistinguishable NT release in response to hypertonic sucrose challenges. The results of this study indicate that RGS2 deficiency leads to changes in presynaptic function that result in changes in short-term synaptic plasticity and further suggests that a similar scenario may likely result in cases of RGS4 deficiency.
Although whether a similar mechanism of alteration in synaptic function occurs in cases of RGS4 deficiency has not been reported, two studies support this possibility by showing that RGS4 has a functional relationship with voltage-gated Ca2+ channel activity analogous to that of RGS2. Rather than exploring effects upon synaptic transmission, however, these two studies that look at RGS4 regulation of voltage-gated Ca2+ channels examine the resulting impact upon action potentials (Saugstad et al., 1998; Ding et al., 2006). One study found that conditions that enhanced expression of RGS4 in medium spiny neurons (MSNs) of the striatum led to the removal of muscarinic GPCR inhibition of voltage-gated Ca2+ channel activity. The enhanced voltage-gated Ca2+ channel activity that resulted from increases in RGS4 expression permitted enhanced Ca2+ currents during APs that recruited activation of Ca2+-activated K+-channels that served to regularize AP rhythms in these striatal neurons. The results of this paper indicate that RGS4 normally serves to reduce voltage-gated Ca2+ channel inhibition and suggest that RGS4 deficiency would lead to reduced voltage-gated Ca2+ activity.
A second study found that the normal suppression of Ca2+-activated K+ currents by mGluR activation in CA1 hippocampal pyramidal neurons could be nearly abolished by inclusion of RGS4 in the recording pipette (Saugstad et al., 1998). Although this study did not directly show that the RGS4 enhancement of Ca2+-activated K+ currents was due to removal of mGluR mediated inhibition of voltage-gated Ca2+ channels, this would seem to be the likely mechanism. Taken together, the results of these two studies supporting RGS4-mediated enhancement of voltage-gated Ca2+ channel activity, although they did not examine synaptic physiology, support the possibility that RGS4 deficiency could lead to a reduced voltage-gated Ca2+ channel activity in presynaptic terminals that would result in reduced NT release and altered short-term synaptic plasticity.
A final study directly explored the impact of RGS4 deficiency upon synaptic plasticity at MSNs in the striatum by employing RGS4-deficient mice (Lerner and Kreitzer, 2012). This study examined an endocannabinoid (eCB) mediated LTD (eCB-LTD) at MSN synapses that is dependent upon mGluR-Gq mediated generation of eCB in the post-synaptic neuron that travels to the presynaptic neuron to inhibit presynaptic NT release by lowering Pr. Normally this LTD pathway can be modulated by the activation of additional postsynaptic GPCRs, but in RGS4-deficient mice this modulation is absent. Through a series of elegant experiments this study showed that this GPCR modulation of LTD occurs through alterations in cAMP/PKA modulation of RGS4 activity that alters the ability of RGS4 to suppress mGluR-dependent production of the eCB retrograde messenger responsible for the LTD. Thus, although by a much more complicated mechanism than described for RGS2, RGS4 deficiency similarly results in alterations in presynaptic NT release probability (Pr). Further, although this study did not directly address changes in ST synaptic plasticity, the inability to modulate presynaptic Pr due to RGS4 deficiency would be predicted to lead to a decreased ability to modulate ST plasticity at this synapse as well.
Taken together, the results of these four studies suggest that the action of RGS4 should normally serve to increase presynaptic NT release probability and that RGS4 deficiency would lead to reductions in Pr causing reduced NT release. While only LT plasticity consequences have been directly explored in RGS4-deficient mice, the results of these studies supporting that presynaptic Pr is a likely target for RGS4 modulation strongly suggest that ST synaptic plasticity—and consequently neural circuit computation—would be strongly impacted by RGS4 deficiency in schizophrenia.
Dysbindin
Reductions in dysbindin expression and function have been implicated in schizophrenia and results from a number of studies point toward a role for dysbindin in regulating presynaptic short-term plasticity by modulation of the probability of neurotransmitter release from synaptic terminals. Although a detailed understanding of dysbindin function remains to be determined, its expression in central synaptic terminals in the mammalian CNS and its association with synaptic vesicles suggests a possible role in the regulation of NT release (Benson et al., 2001; Talbot et al., 2006).
Perhaps the most informative study of dysbindin function was performed at the glutamatergic neuromuscular junction (NMJ) in Drosophila (Dickman and Davis, 2009), a system whose amenability to genetic manipulation permits rapid clarification of the detailed molecular mechanisms of gene action. This study demonstrated that presynaptic dysbindin deficiency led to reduced Ca2+-sensitivity of NT release, enhanced paired-pulse facilitation of post-synaptic responses, enhanced facilitation of post-synaptic responses during trains of stimuli, and a failure of homeostatic upregulation of the number of NT vesicles released per stimulus (quantal content) upon postsynaptic receptor blockade. Each of these results is consistent with a role of dysbindin in enhancing the Pr of presynaptic NT release (and dysbindin deficiency leading to reduced NT release), most likely through an action of dysbindin that enhances the Ca2+ sensitivity of the NT release machinery. This study convincingly illustrated that dysbindin deficiency altered ST plasticity by transforming a modestly facilitating synapse into a strongly facilitating synapse. This finding supports the possibility that dysbindin-deficient synapses in the mammalian CNS would have similar alterations in ST plasticity that would be expected to significantly alter synaptic and neural circuit computation.
A recent study of synaptic transmission onto layer V pyramidal neurons in the mPFC in dysbindin-null mice reported a number of findings consistent with the altered presynaptic function described at the Drosophila NMJ (Saggu et al., 2013). Reduced amplitudes of evoked EPSCs, reduced frequency of mEPSCs, a reduced RRP of synaptic vesicles, reduced SV cycling upon depolarization (as determined by FM1-43 studies), and decreased Ca2+ flux into synaptosomes upon depolarization all consistently point toward a decrease in the presynaptic Pr of NT release in dysbindin-deficient synapses mice. Furthermore, although not reaching statistical significance, this study performed assays to determine Pr more directly and showed a ~35% reduction in presynaptic Pr in dysbindin-deficient mice. Thus, although this study did not directly examine either short-term or long-term plasticity by traditional electrophysiological assays, taken together these findings strongly support a reduced presynaptic Pr due to dysbindin deficiency that would be expected to lead to alterations in short-term plasticity in neural circuits in the mammalian mPFC.
Despite the strong support for dysbindin deficiency leading to decreased presynaptic function through reduced Pr (Dickman and Davis, 2009; Saggu et al., 2013), a recent study in the hippocampus of dysbindin-deficient mice did not see alterations in ST plasticity (Tang et al., 2009). This study of Schaffer collateral input to CA1 in the hippocampus reported a nearly doubled magnitude of LTP at these synapses in dysbindin-deficient mice but both normal paired-pulse facilitation and normal LTD. Yet, taken together, these studies of synaptic function in dysbindin-deficient animals suggest that the most likely role of dysbindin is in regulating presynaptic NT release and that dysbindin deficiency leads to decreases in NT release by reducing presynaptic Pr. Furthermore, as presynaptic Pr is the prime determinant controlling short-term plasticity at synapses, dysbindin deficiency in schizophrenia would be predicted to most significantly alter short-term plasticity and lead to alterations in neural circuit computation that could underlie disease symptomology.
AKT1
Reductions in AKT1 expression and function have been implicated in schizophrenia in a number of recent studies (Emamian et al., 2004; Zhao et al., 2006; Balu et al., 2012). AKT is a protein kinase that has many cytosolic protein targets in a variety signaling pathways both in the CNS as well as in non-neuronal tissues (Yang et al., 2004). AKT1-deficient mice show alterations in the dendritic architecture of mPFC pyramidal neurons as well as working memory deficits that are believed to be representative of analogous deficits found in schizophrenia (Lai et al., 2006). Although the role AKT1 in neuronal and synaptic function remains under-explored, two studies in animal models with reduced AKT1 expression have revealed alterations in synaptic plasticity (Guo and Zhong, 2006; Balu et al., 2012).
While neither of these two studies provides a clear mechanistic explanation of the plasticity alterations observed in AKT1-deficient animals, the study of the glutamatergic synapse at the NMJ of Drosophila provides a more comprehensive analysis (Guo and Zhong, 2006). In this study the authors describe a novel long-term depression (LTD) evoked by high frequency stimulation that quantal analysis revealed to be presynaptic in origin as the quantal size remained unchanged but the quantal content (reflecting the number of released neurotransmitter vesicles per stimulated event) was reduced. Although basic NMJ responses were normal in AKT mutant flies, this novel LTD was significantly reduced in mutant flies with low expression of AKT while normal LTD could be rescued by acute over-expression of WT AKT. Despite the evidence of a presynaptic locus for this LTD, in AKT mutant flies with deficient LTD, measures of baseline presynaptic function such as short-term depression, short-term facilitation, and PTP were found to be normal. Given that these results support that the observed LTD in WT flies was due to reduced presynaptic Pr and that all forms of baseline presynaptic function in AKT-deficient flies were normal—including inferred presynaptic Pr—suggests a role of AKT in the induction of changes in presynaptic Pr during LTD protocols that is reduced or absent in AKT-deficient flies. Interestingly, such a mechanistic deficiency is somewhat similar to the deficient control of presynaptic Pr upon eCB-LTD induction in the striatum of RGS4-deficient mice where RGS deficiency impacts the ability to modulate presynaptic Pr (Lerner and Kreitzer, 2012). Regardless of the details of the mechanism underlying deficient LTD at the NMJ in AKT-deficient flies, an inability to appropriately reduce presynaptic Pr by long-term plasticity mechanisms would consequently lead to an inability to alter presynaptic short-term plasticity normally at AKT-deficient synapses.
In a separate study in AKT1-deficient mice deficits in long-term plasticity were observed at multiple synapses in the hippocampus (Balu et al., 2012). AKT1-deficient mice showed deficits in LTP in CA1 upon Schaffer collateral stimulation (SC-CA1) and deficits in LTP in the dentate gyrus upon medial perforant path stimulation (EC-GC). At both of these sites short-term plasticity of synaptic responses during the tetanus were normal in AKT1-deficient mice suggesting no major alteration in baseline presynaptic short-term plasticity mechanisms. The post-tetanic short-term potentiation, however, although normal at SC-CA1 synapses, was completely absent at EC-GC synapses suggesting a more severe plasticity deficit at the synapses onto granule cells in the dentate gyrus. Despite the profound deficits in synaptic plasticity described in AKT1-deficient mice in this report, given that the mechanisms underlying these deficits were not further experimentally pursued it is difficult to speculate upon the precise role of AKT1 function at these synapses.
Taken together the results of these two studies in AKT-deficient animals indicate that AKT1 may play an significant role in regulating long-term synaptic plasticity and that AKT1 deficiency in schizophrenia may result in similar long-term plasticity alterations that could contribute to disease symptomology.
Autism
With the exception of schizophrenia, perhaps the most relevant studies evaluating synaptic plasticity that employ genetic mouse-models based upon strong candidate risk genes in human neuropsychiatric disease have been carried out in models of autism. Autism spectrum disorder (ASD) is a heterogeneous neurodevelopmental with an approximate 1% prevalence in the population that has a heritability supporting a strong genetic component (Abrahams and Geschwind, 2008). ASD is symptomology is characterized by abnormal reciprocal social interactions, impaired communication (particularly involving language but also non-verbal), and repetitive behaviors (American Psychiatric Association, 2013). Among the diverse array of autism risk genes that have been identified both from syndromes with autistic features as well as though genome analyses of large patient populations are a significant number of genes known to affect synaptic function and plasticity (Zoghbi, 2003; Abrahams and Geschwind, 2008; Südhof, 2008; Silverman et al., 2010; Gauthier and Rouleau, 2012; Iossifov et al., 2012; Neale et al., 2012; O'Roak et al., 2012; Sanders et al., 2012). And, in contrast genetic mouse models of schizophrenia which predominantly display selective alterations in short-term synaptic plasticity, mouse models based on ASD risk genes have shown a more balanced combination of both short- and long-term synaptic plasticity alterations (Arguello and Gogos, 2012).
Functional imaging studies in autism overall support that long-range connections between brain regions (such as from higher-order association areas to the frontal lobe) show reduced functional connectivity while local microcircuitry and short-range connections show evidence of enhanced connectivity (Mundy, 2003; Belmonte et al., 2004; Geschwind and Levitt, 2007; Markram and Markram, 2010; Ecker and Murphy, 2014). Assessment of network activity in autism with EEG and MEG has shown reduction of local microcircuit activity evidenced by reduced gamma oscillations at rest and during sensory processing and facial presentation tasks (Wilson et al., 2007; Sun et al., 2012; but see Orekhova et al., 2007). Furthermore, long-range connectivity between brain regions appears impaired as, despite increased strength of theta rhythms locally, coherence of these rhythms between brain regions was reduced (Murias et al., 2007). That some of these network alterations have also been seen in unaffected relatives of autistics suggests that both local and long-range network dysfunction may represent significant intermediate endophenotypes (Rojas et al., 2011). Finally, in vivo studies of long-term plasticity have indicated that at least a subset of autistics show enhanced LTP- and LTD-like phenomena that are consistent with theories of hyper-plasticity in autism (Markram and Markram, 2010; Oberman et al., 2010).
Although a number of mouse models of autism based on human genetic findings have been made, neuronal and synaptic function have been most thoroughly explored in mice modeled upon Rett syndrome (MECP2 gene deletion) and fragile X syndrome (FMR1 gene deletion). While both of these human disorders are syndromic in nature and manifest a variety of clinical features in addition to ASD symptomology, important insights about synaptic plasticity alterations underlying autism symptomology can still be gleaned from these mouse models despite these potentially complicating factors. Both Fmr1 and Mecp2 null mice demonstrate deficits in long-term memory and learning that are consistent with the possibility of an underlying deficit in long-term synaptic plasticity (Moretti et al., 2006; MacLeod et al., 2010). While there are no reports on working memory (MW)—a short-term memory process thought to rely largely upon short-term synaptic plasticity—in Mecp2 null mice, MW memory may be deficient in Fmr1 null mice although reports are conflicting (Van Dam et al., 2000; Yan et al., 2004; Mongillo et al., 2008; Udagawa et al., 2013).
Both Mecp2 and Fmr1 null mice show alterations in presynaptic short-term plasticity. Decreased paired-pulse facilitation observed in Mecp2 null mice suggests increased initial Pr for neurotransmitter release that shifts synaptic function in the direction of short-term depression (Moretti et al., 2006; Weng et al., 2011). These mice have also shown reduced PTP, a more sustained form of short-term plasticity induced by high-frequency activity (Weng et al., 2011). In Fmr1 null mice, although paired-pulse facilitation is normal, these mice showed strongly increased augmentation—a longer-lasting component of short-term plasticity—and reduced short-term depression that is entirely due to presynaptic mechanisms and were at least partially attributable to enhanced presynaptic Ca2+ influx due to AP broadening and increases in the readily releasable pool of NT vesicles during high frequency activity (Deng et al., 2011, 2013; Wang et al., 2014). These changes also resulted in changes in synaptic information processing as there was a significant enhancement of responses during natural stimulus trains that were most notable during high-frequency epochs of activity (Deng et al., 2011).
Both Mecp2 and Fmr1 null mice show alterations in long-term plasticity. MeCP2 null mice have been shown to have impaired hippocampal LTP and LTD in the hippocampus and impaired LTP in motor and sensory cortex (Asaka et al., 2006; Moretti et al., 2006; Weng et al., 2011). Although Fmr1 null mice display normal hippocampal LTP they have impaired neocortical LTP and show enhanced LTD in both the hippocampus and cerebellum (Godfraind et al., 1996; Paradee et al., 1999; Huber et al., 2002; Li et al., 2002; Koekkoek et al., 2005; Larson et al., 2005; Hayashi et al., 2007).
The limited studies of network activation in mouse models of autism suggest enhanced activity in local microcircuitry. Drug-induced gamma oscillations are enhanced in Mecp2 null mice and, although gamma oscillations have not been assessed in Fmr1 null mice, these mice also display local network dysfunction in the form of enhanced up-states during slow wave oscillations (Hays et al., 2011; McLeod et al., 2013). These enhancements in local network activity in mouse models of autism are consistent with some reports of enhanced high-frequency oscillations observed in humans studies (Orekhova et al., 2007). Coupled with the findings in human studies, these complementary findings in mouse models of autism suggest that significant alterations in both long-term and short-term synaptic plasticity may likely be responsible for the information processing and behavioral dysfunction present in the human disease.
A Strategy to Dissecting Neural Circuit Dysfunction in Genetic Mouse Models of Neuropsychiatric Disease
While a small number of etiologically valid genetic mouse models of schizophrenia and autism have begun to enable exploration of the underlying dysfunction in neural circuits and synaptic plasticity in these diseases, similar progress for most other neuropsychiatric diseases has been limited to date as equally robust predisposing genetic variants necessary to recapitulate disease in animal models have yet to be identified for these diseases. Yet it is both the complex nature of the genetic risk of neuropsychiatric disease as well as our current symptoms-based vantage point that continue to present a number of challenging issues that more broadly complicate and confound the strategic approach to unraveling root causes underlying neural circuit dysfunction in genetic mouse models of neuropsychiatric diseases.
Issue—Genetic Architecture
Genomic studies of neuropsychiatric disease have identified a surprisingly large and diverse array of risk genes. Despite this genetic heterogeneity, the fact that identified risk genes are preferentially involved in synaptic function, growth, and maintenance as well as axonal guidance suggests that genetic risk factors for neuropsychiatric disease may converge upon the common substrate of neural circuit function (Walsh et al., 2008; Glessner et al., 2009; Guilmatre et al., 2009; Akil et al., 2010; Insel et al., 2010; Gilman et al., 2011; O'Dushlaine et al., 2011; Sanders et al., 2011). In addition, despite heterogeneity, a small number of rare but highly penetrant genetic risk variants have been identified that reliably lead to disease that is clinically indistinguishable from the majority of disease cases (Farooqi and O'Rahilly, 2005; Peltonen et al., 2006). Although these highly penetrant genetic variants are usually only responsible for a very small percentage of cases of the broader disease, their high reliability to cause disease renders them invaluable, essential tools in creating mouse models that faithfully recapitulate disease. While a small number of such high-risk genetic variants have been identified in cases of schizophrenia, autism, and AD that have enabled the creation of reliable disease models in mice, for many common neuropsychiatric diseases—e.g., major depression, bipolar disorder, anxiety disorders, attention-deficit hyperactivity disorder—similarly high-risk genetic variants that reliably lead to human disease have yet to be identified thus leaving valid genetic mouse models of these diseases currently out of reach (Pulver et al., 1994; Hardy, 1997; Murphy et al., 1999; Millar et al., 2000; Blackwood et al., 2001; Hatton et al., 2006; Clifford et al., 2007; Ramocki et al., 2009; Vassos et al., 2010; Neul, 2012). While the genetic selective pressures—such as the strong selection pressures against retaining risk genes for socially debilitating and early-onset diseases like autism and schizophrenia that largely preclude successful reproductive transmission—that shape the genetic architecture of disease may be substantially different for diseases such as major depression, it nonetheless seems reasonable to posit (and hope) that high-risk genetic variants for these diseases in fact exist yet have eluded detection and remain to be found. Based upon these considerations, creating reliable genetic mouse models of disease must begin with the identification of highly penetrant genetic variants and in the case of neuropsychiatric diseases in which such genetic variants remain to be identified our understanding of the details of neural circuit dysfunction in these diseases likely must patiently await this first crucial step.
Issue—Overlapping Disease Phenomenology
Further complicating the understanding of the causality between genes and disease, genomic studies have revealed a surprising overlapping genetic risk among multiple neuropsychiatric diseases that raises significant questions as to the validity of the boundaries between these diseases (Craddock and Owen, 2005; Craddock et al., 2006; Friedman et al., 2008; Guilmatre et al., 2009; McCarthy et al., 2009; O'Donovan et al., 2009; Williams et al., 2010; Smoller et al., 2013). That is, in many cases the same genes implicated in increased risk for one neuropsychiatric disease have been shown to predispose to increased risk for multiple other distinct neuropsychiatric diseases. Furthermore, although some genetic variants appear to lead to narrower, better-defined clinical phenotypes, genomic studies have also revealed that in some cases the same genetic variant can be found in patients with different clinical diagnoses (St Clair et al., 1990; Hatton et al., 2006; Bijlsma et al., 2009; McCarthy et al., 2009; Ramocki et al., 2009; Shi et al., 2009; Wang et al., 2009; Karayiorgou et al., 2010; Shinawi et al., 2010). The potential significance of these results is further supported by heritability studies that have shown that within family pedigrees the presence of one neuropsychiatric illness is frequently associated with cross-vulnerability to other neuropsychiatric illnesses (Biederman et al., 1991; Maier et al., 1993; Lichtenstein et al., 2009b, 2010; Rommelse et al., 2010; Sullivan et al., 2012). Furthermore, beyond considering these concerns of genetic promiscuity among distinct neuropsychiatric diseases further suggests that beyond choosing highly penetrant genetic variants, the clearest path to understanding distinct diseases through genetic mouse models may further require carefully choosing among genetic variants for those associated with narrow, well-defined clinical presentations.
Beyond these concerns, however, the additional overlapping symptomology and overlapping responsiveness to medication found between some diseases raises the legitimate possibility that the boundaries between “distinct” diseases may be inherently blurred due to a true shared causation (Milberger et al., 1995; Citrome et al., 2005; Lake and Hurwitz, 2007; O'Donovan et al., 2009; Sinzig et al., 2009; Ivleva et al., 2010; Gros et al., 2012; Zbozinek et al., 2012). For example, while NMDA receptor hypofunction has been implicate in the causation of schizophrenia, recent studies in rodent models support NMDA receptor hypofuction in autism suggesting the possibility of a continuum of related dysfunction between and among distinct diseases (Mohn et al., 1999; Gandal et al., 2012; Saunders et al., 2012; Schmeisser et al., 2012; Won et al., 2012). Taken together, these multiple issues of overlapping disease phenomenology suggest the possibility that the traditional diagnostic criteria used to create distinct boundaries between diseases based upon behavioral manifestations—while clinically useful in disease management—may not be ideally suited as a scientific framework from which to determine underlying disease mechanisms which may instead require the development of more refined and quantifiable measures targeted at directly assessing endpoints related to brain function including the function of neural circuits (Geyer, 2006; Gould and Gottesman, 2006; Benes, 2007; Psychiatric GWAS Consortium Coordinating Committee, 2009; Luck et al., 2011; Smoller et al., 2013).
Issue—Alternative Measures of Function
Unlike traditional physiological diseases such as cardiovascular disease, diabetes, or obesity, the understanding of neuropsychiatric disease processes is currently hampered by the underdevelopment and underutilization of analytic measures that provide simple yet precise quantitative readouts of the function and status of the underlying physiological systems that directly contribute to disease pathogenesis (Gould and Gottesman, 2006). For example, psychiatric diagnosis and evaluation as put forth by the DSM-V is based upon a checklist of behavioral signs and symptoms that are considered either present or absent in a binary manner and are far removed from any direct quantitative measure of underlying brain or neural circuit function (American Psychiatric Association, 2013). Furthermore, most current evaluative measures in neuropsychiatric disease are inherently complex—e.g., evaluation of complex phenomena such as social function, judgment, and language—and do not obviously point to precisely circumscribed loci of neural circuit dysfunction. Due to this complexity, these measures—while both interesting and clinically relevant in terms of monitoring the progression and the functional impact of disease—are not terribly useful as scientific bases for a reductionist approach aimed at revealing fundamental disease mechanisms (Insel et al., 2010). Moreover, many of these complex manifestations seem to be uniquely human in nature do not appear to present obvious translatable equivalents that could readily permit correlative evaluation in mouse models. These considerations highlight the intense need in neuropsychiatric diseases for the development of more simplified and precise parameter assays that directly and quantitatively report upon underlying pathophysiological processes such as neural circuit dysfunction (Gould and Gottesman, 2006; Akil et al., 2010; Insel et al., 2010).
Analysis of disease related endophenotypes may represent the most effective strategy to provide the simplified and precisely quantitative functional measures required to efficiently dissect fundamental disease mechanisms. Endophenotypes are relatively simple, quantifiable, and heritable characteristics present in both individuals diagnosed with disease and in unaffected members within their familial pedigrees and are therefore believed to represent disease vulnerability traits (Gould and Gottesman, 2006). Compared to “top level” symptoms in neuropsychiatric disease that often appear to be uniquely human in nature without clear parallels in mouse models, the more rudimentary nature of endophenotypes present the further advantage that their assays are frequently readily translatable into mouse models (Geyer, 2008; Ecker and Murphy, 2014). In consensus recognition of the need for more quantifiable measures in neuropsychiatric disease, an initial, though partial, effort to address these issues strategically has recently begun to be laid out by the NIMH in the form of their proposed Research Domain Criteria (RDoC) project initiative.
While assessment of endophenotypes frequently provides little direct clinical utility, their quantification is scientifically valuable as endophenotypes are believed to represent the most fundamental consequences of the basic pathological processes underlying the more complex manifestations neuropsychiatric diseases. From the standpoint of assessing suspected neural circuit dysfunction in neuropsychiatric disease, particularly appealing among endophenotypes are the neurophysiological measures which quantify rather stereotyped responses to very simplified stimuli (Turetsky et al., 2007; Luck et al., 2011). These assays, through assessing the dysfunction of simplified neural circuits involved with very rudimentary information processing, present an effective and strategic platform from which to reveal the cellular and molecular details of the neuronal and synaptic alterations underlying neural circuit dysfunction in neuropsychiatric disease. To better appreciate the refined analytical power and broad utility such assays present for guiding a detailed mechanistic understanding of neural circuit dysfunction in neuropsychiatric disease, it is useful to specifically examine the strategic approach to understanding neural circuit dysfunction in mouse models of schizophrenia.
Optimizing the Approach to Neural Circuit Analysis: Schizophrenia as Example
Currently the most prevalent approach to understanding neural circuit dysfunction in genetic mouse models of schizophrenia has involved assessing working memory deficits in these animals coupled with electrophysiological recordings from the underlying brain regions believed to contribute most to working memory performance. While not as striking as the positive symptomology of schizophrenia, the pervasive cognitive deficits in schizophrenia have presented a tractable experimental endophenotype for examining disease-relevant alterations in behavior and neural circuit function in genetic mouse models (Glahn et al., 2003; Heinrichs, 2005; Saperstein et al., 2006; Forbes et al., 2009; Arguello and Gogos, 2010, 2012). As such, assessment of working memory in genetic mouse models of schizophrenia has been commonly employed and any working memory deficits detected have been interpreted as reflecting dysfunction of neural circuits in either the hippocampus or the medial prefrontal cortex (Arguello and Gogos, 2010, 2012). Motivated by such findings these brain regions have then been subsequently probed more directly with electrophysiological assays in attempts to detect and localize neuronal dysfunction within specific neurons or synapses and to determine the precise mechanisms underlying this dysfunction and this approach has successfully revealed—consistent with WM deficits—prominent alterations in short-term synaptic plasticity (Zeng et al., 2001; Kvajo et al., 2008, 2011; Mongillo et al., 2008; Jentsch et al., 2009; Sigurdsson et al., 2010; Cottrell et al., 2013; Fénelon et al., 2013).
Despite the strong validity, disease-relevance, and translational potential of working memory tasks as a framework from which to assess and reverse neuronal and synaptic dysfunction in mouse models, the inherent complexity of such tasks limits their potential to identify and precisely localize underlying neuronal dysfunction (Arguello and Gogos, 2010, 2012). Specifically, during a spatial working memory task information processing in disparate brain regions involving sensory, motor, motivational, volitional, appetitive, and fear and anxiety related neural circuits is likely influencing behavioral performance in addition to the neural circuit activation within the mPFC and hippocampus (Gisquet-Verrier and Delatour, 2006; McNab and Klingberg, 2008; Edin et al., 2009; Baddeley, 2012).
As the ultimate goal of investigations in genetic mouse models of neuropsychiatric disease is to reveal the precise mechanisms of neuronal and synaptic dysfunction underlying the neural circuit dysfunction in these diseases, the optimal assays of in vivo neural circuit function should ideally be as simple and uncomplicated as possible such that any dysfunction observed can be confidently and unambiguously attributed to a distinct neural circuit or brain region that can then be directly targeted with invasive, high-resolution electrophysiological assays. Although finding a functional measure that can be purely attributed to a single, well-defined neural circuitry is a non-trivial hurdle, a number of schizophrenia-related neurophysiological endophenotypes involving sensory evoked responses may come the closest to fitting this requirement (Turetsky et al., 2007; Thaker, 2008). While the study of the neural circuits underlying these endophenotypes has yet to be aggressively pursued, due to their exquisite simplicity these endophenotypes may represent the most tractable and efficient analytical path toward understanding the discrete neuronal and synaptic mechanisms underlying neural circuit dysfunction in well-defined and valid genetic animal models of schizophrenia (Goff et al., 1980; Javitt et al., 1994; Freedman et al., 1996; Swerdlow et al., 2001; Grunwald et al., 2003; Kumari et al., 2003; Schall et al., 2003; Luck et al., 2011).
Specifically, among the many neurophysiological assays showing characteristic alterations in schizophrenics, the sensory evoked response endophenotypes involving PPI of acoustic startle, P50 auditory evoked potential suppression (P50), and acoustic mismatch negativity (MMN) appear to reflect information processing abnormalities within relatively simple and isolated neural circuits (Clements et al., 1998; Bramon et al., 2004; Light and Braff, 2005; Swerdlow et al., 2006; Brockhaus-Dumke et al., 2008). From a reductionist point of view, the most compelling characteristic of these three endophenotype assays is that they involve neural circuit processing that is either entirely or largely preattentive—and thus nearly reflexive—in nature (Naatanen, 1992). These measures thus appear to represent read-outs of rudimentary neural circuit computation with minimal contaminating influences from other neural circuitry that might otherwise confound interpretation of results (Naatanen, 2003; Braff and Light, 2004). As such, these assays of neural circuit function that are largely devoid of the complications of attentional, motivational, or cognitive control may be optimal assays of neural circuit function in mouse models of schizophrenia—through use of analogous assays in rodents that have been developed for each of these measures—as they examine hard-wired, relatively inflexible computational elements that present a greatly simplified system in which to explore the discrete neuronal and synaptic mechanisms underlying neural circuit dysfunction (Swerdlow et al., 2001; Gould and Gottesman, 2006; Metzger et al., 2007; Geyer, 2008; Javitt et al., 2008; Ehrlichman et al., 2009; Luck et al., 2011). Furthermore, as these neurophysiological assays employ either pairs or trains of acoustic stimuli delivered at very brief intervals, these assays bear a striking resemblance to electrophysiological assays that employ similarly brief intervals of stimulation to determine short-term plasticity properties of synapses suggesting the possibility that abnormalities observed in neurophysiological assays may reflect underlying alterations in short-term synaptic plasticity (Freedman et al., 1996; Garrido et al., 2009; Blundell et al., 2010). In summary, while no neural circuit exists in isolation from the influence of other circuits within the brain, the neurophysiological assays of preattentive schizophrenia endophenotypes that activate rudimentary neural circuits come close to this ideal and may represent the optimal assays to guide the elucidation of the synaptic and neuronal mechanisms underlying neural circuit dysfunction in valid genetic mouse models of schizophrenia.
Concluding Remarks
The progress in unraveling the mechanisms underlying neural circuit dysfunction in human neuropsychiatric diseases has been enormously accelerated by the recent advances in human genomics that have enabled the identification of an abundance of candidate genetic risk variants that may be the responsible agents for causing disease symptomology. Specifically, the identification of highly penetrant risk genes for schizophrenia and autism has led the creation of etiologically valid mouse models for these diseases that have enabled detailed investigations directed at identifying synaptic, neuronal, and neural circuit dysfunction. Studies in these mouse models have revealed both neural circuit dysfunction as well as a variety of synaptic plasticity alterations that may be responsible for this dysfunction given the critical role of synaptic plasticity in sculpting information flow within neural circuits. Overall, while mouse models of schizophrenia have shown a preponderance of short-term plasticity alterations consistent with disease symptomology, mouse models of autism appear to present a more balanced combination of both short-term and long-term plasticity alterations that may be consistent with the more profound dysfunction present in the clinical picture of this spectrum of disorders.
These initial successes with schizophrenia and autism strongly suggests that, despite the failure to date to identify highly penetrant genetic risk variants for many of the more common neuropsychiatric diseases such as anxiety disorders and major depression, this strategic approach may represent the necessary pathway forward for these diseases as well. Yet, beyond negotiating the complexities of the genetic architecture of neuropsychiatric diseases, in order to effectively identify neural circuit dysfunction and resolve the underlying neuronal and synaptic mechanisms that lead to it, additional considerations need to be thoughtfully addressed. As a symptomology-based approach to neuropsychiatric disease has revealed significant overlapping disease phenomenology, dissecting the causes of unique forms of neural circuit dysfunction underlying disease will likely require further identification and utilization of more simplified, quantifiable endophenotypes that examine precisely defined parameters more directly and more closely related to alterations in specific brain regions and functions. In addition to the advantage that assays of such endophenotypes are readily translatable to assess neural circuit dysfunction in mouse models of disease, assessing the genetic risk variants contributing to individual endophenotypes—compared to the global risk for disease based upon symptomology—should facilitate dissecting disease mechanisms due to the likely identification of a much smaller and more manageable number of genes responsible for dysfunction. Finally, although our current understanding of even the most fundamental aspects of normal neural circuit function remain rudimentary, the continuing technical advances in in vivo imaging, electrophysiology, and optogenetics are beginning to help reveal new levels of details of normal neural circuit that will enable more sophisticated assessment of neural circuit function in disease. Taking these considerations together, the strategic approach to understanding neural circuit dysfunction in neuropsychiatric disease is rapidly evolving on multiple technical fronts and now seems poised to shortly make major revelations regarding the mechanisms of neural circuit dysfunction in these diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, L. F., and Regehr, W. G. (2004). Synaptic computation. Nature 431, 796–803. doi: 10.1038/nature03010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Achard, S., Salvador, R., Whitcher, B., Suckling, J., and Bullmore, E. (2006). A resilient, low-frequency, small-world human brain functional network with highly connected association cortical hubs. J. Neurosci. 26, 63–72. doi: 10.1523/JNEUROSCI.3874-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Agarwal, A., Zhang, M., Trembak-Duff, I., Unterbarnscheidt, T., Radyushkin, K., Dibaj, P., et al. (2014). Dysregulated expression of neuregulin-1 by cortical pyramidal neurons disrupts synaptic plasticity. Cell Rep. 8, 1130–1145. doi: 10.1016/j.celrep.2014.07.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ahmari, S. E., Spellman, T., Douglass, N. L., Kheirbek, M. A., Simpson, H. B., Deisseroth, K., et al. (2013). Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science 340, 1234–1239. doi: 10.1126/science.1234733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Akil, H., Brenner, S., Kandel, E., Kendler, K. S., King, M. C., Scolnick, E., et al. (2010). Medicine. The future of psychiatric research: genomes and neural circuits. Science 327, 1580–1581. doi: 10.1126/science.1188654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
American Psychiatric Association. (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th Edn. Washington, DC: APA.
Andersson, R. H., Johnston, A., Herman, P. A., Winzer-Serhan, U. H., Karavanova, I., Vullhorst, D., et al. (2012). Neuregulin and dopamine modulation of hippocampal gamma oscillations is dependent on dopamine D4 receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 13118–13123. doi: 10.1073/pnas.1201011109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arguello, P. A., and Gogos, J. A. (2010). Cognition in mouse models of schizophrenia susceptibility genes. Schizophr. Bull. 36, 289–300. doi: 10.1093/schbul/sbp153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arguello, P. A., and Gogos, J. A. (2012). Genetic and cognitive windows into circuit mechanisms of psychiatric disease. Trends Neurosci. 35, 3–13. doi: 10.1016/j.tins.2011.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arguello, P. A., Markx, S., Gogos, J. A., and Karayiorgou, M. (2010). Development of animal models for schizophrenia. Dis. Model. Mech. 3, 22–26. doi: 10.1242/dmm.003996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227. doi: 10.1016/j.nbd.2005.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Austin, C. P., Ky, B., Ma, L., Morris, J. A., and Shughrue, P. J. (2004). Expression of Disrupted-In-Schizophrenia-1, a schizophrenia-associated gene, is prominent in the mouse hippocampus throughout brain development. Neuroscience 124, 3–10. doi: 10.1016/j.neuroscience.2003.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baddeley, A. (2012). Working memory: theories, models, and controversies. Annu. Rev. Psychol. 63, 1–29. doi: 10.1146/annurev-psych-120710-100422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baldessarini, R. J., and Tarazi, F. I. (2006). Pharmacotherapy of Psychosis and Mania; The Pharmacological Basis of Therapeutics, 11e. NewYork, NY: McGraw-Hill.
Balu, D. T., Carlson, G. C., Talbot, K., Kazi, H., Hill-Smith, T. E., Easton, R. M., et al. (2012). Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus 22, 230–240. doi: 10.1002/hipo.20887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barch, D. M. (2009). Neuropsychological abnormalities in schizophrenia and major mood disorders: similarities and differences. Curr. Psychiatry Rep. 11, 313–319. doi: 10.1007/s11920-009-0045-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bartos, M., Vida, I., and Jonas, P. (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 8, 45–56. doi: 10.1038/nrn2044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bearden, C. E., Woodin, M. F., Wang, P. P., Moss, E., McDonald-McGinn, D., Zackai, E., et al. (2001). The neurocognitive phenotype of the 22q11.2 deletion syndrome: selective deficit in visual-spatial memory. J. Clin. Exp. Neuropsychol. 23, 447–464. doi: 10.1076/jcen.23.4.447.1228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belforte, J. E., Zsiros, V., Sklar, E. R., Jiang, Z., Yu, G., Li, Y., et al. (2010). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 13, 76–83. doi: 10.1038/nn.2447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belmonte, M. K., Allen, G., Beckel-Mitchener, A., Boulanger, L. M., Carper, R. A., and Webb, S. J. (2004). Autism and abnormal development of brain connectivity. J. Neurosci. 24, 9228–9231. doi: 10.1523/JNEUROSCI.3340-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benes, F. M. (2007). Searching for unique endophenotypes for schizophrenia and bipolar disorder within neural circuits and their molecular regulatory mechanisms. Schizophr. Bull. 33, 932–936. doi: 10.1093/schbul/sbm064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benson, M. A., Newey, S. E., Martin-Rendon, E., Hawkes, R., and Blake, D. J. (2001). Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J. Biol. Chem. 276, 24232–24241. doi: 10.1074/jbc.M010418200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertram, I., Bernstein, H. G., Lendeckel, U., Bukowska, A., Dobrowolny, H., Keilhoff, G., et al. (2007). Immunohistochemical evidence for impaired neuregulin-1 signaling in the prefrontal cortex in schizophrenia and in unipolar depression. Ann. N.Y. Acad. Sci. 1096, 147–156. doi: 10.1196/annals.1397.080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biederman, J., Faraone, S. V., Keenan, K., Steingard, R., and Tsuang, M. T. (1991). Familial association between attention deficit disorder and anxiety disorders. Am. J. Psychiatry 148, 251–256.
Bijlsma, E. K., Gijsbers, A. C., Schuurs-Hoeijmakers, J. H., van Haeringen, A., Fransen van de Putte, D. E., Anderlid, B. M., et al. (2009). Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur. J. Med. Genet. 52, 77–87. doi: 10.1016/j.ejmg.2009.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bjarnadottir, M., Misner, D. L., Haverfield-Gross, S., Bruun, S., Helgason, V. G., Stefansson, H., et al. (2007). Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/- knock-outs compared with wild-type mice. J. Neurosci. 17, 4519–4529. doi: 10.1523/JNEUROSCI.4314-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blackwood, D. H., Fordyce, A., Walker, M. T., St Clair, D. M., Porteous, D. J., and Muir, W. J. (2001). Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am. J. Hum. Genet. 69, 428–433. doi: 10.1086/321969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blum, B. P., and Mann, J. J. (2002). The GABAergic system in schizophrenia. Int. J. Neuropsychopharm. 5, 159–179. doi: 10.1017/S1461145702002894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blundell, J., Kaeser, P. S., Südhof, T. C., and Powell, C. M. (2010). RIM1alpha and interacting proteins involved in presynaptic plasticity mediate prepulse inhibition and additional behaviors linked to schizophrenia. J. Neurosci. 30, 5326–5333. doi: 10.1523/JNEUROSCI.0328-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonner-Jackson, A., Yodkovik, N., Csernansky, J. G., and Barch, D. M. (2008). Episodic memory in schizophrenia: the influence of strategy use on behavior and brain activation. Psychiatry Res. 164, 1–15. doi: 10.1016/j.pscychresns.2007.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boucher, A. A., Arnold, J. C., Duffy, L., Schofield, P. R., Micheau, J., and Karl, T. (2007). Heterozygous neuregulin 1 mice are more sensitive to the behavioural effects of Delta9-tetrahydrocannabinol. Psychopharmacology (Berl.) 192, 325–336. doi: 10.1007/s00213-007-0721-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bradshaw, N. J., Ogawa, F., Antolin-Fontes, B., Chubb, J. E., Carlyle, B. C., Christie, S., et al. (2008). DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem. Biophys. Res. Commun. 377, 1091–1096. doi: 10.1016/j.bbrc.2008.10.120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braff, D. L., and Light, G. A. (2004). Preattentional and attentional cognitive deficits as targets for treating schizophrenia. Psychopharmacology (Berl.) 174, 75–85. doi: 10.1007/s00213-004-1848-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bramon, E., Rabe-Hesketh, S., Sham, P., Murray, R. M., and Frangou, S. (2004). Meta-analysis of the P300 and P50 waveforms in schizophrenia. Schizophr. Res. 70, 315–329. doi: 10.1016/j.schres.2004.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brandon, N. J., and Sawa, A. (2011). Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 12, 707–722. doi: 10.1038/nrn3120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brennan, A. M., Harris, A. W., and Williams, L. M. (2013). Functional dysconnectivity in schizophrenia and its relationship to neural synchrony. Expert Rev. Neurother. 13, 755–765. doi: 10.1586/14737175.2013.811899
Brockhaus-Dumke, A., Schultze-Lutter, F., Mueller, R., Tendolkar, I., Bechdolf, A., Pukrop, R., et al. (2008). Sensory gating in schizophrenia: P50 and N100 gating in antipsychotic-free subjects at risk, first-episode, and chronic patients. Biol. Psychiatry 64, 376–384. doi: 10.1016/j.biopsych.2008.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, A. S., and McGrath, J. J. (2011). The prevention of schizophrenia. Schizophr. Bull. 37, 257–261. doi: 10.1093/schbul/sbq122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buchanan, R. W., Javitt, D. C., Marder, S. R., Schooler, N. R., Gold, J. M., McMahon, R. P., et al. (2007). The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am. J. Psychiatry 164, 1593–1602. doi: 10.1176/appi.ajp.2007.06081358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carlén, M., Meletis, K., Siegle, J. H., Cardin, J. A., Futai, K., Vierling-Claassen, D., et al. (2012). A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol. Psychiatry 17, 537–548. doi: 10.1038/mp.2011.31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carter, C. S., Barch, D. M., Buchanan, R. W., Bullmore, E., Krystal, J. H., Cohen, J., et al. (2008). Identifying cognitive mechanisms targeted for treatment development in schizophrenia: an overview of the first meeting of the Cognitive Neuroscience Treatment Research to Improve Cognition in Schizophrenia Initiative. Biol. Psychiatry 64, 4–10. doi: 10.1016/j.biopsych.2008.03.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, J., Lipska, B. K., and Weinberger, D. R. (2006). Genetic mouse models of schizophrenia: from hypothesis-based to susceptibility gene-based models. Biol. Psychiatry 59, 1180–1188. doi: 10.1016/j.biopsych.2006.02.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y. J., Johnson, M. A., Lieberman, M. D., Goodchild, R. E., Schobel, S., Lewandowski, N., et al. (2008). Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J. Neurosci. 28, 6872–6883. doi: 10.1523/JNEUROSCI.1815-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y. J., Zhang, M., Yin, D. M., Wen, L., Ting, A., Wang, P., et al. (2010). ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc. Natl. Acad. Sci. U.S.A. 107, 21818–21823. doi: 10.1073/pnas.1010669107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chow, E. W., Watson, M., Young, D. A., and Bassett, A. S. (2006). Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr. Res. 87, 270–278. doi: 10.1016/j.schres.2006.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Citrome, L., Goldberg, J. F., and Stahl, S. M. (2005). Toward convergence in the medication treatment of bipolar disorder and schizophrenia. Harv. Rev. Psychiatry 13, 28–42. doi: 10.1080/10673220590923164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clelland, C. L., Read, L. L., Baraldi, A. N., Bart, C. P., Pappas, C. A., Panek, L. J., et al. (2011). Evidence for association of hyperprolinemia with schizophrenia and a measure of clinical outcome. Schizophr. Res. 131, 139–145. doi: 10.1016/j.schres.2011.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clements, B. A., Geyer, M. A., and Braff, D. L. (1998). Poor P50 suppression among schizophrenia patients and their first-degree biological relatives. Am. J. Psychiatry 155, 1691–1694.
Clifford, S., Dissanayake, C., Bui, Q. M., Huggins, R., Taylor, A. K., and Loesch, D. Z. (2007). Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 37, 738–747. doi: 10.1007/s10803-006-0205-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Collingridge, G. L., Peineau, S., Howland, J. G., and Wang, Y. T. (2010). Long-term depression in the CNS. Nat. Rev. Neurosci. 11, 459–473. doi: 10.1038/nrn2867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Compte, A., Brunel, N., Goldman-Rakic, P. S., and Wang, X. J. (2000). Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical network model. Cereb. Cortex 10, 910–923. doi: 10.1093/cercor/10.9.910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cottrell, J. R., Levenson, J. M., Kim, S. H., Gibson, H. E., Richardson, K. A., Sivula, M., et al. (2013). Working memory impairment in calcineurin knock-out mice is associated with alterations in synaptic vesicle cycling and disruption of high-frequency synaptic and network activity in prefrontal cortex. J. Neurosci. 33, 10938–10949. doi: 10.1523/JNEUROSCI.5362-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Craddock, N., O'Donovan, M. C., and Owen, M. J. (2006). Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr. Bull. 32, 9–16. doi: 10.1093/schbul/sbj033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Craddock, N., and Owen, M. J. (2005). The beginning of the end for the Kraepelinian dichotomy. Br. J. Psychiatry 186, 364–366. doi: 10.1192/bjp.186.5.364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalmau, J., Tüzün, E., Wu, H. Y., Masjuan, J., Rossi, J. E., Voloschin, A., et al. (2007). Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 61, 25–36. doi: 10.1002/ana.21050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daskalakis, Z. J., Christensen, B. K., Fitzgerald, P. B., and Chen, R. (2008). Dysfunctional neural plasticity in patients with schizophrenia. Arch. Gen. Psychiatry 65, 378–385. doi: 10.1001/archpsyc.65.4.378
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deakin, I. H., Law, A. J., Oliver, P. L., Schwab, M. H., Nave, K. A., Harrison, P. J., et al. (2009). Behavioural characterization of neuregulin 1 type I overexpressing transgenic mice. Neuroreport 20, 1523–1528. doi: 10.1097/WNR.0b013e328330f6e7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deakin, I. H., Nissen, W., Law, A. J., Lane, T., Kanso, R., Schwab, M. H., et al. (2012). Transgenic overexpression of the type I isoform of neuregulin 1 affects working memory and hippocampal oscillations but not long-term potentiation. Cereb. Cortex 22, 1520–1529. doi: 10.1093/cercor/bhr223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Pino, I., García-Frigola, C., Dehorter, N., Brotons-Mas, J. R., Alvarez-Salvado, E., Martínez de Lagrán, M., et al. (2013). Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168.
Deng, P. Y., and Klyachko, V. A. (2011). The diverse functions of short-term plasticity components in synaptic computations. Commun. Integr. Biol. 4, 543–548. doi: 10.4161/cib.15870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, P. Y., Rotman, Z., Blundon, J. A., Cho, Y., Cui, J., Cavalli, V., et al. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. doi: 10.1016/j.neuron.2012.12.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, P. Y., Sojka, D., and Klyachko, V. A. (2011). Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J. Neurosci. 31, 10971–10982. doi: 10.1523/JNEUROSCI.2021-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Vries, L., Zheng, B., Fischer, T., Elenko, E., and Farquhar, M. G. (2000). The regulator of G protein signaling family. Annu. Rev. Pharmacol. Toxicol. 40, 235–271. doi: 10.1146/annurev.pharmtox.40.1.235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dias, E. C., Butler, P. D., Hoptman, M. J., and Javitt, D. C. (2011). Early sensory contributions to contextual encoding deficits in schizophrenia. Arch. Gen. Psychiatry 68, 654–664. doi: 10.1001/archgenpsychiatry.2011.17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dickman, D. K., and Davis, G. W. (2009). The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 326, 1127–1130. doi: 10.1126/science.1179685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ding, J., Guzman, J. N., Tkatch, T., Chen, S., Goldberg, J. A., Ebert, P. J., et al. (2006). RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat. Neurosci. 9, 832–842. doi: 10.1038/nn1700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dobrunz, L. E., and Stevens, C. F. (1999). Response of hippocampal synapses to natural stimulation patterns. Neuron 22, 157–166. doi: 10.1016/S0896-6273(00)80687-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drew, L. J., Crabtree, G. W., Markx, S., Stark, K. L., Chaverneff, F., Xu, B., et al. (2011a). The 22q11.2 microdeletion: fifteen years of insights into the genetic and neural complexity of psychiatric disorders. Int. J. Dev. Neurosci. 29, 259–281. doi: 10.1016/j.ijdevneu.2010.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drew, L. J., Stark, K. L., Fénelon, K., Karayiorgou, M., Macdermott, A. B., and Gogos, J. A. (2011b). Evidence for altered hippocampal function in a mouse model of the human 22q11.2 microdeletion. Mol. Cell. Neurosci. 47, 293–305. doi: 10.1016/j.mcn.2011.05.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eastwood, S. L., Burnet, P. W., and Harrison, P. J. (2005). Decreased hippocampal expression of the susceptibility gene PPP3CC and other calcineurin subunits in schizophrenia. Biol. Psychiatry 57, 702–710. doi: 10.1016/j.biopsych.2004.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ecker, C., and Murphy, D. (2014). Neuroimaging in autism–from basic science to translational research. Nat. Rev. Neurol. 10, 82–91. doi: 10.1038/nrneurol.2013.276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ecker, D., Unrath, A., Kassubek, J., and Sabolek, M. (2009). Dopamine Agonists and their risk to induce psychotic episodes in Parkinson's disease: a case-control study. BMC Neurol. 9:23. doi: 10.1186/1471-2377-9-23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edin, F., Klingberg, T., Johansson, P., McNab, F., Tegnér, J., and Compte, A. (2009). Mechanism for top-down control of working memory capacity. Proc. Natl. Acad. Sci. U.S.A. 106, 6802–6807. doi: 10.1073/pnas.0901894106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ehrlichman, R. S., Luminais, S. N., White, S. L., Rudnick, N. D., Ma, N., Dow, H. C., et al. (2009). Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res. 1294, 116–127. doi: 10.1016/j.brainres.2009.07.065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 36, 131–137. doi: 10.1038/ng1296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Esslinger, C., Walter, H., Kirsch, P., Erk, S., Schnell, K., Arnold, C., et al. (2009). Neural mechanisms of a genome-wide supported psychosis variant. Science 324, 605. doi: 10.1126/science.1167768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farooqi, I. S., and O'Rahilly, S. (2005). Monogenic obesity in humans. Annu. Rev. Med. 56, 443–458. doi: 10.1146/annurev.med.56.062904.144924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fazzari, P., Paternain, A. V., Valiente, M., Pla, R., Luján, R., Lloyd, K., et al. (2010). Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464, 1376–1380. doi: 10.1038/nature08928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fénelon, K., Mukai, J., Xu, B., Hsu, P. K., Drew, L. J., Karayiorgou, M., et al. (2011). Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 108, 4447–4452. doi: 10.1073/pnas.1101219108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fénelon, K., Xu, B., Lai, C. S., Mukai, J., Markx, S., Stark, K. L., et al. (2013). The pattern of cortical dysfunction in a mouse model of a schizophrenia-related microdeletion. J. Neurosci. 37, 14825–14839. doi: 10.1523/JNEUROSCI.1611-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Filipović, D., Zlatković, J., Gass, P., and Inta, D. (2013). The differential effects of acute vs. chronic stress and their combination on hippocampal parvalbumin and inducible heat shock protein 70 expression. Neuroscience 236, 47–54. doi: 10.1016/j.neuroscience.2013.01.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fisahn, A., Neddens, J., Yan, L., and Buonanno, A. (2009). Neuregulin-1 modulates hippocampal gamma oscillations: implications for schizophrenia. Cereb. Cortex 19, 612–618. doi: 10.1093/cercor/bhn107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fitzgerald, P. B., Brown, T. L., Marston, N. A., Oxley, T., De Castella, A., Daskalakis, Z. J., et al. (2004). Reduced plastic brain responses in schizophrenia: a transcranial magnetic stimulation study. Schizophr. Res. 71, 17–26. doi: 10.1016/j.schres.2004.01.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forbes, N. F., Carrick, L. A., McIntosh, A. M., and Lawrie, S. M. (2009). Working memory in schizophrenia: a meta-analysis. Psychol. Med. 39, 889–905. doi: 10.1017/S0033291708004558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ford, J. M., Mathalon, D. H., Whitfield, S., Faustman, W. O., and Roth, W. T. (2002). Reduced communication between frontal and temporal lobes during talking in schizophrenia. Biol. Psychiatry 51, 485–492. doi: 10.1016/S0006-3223(01)01335-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ford, J. M., Roach, B. J., Faustman, W. O., and Mathalon, D. H. (2008). Out-of synch and out-of-sorts: dysfunction of motor-sensory communication in schizophrenia. Biol. Psychiatry 63, 736–743. doi: 10.1016/j.biopsych.2007.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frankle, W. G., Lerma, J., and Laruelle, M. (2003). The synaptic hypothesis of schizophrenia. Neuron 39, 205–216. doi: 10.1016/S0896-6273(03)00423-9
Frantseva, M. V., Fitzgerald, P. B., Chen, R., Moller, B., Daigle, M., Daskalakis, Z. J., et al. (2008). Evidence for impaired long-term potentiation in schizophrenia and its relationship to motor skill learning. Cereb. Cortex 18, 990–996. doi: 10.1093/cercor/bhm151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freedman, R., Adler, L. E., Myles-Worsley, M., Nagamoto, H. T., Miller, C., Kisley, M., et al. (1996). Inhibitory gating of an evoked response to repeated auditory stimuli in schizophrenic and normal subjects. Human recordings, computer simulation, and an animal model. Arch. Gen. Psychiatry 53, 1114–1121. doi: 10.1001/archpsyc.1996.01830120052009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedman, J. I., Vrijenhoek, T., Markx, S., Janssen, I. M., van der Vliet, W. A., Faas, B. H., et al. (2008). CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol. Psychiatry 13, 261–266. doi: 10.1038/sj.mp.4002049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gandal, M. J., Anderson, R. L., Billingslea, E. N., Carlson, G. C., Roberts, T. P., and Siegel, S. J. (2012). Mice with reduced NMDA receptor expression: more consistent with autism than schizophrenia? Genes Brain Behav. 11, 740–750. doi: 10.1111/j.1601-183X.2012.00816.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia, R. A., Vasudevan, K., and Buonanno, A. (2000). The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc. Natl. Acad. Sci. U.S.A. 97, 3596–3601. doi: 10.1073/pnas.97.7.3596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garrido, M. I., Kilner, J. M., Stephan, K. E., and Friston, K. J. (2009). The mismatch negativity: a review of underlying mechanisms. Clin. Neurophysiol. 120, 453–463. doi: 10.1016/j.clinph.2008.11.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gauthier, J., and Rouleau, G. A. (2012). De novo mutations in neurological and psychiatric disorders: effects, diagnosis and prevention. Genome Med. 4:71. doi: 10.1186/gm372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gerber, D. J., Hall, D., Miyakawa, T., Demars, S., Gogos, J. A., Karayiorgou, M., et al. (2003). Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin gamma subunit. Proc. Natl. Acad. Sci. U.S.A. 100, 8993–8998. doi: 10.1073/pnas.1432927100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geschwind, D. H., and Levitt, P. (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 17, 103–111. doi: 10.1016/j.conb.2007.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geyer, M. A. (2006). The family of sensorimotor gating disorders: comorbidities or diagnostic overlaps? Neurotox. Res. 10, 211–220. doi: 10.1007/BF03033358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geyer, M. A. (2008). Developing translational animal models for symptoms of schizophrenia or bipolar mania. Neurotox. Res. 14, 71–78. doi: 10.1007/BF03033576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghashghaei, H. T., Weber, J., Pevny, L., Schmid, R., Schwab, M. H., Lloyd, K. C., et al. (2006). The role of neuregulin-ErbB4 interactions on the proliferation and organization of cells in the subventricular zone. Proc. Natl. Acad. Sci. U.S.A. 103, 1930–1935. doi: 10.1073/pnas.0510410103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilman, S. R., Iossifov, I., Levy, D., Ronemus, M., Wigler, M., and Vitkup, D. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. doi: 10.1016/j.neuron.2011.05.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Girard, S. L., Gauthier, J., Noreau, A., Xiong, L., Zhou, S., Jouan, L., et al. (2011). Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863. doi: 10.1038/ng.886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gisquet-Verrier, P., and Delatour, B. (2006). The role of the rat prelimbic/infralimbic cortex in working memory: not involved in the short-term maintenance but in monitoring and processing functions. Neuroscience 141, 585–596. doi: 10.1016/j.neuroscience.2006.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glahn, D. C., Therman, S., Manninen, M., Huttunen, M., Kaprio, J., Lönnqvist, J., et al. (2003). Spatial working memory as an endophenotype for schizophrenia. Biol. Psychiatry 53, 624–626. doi: 10.1016/S0006-3223(02)01641-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glatt, S. J., Faraone, S. V., and Tsuang, M. T. (2003). Association between a functional catechol O-methyltransferase gene polymorphism and schizophrenia: meta-analysis of case-control and family-based studies. Am. J. Psychiatry 160, 469–476. doi: 10.1176/appi.ajp.160.3.469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Godfraind, J. M., Reyniers, E., De Boulle, K., D'Hooge, R., De Deyn, P. P., Bakker, C. E., et al. (1996). Long-term potentiation in the hippocampus of fragile X knockout mice. Am. J. Med. Genet. 64, 246–251. doi: 10.1002/(SICI)1096-8628(19960809)64:2<246::AID-AJMG2>3.0.CO;2-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goes, F. S., Willour, V. L., Zandi, P. P., Belmonte, P. L., MacKinnon, D. F., Mondimore, F. M., et al. (2009). Family-based association study of Neuregulin 1 withpsychotic bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 693–702. doi: 10.1002/ajmg.b.30895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goff, W. R., Williamson, P. D., VanGilder, J. C., Allison, T., and Fisher, T. C. (1980). Neural origins of long latency evoked potentials recorded from the depth and from the cortical surface of the brain in man. Prog. Clin. Neurophysiol. 7, 126–145.
Gogos, J. A., and Gerber, D. J. (2006). Schizophrenia susceptibility genes: emergence of positional candidates and future directions. Trends Pharmacol. Sci. 27, 226–233. doi: 10.1016/j.tips.2006.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gogos, J. A., Santha, M., Takacs, Z., Beck, K. D., Luine, V., Lucas, L. R., et al. (1999). The gene encoding proline dehydrogenase modulates sensorimotor gating in mice. Nat. Genet. 21, 434–439. doi: 10.1038/7777
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gottschalk, W., Pozzo-Miller, L. D., Figurov, A., and Lu, B. (1998). Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J. Neurosci. 18, 6830–6839.
Gould, T. D., and Gottesman, I. I. (2006). Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav. 5, 113–119. doi: 10.1111/j.1601-183X.2005.00186.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Granger, A. J., and Nicoll, R. A. (2013). Expression mechanisms underlying long-term potentiation: a postsynaptic view, 10 years on. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130136. doi: 10.1098/rstb.2013.0136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gros, D. F., Price, M., Magruder, K. M., and Frueh, B. C. (2012). Symptom overlap in posttraumatic stress disorder and major depression. Psychiatry Res. 196, 267–270. doi: 10.1016/j.psychres.2011.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groth, R. D., Dunbar, R. L., and Mermelstein, P. G. (2003). Calcineurin regulation of neuronal plasticity. Biochem. Biophys. Res. Commun. 311, 1159–1171. doi: 10.1016/j.bbrc.2003.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grunwald, T., Boutros, N. N., Pezer, N., von Oertzen, J., Fernández, G., Schaller, C., et al. (2003). Neuronal substrates of sensory gating within the human brain. Biol. Psychiatry 53, 511–519. doi: 10.1016/S0006-3223(02)01673-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gu, Q. (2002). Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience 111, 815–835. doi: 10.1016/S0306-4522(02)00026-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guilmatre, A., Dubourg, C., Mosca, A. L., Legallic, S., Goldenberg, A., Drouin-Garraud, V., et al. (2009). Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry 66, 947–956. doi: 10.1001/archgenpsychiatry.2009.80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, H. F., and Zhong, Y. (2006). Requirement of Akt to mediate long-term synaptic depression in Drosophila. J. Neurosci. 26, 4004–4014. doi: 10.1523/JNEUROSCI.3616-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haenschel, C., Bittner, R. A., Waltz, J., Haertling, F., Wibral, M., Singer, W., et al. (2009). Cortical oscillatory activity is critical for working memory as revealed by deficits in early-onset schizophrenia. J. Neurosci. 29, 9481–9489. doi: 10.1523/JNEUROSCI.1428-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hahn, C. G., Wang, H. Y., Cho, D. S., Talbot, K., Gur, R. E., Berrettini, W. H., et al. (2006). Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 12, 824–828. doi: 10.1038/nm1418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, J., Trent, S., Thomas, K. L., O'Donovan, M. C., and Owen, M. J. (2014). Genetic risk for schizophrenia: convergence on synaptic pathways involved in plasticity. Biol. Psychiatry. doi: 10.1016/j.biopsych.2014.07.011. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, M. H., Taylor, G., Sham, P., Schulze, K., Rijsdijk, F., Picchioni, M., et al. (2011). The early auditory gamma-band response is heritable and a putative endophenotype of schizophrenia. Schizophr. Bull. 37, 778–787. doi: 10.1093/schbul/sbp134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, J., Mark, M. D., Li, X., Xie, M., Waka, S., Rettig, J., et al. (2006). RGS2 determines short-term synaptic plasticity in hippocampal neurons by regulating Gi/o-mediated inhibition of presynaptic Ca2+ channels. Neuron 51, 575–586. doi: 10.1016/j.neuron.2006.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hardy, J. (1997). Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 20, 154–159. doi: 10.1016/S0166-2236(96)01030-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hasan, A., Aborowa, R., Nitsche, M. A., Marshall, L., Schmitt, A., Gruber, O., et al. (2012b). Abnormal bihemispheric responses in schizophrenia patients following cathodal transcranial direct stimulation. Eur. Arch. Psychiatry Clin. Neurosci. 262, 415–423. doi: 10.1007/s00406-012-0298-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hasan, A., Bergener, T., Nitsche, M. A., Strube, W., Bunse, T., Falkai, P., et al. (2013). Impairments of motor-cortex responses to unilateral and bilateral direct current stimulation in schizophrenia. Front. Psychiatry 4:121. doi: 10.3389/fpsyt.2013.00121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hasan, A., Nitsche, M. A., Herrmann, M., Schneider-Axmann, T., Marshall, L., Gruber, O., et al. (2012a). Impaired long-term depression in schizophrenia: a cathodal tDCS. pilot study. Brain Stimul. 5, 475–483. doi: 10.1016/j.brs.2011.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hasan, A., Nitsche, M. A., Rein, B., Schneider-Axmann, T., Guse, B., Gruber, O., et al. (2011). Dysfunctional long-term potentiation-like plasticity in schizophrenia revealed by transcranial direct current stimulation. Behav. Brain Res. 224, 15–22. doi: 10.1016/j.bbr.2011.05.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashimoto, R., Straub, R. E., Weickert, C. S., Hyde, T. M., Kleinman, J. E., and Weinberger, D. R. (2004). Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol. Psychiatry 9, 299–307. doi: 10.1038/sj.mp.4001434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hatton, D. D., Sideris, J., Skinner, M., Mankowski, J., Bailey, D. B. Jr., Roberts, J., et al. (2006). Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. A 140, 1804–1813. doi: 10.1002/ajmg.a.31286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, M. L., Rao, B. S., Seo, J. S., Choi, H. S., Dolan, B. M., Choi, S. Y., et al. (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11489–11494. doi: 10.1073/pnas.0705003104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hays, S. A., Huber, K. M., and Gibson, J. R. (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci. 31, 14223–14234. doi: 10.1523/JNEUROSCI.3157-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heinrichs, R. W. (2005). The primacy of cognition in schizophrenia. Am. Psychol. 60, 229–242. doi: 10.1037/0003-066X.60.3.229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hensch, T. K. (2005). Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6, 877–888. doi: 10.1038/nrn1787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirano, S., Hirano, Y., Maekawa, T., Obayashi, C., Oribe, N., Kuroki, T., et al. (2008). Abnormal neural oscillatory activity to speech sounds in schizophrenia: a magnetoencephalography study. J. Neurosci. 28, 4897–4903. doi: 10.1523/JNEUROSCI.5031-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holthausen, E. A., Wiersma, D., Sitskoorn, M. M., Dingemans, P. M., Schene, A. H., and van den Bosch, R. J. (2003). Long-term memory deficits in schizophrenia: primary or secondary dysfunction? Neuropsychology 17, 539–547. doi: 10.1037/0894-4105.17.4.539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, L. E., Summerfelt, A., Mitchell, B. D., McMahon, R. P., Wonodi, I., Buchanan, R. W., et al. (2008). Sensory gating endophenotype based on its neural oscillatory pattern and heritability estimate. Arch. Gen. Psychiatry 65, 1008–1016. doi: 10.1001/archpsyc.65.9.1008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horiuchi, Y., Ishiguro, H., Koga, M., Inada, T., Iwata, N., Ozaki, N., et al. (2007). Support for association of the PPP3CC gene with schizophrenia. Mol. Psychiatry 12, 891–893. doi: 10.1038/sj.mp.4002019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, W., Zhang, M., Czéh, B., Flügge, G., and Zhang, W. (2010). Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 35, 1693–1707. doi: 10.1038/npp.2010.31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, Y. Z., Won, S., Ali, D. W., Wang, Q., Tanowitz, M., Du, Q. S., et al. (2000). Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron 26, 443–455. doi: 10.1016/S0896-6273(00)81176-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Insel, T., Cuthbert, B., Garvey, M., Heinssen, R., Pine, D. S., Quinn, K., et al. (2010). Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am. J. Psychiatry 167, 748–751. doi: 10.1176/appi.ajp.2010.09091379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inta, D., Meyer-Lindenberg, A., and Gass, P. (2011). Alterations in postnatal neurogenesis and dopamine dysregulation in schizophrenia: a hypothesis. Schizophr. Bull. 37, 674–680. doi: 10.1093/schbul/sbq134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inta, D., Vogt, M. A., Elkin, H., Weber, T., Lima-Ojeda, J. M., Schneider, M., et al. (2014). Phenotype of mice with inducible ablation of GluA1 AMPA receptors during late adolescence: relevance for mental disorders. Hippocampus 24, 424–435. doi: 10.1002/hipo.22236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ivleva, E. I., Morris, D. W., Moates, A. F., Suppes, T., Thaker, G. K., and Tamminga, C. A. (2010). Genetics and intermediate phenotypes of the schizophrenia–bipolar disorder boundary. Neurosci. Biobehav. Rev. 34, 897–921. doi: 10.1016/j.neubiorev.2009.11.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iyengar, S. S., and Mott, D. D. (2008). Neuregulin blocks synaptic strengthening after epileptiform activity in the rat hippocampus. Brain Res. 1208, 67–73. doi: 10.1016/j.brainres.2008.02.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacquet, H., Demily, C., Houy, E., Hecketsweiler, B., Bou, J., Raux, G., et al. (2005). Hyperprolinemia is a risk factor for schizoaffective disorder. Mol. Psychiatry 10, 479–485. doi: 10.1038/sj.mp.4001597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Javitt, D. C., Spencer, K. M., Thaker, G. K., Winterer, G., and Hajós, M. (2008). Neurophysiological biomarkers for drug development in schizophrenia. Nat. Rev. Drug Discov. 7, 68–83. doi: 10.1038/nrd2463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Javitt, D. C., Steinschneider, M., Schroeder, C. E., Vaughan, H. G., and Arezzo, J. C. (1994). Detection of stimulus deviance within primate primary auditory cortex: intracortical mechanisms of mismatch negativity (MMN) generation. Brain Res. 667, 192–200. doi: 10.1016/0006-8993(94)91496-6
Jentsch, J. D., Trantham-Davidson, H., Jairl, C., Tinsley, M., Cannon, T. D., and Lavin, A. (2009). Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology 34, 2601–2608. doi: 10.1038/npp.2009.90
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, L., Emmetsberger, J., Talmage, D. A., and Role, L. W. (2013). Type III neuregulin 1 is required for multiple forms of excitatory synaptic plasticity of mouse cortico-amygdala circuits. J. Neurosci. 33, 9655–9666. doi: 10.1523/JNEUROSCI.2888-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karayiorgou, M., Morris, M. A., Morrow, B., Shprintzen, R. J., Goldberg, R., Borrow, J., et al. (1995). Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc. Natl. Acad. Sci. U.S.A. 92, 7612–7616. doi: 10.1073/pnas.92.17.7612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karayiorgou, M., and Gogos, J. A. (1997). A turning point in schizophrenia genetics. Neuron 19, 967–979. doi: 10.1016/S0896-6273(00)80390-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karayiorgou, M., Simon, T. J., and Gogos, J. A. (2010). 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 11, 402–416. doi: 10.1038/nrn2841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kikuchi, M., Hashimoto, T., Nagasawa, T., Hirosawa, T., Minabe, Y., Yoshimura, M., et al. (2011). Frontal areas contribute to reduced global coordination of resting-state gamma activities in drug-naive patients with schizophrenia. Schizophr. Res. 130, 187–194. doi: 10.1016/j.schres.2011.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J. Y., Liu, C. Y., Zhang, F., Duan, X., Wen, Z., Song, J., et al. (2012). Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 148, 1051–1064. doi: 10.1016/j.cell.2011.12.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kirkpatrick, B., Xu, L., Cascella, N., Ozeki, Y., Sawa, A., and Roberts, R. C. (2006). DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J. Comp. Neurol. 497, 436–450. doi: 10.1002/cne.21007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kirov, G., Pocklington, A. J., Holmans, P., Ivanov, D., Ikeda, M., Ruderfer, D., et al. (2012). De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153. doi: 10.1038/mp.2011.154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R., et al. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352. doi: 10.1016/j.neuron.2005.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koike, H., Arguello, P. A., Kvajo, M., Karayiorgou, M., and Gogos, J. A. (2006). Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3693–3697. doi: 10.1073/pnas.0511189103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krishnan, V., and Nestler, E. J. (2010). Linking molecules to mood: new insight into the biology of depression. Am. J. Psychiatry 167, 1305–1320. doi: 10.1176/appi.ajp.2009.10030434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumari, V., Gray, J. A., Geyer, M. A., ffytche, D., Soni, W., Mitterschiffthaler, M. T., et al. (2003). Neural correlates of tactile prepulse inhibition: a functional MRI study in normal and schizophrenic subjects. Psychiatry Res. 122, 99–113. doi: 10.1016/S0925-4927(02)00123-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kvajo, M., McKellar, H., Arguello, P. A., Drew, L. J., Moore, H., MacDermott, A. B., et al. (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Natl. Acad. Sci. U.S.A. 105, 7076–7081. doi: 10.1073/pnas.0802615105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kvajo, M., McKellar, H., Drew, L. J., Lepagnol-Bestel, A. M., Xiao, L., Levy, R. J., et al. (2011). Altered axonal targeting and short-term plasticity in the hippocampus of Disc1 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 108, 1349–1358. doi: 10.1073/pnas.1114113108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kwon, O. B., Longart, M., Vullhorst, D., Hoffman, D. A., and Buonanno, A. (2005). Neuregulin-1 reverses long-term potentiation at CA1 hippocampal synapses. J. Neurosci. 25, 9378–9383. doi: 10.1523/JNEUROSCI.2100-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lahti, A. C., Weiler, M. A., Tamara Michaelidis, B. A., Parwani, A., and Tamminga, C. A. (2001). Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25, 455–467. doi: 10.1016/S0893-133X(01)00243-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, W. S., Xu, B., Westphal, K. G., Paterlini, M., Olivier, B., Pavlidis, P., et al. (2006). Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc. Natl. Acad. Sci. U.S.A. 103, 16906–16911. doi: 10.1073/pnas.0604994103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lake, C. R., and Hurwitz, N. (2007). Schizoaffective disorder merges schizophrenia and bipolar disorders as one disease–there is no schizoaffective disorder. Curr. Opin. Psychiatry 20, 365–379. doi: 10.1097/YCO.0b013e3281a305ab
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lamprecht, R., and LeDoux, J. (2004). Structural plasticity and memory. Nat. Rev. Neurosci. 5, 45–54. doi: 10.1038/nrn1301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larson, J., Jessen, R. E., Kim, D., Fine, A. K., and du Hoffmann, J. (2005). Agedependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J. Neurosci. 25, 9460–9469. doi: 10.1523/JNEUROSCI.2638-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lawrie, S. M., Buechel, C., Whalley, H. C., Frith, C. D., Friston, K. J., and Johnstone, E. C. (2002). Reduced frontotemporal functional connectivity in schizophrenia associated with auditory hallucinations. Biol. Psychiatry 51, 1008–1011. doi: 10.1016/S0006-3223(02)01316-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Le Be, J. V., and Markram, H. (2006). Spontaneous and evoked synaptic rewiring in the neonatal neocortex. Proc. Natl. Acad. Sci. U.S.A. 103, 13214–13219. doi: 10.1073/pnas.0604691103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lerner, T. N., and Kreitzer, A. C. (2012). RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron 73, 347–359. doi: 10.1016/j.neuron.2011.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lesh, T. A., Niendam, T. A., Minzenberg, M. J., and Carter, C. S. (2011). Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology 36, 316–338. doi: 10.1038/npp.2010.156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, J., Pelletier, M. R., Perez Velazquez, J. L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151. doi: 10.1006/mcne.2001.1085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, K. X., Lu, Y. M., Xu, Z. H., Zhang, J., Zhu, J. M., Zhang, J. M., et al. (2011). Neuregulin 1 regulates excitability of fast-spiking neurons through Kv1.1 and acts in epilepsy. Nat. Neurosci. 15, 267–273. doi: 10.1038/nn.3006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lichtenstein, P., Yip, B. H., Björk, C., Pawitan, Y., Cannon, T. D., Sullivan, P. F., et al. (2009a). Common genetic influences for schizophrenia and bipolar disorder: a population-based study of 2 million nuclear families. Lancet 373, 234–239. doi: 10.1016/S0140-6736(09)60072-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lichtenstein, P., Carlström, E., Råstam, M., Gillberg, C., and Anckarsäter, H. (2010). The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am. J. Psychiatry 167, 1357–1363. doi: 10.1176/appi.ajp.2010.10020223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lichtenstein, P., Yip, B. H., Björk, C., Pawitan, Y., Cannon, T. D., Sullivan, P. F., et al. (2009b). Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373, 234–239. doi: 10.1016/S0140-6736(09)60072-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Light, G. A., and Braff, D. L. (2005). Mismatch negativity deficits are associated with poor functioning in schizophrenia patients. Arch. Gen. Psychiatry 62, 127–136. doi: 10.1001/archpsyc.62.2.127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lisman, J. E., Coyle, J. T., Green, R. W., Javitt, D. C., Benes, F. M., Heckers, S., et al. (2008). Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 31, 234–242. doi: 10.1016/j.tins.2008.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y. L., Fann, C. S., Liu, C. M., Chang, C. C., Yang, W. C., Hung, S. I., et al. (2007). More evidence supports the association of PPP3CC with schizophrenia. Mol. Psychiatry 12, 966–974. doi: 10.1038/sj.mp.4001977
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luck, S. J., Mathalon, D. H., O'Donnell, B. F., Hämäläinen, M. S., Spencer, K. M., Javitt, D. C., et al. (2011). A roadmap for the development and validation of event-related potential biomarkers in schizophrenia research. Biol. Psychiatry 70, 28–34. doi: 10.1016/j.biopsych.2010.09.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, X., He, W., Hu, X., and Yan, R. (2014). Reversible overexpression of bace1-cleaved neuregulin-1 N-terminal fragment induces schizophrenia-like phenotypes in mice. Biol. Psychiatry 76, 120–127. doi: 10.1016/j.biopsych.2013.09.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lynall, M. E., Bassett, D. S., Kerwin, R., McKenna, P. J., Kitzbichler, M., Muller, U., et al. (2010). Functional connectivity and brain networks in schizophrenia. J. Neurosci. 30, 9477–9487. doi: 10.1523/JNEUROSCI.0333-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacLeod, L. S., Kogan, C. S., Collin, C. A., Berry-Kravis, E., Messier, C., and Gandhi, R. (2010). A comparative study of the performance of individuals with fragile X syndrome and Fmr1 knockout mice on Hebb-Williams mazes. Genes Brain Behav. 9, 53–64. doi: 10.1111/j.1601-183X.2009.00534.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maher, B. J., and LoTurco, J. J. (2012). Disrupted-in-schizophrenia (DISC1) functions presynaptically at glutamatergic synapses. PLoS ONE 7:e34053. doi: 10.1371/journal.pone.0034053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maier, W., Lichtermann, D., Minges, J., Hallmayer, J., Heun, R., Benkert, O., et al. (1993). Continuity and discontinuity of affective disorders and schizophrenia. Results of a controlled family study. Arch. Gen. Psychiatry 50, 871–883. doi: 10.1001/archpsyc.1993.01820230041004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Manahan-Vaughan, D., von Haebler, D., Winter, C., Juckel, G., and Heinemann, U. (2008). A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus 18, 125–134. doi: 10.1002/hipo.20367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., et al. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031. doi: 10.1016/j.cell.2008.12.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Markram, H., and Tsodyks, M. (1996). Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature 382, 807–810. doi: 10.1038/382807a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Markram, K., and Markram, H. (2010). The intense world theory - a unifying theory of the neurobiology of autism. Front. Hum. Neurosci. 4:224. doi: 10.3389/fnhum.2010.00224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, S. J., Grimwood, P. D., and Morris, R. G. (2000). Synaptic plasticity and memory: an evaluation of the hypothesis. Annu. Rev. Neurosci. 23, 649–711. doi: 10.1146/annurev.neuro.23.1.649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matveev, V., and Wang, X. J. (2000). Differential short-term synaptic plasticity and transmission of complex spike trains: to depress or to facilitate? Cereb. Cortex 10, 1143–1153. doi: 10.1093/cercor/10.11.1143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCarthy, S. E., Makarov, V., Kirov, G., Addington, A. M., McClellan, J., Yoon, S., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41, 1223–1227. doi: 10.1038/ng.474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McDonald-McGinn, D. M., Tonnesen, M. K., Laufer-Cahana, A., Finucane, B., Driscoll, D. A., Emanuel, B. S., et al., (2001). Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet. Med. 3, 23–29. doi: 10.1097/00125817-200101000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McGuffin, P., Owen, M. J., and Farmer, A. E. (1995). Genetic basis of schizophrenia. Lancet 346, 678–682. doi: 10.1016/S0140-6736(95)92285-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McLeod, F., Ganley, R., Williams, L., Selfridge, J., Bird, A., and Cobb, S. R. (2013). Reduced seizure threshold and altered network oscillatory properties in a mouse model of Rett syndrome. Neuroscience 231, 195–205. doi: 10.1016/j.neuroscience.2012.11.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McNab, F., and Klingberg, T. (2008). Prefrontal cortex and basal ganglia control access to working memory. Nat. Neurosci. 11, 103–107. doi: 10.1038/nn2024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mei, L., and Nave, K. A. (2014). Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49. doi: 10.1016/j.neuron.2014.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Metzger, K. L., Maxwell, C. R., Liang, Y., and Siegel, S. J. (2007). Effects of nicotine vary across two auditory evoked potentials in the mouse. Biol. Psychiatry 61, 23–30. doi: 10.1016/j.biopsych.2005.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meyer-Lindenberg, A. S., Olsen, R. K., Kohn, P. D., Brown, T., Egan, M. F., Weinberger, D. R., et al. (2005). Regionally specific disturbance of dorsolateral prefrontal-hippocampal functional connectivity in schizophrenia. Arch. Gen. Psychiatry 62, 379–386. doi: 10.1001/archpsyc.62.4.379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Milberger, S., Biederman, J., Faraone, S. V., Murphy, J., and Tsuang, M. T. (1995). Attention deficit hyperactivity disorder and comorbid disorders: issues of overlapping symptoms. Am. J. Psychiatry 152, 1793–1799.
Millar, J. K., Pickard, B. S., Mackie, S., James, R., Christie, S., Buchanan, S. R., et al. (2005). DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 310, 1187–1191. doi: 10.1126/science.1112915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423. doi: 10.1093/hmg/9.9.1415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, B. R., and Hen, R. (2015). The current state of the neurogenic theory of depression and anxiety. Curr. Opin. Neurobiol. 30, 51–58. doi: 10.1016/j.conb.2014.08.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Milner, B., Squire, L. R., and Kandel, E. R. (1998). Cognitive neuroscience and the study of memory. Neuron 20, 445–468.
Ming, G. L., and Song, H. (2009). DISC1 partners with GSK3beta in neurogenesis. Cell 136, 990–992. doi: 10.1016/j.cell.2009.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Minzenberg, M. J., Firl, A. J., Yoon, J. H., Gomes, G. C., Reinking, C., and Carter, C. S. (2010). Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology 35, 2590–2599. doi: 10.1038/npp.2010.150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyakawa, T., Leiter, L. M., Gerber, D. J., Gainetdinov, R. R., Sotnikova, T. D., Zeng, H., et al. (2003). Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 100, 8987–8992. doi: 10.1073/pnas.1432926100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Modha, D. S., and Singh, R. (2010). Network architecture of the long-distance pathways in the macaque brain. Proc. Natl. Acad. Sci. U.S.A. 107, 13485–13490. doi: 10.1073/pnas.1008054107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mohn, A. R., Gainetdinov, R. R., Caron, M. G., and Koller, B. H. (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436. doi: 10.1016/S0092-8674(00)81972-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mongillo, G., Barak, O., and Tsodyks, M. (2008). Synaptic theory of working memory. Science 319, 1543–1546. doi: 10.1126/science.1150769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., et al. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327. doi: 10.1523/JNEUROSCI.2623-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morton, W. A. (1999). Cocaine and psychiatric symptoms. Prim. Care Companion J. Clin. Psychiatry 1, 109–113. doi: 10.4088/PCC.v01n0403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mundy, P. (2003). Annotation: the neural basis of social impairments in autism: the role of the dorsal medial-frontal cortex and anterior cingulate system. J. Child Psychol. Psychiatry 44, 793–809. doi: 10.1111/1469-7610.00165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murias, M., Webb, S. J., Greenson, J., and Dawson, G. (2007). Resting state cortical connectivity reflected in EEG coherence in individuals with autism. Biol. Psychiatry 62, 270–273. doi: 10.1016/j.biopsych.2006.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, K. C., Jones, L. A., and Owen, M. J. (1999). High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch. Gen. Psychiatry 56, 940–945. doi: 10.1001/archpsyc.56.10.940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Naatanen, R., (1992). Attention and Brain Function. Hillsdale, NJ: Lawrence Erlbaum Associates, Inc.
Naatanen, R. (2003). Mismatch negativity: clinical research and possible applications. Int. J. Psychophysiol. 48, 179–188. doi: 10.1016/S0167-8760(03)00053-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neale, B. M., Kou, Y., Liu, L., Ma'ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M. (2002). Neurobiology of depression. Neuron 34, 13–25. doi: 10.1016/S0896-6273(02)00653-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neul, J. L. (2012). The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin. Neurosci. 14, 253–262.
Nicoll, R. A., and Schmitz, D. (2005). Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci. 6, 863–876. doi: 10.1038/nrn1786
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Carroll, R. (2000). Cognitive impairment in schizophrenia. Adv. Psychiatr. Treat. 6, 166–168. doi: 10.1192/apt.6.3.161
O'Donovan, M. C., Craddock, N., Norton, N., Williams, H., Peirce, T., Moskvina, V., et al. (2008). Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 40, 1053–1055. doi: 10.1038/ng.201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oberman, L., Ifert-Miller, F., Najib, U., Bashir, S., Woollacott, I., Gonzalez-Heydrich, J., et al. (2010). Transcranial magnetic stimulation provides means to assess cortical plasticity and excitability in humans with fragile X syndrome and autism spectrum disorder. Front. Synaptic Neurosci. 2:26. doi: 10.3389/fnsyn.2010.00026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Donovan, M. C., Craddock, N. J., and Owen, M. J. (2009). Genetics of psychosis; insights from views across the genome. Hum. Genet. 126, 3–12. doi: 10.1007/s00439-009-0703-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Dushlaine, C., Kenny, E., Heron, E., Donohoe, G., Gill, M., Morris, D., et al. (2011). Molecular pathways involved in neuronal cell adhesion and membrane scaffolding contribute to schizophrenia and bipolar disorder susceptibility. Mol. Psychiatry 16, 286–292. doi: 10.1038/mp.2010.7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orekhova, E. V., Stroganova, T. A., Nygren, G., Tsetlin, M. M., Posikera, I. N., Gillberg, C., et al. (2007). Excess of high frequency electroencephalogram oscillations in boys with autism. Biol. Psychiatry 62, 1022–1029. doi: 10.1016/j.biopsych.2006.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Paradee, W., Melikian, H. E., Rasmussen, D. L., Kenneson, A., Conn, P. J., and Warren, S. T. (1999). Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94, 185–192. doi: 10.1016/S0306-4522(99)00285-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parlapani, E., Schmitt, A., Wirths, O., Bauer, M., Sommer, C., Rueb, U., et al. (2010). Gene expression of neuregulin-1 isoforms in different brain regions of elderly schizophrenia patients. World J. Biol. Psychiatry 11, 243–250. doi: 10.3109/15622970802022376
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paterlini, M., Zakharenko, S. S., Lai, W. S., Qin, J., Zhang, H., Mukai, J., et al. (2005). Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat. Neurosci. 8, 1586–1594. doi: 10.1038/nn1562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peltonen, L., Perola, M., Naukkarinen, J., and Palotie, A. (2006). Lessons from studying monogenic disease for common disease. Hum. Mol. Genet. 15, R67–R74. doi: 10.1093/hmg/ddl060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petryshen, T. L., Middleton, F. A., Kirby, A., Aldinger, K. A., Purcell, S., Tahl, A. R., et al. (2005). Support for involvement of neuregulin 1 in schizophrenia pathophysiology. Mol. Psychiatry 10, 366–374. doi: 10.1038/sj.mp.4001608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Phang, J. M., Hu, C. A., and Valle, D. (2001). Disorders of Proline and Hydoxyproline Metabolism; The Metabolic and Molecular and Bases of Inherited Disease, 8e. New York, NY: McGraw-Hill.
Pitcher, G. M., Beggs, S., Woo, R. S., Mei, L., and Salter, M. W. (2008). ErbB4 is a suppressor of long-term potentiation in the adult hippocampus. Neuroreport 19, 139–143. doi: 10.1097/WNR.0b013e3282f3da10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prasad, K. M., Talkowski, M. E., Chowdari, K. V., McClain, L., Yolken, R. H., and Nimgaonkar, V. L. (2010). Candidate genes and their interactions with other genetic/environmental risk factors in the etiology of schizophrenia. Brain Res. Bull. 83, 86–92. doi: 10.1016/j.brainresbull.2009.08.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pratt, K. G., Taft, C. E., Burbea, M., and Turrigiano, G. G. (2008). Dynamics underlying synaptic gain between pairs of cortical pyramidal neurons. Dev. Neurobiol. 68, 143–151. doi: 10.1002/dneu.20577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pulver, A. E., Nestadt, G., Goldberg, R., Shprintzen, R. J., Lamacz, M., Wolyniec, P. S., et al. (1994). Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J. Nerv. Ment. Dis. 182, 476–478. doi: 10.1097/00005053-199408000-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Psychiatric GWAS Consortium Coordinating Committee. (2009). Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am. J. Psychiatry 166, 540–556. doi: 10.1176/appi.ajp.2008.08091354
Ragland, J. D., Gur, R. C., Valdez, J. N., Loughead, J., Elliott, M., Kohler, C., et al. (2005). Levels-of-processing effect on frontotemporal function in schizophrenia during word encoding and recognition. Am. J. Psychiatry 162, 1840–1848. doi: 10.1176/appi.ajp.162.10.1840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramocki, M. B., Peters, S. U., Tavyev, Y. J., Zhang, F., Carvalho, C. M., Schaaf, C. P., et al. (2009). Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 66, 771–782. doi: 10.1002/ana.21715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Randall, A. D., Kurihara, M., Brandon, N. J., and Brown, J. T. (2014). Disrupted in schizophrenia 1 and synaptic function in the mammalian central nervous system. Eur. J. Neurosci. 39, 1068–1073. doi: 10.1111/ejn.12500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ranganath, C., Minzenberg, M. J., and Ragland, J. D. (2000). The cognitive neuroscience of memory function and dysfunction in schizophrenia. Biol. Psychiatry 64, 18–25. doi: 10.1016/j.biopsych.2008.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ransdell, J. L., Nair, S. S., and Schulz, D. J. (2012). Rapid homeostatic plasticity of intrinsic excitability in a central pattern generator network stabilizes functional neural network output. J. Neurosci. 32, 9649–9658. doi: 10.1523/JNEUROSCI.1945-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raux, G., Bumsel, E., Hecketsweiler, B., van Amelsvoort, T., Zinkstok, J., Manouvrier-Hanu, S., et al. (2007). Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum. Mol. Genet. 16, 83–91. doi: 10.1093/hmg/ddl443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reif, A., Fritzen, S., Finger, M., Strobel, A., Lauer, M., Schmitt, A., et al. (2006). Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol. Psychiatry 11, 514–522. doi: 10.1038/sj.mp.4001791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reif, A., Schmitt, A., Fritzen, S., and Lesch, K. P. (2007). Neurogenesis and schizophrenia: dividing neurons in a divided mind? Eur. Arch. Psychiatry Clin. Neurosci. 257, 290–299. doi: 10.1007/s00406-007-0733-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ricoy, U. M., Mao, P., Manczak, M., Reddy, P. H., and Frerking, M. E. (2011). A transgenic mouse model for Alzheimer's disease has impaired synaptic gain but normal synaptic dynamics. Neurosci. Lett. 500, 212–215. doi: 10.1016/j.neulet.2011.06.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ripke, S., Sanders, A. R., Kendler, K. S., Levinson, D. F., Sklar, P., Holmans, P. A., et al. (2011). Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976. doi: 10.1038/ng.940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ripke, S., O'Dushlaine, C., Chambert, K., Moran, J. L., Kähler, A. K., Akterin, S., et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159. doi: 10.1038/ng.2742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodriguez-Murillo, L., Gogos, J. A., and Karayiorgou, M. (2012). The genetic architecture of schizophrenia: new mutations and emerging paradigms. Annu. Rev. Med. 63, 63–80. doi: 10.1146/annurev-med-072010-091100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rojas, D. C., Teale, P. D., Maharajh, K., Kronberg, E., Youngpeter, K., Wilson, L. B., et al. (2011). Transient and steady-state auditory gamma-band responses in first-degree relatives of people with autism spectrum disorder. Mol. Autism 2, 11. doi: 10.1186/2040-2392-2-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rommelse, N. N., Franke, B., Geurts, H. M., Hartman, C. A., and Buitelaar, J. K. (2010). Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. Eur. Child Adolesc. Psychiatry 19, 281–295. doi: 10.1007/s00787-010-0092-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rotman, Z., Deng, P. Y., and Klyachko, V. A. (2011). Short-term plasticity optimizes synaptic information transmission. J. Neurosci. 31, 14800–14809. doi: 10.1523/JNEUROSCI.3231-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roux, F., Wibral, M., Mohr, H. M., Singer, W., and Uhlhaas, P. J. (2012). Gamma-band activity in human prefrontal cortex codes for the number of relevant items maintained in working memory. J. Neurosci. 32, 12411–12420. doi: 10.1523/JNEUROSCI.0421-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saggu, S., Cannon, T. D., Jentsch, J. D., and Lavin, A. (2013). Potential molecular mechanisms for decreased synaptic glutamate release in dysbindin-1 mutant mice. Schizophr. Res. 146, 254–263. doi: 10.1016/j.schres.2013.01.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saperstein, A. M., Fuller, R. L., Avila, M. T., Adami, H., McMahon, R. P., Thaker, G. K., et al. (2006). Spatial working memory as a cognitive endophenotype of schizophrenia: assessing risk for pathophysiological dysfunction. Schizophr. Bull. 32, 498–506. doi: 10.1093/schbul/sbj072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saugstad, J. A., Marino, M. J., Folk, J. A., Hepler, J. R., and Conn, P. J. (1998). RGS4 inhibits signaling by group I metabotropic glutamate receptors. J. Neurosci. 18, 905–913.
Saunders, J. A., Gandal, M. J., Roberts, T. P., and Siegel, S. J. (2012). NMDA antagonist MK801 recreates auditory electrophysiology disruption present in autism and other neurodevelopmental disorders. Behav. Brain Res. 234, 233–237. doi: 10.1016/j.bbr.2012.06.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schall, U., Johnston, P., Todd, J., Ward, P. B., and Michie, P. T. (2003). Functional neuroanatomy of auditory mismatch processing: an event-related fMRI study of duration-deviant oddballs. Neuroimage 20, 729–736. doi: 10.1016/S1053-8119(03)00398-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. doi: 10.1038/nature11015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmitt, A., Malchow, B., Hasan, A., and Falkai, P. (2014). The impact of environmental factors in severe psychiatric disorders. Front. Neurosci. 8:19. doi: 10.3389/fnins.2014.00019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selkoe, D., Mandelkow, E., and Holtzman, D. (2012). Deciphering Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a011460. doi: 10.1101/cshperspect.a011460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shamir, A., Kwon, O. B., Karavanova, I., Vullhorst, D., Leiva-Salcedo, E., Janssen, M. J., et al. (2012). The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J. Neurosci. 32, 2988–2997. doi: 10.1523/JNEUROSCI.1899-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, J., Levinson, D. F., Duan, J., Sanders, A. R., Zheng, Y., Pe'er, I., et al. (2009). Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753–757. doi: 10.1038/nature0819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shibuya, M., Komi, E., Wang, R., Kato, T., Watanabe, Y., Sakai, M., et al. (2010). Measurement and comparison of serum neuregulin1 immunoreactivity in control subjects and patients with schizophrenia: an influence of its genetic polymorphism. J. Neural Transm. 117, 887–895. doi: 10.1007/s00702-010-0418-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shinawi, M., Liu, P., Kang, S. H., Shen, J., Belmont, J. W., Scott, D. A., et al. (2010). Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 47, 332–341. doi: 10.1136/jmg.2009.073015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sigurdsson, T., Stark, K. L., Karayiorgou, M., Gogos, J. A., and Gordon, J. A. (2010). Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature 464, 763–767. doi: 10.1038/nature08855
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silverman, J. L., Yang, M., Lord, C., and Crawley, J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502. doi: 10.1038/nrn2851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sinzig, J., Walter, D., and Doepfner, M. (2009). Attention deficit/hyperactivity disorder in children and adolescents with autism spectrum disorder: symptom or syndrome? J. Atten. Disord. 13, 117–126. doi: 10.1177/1087054708326261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smoller, J. W., Ripke, S., Lee, P. H., Neale, B., Nurnberger, J. I., Santangelo, S., et al. (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. doi: 10.1016/S0140-6736(12)62129-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sobin, C., Kiley-Brabeck, K., Daniels, S., Khuri, J., Taylor, L., Blundell, M., et al. (2005). Neuropsychological characteristics of children with the 22q11 Deletion Syndrome: a descriptive analysis. Child Neuropsychol. 11, 39–53. doi: 10.1080/09297040590911167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spellman, T. J., and Gordon, J. A. (2014). Synchrony in schizophrenia: a window into circuit-level pathophysiology. Curr. Opin. Neurobiol. 30, 17–23. doi: 10.1016/j.conb.2014.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spencer, K. M. (2012). Baseline gamma power during auditory steady-state stimulation in schizophrenia. Front. Hum. Neurosci. 5:190. doi: 10.3389/fnhum.2011.00190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spencer, K. M., Nestor, P. G., Niznikiewicz, M. A., Salisbury, D. F., Shenton, M. E., and McCarley, R. W. (2003). Abnormal neural synchrony in schizophrenia. J. Neurosci. 23, 7407–7411.
Stark, K. L., Xu, B., Bagchi, A., Lai, W. S., Liu, H., Hsu, R., et al. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751–760. doi: 10.1038/ng.138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
St Clair, C. D., Blackwood, D., Muir, W., Carothers, A., Walker, M., Spowart, G., et al. (1990). Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336, 13–16. doi: 10.1016/0140-6736(90)91520-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S., et al. (2002). Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71, 877–892. doi: 10.1086/342734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefansson, H., Ophoff, R. A., Steinberg, S., Andreassen, O. A., Cichon, S., Rujescu, D., et al. (2009). Common variants conferring risk of schizophrenia. Nature 460, 744–747. doi: 10.1038/nature08186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stephan, K. E., Friston, K. J., and Frith, C. D. (2009). Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr. Bull. 35, 509–527. doi: 10.1093/schbul/sbn176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steullet, P., Cabungcal, J. H., Kulak, A., Kraftsik, R., Chen, Y., Dalton, T. P., et al. (2010). Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 30, 2547–2558. doi: 10.1523/JNEUROSCI.3857-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suh, J., Foster, D. J., Davoudi, H., Wilson, M. A., and Tonegawa, S. (2013). Impaired hippocampal ripple-associated replay in a mouse model of schizophrenia. Neuron 80, 484–493. doi: 10.1016/j.neuron.2013.09.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sullivan, P. F., Kendler, K. S., and Neale, M. C. (2003). Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192. doi: 10.1001/archpsyc.60.12.1187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sullivan, P. F., Magnusson, C., Reichenberg, A., Boman, M., Dalman, C., Davidson, M., et al. (2012). Family history of schizophrenia and bipolar disorder as risk factors for autism. Arch. Gen. Psychiatry 69, 1099–1103. doi: 10.1001/archgenpsychiatry.2012.730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, L., Grützner, C., Bölte, S., Wibral, M., Tozman, T., Schlitt, S., et al. (2012). Impaired gamma-band activity during perceptual organization in adults with autism spectrum disorders: evidence for dysfunctional network activity in frontal-posterior cortices. J. Neurosci. 32, 9563–9573. doi: 10.1523/JNEUROSCI.1073-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Swerdlow, N. R., and Geyer, M. A. (1998). Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr. Bull. 24, 285–301. doi: 10.1093/oxfordjournals.schbul.a033326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Swerdlow, N. R., Geyer, M. A., and Braff, D. L. (2001). Neural circuitry of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology 156, 194–215. doi: 10.1007/s002130100799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Swerdlow, N. R., Light, G. A., Cadenhead, K. S., Sprock, J., Hsieh, M. H., and Braff, D. L. (2006). Startle gating deficits in a large cohort of patients with schizophrenia: relationship to medications, symptoms, neurocognition, and level of function. Arch. Gen. Psychiatry 63, 1325–1335. doi: 10.1001/archpsyc.63.12.1325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Talbot, K., Cho, D. S., Ong, W. Y., Benson, M. A., Han, L. Y., Kazi, H. A., et al. (2006). Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum. Mol. Genet. 15, 3041–3054. doi: 10.1093/hmg/ddl246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, T. T., Yang, F., Chen, B. S., Lu, Y., Ji, Y., Roche, K. W., et al. (2009). Dysbindin regulates hippocampal LTP by controlling NMDA receptor surface expression. Proc. Natl. Acad. Sci. U.S.A. 106, 21395–21400. doi: 10.1073/pnas.0910499106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taya, S., Shinoda, T., Tsuboi, D., Asaki, J., Nagai, K., Hikita, T., et al. (2007). DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J. Neurosci. 27, 15–26. doi: 10.1523/JNEUROSCI.3826-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thaker, G. K. (2008). Neurophysiological endophenotypes across bipolar and schizophrenia psychosis. Schizophr. Bull. 34, 760–773. doi: 10.1093/schbul/sbn049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomson, P. A., Christoforou, A., Morris, S. W., Adie, E., Pickard, B. S., Porteous, D. J., et al. (2007). Association of Neuregulin 1 with schizophrenia and bipolar disorder in a second cohort from the Scottish population. Mol. Psychiatry 12, 94–104. doi: 10.1038/sj.mp.4001889
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Todd, K. J., Serrano, A., Lacaille, J. C., and Robitaille, R. (2006). Glial cells in synaptic plasticity. J. Physiol. Paris 99, 75–83. doi: 10.1016/j.jphysparis.2005.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tomari, Y., and Zamore, P. D. (2005). MicroRNA biogenesis: Drosha can't cut it without a partner. Curr. Biol. 15, R61–R64. doi: 10.1016/j.cub.2004.12.057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsodyks, M. V., and Markram, H. (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. U.S.A. 94, 719–723. doi: 10.1073/pnas.94.2.719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turetsky, B. I., Calkins, M. E., Light, G. A., Olincy, A., Radant, A. D., and Swerdlow, N. R. (2007). Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr. Bull. 33, 69–94. doi: 10.1093/schbul/sbl060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turjanski, N., and Lloyd, G. G. (2005). Psychiatric side-effects of medications: recent developments. Adv. Psychiatr. Treat. 11, 58–70. doi: 10.1192/apt.11.1.58
Udagawa, T., Farny, N. G., Jakovcevski, M., Kaphzan, H., Alarcon, J. M., Anilkumar, S., et al. (2013). Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat. Med. 19, 1473–1477. doi: 10.1038/nm.3353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uhlhaas, P. J. (2013). Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Curr. Opin. Neurobiol. 23, 283–290. doi: 10.1016/j.conb.2012.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uhlhaas, P. J., and Singer, W. (2006). Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron 52, 155–168. doi: 10.1016/j.neuron.2006.09.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uhlhaas, P. J., and Singer, W. (2012). Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron 75, 963–980. doi: 10.1016/j.neuron.2012.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Dam, D., D'Hooge, R., Hauben, E., Reyniers, E., Gantois, I., Bakker, C. E., et al. (2000). Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav. Brain Res. 117, 127–136. doi: 10.1016/S0166-4328(00)00296-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Snellenberg, J. X. (2009). Working memory and long-term memory deficits in schizophrenia: is there a common substrate? Psychiatry Res. 174, 89–96. doi: 10.1016/j.pscychresns.2009.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vassos, E., Collier, D. A., Holden, S., Patch, C., Rujescu, D., St Clair, D., et al. (2010). Penetrance for copy number variants associated with schizophrenia. Hum. Mol. Genet. 19, 3477–3481. doi: 10.1093/hmg/ddq259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vilain, J., Galliot, A. M., Durand-Roger, J., Leboyer, M., Llorca, P. M., Schürhoff, F., et al. (2013). Environmental risk factors for schizophrenia: a review. Encephale 39, 19–28. doi: 10.1016/j.encep.2011.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vullhorst, D., Neddens, J., Karavanova, I., Tricoire, L., Petralia, R. S., McBain, C. J., et al. (2009). Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J. Neurosci. 29, 12255–12264. doi: 10.1523/JNEUROSCI.2454-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walker, R. M., Christoforou, A., Thomson, P. A., McGhee, K. A., Maclean, A., Mühleisen, T. W., et al. (2010). Association analysis of Neuregulin 1 candidate regions in schizophrenia and bipolar disorder. Neurosci. Lett. 478, 9–13. doi: 10.1016/j.neulet.2010.04.056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walss-Bass, C., Liu, W., Lew, D. F., Villegas, R., Montero, P., Dassori, A., et al. (2006). A novel missense mutation in the transmembrane domain of neuregulin 1 is associated with schizophrenia. Biol. Psychiatry 60, 548–553. doi: 10.1016/j.biopsych.2006.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X. J. (2010). Neurophysiological and computational principles of cortical rhythms in cognition. Physiol. Rev. 90, 1195–1268. doi: 10.1152/physrev.00035.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X. S., Peng, C. Z., Cai, W. J., Xia, J., Jin, D., Dai, Y., et al. (2014). Activity-dependent regulation of release probability at excitatory hippocampal synapses: a crucial role of fragile X mental retardation protein in neurotransmission. Eur. J. Neurosci. 39, 1602–1612. doi: 10.1111/ejn.12546
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Webb, W. L. Jr., and Gehi, M. (1981). Electrolyte and fluid imbalance: neuropsychiatric manifestations. Psychosomatics 22, 199–203. doi: 10.1016/S0033-3182(81)73532-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Welch, J. M., Lu, J., Rodriguiz, R. M., Trotta, N. C., Peca, J., Ding, J. D., et al. (2007). Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 448, 894–900. doi: 10.1038/nature06104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weng, S. M., McLeod, F., Bailey, M. E., and Cobb, S. R. (2011). Synaptic plasticity deficits in an experimental model of rett syndrome: long-term potentiation saturation and its pharmacological reversal. Neuroscience 180, 314–321. doi: 10.1016/j.neuroscience.2011.01.061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Whitlock, J. R., Heynen, A. J., Shuler, M. G., and Bear, M. F. (2006). Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097. doi: 10.1126/science.1128134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiedholz, L. M., Owens, W. A., Horton, R. E., Feyder, M., Karlsson, R. M., Hefner, K., et al. (2008). Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and “schizophrenia-related” behaviors. Mol. Psychiatry 13, 631–640. doi: 10.1038/sj.mp.4002056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Williams, N. M., Zaharieva, I., Martin, A., Langley, K., Mantripragada, K., Fossdal, R., et al. (2010). Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genomewide analysis. Lancet 376, 1401–1408. doi: 10.1016/S0140-6736(10)61109-9
Wilson, T. W., Rojas, D. C., Reite, M. L., Teale, P. D., and Rogers, S. J. (2007). Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biol. Psychiatry 62, 192–197. doi: 10.1016/j.biopsych.2006.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woo, R. S., Li, X. M., Tao, Y., Carpenter-Hyland, E., Huang, Y. Z., Weber, J., et al. (2007). Neuregulin-1 enhances depolarization-induced GABA release. Neuron 54, 599–610. doi: 10.1016/j.neuron.2007.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woodin, M., Wang, P. P., Aleman, D., McDonald-McGinn, D., Zackai, E., and Moss, E. (2001). Neuropsychological profile of children and adolescents with the 22q11.2 microdeletion. Genet. Med. 3, 34–39. doi: 10.1097/00125817-200101000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, B., Hsu, P. K., Stark, K. L., Karayiorgou, M., and Gogos, J. A. (2013). Derepression of a neuronal inhibitor due to miRNA dysregulation in a schizophrenia-related microdeletion. Cell 152, 262–275. doi: 10.1016/j.cell.2012.11.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, B., Ionita-Laza, I., Roos, J. L., Boone, B., Woodrick, S., Sun, Y., et al. (2012). De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 44, 1365–1369. doi: 10.1038/ng.2446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, B., Roos, J. L., Dexheimer, P., Boone, B., Plummer, B., Levy, S., et al. (2011). Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 43, 864–868. doi: 10.1038/ng.902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, B., Roos, J. L., Levy, S., van Rensburg, E. J., Gogos, J. A., and Karayiorgou, M. (2008). Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 40, 880–885. doi: 10.1038/ng.162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, Q. J., Asafo-Adjei, P. K., Arnold, H. M., Brown, R. E., and Bauchwitz, R. P. (2004). A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 3, 337–359. doi: 10.1111/j.1601-183X.2004.00087.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Z. Z., Tschopp, O., Baudry, A., Dümmler, B., Hynx, D., and Hemmings, B. A. (2004). Physiological functions of protein kinase B/Akt. Biochem. Soc. Trans. 32(Pt 2), 350–354. doi: 10.1042/BST0320350
Yin, D. M., Chen, Y. J., Lu, Y. S., Bean, J. C., Sathyamurthy, A., Shen, C., et al. (2013). Reversal of behavioral deficits and synaptic dysfunction in mice overexpressing neuregulin 1. Neuron 78, 644–657. doi: 10.1016/j.neuron.2013.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yizhar, O., Fenno, L. E., Prigge, M., Schneider, F., Davidson, T. J., O'Shea, D. J., et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. doi: 10.1038/nature10360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zakharenko, S. S., Patterson, S. L., Dragatsis, I., Zeitlin, S. O., Siegelbaum, S. A., Kandel, E. R., et al. (2003). Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39, 975–990. doi: 10.1016/S0896-6273(03)00543-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zbozinek, T. D., Rose, R. D., Wolitzky-Taylor, K. B., Sherbourne, C., Sullivan, G., Stein, M. B., et al. (2012). Diagnostic overlap of generalized anxiety disorder and major depressive disorder in a primary care sample. Depress. Anxiety 29, 1065–1071. doi: 10.1002/da.22026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeng, H., Chattarji, S., Barbarosie, M., Rondi-Reig, L., Philpot, B. D., Miyakawa, T., et al. (2001). Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 107, 617–629. doi: 10.1016/S0092-8674(01)00585-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, Z., Ksiezak-Reding, H., Riggio, S., Haroutunian, V., and Pasinetti, G. M. (2006). Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr. Res. 84, 1–14. doi: 10.1016/j.schres.2006.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, J. J., Lee, S., and Zhou, Z. J. (2004). A developmental switch in the excitability and function of the starburst network in the mammalian retina. Neuron 44, 851–864. doi: 10.1016/j.neuron.2004.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zoghbi, H. Y. (2003). Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302, 826–830. doi: 10.1126/science.1089071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zucker, R. S., and Regehr, W. G. (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405. doi: 10.1146/annurev.physiol.64.092501.114547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: neuropsychiatric diseases, short-term synaptic plasticity, neural circuits, 22q11.2 microdeletion, DISC1, schizophrenia, autism spectrum disorder, risk genes
Citation: Crabtree GW and Gogos JA (2014) Synaptic plasticity, neural circuits, and the emerging role of altered short-term information processing in schizophrenia. Front. Synaptic Neurosci. 6:28. doi: 10.3389/fnsyn.2014.00028
Received: 20 May 2014; Accepted: 22 October 2014;
Published online: 25 November 2014.
Edited by:
Florentin Wörgötter, University Goettingen, GermanyReviewed by:
Peter Kalivas, University of South Carolina, USADragos Inta, Central Institute of Mental Health, Germany
Christoph Nissen, University Medical Center Freiburg, Germany
Copyright © 2014 Crabtree and Gogos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph A. Gogos, Department of Physiology and Cellular Biophysics, Department of Neuroscience, College of Physicians and Surgeons, Columbia University, 630 W. 168th Street, New York, NY 10032, USA e-mail:amFnOTBAY29sdW1iaWEuZWR1