 Jane Dudley-Fraser
Jane Dudley-Fraser Katrin Rittinger
Katrin Rittinger- Molecular Structure of Cell Signalling Laboratory, The Francis Crick Institute, London, United Kingdom
The tripartite motif (TRIM) protein family members have been implicated in a multitude of physiologies and pathologies in different tissues. With diverse functions in cellular processes including regulation of signaling pathways, protein degradation, and transcriptional control, the impact of TRIM dysregulation can be multifaceted and complex. Here, we focus on the cellular and molecular roles of TRIMs identified in the brain in the context of a selection of pathologies including cancer and neurodegeneration. By examining each disease in parallel with described roles in brain development, we aim to highlight fundamental common mechanisms employed by TRIM proteins and identify opportunities for therapeutic intervention.
1 Introduction
The tripartite motif proteins are defined by their eponymous TRIpartite Motif composed of a RING domain, one or two B-box domains, and a coiled-coil domain, which is followed by different C-terminal domains that are used to classify TRIMs into 11 classes (I-XI) (Reymond et al., 2001). The tripartite motif is highly conserved whereas the C-terminal domains vary and are proposed to offer target binding diversity, (Sardiello et al., 2008; Hatakeyama, 2017). TRIM proteins have been linked to the regulation of many cellular functions, including innate immunity, cell-cycle regulation, transcription regulation, and autophagy (Rajsbaum et al., 2014; Hatakeyama, 2017). Although TRIM protein functions have been studied across different tissues [e.g., skeletal muscle (Perera et al., 2012), the heart (Zhang J.-R. et al., 2020), and the digestive system (Chen et al., 2022)] and in multiple disease settings [e.g., immunity (Kirmaier et al., 2010; Vaysburd et al., 2013) and cancer (Hatakeyama, 2011, 2017)], here we will focus on the roles of TRIMs in brain health and disease, which themselves are diverse and extensive, with a multitude of TRIMs implicated across many different brain areas (Figures 1, 2).
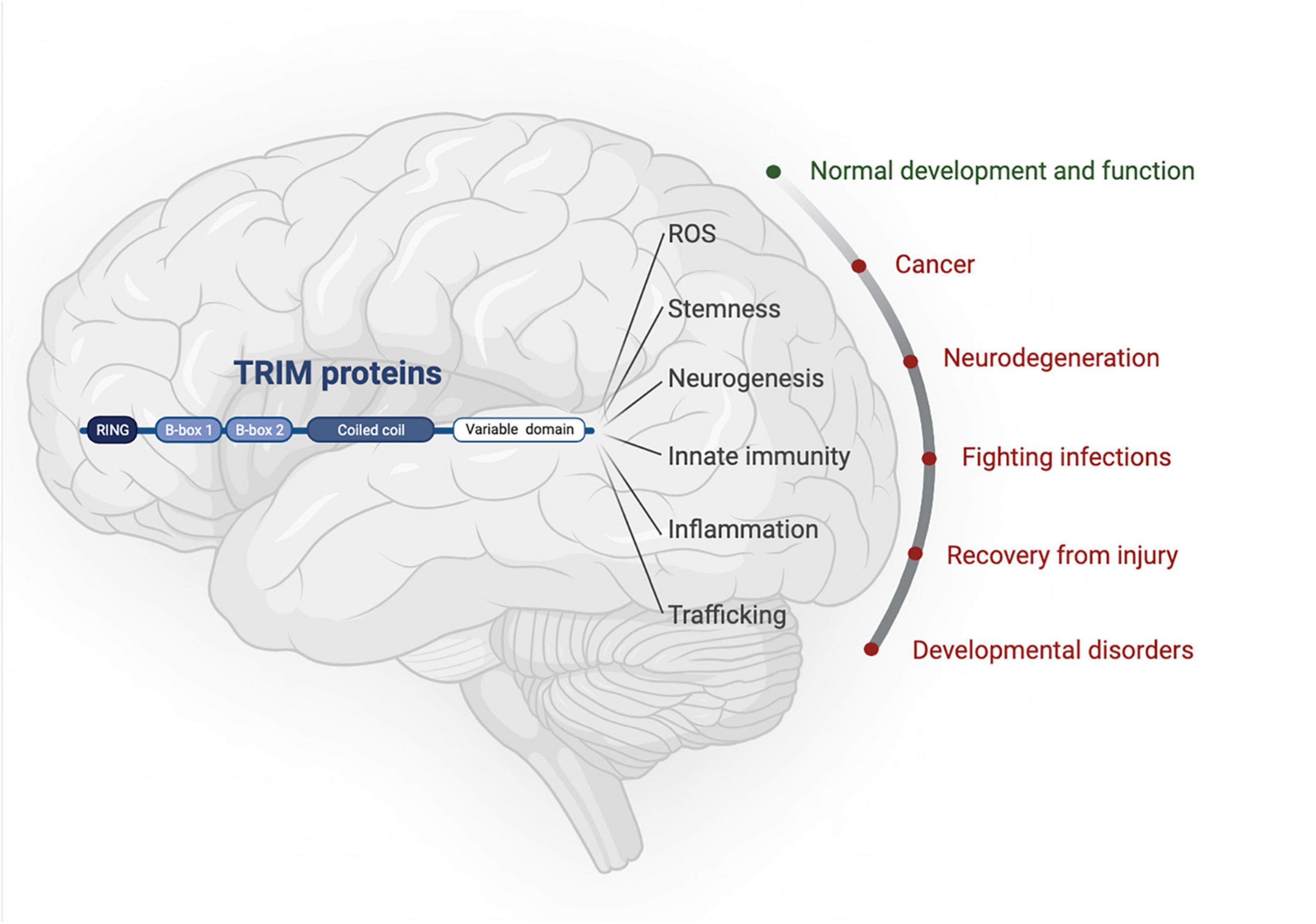
Figure 1. TRIM proteins have roles in normal brain development as well as being implicated in an array of neuropathologies. Created with BioRender.com.
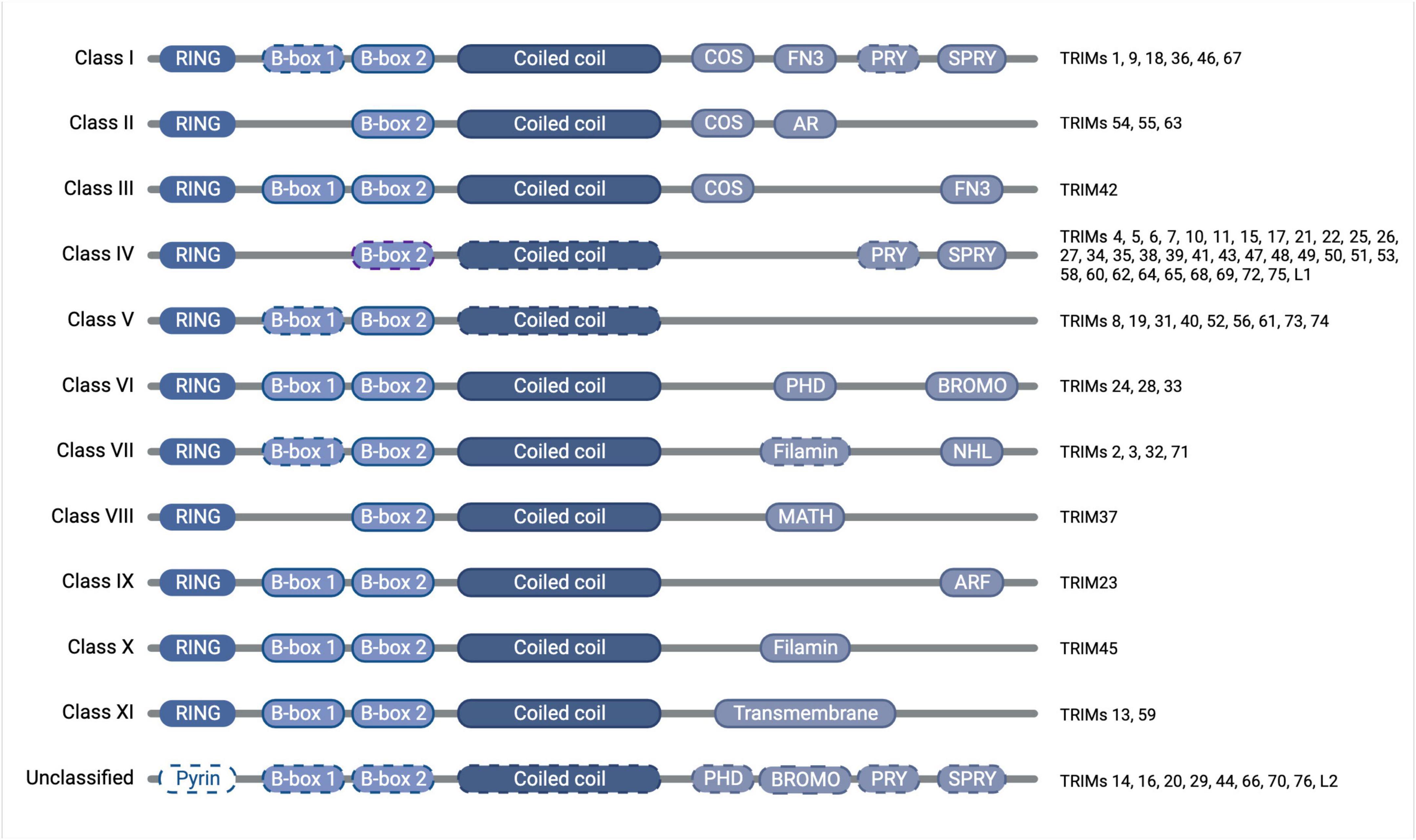
Figure 2. Diagram of TRIM protein domain organization and classification, with dashed outlines denoting where the domain is found only in some of the members of that class. The tripartite motif of the RING domain (E3 ubiquitin ligase catalytic domain), B-box domain(s) (functions somewhat unclear but auto-inhibition and higher-order oligomerisation have been attributed to the B-box domains of some TRIMs), and coiled-coil domain (mediates anti-parallel homodimers) is conserved throughout the family, whereas the C-terminal domains vary and confer divergent functions, which have previously been subjected to thorough phylogenetic analyses (Sardiello et al., 2008; Williams et al., 2019). Created with BioRender.com.
Extensive research has contributed to our current understanding of the roles of each domain of the TRIM proteins. While it is now widely accepted that the coiled-coil domain is responsible for forming antiparallel TRIM homodimers, the function of the B-box domain remains somewhat unclear, though for some TRIMs they have been shown to contribute to auto-inhibition and higher-order oligomerisation (Li and Sodroski, 2008; Sanchez et al., 2014; Dickson et al., 2018). The presence of a conserved RING domain, meanwhile, has led to the assumption that the majority of TRIMs function as E3 ubiquitin ligases. E3 ligases
perform the final stage in the ubiquitination cascade by facilitating the transfer of ubiquitin onto a substrate, subsequent to the sequential action of an E1 ubiquitin-activating enzyme and an E2 ubiquitin-conjugating enzyme (Komander and Rape, 2012). RING-type E3 ligases function as adaptors to bring the substrate together with the E2-ubiquitin conjugate to mediate ubiquitin transfer.
The modification of proteins with ubiquitin or ubiquitin-like proteins [UBLs, e.g., small ubiquitin-like modifier (SUMO)], can be considered as a modular signaling code that is read by specific binding proteins to bring about particular downstream functions (Dikic and Schulman, 2023). Protein ubiquitination largely occurs through reaction with the amino group of lysine (Lys) residues, although N-terminal ubiquitination and even modification of serine and threonine residues have also been described (Wang et al., 2007; Tatham et al., 2013; Mattiroli and Sixma, 2014; Bhogaraju et al., 2016). Targets are often modified with polyubiquitin chains, the architecture of which defines the cellular response, such as degradation through the 26S proteasome induced by Lys48-linked polyubiquitin chains or activation of signaling pathways and autophagy mediated by Lys63-linked chains (Deng et al., 2000; Wang et al., 2001; Hoege et al., 2002; Xu et al., 2009; Lu et al., 2015; Ordureau et al., 2015; Yau and Rape, 2016; Grumati and Dikic, 2018).
Interestingly, there are an increasing number of reports suggesting that some RING-containing TRIM proteins do not exhibit ubiquitin ligase activity in in vitro assays with recombinant proteins (Guimarães and Gomes, 2018; Stevens et al., 2019; Fiorentini et al., 2020; Esposito et al., 2022). Moreover, several TRIM proteins have been ascribed additional functionalities, including SUMO ligase activity, RNA or lipid binding properties, membrane repair and transcriptional regulation, or even repressing the activity of other, ligase-competent TRIMs, largely through interactions via their variable C-terminal domains (Chu and Yang, 2011; Herquel et al., 2011; Kim et al., 2012; Lassot et al., 2018; Williams et al., 2019; Esposito et al., 2022; Randolph et al., 2022; Ma et al., 2023).
2 Brief overview of common pathologies of the brain
Neurodegenerative diseases result in the progressive loss of neurons and cognitive decline, as well as manifesting in a spectrum of other symptoms. Many of these diseases are characterized by inflammation, reactive oxygen species (ROS), and aberrant protein aggregation (e.g., α-synuclein in Parkinson’s disease; amyloid β and tau in Alzheimer’s disease; huntingtin in Huntington’s disease), although precise causes remain elusive (Kumar et al., 2016). Ischemic stroke (i.e., oxygen deprivation) is a major cause of death but the molecular mechanisms and druggable targets are still uncertain, although inflammation, mitochondrial dysfunction, excitotoxicity, and, oxidative stress have been implicated in the resulting neuronal cell loss (Feigin et al., 2010; Ng and Lee, 2019; Shi et al., 2019; Feske, 2021). The brain is responsible for ∼20% of the body’s oxygen demand and requires oxygen levels between 1–5% for normal function as well as to facilitate proper brain development (Silver and Erecińska, 1998; Tomita et al., 2003; Vannucci and Hagberg, 2004).
Brain infections of viruses and bacteria are targeted by microglia, the brain’s resident macrophages, which have also been shown to have roles in normal brain development (Rock et al., 2004; Neumann et al., 2009; Reemst et al., 2016; Carroll et al., 2018). However, innate neuronal and glial cell immunity is also a important line of defense: not only can they generate inflammatory cytokines to trigger the recruitment of specialist immune cells, it is also becoming increasingly understood that neural circuitry exists to control inflammation (Nair and Diamond, 2015; Pavlov and Tracey, 2017). As well as the pathogenic effects of the bacterium or virus themselves, the resulting inflammation can damage neuronal survival and activity. Infections and inflammation can also have an impact on the developing brain, with high prenatal levels of inflammatory cytokines linked with neurological development and various disorders (Zengeler and Lukens, 2021).
Autoimmunity in the brain can similarly lead to complex and devastating loss of brain function, also as a result of aberrant inflammatory signaling (Harris and Hughes, 1985; Karagkouni et al., 2013; Graus et al., 2016).
Brain cancers can fall into different classifications, but over 50% are gliomas, which are subclassified from grade 1 (less malignant) to grade 4 [most malignant, also known as glioblastoma multiforme (GBM)] (Kleihues and Cavenee, 1997). There is a striking unmet clinical need for GBM, which makes up half of glioma cases, with the 5-year survival estimated to still be approximately 5% (Delgado-López and Corrales-García, 2016). Key features of GBM include overactive receptor tyrosine kinase signaling, loss of p53, and stem-like properties that underpin treatment resistance (Venkataramani et al., 2019; Virtuoso et al., 2021; Wang et al., 2021).
Neurological developmental disorders caused by genetic mutations can manifest as wide-ranging, pleiotropic affects including perturbed behavior, motor skills, or learning/intellectual abilities (Parenti et al., 2020). Understanding the molecular pathologies of these conditions can inform fundamental biology as well as clinical management options.
3 The impact of TRIMs on aberrant cell division and cancer in the brain
In this section we outline roles for TRIM proteins according to the ‘Hallmarks of Cancer’ with relation to gliomas and highlight how these, in fact, relate to their functions in normal brain development (Hanahan and Weinberg, 2011).
3.1 Sustaining proliferative signaling
The proteomic and signaling reprogramming required to drive cancer cell growth is tightly interconnected with ubiquitination, as is the stem-like state that confers more malignant properties (Hanahan and Weinberg, 2011; Batlle and Clevers, 2017; Mansour, 2018; Yang L. et al., 2020; Zou et al., 2023). Similarly, during development a rapid expansion of neural stem cells is required to populate the growing brain (Stiles and Jernigan, 2010). It is rational, therefore, that where TRIM proteins are implicated in regulating stemness and differentiation in brain development, they may have parallel roles in gliomagenesis.
TRIM3 (class VII) is the natural place to start this section, having originally been identified as BRAin Tumor (BRAT) in Drosophila melanogaster. Its deletion causes optic neuroblasts and ganglion progenitors to undergo a dramatic expansion without differentiation, resulting in brat mutant brains reaching 10 times their normal size (Gateff et al., 1993; Arama et al., 2000). Therefore, TRIM3 was defined as a tumor suppressor and, indeed, TRIM3 loss of heterozygosity is seen in approximately a quarter of human gliomas, correlating with faster tumor growth, whilst in healthy adults it is highly expressed in the cerebellum (Boulay et al., 2009; Liu et al., 2014). It also implies a pro-differentiation role for TRIM3, which is reinforced by its ability to promote neuronal plasticity via the regulation of γ-actin and motor protein myosin V (El-Husseini and Vincent, 1999; Schreiber et al., 2015). Moreover, TRIM3 is implicated in the trafficking of GABAA receptors in order to generate post-synaptic currents in differentiated cortical neurons (Cheung et al., 2010). In this context, TRIM3 expression is regulated by p53, an interplay that has been further explored in colorectal cancer (Han et al., 2023), but it is unknown whether this is relevant to its role in brain cancers, despite the p53 pathway being dysregulated in over 80% of glioblastoma (GBM) patients (Zhang et al., 2018). TRIM3 suppresses oncogenic C-MYC expression in GBM, resulting in a lower levels of stem cell markers CD133, Nestin, and Nanog, and subsequently reducing GBM neurosphere growth and confirming a pro-differentiation function for TRIM3 (Chen et al., 2014). It would be intriguing to understand whether these effects of TRIM3 are related to target ubiquitination, as it does for another cell-cycle regulator, p21, which is bound and ubiquitinated by TRIM3, thereby repressing cell growth (Liu et al., 2014; Raheja et al., 2014).
Another class VII member, TRIM71, appears to drive a stem-like phenotype and has functional redundancy with C-MYC pathways, as they are interchangeable in the Yamanaka stem cell reprogramming cocktail, although mechanistic details remain elusive (Takahashi and Yamanaka, 2006; Worringer et al., 2014). Despite these links with C-MYC, TRIM71 does not have an established role in gliomagenesis. However, a neural progenitor-related function may be inferred by its documented importance in mouse neural tube closure, in contrast to low levels of TRIM71 expression in adult brains (Maller Schulman et al., 2008; Uhlén et al., 2015). It is also important to understand whether these effects are brought about through the mRNA-binding translation repression function of TRIM71 (Williams et al., 2019), or its ubiquitination activity, which has been shown in a cellular context but appears to be lacking in vitro (Chen Y. et al., 2019; Esposito et al., 2022).
TRIM32 (class VII) is highly expressed in brain tissue, as well as being linked to neuromuscular pathologies (Kudryashova et al., 2009; Kumarasinghe et al., 2021). Specifically, TRIM32 levels are elevated in cortical neurons during development, becoming increasingly expressed in the cortical layers and depleting from the ventricular zone of the embryo over time. Mechanistically, TRIM32 has also been shown to bind and ubiquitinate C-MYC, resulting in its degradation and subsequently affecting transcriptional re-programming and neuronal differentiation (Schwamborn et al., 2009). In an intriguing parallel with normal neurogenesis, in neuroblastoma-initiating cells TRIM32 binds and ubiquitinates another MYC family member, N-MYC, at spindle poles during mitosis to drive asymmetric cell division that eventually results in tumor cell death (Izumi and Kaneko, 2014). TRIM32 also participates in a complex with Let-7a miRNA, the Argonaute components of the RISC complex, and the RNA helicase DDX6 to promote neuronal differentiation (Schwamborn et al., 2009; Nicklas et al., 2015, 2019). These findings may help us understand observations that TRIM32 overexpression promotes a differentiated, less malignant phenotype. In a murine neuroblastoma model TRIM32 enhances differentiation by catalyzing the addition of stabilizing ubiquitin chains (linkage type not defined) to the retinoic acid receptor (RARα), a factor that has well-known roles in neuronal differentiation (Sato et al., 2011; Janesick et al., 2015). Moreover, Wang et al. (2020) showed that TRIM32 also promotes the differentiation of granule neuron progenitor cells during cerebral development by inducing the degradation of SHH effector Gli1, and that loss of this regulation promotes medulloblastoma formation. This, however, can only occur once the TRIM32:PKCζ complex is disrupted, implicating this complex in stem cell maintenance (Hillje et al., 2011). In addition to development and cancer, these findings are also pertinent in limb-girdle muscular dystrophy 2H, an hereditary skeletal muscle disorder caused by TRIM32 mutations, where C-MYC regulation by TRIM32 in myogenic progenitors is implicated (Kudryashova et al., 2009; Nicklas et al., 2012).
Another TRIM that influences C-MYC is TRIM47 (class IV), whose knockdown instead reduces levels of C-MYC as well as β-Catenin and Cyclin-D1 in glioma cells. This attenuates proliferation, epithelial-to-mesenchymal transition markers, and invasive phenotypes, translating to reduced tumor burden in vivo (Chen et al., 2020; Ji et al., 2021). Indeed, TRIM47 expression is higher in GBM and higher grade gliomas, correlating with poorer survival rates overall, although it is also reasonably well expressed in normal brain tissue (Uhlén et al., 2015; Ji et al., 2021). The molecular mechanisms and how this relates to TRIM47 ubiquitination activity is, however, unknown.
TRIM8 (class V) expression levels also correlate with poor clinical outcomes in GBM (Micale et al., 2015). TRIM8 re-localizes from the cytoplasm of healthy neurons to the nucleus in GBM cells to establish a stem-like phenotype, with an increase in malignancy and glioblastoma stem cell markers, such as STAT3, SOX2, Nestin, and Nanog (Zhang C. et al., 2017; Venuto et al., 2019). Mechanistically, TRIM8 ubiquitinates and degrades the STAT3 inhibitor, PIAS3, to promote this pro-stem re-programming (Zhang C. et al., 2017). This is in stark contrast to the role of TRIM8 in development, where it suppresses proliferation and promotes differentiation of neural progenitor cells (Ding et al., 2021). TRIM8 knockdown thereby reduces excitatory synaptic transmission, perhaps giving context to studies showing that TRIM8 truncation mutants can result in early-onset epileptic encephalopathy, a neurodevelopmental disorder characterized by seizures and limited use of language (Sakai et al., 2016; Assoum et al., 2018). Additionally, during mouse embryonic development, TRIM8 localizes to, and therefore may regulate development of, the cerebellum, hippocampus, and cerebral cortex, which all have demonstrated roles in speech, language, and learning, and then continues to be well expressed in adult brains (Uhlén et al., 2015; Sakai et al., 2016). Better understanding of the cellular contexts and molecular mechanisms at play, including any potential ubiquitin ligase activity, may help align these seemingly opposing pro-GBM stemness and anti-neuronal stemness roles for TRIM8 and so inform glioma treatments.
TRIM11 (class IV) also has conflicting roles in neural and gliomagenic stem cells. During mouse embryogenesis, TRIM11 interacts with the neural stem cell regulator PAX6 to effect its proteasomal degradation, presumably via ubiquitination, thereby ablating PAX6-mediated regulation of a suite of neuronal effectors, as well as the expression of TRIM11 itself in an autoregulatory feedback loop (Tuoc and Stoykova, 2008; Sansom et al., 2009). Furthermore, TRIM11 knockdown mice exhibit aggregates of insoluble PAX6 and apoptosis in the cortex. TRIM11 levels, contrastingly, positively correlate with CD133+ and Nestin+ neural stem cell marker staining in GBM cells (Di et al., 2013). TRIM11 knockdown inhibits malignant GBM phenotypes in vitro, correlating with reduced EGFR/MAPK signaling pathway activity, although the relevance of TRIM11 ubiquitin ligase function in this context remains to be explored. Correspondingly, mouse xenograft models with TRIM11 overexpression exhibit stem-like phenotypes and enhanced tumor growth, and, importantly, clinical data shows that TRIM11 expression levels are moderate in normal brains but correlate positively with tumor grade and worse patient prognosis (Di et al., 2013; Uhlén et al., 2015).
TRIM28 (class VI), meanwhile, can function in PAX6-mediated gene expression at sites of H2K9me3 enrichment by forming a complex with it and Pauper long non-coding RNA (lncRNA), with impacts on neural stem cell function and proliferation (Pavlaki et al., 2018). A role for ubiquitin in this process has not been described. Others have shown an alternative role for TRIM28 in establishing H3K9me3 sites, which then repress endogenous retroviruses and transposable elements in neural progenitor cells to maintain stemness, and that without TRIM28 mouse embryos are not viable (Cammas et al., 2000; Fasching et al., 2015; Brattås et al., 2017; Miles et al., 2017; Grassi et al., 2019). Moreover, a co-repressor complex of TRIM28/HATS/DNMT can promote H3K27me3 marks and methylation of the promoter of SIX3, a differentiation-inducing transcription factor, which thereby reduces its expression and promotes a stem-like phenotype (Yu et al., 2020). This is, therefore, in agreement with other studies that implicate TRIM28 in glioma stemness and increased tumor grade (Jovčevska et al., 2017; Peng et al., 2019; Porčnik et al., 2021). TRIM28 can also promote growth of a variety of other cancers and drive resistance to treatments like temozolomide, although recent studies have shown this can be offset by combination treatments with DNA damage response effector inhibitors (e.g., PARP or ATM kinase) (Golding et al., 2012; Gupta et al., 2016; Czerwińska et al., 2017; Yu et al., 2020). In addition to these reports of transcription-based functions, other studies suggest TRIM28 can act as a MAGE protein-dependent ubiquitin ligase or a SUMO E3 ligase, highlighting the need for better context-dependent understanding of this protein (Ivanov et al., 2007; Doyle et al., 2010; Pineda et al., 2015; Stevens et al., 2019).
TRIM33 (class VI) is implicated in neural stem cell and glioma regulation via the TGFβ/SMAD4 and β-Catenin signaling pathways, respectively. In both cases, TRIM33 represses proliferation: murine cortical neural stem cells undergo excessive proliferation and fail to differentiate properly when TRIM33 is knocked out alongside SMAD4, indicative of potential redundancy in the pathway; whereas in human GBM, β-Catenin phosphorylation by PKCδ triggers its ubiquitination by TRIM33 and subsequent degradation, leading to suppression of tumor cell proliferation (Falk et al., 2014; Xue et al., 2015). This is supported by the observation that TRIM33 expression is lower in glioma tissue than normal brain tissue (Uhlén et al., 2015; Xue et al., 2015). However, it would be pertinent to identify the additional factors that can align the described ubiquitination of β-Catenin with the lack of detectable TRIM33 ubiquitin ligase activity in vitro (Stevens et al., 2019).
3.2 Resisting cell death
Recent studies have shown that TRIM17 (class IV) regulates neuronal cell survival or death decisions. TRIM17 expression is highest in the brain, specifically in the basal ganglia, cerebellum, and cortex (Basu-Shrivastava et al., 2021). Conversely, TRIM17 levels are lower in high grade tumors (Xiao et al., 2022). In vitro, TRIM17 overexpression ablates glioma cell line colony formation, aligning with data showing that TRIM17 overexpression in cerebellar neurons induces apoptosis, dependent on its RING domain (Lassot et al., 2010; Xiao et al., 2022). TRIM17-mediated neuronal apoptosis in that context is part of an orchestrated programme required for proper cerebellar developmental morphogenesis and is responsive to neurotrophic factor signaling through the PI3K/Akt/GSK signaling axis, which is, interestingly, also upregulated in glioma (Lassot et al., 2010; Yamaguchi and Miura, 2015). Understanding and harnessing the pro-apoptotic function of TRIM17 may be a powerful tool to fight glioma.
3.3 Evading growth repressors
TRIM45 (class X) is highly expressed in human adult brains, whilst in normal development it has been seen to be required for proper formation of the hypothalamus, hindbrain, and retina in a zebrafish model, via a mechanism that is yet to be uncovered, with ectopic overexpression resulting in aberrant expansion of these tissues (Wang et al., 2004; Choe et al., 2020). This is at odds, however, with the observation that TRIM45 expression levels are reduced in more aggressive gliomas (Zhang J. et al., 2017). On a molecular level, in glioma TRIM45 stabilizes tumor suppressor p53 by modifying it with Lys63-linked ubiquitin chains, thereby occluding its Lys48-linked ubiquitination by MDM2. Understanding how and why TRIM45 exerts seemingly both pro- and anti-proliferative effects may uncover development- or tumourigenic-dependent mechanisms.
3.4 Activating invasion and metastasis
TRIM67 (class I) is implicated in cytoskeletal regulation in both developmental and tumourigenic contexts in the brain. It is one of the most highly expressed TRIMs during cortex development in the late embryogenesis, particularly in neurons, where it is dispensable for proliferation but critical for post-mitotic cell functions and cortex maturation (Boyer et al., 2018, 2019; Bouron and Fauvarque, 2022). TRIM67 knockout mice have impaired spatial memory, cognition, and social functions (Boyer et al., 2018). On a molecular level, it interacts with a range of cytoskeletal, endo- and exocytotic, and synaptic regulators, with which it co-localizes at the axonal periphery and the tips of filopodia (Menon et al., 2021). In the case of the filopodial actin polymerase VASP, TRIM67 antagonizes its non-degradative ubiquitination by TRIM9 (a closely related class I family member) by competitively binding to TRIM9 (Boyer et al., 2019). The knockdown of TRIM67 in this context results in failed filopodia growth and dynamics and corresponding loss of axon turning and branching. Similarly, TRIM67 has also been shown to drive neuronal morphogenesis by regulating the SNAP47-mediated fusion of vesicles to the plasma membrane, thereby expanding the leading edge of the neuron, albeit in a ubiquitin-independent manner (Urbina et al., 2021). Remarkably, although Boyer and colleagues found that TRIM67 expression is largely restricted to neuronal cells in healthy brains, Demirdizen et al. (2022) demonstrated that it becomes aberrantly overexpressed in glial-derived oligodendrogliomas. In this context, TRIM67 promotes membrane protrusion and increased cell motility that can drive tumor growth in mouse models and correlates with alterations in Rho GTPase/ROCK2 pathway signaling (Boyer et al., 2018; Demirdizen et al., 2022). Aberrant expression of TRIM67 in non-neuronal-derived tumors in the brain is also found specifically in brain metastases from breast cancers, again correlating with regulation of invasive properties, as well as DNA damage response markers (Xuan et al., 2022). Why TRIM67 should be important particularly in non-neuronally-derived tumors, despite its neuronal functions in development, is not yet clear. Moreover, it is intriguing that a ubiquitin ligase-dependent mechanism for TRIM67 activity in the brain has not yet been identified.
TRIM37 (class VIII) is a developmentally important protein, with truncation mutations leading to MUscle-LIver-BRain-EYe (MULIBREY) nanism (i.e., individuals with unusually restricted growth). Although gross morphological brain development is normal, patients have motor and speech developmental delay, suggesting a role for TRIM37 in proper neural network formation (Karlberg et al., 2004). MULIBREY patients also experience significantly higher tumor rates and TRIM37 has also been implicated in non-MULIBREY-related cancers (Brigant et al., 2019). In glioma, for example, TRIM37 has been found to have aberrantly high expression (Tang et al., 2018). Knockdown of TRIM37 in this context correlates with reduced PI3K/AKT signaling, migration, and proliferation. If it can be understood why TRIM37 overexpression in glioma and truncation in MULIBREY can similarly lead to tumorigenesis, opportunities to treat both might be identified. The known ubiquitin ligase activity of TRIM37 has not, however, been attributed to any of these effects and might shed light on an explanation in this regard (Kallijärvi et al., 2005).
3.5 Inducing angiogenesis
In healthy brains, TRIM47 is developmentally regulated to facilitate hippocampal synapse development (Sharma and Banerjee, 2022). In adults, however, TRIM47 is more strongly localized to brain blood vessel endothelial cells, which may impact tumor growth by delivering oxygen and nutrients to the expanding tumor mass, thus correlating with increased TRIM47 expression in higher grade gliomas, although this has not yet been explored (Hanahan and Weinberg, 2011; Hao et al., 2019; Ji et al., 2021; Mishra et al., 2022). Moreover, it would be of interest to understand the differential molecular effects, and perhaps ubiquitination targets, that TRIM47 exerts in developing neurons versus blood vessel endothelial cells.
4 Regulation of protein aggregation by TRIMs
It is critical to turn on and off protein degradation during brain development to allow for the formation of different structures, such as axons, but then prevent unchecked accumulation (Saritas-Yildirim and Silva, 2014). When this is not kept under control, proteins can form pathogenic aggregates that lead to neurodegeneration (e.g., α-synuclein in Parkinson’s disease; amyloid β and tau in Alzheimer’s disease; huntingtin in Huntington’s disease) (Kumar et al., 2016). Here, we describe which TRIMs have been implicated in the aggregation of different pathogenic proteins in the brain and how this may be reflected in their roles in developmental regulation of those proteins.
4.1 Tau
TRIM1 and TRIM18 (also known as MID2 and MID1, both class I) are closely related proteins that can interact, localize to microtubules, and interact with cytoskeletal regulators and translation factors. Both are highly expressed in the brain during embryogenesis and are required for proper neural tube closure in Xenopus (Buchner et al., 1999; Suzuki et al., 2010). Moreover, an X-linked disease of midline development, Opitz G/BBB syndrome, is caused by mutations in TRIM18 that result in dysplasia of the corpus callosum and the vermis (the connection between the two lobes of the cerebellum), resulting in intellectual disabilities, as well as hypertelorism, lip-palate-laryngotracheal clefts, and some congenital heart defects (Trockenbacher et al., 2001; De Falco et al., 2003; Pinson et al., 2004; Lancioni et al., 2010). Although causative mutations (found throughout the gene with the exception of the sequence encoding the RING domain) are heterogeneous and lead to a spectrum of clinical phenotypes, dysplasia of midline structures in the brain is a central clinical feature (Pinson et al., 2004; Fontanella et al., 2008; Li et al., 2016). There have also been patients identified with TRIM1 mutations, which suggests a potential overlapping mechanism of action (Li et al., 2016). On a molecular level, mutant TRIM18 protein fails to bind to the α4 subunit of the protein phosphatase PP2A. This results in reduced TRIM18-mediated ubiquitination of PP2A, thereby increasing its activity and the subsequent hypophosphorylation of its downstream microtubule-associated substrates. One such substrate is tau, whose dephosphorylated form stabilizes microtubules. In support of this, TRIM18-deficient neurons have increased axon length and branching propensity, which then disrupt formation of the corpus callosum (Lu et al., 2013). The dysregulation of tau is also an important mechanism in Alzheimer’s (AD) and Huntington’s (HD) diseases, where it can form cytotoxic aggregates in its hyperphosphorylated form, suggesting that TRIM18-mediated degradation of tau phosphatase PP2A may contribute to neurodegeneration, although this requires further study (Schweiger et al., 2017; Rawat et al., 2022).
TRIM11 (class IV), meanwhile, has been attributed roles in the establishment of tauopathies AD and progressive supranuclear palsy (PSP, the most common cause of atypical Parkinsonism). TRIM11 is found in neurons of the cerebellum and basal ganglia in healthy adults and is also is expressed during development to regulate stem-like factors, as described in the section above on cancer, with TRIM11 knockdown resulting in the accumulation of cytotoxic insoluble aggregates of PAX6 (Tuoc and Stoykova, 2008; Jabbari et al., 2018). Similarly, in PSP, TRIM11 mutations increase levels of phosphorylated tau that can then form extensive neurofibrillary tangles (Jabbari et al., 2018; Valentino et al., 2020).
In a recent paper, TRIM11 was seen to be downregulated in the brains of AD patients and disease phenotypes in different mouse tauopathy models could be rescued by TRIM11 overexpression (Zhang et al., 2023). This was suggested to be achieved by: (a) tau SUMOylation by TRIM11, which promotes its degradation via the proteasome (although ubiquitination was not assessed here); and (b) stabilization of monomeric, non-aggregated tau through a chaperone-like function of TRIM11 via an undetermined interface. A better understanding of such molecular mechanisms of TRIMs in neurodegeneration might highlight interesting novel treatment options.
TRIM46 (class I) has been found to be key in axon specification and polarity of neurons in the cerebellum, cortex, and hippocampus (van Beuningen et al., 2015). On closer examination, TRIM46 is seen to localize proximal to the axon in parallel cross-bridged microtubules, or fascicles, a structure which is dubbed the axon initial segment (AIS) (van Beuningen et al., 2015; Fréal et al., 2019; Harterink et al., 2019; Ichinose et al., 2019; Bell et al., 2021; Bell and Zempel, 2022). Here it co-localized with Ankyrin G (ANKG) to scaffold microtubule binding proteins and recruit them to the plasma membrane, thereby facilitating cargo transport to the proximal axon (van Beuningen et al., 2015; Fréal et al., 2019). This is important as selective axonal transport is essential for neuronal polarization and function. During early neuronal differentiation, TRIM46 accumulates at the AIS via the action of KIF3/KAP3 microtubule motors, prior to the establishment of the fasciculated microtubules, requiring properly executed spatiotemporal resolution (Ichinose et al., 2019). Studies from primary neurons suggest that without TRIM46, tau is mis-sorted and improperly trafficked, whilst transformed neurons do not require TRIM46 or ANKG for axonal tau trafficking, hinting at a differentially regulated process (van Beuningen et al., 2015; Bell et al., 2021; Bell and Zempel, 2022). Given the implications of improper trafficking and accumulation of tau in neurodegenerative disease, understanding this potentially developmental distinction may prove vital. Additionally, whether TRIM46 plays simply a scaffolding platform or an active enzymatic function has not been fully explored.
4.2 Huntingtin, amyloid, and ataxin-1
TRIM18-induced PP2A ubiquitination increases the phosphorylation not only of tau, as described above, but also of translational inducers mammalian target of rapamycin (mTOR) and S6, thereby driving overall protein production. Moreover, the TRIM18:PP2A complex interacts with and promotes the translation of certain mRNAs, as well as interacting with several mRNA transport factors (Aranda-Orgillés et al., 2008, 2011; Liu et al., 2011; Krauß et al., 2013; Monteiro et al., 2018). This enhances the translation of pathogenic Huntingtin CAG repeat expansions in HD, as well as amyloid pre-cursor protein (APP) in AD (Müller et al., 2017; Matthes et al., 2018; Monteiro et al., 2018; Heinz et al., 2021). Indeed, elevated TRIM18 expression is observed in the temporal lobe of patients with HD (Heinz et al., 2021). Therefore, the specific depletion or inhibition of TRIM18 may be promising in helping to tackle these diseases.
Research from the Yang lab has shown that TRIM19 (also known as PML, class V) mediates the SUMOylation of poly-Q mutant Ataxin-1 and Huntingtin, thereby triggering their ubiquitination by RNF4 and subsequent clearance from cells (Guo et al., 2014; Chen et al., 2017; Zhu et al., 2020). Interestingly, TRIM11 and TRIM21 can also clear aggregates of Ataxin-1 (Zhu et al., 2020). This correlates with observations that TRIM19 can clear misfolded proteins in the nucleus, thereby preventing neurodegeneration in a polyQ expansion model of spinocerebellar ataxia (Guo et al., 2014). Given the low expression of TRIM19 detected in the brain, the extent to which this defense is employed is uncertain, unless it can be stimulated by specific triggers (Uhlén et al., 2015). In the context of cancer, meanwhile, TRIM19 has been implicated in the clearance of misfolded proteins as part of a pro-tumourigenic anti-oxidant response, in accordance with its role as an oncogenic driver as part of the TRIM19/RARα fusion protein (Chen et al., 2017). Although a developmental role for TRIM19 in the brain remains to be uncovered, SUMOylation [a suggested function of TRIM19 (Chu and Yang, 2011; Guo et al., 2014)] is extensive during brain development, particularly in the hippocampus, and TRIM19 has been implicated in driving the stem-like properties in the context of cancer (Henley et al., 2014; Zhou and Bao, 2014). Connecting these disparate lines of research and mechanisms involving TRIM19 may offer interesting answers for each disease challenge.
4.3 α–synuclein
TRIM41-mediated ZSCAN21 ubiquitination and degradation is inhibited by the competitive binding of TRIM17 to TRIM41 (Lassot et al., 2018). Correspondingly, increased TRIM17 levels correlate with less ZSCAN21 ubiquitination and higher ZSCAN21-induced expression of α-synuclein in PD animal models and patients (Lassot et al., 2018). Furthermore, genetic variants of TRIM17, TRIM41, and ZSCAN21 are significantly associated with familial forms of PD (Farlow et al., 2016; Lassot et al., 2018). The normal function of α-synuclein is to facilitate presynaptic homeostasis and neurotransmitter release, with perturbed α-synuclein regulation observed in autism spectrum disorders that experience synapse dysfunction (Scott and Roy, 2012; Vargas et al., 2017; Morato Torres et al., 2020). ZSCAN21 induces α-synuclein expression in primary neuronal cultures, with α-synuclein expression peaking before birth, and it would be interesting to know whether TRIM17 or TRIM41 also play a role in this context (Raghavan et al., 2004; Dermentzaki et al., 2016). Intriguingly, however, TRIM17, TRIM41, and ZSCAN21 genetic variants have also been linked to autism (Iossifov et al., 2012, 2014; Lim et al., 2017; Satterstrom et al., 2020).
Alternatively, the SUMO E3 ligase activity of TRIM11 has been seen to reduce α-synuclein fibrillar aggregates in PD and facilitate the recruitment of a SUMO-targeted ubiquitin ligases to trigger their clearance (Zhu et al., 2020). Moreover, TRIM11 overexpression can mitigate α-synuclein-mediated pathology, loss of dopaminergic neurons, and lessen PD-related behavioral phenotypes in a mouse model. Connecting all these instances is TRIM11-mediated protein degradation, which may plausibly also be attributable to its enhancement of the proteasome-activating function of USP14 that could subsequently increase overall protein turnover in the cell (Chen et al., 2018). Additional research is needed to unpick these hypotheses and align to the developmental importance of TRIM11 described in the section above.
Although TRIM21 is only expressed at low levels in the brain (Zhang et al., 2014; Uhlén et al., 2015), it is sufficient to clear both α-synuclein and tau aggregates through an antibody-mediated mechanism reminiscent of its well-documented function in clearing viral substrates, implicating it in repressing AD and PD (Mallery et al., 2010; Kondo et al., 2015; McEwan et al., 2017). It is difficult to imagine how this might be relevant during normal development, however, other than in clearing pre-natal infections (see Section 6 “The role of TRIMs in fighting viruses in the brain”).
Meanwhile, α-synuclein and tau SUMOylation by TRIM28 (class VI) results not in their degradation, but instead in their stabilization and re-localization to the nucleus, thereby increasing cytotoxicity and neurodegeneration (Rousseaux et al., 2015, 2016, 2018). The SUMOylation activity of TRIM28 has also been seen to be required for its role as transcriptional repressor, a function which is important in neurogenesis and differentiation (Lagutin et al., 2003; Ivanov et al., 2007; Yu et al., 2020). Indeed, TRIM28 is essential for post-implantation embryogenesis, including for brain development (Cammas et al., 2000; Brattås et al., 2017). Considering these lines of research together suggests that inhibition of TRIM28-mediated SUMOylation as a therapeutic strategy for neurodegeneration may impart counteracting consequences for post-mitotic neuronal fitness and function.
TRIM9 (class I) is predominantly expressed in the cerebellum, hippocampus, and cortex of adult brains, whereas during development expression is highest in the neocortex, dorsal thalamus, midbrain, basal area of the hindbrain, and spinal cord, particularly in regions of proliferation and differentiation (Berti et al., 2002). TRIM9 knockout disrupts hippocampal neuron branching, as well as brain morphogenesis more widely, thereby impairing the development of spatial learning and memory (Winkle et al., 2016; Boyer et al., 2019). In accordance with this observation, TRIM9 levels are lower in the cytoplasm of hippocampal and temporal cortex neurons of PD patients, but are enriched in intracellular Lewy body aggregates (Tanji et al., 2010). It is unclear whether this is a correlative or causative link, however, or what the molecular mechanisms are, and other studies have suggested another non-aggregation-related role for TRIM9 in PD (see Section “5 TRIMs in the regulation of cerebral inflammation”).
4.4 LRRK2
LRRK2 is a cytoskeleton remodelling protein that is crucial in normal neuronal morphogenesis and is one of the most frequently mutated proteins in familial PD, where it both promotes neurotoxic protein aggregation and prevents the clearance of aggregates by autophagy (Jaleel et al., 2007; Parisiadou et al., 2009). TRIM1 is therefore implicated in both neurodevelopment and PD because it can drive the ubiquitin-mediated degradation of wild-type or mutant LRRK2 (Stormo et al., 2022). Given that TRIM1 and TRIM18 are both expressed in the brain and have been shown to interact, it would be interesting to investigate whether their interplay impacts their regulation of tau and LRKK2, respectively.
4.5 Neurofilament
TRIM2 and TRIM3 (class VII), another pair of TRIMs with high sequence homology that can interact (Esposito et al., 2022), also have been implicated in protein aggregation-mediated neuronal pathologies. Although more studies are needed to understand the observed downregulation of TRIM3 in PD patients, which correlates with reduced PI3K/AKT pathway signaling (Dong et al., 2019, 2020), more has been uncovered regarding TRIM2. Specifically, TRIM2 can interact with and ubiquitinate cytoskeletal components, including neurofilament light chain (NF-L) (Ohkawa et al., 2001; Balastik et al., 2008; Khazaei et al., 2011). Mutations in the coiled-coil and NHL domains of TRIM2 that effect its function or stability cause Charcot-Marie-Tooth neuropathy, characterized by progressive early-onset axonal degeneration, particularly in cranial nerves, resulting in a phenotypic spectrum including muscle wasting, facial weakness, and breathing difficulties (Ylikallio et al., 2013; Magri et al., 2020). Mechanistically, TRIM2 mutations prevent the ubiquitination and degradation of NF-L, leading to neuropathic accumulations of neurofilaments in axons (Ylikallio et al., 2013). During development, however, TRIM2-mediated ubiquitination of NF-L is required for normal axonal growth, demonstrating a parallel between neurogenesis and degeneration (Khazaei et al., 2011). In our recent study, we found that TRIM2 and TRIM3 interact at lamellipodia-like membrane protrusions, reminiscent of nascent axons, and cross-regulate one another’s E3 ligase activities (Esposito et al., 2022). In light of this discovery, it will be important to interrogate the interplay of TRIM2 and TRIM3 in mediating neurodegenerative phenotypes.
Although another class VII family member, TRIM32 has also been implicated in neurofilament regulation, its knockout in fact reduces the number of neurofilaments and the diameter of myelinated motor axons, but mice present with a sarcotubular myopathy instead of neurodegeneration (Kudryashova et al., 2009). However, as TRIM32 knockout results in aberrant differentiation into excitatory glutaminergic neurons rather than inhibitory GABAergic neurons, leading to excitotoxicity and reduced overall neuronal numbers in the hippocampus and cortex, it may implicated in neurodegeneration by another means (Hillje et al., 2013; Ntim et al., 2020). Whether these effects can be connected to TRIM32 ubiquitin ligase function remains to be uncovered.
5 TRIMs in the regulation of cerebral inflammation
Inflammation of the brain either in adults or during development can inflict significant damage, resulting in neuronal degeneration or neurodevelopmental defects, respectively (Aktas et al., 2007; Bennet et al., 2018). Understanding how this inflammation is triggered and resolved is therefore critical.
5.1 NF-κB signaling and cytokine release in the brain
As discussed above, TRIM9 (class I) expression is important in brain development, particularly in promoting axonal branching, which may be relevant to axon degeneration in PD. Others have proposed an alternative role for TRIM9 in repressing PD through its inhibition of NF-κB signaling and inflammatory cytokine release, which are known to correlate with PD (Hunot et al., 1997; Kaltschmidt et al., 1997; Tansey and Goldberg, 2010). Mechanistically, TRIM9 binds β-TrCP, a component of the Skp-Cullin-F box (SCF) E3 ligase complex, which blocks SCF-mediated ubiquitination of IκBa and p100, thereby stabilizing them and hence inhibiting NF-κB (Shi et al., 2014). Interestingly, this appears to be a non-ligase-related function for TRIM9. NF-κB suppression by TRIM9 is also important during ischemic stroke, where TRIM9 upregulation in the peri-infarct area is anti-inflammatory and neuroprotective. The ability of TRIM9 to reduce NF-κB signaling may also feed into its other function in promoting axonal guidance during development, albeit in a temporally-dependent fashion, as NF-κB can be inhibitory or stimulatory in driving axonal growth, according to the developmental stage (Gutierrez and Davies, 2011). Understanding the interplay between TRIM9, NF-κB, inflammation, and axonogenesis in more detail may inform not only neuroprotective mechanisms but also treatment options in PD.
TRIM37 (class VIII) expression largely localizes to epithelial tissues in embryos, whereas in adults it is found in central and peripheral nervous systems (Kallijärvi et al., 2006). The importance of TRIM37 in regulating these systems is suggested by the motor and language developmental delays and muscle hypotonicity documented in MULIBREY patients who harbor autosomal recessive TRIM37 mutations (Karlberg et al., 2004). Interestingly, TRIM37 can ubiquitinate and degrade PPARγ, a pro-differentiation regulator of neural stem cells (Kanakasabai et al., 2012). However, this was uncovered in the context of intracerebral hemorrhage, where TRIM37-mediated PPARγ degradation in microglia promotes pro-inflammatory IL-1β release and apoptosis rather than a differentiation process (Han et al., 2019). Taken together, these data suggest that TRIM37 may act as a double-edged sword, capable of driving both development and inflammation in the brain.
Similarly to TRIM37, TRIM47 and TRIM62 (both class IV), are both upregulated and promote inflammation in the hippocampus in an ischemia/reperfusion (I/R) injury model of stroke. Their genetic ablation correspondingly reduces inflammatory signaling and caspase cleavage after I/R injury (Hao et al., 2019; Liu and Lei, 2020). I/R induces TRIM62 ubiquitination with K63-linked chains that are required for its interaction with NLRP3 (a key player in NF-κB pro-inflammatory signaling) (Liu and Lei, 2020). Unfortunately, this cannot be put into wider perspective as, to our knowledge, TRIM62 has not yet been studied in other neurological contexts. TRIM47, however, has been shown to be specifically expressed in blood vessels in the brain, the damage and rupture of which can cause stroke and also correlates with dementia (Marchesi, 2011; Vanlandewijck et al., 2018; Mishra et al., 2022). It would be interesting to understand the seeming discrepancy, however, between observed increased vessel permeability but reduced inflammatory signaling when TRIM47 is ablated, as previous studies would suggest that inflammation would lead to break down of vessel boundaries (Ono et al., 2017). Identifying the molecular mechanisms at play in these different cellular and environmental contexts may help resolve this issue.
TRIM45 (class X) has also been seen to be pro-inflammatory, with I/R triggering TRIM45-driven NF-κB signaling and cytokine production (Xia et al., 2022). This is brought about by the interaction of TRIM45 with TAB2, which it modifies with Lys63-linked poly-ubiquitin chains, promoting the formation of the TAB1/2-TAK1 complex and inducing NF-κB signaling. TRIM45 knockdown, therefore, reduces inflammation and gives more favorable outcomes after I/R. Elevated TRIM45 levels after I/R are echoed by higher expression during development so it would be interesting to understand how TRIM45 functions are determined according to circumstance (Choe et al., 2020).
Likewise, TRIM8 (class V) is pro-inflammatory after I/R- or lipopolysaccharide (LPS)-induced cerebral injury (Bai et al., 2020; Zhao et al., 2020). Upregulated TRIM8 expression after these challenges causes cerebral damage through elevated ROS or cognitive deterioration dependent on NF-κB activity, respectively. This reinforces a previous study documenting Lys63-linked ubiquitination of TAK1 by TRIM8 in response to IL-1β or TNFα stimulation, which drives subsequent NF-κB activation (Li et al., 2011). It would be intriguing to investigate whether the inflammatory responses documented in the brain also depend on this mechanism.
In the context of spinal cord injury, meanwhile, knockout of TRIM32 (class VII) results in elevated pro-inflammatory cytokine production (e.g., IL-1 and IL-10), increased cell proliferation, reduced axon initiation, and delayed recovery of motor functions (Fu et al., 2017). This phenotype finds a parallel in development, where TRIM32 reduces proliferation and promotes differentiation, largely brought about through MYC degradation and enhancing Let-7 miRNA function (Schwamborn et al., 2009). Understanding and harnessing the anti-proliferative, pro-axogenesis function of TRIM32 during development offers an opportunity to identify better treatments after spinal cord injury.
TRIM72 (class IV) has also been implicated in improving recovery from inflammatory neurological damage. By using recombinant TRIM72 protein in combination with umbilical cord-derived stem cells, it is proposed that TRIM72 can alleviate LPS-induced damage of the brain, correlating with reduced pro-inflammatory TLF4/NF-κB signaling (Guan et al., 2019b; Ma et al., 2020). TRIM72 is similarly suggested to serve a neuroprotective role after I/R injury, where it can promote survival signaling through AKT/GSK3β (Yao et al., 2016; Wu et al., 2020). In both cases, however, the specific molecular function of TRIM72 requires further study and, crucially, it is noted that TRIM72 is not expressed in the brain but rather is either exogenously delivered or possibly secreted from muscles and transported through the blood-brain barrier. This diminishes the likelihood that TRIM72 plays a role in normal brain development and function, and indeed one has not yet been described.
5.2 Autoimmune brain inflammation
TRIM21 (class IV) is targeted by autoantibodies in Sjögren’s syndrome, an inflammatory autoimmune condition (Tetsuka et al., 2021). Approximately 5% of Sjögren’s syndrome patients have cerebellar atrophy, with Purkinje cells predominantly affected, consistent with the observation that TRIM21 expression, whilst generally low, is enriched in Purkinje neurons of the hippocampus, cerebral cortex, and cerebellum (Zhang et al., 2014; Uhlén et al., 2015; Tetsuka et al., 2021). It remains to be understood what triggers this attack on Purkinje neurons in only a small proportion of cases.
In mice on a high-fat diet, meanwhile, brain-specific deletion of TRIM13 (class XI) potentiates insulin resistance and metabolic dysfunction, causing systematic inflammation (Qian et al., 2020). Notably, pro-inflammatory cytokine production and inflammation in the cortex, hippocampus, and hypothalamus are observed (Qian et al., 2020). This is supported by a whole-body Trim13–/– mouse model that results in reduced type I interferon (IFN) signaling and curbs the ability of macrophages to respond to viral infection (see Section 6 “The role of TRIMs in fighting viruses in the brain”) (Narayan et al., 2014; Li et al., 2022). As TRIM13 is well expressed in the CNS and proper metabolic regulation and signaling is also critical in neuronal development, such as in neuronal polarization and axogenesis, understanding the intersection between TRIM13, metabolism, and inflammation may offer valuable insights (Williams et al., 2011; Uhlén et al., 2015).
6 The role of TRIMs in fighting viruses in the brain
Perhaps the most well-known role for TRIM proteins is in the innate immune response to infections (van Gent et al., 2018). In this section we outline TRIM-mediated responses to brain viral infection and draw attention to studies from other perspectives that may be interconnected.
6.1 HSV-1
Further to its ability to dampen inflammation in metabolic stress models, TRIM13 (class XI) also curbs NF-κB signaling during viral infection, offering a more permissive environment for replication (Li et al., 2022). This is exemplified in a mouse model of infection by the DNA virus Herpes simplex virus 1 (HSV-1), which accumulates in the brain. In Trim13–/– mice viral load is reduced, corresponding with upregulated NF-κB signaling. Mechanistically, TRIM13 was found to add Lys6 poly-ubiquitin chains to the innate immune signaling trigger STING, which results in it being held in the endoplasmic reticulum and promotes its degradation. An alternative mechanism has also been proposed, albeit to the same effect, for TRIM13 regulation of the RNA-based encephalomyocarditis virus, which causes neurological disease (Carocci and Bakkali-Kassimi, 2012; Narayan et al., 2014). In this model, TRIM13 dampens the activity of the intracellular viral RNA sensor MDA5 to reduce type I IFN production, with the result that Trim13–/– mice can more effectively restrict the virus. TRIM13-mediated regulation of IFNs is a compelling idea to investigate in the context of neurodevelopment and degeneration, where type I IFNs also have been seen to play a key role (Main et al., 2016; Taylor et al., 2018; Hosseini et al., 2020).
In contrast, TRIM41 represses HSV-1 replication in mouse brains by generating a signaling hub for NEMO activity (Yu et al., 2021). TRIM41, which is well-expressed in the brain, interacts with and adds Lys63-linked ubiquitin chains to BCL10, to which NEMO is then recruited and subsequently activates NF-κB and TBK1/IRF3 pathways to induce type I IFNs (Tanaka et al., 2005). This immune regulatory mechanism can be connected with three other observations in development and neurodegeneration: (a) NF-κB has an important role in axon guidance in development (Gutierrez and Davies, 2011), (b) type I IFNs have additionally been implicated in development and PD (Main et al., 2016; Taylor et al., 2018; Hosseini et al., 2020), and (c) the regulation of α-synuclein by TRIM41 is relevant for presynaptic function in both development and PD. Therefore, the regulation of α-synuclein and NF-κB by TRIM41 may have implications for neuronal function across development, neurodegeneration, and infection.
Alternatively, TRIM11 (class IV) restricts HSV-1 infection through the binding and ubiquitination of AIM2, an inflammasome component, after infection, thereby inducing the autophagic degradation of HSV-1 (Liu et al., 2016). This dampens inflammatory responses, such as the production of IL-1β and IL-18. AIM2 has been previously shown to repress dendritic branching but increase axon extension in murine hippocampal neurons during development, with an impact on spatial memory (Chen J. et al., 2019). Whether TRIM11, which is also expressed in the developing brain, can similarly ubiquitinate AIM2 in this capacity, remains to be seen (Tuoc and Stoykova, 2008). It is also interesting to note that TRIM11 both restricts HSV-1 and negatively correlates with AD pathology, given that latent HSV-1 re-activation in the brain has been suggested to increase AD risk (see Section 4.1 “Tau”) (Cairns et al., 2022).
6.2 Japanese encephalitis virus
TRIM21 (class IV) has a well-characterized anti-viral role (Mallery et al., 2010). Conversely, in the context of Japanese Encephalitis Virus (JEV) infection of the brain, TRIM21 appears to support viral replication as it interacts with and downregulates IRF-3 in a RING-dependent manner, thereby reducing virus-restrictive type I IFN signaling (Manocha et al., 2014). TRIM52 (class V), however, ubiquitinates and degrades JEV viral protein NS2A, possibly also supported by its ability to promote NF-κB signaling (Fan et al., 2016, 2017; Zhang P. et al., 2020). Whilst neither of these TRIMs have identifiable roles in brain development, IFN and NF-κB signaling do, as described elsewhere in this article, and their interplay with TRIM proteins remains to be fully explored in disease and developmental contexts.
6.3 Endogenous retroviruses
Viruses of a different kind are linked to TRIM5 and TRIM22 in the context of the brain: human endogenous retroviruses (ERVs). Multiple sclerosis (MS), which some research has suggested may have a link to ERVs, is a progressive condition that results in myelin loss in the nerves of the brain and spinal cord (Hansen et al., 2011; Nexø et al., 2011; Morris et al., 2019). TRIM5 and TRIM22, which have been shown to suppress invading viruses, have genetic variants that correlate with increased MS risk, supporting the concept of a potential viral element in MS development (Pertel et al., 2011; Di Pietro et al., 2013; Nexø et al., 2013). However, the molecular functions of TRIM5 and TRIM22 in this regard are yet to be interrogated. Alternatively, TRIM28 has been implicated in silencing ERVs during neuronal differentiation processes, and which it may be interesting to also assess in the context of MS (Fasching et al., 2015; Brattås et al., 2017).
7 ROS modulation by TRIMs in the brain
Although the etiology of neurodegenerative diseases, such as PD, remains unclear, reactive oxygen species (ROS) and oxidative damage have been implicated (Singh et al., 2019). Interestingly, some TRIMs have been observed to participate in this connection. TRIMs have also been shown to play a role in regulating ROS in other contexts, including ischemic or traumatic injuries and after viral insult, which may connect to their functions in development and neurodegeneration.
7.1 ROS in neurodegeneration
TRIM10 (class IV) appears to have a pathogenic role in PD, where its expression is increased and genetic mutations are associated with disease risk (Witoelar et al., 2017). On a molecular level, TRIM10 ubiquitinates and degrades the phosphatase DUSP6, thereby counteracting DUSP6-mediated ERK activation, ROS suppression, and apoptosis inhibition (Huang et al., 2019). TRIM3 (class VII), meanwhile, upregulates the AKT/PI3K pathway, via an unknown molecular function, thereby dampening ROS, which correlates with a reduction in PD symptoms in a mouse model (Dong et al., 2020). Interestingly, ERK, AKT, and ROS cross-talk has been demonstrated during neurite outgrowth and neuronal apoptosis (Subramaniam et al., 2003; Chang et al., 2004; Myhre et al., 2004; Zeng et al., 2010; Wang et al., 2011; Fu et al., 2020). Being able to position TRIMs in this picture may help contextualize the two contrasting outcomes of development and degeneration that are both stimulated by ERK/AKT/ROS.
As mentioned above, TRIM72 (class IV) is expressed in muscle and not the brain. However, when recombinant TRIM72 protein is administered alongside human umbilical-derived mesenchymal stem cells, oxidative stress is relieved via the activation of NRF2 and neurogenesis is promoted, thereby increasing cognitive function in a mouse model of AD (Ma et al., 2022). This is also relevant for studies of TRIM72 in regulating ROS following brain injury (see Section 7.2 “ROS in ischemic and traumatic brain and spinal injuries”).
7.2 ROS in ischemic and traumatic brain and spinal injuries
The mediation of NRF2 signaling by TRIM72 described above may also be linked to previous work describing a protective and regenerative role for TRIM72 in neurons damaged by H2O2, I/R, or traumatic brain injury (Yao et al., 2016; Guan et al., 2019a). In these contexts, TRIM72 reduces oxidative damage, promotes neuronal proliferation and migration, and therefore alleviates brain oedema and neurological defects. However, this is confounded by studies from heart muscle, where TRIM72 function in cellular membrane repair is hindered by oxidative stress conditions, such as elevated ROS (Cai et al., 2009; Hwang et al., 2011). By connecting research from different tissues, it may be possible to ascertain a deeper understanding of the normal and therapeutic functions of TRIM72.
TRIM32 (class VII), meanwhile, has been described to have both pro- and anti-neuronal regeneration functions. Firstly, after I/R injury, TRIM32 knockdown promotes hippocampal neuron survival through elevated NRF2 pathway activity, which protects cells against ROS-induced apoptosis (Wei et al., 2019). This is in agreement with observations that TRIM32 hinders motor function recovery after traumatic brain injury, which is attributed to increased levels of p53 superfamily member p73 and elevated apoptosis (Zhang Z.-B. et al., 2017). However, these observations are at odds with other reports of TRIM32 promoting recovery from injury. For example, TRIM32 has also been proposed to interact with ERK following spinal cord injury, resulting in improved neuronal differentiation and recovery (Xue et al., 2020). In development, TRIM32 has similarly been shown to have a role in neuronal differentiation (Sato et al., 2011; Nicklas et al., 2015, 2019; Wang et al., 2020). In order to effectively develop new therapies to treat brain injuries, particularly hypoxic damage, it would be important to disentangle how different contexts can dictate whether TRIM32 is either beneficial and detrimental to neuronal cell survival and function.
TRIM31 (class V) supports recovery after I/R by reducing ROS, driving the pentose-phosphate pathway (PPP), and maintaining mitochondrial homeostasis (Zeng et al., 2021). This is brought about by the TRIM31-mediated ubiquitination and subsequent degradation of TIGAR, a PPP inhibitor. Interestingly, neural stem cells are particularly dependent on the PPP, which is intrinsically a reducing system and, therefore, anti-ROS, and TIGAR can inhibit the PPP to drive neural differentiation (Candelario et al., 2013; Zhou et al., 2019). Although TRIM31 has been seen to promote recovery after I/R injury, its potential in regulating neuronal differentiation has not yet been explored.
8 Conclusion
The roles of TRIM proteins in the brain find numerous parallels between pathological states and healthy development, with many common regulatory targets (Table 1). Intriguingly, not only are there shared functions for TRIM targets across development and disease, but there also appears to be a level of redundancy between the TRIMs, with four different TRIMs described to target MYC paralogs and eleven TRIMs capable of impacting the NF-κB pathway. Further studies are now required to understand whether this may reflect cell type- or context-specific expression patterns of these TRIMs or true functional redundancy. Some of the findings summarized above may be understood in greater depth, and any possible discrepancies resolved, by more extensive exploration and comparisons of the appropriate brain cell types and conditions in each context where TRIMs function (Figures 3, 4). Whilst the overall context-dependency of TRIM E3 ubiquitin (or SUMO) ligase activities across different bodily systems would also benefit from further scrutiny, given the multitude of physiologically-relevant putative substrates described here, the brain may prove a valuable model system to assess regulatory mechanisms. However, given the difficulty of accurately modeling this complex organ using in vitro models, coupled with the scarcity of human brain tissue for analysis, this is likely to pose a significant practical challenge.
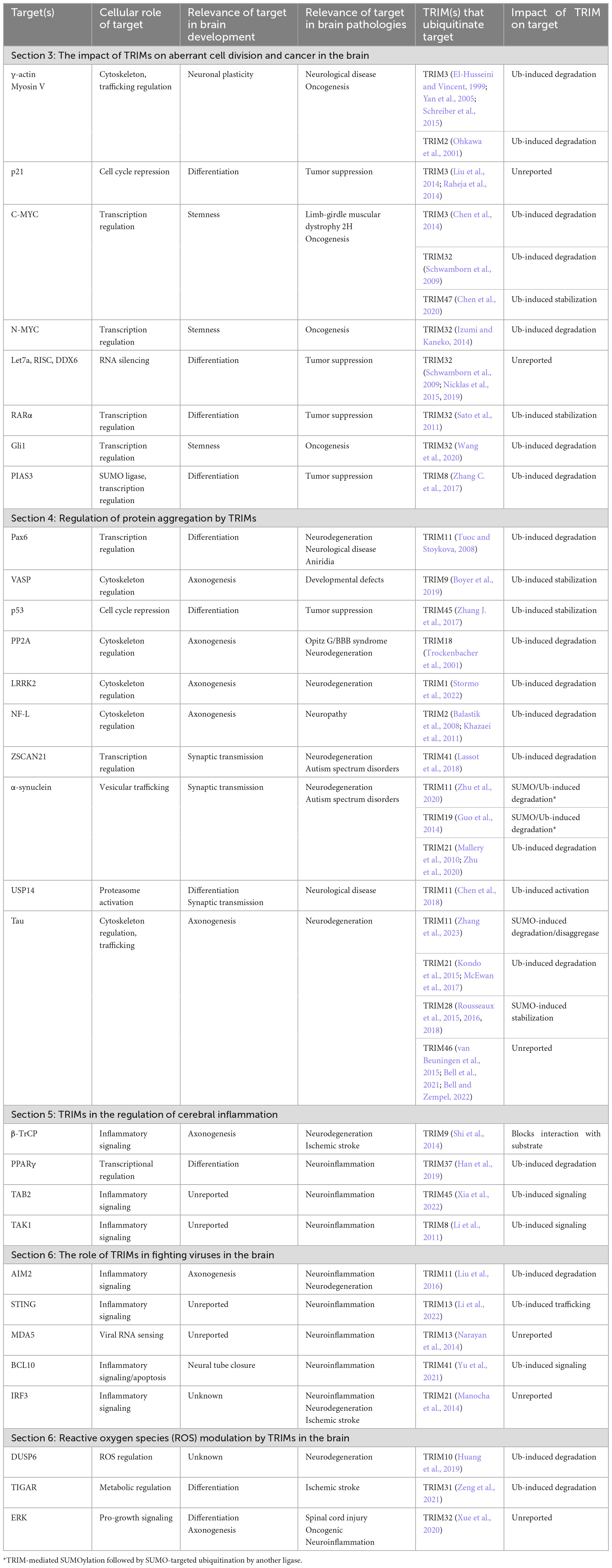
Table 1. Proteins with dual roles in brain development and pathologies are regulated by TRIM family members with varying relationships to ubiquitination, structured according to the sections in the main text.
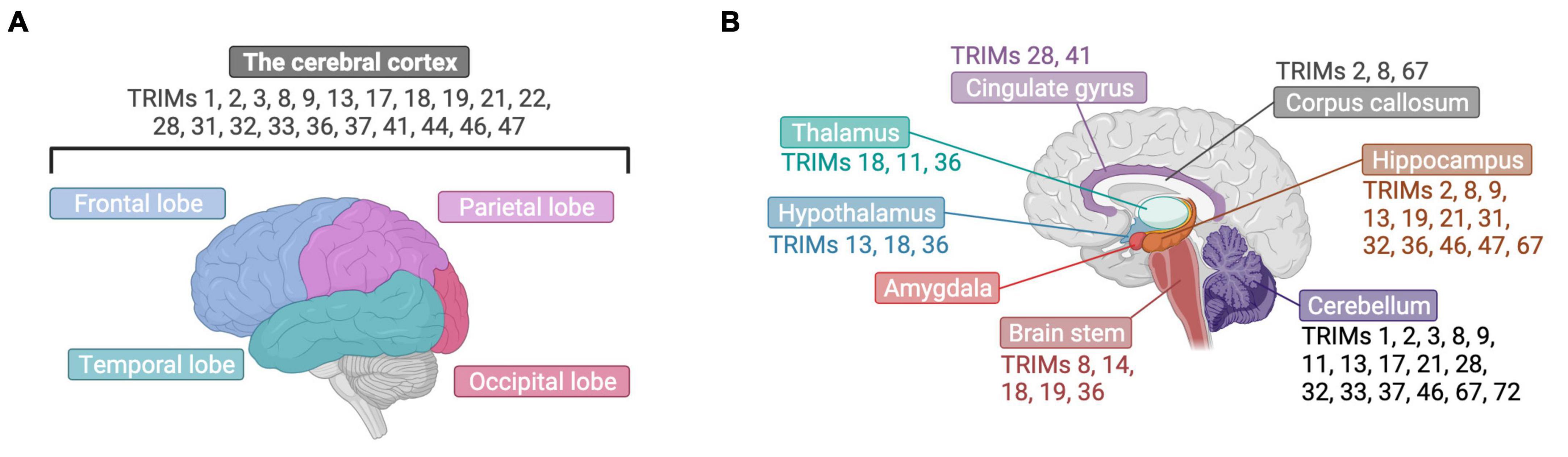
Figure 3. TRIM proteins have been found in varied brain structures. (A) The expression of many TRIMs has been detected in the cerebral cortex, presented here from a lateral view with labeling of its four different cerebral lobes. (B) TRIMs have been found across diverse brain structures, which are highlighted here in brain cut longitudinally and displayed laterally. Created with BioRender.com.
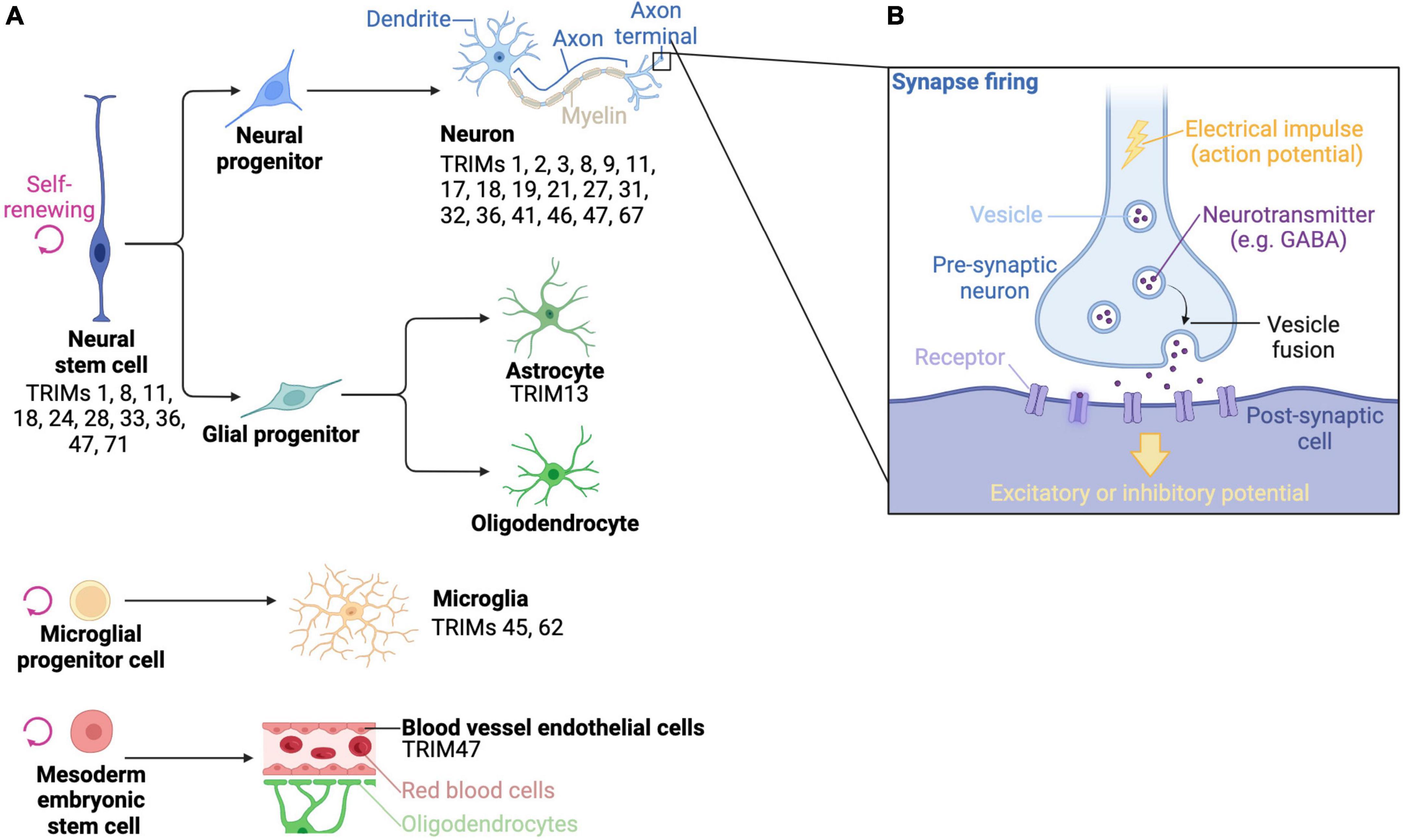
Figure 4. The brain is composed of complex array of cells, which express a variety of TRIM proteins. (A) Stylized drawings of the main differentiated cell types of the brain, as well as the stem cells that they derive from, and which TRIMs have been described in different cell types. (B) A diagram of a how electrical impulses are transmitted from a pre-synaptic neuron to a post-synaptic cell. Created with BioRender.com.
It is tempting to consider the extensive number of TRIMs involved in brain immunity-related processes with respect to evolution. Specifically, the adaptive immune system and the TRIM family expanded greatly during jawed vertebrate evolution, coinciding with the emergence of complex nervous systems, which may suggest a co-dependency between more sophisticated brains and the immune system, as well as potentially TRIM proteins, as has been proposed elsewhere (Meroni, 2012; Nataf, 2017; Yang W. et al., 2020; Kraus et al., 2021).
By highlighting the commonality of different players and pathways in brain development and pathologies we aimed to identify areas for future study as well as therapeutic opportunities. For example, neuronal stemness or differentiation status can be promoted or repressed by different TRIM family members, which is not just important during brain development but also during gliomagenesis, as well as being a factor in recovering from injury. Alternatively, by determining how TRIMs regulate cytoskeletal components during neurogenesis we are able to build up a more complete picture of the molecular dysfunction that leads to neurogeneration.
With regards to therapeutic development, TRIMs have been mostly studied in terms of the E3 ligase function of their RING domains to induce ubiquitin-mediated targeted protein degradation, such as in PROteolysis Targeting Chimera (PROTAC) design (D’Amico et al., 2021). However, it is notable that in many cases described above, ubiquitination is either: (a) not described; (b) does not induce target degradation; or (c) is not relevant to the effect of the TRIM (e.g., the TRIM acts an interaction scaffold) (Table 1). Therefore, rather than employing TRIMs as the ligase in a PROTAC molecule, it may be more appropriate to consider them as the target for degradation.
Moreover, given the difficulty in getting PROTACs across the blood-brain barrier due to their size and chemical properties, it may be necessary to focus on molecular glues to treat neurological pathologies (Farrell and Jarome, 2021). However, given the difficulty of fully understanding the protein-protein interface required for molecular glue prediction and design, and that the majority of molecular glues have been identified by chance, developing such strategies are likely to remain a significant challenge (Kozicka and Thomä, 2021).
Importantly, we hope that by compiling literature across different fields we are able to show that considering data from developmental and disease studies in an integrated and complimentary manner will aid the development of TRIM-based therapeutics for brain pathologies and mitigate unintended side effects (Khan et al., 2020; Békés et al., 2022; Zhang et al., 2022).
Author contributions
JD-F: Writing—original draft, Writing—review and editing. KR: Funding acquisition, Writing—review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (CC2075), the UK Medical Research Council (CC2075), and the Wellcome Trust (CC2075).
Acknowledgments
We are grateful to Mike Devine for his comments on the manuscript. Figures created with BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aktas, O., Ullrich, O., Infante-Duarte, C., Nitsch, R., and Zipp, F. (2007). Neuronal damage in brain inflammation. Arch. Neurol. 64, 185–189. doi: 10.1001/archneur.64.2.185
Arama, E., Dickman, D., Kimchie, Z., Shearn, A., and Lev, Z. (2000). Mutations in the β-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene 19, 3706–3716. doi: 10.1038/sj.onc.1203706
Aranda-Orgillés, B., Rutschow, D., Zeller, R., Karagiannidis, A. I., Köhler, A., Chen, C., et al. (2011). Protein phosphatase 2A (PP2A)-specific ubiquitin ligase MID1 is a sequence-dependent regulator of translation efficiency controlling 3-phosphoinositide-dependent protein kinase-1 (PDPK-1). J. Biol. Chem. 286, 39945–39957. doi: 10.1074/jbc.M111.224451
Aranda-Orgillés, B., Trockenbacher, A., Winter, J., Aigner, J., Köhler, A., Jastrzebska, E., et al. (2008). The opitz syndrome gene product MID1 assembles a microtubule-associated ribonucleoprotein complex. Hum. Genet. 123, 163–176.
Assoum, M., Lines, M. A., Elpeleg, O., Darmency, V., Whiting, S., Edvardson, S., et al. (2018). Further delineation of the clinical spectrum of de novo TRIM8 truncating mutations. Am. J. Med. Genet. A 176, 2470–2478.
Bai, X., Zhang, Y.-L., and Liu, L.-N. (2020). Inhibition of TRIM8 restrains ischaemia-reperfusion-mediated cerebral injury by regulation of NF-κB activation associated inflammation and apoptosis. Exp. Cell Res. 388:111818. doi: 10.1016/j.yexcr.2020.111818
Balastik, M., Ferraguti, F., Pires-da Silva, A., Lee, T. H., Alvarez-Bolado, G., Lu, K. P., et al. (2008). Deficiency in ubiquitin ligase TRIM2 causes accumulation of neurofilament light chain and neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 105, 12016–12021.
Basu-Shrivastava, M., Kozoriz, A., Desagher, S., and Lassot, I. (2021). To ubiquitinate or not to ubiquitinate: TRIM17 in cell life and death. Cells 10:1235.
Batlle, E., and Clevers, H. (2017). Cancer stem cells revisited. Nat. Med. 23, 1124–1134. doi: 10.1038/nm.4409
Békés, M., Langley, D. R., and Crews, C. M. (2022). PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 21, 181–200. doi: 10.1038/s41573-021-00371-6
Bell, M., Bachmann, S., Klimek, J., Langerscheidt, F., and Zempel, H. (2021). Axonal TAU sorting requires the C-terminus of TAU but is independent of ANKG and TRIM46 enrichment at the AIS. Neuroscience 461, 155–171. doi: 10.1016/j.neuroscience.2021.01.041
Bell, M., and Zempel, H. (2022). A simple human cell model for TAU trafficking and tauopathy-related TAU pathology. Neural Regen. Res. 17, 770–772. doi: 10.4103/1673-5374.322450
Bennet, L., Dhillon, S., Lear, C. A., van den Heuij, L., King, V., Dean, J. M., et al. (2018). Chronic inflammation and impaired development of the preterm brain. J. Reprod. Immunol. 125, 45–55. doi: 10.1016/j.jri.2017.11.003
Berti, C., Messali, S., Ballabio, A., Reymond, A., and Meroni, G. (2002). TRIM9 is specifically expressed in the embryonic and adult nervous system. Mech. Dev. 113, 159–162. doi: 10.1016/S0925-4773(02)00013-8
Bhogaraju, S., Kalayil, S., Liu, Y., Bonn, F., Colby, T., Matic, I., et al. (2016). Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell 167, 1636.e13–1649.e13. doi: 10.1016/j.cell.2016.11.019
Boulay, J.-L., Stiefel, U., Taylor, E., Dolder, B., Merlo, A., and Hirth, F. (2009). Loss of heterozygosity of TRIM3 in malignant gliomas. BMC Cancer 9:71. doi: 10.1186/1471-2407-9-71
Bouron, A., and Fauvarque, M.-O. (2022). Genome-wide analysis of genes encoding core components of the ubiquitin system during cerebral cortex development. Mol. Brain 15:72. doi: 10.1186/s13041-022-00958-z
Boyer, N. P., McCormick, L. E., Menon, S., Urbina, F. L., and Gupton, S. L. (2019). A pair of E3 ubiquitin ligases compete to regulate filopodial dynamics and axon guidance. J. Cell Biol. 219:e201902088. doi: 10.1083/jcb.201902088
Boyer, N. P., Monkiewicz, C., Menon, S., Moy, S. S., and Gupton, S. L. (2018). Mammalian TRIM67 functions in brain development and behavior. eNeuro 5:ENEURO.0186-18.2018. doi: 10.1523/eneuro.0186-18.2018
Brattås, P. L., Jönsson, M. E., Fasching, L., Nelander Wahlestedt, J., Shahsavani, M., Falk, R., et al. (2017). TRIM28 controls a gene regulatory network based on endogenous retroviruses in human neural progenitor cells. Cell Rep. 18, 1–11. doi: 10.1016/j.celrep.2016.12.010
Brigant, B., Metzinger-Le Meuth, V., Rochette, J., and Metzinger, L. (2019). TRIMming down to TRIM37: Relevance to inflammation, cardiovascular disorders, and cancer in MULIBREY nanism. Int. J. Mol. Sci. 2:67.
Buchner, G., Montini, E., Andolfi, G., Quaderi, N., Cainarca, S., Messali, S., et al. (1999). MID2, a homologue of the opitz syndrome gene MID1: Similarities in subcellular localization and differences in expression during development. Hum. Mol. Genet. 8, 1397–1407. doi: 10.1093/hmg/8.8.1397
Cai, C., Masumiya, H., Weisleder, N., Matsuda, N., Nishi, M., Hwang, M., et al. (2009). MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64. doi: 10.1038/ncb1812
Cairns, D. M., Itzhaki, R. F., and Kaplan, D. L. (2022). Potential involvement of varicella zoster virus in Alzheimer’s disease via reactivation of quiescent herpes simplex virus type 1. J. Alzheimers Dis. 88, 1189–1200. doi: 10.3233/jad-220287
Cammas, F., Mark, M., Dollé, P., Dierich, A., Chambon, P., and Losson, R. (2000). Mice lacking the transcriptional corepressor TIF1β are defective in early postimplantation development. Development 127, 2955–2963. doi: 10.1242/dev.127.13.2955
Candelario, K. M., Shuttleworth, C. W., and Cunningham, L. A. (2013). Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia inducible factor-1alpha expression. J. Neurochem. 125, 420–429. doi: 10.1111/jnc.12204
Carocci, M., and Bakkali-Kassimi, L. (2012). The encephalomyocarditis virus. Virulence 3, 351–367. doi: 10.4161/viru.20573
Carroll, J. A., Race, B., Williams, K., Striebel, J., and Chesebro, B. (2018). Microglia are critical in host defense against prion disease. J. Virol. 92:e00549-18. doi: 10.1128/jvi.00549-18
Chang, S. H., Poser, S., and Xia, Z. (2004). A novel role for serum response factor in neuronal survival. J. Neurosci. 24, 2277–2285. doi: 10.1523/jneurosci.4868-03.2004
Chen, G., Kong, J., Tucker-Burden, C., Anand, M., Rong, Y., Rahman, F., et al. (2014). Human brat ortholog TRIM3 is a tumor suppressor that regulates asymmetric cell division in glioblastoma. Cancer Res. 74, 4536–4548. doi: 10.1158/0008-5472.CAN-13-3703
Chen, J., Shu, S., Chen, Y., Liu, Z., Yu, L., Yang, L., et al. (2019). AIM2 deletion promotes neuroplasticity and spatial memory of mice. Brain Res. Bull. 152, 85–94. doi: 10.1016/j.brainresbull.2019.07.011
Chen, L., Brewer, M. D., Guo, L., Wang, R., Jiang, P., and Yang, X. (2017). enhanced degradation of misfolded proteins promotes tumorigenesis. Cell Rep. 18, 3143–3154. doi: 10.1016/j.celrep.2017.03.010
Chen, L., Li, M., Li, Q., Xu, M., and Zhong, W. (2020). Knockdown of TRIM47 inhibits glioma cell proliferation, migration and invasion through the inactivation of Wnt/β-catenin pathway. Mol. Cell. Probes 53:101623. doi: 10.1016/j.mcp.2020.101623
Chen, L., Zhu, G., Johns, E. M., and Yang, X. (2018). TRIM11 activates the proteasome and promotes overall protein degradation by regulating USP14. Nat. Commun. 9, 1223–1223. doi: 10.1038/s41467-018-03499-z
Chen, R., Tie, Y., Lu, J., Li, L., Zeng, Z., Chen, M., et al. (2022). Tripartite motif family proteins in inflammatory bowel disease: Mechanisms and potential for interventions. Cell Prolif. 55:e13222. doi: 10.1111/cpr.13222
Chen, Y., Hao, Q., Wang, J., Li, J., Huang, C., Zhang, Y., et al. (2019). Ubiquitin ligase TRIM71 suppresses ovarian tumorigenesis by degrading mutant p53. Cell Death Dis. 10:737. doi: 10.1038/s41419-019-1977-3
Cheung, C. C., Yang, C., Berger, T., Zaugg, K., Reilly, P., Elia, A. J., et al. (2010). Identification of BERP (brain-expressed RING finger protein) as a p53 target gene that modulates seizure susceptibility through interacting with GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 11883–11888. doi: 10.1073/pnas.1006529107
Choe, S., Huh, T.-L., and Rhee, M. (2020). Trim45 is essential to the development of the diencephalon and eye in zebrafish embryos. Anim. Cells Syst. 24, 99–106. doi: 10.1080/19768354.2020.1751281
Chu, Y., and Yang, X. (2011). SUMO E3 ligase activity of TRIM proteins. Oncogene 30, 1108–1116. doi: 10.1038/onc.2010.462
Czerwińska, P., Mazurek, S., and Wiznerowicz, M. (2017). The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 24:63. doi: 10.1186/s12929-017-0374-4
D’Amico, F., Mukhopadhyay, R., Ovaa, H., and Mulder, M. P. C. (2021). Targeting TRIM proteins: A quest towards drugging an emerging protein class. ChemBioChem 22, 2011–2031. doi: 10.1002/cbic.202000787
De Falco, F., Cainarca, S., Andolfi, G., Ferrentino, R., Berti, C., Rodríguez Criado, G., et al. (2003). X-linked Opitz syndrome: Novel mutations in the MID1 gene and redefinition of the clinical spectrum. Am. J. Med. Genet. A 120a, 222–228. doi: 10.1002/ajmg.a.10265
Delgado-López, P. D., and Corrales-García, E. M. (2016). Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 18, 1062–1071. doi: 10.1007/s12094-016-1497-x
Demirdizen, E., Al-Ali, R., Narayanan, A., Sun, X., Varga, J. P., Steffl, B., et al. (2022). TRIM67 drives tumorigenesis in oligodendrogliomas through Rho GTPase-dependent membrane blebbing. Neuro Oncol. 25, 1031–1043. doi: 10.1093/neuonc/noac233
Deng, L., Wang, C., Spencer, E., Yang, L., Braun, A., You, J., et al. (2000). Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361. doi: 10.1016/S0092-8674(00)00126-4
Dermentzaki, G., Paschalidis, N., Politis, P. K., and Stefanis, L. (2016). Complex effects of the ZSCAN21 transcription factor on transcriptional regulation of alpha-synuclein in primary neuronal cultures and in vivo. J. Biol. Chem. 291, 8756–8772. doi: 10.1074/jbc.M115.704973
Di, K., Linskey, M. E., and Bota, D. A. (2013). TRIM11 is overexpressed in high-grade gliomas and promotes proliferation, invasion, migration and glial tumor growth. Oncogene 32, 5038–5047. doi: 10.1038/onc.2012.531
Di Pietro, A., Kajaste-Rudnitski, A., Oteiza, A., Nicora, L., Towers Greg, J., Mechti, N., et al. (2013). TRIM22 inhibits influenza a virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 87, 4523–4533. doi: 10.1128/JVI.02548-12
Dickson, C., Fletcher, A. J., Vaysburd, M., Yang, J.-C., Mallery, D. L., Zeng, J., et al. (2018). Intracellular antibody signalling is regulated by phosphorylation of the Fc receptor TRIM21. eLife 7:e32660. doi: 10.7554/eLife.32660
Dikic, I., and Schulman, B. A. (2023). An expanded lexicon for the ubiquitin code. Nat. Rev. Mol. Cell Biol. 24, 273–287. doi: 10.1038/s41580-022-00543-1
Ding, C., Zhang, C., Kopp, R., Kuney, L., Meng, Q., Wang, L., et al. (2021). Transcription factor POU3F2 regulates TRIM8 expression contributing to cellular functions implicated in schizophrenia. Mol. Psychiatry 26, 3444–3460. doi: 10.1038/s41380-020-00877-2
Dong, W., Luo, B., Qiu, C., Jiang, X., Shen, B., Zhang, L., et al. (2020). TRIM3 attenuates apoptosis in Parkinson’s disease via activating PI3K/AKT signal pathway. Aging 13, 735–749. doi: 10.18632/aging.202181
Dong, W., Qiu, C., Gong, D., Jiang, X., Liu, W., Liu, W., et al. (2019). Proteomics and bioinformatics approaches for the identification of plasma biomarkers to detect Parkinson’s disease. Exp. Ther. Med. 18, 2833–2842. doi: 10.3892/etm.2019.7888
Doyle, J. M., Gao, J., Wang, J., Yang, M., and Potts, P. R. (2010). MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 39, 963–974. doi: 10.1016/j.molcel.2010.08.029
El-Husseini, A. E.-D., and Vincent, S. R. (1999). Cloning and characterization of a novel RING finger protein that interacts with class V myosins. J. Biol. Chem. 274, 19771–19777. doi: 10.1074/jbc.274.28.19771
Esposito, D., Dudley-Fraser, J., Garza-Garcia, A., and Rittinger, K. (2022). Divergent self-association properties of paralogous proteins TRIM2 and TRIM3 regulate their E3 ligase activity. Nat. Commun. 13:7583. doi: 10.1038/s41467-022-35300-7
Falk, S., Joosten, E., Kaartinen, V., and Sommer, L. (2014). Smad4 and Trim33/Tif1γ redundantly regulate neural stem cells in the developing cortex. Cereb. Cortex 24, 2951–2963. doi: 10.1093/cercor/bht149
Fan, W., Liu, T., Li, X., Zhou, Y., Wu, M., Cui, X., et al. (2017). TRIM52: A nuclear TRIM protein that positively regulates the nuclear factor-kappa B signaling pathway. Mol. Immunol. 82, 114–122. doi: 10.1016/j.molimm.2017.01.003
Fan, W., Wu, M., Qian, S., Zhou, Y., Chen, H., Li, X., et al. (2016). TRIM52 inhibits Japanese Encephalitis Virus replication by degrading the viral NS2A. Sci. Rep. 6:33698. doi: 10.1038/srep33698
Farlow, J. L., Robak, L. A., Hetrick, K., Bowling, K., Boerwinkle, E., Coban-Akdemir, Z. H., et al. (2016). Whole-exome sequencing in familial Parkinson disease. JAMA Neurol. 73, 68–75. doi: 10.1001/jamaneurol.2015.3266
Farrell, K., and Jarome, T. J. (2021). Is PROTAC technology really a game changer for central nervous system drug discovery? Expert Opin. Drug Discov. 16, 833–840. doi: 10.1080/17460441.2021.1915979
Fasching, L., Kapopoulou, A., Sachdeva, R., Petri, R., Jönsson, M. E., Männe, C., et al. (2015). TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 10, 20–28. doi: 10.1016/j.celrep.2014.12.004
Feigin, V. L., Barker-Collo, S., Krishnamurthi, R., Theadom, A., and Starkey, N. (2010). Epidemiology of ischaemic stroke and traumatic brain injury. Best Pract. Res. Clin. Anaesthesiol. 24, 485–494. doi: 10.1016/j.bpa.2010.10.006
Fiorentini, F., Esposito, D., and Rittinger, K. (2020). Does it take two to tango? RING domain self-association and activity in TRIM E3 ubiquitin ligases. Biochem. Soc. Trans. 48, 2615–2624. doi: 10.1042/bst20200383
Fontanella, B., Russolillo, G., and Meroni, G. (2008). MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum. Mutat. 29, 584–594. doi: 10.1002/humu.20706
Fréal, A., Rai, D., Tas, R. P., Pan, X., Katrukha, E. A., van de Willige, D., et al. (2019). Feedback-driven assembly of the axon initial segment. Neuron 104, 305.e8–321.e8. doi: 10.1016/j.neuron.2019.07.029
Fu, Q., Zou, M.-M., Zhu, J.-W., Zhang, Y., Chen, W.-J., Cheng, M., et al. (2017). TRIM32 affects the recovery of motor function following spinal cord injury through regulating proliferation of glia. Oncotarget 8, 45380–45390.
Fu, S.-C., Liu, J.-M., Lee, K.-I., Tang, F.-C., Fang, K.-M., Yang, C.-Y., et al. (2020). Cr(VI) induces ROS-mediated mitochondrial-dependent apoptosis in neuronal cells via the activation of Akt/ERK/AMPK signaling pathway. Toxicol. Vitro 65:104795. doi: 10.1016/j.tiv.2020.104795
Gateff, E., Löffler, T., and Wismar, J. (1993). A temperature-sensitive brain tumor suppressor mutation of Drosophila melanogaster: Developmental studies and molecular localization of the gene. Mech. of Dev. 41, 15–31. doi: 10.1016/0925-4773(93)90052-Y
Golding, S. E., Rosenberg, E., Adams, B. R., Wignarajah, S., Beckta, J. M., O’Connor, M. J., et al. (2012). Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle 11, 1167–1173. doi: 10.4161/cc.11.6.19576
Grassi, D. A., Jönsson, M. E., Brattås, P. L., and Jakobsson, J. (2019). TRIM28 and the control of transposable elements in the brain. Brain Res. 1705, 43–47. doi: 10.1016/j.brainres.2018.02.043
Graus, F., Titulaer, M. J., Balu, R., Benseler, S., Bien, C. G., Cellucci, T., et al. (2016). A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 15, 391–404. doi: 10.1016/S1474-4422(15)00401-9
Grumati, P., and Dikic, I. (2018). Ubiquitin signaling and autophagy. J. Biol. Chem. 293, 5404–5413. doi: 10.1074/jbc.TM117.000117
Guan, F., Zhou, X., Li, P., Wang, Y., Liu, M., Li, F., et al. (2019b). MG53 attenuates lipopolysaccharide-induced neurotoxicity and neuroinflammation via inhibiting TLR4/NF-κB pathway in vitro and in vivo. Prog. Neuro Psychopharmacol. Biol. Psychiatry 95:109684. doi: 10.1016/j.pnpbp.2019.109684
Guan, F., Huang, T., Wang, X., Xing, Q., Gumpper, K., Li, P., et al. (2019a). The TRIM protein Mitsugumin 53 enhances survival and therapeutic efficacy of stem cells in murine traumatic brain injury. Stem Cell Res. Ther. 10:352. doi: 10.1186/s13287-019-1433-4
Guimarães, D. S., and Gomes, M. D. (2018). Expression, purification, and characterization of the TRIM49 protein. Protein Exp. Purificat. 143, 57–61. doi: 10.1016/j.pep.2017.10.014
Guo, L., Giasson, B. I., Glavis-Bloom, A., Brewer, M. D., Shorter, J., Gitler, A. D., et al. (2014). A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 55, 15–30. doi: 10.1016/j.molcel.2014.04.030
Gupta, S. K., Kizilbash, S. H., Carlson, B. L., Mladek, A. C., Boakye-Agyeman, F., Bakken, K. K., et al. (2016). Delineation of MGMT hypermethylation as a biomarker for veliparib-mediated temozolomide-sensitizing therapy of glioblastoma. J. Nat. Cancer Inst. 108:djv369. doi: 10.1093/jnci/djv369
Gutierrez, H., and Davies, A. M. (2011). Regulation of neural process growth, elaboration and structural plasticity by NF-κB. Trends Neurosci. 34, 316–325. doi: 10.1016/j.tins.2011.03.001
Han, C., Xia, X., Jiao, S., Li, G., Ran, Q., and Yao, S. (2019). Tripartite motif containing protein 37 involves in thrombin stimulated BV-2 microglial cell apoptosis and interleukin 1β release. Biochem. Biophys. Res. Commun. 516, 1252–1257. doi: 10.1016/j.bbrc.2019.06.158
Han, Y., Lu, S., Song, C., Xuan, Y., Zhang, M., and Cai, H. (2023). Dual roles of TRIM3 in colorectal cancer by retaining p53 in the cytoplasm to decrease its nuclear expression. Cell Death Discov. 9:85. doi: 10.1038/s41420-023-01386-1
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hansen, B., Oturai, A. B., Harbo, H. F., Celius, E. G., Nissen, K. K., Laska, M. J., et al. (2011). Genetic association of multiple sclerosis with the marker rs391745 near the endogenous retroviral locus HERV-Fc1: Analysis of disease subtypes. PLoS One 6:e26438. doi: 10.1371/journal.pone.0026438
Hao, M.-Q., Xie, L.-J., Leng, W., and Xue, R.-W. (2019). Trim47 is a critical regulator of cerebral ischemia-reperfusion injury through regulating apoptosis and inflammation. Biochem. Biophys. Res. Commun. 515, 651–657. doi: 10.1016/j.bbrc.2019.05.065
Harris, E. N., and Hughes, G. R. V. (1985). Cerebral disease in systemic lupus erythematosus. Springer Semin. Immunopathol. 8, 251–266. doi: 10.1007/BF00197299
Harterink, M., Vocking, K., Pan, X., Soriano Jerez, E. M., Slenders, L., Fréal, A., et al. (2019). TRIM46 organizes microtubule fasciculation in the axon initial segment. J. Neurosci. 39, 4864–4873. doi: 10.1523/jneurosci.3105-18.2019
Hatakeyama, S. (2017). TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 42, 297–311. doi: 10.1016/j.tibs.2017.01.002
Heinz, A., Schilling, J., van Roon-Mom, W., and Krauß, S. (2021). The MID1 protein: A promising therapeutic target in Huntington’s disease. Front. Genet. 12:761714. doi: 10.3389/fgene.2021.761714
Henley, J. M., Craig, T. J., and Wilkinson, K. A. (2014). Neuronal SUMOylation: Mechanisms, physiology, and roles in neuronal dysfunction. Physiol. Rev. 94, 1249–1285. doi: 10.1152/physrev.00008.2014
Herquel, B., Ouararhni, K., Khetchoumian, K., Ignat, M., Teletin, M., Mark, M., et al. (2011). Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc. Natl. Acad. Sci. U.S.A. 108, 8212–8217. doi: 10.1073/pnas.1101544108
Hillje, A. L., Pavlou, M. A. S., Beckmann, E., Worlitzer, M. M. A., Bahnassawy, L., Lewejohann, L., et al. (2013). TRIM32-dependent transcription in adult neural progenitor cells regulates neuronal differentiation. Cell Death Dis. 4:e976. doi: 10.1038/cddis.2013.487
Hillje, A.-L., Worlitzer, M. M. A., Palm, T., and Schwamborn, J. C. (2011). Neural stem cells maintain their stemness through protein kinase C ζ-mediated inhibition of TRIM32. Stem Cells 29, 1437–1447. doi: 10.1002/stem.687
Hoege, C., Pfander, B., Moldovan, G.-L., Pyrowolakis, G., and Jentsch, S. (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141. doi: 10.1038/nature00991
Hosseini, S., Michaelsen-Preusse, K., Grigoryan, G., Chhatbar, C., Kalinke, U., and Korte, M. (2020). Type I interferon receptor signaling in astrocytes regulates hippocampal synaptic plasticity and cognitive function of the healthy CNS. Cell Rep. 31:107666. doi: 10.1016/j.celrep.2020.107666
Huang, Q., Zhu, X., and Xu, M. (2019). Silencing of TRIM10 alleviates apoptosis in cellular model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 518, 451–458. doi: 10.1016/j.bbrc.2019.08.041
Hunot, S., Brugg, B., Ricard, D., Michel, P. P., Muriel, M.-P., Ruberg, M., et al. (1997). Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 94, 7531–7536. doi: 10.1073/pnas.94.14.7531
Hwang, M., Ko, J.-K., Weisleder, N., Takeshima, H., and Ma, J. (2011). Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am. J. Physiol. Cell Physiol. 301, C106–C114. doi: 10.1152/ajpcell.00382.2010
Ichinose, S., Ogawa, T., Jiang, X., and Hirokawa, N. (2019). The spatiotemporal construction of the axon initial segment via KIF3/KAP3/TRIM46 transport under MARK2 signaling. Cell Rep. 28, 2413.e7–2426.e7. doi: 10.1016/j.celrep.2019.07.093
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). de novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Ivanov, A. V., Peng, H., Yurchenko, V., Yap, K. L., Negorev, D. G., Schultz, D. C., et al. (2007). PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 28, 823–837. doi: 10.1016/j.molcel.2007.11.012
Izumi, H., and Kaneko, Y. (2014). Trim32 facilitates degradation of MYCN on spindle poles and induces asymmetric cell division in human neuroblastoma cells. Cancer Res. 74, 5620–5630. doi: 10.1158/0008-5472.Can-14-0169
Jabbari, E., Woodside, J., Tan, M. M. X., Shoai, M., Pittman, A., Ferrari, R., et al. (2018). Variation at the TRIM11 locus modifies progressive supranuclear palsy phenotype. Ann. Neurol. 84, 485–496. doi: 10.1002/ana.25308
Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A., et al. (2007). LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 405, 307–317. doi: 10.1042/BJ20070209
Janesick, A., Wu, S. C., and Blumberg, B. (2015). Retinoic acid signaling and neuronal differentiation. Cell. Mol. Life Sci. 72, 1559–1576. doi: 10.1007/s00018-014-1815-9
Ji, B., Liu, L., Guo, Y., Ming, F., Jiang, J., Li, F., et al. (2021). Upregulated tripartite motif 47 could facilitate glioma cell proliferation and metastasis as a tumorigenesis promoter. Comput. Math. Methods Med. 2021:5594973. doi: 10.1155/2021/5594973
Jovčevska, I., Zupanec, N., Urlep, Ž, Vranič, A., Matos, B., Stokin, C. L., et al. (2017). Differentially expressed proteins in glioblastoma multiforme identified with a nanobody-based anti-proteome approach and confirmed by OncoFinder as possible tumor-class predictive biomarker candidates. Oncotarget 8, 44141–44158. doi: 10.18632/oncotarget.17390
Kallijärvi, J., Hämäläinen, R. H., Karlberg, N., Sainio, K., and Lehesjoki, A.-E. (2006). Tissue expression of the mulibrey nanism-associated Trim37 protein in embryonic and adult mouse tissues. Histochem. Cell Biol. 126, 325–334. doi: 10.1007/s00418-006-0162-9
Kallijärvi, J., Lahtinen, U., Hämäläinen, R., Lipsanen-Nyman, M., Palvimo, J. J., and Lehesjoki, A.-E. (2005). TRIM37 defective in mulibrey nanism is a novel RING finger ubiquitin E3 ligase. Exp. Cell Res. 308, 146–155. doi: 10.1016/j.yexcr.2005.04.001
Kaltschmidt, B., Uherek, M., Volk, B., Baeuerle, P. A., and Kaltschmidt, C. (1997). Transcription factor NF-κB is activated in primary neurons by amyloid β peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94, 2642–2647. doi: 10.1073/pnas.94.6.2642
Kanakasabai, S., Pestereva, E., Chearwae, W., Gupta, S. K., Ansari, S., and Bright, J. J. (2012). PPARγ agonists promote oligodendrocyte differentiation of neural stem cells by modulating stemness and differentiation genes. PLoS One 7:e50500. doi: 10.1371/journal.pone.0050500
Karagkouni, A., Alevizos, M., and Theoharides, T. C. (2013). Effect of stress on brain inflammation and multiple sclerosis. Autoimmun. Rev. 12, 947–953. doi: 10.1016/j.autrev.2013.02.006
Karlberg, N., Jalanko, H., Perheentupa, J., and Lipsanen-Nyman, M. (2004). Mulibrey nanism: Clinical features and diagnostic criteria. J. Med. Genet. 41, 92–98. doi: 10.1136/jmg.2003.014118
Khan, S., He, Y., Zhang, X., Yuan, Y., Pu, S., Kong, Q., et al. (2020). PROteolysis TArgeting chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 39, 4909–4924. doi: 10.1038/s41388-020-1336-y
Khazaei, M. R., Bunk, E. C., Hillje, A.-L., Jahn, H. M., Riegler, E. M., Knoblich, J. A., et al. (2011). The E3-ubiquitin ligase TRIM2 regulates neuronal polarization. J. Neurochem. 117, 29–37. doi: 10.1111/j.1471-4159.2010.06971.x
Kim, S., Seo, J., Ko, Y. G., Huh, Y. D., and Park, H. (2012). Lipid-binding properties of TRIM72. BMB Rep. 45, 26–31. doi: 10.5483/bmbrep.2012.45.1.26
Kirmaier, A., Wu, F., Newman, R. M., Hall, L. R., Morgan, J. S., O’Connor, S., et al. (2010). TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8:e1000462. doi: 10.1371/journal.pbio.1000462
Kleihues, P., and Cavenee, W. K. (1997). Pathology and genetics of tumours of the nervous system. Oxford: Oxford University Press.
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. doi: 10.1146/annurev-biochem-060310-170328
Kondo, A., Shahpasand, K., Mannix, R., Qiu, J., Moncaster, J., Chen, C.-H., et al. (2015). Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436. doi: 10.1038/nature14658
Kozicka, Z., and Thomä, N. H. (2021). Haven’t got a glue: Protein surface variation for the design of molecular glue degraders. Cell Chem. Biol. 28, 1032–1047. doi: 10.1016/j.chembiol.2021.04.009
Kraus, A., Buckley, K. M., and Salinas, I. (2021). Sensing the world and its dangers: An evolutionary perspective in neuroimmunology. eLife 10:66706. doi: 10.7554/eLife.66706
Krauß, S., Griesche, N., Jastrzebska, E., Chen, C., Rutschow, D., Achmüller, C., et al. (2013). Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1–PP2A protein complex. Nat. Commun. 4:1511. doi: 10.1038/ncomms2514
Kudryashova, E., Wu, J., Havton, L. A., and Spencer, M. J. (2009). Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 18, 1353–1367. doi: 10.1093/hmg/ddp036
Kumar, V., Sami, N., Kashav, T., Islam, A., Ahmad, F., and Hassan, M. I. (2016). Protein aggregation and neurodegenerative diseases: From theory to therapy. Eur. J. Med. Chem. 124, 1105–1120. doi: 10.1016/j.ejmech.2016.07.054
Kumarasinghe, L., Xiong, L., Garcia-Gimeno, M. A., Lazzari, E., Sanz, P., and Meroni, G. (2021). TRIM32 and malin in neurological and neuromuscular rare diseases. Cells 10:820.
Lagutin, O. V., Zhu, C. C., Kobayashi, D., Topczewski, J., Shimamura, K., Puelles, L., et al. (2003). Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 17, 368–379. doi: 10.1101/gad.1059403
Lancioni, A., Pizzo, M., Fontanella, B., Ferrentino, R., Napolitano, L. M., De Leonibus, E., et al. (2010). Lack of Mid1, the mouse ortholog of the Opitz syndrome gene, causes abnormal development of the anterior cerebellar vermis. J. Neurosci. 30, 2880–2887. doi: 10.1523/jneurosci.4196-09.2010
Lassot, I., Mora, S., Lesage, S., Zieba, B. A., Coque, E., Condroyer, C., et al. (2018). The e3 ubiquitin ligases TRIM17 and TRIM41 modulate α-synuclein expression by regulating ZSCAN21. Cell Rep. 25, 2484.e9–2496.e9. doi: 10.1016/j.celrep.2018.11.002
Lassot, I., Robbins, I., Kristiansen, M., Rahmeh, R., Jaudon, F., Magiera, M. M., et al. (2010). Trim17, a novel E3 ubiquitin-ligase, initiates neuronal apoptosis. Cell Death Differ. 17, 1928–1941. doi: 10.1038/cdd.2010.73
Li, B., Zhou, T., and Zou, Y. (2016). Mid1/Mid2 expression in craniofacial development and a literature review of X-linked Opitz syndrome. Mol. Genet. Genomic Med. 4, 95–105. doi: 10.1002/mgg3.183
Li, Q., Yan, J., Mao, A.-P., Li, C., Ran, Y., Shu, H.-B., et al. (2011). Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β–triggered NF-κB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. U.S.A. 108, 19341–19346. doi: 10.1073/pnas.1110946108
Li, X., and Sodroski, J. (2008). The TRIM5α B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J. Virol. 82, 11495–11502.
Li, X., Yu, Z., Fang, Q., Yang, M., Huang, J., Li, Z., et al. (2022). The transmembrane endoplasmic reticulum–associated E3 ubiquitin ligase TRIM13 restrains the pathogenic-DNA–triggered inflammatory response. Sci. Adv. 8:eabh0496. doi: 10.1126/sciadv.abh0496
Lim, E. T., Uddin, M., De Rubeis, S., Chan, Y., Kamumbu, A. S., Zhang, X., et al. (2017). Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 20, 1217–1224. doi: 10.1038/nn.4598
Liu, E., Knutzen, C. A., Krauss, S., Schweiger, S., and Chiang, G. G. (2011). Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc. Nal. Acad. Sci. U.S.A. 108, 8680–8685. doi: 10.1073/pnas.1100131108
Liu, T., Tang, Q., Liu, K., Xie, W., Liu, X., Wang, H., et al. (2016). TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep. 16, 1988–2002. doi: 10.1016/j.celrep.2016.07.019
Liu, X., and Lei, Q. (2020). TRIM62 knockout protects against cerebral ischemic injury in mice by suppressing NLRP3-regulated neuroinflammation. Biochem. Biophys. Res. Commun. 529, 140–147. doi: 10.1016/j.bbrc.2020.06.014
Liu, Y., Raheja, R., Yeh, N., Ciznadija, D., Pedraza, A. M., Ozawa, T., et al. (2014). TRIM3, a tumor suppressor linked to regulation of p21(Waf1/Cip1). Oncogene 33, 308–315. doi: 10.1038/onc.2012.596
Lu, T., Chen, R., Cox, T. C., Moldrich, R. X., Kurniawan, N., Tan, G., et al. (2013). X-linked microtubule-associated protein, Mid1, regulates axon development. Proc. Natl. Acad. Sci. U.S.A. 110, 19131–19136. doi: 10.1073/pnas.1303687110
Lu, Y., Lee, B.-H., King, R. W., Finley, D., and Kirschner, M. W. (2015). Substrate degradation by the proteasome: A single-molecule kinetic analysis. Science 348:1250834. doi: 10.1126/science.1250834
Ma, S., Wang, Y., Zhou, X., Li, Z., Zhang, Z., Wang, Y., et al. (2020). MG53 protects hUC-MSCs against Inflammatory damage and synergistically enhances their efficacy in neuroinflammation injured brain through inhibiting NLRP3/Caspase-1/IL-1β axis. ACS Chem. Neurosci. 11, 2590–2601. doi: 10.1021/acschemneuro.0c00268
Ma, S., Zhou, X., Wang, Y., Li, Z., Wang, Y., Shi, J., et al. (2022). MG53 protein rejuvenates hUC-MSCs and facilitates their therapeutic effects in AD mice by activating Nrf2 signaling pathway. Redox Biol. 53:102325. doi: 10.1016/j.redox.2022.102325
Ma, Y., Ding, L., Li, Z., and Zhou, C. (2023). Structural basis for TRIM72 oligomerization during membrane damage repair. Nat. Commun. 14:1555. doi: 10.1038/s41467-023-37198-1
Magri, S., Danti, F. R., Balistreri, F., Baratta, S., Ciano, C., Pagliano, E., et al. (2020). Expanding the phenotypic spectrum of TRIM2-associated charcot-marie-tooth disease. J. Peripher. Nerv. Syst. 25, 429–432. doi: 10.1111/jns.12410
Main, B. S., Zhang, M., Brody, K. M., Ayton, S., Frugier, T., Steer, D., et al. (2016). Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson’s disease. Glia 64, 1590–1604. doi: 10.1002/glia.23028
Maller Schulman, B., Liang, X., Stahlhut, C., DelConte, C., Stefani, G., and Slack, F. J. (2008). The let-7 microRNA target gene, Mlin41/Trim71 is required for mouse embryonic survival and neural tube closure. Cell Cycle 7, 3935–3942. doi: 10.4161/cc.7.24.7397
Mallery, D. L., McEwan, W. A., Bidgood, S. R., Towers, G. J., Johnson, C. M., and James, L. C. (2010). Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. U.S.A. 107, 19985–19990. doi: 10.1073/pnas.1014074107
Manocha, G. D., Mishra, R., Sharma, N., Kumawat, K. L., Basu, A., and Singh, S. K. (2014). Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J. Neuroinflamm. 11:24. doi: 10.1186/1742-2094-11-24
Mansour, M. A. (2018). Ubiquitination: Friend and foe in cancer. Int. J. Biochem. Cell Biol. 101, 80–93. doi: 10.1016/j.biocel.2018.06.001
Marchesi, V. T. (2011). Alzheimer’s dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: Implications for early detection and therapy. FASEB J. 25, 5–13. doi: 10.1096/fj.11-0102ufm
Matthes, F., Hettich, M. M., Schilling, J., Flores-Dominguez, D., Blank, N., Wiglenda, T., et al. (2018). Inhibition of the MID1 protein complex: A novel approach targeting APP protein synthesis. Cell Death Discov. 4:4. doi: 10.1038/s41420-017-0003-8
Mattiroli, F., and Sixma, T. K. (2014). Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat. Struct. Mol. Biol. 21, 308–316. doi: 10.1038/nsmb.2792
McEwan, W. A., Falcon, B., Vaysburd, M., Clift, D., Oblak, A. L., Ghetti, B., et al. (2017). Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. U.S.A. 114, 574–579. doi: 10.1073/pnas.1607215114
Menon, S., Goldfarb, D., Ho, C. T., Cloer, E. W., Boyer, N. P., Hardie, C., et al. (2021). The TRIM9/TRIM67 neuronal interactome reveals novel activators of morphogenesis. Mol. Biol. Cell 32, 314–330. doi: 10.1091/mbc.E20-10-0622
Meroni, G. (2012). Genomics and evolution of the TRIM gene family. Adv. Exp. Med. Biol. 770, 1–9. doi: 10.1007/978-1-4614-5398-7_1
Micale, L., Fusco, C., Fontana, A., Barbano, R., Augello, B., De Nittis, P., et al. (2015). TRIM8 downregulation in glioma affects cell proliferation and it is associated with patients survival. BMC Cancer 15:470. doi: 10.1186/s12885-015-1449-9
Miles, D. C., de Vries, N. A., Gisler, S., Lieftink, C., Akhtar, W., Gogola, E., et al. (2017). TRIM28 Is an epigenetic barrier to induced pluripotent stem cell reprogramming. Stem Cells 35, 147–157. doi: 10.1002/stem.2453
Mishra, A., Duplaà, C., Vojinovic, D., Suzuki, H., Sargurupremraj, M., Zilhão, N. R., et al. (2022). Gene-mapping study of extremes of cerebral small vessel disease reveals TRIM47 as a strong candidate. Brain 145, 1992–2007. doi: 10.1093/brain/awab432
Monteiro, O., Chen, C., Bingham, R., Argyrou, A., Buxton, R., Pancevac Jönsson, C., et al. (2018). Pharmacological disruption of the MID1/α4 interaction reduces mutant Huntingtin levels in primary neuronal cultures. Neurosci. Lett. 673, 44–50. doi: 10.1016/j.neulet.2018.02.061
Morato Torres, C. A., Wassouf, Z., Zafar, F., Sastre, D., Outeiro, T. F., and Schüle, B. (2020). The role of alpha-synuclein and other Parkinson’s genes in neurodevelopmental and neurodegenerative disorders. Int. J. Mol. Sci. 21:5724.
Morris, G., Maes, M., Murdjeva, M., and Puri, B. K. (2019). Do human endogenous retroviruses contribute to multiple sclerosis, and if so. How? Mol. Neurobiol. 56, 2590–2605. doi: 10.1007/s12035-018-1255-x
Müller, U. C., Deller, T., and Korte, M. (2017). Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 18, 281–298. doi: 10.1038/nrn.2017.29
Myhre, O., Sterri, S. H., Bogen, I. L., and Fonnum, F. (2004). Erk1/2 phosphorylation and reactive oxygen species formation via nitric oxide and Akt-1/Raf-1 crosstalk in cultured rat cerebellar granule cells exposed to the organic solvent 1,2,4-trimethylcyclohexane. Toxicol. Sci. 80, 296–303. doi: 10.1093/toxsci/kfh166
Nair, S., and Diamond, M. S. (2015). Innate immune interactions within the central nervous system modulate pathogenesis of viral infections. Curr. Opin. Immunol. 36, 47–53. doi: 10.1016/j.coi.2015.06.011
Narayan, K., Waggoner, L., Pham, S. T., Hendricks, G. L., Waggoner, S. N., Conlon, J., et al. (2014). TRIM13 Is a negative regulator of MDA5-mediated type i interferon production. J. Virol. 88, 10748–10757. doi: 10.1128/JVI.02593-13
Nataf, S. (2017). Evolution, immunity and the emergence of brain superautoantigens. F1000Res. 6:171.
Neumann, H., Kotter, M. R., and Franklin, R. J. (2009). Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 132(Pt 2), 288–295. doi: 10.1093/brain/awn109
Nexø, B. A., Christensen, T., Frederiksen, J., Møller-Larsen, A., Oturai, A. B., Villesen, P., et al. (2011). The etiology of multiple sclerosis: Genetic evidence for the involvement of the human endogenous retrovirus HERV-Fc1. PLoS One 6:e16652. doi: 10.1371/journal.pone.0016652
Nexø, B. A., Hansen, B., Nissen, K. K., Gundestrup, L., Terkelsen, T., Villesen, P., et al. (2013). Restriction genes for retroviruses influence the risk of multiple sclerosis. PLoS One 8:e74063. doi: 10.1371/journal.pone.0074063
Ng, S. Y., and Lee, A. Y. W. (2019). Traumatic brain injuries: Pathophysiology and potential therapeutic targets. Front. Cell. Neurosci. 13:528. doi: 10.3389/fncel.2019.00528
Nicklas, S., Hillje, A. L., Okawa, S., Rudolph, I. M., Collmann, F. M., van Wuellen, T., et al. (2019). A complex of the ubiquitin ligase TRIM32 and the deubiquitinase USP7 balances the level of c-Myc ubiquitination and thereby determines neural stem cell fate specification. Cell Death Differ. 26, 728–740. doi: 10.1038/s41418-018-0144-1
Nicklas, S., Okawa, S., Hillje, A.-L., González-Cano, L., del Sol, A., and Schwamborn, J. C. (2015). The RNA helicase DDX6 regulates cell-fate specification in neural stem cells via miRNAs. Nucleic Acids Res. 43, 2638–2654. doi: 10.1093/nar/gkv138
Nicklas, S., Otto, A., Wu, X., Miller, P., Stelzer, S., Wen, Y., et al. (2012). TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PLoS One 7:e30445. doi: 10.1371/journal.pone.0030445
Ntim, M., Li, Q.-F., Zhang, Y., Liu, X.-D., Li, N., Sun, H.-L., et al. (2020). TRIM32 deficiency impairs synaptic plasticity by excitatory-inhibitory imbalance via notch pathway. Cereb. Cortex 30, 4617–4632. doi: 10.1093/cercor/bhaa064
Ohkawa, N., Kokura, K., Matsu-ura, T., Obinata, T., Konishi, Y., and Tamura, T.-A. (2001). Molecular cloning and characterization of neural activity-related RING finger protein (NARF): A new member of the RBCC family is a candidate for the partner of myosin V. J. Neurochem. 78, 75–87. doi: 10.1046/j.1471-4159.2001.00373.x
Ono, S., Egawa, G., and Kabashima, K. (2017). Regulation of blood vascular permeability in the skin. Inflammat. Regen. 37:11. doi: 10.1186/s41232-017-0042-9
Ordureau, A., Heo, J.-M., Duda, D. M., Paulo, J. A., Olszewski, J. L., Yanishevski, D., et al. (2015). Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. U.S.A. 112, 6637–6642. doi: 10.1073/pnas.1506593112
Parenti, I., Rabaneda, L. G., Schoen, H., and Novarino, G. (2020). Neurodevelopmental disorders: From genetics to functional pathways. Trends Neurosci. 43, 608–621. doi: 10.1016/j.tins.2020.05.004
Parisiadou, L., Xie, C., Cho, H. J., Lin, X., Gu, X.-L., Long, C.-X., et al. (2009). Phosphorylation of Ezrin/Radixin/Moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 29, 13971–13980. doi: 10.1523/jneurosci.3799-09.2009
Pavlaki, I., Alammari, F., Sun, B., Clark, N., Sirey, T., Lee, S., et al. (2018). The long non-coding RNA Paupar promotes KAP1-dependent chromatin changes and regulates olfactory bulb neurogenesis. EMBO J. 37:e98219. doi: 10.15252/embj.201798219
Pavlov, V. A., and Tracey, K. J. (2017). Neural regulation of immunity: Molecular mechanisms and clinical translation. Nat. Neurosci. 20, 156–166. doi: 10.1038/nn.4477
Peng, Y., Zhang, M., Jiang, Z., and Jiang, Y. (2019). TRIM28 activates autophagy and promotes cell proliferation in glioblastoma. Onco Targets Ther. 12, 397–404. doi: 10.2147/ott.S188101
Perera, S., Mankoo, B., and Gautel, M. (2012). Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. J. Muscle Res. Cell Motil. 33, 107–122. doi: 10.1007/s10974-012-9288-7
Pertel, T., Hausmann, S., Morger, D., Züger, S., Guerra, J., Lascano, J., et al. (2011). TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365. doi: 10.1038/nature09976
Pineda, C. T., Ramanathan, S., Fon, T. K., Weon, J. L., Potts, M. B., Ou, Y.-H., et al. (2015). Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728. doi: 10.1016/j.cell.2015.01.034
Pinson, L., Augé, J., Audollent, S., Mattéi, G., Etchevers, H., Gigarel, N., et al. (2004). Embryonic expression of the human MID1 gene and its mutations in opitz syndrome. J. Med. Genet. 41, 381–386. doi: 10.1136/jmg.2003.014829
Porčnik, A., Novak, M., Breznik, B., Majc, B., Hrastar, B., Šamec, N., et al. (2021). TRIM28 selective nanobody reduces glioblastoma stem cell invasion. Molecules 26:5141.
Qian, Y., Lei, G., and Wen, L. (2020). Brain-specific deletion of TRIM13 promotes metabolic stress-triggered insulin resistance, glucose intolerance, and neuroinflammation. Biochem. Biophys. Res. Commun. 527, 138–145. doi: 10.1016/j.bbrc.2020.03.076
Raghavan, R., Kruijff, L. D., Sterrenburg, M. D., Rogers, B. B., Hladik, C. L., and White, C. L. (2004). Alpha-synuclein expression in the developing human brain. Pediatr. Dev. Pathol. 7, 506–516. doi: 10.1007/s10024-003-7080-9
Raheja, R., Liu, Y., Hukkelhoven, E., Yeh, N., and Koff, A. (2014). The ability of TRIM3 to induce growth arrest depends on RING-dependent E3 ligase activity. Biochem. J. 458, 537–545. doi: 10.1042/bj20131288
Rajsbaum, R., García-Sastre, A., and Versteeg, G. A. (2014). TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 426, 1265–1284. doi: 10.1016/j.jmb.2013.12.005
Randolph, K., Hyder, U., and D’Orso, I. (2022). KAP1/TRIM28: Transcriptional activator and/or repressor of viral and cellular programs? Front. Cell. Infect. Microbiol. 12:834636. doi: 10.3389/fcimb.2022.834636
Rawat, P., Sehar, U., Bisht, J., Selman, A., Culberson, J., and Reddy, P. H. (2022). Phosphorylated Tau in Alzheimer’s disease and other tauopathies. Int. J. Mol. Sci. 23:12841.
Reemst, K., Noctor, S. C., Lucassen, P. J., and Hol, E. M. (2016). The indispensable roles of microglia and astrocytes during brain development. Front. Hum. Neurosci. 10:566. doi: 10.3389/fnhum.2016.00566
Reymond, A., Meroni, G., Fantozzi, A., Merla, G., Cairo, S., Luzi, L., et al. (2001). The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151. doi: 10.1093/emboj/20.9.2140
Rock, R. B., Gekker, G., Hu, S., Sheng, W. S., Cheeran, M., Lokensgard, J. R., et al. (2004). Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 17, 942–964. doi: 10.1128/cmr.17.4.942-964.2004
Rousseaux, M. W. C., de Haro, M., Lasagna-Reeves, C. A., De Maio, A., Jafar-Nejad, P., Park, J., et al. (2015). TRIM28 regulates the stability and toxicity of alpha-synuclein and tau through a common mechanism. J. Neurol. Sci. 357, e285–e286. doi: 10.1016/j.jns.2015.08.996
Rousseaux, M. W. C., de Haro, M., Lasagna-Reeves, C. A., De Maio, A., Park, J., Jafar-Nejad, P., et al. (2016). TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. eLife 5:e19809. doi: 10.7554/eLife.19809
Rousseaux, M. W. C., Revelli, J.-P., Vaìzquez-Veìlez, G. E., Kim, J.-Y., Craigen, E., Gonzales, K., et al. (2018). Depleting Trim28 in adult mice is well tolerated and reduces levels of α-synuclein and tau. eLife 7:e36768. doi: 10.7554/eLife.36768
Sakai, Y., Fukai, R., Matsushita, Y., Miyake, N., Saitsu, H., Akamine, S., et al. (2016). De novo truncating mutation of TRIM8 causes early-onset epileptic encephalopathy. Ann. Hum. Genet. 80, 235–240. doi: 10.1111/ahg.12157
Sanchez, J. G., Okreglicka, K., Chandrasekaran, V., Welker, J. M., Sundquist, W. I., and Pornillos, O. (2014). The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. U.S.A. 111, 2494–2499. doi: 10.1073/pnas.1318962111
Sansom, S. N., Griffiths, D. S., Faedo, A., Kleinjan, D.-J., Ruan, Y., Smith, J., et al. (2009). The level of the transcription factor pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 5:e1000511. doi: 10.1371/journal.pgen.1000511
Sardiello, M., Cairo, S., Fontanella, B., Ballabio, A., and Meroni, G. (2008). Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 8:225. doi: 10.1186/1471-2148-8-225
Saritas-Yildirim, B., and Silva, E. M. (2014). The role of targeted protein degradation in early neural development. Genesis 52, 287–299. doi: 10.1002/dvg.22771
Sato, T., Okumura, F., Kano, S., Kondo, T., Ariga, T., and Hatakeyama, S. (2011). TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J. Cell Sci. 124(Pt 20), 3492–3502. doi: 10.1242/jcs.088799
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J.-Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568.e23–584.e23. doi: 10.1016/j.cell.2019.12.036
Schreiber, J., Végh, M. J., Dawitz, J., Kroon, T., Loos, M., Labonté, D., et al. (2015). Ubiquitin ligase TRIM3 controls hippocampal plasticity and learning by regulating synaptic γ-actin levels. J. Cell Biol. 211, 569–586. doi: 10.1083/jcb.201506048
Schwamborn, J. C., Berezikov, E., and Knoblich, J. A. (2009). The TRIM-NHL protein TRIM32 activates MicroRNAs and prevents self-renewal in mouse neural progenitors. Cell 136, 913–925. doi: 10.1016/j.cell.2008.12.024
Schweiger, S., Matthes, F., Posey, K., Kickstein, E., Weber, S., Hettich, M. M., et al. (2017). Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 7:13753. doi: 10.1038/s41598-017-12974-4
Scott, D., and Roy, S. (2012). α-synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 32, 10129–10135. doi: 10.1523/jneurosci.0535-12.2012
Sharma, G., and Banerjee, S. (2022). Activity-regulated E3 ubiquitin ligase TRIM47 modulates excitatory synapse development. Front. Mol. Neurosci. 15:943980. doi: 10.3389/fnmol.2022.943980
Shi, K., Tian, D.-C., Li, Z.-G., Ducruet, A. F., Lawton, M. T., and Shi, F.-D. (2019). Global brain inflammation in stroke. Lancet Neurol. 18, 1058–1066. doi: 10.1016/S1474-4422(19)30078-X
Shi, M., Cho, H., Inn, K.-S., Yang, A., Zhao, Z., Liang, Q., et al. (2014). Negative regulation of NF-κB activity by brain-specific TRIpartite Motif protein 9. Nat. Commun. 5:4820. doi: 10.1038/ncomms5820
Silver, I., and Erecińska, M. (1998). Oxygen and ion concentrations in normoxic and hypoxic brain cells. Adv. Exp. Med. Biol. 454, 7–16. doi: 10.1007/978-1-4615-4863-8_2
Singh, A., Kukreti, R., Saso, L., and Kukreti, S. (2019). Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 24:1583.
Stevens, R. V., Esposito, D., and Rittinger, K. (2019). Characterisation of class VI TRIM RING domains: linking RING activity to C-terminal domain identity. Life Sci. Allian. 2:e201900295. doi: 10.26508/lsa.201900295
Stiles, J., and Jernigan, T. L. (2010). The basics of brain development. Neuropsychol. Rev. 20, 327–348. doi: 10.1007/s11065-010-9148-4
Stormo, A. E. D., Shavarebi, F., FitzGibbon, M., Earley, E. M., Ahrendt, H., Lum, L. S., et al. (2022). The E3 ligase TRIM1 ubiquitinates LRRK2 and controls its localization, degradation, and toxicity. J. Cell Biol. 221:202010065. doi: 10.1083/jcb.202010065
Subramaniam, S., Strelau, J., and Unsicker, K. (2003). Growth differentiation factor-15 prevents low potassium-induced cell death of cerebellar granule neurons by differential regulation of akt and erk pathways. J. Biol. Chem. 278, 8904–8912. doi: 10.1074/jbc.M210037200
Suzuki, M., Hara, Y., Takagi, C., Yamamoto, T. S., and Ueno, N. (2010). MID1 and MID2 are required for Xenopus neural tube closure through the regulation of microtubule organization. Development 137, 2329–2339. doi: 10.1242/dev.048769
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tanaka, M., Fukuda, Y., Mashima, K., and Hanai, R. (2005). Intracellular localization and domain organization of human TRIM41 proteins. Mol. Biol. Rep. 32, 87–93. doi: 10.1007/s11033-004-6613-2
Tang, S. L., Gao, Y. L., and Wen-Zhong, H. (2018). Knockdown of TRIM37 suppresses the proliferation, migration and invasion of glioma cells through the inactivation of PI3K/Akt signaling pathway. Biomed. Pharmacother. 99, 59–64. doi: 10.1016/j.biopha.2018.01.054
Tanji, K., Kamitani, T., Mori, F., Kakita, A., Takahashi, H., and Wakabayashi, K. (2010). TRIM9, a novel brain-specific E3 ubiquitin ligase, is repressed in the brain of Parkinson’s disease and dementia with Lewy bodies. Neurobiol. Dis. 38, 210–218. doi: 10.1016/j.nbd.2010.01.007
Tansey, M. G., and Goldberg, M. S. (2010). Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518. doi: 10.1016/j.nbd.2009.11.004
Tatham, M. H., Plechanovová, A., Jaffray, E. G., Salmen, H., and Hay, R. T. (2013). Ube2W conjugates ubiquitin to α-amino groups of protein N-termini. Biochem. J. 453, 137–145. doi: 10.1042/BJ20130244
Taylor, J. M., Moore, Z., Minter, M. R., and Crack, P. J. (2018). Type-I interferon pathway in neuroinflammation and neurodegeneration: Focus on Alzheimer’s disease. J. Neural Trans. 125, 797–807. doi: 10.1007/s00702-017-1745-4
Tetsuka, S., Suzuki, T., Ogawa, T., Hashimoto, R., and Kato, H. (2021). Anti-Ro/SSA antibodies may be responsible for cerebellar degeneration in Sjogren’s syndrome. J. Clin. Med. Res. 13, 113–120. doi: 10.14740/jocmr4429
Tomita, S., Ueno, M., Sakamoto, M., Kitahama, Y., Ueki, M., Maekawa, N., et al. (2003). Defective brain development in mice lacking the Hif-1α gene in neural cells. Mol. Cell. Biol. 23, 6739–6749. doi: 10.1128/MCB.23.19.6739-6749.2003
Trockenbacher, A., Suckow, V., Foerster, J., Winter, J., Krauss, S., Ropers, H. H., et al. (2001). MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 29, 287–294. doi: 10.1038/ng762
Tuoc, T. C., and Stoykova, A. (2008). Trim11 modulates the function of neurogenic transcription factor Pax6 through ubiquitin–proteosome system. Genes Dev. 22, 1972–1986. doi: 10.1101/gad.471708
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Urbina, F. L., Menon, S., Goldfarb, D., Edwards, R., Ben Major, M., Brennwald, P., et al. (2021). TRIM67 regulates exocytic mode and neuronal morphogenesis via SNAP47. Cell Rep. 34:108743. doi: 10.1016/j.celrep.2021.108743
Valentino, R. R., Koga, S., Heckman, M. G., Brushaber, D. E., Diehl, N. N., Walton, R. L., et al. (2020). Association of tripartite motif containing 11 rs564309 with tau pathology in progressive supranuclear Palsy. Mov. Disord. 35, 890–894. doi: 10.1002/mds.28010
van Beuningen, S. F. B., Will, L., Harterink, M., Chazeau, A., van Battum, E. Y., Frias, C. P., et al. (2015). TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron 88, 1208–1226. doi: 10.1016/j.neuron.2015.11.012
van Gent, M., Sparrer, K. M. J., and Gack, M. U. (2018). TRIM proteins and their roles in antiviral host defenses. Annu. Rev. Virol. 5, 385–405. doi: 10.1146/annurev-virology-092917-043323
Vanlandewijck, M., He, L., Mäe, M. A., Andrae, J., Ando, K., Del Gaudio, F., et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. doi: 10.1038/nature25739
Vannucci, S. J., and Hagberg, H. (2004). Hypoxia–ischemia in the immature brain. J. Exp. Biol. 207, 3149–3154. doi: 10.1242/jeb.01064
Vargas, K. J., Schrod, N., Davis, T., Fernandez-Busnadiego, R., Taguchi, Y. V., Laugks, U., et al. (2017). Synucleins have multiple effects on presynaptic architecture. Cell Rep. 18, 161–173. doi: 10.1016/j.celrep.2016.12.023
Vaysburd, M., Watkinson, R. E., Cooper, H., Reed, M., O’Connell, K., Smith, J., et al. (2013). Intracellular antibody receptor TRIM21 prevents fatal viral infection. Proc. Natl. Acad. Sci. U.S.A. 110, 12397–12401. doi: 10.1073/pnas.1301918110
Venkataramani, V., Tanev, D. I., Strahle, C., Studier-Fischer, A., Fankhauser, L., Kessler, T., et al. (2019). Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538. doi: 10.1038/s41586-019-1564-x
Venuto, S., Castellana, S., Monti, M., Appolloni, I., Fusilli, C., Fusco, C., et al. (2019). TRIM8-driven transcriptomic profile of neural stem cells identified glioma-related nodal genes and pathways. Biochim. Biophys. Acta Gen. Subj. 1863, 491–501. doi: 10.1016/j.bbagen.2018.12.001
Virtuoso, A., Giovannoni, R., De Luca, C., Gargano, F., Cerasuolo, M., Maggio, N., et al. (2021). The Glioblastoma microenvironment: Morphology, metabolism, and molecular signature of glial dynamics to discover metabolic rewiring sequence. Int. J. Mol. Sci. 22:3301.
Wang, C., Deng, L., Hong, M., Akkaraju, G. R., Inoue, J.-I., and Chen, Z. J. (2001). TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351. doi: 10.1038/35085597
Wang, M., Luo, W., Zhang, Y., Yang, R., Li, X., Guo, Y., et al. (2020). Trim32 suppresses cerebellar development and tumorigenesis by degrading Gli1/sonic hedgehog signaling. Cell Death Differ. 27, 1286–1299. doi: 10.1038/s41418-019-0415-5
Wang, X., Herr, R. A., Chua, W.-J., Lybarger, L., Wiertz, E. J. H. J., and Hansen, T. H. (2007). Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 177, 613–624. doi: 10.1083/jcb.200611063
Wang, X., Wang, Z., Yao, Y., Li, J., Zhang, X., Li, C., et al. (2011). Essential role of ERK activation in neurite outgrowth induced by α-lipoic acid. Biochim. Biophys. Acta Mol. Cell Res. 1813, 827–838. doi: 10.1016/j.bbamcr.2011.01.027
Wang, Y., Li, Y., Qi, X., Yuan, W., Ai, J., Zhu, C., et al. (2004). TRIM45, a novel human RBCC/TRIM protein, inhibits transcriptional activities of ElK-1 and AP-1. Biochem. Biophys. Res. Commun. 323, 9–16. doi: 10.1016/j.bbrc.2004.08.048
Wang, Z., Zhang, H., Xu, S., Liu, Z., and Cheng, Q. (2021). The adaptive transition of glioblastoma stem cells and its implications on treatments. Signal Trans. Target. Ther. 6:124. doi: 10.1038/s41392-021-00491-w
Wei, L., Zhang, J.-S., Ji, S.-F., Xu, H., Zhao, Z.-H., Zhang, L., et al. (2019). Knockdown of TRIM32 protects hippocampal neurons from oxygen–glucose deprivation-induced injury. Neurochem. Res. 44, 2182–2189. doi: 10.1007/s11064-019-02857-7
Williams, F. P., Haubrich, K., Perez-Borrajero, C., and Hennig, J. (2019). Emerging RNA-binding roles in the TRIM family of ubiquitin ligases. Biol. Chem. 400, 1443–1464. doi: 10.1515/hsz-2019-0158
Williams, T., Courchet, J., Viollet, B., Brenman, J. E., and Polleux, F. (2011). AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc. Natl. Acad. Sci. U.S.A. 108, 5849–5854. doi: 10.1073/pnas.1013660108
Winkle, C. C., Olsen, R. H. J., Kim, H., Moy, S. S., Song, J., and Gupton, S. L. (2016). Trim9 deletion alters the morphogenesis of developing and adult-born hippocampal neurons and impairs spatial learning and memory. J. Neurosci. 36, 4940–4958. doi: 10.1523/jneurosci.3876-15.2016
Witoelar, A., Jansen, I. E., Wang, Y., Desikan, R. S., Gibbs, J. R., Blauwendraat, C., et al. (2017). Genome-wide pleiotropy between parkinson disease and autoimmune diseases. JAMA Neurol. 74, 780–792. doi: 10.1001/jamaneurol.2017.0469
Worringer, K. A., Rand, T. A., Hayashi, Y., Sami, S., Takahashi, K., Tanabe, K., et al. (2014). The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 14, 40–52. doi: 10.1016/j.stem.2013.11.001
Wu, L., Xiong, X., Wu, X., Ye, Y., Jian, Z., Zhi, Z., et al. (2020). Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front. Mol. Neurosci. 13:28. doi: 10.3389/fnmol.2020.00028
Xia, Q., Zhan, G., Mao, M., Zhao, Y., and Li, X. (2022). TRIM45 causes neuronal damage by aggravating microglia-mediated neuroinflammation upon cerebral ischemia and reperfusion injury. Exp. Mol. Med. 54, 180–193. doi: 10.1038/s12276-022-00734-y
Xiao, S., Yu, J., Yuan, X., and Chen, Q. (2022). Identification of a tripartite motif family gene signature for predicting the prognosis of patients with glioma. Am. J. Transl. Res. 14, 1535–1550.
Xu, P., Duong, D. M., Seyfried, N. T., Cheng, D., Xie, Y., Robert, J., et al. (2009). Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145. doi: 10.1016/j.cell.2009.01.041
Xuan, Z., Ma, T., Qin, Y., and Guo, Y. (2022). Role of ultrasound imaging in the prediction of TRIM67 in brain metastases from breast cancer. Front. Neurol. 13:889106. doi: 10.3389/fneur.2022.889106
Xue, J., Chen, Y., Wu, Y., Wang, Z., Zhou, A., Zhang, S., et al. (2015). Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nat. Commun. 6:6156. doi: 10.1038/ncomms7156
Xue, W., Zhao, Y., Xiao, Z., Wu, X., Ma, D., Han, J., et al. (2020). Epidermal growth factor receptor-extracellular-regulated kinase blockade upregulates TRIM32 signaling cascade and promotes neurogenesis after spinal cord injury. Stem Cells 38, 118–133. doi: 10.1002/stem.3097
Yamaguchi, Y., and Miura, M. (2015). Programmed cell death in neurodevelopment. Dev. Cell 32, 478–490. doi: 10.1016/j.devcel.2015.01.019
Yan, Q., Sun, W., Kujala, P., Lotfi, Y., Vida, T. A., and Bean, A. J. (2005). CART: An Hrs/Actinin-4/BERP/Myosin V protein complex required for efficient receptor recycling. Mol. Biol. Cell 16, 2470–2482. doi: 10.1091/mbc.e04-11-1014
Yang, L., Shi, P., Zhao, G., Xu, J., Peng, W., Zhang, J., et al. (2020). Targeting cancer stem cell pathways for cancer therapy. Signal Trans. Targeted Ther. 5:8. doi: 10.1038/s41392-020-0110-5
Yang, W., Gu, Z., Zhang, H., and Hu, H. (2020). To TRIM the immunity: From innate to adaptive immunity. Front. Immunol. 11:2157. doi: 10.3389/fimmu.2020.02157
Yao, Y., Zhang, B., Zhu, H., Li, H., Han, Y., Chen, K., et al. (2016). MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 7, 22474–22485. doi: 10.18632/oncotarget.7965
Yau, R., and Rape, M. (2016). The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586. doi: 10.1038/ncb3358
Ylikallio, E., Pöyhönen, R., Zimon, M., De Vriendt, E., Hilander, T., Paetau, A., et al. (2013). Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum. Mol. Genet. 22, 2975–2983. doi: 10.1093/hmg/ddt149
Yu, Z., Feng, J., Wang, W., Deng, Z., Zhang, Y., Xiao, L., et al. (2020). The EGFR-ZNF263 signaling axis silences SIX3 in glioblastoma epigenetically. Oncogene 39, 3163–3178. doi: 10.1038/s41388-020-1206-7
Yu, Z., Li, X., Yang, M., Huang, J., Fang, Q., Jia, J., et al. (2021). TRIM41 is required to innate antiviral response by polyubiquitinating BCL10 and recruiting NEMO. Signal Trans. Target. Ther. 6:90. doi: 10.1038/s41392-021-00477-8
Zeng, S., Zhao, Z., Zheng, S., Wu, M., Song, X., Li, Y., et al. (2021). The E3 ubiquitin ligase TRIM31 is involved in cerebral ischemic injury by promoting degradation of TIGAR. Redox Biol. 45:102058. doi: 10.1016/j.redox.2021.102058
Zeng, X. J., Yu, S. P., Zhang, L., and Wei, L. (2010). Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Exp. Res. 316, 1773–1783. doi: 10.1016/j.yexcr.2010.02.005
Zengeler, K. E., and Lukens, J. R. (2021). Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 21, 454–468. doi: 10.1038/s41577-020-00487-7
Zhang, C., Mukherjee, S., Tucker-Burden, C., Ross, J. L., Chau, M. J., Kong, J., et al. (2017). TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 11, 280–294. doi: 10.1002/1878-0261.12034
Zhang, J., Zhang, C., Cui, J., Ou, J., Han, J., Qin, Y., et al. (2017). TRIM45 functions as a tumor suppressor in the brain via its E3 ligase activity by stabilizing p53 through K63-linked ubiquitination. Cell Death Dis. 8:e2831. doi: 10.1038/cddis.2017.149
Zhang, J.-R., Li, X.-X., Hu, W.-N., and Li, C.-Y. (2020). Emerging role of TRIM family proteins in Cardiovascular disease. Cardiology 145, 390–400. doi: 10.1159/000506150
Zhang, P., Wu, Y., Li, R., Lv, H., and Yu, B. (2020). Tripartite motif containing 52 positively regulates NF-κB signaling by promoting IκBα ubiquitination in lipopolysaccharide-treated microglial cell activation. Med. Sci. Monit. 26, e925356. doi: 10.12659/msm.925356
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/jneurosci.1860-14.2014
Zhang, Y., Dube, C., Gibert, M., Cruickshanks, N., Wang, B., Coughlan, M., et al. (2018). The p53 pathway in glioblastoma. Cancers 10:297.
Zhang, Y., Zhang, W., Zheng, L., and Guo, Q. (2022). The roles and targeting options of TRIM family proteins in tumor. Front. Pharmacol. 13:999380. doi: 10.3389/fphar.2022.999380
Zhang, Z.-B., Xiong, L.-L., Lu, B.-T., Zhang, H.-X., Zhang, P., and Wang, T.-H. (2017). Suppression of Trim32 enhances motor function repair after traumatic brain injury associated with antiapoptosis. Cell Trans. 26, 1276–1285. doi: 10.1177/0963689717716510
Zhang, Z.-Y., Harischandra, D. S., Wang, R., Ghaisas, S., Zhao, J. Y., McMonagle, T. P., et al. (2023). TRIM11 protects against tauopathies and is down-regulated in Alzheimer’s disease. Science 381:eadd6696. doi: 10.1126/science.add6696
Zhao, W., Zhang, X., Chen, Y., Shao, Y., and Feng, Y. (2020). Downregulation of TRIM8 protects neurons from oxygen–glucose deprivation/re-oxygenation-induced injury through reinforcement of the AMPK/Nrf2/ARE antioxidant signaling pathway. Brain Res. 1728:146590. doi: 10.1016/j.brainres.2019.146590
Zhou, W., and Bao, S. (2014). PML-mediated signaling and its role in cancer stem cells. Oncogene 33, 1475–1484. doi: 10.1038/onc.2013.111
Zhou, W., Zhao, T., Du, J., Ji, G., Li, X., Ji, S., et al. (2019). TIGAR promotes neural stem cell differentiation through acetyl-CoA-mediated histone acetylation. Cell Death Dis. 10:198. doi: 10.1038/s41419-019-1434-3
Zhu, G., Harischandra, D. S., Ghaisas, S., Zhang, P., Prall, W., Huang, L., et al. (2020). TRIM11 prevents and reverses protein aggregation and rescues a mouse model of Parkinson’s disease. Cell Rep. 33:108418. doi: 10.1016/j.celrep.2020.108418
Keywords: TRIM, ubiquitin, brain, neurodevelopment, neurodegeneration, glioma, infection, inflammation
Citation: Dudley-Fraser J and Rittinger K (2023) It’s a TRIM-endous view from the top: the varied roles of TRIpartite Motif proteins in brain development and disease. Front. Mol. Neurosci. 16:1287257. doi: 10.3389/fnmol.2023.1287257
Received: 18 September 2023; Accepted: 13 November 2023;
Published: 05 December 2023.
Edited by:
Germana Meroni, University of Trieste, ItalyReviewed by:
Solange Desagher, UMR5535 Institut de Génétique Moléculaire de Montpellier (IGMM), FranceJulien Licchesi, University of Bath, United Kingdom
Copyright © 2023 Dudley-Fraser and Rittinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jane Dudley-Fraser, amFuZS5kdWRsZXlAY3JpY2suYWMudWs=; Katrin Rittinger, a2F0cmluLnJpdHRpbmdlckBjcmljay5hYy51aw==