
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 05 October 2023
Sec. Molecular Signalling and Pathways
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1275924
This article is part of the Research TopicThe Legacy of Dr. Rita Levi-Montalcini: Advances in Neurotrophic Factors in Brain Disease Development and TreatmentView all 7 articles
 Marina Cefis1,2*
Marina Cefis1,2* Remi Chaney2Julien Wirtz2
Remi Chaney2Julien Wirtz2 Alexandre Méloux2
Alexandre Méloux2 Aurore Quirié2Clémence Leger2
Aurore Quirié2Clémence Leger2 Anne Prigent-Tessier2
Anne Prigent-Tessier2 Philippe Garnier2,3*
Philippe Garnier2,3*Accumulating evidence supports that physical exercise (EX) is the most effective non-pharmacological strategy to improve brain health. EX prevents cognitive decline associated with age and decreases the risk of developing neurodegenerative diseases and psychiatric disorders. These positive effects of EX can be attributed to an increase in neurogenesis and neuroplastic processes, leading to learning and memory improvement. At the molecular level, there is a solid consensus to involve the neurotrophin brain-derived neurotrophic factor (BDNF) as the crucial molecule for positive EX effects on the brain. However, even though EX incontestably leads to beneficial processes through BDNF expression, cellular sources and molecular mechanisms underlying EX-induced cerebral BDNF overproduction are still being elucidated. In this context, the present review offers a summary of the different molecular mechanisms involved in brain’s response to EX, with a specific focus on BDNF. It aims to provide a cohesive overview of the three main mechanisms leading to EX-induced brain BDNF production: the neuronal-dependent overexpression, the elevation of cerebral blood flow (hemodynamic hypothesis), and the exerkine signaling emanating from peripheral tissues (humoral response). By shedding light on these intricate pathways, this review seeks to contribute to the ongoing elucidation of the relationship between EX and cerebral BDNF expression, offering valuable insights into the potential therapeutic implications for brain health enhancement.
Physical exercise (EX) is the most efficient non-pharmacological strategy to improve health and prevent pathologies. Being physically active reduces not only the risk of developing cardiovascular, metabolic and chronic diseases but also brain disorders. Indeed, numerous studies reported the beneficial effects of EX across a lifespan on cognitive functions and neuroplastic mechanisms (Voss et al., 2011). EX reduces the prevalence and incidence of stroke, neurodegenerative diseases (e.g., Alzheimer’s, Parkinson’s diseases [AD, PD]) and mental disorders such as depression, schizophrenia and addiction (Dunn et al., 2005; Sofi et al., 2011; Paillard et al., 2015; Xu et al., 2023). Accumulating evidence supports that EX improves learning, memory, executive functions, attention in children (Chaddock et al., 2010; de Greeff et al., 2018) and adulthoods (Colcombe et al., 2006; Erickson et al., 2011; Sofi et al., 2011; Ludyga et al., 2020; Cheval et al., 2023). Moreover, EX has been linked to a decrease in stress and anxiety, as well as an improvement in emotional stability and sleep quality resulting in improved psychological well-being (Mandolesi et al., 2018).
In more details, the positive effects of EX on the brain may be explained by an increase in hippocampal neurogenesis, an enhancement of long-term potentiation (LTP) and the regulation of synaptic plasticity (van Praag et al., 1999; Farmer et al., 2004; Cefis et al., 2019). It has also been demonstrated that EX induces cerebral angiogenesis by increasing the density and sprouting of new capillaries from pre-existing vessels (Black et al., 1990; Ding et al., 2004). From animal and human studies, there is a consensus involving the neurotrophin brain-derived neurotrophic factor (BDNF) as responsible for the positive effects of EX on the brain. This neurotrophin is widely produced in the brain, where it plays a crucial role in neurogenesis, synaptic plasticity, angiogenesis and exerts neuroprotective effects (Korte et al., 1995; Bartoletti et al., 2002; Kim et al., 2004). The crucial role of BDNF is unquestionable since in animal studies, anti-BDNF strategies negate EX-associated cognitive benefits (Vaynman et al., 2003, 2004; Liu et al., 2008), while in humans, the val66met polymorphism (single nucleotide polymorphism in bdnf gene corresponding to a valine-to-methionine substitution), which is associated with a defect in activity-dependent regulated secretion, attenuates the cognitive advantage of EX (Egan et al., 2003; Erickson et al., 2012; Hopkins et al., 2012). The synergic interrelation between neuronal activity and synaptic plasticity designates BDNF as an ideal mediator of cellular and molecular mechanisms underlying cognitive and memory improvements induced by EX. However, even though BDNF involvement is incontestable, molecular mechanisms underlying EX-induced cerebral BDNF overproduction are not entirely delineated.
In this context, this review aims to provide a comprehensive overview of scientific evidence on the different molecular mechanisms explaining how EX leads to cerebral BDNF upregulation. It is important to underscore that within the context of this review, the term “EX” has been utilized inclusively, encompassing a wide range of physical activities, irrespective of their specific type, duration, or intensity while these factors influence the cerebral expression of BDNF (Walsh et al., 2020; Fernandez-Rodriguez et al., 2022). After a brief description of BDNF metabolism and signaling pathways, this manuscript will focus on the different mechanisms responsible for EX-induced cerebral BDNF production. Hence, the increase in neuronal expression through activity-dependent mechanisms, the contribution of endothelial cells through cerebral blood flow (CBF) elevation (hemodynamic hypothesis) and the recent mechanisms involving humoral factors (exerkines) originating from peripheral tissues will be detailed and provided in Figure 1A. A better comprehension of the molecular mechanisms that lead to cerebral BDNF upregulation is required, not only because it could help to promote exercise prescription for brain health, but also since the modulation of these mechanisms could be an attractive possibility for the prevention and treatment of various brain pathologies.
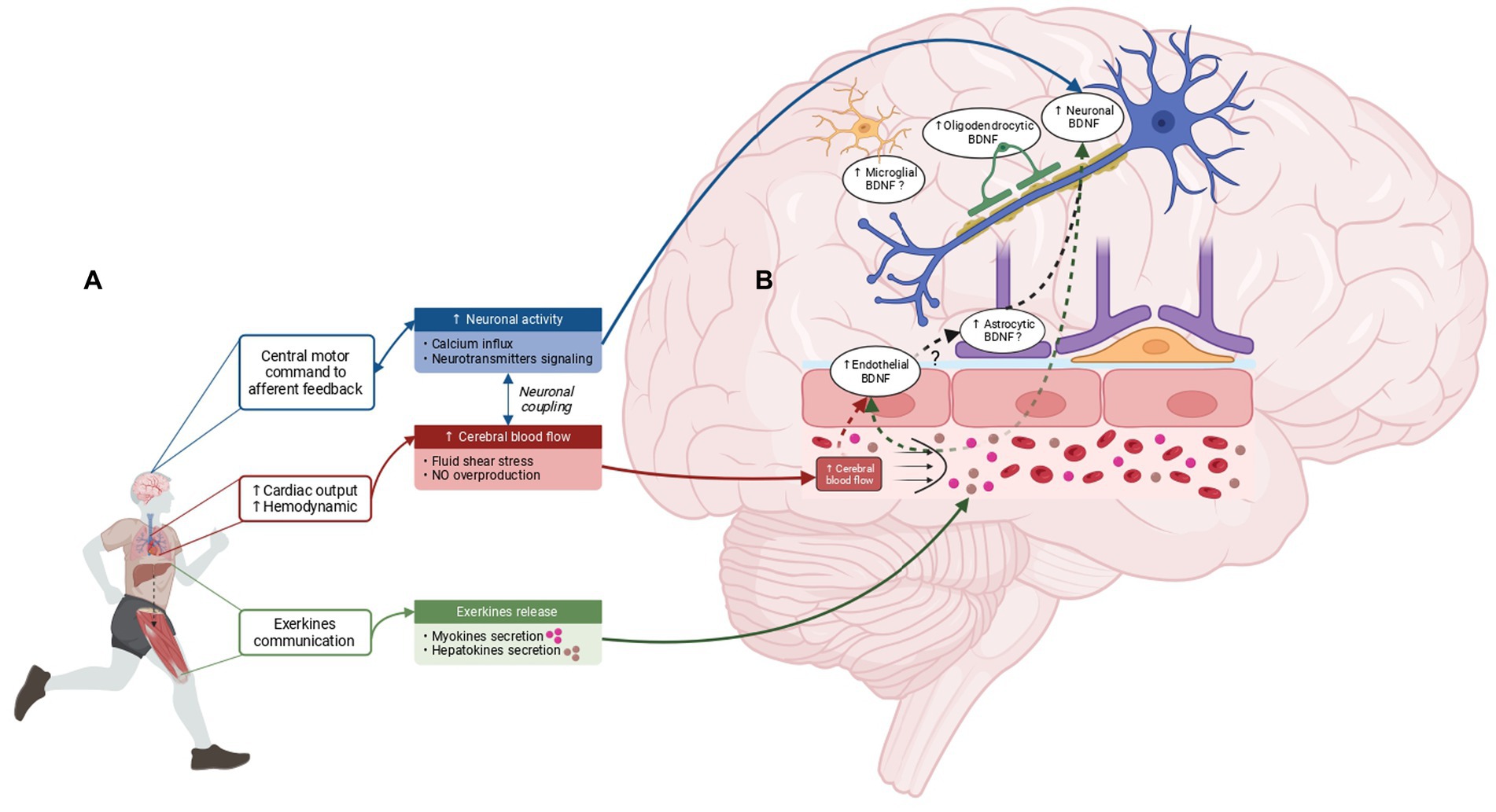
Figure 1. Mechanisms underlying EX-induced brain BDNF overproduction and cerebral BDNF-expressing cells in response to EX. (A) Cerebral BDNF increase in response to EX is thought to be driven by three main mechanisms: the increase in neuronal activity, the elevation of CBF and the release of exerkines from peripheral tissues. The brain detects EX through central motor control and afferent feedback which trigger an increase in neuronal activity involving neurotransmitter signaling and Ca2+ influx. In addition, EX leads to an increase in hemodynamic response subsequent to the cardiac output augmentation resulting in elevated cerebral blood flow and an increase in fluid shear stress. Of note, CBF elevation is also contingent upon neuronal coupling. Finally, peripheral tissues such as the liver and the skeletal muscle perceive EX and release exerkines into the bloodstream, capable of signaling to the brain and inducing cerebral BDNF increase. (B) BDNF overexpression in response to EX is not limited to neuronal cells. Studies indicate that BDNF overexpression is induced in endothelial cells and oligodendrocytes, while the involvement of microglia and astrocytes as potential sources of BDNF in response to EX requires further investigation. Created with BioRender.com.
Initially discovered in the porcine brain by Barde et al. (1982), BDNF is a member of the neurotrophic factor family expressed by the neurons of the central nervous system (CNS) predominantly in the cortex and hippocampus (Ernfors et al., 1990; Hofer et al., 1990). In addition to neurons, many cells have been shown to express BDNF in the brain, such as astrocytes, microglia (Dougherty et al., 2000), pericytes (Ishitsuka et al., 2012) and endothelial cells (Leventhal et al., 1999; Nakahashi et al., 2000). Cerebral endothelial cells (CEC) are particularly noteworthy, as they synthesize 50 times more BDNF than primary cortical neuron cultures (Guo et al., 2008). Consistently, data reported that the in vivo removal of cerebral endothelium using a detergent (3-[(3-cholamidopropyl) dimethylammonio]-1-propane sulphonate, CHAPS), halved the cerebral BDNF content (Monnier et al., 2017b). Contrary to what its name may suggest, BDNF is also expressed in the cardiovascular system (heart, vessels) (Timmusk et al., 1993; Meuchel et al., 2011; Quirie et al., 2012; Prigent-Tessier et al., 2013), lungs, thymus, spleen (Aid et al., 2007), skeletal muscles (Cefis et al., 2022), and blood (Rosenfeld et al., 1995).
BDNF is initially synthesized in the endoplasmic reticulum as a precursor form, the pre-pro-BDNF that is subsequently transformed into pro-BDNF by the removal of the signal peptide. After N-terminal glycosylation of the N123 residue in the pro-domain, pro-BDNF is then either proteolytically cleaved intracellularly (i.e., by furin and pro-protein convertase 7 in Golgi but also by pro-protein convertase 1/3 in secretion vesicles) or extracellularly (i.e., by tissue plasminogen activator [tPA]/plasmin and matrix metalloproteinases) in mature BDNF (Mowla et al., 2001). Of note, extracellular processing of pro-BDNF is still a matter of debate since evidence came from in vitro studies using tagged pro-BDNF that do not reflect physiological conditions (Pang et al., 2004; Nagappan et al., 2009). BDNF is then secreted by either a constitutive or a regulated pathway, the latter involving the interaction of the pro-domain with the sortilin receptor (Chen et al., 2005). According to Aid and colleague’s data, bdnf gene consists in a common 3′ exon that encodes the entire pro-BDNF protein and at least eight 5′ non-coding exons (exons I-VIII). Each 5′ non-coding exon is spliced into the coding exon that encodes a similar BDNF protein product (Aid et al., 2007). Thus, the gene encoding for BDNF protein leads to multiple transcripts according to the use of alternative promoters and of the different splicing and polyadenylation sites. Although the significance of this complex organization is still obscure, it could afford for a distinct tissue-specific expression and regulation under specific physiological conditions (Timmusk et al., 1993; Aid et al., 2007). Concerning neuronal activity-dependent regulation of BDNF transcription, exons I and IV have been shown to be the most upregulated (Tao et al., 1998; Pruunsild et al., 2011; Zheng et al., 2011). Besides, promoters I- and IV-dependent bdnf transcription are both increased in response to EX (Intlekofer et al., 2013; Luft et al., 2022).
BDNF acts through two different plasma membrane receptors, the tropomyosin-related kinase B (TrkB) receptor and pan75 neurotrophin receptor (p75NTR), the affinity being much higher for TrkB than p75NTR (Chao, 2003) with its positive action being attributed to TrkB activation only. Conversely, the p75NTR is the preferential receptor for pro-BDNF and its activation mainly leads to the activation of pro-apoptotic pathway (Friedman, 2000; Beattie et al., 2002). These data showed the yin and yang effects of this neurotrophin and highlighted the importance of the balance between the two forms for the proper functioning of BDNF (for review Lu et al., 2005). In neurons, TrkB receptors include the full-length (FL) form with a tyrosine kinase domain and the truncated forms (T1, T2, T3) devoid of the kinase domains (Stoilov et al., 2002; Fenner, 2012). All these isoforms share the same extracellular pattern and binding affinity for BDNF. The binding of BDNF to the TrkB-FL receptor induces its dimerization and auto-phosphorylation of different tyrosine residues at the cytoplasmic domain leading to the activation of three major signaling pathways involving the phospholipase Cγ (PLC-γ), the phosphatidylinositol-3 kinase (PI3K) and the mitogen-activated protein kinase (MAPK) pathways that trigger neurite outgrowth, cell differentiation, neuronal survival and synaptic plasticity (Huang and Reichardt, 2003; Reichardt, 2006; Fenner, 2012). For the major truncated form, it has been shown that when TrkB-FL and -T1 are co-expressed, TrkB-T1 can repress the TrkB-FL signaling (Fenner, 2012). Furthermore, BDNF binding to TrkB-T1 could induce both its own signalization (Rose et al., 2003), internalization and/or release (Alderson et al., 2000). Of note, also poorly documented, TrkB expression is not restricted to neurons but also expressed in different cells of the neurovascular unit such as endothelial cells (TrkB-FL and truncated) (Kim et al., 2004; Pedard et al., 2017) and astrocytes which in addition to p75NTR (Rudge et al., 1994), expressed almost exclusively the truncated isoform, TrkB-T1 (Rose et al., 2003).
The cerebral cells responding to EX by an increase of cerebral BDNF expression are illustrated and summarized in Figure 1B.
As an immediate-early gene, transcription of new BDNF mRNA occurs rapidly without the requirement of new protein synthesis in a process involving post-translational modification of pre-existing transcription factors. Using in situ hybridization, Neeper et al. (1995) were the first to report that voluntary wheel running increases BDNF mRNA in hippocampal and cortical neurons of rats. Since this initial finding, many studies have confirmed this discovery using different types, intensities and durations of EX (Oliff et al., 1998; Berchtold et al., 2005; Griffin et al., 2009; Cefis et al., 2019; Pedard et al., 2019). The mechanism regulating bdnf gene expression is intriguing since synaptic activation regulates the neuronal synthesis and secretion of BDNF, which in turn modifies synaptic morphology and efficacy (Poo, 2001). This review only focuses on biological mechanisms induced by EX that led to transcriptional upregulation.
Initial investigations on endothelial BDNF expression have demonstrated its constitutive synthesis, release and regulation (Leventhal et al., 1999) by both peripheral (Nakahashi et al., 2000) and CEC (Bayas et al., 2002; Takeo et al., 2003) in culture. Secreted BDNF is bioactive since it exerts a neuroprotective effect by promoting neuronal growth and survival (Guo et al., 2008). In vivo studies have shown the presence of BDNF in the vascular endothelium both at peripheral and cerebral levels, with a significant part of cerebral BDNF corresponding to BDNF expressed by the cerebral endothelium (Monnier et al., 2017b; Cefis et al., 2020; Totoson et al., 2021). Moreover, endothelial BDNF expression was significantly higher after EX in the aorta, vein and cerebral microvessel fractions (Monnier et al., 2017a; Cefis et al., 2020). Conversely, pathologies associated with endothelial dysfunction, such as diabetes (Navaratna et al., 2011), cerebral ischemia (Bejot et al., 2011), high blood pressure (Monnier et al., 2017b) or rheumatoid arthritis (Pedard et al., 2017) decrease cerebral BDNF expression.
The question of whether astrocytes serve as a source of BDNF in response to EX remains to be clarified. Research has demonstrated that EX stimulates astrocyte proliferation (Uda et al., 2006), as well as morphological changes in an AD mouse model (Rodriguez et al., 2013). In addition, studies have suggested that astrocytes overexpressing BDNF can promote hippocampal neurogenesis (Quesseveur et al., 2013) and their own remodeling (Holt et al., 2019). Besides, EX was reported to elongate astrocytic projections in the dentate gyrus and to increase TrkB expression in GFAP-positive cells (Fahimi et al., 2017) while in a PD mouse model, Palasz et al. (2019) showed that treadmill training protects neurons through increase in astrocyte-derived BDNF. Given the central role of these cells in maintaining CNS homeostasis, in modulating synaptic transmission and their function as gatekeepers of the blood–brain barrier (BBB) (Li et al., 2021), the observed astrocytic BDNF–TrkB expression following EX may reflect a complex interplay between endothelial and neuronal cells, with astrocytes acting as a bridge, as we propose in the hemodynamic hypothesis section. Further investigation is needed to unravel the intricacies of astrocytic involvement in EX-induced cerebral BDNF production.
In addition to astrocytes, EX has been reported to promote oligodendrogenesis (For review, Guo et al., 2020). After EX, oligodendrocyte proliferation has been observed in spinal cord and hippocampus of healthy mice (Krityakiarana et al., 2010; Matsumoto et al., 2011) and in various diseases models including multiple sclerosis (Guo et al., 2020) or chronic cerebral hypoperfusion (Jiang et al., 2017). In addition, oligodendrocytes modulate synaptic transmission through the secretion of BDNF (Jang et al., 2019). Consistently, glutamate transmission and vesicular release were decreased in a model of mice with conditional deletion for BDNF in oligodendrocytes while these effects were offsetted with the application of BDNF or TrkB agonist (7,8-DHF). Taken together, these data suggest that oligodendrocytes are involved as a cellular source in EX-induced cerebral BDNF expression.
Concerning microglial cells, EX has been shown to modulate microglial activation. Thus, Mee-Inta et al. (2019) have highlighted the regulatory effect of EX on microglial activation, leading to an increase in anti-inflammatory factors and a decrease in pro-inflammatory factors. Additionally, in a mouse model of AD submitted to long-term EX, microglial activation was decreased in cerebral cortex and hippocampus, accompanied by an increase of BDNF-positive cells (Xiong et al., 2015). While there is evidence suggesting that microglia can produce BDNF (Madinier et al., 2009) and that microglia promote learning and memory through BDNF signaling (Parkhurst et al., 2013), a recent article suggests that BDNF expressed by microglia is not produced in sufficient amount to modulate neuronal function (Honey et al., 2022). Therefore, further investigations are needed to fully elucidate the involvement of microglia in the cellular expression of BDNF in response to EX.
All the mechanisms described in sections a-e are summarized in Figure 2.
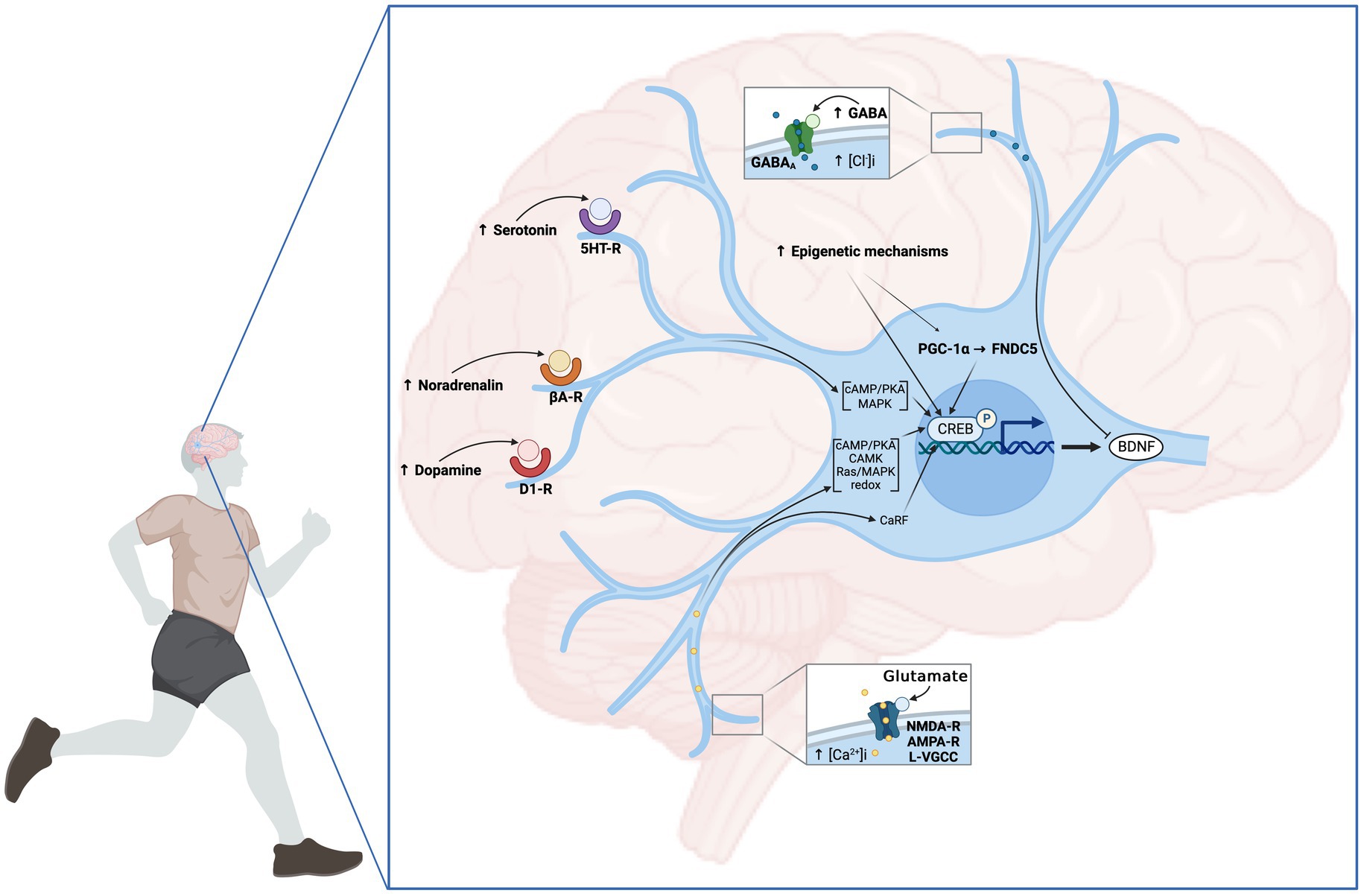
Figure 2. Neuronal mechanisms involved in EX-induced BDNF increase. From the central motor control to the afferent feedback, EX induces an increase in neuronal activity and the release of neurotransmitters leading to the upregulation of bdnf gene expression. The modulation of neuronal activity is primarily promoted by the elevation of Ca2+ influx through membrane L-VGCC. In addition to membrane depolarization, glutamatergic transmission mediated by AMPA and NMDA receptors plays a preponderant role in this process. Additionally, signaling from dopaminergic, noradrenergic and serotoninergic transmitters contributes to the upregulation of neuronal bdnf expression through D1-like receptor (D1-R), βadrenergic receptor (βA-R), and serotonin receptor (5HT-R), respectively. Conversely, GABA negatively affects hippocampal BDNF expression via GABAA receptors. Multiple Ca2+-dependent mechanisms have been proposed as responsible for CREB phosphorylation including Ca2+-sensitive adenylate cyclase/PKA, Ca2+/CAMK, Ras/MAPK and the redox signaling. Another mechanism leading to neuronal BDNF expression is associated with the activation of neuronal PGC-1α/FNDC5/Irisin pathway. Finally, epigenetics mechanisms involving HAT, DNMT, HDAC activities and miRNA, particularly mi138 were demonstrated. Created with BioRender.com.
Neuronal activity-regulated gene expression has been proposed to be a key mediator for experience-dependent changes in the nervous system (Katz and Shatz, 1996). From initial studies showing that EX, from the central motor control to the afferent feedback, induced electrical activity and sustained hippocampal activation (Vanderwolf, 1969; Czurko et al., 1999), the mechanism by which EX induces neuronal BDNF synthesis has been well investigated. From electrical to chemical level, Ca2+ influx is critical in transmitting depolarization status and triggering synaptic activity. Thus, using physiologically relevant stimulation patterns of hippocampal neurons, Balkowiec and Katz (2002) showed that BDNF production needed not only Ca2+ influx but also mobilization from intracellular Ca2+ stores. Once initiated, Ca2+-dependent signaling cascades trigger activity-induced changes in gene expression and protein synthesis (Ghosh and Greenberg, 1995). Using the induction of BDNF mRNA, West et al. (2001) have shown how Ca2+ induces neuronal gene expression. Membrane depolarization and neurotransmitter binding lead to ligand-gated and voltage-gated Ca2+ channels (L-VGCC) at the cell membrane, triggering the influx of extracellular Ca2+ into the cell. In terms of ligand-gated channels responsible for Ca2+ entry into neurons after EX, different mechanisms have been proposed involving preponderantly glutamatergic transmission through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate-type (NMDA) receptors (Kitamura et al., 2003; Vaynman et al., 2003; Nishijima et al., 2012). As stated above, promoter IV-dependent bdnf transcription, one of the main promoters driving neuronal activity-dependent BDNF expression, is the most thoroughly investigated (Tao et al., 2002). Using a mouse model with impaired activity-dependent bdnf expression through mutagenesis, Hong et al. (2008) revealed the existence of three Ca2+-responsive elements (CaREs) able to drive activity-dependent induction of promoter IV. Among the three DNA sequences that are cooperatively recruited for Ca2+-induced bdnf promoter IV expression, Tao et al. (2002) have shown that Ca2+-response factor (CaRF) is a regulator of activity-dependent bdnf gene expression through CaRE1 and that the transcriptional activity of CaRF is dependent on Ca2+ influx in a neuron selective manner. CaRE3 has also received specific attention. Indeed, once bounds by CREB (cAMP response element-binding protein), this latter is phosphorylated on serine 133 (Ma et al., 2014) and then induces BDNF transcription (Tao et al., 1998; Shen et al., 2001). Multiple Ca2+-dependent mechanisms have been proposed to be responsible for CREB phosphorylation at the upstream steps of CREB activation. These mechanisms included the Ca2+-sensitive adenylate cyclase/PKA (Shen et al., 2001), the Ca2+/calmodulin-activated kinase (CAMK) (Sun et al., 1994; Bito et al., 1996), Ras/MAPK (Wu et al., 2001) and the redox signaling (Radak et al., 2016) systems. Since BDNF has been shown to subsequently interact with NMDA receptors, CAMK and MAPK (Vaynman et al., 2003; Nakata and Nakamura, 2007), EX-induced changes in neuronal activity-dependent gene induction may use BDNF itself as a perpetuating loop.
In addition to glutamate, signaling from cholinergic, noradrenergic and serotoninergic transmitters are involved in cerebral bdnf gene expression (Garcia et al., 2003; Ivy et al., 2003; Chen et al., 2007). The cortical acetylcholine (ACh) content is increased in rats following a short period of walking, suggesting that ACh is released even at low-intensity of EX (Kurosawa et al., 1993). Conversely, cholinergic transmission impairment is associated with a downregulation of the hippocampal BDNF pathway in rats submitted to Morris Water Maze training that could be reversed by galantamine, a selective ACh esterase inhibitor (Gil-Bea et al., 2011). Interestingly, the blockade of noradrenergic signaling blunts the EX-effect on bdnf gene transcription (Garcia et al., 2003; Ivy et al., 2003) and is reversed when animals received reboxetine, a selective noradrenalin reuptake inhibitor (Russo-Neustadt et al., 2004). Mechanistically, the binding of noradrenalin to adrenergic receptor activates the phosphorylation cascades and induces BDNF expression in hippocampal neurons (Chen et al., 2007). Dopamine (DA) has also been reported to be upregulated by EX and to induce an increase in serum Ca2+ levels, which can enhance DA synthesis in the brain (Sutoo and Akiyama, 2003). There is evidence showing that DA receptor activation leads to BDNF expression probably through D1-like receptor activation (Kuppers and Beyer, 2001; Williams and Undieh, 2009). In addition, serotonin (5-HT) signaling may also be involved in EX-induced bdnf expression but to a lesser extent since 5-HT2A/C blockade minimally altered EX-induced BDNF mRNA activation (Ivy et al., 2003; Russo-Neustadt et al., 2004). Finally, gamma-aminobutyric acid (GABA), the primary inhibitory neurotransmitter of the CNS, has been shown to decrease hippocampal BDNF expression and to impair learning and memory (Kim et al., 2012) while bicuculline, a GABAA receptor antagonist, enhances memory consolidation by increasing hippocampal BDNF levels (Kim et al., 2012). Interestingly, the anxiolytic effect of a 3-week running protocol in mice was blocked by the infusion of bicuculline in the ventral hippocampus. These observed results suggest that GABAergic transmission in the hippocampus, in response to EX, may play an important role in dampening excitatory circuits that might otherwise trigger an anxious response. These findings demonstrate a complex and finely tuned adaptation (Schoenfeld et al., 2013).
Primarily discovered as a secreted factor from skeletal muscle, irisin is regulated by the peroxisome proliferator activator receptor γ coactivator-1α (PGC-1α) pathway, cleaved from fibronectin type III domain-containing protein 5 (FNDC5) and released in blood circulation (Bostrom et al., 2012). In addition to its peripheral expression, FNDC5/irisin is also produced in different brain regions (Dun et al., 2013) and Wrann et al. (2013) were the first showing that EX increases hippocampal BDNF expression through the PGC-1α/FNDC5 pathway. Indeed, the authors reported that the knockdown of PGC-1α reduced FNDC5 expression in the brain. Using primary cortical neurons, forced expression of FNDC5 increases BDNF levels whereas RNA interference directed against FNDC5 reduces its expression. Consistent with these findings, another study in mice has reported that the val66met polymorphism is associated with reduced expression of brain FNDC5 and BDNF following EX (Ieraci et al., 2016). Additionally, in an AD mouse model, hippocampal neurogenesis and cognition are improved by EX through both FNDC5 and BDNF upregulation (Choi et al., 2018). Interestingly, data reported that the exposition of ex vivo human cortical slices to recombinant irisin leads to the activation of the cAMP–PKA–CREB memory pathway (Lourenco et al., 2019).
Among epigenetic mechanisms, DNA methylation and histone modifications through methylation or acetylation are the most studied processes affecting gene expression. Numerous studies have shown that EX can act as an epigenetic modulator of brain plasticity and cognition influencing the activity of enzymes responsible for methylation (DNA methyltransferase, DNMT), histone acetylation (histone acetyltransferase, HAT) or deacetylation (histone deacetylase, HDAC). Thus, various studies have demonstrated that different models of EX lead to the modulation of HAT, DNMT, and HDAC activities at BDNF promoters (Gomez-Pinilla et al., 2011; Intlekofer et al., 2013; Sleiman et al., 2016). These changes ultimately result in an increase in bdnf gene transcription associated with cognitive improvement. For example, the research conducted by Gomez-Pinilla et al. (2011) demonstrated that EX has the capability to decrease methylation of CpG sites. This reduction occurs through the dissociation of methyl-CpG-binding protein 2 due to its phosphorylation, leading to enhanced bdnf transcription (Gomez-Pinilla et al., 2011). Additionally, Ionescu-Tucker et al. (2021) have recently highlighted that EX counteracts with the repressive histone modification trimethylated histone 3, lysine 9 (H3K9me3) at bdnf promoter, increasing BDNF expression in aged mice. Additionally, evidence suggests the role of small non-coding mRNAs on transcriptional gene silencing. Using transgenic mice in which Dicer which plays a pivotal role in the initiation of RNA silencing was inactivated, the overall decrease in hippocampal miRNAs was associated with a higher BDNF level leading to learning and memory improvements (Konopka et al., 2010). Using profiling array data, the authors identified two miRNAs (miRNA-138 and miRNA-384-5p) that potentially target BDNF mRNA. miRNA-138 was shown to act specifically on sirtuin 1 (SIRT-1), which regulates the acetylation status of PGC-1α responsible for bdnf gene expression through FNDC5 as stated in the preceding paragraph (Li et al., 2020). On the other hand, it has been reported that EX increases hippocampal SIRT-1 activity that downregulated miRNA-134 known to repress the translation of CREB and consequently that of BDNF (Gao et al., 2010). Other miRNAs have been proposed as regulators of BDNF (Varendi et al., 2014) and it has been reported that EX modifies the levels of many miRNAs in the brain (Cosin-Tomas et al., 2014; Pan-Vazquez et al., 2015). However, whether EX modulates miRNAs that directly affect cerebral bdnf gene expression requires further clarification.
EX is known to increase CBF in sensorimotor region and areas involved in cognitive capacities such as the prefrontal cortex, hippocampus and amygdala (Nishijima and Soya, 2006; Endo et al., 2013; Matsukawa et al., 2015). Considering the established synthesis of tPA by endothelial cells and its release into the bloodstream in response to different stimuli, including EX (Ding et al., 2011), a hypothetical mechanism can be proposed. Specifically, the elevation of CBF and the subsequent increase in fluid shear stress (FSS) induced by EX may result in an increase in tPA activity, which could then influence BDNF processing by cleaving pro-BDNF into its mature form through the conversion of plasminogen into active protease plasmin. Alternatively, tPA by facilitating neuronal NMDA receptors activation (Nicole et al., 2001; Anfray et al., 2020), could trigger Ca2+ influx and bdnf gene expression as stated in a precedent section. This notion is supported by the findings of Ding, Ying, and Gomez-Pinilla et al. (2011), who demonstrated that tPA-blocking strategies effectively abolished the increase in both pro- and mature BDNF in rat hippocampus, as well as the effect of EX on TrkB signaling and synaptic plasticity. The hypothesis that tPA produced by endothelial cells in response to FSS-related EX plays a key role in BDNF processing is plausible, particularly in light of evidence showing that blood tPA can cross the BBB via low-density lipoprotein receptor-related protein (LRP)-mediated transcytosis (Benchenane et al., 2005). Furthermore, considering the expression of NMDA and LRP receptors by endothelial cells (Hogan-Cann et al., 2019; Lu et al., 2019; Anfray et al., 2020), it is conceivable to postulate that tPA may function as an autocrine molecule, triggering the production of BDNF within these cells. To investigate these different hypotheses, the implementation of an endothelial tPA knockout (KO) model subjected to EX could provide valuable insights.
Strong evidence in favor of the hemodynamic hypothesis is also provided when strategies to reduce CBF were applied. One-week daily EX in rats with unilateral carotid occlusion (irreversible clamping) did not increase cortical BDNF levels in ipsilateral compared to exercised rats without carotid occlusion (Banoujaafar et al., 2014). Mechanistically, EX induces elevation of CBF, increases FSS which in turn stimulates endothelial nitric oxide synthase (eNOS) and the subsequent production of endothelial NO (Rubanyi et al., 1986). Interestingly, pharmacological inhibition of NO production by L-NAME (N-nitro-L-arginine methyl ester) (Chen et al., 2006) prevents the enhancement of BDNF mRNA occurring with EX while genetic deletion of eNOS abolishes the positive effects of EX on ischemic stroke (Endres et al., 2003). Accordingly, using an FSS-dependent increase in endothelial NO production, data demonstrate in Human umbilical vein endothelial cells subjected to different flows that endothelial BDNF expression was proportional to shear stress intensity (Prigent-Tessier et al., 2013). The hypothesis of NO regulation is also supported by a strong correlation, in cerebral microvessels, between BDNF protein levels and the activated form of eNOS (p-eNOS) phosphorylated at serine 1,177. Additionally, a positive association was also found between the elevation of p-eNOS and p-TrkB activation, in response to EX (Cefis et al., 2020). Besides, Monnier et al. (2017b) have shown that exposure of cerebral microvessel-enriched fractions or hippocampus sections from spontaneous hypertensive rats (SHR) to slow-releasing NO donor (glycerol trinitrate) increased endothelial BDNF production. Importantly, FSS-related EX-dependent response was prevented by TrkB inhibition (Wang et al., 2018). Collectively, these data and the one showing that NO controls t-PA release by human endothelial cells (Giannarelli et al., 2007) provide strong arguments suggesting that NO may be the intermediate between FSS and endothelial BDNF synthesis and maturation at the cerebral level.
Finally, the precise role of endothelial BDNF in the brain remains speculative, but there is compelling evidence suggesting its involvement in cerebral vessel vasodilation (Santhanam et al., 2010; Bordy et al., 2020), cerebral angiogenesis (Kim et al., 2004) and neuroplastic processes (Marie et al., 2018). The presence of endothelial TrkB-FL receptors suggests that endothelial BDNF may exert an autocrine effect by promoting NO production, which could then diffuse from endothelial cells to neurons, enhancing LTP and neuroplasticity (Hopper and Garthwaite, 2006). Alternatively, endothelium-derived BDNF may directly bind to neuronal TrkB-FL receptors and trigger neuroplastic pathways. The proximity between synapses and capillaries supports this hypothesis, although it still needs to be demonstrated. Finally, as mentioned above, endothelial BDNF may promote neuroplasticity through an astrocyte-dependent mechanism. Astrocytes have been implicated in the recycling of extracellular BDNF proteins, suggesting their involvement in facilitating BDNF availability. Two potential mechanisms can be proposed in this regard. A first mechanism may involve internalization of pro-BDNF through p75NTR -dependent endocytosis which is subsequently re-secreted by astrocytes in its mature form (Bergami et al., 2008; Vignoli et al., 2016). The second mechanism implies BDNF release by CEC into the perivascular space. The nearby astrocytes end-feet able to internalize it through TrkB-T1 receptor could then transfer BDNF to neighboring neurons via transcytosis or a related mechanism. Recent research by Han et al. (2021) showing in astrocyte recycling, the re-secretion of TrkB-endocytic BDNF substantiates this theory. Consequently, the released BDNF from astrocytes into the extracellular space could bind to neuronal TrkB-FL triggering neuroplastic signaling pathway. Ultimately, the utilization of molecular strategies aimed at specifically inhibiting endothelial BDNF could serve as a potent tool to elucidate its precise role, especially in the context of neuroplasticity. All mechanisms proposed in the present section are summarized in Figure 3.
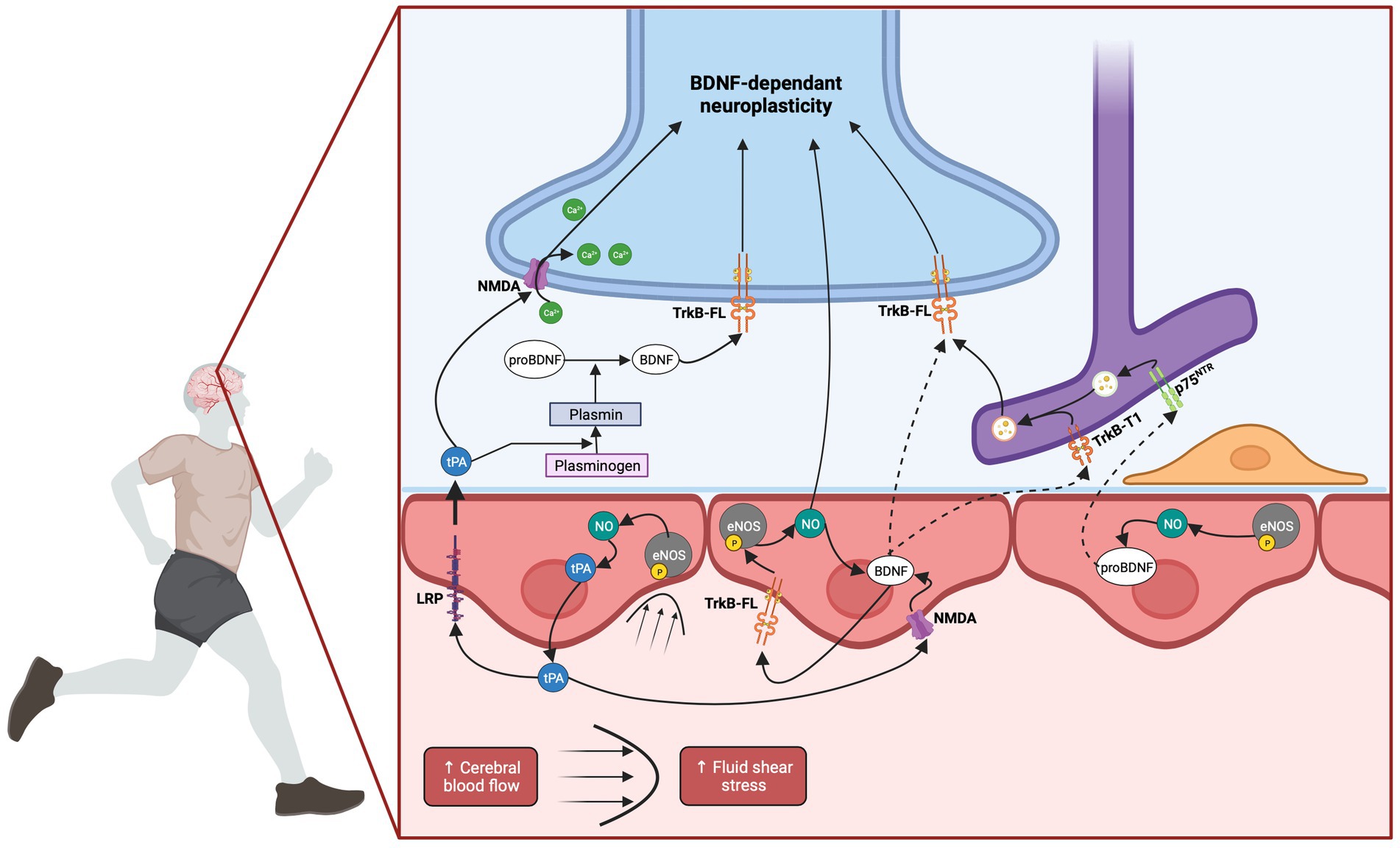
Figure 3. Hemodynamic hypothesis: involvement of cerebral blood flow in EX-induced BDNF increase. EX induces an increase in CBF, leading to elevated FSS. This mechanical stimulation triggers the activation of eNOS through its phosphorylation, resulting in the production of NO. The released NO has the potential to diffuse from endothelial cells to neurons, initiating neuronal BDNF expression. Additionally, endothelial NO has been demonstrated to induce BDNF expression in endothelial cells themselves. Endothelium-derived BDNF may directly bind to neuronal TrkB-FL receptors thus activating neuroplastic pathways. The proximity between synapses and capillaries supports this hypothesis. Alternatively, endothelium-derived BDNF could act in an autocrine manner, amplifying the NO response through endothelial TrkB-FL receptors. In parallel, astrocytes, through their nearby end-feet, might internalize either BDNF or pro-BDNF through their TrkB-T1 or p75NTR receptors, respectively. Subsequently, they could re-secrete BDNF which might be transferred to neighboring neurons. Finally, the elevation of CBF and subsequent NO production could trigger tPA release. tPA has the potential to cross the BBB via LRP-mediated transcytosis. It could then facilitate neuronal NMDA receptor activation or influence the processing of pro-BDNF to BDNF, through the conversion of plasminogen into plasmin. Alternatively, tPA might facilitate endothelial NMDA receptor activation which could potentially contribute to endothelial BDNF expression. Created with BioRender.com.
In the present section, our focus lies on elucidating the various humoral factors that have the potential to modulate brain BDNF levels in the context of liver- or muscle-brain crosstalk after EX. All mechanisms described in the following sections are summarized in Figure 4. We have assigned these factors into liver- or muscle-brain sections based on preliminary findings and their prominent expression in these organs. However, it should be emphasized that several molecules which will be expounded upon herein are known to be produced and secreted by various sources including the brain. In addition, while we have focused on molecules produced by the liver (Figure 4A) and skeletal muscle (Figure 4B), it should be noted that EX results in adaptations of many organ systems including adipose tissue and bone which could also contribute to the humoral pathway.

Figure 4. Peripheral mechanisms involved in EX-induced BDNF increase. Many organs respond to EX through the release of exerkines. Among them, the liver and the skeletal muscle have received particular attention and several exerkines originating from these organs have the potential to influence brain BDNF expression. (A) In response to EX, the liver secretes exerkines named hepatokines. Studies have reported that β-HB can cross the BBB and function as a signaling molecule promoting hippocampal BDNF expression by HDAC inhibition. In addition, IGF-1 can cross the BBB through IGF-1 receptor binding. Inhibition of IGF-1 signaling through blocking antibodies prevented EX-induced hippocampal BDNF expression. FGF-21 is also secreted by the liver in response to EX, can enter the brain and improve cognition possibly through an increase in brain BDNF expression since intraperitoneal administration of FGF-21 was associated with BDNF upregulation in aged mice. Recent research has unveiled Gpld1 as a newly discovered hepatokine that appears to correlate with cognitive performance in mice undergoing EX. Moreover, overexpression of Gpld1 in the liver has been associated with a significant rise in hippocampal BDNF expression and an enhancement of neurogenesis. Although Gpld1 does not cross the BBB, data suggest that this enzyme may be involved in coagulation as well as in the complement system cascades of molecules downstream of GPI-anchored substrate. (B) Several molecules named myokines are secreted by skeletal muscles in response to EX and have been shown to promote brain BDNF expression. Lactate that is significantly produced and released during EX can cross the BBB via MCT transporters. The pharmacological blockade of MCT in mice submitted to EX abolished hippocampal bdnf gene expression while intraperitoneal administration of lactate elicited a hippocampal BDNF increase comparable to that observed in trained mice. Lactate effect on hippocampal bdnf gene expression is thought to be dependent on the potentiation of NMDA glutamatergic transmission and upregulation of SIRT-1 activity fostering the PGC-1α/FNDC5/BDNF pathway. The myokine CTSB is also released by skeletal muscle during EX though an AMPK-dependent mechanism. In vivo experiments provide evidence that CTSB can cross the BBB and elicit BDNF expression while EX in CTSB KO mice failed to enhance neurogenesis and improve spatial memory. Although the role of skeletal muscle as a source of blood BDNF remains a topic of debate, recent studies using NMES as a model of muscle contraction seem to support this hypothesis. The BBB crossing of circulating BDNF is uncertain but exosomal transport might be involved. Additionally, peripheral delivery of BDNF has been shown to induce neurogenesis and increase hippocampal BDNF levels. During muscle contraction, the increased calcium signaling enhances PGC-1α activation which leads to an increase in FNDC5, a transmembrane protein that is cleaved during EX and released in the bloodstream as irisin. Data reported that irisin could cross or signal at the BBB, potentially via its recently discovered binding to activated integrin αVβ5 receptors. In an AD mice model, peripheral delivery of FNDC5/irisin rescued memory impairment and synaptic plasticity deficits through mechanisms dependent on cerebral BDNF. Conversely, the blockade of peripheral FNDC5/irisin attenuates the effect of EX on LTP and memory tests. PGC-1α activation during EX also stimulates the expression of KAT enzymes within skeletal muscle. This enzyme catalyzes the conversion of KYN, a neurotoxic metabolite that can cross the BBB and lead to depression to KYNA which is unable to cross the BBB. Mice with muscle-specific overexpression of PGC-1α were resilient to stress-induced depression and did not display decreased hippocampal bdnf gene expression while peripheral KYN administration induced depressive behavior in wild-type but not in transgenic animals. This last mechanism illustrates the crosstalk between peripheral organs, as KYN is a metabolite of tryptophan produced in the liver. Created with BioRender.com.
The ketone bodies acetoacetate and β-hydroxybutyrate (β-HB) serve as fuel substrates that are upregulated in metabolic changes such as caloric restriction (Lin et al., 2015), ketogenic diets, fasting and EX (Sleiman et al., 2016; Evans et al., 2017). It has been reported that β-HB can cross the BBB and function as a signaling molecule promoting BDNF expression after EX (Sleiman et al., 2016). Indeed, these authors demonstrated that hippocampal β-HB contents were significantly increased in mice after 30 days of voluntary wheel running. Furthermore, exposing both cortical neuron cultures and mice hippocampus slices to β-HB led to an overexpression of bdnf gene. In vivo, intraventricular administration of β-HB in mice induced a significant increase in hippocampal bdnf gene expression. Consistently, a positive correlation between plasmatic β-HB levels and hippocampal BDNF contents was highlighted in mice after 6 weeks of voluntary running (Marosi et al., 2016). Interestingly, in the absence of EX, a similar increase in hippocampal BDNF was observed after in vivo β-HB administration in mice with normal diet (Hu et al., 2018) while infusion of β-HB attenuates motor deficits in mouse models of Huntington’s disease and protects neurons in models of AD and PD (Kashiwaya et al., 2000). Additionally, a ketogenic diet seems to play neuroprotective roles during cerebral ischemia, neurodegenerative diseases and enhanced memory processes in aged mice (Suzuki et al., 2002; Gano et al., 2014; Paoli et al., 2014; Newman et al., 2017). At the molecular level, β-HB acts directly on bdnf gene promoter by HDAC inhibition. Indeed, β-HB exposure of cultured primary neurons decreases HDAC 2/3 activity on the bdnf promoter. Besides, the inactivation of HDAC3 by BRD3308 (an inhibitor) or short hairpin RNA strategy, increases bdnf transcript expression (Sleiman et al., 2016).
Structurally related to pro-insulin, Insulin like growth factor 1 (IGF-1) is a polypeptide hormone primarily identified in the liver, which can be transported to other tissues and act as an important mediator of body growth and tissue remodelling (Laron, 2001). Although bone and skeletal muscle can produce IGF-1 (Yakar et al., 2010), the major peripheral source of IGF-1 is believed to be the liver as demonstrated by a 75% decrease in plasma concentration in liver-specific IGF-1 deficient mice (Yakar et al., 1999). IGF-1 is involved not only in the growth and development of the brain during early life, but also in its maintenance and plasticity. Once secreted in the systemic circulation, IGF-1 can cross the BBB (Pan and Kastin, 2000) by binding to IGF-1 receptors in a mechanism driven by neuronal activity (Trejo et al., 2001; Nishijima et al., 2010). Data indicated that IGF-1 mediates neurogenesis (Ding et al., 2006), synaptogenesis (O’Kusky et al., 2000), vessels growth (Lopez-Lopez et al., 2004), neuroprotection and neuroplasticity (Ding et al., 2006). In rats, intracarotid injection of IGF-1 induced BDNF mRNA expression in the hippocampus (Carro et al., 2000). In addition, the specific inhibition of IGF-1 receptors in the hippocampus using latex microbeads containing alphaIR3 antibody in trained rats, prevented EX-induced enhancement in memory recall and significantly decreased both pro-BDNF and BDNF expressions (Ding et al., 2006). Similarly, using antiserum raised against IGF-1, Chen and Russo-Neustadt (2007) reported that the blockade of the uptake of peripheral IGF-1 reversed the increase in BDNF mRNA and protein expressions elicited by EX.
Using liver specific Fibroblast growth factor 21 (FGF21) knockout mice, Markan et al. (2014) demonstrated that FGF21 is preferentially expressed in the liver. FGF21 functions as a metabolic regulator (Kharitonenkov et al., 2005) capable of preventing insulin resistance, increasing fatty acid oxidation (Badman et al., 2007) and weight loss in obese animal models (Jimenez et al., 2018) and humans (Gaich et al., 2013). EX induces an increase in FGF21 expression in mice and humans (Kim et al., 2013) which seems to be dependent on the rise in glucagon-to-insulin ratio since prevention of this increase during EX blunts EX-induced increase in FGF21 (Hansen et al., 2016). In addition to its beneficial effects on the whole body, FGF21 may also exert positive effects on cognition in response to EX. In this regard, FGF21 has been shown to enter the brain (Hsuchou et al., 2007), to improve cognition by restoring synaptic plasticity in obese-insulin-resistant male rats (Sa-Nguanmoo et al., 2016) and to be neuroprotective in a mouse model of aging (Yu et al., 2015). Moreover, data have shown that the intraperitoneal administration of FGF21 in aged mice was associated with activation of the AMPK pathway associated with an increase in cerebral BDNF levels and a significant improvement in Morris water maze assessment (Kang et al., 2020).
Recently, using an elegant plasma transfer strategy, Horowitz et al. (2020) showed that plasma obtained from both aged and mature exercised mice could alleviate hippocampal impairments when transferred to naïve aged mice. Among the 12 factors that were found to be increased in trained mice, the authors focused on Gpld1. They confirmed that this enzyme was predominantly expressed in the liver (Maguire and Gossner, 1995) and that liver Gpld1 expression was increased after EX whereas no change was observed in muscle and hippocampus. Furthermore, Gpld1 plasma contents were significantly enhanced by EX in both mature and aged mice with a significant correlation between liver-derived Gpld1 and cognitive performances in the radial arm water-maze. In addition, they reported that liver overexpression of Gpld1 was associated with a significant increase in hippocampal BDNF expression and an enhancement of neurogenesis markers in the dentate gyrus. These cellular and molecular changes were consistent with an improvement of cognitive function since these mice showed better spatial and recognition memory (Horowitz et al., 2020). Although the precise mechanism underlying the connection between this Glycosylphosphatidylinositol (GPI) hydrolyzing enzyme and the improvement of hippocampal-dependent learning and memory is not fully delineated by the authors, Horowitz and colleagues reported using a catalytically inactive mutant, that the enzymatic activity was necessary for its effects. The authors suggest that this enzyme may be involved in coagulation, as well as in the complement system cascades of molecules downstream of GPI-anchored substrate. Accordingly, in a similar experimental design using plasma transfer from exercising mice, complement and coagulation factor such as clusterin, largely produced by hepatocytes, was recently reported to reduce hippocampal inflammation and promote neurogenesis and cognition although BDNF expression was not assessed in this study (De Miguel et al., 2021).
Over an extended period, the term “exercise factor” served to depict molecules originating from contracting muscles, potentially orchestrating metabolic and physiological effects on central or peripheral organs. Among these factors, cytokines were the first reported in the literature (Sprenger et al., 1992) and it was through studies showing that muscle IL-6 can act as a humoral factor operating in an autocrine, paracrine, or endocrine manner that the neologism “myokines” was introduced (Pedersen et al., 2003). Although it would have been logical to initiate this muscle-brain crosstalk section with a paragraph dedicated to cytokines, the evidence of a direct impact of these molecules on cerebral BDNF production remains inconclusive despite it has been demonstrated that IL-6 can cross the BBB (Banks et al., 1994). The same lack of direct evidence exists for IL-10 or TNF-α even though studies have reported a similar pattern of variation with BDNF in the hippocampus (Noga et al., 2007; Cabral-Santos et al., 2016; Jahangiri et al., 2019). In contrast, several myokines have been identified, bolstered by compelling scientific rationale, establishing a causative link to brain BDNF expression.
Lactate is a metabolite largely produced during EX and mainly released from skeletal muscles. Lactate can cross the BBB via monocarboxylate transporters (MCT) binding on neurogliovascular unit (including astrocytes, neurons, endothelial cells and pericytes) (Proia et al., 2016) and participate to neuroplastic processes such as neurogenesis (Lev-Vachnish et al., 2019), neuronal excitability (Skwarzynska et al., 2023) and LTP (Suzuki et al., 2011). Recently, lactate has been identified as an appealing candidate for inducing cerebral BDNF expression in response to EX. Interestingly, using intraperitoneal infusion of the lactate MCT inhibitor (AR-C155858) in mice submitted to voluntary EX, El Hayek et al. (2019) demonstrated that hippocampal bdnf gene expression was completely abolished. Conversely, intraperitoneal administration of lactate in mice elicited a hippocampal BDNF increase similar to that obtained in trained mice. The authors also reported that the improvement in learning/memory performance was dependent on BDNF as co-administration of lactate and a TrkB antagonist (CEP701) prevented this effect. Although the interaction between lactate and cerebral BDNF levels is not fully elucidated, some mechanisms have been proposed [For review (Muller et al., 2020)]. Firstly, lactate promoted plasticity-related gene expressions (bdnf, Arc, Zif268 and c-fos) in neuronal cultures of mouse neocortex by potentiating neuronal NMDA glutamatergic transmission (Yang et al., 2014). Secondly, peripheral administration of lactate induced SIRT-1 activity, thereby fostering the cerebral PGC-1α/FNDC5/BDNF pathway (El Hayek et al., 2019). Conversely, inhibition of SIRT-1 with sirtinol administration or RNA interference impeded both hippocampal bdnf gene induction and the lactate- and EX-mediated improvement of learning/memory (El Hayek et al., 2019).
Recent research has provided compelling evidence suggesting that the activation of the PGC-1α FNDC5/irisin pathway in skeletal muscles is also involved in EX-induced cerebral plasticity (Lourenco et al., 2019). Using AD mice models, the authors demonstrated that FNDC5 adenovirus delivery through the tail vein or intra-cerebrovascular injection of FNDC5 rescued memory impairment and synaptic plasticity mechanisms dependent on cerebral BDNF. Conversely, the blockade of peripheral FNDC5/irisin attenuates the effect of EX on LTP and memory tests. Although this blockade was not directly linked to a decrease in brain BDNF expression in this study, these data support the role of peripheral FNDC5/irisin on physiological memory process. In this regard, in the context of hypertension, EX-induced enhanced BDNF expression was reported to be dependent on peripheral but not central FNDC5 (Wang et al., 2019). Furthermore, based on the premise that irisin is secreted by the skeletal muscle, Islam et al. (2021) have provided evidence that irisin can cross the BBB. Indeed, through the injection of fluorescently labeled irisin into the bloodstream of mice and subsequent confocal microscopy analysis, the authors successfully detected the distribution of labeled irisin within the cerebral region. Then, they have shown that peripheral delivery of irisin improved learning and memory in young and old mice and significantly reduced the buildup of beta-amyloid plaques in a mouse model of AD. Collectively, these findings support the hypothesis that FNDC5/irisin may cross the BBB or induce another factor that triggers cerebral BDNF expression, possibly through CREB activation, as previously stated. Accordingly, a very recent study reported that irisin binds to activated integrin αVβ5 receptors, which are abundantly expressed on brain endothelial cells (Mu et al., 2023). However, while FNDC5/irisin appears as a very attractive myokine, it is important to note that the detection of this hormone by commercial antibodies reveals an important cross-reactivity (Albrecht et al., 2015). Besides, the translatability of the studies from animals to humans may encounter a major pitfall since the transcription pattern of fndc5 gene is not conserved from rodent to human resulting in very low translation efficiency (Raschke et al., 2013; Albrecht et al., 2020).
Another PGC-1α dependent mechanism involves the conversion of Kynurenine (KYN) to kynurenic acid (KYNA). Indeed, tryptophan, primarily derived from the liver, is metabolized to KYN through indolamine-2,3-dioxygenase (IDO) (Martin et al., 2020). Interestingly, KYN has been reported to cross the BBB whereas KYNA cannot (Fukui et al., 1991) and was shown to be neurotoxic (Sundaram et al., 2020) leading to depression and neurodegenerative disorders (Myint et al., 2007). Conversely, KYNA which is produced from KYN via muscle kynurenine aminotransferases (KAT) activity (Schwarcz et al., 2012), is neuroprotective (Foster et al., 1984) and cognitive performance were positively associated with KYNA concentrations in plasma of AD patients (Gulaj et al., 2010). Therefore, maintaining a well-balanced ratio between these two metabolites appears to be crucial. Following EX, the activation of the PGC-1α pathway stimulates the expression of KAT enzymes within skeletal muscle. Consequently, data also reported that EX increases plasma KYNA levels both in rodents (Agudelo et al., 2014) and humans (Lewis et al., 2010) while an increase in KYN was shown in a model of stress-induced depression (Agudelo et al., 2014). Interestingly, mice with muscle-specific overexpression of PGC-1α were resilient to stress-induced depression compared to wild-type littermates and did not exhibit decreased hippocampal bdnf gene expression while peripheral KYN administration induced depressive behavior in wild-type but not in transgenic animals (Agudelo et al., 2014). Consistent with these results, in a murine model of PD, IDO inhibition was associated with reduced oxidative stress, lower impairment in coordination and locomotion and restoration of striatal BDNF levels (Sodhi et al., 2021). Collectively, these data emphasize the importance of muscle KYN-to-KYNA conversion in facilitating BDNF-induced cognitive improvement after EX.
Cathepsin-B (CTSB), a lysosomal cysteine protease ubiquitously expressed was identified in 2016 as a myokine by Moon et al. (2016). Using proteomic analysis of the culture media from L6 myoblast cells treated with the AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) to mimic the effects of EX, the authors reported an increase in CTSB. To validate CTSB as a candidate myokine, they further showed that the rise in plasma CTSB levels in voluntary running mice was concomitant with an increase in CTSB mRNA and protein expressions in gastrocnemius muscles after 30 days. In addition, similar increases in CTSB plasma levels were observed in Rhesus monkeys and humans after four months of treadmill running. To delineate the underlying mechanisms, Moon et al. (2016) conducted studies in CTSB KO mice showing that compared to wild-type control mice, EX in CTSB KO mice failed to enhance neurogenesis and improve spatial memory. Additionally, intravenous injection of CTSB in CTSB KO mice led to a significant increase in CTSB levels in the blood and brain tissues, indicating that CTSB can cross the BBB. Interestingly, these authors demonstrated that recombinant CTSB administration induced the increase in both doublecortin and BDNF mRNA levels in adult hippocampal progenitor cells. These data are consistent with a previous study showing in contrast that inhibition of cathepsins B and L reduced kainate-induced BDNF mRNA expression in cultured hippocampal slices (Bednarski et al., 1998). Although these data support a causative link between CTSB and cerebral BDNF expressions, the complex interplay between them should be interpreted considering the origin of CTSB and the specific physiological context. Indeed, recent studies have suggested that microglia-derived CTSB may be a key driver in inflammatory brain diseases and aging (Nakanishi, 2020) while genetic CTSB deletion prevented cognitive impairments, reduced amyloid peptides, brain damage and pro-inflammatory factors, in animal models of traumatic brain injury, AD and aging (Hook et al., 2022).
BDNF is produced in skeletal muscle by various cell types, including myofibers, satellite cells, motoneurons and endothelial cells (Mousavi and Jasmin, 2006; Matthews et al., 2009; Cefis et al., 2022). BDNF has been found to play an important role in muscle growth, function, regeneration (Mousavi and Jasmin, 2006; Clow and Jasmin, 2010) and metabolism (Matthews et al., 2009; Yang et al., 2019). In response to EX, a significant increase in BDNF mRNA and/or protein levels has been observed in skeletal muscles (Cuppini et al., 2007; Ogborn and Gardiner, 2010; Yu et al., 2017). According to the definition of myokines, BDNF could be classified as one (Matthews et al., 2009; Yang et al., 2019). However, whether it may be tempting to believe that skeletal muscle could be a primary source of circulating BDNF levels remains a subject of debate. Although the muscle cells produce BDNF, the precise mechanism of its release into the bloodstream remains uncertain, and there is supportive evidence from various studies indicating that circulating BDNF predominantly originates from the brain (Rasmussen et al., 2009). Thus, using prolonged EX, Rasmussen and collaborators demonstrated in Human that the increase in circulating BDNF originated from the brain, as evidenced by the difference between plasmatic BDNF from arterial and jugular origins (Rasmussen et al., 2009). Besides, in skeletal muscle, a study using electroporation to overproduce BDNF in mice failed to increase circulating BDNF levels, suggesting that muscle derived-BDNF may act only in an auto/paracrine manner (Matthews et al., 2009). However, recent evidences have shown that neuromuscular electric stimulation (NMES) increases circulating BDNF levels both in humans (Miyamoto et al., 2018; Kimura et al., 2019) and animals (Fulgenzi et al., 2020). Additionally, it has been recently demonstrated for the first time that human myocytes produced and secreted biologically active BDNF (Fulgenzi et al., 2020). These outcomes reinforce the idea that skeletal muscle could be a potential source of circulating BDNF in humans and animals, but a further question still needs to be clarified: Is BDNF released from the skeletal muscle into bloodstream can directly interact with the brain?
To address this question, particular attention should be provided to extracellular vesicles, especially exosomes. Based on the provided references, BDNF can be transported within exosomes, (Yuan et al., 2017; Gelle et al., 2021) and it has been observed that muscle contraction induced their release (Watanabe et al., 2022). At the cerebral level, it was recently suggested that BDNF transported in exosomes could allow sustained and specific release of BDNF in the brain (Yuan et al., 2017), a mechanism that could potentially extend to BDNF derived from skeletal muscle. In addition, the transport by exosomes offers a considerable advantage since exosomes can cross the BBB (Yuan et al., 2017; D’Anca et al., 2019). Indeed, at the peripheral level, exosomes could allow BDNF, which has a short half-life, to evade the catabolic processes. Finally, even if modulation of neuroplasticity by skeletal muscle-derived BDNF has yet to be demonstrated, peripheral delivery of BDNF has been shown to induce neurogenesis and increase BDNF levels in hippocampus, leading to antidepressant and anxiolytic effects in mice (Schmidt and Duman, 2010). Taken together, these findings support the hypothesis that muscle-derived BDNF, may cross the BBB and positively act in the brain following EX. Further in vivo studies are needed to provide more conclusive evidence in support of this hypothesis.
As the most downstream factor mediating EX-induced brain health, manipulating BDNF content in the brain has emerged as a promising strategy for mitigating cognitive deficits, addressing neurodegenerative disorders, counteracting age-related cognitive impairment, and promoting overall brain health. EX has been identified as a potent and robust non-pharmacological intervention for enhancing cognitive function and limiting cognitive deficits through increased brain BDNF levels. Recent research has shed light on two additional pathways, namely hemodynamic and humoral, which complement the well-established role of neuronal activity in modulating brain BDNF levels. Notably, these pathways offer more feasible and achievable pharmacological strategies for intensifying brain BDNF expression compared to the neuronal activity pathway, as they circumvent the challenges of drug delivery across the BBB and potential drug-related side effects on neurotransmission.
However, the question of the relative contribution of each of these mechanisms to the beneficial effects of EX-induced brain BDNF increase remains complex and challenging to answer definitively. It is hypothesized that each pathway plays an essential role, as strategies that dampen any of these mechanisms individually have been shown to result in defective BDNF expression associated with cognitive impairment. It is plausible that these distinct mechanisms exist to offer a differential and persistent age-dependent temporal response, or to compensate for the deficiencies of one pathway with the others. Moreover, evidence suggests that these mechanisms are intricately interconnected, as neuronal activation is coupled with CBF regulation, and irisin, one of the well-studied humoral factors, has been found to modulate both vasorelaxation and neuronal activity. To understand the contributive part of the different pathways involved, employing transgenic mice with selective endothelial deletion of tPA or BDNF could provide valuable insights into the neuroplastic consequences of the hemodynamic pathway. Similarly, conducting studies that compare NMES to conventional models of EX would enable the isolation of the humoral muscle-brain crosstalk from the broader whole-body response. Indeed, this strategy would overcome an experimental bias as current studies investigating the muscle-brain dialogue are conducted following conventional protocols which engender a pre-dialogue between peripheral tissues that are also responsive to EX. The conversion of KYN into KYNA or the production of irisin by adipose tissue are some examples.
In conclusion, manipulating brain BDNF content through non-pharmacological interventions such as EX has emerged as a promising strategy to enhance cognitive function and mitigate cognitive deficits. Recent findings on hemodynamic and humoral pathways provide additional insights into the mechanisms through which EX induces brain BDNF overproduction. Further research is warranted to better understand the contribution of each pathway to the beneficial effects of EX on brain health and cognitive function. In addition, due to a wide range of EX types and the complexity of their classification, it is crucial to keep in mind that the contribution of the pathways described could be contingent upon EX type (e.g., resistance, aerobic), duration, intensity, frequency. Although this aspect has not been addressed in the manuscript, we encourage readers to consider this remark. Altogether, the findings on hemodynamic and humoral pathways will undoubtedly have implications for clinicians aiming to promote EX interventions or for scientists seeking to develop alternative strategies to mimic EX effects, particularly in populations who may face challenges with regular EX adherence. By unraveling the intricate interplay of these mechanisms, the scientific community can unlock the full potential of brain BDNF modulation for cognitive health and neuroprotection in various clinical and research settings.
MC: Writing – original draft, Writing – review & editing. RC: Writing – original draft, Writing – review & editing. JW: Writing – review & editing. AM: Writing – review & editing. AQ: Writing – review & editing. CL: Writing – review & editing. AP-T: Writing – original draft, Writing – review & editing. PG: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. MC is was supported by a postdoctoral fellowship from the Fonds de Recherche en Santé du Québec. RC is was supported by a Ph.D. scholarship from the Fondation pour la Recherche Médicale. This work has been support by INSERM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agudelo, L. Z., Femenia, T., Orhan, F., Porsmyr-Palmertz, M., Goiny, M., Martinez-Redondo, V., et al. (2014). Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cells 159, 33–45. doi: 10.1016/j.cell.2014.07.051
Aid, T., Kazantseva, A., Piirsoo, M., Palm, K., and Timmusk, T. (2007). Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 85, 525–535. doi: 10.1002/jnr.21139
Albrecht, E., Norheim, F., Thiede, B., Holen, T., Ohashi, T., Schering, L., et al. (2015). Irisin - a myth rather than an exercise-inducible myokine. Sci. Rep. 5:8889. doi: 10.1038/srep08889
Albrecht, E., Schering, L., Buck, F., Vlach, K., Schober, H. C., Drevon, C. A., et al. (2020). Irisin: still chasing shadows. Mol. Metab. 34, 124–135. doi: 10.1016/j.molmet.2020.01.016
Alderson, R. F., Curtis, R., Alterman, A. L., Lindsay, R. M., and DiStefano, P. S. (2000). Truncated TrkB mediates the endocytosis and release of BDNF and neurotrophin-4/5 by rat astrocytes and schwann cells in vitro. Brain Res. 871, 210–222. doi: 10.1016/S0006-8993(00)02428-8
Anfray, A., Drieu, A., Hingot, V., Hommet, Y., Yetim, M., Rubio, M., et al. (2020). Circulating tPA contributes to neurovascular coupling by a mechanism involving the endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 40, 2038–2054. doi: 10.1177/0271678X19883599
Badman, M. K., Pissios, P., Kennedy, A. R., Koukos, G., Flier, J. S., and Maratos-Flier, E. (2007). Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437. doi: 10.1016/j.cmet.2007.05.002
Balkowiec, A., and Katz, D. M. (2002). Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J. Neurosci. 22, 10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002
Banks, W. A., Kastin, A. J., and Gutierrez, E. G. (1994). Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci. Lett. 179, 53–56. doi: 10.1016/0304-3940(94)90933-4
Banoujaafar, H., Van Hoecke, J., Mossiat, C. M., and Marie, C. (2014). Brain BDNF levels elevation induced by physical training is reduced after unilateral common carotid artery occlusion in rats. J. Cereb. Blood Flow Metab. 34, 1681–1687. doi: 10.1038/jcbfm.2014.133
Barde, Y. A., Edgar, D., and Thoenen, H. (1982). Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1, 549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x
Bartoletti, A., Cancedda, L., Reid, S. W., Tessarollo, L., Porciatti, V., Pizzorusso, T., et al. (2002). Heterozygous knock-out mice for brain-derived neurotrophic factor show a pathway-specific impairment of long-term potentiation but normal critical period for monocular deprivation. J. Neurosci. 22, 10072–10077. doi: 10.1523/JNEUROSCI.22-23-10072.2002
Bayas, A., Hummel, V., Kallmann, B. A., Karch, C., Toyka, K. V., and Rieckmann, P. (2002). Human cerebral endothelial cells are a potential source for bioactive BDNF. Cytokine 19, 55–58. doi: 10.1006/cyto.2002.0892
Beattie, M. S., Harrington, A. W., Lee, R., Kim, J. Y., Boyce, S. L., Longo, F. M., et al. (2002). ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36, 375–386. doi: 10.1016/S0896-6273(02)01005-X
Bednarski, E., Lauterborn, J. C., Gall, C. M., and Lynch, G. (1998). Lysosomal dysfunction reduces brain-derived neurotrophic factor expression. Exp. Neurol. 150, 128–135. doi: 10.1006/exnr.1997.6747
Bejot, Y., Mossiat, C., Giroud, M., Prigent-Tessier, A., and Marie, C. (2011). Circulating and brain BDNF levels in stroke rats. Relevance to clinical studies. PLoS One 6:e29405. doi: 10.1371/journal.pone.0029405
Benchenane, K., Berezowski, V., Ali, C., Fernandez-Monreal, M., Lopez-Atalaya, J. P., Brillault, J., et al. (2005). Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein receptor-related protein-mediated transcytosis. Circulation 111, 2241–2249. doi: 10.1161/01.CIR.0000163542.48611.A2
Berchtold, N. C., Chinn, G., Chou, M., Kesslak, J. P., and Cotman, C. W. (2005). Exercise primes a molecular memory for brain-derived neurotrophic factor protein induction in the rat hippocampus. Neuroscience 133, 853–861. doi: 10.1016/j.neuroscience.2005.03.026
Bergami, M., Santi, S., Formaggio, E., Cagnoli, C., Verderio, C., Blum, R., et al. (2008). Uptake and recycling of pro-BDNF for transmitter-induced secretion by cortical astrocytes. J. Cell Biol. 183, 213–221. doi: 10.1083/jcb.200806137
Bito, H., Deisseroth, K., and Tsien, R. W. (1996). CREB phosphorylation and dephosphorylation: a ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cells 87, 1203–1214. doi: 10.1016/S0092-8674(00)81816-4
Black, J. E., Isaacs, K. R., Anderson, B. J., Alcantara, A. A., and Greenough, W. T. (1990). Learning causes synaptogenesis, whereas motor activity causes angiogenesis, in cerebellar cortex of adult rats. Proc. Natl. Acad. Sci. U. S. A. 87, 5568–5572. doi: 10.1073/pnas.87.14.5568
Bordy, R., Quirie, A., Marie, C., Wendling, D., Totoson, P., and Demougeot, C. (2020). Vascular arginase is a relevant target to improve cerebrovascular endothelial dysfunction in rheumatoid arthritis: evidence from the model of adjuvant-induced arthritis. Transl. Stroke Res. 11, 4–15. doi: 10.1007/s12975-019-00699-7
Bostrom, P., Wu, J., Jedrychowski, M. P., Korde, A., Ye, L., Lo, J. C., et al. (2012). A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468. doi: 10.1038/nature10777
Cabral-Santos, C., Castrillon, C. I., Miranda, R. A., Monteiro, P. A., Inoue, D. S., Campos, E. Z., et al. (2016). Corrigendum: inflammatory cytokines and BDNF response to high-intensity intermittent exercise: effect the exercise volume. Front. Physiol. 7:662. doi: 10.3389/fphys.2016.00662
Carro, E., Nunez, A., Busiguina, S., and Torres-Aleman, I. (2000). Circulating insulin-like growth factor I mediates effects of exercise on the brain. J. Neurosci. 20, 2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000
Cefis, M., Chaney, R., Quirie, A., Santini, C., Marie, C., Garnier, P., et al. (2022). Endothelial cells are an important source of BDNF in rat skeletal muscle. Sci. Rep. 12:311. doi: 10.1038/s41598-021-03740-8
Cefis, M., Prigent-Tessier, A., Quirie, A., Pernet, N., Marie, C., and Garnier, P. (2019). The effect of exercise on memory and BDNF signaling is dependent on intensity. Brain Struct. Funct. 224, 1975–1985. doi: 10.1007/s00429-019-01889-7
Cefis, M., Quirie, A., Pernet, N., Marie, C., Garnier, P., and Prigent-Tessier, A. (2020). Brain-derived neurotrophic factor is a full endothelium-derived factor in rats. Vasc. Pharmacol. 128-129:106674. doi: 10.1016/j.vph.2020.106674
Chaddock, L., Erickson, K. I., Prakash, R. S., Kim, J. S., Voss, M. W., Vanpatter, M., et al. (2010). A neuroimaging investigation of the association between aerobic fitness, hippocampal volume, and memory performance in preadolescent children. Brain Res. 1358, 172–183. doi: 10.1016/j.brainres.2010.08.049
Chao, M. V. (2003). Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309. doi: 10.1038/nrn1078
Chen, M. J., Ivy, A. S., and Russo-Neustadt, A. A. (2006). Nitric oxide synthesis is required for exercise-induced increases in hippocampal BDNF and phosphatidylinositol 3′ kinase expression. Brain Res. Bull. 68, 257–268. doi: 10.1016/j.brainresbull.2005.08.013
Chen, M. J., Nguyen, T. V., Pike, C. J., and Russo-Neustadt, A. A. (2007). Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell. Signal. 19, 114–128. doi: 10.1016/j.cellsig.2006.05.028
Chen, M. J., and Russo-Neustadt, A. A. (2007). Running exercise- and antidepressant-induced increases in growth and survival-associated signaling molecules are IGF-dependent. Growth Factors 25, 118–131. doi: 10.1080/08977190701602329
Chen, Z. Y., Ieraci, A., Teng, H., Dall, H., Meng, C. X., Herrera, D. G., et al. (2005). Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J. Neurosci. 25, 6156–6166. doi: 10.1523/JNEUROSCI.1017-05.2005
Cheval, B., Darrous, L., Choi, K. W., Klimentidis, Y. C., Raichlen, D. A., Alexander, G. E., et al. (2023). Genetic insights into the causal relationship between physical activity and cognitive functioning. Sci. Rep. 13:5310. doi: 10.1038/s41598-023-32150-1
Choi, S. H., Bylykbashi, E., Chatila, Z. K., Lee, S. W., Pulli, B., Clemenson, G. D., et al. (2018). Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 361. doi: 10.1126/science.aan8821
Clow, C., and Jasmin, B. J. (2010). Brain-derived neurotrophic factor regulates satellite cell differentiation and skeltal muscle regeneration. Mol. Biol. Cell 21, 2182–2190. doi: 10.1091/mbc.e10-02-0154
Colcombe, S. J., Erickson, K. I., Scalf, P. E., Kim, J. S., Prakash, R., McAuley, E., et al. (2006). Aerobic exercise training increases brain volume in aging humans. J. Gerontol. A Biol. Sci. Med. Sci. 61, 1166–1170. doi: 10.1093/gerona/61.11.1166
Cosin-Tomas, M., Alvarez-Lopez, M. J., Sanchez-Roige, S., Lalanza, J. F., Bayod, S., Sanfeliu, C., et al. (2014). Epigenetic alterations in hippocampus of SAMP8 senescent mice and modulation by voluntary physical exercise. Front. Aging Neurosci. 6:51. doi: 10.3389/fnagi.2014.00051
Cuppini, R., Sartini, S., Agostini, D., Guescini, M., Ambrogini, P., Betti, M., et al. (2007). Bdnf expression in rat skeletal muscle after acute or repeated exercise. Arch. Ital. Biol. 145, 99–110. doi: 10.4449/aib.v145i2.868
Czurko, A., Hirase, H., Csicsvari, J., and Buzsaki, G. (1999). Sustained activation of hippocampal pyramidal cells by ‘space clamping’ in a running wheel. Eur. J. Neurosci. 11, 344–352. doi: 10.1046/j.1460-9568.1999.00446.x
D’Anca, M., Fenoglio, C., Serpente, M., Arosio, B., Cesari, M., Scarpini, E. A., et al. (2019). Exosome determinants of physiological aging and age-related neurodegenerative diseases. Front. Aging Neurosci. 11:232. doi: 10.3389/fnagi.2019.00232
de Greeff, J. W., Bosker, R. J., Oosterlaan, J., Visscher, C., and Hartman, E. (2018). Effects of physical activity on executive functions, attention and academic performance in preadolescent children: a meta-analysis. J. Sci. Med. Sport 21, 501–507. doi: 10.1016/j.jsams.2017.09.595
De Miguel, Z., Khoury, N., Betley, M. J., Lehallier, B., Willoughby, D., Olsson, N., et al. (2021). Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 600, 494–499. doi: 10.1038/s41586-021-04183-x
Ding, Q., Vaynman, S., Akhavan, M., Ying, Z., and Gomez-Pinilla, F. (2006). Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 140, 823–833. doi: 10.1016/j.neuroscience.2006.02.084
Ding, Q., Ying, Z., and Gomez-Pinilla, F. (2011). Exercise influences hippocampal plasticity by modulating brain-derived neurotrophic factor processing. Neuroscience 192, 773–780. doi: 10.1016/j.neuroscience.2011.06.032
Ding, Y., Li, J., Luan, X., Ding, Y. H., Lai, Q., Rafols, J. A., et al. (2004). Exercise pre-conditioning reduces brain damage in ischemic rats that may be associated with regional angiogenesis and cellular overexpression of neurotrophin. Neuroscience 124, 583–591. doi: 10.1016/j.neuroscience.2003.12.029
Dougherty, K. D., Dreyfus, C. F., and Black, I. B. (2000). Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol. Dis. 7, 574–585. doi: 10.1006/nbdi.2000.0318
Dunn, A. L., Trivedi, M. H., Kampert, J. B., Clark, C. G., and Chambliss, H. O. (2005). Exercise treatment for depression: efficacy and dose response. Am. J. Prev. Med. 28, 1–8. doi: 10.1016/j.amepre.2004.09.003
Dun, S. L., Lyu, R. M., Chen, Y. H., Chang, J. K., Luo, J. J., and Dun, N. J. (2013). Irisin-immunoreactivity in neural and non-neural cells of the rodent. Neuroscience 240, 155–162. doi: 10.1016/j.neuroscience.2013.02.050
Egan, M. F., Kojima, M., Callicott, J. H., Goldberg, T. E., Kolachana, B. S., Bertolino, A., et al. (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cells 112, 257–269. doi: 10.1016/S0092-8674(03)00035-7
El Hayek, L., Khalifeh, M., Zibara, V., Abi Assaad, R., Emmanuel, N., Karnib, N., et al. (2019). Lactate mediates the effects of exercise on learning and memory through SIRT1-dependent activation of hippocampal brain-derived neurotrophic factor (BDNF). J. Neurosci. 39, 2369–2382. doi: 10.1523/JNEUROSCI.1661-18.2019
Endo, K., Matsukawa, K., Liang, N., Nakatsuka, C., Tsuchimochi, H., Okamura, H., et al. (2013). Dynamic exercise improves cognitive function in association with increased prefrontal oxygenation. J. Physiol. Sci. 63, 287–298. doi: 10.1007/s12576-013-0267-6
Endres, M., Gertz, K., Lindauer, U., Katchanov, J., Schultze, J., Schrock, H., et al. (2003). Mechanisms of stroke protection by physical activity. Ann. Neurol. 54, 582–590. doi: 10.1002/ana.10722
Erickson, K. I., Miller, D. L., and Roecklein, K. A. (2012). The aging hippocampus: interactions between exercise, depression, and BDNF. Neuroscientist 18, 82–97. doi: 10.1177/1073858410397054
Erickson, K. I., Voss, M. W., Prakash, R. S., Basak, C., Szabo, A., Chaddock, L., et al. (2011). Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. U. S. A. 108, 3017–3022. doi: 10.1073/pnas.1015950108
Ernfors, P., Ibanez, C. F., Ebendal, T., Olson, L., and Persson, H. (1990). Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc. Natl. Acad. Sci. U. S. A. 87, 5454–5458. doi: 10.1073/pnas.87.14.5454
Evans, M., Cogan, K. E., and Egan, B. (2017). Metabolism of ketone bodies during exercise and training: physiological basis for exogenous supplementation. J. Physiol. 595, 2857–2871. doi: 10.1113/JP273185
Fahimi, A., Baktir, M. A., Moghadam, S., Mojabi, F. S., Sumanth, K., McNerney, M. W., et al. (2017). Physical exercise induces structural alterations in the hippocampal astrocytes: exploring the role of BDNF-TrkB signaling. Brain Struct. Funct. 222, 1797–1808. doi: 10.1007/s00429-016-1308-8
Farmer, J., Zhao, X., van Praag, H., Wodtke, K., Gage, F. H., and Christie, B. R. (2004). Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 124, 71–79. doi: 10.1016/j.neuroscience.2003.09.029
Fenner, B. M. (2012). Truncated TrkB: beyond a dominant negative receptor. Cytokine Growth Factor Rev. 23, 15–24. doi: 10.1016/j.cytogfr.2012.01.002
Fernandez-Rodriguez, R., Alvarez-Bueno, C., Martinez-Ortega, I. A., Martinez-Vizcaino, V., Mesas, A. E., and Notario-Pacheco, B. (2022). Immediate effect of high-intensity exercise on brain-derived neurotrophic factor in healthy young adults: a systematic review and meta-analysis. J. Sport Health Sci. 11, 367–375. doi: 10.1016/j.jshs.2021.08.004
Foster, A. C., Vezzani, A., French, E. D., and Schwarcz, R. (1984). Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci. Lett. 48, 273–278. doi: 10.1016/0304-3940(84)90050-8
Friedman, W. J. (2000). Neurotrophins induce death of hippocampal neurons via the p75 receptor. J. Neurosci. 20, 6340–6346. doi: 10.1523/JNEUROSCI.20-17-06340.2000
Fukui, S., Schwarcz, R., Rapoport, S. I., Takada, Y., and Smith, Q. R. (1991). Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J. Neurochem. 56, 2007–2017. doi: 10.1111/j.1471-4159.1991.tb03460.x
Fulgenzi, G., Hong, Z., Tomassoni-Ardori, F., Barella, L. F., Becker, J., Barrick, C., et al. (2020). Novel metabolic role for BDNF in pancreatic beta-cell insulin secretion. Nat. Commun. 11:1950. doi: 10.1038/s41467-020-15833-5
Gaich, G., Chien, J. Y., Fu, H., Glass, L. C., Deeg, M. A., Holland, W. L., et al. (2013). The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 18, 333–340. doi: 10.1016/j.cmet.2013.08.005
Gano, L. B., Patel, M., and Rho, J. M. (2014). Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 55, 2211–2228. doi: 10.1194/jlr.R048975
Gao, J., Wang, W. Y., Mao, Y. W., Graff, J., Guan, J. S., Pan, L., et al. (2010). A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466, 1105–1109. doi: 10.1038/nature09271
Garcia, C., Chen, M. J., Garza, A. A., Cotman, C. W., and Russo-Neustadt, A. (2003). The influence of specific noradrenergic and serotonergic lesions on the expression of hippocampal brain-derived neurotrophic factor transcripts following voluntary physical activity. Neuroscience 119, 721–732. doi: 10.1016/S0306-4522(03)00192-1
Gelle, T., Samey, R. A., Plansont, B., Bessette, B., Jauberteau-Marchan, M. O., Lalloue, F., et al. (2021). BDNF and pro-BDNF in serum and exosomes in major depression: evolution after antidepressant treatment. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 109:110229. doi: 10.1016/j.pnpbp.2020.110229
Ghosh, A., and Greenberg, M. E. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268, 239–247. doi: 10.1126/science.7716515
Giannarelli, C., De Negri, F., Virdis, A., Ghiadoni, L., Cipriano, A., Magagna, A., et al. (2007). Nitric oxide modulates tissue plasminogen activator release in normotensive subjects and hypertensive patients. Hypertension 49, 878–884. doi: 10.1161/01.HYP.0000260471.16113.d8
Gil-Bea, F. J., Solas, M., Mateos, L., Winblad, B., Ramirez, M. J., and Cedazo-Minguez, A. (2011). Cholinergic hypofunction impairs memory acquisition possibly through hippocampal arc and BDNF downregulation. Hippocampus 21, 999–1009. doi: 10.1002/hipo.20812
Gomez-Pinilla, F., Zhuang, Y., Feng, J., Ying, Z., and Fan, G. (2011). Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur. J. Neurosci. 33, 383–390. doi: 10.1111/j.1460-9568.2010.07508.x
Griffin, E. W., Bechara, R. G., Birch, A. M., and Kelly, A. M. (2009). Exercise enhances hippocampal-dependent learning in the rat: evidence for a BDNF-related mechanism. Hippocampus 19, 973–980. doi: 10.1002/hipo.20631
Gulaj, E., Pawlak, K., Bien, B., and Pawlak, D. (2010). Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 55, 204–211. doi: 10.2478/v10039-010-0023-6
Guo, L. Y., Lozinski, B., and Yong, V. W. (2020). Exercise in multiple sclerosis and its models: focus on the central nervous system outcomes. J. Neurosci. Res. 98, 509–523. doi: 10.1002/jnr.24524
Guo, S., Kim, W. J., Lok, J., Lee, S. R., Besancon, E., Luo, B. H., et al. (2008). Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. U. S. A. 105, 7582–7587. doi: 10.1073/pnas.0801105105
Han, J., Yoon, S., and Park, H. (2021). Endocytic BDNF secretion regulated by Vamp3 in astrocytes. Sci. Rep. 11:21203. doi: 10.1038/s41598-021-00693-w
Hansen, J. S., Pedersen, B. K., Xu, G., Lehmann, R., Weigert, C., and Plomgaard, P. (2016). Exercise-induced secretion of FGF21 and Follistatin are blocked by pancreatic clamp and impaired in type 2 diabetes. J. Clin. Endocrinol. Metab. 101, 2816–2825. doi: 10.1210/jc.2016-1681
Hofer, M., Pagliusi, S. R., Hohn, A., Leibrock, J., and Barde, Y. A. (1990). Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 9, 2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x
Hogan-Cann, A. D., Lu, P., and Anderson, C. M. (2019). Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc. Natl. Acad. Sci. U. S. A. 116, 10229–10231. doi: 10.1073/pnas.1902647116
Holt, L. M., Hernandez, R. D., Pacheco, N. L., Torres Ceja, B., Hossain, M., and Olsen, M. L. (2019). Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. eLife 8:e44667. doi: 10.7554/eLife.44667
Honey, D., Wosnitzka, E., Klann, E., and Weinhard, L. (2022). Analysis of microglial BDNF function and expression in the motor cortex. Front. Cell. Neurosci. 16:961276. doi: 10.3389/fncel.2022.961276
Hong, E. J., McCord, A. E., and Greenberg, M. E. (2008). A biological function for the neuronal activity-dependent component of BDNF transcription in the development of cortical inhibition. Neuron 60, 610–624. doi: 10.1016/j.neuron.2008.09.024
Hook, G., Reinheckel, T., Ni, J., Wu, Z., Kindy, M., Peters, C., et al. (2022). Cathepsin B gene knockout improves behavioral deficits and reduces pathology in models of neurologic disorders. Pharmacol. Rev. 74, 600–629. doi: 10.1124/pharmrev.121.000527
Hopkins, M. E., Davis, F. C., Vantieghem, M. R., Whalen, P. J., and Bucci, D. J. (2012). Differential effects of acute and regular physical exercise on cognition and affect. Neuroscience 215, 59–68. doi: 10.1016/j.neuroscience.2012.04.056
Hopper, R. A., and Garthwaite, J. (2006). Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J. Neurosci. 26, 11513–11521. doi: 10.1523/JNEUROSCI.2259-06.2006
Horowitz, A. M., Fan, X., Bieri, G., Smith, L. K., Sanchez-Diaz, C. I., Schroer, A. B., et al. (2020). Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 369, 167–173. doi: 10.1126/science.aaw2622
Hsuchou, H., Pan, W., and Kastin, A. J. (2007). The fasting polypeptide FGF21 can enter brain from blood. Peptides 28, 2382–2386. doi: 10.1016/j.peptides.2007.10.007
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642. doi: 10.1146/annurev.biochem.72.121801.161629
Hu, E., Du, H., Zhu, X., Wang, L., Shang, S., Wu, X., et al. (2018). Beta-hydroxybutyrate promotes the expression of BDNF in hippocampal neurons under adequate glucose supply. Neuroscience 386, 315–325. doi: 10.1016/j.neuroscience.2018.06.036
Ieraci, A., Madaio, A. I., Mallei, A., Lee, F. S., and Popoli, M. (2016). Brain-derived neurotrophic factor Val66Met human polymorphism impairs the beneficial exercise-induced neurobiological changes in mice. Neuropsychopharmacology 41, 3070–3079. doi: 10.1038/npp.2016.120
Intlekofer, K. A., Berchtold, N. C., Malvaez, M., Carlos, A. J., McQuown, S. C., Cunningham, M. J., et al. (2013). Exercise and sodium butyrate transform a subthreshold learning event into long-term memory via a brain-derived neurotrophic factor-dependent mechanism. Neuropsychopharmacology 38, 2027–2034. doi: 10.1038/npp.2013.104
Ionescu-Tucker, A., Butler, C. W., Berchtold, N. C., Matheos, D. P., Wood, M. A., and Cotman, C. W. (2021). Exercise reduces H3K9me3 and regulates brain derived neurotrophic factor and GABRA2 in an age dependent manner. Front. Aging Neurosci. 13:798297. doi: 10.3389/fnagi.2021.798297
Ishitsuka, K., Ago, T., Arimura, K., Nakamura, K., Tokami, H., Makihara, N., et al. (2012). Neurotrophin production in brain pericytes during hypoxia: a role of pericytes for neuroprotection. Microvasc. Res. 83, 352–359. doi: 10.1016/j.mvr.2012.02.009
Islam, M. R., Valaris, S., Young, M. F., Haley, E. B., Luo, R., Bond, S. F., et al. (2021). Exercise hormone irisin is a critical regulator of cognitive function. Nat. Metab. 3, 1058–1070. doi: 10.1038/s42255-021-00438-z
Ivy, A. S., Rodriguez, F. G., Garcia, C., Chen, M. J., and Russo-Neustadt, A. A. (2003). Noradrenergic and serotonergic blockade inhibits BDNF mRNA activation following exercise and antidepressant. Pharmacol. Biochem. Behav. 75, 81–88. doi: 10.1016/S0091-3057(03)00044-3
Jahangiri, Z., Gholamnezhad, Z., and Hosseini, M. (2019). The effects of exercise on hippocampal inflammatory cytokine levels, brain oxidative stress markers and memory impairments induced by lipopolysaccharide in rats. Metab. Brain Dis. 34, 1157–1169. doi: 10.1007/s11011-019-00410-7
Jang, M., Gould, E., Xu, J., Kim, E. J., and Kim, J. H. (2019). Oligodendrocytes regulate presynaptic properties and neurotransmission through BDNF signaling in the mouse brainstem. eLife 8:e42156. doi: 10.7554/eLife.42156
Jiang, T., Zhang, L., Pan, X., Zheng, H., Chen, X., Li, L., et al. (2017). Physical exercise improves cognitive function together with microglia phenotype modulation and Remyelination in chronic cerebral Hypoperfusion. Front. Cell. Neurosci. 11:404. doi: 10.3389/fncel.2017.00404
Jimenez, V., Jambrina, C., Casana, E., Sacristan, V., Munoz, S., Darriba, S., et al. (2018). FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 10:e8y. doi: 10.15252/emmm.201708791
Kang, K., Xu, P., Wang, M., Chunyu, J., Sun, X., Ren, G., et al. (2020). FGF21 attenuates neurodegeneration through modulating neuroinflammation and oxidant-stress. Biomed. Pharmacother. 129:110439. doi: 10.1016/j.biopha.2020.110439
Kashiwaya, Y., Takeshima, T., Mori, N., Nakashima, K., Clarke, K., and Veech, R. L. (2000). D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 97, 5440–5444. doi: 10.1073/pnas.97.10.5440
Katz, L. C., and Shatz, C. J. (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138. doi: 10.1126/science.274.5290.1133
Kharitonenkov, A., Shiyanova, T. L., Koester, A., Ford, A. M., Micanovic, R., Galbreath, E. J., et al. (2005). FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635. doi: 10.1172/JCI23606
Kim, D. H., Kim, J. M., Park, S. J., Cai, M., Liu, X., Lee, S., et al. (2012). GABA(A) receptor blockade enhances memory consolidation by increasing hippocampal BDNF levels. Neuropsychopharmacology 37, 422–433. doi: 10.1038/npp.2011.189
Kim, H., Li, Q., Hempstead, B. L., and Madri, J. A. (2004). Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J. Biol. Chem. 279, 33538–33546. doi: 10.1074/jbc.M404115200
Kim, K. H., Kim, S. H., Min, Y. K., Yang, H. M., Lee, J. B., and Lee, M. S. (2013). Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS One 8:e63517. doi: 10.1371/journal.pone.0063517
Kimura, T., Kaneko, F., Iwamoto, E., Saitoh, S., and Yamada, T. (2019). Neuromuscular electrical stimulation increases serum brain-derived neurotrophic factor in humans. Exp. Brain Res. 237, 47–56. doi: 10.1007/s00221-018-5396-y
Kitamura, T., Mishina, M., and Sugiyama, H. (2003). Enhancement of neurogenesis by running wheel exercises is suppressed in mice lacking NMDA receptor epsilon 1 subunit. Neurosci. Res. 47, 55–63. doi: 10.1016/S0168-0102(03)00171-8
Konopka, W., Kiryk, A., Novak, M., Herwerth, M., Parkitna, J. R., Wawrzyniak, M., et al. (2010). MicroRNA loss enhances learning and memory in mice. J. Neurosci. 30, 14835–14842. doi: 10.1523/JNEUROSCI.3030-10.2010
Korte, M., Carroll, P., Wolf, E., Brem, G., Thoenen, H., and Bonhoeffer, T. (1995). Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. U. S. A. 92, 8856–8860. doi: 10.1073/pnas.92.19.8856
Krityakiarana, W., Espinosa-Jeffrey, A., Ghiani, C. A., Zhao, P. M., Topaldjikian, N., Gomez-Pinilla, F., et al. (2010). Voluntary exercise increases oligodendrogenesis in spinal cord. Int. J. Neurosci. 120, 280–290. doi: 10.3109/00207450903222741
Kuppers, E., and Beyer, C. (2001). Dopamine regulates brain-derived neurotrophic factor (BDNF) expression in cultured embryonic mouse striatal cells. Neuroreport 12, 1175–1179. doi: 10.1097/00001756-200105080-00025
Kurosawa, M., Okada, K., Sato, A., and Uchida, S. (1993). Extracellular release of acetylcholine, noradrenaline and serotonin increases in the cerebral cortex during walking in conscious rats. Neurosci. Lett. 161, 73–76. doi: 10.1016/0304-3940(93)90143-9
Laron, Z. (2001). Insulin-like growth factor 1 (IGF-1): a growth hormone. Mol. Pathol. 54, 311–316. doi: 10.1136/mp.54.5.311
Leventhal, C., Rafii, S., Rafii, D., Shahar, A., and Goldman, S. A. (1999). Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol. Cell. Neurosci. 13, 450–464. doi: 10.1006/mcne.1999.0762
Lev-Vachnish, Y., Cadury, S., Rotter-Maskowitz, A., Feldman, N., Roichman, A, Illouz, T., et al. (2019). L-Lactate Promotes Adult Hippocampal Neurogenesis. Front Neurosci. 24: 403. doi: 10.3389/fnins.2019.00403
Lewis, G. D., Farrell, L., Wood, M. J., Martinovic, M., Arany, Z., Rowe, G. C., et al. (2010). Metabolic signatures of exercise in human plasma. Sci. Transl. Med. 2:33ra37. doi: 10.1126/scitranslmed.3001006
Li, C., Wang, F., Miao, P., Yan, L., Liu, S., Wang, X., et al. (2020). miR-138 increases depressive-like behaviors by targeting SIRT1 in Hippocampus. Neuropsychiatr. Dis. Treat. 16, 949–957. doi: 10.2147/NDT.S237558
Li, F., Geng, X., Yun, H. J., Haddad, Y., Chen, Y., and Ding, Y. (2021). Neuroplastic effect of exercise through astrocytes activation and cellular crosstalk. Aging Dis. 12, 1644–1657. doi: 10.14336/AD.2021.0325
Lin, A. L., Zhang, W., Gao, X., and Watts, L. (2015). Caloric restriction increases ketone bodies metabolism and preserves blood flow in aging brain. Neurobiol. Aging 36, 2296–2303. doi: 10.1016/j.neurobiolaging.2015.03.012
Liu, Y. F., Chen, H. I., Yu, L., Kuo, Y. M., Wu, F. S., Chuang, J. I., et al. (2008). Upregulation of hippocampal TrkB and synaptotagmin is involved in treadmill exercise-enhanced aversive memory in mice. Neurobiol. Learn. Mem. 90, 81–89. doi: 10.1016/j.nlm.2008.02.005
Lopez-Lopez, C., LeRoith, D., and Torres-Aleman, I. (2004). Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc. Natl. Acad. Sci. U. S. A. 101, 9833–9838. doi: 10.1073/pnas.0400337101
Lourenco, M. V., Frozza, R. L., de Freitas, G. B., Zhang, H., Kincheski, G. C., Ribeiro, F. C., et al. (2019). Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 25, 165–175. doi: 10.1038/s41591-018-0275-4
Lu, B., Pang, P. T., and Woo, N. H. (2005). The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 6, 603–614. doi: 10.1038/nrn1726
Ludyga, S., Gerber, M., Puhse, U., Looser, V. N., and Kamijo, K. (2020). Systematic review and meta-analysis investigating moderators of long-term effects of exercise on cognition in healthy individuals. Nat. Hum. Behav. 4, 603–612. doi: 10.1038/s41562-020-0851-8
Luft, C., da Costa, M. S., Antunes, G. L., de Oliveira, J. R., and Donadio, M. V. F. (2022). The role of maternal exercise on placental, behavioral and genetic alterations induced by prenatal stress. Neurochem. Int. 158:105384. doi: 10.1016/j.neuint.2022.105384
Lu, L., Hogan-Cann, A. D., Globa, A. K., Lu, P., Nagy, J. I., Bamji, S. X., et al. (2019). Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 39, 481–496. doi: 10.1177/0271678X17734100
Madinier, A., Bertrand, N., Mossiat, C., Prigent-Tessier, A., Beley, A., Marie, C., et al. (2009). Microglial involvement in neuroplastic changes following focal brain ischemia in rats. PLoS One 4:e8101. doi: 10.1371/journal.pone.0008101
Maguire, G. A., and Gossner, A. (1995). Glycosyl phosphatidyl inositol phospholipase D activity in human serum. Ann. Clin. Biochem. 32, 74–78. doi: 10.1177/000456329503200107
Ma, H., Groth, R. D., Cohen, S. M., Emery, J. F., Li, B., Hoedt, E., et al. (2014). gammaCaMKII shuttles ca(2)(+)/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cells 159, 281–294. doi: 10.1016/j.cell.2014.09.019
Mandolesi, L., Polverino, A., Montuori, S., Foti, F., Ferraioli, G., Sorrentino, P., et al. (2018). Effects of physical exercise on cognitive functioning and wellbeing: biological and psychological benefits. Front. Psychol. 9:509. doi: 10.3389/fpsyg.2018.00509
Marie, C., Pedard, M., Quirie, A., Tessier, A., Garnier, P., Totoson, P., et al. (2018). Brain-derived neurotrophic factor secreted by the cerebral endothelium: a new actor of brain function? J. Cereb. Blood Flow Metab. 38, 935–949. doi: 10.1177/0271678X18766772
Markan, K. R., Naber, M. C., Ameka, M. K., Anderegg, M. D., Mangelsdorf, D. J., Kliewer, S. A., et al. (2014). Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63, 4057–4063. doi: 10.2337/db14-0595
Marosi, K., Kim, S. W., Moehl, K., Scheibye-Knudsen, M., Cheng, A., Cutler, R., et al. (2016). 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 139, 769–781. doi: 10.1111/jnc.13868
Martin, K. S., Azzolini, M., and Lira Ruas, J. (2020). The kynurenine connection: how exercise shifts muscle tryptophan metabolism and affects energy homeostasis, the immune system, and the brain. Am. J. Physiol. Cell Physiol. 318, C818–C830. doi: 10.1152/ajpcell.00580.2019
Matsukawa, K., Ishii, K., Liang, N., Endo, K., Ohtani, R., Nakamoto, T., et al. (2015). Increased oxygenation of the cerebral prefrontal cortex prior to the onset of voluntary exercise in humans. J. Appl. Physiol. 1985, 452–462. doi: 10.1152/japplphysiol.00406.2015
Matsumoto, Y., Tsunekawa, Y., Nomura, T., Suto, F., Matsumata, M., Tsuchiya, S., et al. (2011). Differential proliferation rhythm of neural progenitor and oligodendrocyte precursor cells in the young adult hippocampus. PLoS One 6:e27628. doi: 10.1371/journal.pone.0027628
Matthews, V. B., Astrom, M. B., Chan, M. H., Bruce, C. R., Krabbe, K. S., Prelovsek, O., et al. (2009). Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52, 1409–1418. doi: 10.1007/s00125-009-1364-1
Mee-Inta, O., Zhao, Z. W., and Kuo, Y. M. (2019). Physical exercise inhibits inflammation and microglial activation. Cells 8:691. doi: 10.3390/cells8070691
Meuchel, L. W., Thompson, M. A., Cassivi, S. D., Pabelick, C. M., and Prakash, Y. S. (2011). Neurotrophins induce nitric oxide generation in human pulmonary artery endothelial cells. Cardiovasc. Res. 91, 668–676. doi: 10.1093/cvr/cvr107
Miyamoto, T., Iwakura, T., Matsuoka, N., Iwamoto, M., Takenaka, M., Akamatsu, Y., et al. (2018). Impact of prolonged neuromuscular electrical stimulation on metabolic profile and cognition-related blood parameters in type 2 diabetes: A randomized controlled cross-over trial. Diabetes Res. Clin. Pract. 142, 37–45. doi: 10.1016/j.diabres.2018.05.032
Monnier, A., Garnier, P., Quirie, A., Pernet, N., Demougeot, C., Marie, C., et al. (2017a). Effect of short-term exercise training on brain-derived neurotrophic factor signaling in spontaneously hypertensive rats. J. Hypertens. 35, 279–290. doi: 10.1097/HJH.0000000000001164
Monnier, A., Prigent-Tessier, A., Quirie, A., Bertrand, N., Savary, S., Gondcaille, C., et al. (2017b). Brain-derived neurotrophic factor of the cerebral microvasculature: a forgotten and nitric oxide-dependent contributor of brain-derived neurotrophic factor in the brain. Acta Physiol (Oxf.) 219, 790–802. doi: 10.1111/apha.12743
Moon, H. Y., Becke, A., Berron, D., Becker, B., Sah, N., Benoni, G., et al. (2016). Running-induced systemic Cathepsin B secretion is associated with memory function. Cell Metab. 24, 332–340. doi: 10.1016/j.cmet.2016.05.025
Mousavi, K., and Jasmin, B. J. (2006). BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J. Neurosci. 26, 5739–5749. doi: 10.1523/JNEUROSCI.5398-05.2006
Mowla, S. J., Farhadi, H. F., Pareek, S., Atwal, J. K., Morris, S. J., Seidah, N. G., et al. (2001). Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J. Biol. Chem. 276, 12660–12666. doi: 10.1074/jbc.M008104200
Mu, A., Wales, T. E., Zhou, H., Draga-Coleta, S. V., Gorgulla, C., Blackmore, K. A., et al. (2023). Irisin acts through its integrin receptor in a two-step process involving extracellular Hsp90alpha. Mol. Cell 83:e12
Muller, P., Duderstadt, Y., Lessmann, V., and Muller, N. G. (2020). Lactate and BDNF: key mediators of exercise induced neuroplasticity? J. Clin. Med. 9:1136. doi: 10.3390/jcm9041136
Myint, A. M., Kim, Y. K., Verkerk, R., Scharpe, S., Steinbusch, H., and Leonard, B. (2007). Kynurenine pathway in major depression: evidence of impaired neuroprotection. J. Affect. Disord. 98, 143–151. doi: 10.1016/j.jad.2006.07.013
Nagappan, G., Zaitsev, E., Senatorov, V. V. Jr., Yang, J., Hempstead, B. L., and Lu, B. (2009). Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc. Natl. Acad. Sci. U. S. A. 106, 1267–1272. doi: 10.1073/pnas.0807322106
Nakahashi, T., Fujimura, H., Altar, C. A., Li, J., Kambayashi, J., Tandon, N. N., et al. (2000). Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett. 470, 113–117. doi: 10.1016/S0014-5793(00)01302-8
Nakanishi, H. (2020). Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen. Res. 15, 25–29. doi: 10.4103/1673-5374.264444
Nakata, H., and Nakamura, S. (2007). Brain-derived neurotrophic factor regulates AMPA receptor trafficking to post-synaptic densities via IP3R and TRPC calcium signaling. FEBS Lett. 581, 2047–2054. doi: 10.1016/j.febslet.2007.04.041
Navaratna, D., Guo, S. Z., Hayakawa, K., Wang, X., Gerhardinger, C., and Lo, E. H. (2011). Decreased cerebrovascular brain-derived neurotrophic factor-mediated neuroprotection in the diabetic brain. Diabetes 60, 1789–1796. doi: 10.2337/db10-1371
Neeper, S. A., Gomez-Pinilla, F., Choi, J., and Cotman, C. (1995). Exercise and brain neurotrophins. Nature 373:109. doi: 10.1038/373109a0
Newman, J. C., Covarrubias, A. J., Zhao, M., Yu, X., Gut, P., Ng, C. P., et al. (2017). Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 26:e8. doi: 10.1016/j.cmet.2017.08.004
Nicole, O., Docagne, F., Ali, C., Margaill, I., Carmeliet, P., MacKenzie, E. T., et al. (2001). The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat. Med. 7, 59–64. doi: 10.1038/83358
Nishijima, T., Okamoto, M., Matsui, T., Kita, I., and Soya, H. (2012). Hippocampal functional hyperemia mediated by NMDA receptor/NO signaling in rats during mild exercise. J. Appl. Physiol. 1985, 197–203. doi: 10.1152/japplphysiol.00763.2011
Nishijima, T., Piriz, J., Duflot, S., Fernandez, A. M., Gaitan, G., Gomez-Pinedo, U., et al. (2010). Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 67, 834–846. doi: 10.1016/j.neuron.2010.08.007
Nishijima, T., and Soya, H. (2006). Evidence of functional hyperemia in the rat hippocampus during mild treadmill running. Neurosci. Res. 54, 186–191. doi: 10.1016/j.neures.2005.11.005
Noga, O., Peiser, M., Altenahr, M., Knieling, H., Wanner, R., Hanf, G., et al. (2007). Differential activation of dendritic cells by nerve growth factor and brain-derived neurotrophic factor. Clin. Exp. Allergy 37, 1701–1708. doi: 10.1111/j.1365-2222.2007.02832.x
Ogborn, D. I., and Gardiner, P. F. (2010). Effects of exercise and muscle type on BDNF, NT-4/5, and TrKB expression in skeletal muscle. Muscle Nerve 41, 385–391. doi: 10.1002/mus.21503
O’Kusky, J. R., Ye, P., and D’Ercole, A. J. (2000). Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J. Neurosci. 20, 8435–8442. doi: 10.1523/JNEUROSCI.20-22-08435.2000
Oliff, H. S., Berchtold, N. C., Isackson, P., and Cotman, C. W. (1998). Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Brain Res. Mol. Brain Res. 61, 147–153. doi: 10.1016/S0169-328X(98)00222-8
Paillard, T., Rolland, Y., and de Souto Barreto, P. (2015). Protective effects of physical exercise in Alzheimer’s disease and Parkinson’s disease: a narrative review. J. Clin. Neurol. 11, 212–219. doi: 10.3988/jcn.2015.11.3.212
Palasz, E., Niewiadomski, W., Gasiorowska, A., Mietelska-Porowska, A., and Niewiadomska, G. (2019). Neuroplasticity and neuroprotective effect of treadmill training in the chronic mouse model of Parkinson’s disease. Neural Plast. 2019:8215017. doi: 10.1155/2019/8215017
Pang, P. T., Teng, H. K., Zaitsev, E., Woo, N. T., Sakata, K., Zhen, S., et al. (2004). Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306, 487–491. doi: 10.1126/science.1100135
Pan-Vazquez, A., Rye, N., Ameri, M., McSparron, B., Smallwood, G., Bickerdyke, J., et al. (2015). Impact of voluntary exercise and housing conditions on hippocampal glucocorticoid receptor, miR-124 and anxiety. Mol. Brain 8:40. doi: 10.1186/s13041-015-0128-8
Pan, W., and Kastin, A. J. (2000). Interactions of IGF-1 with the blood-brain barrier in vivo and in situ. Neuroendocrinology 72, 171–178. doi: 10.1159/000054584
Paoli, A., Bianco, A., Damiani, E., and Bosco, G. (2014). Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed. Res. Int. 2014:474296. doi: 10.1155/2014/474296
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. 3rd, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cells 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Pedard, M., Cefis, M., Ennequin, G., Quirie, A., Garnier, P., Prigent-Tessier, A., et al. (2019). Brain-derived neurotrophic factor pathway after downhill and uphill training in rats. Med. Sci. Sports Exerc. 51, 27–34. doi: 10.1249/MSS.0000000000001771
Pedard, M., Quirie, A., Garnier, P., Tessier, A., Demougeot, C., and Marie, C. (2017). The cerebral brain-derived neurotrophic factor pathway, either neuronal or endothelial, is impaired in rats with adjuvant-induced arthritis. Connection with endothelial dysfunction. Front. Physiol. 8:1125. doi: 10.3389/fphys.2017.01125
Pedersen, B. K., Steensberg, A., Fischer, C., Keller, C., Keller, P., Plomgaard, P., et al. (2003). Searching for the exercise factor: is IL-6 a candidate? J. Muscle Res. Cell Motil. 24, 113–119. doi: 10.1023/A:1026070911202
Poo, M. M. (2001). Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2, 24–32. doi: 10.1038/35049004
Prigent-Tessier, A., Quirie, A., Maguin-Gate, K., Szostak, J., Mossiat, C., Nappey, M., et al. (2013). Physical training and hypertension have opposite effects on endothelial brain-derived neurotrophic factor expression. Cardiovasc. Res. 100, 374–382. doi: 10.1093/cvr/cvt219
Proia, P., Di Liegro, C. M., Schiera, G., Fricano, A., and Di Liegro, I. (2016). Lactate as a metabolite and a regulator in the central nervous system. Int. J. Mol. Sci. 17:1450. doi: 10.3390/ijms17091450
Pruunsild, P., Sepp, M., Orav, E., Koppel, I., and Timmusk, T. (2011). Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene. J. Neurosci. 31, 3295–3308. doi: 10.1523/JNEUROSCI.4540-10.2011
Quesseveur, G., David, D. J., Gaillard, M. C., Pla, P., Wu, M. V., Nguyen, H. T., et al. (2013). BDNF overexpression in mouse hippocampal astrocytes promotes local neurogenesis and elicits anxiolytic-like activities. Transl. Psychiatry 3:e253. doi: 10.1038/tp.2013.30
Quirie, A., Hervieu, M., Garnier, P., Demougeot, C., Mossiat, C., Bertrand, N., et al. (2012). Comparative effect of treadmill exercise on mature BDNF production in control versus stroke rats. PLoS One 7:e44218. doi: 10.1371/journal.pone.0044218
Radak, Z., Suzuki, K., Higuchi, M., Balogh, L., Boldogh, I., and Koltai, E. (2016). Physical exercise, reactive oxygen species and neuroprotection. Free Radic. Biol. Med. 98, 187–196. doi: 10.1016/j.freeradbiomed.2016.01.024
Raschke, S., Elsen, M., Gassenhuber, H., Sommerfeld, M., Schwahn, U., Brockmann, B., et al. (2013). Evidence against a beneficial effect of irisin in humans. PLoS One 8:e73680. doi: 10.1371/journal.pone.0073680
Rasmussen, P., Brassard, P., Adser, H., Pedersen, M. V., Leick, L., Hart, E., et al. (2009). Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 94, 1062–1069. doi: 10.1113/expphysiol.2009.048512
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 361, 1545–1564. doi: 10.1098/rstb.2006.1894
Rodriguez, J. J., Terzieva, S., Olabarria, M., Lanza, R. G., and Verkhratsky, A. (2013). Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis. 4:e678. doi: 10.1038/cddis.2013.194
Rose, C. R., Blum, R., Pichler, B., Lepier, A., Kafitz, K. W., and Konnerth, A. (2003). Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature 426, 74–78. doi: 10.1038/nature01983
Rosenfeld, R. D., Zeni, L., Haniu, M., Talvenheimo, J., Radka, S. F., Bennett, L., et al. (1995). Purification and identification of brain-derived neurotrophic factor from human serum. Protein Expr. Purif. 6, 465–471. doi: 10.1006/prep.1995.1062
Rubanyi, G. M., Romero, J. C., and Vanhoutte, P. M. (1986). Flow-induced release of endothelium-derived relaxing factor. Am. J. Phys. 250, H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145
Rudge, J. S., Li, Y., Pasnikowski, E. M., Mattsson, K., Pan, L., Yancopoulos, G. D., et al. (1994). Neurotrophic factor receptors and their signal transduction capabilities in rat astrocytes. Eur. J. Neurosci. 6, 693–705. doi: 10.1111/j.1460-9568.1994.tb00981.x
Russo-Neustadt, A. A., Alejandre, H., Garcia, C., Ivy, A. S., and Chen, M. J. (2004). Hippocampal brain-derived neurotrophic factor expression following treatment with reboxetine, citalopram, and physical exercise. Neuropsychopharmacology 29, 2189–2199. doi: 10.1038/sj.npp.1300514
Sa-Nguanmoo, P., Tanajak, P., Kerdphoo, S., Satjaritanun, P., Wang, X., Liang, G., et al. (2016). FGF21 improves cognition by restored synaptic plasticity, dendritic spine density, brain mitochondrial function and cell apoptosis in obese-insulin resistant male rats. Horm. Behav. 85, 86–95. doi: 10.1016/j.yhbeh.2016.08.006
Santhanam, A. V., Smith, L. A., and Katusic, Z. S. (2010). Brain-derived neurotrophic factor stimulates production of prostacyclin in cerebral arteries. Stroke 41, 350–356. doi: 10.1161/STROKEAHA.109.564492
Schmidt, H. D., and Duman, R. S. (2010). Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology 35, 2378–2391. doi: 10.1038/npp.2010.114
Schoenfeld, T. J., Rada, P., Pieruzzini, P. R., Hsueh, B., and Gould, E. (2013). Physical exercise prevents stress-induced activation of granule neurons and enhances local inhibitory mechanisms in the dentate gyrus. J. Neurosci. 33, 7770–7777. doi: 10.1523/JNEUROSCI.5352-12.2013
Schwarcz, R., Bruno, J. P., Muchowski, P. J., and Wu, H. Q. (2012). Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477. doi: 10.1038/nrn3257
Shen, H., Tong, L., Balazs, R., and Cotman, C. W. (2001). Physical activity elicits sustained activation of the cyclic AMP response element-binding protein and mitogen-activated protein kinase in the rat hippocampus. Neuroscience 107, 219–229. doi: 10.1016/S0306-4522(01)00315-3
Skwarzynska, D., Sun, H., Williamson, J., Kasprzak, I., and Kapur, J. (2023). Glycolysis regulates neuronal excitability via lactate receptor, HCA1R. Brain 146, 1888–1902. doi: 10.1093/brain/awac419
Sleiman, S. F., Henry, J., Al-Haddad, R., El Hayek, L., Abou Haidar, E., Stringer, T., et al. (2016). Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. eLife 5:e15092. doi: 10.7554/eLife.15092
Sodhi, R. K., Bansal, Y., Singh, R., Saroj, P., Bhandari, R., Kumar, B., et al. (2021). IDO-1 inhibition protects against neuroinflammation, oxidative stress and mitochondrial dysfunction in 6-OHDA induced murine model of Parkinson’s disease. Neurotoxicology 84, 184–197. doi: 10.1016/j.neuro.2021.03.009
Sofi, F., Valecchi, D., Bacci, D., Abbate, R., Gensini, G. F., Casini, A., et al. (2011). Physical activity and risk of cognitive decline: a meta-analysis of prospective studies. J. Intern. Med. 269, 107–117. doi: 10.1111/j.1365-2796.2010.02281.x
Sprenger, H., Jacobs, C., Nain, M., Gressner, A. M., Prinz, H., Wesemann, W., et al. (1992). Enhanced release of cytokines, interleukin-2 receptors, and neopterin after long-distance running. Clin. Immunol. Immunopathol. 63, 188–195. doi: 10.1016/0090-1229(92)90012-D
Stoilov, P., Castren, E., and Stamm, S. (2002). Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochem. Biophys. Res. Commun. 290, 1054–1065. doi: 10.1006/bbrc.2001.6301
Sundaram, G., Lim, C. K., and Brew, B. J. (2020). Kynurenine pathway modulation reverses the experimental autoimmune encephalomyelitis mouse disease progression. J Neuroinflammation 17, 176. doi: 10.1186/s12974-020-01844-y
Sun, P., Enslen, H., Myung, P. S., and Maurer, R. A. (1994). Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 8, 2527–2539. doi: 10.1101/gad.8.21.2527
Sutoo, D., and Akiyama, K. (2003). Regulation of brain function by exercise. Neurobiol. Dis. 13, 1–14. doi: 10.1016/S0969-9961(03)00030-5
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cells 144, 810–823. doi: 10.1016/j.cell.2011.02.018
Suzuki, M., Suzuki, M., Kitamura, Y., Mori, S., Sato, K., Dohi, S., et al. (2002). Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn. J. Pharmacol. 89, 36–43. doi: 10.1254/jjp.89.36
Takeo, C., Nakamura, S., Tanaka, T., Uchida, D., Noguchi, Y., Nagao, T., et al. (2003). Rat cerebral endothelial cells express trk C and are regulated by neurotrophin-3. Biochem. Biophys. Res. Commun. 305, 400–406. doi: 10.1016/S0006-291X(03)00770-8
Tao, X., Finkbeiner, S., Arnold, D. B., Shaywitz, A. J., and Greenberg, M. E. (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726. doi: 10.1016/S0896-6273(00)81010-7
Tao, X., West, A. E., Chen, W. G., Corfas, G., and Greenberg, M. E. (2002). A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron 33, 383–395. doi: 10.1016/S0896-6273(01)00561-X
Timmusk, T., Palm, K., Metsis, M., Reintam, T., Paalme, V., Saarma, M., et al. (1993). Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 10, 475–489. doi: 10.1016/0896-6273(93)90335-O
Totoson, P., Santini, C., Prigent-Tessier, A., Marie, C., and Demougeot, C. (2021). Endothelial TrkB receptor activation controls vascular tone of rat middle cerebral artery. Vasc. Pharmacol. 141:106930. doi: 10.1016/j.vph.2021.106930
Trejo, J. L., Carro, E., and Torres-Aleman, I. (2001). Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J. Neurosci. 21, 1628–1634. doi: 10.1523/JNEUROSCI.21-05-01628.2001
Uda, M., Ishido, M., Kami, K., and Masuhara, M. (2006). Effects of chronic treadmill running on neurogenesis in the dentate gyrus of the hippocampus of adult rat. Brain Res. 1104, 64–72. doi: 10.1016/j.brainres.2006.05.066
Vanderwolf, C. H. (1969). Hippocampal electrical activity and voluntary movement in the rat. Electroencephalogr. Clin. Neurophysiol. 26, 407–418. doi: 10.1016/0013-4694(69)90092-3
van Praag, H., Christie, B. R., Sejnowski, T. J., and Gage, F. H. (1999). Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. U. S. A. 96, 13427–13431. doi: 10.1073/pnas.96.23.13427
Varendi, K., Kumar, A., Harma, M. A., and Andressoo, J. O. (2014). miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell. Mol. Life Sci. 71, 4443–4456. doi: 10.1007/s00018-014-1628-x
Vaynman, S., Ying, Z., and Gomez-Pinilla, F. (2003). Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience 122, 647–657. doi: 10.1016/j.neuroscience.2003.08.001
Vaynman, S., Ying, Z., and Gomez-Pinilla, F. (2004). Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 20, 2580–2590. doi: 10.1111/j.1460-9568.2004.03720.x
Vignoli, B., Battistini, G., Melani, R., Blum, R., Santi, S., Berardi, N., et al. (2016). Peri-synaptic glia recycles brain-derived neurotrophic factor for LTP stabilization and memory retention. Neuron 92, 873–887. doi: 10.1016/j.neuron.2016.09.031
Voss, M. W., Nagamatsu, L. S., Liu-Ambrose, T., and Kramer, A. F. (2011). Exercise, brain, and cognition across the life span. J. Appl. Physiol. 1985, 1505–1513. doi: 10.1152/japplphysiol.00210.2011
Walsh, E. I., Smith, L., Northey, J., Rattray, B., and Cherbuin, N. (2020). Towards an understanding of the physical activity-BDNF-cognition triumvirate: A review of associations and dosage. Ageing Res. Rev. 60:101044. doi: 10.1016/j.arr.2020.101044
Wang, B. L., Jin, H., Han, X. Q., Xia, Y., and Liu, N. F. (2018). Involvement of brain-derived neurotrophic factor in exercise-induced cardioprotection of post-myocardial infarction rats. Int. J. Mol. Med. 42, 2867–2880. doi: 10.3892/ijmm.2018.3841
Wang, K., Song, F., Xu, K., Liu, Z., Han, S., Li, F., et al. (2019). Irisin attenuates Neuroinflammation and prevents the memory and cognitive deterioration in Streptozotocin-induced diabetic mice. Mediat. Inflamm. 2019:1567179. doi: 10.1155/2019/1567179
Watanabe, S., Sudo, Y., Makino, T., Kimura, S., Tomita, K., Noguchi, M., et al. (2022). Skeletal muscle releases extracellular vesicles with distinct protein and microRNA signatures that function in the muscle microenvironment. PNAS Nexus 1:173. doi: 10.1093/pnasnexus/pgac173
West, A. E., Chen, W. G., Dalva, M. B., Dolmetsch, R. E., Kornhauser, J. M., Shaywitz, A. J., et al. (2001). Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U. S. A. 98, 11024–11031. doi: 10.1073/pnas.191352298
Williams, S. N., and Undieh, A. S. (2009). Dopamine D1-like receptor activation induces brain-derived neurotrophic factor protein expression. Neuroreport 20, 606–610. doi: 10.1097/WNR.0b013e32832a0a98
Wrann, C. D., White, J. P., Salogiannnis, J., Laznik-Bogoslavski, D., Wu, J., Ma, D., et al. (2013). Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 18, 649–659. doi: 10.1016/j.cmet.2013.09.008
Wu, G. Y., Deisseroth, K., and Tsien, R. W. (2001). Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. U. S. A. 98, 2808–2813. doi: 10.1073/pnas.051634198
Xiong, J. Y., Li, S. C., Sun, Y. X., Zhang, X. S., Dong, Z. Z., Zhong, P., et al. (2015). Long-term treadmill exercise improves spatial memory of male APPswe/PS1dE9 mice by regulation of BDNF expression and microglia activation. Biol. Sport 32, 295–300. doi: 10.5604/20831862.1163692
Xu, L., Liu, R., Qin, Y., and Wang, T. (2023). Brain metabolism in Alzheimer’s disease: biological mechanisms of exercise. Transl. Neurodegener. 12:33. doi: 10.1186/s40035-023-00364-y
Yakar, S., Courtland, H. W., and Clemmons, D. (2010). IGF-1 and bone: new discoveries from mouse models. J. Bone Miner. Res. 25, 2543–2552. doi: 10.1002/jbmr.234
Yakar, S., Liu, J. L., Stannard, B., Butler, A., Accili, D., Sauer, B., et al. (1999). Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. U. S. A. 96, 7324–7329. doi: 10.1073/pnas.96.13.7324
Yang, J., Ruchti, E., Petit, J. M., Jourdain, P., Grenningloh, G., Allaman, I., et al. (2014). Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. U. S. A. 111, 12228–12233. doi: 10.1073/pnas.1322912111
Yang, X., Brobst, D., Chan, W. S., Tse, M. C. L., Herlea-Pana, O., Ahuja, P., et al. (2019). Muscle-generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci. Signal. 12. doi: 10.1126/scisignal.aau1468
Yuan, D., Zhao, Y., Banks, W. A., Bullock, K. M., Haney, M., Batrakova, E., et al. (2017). Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 142, 1–12. doi: 10.1016/j.biomaterials.2017.07.011
Yu, T., Chang, Y., Gao, X. L., Li, H., and Zhao, P. (2017). Dynamic expression and the role of BDNF in exercise-induced skeletal muscle regeneration. Int. J. Sports Med. 38, 959–966. doi: 10.1055/s-0043-118343
Yu, Y., Bai, F., Wang, W., Liu, Y., Yuan, Q., Qu, S., et al. (2015). Fibroblast growth factor 21 protects mouse brain against D-galactose induced aging via suppression of oxidative stress response and advanced glycation end products formation. Pharmacol. Biochem. Behav. 133, 122–131. doi: 10.1016/j.pbb.2015.03.020
Keywords: physical exercise, BDNF, exerkine, cognition, muscle, neuronal activity, cerebral blood flow, hemodynamic
Citation: Cefis M, Chaney R, Wirtz J, Méloux A, Quirié A, Leger C, Prigent-Tessier A and Garnier P (2023) Molecular mechanisms underlying physical exercise-induced brain BDNF overproduction. Front. Mol. Neurosci. 16:1275924. doi: 10.3389/fnmol.2023.1275924
Edited by:
George Smith, Temple University, United StatesReviewed by:
Marta Nowacka-Chmielewska, Jerzy Kukuczka Academy of Physical Education in Katowice, PolandCopyright © 2023 Cefis, Chaney, Wirtz, Méloux, Quirié, Leger, Prigent-Tessier and Garnier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Cefis, Q2VmaXMubWFyaW5hQGNvdXJyaWVyLnVxYW0uY2E=; Philippe Garnier, UGhpbGlwcGUuR2FybmllckBpdXQtZGlqb24udS1ib3VyZ29nbmUuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.