



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Synaptic Neurosci. , 30 July 2010
Volume 2 - 2010 | https://doi.org/10.3389/fnsyn.2010.00034
This article is part of the Research Topic Spike-timing dependent plasticity View all 33 articles
Spike-timing dependent plasticity (STDP) has been studied extensively in a variety of animal models during the past decade but whether it can be studied at the systems level of the human cortex has been a matter of debate. Only recently newly developed non-invasive brain stimulation techniques such as transcranial magnetic stimulation (TMS) have made it possible to induce and assess timing dependent plasticity in conscious human subjects. This review will present a critical synopsis of these experiments, which suggest that several of the principal characteristics and molecular mechanisms of TMS-induced plasticity correspond to those of STDP as studied at a cellular level. TMS combined with a second phasic stimulation modality can induce bidirectional long-lasting changes in the excitability of the stimulated cortex, whose polarity depends on the order of the associated stimulus-evoked events within a critical time window of tens of milliseconds. Pharmacological evidence suggests an NMDA receptor mediated form of synaptic plasticity. Studies in human motor cortex demonstrated that motor learning significantly modulates TMS-induced timing dependent plasticity, and, conversely, may be modulated bidirectionally by prior TMS-induced plasticity, providing circumstantial evidence that long-term potentiation-like mechanisms may be involved in motor learning. In summary, convergent evidence is being accumulated for the contention that it is now possible to induce STDP-like changes in the intact human central nervous system by means of TMS to study and interfere with synaptic plasticity in neural circuits in the context of behavior such as learning and memory.
Spike-timing dependent plasticity (STDP) has been studied extensively in a variety of model systems, ranging from cultured neurons (Bi and Poo, 1998) and cortical slice preparations (Magee and Johnston, 1997; Markram et al., 1997) to intact animals (Zhang et al., 1998; Jacob et al., 2007), but whether or not timing dependent plasticity can be studied at the systems level of the human cortex has been a matter of debate. Human models, however, may contribute particularly relevant information as they may aid in translating knowledge about synaptic plasticity derived from animal studies into diagnostic or therapeutic progress for patients and inform further experimental animal studies.
Transcranial magnetic stimulation (TMS) is a painless non-invasive brain stimulation technique which allows for activation of the intact human cortex (Barker et al., 1985). Stimulation of the primary motor cortex (M1) activates pyramidal output neurons, most likely transsynaptically via a chain of interneurons (Amassian et al., 1987; Ziemann and Rothwell, 2000; Di Lazzaro et al., 2004; Di Lazzaro et al., 2008). Pyramidal neurons in the motor cortex or interneurons within the same microcolumn projecting onto them receive somatosensory input at short latency and with high topographical specificity via afferents from somatosensory cortex (Rosen and Asanuma, 1972; Caria et al., 1997). Thus, pairing of TMS of M1 conjointly with an afferent input to the motor cortex (such as somatosensory information by peripheral nerve stimulation) may result in convergent activation of neural elements in the motor cortex. Based on Hebb’s theoretical framework of synaptic plasticity (Hebb, 1949), it was hypothesized that this form of associative stimulation may induce timing dependent plasticity at the systems level in conscious humans.
In the original experiments of paired associative stimulation (PAS) of the human cortex, PAS consists of electrical stimulation of the median nerve at the wrist (MNS) followed by TMS of the contralateral M1 (Figures 1A–C) (Stefan et al., 2000). Excitability of the corticospinal system is probed by TMS over the optimal scalp position for eliciting motor evoked potentials (MEPs) in a resting intrinsic hand muscle (M. abductor pollicis brevis, APB) (Figure 1D). The interval between MNS and TMS was chosen to be 25 ms (PAS25). The first component (N20) of the median nerve somatosensory-evoked potential (MN-SSEP) typically arrives in the primary somatosensory cortex (S1) at around 20 ms (Figure 1D) (Allison et al., 1991). Taking into account some additional milliseconds for the MNS signal to be relayed from S1 to M1, it is thought that the afferent signal evoked by MNS arrives in M1 nearly synchronously, or even shortly before transsynaptic excitation of corticospinal neurons by the TMS pulse. MNS intensity is set at three times the perceptual sensory threshold. This intensity is generally subthreshold for activation of corticospinal neurons in contralateral M1, as evidenced by the absence of long latency electromyographic reflex activity in the target muscle, which typically requires MNS intensities close to the motor threshold in addition to voluntary muscle activation (Deuschl et al., 1985; Deuschl and Lücking, 1990). In contrast, TMS intensity is adjusted to result in action potentials in corticospinal neurons as indicated by elicitation of MEPs of, on average, 1 mV peak-to-peak amplitude. Ninety pairs of MNS and TMS applied at a frequency of 0.05 Hz over 30 min significantly increase MEP amplitudes in the resting APB muscle (Figure 1E) (Stefan et al., 2000). This effect is critically dependent on the interval between MNS and TMS because an interstimulus interval (ISI) of only 10 ms (PAS10) results in depression of MEPs (Figure 1F) (Wolters et al., 2003), while much longer ISIs of 100, 525, and 5000 ms have no effect (Stefan et al., 2000). These experiments demonstrate a temporally asymmetric Hebbian-like rule governing PAS-induced changes of excitability in the human motor cortex.
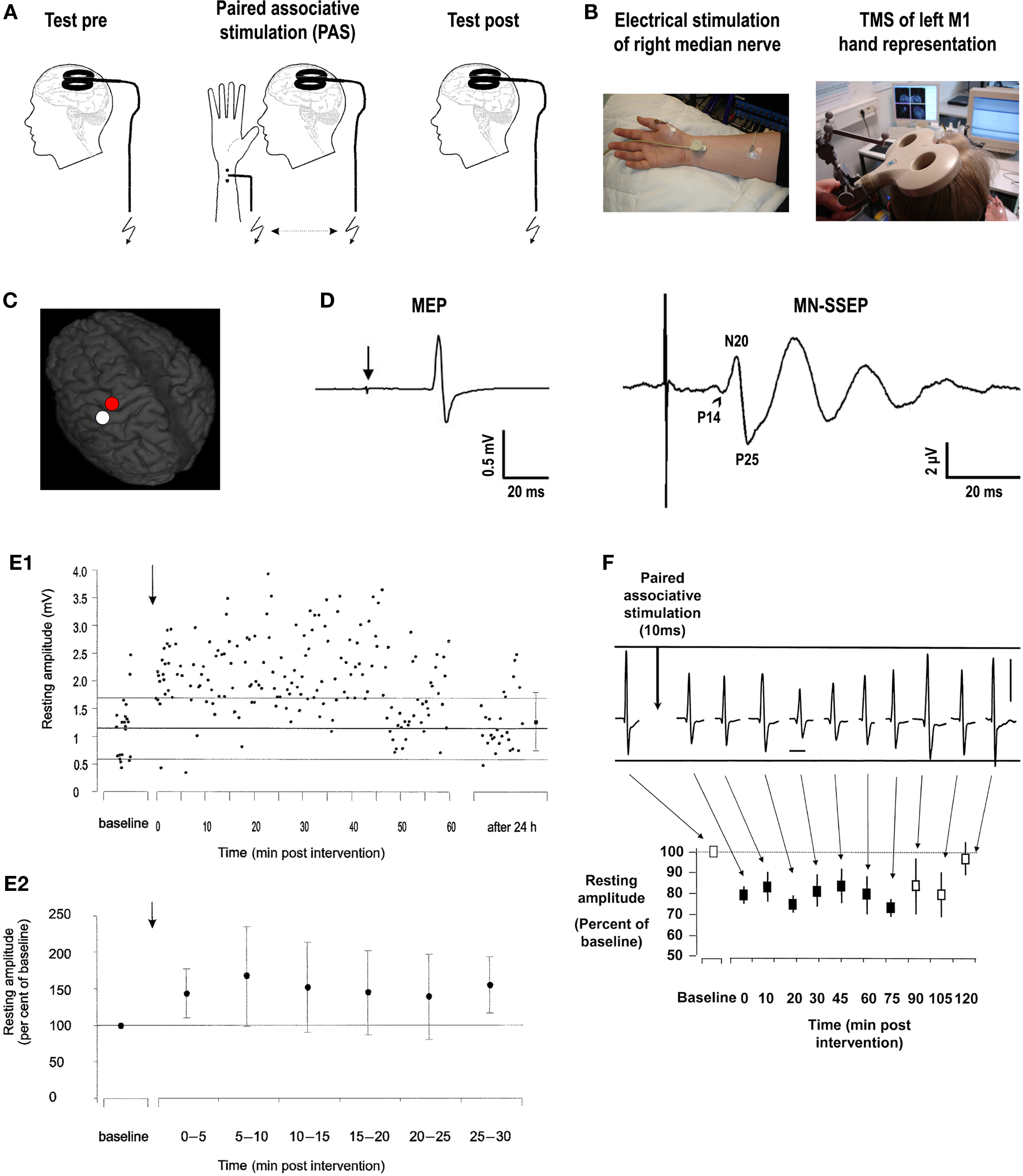
Figure 1. Paired associative stimulation induces timing dependent plasticity in the human primary motor cortex. (A) Experimental design. Paired associative stimulation (PAS) consisted of electrical right median nerve stimulation (MNS) and transcranial magnetic stimulation (TMS) of the left primary motor cortex (M1) over the optimal site for eliciting motor evoked potentials (MEPs) in a muscle of the right hand (M. abductor pollicis brevis, APB). Ninety pairs of stimuli were applied with a constant interstimulus interval (ISI) at a frequency of 0.05 Hz. Corticospinal excitability was probed before and after PAS by MEP amplitude in the right APB elicited by single-pulse TMS (adapted from Figure 1, Stefan et al., 2000. Permission pending). (B) Experimental setup (see text for explanations). (C) Stimulation site of TMS superimposed on a 3-dimensional structural MRI (red dot over left precentral gyrus). The white dot over the left postcentral gyrus indicates the primary somatosensory cortex (S1) hand representation. (D) Illustrative examples of MEP in the target muscle (APB) elicited by single-pulse TMS over M1 (red dot in B) at rest (left; arrow, time of TMS pulse delivery) and median nerve somatosensory-evoked potential (MN-SSEP) recorded from scalp electrodes overlying S1 (white dot in B), respectively. P14, P14-potential (positive deflection ∼14 ms after MNS, arrowhead); N20, N20-potential (negative deflection ∼20 ms after MNS); P25, P25-potential (positive deflection ∼25 ms after MNS). (E) PAS with an ISI of 25ms between MNS and TMS of M1 (PAS25) induces long-lasting increases in MEPs. (E1) MEP amplitudes before and after PAS25 (arrow) from a representative subject at rest (resting amplitude, y-axis). The thick horizontal line indicates the mean resting MEP amplitude at baseline prior to PAS25, and the thin lines indicate one standard deviation. Note persistence of increased excitability for 60 min and return to baseline 24 h after PAS25 (adapted from Figure 3B, Stefan et al., 2000. Permission pending). (E2) Time course of resting MEP amplitudes after PAS25 (arrow) normalized to baseline before PAS25. Data are means from 11 subjects (±S.D.) and are binned to 5 min epochs after PAS25. MEP amplitudes after PAS25 are significantly increased compared to baseline for each of the six epochs (adapted from Figure 3A, Stefan et al., 2000. Permission pending). (F) PAS with an ISI of 10 ms between MNS and TMS of M1 (PAS10) induces long-lasting decreases in MEPs. Recordings from a representative subject (vertical bar, 1 mV; horizontal bar, 20 ms) and time course of mean MEP amplitudes at rest normalized to baseline before PAS10 (resting amplitude, y-axis) from 10 subjects (±S.E.M.) are shown. Filled squares indicate significant MEP amplitude decreases (adapted from Figure 3, Wolters et al., 2003. Permission pending).
The physiological properties of this potentiation and depression of MEPs comprise inducibility within 30 min, duration of 30–60 min minimum, reversibility within 24 h and topographical specificity with respect to muscles with cortical representations not receiving dual and synchronous input from MNS and TMS (Stefan et al., 2000; Wolters et al., 2003). PAS most likely does not alter GABAAergic inhibition in motor cortex (Stefan et al., 2002), as indexed by short-latency paired-pulse TMS (Kujirai et al., 1993; Ziemann et al., 1996). Thus, timing dependent PAS effects are very likely not associated with GABAAergic disinhibition of the motor cortex. Experimental studies and modeling of PAS-induced changes in MEP amplitude as a function of the interstimulus interval between the associative stimuli revealed dependence on near-synchronicity of arrival of the two stimulus-induced events in M1, with a critical window in the order of tens of milliseconds and a relatively steep transition phase from depressant to facilitatory PAS effects (Figure 2) (Wolters et al., 2003, 2005). MEP amplitudes increase when suprathreshold TMS-induced activation of pyramidal neurons presumably follows subthreshold activation of these neural elements by MNS by a few milliseconds; reversing the sequence of these events decreases MEP size (Wolters et al., 2003). These findings are reminiscent of spike-timing dependent long-term potentiation (LTP) and long-term depression (LTD) windows for excitatory-to-excitatory connections as observed in animal experiments (Caporale and Dan, 2008).
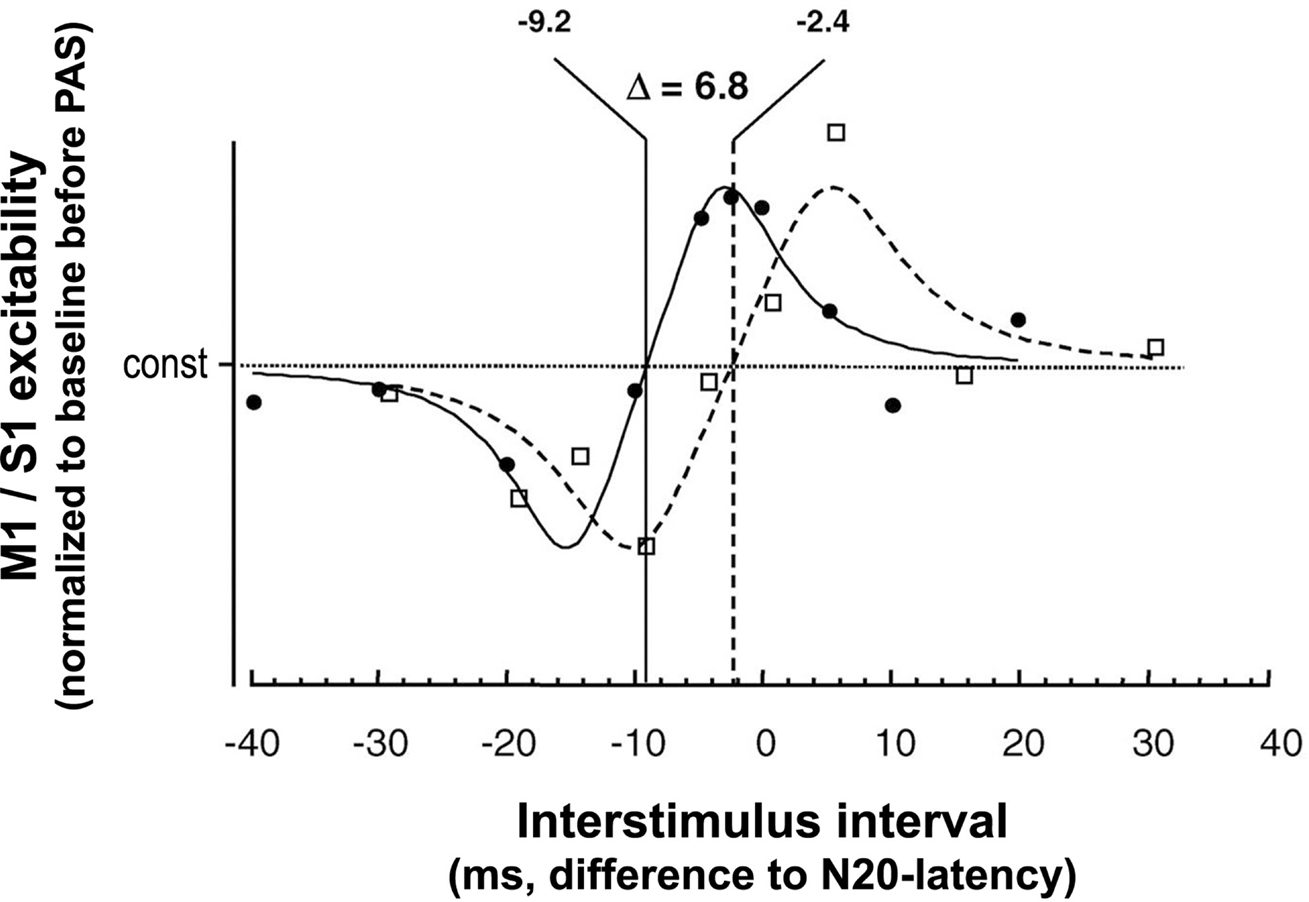
Figure 2. Time window of PAS-induced timing dependent LTP- and LTD-like plasticity in human sensorimotor cortex. X-axis, interstimulus intervals (in ms) between MNS and TMS are given with respect to the individual N20-latency of the median nerve somatosensory-evoked potential (MN-SSEP). Y-axis, M1 and S1 excitability as determined by MEP amplitude and the P25 component of the MN-SSEP, respectively, normalized to baseline before PAS. Fitted data are scaled to their relative extremes. □, data from PAS of the primary motor cortex (M1), Wolters et al. (2003); •, data from PAS of the primary somatosensory cortex (S1), Wolters et al. (2005). In S1, LTD-like plasticity turns to LTP-like plasticity at an ISI 6.8 ms shorter than in M1 (modified from Figure 6 from Wolters et al., 2005. Permission pending). Continuous and dashed lines indicate modeling of the function relating the PAS-induced magnitude of the P25 and MEP amplitude change, respectively, to the interval between the stimulation modalities. Modeling was done using commercially available software (DataFit program, Version 8.0, Oakdale Engineering, Oakdale, PA, USA). The following function was found empirically y = a + (x/b − c)/((x/b − c)4 + d). “const” in the figure corresponds to “a” in the model. A similar function was found, if a was set to 1.
Subsequent studies have modified the original LTP-like plasticity inducing PAS protocol (Stefan et al., 2000) by using a slightly shorter interval between MNS and TMS of 21.5 ms (PAS21.5) (Weise et al., 2006) or by setting the interval to the individual N20 latency (PASN20) (Ziemann et al., 2004) or to N20 + 2 ms (PASN20 + 2) (Müller et al., 2007; Jung and Ziemann, 2009). All these protocols result in a significant and long-term increase of MEPs because the temporal order of events in motor cortex, i.e., arrival of the MNS input within a few milliseconds before or synchronously with the TMS pulse, is obeyed. A form of rapid-rate paired associative stimulation with a stimulation rate of 5 Hz induces timing dependent and long-lasting (up to 6 h) MEP increases within only 2 min providing evidence for rapid induction of PAS-induced LTP-like plasticity (Quartarone et al., 2006). The facilitatory PAS effect on MEPs has been shown to be dose-dependent, i.e., the magnitude and duration of MEP increases scale with the number of stimulus pairs (Nitsche et al., 2007). LTP-like PAS effects saturate at a level of around 160–170% of MEP baseline values (Wolters et al., 2003; Stefan et al., 2004; Nitsche et al., 2007).
The following lines of evidence suggest that the site of action of PAS-induced plasticity is at the level of the cortex: (i) PAS does not change the magnitude of F-waves, an index of spinal motor neuron excitability tested by median nerve stimulation, at the same time when MEP amplitudes were increased (Stefan et al., 2000). (ii) MEPs induced by electrical brainstem stimulation, which excites corticospinal axons directly at the level of the craniocervical junction, downstream of the cortex (Ugawa et al., 1991), remain unchanged after PAS (Stefan et al., 2000). (iii) The cortical silent period of electromyographic activity in the contracting APB muscle, a TMS measure of motor cortical inhibition (Inghilleri et al., 1993), is prolonged by PAS (Stefan et al., 2000) (discussed below in more detail). (iv) Epidural recordings of descending corticospinal activity evoked by TMS demonstrate PAS-induced changes of later descending volleys (Di Lazzaro et al., 2009a,b), which reflect the intracortical transsynaptic activation of pyramidal neurons by TMS (Ziemann and Rothwell, 2000; Di Lazzaro et al., 2004). (v) PAS-experiments targeting the primary somatosensory cortex (reviewed in more detail below) provided evidence for selective modulation of components of somatosensory evoked potentials known to be generated exclusively cortically in strictly confined cortical regions (Wolters et al., 2005; Litvak et al., 2007). (vi) Reversing the direction of the induced current in the brain, which likely leads to preferential activation of intracortical elements located in upper cortical layers with synaptic connection onto corticospinal neurons, allows to decrease the stimulus intensity to below the threshold for activation of corticospinal descending action potentials (Kujirai et al., 2006). (vii) Finally, PAS interferes in a highly specific manner with volitional preparatory cortical motor activity, as measured by changes in movement-related cortical potentials (MRCPs) in EEG recordings, affecting MRCPs only of those movements targeted by PAS (Lu et al., 2009).
Pharmacological studies showed that both PAS-induced timing dependent LTP- and LTD-like plasticity is dependent on N-methyl-d-aspartate (NMDA) receptor activation because the NMDA receptor antagonist dextromethorphan blocks both forms of plasticity (Stefan et al., 2002; Wolters et al., 2003). Moreover, PAS-induced LTD-like plasticity is blocked by the L-type voltage-gated calcium channel antagonist nimodipine (Wolters et al., 2003). These findings are in line with in vitro data indicating a role for NMDA receptors as coincidence detectors of pre- and postsynaptic activity during both spike-timing dependent LTD and LTP induction, while further mechanisms, likely involving Ca2+ influx through voltage-gated Ca2+ channels, additionally operate in spike-timing dependent LTD (Bi and Poo, 1998).
Further studies tested whether PAS induces timing dependent Hebbian-like associative plasticity in other cortical areas, such as S1. Here, PAS consisted of pairing MNS and TMS of S1 (Figure 1C). Excitability of the somatosensory cortex was probed by median nerve somatosensory-evoked cortical potentials (MN-SSEPs) (Figure 1D). These experiments demonstrated timing dependent bidirectional changes of the P25 cortical component of the MN-SSEP (Wolters et al., 2005), with associated behavioral changes in two-point discrimination thresholds (Litvak et al., 2007). Source modeling revealed that the changes in MN-SSEPs are best explained by synaptic modification of superficial layers 2/3 of Brodmann area 3b (Litvak et al., 2007). These findings are consistent with animal data showing that STDP can be induced at excitatory vertical inputs from layer 4 onto layer 2/3 pyramidal neurons in rat barrel cortex with behavioral relevance (Feldman, 2000). The N20 and P14 components of the MN-SSEP, which are thought to be generated in deeper cortical layers and subcortically in the thalamus, respectively, remain unchanged (Wolters et al., 2005). As for PAS of the motor system, PAS effects in the somatosensory cortex are rapidly inducible, long-lasting, yet reversible, topographically specific both for the peripheral and the transcranial stimulation, and confined to a narrow window of effective ISIs between MNS and TMS (Wolters et al., 2005; Litvak et al., 2007). Modeling the modulation of the P25 and MEP amplitude as a function of the ISI between MNS and TMS over S1 and M1, respectively, showed that the timing and widths of the time windows in which LTP- and LTD-like effects can be induced by PAS are quite similar in S1 and M1, with S1 leading M1 by ∼7 ms (Figure 2) (Wolters et al., 2005). This delay of the turning point from LTD- to LTP-like plasticity by ∼7 ms in S1 vs. M1 may be just sufficient for polysynaptic propagation of afferent somatosensory signals from S1 to M1 (Wolters et al., 2005) via relays in Brodmann areas 1 and 2 (Jones, 1986). In contrast, first evidence suggests that human hippocampal synapses change their efficacy in response to timed pre- and postsynaptic activity according to a significantly wider and left-shifted STDP rule in comparison to PAS-induced LTD- and LTP-like plasticity in human sensorimotor cortex (Testa-Silva et al., 2010). However, these findings cannot be directly compared to the PAS data reviewed above, as Testa-Silva and colleagues employed a different experimental approach (ex vivo in vitro vs. in vivo), a different plasticity induction protocol (electrical pairing vs. paired associative electrical peripheral nerve stimulation and TMS) and studied a different tissue substrate (allocortex from drug-resistant [i.e. with a history of drug therapy] epilepsy patients vs. neocortex in healthy human subjects).
In summary, these experiments provide convergent physiological and pharmacological evidence for the contention that PAS-induced timing dependent bidirectional changes of localized neural activity in the human sensorimotor cortex may represent STDP of synaptic efficacy or a closely related mechanism as studied at a cellular level. However, in the absence of invasive neuronal recordings any hypothesis about the precise nature or location of the cellular correlates of PAS-induced plasticity remains, to some extent, speculative. Specifically, a contribution of non-synaptic timing dependent modifications of intrinsic neuronal (Ganguly et al., 2000; Li et al., 2004) and/or local dendritic (Daoudal et al., 2002; Wang et al., 2003; Frick et al., 2004) excitability cannot be ruled out. Homeostatic plasticity such as homeostatic synaptic scaling (Nelson and Turrigiano, 2008) has been shown not only to globally adjust synaptic strength to chronic alterations in network activity (Turrigiano et al., 1998), but also to rapidly tune the efficacy of individual synapses (Pozo and Goda, 2010). Therefore, a full physiological explanation of PAS effects may also include activity-dependent (intrinsic and synaptic) homeostatic plasticity (Nelson and Turrigiano, 2008; Shah et al., 2010) in addition to synaptic STDP-like mechanisms. Further complexity in interpreting PAS effects at a cellular or even molecular level derives from the complex interactions between Hebbian and non-Hebbian forms of plasticity, the temporal and spatial aspects of which are just beginning to emerge (Nelson and Turrigiano, 2008). Finally, TMS-induced modifications of neural activity are coupled to alterations in cerebral hemodynamics (Allen et al., 2007). However, the experiments by Allen and coworkers indicated that TMS-induced changes in hemodynamics are secondary to changes in spontaneous or evoked neural activity, thus being compatible with the notion that PAS effects may be largely explained by terms of neural plasticity.
To keep synaptic weights in a physiological range and maintain overall neuronal and network activity, modifications of synaptic strength need to be carefully controlled. Metaplasticity refers to an activity-dependent mechanism which manifests as a change in the ability to induce subsequent synaptic plasticity (Abraham and Bear, 1996). PAS was used to induce timing dependent LTP-like plasticity (PASN20 + 2) in the primary motor cortex (M1) and study its regulation by prior activity in the stimulated neural pathway (Müller et al., 2007). In line with previous studies, this study showed that PASN20 + 2 leads to a long-lasting increase in MEP amplitudes in the target muscle (APB) when applied to a naïve M1, but this LTP-like effect is occluded or even reversed to MEP depression when applied subsequent to a prior PASN20 + 2 priming protocol (Figure 3A) (Müller et al., 2007). In contrast, priming PASN20 + 2 by an LTD-like plasticity inducing PAS protocol (PASN20 − 5) leads to MEP facilitation similar to the naïve condition or when compared to priming with a control PAS protocol (PASCtrl), which does not change MEP amplitude (Figure 3A). Control experiments ruled out that the suppressive interaction between two consecutive LTP-like plasticity inducing PASN20 + 2 protocols is simply caused by a ceiling effect due to increased excitability of the stimulated cortex after the first PASN20 + 2 intervention (Figure 3A). These findings suggest that homeostatic metaplasticity governs timing dependent PAS-induced plasticity in the human M1, most likely homosynaptically (Müller et al., 2007). Homeostatic mechanisms control PAS-induced plasticity in M1 even when priming involves an afferent input to the motor cortex such as the projection from dorsal premotor cortex to M1 (Pötter-Nerger et al., 2009). Due to the non-invasive nature of these studies the underlying cellular mechanisms remain unknown, but findings argue in favor of a global homeostatic integration of synaptic inputs within M1, consistent with homeostatic mechanisms formalized in the Bienenstock–Cooper–Munro theory of bidirectional synaptic plasticity (Bienenstock et al., 1982). Homeostatic metaplasticity and associated behavioral performance changes were also demonstrated in PAS experiments targeting the primary somatosensory cortex in humans (Bliem et al., 2008), indicating its generalizable occurrence in and across different regions of human cortex (Figure 3B).
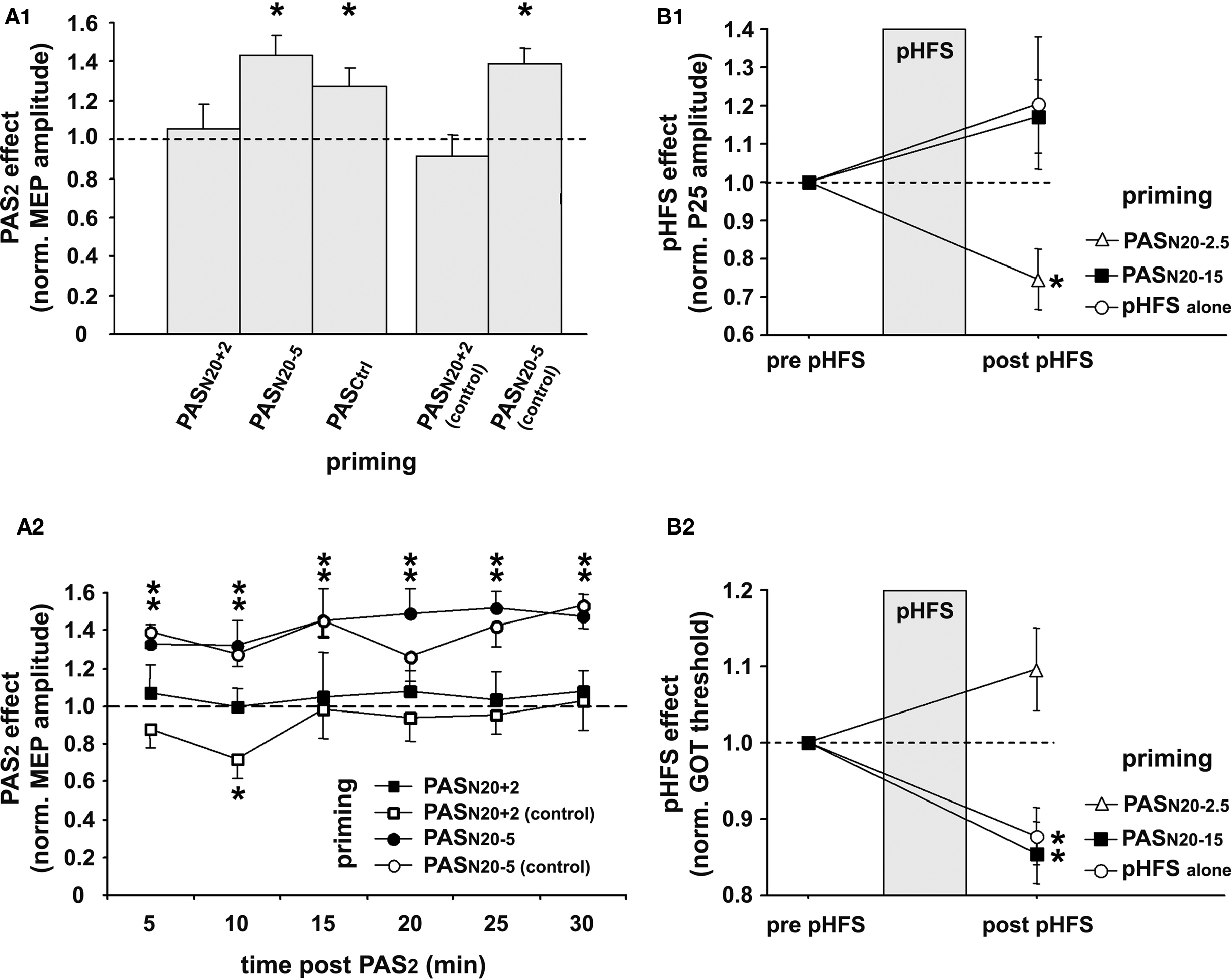
Figure 3. Homeostatic metaplasticity governs timing dependent LTP-like plasticity in human primary motor (M1) and somatosensory (S1) cortex. (A) Homeostatic metaplasticity in M1. (A1) PAS was used to induce timing dependent LTP-like plasticity (PASN20 + 2), LTD-like plasticity (PASN20 − 5), or no change (PASCtrl) in the naïve M1 (data not shown). A second PAS protocol (PAS2) 30 min after the first PAS intervention (PAS1) always consisted of PASN20 + 2. PAS2 induced significant long-lasting MEP increases if primed by PASN20 − 5 or PASCtrl, but this effect was completely suppressed if primed by PASN20 + 2. PASN20 + 2 (control) and PASN20 − 5 (control) refer to control experiments, in which MEP amplitudes after priming with PASN20 + 2 or PASN20 − 5 were carefully re-adjusted to match those at baseline. The PAS2 effects are shown as MEP amplitude after PAS2 normalized to MEP amplitude after PAS1 but before PAS2. (A2) Time course over 30 min of PAS2 effects primed by PASN20 + 2 (filled squares), PASN20 + 2 (control) (open squares), PASN20 − 5 (filled circles), or PASN20 − 5 (control) (open circles). Data are means ± S.E.M. of 11 subjects; asterisks indicate a significant difference to 1.0. (B) Homeostatic metaplasticity in S1. Changes in somatosensory cortical excitability (P25 amplitude of MN-SSEP, (B1)) and in tactile spatial discrimination (grating orientation task [GOT] threshold, (B2)) induced by peripheral high frequency stimulation (pHFS) of the median nerve alone (open circles), or by pHFS primed by PASN20 − 2.5 (open triangle) or PASN20 − 15 (filled square). PASN20 − 2.5 and PASN20 − 15 were shown to induce LTP- and LTD-like plasticity, respectively, in the naïve human S1 (cf. Figure 2, and Wolters et al., 2005). Data are mean ± S.E.M normalized to pre pHFS; asterisks indicate a significant difference to 1.0 (modified, with permission, from Figure 3, Müller et al., 2007, and Figure 5, Bliem et al., 2008).
Metaplasticity was also demonstrated in studies in which facilitatory or depressant PAS was applied after voluntary activation (Figure 4) (Ziemann et al., 2004; Stefan et al., 2006; Rosenkranz et al., 2007b). Repeated fastest possible thumb abduction movements or isometric thumb abductions result in learning, defined by an increase in the maximum peak acceleration of the practiced movement, or by an increase of successful force production, and occlude subsequent PAS-induced LTP-like plasticity, or even reverse it to LTD-like plasticity (Figure 4A), whereas subsequent PAS-induced LTD-like plasticity is either enhanced (Figure 4B) or remains unchanged (Ziemann et al., 2004; Stefan et al., 2006; Rosenkranz et al., 2007b). In contrast, interactions between motor learning and PAS-induced LTP-like plasticity are not observed, when PAS is applied with a delay of 6 h after motor learning, or if PAS is applied to the motor cortex ipsilateral to the training hand (Stefan et al., 2006).
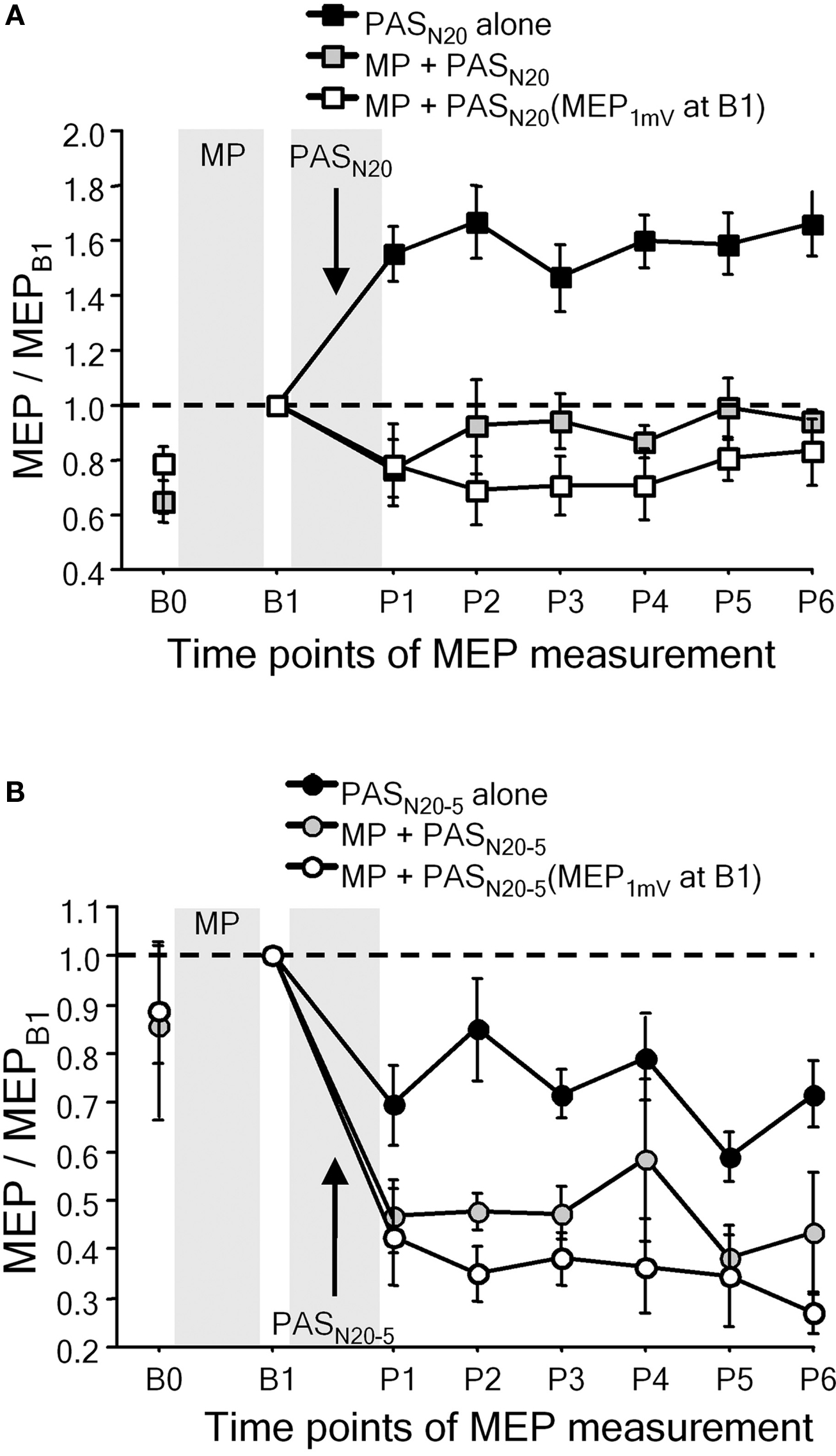
Figure 4. Motor learning involves LTP-like mechanisms in human primary motor cortex. (A) Interactions between motor practice (MP) and timing dependent LTP-like plasticity induced by PASN20. The PASN20-induced long-lasting MEP increase (PASN20 alone, filled square) was completely suppressed by MP associated motor learning immediately prior to PAS (gray squares). White squares refer to a control experiment in which any increase in MEP amplitude induced by MP was adjusted for by a reduction in TMS intensity (reinstallation of baseline MEP amplitude (MEP1mV) at time point B1) (adapted, with permission, from Figure 4, Ziemann et al., 2004). (B) Interactions between MP and timing dependent LTD-like plasticity induced by PASN20 − 5. Note that motor learning enhanced the lasting decrease in MEP amplitude induced by PASN20 − 5 alone (adapted, with permission, from Figure 5, Ziemann et al., 2004).
Which aspect of motor training with the intention to acquire motor skill is relevant for the temporary suppression of PAS-induced timing dependent LTP-like plasticity or even its reversal to LTD-like plasticity? One hypothesis would be that voluntary activity per se without learning or skill acquisition is sufficient to act as an effective priming event. At least two arguments provide evidence against this notion to be the sole explanation for training-induced modulation of PAS effects (Ziemann et al., 2004; Stefan et al., 2006; Rosenkranz et al., 2007b): (i) When the same number of thumb abduction movements is performed slowly, this does not result in motor learning, and this form of motor practice does not alter subsequent PAS-induced plasticity (Ziemann et al., 2004). (ii) In another study (Rosenkranz et al., 2007b), subjects were instructed to optimize acceleration of fast thumb abduction movements during multiple training sessions performed over 5 days (Rosenkranz et al., 2007b). PAS was used to induce LTP-like plasticity, and excitability of M1 was tested by measuring input–output curves of MEPs. Task performance improved continuously over 5 days of training (Rosenkranz et al., 2007b). After practice on day 1, the PAS effect reversed from LTP-like to LTD-like plasticity in line with previous studies (Ziemann et al., 2004; Stefan et al., 2006). In contrast, on day 5 PAS-induced LTP-like plasticity was no longer influenced by the preceding motor practice but showed the same magnitude as at baseline before the first practice on day 1 despite a persistent practice-induced enhancement of the MEP input–output curve (Rosenkranz et al., 2007b). This differential modulation of timing dependent LTP-like plasticity by motor practice is consistent with the notion that not activity per se, but initial motor learning may trigger LTP of existing synaptic connections. Continued skill learning may induce synaptogenesis and motor cortical reorganization with susceptibility to LTP induction being fully restored (Rosenkranz et al., 2007b). These findings support the view that motor learning may be associated in its initial phase with LTP-like mechanisms in human M1.
The magnitude of PAS-induced LTP-like plasticity is highly variable interindividually (Ridding and Ziemann, 2010) and PAS effects decrease significantly with age (Müller-Dahlhaus et al., 2008; Tecchio et al., 2008; Fathi et al., 2010). Likewise, professional musicians show a wider modification range of PAS-induced timing dependent plasticity than non-musicians, as demonstrated by a significantly enhanced increase and decrease of MEP amplitudes after PAS protocols to induce LTP- and LTD-like plasticity, respectively (Rosenkranz et al., 2007a). Similarly enhanced plasticity was noted in physically active individuals (Cirillo et al., 2009). The mechanisms of these findings are not clear, but interindividual differences in the expression level of key neural signals for synaptic plasticity, such as brain-derived neurotrophic factor (BDNF), which was shown to influence experience-dependent and PAS-induced motor cortical plasticity (Kleim et al., 2006; Cheeran et al., 2008), may, among others, account for the observed broad variation of timing dependent PAS effects between subjects. Moreover, LTP-like PAS effects were shown to be critically dependent on the subject’s attention to the stimulation (Stefan et al., 2004). Direct evidence for a role of neuromodulators such as dopamine, norepinephrine, and acetylcholine in shaping associative plasticity in the human cortex comes from pharmacological studies (Ziemann et al., 2006; Kuo et al., 2007, 2008). Likewise, cortisol and GABABergic input may suppress PAS-induced LTP-like plasticity (McDonnell et al., 2007; Sale et al., 2008). These findings are consistent with data from animal experiments showing that STDP is substantially regulated by neuromodulatory and inhibitory input (Caporale and Dan, 2008).
The cortical silent period of electromyographic activity in the contracting APB muscle, a TMS measure of motor cortical inhibition (Inghilleri et al., 1993), is prolonged by PAS (Stefan et al., 2000). PAS may also modulate other aspects of intracortical inhibition such as long-latency inhibition, which refers to the suppression of the MEP evoked by single-pulse TMS when conditioned by either peripheral afferent or magnetic cortical stimulation applied at long (>100 ms) intervals (Russmann et al., 2009). Recent studies utilizing associative stimulation of homologous areas of left and right human M1 demonstrated an interstimulus interval-specific increase of MEP in the conditioned M1 (Koganemaru et al., 2009; Rizzo et al., 2009a). This change in corticospinal excitability by cortico-cortical PAS is associated with a modulation in interhemispheric inhibition, the suppression of one primary motor cortex by the contralateral homologous M1 (Rizzo et al., 2009a). These studies indicate that PAS may influence inhibitory actions, but whether this occurs through an effect on synapses from inhibitory interneurons onto pyramidal cells or indirectly via modulation of excitatory connections, has not been established unequivocally. The studies by Rizzo et al. (2009a) and by Koganemaru et al. (2009) also suggest that timing dependency may govern plasticity between interconnected cortical areas. How this shapes functional and effective connectivity in health and disease is currently not known.
Inducing timing dependent bidirectional plasticity in the human cortex by PAS offers the possibility to assess Hebbian-like associative plasticity on the systems level in patients with neuropsychiatric disorders. PAS-induced facilitation of corticospinal excitability is reduced or absent in a variety of neuropsychiatric disorders, consistent with observations of impaired synaptic plasticity in animal models of these disorders. In Parkinson’s disease (PD), where degeneration of the nigrostriatal dopaminergic projection occurs, two studies showed that PAS with an interstimulus interval between MNS and TMS to induce LTP-like plasticity does not increase MEP amplitudes in the APB muscle of the more affected side in patients off medication (Morgante et al., 2006; Ueki et al., 2006) while another study reported stronger PAS-induced timing dependent LTP-like plasticity and loss of topographical specificity in PD patients off therapy in comparison to a control group (Bagnato et al., 2006). Reduced plasticity could be restored in a subgroup of patients by dopamine replacement therapy (Morgante et al., 2006; Ueki et al., 2006). Reduced plasticity was also found in two other disorders that have been linked with abnormal dopaminergic transmission: restless legs syndrome (Rizzo et al., 2009b) and schizophrenia (Frantseva et al., 2008). Qualitatively different observations were made in writer’s cramp, a neurological disorder characterized by excessive and inappropriate muscular activation during writing. Both PAS-induced LTD- and LTP-like plasticity were enhanced in magnitude and duration in M1 (Quartarone et al., 2003; Weise et al., 2006) and S1 (Tamura et al., 2009). Moreover, in dystonic patients PAS may induce changes in MEP amplitudes in muscle representations of the functional surround of the representation receiving associative stimulation (Weise et al., 2006), suggesting a loss of the topographical specificity of PAS-induced timing dependent plasticity (Figure 5).
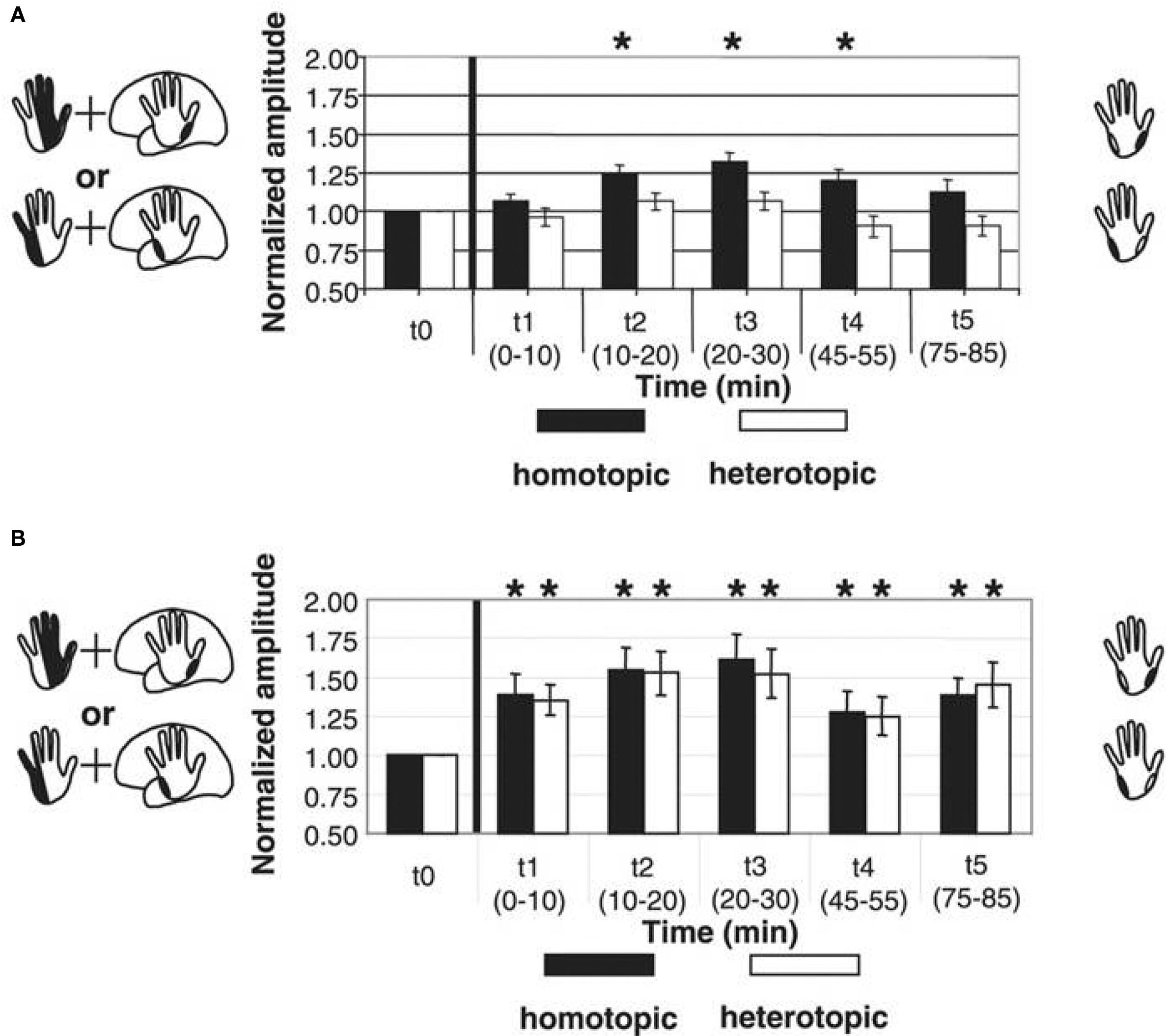
Figure 5. Altered timing dependent LTP-like plasticity in human primary motor cortex (M1) in patients with writer’s cramp. PAS consisted of either electrical median nerve (MN) stimulation combined with TMS over the M. abductor pollicis brevis (APB) M1 representation [upper left scheme in (A) and (B)] or ulnar nerve (UN) stimulation combined with TMS over the M. abductor digiti minimi (ADM) M1 representation [lower left scheme in (A) and (B)]. PAS-induced changes in MEP amplitude were assessed from both muscles, which served as a homotopic target (black in the right schemes) stimulated by PAS (APB in MN-PAS, ADM in UN-PAS) or heterotopic target not stimulated by PAS (APB in UN-PAS, ADM in MN-PAS). (A) In healthy controls, combined data from MN-PAS and UN-PAS with an ISI of 21.5 ms between electrical peripheral nerve and magnetic cortex stimulation (PAS21.5) revealed MEP amplitude increases in homotopically stimulated muscle representations only (black bars) (adapted from Figure 2C, Weise et al., 2006. Permission pending). (B) In contrast, in patients with writer’s cramp, following PAS21.5 MEP amplitudes increased in both homotopically (black bars) and heterotopically (white bars) stimulated muscle representations. Asterisks indicate a significant difference from baseline (adapted from Figure 3C, Weise et al., 2006. Permission pending).
Plasticity mechanisms including activity-dependent rewiring and synaptic strengthening provide the physiological basis for long term functional recovery, for instance after stroke (Murphy and Corbett, 2009). Non-invasive brain stimulation techniques such as PAS may be applied to modulate cellular mechanisms of synaptic plasticity. In support of this notion, motor learning in healthy subjects can be bidirectionally modulated by prior PAS (Figure 6) (Jung and Ziemann, 2009). Motor learning, indexed by an increase in peak acceleration of the trained rapid thumb flexion movements, is enhanced by induction of timing dependent LTD-like plasticity (PASN20-5) 90 min prior to motor practice, but suppressed if primed by LTP-like plasticity inducing PASN20 + 2 (Figure 6B) (Jung and Ziemann, 2009). These findings are in line with homeostatic metaplasticity rules regulating LTP-like processes such as motor learning in human M1 reviewed above (Ziemann et al., 2004; Stefan et al., 2006; Müller et al., 2007), and provide proof-of-principle that modulation of Hebbian-like associative plasticity by PAS may translate into behavioral performance gains. Additionally, the study by Jung and Ziemann (2009) reveals that motor learning immediately following PAS shows partly non-homeostatic interactions, i.e., motor learning is enhanced if primed by PASN20 + 2 (although to a lesser extent than after PASN20 − 5) (Figure 6A) (Jung and Ziemann, 2009). This suggests that homeostatic metaplasticity is fully expressed only if there is a sufficient delay between priming protocol and the subsequent learning process. The underlying mechanisms of this non-homeostatic interaction can only be speculated upon. They include blockade of immediately subsequent LTD-like processes to prevent erasure of the just induced LTP-like plasticity (cf. Peineau et al., 2007) and facilitation of learning if the priming LTP-like plasticity did not saturate the synaptic modification range (cf. Berger, 1984).
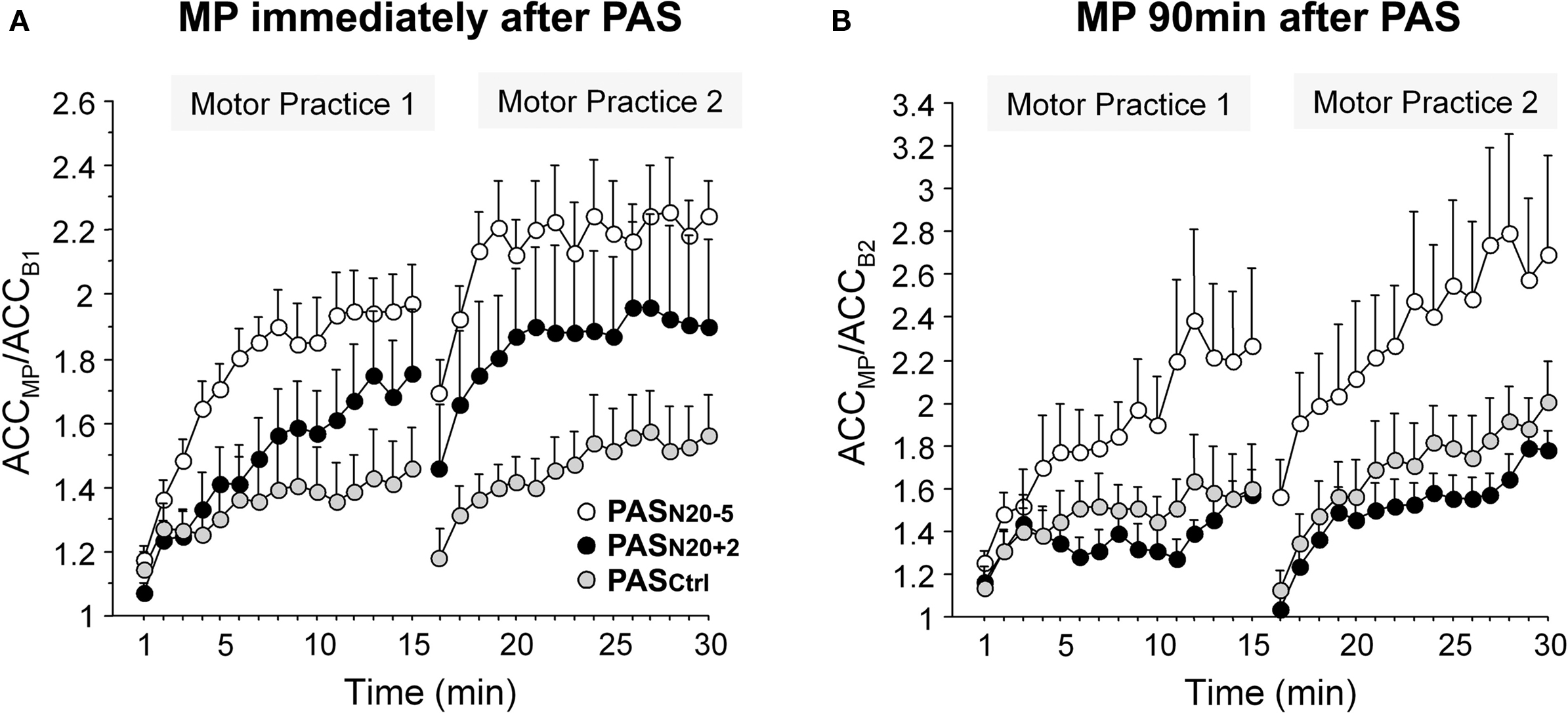
Figure 6. Bidirectional modulation of motor learning in healthy subjects by prior PAS-induced timing dependent LTP- and LTD-like plasticity. Motor learning was indexed by an increase in peak acceleration (ACC) of the trained thumb flexion movement during two blocks of 15 min motor practice (MP) normalized to baseline peak ACC before MP. (A) MP immediately following PASN20 + 2 (black circles) or PASN20 − 5 (white circles) resulted in an enhancement of learning compared with priming by PASCtrl (gray circles, control PAS condition without induction of LTP/LTD-like plasticity). (B) In contrast, MP 90 min after PASN20 − 5 enhanced motor learning, whereas MP following PASN20 + 2 showed a trend toward depression of learning with respect to the PASCtrl condition. Note different scales of y-axes in (A) and (B). Mean ± S.E.M. of nine subjects (adapted, with permission, from Figure 3 in Jung and Ziemann, 2009).
First evidence suggests that PAS can induce timing dependent plasticity in patients with chronic stroke, but whether this translates into behavioral performance gains is currently not known (Jayaram and Stinear, 2009). Several other important questions remain open: what is the best time window for priming interventions (e.g., PAS) to facilitate LTP-dependent processes such as motor learning? Which patients benefit most/at all, as these mechanisms may be altered in brain disease (Quartarone et al., 2005)? How do priming effects change during repeated interventional sessions (Rosenkranz et al., 2007b)? And what is the role of neuromodulatory pharmaceuticals in the framework of stimulation-induced timing dependent plasticity? Clearly, further studies are needed to address and clarify these issues. Only then plasticity-inducing non-invasive brain stimulation techniques such as PAS can be fully exploited to purposefully modulate motor, sensory and cognitive functions in humans.
Paired associative stimulation (PAS) of the human sensorimotor cortex by electrical peripheral nerve and conjoint TMS may induce timing dependent bidirectional long-lasting excitability changes in the stimulated cortex. Pharmacological and physiological evidence suggests that the after-effects of this non-invasive brain stimulation paradigm may be related to STDP. Thus, PAS may serve as an example of translation of studies on synaptic physiology on a cellular level to the systems physiology level of the human brain. PAS was extensively used to assess cortical plasticity in healthy human subjects and provided circumstantial evidence that motor learning involves LTP-like mechanisms, and that homeostatic metaplasticity regulates sensorimotor cortical plasticity. Moreover, PAS experiments may offer some new insight into the pathophysiology of a variety of neuropsychiatric diseases. PAS-induced modulation of synaptic plasticity in the context of behavior may be used purposefully in order to promote motor, sensory and cognitive performance in humans.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abraham, W. C., and Bear, M. F. (1996). Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 19, 126–130.
Allen, E. A., Pasley, B. N., Duong, T., and Freeman, R. D. (2007). Transcranial magnetic stimulation elicits coupled neural and hemodynamic consequences. Science 317, 1918–1921.
Allison, T., McCarthy, G., Wood, C. C., and Jones, S. J. (1991). Potentials evoked in human and monkey cerebral cortex by stimulation of the median nerve. A review of scalp and intracranial recordings. Brain 114(Pt 6), 2465–2503.
Amassian, V. E., Stewart, M., Quirk, G. J., and Rosenthal, J. L. (1987). Physiological basis of motor effects of a transient stimulus to cerebral cortex. Neurosurgery 20, 74–93.
Bagnato, S., Agostino, R., Modugno, N., Quartarone, A., and Berardelli, A. (2006). Plasticity of the motor cortex in Parkinson’s disease patients on and off therapy. Mov. Disord. 21, 639–645.
Barker, A. T., Jalinous, R., and Freeston, I. L. (1985). Non-invasive magnetic stimulation of human motor cortex. Lancet 1, 1106–1107.
Berger, T. W. (1984). Long-term potentiation of hippocampal synaptic transmission affects rate of behavioral learning. Science 224, 627–630.
Bi, G. Q., and Poo, M. M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bienenstock, E. L., Cooper, L. N., and Munro, P. W. (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48.
Bliem, B., Müller-Dahlhaus, J. F., Dinse, H. R., and Ziemann, U. (2008). Homeostatic metaplasticity in the human somatosensory cortex. J. Cogn. Neurosci. 20, 1517–1528.
Caporale, N., and Dan, Y. (2008). Spike timing-dependent plasticity: a Hebbian learning rule. Annu. Rev. Neurosci. 31, 25–46.
Caria, M. A., Kaneko, T., Kimura, A., and Asanuma, H. (1997). Functional organization of the projection from area 2 to area 4gamma in the cat. J. Neurophysiol. 77, 3107–3114.
Cheeran, B., Talelli, P., Mori, F., Koch, G., Suppa, A., Edwards, M., Houlden, H., Bhatia, K., Greenwood, R., and Rothwell, J. C. (2008). A common polymorphism in the brain-derived neurotrophic factor gene (BDNF) modulates human cortical plasticity and the response to rTMS. J. Physiol. 586, 5717–5725.
Cirillo, J., Lavender, A. P., Ridding, M. C., and Semmler, J. G. (2009). Motor cortex plasticity induced by paired associative stimulation is enhanced in physically active individuals. J. Physiol. 587, 5831–5842.
Daoudal, G., Hanada, Y., and Debanne, D. (2002). Bidirectional plasticity of excitatory postsynaptic potential (EPSP)-spike coupling in CA1 hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 14512–14517.
Deuschl, G., and Lücking, C. H. (1990). Physiology and clinical applications of hand muscle reflexes. Electroencephalogr. Clin. Neurophysiol. Suppl. 41, 84–101.
Deuschl, G., Schenck, E., and Lücking, C. H. (1985). Long-latency responses in human thenar muscles mediated by fast conducting muscle and cutaneous afferents. Neurosci. Lett. 55, 361–366.
Di Lazzaro, V., Dileone, M., Pilato, F., Profice, P., Oliviero, A., Mazzone, P., Insola, A., Capone, F., Ranieri, F., and Tonali, P. A. (2009a). Associative motor cortex plasticity: direct evidence in humans. Cereb. Cortex 19, 2326–2330.
Di Lazzaro, V., Dileone, M., Profice, P., Pilato, F., Oliviero, A., Mazzone, P., Di Iorio, R., Capone, F., Ranieri, F., Florio, L., and Tonali, P. A. (2009b). LTD-like plasticity induced by paired associative stimulation: direct evidence in humans. Exp. Brain Res. 194, 661–664.
Di Lazzaro, V., Oliviero, A., Pilato, F., Saturno, E., Dileone, M., Mazzone, P., Insola, A., Tonali, P. A., and Rothwell, J. C. (2004). The physiological basis of transcranial motor cortex stimulation in conscious humans. Clin. Neurophysiol. 115, 255–266.
Di Lazzaro, V., Ziemann, U., and Lemon, R. N. (2008). State of the art: physiology of transcranial motor cortex stimulation. Brain Stimul. 1, 345–362.
Fathi, D., Ueki, Y., Mima, T., Koganemaru, S., Nagamine, T., Tawfik, A., and Fukuyama, H. (2010). Effects of aging on the human motor cortical plasticity studied by paired associative stimulation. Clin. Neurophysiol. 121, 90–93.
Feldman, D. E. (2000). Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27, 45–56.
Frantseva, M. V., Fitzgerald, P. B., Chen, R., Moller, B., Daigle, M., and Daskalakis, Z. J. (2008). Evidence for impaired long-term potentiation in schizophrenia and its relationship to motor skill learning. Cereb. Cortex 18, 990–996.
Frick, A., Magee, J., and Johnston, D. (2004). LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci. 7, 126–135.
Ganguly, K., Kiss, L., and Poo, M. (2000). Enhancement of presynaptic neuronal excitability by correlated presynaptic and postsynaptic spiking. Nat. Neurosci. 3, 1018–1026.
Inghilleri, M., Berardelli, A., Cruccu, G., and Manfredi, M. (1993). Silent period evoked by transcranial stimulation of the human cortex and cervicomedullary junction. J. Physiol. 466, 521–534.
Jacob, V., Brasier, D. J., Erchova, I., Feldman, D., and Shulz, D. E. (2007). Spike timing-dependent synaptic depression in the in vivo barrel cortex of the rat. J. Neurosci. 27, 1271–1284.
Jayaram, G., and Stinear, J. W. (2009). The effects of transcranial stimulation on paretic lower limb motor excitability during walking. J. Clin. Neurophysiol. 26, 272–279.
Jones, E. G. (1986). “Connectivity of the primate sensory-motor cortex,” in Cerebral Cortex, eds E. G. Jones and A. Peters (New York: Plenum Press), pp 113–183.
Jung, P., and Ziemann, U. (2009). Homeostatic and nonhomeostatic modulation of learning in human motor cortex. J. Neurosci. 29, 5597–5604.
Kleim, J. A., Chan, S., Pringle, E., Schallert, K., Procaccio, V., Jimenez, R., and Cramer, S. C. (2006). BDNF val66met polymorphism is associated with modified experience-dependent plasticity in human motor cortex. Nat. Neurosci. 9, 735–737.
Koganemaru, S., Mima, T., Nakatsuka, M., Ueki, Y., Fukuyama, H., and Domen, K. (2009). Human motor associative plasticity induced by paired bihemispheric stimulation. J. Physiol. 587, 4629–4644.
Kujirai, T., Caramia, M. D., Rothwell, J. C., Day, B. L., Thompson, P. D., Ferbert, A., Wroe, S., Asselman, P., and Marsden, C. D. (1993). Corticocortical inhibition in human motor cortex. J. Physiol. 471, 501–519.
Kujirai, K., Kujirai, T., Sinkjaer, T., and Rothwell, J. C. (2006). Associative plasticity in human motor cortex during voluntary muscle contraction. J. Neurophysiol. 96, 1337–1346.
Kuo, M. F., Grosch, J., Fregni, F., Paulus, W., and Nitsche, M. A. (2007). Focusing effect of acetylcholine on neuroplasticity in the human motor cortex. J. Neurosci. 27, 14442–14447.
Kuo, M. F., Paulus, W., and Nitsche, M. A. (2008). Boosting focally-induced brain plasticity by dopamine. Cereb. Cortex 18, 648–651.
Li, C. Y., Lu, J. T., Wu, C. P., Duan, S. M., and Poo, M. M. (2004). Bidirectional modification of presynaptic neuronal excitability accompanying spike timing-dependent synaptic plasticity. Neuron 41, 257–268.
Litvak, V., Zeller, D., Oostenveld, R., Maris, E., Cohen, A., Schramm, A., Gentner, R., Zaaroor, M., Pratt, H., and Classen, J. (2007). LTP-like changes induced by paired associative stimulation of the primary somatosensory cortex in humans: source analysis and associated changes in behaviour. Eur. J. Neurosci. 25, 2862–2874.
Lu, M. K., Bliem, B., Jung, P., Arai, N., Tsai, C. H., and Ziemann, U. (2009). Modulation of preparatory volitional motor cortical activity by paired associative transcranial magnetic stimulation. Hum. Brain Mapp. 30, 3645–3656.
Magee, J. C., and Johnston, D. (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275, 209–213.
Markram, H., Lubke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
McDonnell, M. N., Orekhov, Y., and Ziemann, U. (2007). Suppression of LTP-like plasticity in human motor cortex by the GABA(B) receptor agonist baclofen. Exp. Brain Res. 180 181–186.
Morgante, F., Espay, A. J., Gunraj, C., Lang, A. E., and Chen, R. (2006). Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain 129, 1059–1069.
Müller, J. F., Orekhov, Y., Liu, Y., and Ziemann, U. (2007). Homeostatic plasticity in human motor cortex demonstrated by two consecutive sessions of paired associative stimulation. Eur. J. Neurosci. 25, 3461–3468.
Müller-Dahlhaus, J. F., Orekhov, Y., Liu, Y., and Ziemann, U. (2008). Interindividual variability and age-dependency of motor cortical plasticity induced by paired associative stimulation. Exp. Brain Res. 187, 467–475.
Murphy, T. H., and Corbett, D. (2009). Plasticity during stroke recovery: from synapse to behaviour. Nat. Rev. Neurosci. 10, 861–872.
Nitsche, M. A., Roth, A., Kuo, M. F., Fischer, A. K., Liebetanz, D., Lang, N., Tergau, F., and Paulus, W. (2007). Timing-dependent modulation of associative plasticity by general network excitability in the human motor cortex. J. Neurosci. 27, 3807–3812.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Pötter-Nerger, M., Fischer, S., Mastroeni, C., Groppa, S., Deuschl, G., Volkmann, J., Quartarone, A., Munchau, A., and Siebner, H. R. (2009). Inducing homeostatic-like plasticity in human motor cortex through converging corticocortical inputs. J. Neurophysiol. 102, 3180–3190.
Pozo, K., and Goda, Y. (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351.
Quartarone, A., Bagnato, S., Rizzo, V., Siebner, H. R., Dattola, V., Scalfari, A., Morgante, F., Battaglia, F., Romano, M., and Girlanda, P. (2003). Abnormal associative plasticity of the human motor cortex in writer’s cramp. Brain 126, 2586–2596.
Quartarone, A., Rizzo, V., Bagnato, S., Morgante, F., Sant’angelo, A., Girlanda, P., and Roman Siebner, H. (2006). Rapid-rate paired associative stimulation of the median nerve and motor cortex can produce long-lasting changes in motor cortical excitability in humans. J. Physiol. 575, 657–670.
Quartarone, A., Rizzo, V., Bagnato, S., Morgante, F., Sant’Angelo, A., Romano, M., Crupi, D., Girlanda, P., Rothwell, J. C., and Siebner, H. R. (2005). Homeostatic-like plasticity of the primary motor hand area is impaired in focal hand dystonia. Brain 128, 1943–1950.
Ridding, M. C., and Ziemann, U. (2010). Determinants of the induction of cortical plasticity by non-invasive brain stimulation in healthy subjects. J. Physiol. 588, 2291–2304.
Rizzo, V., Siebner, H. S., Morgante, F., Mastroeni, C., Girlanda, P., and Quartarone, A. (2009a). Paired associative stimulation of left and right human motor cortex shapes interhemispheric motor inhibition based on a Hebbian mechanism. Cereb. Cortex 19, 907–915.
Rizzo, V., Arico, I., Mastroeni, C., Morgante, F., Liotta, G., Girlanda, P., Silvestri, R., and Quartarone, A. (2009b). Dopamine agonists restore cortical plasticity in patients with idiopathic restless legs syndrome. Mov. Disord. 24, 710–715.
Rosen, I., and Asanuma, H. (1972). Peripheral afferent inputs to the forelimb area of the monkey motor cortex: input–output relations. Exp. Brain Res. 14, 257–273.
Rosenkranz, K., Williamon, A., and Rothwell, J. C. (2007a). Motorcortical excitability and synaptic plasticity is enhanced in professional musicians. J. Neurosci. 27, 5200–5206.
Rosenkranz, K., Kacar, A., and Rothwell, J. C. (2007b). Differential modulation of motor cortical plasticity and excitability in early and late phases of human motor learning. J. Neurosci. 27, 12058–12066.
Russmann, H., Lamy, J. C., Shamim, E. A., Meunier, S., and Hallett, M. (2009). Associative plasticity in intracortical inhibitory circuits in human motor cortex. Clin Neurophysiol 120, 1204–1212.
Sale, M. V., Ridding, M. C., and Nordstrom, M. A. (2008). Cortisol inhibits neuroplasticity induction in human motor cortex. J. Neurosci. 28, 8285–8293.
Shah, M. M., Hammond, R. S., and Hoffman, D. A. (2010). Dendritic ion channel trafficking and plasticity. Trends Neurosci. 33, 307–316.
Stefan, K., Kunesch, E., Cohen, L. G., Benecke, R., and Classen, J. (2000). Induction of plasticity in the human motor cortex by paired associative stimulation. Brain 123 Pt 3, 572–584.
Stefan, K., Kunesch, E., Benecke, R., Cohen, L. G., and Classen, J. (2002). Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J. Physiol. 543, 699–708.
Stefan, K., Wycislo, M., and Classen, J. (2004). Modulation of associative human motor cortical plasticity by attention. J. Neurophysiol. 92, 66–72.
Stefan, K., Wycislo, M., Gentner, R., Schramm, A., Naumann, M., Reiners, K., and Classen, J. (2006). Temporary occlusion of associative motor cortical plasticity by prior dynamic motor training. Cereb. Cortex 16, 376–385.
Tamura, Y., Ueki, Y., Lin, P., Vorbach, S., Mima, T., Kakigi, R., and Hallett, M. (2009). Disordered plasticity in the primary somatosensory cortex in focal hand dystonia. Brain 132, 749–755.
Tecchio, F., Zappasodi, F., Pasqualetti, P., De Gennaro, L., Pellicciari, M. C., Ercolani, M., Squitti, R., and Rossini, P. M. (2008). Age dependence of primary motor cortex plasticity induced by paired associative stimulation. Clin Neurophysiol 119, 675–682.
Testa-Silva, G., Verhoog, M. B., Goriounova, N. A., Loebel, A., Hjorth, J., Baayen, J. C., De Kock, C. P. J., and Mansvelder, H. D. (2010). Human synapses show a wide temporal window for spike-timing-dependent plasticity. Front. Syn. Neurosci. 2:12. doi: 10.3389/fnsyn.2010.00012.
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896.
Ueki, Y., Mima, T., Kotb, M. A., Sawada, H., Saiki, H., Ikeda, A., Begum, T., Reza, F., Nagamine, T., and Fukuyama, H. (2006). Altered plasticity of the human motor cortex in Parkinson’s disease. Ann. Neurol. 59, 60–71.
Ugawa, Y., Rothwell, J. C., Day, B. L., Thompson, P. D., and Marsden, C. D. (1991). Percutaneous electrical stimulation of corticospinal pathways at the level of the pyramidal decussation in humans. Ann. Neurol. 29, 418–427.
Wang, Z., Xu, N. L., Wu, C. P., Duan, S., and Poo, M. M. (2003). Bidirectional changes in spatial dendritic integration accompanying long-term synaptic modifications. Neuron 37, 463–472.
Weise, D., Schramm, A., Stefan, K., Wolters, A., Reiners, K., Naumann, M., and Classen, J. (2006). The two sides of associative plasticity in writer’s cramp. Brain 129, 2709–2721.
Wolters, A., Sandbrink, F., Schlottmann, A., Kunesch, E., Stefan, K., Cohen, L. G., Benecke, R., and Classen, J. (2003). A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J. Neurophysiol. 89, 2339–2345.
Wolters, A., Schmidt, A., Schramm, A., Zeller, D., Naumann, M., Kunesch, E., Benecke, R., Reiners, K., and Classen, J. (2005). Timing-dependent plasticity in human primary somatosensory cortex. J. Physiol. 565, 1039–1052.
Zhang, L. I., Tao, H. W., Holt, C. E., Harris, W. A., and Poo, M. (1998). A critical window for cooperation and competition among developing retinotectal synapses. Nature 395, 37–44.
Ziemann, U., Ilic, T. V., Pauli, C., Meintzschel, F., and Ruge, D. (2004). Learning modifies subsequent induction of long-term potentiation-like and long-term depression-like plasticity in human motor cortex. J. Neurosci. 24, 1666–1672.
Ziemann, U., Lonnecker, S., Steinhoff, B. J., and Paulus, W. (1996). Effects of antiepileptic drugs on motor cortex excitability in humans: a transcranial magnetic stimulation study. Ann. Neurol. 40, 367–378.
Ziemann, U., Meintzschel, F., Korchounov, A., and Ilic, T. V. (2006). Pharmacological modulation of plasticity in the human motor cortex. Neurorehabil. Neural. Repair 20, 243–251.
Keywords: spike-timing dependent plasticity, long-term potentiation, long-term depression, paired associative stimulation, transcranial magnetic stimulation, human, cortex, translational neuroscience
Citation: Müller-Dahlhaus F, Ziemann U and Classen J (2010) Plasticity resembling spike-timing dependent synaptic plasticity: the evidence in human cortex. Front. Syn. Neurosci. 2:34. doi: 10.3389/fnsyn.2010.00034
Received: 02 February 2010;
Paper pending published: 19 February 2010;
Accepted: 11 July 2010;
Published online: 30 July 2010
Edited by:
Per Jesper Sjöström, University College London, UKReviewed by:
Huibert D. Mansvelder, VU University, NetherlandsCopyright: © 2010 Müller-Dahlhaus, Ziemann and Classen. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ulf Ziemann, Department of Neurology, Goethe University Frankfurt, Schleusenweg 2-16, 60528 Frankfurt am Main, Germany; JosephClassen, Department of Neurology, University of Leipzig, Liebigstraße 20, 04103 Leipzig, Germany.e-mail:dS56aWVtYW5uQGVtLnVuaS1mcmFua2Z1cnQuZGU=;am9zZXBoLmNsYXNzZW5AbWVkaXppbi51bmktbGVpcHppZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.