 Raven M. A. Fisher
Raven M. A. Fisher Mariana P. Torrente
Mariana P. Torrente
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 13 September 2024
Sec. Brain Disease Mechanisms
Volume 17 - 2024 | https://doi.org/10.3389/fnmol.2024.1456052
This article is part of the Research TopicEpigenetic Modifications in Neurological and Cognitive DisordersView all 7 articles
Alzheimer’s disease (AD), Parkinson’s disease (PD), Frontotemporal Dementia (FTD), and Amyotrophic lateral sclerosis (ALS) are complex and fatal neurodegenerative diseases. While current treatments for these diseases do alleviate some symptoms, there is an imperative need for novel treatments able to stop their progression. For all of these ailments, most cases occur sporadically and have no known genetic cause. Only a small percentage of patients bear known mutations which occur in a multitude of genes. Hence, it is clear that genetic factors alone do not explain disease occurrence. Chromatin, a DNA-histone complex whose basic unit is the nucleosome, is divided into euchromatin, an open form accessible to the transcriptional machinery, and heterochromatin, which is closed and transcriptionally inactive. Protruding out of the nucleosome, histone tails undergo post-translational modifications (PTMs) including methylation, acetylation, and phosphorylation which occur at specific residues and are connected to different chromatin structural states and regulate access to transcriptional machinery. Epigenetic mechanisms, including histone PTMs and changes in chromatin structure, could help explain neurodegenerative disease processes and illuminate novel treatment targets. Recent research has revealed that changes in histone PTMs and heterochromatin loss or gain are connected to neurodegeneration. Here, we review evidence for epigenetic changes occurring in AD, PD, and FTD/ALS. We focus specifically on alterations in the histone PTMs landscape, changes in the expression of histone modifying enzymes and chromatin remodelers as well as the consequences of these changes in heterochromatin structure. We also highlight the potential for epigenetic therapies in neurodegenerative disease treatment. Given their reversibility and pharmacological accessibility, epigenetic mechanisms provide a promising avenue for novel treatments. Altogether, these findings underscore the need for thorough characterization of epigenetic mechanisms and chromatin structure in neurodegeneration.
As human life spans have continued to increase over the last few decades, neurodegenerative diseases have dramatically risen in prevalence (Checkoway et al., 2011). Alzheimer’s disease (AD), Parkinson’s disease (PD), Frontotemporal dementia (FTD), and Amyotrophic lateral sclerosis (ALS) are major neurodegenerative disorders (NDs) affecting different neuron types and leading to a plethora of symptoms. Interestingly, in all these disorders, the majority of cases are considered sporadic and are presumed to arise from environmental factors either acting alone or in conjunction with various dysregulated genes (Spencer et al., 2022; Dilliott et al., 2021). A small proportion of cases -often termed familial- are linked to genetic mutations occurring in a large number of genes. Notably, in many instances, these genetic mutations lead to the misfolding and aggregation of specific proteins such as amyloid precursor protein (APP) in AD, -synuclein (SNCA) in PD and Chromosome 9 open reading frame 72 (C9orf72) in FTD/ALS (Schellenberg and Montine, 2012; Stefanis, 2012; Balendra and Isaacs, 2018). As genetics alone does not explain disease etiology, epigenetics could reveal pathological mechanisms and novel treatment avenues.
Epigenetics involves heritable alterations to phenotype that do not result from a change in DNA sequence (Shahid et al., 2023). Eukaryotic DNA folds and condenses itself around a histone protein octamer consisting of two H2A-H2B dimers and a H3–H4 tetramer (Jenuwein and Allis, 2001). This DNA and histone complex constitutes the nucleosome, the basic repeating unit of chromatin (Shahid et al., 2023; Jenuwein and Allis, 2001; Chereji et al., 2018). In addition to DNA methylation and non-coding RNA action, epigenetic mechanisms include histone post-translational modifications as well as chromatin remodeling, invoking changes in nucleosomal positioning and chromatin structure (Hwang et al., 2017; Bannister and Kouzarides, 2011).
Protruding out of the nucleosome, the tails of histone proteins can undergo various post-translational modifications (PTMs) including acetylation, methylation, and phosphorylation as well as ubiquitylation and SUMOylation (Bannister and Kouzarides, 2011). Other modifications such as mono-ADP-ribosylation, formylation, citrullination, and crotonylation are also possible. Although these other modifications are categorized, functional roles and mechanisms are not fully understood (Zhao and Garcia, 2015). Histone modifications impact DNA-templated processes not only by modulating the physical interaction between histones and DNA but also by serving as binding platforms for other proteins (Shahid et al., 2023; Jenuwein and Allis, 2001). In this way, PTMs comprise a ‘code’ through which they largely regulate gene transcription. Each modification is site-specific and connected to distinct functions. For example, methylation can be linked to transcriptional repression or activation depending on which site is modified (Jenuwein and Allis, 2001; Peterson and Laniel, 2004). Furthermore, ubiquitylation and SUMOylation have also been linked to gene silencing, transcriptional repression as well as DNA repair at certain sites (Bannister and Kouzarides, 2011; Zhao and Garcia, 2015). Accordingly, the enzymes responsible for adding, removing, and recognizing these PTMs are referred to as the writers, erasers, and readers of this code, respectively (Rothbart and Strahl, 2014). For instance, acetylation marks are written by histone acetyltransferases (HATs) such as Gcn5/PCAF, erased by histone deacetylases (HDAC) such as HDAC 1, and read by proteins that contain bromodomains such as BRD4 (Marmorstein and Trievel, 2009; Saha and Pahan, 2006; Marmorstein and Zhou, 2014). Similarly, methylation marks are written by histone methyltransferases (HMTs) like SET7, erased by histone demethylases such as Lysine Specific Demethylase 1 (LSD1), and read by proteins comprising chromodomains such as CHD1 (Teperino et al., 2010; Yun et al., 2011). Phosphorylation marks are written by kinases such as Aurora B Kinase, erased by phosphatases such as Repo-man/PP1, and read by phospho-serine adaptor molecules like 14–3-3 proteins (Peterson and Laniel, 2004; Gil and Vagnarelli, 2019; Winter et al., 2008). One well-characterized ubiquitylation mark, H2AK119ub, is linked to gene repression (Bannister and Kouzarides, 2011). This modification is written by ubiquitin ligase, RING1B, and erased by deubiquitinase, BAP1 (Zhao and Garcia, 2015; Barbour et al., 2020).
Chromatin typically assumes two states which are defined by their accessibility to transcriptional machinery and are directly linked to particular histone PTMs: euchromatin and heterochromatin. On one hand, euchromatin is open and transcriptionally active; it is linked to acetylation at Lysine 9 and 14 (H3K9ac and H3K14ac) as well as phosphorylation at Serine 10 on H3 (H3S10ph) specifically during interphase (Torrente et al., 2011; Komar and Juszczynski, 2020). Euchromatin is typically found within active cis-regulatory elements such as promoters and enhancers (Morrison and Thakur, 2021).
Conversely, heterochromatin is closed, compact, and transcriptionally silent (Shahid et al., 2023). It is further divided into either constitutive heterochromatin or facultative heterochromatin. Facultative heterochromatin (fHC) assembles around highly regulated genes during development and is cell type-specific (Penagos-Puig and Furlan-Magaril, 2020). fHC is characterized by the presence of polycomb group proteins and tri-methylation of Histone H3 at Lysine 27 (H3K27me3) as well as monoubiquitylation of H2A at Lysine 119 (H2AK119ub) (Morrison and Thakur, 2021). H3K27me3 is installed by polycomb repressive complex 2 (PRC2), a set-domain containing methyltransferase (Millán-Zambrano et al., 2022; Laugesen et al., 2016). PRC2 subunits EED and SUZ12 allow the protein complex to also bind to H3K27me3 and propagate fHC. This mark is removed by various demethylases such as KDM6A, KDM6C, and KDM6B (Hyun et al., 2017). Additionally, H2AK119 ubiquitination is catalyzed by polycomb repressive complex 1 (PRC1) which has the ability to phase separate and can cause chromatin compaction without H2Aub (Barbour et al., 2020; Eeftens et al., 2021). Monoubiquitylation of H2A is removed by the deubiquitinase, BAP1 (Barbour et al., 2020). One noteworthy reader of H2AK119ub, JARID2, promotes PRC2 methylation of H3K27me3 linking these two fHC PTMs in a positive feedback mechanism (Barbour et al., 2020; Eeftens et al., 2021).
Constitutive heterochromatin (cHC) comprises a static condensed state that is conserved in different cell types and propagated throughout life once it is established (Morrison and Thakur, 2021). Tri-methylation on H3 at Lysine 9 (H3K9me3) is a key mark characterizing cHC. H3K9me3 is installed by methyltransferases SUV39H1 and SUV39H2. JHDM3/KDM4 are responsible for demethylating this mark (Hyun et al., 2017). Heterochromatin protein 1 (HP1), which binds H3K9me3, is necessary for constitutive heterochromatin condensation. HP1 can compact chromatin into phase-separated liquid condensates (Larson et al., 2017). Acting opposite to H3K9me3, phosphorylation of H3 at Serine 10 (H3S10ph) occludes HP1 binding to H3K9me3 and ejects HP1 from chromatin in a “binary switch” mechanism (Heinrichs, 2005). Indeed, H3S10ph is a key factor in cell cycle regulation and chromatin dynamics due to its ability to kick out HP1 during mitosis (Komar and Juszczynski, 2020).
In conjunction with chromatin, nuclear lamina is necessary for nuclear shape and DNA integrity. Nuclear lamina is found within the nuclear envelope and is comprised of intermediate filament proteins known as Lamin (Carollo and Barra, 2023). Due to their interaction with chromatin, Lamin proteins are important genomic organization factors. Nuclear Lamins, enriched in Lamin proteins B1 and B2, interact with specific chromosomal regions known as Lamin-associated domains (LADs) which are typically consistent with heterochromatin regions (Carollo and Barra, 2023; Maji et al., 2020; van Steensel and Belmont, 2017). Lamin B1 decreases are indicative of heterochromatin decreases (Stephens et al., 2018). In this way, Lamins are thoroughly connected to heterochromatin and indirectly to histone PTMs. Interestingly, Lamin tail domains binding to H2A and H2B are evolutionarily conserved (Goldberg et al., 1999). Alterations in Lamin expression and protein levels, histone PTMs, and chromatin lead to abnormal nuclear morphology and nuclear blebs which are connected to a multitude of diseases (Stephens et al., 2018).
Recent evidence points to the dysregulation of histone PTMs and alterations in heterochromatin playing a notable role in ND pathways (Wang et al., 2021; Song et al., 2023; Lee et al., 2018; Bennett et al., 2019; Chen et al., 2018; Kabir et al., 2023; Gräff et al., 2012; Zimmer-Bensch, 2020; Cobos et al., 2019). Methylation, acetylation, phosphorylation, and ubiquitylation of histone tails have thoroughly been implicated in various ND’s. The role of other modifications such as citrullination, formylation, and crotonylation is yet to be fully understood in this context. Histone PTMs are an attractive drug target given their dynamic nature and accessibility to pharmaceutical intervention. Here, we review changes on the histone PTM and heterochromatin landscape in AD, PD, and FTD/ALS and underscore the potential for novel neurodegeneration therapeutics targeting these epigenetic mechanisms. These findings highlight the need for comprehensive characterization of epigenetic mechanisms and chromatin structure alterations in ND research. We note that while aging and environmental exposures are also linked to epigenetics alterations and ND occurrence (Mir et al., 2023), we will not discuss these factors here. Instead, for an in-depth exploration of these topics, we refer the reader to several thorough reviews (Hou et al., 2019; Lee J.-H. et al., 2020; Cannon and Greenamyre, 2011; Chin-Chan et al., 2015).
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by neuritic plaques and neurofibrillary tangles in the medial temporal lobe and neocortical parts of the brain (Breijyeh and Karaman, 2020). AD has been long associated with misfolding and aggregation of amyloid-beta peptides and Tau proteins (Penney et al., 2020; Gerrish et al., 2012; Bloom, 2014). Most cases of AD are sporadic, for which aging is the greatest risk factor, and occur later in life than the rarer familial forms (Bali et al., 2012; Lozupone et al., 2023). Hence, genetic mutations do not explain most AD cases. Furthermore, the pathological mechanisms by which amyloid-beta and Tau aggregation lead to neurodegeneration remain unknown. Mounting evidence implicates epigenetic mechanisms in AD. Below, we survey recent findings connecting sporadic AD as well as Amyloid-beta and Tau AD to epigenetic mechanisms and heterochromatin structure.
Histone PTMs are connected to neural plasticity and a variety of other important neuronal factors (Farrelly and Maze, 2019). Highlighting a potential link to AD, histone acetylation is linked to synaptic plasticity and memory formation (Guan et al., 2009; Guan et al., 2002). Furthermore, recent evidence links epigenetic alterations to AD cellular dysfunction suggesting a pathological role for epigenetic mechanisms. For instance, cortical neurons derived from sporadic AD-induced pluripotent stem cells (iPSC) displayed reduced BMI1 mRNA and protein levels (Flamier et al., 2018). BMI1 is a major component of PRC1, a H2AK119ub ubiquitin ligase complex connected to fHC (Gray et al., 2016). Interestingly, BMI1 levels were unaffected in familial AD models suggesting this change is independent of amyloid and tau toxicity (Flamier et al., 2018). BMI1 forms a complex with RING1, an E3 ligase subunit of PRC1, and is necessary for enzymatic function. The exact role of BMI1 in the PRC1 complex is not fully understood (Gray et al., 2016). Additionally, temporal cortices from sporadic AD brains displayed increased H3K9me3 levels leading to altered expression of BDNF (Brain-derived neurotrophic factor), HIST2H2BE (Histone H2B type 2E), and SYT12 (Synaptotagmin 12). BDNF and SYT12 mRNA levels were significantly reduced while HIST2H2BE mRNA levels were increased (Lee M. Y. et al., 2020). Histone variant H2BE is typically expressed in olfactory neurons (Santoro and Dulac, 2012). Notably, olfactory impairment and dysfunction, which could be connected to overexpression of H2BE, have been considered early indicators of neurodegeneration (Bhatia-Dey and Heinbockel, 2021). Lastly, ATAC-seq and RNA-seq studies on APOE carriers, a main genetic risk factor for age-linked sporadic AD, revealed overall increased chromatin accessibility within peripheral immune cells when compared to healthy controls (Ramakrishnan et al., 2024). Together, the reduction of BMI1 expression in cortical neurons and increased chromatin accessibility in peripheral immune cells of APOE carriers implicate heterochromatin changes in sporadic AD. Decreases in BMI1 levels coupled with increases in H3K9me3 levels suggest fHC decondensation and cHC hypercondensation, respectively (Flamier et al., 2018; Lee M. Y. et al., 2020). Overall, these results suggest that heterochromatin alterations are associated with sporadic AD pathology. However, additional research is needed to definitively establish this association.
Amyloid-beta (A is a component of the amyloid precursor protein (APP) and plays a key role in the pathogenesis of AD (Chen et al., 2017; Hampel et al., 2021). Mutations in APP lead to A protein aggregation, misfolding, and plaque formation (Hampel et al., 2021). A mutations have been connected to changes in the epigenetic landscape. In APP mice models, the euchromatic marks H4K12ac and H4K5ac are repressed by APP leading to downregulation of immediate early genes, Egr1, c-Fos, and Bdnf. These genes are involved in memory formation, neuronal plasticity, and neuronal growth and survival, respectively (Hendrickx et al., 2014). Of note, inhibition of BRD4, an acetylation reader, increased A levels in H4 cells expressing APP, indicating a role for histone acetylation in A pathology (Zhang et al., 2022). BRD4 has a bromodomain which enables it to bind to H4K5ac through a conserved mechanism that is not yet fully understood (Marmorstein and Zhou, 2014). Moreover, in mice hippocampal neurons, treatment with amyloid-beta oligomers (A ) increased HDAC2 levels (Gräff et al., 2012). HDACs remove acetyl groups and the negative charge from histones which promotes chromatin condensation (Teijido and Cacabelos, 2018). Interestingly, HDAC2 is responsible for deacetylating H4K5 and H4K12 further implicating Histone H4 acetylation in the pathological mechanism of A (Hendrickx et al., 2014). In another study involving mice models overexpressing mutant APP, H3K14ac and H3K9me2 levels were increased in the neocortices of the brain linking increased transcriptional activity with A toxicity. Furthermore, both histone hypermethylation and hyperacetylation were found in adult murine neurons overexpressing A (Lithner et al., 2013). Additionally, H3K9ac levels were decreased in S. cerevisiae overexpressing amyloid- 1–40 (Hugais et al., 2021). H3K9ac has been implicated in memory formation and its decrease is common in aging brains (Currais et al., 2019). Lastly, increased H3S10ph levels were found in the cytoplasm of AD patient neurons compared to patient controls, providing a link between cell cycle re-entry and AD (Ogawa et al., 2003). Overall, A overexpression and APP mutation are associated with variations in histone acetylation and methylation levels. Increased H3K14ac, H3S10ph, and H3K9me2 appear to counteract each other’s transcriptional effect but ultimately indicate genomic instability and suggest heterochromatin loss (Zheng et al., 2019; Booth and Brunet, 2016). Downregulation of Egr1, c-Fos, and BDNF suggests decreased transcriptional activity at specific genomic loci, while epigenetic changes suggest global heterochromatin loss. Interestingly, hypoacetylation of H4 coupled with increased HDAC2 expression suggests a connection between A pathology and DNA replication. Aside from playing a role in transcription, acetylation of H4 has been linked to chromatin decompaction during DNA replication (Vettese-Dadey et al., 1996; Ruan et al., 2015). Further studies looking at distinct cell types and specific genetic loci will illustrate the difference between local and global epigenetic alterations. While these observations provide strong evidence for a link between A pathology and epigenetics, further research is needed to uncover functional and mechanistic details.
Mutations in the MAPT gene, encoding for the Tau protein, are thoroughly implicated in various neurodegenerative diseases including AD (Gerrish et al., 2012; Leveille et al., 2021). These diseases have been categorized as tauopathies as Tau aggregation is a major feature of their disease mechanism (Leveille et al., 2021). Tau -a microtubule-associated protein- becomes hyperphosphorylated and causes neurofibrillary tangles in AD (Maina et al., 2018). Tau acts in a concerted manner with A where aggregation of A leads to Tau hyperphosphorylation and mislocalization of Tau leads to increases in A (Leveille et al., 2021). Several lines of evidence tie Tau to chromatin processes. Remarkably, Tau has been found to localize with cHC and be necessary for cHC stability (Sjöberg et al., 2006). Additionally, in SH-SY5Y cells, the knockdown of Tau led to decreased H3K9me2 and H3K9me3 levels further supporting a link between Tau expression and cHC stability (Maina et al., 2018). In murine neurons, the knockout of Tau protein causes disrupted H3K9me3 levels within chromocenters, interphase-occurring heterochromatin dense masses (Mansuroglu et al., 2016; Gerbi, 2018). In the same study, AD brains displayed an altered distribution of H3K9me3 in the presence of pathological phosphorylated Tau (pTau) showing that pTau causes the same epigenetic response as Tau protein loss (Mansuroglu et al., 2016; El Hajjar et al., 2019). Additionally, in postmortem AD patient brains and Tau mice models, the euchromatic mark H3K4me3 was significantly increased leading to the upregulation of genes commonly implicated in neurodegeneration such as Sgk1 (serum and glucocorticoid-regulated kinase 1) which is involved in memory formation and neuronal plasticity (Cao et al., 2020; Lang et al., 2010). Levels of the methyltransferases KMT2C/2D and SETD1A/1B were found also to be increased in AD excitatory neurons (Cao et al., 2020). Histone methyltransferases (HMTs) catalyze the transfer of methyl groups from (S-adenosylmethionine) SAM to lysine or arginine residues. Among HMTs, lysine methyltransferases (KMTs) are site-specific and contain SET domains (Bannister and Kouzarides, 2011). Increased H3K4me3 KMTs and increased PTM levels suggest that heterochromatin loss is linked to Tau AD.
Tau AD has also been connected to Lamin dysfunction. In Drosophila expressing disease-associated Tau mutants, Lamin protein levels were decreased leading to altered nuclear morphology (Frost et al., 2016). Moreover, Drosophila expressing a loss of function Lam allele recapitulated features of pathological Tau including decreased H3K9me2 and HP1 levels (Frost et al., 2016; Frost et al., 2014). In AD patient brains, Lamin B protein levels were decreased (Frost et al., 2016). In contrast, AD patient hippocampal neurons from all disease stages displayed high expression of Lamin A while Lamin B2 was dysregulated by nucleoplasm accumulation (Gil et al., 2020). In agreement with the notion that high levels of Lamin A are pathological, lower levels of Lamin A and mRNA levels of its respective gene, LMNA, are hypothesized to be protective against AD pathology (Méndez-López et al., 2019). Additionally, heterochromatin-dense chromocenters are lost in Lam mutant Drosophila neurons and AD patient brains (El Hajjar et al., 2019). The same study revealed increased H3S10ph levels in Drosophila neurons by immunostaining (Frost et al., 2016). Overall, Lamin dysfunction -induced either by mutation or pathogenic Tau- leads to epigenetic alterations suggesting heterochromatin loss. Decreased repressive PTMs and increased active PTMs occurring in AD indicate reduced heterochromatin stability. Tau pathology is specifically linked to cHC changes. Tau toxicity connects to decreases in Lamin and repressive histone PTM levels which play an important role in AD pathogenesis. Additional epigenetic investigations are imperative in this context. A graphic summary of the epigenetic alterations in AD discussed here is presented in Figure 1.
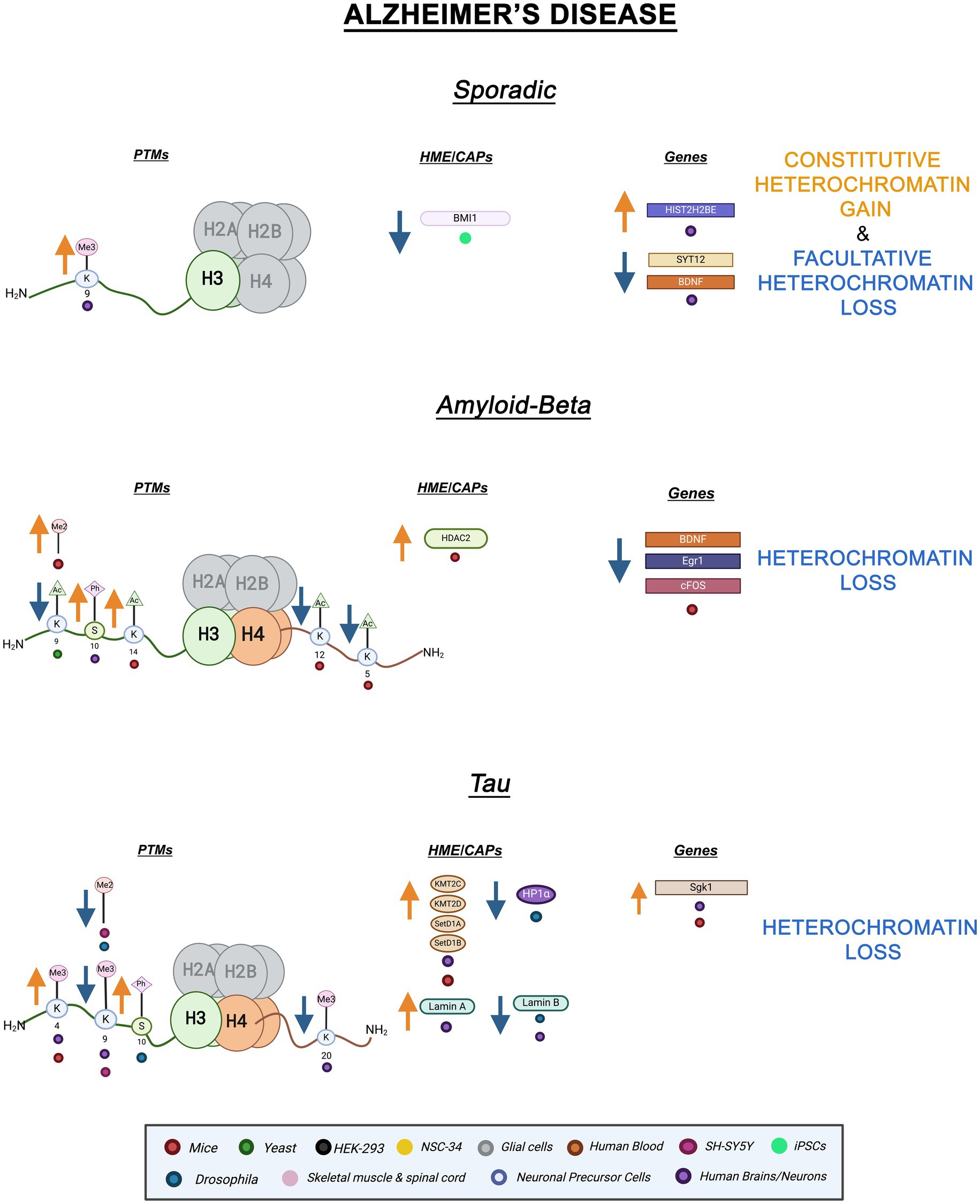
Figure 1. Epigenetic alterations linked to sporadic and familial AD. Columns indicate histone post-translational modifications (PTMs), genes, histone-modifying enzymes (HMEs), and chromatin-associated proteins (CAPs) altered in AD. Yellow arrows denote increases in the levels of proteins, genes, or histone post-translational modifications, while blue arrows denote protein, gene, or post-translational modification level decreases. Colored dots indicate the model system as depicted in the legend. Global Heterochromatin gain is indicated in yellow font, while global heterochromatin loss is indicated in blue font. Figure created with Biorender.com.
Although AD is the most common neurodegenerative disease, there is no cure; recent treatments, although now able to slow disease progression, still produce adverse effects (Knopman and Hershey, 2023). Older therapies including acetylcholinesterase inhibitors such as Donepezil and Rivastigmine aid in improving impaired cognition (Breijyeh and Karaman, 2020). New drugs such as Lecanemab, an anti-amyloid antibody, have shown success with amyloid removal in clinical trials (Knopman and Hershey, 2023). Donanemab, another anti-amyloid antibody, has slowed disease progression in clinical trials but does not reverse disease progression. Interestingly, Donanemab is more effective in patients with low Tau levels indicating a need for Tau-targeting therapies (Reardon, 2024). Despite these advances, there is room for improvement in AD treatment. Epigenetic therapies could provide great benefits to patients in the treatment and even prevention of AD.
Potential epigenetic therapies for AD are already under investigation. Targeting histone methyltransferases has been recently explored as a treatment for many neurodegenerative diseases and cancer (Marzochi et al., 2023). Treating Tau mutant mice and 5xFAD mice with WDR5-0103, a SET1/MLL family HMT inhibitor, restored H3K4me3 levels as well as synaptic function and memory (Cao et al., 2020). Additionally, treatment of FAD mice with the EHMT1/2 inhibitor, BIX01294, ameliorated memory loss and restored H3K9me2 levels (Zheng et al., 2019).
As dysregulation of acetylation marks is a key part of AD pathology, histone deacetylase inhibitors (HDACi) are some of the most promising novel epigenetic treatments. Some HDACis are already used therapeutically in other diseases such as cancer (Eckschlager et al., 2017). HDACIs lead to increased levels of histone acetylation which decreases the positive charge on histones and loosens their association with the negatively charged DNA backbone. This leads to increased chromatin accessibility and transcriptional activity (Carrier, 2013; Gallinari et al., 2007). Multiple studies in mice have shown the benefits of treatment with HDAC inhibitors. Although AD is associated with epigenetic changes mostly consistent with global heterochromatin loss, HDACIs can restore downregulated genes due to local decreases in transcription. For example, treatment of murine APP+/+ cortical neurons with the HDAC inhibitor Trichostatin (TSA) overcomes the inhibitory effects of APP and induces the transcription of immediate early gene Egr1, early growth response gene necessary for synaptic plasticity and long-term potentiation (Hendrickx et al., 2013). Moreover, treatment of APP/PS1 mice with BG45, another class I HDACi, led to decreased phosphorylated Tau expression and neuronal loss (Han et al., 2022). Another study in triple transgenic AD mice and primary cortical neurons treated with the HDACi Sulforaphane showed enhanced expression of BDNF and increased global H3 and H4 acetylation levels (Kim et al., 2017). Lastly, in presenilin 1 and 2 double knockout mice, treatment with the HDACi sodium butyrate reduced Tau hyperphosphorylation. This treatment also resulted in increased H3 acetylation and alleviated loss of contextual memory (Cao et al., 2018). In ApoE3/E4 patient astrocytes, a sporadic AD model, treatment with class I HDACis MS275 and CI994 increased mRNA expression of apoE and increased astrocytic protein secretion (Dresselhaus et al., 2018). While these findings are encouraging, HDAC inhibitors tend to lack selectivity and hence cause undesirable side effects. Of note, most epigenetic-targeting drugs -termed epidrugs- produce off-target effects due to a lack of specificity (Feehley et al., 2023). The development of highly selective and potent HDAC inhibitors with minimal off-target effects is required for the successful translation of these to the clinic (Yang et al., 2017). Combinations of different epidrugs can aid in overcoming adverse effects (Feehley et al., 2023; Majchrzak-Celińska et al., 2021). Nevertheless, HDAC inhibition remains a promising strategy for AD therapy.
Parkinson’s disease (PD) is a progressive neurodegenerative disorder affecting dopaminergic neurons in the substantia nigra (Iarkov et al., 2020). PD usually manifests with tremors and motor problems such as muscular rigidity and gait instability (Song et al., 2023). Much like AD, PD is mostly sporadic with influences from complex environmental factors such as stress and aging (Chai and Lim, 2013). Familial PD is linked to mutations in many genes including SNCA and PRKN which encode for α-synuclein ( -SYN) and Parkin, respectively (Stefanis, 2012; Castelo Rueda et al., 2021). Akin to AD, the mechanisms linking protein aggregation to cell death remain incompletely characterized. Epigenetic factors have been thoroughly implicated in all forms of PD. Chromatin processes could potentially reveal yet undiscovered PD mechanisms.
Aside from environmental factors such as heavy metal exposure and drug use (Ball et al., 2019), sporadic PD has been associated with certain mutations in genes such as SNCA and LRRK2 (leucine-rich repeat kinase 2) (Chai and Lim, 2013; Tolosa et al., 2020). SETD1A, a H3K4 methyltransferase implicated in neurodevelopmental and neuropsychiatric disorders, has been identified as a sporadic PD risk gene but its pathological involvement is not yet understood (Spataro et al., 2015; Witoelar et al., 2017; Chong et al., 2022; Booms et al., 2024). In sporadic PD iPSC-derived dopaminergic neurons, H3K4me3 – a euchromatin mark- is enriched at the SNCA promoter (Guhathakurta et al., 2021). Moreover, in sporadic PD neuronal precursor cells (NPCs) and dopaminergic neurons, mitochondrial dysfunction leads to the reduction of H3K9me3 and H3K27me3 levels due to metabolic dysregulation (Schmidt et al., 2023). In peripheral whole blood cells of sporadic PD patients, transcriptomics revealed differential expression of CBX5, the gene encoding HP1 . CBX5 mRNA levels were decreased in PD patients when compared to healthy controls. Expression of HP1’s interacting partner, DNMT3A, and additional chromatin interacting proteins such as methyltransferase, Enhancer of zeste homolog 1 (Ezh1) were also decreased in PD patient blood cells (Calligaris et al., 2015). Furthermore, in a sporadic PD-like mice model, En1+/−, there was a reduction in DAPI-dense regions of heterochromatin, and a decrease in heterochromatin marks H3K27me3 and H3K9me3 (Rekaik et al., 2015).
As in AD, PD is linked to Lamin, but this link is not yet fully understood. In sporadic PD, substantia nigra pars compacta astrocytes displayed reduced nuclear Lamin B1 protein levels (Chinta et al., 2018). Decreased Lamin levels suggest a connection between heterochromatin instability and PD. Overall, these alterations implicate heterochromatin loss, both cHC and fHC, in sporadic PD pathology.
-SYN is a member of the synuclein protein family which predominately functions as neuronal proteins, localizing at the presynaptic terminals (Stefanis, 2012). Mutations in SNCA lead to misfolding and aggregation of -SYN (Stefanis, 2012; Song et al., 2023). These aggregates form Lewy bodies, cytoplasmic inclusions found in the substantia nigra that are a hallmark of PD (Song et al., 2023; Gomperts, 2016). -SYN overexpression has been connected to changes in histone methylation and heterochromatin composition. Remarkably, both transgenic Drosophila and SH-SY5Y -SYN overexpression models display enhanced H3K9me2 levels and HP1 expression (Sugeno et al., 2016). In the same study, -SYN expression increased EHMT2 and SUV39H2 (H3K9 methyltransferases) levels while EZH1 (H3K27 methyltransferase homolog of EZH2) levels were slightly decreased (Sugeno et al., 2016; Margueron et al., 2008). Additionally, differentiated SH-SY5Y cells overexpressing -SYN displayed increased protein levels of lysine demethylases KDM1B, KDM4B, and KDM4C which target H3K4 and H3K9 methylation, respectively (Hyun et al., 2017; Sugeno et al., 2016). Histone demethylases (HDMs) have different catalytic mechanisms and specificity. Interestingly, KDM1 (LSD1) mechanism utilizes amine oxidation to demethylate H3K4 mono- and di-methylated residues. It is also inactive against trimethylated residues. On the other hand, KDM4B (JMJD2B) undergoes hydrogen bonding within its binding pocket and exhibits specificity for H3K9 di and tri-methylation (Marmorstein and Trievel, 2009). The site and modification specificity of HDMs makes them an attractive epigenetic target.
-SYN expression has also been linked to disturbances in histone acetylation. In murine dopaminergic neurons, cytosolic -SYN expression reduces the activity of histone acetyltransferase p300 (Jin et al., 2011). Interestingly, p300 is responsible for acetylating a variety of proteins, including Smad1, a major intracellular effector of Bone morphogenic proteins (BMP), which play a key role in neurogenesis (Pearson et al., 1999). HATs utilize different catalytic mechanisms depending on cellular context. Of note, distinctly from other HATs, p300 uses a tyrosine residue for acid catalysis (Marmorstein and Trievel, 2009). The variation in mechanisms of histone acetylation may allow for enhanced specificity for epigenetic drugs. Furthermore, in S. cerevisiae overexpressing -SYN, H3K36me2, and H2BT129ph levels are significantly decreased. In yeast, these PTMs are responsible for transcriptional elongation and DNA damage, respectively (Chen et al., 2018). Altogether, these findings show a link between -SYN expression histone modification and heterochromatin dysregulation in various PD models. Increased repressive PTMs and methyltransferase levels as well as decreased activating PTMs suggest heterochromatin gain, specifically cHC, in -SYN pathology. Nonetheless, this has not been directly shown yet.
Parkin, a ubiquitin ligase, is implicated in various cellular processes such as inflammation and mitochondrial biogenesis (Castelo Rueda et al., 2021). Mutations in the gene, PRKN, are the most common cause of early-onset PD (Castelo Rueda et al., 2023). Parkin also has neuroprotective properties and regulates the accumulation of -SYN (Jęśko et al., 2019). The role of epigenetic factors such as miRNA and DNA methylation have been explored within the context of PRKN mutations, but histone PTMs and chromatin structure changes remain mostly uncharacterized (Tsalenchuk et al., 2023). Emerging evidence points to a role for histone PTMs in Parkin PD. For instance, genome-wide hyperacetylation of H3K27 coupled to a decrease in class III HDAC, SIRT1, activity and increased histone acetyltransferase, p300, activity were revealed within the PRKN gene in PD brains (Toker et al., 2021; Milazzo et al., 2020). Histone hypoacetylation and decreased p300 activity have been found in SNCA PD models as well as other neurodegenerative diseases (Beaver et al., 2020; Valor et al., 2013). Both hypo- and hyperacetylation can alter homeostasis and proteostasis leading to disease pathology (Kabir et al., 2023). PRKN-linked hyperacetylation on H3K27 suggests an increase in transcriptional activation and a loss of fHC, hinting at an important mechanistic role for H3K27ac in PD pathology. Figure 2 illustrates the epigenetic alterations linked to familial and sporadic PD discussed here.
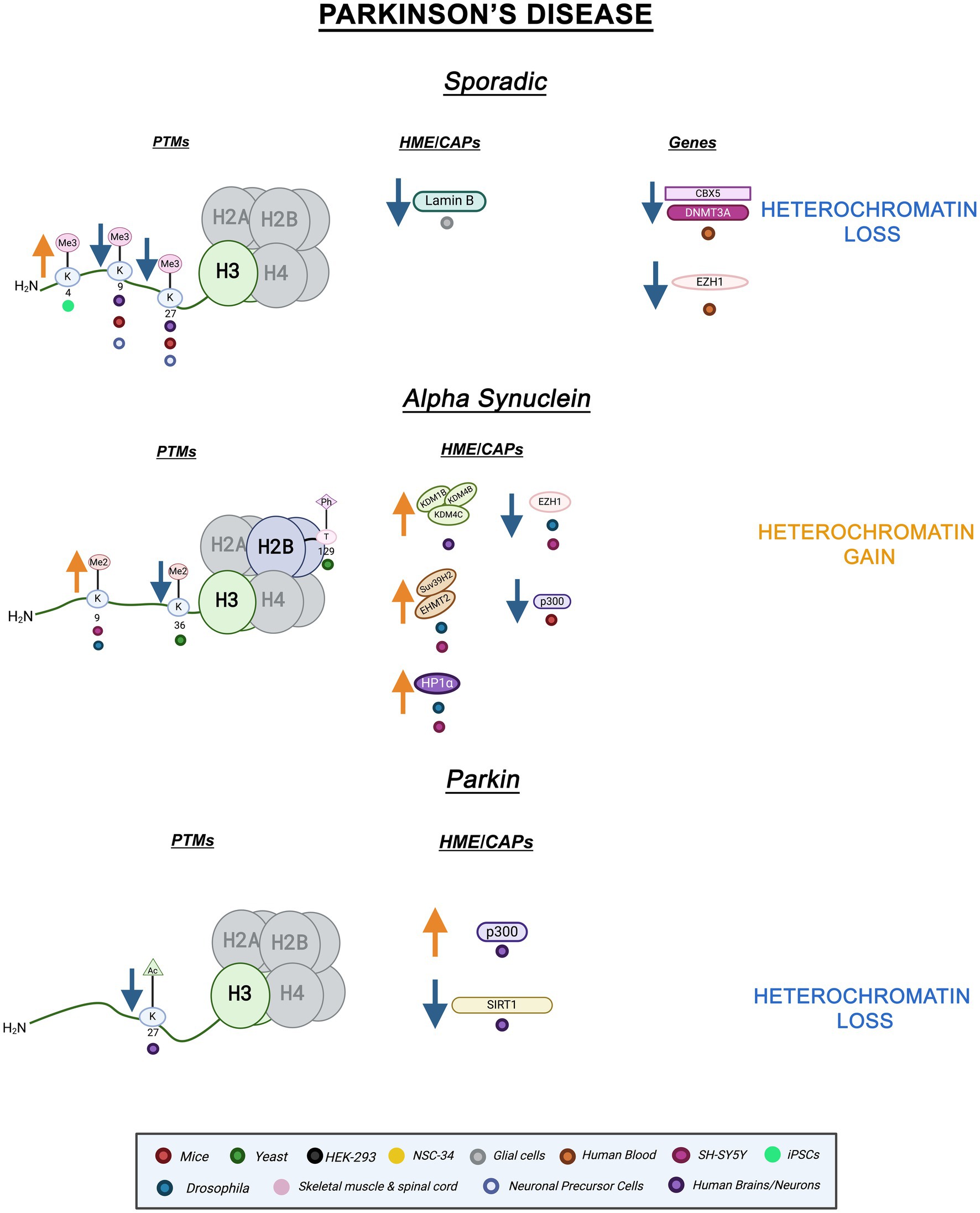
Figure 2. Epigenetic alterations are linked to sporadic and familial PD. Columns indicate histone post-translational modifications (PTMs), genes, histone-modifying enzymes (HMEs) and chromatin-associated proteins (CAPs) altered in PD. Columns are omitted when no changes are revealed. Yellow arrows denote increases in the levels of proteins, genes, or histone post-translational modifications, while blue arrows denote protein, gene, or post-translational modification level decreases. Colored dots indicate the model system as depicted in the legend. Global heterochromatin gain is indicated in yellow font, while global heterochromatin loss is indicated in blue font. Figure created with Biorender.com.
Akin to AD, PD lacks an effective cure. To promote dopamine synthesis, treatment of PD currently involves the dopamine precursor, L-DOPA. Unfortunately, L-DOPA leads to side effects such as motor complications, emotional and psychiatric disturbances (Fox and Lang, 2008; Salat and Tolosa, 2013). For those reasons, novel treatments and preventative measures for PD are quite needed.
As expression of -SYN is modulated by histone methylation, this modification is an attractive therapeutic target (Guhathakurta et al., 2021). As mentioned previously, KMTs and KDMs are already in the clinic for cancer therapy (Marzochi et al., 2023; Morera et al., 2016). Treating SH-SY5Y cells overexpressing -SYN with the histone-lysine n-methyltransferase 2 (EHMT2) inhibitor, UNC0638, restored expression levels of L1CAM and Snap25, genes repressed by increased H3K9me2 (Sugeno et al., 2016). Treatment of PD mice with GSK-J4, a histone demethylase inhibitor able to cross the blood–brain barrier, rescued H3K27me3 alterations conferring neuroprotective effects. SH-SY5Y cells also display increased H3K27me3 after GSK-J4 treatment (Mu et al., 2020). Furthermore, in sporadic PD brains and iPSC models, CRISPR-Cas9 recruited Jarid1A, a histone demethylase, was able to reduce -SYN, and restore H3K4me3 levels at the SNCA locus (Guhathakurta et al., 2021).
HDACi treatment ameliorates -SYN toxicity. Both transgenic Drosophila and SH-SY5Y cells treated with sodium butyrate displayed reduced -SYN toxicity (Kontopoulos et al., 2006). Histone acetyltransferase (HAT) modulators -activators of p300 specifically- could alleviate the reduction of p300 activity triggered by -SYN expression (Jin et al., 2011); however, these compounds are not yet ready for clinical translation due to poor solubility and permeability (Teijido and Cacabelos, 2018; Jin et al., 2011). Epigenetic treatments remain unexplored for PRKN-associated PD. Nevertheless, the alterations in histone post-translational modifications reviewed above suggest promising therapeutic targets in this context.
Frontotemporal dementia (FTD) is characterized by degeneration of neurons in the frontal and temporal lobes of the brain. One of the most common dementias, FTD manifests with personality and behavioral changes as well as language difficulties (Khan and De Jesus, 2023). FTD exists on a disease continuum with amyotrophic lateral sclerosis (ALS), a neurodegenerative disease affecting motor neurons throughout the motor cortex, brainstem, and spinal cord (Mead et al., 2023). ALS symptoms typically include progressive muscular weakness (Abramzon et al., 2020). FTD/ALS has clinical and pathological overlap with patients developing symptoms attributed to either disease or both diseases (Cividini et al., 2022).
Much like other neurodegenerative diseases, most cases of FTD/ALS are sporadic. Familial FTD/ALS is linked to mutations in many genes (Abramzon et al., 2020). SOD1, TARDBP, FUS, and C9orf72 are some of the key genes implicated in FTD/ALS. Mutations in each of these genes lead to aggregation and misfolding of their respective proteins (Gendron et al., 2013; Schwartz et al., 2014; Starr and Sattler, 2018). A growing body of data links epigenetic mechanisms, including histone PTM and heterochromatin alterations, to FTD/ALS pathology.
Epigenetic mechanisms are associated with both sporadic and familial FTD/ALS (Paez-Colasante et al., 2015). For instance, peripheral blood mononuclear cells (PBMCs) from sporadic ALS patients displayed altered ATAC-seq profiles revealing increased chromatin accessibility compared to healthy controls. The differentially accessible genes were enriched in those involved in neuronal health and function (Kühlwein et al., 2023). In sporadic ALS postmortem brains and spinal cords, HDAC2 mRNA was significantly increased while HDAC11 mRNA was decreased (Janssen et al., 2010). HDAC2 is a class I HDAC that can deacetylate lysine residues on histone tails and non-histone targets. On the other hand, HDAC11 is a class IV HDAC associated with antigen-specific T-cell response and is not as understood as the other HDACs (Gallinari et al., 2007; Milazzo et al., 2020; Janssen et al., 2010). Increased HDAC2 and decreased HDAC11 suggest reduced transcriptional accessibility and immune response, respectively (Janssen et al., 2010). Interestingly, post-mortem sporadic ALS spinal motor neurons showed depleted chromatin remodeling complex Brg1/Brm Associated Factor (BAF). The BAF complex interacts with acetylated histones through its tandem PHD finger domain (Bannister and Kouzarides, 2011). BAF loss was also observed in familial and sporadic cases with C9orf72 mutations hinting at its importance in disease processes (Tibshirani et al., 2017). DNA methylation changes were observed in sporadic FTD, but histone PTMs and chromatin structure have not been thoroughly examined in this context (Galimberti et al., 2013). Overall, while there is evidence for heterochromatin loss in sporadic ALS, additional research is needed to definitively ascertain this feature (Fisher et al., 2023).
Mutations in the Cu/Zn superoxide dismutase-1 (SOD1) gene is one of the most common mutations in both familial and sporadic ALS (Parobkova and Matej, 2021). These mutations lead to protein aggregation and mitochondrial mislocalization, but the exact pathological mechanism is not yet understood (Parobkova and Matej, 2021; Berdyński et al., 2022). SH-SY5Y cells expressing SOD1 mutants G93A and H80R as well as SOD1 G39A transgenic mice showed decreased H3S10ph-K14ac, a combinatory euchromatic mark, and H3K4me2, another modification connected to gene transcription. Importantly, the reduction in the levels of H3S10ph-K14ac was directly linked to increases in SOD1 expression (Masala et al., 2018). In another study using the same SOD1 murine model, DNMT3A – a DNA methylase – levels were reduced in both skeletal muscle and spinal cord mitochondria (Wong et al., 2013). Of note, HP1𝛼 has long been known to interact and associate with DNMTs presumably linking SOD1 proteinopathy with heterochromatin alterations (Fuks et al., 2003). Furthermore, in SH-SY5Y cells overexpressing SOD1 G93A, mutant SOD1 was found to strongly associate with chromatin. While wild-type SOD1 also interacts with chromatin, mutant SOD1 has higher affinity suggesting interactions with DNA and nucleosomes are increased by pathogenic mutations (Barbosa et al., 2010). Lastly, NSC-34 cells overexpressing SOD1 G93A showed increased immunoreactivity and protein levels of lysine-specific histone demethylase-1 (LSD1) and decreased immunoreactivity and protein levels of H3K4me2 (Choi et al., 2022). Altogether, overexpression of pathogenic SOD1 mutants revealed decreases in transcriptionally active histone PTMs, H3S10ph-K14ac, and H3K4me2 suggesting a role for heterochromatin gain and loss of transcriptional activity within SOD1 pathology. These results lay the foundation for further epigenetic studies in SOD1 models that will definitively link heterochromatin changes to SOD1 pathology.
Mutations in TARDBP, which encodes for TDP-43 -a gene expression regulator- lead to protein mislocalization, dysfunction, and nuclear depletion (Gendron et al., 2013; Yan et al., 2014; Liu et al., 2019). The overwhelming majority of FTD/ALS cases, including both sporadic and familial cases, display TDP-43 inclusions (Fisher et al., 2023; Ducharme et al., 2024). In a TDP-43 mutant murine model, TDP-43 cytoplasmic overexpression led to differential expression and aberrant polyadenylation of histones. Canonical histone genes were upregulated while variant histone genes were downregulated (Amlie-Wolf et al., 2015). Downregulated histone variants suggest the prevention of critical histone-swapping events (Henikoff and Smith, 2015). Interestingly, in the frontal cortex of FTD patients cytoplasmic TDP-43 aggregates led to reduced levels of CHD2, an ATP-dependent chromatin remodeling factor. Additionally, in the same study, HEK293 cells overexpressing TDP-43 displayed reduced nucleosome clearance and decreased expression of various heat shock genes such as HSPA1A and HSPA6, suggesting a role for TDP-43 in regulating chromatin dynamics (Berson et al., 2017). Furthermore, in FTD murine models, TDP-43 pathogenesis has been linked to reduced HDAC1 (class I deacetylase) activity (Han et al., 2022; Wu et al., 2020). Our own work has revealed histone PTM perturbations in yeast FTD/ALS models. In yeast overexpressing TDP-43, H3K36me3 levels decrease, while H4K12ac and H4K16ac levels increase (Chen et al., 2018). As previously mentioned, methylation of H3K36 is involved in transcriptional elongation as well as chromatin structural maintenance of coding regions while H4K16ac is involved in transcriptional activation. Together, H3K36me3 decreases and H4K16ac increases are indicative of enhanced transcriptional activity and decreased chromatin stability (Chen et al., 2018). Altogether, TDP-43 proteinopathy leads to alterations in histone PTMs and chromatin modifiers indicative of genomic instability and heterochromatin breakdown.
Mutations in FUS, an RNA-binding protein, are also notably implicated in FTD/ALS. Cytoplasmic and nuclear FUS aggregates occur in the motor and cortical neurons of FTD/ALS patients (Schwartz et al., 2014; Mackenzie et al., 2011). Mislocalization of nuclear FUS into cytoplasmic inclusions has been linked to epigenetic dysregulation in FTD/ALS. In mice motor neurons, when FUS mislocalizes to the cytoplasm, protein arginine methyltransferase 1 (PRMT1) is depleted from the nucleus. PRMT1 is responsible for di-methylating arginine 3 on histone H4 (H4R3me2) which is a mark typically associated with transcriptional activity (Tibshirani et al., 2015). Arginine methyltransferases utilize similar catalytic mechanisms to lysine methyltransferases. Interestingly, these enzymes exhibit some specificity in how SAM and the histone come in contact with the catalytic site. This suggests selectivity that can be helpful for epigenetic targeting (Bannister and Kouzarides, 2011). In the same study, H4R3me2 was revealed to be decreased in motor neurons displaying cytoplasmic FUS. Combinatory marks H3K9ac-K14ac were also decreased (Tibshirani et al., 2015). Loss of these transcriptionally active PTMs, especially H4R3me2, has been thoroughly implicated in chromatin hypercondensation and increased heterochromatin formation (Huang et al., 2005). In yeast overexpressing FUS, we find decreases in H3K14ac, H3K56ac, H4R3me2, and H3S10ph levels which are all linked to transcriptional activation (Chen et al., 2018). Additionally, H3K56ac plays a role in DNA replication and genomic stability therefore its decrease would link FUS mislocalization with genomic instability and errors in replication (Yuan et al., 2009). Overall, pathogenic FUS leads to decreases in activation-linked PTMs and histone-interacting enzymes. These findings suggest a connection between FUS pathology, transcriptional silencing, and potentially heterochromatin gain.
C9orf72, the most common genetic cause of FTD/ALS, pathologically includes a hexanucleotide repeat expansion (G4C2) which leads to neurodegeneration through several mechanisms (Zhu et al., 2020). One such mechanism invokes the production of five toxic dipeptide repeat proteins (DPRs) via repeat-associated non-AUG translation: proline-alanine (PA), glycine-proline (GP), glycine–alanine (GA), glycine-arginine (GR), and proline-arginine (PR) (Taylor et al., 2016). DPRs aggregate and ultimately cause neuronal death (Freibaum and Taylor, 2017).
Expanding evidence links DPR pathology with epigenetic alterations. Histone trimethylation causes reduced C9orf72 mRNA levels in both the frontal cortex and cerebella of c9 FTD/ALS patients. Of note, mutant C9orf72 gene binding to H3K9me3 and H3K27me3 was detected in the blood of C9 FTD/ALS patients (Belzil et al., 2013). This suggests epigenetic alterations could be useful as diagnostic biomarkers. Frontal cortices and cerebellum of repeat expansion carriers also revealed increased H3K79me3 (Belzil et al., 2013).
Remarkably, heterochromatin alterations have been directly linked to poly PR toxicity. Upon poly PR expression in mice, this DPR localizes to heterochromatin and causes an increase in both the heterochromatic mark H3K27me3 and the euchromatic mark H3K4me3. Remarkably, poly PR also decreases HP1 expression in mice and alters HP1 phase separation properties in vitro (Zhang et al., 2019). Additionally, C9BAC mice astrocytes show depletion of the heterochromatin mark H3K9me3 (Jury et al., 2020). Very recently, epigenomic studies of c9ALS patients have revealed increased H3K27ac, an activating histone mark. Furthermore, c9ALS patients displayed overall increased chromatin accessibility in non-neuronal cells such as glia and astrocytes at distal regions of differentially expressed ALS genes, such as ERGIC1 and MYOE1, suggesting heterochromatin decondensation (Li et al., 2023). In c9FTD/ALS patient brains, EZH2 expression was decreased when compared to healthy controls. Notably, EZH2 was revealed to form insoluble aggregates in c9 brains suggesting a mechanism for decreased protein levels (Wang et al., 2019). In C9orf72 Drosophila, C9 HRE’s lead to redistribution of proteins due to errors in nucleocytoplasmic transport (Ortega et al., 2020). Histones were found to be enriched in the nucleus as expected but interestingly, PRMT1 was cytosolically enriched evidencing problems with nuclear transport (Ortega et al., 2020; Herrmann and Fackelmayer, 2009). Conversely to AD and PD, altered nuclear morphology due to changes in Lamin expression and invaginations are not pathological in c9FTD/ALS. In C9 FTD/ALS iPSCs, nuclear Lamin B1 invaginations were revealed to occur with age, independently of C9 HREs, and occur similarly in healthy and disease samples (Coyne and Rothstein, 2021).
All in all, changes in active and repressive PTMs coupled with disruption of HP1 and EZH2 suggest heterochromatin decondensation is a key factor in c9FTD/ALS. These results suggest cHC loss but fHC involvement should not be ruled out. EZH2 and Suz12, subunits of PRC2, are necessary for HP1 stability and might affect cHC formation and propagation (Boros et al., 2014). Altogether, both hypercondensation and decondensation of heterochromatin are implicated in FTD/ALS depending on gene mutation. A graphic summary of the epigenetic alterations in FTD/ALS covered in this review is presented in Figure 3. Additionally, Table 1 summarizes all the ND-relevant epigenetic factors including histone-modifying enzymes and chromatin remodeling complexes presented here.
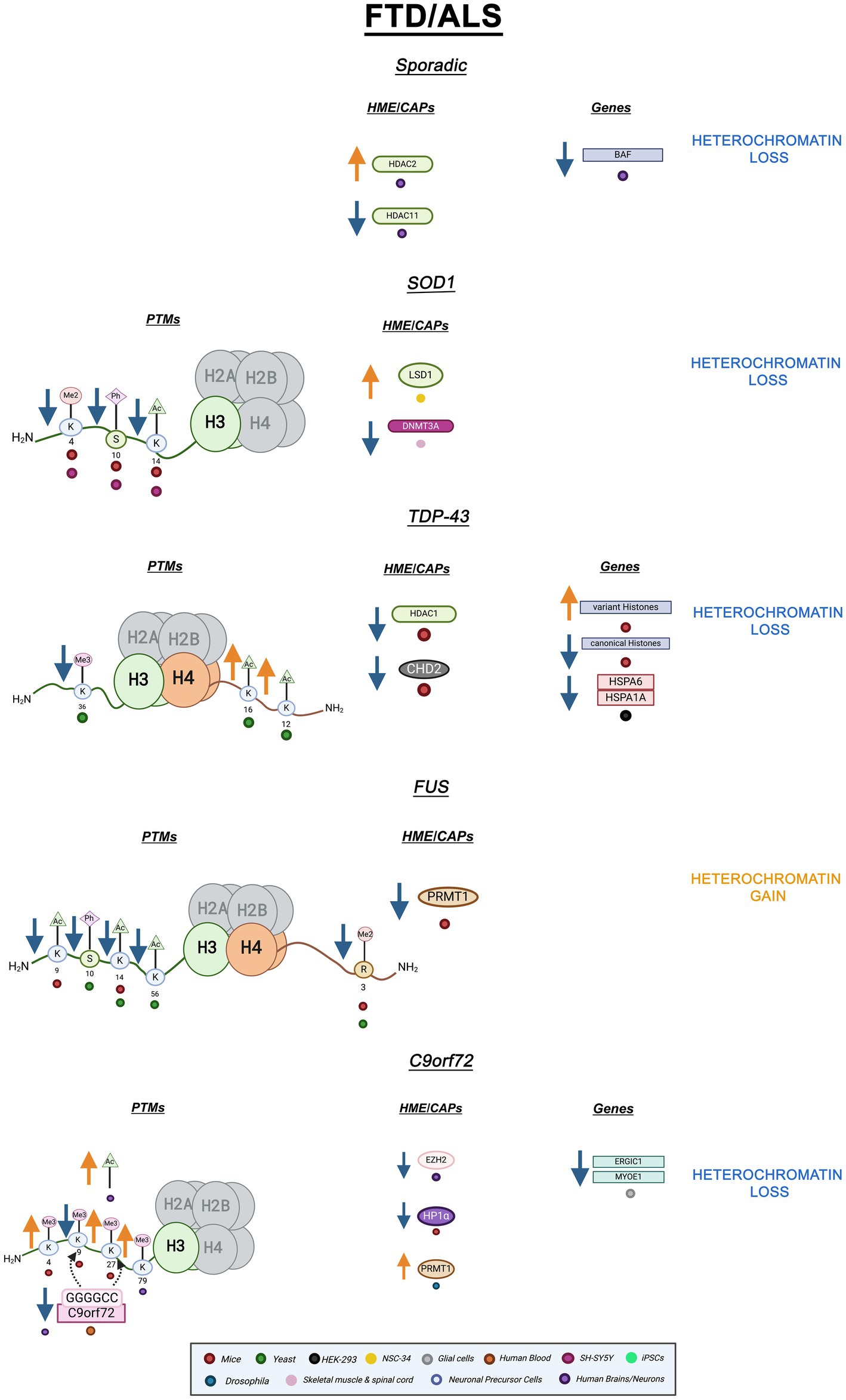
Figure 3. Epigenetic alterations are linked to sporadic and familial FTD/ALS. Columns indicate histone post-translational modifications (PTMs), genes, histone-modifying enzymes (HMEs), and chromatin-associated proteins (CAPs) altered in PD. Columns are omitted when no changes are revealed. Yellow arrows denote increases in the levels of proteins, genes, or histone post-translational modifications, while blue arrows denote protein, gene, or post-translational modification level decreases. Black arrows denote binding interactions. Colored dots indicate the model system as depicted in the legend. Global heterochromatin gain is indicated in yellow font, while global heterochromatin loss is indicated in blue font. Figure created with Biorender.com.
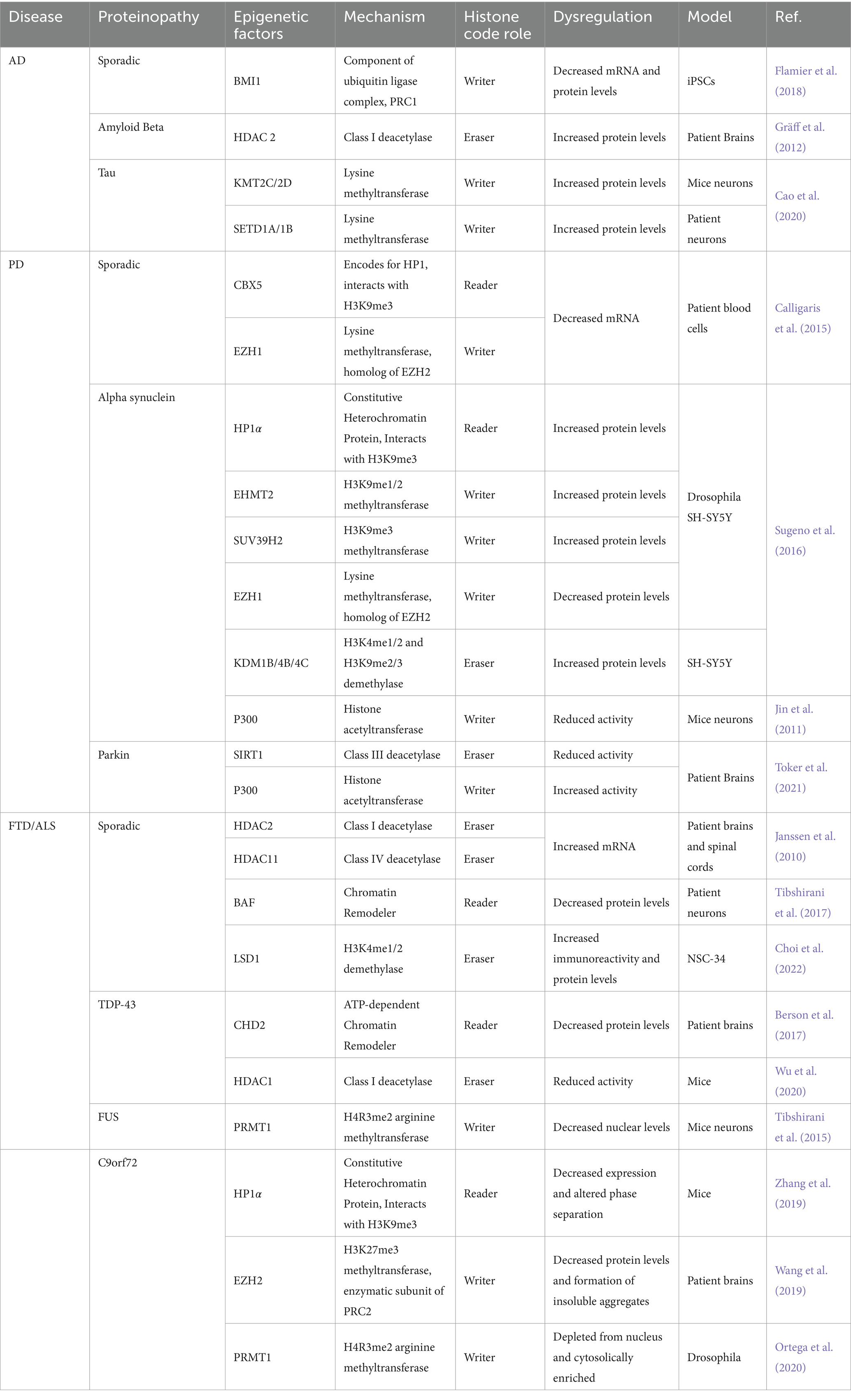
Table 1. Histone interacting proteins involved in AD, PD, and FTD/ALS.
Akin to other NDs, FTD/ALS is incurable but there are a few treatments available. Edaravone is an intravenous drug approved for ALS known to work against oxidative stress. It is found to be most effective during the early stages of the disease (Cho and Shukla, 2020). Riluzole, an FDA-approved ALS treatment, has provided patients with a slight increase in survival rates. Tofersen, an antisense oligonucleotide gene therapy, significantly reduces SOD1 mutant proteins in motor neurons and slows disease progression making it a very promising new therapy (Saini and Chawla, 2024). For FTD, antidepressants such as trazodone and NMDA receptor antagonists are typically used as symptomatic treatments (Mollah et al., 2024). Recent promising therapies for FTD include Latozinemab, targeting GRN-FTD, another FTD-associated gene, reduced progranulin levels (Ward et al., 2024; Crescioli et al., 2024). Although improved therapies have been developed in the last few years, treatment options able to stop FTD/ALS progression are still desperately needed (Mead et al., 2023).
Epigenetic treatments have begun to be utilized in FTD/ALS. Sodium phenylbutyrate is a histone deacetylase inhibitor which improves dysregulation of histone acetylation levels after treatment (Cudkowicz et al., 2009). Approved by the FDA in 2023 as a treatment for ALS, Relyvrio contains sodium phenylbutyrate and taurursodiol and reduces neuronal death in experimental models and clinical trials with only gastrointestinal problems as side effects (Beninger, 2023; Paganoni et al., 2020). However, it was pulled from the market in 2024 due to a lack of efficacy. Curiously, epigenetic channels are not thought to be involved in its mechanism of action. By restoring histone acetylation levels, TSA bypasses the toxic effect of FUS aggregation in yeast (Bennett et al., 2021). Additionally, TSA treatment improved motor neuron survival in SOD1 mice (Yoo and Ko, 2011). In SH-SY5Y cells expressing mutant TDP-43, treatment with the HDACis 4-phenylbutyrate, TSA, and sodium butyrate all increased cell survival and decreased TDP-43 toxicity (Sanna et al., 2020). Transgenic FUS+/+ mice treated with the HDACi ACY-738 displayed extended life, slowed disease progression, and restored H3K9ac and H3K14ac levels (Rossaert et al., 2019).
In SOD1 mice models, treatment with spermidine led to changes in chromatin structure and remodeling as well as changes in gene expression by targeting lysine-specific histone demethylase 1 (LSD1), the demethylase responsible for removing methyl groups from H3K4me2, leading to prolonged survival (Choi et al., 2022). Lastly, treatment with an inhibitor of BRD4 -bromodomain containing histone acetylation reader- JQ1, was shown to reduce cytotoxicity of poly-PR by blocking nuclear aggregation and leading to histone cytoplasmic accumulation in Hela cells treated with (PR)20 (Chen et al., 2024). Targeting epigenetic factors provides promising drug targets to restore altered histone acetylation and methylation and restore the expression of several histone-interacting proteins. These results highlight the importance of the inclusion of epigenetic mechanisms in neurodegeneration research. Further interrogation of these mechanisms will allow for discovery of attractive targets for chemical interference within these complex disease pathways. Epigenetic drugs remain a promising avenue to alter pathological manifestations of NDs that will hopefully halt disease progression. Table 2 summarizes all epigenetic treatments included in this review.
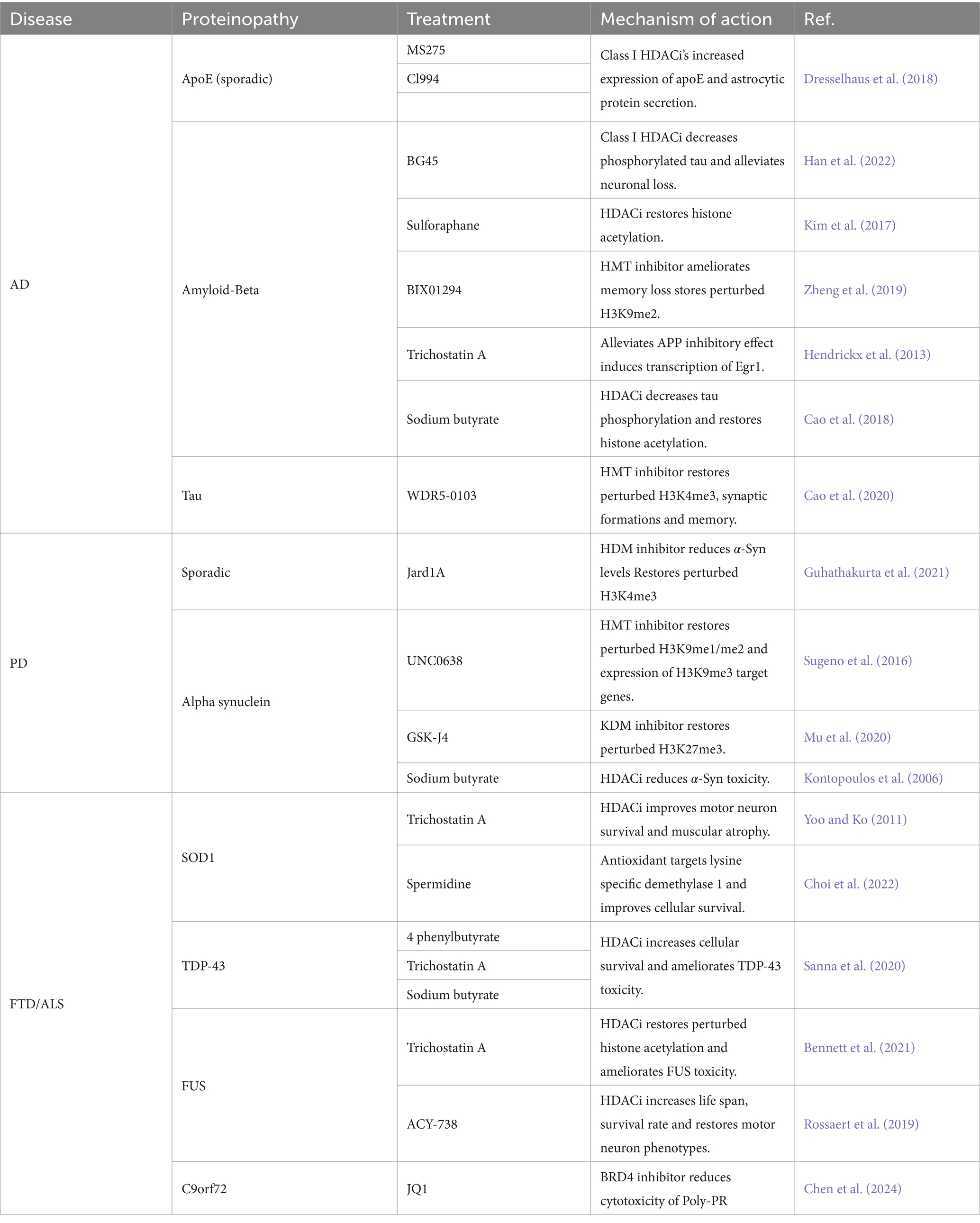
Table 2. Potential epigenetic treatments for neurodegenerative diseases.
Changes to the epigenetic landscape including histone PTM perturbations, alterations in the expression of histone-modifying enzymes (HMEs) and chromatin remodelers, as well as heterochromatin loss and gain are all implicated in neurodegeneration and provide a multitude of epigenetic targets. Altogether, the findings reviewed here illuminate the complexity and importance of the epigenetic landscape in NDs. The changes in histone PTMs and HMEs linked to heterochromatin, whether it be a heterochromatic loss (decondensation) or gain (hypercondensation), occur in the context of different genetic mutations in AD, PD, and FTD/ALS. Loss of heterochromatin may allow for cell cycle reentry leading to neurodegeneration (Frost et al., 2014; Wu et al., 2024). Post-mitotic neurons in the brain re-enter the cell cycle and reveal increased expression of a variety of ND risk genes. Increased cell cycle re-entry is typically observed in AD and PD pathology and is not a consequence of aging, connecting this occurrence with ND pathways (Wu et al., 2024).
It is important to note that while certain NDs appear to be associated with global heterochromatin loss, global chromatin changes do not necessarily mirror local changes at specific genomic loci. Therefore, certain genes can be downregulated even when heterochromatin loss indicates global increased transcriptional activity. For example, in A linked AD, decreased expression of BDNF, Egr1, and c-Fos occurs even though there is a global heterochromatin loss. Differential histone PTM changes may be occurring at specific gene loci. Epigenomic techniques such as ChIP-seq can give more insight by linking global changes to specific genes (Zheng et al., 2019). Additionally, it is also possible that differential gene expression linked to epigenomic changes occurs in different cell types. Single-cell epigenomic investigations can aid in uncovering localized changes and lead to the development of more precise therapies. Moreover, open chromatin caused by heterochromatin loss is connected to increased DNA damage which plays a role in cell cycle re-entry of neurons suggesting a link between heterochromatin loss and neurodegeneration (Kim and Tsai, 2009). Additionally, heterochromatin hypercondensation may contribute mechanistically through repression and downregulation of genes with important cellular roles (Lee M. Y. et al., 2020). As discussed here, both hypercondensation and decondensation are connected to pathological manifestation of these diseases.
Altogether, these findings underscore the crucial need for the inclusion of epigenetic and chromatin structure studies in neurodegeneration. Whether these epigenetic mechanisms are causative or downstream effects of disease pathology, they nevertheless provide a promising avenue for novel treatments to impact cellular outcomes and survival. Epigenetic targets are attractive because while they are heritable, they are also pharmaceutically accessible and reversible. Due to their dynamic nature, they also have the potential to be used in diagnostics (Kelly et al., 2010). In conjunction with epidrugs, epigenetic editing offers a promising novel therapeutic strategy. Due to its precision and specificity, epigenetic editing bypasses the off-target effects usually associated with epidrugs (Ueda et al., 2023). The development of CRISPR-Cas9 has increased the versatility of epigenetic editing allowing for more epigenetic targets (Ueda et al., 2023; Liu and Jaenisch, 2019; Nakamura et al., 2021). For example, CRISPR-Cas9 can potentially be used to study and target other epigenomic factors such as HP1 phase separation and epitranscriptomic alterations (Nakamura et al., 2021). In addition to CRISPR technologies, zinc finger proteins (ZFP) and TALENs approaches possess the ability to fuse with epigenetic modifiers and allow for site specificity (Ueda et al., 2023). Although epigenetic editing expands the reach of epigenetic therapies, there are still many important considerations. CRISPR/Cas9 systems can induce background mutations, R-loop formations, and even cleave DNA (Ueda et al., 2023). Further work is necessary to understand the implications of utilizing epigenetic editing and to develop better technologies with less off-target effects (Sasaki-Honda et al., 2023).
While the results reviewed here illustrate the importance of epigenetic mechanisms in disease, a multitude of questions still remain. Are heterochromatin and histone PTM changes linked to specific genetic mutations recapitulated within different cell types? Can heterochromatin and histone PTM alterations be utilized as both diagnostic biomarkers and therapeutic targets? Uncovering disease pathways will be pivotal in understanding how heterochromatin changes arise or whether these changes are linked to pathological manifestations. Additional investigations into epigenomic and transcriptomic profiles of different cell types such as glial cells and diverse neuronal subtypes will help paint a full picture. Furthermore, continued investigations in sporadic models will also aid in further uncovering the differences and similarities between sporadic and familial diseases. Although this research area is full of promise, a more precise understanding of the epigenetic mechanisms at play in neurodegeneration is necessary to develop improved, clinically applicable epigenetic therapies.
RF: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing. MT: Conceptualization, Data curation, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Brooklyn College, The Professional Staff Congress of CUNY, and an NIH NINDS Research Enhancement Award (1R15NS125394–01) supported MT. The Graduate Center of CUNY, Brooklyn College and an NIH NINDS and NIA AD/ADRD Research Supplement to Promote Diversity (3R15NS125394–01S1) supported RF.
We thank Dr. Samantha Cobos, Rania Frederic, Arefa Yeasmin, and Zoe Jalkut for their critical review of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abramzon, Y. A., Fratta, P., Traynor, B. J., and Chia, R. (2020). The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 14:42. doi: 10.3389/fnins.2020.00042
Amlie-Wolf, A., Ryvkin, P., Tong, R., Dragomir, I., Suh, E., Xu, Y., et al. (2015). Transcriptomic changes due to cytoplasmic TDP-43 expression reveal dysregulation of histone transcripts and nuclear chromatin. PLoS One 10:e0141836. doi: 10.1371/journal.pone.0141836
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Bali, J., Gheinani, A. H., Zurbriggen, S., and Rajendran, L. (2012). Role of genes linked to sporadic Alzheimer’s disease risk in the production of β-amyloid peptides. Proc. Natl. Acad. Sci. 109, 15307–15311. doi: 10.1073/pnas.1201632109
Ball, N., Teo, W.-P., Chandra, S., and Chapman, J. (2019). Parkinson’s disease and the environment. Front. Neurol. 10:218. doi: 10.3389/fneur.2019.00218
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Barbosa, L. F., Cerqueira, F. M., Macedo, A. F. A., Garcia, C. C. M., Angeli, J. P. F., Schumacher, R. I., et al. (2010). Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1802, 462–471. doi: 10.1016/j.bbadis.2010.01.011
Barbour, H., Daou, S., Hendzel, M., and Affar, E. B. (2020). Polycomb group-mediated histone H2A monoubiquitination in epigenome regulation and nuclear processes. Nat. Commun. 11:5947. doi: 10.1038/s41467-020-19722-9
Beaver, M., Bhatnagar, A., Panikker, P., Zhang, H., Snook, R., Parmar, V., et al. (2020). Disruption of Tip60 HAT mediated neural histone acetylation homeostasis is an early common event in neurodegenerative diseases. Sci. Rep. 10:18265. doi: 10.1038/s41598-020-75035-3
Belzil, V. V., Bauer, P. O., Prudencio, M., Gendron, T. F., Stetler, C. T., Yan, I. K., et al. (2013). Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 126, 895–905. doi: 10.1007/s00401-013-1199-1
Beninger, P. (2023). Sodium phenylbutyrate/taurursodiol. Clin. Ther. 45, 921–922. doi: 10.1016/j.clinthera.2023.06.016
Bennett, S. A., Cobos, S. N., Mirzakandova, M., Fallah, M., Son, E., Angelakakis, G., et al. (2021). Trichostatin a relieves growth suppression and restores histone acetylation at specific sites in a FUS ALS/FTD yeast model. Biochemistry 60, 3671–3675. doi: 10.1021/acs.biochem.1c00455
Bennett, S. A., Tanaz, R., Cobos, S. N., and Torrente, M. P. (2019). Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl. Res. J. Lab. Clin. Med. 204, 19–30. doi: 10.1016/j.trsl.2018.10.002
Berdyński, M., Miszta, P., Safranow, K., Andersen, P. M., Morita, M., Filipek, S., et al. (2022). SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep. 12:103. doi: 10.1038/s41598-021-03891-8
Berson, A., Sartoris, A., Nativio, R., Van Deerlin, V., Toledo, J. B., Porta, S., et al. (2017). TDP-43 promotes neurodegeneration by impairing chromatin remodeling. Curr. Biol. CB 27, 3579–3590.e6. doi: 10.1016/j.cub.2017.10.024
Bhatia-Dey, N., and Heinbockel, T. (2021). The olfactory system as marker of neurodegeneration in aging, neurological and neuropsychiatric disorders. Int. J. Environ. Res. Public Health 18:6976. doi: 10.3390/ijerph18136976
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71:505. doi: 10.1001/jamaneurol.2013.5847
Booms, A., Pierce, S. E., Van Der Schans, E. J. C., and Coetzee, G. A. (2024). Parkinson’s disease risk enhancers in microglia. iScience 27:108921. doi: 10.1016/j.isci.2024.108921
Booth, L. N., and Brunet, A. (2016). The aging epigenome. Mol. Cell 62, 728–744. doi: 10.1016/j.molcel.2016.05.013
Boros, J., Arnoult, N., Stroobant, V., Collet, J.-F., and Decottignies, A. (2014). Polycomb repressive complex 2 and H3K27me3 cooperate with H3K9 methylation to maintain heterochromatin protein 1α at chromatin. Mol. Cell. Biol. 34, 3662–3674. doi: 10.1128/MCB.00205-14
Breijyeh, Z., and Karaman, R. (2020). Comprehensive review on Alzheimer’s disease: causes and treatment. Mol. Basel Switz. 25:5789. doi: 10.3390/molecules25245789
Calligaris, R., Banica, M., Roncaglia, P., Robotti, E., Finaurini, S., Vlachouli, C., et al. (2015). Blood transcriptomics of drug-naïve sporadic Parkinson’s disease patients. BMC Genomics 16:876. doi: 10.1186/s12864-015-2058-3
Cannon, J. R., and Greenamyre, J. T. (2011). The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 124, 225–250. doi: 10.1093/toxsci/kfr239
Cao, Q., Wang, W., Williams, J. B., Yang, F., Wang, Z.-J., and Yan, Z. (2020). Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci. Adv. 6:eabc8096. doi: 10.1126/sciadv.abc8096
Cao, T., Zhou, X., Zheng, X., Cui, Y., Tsien, J. Z., Li, C., et al. (2018). Histone deacetylase inhibitor alleviates the neurodegenerative phenotypes and histone dysregulation in presenilins-deficient mice. Front. Aging Neurosci. 10:137. doi: 10.3389/fnagi.2018.00137
Carollo, P. S., and Barra, V. (2023). Chromatin epigenetics and nuclear lamina keep the nucleus in shape: examples from natural and accelerated aging. Biol. Cell. 115:2200023. doi: 10.1111/boc.202200023
Carrier, F. (2013). Chromatin modulation by histone deacetylase inhibitors: impact on cellular sensitivity to ionizing radiation. Mol. Cell. Pharmacol. 5, 51–59.
Castelo Rueda, M. P., Raftopoulou, A., Gögele, M., Borsche, M., Emmert, D., Fuchsberger, C., et al. (2021). Frequency of heterozygous Parkin (PRKN) variants and penetrance of Parkinson’s disease risk markers in the population-based CHRIS cohort. Front. Neurol. 12:706145. doi: 10.3389/fneur.2021.706145
Castelo Rueda, M. P., Zanon, A., Gilmozzi, V., Lavdas, A. A., Raftopoulou, A., Delcambre, S., et al. (2023). Molecular phenotypes of mitochondrial dysfunction in clinically non-manifesting heterozygous PRKN variant carriers. Npj Park. Dis. 9:65. doi: 10.1038/s41531-023-00499-9
Chai, C., and Lim, K.-L. (2013). Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genomics 14, 486–501. doi: 10.2174/1389202914666131210195808
Checkoway, H., Lundin, J. I., and Kelada, S. N. (2011). Neurodegenerative diseases. IARC Sci. Publ. 163, 407–419.
Chen, K., Bennett, S. A., Rana, N., Yousuf, H., Said, M., Taaseen, S., et al. (2018). Neurodegenerative disease proteinopathies are connected to distinct histone post-translational modification landscapes. ACS Chem. Neurosci. 9, 838–848. doi: 10.1021/acschemneuro.7b00297
Chen, M., Guo, X., Guo, J., Shi, C., Wu, Y., Chen, L., et al. (2024). Cytoplasmic accumulation of histones induced by BET inhibition protects cells from C9orf72 poly(PR)-induced cell death. Adv. Biol. 8:e2300334. doi: 10.1002/adbi.202300334
Chen, G., Xu, T., Yan, Y., Zhou, Y., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Chereji, R. V., Ramachandran, S., Bryson, T. D., and Henikoff, S. (2018). Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biol. 19:19. doi: 10.1186/s13059-018-1398-0
Chin-Chan, M., Navarro-Yepes, J., and Quintanilla-Vega, B. (2015). Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 9:124. doi: 10.3389/fncel.2015.00124
Chinta, S. J., Woods, G., Demaria, M., Rane, A., Zou, Y., McQuade, A., et al. (2018). Cellular senescence is induced by the environmental neurotoxin Paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940. doi: 10.1016/j.celrep.2017.12.092
Cho, H., and Shukla, S. (2020). Role of Edaravone as a treatment option for patients with amyotrophic lateral sclerosis. Pharmaceuticals 14:29. doi: 10.3390/ph14010029
Choi, S.-H., Yousefian-Jazi, A., Hyeon, S. J., Nguyen, P. T. T., Chu, J., Kim, S., et al. (2022). Modulation of histone H3K4 dimethylation by spermidine ameliorates motor neuron survival and neuropathology in a mouse model of ALS. J. Biomed. Sci. 29:106. doi: 10.1186/s12929-022-00890-3
Chong, Z.-S., Khong, Z. J., Tay, S. H., and Ng, S.-Y. (2022). Metabolic contributions to neuronal deficits caused by genomic disruption of schizophrenia risk gene SETD1A. Schizophrenia 8:115. doi: 10.1038/s41537-022-00326-9
Cividini, C., Basaia, S., Spinelli, E. G., Canu, E., Castelnovo, V., Riva, N., et al. (2022). Amyotrophic lateral sclerosis–frontotemporal dementia: shared and divergent neural correlates across the clinical spectrum. Neurology 98, e402–e415. doi: 10.1212/WNL.0000000000013123
Cobos, S. N., Bennett, S. A., and Torrente, M. P. (2019). The impact of histone post-translational modifications in neurodegenerative diseases. Biochim. Biophys. Acta Mol. basis Dis. 1865, 1982–1991. doi: 10.1016/j.bbadis.2018.10.019
Coyne, A. N., and Rothstein, J. D. (2021). Nuclear lamina invaginations are not a pathological feature of C9orf72 ALS/FTD. Acta Neuropathol. Commun. 9:45. doi: 10.1186/s40478-021-01150-5
Crescioli, S., Kaplon, H., Chenoweth, A., Wang, L., Visweswaraiah, J., and Reichert, J. M. (2024). Antibodies to watch in 2024. MAbs 16:2297450. doi: 10.1080/19420862.2023.2297450
Cudkowicz, M. E., Andres, P. L., Macdonald, S. A., Bedlack, R. S., Choudry, R., Brown, R. H., et al. (2009). Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph. Lateral Scler 10, 99–106. doi: 10.1080/17482960802320487
Currais, A., Huang, L., Goldberg, J., Petrascheck, M., Ates, G., Pinto-Duarte, A., et al. (2019). Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 8:e47866. doi: 10.7554/eLife.47866
Dilliott, A. A., Abdelhady, A., Sunderland, K. M., Farhan, S. M. K., Abrahao, A., Binns, M. A., et al. (2021). Contribution of rare variant associations to neurodegenerative disease presentation. Npj Genomic Med. 6:80. doi: 10.1038/s41525-021-00243-3
Dresselhaus, E., Duerr, J. M., Vincent, F., Sylvain, E. K., Beyna, M., Lanyon, L. F., et al. (2018). Class I HDAC inhibition is a novel pathway for regulating astrocytic apoE secretion. PLoS One 13:e0194661. doi: 10.1371/journal.pone.0194661
Ducharme, S., Pijnenburg, Y., Rohrer, J. D., Huey, E., Finger, E., and Tatton, N. (2024). Identifying and diagnosing TDP-43 neurodegenerative diseases in psychiatry. Am. J. Geriatr. Psychiatry 32, 98–113. doi: 10.1016/j.jagp.2023.08.017
Eckschlager, T., Plch, J., Stiborova, M., and Hrabeta, J. (2017). Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 18:1414. doi: 10.3390/ijms18071414
Eeftens, J. M., Kapoor, M., Michieletto, D., and Brangwynne, C. P. (2021). Polycomb condensates can promote epigenetic marks but are not required for sustained chromatin compaction. Nat. Commun. 12:5888. doi: 10.1038/s41467-021-26147-5
El Hajjar, J., Chatoo, W., Hanna, R., Nkanza, P., Tétreault, N., Tse, Y. C., et al. (2019). Heterochromatic genome instability and neurodegeneration sharing similarities with Alzheimer’s disease in old Bmi1+/− mice. Sci. Rep. 9:594. doi: 10.1038/s41598-018-37444-3
Farrelly, L. A., and Maze, I. (2019). An emerging perspective on ‘histone code’ mediated regulation of neural plasticity and disease. Curr. Opin. Neurobiol. 59, 157–163. doi: 10.1016/j.conb.2019.07.001
Feehley, T., O’Donnell, C. W., Mendlein, J., Karande, M., and McCauley, T. (2023). Drugging the epigenome in the age of precision medicine. Clin. Epigenetics 15:6. doi: 10.1186/s13148-022-01419-z
Fisher, E. M. C., Greensmith, L., Malaspina, A., Fratta, P., Hanna, M. G., Schiavo, G., et al. (2023). Opinion: more mouse models and more translation needed for ALS. Mol. Neurodegener. 18:30. doi: 10.1186/s13024-023-00619-2
Flamier, A., El Hajjar, J., Adjaye, J., Fernandes, K. J., Abdouh, M., and Bernier, G. (2018). Modeling late-onset sporadic Alzheimer’s disease through BMI1 deficiency. Cell Rep. 23, 2653–2666. doi: 10.1016/j.celrep.2018.04.097
Fox, S. H., and Lang, A. E. (2008). Levodopa-related motor complications--phenomenology. Mov. Disord. Off. J. Mov. Disord. Soc. 23, S509–S514. doi: 10.1002/mds.22021
Freibaum, B. D., and Taylor, J. P. (2017). The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci. 10:35. doi: 10.3389/fnmol.2017.00035
Frost, B., Bardai, F. H., and Feany, M. B. (2016). Lamin dysfunction mediates neurodegeneration in Tauopathies. Curr. Biol. 26, 129–136. doi: 10.1016/j.cub.2015.11.039
Frost, B., Hemberg, M., Lewis, J., and Feany, M. B. (2014). Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 17, 357–366. doi: 10.1038/nn.3639
Fuks, F., Hurd, P. J., Deplus, R., and Kouzarides, T. (2003). The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 31, 2305–2312. doi: 10.1093/nar/gkg332
Galimberti, D., D’Addario, C., Dell’Osso, B., Fenoglio, C., Marcone, A., Cerami, C., et al. (2013). Progranulin gene (GRN) promoter methylation is increased in patients with sporadic frontotemporal lobar degeneration. Neurol. Sci. 34, 899–903. doi: 10.1007/s10072-012-1151-5
Gallinari, P., Marco, S. D., Jones, P., Pallaoro, M., and Steinkühler, C. (2007). HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 17, 195–211. doi: 10.1038/sj.cr.7310149
Gendron, T. F., Rademakers, R., and Petrucelli, L. (2013). TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J. Alzheimers Dis. JAD 33, S35–S45. doi: 10.3233/JAD-2012-129036
Gerrish, A., Russo, G., Richards, A., Moskvina, V., Ivanov, D., Harold, D., et al. (2012). The role of variation at AβPP, PSEN1, PSEN2, and MAPT in late onset Alzheimer’s disease. J. Alzheimers Dis. JAD 28, 377–387. doi: 10.3233/JAD-2011-110824
Gil, L., Niño, S. A., Chi-Ahumada, E., Rodríguez-Leyva, I., Guerrero, C., Rebolledo, A. B., et al. (2020). Perinuclear Lamin a and Nucleoplasmic Lamin B2 characterize two types of hippocampal neurons through Alzheimer’s disease progression. Int. J. Mol. Sci. 21:1841. doi: 10.3390/ijms21051841
Gil, R. S., and Vagnarelli, P. (2019). Protein phosphatases in chromatin structure and function. Biochim. Biophys. Acta Mol. Cell Res. 1866, 90–101. doi: 10.1016/j.bbamcr.2018.07.016
Goldberg, M., Harel, A., Brandeis, M., Rechsteiner, T., Richmond, T. J., Weiss, A. M., et al. (1999). The tail domain of Lamin Dm 0 binds histones H2A and H2B. Proc. Natl. Acad. Sci. 96, 2852–2857. doi: 10.1073/pnas.96.6.2852
Gomperts, S. N. (2016). Lewy body dementias: dementia with Lewy bodies and Parkinson disease dementia. Contin. Lifelong Learn. Neurol. 22, 435–463. doi: 10.1212/CON.0000000000000309
Gräff, J., Rei, D., Guan, J.-S., Wang, W.-Y., Seo, J., Hennig, K. M., et al. (2012). An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. doi: 10.1038/nature10849
Gray, F., Cho, H. J., Shukla, S., He, S., Harris, A., Boytsov, B., et al. (2016). BMI1 regulates PRC1 architecture and activity through homo- and hetero-oligomerization. Nat. Commun. 7:13343. doi: 10.1038/ncomms13343
Guan, Z., Giustetto, M., Lomvardas, S., Kim, J.-H., Miniaci, M. C., Schwartz, J. H., et al. (2002). Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111, 483–493. doi: 10.1016/S0092-8674(02)01074-7
Guan, J.-S., Haggarty, S. J., Giacometti, E., Dannenberg, J.-H., Joseph, N., Gao, J., et al. (2009). HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. doi: 10.1038/nature07925
Guhathakurta, S., Kim, J., Adams, L., Basu, S., Song, M. K., Adler, E., et al. (2021). Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol. Med. 13:e12188. doi: 10.15252/emmm.202012188
Hampel, H., Hardy, J., Blennow, K., Chen, C., Perry, G., Kim, S. H., et al. (2021). The amyloid-β pathway in Alzheimer’s disease. Mol. Psychiatry 26, 5481–5503. doi: 10.1038/s41380-021-01249-0
Han, Y., Chen, L., Liu, J., Chen, J., Wang, C., Guo, Y., et al. (2022). A class I HDAC inhibitor rescues synaptic damage and neuron loss in APP-transfected cells and APP/PS1 mice through the GRIP1/AMPA pathway. Molecules 27:4160. doi: 10.3390/molecules27134160
Hendrickx, A., Pierrot, N., Tasiaux, B., Schakman, O., Brion, J.-P., Kienlen-Campard, P., et al. (2013). Epigenetic induction of EGR-1 expression by the amyloid precursor protein during exposure to novelty. PLoS One 8:e74305. doi: 10.1371/journal.pone.0074305
Hendrickx, A., Pierrot, N., Tasiaux, B., Schakman, O., Kienlen-Campard, P., De Smet, C., et al. (2014). Epigenetic regulations of immediate early genes expression involved in memory formation by the amyloid precursor protein of Alzheimer disease. PLoS One 9:e99467. doi: 10.1371/journal.pone.0099467
Henikoff, S., and Smith, M. M. (2015). Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 7:a019364. doi: 10.1101/cshperspect.a019364
Herrmann, F., and Fackelmayer, F. O. (2009). Nucleo-cytoplasmic shuttling of protein arginine methyltransferase 1 (PRMT1) requires enzymatic activity. Genes Cells 14, 309–317. doi: 10.1111/j.1365-2443.2008.01266.x
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Huang, S., Litt, M., and Felsenfeld, G. (2005). Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 19, 1885–1893. doi: 10.1101/gad.1333905
Hugais, M. M., Cobos, S. N., Bennett, S. A., Paredes, J., Foran, G., and Torrente, M. P. (2021). Changes in histone H3 acetylation on lysine 9 accompany Aβ 1-40 overexpression in an Alzheimer’s disease yeast model. MicroPublication Biol. 2021:492. doi: 10.17912/micropub.biology.000492
Hwang, J.-Y., Aromolaran, K. A., and Zukin, R. S. (2017). The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 18, 347–361. doi: 10.1038/nrn.2017.46
Hyun, K., Jeon, J., Park, K., and Kim, J. (2017). Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49:e324. doi: 10.1038/emm.2017.11
Iarkov, A., Barreto, G. E., Grizzell, J. A., and Echeverria, V. (2020). Strategies for the treatment of Parkinson’s disease: beyond dopamine. Front. Aging Neurosci. 12:4. doi: 10.3389/fnagi.2020.00004
Janssen, C., Schmalbach, S., Boeselt, S., Sarlette, A., Dengler, R., and Petri, S. (2010). Differential histone deacetylase mRNA expression patterns in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 573–581. doi: 10.1097/NEN.0b013e3181ddd404
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jęśko, H., Lenkiewicz, A. M., Wilkaniec, A., and Adamczyk, A. (2019). The interplay between parkin and alpha-synuclein; possible implications for the pathogenesis of Parkinson’s disease. Acta Neurobiol. Exp. 79, 276–289. doi: 10.21307/ane‑2019‑026
Jin, H., Kanthasamy, A., Ghosh, A., Yang, Y., Anantharam, V., and Kanthasamy, A. G. (2011). α-Synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 31, 2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011
Jury, N., Abarzua, S., Diaz, I., Guerra, M. V., Ampuero, E., Cubillos, P., et al. (2020). Widespread loss of the silencing epigenetic mark H3K9me3 in astrocytes and neurons along with hippocampal-dependent cognitive impairment in C9orf72 BAC transgenic mice. Clin. Epigenetics 12:32. doi: 10.1186/s13148-020-0816-9
Kabir, F., Atkinson, R., Cook, A. L., Phipps, A. J., and King, A. E. (2023). The role of altered protein acetylation in neurodegenerative disease. Front. Aging Neurosci. 14:1025473. doi: 10.3389/fnagi.2022.1025473
Kelly, T. K., De Carvalho, D. D., and Jones, P. A. (2010). Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 28, 1069–1078. doi: 10.1038/nbt.1678
Khan, I., and De Jesus, O. (2023) Frontotemporal Lobe Dementia. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2023 [cited 2023 Oct 18]. Available at: http://www.ncbi.nlm.nih.gov/books/NBK559286/ (Accessed October 18, 2023).
Kim, J., Lee, S., Choi, B.-R., Yang, H., Hwang, Y., Park, J. H. Y., et al. (2017). Sulforaphane epigenetically enhances neuronal BDNF expression and TrkB signaling pathways. Mol. Nutr. Food Res. 61:194. doi: 10.1002/mnfr.201600194
Kim, D., and Tsai, L. (2009). Linking cell cycle reentry and DNA damage in neurodegeneration. Ann. N. Y. Acad. Sci. 1170, 674–679. doi: 10.1111/j.1749-6632.2009.04105.x
Knopman, D. S., and Hershey, L. (2023). Implications of the approval of Lecanemab for Alzheimer disease patient care: incremental step or paradigm shift? Neurology 101, 610–620. doi: 10.1212/WNL.0000000000207438
Komar, D., and Juszczynski, P. (2020). Rebelled epigenome: histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin. Epigenetics 12:147. doi: 10.1186/s13148-020-00941-2
Kontopoulos, E., Parvin, J. D., and Feany, M. B. (2006). α-Synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023. doi: 10.1093/hmg/ddl243
Kühlwein, J. K., Ruf, W. P., Kandler, K., Witzel, S., Lang, C., Mulaw, M. A., et al. (2023). ALS is imprinted in the chromatin accessibility of blood cells. Cell. Mol. Life Sci. CMLS 80:131. doi: 10.1007/s00018-023-04769-w
Lang, F., Strutz-Seebohm, N., Seebohm, G., and Lang, U. E. (2010). Significance of SGK1 in the regulation of neuronal function. J. Physiol. 588, 3349–3354. doi: 10.1113/jphysiol.2010.190926
Larson, A. G., Elnatan, D., Keenen, M. M., Trnka, M. J., Johnston, J. B., Burlingame, A. L., et al. (2017). Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240. doi: 10.1038/nature22822
Laugesen, A., Højfeldt, J. W., and Helin, K. (2016). Role of the polycomb repressive complex 2 (PRC2) in transcriptional regulation and Cancer. Cold Spring Harb. Perspect. Med. 6:a026575. doi: 10.1101/cshperspect.a026575
Lee, J.-H., Kim, E. W., Croteau, D. L., and Bohr, V. A. (2020). Heterochromatin: an epigenetic point of view in aging. Exp. Mol. Med. 52, 1466–1474. doi: 10.1038/s12276-020-00497-4
Lee, J. H., Kim, J.-H., Kim, S., Cho, K. S., and Lee, S. B. (2018). Chromatin changes associated with neuronal maintenance and their pharmacological application. Curr. Neuropharmacol. 16, 118–125. doi: 10.2174/1570159X15666170601124220
Lee, M. Y., Lee, J., Hyeon, S. J., Cho, H., Hwang, Y. J., Shin, J., et al. (2020). Epigenome signatures landscaped by histone H3K9me3 are associated with the synaptic dysfunction in Alzheimer’s disease. Aging Cell 19:e13153. doi: 10.1111/acel.13153
Leveille, E., Ross, O. A., and Gan-Or, Z. (2021). Tau and MAPT genetics in tauopathies and synucleinopathies. Parkinsonism Relat. Disord. 90, 142–154. doi: 10.1016/j.parkreldis.2021.09.008
Li, J., Jaiswal, M. K., Chien, J.-F., Kozlenkov, A., Jung, J., Zhou, P., et al. (2023). Divergent single cell transcriptome and epigenome alterations in ALS and FTD patients with C9orf72 mutation. Nat. Commun. 14:5714. doi: 10.1038/s41467-023-41033-y
Lithner, C. U., Lacor, P. N., Zhao, W.-Q., Mustafiz, T., Klein, W. L., Sweatt, J. D., et al. (2013). Disruption of neocortical histone H3 homeostasis by soluble Aβ: implications for Alzheimer’s disease. Neurobiol. Aging 34, 2081–2090. doi: 10.1016/j.neurobiolaging.2012.12.028
Liu, X. S., and Jaenisch, R. (2019). Editing the epigenome to tackle brain disorders. Trends Neurosci. 42, 861–870. doi: 10.1016/j.tins.2019.10.003
Liu, E. Y., Russ, J., Cali, C. P., Phan, J. M., Amlie-Wolf, A., and Lee, E. B. (2019). Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep. 27, 1409–1421.e6. doi: 10.1016/j.celrep.2019.04.003
Lozupone, M., Dibello, V., Sardone, R., Castellana, F., Zupo, R., Lampignano, L., et al. (2023). The impact of apolipoprotein E (APOE) epigenetics on aging and sporadic Alzheimer’s disease. Biology 12:1529. doi: 10.3390/biology12121529
Mackenzie, I. R. A., Ansorge, O., Strong, M., Bilbao, J., Zinman, L., Ang, L.-C., et al. (2011). Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 122, 87–98. doi: 10.1007/s00401-011-0838-7
Maina, M. B., Bailey, L. J., Wagih, S., Biasetti, L., Pollack, S. J., Quinn, J. P., et al. (2018). The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 6:70. doi: 10.1186/s40478-018-0565-6
Majchrzak-Celińska, A., Warych, A., and Szoszkiewicz, M. (2021). Novel approaches to epigenetic therapies: from drug combinations to epigenetic editing. Genes 12:208. doi: 10.3390/genes12020208
Maji, A., Ahmed, J. A., Roy, S., Chakrabarti, B., and Mitra, M. K. (2020). A Lamin-associated chromatin model for chromosome organization. Biophys. J. 118, 3041–3050. doi: 10.1016/j.bpj.2020.05.014
Mansuroglu, Z., Benhelli-Mokrani, H., Marcato, V., Sultan, A., Violet, M., Chauderlier, A., et al. (2016). Loss of tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 6:33047. doi: 10.1038/srep33047
Margueron, R., Li, G., Sarma, K., Blais, A., Zavadil, J., Woodcock, C. L., et al. (2008). Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32, 503–518. doi: 10.1016/j.molcel.2008.11.004
Marmorstein, R., and Trievel, R. C. (2009). Histone modifying enzymes: structures, mechanisms, and specificities. Biochim. Biophys. Acta 1789, 58–68. doi: 10.1016/j.bbagrm.2008.07.009
Marmorstein, R., and Zhou, M.-M. (2014). Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 6:762. doi: 10.1101/cshperspect.a018762
Marzochi, L. L., Cuzziol, C. I., Nascimento Filho, C. H. V. D., dos Santos, J. A., Castanhole-Nunes, M. M. U., Pavarino, É. C., et al. (2023). Use of histone methyltransferase inhibitors in cancer treatment: a systematic review. Eur. J. Pharmacol. 944:175590. doi: 10.1016/j.ejphar.2023.175590
Masala, A., Sanna, S., Esposito, S., Rassu, M., Galioto, M., Zinellu, A., et al. (2018). Epigenetic changes associated with the expression of amyotrophic lateral sclerosis (ALS) causing genes. Neuroscience 390, 1–11. doi: 10.1016/j.neuroscience.2018.08.009
Mead, R. J., Shan, N., Reiser, H. J., Marshall, F., and Shaw, P. J. (2023). Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 22, 185–212. doi: 10.1038/s41573-022-00612-2
Méndez-López, I., Blanco-Luquin, I., Sánchez-Ruiz De Gordoa, J., Urdánoz-Casado, A., Roldán, M., Acha, B., et al. (2019). Hippocampal LMNA gene expression is increased in late-stage Alzheimer’s disease. Int. J. Mol. Sci. 20:878. doi: 10.3390/ijms20040878
Milazzo, G., Mercatelli, D., Di Muzio, G., Triboli, L., De Rosa, P., Perini, G., et al. (2020). Histone deacetylases (HDACs): evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes 11:556. doi: 10.3390/genes11050556
Millán-Zambrano, G., Burton, A., Bannister, A. J., and Schneider, R. (2022). Histone post-translational modifications — cause and consequence of genome function. Nat. Rev. Genet. 23, 563–580. doi: 10.1038/s41576-022-00468-7
Mir, F. A., Amanullah, A., Jain, B. P., Hyderi, Z., and Gautam, A. (2023). Neuroepigenetics of ageing and neurodegeneration-associated dementia: an updated review. Ageing Res. Rev. 91:102067. doi: 10.1016/j.arr.2023.102067
Mollah, S. A., Nayak, A., Barhai, S., and Maity, U. (2024). A comprehensive review on frontotemporal dementia: its impact on language, speech and behavior. Dement. Neuropsychol. 18:e20230072. doi: 10.1590/1980-5764-DN-2023-0072
Morera, L., Lübbert, M., and Jung, M. (2016). Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 8:57. doi: 10.1186/s13148-016-0223-4
Morrison, O., and Thakur, J. (2021). Molecular complexes at Euchromatin, heterochromatin and centromeric chromatin. Int. J. Mol. Sci. 22:6922. doi: 10.3390/ijms22136922
Mu, M.-D., Qian, Z.-M., Yang, S.-X., Rong, K.-L., Yung, W.-H., and Ke, Y. (2020). Therapeutic effect of a histone demethylase inhibitor in Parkinson’s disease. Cell Death Dis. 11:927. doi: 10.1038/s41419-020-03105-5
Nakamura, M., Gao, Y., Dominguez, A. A., and Qi, L. S. (2021). CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 23, 11–22. doi: 10.1038/s41556-020-00620-7
Ogawa, O., Zhu, X., Lee, H.-G., Raina, A., Obrenovich, M. E., Bowser, R., et al. (2003). Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta Neuropathol. 105, 524–528. doi: 10.1007/s00401-003-0684-3
Ortega, J. A., Daley, E. L., Kour, S., Samani, M., Tellez, L., Smith, H. S., et al. (2020). Nucleocytoplasmic proteomic analysis uncovers eRF1 and nonsense-mediated decay as modifiers of ALS/FTD C9orf72 toxicity. Neuron 106, 90–107.e13. doi: 10.1016/j.neuron.2020.01.020
Paez-Colasante, X., Figueroa-Romero, C., Sakowski, S. A., Goutman, S. A., and Feldman, E. L. (2015). Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 11, 266–279. doi: 10.1038/nrneurol.2015.57
Paganoni, S., Macklin, E. A., Hendrix, S., Berry, J. D., Elliott, M. A., Maiser, S., et al. (2020). Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 383, 919–930. doi: 10.1056/NEJMoa1916945
Parobkova, E., and Matej, R. (2021). Amyotrophic lateral sclerosis and frontotemporal lobar degenerations: similarities in genetic background. Diagnostics 11:509. doi: 10.3390/diagnostics11030509
Pearson, K. L., Hunter, T., and Janknecht, R. (1999). Activation of Smad1-mediated transcription by p300/CBP. Biochim. Biophys. Acta 1489, 354–364. doi: 10.1016/s0167-4781(99)00166-9
Penagos-Puig, A., and Furlan-Magaril, M. (2020). Heterochromatin as an important driver of genome organization. Front. Cell Dev. Biol. 8:579137. doi: 10.3389/fcell.2020.579137
Penney, J., Ralvenius, W. T., and Tsai, L.-H. (2020). Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 25, 148–167. doi: 10.1038/s41380-019-0468-3
Peterson, C. L., and Laniel, M.-A. (2004). Histones and histone modifications. Curr. Biol. 14, R546–R551. doi: 10.1016/j.cub.2004.07.007
Ramakrishnan, A., Piehl, N., Simonton, B., Parikh, M., Zhang, Z., Teregulova, V., et al. (2024). Epigenetic dysregulation in Alzheimer’s disease peripheral immunity. Neuron 112, 1235–1248.e5. doi: 10.1016/j.neuron.2024.01.013
Reardon, S. (2024). Alzheimer’s drug with modest benefits wins backing of FDA advisers. Nature. [Epubh ahead of preprint]. doi: 10.1038/d41586-024-01726-w
Rekaik, H., Blaudin de Thé, F.-X., Fuchs, J., Massiani-Beaudoin, O., Prochiantz, A., and Joshi, R. L. (2015). Engrailed homeoprotein protects mesencephalic dopaminergic neurons from oxidative stress. Cell Rep. 13, 242–250. doi: 10.1016/j.celrep.2015.08.076
Rossaert, E., Pollari, E., Jaspers, T., Van Helleputte, L., Jarpe, M., Van Damme, P., et al. (2019). Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol. Commun. 7:107. doi: 10.1186/s40478-019-0750-2
Rothbart, S. B., and Strahl, B. D. (2014). Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta BBA Gene Regul. Mech. 1839, 627–643. doi: 10.1016/j.bbagrm.2014.03.001
Ruan, K., Yamamoto, T. G., Asakawa, H., Chikashige, Y., Kimura, H., Masukata, H., et al. (2015). Histone H4 acetylation required for chromatin decompaction during DNA replication. Sci. Rep. 5:12720. doi: 10.1038/srep12720
Saha, R. N., and Pahan, K. (2006). HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13, 539–550. doi: 10.1038/sj.cdd.4401769
Saini, A., and Chawla, P. A. (2024). Breaking barriers with tofersen: enhancing therapeutic opportunities in amyotrophic lateral sclerosis. Eur. J. Neurol. 31:e16140. doi: 10.1111/ene.16140
Salat, D., and Tolosa, E. (2013). Levodopa in the treatment of Parkinson’s disease: current status and new developments. J. Parkinsons Dis. 3, 255–269. doi: 10.3233/JPD-130186
Sanna, S., Esposito, S., Masala, A., Sini, P., Nieddu, G., Galioto, M., et al. (2020). HDAC1 inhibition ameliorates TDP-43-induced cell death in vitro and in vivo. Cell Death Dis. 11:369. doi: 10.1038/s41419-020-2580-3
Santoro, S. W., and Dulac, C. (2012). The activity-dependent histone variant H2BE modulates the life span of olfactory neurons. eLife 1:e00070. doi: 10.7554/eLife.00070
Sasaki-Honda, M., Akatsuka, K., and Sawai, T. (2023). Is epigenome editing non-inheritable? Implications for ethics and the regulation of human applications. Stem Cell Rep. 18, 2005–2009. doi: 10.1016/j.stemcr.2023.10.003
Schellenberg, G. D., and Montine, T. J. (2012). The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 124, 305–323. doi: 10.1007/s00401-012-0996-2
Schmidt, S., Stautner, C., Vu, D. T., Heinz, A., Regensburger, M., Karayel, O., et al. (2023). A reversible state of hypometabolism in a human cellular model of sporadic Parkinson’s disease. Nat. Commun. 14:7674. doi: 10.1038/s41467-023-42862-7
Schwartz, J. C., Podell, E. R., Han, S. S. W., Berry, J. D., Eggan, K. C., and Cech, T. R. (2014). FUS is sequestered in nuclear aggregates in ALS patient fibroblasts. Mol. Biol. Cell 25, 2571–2578. doi: 10.1091/mbc.E14-05-1007
Shahid, Z., Simpson, B., Miao, K. H., and Singh, G. (2023). “Genetics, histone code” in StatPearls (Treasure Island (FL): StatPearls Publishing).
Sjöberg, M. K., Shestakova, E., Mansuroglu, Z., Maccioni, R. B., and Bonnefoy, E. (2006). Tau protein binds to pericentromeric DNA: a putative role for nuclear tau in nucleolar organization. J. Cell Sci. 119, 2025–2034. doi: 10.1242/jcs.02907
Song, H., Chen, J., Huang, J., Sun, P., Liu, Y., Xu, L., et al. (2023). Epigenetic modification in Parkinson’s disease. Front. Cell Dev. Biol. 11:1123621. doi: 10.3389/fcell.2023.1123621
Spataro, N., Calafell, F., Cervera-Carles, L., Casals, F., Pagonabarraga, J., Pascual-Sedano, B., et al. (2015). Mendelian genes for Parkinson’s disease contribute to the sporadic forms of the disease†. Hum. Mol. Genet. 24, 2023–2034. doi: 10.1093/hmg/ddu616
Spencer, P. S., Salama, M., and Kisby, G. E. (2022). “Mechanisms underlying long-latency neurodegenerative diseases of environmental origin” in Handbook of neurotoxicity. ed. R. M. Kostrzewa (Cham: Springer International Publishing), 71–93.
Starr, A., and Sattler, R. (2018). Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 1693, 98–108. doi: 10.1016/j.brainres.2018.02.011
Stefanis, L. (2012). α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Stephens, A. D., Liu, P. Z., Banigan, E. J., Almassalha, L. M., Backman, V., Adam, S. A., et al. (2018). Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 29, 220–233. doi: 10.1091/mbc.E17-06-0410
Sugeno, N., Jäckel, S., Voigt, A., Wassouf, Z., Schulze-Hentrich, J., and Kahle, P. J. (2016). α-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 6:36328. doi: 10.1038/srep36328
Taylor, J. P., Brown, R. H., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Teijido, O., and Cacabelos, R. (2018). Pharmacoepigenomic interventions as novel potential treatments for Alzheimer’s and Parkinson’s diseases. Int. J. Mol. Sci. 19:3199. doi: 10.3390/ijms19103199
Teperino, R., Schoonjans, K., and Auwerx, J. (2010). Histone methyl-transferases and demethylases; can they link metabolism and transcription? Cell Metab. 12, 321–327. doi: 10.1016/j.cmet.2010.09.004
Tibshirani, M., Tradewell, M. L., Mattina, K. R., Minotti, S., Yang, W., Zhou, H., et al. (2015). Cytoplasmic sequestration of FUS/TLS associated with ALS alters histone marks through loss of nuclear protein arginine methyltransferase 1. Hum. Mol. Genet. 24, 773–786. doi: 10.1093/hmg/ddu494
Tibshirani, M., Zhao, B., Gentil, B. J., Minotti, S., Marques, C., Keith, J., et al. (2017). Dysregulation of chromatin remodelling complexes in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26, 4142–4152. doi: 10.1093/hmg/ddx301
Toker, L., Tran, G. T., Sundaresan, J., Tysnes, O.-B., Alves, G., Haugarvoll, K., et al. (2021). Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson’s disease brain. Mol. Neurodegener. 16:31. doi: 10.1186/s13024-021-00450-7
Tolosa, E., Vila, M., Klein, C., and Rascol, O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107. doi: 10.1038/s41582-019-0301-2
Torrente, M. P., Zee, B. M., Young, N. L., Baliban, R. C., LeRoy, G., Floudas, C. A., et al. (2011). Proteomic interrogation of human chromatin. PLoS One 6:e24747. doi: 10.1371/journal.pone.0024747
Tsalenchuk, M., Gentleman, S. M., and Marzi, S. J. (2023). Linking environmental risk factors with epigenetic mechanisms in Parkinson’s disease. Npj Park. Dis. 9:123. doi: 10.1038/s41531-023-00568-z
Ueda, J., Yamazaki, T., and Funakoshi, H. (2023). Toward the development of epigenome editing-based therapeutics: potentials and challenges. Int. J. Mol. Sci. 24:4778. doi: 10.3390/ijms24054778
Valor, L. M., Viosca, J., Lopez-Atalaya, J. P., and Barco, A. (2013). Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 19, 5051–5064. doi: 10.2174/13816128113199990382
van Steensel, B., and Belmont, A. S. (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. doi: 10.1016/j.cell.2017.04.022
Vettese-Dadey, M., Grant, P. A., Hebbes, T. R., Crane-Robinson, C., Allis, C. D., and Workman, J. L. (1996). Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 15, 2508–2518. doi: 10.1002/j.1460-2075.1996.tb00608.x
Wang, X., Goodrich, K. J., Conlon, E. G., Gao, J., Erbse, A. H., Manley, J. L., et al. (2019). C9orf72 and triplet repeat disorder RNAs: G-quadruplex formation, binding to PRC2 and implications for disease mechanisms. RNA 25, 935–947. doi: 10.1261/rna.071191.119
Wang, L., Yu, C.-C., Liu, X.-Y., Deng, X.-N., Tian, Q., and Du, Y.-J. (2021). Epigenetic modulation of microglia function and phenotypes in neurodegenerative diseases. Neural Plast. 2021, 9912686–9912613. doi: 10.1155/2021/9912686
Ward, M., Carter, L. P., Huang, J. Y., Maslyar, D., Budda, B., Paul, R., et al. (2024). Phase 1 study of latozinemab in progranulin-associated frontotemporal dementia. Alzheimers Dement. Transl. Res. Clin. Interv. 10:e12452. doi: 10.1002/trc2.12452
Winter, S., Simboeck, E., Fischle, W., Zupkovitz, G., Dohnal, I., Mechtler, K., et al. (2008). 14-3-3 proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J. 27, 88–99. doi: 10.1038/sj.emboj.7601954
Witoelar, A., Jansen, I. E., Wang, Y., Desikan, R. S., Gibbs, J. R., Blauwendraat, C., et al. (2017). Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. 74, 780–792. doi: 10.1001/jamaneurol.2017.0469
Wong, M., Gertz, B., Chestnut, B., and Martin, L. (2013). Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell. Neurosci. 7:279. doi: 10.3389/fncel.2013.00279
Wu, C., Jin, L., Wang, I., Wei, W., Ho, P., Liu, Y., et al. (2020). HDAC1 dysregulation induces aberrant cell cycle and DNA damage in progress of TDP-43 proteinopathies. EMBO Mol. Med. 12:e10622. doi: 10.15252/emmm.201910622
Wu, D., Sun, J. K.-L., and Chow, K. H.-M. (2024). Neuronal cell cycle reentry events in the aging brain are more prevalent in neurodegeneration and lead to cellular senescence. PLoS Biol. 22:e3002559. doi: 10.1371/journal.pbio.3002559
Yan, S., Wang, C.-E., Wei, W., Gaertig, M. A., Lai, L., Li, S., et al. (2014). TDP-43 causes differential pathology in neuronal versus glial cells in the mouse brain. Hum. Mol. Genet. 23, 2678–2693. doi: 10.1093/hmg/ddt662
Yang, S., Zhang, R., Wang, G., and Zhang, Y. (2017). The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 6:19. doi: 10.1186/s40035-017-0089-1
Yoo, Y.-E., and Ko, C.-P. (2011). Treatment with trichostatin a initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 231, 147–159. doi: 10.1016/j.expneurol.2011.06.003
Yuan, J., Pu, M., Zhang, Z., and Lou, Z. (2009). Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle Georget. Tex 8, 1747–1753. doi: 10.4161/cc.8.11.8620
Yun, M., Wu, J., Workman, J. L., and Li, B. (2011). Readers of histone modifications. Cell Res. 21, 564–578. doi: 10.1038/cr.2011.42
Zhang, S., Bai, P., Lei, D., Liang, Y., Zhen, S., Bakiasi, G., et al. (2022). Degradation and inhibition of epigenetic regulatory protein BRD4 exacerbate Alzheimer’s disease-related neuropathology in cell models. J. Biol. Chem. 298:101794. doi: 10.1016/j.jbc.2022.101794
Zhang, Y.-J., Guo, L., Gonzales, P. K., Gendron, T. F., Wu, Y., Jansen-West, K., et al. (2019). Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363:eaav2606. doi: 10.1126/science.aav2606
Zhao, Y., and Garcia, B. A. (2015). Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 7:a025064. doi: 10.1101/cshperspect.a025064
Zheng, Y., Liu, A., Wang, Z.-J., Cao, Q., Wang, W., Lin, L., et al. (2019). Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 142, 787–807. doi: 10.1093/brain/awy354
Zhu, Q., Jiang, J., Gendron, T. F., McAlonis-Downes, M., Jiang, L., Taylor, A., et al. (2020). Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 23, 615–624. doi: 10.1038/s41593-020-0619-5
Keywords: histone post translational modification, epigenetics, neurodegeneration, heterochromatin, chromatin
Citation: Fisher RMA and Torrente MP (2024) Histone post-translational modification and heterochromatin alterations in neurodegeneration: revealing novel disease pathways and potential therapeutics. Front. Mol. Neurosci. 17:1456052. doi: 10.3389/fnmol.2024.1456052
Edited by:
Mayur Doke, University of Miami Health System, United StatesReviewed by:
Amrutha Swaminathan, Indian Institute of Science Education and Research, Thiruvananthapuram, IndiaCopyright © 2024 Fisher and Torrente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana P. Torrente, bWFyaWFuYS50b3JyZW50ZUBicm9va2x5bi5jdW55LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.