 Shishi Luo
Shishi Luo Danling Wang
Danling Wang Zhuohua Zhang1,2,4*
Zhuohua Zhang1,2,4*- 1Institute for Future Sciences, Hengyang Medical School, University of South China, Hengyang, Hunan, China
- 2Key Laboratory of Rare Pediatric Diseases, Ministry of Education, Hengyang, Hunan, China
- 3The Affiliated Changsha Central Hospital, Hengyang Medical School, University of South China, Changsha, Hunan, China
- 4Institute of Molecular Precision Medicine, Xiangya Hospital, Key Laboratory of Molecular Precision Medicine of Hunan Province and Center for Medical Genetics, Hunan Key Laboratory of Medical Genetics, Central South University, Changsha, Hunan, China
Parkinson’s disease (PD) is the second most common neurodegenerative disease with currently no cure. Most PD cases are sporadic, and about 5–10% of PD cases present a monogenic inheritance pattern. Mutations in more than 20 genes are associated with genetic forms of PD. Mitochondrial dysfunction is considered a prominent player in PD pathogenesis. Post-translational modifications (PTMs) allow rapid switching of protein functions and therefore impact various cellular functions including those related to mitochondria. Among the PD-associated genes, Parkin, PINK1, and LRRK2 encode enzymes that directly involved in catalyzing PTM modifications of target proteins, while others like α-synuclein, FBXO7, HTRA2, VPS35, CHCHD2, and DJ-1, undergo substantial PTM modification, subsequently altering mitochondrial functions. Here, we summarize recent findings on major PTMs associated with PD-related proteins, as enzymes or substrates, that are shown to regulate important mitochondrial functions and discuss their involvement in PD pathogenesis. We will further highlight the significance of PTM-regulated mitochondrial functions in understanding PD etiology. Furthermore, we emphasize the potential for developing important biomarkers for PD through extensive research into PTMs.
1 Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disease, affecting more than 10 million people worldwide (Kalia and Lang, 2015). Pathologically, PD is characterized by the progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and the accumulation of aggregated α-synuclein in the form of intracellular inclusion called Lewy Body (LB) (Tanner, 1992). The manifestations of PD are chronic and progressive, with the main symptoms involving movement dysfunctions such as tremor, tonicity, bradykinesia, and postural instability. Many patients also experience prodromal symptoms, including non-motor disturbances such as constipation, hyposmia, and mood disorders (Martinez-Martin et al., 2007; Lajoie et al., 2021). Currently, there is no cure for PD. The available treatments only alleviate the movement symptoms with little effects on disease progression.
Most PD cases are sporadic, or idiopathic, with age and environmental exposures (such as pesticides, herbicides, heavy metal, and head injury) being the main known risk factors (Goldman, 2014; Ascherio and Schwarzschild, 2016). About 15% PD cases have a familial history, and about 5–10% of PD patients present a monogenic inheritance pattern (Hernandez et al., 2016). Mutations in at least 20 genes are identified to be linked with familiar PD (Table 1). From which, dominantly associated genes such as SNCA (α-synuclein), LRRK2, and VPS35, as well as recessively associated genes like PRKN (Parkin), DJ-1, GBA, PINK1, ATP13A2, and FBXO7, are identified (Polymeropoulos et al., 1997; Kitada et al., 1998; Bonifati et al., 2003; Lwin et al., 2004; Valente et al., 2004; Gilks et al., 2005; Ramirez et al., 2006; Di Fonzo et al., 2009; Vilarino-Guell et al., 2011; Funayama et al., 2015). More than additional 90 genetic risk loci are shown to be associated with idiopathic PD (Bekris et al., 2010; Nalls et al., 2019). Individuals with genetic changes of those genes likely predispose to PD.
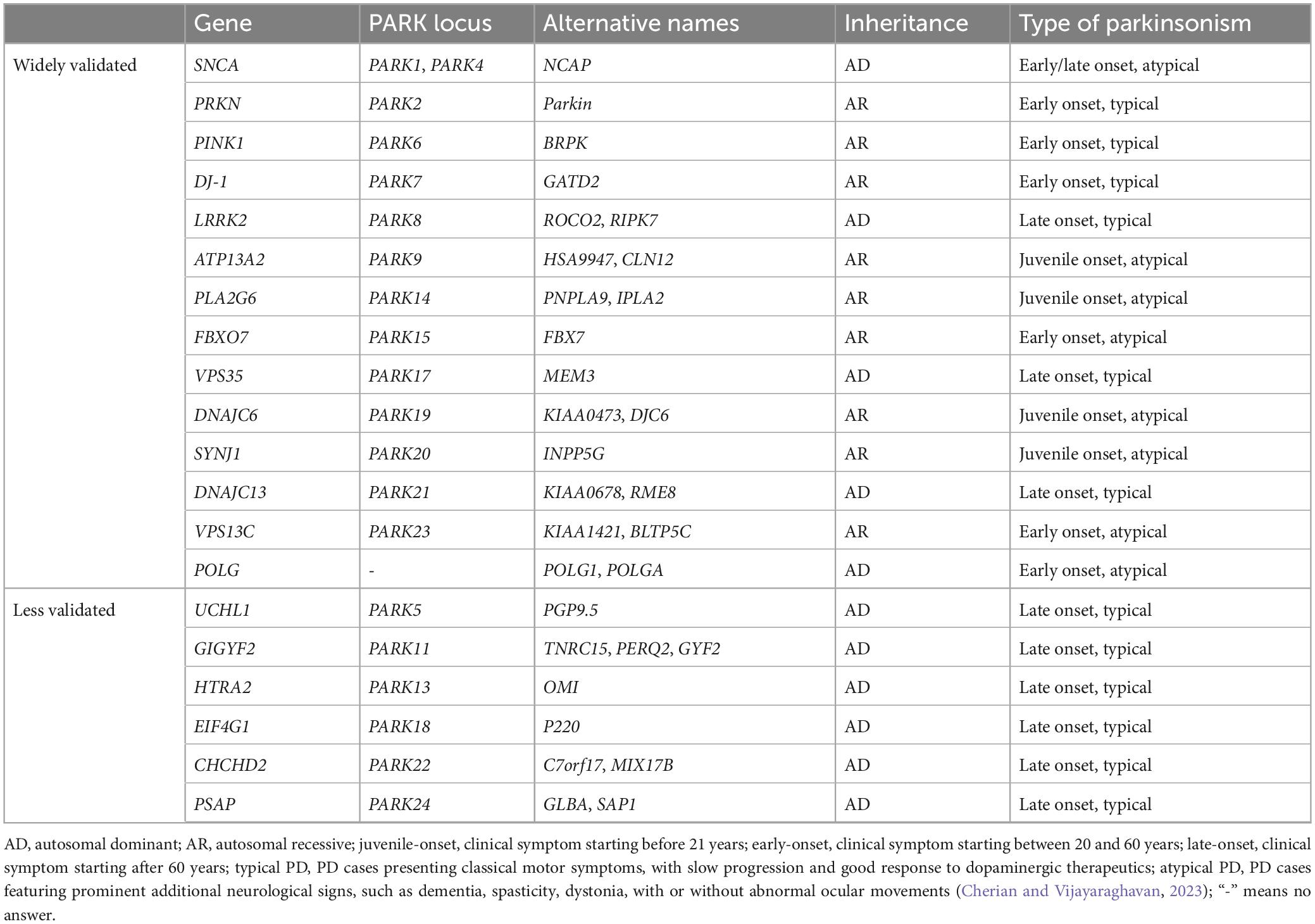
Table 1. Genes associated with familiar PD.
Human genetics have significantly contributed to our understanding of the molecular mechanisms of PD pathogenesis. Several key pathways, including those related to protein misfolding and aggregation, the ubiquitin-proteasomal system, autophagy, mitochondrial dysfunction, lysosomal abnormality, and vesicle trafficking, are revealed to PD (Henchcliffe and Beal, 2008; Meredith et al., 2009; Ebrahimi-Fakhari et al., 2012; Hunn et al., 2015; De Virgilio et al., 2016; Blumenreich et al., 2020). Of particular, mitochondrial dysfunction has long been implied in PD pathogenesis. Methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP), a mitochondrial complex I inhibitor, induces Parkinsonism in both human and animals (Langston et al., 1983). Multiple pesticides and herbicides are shown to induce parkinsonism via mitochondria-mediated mechanisms (Chen et al., 2017). A number of PD pathogenic monogenetic genes either encode mitochondrial proteins or regulate mitochondrial functions (Nicoletti et al., 2021). Genome-wide association studies (GWAS) also indicate that mitochondria-related processes are involved in both familiar and sporadic forms of PD (Billingsley et al., 2019).
Post-translational modifications (PTMs) refer to covalent chemical alterations of a protein after its synthesis. More than 400 types of PTMs are identified (Khoury et al., 2011). Some widely studied PTMs include phosphorylation, ubiquitination, methylation, acetylation, SUMOylation, etc. PTMs increase functional diversity of the modified protein, therefore modulate almost every aspect of cellular processes, including mitochondrial functions (Karve and Cheema, 2011; Stram and Payne, 2016). Among the PD-associated genes, Parkin, PINK1, and LRRK2 encode enzymes that directly catalyze the PTM of target proteins while they are PTM modified themselves (Trempe et al., 2013; Eiyama and Okamoto, 2015; Taylor and Alessi, 2020). PD-related proteins like α-synuclein, FBXO7, HTRA2, VPS35, CHCHD2, and DJ-1 either participate in or are heavily modified by PTMs (Mitsumoto et al., 2001; Plun-Favreau et al., 2007; Kang et al., 2012; Burchell et al., 2013; Tang et al., 2015; Meng et al., 2017). In this review, we aim to summarize the recent findings on major PTMs associated with PD-related proteins, either as enzymes or substrates, that have been shown to play significant roles in various mitochondrial functions, hence, PD pathogenesis.
2 Ubiquitination: regulating mitochondrial functions in Parkinson’s disease
Ubiquitination is a PTM characterized by the covalent binding of ubiquitin (Ub) molecule to a specific target protein. This essential PTM primarily determines protein stability by marking the target protein for proteasomal degradation. However, it has also been reported to be able to enhance protein stability and regulate protein activities. Ubiquitination plays an essential role in maintaining normal mitochondrial functions. Dysregulated ubiquitination can cause mitochondrial dysfunctions. While the accumulation of misfolded and ubiquitinated α-synuclein has long been recognized as the toxic factor in PD brain pathology, evidence implicating the direct involvement of ubiquitination dysregulation in mitochondrial dysfunction and thus PD pathology emerged from the intensive study of PD associated Parkin and PINK1. As non-mitochondrial proteins, other PD-related proteins, such as VPS35, FBXO7, and LRRKs, are found to regulate mitochondrial functions by modifying ubiquitination process (Table 2).
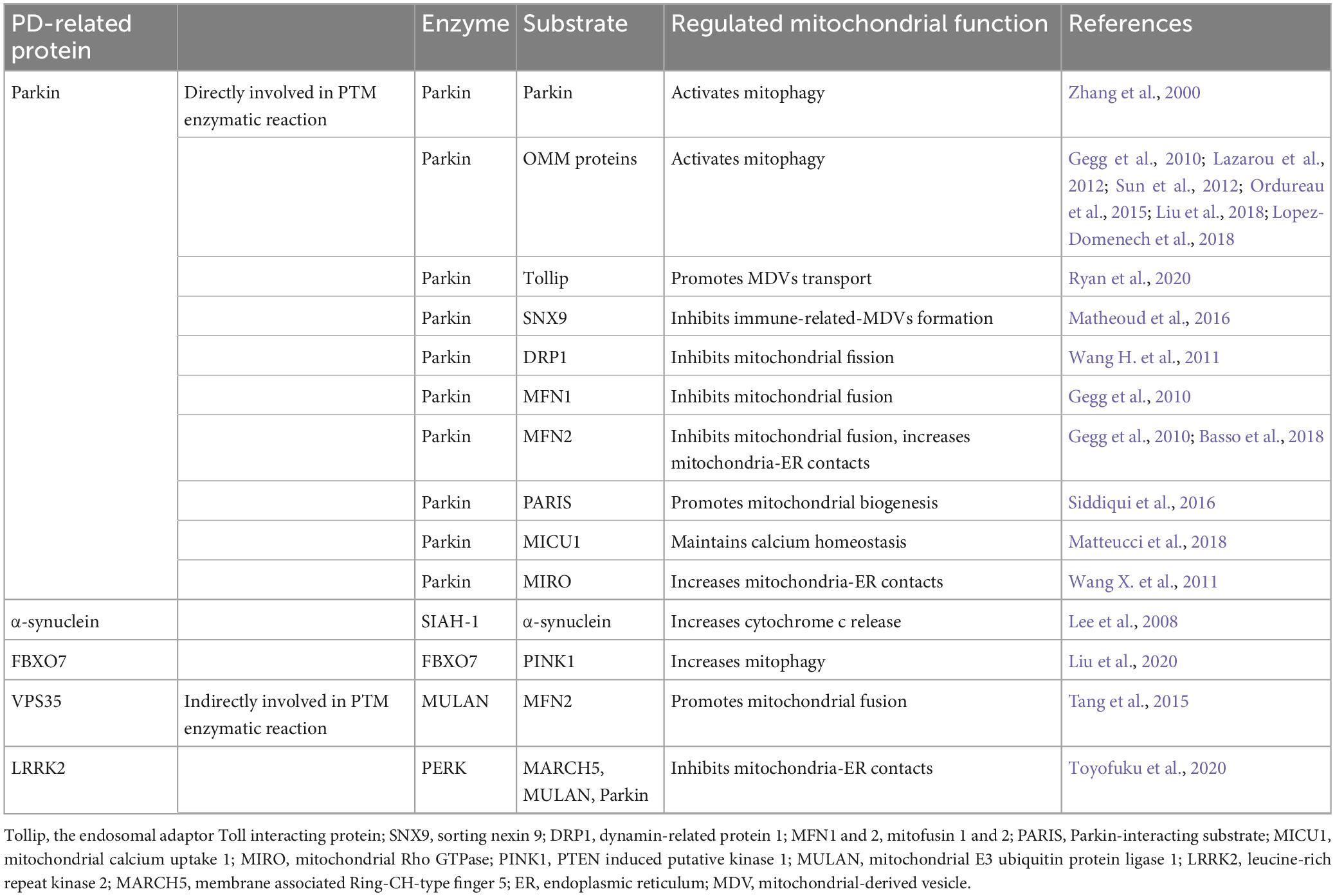
Table 2. Ubiquitination of PD-related proteins and mitochondrial dysfunction.
2.1 Parkin-mediated ubiquitination and mitophagy
Mutations in Parkin gene are the most common genetic cause of juvenile-onset and early-onset PD (EOPD), defined by the appearance of PD symptoms in teens and before the age of 60 (Kitada et al., 1998). The Parkin gene encodes Parkin, an E3 ubiquitin ligase belonging to the RING-between-RING (RBR) family, which accepts Ub from the E2 enzyme and transfers it onto the target protein (Imai et al., 2000; Shimura et al., 2000; Zhang et al., 2000). By ubiquitinating various substrates, Parkin plays a central role in maintaining mitochondrial function and homeostasis (Palacino et al., 2004; Narendra et al., 2008).
The groundbreaking study illustrating Parkin’s role in mitophagy, the biological pathway that selectively eliminates defective mitochondria, was conducted by the Youle laboratory (Narendra et al., 2008). Later studies collectively reveal that Parkin-mediated mitophagy consists of three major steps: (1) initiation, the activation and translocation of Parkin onto the damaged mitochondria (Narendra et al., 2008; Matsuda et al., 2010); (2) priming, the cascade ubiquitination of various targets on the outer mitochondrial membrane (OMM) by Parkin (Tanaka et al., 2010; Chan et al., 2011; Sarraf et al., 2013); and (3) finishing, the lysosomal sequestration and degradation of heavily ubiquitinated mitochondria (Karbowski and Youle, 2011). Evidence suggests that the recruitment of Parkin to the damaged mitochondria is mediated by the binding of Parkin to the mitochondria-situated phosphorylated-Ub (phospho-Ub, pUb), which is formed by the kinase activity of PINK1 (PTEN-induced putative kinase 1) (Xiong et al., 2009; Kane et al., 2014; Sauve et al., 2015). During the priming stage, Parkin ubiquitinates multiple proteins, showing a preference for OMM proteins including Parkin itself, MFN1 and 2 (mitofusin 1 and 2), MIRO1 and 2 (mitochondrial Rho GTPase 1 and 2), VDAC1-3 (voltage-dependent anion channel 1–3), CISD1 (CDGSH iron-sulfur domain 1), and TOM 20, 40, and 70 (translocases of the outer membrane 20, 40, and 70) (Gegg et al., 2010; Lazarou et al., 2012; Sun et al., 2012; Ordureau et al., 2015; Liu et al., 2018; Lopez-Domenech et al., 2018). Ubiquitination of OMM proteins further promotes the recruitment of Ub-binding autophagy receptors such as P62/SQSTM1 (sequestosome 1), OPTN (optineurin), NDP52/CALCOCO2 (calcium-binding and coiled-coil domain-containing protein 2), and NBR1 (neighbor of Brca1). In turn, these receptors elicit the targeting of the damaged mitochondria to LC3-positive phagophores for lysosomal degradation (Figure 1; Heo et al., 2015; Ordureau et al., 2018; Padman et al., 2019).
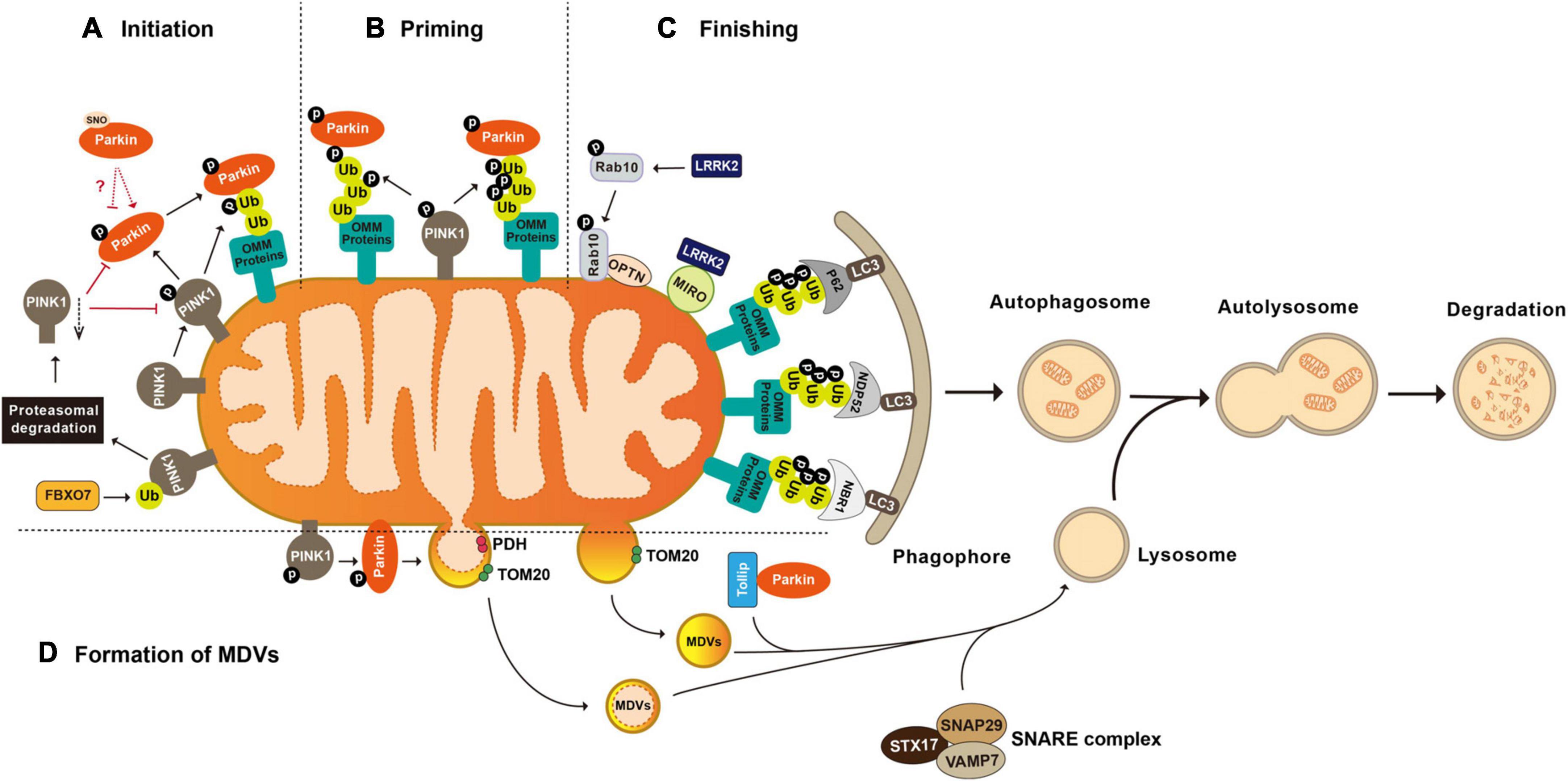
Figure 1. Post-translational modifications in the regulation of mitophagy and mitochondrial-derived vesicles formation. (A) Initiation of mitophagy. Parkin is activated and translocated onto the damaged mitochondria. PINK1 mediated phosphorylation of Parkin and mitochondria situated Ub is essential for Parkin activation. FBXO7 negatively regulates mitophagy activity by enhancing the ubiquitination of PINK1. (B) Priming of mitophagy. After activation, Parkin ubiquitinates a large number of mitochondrial proteins, resulting in increased Ub signal and successively increased pUb signals mediated by PINK1. (C) Finishing of mitophagy. Ubiquitination of OMM proteins further promotes the recruitment of Ub binding autophagy receptors such as P62/SQSTM1, OPTN, NDP52/CALCOCO2, and NBR1 to target the damaged mitochondria to LC3 positive phagophores for lysosomal degradation. LRRK2 mediated phosphorylation regulates mitophagy through its substrates MIRO and Rab10. (D) Formation of mitochondrial-derived vesicles (MDVs). Parkin mediated ubiquitination and PINK1 mediated phosphorylation are required by the formation of PDH+ MDVs. Parkin and Tollip facilitate the transport of TOM20+ MDVs to the endo lysosomal compartment with the help of the STX17 SNAP29 VAMP7 SNARE complex. FBXO7, F-box protein 7; PINK1, PTEN induced putative kinase 1; Ub, ubiquitin; P62/SQSTM 1, sequestosome 1; NDP52/CALCOCO2, calcium binding and coiled coil domain containing protein 2; NBR1, neighbor of Brca1; OPTN, optineurin; MIRO, mitochondrial Rho GTPase; LRRK2, leucine-rich repeat kinase 2; LC3, microtuble-associated protein light chain 3; PDH, pyruvate dehydrogenase; and TOM20, translocases of the outer membrane 20.
Unlike many other E3 ligases that exhibit stringent substrate specificity, Parkin appears to be loose on substrate selectivity. In addition to the well-studied Parkin substrates, recent proteomic analyses have revealed that Parkin ubiquitinates an extensive large number of proteins (Rose et al., 2016; Martinez et al., 2017). After treatment with the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) for extended hours, most OMM proteins are ubiquitinated without necessarily having functional consequences (Chan et al., 2011; Sarraf et al., 2013). In general, Parkin is activated by PINK1-mediated phosphorylation of its N-terminal ubiquitin-like domain. With the help of PINK1, ubiquitination on the OMM by Parkin leads to more pUb and consecutively greater Parkin recruitment and activation, creating a feed-forward loop to amplify the Parkin-mediated ubiquitination to the maximum (Ordureau et al., 2014). Using artificial mitochondria-targeted proteins, Koyano et al. (2019) have found that the substrate specificity of Parkin is not determined by the amino-acid sequence within the substrate, but rather by the presence of pUb on the target protein (Durcan et al., 2012).
Proteomic analyses of purified mitochondria have revealed that Parkin produces a mixture of canonical and non-canonical Ub chains on damaged mitochondria. In general, Ub chains can form through any of the seven lysine (K6, K11, K27, K29, K33, K48, and K63) and the N-terminal methionine (Met1) (Kulathu and Komander, 2012; Akutsu et al., 2016). In vitro, Parkin ubiquitinates mitochondrial proteins with K6-, K11-, K48-, and K63-linked Ub chains to signal damaged mitochondria for mitophagy (Ordureau et al., 2014). Canonical K48-linked Ub chains are crucial for the proteasomal targeting and degradation of modified proteins (Manohar et al., 2019), while K63-linked Ub chains activate the autophagic machinery through recruiting autophagy adaptors like HDAC6 (histone deacetylase 6) and P62 (Seibenhener et al., 2004; Olzmann et al., 2007). Intriguingly, Parkin catalyzes certain degrees of K6- and K11-linked Ub chains on OMM proteins. Activity of Parkin-mediated mitophagy is impaired when either K6 or K11 Ub-linkage was inhibited by the expression of mutant Ub K6R or K11R, suggesting that K6- and K11-linked Ub chains positively regulate the mitophagy process (Durcan et al., 2014; Cunningham et al., 2015). Therefore, Parkin is considered a rather promiscuous E3 ligase that, once activated, ubiquitinates a broad range of proteins with a wide spectrum of Ub chains and amplifies the Ub signals via a positive-feedback manner to maximally ubiquitinated proteins on the damaged mitochondria.
Over 200 missense variants are identified in the Parkin gene, but only a small fraction has been clearly annotated to be pathogenic. Integrating clinical evidence with in vitro mitophagy activity, Yi et al. (2019) has conducted a systematic analysis of 51 Parkin variants, finding a correlation between the degree of mitophagy defect and the level of clinical manifestation. Among them, 13 Parkin variants are classified as pathogenic or likely pathogenic that all display severe mitophagy defects. Those variants designated as non-pathogenic show normal or near-normal mitophagy function (Yi et al., 2019). Likewise, Broadway et al. (2022) have investigated 10 rare Parkin mutants and found 7 with impaired mitophagy activity. These studies suggest that Parkin mediated mitophagy plays important roles in PD pathogenesis.
2.2 Parkin-mediated ubiquitination and mitochondrial-derived vesicles
In addition to mitophagy, an alternative mitochondrial quality control mechanism is via mitochondrial-derived vesicles (MDVs) (Cadete et al., 2016). With this, mitochondria transfer their defective mitochondrial contents to destination organelles, such as lysosomes, peroxisomes, and multivesicular bodies, for degradation via releasing vesicles with 70–150 nm in diameter (Futter et al., 1996; Soubannier et al., 2012a; Vasam et al., 2021; Rosina et al., 2022). Depending on the source of the mitochondrial stress, MDVs derive from the inner mitochondrial membrane (IMM) as a double-membrane structure or from the OMM as a single-membrane vesicle (Konig et al., 2021; Popov, 2022; Heyn et al., 2023). Whether Parkin plays a preferential role in the formation of MDVs remains to be elucidated. In vitro, the formation of MDVs carrying inner membrane marker PDH (pyruvate dehydrogenase) is dependent on Parkin activity (McLelland et al., 2014; Ge et al., 2020). Targeting of Parkin-dependent MDVs to the later endo-lysosomal compartments is mediated by the ternary SNARE protein complex composed of STX17 (syntaxin 17), SNAP29 (synaptosome associated protein 29), and VAMP7 (vesicle-associated membrane protein 7) (Soubannier et al., 2012a; Juhasz, 2016; McLelland et al., 2016). Unlike those PDH-positive MDVs, the presence of MDVs containing only OMM marker like Tom20 is not affected by loss-of-function Parkin mutants T240R or C431S, indicating the independence of Parkin E3 ligase activity (Soubannier et al., 2012b). However, Parkin was found to coordinate with Tollip (the endosomal adaptor Toll interacting protein) to facilitate the transport of these single-membrane MDVs to the endo-lysosomal compartment, with the help of the STX17-SNAP29-VAMP7 SNARE complex (Figure 1; Ryan et al., 2020). In contrast, Parkin seems to inhibit the formation of a subtype of MDVs that plays important roles in immune cells related mitochondrial antigen presentation (MitAP) through triggering ubiquitination and proteasomal degradation of SNX9 (sorting nexin 9) (Matheoud et al., 2016). Thus, Parkin-mediated ubiquitination potentially link between mitochondrial dysfunction and neuroinflammation in PD (Sliter et al., 2018). Consistently, Parkin-null mice show excessive inflammation due to STING-dependent (stimulator-of-interferon-genes dependent) pro-inflammation activity and increased vulnerability to inflammation-induced degeneration (Frank-Cannon et al., 2008; Sliter et al., 2018).
2.3 Parkin-mediated ubiquitination and other mitochondrial functions
2.3.1 Parkin-mediated ubiquitination and mitochondrial dynamics
Mitochondria undergo constant fission-and-fusion in reflection of their functional statuses (Lewis and Lewis, 1914; Chen and Chan, 2017; Kraus et al., 2021). Increased mitochondrial fission results in small and round mitochondria to probably facilitate the mitochondrial trafficking within cells, while enhanced mitochondrial fusion leads to elongated mitochondria for efficient ATP production (Martini and Passos, 2023). Certain mitochondrial damages promote fission activity leading to the separation of depolarized mitochondria to facilitate their later removal by mitophagy or other mitochondrial quality control mechanisms (Han et al., 2020; da Silva Rosa et al., 2021; Cai et al., 2022).
Parkin is responsible for the ubiquitination and proteasomal degradation of several bona fide players in mitochondrial fission-and-fusion pathways, including the fission mediator DRP1 (dynamin-related protein 1) and fusion mediators MFN1 and MFN2 (Narendra et al., 2008; Yamada et al., 2018, 2019; Ham et al., 2020). Parkin interacts with DRP1 through its second RING domain and subsequently ubiquitinates DRP1, leading to its proteasomal degradation. Both the deficiency of Parkin and expression of the PD-associated Parkin mutant C431F inhibit ubiquitination and degradation of DRP1, resulting in mitochondrial fragmentation (Figure 2; Lutz et al., 2009; Wang H. et al., 2011). In Drosophila, overexpression of DRP1 rescues muscle degeneration and mitochondrial abnormalities in PINK1–/– or Parkin–/– mutants (Deng et al., 2008). In vitro, Parkin also ubiquitinates MFN-1 and MFN-2. Ubiquitination of MFN-1 and MFN-2 is reduced in either cell lines with Parkin deficiency or fibroblasts derived from PD patients harboring Parkin mutations (Gegg et al., 2010; Rakovic et al., 2011). In Drosophila, accumulated Marf (Drosophila homolog of MFN), along with reduced ubiquitinated Marf and increased non-ubiquitinated Marf, is observed in Parkin mutant flies (Poole et al., 2010; Ziviani et al., 2010). Thus, Parkin participates in regulation of mitochondrial dynamics.
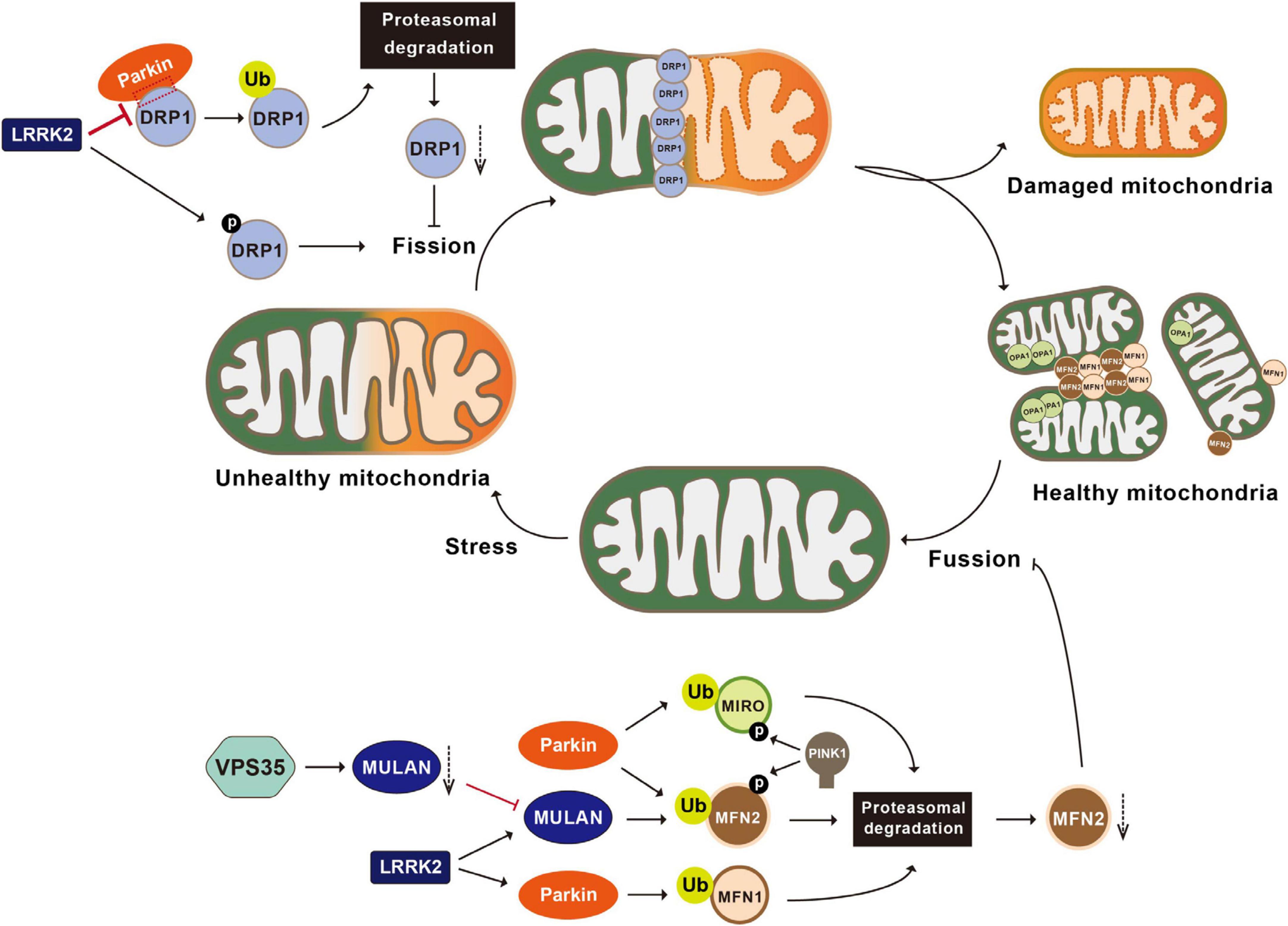
Figure 2. Post-translational modifications in the regulation of mitochondrial dynamics. Mitochondria undergo constant fusion (joining two mitochondria together with the help of MFN1, MFN2, and OPA1) and fission (separating one mitochondrion into two mitochondria with the help of DRP1). Parkin ubiquitinates DRP1, leading to the proteasomal degradation of DRP1 and inhibiting mitochondrial fragmentation. LRRK2 phosphorylates DRP1, impairs the interactions between Parkin and DRP1, and activates MULAN and Parkin’s E3 ligase activities, therefore regulating mitochondrial fragmentation. VPS35 regulates the degradation of the MULAN, thereby inhibiting MUL1 mediated ubiquitination and degradation of MFN2 and promoting mitochondrial fusion. Parkin ubiquitinates MFN1, MFN2 and MIRO, leading to their degradation via the proteasomal system and inhibiting mitochondrial fusion. PINK1 phosphorylates MIRO and activates Parkin mediated ubiquitination and degradation of MIRO. MFN1, mitofusin 1; MFN2, mitofusin 2; OPA1, optic atrophy 1; LRRK2, leucine-rich repeat kinase 2; VPS35, vacuolar protein sorting 35; MULAN, mitochondrial E3 ubiquitin protein ligase; MIRO, mitochondrial Rho GTPase.
2.3.2 Parkin-mediated ubiquitination and mitochondrial biogenesis
Another mechanism cells employ to cope with mitochondrial damage is to generate new mitochondria, a process known as mitochondrial biogenesis (Ivankovic et al., 2016; Popov, 2020). Mitochondrial biogenesis is governed by the master regulator PGC1-α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha). PGC1-α binds and actives nuclear transcription factors NRF-1 and 2 (nuclear respiratory factor 1 and 2) to increase transcription and therefore expression of proteins for mitochondrial biogenesis (Scarpulla, 2008; Li et al., 2011, 2021). PARIS (Parkin-interacting substrate) is a zinc finger protein that binds to and represses PGC-1α. Parkin interacts with PARIS and tags it with Ub chains for proteasomal degradation (Shin et al., 2011; Siddiqui et al., 2015; Stevens et al., 2015; Lee et al., 2017). Parkin deficiency leads to accumulation of PARIS, downregulation of PGC-1α, and selective loss of SNpc DA neurons. All of these are rescued by PGC-1α expression (Shin et al., 2011; Stevens et al., 2015; Siddiqui et al., 2016). These observations suggest that Parkin-mediated ubiquitination plays an important regulatory role in the PGC-1α-mediated mitochondrial biogenesis by ubiquitinating PARIS (Figure 3).
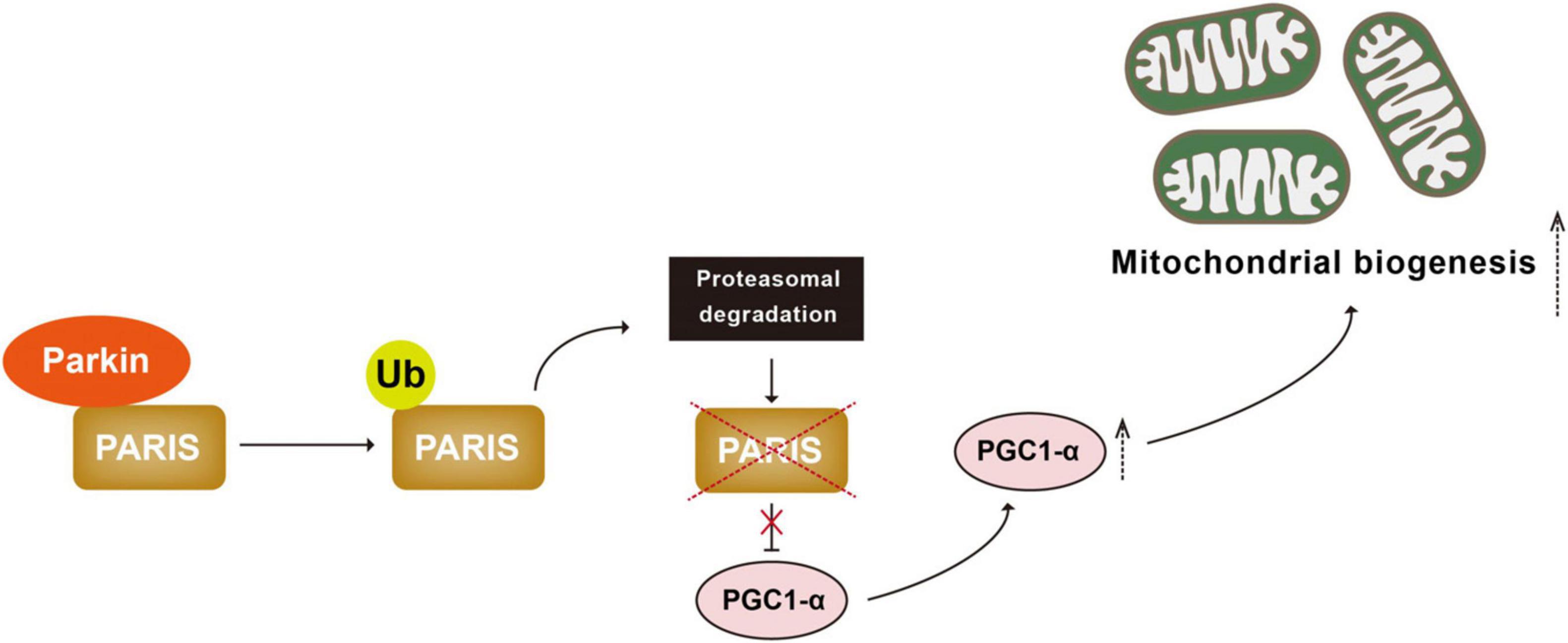
Figure 3. Parkin-mediated ubiquitination regulates mitochondrial biogenesis. PARIS binds to and represses PGC 1α, the master regulator of mitochondrial biogenesis. Parkin interacts with and ubiquitinated PARIS, leading to its proteasomal degradation and promoting mitochondrial biogenesis. PARIS, Parkin-interacting substrate; PGC 1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha.
2.3.3 Parkin-mediated ubiquitination and calcium homeostasis
Recent studies suggest that Parkin regulates calcium homeostasis by ubiquitinating a range of tethering proteins involved in mitochondria-endoplasmic reticulum contact sites (MERCs), such as MFN1, MFN2, MIRO1, and MICU (Figure 4). Parkin in calcium regulation was initially reported by Sandebring et al., 2009. With epidermal growth factor (EGF) stimulation, expression of both PD-associated Parkin mutants (R42P and G328E) or Parkin knockdown results in increased PLCγ1 (phospholipase C gamma1) and elevated basal cytosolic Ca2+ levels. These phenotypes are completely reversed with the treatment of PLC-inhibitor neomycin (Sandebring et al., 2009). A later study demonstrates that glutamate excitotoxicity in neuronal cells triggers Parkin accumulation on the ER and MERCs, suggesting a role of Parkin in the mitochondria-ER crosstalk (Van Laar et al., 2015). In Hela cells that do not express endogenous Parkin, overexpression of Parkin, but not the Parkin mutant lacking Ubl domain, enhances ER-mitochondrial tethering and increases agonist-induced Ca2+ transients (Cali et al., 2013). Conversely, Parkin knockdown impairs mitochondrial Ca2+ transfer and reduces mitochondria-ER contacts, suggesting that Parkin may enhance mitochondria-ER Ca2+ transfer by maintaining MERCs (Cali et al., 2013). However, confounding evidences are observed in fibroblasts derived from PD patients carrying Parkin mutations and in Parkin–/– mice. Loss of Parkin results in close proximity between ER and mitochondria, leading to increased mitochondria-ER Ca2+ transfer, while overexpression of Parkin could restore the cytosolic Ca2+ transient to normal (Gautier et al., 2016).
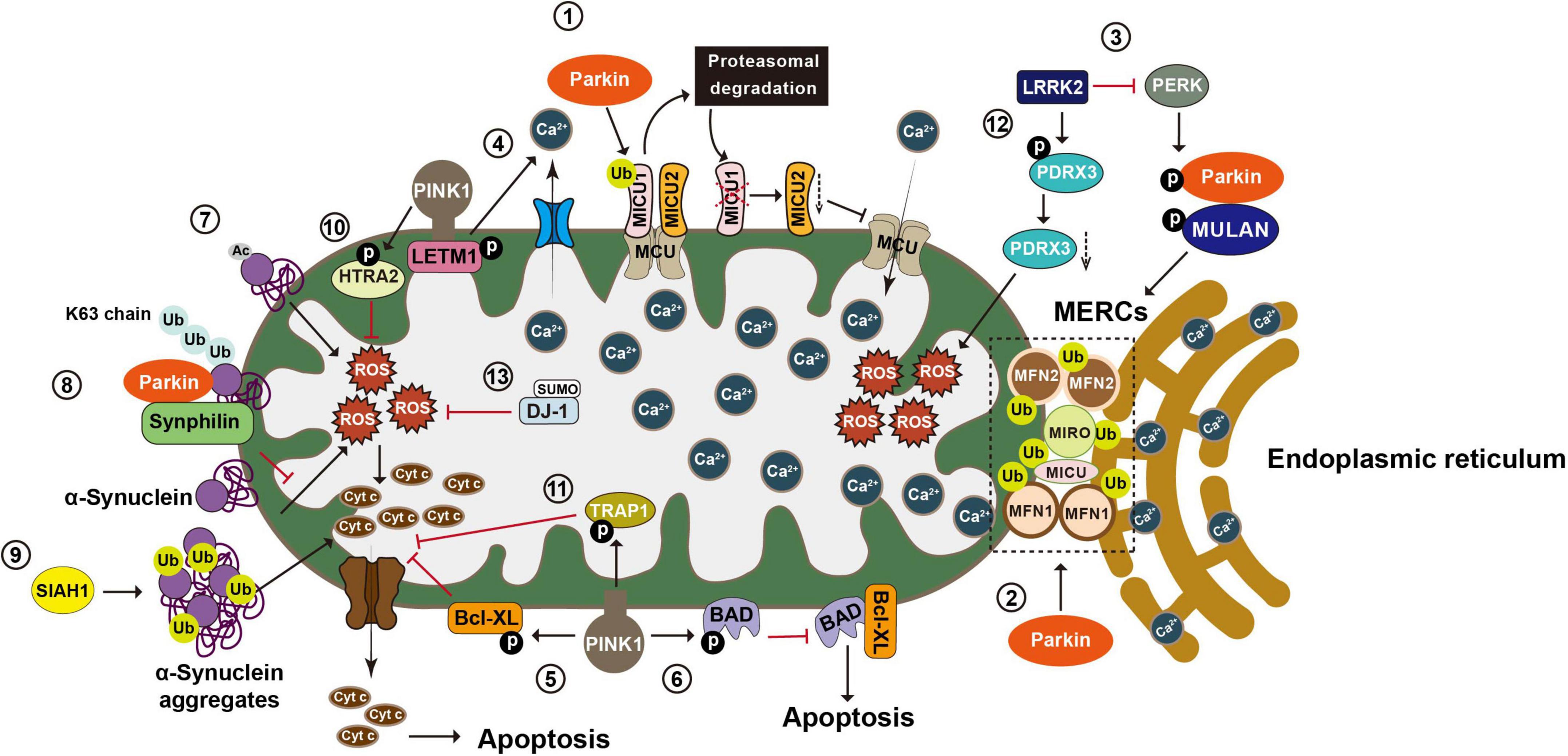
Figure 4. Post-translational modifications in the regulation of calcium homeostasis, oxidative stress, and apoptosis. Mitochondria play an essential role in maintaining Ca2+ homeostasis, regulating oxidative stress, and integrating apoptosis signals. (1) Mitochondria take up Ca2+ through the MCU complex, which is positively and negatively regulated by MICU1 and 2, respectively. Parkin regulates mitochondrial calcium homeostasis by directly ubiquitinating MICU1, leading to its proteasomal degradation, and indirectly affecting MICU2’s protein level. (2) Parkin also regulates calcium homeostasis by ubiquitinating tethering proteins involved in mitochondria endoplasmic reticulum contact sites (MERCs), such as MFN1, MFN2, MIRO1, and MICU. (3) LRRK2 blocks PERK mediated phosphorylation and activation of Parkin and MULAN, causing increased degradation of MERCS tethering proteins and reduced ER mitochondrial contacts. (4) PINK1 phosphorylates LETM1, leading to increased calcium release and facilitating calcium transport in mitochondria. (5) PINK1 phosphorylates Bcl XL and prevents its pro apoptotic cleavage. (6) PINK1 also phosphorylates BAD and prevents the formation of pro apoptotic Bcl XL BAD complex. (7) N α acetylation of α synuclein induces mitochondrial dysfunction. (8) Parkin increases K63-linked Ub chains on α synuclein, leading to the recruitment of Synphilin 1 to inhibit α synuclein toxicity. (9) SIAH-1 facilitates ubiquitination of α synuclein, increasing α synuclein’s insolubility and exacerbating its aggregation associated toxicity. (10) PINK1 mediated phosphorylation enhances the proteolytic activity of HTRA2 and protects cells against mitochondrial stress. (11) Phosphorylation of TRAP1 by PINK1 inhibits cytochrome c release and reduces cell death during oxidative stress. (12) LRRK2 phosphorylates PRDX3 and decreases its peroxidase activity. (13) SUMOylation of DJ 1 is crucial for of DJ 1’s anti-ROS activity. MCU, mitochondrial calcium uniporter; MICU1, mitochondrial calcium uptake 1; MICU2, mitochondrial calcium uptake 2; MFN1, mitofusin 1; MFN2, mitofusin 2; MIRO1, mitochondrial Rho GTPase 1; LRRK2, leucine-rich repeat kinase 2; PERK, protein kinase RNA-like ER kinase; LETM1, leucine zipper-EF-hand-containing transmembrane protein 1; ER, endoplasmic reticulum; Bcl XL, B-cell lymphoma-extra large; BAD, BCL2 associated agonist of cell death; Ub, ubiquitin; SIAH-1, seven in absentia homolog-1; HTRA2, high-temperature requirement serine protease A2; TRAP1, TNF receptor-associated protein 1; PRDX3, peroxiredoxin 3; ROS, reactive oxygen species.
Mitochondria take up Ca2+ through the mitochondrial calcium uniporter (MCU) complex that are composed of four core components, including the pore-forming subunit MCU, gatekeeping subunits MICU1 and 2 (mitochondrial calcium uptake 1 and 2), and the auxiliary subunit EMRE (essential mitochondrial regulator) (Sancak et al., 2013; Raffaello et al., 2013). MICU1 and 2, respectively, act as positive and negative regulators of the MCU complex (Plovanich et al., 2013). Under basal conditions, Parkin ubiquitinates MICU1, leading to its rapid degradation via the proteasomal system. Given that MICU2 stability is strictly dependent on MICU1, Parkin-mediated ubiquitination also indirectly regulates the amount of MICU2 (Matteucci et al., 2018). By maintaining appropriate levels of MICUs, Parkin plays a regulatory role in mitochondrial calcium homeostasis. Additionally, Parkin exerts its effects on ER-mitochondrial tethering via ubiquitinating of MFN2. ER located MFN2 forms a homotypic or heterotypic complex with either mitochondria-located MFN2 or mitochondria-located MFN1, thereby bridging mitochondria and ER (de Brito and Scorrano, 2008). Parkin ubiquitinates MFN2 on K416 in the HR1 domain, which in turn positively affects the physical and functional interactions between mitochondria and ER. Parkin-resistant MFN2 mutant K416R loses the ability to restore the decreased mitochondria-ER interactions in Parkin deficient cells and fibroblasts carrying PD-associated Parkin mutants (Basso et al., 2018). MIRO is another substrate of Parkin that regulates mitochondria-ER contacts. In yeast cells, yeast MIRO (Gem1p) deficiency leads to significant decrease of MERCs (Figure 4; Wang X. et al., 2011).
2.4 Ubiquitination of other PD-associated proteins and mitochondrial functions
2.4.1 Ubiquitination of α-synuclein and mitochondrial functions
α-Synuclein, a principal component of LB, is a key protein involved both genetically and pathologically in PD. Mutations in the α-synuclein gene cause familiar forms of PD. The polymorphisms of α-synuclein confer a relatively increased risk of developing idiopathic PD. Since 1997, point mutations and duplication of α-synuclein gene have been inventoried with autosomal dominant PD, suggesting a gain-of-function mechanism (Polymeropoulos et al., 1997; Fuchs et al., 2007; Elia et al., 2013).
α-Synuclein is predominantly found in the presynaptic terminals of the central nervous system and is implicated in regulating synaptic plasticity and neurotransmitter release (Burre et al., 2010; Venda et al., 2010). Compelling evidence suggests that mitochondrial functions, such as cytochrome c release, calcium homeostasis, ATP production, and mitochondrial fission-and-fusion balance, are also directly regulated by α-synuclein. Remarkably, α-synuclein is found in all mitochondrial compartments and can selectively be localized to mitochondria under stress conditions (Ulmer et al., 2005; Li et al., 2007; Parihar et al., 2008; Zhang et al., 2008; Devi and Anandatheerthavarada, 2010; Subramaniam et al., 2014). The N-terminal 32 amino-acid sequence of α-synuclein is critical for its mitochondrial translocation. α-Synuclein undergoes multiple PTMs, including ubiquitination. α-Synuclein is a target of Parkin upon mitochondrial stress. Under basal conditions, no interaction between α-synuclein and Parkin is detected. With the treatment of CCCP or rotenone, Parkin forms a complex with α-synuclein and catalyzes formation of K63-linked Ub chains to recruit Synphilin 1, a negative regulator of α-synuclein toxicity, to the α-synuclein-Parkin complex. Therefore, Parkin-mediated ubiquitination may negatively regulate α-synuclein’s response to the mitochondrial stress (Norris et al., 2015). Consistently, α-synuclein is the substrate of RING-type E3 ligase SIAH-1 (seven in absentia homolog-1). SIAH-1 facilitates the mono- and poly-ubiquitination of α-synuclein to increase α-synuclein insolubility and exacerbate its aggregation-associated toxicity (Figure 4). Cells overexpressing WT α-synuclein or PD-associated mutant α-synuclein A53T show exacerbated cytochrome c release and apoptosis with increased of SIAH-1 activity (Lee et al., 2008).
Ubiquitinated α-synuclein is predominately found in LBs and has been demonstrated to contribute to the misfolding of α-synuclein, mitochondrial dysfunction, and neuronal death (Spillantini et al., 1997; Devi et al., 2008; Choubey et al., 2011). Despite these associations, efforts to establish α-synuclein as a biomarker for PD diagnosis or prognosis primarily focus on three species of α-synuclein-total α-synuclein, phosphorylated α-synuclein, and the oligomeric form of α-synuclein-in tissues and fluids like blood components, CSF, saliva, and extracellular vesicle (EVs) (Hong et al., 2010; Tokuda et al., 2010; Mollenhauer et al., 2011, 2013; Foulds et al., 2012; Wang Y. et al., 2012; van Steenoven et al., 2018; Vivacqua et al., 2019; Stuendl et al., 2021). Monoubiquitinated and polyubiquitinated α-synuclein are able to be detected in plasma using a polyclonal anti-ubiquitin antibody (FL-76) (Foulds et al., 2011). It will of interest in studying whether this is useful for PD diagnosis.
2.4.2 FBXO7-regulated ubiquitination and mitochondrial functions
Mutations in FBXO7 cause autosomal recessive EOPD (Shojaee et al., 2008). Being a F-box domain-containing protein, FBXO7 (F-box protein 7) acts as an adapter for Skp1-Cullin-F-box (SCF) type E3-ubiquitin ligase (Skowyra et al., 1997). Emerging evidence suggests that FBXO7 participates in the maintenance of mitochondrial homeostasis through facilitating ubiquitination modification of key proteins in the mitochondrial quality control pathways. Overexpression of FBXO7 enhances Parkin recruitment onto damaged mitochondria, suggesting a role of FBXO7 in mitophagy process (Burchell et al., 2013). It seems that FBXO7 negatively regulates mitophagy activity by enhancing the ubiquitination of PINK1, thereby, suppressing Parkin E3 ligase activity (Figure 1; Liu et al., 2020). Furthermore, ubiquitination of MFN1 was also found to be significantly reduced in both fibroblasts derived from patients harboring homozygous FBXO7 R378G mutation and in SH-SY5Y cells with FBXO7 deficiency, which is restored by wild-type FBXO7. However, a recent study reports that FBXO7 is dispensable in the Parkin-mediated mitophagy process. Accumulation of pUb, recruitment of Parkin onto mitochondria, and mitophagy flux are all barely affected in FBXO7–/– cells. Moreover, increased pUb foci were detected in FBXO7–/– cells than that in control cells after treating with mitochondrial targeted HSP90 (heat shock protein 90) inhibitor Gamitrinib-TPP (Kraus et al., 2023). It is possible that FBXO7 regulates mitochondrial functions differentially depending on conditions. Nonetheless, the regulation is likely one via ubiquitination modification.
2.4.3 VPS35-regulated ubiquitination and mitochondrial functions
VPS35 (vacuolar protein sorting 35) is a critical component of the retromer complex that is important for the retrograde transport of transmembrane protein-cargo from endosomes to the trans-Golgi network (TGN) (Hierro et al., 2007). Mutations in VPS35 have recently been identified as the cause of familiar PD. A single missense mutation in VPS35, c.1858G > A (p.D620N), has been unambiguously identified to segregate with late-onset PD (LOPD) in an autosomal dominant manner (Zimprich et al., 2011). Studies suggest that VPS35 can indirectly participate in mitochondrial dynamics via the alteration of ubiquitination events. Using a transgenic mouse model, Tang et al. (2015) revealed that VPS35 regulates the trafficking and degradation of the MULAN (mitochondrial E3 ubiquitin protein ligase 1), thereby inhibiting MUL1-mediated ubiquitination and degradation of MFN2 and promoting mitochondrial fusion (Figure 2). Consistently, selective deletion of the VPS35 in DA neuron results in mitochondrial fragmentation and PD-relevant pathologies in VPS35± mice (Tang et al., 2015).
2.4.4 LRRK2-regulated ubiquitination and mitochondrial functions
Another PD-associated protein that indirectly participate in mitochondrial dynamics through ubiquitination modification is LRRK2 (leucine-rich repeat kinase 2). LRRK2, also known as dardarin (from the Basque word “dardara” that means trembling) and PARK8 (from early identified association with PD), is a large multifunctional kinase. Variants of this gene are associated with increased risk of PD and Crohn’s disease (Funayama et al., 2002). LRRK2, through its N-terminal domain, interacts with mitochondrial membrane-binding E3 ubiquitin ligases MARCH5 (membrane associated Ring-CH-type finger 5), MULAN, and Parkin. The kinase activity of LRRK2 is required for activation of these E3 ligase (Figure 2). No evidence of direct phosphorylation of these E3 ligases by LRRK2 has been found in in vitro assays, but screening assay using siRNA library revealed that PERK (protein kinase RNA-like ER kinase) is the kinase that directly phosphorylates and activates these E3 ligases. Via binding to these E3 ligases, LRRK2 WT blocks the PERK-mediated phosphorylation and activation of these E3 ligases. PD-associated LRRK2 mutant G2019S has decreased binding activity to these E3 ligases, resulting in increased phosphorylation and activation of E3 ubiquitin ligases by PERK, which consequently causes increased degradation of MERCS tethering proteins and reduced ER-mitochondrial contacts (Figure 4; Toyofuku et al., 2020).
3 Regulation of mitochondrial functions by phosphorylation in Parkinson’s disease
Protein phosphorylation, a prevalent PTM mediated by kinases, involves the covalent attachment of a phosphate group to an amino acid residue like serine (S), threonine (T), or tyrosine (Y). This dynamic modification offers a swift mechanism to alter protein function, thus playing crucial roles in regulation of various cellular pathways. Emerging evidence indicates that aberrant phosphorylation of proteins impacts mitochondrial functions, such as mitochondrial dynamics, mitophagy, MDV formation, mitochondrial respiratory activity, and calcium homeostasis. Notably, PD-associated kinase proteins, such as PINK1 and LRRK2, have been found to cause mitochondrial dysfunction through their dysregulated phosphorylation activity, ultimately leading to PD pathogenesis. Therefore, we aim to provide an overview elucidating how PINK1 and LRRK2 modulate a range of mitochondrial functions via their kinase activity, outlining their implications in the pathogenesis of PD through protein phosphorylation (Table 3).
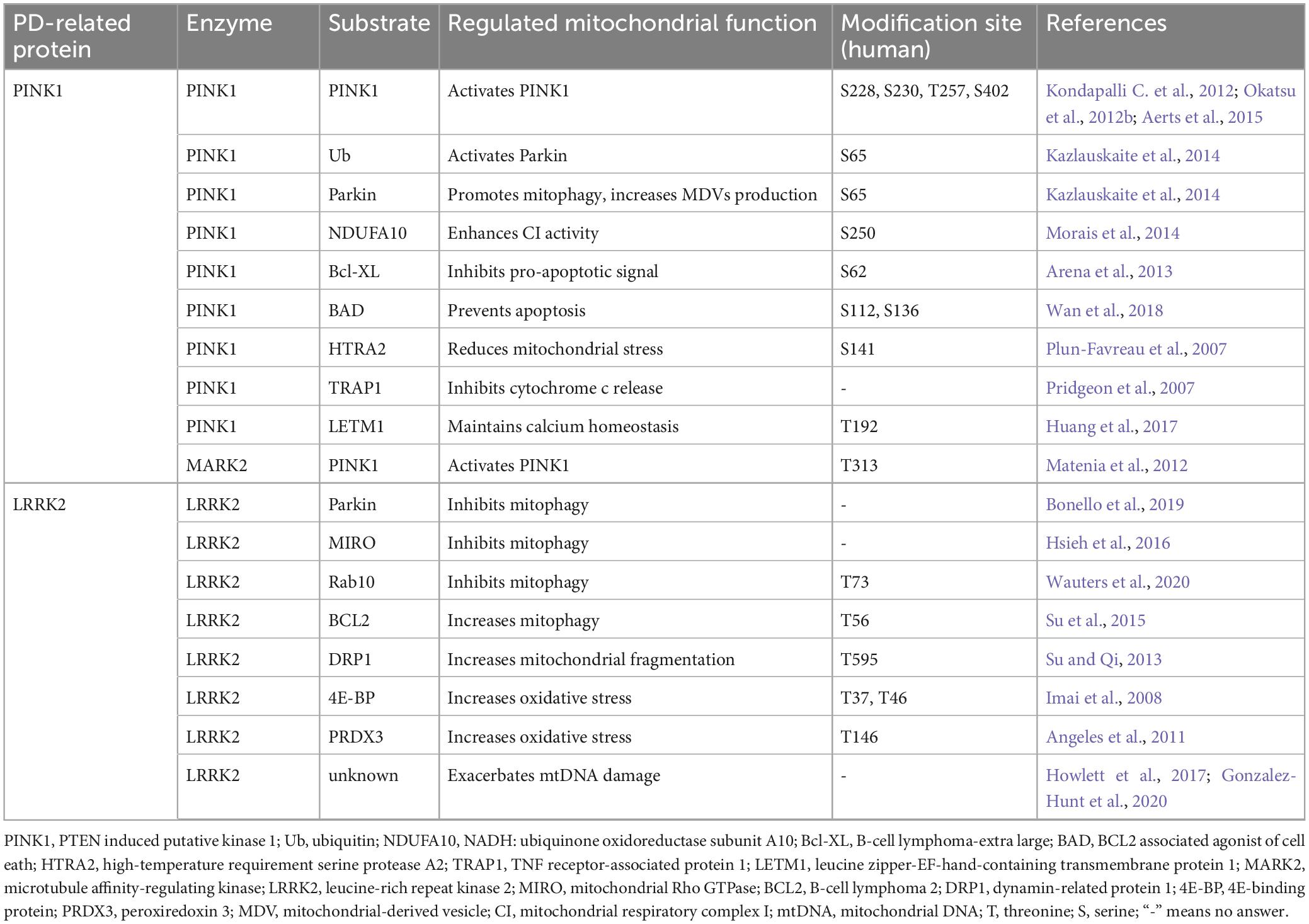
Table 3. Phosphorylation of PD-related proteins and mitochondrial dysfunction.
3.1 PINK1-mediated phosphorylation and mitophagy
Mutations in PINK1 are the second most common cause of EOPD, accounting for 1–9% PD patients. More than 300 PINK1 variants have been identified from PD patients (Ma et al., 2021; Vizziello et al., 2021). The PINK1 gene encodes a 581 amino acid protein, which contains an N-terminal mitochondrial targeting sequence (MTS), a transmembrane domain (TMD), and a highly conserved S/T kinase domain (Cardona et al., 2011). Under normal conditions, PINK1 is targeted to mitochondria through its MTS via the TOM (translocase of the outer membrane) and TIM (translocase of the inner membrane) complexes (Lazarou et al., 2012). During the translocation process, PINK1 undergoes consecutive cleavages by MPP (mitochondrial processing peptidase) and PARL (presenilin-associated rhomboid-like protease) (Jin et al., 2010; Deas et al., 2011; Greene et al., 2012), and the cleaved 52-kDa PINK1 is retro-translocated into the cytosol and undergoes rapid turnover by the proteasome via the N-end rule pathway (Whitworth et al., 2008; Greene et al., 2012). Therefore, PINK1 is normally maintained at a very low steady-state level. With damaged mitochondria, PINK1’s mitochondrial import is inhibited by the reduced mitochondrial membrane potential, which leads to the accumulation of full-length PINK1 on the OMM (Jin and Youle, 2013).
The landmark studies suggesting that both Parkin and PINK1 function through a common pathway to regulate mitochondrial function were from a series of Drosophila research (Greene et al., 2003; Clark et al., 2006; Park et al., 2006). Not only do PINK1–/– and Parkin–/– mutant flies exhibit similar degenerative phenotypes in neuron and muscle cells due to mitochondrial abnormalities, but overexpression of Parkin also rescues the PINK1–/– phenotype, not vice versa, suggesting PINK1 acts upstream of Parkin in a linear pathway (Yang et al., 2006). Later on, more studies gradually unveil the models of how PINK1 and Parkin work together to regulate various mitochondrial functions (Durcan and Fon, 2015; Mouton-Liger et al., 2017).
PINK1-mediated phosphorylation plays essential roles at multiple steps of the mitophagy process, largely interacting with Parkin-mediated ubiquitination modification (Ordureau et al., 2014). First, autophosphorylation of PINK1 is important for its own activation, coinciding with its accumulation on the OMM (Kondapalli C. et al., 2012). PINK1 activity is determined by autophosphorylation at residues S228, S230, T257, and S402 (Kondapalli C. et al., 2012; Okatsu et al., 2012b; Aerts et al., 2015; Rasool et al., 2022). Although conflicting results regarding the regulatory role of autophosphorylation residues in PINK1are reported, autophosphorylation at S228 is important for the subsequent phosphorylation of PINK1 substrates in cells (Okatsu et al., 2012a; Aerts et al., 2015; Kumar et al., 2017; Rasool et al., 2018). Furthermore, the equivalent site of human PINK1 S228 is confirmed to be autophosphorylated in multiple PINK1 homologs, including S346 of Drosophila PINK1, S205 of Tribolium PINK1, and S202 of Pediculus PINK1 (Rasool et al., 2018). Drosophila carrying the PINK1 S346A mutant displays similar mitochondrial defects to those observed in PINK1–/– mutant flies (Clark et al., 2006). Second, PINK1 phosphorylates Parkin at S65 of the ubiquitin domain to induce recruitment of Parkin to mitochondria and the release of Parkin E3 ligase activity. PINK1-mediated phosphorylation is essential for Parkin activation (Xiong et al., 2009; Kondapalli K. C. et al., 2012; Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Wauer et al., 2015a). In the absence of an activation signal, Parkin stays in the cytosol in an auto-inhibited structure due to the inhibitory intradomain contacts (Trempe and Gehring, 2023). PINK1 phosphorylates Ub on S65 (Kazlauskaite et al., 2014; Koyano et al., 2014; Wauer et al., 2015b). pUb serves as a receptor for Parkin to bind. Upon pUb binding, Parkin Ubl domain becomes more accessible by PINK1, leading to the subsequent phosphorylation of Ubl S65 (Durcan and Fon, 2015). Phosphorylation of Parkin by PINK1 further dissociates the inhibitory intradomain-contacting inside Parkin, resulting in the full activation of Parkin’s enzymatic activity (Trempe and Gehring, 2023). Third, PINK1-mediated phosphorylation amplifies the Parkin-mediated ubiquitination signal. After activation, Parkin ubiquitinates a large number of mitochondrial proteins and thereby produces increased Ub substrates for PINK1 to generate pUb signals, which successively initiate more Parkin recruitment and greater Parkin activation, creating a feed-forward loop to reach a maximal of Parkin-mediated ubiquitination (Figure 1; Ordureau et al., 2015; Koyano et al., 2019). On a relevant note, MARK2 (microtubule affinity-regulating kinase) is identified as an activating kinase of PINK1. MARK2 phosphorylates PINK1 at residue T313 that coincidentally is a residue frequently mutated to a non-phosphorylatable form T313M in PD cases (Tang et al., 2006). The expression of PINK1 T313M causes severe toxicity and abnormal mitochondrial accumulation in cells, also suggesting the mitochondrial consequence of the PINK1 activity (Matenia et al., 2012).
PINK1-mediated phosphorylation is a potential biomarker for PD diagnosis (Chin and Li, 2016). An antibody designed for pS65 on Ub (pS65-Ub) reveals that a rapid accumulation of pS65-Ub signal in mitochondria following mitochondrial damage. The presence of pS65-Ub positive granule also increases in cells derived from aging individuals and sporadic PD cases (Fiesel et al., 2015). Furthermore, a patent filed by Geldenhuys et al. (2014) details the use of PINK1 T257 autophosphorylation and PINK1-mediated phosphorylation of Parkin at S56 in serum and CSF as diagnostic measures. Additional efforts will be required to evaluate their clinical applicability.
Of the reported PD-associated PINK1 mutations, about 30 of them are defined as “pathogenic” or “likely pathogenic,” causing similar clinical symptoms as in the cases caused by Parkin mutations (Richards et al., 2015; Ellard et al., 2020). Recently, Ma et al. (2021) analyzed 50 PINK1 variants and found most these pathogenic variants cause a significant decrease in mitophagy activity. However, these consist of only a small fraction of identified pathogenic PINK1. Further investigation is still needed to understand PINK1-phosphorylation regulated mitophagy in PD pathogenesis (Lin and Kang, 2008).
3.2 PINK1-mediated phosphorylation and mitochondrial-derived vesicles
PINK1, along with Parkin, are identified as key regulators of the MDV pathways that plays important roles in the regulation of mitochondrial turnover and MitAP production (Matheoud et al., 2016). PINK1 was initially found to be required for the formation of MDVs that deliver damaged mitochondrial portion to the lysosome for degradation (Sugiura et al., 2014; Pickrell and Youle, 2015). Ramirez et al. (2022) have recently reported that cannabidiol (CBD) activates PINK1 and Parkin in a dose-dependent manner, leading to elevated production of MDVs. CBD causes PINK1 accumulation on the mitochondrial out membrane to activate Parkin to promote the generation of MDVs (Figure 1). However, detailed mechanism underlying PINK1-regulated MDV formation remains unknown. It is possible that PINK1 participates in recognition of damaged sites of mitochondria and promotes segregation of damaged part of mitochondria. Consistent with this notion, PINK1 is shown to phosphorylate DRP1at S616 to activate fission, a potential mechanism to separate damaged and health portion of a mitochondrion (Han et al., 2020).
3.3 PINK1-mediated phosphorylation and other mitochondrial functions
3.3.1 PINK1-mediated phosphorylation and mitochondrial dynamics
PINK1 is implicated in the phosphorylation of mitochondrial proteins that are important for the regulation of mitochondrial dynamics. First, PINK1 phosphorylates a group of key players in the mitochondrial dynamic pathways. The dynamin-related GTPase DRP1 is a crucial factor of the mitochondrial fission machinery, and its activity is regulated by phosphorylation (Yu et al., 2019; Han et al., 2020). Around 10 residues of DRP1 are able to be phosphorylated. Phosphorylation of S616 and S637 is extensively studied (Kim et al., 2016; Cha et al., 2021). After recruiting to the OMM, DRP1 is phosphorylated by PKA (protein kinase A) at S637, inhibiting DRP1 GTPase activity and suppressing its translocation to the mitochondria, and thereby impeding mitochondrial fission (Cereghetti et al., 2008). DRP1S616 was initially found to be phosphorylated by Cdk1/cyclin B, resulting in mitochondrial fragmentation (Pryde et al., 2016). We recently demonstrate that PINK1 directly phosphorylate DRP1S616 site to regulate mitochondrial fission that is independent of Parkin and autophagy activity (Han et al., 2020). MFN2 is also a substrate of PINK1. PINK1 phosphorylates MFN2 at residues T111 and S442, resulting in increased ubiquitination and proteasomal degradation of MFN2, leading to eventual mitochondrial fusion via Parkin mediated mechanism (Chen and Dorn, 2013; Tsai et al., 2014). Likewise, PINK1 phosphorylates MIRO1 at residue S156, in turn activates Parkin-mediated ubiquitination and degradation of MIRO1, therefore, inhibits axonal transport of mitochondria (Figure 2; Wang X. et al., 2011).
3.3.2 PINK1-mediated phosphorylation and mitochondrial respiratory activity
Both PINK1 deficiency and PD-associated PINK1 mutants impair functions of mitochondrial respiratory complex I (CI) (Morais et al., 2009, 2014). NDUFA10 (NADH: ubiquinone oxidoreductase subunit A10) is an auxiliary subunit of CI. Although it is still unclear whether PINK1 directly phosphorylates NDUFA10, phosphoproteomic analysis reveals abolished phosphorylation of NDUFA10 at residue S250 in PINK1 knockout (KO) mice. Furthermore, both WT NDUFA10 and the phosphomimetic NDUFA10 mutant (S250D) enhance CI activity and rescue PINK1 deficiency-induced mitochondrial damage in mouse and cellular models. In contrast, the phosphorylation deficient mutant NDUFA10 S250A fails to rescue the PINK1 deficiency-related phenotypes (Morais et al., 2014). These results suggest a crucial role of phosphorylated NDUFA10 in the regulation of mitochondrial bioenergetics. Consistently, NDUFA10 improves PINK1 knockdown (KD)-induced mitochondrial hyperfusion in Drosophila by increasing CI activity (Pogson et al., 2014).
3.3.3 PINK1-mediated phosphorylation and apoptosis
Mitochondria hold a central position in the apoptosis process. PINK1-mediated phosphorylation is found to prevent mitochondria-mediated cell death in multiple ways. PINK1 phosphorylates Bcl-XL (B-cell lymphoma-extra large) and prevents its pro-apoptotic cleavage. Bcl-XL has an anti-apoptotic activity by protecting the mitochondrial membrane potential (Δψ) and preventing cytochrome c release via its binding to and inhibition of VDACs. The N-terminal BH4 domain of the Bcl-XL is essential for this apoptosis inhibition activity (Shimizu et al., 2000). Upon mitochondrial depolarization, PINK1 interacts with Bcl-XL and phosphorylates it at residue S62, leading to the resistance of Bcl-XL to the cleavage of its N-terminal and the reduction of pro-apoptotic signal (Arena et al., 2013). BAD (BCL2 associated agonist of cell death) is a BH3-only protein. Phosphorylation of BAD at residue S112 inhibits its ability to form pro-apoptotic complex with Bcl-XL on the OMM (Hirai and Wang, 2001). Upon CCCP treatment, PINK1 phosphorylates BAD at residues S112 and S136, preventing the formation of pro-apoptotic Bcl-XL-BAD complex, leading to cell survival (Figure 4; Wan et al., 2018).
HTRA2 (high-temperature requirement serine protease A2) is implicated in the pathogenesis of PD and other neurodegenerative conditions. Mutations in HTRA2 are a risk factor for sporadic PD cases. As a mitochondrial serine protease, HTRA2 functions in mitochondrial quality control and apoptosis. During apoptosis, HTRA2 is released into the cytosol and facilitates apoptosis by antagonizing IAPs (inhibitors of apoptosis). Interestingly, HTRA2 activation is dependent on the direct phosphorylation of its S141 by PINK1. PINK1-mediated S141 phosphorylation enhances the proteolytic activity of HTRA2 and protects cells against mitochondrial stress (Figure 4; Plun-Favreau et al., 2007). HTRA2 variants, A141S and P143A, identified in sporadic PD patients are in close proximity to the S142, suggesting they may contribute to PD by interfering with PINK1-mediated phosphorylation and HTRA2 activation (Strauss et al., 2005; Lin et al., 2011). Genetic studies in Drosophila further demonstrated the functional interaction between PINK1 and HTRA2. These studies collectively suggest that HTRA2, in parallel with Parkin, acts downstream of the PINK1 to maintain mitochondrial integrity (Whitworth et al., 2008; Tain et al., 2009). Consistently, reduced HTRA2 phosphorylation is observed in brains of PD patients carrying PINK1 mutations (Plun-Favreau et al., 2007).
TRAP1 (TNF receptor-associated protein 1), also known as HSP75 (heat shock protein 75), is a mitochondrial chaperone protein. PINK1 binds to TRAP1 on mitochondria and phosphorylates TRAP1 in the mitochondrial intermembrane space (IMS). Phosphorylation of TRAP1 by PINK1 inhibits cytochrome c release and reduces cell death during oxidative stress. PD-associated PINK1 mutants G309D and L347P, both with reduced kinase activity, diminish the TRAP1 phosphorylation and result in increased apoptosis upon mitochondrial oxidative stress (Pridgeon et al., 2007). In Drosophila, TRAP1 deficiency results in mitochondrial dysfunction and vulnerability to various mitochondrial stress. Overexpression of human TRAP1 rescues PINK1-deficiency induced mitochondrial abnormalities (Costa et al., 2013; Zhang et al., 2013).
3.3.4 PINK1-mediated phosphorylation and calcium homeostasis
Mitochondria are both major effectors and essential regulators of intracellular Ca2+ levels. The amount of Ca2+ retained inside the mitochondrial matrix is regulated by mitochondrial Ca2+ transient, which includes Ca2+ influx mediated by MCU and mitochondrial Ca2+ efflux mediated by Na+/Ca2+ and H+/Ca2+ antiporters (Szabadkai et al., 2006). PINK1 deficiency leads to dysfunction of the Na+/Ca2+ exchanger and causes mitochondrial calcium overload (Gandhi et al., 2009). Studies reveal that LETM1 (leucine zipper-EF-hand-containing transmembrane protein 1) is a mitochondrial H+/Ca2+ antiporter situated on the IMM. PINK1 interacts with LETM1 and directly phosphorylates it at residue T192, leading to increased calcium release in liposomes and facilitating calcium transport in mitochondria (Figure 4). Both PINK1 deficiency and PD-associated mutant PINK1 Q456X significantly reduce LETM1 phosphorylation, causing mitochondrial calcium-transport dysfunction and neuronal death (Huang et al., 2017).
3.4 LRRK2-mediated phosphorylation and mitochondrial functions
3.4.1 LRRK2-mediated phosphorylation and mitophagy
Mutations in LRRK2 are the most common cause of autosomal dominant LOPD (Zimprich et al., 2004). LRRK2 encodes a 286-kDa protein containing multiple domains, including a leucine-rich repeat (LRR), a Ras of complex protein (ROC) GTPase domain, a mitogen-activated kinase domain, and WD40 domains. Therefore, LRRK2 is a bienzymatic protein with both GTPase and kinase activities. Six pathogenic mutations have been identified in LRRK2, including R1441C/G, N1437H, Y1699C, G2019S, and I2020T (Ross et al., 2011). The most common LRRK2 mutation, G2019S, is located right in the kinase domain. This mutation increases LRRK2 kinase activity toward itself and other substrates (Ross et al., 2011). Thus, the increased kinase activity of LRRK2 is considered important in the pathogenesis of PD.
While majority of LRRK2 is located at cytoplasm, a portion of LRRK2 is associated with the OMM (West et al., 2005; Biskup et al., 2006). iPSC-derived neural cells bearing LRRK2 G2019S and fibroblasts derived from PD patients carrying LRRK2 G2019S show mitochondrial impairment, suggesting that LRRK2 pathogenesis might involve mitochondrial dysfunction (Mortiboys et al., 2010; Sanders et al., 2014). In vitro, expression of LRRK2 increases mitochondrial clustering and reduced mitochondrial clearance upon CCCP treatment. In contrast, LRRK2 G2019S further exacerbated the damaging effects (Hsieh et al., 2016). Likewise, reduced mitochondrial autophagy in DA neurons and astrocytes in LRRK2 G2019S mouse brain that is rescued by treatment with LRRK2 kinase inhibitor GSK3357679A (Singh et al., 2021). Several possible mechanisms for the negative regulation of mitophagy by LRRK2 kinase activity are proposed: (1) LRRK2 impairs the interactions between Parkin and DRP1 and their mitochondrial targets in a kinase-dependent manner (Bonello et al., 2019); (2) LRRK2 interacts with MIRO, and the LRRK2 G2019S mutant prevents proteasomal degradation of MIRO, leading to delayed mitophagy (Hsieh et al., 2016); (3) LRRK2 phosphorylates Rab10 on the residue T73, and PD-associated LRRK2 mutants (G2019S and R1441C) impair Rab10 mitochondrial localization and disrupts its interaction with OPTN, resulting in impaired mitochondrial autophagy via a kinase activity related manner (Figures 1, 2; Wauters et al., 2020). In contrast, a study suggests a positive regulation of mitophagy by LRRK2’s kinase activity. LRRK2 G2019S phosphorylates BCL2 at residue T56, leading to the loss of Δψ and triggering excessive mitophagy via the recruitment of P62 to the mitochondria, (Su et al., 2015).
Recognizing the crucial role of elevated LRRK2 kinase activity in PD pathogenesis, multiple research groups have undertaken studies to quantitatively assess LRRK2-related phosphorylation across various tissues and biofluids, exploring their potential of being used as a PD diagnosis (Delbroek et al., 2013; Wang et al., 2017; Padmanabhan et al., 2020; Vissers et al., 2023). Collectively, the expressional level of total LRRK2 (tLRRK2), phosphorylation of LRRK2 at S1292 or S935 (pS1292-LRRK2 or pS935-LRRK2), and phosphorylation of the LRRK2 substrate Rab10 at T73 (pT73-Rab10) have been the focus of extensive study. The observed changes of tLRRK2 and LRRK2-associated phosphorylation in PD cases vary among brain regions, tissues, and cell types (Rideout et al., 2020). Increased tLRRK2 in the frontal cortex in sporadic PD cases, conflicting changes of tLRRK2 l in cerebrospinal fluid (CSF) in sporadic PD cases, elevated pS1292-LRRK2 in urinary EVs in but deceased pS935-LRRK2 in PBMCs among LRRK2-G2019S carriers, and increased pT73-Rab10 in neutrophils in idiopathic PD and LRRK2-G2019S carriers have been reported (Cho et al., 2013; Fraser et al., 2016; Fan et al., 2018; Mabrouk et al., 2020; Padmanabhan et al., 2020).
Remarkably, the highest expression of LRRK2 is not observed in neurons but in peripheral blood mononuclear cells (PBMCs) (Thevenet et al., 2011). Particularly, certain cell types in PMBCs show increased LRRK2 expression in PD patients compared to healthy controls, including B cells, T cells, CD16+ monocytes, neutrophiles, but not mixed PBMCs (Cook et al., 2017; Atashrazm et al., 2019). An LRRK2 inhibitor MLi225 significantly reduced pS935-LRRK2 and pT73-Rab10 in both neutrophils and mixed PBMC (Atashrazm et al., 2019). Although pS935-LRRK2 does not directly reflect LRRK2 kinase activity like pS1292-LRRK2, it is sensitive to dephosphorylation caused by LRRK2 kinase inhibitor (Delbroek et al., 2013; Lobbestael et al., 2013). Therefore, pS935-LRRK2 and pT73-Rab10 are considered potential pharmacodynamics marker in clinical trials of LRRK2 kinase inhibitors.
3.4.2 LRRK2-mediated phosphorylation and other mitochondrial functions
LRRK2 regulates mitochondrial dynamics via DRP1 in a kinase-dependent manner. In BV2 microglia cells and primary cultured microglia cells, treatment of lipopolysaccharide (LPS) activates microglia, resulting in enhanced mitochondrial fragmentation (Ho et al., 2018). Interestingly, LPS-induced mitochondrial fragmentation can be reversed by LRRK2 kinase inhibitor GSK2578215A. Results suggest an important role of LRRK2 kinase activity in regulating mitochondrial dynamics in microglia (Ho et al., 2018). Consistently, overexpression of WT LRRK2 or LRRK2 G2019S in cells results in apparent mitochondrial fragmentation, while overexpression of LRRK2 kinase-dead mutant D1994A does not cause such phenotype (Wang X. et al., 2012; Perez Carrion et al., 2018). LRRK2 directly interacts with and phosphorylates DRP1. PD-associated LRRK2 G2019S and R1441C mutants further enhance this interaction (Su and Qi, 2013; Stafa et al., 2014). LRRK2 G2019S phosphorylates DRP1 at residue T595 (Su and Qi, 2013). In fibroblasts derived from PD patients carrying LRRK2 G2019S, both the selective DRP1-inhibitor P110 or the expression of non-phosphorylatable mutant DRP1 T595A reverse the mitochondrial fragmentation and improve mitochondrial quality, indicating that LRRK2 G2019S-induced mitochondrial fragmentation is possible via a mechanism related to DRP1 T595 phosphorylation (Figure 2; Su and Qi, 2013).
Cells expressing PD associated LRRK2 mutants increase susceptibility to oxidative stress, suggesting that increased LRRK2 kinase activity might interfere antioxidant defense mechanism (Heo et al., 2010; Nguyen et al., 2011; Bahnassawy et al., 2013; Kim et al., 2019). LRRK2 phosphorylates 4E-BP (4E-binding protein) at residues T37/T46, both in vitro and in vivo. 4E-BP is a eukaryotic translation initiation factor regulating overall protein translation in cells that is crucial for cell survival under stress conditions (Haghighat et al., 1995; Tettweiler et al., 2005). Drosophila LRRK2 also phosphorylates 4E-BP, attenuating the resistance to oxidative stress via a 4E-BP phosphorylation-dependent manner (Imai et al., 2008). Therefore, LRRK2 kinase activity regulates cell response to oxidative stress through a 4E-BP mediated pathway. Studies also suggest that LRRK2 affects antioxidant defense mechanism through its phosphorylation of PRDX3 (peroxiredoxin 3). PRDX3 is a mitochondrial antioxidant of the thioredoxin-peroxidase family, efficiently scavenging peroxides and controlling the level of reactive oxygen species (ROS) in mitochondria (Fujii and Ikeda, 2002). In vitro, LRRK2 interacts with PRDX3 and potentially phosphorylates PRDX3 at residue T146. PD-associated mutant LRRK2 G2019S enhances its interaction with and causes decreased peroxidase activity of PRDX3 along with increased cell death (Figure 4; Angeles et al., 2011). Consistently, Drosophila expressing LRRK2 G2019S show reduced PRDX3 peroxidase activity and exacerbated oxidative stress (Angeles et al., 2014).
Furthermore, LRRK2 kinase activity is associated with increased mitochondrial DNA (mtDNA) damage. In various cellular models, including iPSC-derived neural cells, immune cells and fibroblasts, PD-associated mutant LRRK2 G2019S increases mtDNA damage that is abrogated by either gene editing to correct the G2019S mutation or by treatment with LRRK2 kinase inhibitors (Howlett et al., 2017; Gonzalez-Hunt et al., 2020).
Together, LRRK2-mediated phosphorylation regulates mitochondrial functions. Elevated LRRK2 kinase activity could contribute PD pathogenesis through impairing mitochondria.
4 Other post-translational- modifications: regulating mitochondrial functions in Parkinson’s disease
In addition to ubiquitination and phosphorylation, multiple other PTMs regulates mitochondrial functions and is implicated in the PD pathogenesis. In this context, we will provide a summary of recent research findings on how PD-related proteins affect mitochondrial functions through SUMOylation, acetylation, or s-nitrosylation, to elucidate their involvement in the development of PD (Table 4).
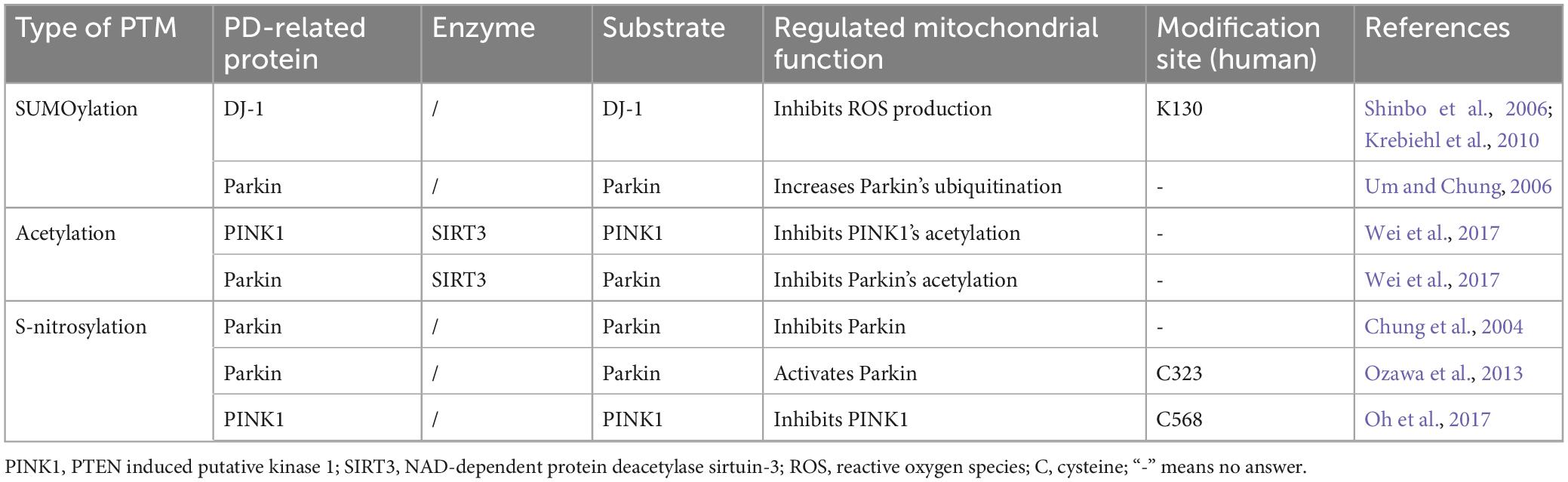
Table 4. Other PTMs of PD-related proteins and mitochondrial dysfunction.
4.1 SUMOylation-regulated mitochondrial function and Parkinson’s disease
SUMOylation refers to the PTM that covalently attaches small ubiquitin-like modifier (SUMO) to lysine residues on the substrate protein. SUMOylation occurs through multiple steps of enzymatic reactions, very similar to those in the ubiquitination process but with different specific enzymes, causing biochemical and functional changes of the target protein. SUMOylation modifies a broad range of proteins and regulates a diversity of biological processes, such as chromatin remodeling, transcription, and mitochondrial dynamics. SUMOylation regulates mitochondrial dynamics through a number of proteins that are either directly encoded by or in close functional-relationship with PD-associated genes (Guerra de Souza et al., 2016).
Mutations in DJ-1 cause autosomal recessive forms of PD. Being a peroxiredoxin-like peroxidase, DJ-1 helps maintain mitochondrial function during oxidative stress as a sensor of damage and a regulator of CI activity (Taira et al., 2004; Andres-Mateos et al., 2007). DJ-1 deficiency results in increased production of ROS and decreased Δψ in cellular and mouse models (Krebiehl et al., 2010). SUMOylation plays important roles in regulating DJ-1 function. SUMOylation of DJ-1 K130 is crucial for the full activity of DJ-1 (Figure 4). The PD-associated DJ-1 L166P mutant becomes improperly SUMOylated and hence more insoluble, leading to its aggregation in mitochondria, ultimately suppresses its proteasomal degradation (Shinbo et al., 2006). DJ-1 is not only an effector of SUMOylation but also suppresses SUMOylating of other proteins at the global level through its interaction with key proteins of the SUMOylation machinery. For example, DJ-1 inhibits the SUMOylation of PSF (pyrimidine tract-binding protein-associated splicing factor), consequently reducing PSF-mediated apoptosis. The PD-associated pathogenic DJ-1 mutant L166P causes accumulation of high-molecular-weight SUMOylated PSF (Zhong et al., 2006). Together, these findings suggest that SUMOylation is involved in DJ-1’s regulation on mitochondria-associated oxidative stress and apoptosis.
Several biomarker studies have aimed to identify and quantify different DJ-1 species levels in PD patients compared to healthy controls. Presently, these studies primarily concentrate on the measurement of total DJ-1 in CSF, DJ-1 isoforms in whole blood, or oxidized DJ-1 in blood or urine (Waragai et al., 2006; Hong et al., 2010; Lin et al., 2012; Gui et al., 2015; Saito, 2017; Jang et al., 2018). Until now, the reliable detection of SUMOylated DJ-1 in various tissues or biofluids, and its potential use as an indicator for PD disease progression, stays unexplored. Similar to the challenge faced in using ubiquitinated proteins as biomarkers, the obstacle here may also be attributed to the lack of specific antibodies targeting SUMOylated DJ-1 (Magalhaes and Lashuel, 2022).
It’s noteworthy that not only is proper SUMOylation essential for the solubility and activity of DJ-1 protein, but also is the PKA induced phosphorylation at the T154 residue of DJ-1 (Ko et al., 2019). Currently, there is no evidence showing crosstalk between the T154 phosphorylation and the K130 SUMOylation of DJ-1. However, it is an intriguing question worth investigating.
Parkin has also been reported to selectively interact with SUMO-1 (small ubiquitin like modifier 1), both in vitro and in vivo, resulting in increased ubiquitination and nuclear translocation of Parkin. Therefore, SUMOylation might play a role in Parkin-mediated ubiquitination and its relevance to PD pathogenesis (Um and Chung, 2006). In agreement with this, DRP1 is a target of all SUMO isoforms with various functional-consequences. SUMOylation of DRP1 by SUMO-1 enhances its association with mitochondria, promotes mitochondrial fragmentation, and increases apoptosis (Wasiak et al., 2007). In contrast, SUMOylation of DRP1 by SUMO-2/3 decreases its mitochondrial localization and reduces apoptosis under stress conditions (Guo et al., 2013). Collectively, these studies underscore the significance of SUMO-regulated mitochondrial functions in the pathogenesis of PD.
4.2 Acetylation-regulated mitochondrial function and Parkinson’s disease
Protein acetylation refers to the transfer of an acetyl group (CH3CO) from acetyl-CoA to either the ε-amino group (NH3+) of lysine residues (ε-lysine acetylation) or to the N-terminal amino acid of a protein (N-α-acetylation). N-α-acetylation is an irreversible reaction catalyzed by N-terminal acetyltransferases (NATs), while l ε-lysine acetylation is a reversible modification tightly regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Drazic et al., 2016). Recent evidence indicates that acetylation of PD-associated proteins might have important functional-consequences in mitochondria, although the detailed mechanism remains largely unknown.
In vitro, N-α-acetylation affects the secondary structure of α-synuclein, leading to the oligomeric form with a partial α-helical structure (Kang et al., 2012). In vivo, decreased Δψ and increased ROS level are detected in the mouse brain overexpressing predominantly N-terminally acetylated α-synuclein (Sarafian et al., 2013), suggesting that N-α-acetylation of α-synuclein cause mitochondrial dysfunction and have pathological implications in PD. Consistently, knockdown of deacetylase SIRT3 (NAD-dependent protein deacetylase sirtuin-3) in SH-SY5Y cells significantly increases rotenone-induced α-synuclein accumulation and reduces the activities of SOD (superoxide dismutase) and GSH (glutathione), leading to increased ROS generation and damaged mitochondria (Zhang et al., 2016). SIRT3 is also reported to be inversely related to the acetylation of Parkin and PINK1—acetylated PINK1 and Parkin are increased with knockdown of SIRT3 but decreased with overexpression of SIRT3 (Wei et al., 2017). PKAN (pantothenate kinase-associated neurodegeneration) is the enzyme catalyzing the first and rate-limiting step of CoA synthesis (Leonardi et al., 2005). A recently study reported that Fbl (Fumble, Drosophila homolog of PANK2), functioning downstream of PINK1, regulates acetylation of Ref(2)P (Drosophila homolog of P62) and promotes mitophagy activity (Huang et al., 2022).
4.3 S-nitrosylation-regulated mitochondrial function and Parkinson’s disease
S-nitrosylation involves the covalent attachment of a nitro oxide group (-NO) to the thiol side chain of a cysteine residue within a protein (Hess and Stamler, 2012). Like other PTMs, s-nitrosylation has emerged as an important regulator of various classes of proteins. S-nitrosylation plays a role in PD-related mitochondrial pathology through its modification of Parkin and PINK1. The initial two studies showing Parkin could be s-nitrosylated were both published in 2004, yielding contrary conclusions. Chung et al. (2004) reported that s-nitrosylation of Parkin inhibits its E3 ligase activity, resulting in decreased ubiquitination of Parkin substrates, including Parkin itself and Synphilin-1. While Yao et al. (2004) found that s-nitrosylation of Parkin stimulates its E3 ligase activity, leading to increased self-ubiquitination. Later on, Ozawa et al. (2013) reported that Parkin is predominantly s-nitrosylated at residue C323 resulting in activation of Parkin’s E3 ligase activity and induces mitochondrial degradation. Interestingly, s-nitrosylation of Parkin is regulated by DJ-1, another PD-associated protein that has been found in the same complex with Parkin and PINK1 (Tang et al., 2006; Xiong et al., 2009). Loss-of-function of DJ-1 results in decreased s-nitrosylation of Parkin, along with increased mitochondrial depolarization and cell death (Ozawa et al., 2020), adding additional evidence for the long-observed functional interaction between Parkin, PINK1, and DJ-1. Intriguingly, s-nitrosylation has also been observed with PINK1 at residue C568 that negatively regulates PINK1 kinase activity, therefore reducing PINK1-dependent phosphorylation and activation of Parkin (Oh et al., 2017). However, the detailed mechanisms of how s-nitrosylation regulates Parkin and PINK1 mediated mitophagy, as well as whether DJ-1 is also required for PINK1’s s-nitrosylation, remain unclear and require further study to elucidate.
5 Perspectives and conclusion
Mounting evidence indicates involvement of PTMs-regulated mitochondrial functions in the PD etiology. A major challenge in the field is to distinguish physiological functions from the pathological roles within these pathways implicated in PD. For example, understanding how PINK1/Parkin- and BNIP3-regulated mitophagy contributes to PD pathogenesis is crucial. It is well known that PINK1 and Parkin can function both collaboratively and independently regulating mitochondrial functions, but which function of PINK1 and Parkin is critical for PD? Moreover, since the deletion of PINK1, Parkin, or both does not result in significant DA neurodegeneration in mouse models, are there other factors important for DA neurodegeneration in patients carrying PINK1 and Parkin mutations?
Phosphorylated tau protein has recently been demonstrated as a valuable biomarker of Alzheimer’s disease at a systemic level (Karikari et al., 2020; Teunissen et al., 2022). A deeper understanding of the PD-related, specific alterations of PTMs-especially those with significant consequences on mitochondrial functions—may lead to candidates for the long sought-after biomarkers for PD. However, the development of PD biomarkers based on PTMs is currently in its early stages.
Author contributions
SL: Writing – original draft, Writing – review & editing. DW: Writing – original draft, Writing – review & editing. ZZ: Conceptualization, Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (8171101313, 81842044, 31730036, 81429002, and 31330031), the Discipline Innovative Engineering Plan (111 Program) of China (B13036), the Department of Science and Technology of Hunan Province (2021SK1010, 2016TP1006, 2018SK1030, 2022WZ1027, and 2021SK1014), the Department of Science and Technology of Changsha City (KC1702038), and the Education Department Program of Hunan Province (HNJG-2020-0440).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aerts, L., Craessaerts, K., De Strooper, B., and Morais, V. A. (2015). PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 290, 2798–2811. doi: 10.1074/jbc.M114.620906
Akutsu, M., Dikic, I., and Bremm, A. (2016). Ubiquitin chain diversity at a glance. J. Cell Sci. 129, 875–880. doi: 10.1242/jcs.183954
Andres-Mateos, E., Perier, C., Zhang, L., Blanchard-Fillion, B., Greco, T. M., Thomas, B., et al. (2007). DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 14807–14812. doi: 10.1073/pnas.0703219104
Angeles, D. C., Gan, B. H., Onstead, L., Zhao, Y., Lim, K. L., Dachsel, J., et al. (2011). Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 32, 1390–1397. doi: 10.1002/humu.21582
Angeles, D. C., Ho, P., Chua, L. L., Wang, C., Yap, Y. W., Ng, C., et al. (2014). Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila. Hum. Mol. Genet. 23, 3157–3165. doi: 10.1093/hmg/ddu026
Arena, G., Gelmetti, V., Torosantucci, L., Vignone, D., Lamorte, G., De Rosa, P., et al. (2013). PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 20, 920–930. doi: 10.1038/cdd.2013.19
Ascherio, A., and Schwarzschild, M. A. (2016). The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 15, 1257–1272. doi: 10.1016/S1474-4422(16)30230-7
Atashrazm, F., Hammond, D., Perera, G., Bolliger, M. F., Matar, E., Halliday, G. M., et al. (2019). LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov. Disord. 34, 406–415. doi: 10.1002/mds.27601
Bahnassawy, L., Nicklas, S., Palm, T., Menzl, I., Birzele, F., Gillardon, F., et al. (2013). The parkinson’s disease-associated LRRK2 mutation R1441G inhibits neuronal differentiation of neural stem cells. Stem Cells Dev. 22, 2487–2496. doi: 10.1089/scd.2013.0163
Basso, V., Marchesan, E., Peggion, C., Chakraborty, J., von Stockum, S., Giacomello, M., et al. (2018). Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 138, 43–56. doi: 10.1016/j.phrs.2018.09.006
Bekris, L. M. I, Mata, F., and Zabetian, C. P. (2010). The genetics of Parkinson disease. J. Geriatr. Psychiatry Neurol. 23, 228–242. doi: 10.1177/0891988710383572
Billingsley, K. J., Barbosa, I. A., Bandres-Ciga, S., Quinn, J. P., Bubb, V. J., Deshpande, C., et al. (2019). Mitochondria function associated genes contribute to Parkinson’s disease risk and later age at onset. NPJ Park. Dis. 5:8. doi: 10.1038/s41531-019-0080-x
Biskup, S., Moore, D. J., Celsi, F., Higashi, S., West, A. B., Andrabi, S. A., et al. (2006). Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 60, 557–569. doi: 10.1002/ana.21019
Blumenreich, S., Barav, O. B., Jenkins, B. J., and Futerman, A. H. (2020). Lysosomal storage disorders shed light on lysosomal dysfunction in Parkinson’s disease. Int. J. Mol. Sci. 21:4966. doi: 10.3390/ijms21144966
Bonello, F., Hassoun, S. M., Mouton-Liger, F., Shin, Y. S., Muscat, A., Tesson, C., et al. (2019). LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 28, 1645–1660. doi: 10.1093/hmg/ddz004
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Broadway, B. J., Boneski, P. K., Bredenberg, J. M., Kolicheski, A., Hou, X., Soto-Beasley, A. I., et al. (2022). Systematic functional analysis of PINK1 and PRKN coding variants. Cells 11:2426. doi: 10.3390/cells11152426
Burchell, V. S., Nelson, D. E., Sanchez-Martinez, A., Delgado-Camprubi, M., Ivatt, R. M., Pogson, J. H., et al. (2013). The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 16, 1257–1265. doi: 10.1038/nn.3489
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Cadete, V. J., Deschenes, S., Cuillerier, A., Brisebois, F., Sugiura, A., Vincent, A., et al. (2016). Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 594, 5343–5362. doi: 10.1113/JP272703
Cai, C., Wu, F., He, J., Zhang, Y., Shi, N., Peng, X., et al. (2022). Mitochondrial quality control in diabetic cardiomyopathy: From molecular mechanisms to therapeutic strategies. Int. J. Biol. Sci. 18, 5276–5290. doi: 10.7150/ijbs.75402
Cali, T., Ottolini, D., Negro, A., and Brini, M. (2013). Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508. doi: 10.1016/j.bbadis.2013.01.004
Cardona, F., Sanchez-Mut, J. V., Dopazo, H., and Perez-Tur, J. (2011). Phylogenetic and in silico structural analysis of the Parkinson disease-related kinase PINK1. Hum. Mutat. 32, 369–378. doi: 10.1002/humu.21444
Cereghetti, G. M., Stangherlin, A., Martins de Brito, O., Chang, C. R., Blackstone, C., Bernardi, P., et al. (2008). Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808. doi: 10.1073/pnas.0808249105
Cha, Y., Kim, T., Jeon, J., Jang, Y., Kim, P. B., Lopes, C., et al. (2021). SIRT2 regulates mitochondrial dynamics and reprogramming via MEK1-ERK-DRP1 and AKT1-DRP1 axes. Cell Rep. 37:110155. doi: 10.1016/j.celrep.2021.110155
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chen, H., and Chan, D. C. (2017). Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 26, 39–48. doi: 10.1016/j.cmet.2017.05.016
Chen, T., Tan, J., Wan, Z., Zou, Y., Afewerky, H. K., Zhang, Z., et al. (2017). Effects of commonly used pesticides in china on the mitochondria and ubiquitin-proteasome system in Parkinson’s disease. Int. J. Mol. Sci. 18:2507. doi: 10.3390/ijms18122507
Chen, Y., and Dorn, G. W. II (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Cherian, A., K, P. D., and Vijayaraghavan, A. (2023). Parkinson’s disease - genetic cause. Curr. Opin. Neurol. 36, 292–301. doi: 10.1097/WCO.0000000000001167
Chin, L. S., and Li, L. (2016). Ubiquitin phosphorylation in Parkinson’s disease: Implications for pathogenesis and treatment. Transl. Neurodegener. 5:1. doi: 10.1186/s40035-015-0049-6
Cho, H. J., Liu, G., Jin, S. M., Parisiadou, L., Xie, C., Yu, J., et al. (2013). MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 22, 608–620. doi: 10.1093/hmg/dds470
Choubey, V., Safiulina, D., Vaarmann, A., Cagalinec, M., Wareski, P., Kuum, M., et al. (2011). Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 286, 10814–10824. doi: 10.1074/jbc.M110.132514
Chung, K. K., Thomas, B., Li, X., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2004). S-nitrosylation of parkin regulates ubiquitination and compromises Parkin’s protective function. Science 304, 1328–1331. doi: 10.1126/science.1093891
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Cook, D. A., Kannarkat, G. T., Cintron, A. F., Butkovich, L. M., Fraser, K. B., Chang, J., et al. (2017). LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Park. Dis. 3:11. doi: 10.1038/s41531-017-0010-8
Costa, A. C., Loh, S. H., and Martins, L. M. (2013). Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson’s disease. Cell Death Dis. 4:e467. doi: 10.1038/cddis.2012.205
Cunningham, C. N., Baughman, J. M., Phu, L., Tea, J. S., Yu, C., Coons, M., et al. (2015). USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 17, 160–169. doi: 10.1038/ncb3097
da Silva Rosa, S. C., Martens, M. D., Field, J. T., Nguyen, L., Kereliuk, S. M., Hai, Y., et al. (2021). BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 17, 2257–2272. doi: 10.1080/15548627.2020.1821548
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Virgilio, A., Greco, A., Fabbrini, G., Inghilleri, M., Rizzo, M. I., Gallo, A., et al. (2016). Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 15, 1005–1011. doi: 10.1016/j.autrev.2016.07.022
Deas, E., Plun-Favreau, H., Gandhi, S., Desmond, H., Kjaer, S., Loh, S. H., et al. (2011). PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879. doi: 10.1093/hmg/ddq526
Delbroek, L., Van Kolen, K., Steegmans, L., da Cunha, R., Mandemakers, W., Daneels, G., et al. (2013). Development of an enzyme-linked immunosorbent assay for detection of cellular and in vivo LRRK2 S935 phosphorylation. J. Pharm. Biomed. Anal. 76, 49–58. doi: 10.1016/j.jpba.2012.12.002
Deng, H., Dodson, M. W., Huang, H., and Guo, M. (2008). The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 105, 14503–14508. doi: 10.1073/pnas.0803998105
Devi, L., and Anandatheerthavarada, H. K. (2010). Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochim. Biophys. Acta 1802, 11–19. doi: 10.1016/j.bbadis.2009.07.007
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Di Fonzo, A., Dekker, M. C., Montagna, P., Baruzzi, A., Yonova, E. H., Correia Guedes, L., et al. (2009). FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72, 240–245. doi: 10.1212/01.wnl.0000338144.10967.2b
Drazic, A., Myklebust, L. M., Ree, R., and Arnesen, T. (2016). The world of protein acetylation. Biochim. Biophys. Acta 1864, 1372–1401. doi: 10.1016/j.bbapap.2016.06.007
Durcan, T. M., and Fon, E. A. (2015). The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 29, 989–999. doi: 10.1101/gad.262758.115
Durcan, T. M., Kontogiannea, M., Bedard, N., Wing, S. S., and Fon, E. A. (2012). Ataxin-3 deubiquitination is coupled to Parkin ubiquitination via E2 ubiquitin-conjugating enzyme. J. Biol. Chem. 287, 531–541. doi: 10.1074/jbc.M111.288449
Durcan, T. M., Tang, M. Y., Perusse, J. R., Dashti, E. A., Aguileta, M. A., McLelland, G. L., et al. (2014). USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 33, 2473–2491. doi: 10.15252/embj.201489729
Ebrahimi-Fakhari, D., Wahlster, L., and McLean, P. J. (2012). Protein degradation pathways in Parkinson’s disease: Curse or blessing. Acta Neuropathol. 124, 153–172. doi: 10.1007/s00401-012-1004-6
Eiyama, A., and Okamoto, K. (2015). PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 33, 95–101. doi: 10.1016/j.ceb.2015.01.002
Elia, A. E., Petrucci, S., Fasano, A., Guidi, M., Valbonesi, S., Bernardini, L., et al. (2013). Alpha-synuclein gene duplication: Marked intrafamilial variability in two novel pedigrees. Mov. Disord. 28, 813–817. doi: 10.1002/mds.25518
Ellard, S., Colclough, K., Patel, K. A., and Hattersley, A. T. (2020). Prediction algorithms: Pitfalls in interpreting genetic variants of autosomal dominant monogenic diabetes. J. Clin. Invest. 130, 14–16. doi: 10.1172/JCI133516
Fan, Y., Howden, A. J. M., Sarhan, A. R., Lis, P., Ito, G., Martinez, T. N., et al. (2018). Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem. J. 475, 23–44. doi: 10.1042/BCJ20170803
Fiesel, F. C., Ando, M., Hudec, R., Hill, A. R., Castanedes-Casey, M., Caulfield, T. R., et al. (2015). (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 16, 1114–1130. doi: 10.15252/embr.201540514
Foulds, P. G., Mitchell, J. D., Parker, A., Turner, R., Green, G., Diggle, P., et al. (2011). Phosphorylated alpha-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson’s disease. FASEB J. 25, 4127–4137. doi: 10.1096/fj.10-179192
Foulds, P. G., Yokota, O., Thurston, A., Davidson, Y., Ahmed, Z., Holton, J., et al. (2012). Post mortem cerebrospinal fluid alpha-synuclein levels are raised in multiple system atrophy and distinguish this from the other alpha-synucleinopathies, Parkinson’s disease and Dementia with Lewy bodies. Neurobiol. Dis. 45, 188–195. doi: 10.1016/j.nbd.2011.08.003
Frank-Cannon, T. C., Tran, T., Ruhn, K. A., Martinez, T. N., Hong, J., Marvin, M., et al. (2008). Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 28, 10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008
Fraser, K. B., Rawlins, A. B., Clark, R. G., Alcalay, R. N., Standaert, D. G., Liu, N., et al. (2016). Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Mov. Disord. 31, 1543–1550. doi: 10.1002/mds.26686
Fuchs, J., Nilsson, C., Kachergus, J., Munz, M., Larsson, E. M., Schule, B., et al. (2007). Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68, 916–922. doi: 10.1212/01.wnl.0000254458.17630.c5
Fujii, J., and Ikeda, Y. (2002). Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 7, 123–130. doi: 10.1179/135100002125000352
Funayama, M., Hasegawa, K., Kowa, H., Saito, M., Tsuji, S., and Obata, F. (2002). A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 51, 296–301. doi: 10.1002/ana.10113
Funayama, M., Ohe, K., Amo, T., Furuya, N., Yamaguchi, J., Saiki, S., et al. (2015). CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome-wide linkage and sequencing study. Lancet Neurol. 14, 274–282. doi: 10.1016/S1474-4422(14)70266-2
Futter, C. E., Pearse, A., Hewlett, L. J., and Hopkins, C. R. (1996). Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J. Cell Biol. 132, 1011–1023. doi: 10.1083/jcb.132.6.1011
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Gautier, E. F., Ducamp, S., Leduc, M., Salnot, V., Guillonneau, F., Dussiot, M., et al. (2016). Comprehensive proteomic analysis of human erythropoiesis. Cell Rep. 16, 1470–1484. doi: 10.1016/j.celrep.2016.06.085
Ge, P., Dawson, V. L., and Dawson, T. M. (2020). PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 15:20. doi: 10.1186/s13024-020-00367-7
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Geldenhuys, W. J., Abdelmagid, S. M., Gallegos, P. J., and Safadi, F. F. (2014). Parkinson’s disease biomarker: A patent evaluation of WO2013153386. Expert Opin. Ther. Pat. 24, 947–951. doi: 10.1517/13543776.2014.931375
Gilks, W. P., Abou-Sleiman, P. M., Gandhi, S., Jain, S., Singleton, A., Lees, A. J., et al. (2005). A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365, 415–416. doi: 10.1016/S0140-6736(05)17830-1
Goldman, S. M. (2014). Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 54, 141–164. doi: 10.1146/annurev-pharmtox-011613-135937
Gonzalez-Hunt, C. P., Thacker, E. A., Toste, C. M., Boularand, S., Deprets, S., Dubois, L., et al. (2020). Mitochondrial DNA damage as a potential biomarker of LRRK2 kinase activity in LRRK2 Parkinson’s disease. Sci. Rep. 10:17293. doi: 10.1038/s41598-020-74195-6
Greene, A. W., Grenier, K., Aguileta, M. A., Muise, S., Farazifard, R., Haque, M. E., et al. (2012). Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385. doi: 10.1038/embor.2012.14
Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B., and Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 4078–4083. doi: 10.1073/pnas.0737556100
Guerra de Souza, A. C., Prediger, R. D., and Cimarosti, H. (2016). SUMO-regulated mitochondrial function in Parkinson’s disease. J. Neurochem. 137, 673–686. doi: 10.1111/jnc.13599
Gui, Y., Liu, H., Zhang, L., Lv, W., and Hu, X. (2015). Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 6, 37043–37053. doi: 10.18632/oncotarget.6158
Guo, C., Hildick, K. L., Luo, J., Dearden, L., Wilkinson, K. A., and Henley, J. M. (2013). SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 32, 1514–1528. doi: 10.1038/emboj.2013.65
Haghighat, A., Mader, S., Pause, A., and Sonenberg, N. (1995). Repression of cap-dependent translation by 4E-binding protein 1: Competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 14, 5701–5709. doi: 10.1002/j.1460-2075.1995.tb00257.x
Ham, S. J., Lee, D., Yoo, H., Jun, K., Shin, H., and Chung, J. (2020). Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 117, 4281–4291. doi: 10.1073/pnas.1909814117
Han, H., Tan, J., Wang, R., Wan, H., He, Y., Yan, X., et al. (2020). PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 21:e48686. doi: 10.15252/embr.201948686
Henchcliffe, C., and Beal, M. F. (2008). Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 4, 600–609. doi: 10.1038/ncpneuro0924
Heo, H. Y., Park, J. M., Kim, C. H., Han, B. S., Kim, K. S., and Seol, W. (2010). LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp. Cell Res. 316, 649–656. doi: 10.1016/j.yexcr.2009.09.014
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Hernandez, D. G., Reed, X., and Singleton, A. B. (2016). Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 139 Suppl 1, (Suppl. 1), 59–74. doi: 10.1111/jnc.13593
Hess, D. T., and Stamler, J. S. (2012). Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 287, 4411–4418. doi: 10.1074/jbc.R111.285742
Heyn, J., Heuschkel, M. A., and Goettsch, C. (2023). Mitochondrial-derived vesicles-link to extracellular vesicles and implications in cardiovascular disease. Int. J. Mol. Sci. 24:2637. doi: 10.3390/ijms24032637
Hierro, A., Rojas, A. L., Rojas, R., Murthy, N., Effantin, G., Kajava, A. V., et al. (2007). Functional architecture of the retromer cargo-recognition complex. Nature 449, 1063–1067. doi: 10.1038/nature06216
Hirai, I., and Wang, H. G. (2001). Survival-factor-induced phosphorylation of Bad results in its dissociation from Bcl-x(L) but not Bcl-2. Biochem J 359(Pt. 2), 345–352. doi: 10.1042/0264-6021:3590345
Ho, D. H., Je, A. R., Lee, H., Son, I., Kweon, H. S., Kim, H. G., et al. (2018). LRRK2 kinase activity induces mitochondrial fission in microglia via Drp1 and modulates neuroinflammation. Exp. Neurobiol. 27, 171–180. doi: 10.5607/en.2018.27.3.171
Hong, Z., Shi, M., Chung, K. A., Quinn, J. F., Peskind, E. R., Galasko, D., et al. (2010). DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133(Pt. 3), 713–726. doi: 10.1093/brain/awq008
Howlett, E. H., Jensen, N., Belmonte, F., Zafar, F., Hu, X., Kluss, J., et al. (2017). LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum. Mol. Genet. 26, 4340–4351. doi: 10.1093/hmg/ddx320
Hsieh, C. H., Shaltouki, A., Gonzalez, A. E., Bettencourt da Cruz, A., Burbulla, L. F., St Lawrence, E., et al. (2016). Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19, 709–724. doi: 10.1016/j.stem.2016.08.002
Huang, E., Qu, D., Huang, T., Rizzi, N., Boonying, W., Krolak, D., et al. (2017). PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 8:1399. doi: 10.1038/s41467-017-01435-1
Huang, Y., Wan, Z., Tang, Y., Xu, J., Laboret, B., Nallamothu, S., et al. (2022). Pantothenate kinase 2 interacts with PINK1 to regulate mitochondrial quality control via acetyl-CoA metabolism. Nat. Commun. 13:2412. doi: 10.1038/s41467-022-30178-x
Hunn, B. H., Cragg, S. J., Bolam, J. P., Spillantini, M. G., and Wade-Martins, R. (2015). Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 38, 178–188. doi: 10.1016/j.tins.2014.12.009
Imai, Y., Gehrke, S., Wang, H. Q., Takahashi, R., Hasegawa, K., Oota, E., et al. (2008). Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 27, 2432–2443. doi: 10.1038/emboj.2008.163
Imai, Y., Soda, M., and Takahashi, R. (2000). Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 275, 35661–35664. doi: 10.1074/jbc.C000447200
Ivankovic, D., Chau, K. Y., Schapira, A. H., and Gegg, M. E. (2016). Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 136, 388–402. doi: 10.1111/jnc.13412
Jang, J., Jeong, S., Lee, S. I., Seol, W., Seo, H., Son, I., et al. (2018). Oxidized DJ-1 levels in urine samples as a putative biomarker for Parkinson’s disease. Park. Dis. 2018:1241757. doi: 10.1155/2018/1241757
Jin, S. M., and Youle, R. J. (2013). The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757. doi: 10.4161/auto.26122
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Juhasz, G. (2016). A mitochondrial-derived vesicle HOPS to endolysosomes using Syntaxin-17. J. Cell Biol. 214, 241–243. doi: 10.1083/jcb.201607024
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kang, L., Moriarty, G. M., Woods, L. A., Ashcroft, A. E., Radford, S. E., and Baum, J. (2012). N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 21, 911–917. doi: 10.1002/pro.2088
Karbowski, M., and Youle, R. J. (2011). Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 23, 476–482. doi: 10.1016/j.ceb.2011.05.007
Karikari, T. K., Pascoal, T. A., Ashton, N. J., Janelidze, S., Benedet, A. L., Rodriguez, J. L., et al. (2020). Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 19, 422–433. doi: 10.1016/S1474-4422(20)30071-5
Karve, T. M., and Cheema, A. K. (2011). Small changes huge impact: The role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids 2011:207691. doi: 10.4061/2011/207691
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/BJ20140334
Khoury, G. A., Baliban, R. C., and Floudas, C. A. (2011). Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 1:190. doi: 10.1038/srep00090
Kim, D. I., Lee, K. H., Gabr, A. A., Choi, G. E., Kim, J. S., Ko, S. H., et al. (2016). Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 1863, 2820–2834. doi: 10.1016/j.bbamcr.2016.09.003
Kim, J., Pajarillo, E., Rizor, A., Son, D. S., Lee, J., Aschner, M., et al. (2019). LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS One 14:e0210248. doi: 10.1371/journal.pone.0210248
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Ko, Y. U., Kim, S. J., Lee, J., Song, M. Y., Park, K. S., Park, J. B., et al. (2019). Protein kinase A-induced phosphorylation at the Thr154 affects stability of DJ-1. Park. Relat. Disord. 66, 143–150. doi: 10.1016/j.parkreldis.2019.07.029
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Kondapalli, K. C., Kallay, L. M., Muszelik, M., and Rao, R. (2012). Unconventional chemiosmotic coupling of NHA2, a mammalian Na+/H+ antiporter, to a plasma membrane H+ gradient. J. Biol. Chem. 287, 36239–36250. doi: 10.1074/jbc.M112.403550
Konig, T., Nolte, H., Aaltonen, M. J., Tatsuta, T., Krols, M., Stroh, T., et al. (2021). MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 23, 1271–1286. doi: 10.1038/s41556-021-00798-4
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Koyano, F., Yamano, K., Kosako, H., Tanaka, K., and Matsuda, N. (2019). Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J. Biol. Chem. 294, 10300–10314. doi: 10.1074/jbc.RA118.006302
Kraus, F., Goodall, E. A. I, Smith, R., Jiang, Y., Paoli, J. C., Adolf, F., et al. (2023). PARK15/FBXO7 is dispensable for PINK1/Parkin mitophagy in iNeurons and HeLa cell systems. EMBO Rep. 24:e56399. doi: 10.15252/embr.202256399
Kraus, F., Roy, K., Pucadyil, T. J., and Ryan, M. T. (2021). Function and regulation of the divisome for mitochondrial fission. Nature 590, 57–66. doi: 10.1038/s41586-021-03214-x
Krebiehl, G., Ruckerbauer, S., Burbulla, L. F., Kieper, N., Maurer, B., Waak, J., et al. (2010). Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One 5:e9367. doi: 10.1371/journal.pone.0009367
Kulathu, Y., and Komander, D. (2012). Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 13, 508–523. doi: 10.1038/nrm3394
Kumar, A., Tamjar, J., Waddell, A. D., Woodroof, H. I., Raimi, O. G., Shaw, A. M., et al. (2017). Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. Elife 6:e29985. doi: 10.7554/eLife.29985
Lajoie, A. C., Lafontaine, A. L., and Kaminska, M. (2021). The spectrum of sleep disorders in Parkinson disease: A review. Chest 159, 818–827. doi: 10.1016/j.chest.2020.09.099
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. doi: 10.1126/science.6823561
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Lee, J. T., Wheeler, T. C., Li, L., and Chin, L. S. (2008). Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 17, 906–917. doi: 10.1093/hmg/ddm363
Lee, Y., Stevens, D. A., Kang, S. U., Jiang, H., Lee, Y. I., Ko, H. S., et al. (2017). PINK1 primes parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep. 18, 918–932. doi: 10.1016/j.celrep.2016.12.090
Leonardi, R., Zhang, Y. M., Rock, C. O., and Jackowski, S. (2005). Coenzyme A: Back in action. Prog. Lipid Res. 44, 125–153. doi: 10.1016/j.plipres.2005.04.001
Lewis, M. R., and Lewis, W. H. (1914). Mitochondria in tissue culture. Science 39, 330–333. doi: 10.1126/science.39.1000.330
Li, L., Muhlfeld, C., Niemann, B., Pan, R., Li, R., Hilfiker-Kleiner, D., et al. (2011). Mitochondrial biogenesis and PGC-1alpha deacetylation by chronic treadmill exercise: Differential response in cardiac and skeletal muscle. Basic Res. Cardiol. 106, 1221–1234. doi: 10.1007/s00395-011-0213-9
Li, W. W., Yang, R., Guo, J. C., Ren, H. M., Zha, X. L., Cheng, J. S., et al. (2007). Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18, 1543–1546. doi: 10.1097/WNR.0b013e3282f03db4
Li, Y., Feng, Y. F., Liu, X. T., Li, Y. C., Zhu, H. M., Sun, M. R., et al. (2021). Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 38:101771. doi: 10.1016/j.redox.2020.101771
Lin, C. H., Chen, M. L., Chen, G. S., Tai, C. H., and Wu, R. M. (2011). Novel variant Pro143Ala in HTRA2 contributes to Parkinson’s disease by inducing hyperphosphorylation of HTRA2 protein in mitochondria. Hum. Genet. 130, 817–827. doi: 10.1007/s00439-011-1041-6
Lin, W., and Kang, U. J. (2008). Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 106, 464–474. doi: 10.1111/j.1471-4159.2008.05398.x
Lin, X., Cook, T. J., Zabetian, C. P., Leverenz, J. B., Peskind, E. R., Hu, S. C., et al. (2012). DJ-1 isoforms in whole blood as potential biomarkers of Parkinson disease. Sci. Rep. 2:954. doi: 10.1038/srep00954
Liu, W., Duan, X., Fang, X., Shang, W., and Tong, C. (2018). Mitochondrial protein import regulates cytosolic protein homeostasis and neuronal integrity. Autophagy 14, 1293–1309. doi: 10.1080/15548627.2018.1474991
Liu, Y., Lear, T. B., Verma, M., Wang, K. Z., Otero, P. A., McKelvey, A. C., et al. (2020). Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight 5:e131834. doi: 10.1172/jci.insight.131834
Lobbestael, E., Zhao, J. I, Rudenko, N., Beylina, A., Gao, F., Wetter, J., et al. (2013). Identification of protein phosphatase 1 as a regulator of the LRRK2 phosphorylation cycle. Biochem. J. 456, 119–128. doi: 10.1042/BJ20121772
Lopez-Domenech, G., Covill-Cooke, C., Ivankovic, D., Halff, E. F., Sheehan, D. F., Norkett, R., et al. (2018). Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 37, 321–336. doi: 10.15252/embj.201696380
Lutz, A. K., Exner, N., Fett, M. E., Schlehe, J. S., Kloos, K., Lammermann, K., et al. (2009). Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951. doi: 10.1074/jbc.M109.035774
Lwin, A., Orvisky, E., Goker-Alpan, O., LaMarca, M. E., and Sidransky, E. (2004). Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 81, 70–73. doi: 10.1016/j.ymgme.2003.11.004
Ma, K. Y., Fokkens, M. R., van Laar, T., and Verbeek, D. S. (2021). Systematic analysis of PINK1 variants of unknown significance shows intact mitophagy function for most variants. NPJ Park. Dis. 7:113. doi: 10.1038/s41531-021-00258-8
Mabrouk, O. S., Chen, S., Edwards, A. L., Yang, M., Hirst, W. D., and Graham, D. L. (2020). Quantitative measurements of LRRK2 in human cerebrospinal fluid demonstrates increased levels in G2019S patients. Front. Neurosci. 14:526. doi: 10.3389/fnins.2020.00526
Magalhaes, P., and Lashuel, H. A. (2022). Opportunities and challenges of alpha-synuclein as a potential biomarker for Parkinson’s disease and other synucleinopathies. NPJ Park. Dis 8:93. doi: 10.1038/s41531-022-00357-0
Manohar, S., Jacob, S., Wang, J., Wiechecki, K. A., Koh, H. W. L., Simoes, V., et al. (2019). polyubiquitin chains linked by lysine residue 48 (K48) selectively target oxidized proteins in vivo. Antioxid. Redox Signal. 31, 1133–1149. doi: 10.1089/ars.2019.7826
Martinez, A., Lectez, B., Ramirez, J., Popp, O., Sutherland, J. D., Urbe, S., et al. (2017). Quantitative proteomic analysis of Parkin substrates in Drosophila neurons. Mol. Neurodegener. 12:29. doi: 10.1186/s13024-017-0170-3
Martinez-Martin, P., Schapira, A. H., Stocchi, F., Sethi, K., Odin, P., MacPhee, G., et al. (2007). Prevalence of nonmotor symptoms in Parkinson’s disease in an international setting; study using nonmotor symptoms questionnaire in 545 patients. Mov. Disord. 22, 1623–1629. doi: 10.1002/mds.21586
Martini, H., and Passos, J. F. (2023). Cellular senescence: All roads lead to mitochondria. FEBS J. 290, 1186–1202. doi: 10.1111/febs.16361
Matenia, D., Hempp, C., Timm, T., Eikhof, A., and Mandelkow, E. M. (2012). Microtubule affinity-regulating kinase 2 (MARK2) turns on phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) at Thr-313, a mutation site in Parkinson disease: Effects on mitochondrial transport. J. Biol. Chem. 287, 8174–8186. doi: 10.1074/jbc.M111.262287
Matheoud, D., Sugiura, A., Bellemare-Pelletier, A., Laplante, A., Rondeau, C., Chemali, M., et al. (2016). Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 166, 314–327. doi: 10.1016/j.cell.2016.05.039
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Matteucci, A., Patron, M., Vecellio Reane, D., Gastaldello, S., Amoroso, S., Rizzuto, R., et al. (2018). Parkin-dependent regulation of the MCU complex component MICU1. Sci. Rep. 8:14199. doi: 10.1038/s41598-018-32551-7
McLelland, G. L., Lee, S. A., McBride, H. M., and Fon, E. A. (2016). Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 214, 275–291. doi: 10.1083/jcb.201603105
McLelland, G. L., Soubannier, V., Chen, C. X., McBride, H. M., and Fon, E. A. (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. doi: 10.1002/embj.201385902
Meng, H., Yamashita, C., Shiba-Fukushima, K., Inoshita, T., Funayama, M., Sato, S., et al. (2017). Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 8:15500. doi: 10.1038/ncomms15500
Meredith, G. E., Totterdell, S., Beales, M., and Meshul, C. K. (2009). Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol. 219, 334–340. doi: 10.1016/j.expneurol.2009.06.005
Mitsumoto, A., Nakagawa, Y., Takeuchi, A., Okawa, K., Iwamatsu, A., and Takanezawa, Y. (2001). Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic. Res. 35, 301–310. doi: 10.1080/10715760100300831
Mollenhauer, B., Locascio, J. J., Schulz-Schaeffer, W., Sixel-Doring, F., Trenkwalder, C., and Schlossmacher, M. G. (2011). alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol. 10, 230–240. doi: 10.1016/S1474-4422(11)70014-X
Mollenhauer, B., Trautmann, E., Taylor, P., Manninger, P., Sixel-Doring, F., Ebentheuer, J., et al. (2013). Total CSF alpha-synuclein is lower in de novo Parkinson patients than in healthy subjects. Neurosci. Lett. 532, 44–48. doi: 10.1016/j.neulet.2012.11.004
Morais, V. A., Haddad, D., Craessaerts, K., De Bock, P. J., Swerts, J., Vilain, S., et al. (2014). PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344, 203–207. doi: 10.1126/science.1249161
Morais, V. A., Verstreken, P., Roethig, A., Smet, J., Snellinx, A., Vanbrabant, M., et al. (2009). Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 1, 99–111. doi: 10.1002/emmm.200900006
Mortiboys, H., Johansen, K. K., Aasly, J. O., and Bandmann, O. (2010). Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75, 2017–2020. doi: 10.1212/WNL.0b013e3181ff9685
Mouton-Liger, F., Jacoupy, M., Corvol, J. C., and Corti, O. (2017). PINK1/parkin-dependent mitochondrial surveillance: From pleiotropy to Parkinson’s disease. Front. Mol. Neurosci. 10:120. doi: 10.3389/fnmol.2017.00120
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/S1474-4422(19)30320-5
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Nicoletti, V., Palermo, G., Del Prete, E., Mancuso, M., and Ceravolo, R. (2021). Understanding the multiple role of mitochondria in Parkinson’s disease and related disorders: Lesson from genetics and protein-interaction network. Front. Cell Dev. Biol. 9:636506. doi: 10.3389/fcell.2021.636506
Norris, K. L., Hao, R., Chen, L. F., Lai, C. H., Kapur, M., Shaughnessy, P. J., et al. (2015). Convergence of Parkin, PINK1, and alpha-Synuclein on Stress-induced Mitochondrial Morphological Remodeling. J. Biol. Chem. 290, 13862–13874. doi: 10.1074/jbc.M114.634063
Oh, C. K., Sultan, A., Platzer, J., Dolatabadi, N., Soldner, F., McClatchy, D. B., et al. (2017). S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 21, 2171–2182. doi: 10.1016/j.celrep.2017.10.068
Okatsu, K., Iemura, S., Koyano, F., Go, E., Kimura, M., Natsume, T., et al. (2012a). Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem. Biophys. Res. Commun. 428, 197–202. doi: 10.1016/j.bbrc.2012.10.041
Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., et al. (2012b). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3:1016. doi: 10.1038/ncomms2016
Olzmann, J. A., Li, L., Chudaev, M. V., Chen, J., Perez, F. A., Palmiter, R. D., et al. (2007). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038. doi: 10.1083/jcb.200611128
Ordureau, A., Heo, J. M., Duda, D. M., Paulo, J. A., Olszewski, J. L., Yanishevski, D., et al. (2015). Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. U.S.A. 112, 6637–6642. doi: 10.1073/pnas.1506593112
Ordureau, A., Paulo, J. A., Zhang, W., Ahfeldt, T., Zhang, J., Cohn, E. F., et al. (2018). Dynamics of PARKIN-dependent mitochondrial ubiquitylation in induced neurons and model systems revealed by digital snapshot proteomics. Mol. Cell 70, 211–227e8. doi: 10.1016/j.molcel.2018.03.012
Ordureau, A., Sarraf, S. A., Duda, D. M., Heo, J. M., Jedrychowski, M. P., Sviderskiy, V. O., et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360–375. doi: 10.1016/j.molcel.2014.09.007
Ozawa, K., Komatsubara, A. T., Nishimura, Y., Sawada, T., Kawafune, H., Tsumoto, H., et al. (2013). S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 3:2202. doi: 10.1038/srep02202
Ozawa, K., Tsumoto, H., Miura, Y., Yamaguchi, J., Iguchi-Ariga, S. M. M., Sakuma, T., et al. (2020). DJ-1 is indispensable for the S-nitrosylation of Parkin, which maintains function of mitochondria. Sci. Rep. 10:4377. doi: 10.1038/s41598-020-61287-6
Padman, B. S., Nguyen, T. N., Uoselis, L., Skulsuppaisarn, M., Nguyen, L. K., and Lazarou, M. (2019). LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 10:408. doi: 10.1038/s41467-019-08335-6
Padmanabhan, S., Lanz, T. A., Gorman, D., Wolfe, M., Joyce, A., Cabrera, C., et al. (2020). An assessment of LRRK2 serine 935 phosphorylation in human peripheral blood mononuclear cells in idiopathic Parkinson’s disease and G2019S LRRK2 cohorts. J. Park. Dis. 10, 623–629. doi: 10.3233/JPD-191786
Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Wacker, M., et al. (2004). Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 279, 18614–18622. doi: 10.1074/jbc.M401135200
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol. Life Sci. 65, 1272–1284. doi: 10.1007/s00018-008-7589-1
Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. doi: 10.1038/nature04788
Perez Carrion, M., Pischedda, F., Biosa, A., Russo, I., Straniero, L., Civiero, L., et al. (2018). The LRRK2 variant E193K prevents mitochondrial fission upon MPP+ treatment by altering LRRK2 binding to DRP1. Front. Mol. Neurosci. 11:64. doi: 10.3389/fnmol.2018.00064
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Plovanich, M., Bogorad, R. L., Sancak, Y., Kamer, K. J., Strittmatter, L., Li, A. A., et al. (2013). MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8:e55785. doi: 10.1371/journal.pone.0055785
Plun-Favreau, H., Klupsch, K., Moisoi, N., Gandhi, S., Kjaer, S., Frith, D., et al. (2007). The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 9, 1243–1252. doi: 10.1038/ncb1644
Pogson, J. H., Ivatt, R. M., Sanchez-Martinez, A., Tufi, R., Wilson, E., Mortiboys, H., et al. (2014). The complex I subunit NDUFA10 selectively rescues Drosophila pink1 mutants through a mechanism independent of mitophagy. PLoS Genet. 10:e1004815. doi: 10.1371/journal.pgen.1004815
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Poole, A. C., Thomas, R. E., Yu, S., Vincow, E. S., and Pallanck, L. (2010). The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5:e10054. doi: 10.1371/journal.pone.0010054
Popov, L. D. (2020). Mitochondrial biogenesis: An update. J. Cell Mol. Med. 24, 4892–4899. doi: 10.1111/jcmm.15194
Popov, L. D. (2022). Mitochondrial-derived vesicles: Recent insights. J. Cell Mol. Med. 26, 3323–3328. doi: 10.1111/jcmm.17391
Pridgeon, J. W., Olzmann, J. A., Chin, L. S., and Li, L. (2007). PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5:e172. doi: 10.1371/journal.pbio.0050172
Pryde, K. R., Smith, H. L., Chau, K. Y., and Schapira, A. H. (2016). PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 213, 163–171. doi: 10.1083/jcb.201509003
Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., et al. (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376. doi: 10.1038/emboj.2013.157
Rakovic, A., Grunewald, A., Kottwitz, J., Bruggemann, N., Pramstaller, P. P., Lohmann, K., et al. (2011). Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One 6:e16746. doi: 10.1371/journal.pone.0016746
Ramirez, A., Heimbach, A., Grundemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi: 10.1038/ng1884
Ramirez, A., Old, W., Selwood, D. L., and Liu, X. (2022). Cannabidiol activates PINK1-Parkin-dependent mitophagy and mitochondrial-derived vesicles. Eur. J. Cell Biol. 101:151185. doi: 10.1016/j.ejcb.2021.151185
Rasool, S., Soya, N., Truong, L., Croteau, N., Lukacs, G. L., and Trempe, J. F. (2018). PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 19:e44981. doi: 10.15252/embr.201744981
Rasool, S., Veyron, S., Soya, N., Eldeeb, M. A., Lukacs, G. L., Fon, E. A., et al. (2022). Mechanism of PINK1 activation by autophosphorylation and insights into assembly on the TOM complex. Mol. Cell 82, 44–59.e6. doi: 10.1016/j.molcel.2021.11.012
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rideout, H. J., Chartier-Harlin, M. C., Fell, M. J., Hirst, W. D., Huntwork-Rodriguez, S., Leyns, C. E. G., et al. (2020). The current state-of-the art of LRRK2-based biomarker assay development in Parkinson’s disease. Front. Neurosci. 14:865. doi: 10.3389/fnins.2020.00865
Rose, C. M., Isasa, M., Ordureau, A., Prado, M. A., Beausoleil, S. A., Jedrychowski, M. P., et al. (2016). Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst. 3, 395–403.e4. doi: 10.1016/j.cels.2016.08.009
Rosina, M., Ceci, V., Turchi, R., Chuan, L., Borcherding, N., Sciarretta, F., et al. (2022). Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue. Cell Metab. 34, 533–548.e12. doi: 10.1016/j.cmet.2022.02.016
Ross, O. A., Soto-Ortolaza, A. I., Heckman, M. G., Aasly, J. O., Abahuni, N., Annesi, G., et al. (2011). Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: A case-control study. Lancet Neurol. 10, 898–908. doi: 10.1016/S1474-4422(11)70175-2
Ryan, T. A., Phillips, E. O., Collier, C. L., Jb Robinson, A., Routledge, D., Wood, R. E., et al. (2020). Tollip coordinates Parkin-dependent trafficking of mitochondrial-derived vesicles. EMBO J. 39:e102539. doi: 10.15252/embj.2019102539
Saito, Y. (2017). DJ-1 as a biomarker of Parkinson’s disease. Adv. Exp. Med. Biol. 1037, 149–171. doi: 10.1007/978-981-10-6583-5_10
Sancak, Y., Markhard, A. L., Kitami, T., Kovacs-Bogdan, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. doi: 10.1126/science.1242993
Sandebring, A., Dehvari, N., Perez-Manso, M., Thomas, K. J., Karpilovski, E., Cookson, M. R., et al. (2009). Parkin deficiency disrupts calcium homeostasis by modulating phospholipase C signalling. FEBS J. 276, 5041–5052. doi: 10.1111/j.1742-4658.2009.07201.x
Sanders, L. H., Laganiere, J., Cooper, O., Mak, S. K., Vu, B. J., Huang, Y. A., et al. (2014). LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: Reversal by gene correction. Neurobiol. Dis. 62, 381–386. doi: 10.1016/j.nbd.2013.10.013
Sarafian, T. A., Ryan, C. M., Souda, P., Masliah, E., Kar, U. K., Vinters, H. V., et al. (2013). Impairment of mitochondria in adult mouse brain overexpressing predominantly full-length, N-terminally acetylated human alpha-synuclein. PLoS One 8:e63557. doi: 10.1371/journal.pone.0063557
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi: 10.1038/nature12043
Sauve, V., Lilov, A., Seirafi, M., Vranas, M., Rasool, S., Kozlov, G., et al. (2015). A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 34, 2492–2505. doi: 10.15252/embj.201592237
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638. doi: 10.1152/physrev.00025.2007
Seibenhener, M. L., Babu, J. R., Geetha, T., Wong, H. C., Krishna, N. R., and Wooten, M. W. (2004). Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol. 24, 8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004
Shimizu, S., Konishi, A., Kodama, T., and Tsujimoto, Y. (2000). BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 3100–3105. doi: 10.1073/pnas.97.7.3100
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. doi: 10.1038/77060
Shin, J. H., Ko, H. S., Kang, H., Lee, Y., Lee, Y. I., Pletinkova, O., et al. (2011). PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702. doi: 10.1016/j.cell.2011.02.010
Shinbo, Y., Niki, T., Taira, T., Ooe, H., Takahashi-Niki, K., Maita, C., et al. (2006). Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ. 13, 96–108. doi: 10.1038/sj.cdd.4401704
Shojaee, S., Sina, F., Banihosseini, S. S., Kazemi, M. H., Kalhor, R., Shahidi, G. A., et al. (2008). Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am. J. Hum. Genet. 82, 1375–1384. doi: 10.1016/j.ajhg.2008.05.005
Siddiqui, A., Bhaumik, D., Chinta, S. J., Rane, A., Rajagopalan, S., Lieu, C. A., et al. (2015). Mitochondrial quality control via the PGC1alpha-TFEB signaling pathway is compromised by Parkin Q311X mutation but independently restored by rapamycin. J. Neurosci. 35, 12833–12844. doi: 10.1523/JNEUROSCI.0109-15.2015
Siddiqui, A., Rane, A., Rajagopalan, S., Chinta, S. J., and Andersen, J. K. (2016). Detrimental effects of oxidative losses in parkin activity in a model of sporadic Parkinson’s disease are attenuated by restoration of PGC1alpha. Neurobiol. Dis. 93, 115–120. doi: 10.1016/j.nbd.2016.05.009
Singh, F., Prescott, A. R., Rosewell, P., Ball, G., Reith, A. D., and Ganley, I. G. (2021). Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. Elife 10:e67604. doi: 10.7554/eLife.67604
Skowyra, D., Craig, K. L., Tyers, M., Elledge, S. J., and Harper, J. W. (1997). F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91, 209–219. doi: 10.1016/s0092-8674(00)80403-1
Sliter, D. A., Martinez, J., Hao, L., Chen, X., Sun, N., Fischer, T. D., et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. doi: 10.1038/s41586-018-0448-9
Soubannier, V., McLelland, G. L., Zunino, R., Braschi, E., Rippstein, P., Fon, E. A., et al. (2012a). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. doi: 10.1016/j.cub.2011.11.057
Soubannier, V., Rippstein, P., Kaufman, B. A., Shoubridge, E. A., and McBride, H. M. (2012b). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7:e52830. doi: 10.1371/journal.pone.0052830
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stafa, K., Tsika, E., Moser, R., Musso, A., Glauser, L., Jones, A., et al. (2014). Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 23, 2055–2077. doi: 10.1093/hmg/ddt600
Stevens, D. A., Lee, Y., Kang, H. C., Lee, B. D., Lee, Y. I., Bower, A., et al. (2015). Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. U.S.A. 112, 11696–11701. doi: 10.1073/pnas.1500624112
Stram, A. R., and Payne, R. M. (2016). Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell Mol. Life Sci. 73, 4063–4073. doi: 10.1007/s00018-016-2280-4
Strauss, K. M., Martins, L. M., Plun-Favreau, H., Marx, F. P., Kautzmann, S., Berg, D., et al. (2005). Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 14, 2099–2111. doi: 10.1093/hmg/ddi215
Stuendl, A., Kraus, T., Chatterjee, M., Zapke, B., Sadowski, B., Moebius, W., et al. (2021). alpha-Synuclein in plasma-derived extracellular vesicles is a potential biomarker of Parkinson’s disease. Mov. Disord. 36, 2508–2518. doi: 10.1002/mds.28639
Su, Y. C., and Qi, X. (2013). Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 22, 4545–4561. doi: 10.1093/hmg/ddt301
Su, Y. C., Guo, X., and Qi, X. (2015). Threonine 56 phosphorylation of Bcl-2 is required for LRRK2 G2019S-induced mitochondrial depolarization and autophagy. Biochim. Biophys. Acta 1852, 12–21. doi: 10.1016/j.bbadis.2014.11.009
Subramaniam, S. R., Vergnes, L., Franich, N. R., Reue, K., and Chesselet, M. F. (2014). Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 70, 204–213. doi: 10.1016/j.nbd.2014.06.017
Sugiura, A., McLelland, G. L., Fon, E. A., and McBride, H. M. (2014). A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 33, 2142–2156. doi: 10.15252/embj.201488104
Sun, Y., Vashisht, A. A., Tchieu, J., Wohlschlegel, J. A., and Dreier, L. (2012). Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 287, 40652–40660. doi: 10.1074/jbc.M112.419721
Szabadkai, G., Simoni, A. M., Bianchi, K., De Stefani, D., Leo, S., Wieckowski, M. R., et al. (2006). Mitochondrial dynamics and Ca2+ signaling. Biochim. Biophys. Acta 1763, 442–449. doi: 10.1016/j.bbamcr.2006.04.002
Tain, L. S., Chowdhury, R. B., Tao, R. N., Plun-Favreau, H., Moisoi, N., Martins, L. M., et al. (2009). Drosophila HtrA2 is dispensable for apoptosis but acts downstream of PINK1 independently from Parkin. Cell Death Differ. 16, 1118–1125. doi: 10.1038/cdd.2009.23
Taira, T., Saito, Y., Niki, T., Iguchi-Ariga, S. M., Takahashi, K., and Ariga, H. (2004). DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218. doi: 10.1038/sj.embor.7400074
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tang, B., Xiong, H., Sun, P., Zhang, Y., Wang, D., Hu, Z., et al. (2006). Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson’s disease. Hum. Mol. Genet. 15, 1816–1825. doi: 10.1093/hmg/ddl104
Tang, F. L., Liu, W., Hu, J. X., Erion, J. R., Ye, J., Mei, L., et al. (2015). VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 12, 1631–1643. doi: 10.1016/j.celrep.2015.08.001
Tanner, C. M. (1992). Occupational and environmental causes of parkinsonism. Occup. Med. 7, 503–513.
Taylor, M., and Alessi, D. R. (2020). Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 63, 102–113. doi: 10.1016/j.ceb.2020.01.001
Tettweiler, G., Miron, M., Jenkins, M., Sonenberg, N., and Lasko, P. F. (2005). Starvation and oxidative stress resistance in Drosophila are mediated through the eIF4E-binding protein, d4E-BP. Genes Dev. 19, 1840–1843. doi: 10.1101/gad.1311805
Teunissen, C. E. I, Verberk, M. W., Thijssen, E. H., Vermunt, L., Hansson, O., Zetterberg, H., et al. (2022). Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 21, 66–77. doi: 10.1016/S1474-4422(21)00361-6
Thevenet, J., Pescini Gobert, R., Hooft van Huijsduijnen, R., Wiessner, C., and Sagot, Y. J. (2011). Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One 6:e21519. doi: 10.1371/journal.pone.0021519
Tokuda, T., Qureshi, M. M., Ardah, M. T., Varghese, S., Shehab, S. A., Kasai, T., et al. (2010). Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75, 1766–1772. doi: 10.1212/WNL.0b013e3181fd613b
Toyofuku, T., Okamoto, Y., Ishikawa, T., Sasawatari, S., and Kumanogoh, A. (2020). LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 39:e100875. doi: 10.15252/embj.2018100875
Trempe, J. F., and Gehring, K. (2023). Structural mechanisms of mitochondrial quality control mediated by PINK1 and Parkin. J. Mol. Biol. 435:168090. doi: 10.1016/j.jmb.2023.168090
Trempe, J. F., Sauve, V., Grenier, K., Seirafi, M., Tang, M. Y., Menade, M., et al. (2013). Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455. doi: 10.1126/science.1237908
Tsai, P. I., Course, M. M., Lovas, J. R., Hsieh, C. H., Babic, M., Zinsmaier, K. E., et al. (2014). PINK1-mediated phosphorylation of Miro inhibits synaptic growth and protects dopaminergic neurons in Drosophila. Sci. Rep. 4:6962. doi: 10.1038/srep06962
Ulmer, T. S., Bax, A., Cole, N. B., and Nussbaum, R. L. (2005). Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 280, 9595–9603. doi: 10.1074/jbc.M411805200
Um, J. W., and Chung, K. C. (2006). Functional modulation of parkin through physical interaction with SUMO-1. J. Neurosci. Res. 84, 1543–1554. doi: 10.1002/jnr.21041
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Van Laar, V. S., Roy, N., Liu, A., Rajprohat, S., Arnold, B., Dukes, A. A., et al. (2015). Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 74, 180–193. doi: 10.1016/j.nbd.2014.11.015
van Steenoven, I., Majbour, N. K., Vaikath, N. N., Berendse, H. W., van der Flier, W. M., van de Berg, W. D. J., et al. (2018). alpha-Synuclein species as potential cerebrospinal fluid biomarkers for dementia with lewy bodies. Mov. Disord. 33, 1724–1733. doi: 10.1002/mds.111
Vasam, G., Nadeau, R., Cadete, V. J. J., Lavallee-Adam, M., Menzies, K. J., and Burelle, Y. (2021). Proteomics characterization of mitochondrial-derived vesicles under oxidative stress. FASEB J. 35:e21278. doi: 10.1096/fj.202002151R
Venda, L. L., Cragg, S. J., Buchman, V. L., and Wade-Martins, R. (2010). Alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 33, 559–568. doi: 10.1016/j.tins.2010.09.004
Vilarino-Guell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167. doi: 10.1016/j.ajhg.2011.06.001
Vissers, M., Troyer, M. D., Thijssen, E., Pereira, D. R., Heuberger, J. A., Groeneveld, G. J., et al. (2023). A leucine-rich repeat kinase 2 (LRRK2) pathway biomarker characterization study in patients with Parkinson’s disease with and without LRRK2 mutations and healthy controls. Clin. Transl. Sci. 16, 1408–1420. doi: 10.1111/cts.13541
Vivacqua, G., Suppa, A., Mancinelli, R., Belvisi, D., Fabbrini, A., Costanzo, M., et al. (2019). Salivary alpha-synuclein in the diagnosis of Parkinson’s disease and Progressive Supranuclear Palsy. Park. Relat. Disord. 63, 143–148. doi: 10.1016/j.parkreldis.2019.02.014
Vizziello, M., Borellini, L., Franco, G., and Ardolino, G. (2021). Disruption of mitochondrial homeostasis: The role of PINK1 in Parkinson’s disease. Cells 10:3022. doi: 10.3390/cells10113022
Wan, H., Tang, B., Liao, X., Zeng, Q., Zhang, Z., and Liao, L. (2018). Analysis of neuronal phosphoproteome reveals PINK1 regulation of BAD function and cell death. Cell Death Differ. 25, 904–917. doi: 10.1038/s41418-017-0027-x
Wang, H., Song, P., Du, L., Tian, W., Yue, W., Liu, M., et al. (2011). Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 286, 11649–11658. doi: 10.1074/jbc.M110.144238
Wang, S., Liu, Z., Ye, T., Mabrouk, O. S., Maltbie, T., Aasly, J., et al. (2017). Elevated LRRK2 autophosphorylation in brain-derived and peripheral exosomes in LRRK2 mutation carriers. Acta Neuropathol. Commun. 5:86. doi: 10.1186/s40478-017-0492-y
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Wang, X., Yan, M. H., Fujioka, H., Liu, J., Wilson-Delfosse, A., Chen, S. G., et al. (2012). LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 21, 1931–1944. doi: 10.1093/hmg/dds003
Wang, Y., Shi, M., Chung, K. A., Zabetian, C. P., Leverenz, J. B., Berg, D., et al. (2012). Phosphorylated alpha-synuclein in Parkinson’s disease. Sci. Transl. Med. 4:121ra20. doi: 10.1126/scitranslmed.3002566
Waragai, M., Wei, J., Fujita, M., Nakai, M., Ho, G. J., Masliah, E., et al. (2006). Increased level of DJ-1 in the cerebrospinal fluids of sporadic Parkinson’s disease. Biochem. Biophys. Res. Commun. 345, 967–972. doi: 10.1016/j.bbrc.2006.05.011
Wasiak, S., Zunino, R., and McBride, H. M. (2007). Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 177, 439–450. doi: 10.1083/jcb.200610042
Wauer, T., Simicek, M., Schubert, A., and Komander, D. (2015a). Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374. doi: 10.1038/nature14879
Wauer, T., Swatek, K. N., Wagstaff, J. L., Gladkova, C., Pruneda, J. N., Michel, M. A., et al. (2015b). Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 34, 307–325. doi: 10.15252/embj.201489847
Wauters, F., Cornelissen, T., Imberechts, D., Martin, S., Koentjoro, B., Sue, C., et al. (2020). LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16, 203–222. doi: 10.1080/15548627.2019.1603548
Wei, T., Huang, G., Gao, J., Huang, C., Sun, M., Wu, J., et al. (2017). Sirtuin 3 deficiency accelerates hypertensive cardiac remodeling by impairing angiogenesis. J. Am. Heart Assoc. 6:e006114. doi: 10.1161/JAHA.117.006114
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., et al. (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847. doi: 10.1073/pnas.0507360102
Whitworth, A. J., Lee, J. R., Ho, V. M., Flick, R., Chowdhury, R., and McQuibban, G. A. (2008). Rhomboid-7 and HtrA2/Omi act in a common pathway with the Parkinson’s disease factors Pink1 and Parkin. Dis. Model. Mech. 1, 168–74; discussion 173. doi: 10.1242/dmm.000109
Xiong, H., Wang, D., Chen, L., Choo, Y. S., Ma, H., Tang, C., et al. (2009). Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest. 119, 650–660. doi: 10.1172/JCI37617
Yamada, T., Dawson, T. M., Yanagawa, T., Iijima, M., and Sesaki, H. (2019). SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 15, 2012–2018. doi: 10.1080/15548627.2019.1643185
Yamada, T., Murata, D., Adachi, Y., Itoh, K., Kameoka, S., Igarashi, A., et al. (2018). Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 28, 588–604.e5. doi: 10.1016/j.cmet.2018.06.014
Yang, Y., Kovacs, M., Sakamoto, T., Zhang, F., Kiehart, D. P., and Sellers, J. R. (2006). Dimerized Drosophila myosin VIIa: A processive motor. Proc. Natl. Acad. Sci. U.S.A. 103, 5746–5751. doi: 10.1073/pnas.0509935103
Yao, D., Gu, Z., Nakamura, T., Shi, Z. Q., Ma, Y., Gaston, B., et al. (2004). Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. U.S.A. 101, 10810–10814. doi: 10.1073/pnas.0404161101
Yi, W., MacDougall, E. J., Tang, M. Y., Krahn, A. I., Gan-Or, Z., Trempe, J. F., et al. (2019). The landscape of Parkin variants reveals pathogenic mechanisms and therapeutic targets in Parkinson’s disease. Hum. Mol. Genet. 28, 2811–2825. doi: 10.1093/hmg/ddz080
Yu, R., Liu, T., Ning, C., Tan, F., Jin, S. B., Lendahl, U., et al. (2019). The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 294, 17262–17277. doi: 10.1074/jbc.RA119.008202
Zhang, J. Y., Deng, Y. N., Zhang, M., Su, H., and Qu, Q. M. (2016). SIRT3 acts as a neuroprotective agent in rotenone-induced parkinson cell model. Neurochem. Res. 41, 1761–1773. doi: 10.1007/s11064-016-1892-2
Zhang, L., Karsten, P., Hamm, S., Pogson, J. H., Muller-Rischart, A. K., Exner, N., et al. (2013). TRAP1 rescues PINK1 loss-of-function phenotypes. Hum. Mol. Genet. 22, 2829–2841. doi: 10.1093/hmg/ddt132
Zhang, L., Zhang, C., Zhu, Y., Cai, Q., Chan, P. Uéda, K., et al. (2008). Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: An immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 1244, 40–52. doi: 10.1016/j.brainres.2008.08.067
Zhang, Y., Gao, J., Chung, K. K., Huang, H., Dawson, V. L., and Dawson, T. M. (2000). Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. U.S.A. 97, 13354–13359. doi: 10.1073/pnas.240347797
Zhong, N., Kim, C. Y., Rizzu, P., Geula, C., Porter, D. R., Pothos, E. N., et al. (2006). DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of pyrimidine tract-binding protein-associated splicing factor. J. Biol. Chem. 281, 20940–20948. doi: 10.1074/jbc.M601935200
Zimprich, A., Benet-Pages, A., Struhal, W., Graf, E., Eck, S. H., Offman, M. N., et al. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175. doi: 10.1016/j.ajhg.2011.06.008
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. doi: 10.1016/j.neuron.2004.11.005
Keywords: Parkinson’s disease, post-translational modification (PTM), mitochondrial function, ubiquitination, phosphorylation, acetylation, SUMOylation, s-nitrosylation
Citation: Luo S, Wang D and Zhang Z (2024) Post-translational modification and mitochondrial function in Parkinson’s disease. Front. Mol. Neurosci. 16:1329554. doi: 10.3389/fnmol.2023.1329554
Received: 29 October 2023; Accepted: 21 December 2023;
Published: 11 January 2024.
Edited by:
Judit Symmank, University Hospital Jena, GermanyReviewed by:
Claudia Crosio, University of Sassari, ItalyMariaelena Repici, Aston University, United Kingdom
Copyright © 2024 Luo, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuohua Zhang, emhhbmd6aHVvaHVhQHNrbG1nLmVkdS5jbg==; Danling Wang, ZGFubGluZ3dhbmdAdXNjLmVkdS5jbg==