 Jaime Eugenín
Jaime Eugenín Laura Eugenín-von Bernhardi
Laura Eugenín-von Bernhardi Rommy von Bernhardi
Rommy von Bernhardi- 1Facultad de Química y Biología, Departamento de Biología, Universidad de Santiago de Chile, USACH, Santiago, Chile
- 2Complejo Asistencial Hospital Dr. Sótero del Río, Santiago, Chile
- 3Facultad de Ciencias para el Cuidado de la Salud, Universidad San Sebastián, Santiago, Chile
The chemokine fractalkine (FKN, CX3CL1), a member of the CX3C subfamily, contributes to neuron–glia interaction and the regulation of microglial cell activation. Fractalkine is expressed by neurons as a membrane-bound protein (mCX3CL1) that can be cleaved by extracellular proteases generating several sCX3CL1 forms. sCX3CL1, containing the chemokine domain, and mCX3CL1 have high affinity by their unique receptor (CX3CR1) which, physiologically, is only found in microglia, a resident immune cell of the CNS. The activation of CX3CR1contributes to survival and maturation of the neural network during development, glutamatergic synaptic transmission, synaptic plasticity, cognition, neuropathic pain, and inflammatory regulation in the adult brain. Indeed, the various CX3CL1 forms appear in some cases to serve an anti-inflammatory role of microglia, whereas in others, they have a pro-inflammatory role, aggravating neurological disorders. In the last decade, evidence points to the fact that sCX3CL1 and mCX3CL1 exhibit selective and differential effects on their targets. Thus, the balance in their level and activity will impact on neuron–microglia interaction. This review is focused on the description of factors determining the emergence of distinct fractalkine forms, their age-dependent changes, and how they contribute to neuroinflammation and neurodegenerative diseases. Changes in the balance among various fractalkine forms may be one of the mechanisms on which converge aging, chronic CNS inflammation, and neurodegeneration.
1. Introduction
The aging of the population is a major challenge in public health because aging is the main risk factor for many chronic diseases (Liguori et al., 2018), including neurodegenerative diseases (Hou et al., 2019). Additionally, demographic projections show that the elderly population is growing at an unprecedented rate (Partridge et al., 2018). A relevant feature of physiological aging is a low-grade chronic systemic inflammation. This has been called “inflamm-aging,” a condition that increases, per se, the mortality and morbidity among older adults (Franceschi et al., 2000).
Several age-related changes in neural function are associated with glial dysregulation (von Bernhardi, 2007). Age-related changes in glial cells functions can exert beneficial or detrimental influence onto neuronal circuits and, therefore, modify the course of the aging process (Lynch et al., 2010). However, the role of glial cells on the function/dysfunction of the aged central nervous system (CNS) is still poorly understood. For instance, glia may contribute to the age-related chronic inflammatory state of the nervous system (Franceschi et al., 2000, 2007; Franceschi, 2007). That is, glia can contribute to create a “pathological environment” providing deleterious factors that acting synergistically, can favor neuronal death and neurodegeneration (von Bernhardi and Eugenin, 2012). These changes can be crucial in cognitive impairment, failure of vital functions, and they can be part of the pathogenesis of highly prevalent CNS pathologies like neurodegenerative and neuropsychiatric diseases (Wu et al., 2015; Hong et al., 2016).
Therefore, it is not a surprise that changes or impairment of neuron-glial signaling will impact neuronal and glial properties at multiple levels, including synapse integrity and plasticity, network excitability, up to animal behavior (Wu et al., 2015). Here we will discuss age-dependent changes in fractalkine (FKN, CX3CL1) signaling, on the balance among soluble (sCX3CL1) vs. membrane-bound CX3CL1 (mCX3CL1) forms, and how changes in this balance contributes to switching microglia from an anti-inflammatory to a pro-inflammatory status, a key mechanism likely involved in chronic CNS inflammation and neurodegenerative diseases.
1.1. Aging
Aging affects cells, organs, and functions in various ways, particularly in the CNS (Ramirez et al., 2008; von Bernhardi et al., 2019). Aging is characterized by a low-grade inflammatory condition and the progressive deterioration of various physiological functions of the organism, being recognized as the most robust risk factor for neurodegenerative diseases like Alzheimer’s disease (AD) (Guerreiro and Bras, 2015). The onset of the changes characteristic of diseases associated with aging is unclear (Lynch and Smith, 2005). Often, neurodegenerative diseases develop progressively, for many years before their clinical manifestations. For instance, in a patient diagnosed with AD at age 75, pathophysiological changes started when the person was 50–55 years old. Aged individuals have also changes on the immune system, known as immune senescence (Jones et al., 2010), characterized by decreased adaptive immunity, making the subject more susceptible to infections and tumors, and exacerbated innate immune function (von Bernhardi et al., 2019) favoring chronic neuroinflammation, which appears to be also associated with the appearance of neurodegenerative pathologies (Block et al., 2007; von Bernhardi, 2007; Gao and Hong, 2008).
1.2. Age-dependent changes of brain cells
A functional hallmark of brain aging (Gonzales et al., 2022), often occurring in the absence of neurological disease, is cognitive impairment, especially some memory and learning deficits. Although cell loss is minimal in most brain regions, in regions such as the hippocampus it may reach from 10 to 60% (Coleman and Flood, 1987). Functional decline can also be the result of exacerbated synapse pruning depending on the dysregulation of astrocytes and microglia (Paolicelli et al., 2011; Faust et al., 2021).
1.2.1. Neurons
Aging in humans is associated with a reduction in total brain volume (Hedman et al., 2012), being the reduction of gray matter volume more pronounced than that of white matter (Ge et al., 2002). The annual rate of volume loss is about 0.2% after 35 years of age, which increases to 0.5% at age of 60, becoming over 0.5% after 60 (Hedman et al., 2012). Age-dependent cortical thinning, which is observed across most brain regions, is more accentuated in the frontal lobe (Raz et al., 1997; Salat et al., 2004). Although decline in hippocampus CA1 region, hilus, and subiculum, volumes appear to be a consequence of neuronal loss, in other regions, like the frontal cortex, the number of neurons are relatively preserved (August et al., 2022).
The brain undergoes a myriad of changes during aging that can lead to an atrophic aged brain. Neurons show dendrite retraction, especially in the prefrontal cortex and hippocampus. With increasing age, dendrites shrink, their branching become less complex, and lose spines (Dickstein et al., 2013), affecting inter-neuronal connections. There are also age-dependent changes in the synthesis of neurotransmitters. For instance, older people brains synthesize less dopamine, and have fewer receptors (Yin et al., 2014). Older adults with mild cognitive impairment appear to have less serotonin (Yin et al., 2014), which could potentiate memory loss. Functional effects are especially relevant for processes that require a high degree of synaptic plasticity. The number of synapses is reduced, which can affect learning and memory, facilitating cognitive decline. Although synaptic changes are selective and mild, they appear to affect cognitive decline beyond their changes in structure and neurotransmitters. The myelin wrapping of axons thins out (Bender et al., 2016), reducing the speed of propagation of action potentials, and impacting neuronal communication. Neurogenesis also declines with age (Li Puma et al., 2020); interestingly, in humans and rodents, physiological strategies that boost neurogenesis, such as regular exercise, can improve cognitive function (Ma et al., 2017). Studies on adult hippocampal neurogenesis in AD mouse models show a severe decline of neurogenesis before cognitive impairment (Li Puma et al., 2020).
Neurons are post-mitotic cells, arrested in the G0 phase of the cell cycle. Therefore, recognition of neuronal senescence cannot be performed on basis of proliferation arrest. A growing body of evidence indicates that cell senescence-like changes in neurons can be observed in aging brains. These characteristics involve shortening of telomeres (Ain et al., 2018), senescence-associated secretory phenotype (SASP), altered morphology and proteostasis, propensity to undergo apoptosis, autophagy impairment, accumulation of lipid droplets, increased activity of senescence-associated-b-galactosidase (SA-b-gal), and epigenetic alterations, including DNA methylation, chromatin remodeling, and histone post-translational modifications that affect gene expression (Di Micco et al., 2021; Sikora et al., 2021; Sahu et al., 2022).
1.2.2. Oligodendrocytes
Oligodendrocytes are the myelinating cells of the CNS, generated from oligodendrocyte progenitor cells (OPCs), also known as NG2 cells, because they express the proteoglycan, NG2 (Bradl and Lassmann, 2010; Kuhn et al., 2019). The density of NG2(+) cells in white matter is 1.5-fold higher than that in gray matter (Dawson et al., 2003) and their morphology in the mature CNS vary from region to region (Hughes et al., 2013). NG2 cells maintain their migrating, proliferating, and differentiating capacities throughout life to generate oligodendrocytes and regenerate myelin (Young et al., 2013). In the white matter of the adult mouse brain, NG2 cells are continuously generating mature, myelinating oligodendrocytes, whereas in the gray matter, they mostly generate postmitotic NG2 cells (Dimou et al., 2008). In adult mice, NG2 cells proliferate and differentiate in response to several stimuli such as voluntary physical activity in a brain region-dependent manner. Furthermore, the physical activity-associated cognitive improvement is abolished when NG2 cells differentiation is prevented (Eugenin von Bernhardi and Dimou, 2022).
In addition to increasing the speed of propagation of the action potential, oligodendrocytes provide lactate and pyruvate as metabolic support for neurons (Lee et al., 2012; Franklin and Ffrench-Constant, 2017; Rawji et al., 2023). These functions are lost with the loss of oligodendrocytes observed in demyelinating diseases like multiple sclerosis (MS) resulting in lack of trophic support, leading to slowing of axonal conduction of action potentials, and irreversible degeneration of demyelinated axons (Franklin and Ffrench-Constant, 2017).
The decline in white matter volume with normal aging (Taubert et al., 2020), is thought to contribute to age-dependent cognitive impairment. In aging rodent and nonhuman primates, electron microscopy revealed myelin disintegration and decompaction with accumulation of electron-dense cytoplasm within some sheaths, suggestive of myelin breakdown (Peters and Sethares, 2003; Pannese, 2011). Some myelinated nerve fibers in white matter degenerate and disappear, whereas others show degeneration restricted to myelin sheaths, but axons remain intact (Peters, 2009). Remyelination of these axons results in thinner myelin sheaths with shorter internodes than that observed in young fibers (Peters and Sethares, 2003). This remyelination depends on oligodendrogenesis in the spinal cord, where half of newly generated cells expressed NG2 (Lasiene et al., 2009). Multiphoton live imaging of the upper layers of the cortex in mice has confirmed the occurrence of degeneration and decrease of internodes with advancing age (Hill et al., 2018).
Single-cell RNA sequencing revealed age-associated transcriptomic changes in mice brains. Aging oligodendrocytes show downregulation of genes encoding myelin proteins (Mog, Plp, and Cnp) and cholesterol synthesis pathway (Hmgcs1). By contrast, aging oligodendrocytes upregulate genes involved in ribosome biogenesis (Rpl6, Rps29, and Rpl23a) and immune-response (C4b and Il33) (Ximerakis et al., 2019; Rawji et al., 2023).
Adult rodent NG2 cells progressively loss their differentiation and proliferation potential which results in the slowing of remyelination capacity with aging (Neumann et al., 2019). They fail to be recruited into the lesion area and they are not able to differentiate into oligodendrocytes (Sim et al., 2002). This loss of function with aging is relevant for understanding that in chronically demyelinated MS lesions there is a decrease of the number of NG2 cells and their differentiation into oligodendrocytes (Neumann et al., 2019). Furthermore, NG2 cells microenvironment stiffens with age, and this stiffness appears to be sufficient to cause age-related loss of function of NG2 cells (Segel et al., 2019). Bulk RNA sequencing analysis revealed that the decline in functional capacity is associated with hallmarks of stem cell aging, such as mitochondrial dysfunction, decreased metabolic function, inflammasome signaling, increased DNA damage, dysregulated nutrient sensing, autophagy, and the unfolded protein response (Neumann et al., 2019; Rawji et al., 2023). Proteomic analysis reveals that the amount of myelin-associated proteins, and proteins associated with oxidative phosphorylation, inflammatory responses and actin cytoskeletal structure are upregulated by aging (de la Fuente et al., 2020). By contrast, enzymes related with transcription factors, cell cycle proteins, and biosynthesis of cholesterol, essential for the production of myelin, are downregulated (de la Fuente et al., 2020). Interestingly, regenerative capacity of aged NG2 cells can be enhanced with youthful systemic milieu containing monocytes, fasting, metformin treatment, or reduction of stiffness of the extracellular matrix. All these stimuli will improve remyelination in aged animals (Neumann et al., 2019; Segel et al., 2019).
1.2.3. Astrocytes
Astrocytes, not only support neurons, but serve also a wide range of functions that involve ion buffering, water and ion homeostasis, neurotransmitter recycling, formation, maturation, maintenance, pruning and remodeling of synapses, blood–brain barrier formation, inflammation regulation, and interoception. They are located in close contact with all nervous system structures to accomplish metabolic and homeostatic functions indispensable for the proper functioning of neuronal circuits in the brain. They extend processes that occupy non-overlapping domains, into the vicinity of synaptic clefts. They enwrap presynaptic and postsynaptic neuronal regions, forming the so called “tripartite synapse.” In response to synaptic activity (Gomez-Gonzalo et al., 2017) or specific stimuli like hypercapnic acidosis in the brainstem (Beltran-Castillo et al., 2017), they show a calcium dependent release of gliotransmitters (Glu, Gaba, D-serine, ATP) which regulate the excitability and the efficacy of synapses (Araque et al., 2014). Since one astrocyte can contact thousands of synapses, they could recruit and enhance the activity of distant neurons and synapses in brain circuits (Halassa and Haydon, 2010). Together with microglia, astrocytes contribute also to inflammatory processes (Giovannoni and Quintana, 2020).
Astrocyte phenotype changes in response to brain injury, ischemia, infection, neuroinflammation or neurodegenerative diseases, which leads to “reactive astrocytosis.” Reactive astrocytosis involves morphological changes such as hypertrophy, increased astrocyte proliferation, up- and down-regulation of several genes, and particularly, after acute CNS trauma, are associated with glial scar formation (Liddelow et al., 2017; Lopez-Teros et al., 2022). Reactive astrocytosis englobes different astrocyte responses depending on the nature of insults (Zamanian et al., 2012). For example, neuroinflammation and stroke ischemia in mice result in upregulation of genes, whereas 50% are upregulated by both insults, the rest of genes are specific for each injury type. In brain inflammation, reactive astrocytes upregulate complement cascade which are deleterious for synapses, and compatible with a neurotoxic phenotype. By contrast, in ischemia, reactive astrocytes upregulate many neurotrophic factors suggesting a neuroprotective phenotype (Zamanian et al., 2012; Liddelow et al., 2017). As observed in cytotoxic microglia,. inflammatory astrocytes lose several astrocyte properties (Kigerl et al., 2009; Zamanian et al., 2012; Liddelow et al., 2017) such as promotion of neuronal survival, outgrowth, synaptic functions, and phagocytosis. They can induce death of neurons and oligodendrocytes in vitro, and of axotomized neurons in vivo (Liddelow et al., 2017). Increased numbers of activated astrocytes are also observed in human neurodegenerative diseases (Liddelow et al., 2017).
As part of astrocyte/microglia interaction, microglia modulate astrocytes (Liddelow et al., 2017). In wild-type (WT) mice, LPS injection activates astrocytes, whereas in Csf1r−/− mice, which lack microglia cells, LPS fails to the activation of astrocytes (Liddelow et al., 2017). Furthermore, activated microglia cells induce differentiation of astrocytes by secreting interleukin 1α (IL1α), tumor necrosis factor α (TNFα), and complement 1q (C1q), which together are necessary and sufficient to induce the activation (Liddelow et al., 2017). It is worth noting that C1q is part of the first component of the C1 complex, which is bound by antigen/antibody complexes, neuronal blebs, apoptotic cells, fibrillary β amyloid (Aβ) or phospho-tau, to activate the classical pathway of complement. C1q appears to have a role of tagging weak synapses to be engulfed by microglia (Gomez-Arboledas et al., 2021).
Reactive astrocytes can lead to beneficial or detrimental effects. For instance, they can produce a glial scar as a defensive barrier against inflammatory cells or pathogens, promote neuronal survival, detect signals of brain damage, or secrete cytokines and chemokines. However, they also can inhibit cell migration and axonal regeneration (Escartin et al., 2021; Lopez-Teros et al., 2022). The astrocyte’s responses to brain aging are heterogenous. Proliferation-competent glial cells can undergo senescence both in vitro and in vivo, and contribute to neuroinflammation in the aging brain. Astrocytes can become reactive or senescent with aging, depending on stressful stimuli, contributing to the loss of cognitive function through inflammatory mediators (Cohen and Torres, 2019; Escartin et al., 2021; Lopez-Teros et al., 2022; Gaspar-Silva et al., 2023). Astrocyte senescence implies permanent cell cycle arrest, increased cell size, and several characteristics that involve a secretory profile called senescent associated secretory phenotype (SAPS) (Rodier and Campisi, 2011), the presence of DNA damage or “scars,” changes in heterochromatin called Senescence-Associated Heterochromatin Foci (SAHF) (Narita et al., 2003), increment of b-galactosidase enzyme activity (Dimri et al., 1995), lipofuscin accumulation, and a decrease in lamin B1 (Shimi et al., 2011).
In humans, astrocyte- and oligodendrocyte-specific genes, but not neuron-specific genes, show significant age-dependent changes in their regional expression patterns, particularly in the hippocampus and substantia nigra. By contrast, microglia- and endothelial-specific genes increase in all brain regions (Soreq et al., 2017). A hallmark of brain physiological aging, the increase in astrocyte reactivity, assessed through the increase in glial fibrillary acidic protein (GFAP) labeling, is mainly observed in the hippocampus, frontal, temporal, and entorhinal cortex in humans (Nichols et al., 1993; Porchet et al., 2003). Similar findings have been observed in aged mice and rats (Nichols et al., 1993; Rodriguez et al., 2014; Boisvert et al., 2018). In mice, astrocytes undergo various age-dependent morphological and molecular changes in specific brain regions (Boisvert et al., 2018). Such changes involve astrocyte hypertrophy, rearrangement of cytoskeleton, GFAP upregulation, and inflammatory phenotype. Aged astrocytes from the hypothalamus and cerebellum, but not from the cortex, show significant increase in the expression of genes associated with inflammatory response, astrocyte reactivity (GFAP and Serpin3n), synapse elimination pathways (complement C3 and C4b), and downregulation of cholesterol synthesis enzymes (Boisvert et al., 2018).
RNA sequencing (RNAseq) analysis of the differentially expressed astrocytes genes along the mouse lifespan revealed that in aged astrocytes there is a significant increase in the proportion of reactive astrocytes exhibiting a neuroinflammatory phenotype (Clarke et al., 2018) in a brain region-dependent phenomenon. Hippocampal and striatal astrocytes upregulate a higher number of reactive astrocyte genes than that observed in cortical astrocytes. Furthermore, the LPS-induced transformation of astrocytes is increased by aging. Likely, the aging effect may be associated to the 300-fold increase in C1q observed in aging mouse and human brains (Stephan et al., 2013) and the upregulation of inflammatory genes in aging microglia, in special because the aging-dependent upregulation of reactive astrocyte genes was significantly reduced in mice lacking microglia-secreted cytokines (IL1α, TNF, and C1q) (Clarke et al., 2018).
These results show that microglia play a role in astrocyte activation. Aged astrocytes contribute to create an environment favoring synapse elimination and neuronal damage, likely promoting aging-associated cognitive decline (Boisvert et al., 2018). In fact, astrocytes undergo age-dependent changes in gene expression that make specific brain regions more vulnerable to age-associated synapse loss and neuroinflammation. If astrocyte heterogeneity defines the susceptibility of different brain regions to certain insults, and the way brain regions age are still open questions.
1.2.4. Microglia
In contrast to neurons and astrocytes, microglia have mesodermal origin. Microglia are derived from myeloid precursor cells from the yolk sac, which differentiate in tissue-resident macrophage precursors that migrate into the CNS where they finally become resident microglia at an early embryonic state.
The microglia, as “resident macrophages” (Rivest, 2009), constitute the main defense system of the CNS (Tremblay et al., 2011). Microglia are the main contributors to synaptic pruning in the brain, although astrocytes can participate in this process, particularly, during development and early postnatal life. In adult animals, failure in microglia regulation of pruning can exacerbate synaptic loss leading to memory deficits observed in neurodegenerative diseases (Wu et al., 2015; Hong et al., 2016; Liddelow et al., 2017; Gomez-Arboledas et al., 2021).
Microglia surveil systematically the nervous tissue and detect a wide spectrum of signals associated with autoimmune damage, infections, ischemia, trauma, and toxins (Rivest, 2009). Depending on the nature of these signals, they can trigger an integrative microglial response to maintain brain homeostasis, modifying their morphology and functional properties, phagocytizing, and degrading potentially harmful endogenous and exogenous compounds. Thus, surveillance microglia can adopt an activated phenotype, which can be pro- or anti-inflammatory. Depending on their phenotype, microglia are able to release a broad spectrum of molecules including inflammatory cytokines, such as interleukin 1β (IL1β), interleukin 6 (IL6), tumor necrosis factor α (TNFα), and interferon γ (IFNγ), reactive oxygen species (ROS) (Qin et al., 2005; Block et al., 2007; Li et al., 2007; Kettenmann et al., 2011; Welser-Alves and Milner, 2013; Hickman et al., 2018), and nitric oxide (NO) (von Bernhardi and Eugenin, 2004; Li et al., 2007; Nakajima et al., 2007; Sierra et al., 2007; Tichauer and von Bernhardi, 2012; Welser-Alves and Milner, 2013; von Bernhardi et al., 2015). Interestingly, microglia can modulate regulatory cytokines such as interleukin 10 (IL10) and transforming growth factor β (TGFβ), among other trophic factors. In addition to be the main source of inflammatory molecules, they also release gliotransmitters (Glu, ATP and D-serine) in the CNS (Imura et al., 2013; Beltran-Castillo et al., 2018; Dos-Santos-Pereira et al., 2018).
Microglia undergo senescence, showing characteristic morphological and functional features (Paolicelli et al., 2022), that differ from those found in activated or quiescent microglia (Hart et al., 2012; Spittau, 2017; Wendimu and Hooks, 2022). A feature of abnormal aged dystrophic microglia is the reduction or absence of cytoplasmic processes leading, occasionally, to the formation of spheroids, a striking sign of dystrophia with partial fragmentation of their cytoplasm (Mecca et al., 2018; Edler et al., 2021; Javanmehr et al., 2022; Munoz-Castro et al., 2022; Paolicelli et al., 2022; St-Pierre et al., 2022). Double immunofluorescence, against ionized calcium binding adaptor molecule 1(Iba1)/cluster of differentiation 68 (CD68) and Iba-1/major histocompatibility complex (MHC) class II to identify microglia, together with electron microscopy, revealed that isolated Iba-1(+) fragments persisted connected to each other by CD68(+) or MHCII(+) segments of the microglial process. That is, apparent fragments by light microscopy, indeed still are connected to the microglial soma when observed by electron microscopy (Tischer et al., 2016).
Since microglia is the main generator of inflammatory cytokines and oxidative mediators in the CNS (Pawate et al., 2004; Qin et al., 2005; Hayashi et al., 2008), changes in the regulation of aged microglia may lead microglial activation from neuroprotective to deleterious (von Bernhardi et al., 2015). Hence, microglial dysregulation arises as a key element for inducing chronic neuroinflammation (Tremblay et al., 2011). On the other hand, neuroinflammation promotes neurotoxicity by inducing cytotoxic activation of microglia (Ramirez et al., 2008), inhibition of Aβ clearance (von Bernhardi, 2007), and synergistic deleterious effects promoting neuronal death (Nguyen et al., 2002; von Bernhardi and Eugenin, 2012). Thus, as a global effect of aging, microglia switch from a neuroprotective to a more cytotoxic phenotype (Banati et al., 1993; von Bernhardi et al., 2010, 2015, 2019; Streit et al., 2021). Accordingly, microglia from older individuals present morphological evidence of activation compared with young ones (Streit et al., 2004; von Bernhardi, 2007; von Bernhardi et al., 2010, 2011), elevated basal levels of inflammatory cytokines, such as IL6 and IL1β (Ye and Johnson, 1999; Sierra et al., 2007), decreased signaling by suppressor of mothers against decapentaplegic 3 (Smad3)-TGFβ in inflammation (Tichauer and von Bernhardi, 2012), decreased Aβ-induced phagocytosis (Floden and Combs, 2011), and increased ROS production (Tichauer and von Bernhardi, 2012) and oxidative stress (von Bernhardi and Eugenin, 2012; Tichauer et al., 2014). This microglia activation has been described in the aging CNS (von Bernhardi, 2007; von Bernhardi et al., 2010; Villeda et al., 2014), as well as, in various pathologies, including cerebrovascular disease and AD (Hickman et al., 2008; Murgas et al., 2012; von Bernhardi and Eugenin, 2012; Trougakos, 2019). Changes in the expression of receptors relevant for cellular communication and inflammatory activation (Gu et al., 2019) underly microglial dysfunction (Tarkowski et al., 2002; Tse and Herrup, 2017).
Microglial toxicity is modulated by astrocytes through a cross-regulation that includes TGFβ and IL1β (Tichauer et al., 2007; Orellana et al., 2013), the neuroprotective response being especially conspicuous in acidic microenvironments (Uribe-San Martin et al., 2009). Hypercapnic acidification has also effects on phagocytosis (Eugenin et al., 2016) and synaptic function, inducing the release of D-serine by astrocytes (Beltran-Castillo et al., 2017, 2018), which could also participate in aging. Multiple changes associated with glial dysregulation in aging (von Bernhardi, 2007; Hickman et al., 2018) result in the impairment of neuronal function, including the production of soluble mediators by activated cells in aging (Conboy et al., 2005, 2013; Villeda et al., 2014), which affect synaptic function (Johnson-Venkatesh and Umemori, 2010; Schafer et al., 2013; Cerpa et al., 2016), and the regulation of cellular activation by TGFβ and mTOR, the mammalian target of rapamycin (Flores and von Bernhardi, 2012; Herrera-Molina et al., 2012; Switon et al., 2017).
The development of methodology to cultivate microglia from aged mice (von Bernhardi et al., 2011), allowed us to determine that Aβ phagocytosis declines in microglia obtained from adult and aged mice (Alarcon et al., 2005; Cornejo and von Bernhardi, 2013). The reduced uptake is related to the decreased expression of Scavenger Receptor A (SRA), which is relevant for Aβ phagocytosis and appears to mediate the enhancement of Aβ cytotoxicity (Murgas et al., 2012), and for shaping glial inflammatory activation by regulating the secretion of cytokines, ROS and reactive nitrogen species, and the activation of signaling pathways associated with inflammation (Godoy et al., 2012; Murgas et al., 2014). SRA expression is regulated by TGFβ-Smad signaling (Cornejo et al., 2018), which is altered in aging and AD (Tichauer et al., 2014).
Recently, it has been proposed that a subpopulation of microglia can have a protective effect in neurodegenerative diseases, in particular AD and MS (Keren-Shaul et al., 2017; Krasemann et al., 2017). Using single cell transcriptomics, Keren-Shaul et al. (2017) recognized a microglia sub-population, which was denominated disease associated microglia (DAM), also known as activated response microglia (ARM) or microglial neurodegenerative phenotype (MGnD) (Keren-Shaul et al., 2017; Sobue et al., 2023). DAM microglia were found near Aβ plaques in human and mice brains showing intracellular phagocytosed Aβ, suggesting their capacity to restrict Aβ plaque formation by degrading Aβ (Keren-Shaul et al., 2017). Physiologically, DAM activation is achieved via a 2-step process. The first step is a triggering receptor expressed on myeloid cells 2 (TREM2)- independent process, in which microglia transits toward to a stage 1 DAM, characterized by a reduced expression of homeostatic microglia checkpoint genes such as Cx3cr1 and P2ry12/P2ry12, and upregulation of B2m, and AD-associated genes such as Tyrobp and Apoe (Xu et al., 2022). The second step is a TREM2-dependent process, in which stage 1 DAM cells are transformed into stage 2 DAM cells, characterized by the upregulation of Cst7, Lpl, and Trem2 genes (Xu et al., 2022). It is thought that the inhibition of some of these microglia specific inhibitory checkpoints (such as Cx3cr1), which disinhibits DAM activation, could be an important therapeutic target (Keren-Shaul et al., 2017). Another study identified a similar subpopulation of microglia, named neurodegeneration-associated microglia (MGnD) controlled by TREM2-APOE pathway and post-transcriptionally regulated by microRNA(miR)-155 in the surrounding of plaques (Krasemann et al., 2017). TREM2 activates APOE pathway, which transforms a homeostatic into a neurodegenerative microglia phenotype after phagocytosis of apoptotic neurons. Interestingly, targeting the TREM2-APOE pathway can restore the homeostatic phenotype of microglia in amyotrophic lateral sclerosis (ALS) and AD mice models, and prevented neuronal loss in an acute neurodegeneration model (Krasemann et al., 2017).
1.3. The CX3CL1/CX3CR1 axis
The chemokine fractalkine is found as an anchored transmembrane protein, with its chemokine domain bound to a mucin stalk, which endows it with a cell adhesion function (Bazan et al., 1997; Figure 1). CX3CL1 is composed of 373 amino acids (aa), that conform four domains: chemokine domain (CKD, 76 aa), mucin stalk domain (MS, 241 aa), containing 17 mucin repeats with glycosylation-dependent stiffness to present the CKD away from the membrane and reduce the CX3CL1 diffusion within the membrane, transmembrane domain (TM, 19 aa), involved in the aggregation of several CX3CL1 molecules to strengthen adhesion, and cytosolic domain (CD, 37 aa) that provides anchoring to cytoskeletal proteins increasing fractalkine adhesion with its receptor (Ostuni et al., 2014, 2020; Rivas-Fuentes et al., 2021; Figure 1).
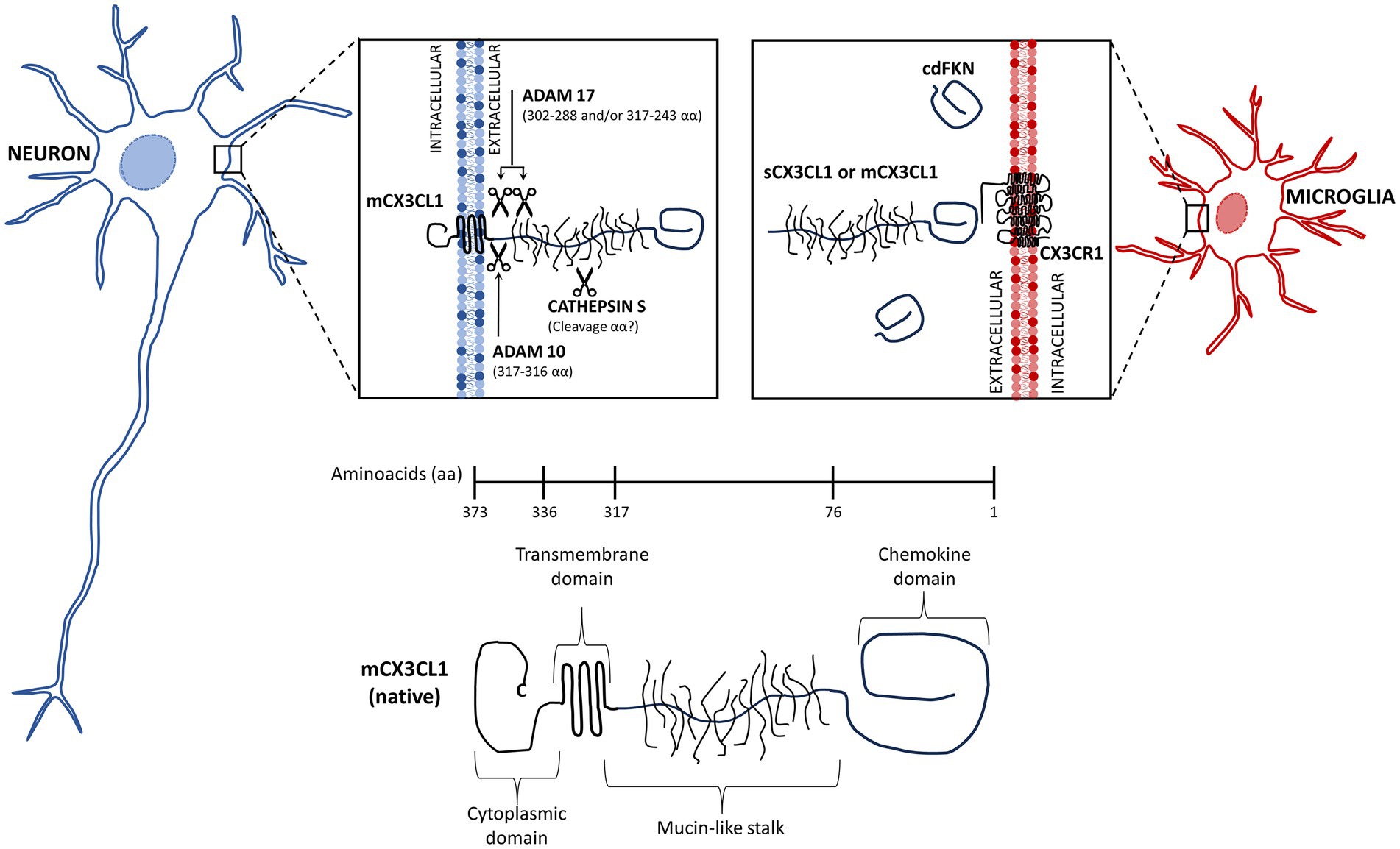
Figure 1. Membrane-bound and soluble fractalkine. Fractalkine (CX3CL1) is synthesized in neurons as a precursor that is rapidly transported to the cell surface, where it is incorporated as a transmembrane protein (mCX3CL1, native). We illustrate the mCX3CL1 molecule indicating its chemokine, mucine-like stalk, transmembrane, and cytoplasmic domains. At the cellular surface, mCX3CL1 is targeted for metalloproteinase-dependent cleavage that releases soluble fractalkine (sCX3CL1) containing most of the mucine-like stalk and the chemokine domain. Key proteases contributing to CX3CL1 cleavage include TNFα converting enzyme (TACE; ADAM17), ADAM10, and cathepsin S. Physiological CX3CL1 cleavage occurs at different sites depending on the protease. sCX3CL1 forms do not differ much in their functional effects. Both CX3CL1 types, mCX3C1L and sCX3CL1, bind the CX3CR1 located in microglia. A CX3CL1 form restricted to the chemokine domain (cdCX3CL1) has been artificially generated.
In the CNS, CX3CL1 is abundant and constitutively expressed by neurons, whereas its expression by astrocytes is induced by TNFα and IFNγ (Yoshida et al., 2001). The CX3CL1 anchored to the neuron membrane is known as the “membrane bound CX3CL1” (mCX3CL1). However, in an inflammatory environment, it undergoes cleavage by metalloproteases (ADAM 10 and ADAM 17) and other proteases, releasing soluble forms (sCX3CL1) (Jones et al., 2010). All forms of fractalkine bind to the sole receptor CX3CR1, a 7-transmembrane receptor coupled to heterotrimeric G protein (GPCRs) found constitutively in microglia (Harrison et al., 1998; Williams et al., 2014; Szepesi et al., 2018), whereas in astrocytes it is expressed only under inflammatory conditions (Rivas-Fuentes et al., 2021).
The binding of CX3CL1 with CX3CR1 dissociates the α subunit from the βγ complex of the associated-G protein, activating several signaling pathways and intracellular Ca2+ mobilization (Harrison et al., 1998; Boddeke et al., 1999), phosphoinositide 3-kinase (PI3K) and mitogen activated protein kinases (MAPK), such as c-Jun N-terminal kinase (JNK), extracellular-signed regulated kinase (ERK) 1/2, p38-mitogen activated protein kinase (p38 MAPK), protein kinase B (PKB, also called AKT Ser-473 Thr-308), proto-oncogene tyrosine-protein kinase Src (c-Src), and endothelial nitric oxide synthetase (eNOS) (Meucci et al., 2000; Cambien et al., 2001; Kansra et al., 2001; Deiva et al., 2004; Lee et al., 2006; Volin et al., 2007; Yang et al., 2007), which contribute to cellular responses such as migration, survival and apoptosis resistance.
The CX3CL1/CX3CR1 axis contributes to brain functions along the whole lifespan. During development, CX3CL1/CX3CR1 affects survival, the maturation of neuronal networks, microglial recruitment and pruning (Paolicelli et al., 2011; Ueno et al., 2013), and functional maturation of synapses (Hoshiko et al., 2012). In the adult brain, CX3CL1/CX3CR1 signaling regulates glutamatergic synaptic transmission and plasticity (Bertollini et al., 2006; Ragozzino et al., 2006; Maggi et al., 2009), as well as cognitive functions (Maggi et al., 2011; Rogers et al., 2011; Sheridan et al., 2014).
Several experiments have been performed in mouse models that control the expression of fractalkine and its receptor: Cx3cl1-mCherry, in which exon 1 of Cx3cl1 is replaced with an mCherry fluorescent protein. mCherry fluorescence is observed in mature neurons in the hippocampus, striatum, and cortical layer II and in epithelial cell layers. Cx3cr1+/GFP knock-in (Kin) mice, in which the Cx3cr1 gene was replaced by a green fluorescent protein (GFP) reporter gene, that is, the heterozygote Cx3cr1 + /GFP mice express GFP in cells that retain receptor function; this Kin model allows to generate a Cx3cr1−/− mouse through a Cx3cr1GFP/GFP double Kin mouse model. Cx3cr1-Cre mice express Cre recombinase under the direction of the Cx3cr1 promoter in the mononuclear phagocyte system. These mice do not express endogenous Cx3cr1. Cx3cr1CreER Kin/knock-out (KO) mice express a tamoxifen-inducible Cre recombinase under the direction of the Cx3cr1 promoter in the mononuclear phagocyte system. Insertion of the Cre-ER fusion protein KO endogenous CX3CR1 expression.
1.4. CX3CL1/CX3CR1 axis and inflammation
In vitro and in vivo experimental results suggest that activation of the CX3CL1/CX3CR1 axis has anti-inflammatory effects. CX3CL1 inhibits the bacterial endotoxin lipopolysaccharide (LPS)-induced release of TNFα, IL6, and IL1β by microglia in culture (Zujovic et al., 2000; Mizuno et al., 2003). Furthermore, LPS-induced TNFα secretion was enhanced by CX3CL1 immune neutralization (Zujovic et al., 2000). Accordingly, neutralizing anti-CX3CL1 antibodies enhance acute brain inflammation induced by the intracerebroventricular (ICV) injection of LPS (Zujovic et al., 2001). In fact, Cx3cr1−/− mice show an increased microglial IL1β expression, neurotoxicity, and higher mortality induced by repeated intraperitoneal (IP) injections of LPS than those observed in Cx3cr1+/− mice (Cardona et al., 2006).
LPS treatment downregulates, in turn, the expression of CX3CR1 in rat and mouse microglia cell cultures (Zujovic et al., 2000; Inoue et al., 2021). Fractalkine applied before LPS, inhibits the increase of LPS-induced NO. The overexpression of CX3CR1 decreased the LPS-induced NO production even without the application of exogenous CX3CL1 (Inoue et al., 2021).
Similarly, in in vivo experiments, a reduction of CX3CR1 mRNA is observed 4 h after the IP injection of LPS in adult and aged mice microglia. The LPS-induced reduction of CX3CR1 mRNA is observed for up to 24 h on aged mice microglia. Interestingly, CX3CR1 downregulation was associated with a prolonged microglial activation, reduction of TGFβ, and persistent signs of sickness behavior (Wynne et al., 2010). Given that 4 h of TGFβ treatment of BV2 microglia increases CX3CR1 mRNA and decreases IL1β mRNA (Wynne et al., 2010), it appears that the CX3CL1/CX3CR1 axis and TGFβ pathway are under reciprocal regulation to generate a more intense anti-inflammatory response.
CX3CL1 treatment is also effective for attenuating inflammatory activation. Its protective effects have been reported on microglia activation and behavioral performance after radiation-induced brain injury (RIBI) (Wang et al., 2021). Irradiation of BV2 microglia in culture induces inflammatory activation and increase of pro-inflammatory cytokines mediated by activation of the microglial nuclear factor κB (NFκB) pathway (Xue et al., 2014; Dong et al., 2015). RIBI activates also microglia NFκB in mice, increasing mRNA and protein expression of TNFα and IL1β, associated with hippocampal neurogenesis impairment and memory deficit evaluated with Morris water maze, in which the mouse must localize a submerged platform guided by visual and spatial cues allowing to test memory, learning, and spatial working (Dong et al., 2015). Exogenous CX3CL1 reduced the RIBI-induced increase in IL1β and TNFα, facilitates inflammatory microglial to change into an anti-inflammatory phenotype, and improves the performance of irradiated mice in the Morris water maze (Wang et al., 2021).
The intracellular signaling pathways activated by CX3CL1/CX3CR1 axis affect neuroprotection. Activation of CX3CR1 activates MAPKs. Activation of ERK, p38-MAPK, and JNK, through the activation of the stress-activated protein kinase-1 (MSK1) (Bachstetter et al., 2011; Galan-Ganga et al., 2019), activate NFκB, which increases the expression of inflammatory mediators (Zujovic et al., 2000; Galan-Ganga et al., 2019; Figure 2). By contrast, the activation of the AKT/ERK signaling pathway induces factor 2 related to nuclear factor E2 (Nrf2) that translocate to the nucleus and increases the transcription of various antioxidant and cytoprotective genes such as antioxidant response element (ARE) and heme oxygenase-1 (HO-1), which increase the phagocytic and anti-inflammatory capacity of microglia (Lastres-Becker et al., 2014; Castro-Sanchez et al., 2019; Li et al., 2019; Trougakos, 2019; Figure 2). Hence, as a summary, activation of CX3CL1/CX3CR1 axis can activate NFκB (Galan-Ganga et al., 2019; Liu et al., 2019), increasing production of inflammatory cytokines by microglia, and enhance the Nrf2 activation, increasing antioxidant and anti-inflammatory effects (Lastres-Becker et al., 2014; Castro-Sanchez et al., 2019; Li et al., 2019; Trougakos, 2019).
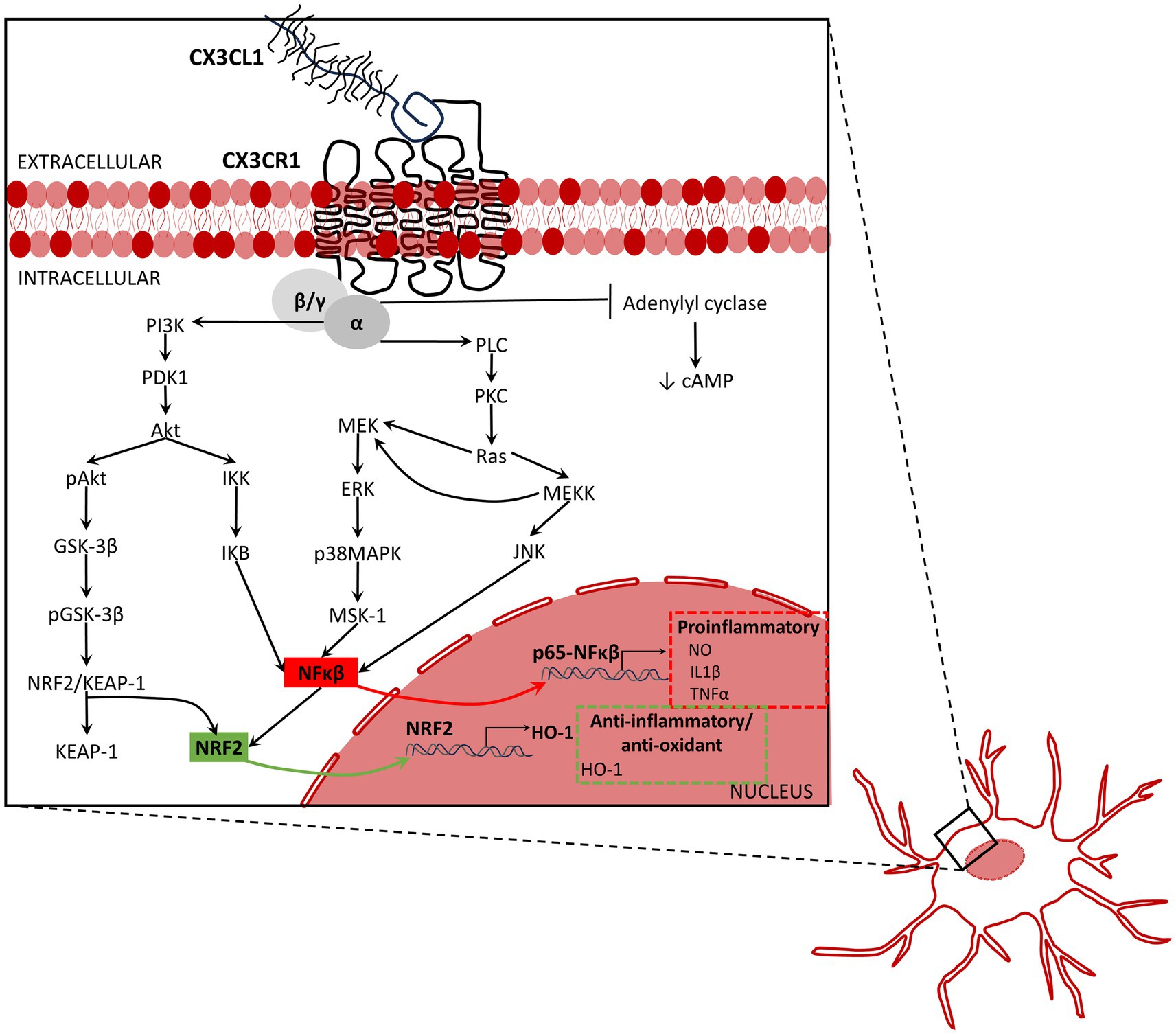
Figure 2. Inflammation associated signaling activated by fractalkine. The activation of the CX3CL1/CX3CR1 axis activates the mitogen-activated protein kinases (MAPKs) including the extracellular signal-regulated kinases (ERK), p38-MAPK, and c-Jun NH(2)-terminal kinase (JNK). MAPKS activate the stress-activated protein kinase-1 (MSK1), and consequently, activate NFκB pathway, and therefore, the production and release of inflammatory mediators. By contrast, the activation of the AKT/ERK signaling pathway induces factor 2 related to nuclear factor E2 (Nrf2) to be translocated into the nucleus leading to increased transcription of various antioxidant and cytoprotective genes such as antioxidant response element (ARE) and heme oxygenase-1 (HO-1), which increase the phagocytic and anti-inflammatory capacity of microglia. To summarize, activation of CX3CL1/CX3CR1 axis increases NFκB (Galan-Ganga et al., 2019; Liu et al., 2019), promotes release of pro-inflammatory cytokines by microglia, and enhances the Nrf2 activation that increases anti-oxidant and anti-inflammatory response (Lastres-Becker et al., 2014; Castro-Sanchez et al., 2019; Li et al., 2019; Trougakos, 2019). Akt, protein kinase B; ERK, extracellular signal-regulated kinases; GSK-3β, glycogen synthase kinase-3β; HO-1, heme oxygenase 1; IKB, Ikappa B protein; IKK, IkappaB kinase; IL1β, interleukin-1β; JNK, c-Jun N-terminal kinase; KEAP-1, Kelch-like ECH associating protein-1; MEK, mitogen-activated protein kinase kinase; MEKK, MEK kinase or mitogen activated protein (MAP) kinase kinase kinase; MSK-1, mitogen- and stress-activated kinase 1; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; NRF2, nuclear factor erythroid 2-related factor 2; NRF2/KEAP-1, nuclear factor erythroid 2-related factor 2/Kelch-like ECH associating protein-1;NO, nitric oxide; pAkt, phosphorylated AKT; pGSK-3β, phosphorylated glycogen synthase kinase-3β; PDK1, protein 3-phosphoinositide-dependent protein kinase-1; PI3K, phosphoinositide 3-kinase, also called phosphatidylinositol 3-kinase; PKC, protein kinase-C; PLC, phospholipase C p38MAPK, p38 mitogen-activated protein kinase; Ras, rat sarcoma virus small-GTPase; TNFα, tumor necrosis factor-α.
Activation of CX3CL1/CX3CR1 does not lead always to neuroprotection, depending on the triggering inflammatory-like processes. After transient occlusion of the middle cerebral artery (MCAO), Cx3cr1GFP/GFP mice exhibited reduced areas of brain infarcts, reduced number of apoptotic cells and infiltrating leucocytes, and reduced blood–brain barrier (BBB) damage compared with Cx3cr1+/GFP and Cx3cr1+/+ mice. Moreover, Cx3cr1GFP/GFP mice expressed less IL1β, IL1Ra, and TNFα mRNAs and showed better functional outcomes than the other groups (Denes et al., 2008). The disruption of the CX3CL1/CX3CR1 axis in permanent MCAO upregulate CX3CR1 in neurons in the striatum and the hippocampus, which was associated with an increased apoptotic-like neuronal morphology and increased number of caspase 3(+) neurons (Wang et al., 2018). By contrast, ischemia-induced apoptotic neuronal cell death was decreased in CX3CR1 KO mice (Wang et al., 2018). That is, CX3CR1 deletion can endow neuroprotection in mouse models of brain ischemia (Denes et al., 2008; Wang et al., 2018).
The CX3CL1/CX3R1 axis is regulated by inflammatory cytokines such as TGFβ, IFNγ, TNFα, or IL1β (Rivas-Fuentes et al., 2021). In newborn rat microglia cultures, 1–10 ng/mL TGFβ1 for 16 h increased CX3CR1 mRNA and protein level (Chen et al., 2002), and reduce the activation of microglial ERK1/2 and p38 MAPK induced by a 10 min CX3CL1 stimulation (Chen et al., 2002).
1.5. The impact of CX3CL1/CX3CR1 axis on neurodegenerative diseases
Although, the absence of CX3CR1, per se, does not result in microglia activation or neurodegeneration, the impairment of the CX3CL1/CX3CR1 signaling by deletion of the Cx3cr1 gene results in increased neurotoxicity in mouse models of LPS-induced systemic inflammation, Parkinson’s disease (PD), AD, and ALS (Cardona et al., 2006; Lee et al., 2010; Cho et al., 2011). Conversely, activation of CX3CL1/CX3CR1 axis can promote neural protection not only in different inflammatory conditions but also in pathological processes leading to neurodegenerative diseases and their associated inflammation.
1.5.1. Amyotrophic lateral sclerosis
In murine model of ALS, in which human superoxide dismutase 1 (SOD1) with the G93A mutation is expressed under control of the cistronic human SOD1 promotor, the lack of expression of CX3CR1 accelerates ALS-like disease progression leading to a rapid and increased neuronal cell death (Boillee et al., 2006; Cardona et al., 2006; Liu et al., 2019). In this murine model of ALS, the absence of CX3CR1, increases the activation of NFκB, and impairs the autophagy-lysosome degradation pathway and the autophagosome maturation (Liu et al., 2019) resulting in an intense damage of motoneurons. Thus, the CX3CL1/CX3CR1 pathway has anti-inflammatory and neuroprotective effects and appears to play an important role in maintaining autophagy activity.
The Cx3cr1 human polymorphism I249/M280 is present in about 20% of the population. The protein product of this polymorphism exhibits reduced affinity for CX3CL1 resulting in a reduced regulatory effect on microglia. Accordingly, the hypofunctional variant of CX3CR1 has been associated with a reduction of the life span of ALS patients (Lopez-Lopez et al., 2014). Thus, ALS patients, who carry one or two copies of the CX3CR1-Val249Ile allele, have a rapid course of the disease and a shorter survival than patients who are carriers of the WT Cx3cr1 (Lopez-Lopez et al., 2014).
1.5.2. Multiple sclerosis
MS is a chronic, inflammatory, predominantly immune-mediated disorder of the CNS showing focal lesions in white matter of the brain and spinal cord, characterized by prominent demyelination ensuing axonal damage and glial scar formation, leading to loss of motor and sensory function (Karussis, 2014; Oh et al., 2018; Dobson and Giovannoni, 2019; Absinta et al., 2020). In MS patients, Cx3cr1-Val249Ile polymorphisms revealed that Cx3cr1 Ile249 Thr280 haplotype could endow a protective effect by impairing the switch of MS from the relapsing–remitting type (RRMS) into the secondary progressive type (SPMS) (Stojkovic et al., 2012; Arli et al., 2013). Patients having the variant in both alleles (homozygosity) have a higher risk for disability (Arli et al., 2013).
Current immune therapy is useful for ameliorating RRMS, but not for preventing its progressive forms (Oh et al., 2018; Dobson and Giovannoni, 2019; Absinta et al., 2020). Among several processes, remyelination is required for normalization of neural functions (Kotter et al., 2011; Lampron et al., 2015; Rivest, 2015; Mendiola et al., 2022). Unfortunately, remyelination in MS fails or is incomplete (Oh et al., 2018; Dobson and Giovannoni, 2019).
Chronic microglia activation is related with MS disease progression (Absinta et al., 2020). Besides, a switch in microglia from a pro-inflammatory to a regulatory phenotype is associated with the beginning of remyelination (Miron et al., 2013). The regulatory phenotype favors oligodendrocyte differentiation and is observed in injured CNS regions of aged mice in which remyelination is enhanced, like in MS lesions (Miron et al., 2013).
It has been suggested that CX3CL1/CX3CR1 axis is a relevant regulator of the clearance of myelin debris by microglia (Lampron et al., 2015; Rivest, 2015; Mendiola et al., 2022). Experimental autoimmune-independent demyelination without BBB disruption followed by complete remyelination can be attained with cuprizone, a copper chelating toxin that induces apoptosis of oligodendrocytes and can be administered through the diet (Lampron et al., 2015; Rivest, 2015; Mendiola et al., 2022). In WT mice, cuprizone induces massive demyelination as consequence of oligodendrocytes apoptosis during the first 3 weeks (wk.) accompanied by recruitment of NG2 cells, astrogliosis, and microgliosis (Hiremath et al., 1998; Gudi et al., 2014). Cuprizone removal from diet is followed by complete remyelination within 1–3 wk. In CX3CR1−/− mice, inflammatory response was like that observed in cuprizone-challenged WT mice. However, cuprizone-induced demyelination was followed by uncomplete remyelination. Microglia in CX3CR1−/− mice exhibited an impaired migration into the corpus callosum and phagocytosis, and consequently, persistent myelin debris and defective axonal remyelination characterized by aberrant myelin patterns. Unlike WT microglia that showed abundant phagocytic inclusions, CX3CR1−/− microglia lack them (Lampron et al., 2015). Induction of experimental autoimmune encephalomyelitis (EAE) on a mouse model of CX3CR1 hypofunction by replacement of the normal mo Cx3cr1 locus for the hu Cx3cr1-I249/M280 variant, revealed exacerbated functional signs of EAE, with more severe inflammation and neuronal loss (Cardona et al., 2018).
Transgenic mice expressing the polymorphic hu Cx3cr1-I249/M280 variant, and fractalkine-deficient (Cx3cl1−/−) mice showed exacerbated cuprizone-induced demyelination in the anterior corpus callosum 4 w, compared with that observed in CX3CR1−/− deficient (Cx3cr1GFP/GFP) and WT mice (Mendiola et al., 2022). Microgliosis and CD68 (phagocytic microglia marker) were similar in hu Cx3cr1-I249/M280, Cx3cl1−/−, and Cx3cr1GFP/GFP mice, but higher than those in WT mice after cuprizone treatment (Mendiola et al., 2022). Notoriously, after 1 wk. removal of cuprizone, only Cx3cr1GFP/GFP and WT mice showed significant remyelination (Mendiola et al., 2022). WT mice showed significant remyelination at the anterior and posterior corpus callosum. In hu Cx3cr1-I249/M280 mice, significant remyelination was only observed in the anterior corpus callosum, whereas in Cx3cr1GFP/GFP mice, only in the posterior corpus callosum. By contrast, Cx3cl1−/−mice did not show significant early remyelination (Mendiola et al., 2022).
Using EAE as a rat model of MS, it was shown that 12 days after the inoculation of myelin basic protein (MBP), CX3CL1 and CX3CR1 mRNA and protein levels were increased in dorsal root ganglia (DRG) and spinal cord (Zhu et al., 2013). The increase was associated with thermal sensory abnormalities, suggestive of neuropathic pain, and correlated with neurological impairment (Zhu et al., 2013). These results are indicative of the role of CX3CL1/CX3CR1 axis as a critical pathway involved in the MS-induced neuropathy.
1.5.3. Retinal neurodegeneration
Studies in humans of the genotype distribution of CX3CL1 variants and their association with disease, also show a potential link between CX3CR1-Thr280Met, a variant associated with cell migration deficit, and a higher risk of human age-dependent macular degeneration (AMD) (Tuo et al., 2004; Chan et al., 2005; Combadiere et al., 2007). Evidence supporting a role for CX3CR1-Val249Ile variant in AMD is controversial (Combadiere et al., 2007). Homozygosity for the Thr280Met allele is a more consistent finding in AMD than of other diseases (Combadiere et al., 2007). In the macula of AMD, photoreceptors show signs of degeneration, retinal pigment epithelium is disrupted and cells expressing CX3CR1 are found in the outer retina, in the subretinal space, in the perivascular vicinity, and in choroidal neovascularization. In addition, deposits of CX3CR1 were found in drusen spots (Combadiere et al., 2007). Because they show a reduced affinity for CX3CL1, deletion of CX3CR1 has been experimentally used to simulate the main effect of these variants. Interestingly, aged albino mice with Cx3cr1 deletion exhibit, in regions of retinal degeneration, accumulation of microglia in the subretinal space associated with drusen-like yellowish-white dots, and increased choroidal neovascularization, histological findings like those found in patients with AMD (Combadiere et al., 2007).
Retinitis pigmentosa is the more common non-syndromic inherited retinal dystrophy that include several heterogeneous retinal neurodegenerative conditions in which mutations in photoreceptor or retinal pigment epithelium genes result in progressive degeneration of photoreceptors (Hartong et al., 2006; Murro et al., 2023). Retinitis pigmentosa is characterized by a primary degeneration of rod photoreceptors that progresses to the loss of cone photoreceptors, followed by an aberrant remodeling of retina resulting in disconnection of neural retina from photoreceptors, associated with changes from the molecular to tissue levels (Hartong et al., 2006; Zieger et al., 2014; Murro et al., 2023).
Retinal degeneration 10 (rd10) is a mouse model of autosomal recessive retinitis pigmentosa, consisting in the spontaneous mutation of the rod-phosphodiesterase-6b (Pde6b) gene, that leads to rod degeneration and later to cone degeneration (Chang et al., 2002). Analysis of mRNA and protein levels of CX3CL1 revealed that in both WT and rd10 mice, a 100 kDa membrane bound CX3CL1 form and a cleaved soluble 85 kDa CX3CL1 form were present at the postnatal day 5 (P5). At P10, a 95 kDa form is accumulated, whereas the 85-kDa form is decreased. In older animals, the 95 kDa form became principal in wt retina, whereas in rd10 retinas, there is a significant increase of soluble 85 kDa form. Retinas of rd10 mice had significantly lower levels of total CX3CL1 protein (from P10 onwards) and lower CX3CL1 mRNA levels (from P14) than those observed in WT animals. In situ hybridization histochemistry and immunofluorescence using transgenic Cx3cl1cherry mice showed that neurons of the inner retina layers were the main sites of fractalkine synthesis in WT and rd10 mice (Zieger et al., 2014). In the rd10 mouse model, CX3CL1 was detected in apoptotic photoreceptors, suggesting a potential role of this chemokine on the recruitment of microglia into the outer nuclear layer of the retina (Makabe et al., 2020).
Microglia have been implicated in many degenerative eye disorders, including retinitis pigmentosa (Wang and Cepko, 2022), in which activated microglia phagocytose death photoreceptors and their debris and release pro-inflammatory factors that contribute to retinal degeneration (Zabel et al., 2016). In fact, in CX3CR1 deficient (Cx3cr1GFP/GFP) rd10 mice, microglial infiltration into the photoreceptor layer was greater, and the photoreceptor atrophy and apoptosis were higher than those observed in heterozygous Cx3cr1GFP/+ rd10 littermates. Furthermore, CX3CR1 deficient microglia showed increased phagocytosis and increased expression of inflammatory cytokines associated with the increase of activation markers. Activation of CX3CL1/CX3CR1 axis in the rd10 retina via exogenous intravitreal delivery of recombinant CX3CL1 decreased microglial infiltration, phagocytosis, and activation together with reduction of morphological damage and improvement of photoreceptors functionality (Zabel et al., 2016).
Furthermore, synthetic progestin ‘norgestrel’ has been proven as a neuroprotective agent in the retinitis pigmentosa, likely throughout the increase of growth factors, such as basic fibroblast growth factor (bFGF) and leukemia inhibitory factor (LIF) in the retina, favoring the upregulation of pro-survival and downregulation of apoptotic pathways and reduction of microglial pro-inflammatory activity (Doonan et al., 2011; Roche et al., 2016). In primary cultures, rd10 microglia promote neuronal cell death. Norgestrel, in contrast, reduce pro-inflammatory activation and prevent neuronal cell death, reducing the expression of cytokine, chemokine, and danger-associated molecular pattern molecule (DAMP) in the rd10 retina. Furthermore, norgestrel upregulates CX3CL1/CX3CR1 signaling in the rd10 mouse, 1,000-fold at the RNA level (Roche et al., 2016). Norgestrel’s neuroprotection would be mediated by its actions on photoreceptors, which are induced to release CX3CL1, and this chemokine in turn, would refrain harmful microglia activity (Roche et al., 2017).
1.5.4. Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the main cause of dementia. Clinical symptoms include memory loss, learning difficulties, and problem-solving impairment. The main risk factor is aging; approximately 5–8% individuals older than 65 are affected, increasing to 35–50% in adults older than 85. The prevalence of AD for women is higher than in men by 19%. Risk factors associated with development of AD involve genetic predisposition (familial early-onset forms), allele ApoE-4 for apolipoprotein E, age, sedentarism, hypertension, diabetes, and metabolic syndrome, among others.
AD is characterized by the extracellular accumulation of Aβ1-42 (senile plaques) and intraneuronal aggregates as neurofibrillary tangles (NFTs) of hyperphosphorylated microtubule associated protein tau (MAPT) (Jellinger, 2020). The time course of AD progression indicates that Aβ deposition begins around 25 years before the onset of clinical diagnoses of AD, and is followed by neurofibrillary tangles (NFT) formation (Saito and Saido, 2018; Jellinger, 2020; Yin et al., 2021). Aβ is generated from the amyloid precursor protein (APP) cleavage by a two-step process: the β-site APP cleaving enzyme 1 (BACE1, or β-secretase), cleaves APP releasing a globular extracellular soluble protein and the membrane-anchored C-terminal fragment; later, γ-secretase (Presenilin) subsequently cleaves this fragment to excise Aβ (a peptide mostly of 39–43 amino acids). In addition to the protein aggregates, AD is characterized by the activation of microglia and astrocytes, neuroinflammation, and the progressive degeneration of neurons, with a predominant early damage of the entorhinal cortex and the hippocampus.
The participation of the CX3CL1/CX3CR1 axis on the course of AD differ in different AD mouse models. In APP/PS1 mice, a well-known mouse model for AD, the level of CX3CR1 does not differ, but the level of CX3CL1 is slightly reduced compared with WT littermates. In APP/PS1 mice, Cx3cr1 deletion reduces plaques, either because an increased clearance or a reduced deposition of Aβ plaques. However, rTg4510 mice, an AD model with inducible overexpression of human mutant tau (P301L), show a 5-fold increase in CX3CR1 and a slight increase in CX3CL1 levels compared with the WT littermates (Nash et al., 2013). In the hu Tau model for AD, Cx3cr1−/− accelerates the onset of tauopathy and behavioral deficits (Bhaskar et al., 2010; Lee et al., 2010; Cho et al., 2011). Furthermore, the increased presence of sCX3CL1 ameliorates the severity of tauopathy in rTg4510 mice. Three months before rTg4510 mice show significant neuron loss, mice were injected with recombinant adeno associated virus (rAAV), expressing sCX3CL1, into both hippocampi. Three months after viral injection, 6 months old mice expressing sCX3CL1 show a significant decrease in tau pathology, an increased density of NeuN(+) cells, reduced hippocampus volume loss, and reduced microglia compared with the control condition (Nash et al., 2013). Memory and learning performance of WT and rTg4510 mice were assessed in the radial arm water maze. The injection of rAAV coding for sCX3CL1 did not significantly improve the behavioral deficit observed in the rTg4510 mice compared with non-transgenic mice (Nash et al., 2013).
To reveal the effect of CX3CL1/CX3CR1 axis on Aβ-induced toxicity, neuron–microglia primary cultures were exposed to hu Aβ1-42. WT neurons released CX3CL1 in response to hu Aβ1-42. Administration of 2 μM hu Aβ1-42 in microglia-depleted Cx3cr1−/− mixed cortical and hippocampal cultures produced lower release of LDH revealing decreased cell death than that observed in WT cultures (Dworzak et al., 2015). Furthermore, Aβ1–42 treatment impaired synaptic transmission, decreasing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-dependent miniature excitatory postsynaptic currents (mEPSCs), suggesting both a pre- and post-synaptic compromise (Dworzak et al., 2015).
Conversely, cell loss in the purified microglia cell culture from Cx3cr1−/− mice were higher than the loss observed in WT microglia cultures. The opposite cytotoxic response induced by Aβ observed in Cx3cr1−/− microglia and neurons suggest that CX3CR1 endows each cell type with different effector functions.
Combining Cx3cr1 deletion, a triple-transgenic mouse model of AD (3xTg-AD: PS1M146V Kin, transgenic APPSwe and tauP301L), and two-photon microscopy, it was possible to evaluate simultaneously neuronal death and microglial cell behavior during a 28-d imaging in living mice (Fuhrmann et al., 2010). A significant 2% neuronal loss in cortical layer III was observed in 4-6-month-old 3xTg-Cx3cr1+/− mice. By contrast, Cx3cr1+/−, Cx3cr1−/− or 3xTg-Cx3cr1−/− mice, did not exhibit neuron loss over the same imaging period (Fuhrmann et al., 2010). Thus, Cx3cr1 deletion appears to be neuroprotective. Additionally, in 3xTg-Cx3cr1+/− mice, microglia, showing an increased migration, were recruited to the sites of neuron loss before the death of neurons. Interestingly, microglia Aβ-phagocytosing activity was not modified by Cx3cr1 deletion (Fuhrmann et al., 2010). This suggests that expression of CX3CR1 by microglia is required for neuron elimination in the context of an AD model (Fuhrmann et al., 2010), but the cellular mechanisms underlying such neuron–microglia interaction remain unknown.
Deletion of Cx3cr1 also appears to be neuroprotective in other AD mice models. For instance, in APP/PS1 and R1.40 mice, APP/PS1 mice co-express hu APP carrying the K670M/N671L familial AD mutation and presenilin 1 with the L166P familial AD mutation leading to primarily Aβ1-42 oligomer aggregation. The R1.40 transgene is a full genomic copy of hu APP carrying the K670M/N671L familial AD mutation that leads to gradual deposition of Aβ1-40 oligomer. In 4-month-old APP/PS1 mice and in 20-24-month-old R1.40 mice, Cx3cr− deletion reduced Aβ deposition and augmented microglia accumulation around Aβ deposits. In both models, Cx3cr1 deletion reduced immunodetection of CD68(+) microglia, whereas qRT-PCR revealed that CX3CR1 deficiency reduced TNFα and the chemokine (C-C motif) ligand 2 (CCL2) mRNA levels, and increased IL1β mRNA. In addition, in both AD models, in vitro and in vivo microglial phagocytosis assessment revealed enhanced Aβ uptake (Lee et al., 2010), suggestive of enhanced Aβ clearance and reduced inflammation (Lee et al., 2010).
In the brain of APP/PS1 mice heterozygous for Cx3cr1 (APP/PS1- Cx3cr1+/−), Aβ level and senile-like plaque deposition are reduced in comparison with age-matched APP/PS1 mice. Reduced Aβ level in the brain was associated with high level of the neuronal-expressed Aβ-degrading enzymes (insulysin and matrix metalloproteinase 9) and with improved performance in the Barnes Maze cognitive test (Hickman et al., 2019). Barnes maze assesses memory and spatial learning placing a rodent on a circular arena, where, moved by its instinctive aversion to open spaces and its natural preference for dark and sheltered spaces, mice select 1 out of 20 equally distributed holes on the arena periphery, which is the unique hole equipped with the animal’s home cage.
1.5.5. Parkinson’s disease
PD is the second most common neurodegenerative disease after AD, affecting about 1% of the population older than 60 years (de Lau and Breteler, 2006). PD is characterized by the gradual loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), the remaining SNpc neurons can show intracellular protein aggregates called Lewy bodies, which are mainly constituted by α-synuclein (SNCA). The cellular pathogenesis of PD is associated with mitochondrial dysfunction, oxidative stress, neuroinflammation, proteasomal dysfunction, impaired autophagy, increased protein aggregation and enhanced apoptosis (Shan et al., 2011; Angelopoulou et al., 2020). PD patients exhibit a constellation of motor and non-motor signs and symptoms. Rest tremor, rigidity, bradykinesia, and loss of postural reflexes are considered as cardinal signs (Jankovic, 2008). Motor signs also include, among others, shuffling gait with decreased arm swing, difficulty arising from chair, turning in bed, hypomimia affecting facial gestural expression and communication, dysarthria, dysphagia, micrography, glabellar reflex, blepharospasm, dystonia, and sialorrhea. Non-motor signs and symptoms include cognitive impairment, bradyphrenia, tip-of-the-tongue (word finding) phenomenon, depression, apathy, anhedonia, fatigue, other behavioral and psychiatric problems including dementia, anosmia, ageusia, pain (shoulder, back), paresthesia, dysautonomia (orthostatic hypotension, constipation, urinary and sexual dysfunction, abnormal sweating, seborrhea), weight loss, REM sleep disorders, sleep fragmentation, and restless legs syndrome (Jankovic, 2008). Current therapies provide symptomatic relief, but fail to stop disease progression (Angelopoulou et al., 2020).
Administration of 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine (MPTP), or its active metabolite, 1-methyl-4-phenylpyridinium (MPP+), has been extensively used to model PD in non-human primates and rodents. In mice receiving IP MPTP injection, Cx3cl1 or Cx3cr1 deletion worsened the dopaminergic neuronal loss and increased microglial activation in the SNpc (Cardona et al., 2006).
The effects induced by IP injection of MPTP varies on different mice strains. For instance, IP injection of MPTP in C57BL/6 mice, but not into BALB/c (BALB) mice, induced behavioral dysfunction, activated microglia/astrocytes, and increase of IL10, IL12, IL13, IFNγ, and CCL2 in cerebrospinal fluid (CSF). It has been proposed that differences in the immunological background affect the outcome. B6 mice show a bias toward Th1 acquired immune responses, whereas BALB mice toward Th2, which are characterized by their distinct cytokine profiles (Yasuda et al., 2008). IL2, IFNγ and TNFα are Th1 cytokines and IL4, IL5, IL6, IL10, and IL13 is Th2 cytokines. Activation of Th1 promotes cell-mediated immunity and activation of Th2 promotes humoral immunity (Yasuda et al., 2008).
Unilateral injection of MPP+ into the rat SN increase the expression of CX3CL1, and CX3CR1 in the ipsilateral (injected) but not in the contralateral SN (Shan et al., 2011). In contrast, both proteins are weakly expressed in sham animals. On the other hand, unilateral injection of CX3CL1 into the rat SN increased microglial activation, dopaminergic neuron loss and motor dysfunction. After 14 days of MPP+ treatment, almost all CX3CR1(+) cells in the injected SN expressed CD11b, also called integrin alpha-M, ITGAM, or integrin alpha-X, ITGAX, is commonly used to identify macrophages and microglia and showed an activated microglia-like phenotype. Furthermore, daily ICV injection of anti-CX3CR1 neutralizing antibodies for a week after injection of MPP+ reduced, in a dose-dependent way, the MPP + -induced rotation behavior. In addition, microglia activation, dopaminergic degeneration, and CX3CL1-induced behavior abnormalities induced by the injection of CX3CL1 into the SN were prevented by ICV administration of anti-CX3CR1 neutralizing antibody 6 and 1 h before CX3CL1 injection (Shan et al., 2011). Moreover, ICV administration of minocycline, a selective microglia inhibitor, prevented CX3CL1-induced rotation behavior and reduced dopaminergic neuron loss in a dose-dependent way (Shan et al., 2011), supporting the involvement of microglia in the dopaminergic toxicity induced by MPP+ and CX3CL1.
Mice with a Cx3cr1 deletion have reduced dopamine levels in the striatum, independently of the neurotoxin administration. After 7 days of intranasal inoculation of MPTP, mice of various genotypes (Cx3cr1+/+, Cx3cr1+/−, Cx3cr1−/−) showed a reduction in tyrosine hydroxylase (TH)- and dopamine transporter (DAT)-positive cells in the striatum. Whereas, in the SN, Cx3cr1+/+ and Cx3cr1+/− genotypes showed a reduction of TH-positive cells, but in Cx3cr1−/− mice, no change of TH- or DAT-positive cells after intranasal MPTP inoculation was observed (Tristao et al., 2016).
The effects of Cx3cr1 deletion on MPPT-treated mice do not depend only on the brain region analyzed (SN vs. striatum), but also depend on the via of MPPT administration (Tristao et al., 2016). After 3 and 7 days of intranasal MPPT, Cx3cr1 deletion did not affect microglial activation and astrogliosis in the striatum. Although, Cx3cr1 deletion at day 3 did not affect the loss of dopamine neurons in the striatum, it showed neuroprotective effects on day 7. In contrast, after intraperitoneal MPTP treatment, Cx3cr1 deletion did not affect dopaminergic degeneration or astrogliosis but increased microglial activation in the striatum and SN (Tristao et al., 2016).
Another model of PD consists in the unilateral stereotaxic administration of the catecholaminergic neurotoxin 6-hydroxydopamine (6 − OHDA), in the rat striatum or SN. 6 − OHDA does not cross the BBB and induces dopaminergic degeneration essentially by increasing ROS and inflammation (Blesa et al., 2012). The administration of recombinant CX3CL1 via an osmotic minipump for 28 days starting 7 days after the 6-OHDA injection into the same striatum, prevented activation of microglia, and showed neuroprotective effect, reducing the loss of striatum neurons that resulted in the reduction in volume of the striatum. Application of CX3CL1 also reduced dopaminergic cell loss and inflammation in the SN (Pabon et al., 2011).
Transgenic PD models have been developed by overexpression of mutant genes for autosomal dominant genes such as α-synuclein and leucine rich repeat kinase 2 (LRRK2) and KO or Kdown models for autosomal recessive genes, such as Parkin, DJ-1, phosphatase and tensin homolog (PTEN)-induced novel kinase 1 (PINK1) (Dawson et al., 2010; Blesa et al., 2012; Angelopoulou et al., 2020). Increase of α-synuclein promotes neuroinflammation, astrocytosis, microgliosis, activation of microglia, and NFκB activation, leading to an increased release of inflammatory cytokines, promoting neuroinflammation and worsening dopaminergic neurodegeneration (Angelopoulou et al., 2020). Microglia activation results in an increased phagocytosis of α-synuclein, which exacerbates the production of ROS and inflammatory activation (Theodore et al., 2008; Angelopoulou et al., 2020).
CX3CL1 has diverse effects on neuroinflammation, and degeneration in α-synuclein models of PD (Angelopoulou et al., 2020). CX3CL1 can be neuroprotective, inhibiting motor impairment and dopaminergic cell loss in the SN and striatum of rats injected with rAAVs coding for overexpressed hu α-synuclein (Nash et al., 2015). Overexpression of hu α-synuclein or hu α-synuclein A53T in WT (Cx3cr1+/+) or null (Cx3cr1−/−) mice display very low levels of CX3CL1. Interestingly, overexpression of hu α-synuclein induced microgliosis, neuroinflammation, and dopaminergic neuronal death in SN, which are of similar magnitude in Cx3cr1+/+ or Cx3cr1−/− mice. However, the overexpression of hu α-synuclein A53T resulted in an exacerbated neurodegeneration compared to hu α-synuclein overexpression. Moreover, α-synuclein A53T-induced neurodegeneration was enhanced in Cx3cr1−/− mice (Castro-Sanchez et al., 2018).
1.6. CX3CL1/CX3CR1 axis and aging
CX3CL1 expression is reduced in the brain of aged rats, being associated to an age-related increase of microglial cell activation (Lyons et al., 2009). Levels of sCX3CL1 decrease with aging, which could lead to an enhanced inflammation, deficits in synaptic remodeling, and eventually to cognitive impairment (Winter et al., 2020). Consistent with this, treatment of aged rats with CX3CL1 blunt the age-related increase in microglial activation (Lyons et al., 2009). Aged (18–22 m) BALB/c mice show a reduced level of basal total CX3CL1 in cortex and hippocampus and the level of CX3CL1 is unchanged 24 h after LPS IP injection, compared with the response observed in young (3–6 m) mice (Wynne et al., 2010). CX3CR1 level in old mice is also lower than in young ones.
1.6.1. CX3CL1 cleavage and aging
The native 95 kDa membrane-bound CX3CL1 (mCX3CL1) can be transformed into a 65 kDa chemotactic glycoprotein (sCX3CL1, which contains the extracellular N-terminal CKD and MS domains), by metalloproteinases such as the TNFα converting enzyme (TACE, ADAM17) (Garton et al., 2001; Tsou et al., 2001), ADAM10 (α-secretase) (Hundhausen et al., 2003, 2007), matrix metalloproteinase 2 (MMP-2) (Bourd-Boittin et al., 2009) or the Cathepsin S (CatS) produced by microglia (Clark et al., 2009). CX3CL1 is also a substrate for β-secretase (BACE1), and γ-secretase, involved in the APP cleavage to generate Aβ (Fan et al., 2019).
Age-dependent changes in CX3CL1 shedding can be the result of changes in the amount of protease available to process mCX3CL1, as consequence of changes in their synthesis or degradation. Alternatively, age-dependent changes in the enzymatic efficiency of proteases may be caused by changes in the amount of endogenous inhibitors or enzyme enhancers (Postina, 2012; Mathews and Levy, 2016; Shi et al., 2018; Sharma et al., 2022).
ADAM10 and ADAM17 are the best characterized members of the disintegrin and metalloprotease (ADAM) family (Black and White, 1998) involved in the processing of membrane-associated proteins such cytokine receptors, chemokines, adhesion molecules, and growth factors. Their targets include TNFα, TGFβ, Notch, CX3CL1, APP, the low-density lipoprotein receptor-related protein 1 (LRP1), and the anti-aging protein α-klotho (Blobel, 1997; Peschon et al., 1998; Borrell-Pages et al., 2003; Liu et al., 2009; Chen et al., 2014; Brifault et al., 2017).
The enzymes, ADAM 10, and ADAM 17 are implicated in the α-secretase non-amyloidogenic APP processing, leading to the production of soluble APP (APPsα), which exhibits properties of neuroprotection, memory-enhancer, and regulation of neuronal excitability, synaptogenesis, and synaptic plasticity (Turner et al., 2003; Reinhard et al., 2005; Zheng and Koo, 2006). Mass spectrometric analysis revealed that 23-month-old rats showed a reduction of soluble APPsα in cisternal CSF compared with that observed in 3- and 13-month-old animals (Anderson et al., 1999). The reduction of APPsα has a positive correlation (r = 0.52–0.57, p < 0.001) with the performance in spatial memory tasks observed in young and aged rats (Anderson et al., 1999). Accordingly, ADAM 17 activity in cortex and hippocampus changes with aging (Bertoldi et al., 2017). Aged rats show a reduced ADAM 17 activity, whereas the amyloidogenic BACE activity is like that in young rats (Bertoldi et al., 2017). This age-dependent reduction in ADAM 17 activity is accompanied with a reduced performance in aversive memory test (Bertoldi et al., 2017). By contrast, daily moderate treadmill exercise for two weeks had not impact on ADAM 17 and BACE activities at various ages (Bertoldi et al., 2017).
CatS is a member of the family of cysteine lysosomal proteases preferentially expressed in macrophages and microglia and released from them in response to neurotrophic factors and inflammatory mediators. It plays a role, together with ADAM 17 and ADAM10, in CX3CL1 shedding (Petanceska et al., 1996; Liuzzo et al., 1999a,b; Turk et al., 2000; Nakanishi, 2003; Wendt et al., 2008).
Age-dependent changes in the protein expression of CatS has been detected by Western Blot in cerebral cortex, cerebellum, brainstem, and spinal cord. In 1-wk-old mice, only pro- CatS is found. Starting at 6 months, age-dependent upregulation of prepro-form, pro-form, and mature form of CatS is evidenced (Wendt et al., 2008). Furthermore, analysis of gene expression by DNA microarrays reveals that CatS gene expression was higher in 25-month-old mice than that observed in 5-month-old mice (Park et al., 2009).
Double immunofluorescent labeling revealed that most CatS positive cells in the CNS are microglia (PT66), followed by astrocytes (GFAP) and a few neurons (Neu) or endothelial cells (von Willebrand factor). Aging upregulates the number and the labeling intensity of CatS positive cells. The number of glia-like cells as well as neurons expressing CatS in cortex and brainstem clearly increases in the aged mouse brain (Wendt et al., 2008).
Age-dependent changes in the activity of CatS are not only due to the increased, but also to age-dependent changes of the level of the cysteine-protease inhibitor cystatin C (CysC, also known as γ-trace), an endogenous inhibitor of CatS, a member of the endogenous cysteine-protease inhibitors (Barrett et al., 1986; Mathews and Levy, 2016), which is found in neurons, astrocytes, endothelial, and microglia cells (Yasuhara et al., 1993; Palm et al., 1995; Miyake et al., 1996). Besides of inhibiting CatS, CysC also inhibits the cysteine-proteases cathepsin (Cat) B, Cat H, Cat K, and Cat L (Turk et al., 2008). In turn, CysC is targeted and inactivated by aspartyl-protease Cat D and the serine-protease elastase (Lenarcic et al., 1991; Abrahamson et al., 1991a,b).
Hu CysC is a 120 aa protein which is preceded by a26 aa amino terminal that endows it with the capacity of being secreted (Turk and Bode, 1991; Turk et al., 1997). The novo synthesized CysC can access the endosomal-lysosomal compartment via an intracellular pathway or most of it is secreted to the extracellular compartment (Zucker-Franklin et al., 1987; Chapman et al., 1990; Barka et al., 1992; Tavera et al., 1992; Paraoan et al., 2001) and, only a fraction remains within exosomes (Ghidoni et al., 2011). Secreted CysC can be found in cell surfaces or in the extracellular matrix (Calkins and Sloane, 1995; Sastre et al., 2004; Kolodziejczyk et al., 2010), where it can inhibit the activity of released cathepsins, or alternatively, it can be internalized into other cells (Merz et al., 1997; Ekstrom et al., 2008). CysC is internalized via endocytosis, allowing its access into the endosomal-lysosomal compartment (Merz et al., 1997; Pierre and Mellman, 1998; Ekstrom et al., 2008) where it can inhibit lysosomal cathepsins and be degraded by aspartyl-protease Cat D (Wallin et al., 2013).
CysC secretion appears to be age-dependent since CysC levels in serum correlates with age in dogs, cats, and humans (Ohara et al., 2012; Ghys et al., 2015; Iwasa et al., 2022). Changes of CysC brain expression and secretion are found in several neurological disorders, and also correlates with animal models of neurodegeneration (Mathews and Levy, 2016). However, up to date, conclusive studies on age-dependent changes in CysC expression are lacking.
1.7. Differential effects of CX3CL1 forms
As described, sCX3CL1 with different sizes are the result of cleavage of the mCX3CL1 by proteases (Bazan et al., 1997; Figures 1, 3). Notoriously, both variants showed selective functions (Bazan et al., 1997). sCX3CL1 have a potent chemoattractant activity for T cells and monocytes, favoring cellular migration and recruitment, whereas the mCX3CL1 endowed primary endothelial cells with recruitment and strong adhesion to leukocytes (Bazan et al., 1997; Imai et al., 1997; Hermand et al., 2008; Ostuni et al., 2014). However, mCX3CL1 and sCX3CL1 variants can also share functions, for instance, they can reduce the expression of microglia pro-inflammatory genes, like NOS, IL1β, TNFα, and IL6, attenuate LPS-induced microglial cell activation in vitro, the age-related increase in microglial cell activation, and reduce neuronal death induced by microglia activated by IFNγ or LPS (Zujovic et al., 2000; Mizuno et al., 2003; Lyons et al., 2009).
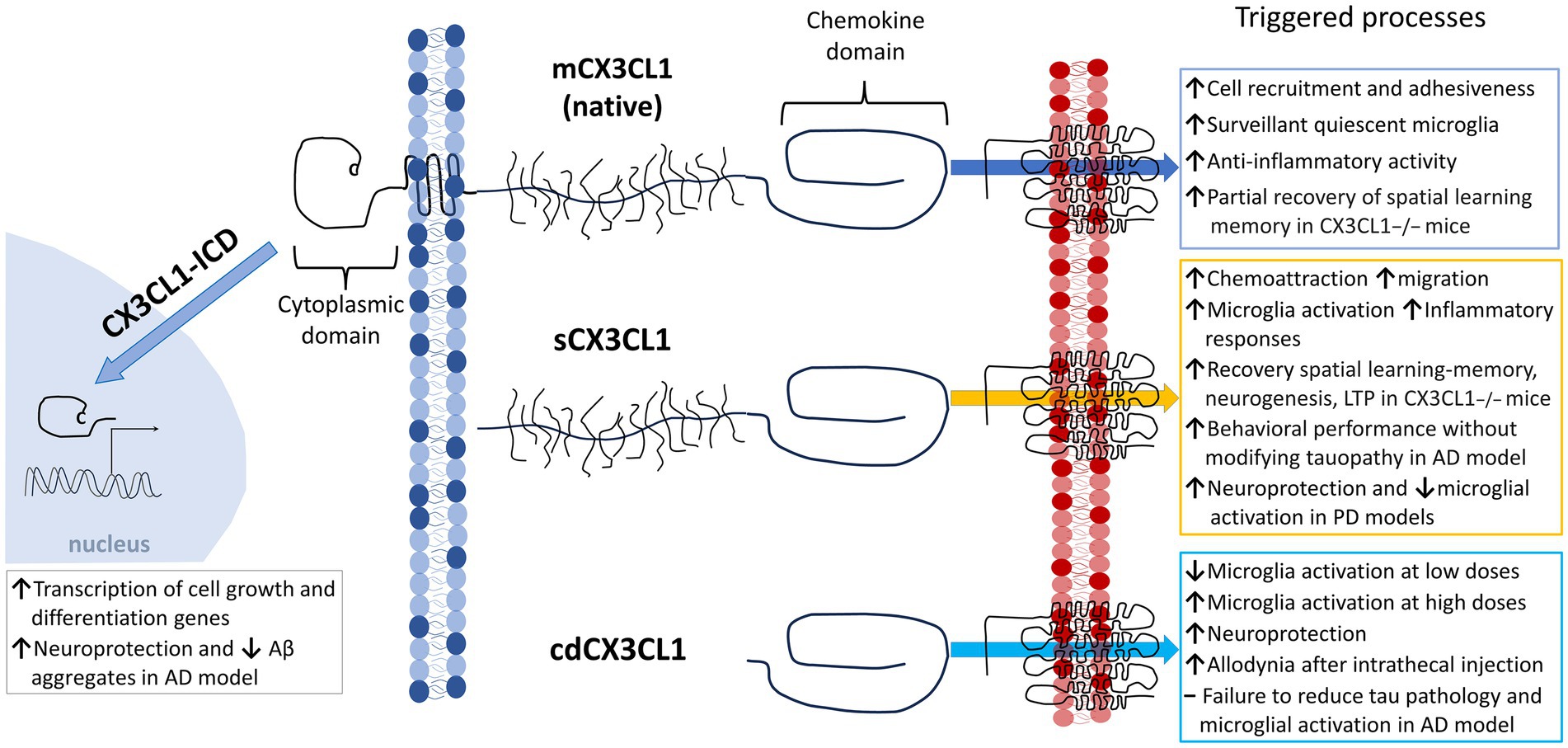
Figure 3. Differential functions of the various Fractalkine forms. Scheme illustrating different processes triggered by the activation of microglia by CX3CR1 binding mCX3CL1, sCX3CL1, and cdCX3CL1. Newly reported CX3CL1-ICD proposed functions are included. Within sCX3CL1s, we considered fragments resulting from ectodomain shedding or recombinant synthesis based on the action of proteases. Note that cdCX3CL1 is considered to be independent from sCX3CL1.
What is striking are the findings that binding of mCX3CL1 with CX3CR1 acts as a cell–cell adhesion molecule and has anti-inflammatory activity, keeping the microglia in a state of surveillance (Ludwig and Mentlein, 2008; Lee et al., 2014; Williams et al., 2014; Mecca et al., 2018; Figure 3). By contrast, the binding of sCX3CL1 with CX3CR1 can induce inflammatory activation of microglia, having a proinflammatory and a chemoattractant effect for microglia and other innate immune cells (Jones et al., 2010). Differential functions between mCX3CL1 and sCX3CL1 were observed in the APP/PS1 mouse model, in which Cx3cl1-deficient APP/PS1 animals exhibited reduced Aβ deposition but enhanced neuronal tau phosphorylation (Lee et al., 2014). Surprisingly, reduced Aβ deposition and enhanced neuronal tau phosphorylation were unmodified by the transgenic expression of sCX3CL1 isoforms, suggesting that mCX3CL1 regulates both processes (Lee et al., 2014).
Physiologically, the CX3CL1/CX3CR1 axis appears to be involved in the regulation of cognition and memory (Rogers et al., 2011). For instance, it has been reported that sCX3CL1 is beneficial in rescuing memory, whereas the mCX3CL1 lacks this effect (Winter et al., 2020). Cx3cl1−/− mice exhibit a significant cognitive impairment in a fear conditioning task for long-term memory and in the Barnes maze task for assessing spatial learning and memory (Winter et al., 2020). These deficits correlate with reduced hippocampal neurogenesis and LTP. After 2 months of the injection of rAAV expressing a mutated, obligate membrane-bound form of mCX3CL1 or a sCX3CL1 in Cx3cl1−/− mice, CX3CL1 was detected by ELISA in brain tissue. In Cx3cl1−/− mice injected with rAAV expressing mCX3CL1, CX3CL1 level was like that of WT mice, whereas in mice injected with AAV expressing sCX3CL1, high levels of CX3CL1 (almost doubling WT level) were observed. The Cx3cl1−/− mice treated with rAAV expressing mCX3CL1 showed an improved performance in the accelerating rotarod test compared to WT mice, whereas those injected with rAAV expressing sCX3CL1, showed similar motor performance to WT. Remarkable, treatment with AAV-mCX3CL1 partially restored spatial learning and memory in Cx3cl1−/− mice, but did not rescue long-term memory, or neurogenesis (Winter et al., 2020). In contrast, treatment with AAV-sCX3CL1 partially corrected changes in both cognitive and motor function and restored neurogenesis and LTP to levels like WT animals (Winter et al., 2020). These results could indicate that treatments based on restoration of the level of sCX3CL1 may be a viable therapeutic target for dysfunctions in aging and disease. How much of these differences rely on the CX3CL1 levels attained by each kind of transgenic or in the functional capacity of mutated mCX3CL1 protein in the experiments (Winter et al., 2020) remain as open questions.
To evaluate the impact of sCX3CL1 forms upon pathological hallmarks of AD, Lee (Lee et al., 2014) used a mouse line expressing exclusively sCX3CL1, obtained by introducing an artificial chromosome transgene encoding truncated CX3CL1 (SolTg) to Cx3cl1−/− mice. These Cx3cl1−/−; SolTg and Cx3cl1−/−mice were mated with APP/PS1 mice, resulting in APP/PS1; Cx3cl1−/−; SolTg and APP/PS1; Cx3cl1−/− mouse lines, respectively. As expected, CX3CL1-deficient APP/PS1 mice, as previously reported for CX3CR1-deficient APP/PS1 mice (Lee et al., 2010), also exhibited reduced Aβ deposition on cortex and hippocampus compared with APP/PS1 mice. However, APP/PS1;Cx3cl1−/−;SolTg mice brains also exhibited reduced Aβ deposition with levels undistinguishable from those in Cx3cl1−/−- and Cx3cl1−/−- deficient APP/PS1. Surprisingly, despite reduced Aβ deposition, Cx3cl1−/−-deficient APP/PS1 mice demonstrated elevated phosphorylated tau levels (Lee et al., 2014). In fact, Lee et al. (2014) proposed that the absence of mCX3CL1 reduces Aβ deposition via p38 MAPK-mediated activation of microglia and the SRA (macrophage scavenger receptor 1), (encoded by msr1)-dependent phagocytosis (Lee et al., 2014; Cornejo et al., 2018). Nevertheless, the assignation of mCX3CL1 functions by subtraction or default should be taken with caution, because no direct manipulation of mCX3CL1 was performed and a measurement of the level of sCX3CL1 in the APP/PS1 Cx3cl1−/−;SolTg mice were not performed. If the amount, regulation, or activity of metalloproteases are different in APP/PS1 Cx3cl1−/−;SolTg mice, the level of CX3CL1 active fragments could change.
Disruption of CX3CL1 signaling has shown to have opposite effects on Aβ and tau aggregation on mouse AD models. The sCX3CL1 overexpression in the hippocampus does not affect Aβ deposits and microglia activation found in the rTg4510 mouse model (Nash et al., 2013). Conversely, as described in the section of neurodegenerative diseases, Nash et al. (2013) show that the restricted focalized overexpression of sCX3CL1 in the hippocampus of the rTg4510 mouse model can ameliorate tauopathy, without significant improvement in performance in the radial arm water maze (Nash et al., 2013). Notably, through a change in the methodological strategy, 5-month-old rTg4510 mice received ICV injections with serotype 4 AAV (AAV4) coding for sCX3CL1. This AAV4 has the capacity of easily infect cells lining the ventricular system in the CNS. Thus, sCX3CL1 was overexpressed by cells lining the ventricles, which was associated with significantly improved cognitive performance on the mouse recognition and radial arm water maze (Finneran et al., 2019). Behavioral benefits were attained without reduction in tau hyperphosphorylation, hippocampal atrophy, or microglial cell number (Finneran et al., 2019).
Recently, it was reported that recombinant full-length hu sCX3CL1 and the small soluble fragment constituted by its chemokine domain (cdCX3CL1) show, depending on the assay, functional differences (Finneran et al., 2023). Both CX3CL1 isoforms show modest differences in their receptor affinity but similar potency and efficacy using a cell-based assay of CX3CR1-dependent reduction in forskolin-induced cAMP.
By contrast, sCX3CL1 show a 10-fold higher potency than that of cdCX3CL1 in the β-arrestin recruitment assay. Furthermore, sCX3CL1 is also 10-fold more potent than cdCX3CL1 in decreasing LPS-induced TNFα release by microglia, which suggests that CX3CR1 signaling through β-arrestin may be involved in the anti-inflammatory effect of CX3CL1. When recombinant sCX3CL1 is administered at concentrations above normal basal plasma level, it induces an increase in the inflammatory microglial activation. The opposite effect is observed with cdCX3CL1, but it is 10-fold less potent (Finneran et al., 2023).
Differential effects elicited by sCX3CL1 and cdCX3CL1 has also been reported on neuropathic pain. In response to nerve injury, microglial activation and CX3CL1 expression are increased. CX3CR1 antagonists can relieve nerve injury-induced pain in rats (Milligan et al., 2004; Sun et al., 2013; Sessler et al., 2021). Furthermore, intrathecal administration of cdCX3CL1 but not sCX3CL1 enhance mechanical allodynia, whereas inhibition of CatS has antihyperalgesic and antiallodynic effects in a neuropathic pain rat models and reduced microglia activation in the spinal cord (Clark et al., 2007). In fact, intrathecal administration in the spinal cord of recombinant CatS induce hyperalgesia and allodynia in naïve rats (Clark et al., 2007). CatS generates a 55 KDa sCX3CL1 (Fonovic et al., 2013) shorter than that generated by ADAM10/17 (about 85 KDa) (Roche et al., 2016) and similar in size to the 76 aa of the chemokine domain.
To evaluate the effect of cdCX3CL1 on tau pathology and its behavioral impact, a mouse line (CX3CL1105Δ) was developed. CX3CL1105Δ expresses exclusively cdCX3CL1, lacking the mucin stalk, and overexpressing hu Tau protein for developing tau neurofibrillary pathology (Bemiller et al., 2018). Despite of being overexpressed, cdCX3CL1 fails to reduce tau pathology and microglial activation (Bemiller et al., 2018). In addition, expression of CX3CR1 in the microglial cell membrane is significantly reduced in CX3CL1105Δ mice, mimicking a CX3CR1 deficiency, suggesting that cdCX3CL1 overexpression downregulates CX3CR1 expression by microglia and increases tau pathology. Similar results were observed using other AD mice models carrying the APP/PS1 double mutation and expressing only the chemokine domain of CX3CL1 (CX3CL1105Δ). APP/PS1:CX3CL1105Δ mice presented a more severe pathological phenotype and time course than that observed in APP/PS1 mice lacking the entire gene (APP/PS1:Cx3cl1−/−) (Lee et al., 2014).
Additionally, recent reports show that successive cleavage of mCX3CL1 by β- and γ-secretases, release the intracellular domain of CX3CL1 (CX3CL1-ICD), which translocate to the cell nucleus and regulates the transcription of several genes important for cell growth and differentiation (Fan et al., 2019). Interestingly, Tg-CX3CL1-ICD mice did not exhibit overgrowth, whereas 5xFAD, a mouse model of AD that overexpresses CX3CL1-ICD exhibits reduced Aβ deposition and neuronal loss, the latter, likely due, to enhanced neurogenesis. These results demonstrate a beneficial effect of the CX3CL1-ICD in an AD mouse model independent of the activation of CX3CR1 (Fan et al., 2019).
The role of soluble and membrane-bound CX3CL1 has also been examined in PD mouse models (Morganti et al., 2012; Nash et al., 2015). SNpc injections of AAV vectors for sCX3CL1, a mutant mCX3CL1 (with mutations R337A + R338A that abolish ADAM10/17 cleavage into sCX3CL1) or a control vector expressing GFP were performed in 3-4-month-old Cx3cl1−/− mice. Six weeks after the viral vector injection, mice received an intraperitoneal MPTP injection to induce PD-like disorder (Morganti et al., 2012). The soluble, but not the mCX3CL1 reduced the MPTP-induced motor impairment, dopaminergic neurons loss, and microglial activation in the SNpc. These results were associated to the decreased release of inflammatory cytokines (TNFα and IL1β) and microglial expression of CD68 and CD11b (Morganti et al., 2012). These results suggest that in PD, the ADAM10/17 cleavage variant sCX3CL1 (aa 1–336) appears to be the one that confers neuroprotection and not the mCX3CL1.
The role of various molecular forms of CX3CL1 was also studied in a rat model of PD induced by SNpc injection of rAAV coding for hu α-synuclein in rats (Nash et al., 2015). Rats that received a co-injection of viral vectors coding for hu α-synuclein and sCX3CL1 exhibited reduced loss of tyrosine hydroxylase and Neu-N labeling in the SNpc, indicative of neuroprotection of dopaminergic neurons. Co-injection of viral vectors carrying other fractalkine forms showed a loss of dopaminergic neurons comparable to those observed in controls receiving injection of huα-synuclein alone or coinfected with the control viral vector coding for GFP. Similar neuroprotective effect was observed in animals expressing fractalkine forms in astrocytes, thanks to AAV constructs containing the fibrillary acid protein (GFAP) promoter (Nash et al., 2015).
2. Conclusion
Microglia are essential components of the homeostatic response of the CNS. They are constantly under the influence of “off” and “on” signals involved in the dialog between microglia and other CNS cells, both in physiological and pathological settings. ‘Off signals” tend to maintain microglia in a surveillant status and include the CX3CL1/CX3CR1 and CD200/CD200R axes, with ligands presented by neurons and their receptors by microglia, soluble mediators like TGFβ, IL10, IL34, and colony stimulating factor 1 (CSF1), which are secreted by healthy neurons and their receptors, TGFβR and CSF1R, are present, among many cells, in microglia, as well as receptors for different neurotransmitters (glutamate, GABA, acetylcholine and noradrenaline) (Manich et al., 2019). Thus, in various models of neuroinflammation and neurodegeneration, impairment of the CX3CL1/CX3CR1 or CD200/CD200R axes, or similarly, when microglial receptors for TGFβ or CSF1 are deleted, microglia activation become more neurotoxic (Hoek et al., 2000; Cardona et al., 2006; Buttgereit et al., 2016). On the other hand, “on” signals that activate microglia, include soluble factors like cytokines/chemokines, trophic factors, having receptors on activated microglia. In addition, microglia present several membrane-bound receptors that recognize and bind molecules expressed by damaged neurons (Manich et al., 2019). Thus, a constellation of molecules regulates microglial function through numerous molecules. The fact that fractalkine axis is only one among multiple molecular systems contributing to the regulation of microglia, should be a warning note for interpreting negative results obtained through the manipulation of a single component of the CX3CL1/CX3CR1 axis. It is possible that homeostatic changes in other system could counterbalance the effects of CX3CL1 or CX3CR1 deletion.
Current evidence stress the importance of the various forms of CX3CL1 exhibiting regulatory roles on several physiological processes that include neuronal cell migration, synaptic pruning, synaptic maturation, microglial activation, neuroprotection, and cognitive functions. Dual pro- and anti-inflammatory effects relying on different CX3CL1 forms may represent a strategy for achieving brain homeostasis under various environmental conditions. Therefore, the regulation of both the level of CX3CL1 and the balance among its various forms, appears as a fine-tuning leading to the regulation of microglial functional state and the release of pro- or anti-inflammatory cytokines in response to the microenvironment challenges.
Thus, the search for identifying functional forms of CX3CL1 has yield several forms that could be separated in two categories, mCX3CL1 anchored to the neural plasma membrane and sCX3CL1, resulting from the cleavage of mCX3CL1 by metalloproteinases ADAM17, ADAM10 (α-secretase), CatS, matrix metalloproteinase 2 (MMP-2), or secretases involved in the Aβ processing. Each protease helps to generate a different sCX3CL1 form with a different size and apparently some functional properties, in addition to some newly found additions like the intracellular regions or the chemokine domain, which still require additional scrutiny.
Most of sCX3CL1 and mCX3CL1forms have in common the chemokine domain. However, the human full-length recombinant sCX3CL1 is 10-fold more potent in biological assays, in reducing LPS-mediated TNFα release, and for increasing inflammatory activation of microglia than cdCX3CL1 (Finneran et al., 2023). On the other hand, intrathecal administration of cdCX3CL1 but not sCX3CL1 enhanced mechanical allodynia and spinal cord microglia activation (Clark et al., 2007). The main differences in potency can be explained by conformational changes in the chemokine domain when it is separated from the mucine-like stalk domain, whereas the major allodynia may be the consequence of a better access of smaller proteins into the spinal cord structures. Nevertheless, up to now, it is not understood how different CX3CL1 forms acting on a sole CX3CR1 could trigger different, even opposite cellular responses, yet. Why the mCX3CL1 has, in general, an anti-inflammatory effect, whereas binding to the sCX3CL1 results in an inflammatory deleterious activation? A possible explanation to this question may be related to the fact that sCX3CL1 lose the property of physical anchoring between neurons and microglia, property that is only observed for the mCX3CL1. Such intimate cell-to-cell interaction could promote neuron–glia interactions required by other trophic factors and membrane-bound proteins like CD200 (Zhang et al., 2018) facilitating the activation of parallel intracellular pathways. To answer this puzzling and central question will allow us to understand the regulatory role of CX3CL1 in the cytotoxic activation leading to neurodegeneration.
Recently, the rule that all CX3CL1 forms perform their functions by binding CX3CR1 has been challenged by at least one exception, the intracellular domain of fractalkine, CX3CL1-ICD. CX3CL1-ICD is generated through the successive cleavage of mCX3CL1 by β- and γ-secretases. After being released in the cytoplasm, it is translocated into the cell nucleus for regulating genes important for cell growth and differentiation. This means that the fractalkine system has a wider spectrum of functions than originally thought.
2.1. Outlook
Worth emphasizing, the CX3CL1/CX3CR1 axis is under the influence of regulatory factors affected by the functional status of the brain, being modified by aging and inflammatory activation. Aging is associated with neuroinflammation and is the main risk factor for most neurodegenerative diseases. It has been established that the level expression of CX3CL1 and CX3CR1, the level of proteases and their inhibitors, and the microglial reactivity are all affected by aging. More studies are needed to improve our comprehension of aging effects and their impact on the activity ratios among different forms of CX3CL1.
The potential therapeutic role of the CX3CL1/CX3CR1 axis has been explored in animal models with controversial results. Further study requires a more complete and extensive description of the multiple factors associated with the experimental manipulation. The lack of an exhaustive evaluation of the functional properties of genetic constructs could mask an artefactual loss of affinity or steric impairment for an efficient binding, among several additional considerations that should be considered.
Author contributions
JE conducted literature search and wrote the manuscript. LE-vB drew the figures and conducted literature search. RB conducted literature search, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The current study was supported by grants Fondo Nacional del Desarrollo de la Ciencia y Tecnología (FONDECYT) 1211359 (JE) y 1221028 (RB); ANID Redes ‑190187 (RB).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrahamson, M., Buttle, D. J., Mason, R. W., Hansson, H., Grubb, A., Lilja, H., et al. (1991a). Regulation of cystatin C activity by serine proteinases. Biomed. Biochim. Acta 50, 587–593.
Abrahamson, M., Mason, R. W., Hansson, H., Buttle, D. J., Grubb, A., and Ohlsson, K. (1991b). Human cystatin C. role of the N-terminal segment in the inhibition of human cysteine proteinases and in its inactivation by leucocyte elastase. Biochem. J. 273, 621–626. doi: 10.1042/bj2730621
Absinta, M., Lassmann, H., and Trapp, B. D. (2020). Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 33, 277–285. doi: 10.1097/WCO.0000000000000818
Ain, Q., Schmeer, C., Penndorf, D., Fischer, M., Bondeva, T., Forster, M., et al. (2018). Cell cycle-dependent and -independent telomere shortening accompanies murine brain aging. Aging (Albany NY) 10, 3397–3420. doi: 10.18632/aging.101655
Alarcon, R., Fuenzalida, C., Santibanez, M., and von Bernhardi, R. (2005). Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J. Biol. Chem. 280, 30406–30415. doi: 10.1074/jbc.M414686200
Anderson, J. J., Holtz, G., Baskin, P. P., Wang, R., Mazzarelli, L., Wagner, S. L., et al. (1999). Reduced cerebrospinal fluid levels of alpha-secretase-cleaved amyloid precursor protein in aged rats: correlation with spatial memory deficits. Neuroscience 93, 1409–1420. doi: 10.1016/s0306-4522(99)00244-4
Angelopoulou, E., Paudel, Y. N., Shaikh, M. F., and Piperi, C. (2020). Fractalkine (CX3CL1) signaling and neuroinflammation in Parkinson's disease: potential clinical and therapeutic implications. Pharmacol. Res. 158:104930. doi: 10.1016/j.phrs.2020.104930
Araque, A., Carmignoto, G., Haydon, P. G., Oliet, S. H., Robitaille, R., and Volterra, A. (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. doi: 10.1016/j.neuron.2014.02.007
Arli, B., Irkec, C., Menevse, S., Yilmaz, A., and Alp, E. (2013). Fractalkine gene receptor polymorphism in patients with multiple sclerosis. Int. J. Neurosci. 123, 31–37. doi: 10.3109/00207454.2012.723079
August, I., Semendeferi, K., and Marchetto, M. C. (2022). Brain aging, Alzheimer's disease, and the role of stem cells in primate comparative studies. J. Comp. Neurol. 530, 2940–2953. doi: 10.1002/cne.25394
Bachstetter, A. D., Xing, B., de Almeida, L., Dimayuga, E. R., Watterson, D. M., and Van Eldik, L. J. (2011). Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta). J. Neuroinflammation 8:79. doi: 10.1186/1742-2094-8-79
Banati, R. B., Gehrmann, J., Schubert, P., and Kreutzberg, G. W. (1993). Cytotoxicity of microglia. Glia 7, 111–118. doi: 10.1002/glia.440070117
Barka, T., van der Noen, H., and Patil, S. (1992). Cysteine proteinase inhibitor in cultured human medullary thyroid carcinoma cells. Lab. Investig. 66, 691–700.
Barrett, A. J., Fritz, H., Grubb, A., Isemura, S., Jarvinen, M., Katunuma, N., et al. (1986). Nomenclature and classification of the proteins homologous with the cysteine-proteinase inhibitor chicken cystatin. Biochem. J. 236:312. doi: 10.1042/bj2360312
Bazan, J. F., Bacon, K. B., Hardiman, G., Wang, W., Soo, K., Rossi, D., et al. (1997). A new class of membrane-bound chemokine with a CX3C motif. Nature 385, 640–644. doi: 10.1038/385640a0
Beltran-Castillo, S., Eugenin, J., and von Bernhardi, R. (2018). Impact of aging in microglia-mediated D-serine balance in the CNS. Mediat. Inflamm. 2018:7219732. doi: 10.1155/2018/7219732
Beltran-Castillo, S., Olivares, M. J., Contreras, R. A., Zuniga, G., Llona, I., von Bernhardi, R., et al. (2017). D-serine released by astrocytes in brainstem regulates breathing response to CO2 levels. Nat. Commun. 8:838. doi: 10.1038/s41467-017-00960-3
Bemiller, S. M., Maphis, N. M., Formica, S. V., Wilson, G. N., Miller, C. M., Xu, G., et al. (2018). Genetically enhancing the expression of chemokine domain of CX(3)CL1 fails to prevent tau pathology in mouse models of tauopathy. J. Neuroinflammation 15:278. doi: 10.1186/s12974-018-1310-6
Bender, A. R., Volkle, M. C., and Raz, N. (2016). Differential aging of cerebral white matter in middle-aged and older adults: a seven-year follow-up. NeuroImage 125, 74–83. doi: 10.1016/j.neuroimage.2015.10.030
Bertoldi, K., Cechinel, L. R., Schallenberger, B., Meireles, L., Basso, C., Lovatel, G. A., et al. (2017). Aging process alters hippocampal and cortical secretase activities of Wistar rats. Behav. Brain Res. 317, 374–381. doi: 10.1016/j.bbr.2016.09.066
Bertollini, C., Ragozzino, D., Gross, C., Limatola, C., and Eusebi, F. (2006). Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology 51, 816–821. doi: 10.1016/j.neuropharm.2006.05.027
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Black, R. A., and White, J. M. (1998). ADAMs: focus on the protease domain. Curr. Opin. Cell Biol. 10, 654–659. doi: 10.1016/s0955-0674(98)80042-2
Blesa, J., Phani, S., Jackson-Lewis, V., and Przedborski, S. (2012). Classic and new animal models of Parkinson's disease. J. Biomed. Biotechnol. 2012:845618. doi: 10.1155/2012/845618
Blobel, C. P. (1997). Metalloprotease-disintegrins: links to cell adhesion and cleavage of TNF alpha and notch. Cells 90, 589–592. doi: 10.1016/s0092-8674(00)80519-x
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Boddeke, E. W., Meigel, I., Frentzel, S., Biber, K., Renn, L. Q., and Gebicke-Harter, P. (1999). Functional expression of the fractalkine (CX3C) receptor and its regulation by lipopolysaccharide in rat microglia. Eur. J. Pharmacol. 374, 309–313. doi: 10.1016/s0014-2999(99)00307-6
Boillee, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22, 269–285. doi: 10.1016/j.celrep.2017.12.039
Borrell-Pages, M., Rojo, F., Albanell, J., Baselga, J., and Arribas, J. (2003). TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 22, 1114–1124. doi: 10.1093/emboj/cdg111
Bourd-Boittin, K., Basset, L., Bonnier, D., L'Helgoualc'h, A., Samson, M., and Theret, N. (2009). CX3CL1/fractalkine shedding by human hepatic stellate cells: contribution to chronic inflammation in the liver. J. Cell. Mol. Med. 13, 1526–1535. doi: 10.1111/j.1582-4934.2009.00787.x
Bradl, M., and Lassmann, H. (2010). Oligodendrocytes: biology and pathology. Acta Neuropathol. 119, 37–53. doi: 10.1007/s00401-009-0601-5
Brifault, C., Gilder, A. S., Laudati, E., Banki, M., and Gonias, S. L. (2017). Shedding of membrane-associated LDL receptor-related protein-1 from microglia amplifies and sustains neuroinflammation. J. Biol. Chem. 292, 18699–18712. doi: 10.1074/jbc.M117.798413
Buttgereit, A., Lelios, I., Yu, X., Vrohlings, M., Krakoski, N. R., Gautier, E. L., et al. (2016). Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406. doi: 10.1038/ni.3585
Calkins, C. C., and Sloane, B. F. (1995). Mammalian cysteine protease inhibitors: biochemical properties and possible roles in tumor progression. Biol. Chem. Hoppe Seyler 376, 71–80.
Cambien, B., Pomeranz, M., Schmid-Antomarchi, H., Millet, M. A., Breittmayer, V., Rossi, B., et al. (2001). Signal transduction pathways involved in soluble fractalkine-induced monocytic cell adhesion. Blood 97, 2031–2037. doi: 10.1182/blood.v97.7.2031
Cardona, S. M., Kim, S. V., Church, K. A., Torres, V. O., Cleary, I. A., Mendiola, A. S., et al. (2018). Role of the Fractalkine receptor in CNS autoimmune inflammation: new approach utilizing a mouse model expressing the human CX3CR1(I249/M280) variant. Front. Cell. Neurosci. 12:365. doi: 10.3389/fncel.2018.00365
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924. doi: 10.1038/nn1715
Castro-Sanchez, S., Garcia-Yague, A. J., Kugler, S., and Lastres-Becker, I. (2019). CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: implications in tauopathies. Redox Biol. 22:101118. doi: 10.1016/j.redox.2019.101118
Castro-Sanchez, S., Garcia-Yague, A. J., Lopez-Royo, T., Casarejos, M., Lanciego, J. L., and Lastres-Becker, I. (2018). Cx3cr1-deficiency exacerbates alpha-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson's disease. Glia 66, 1752–1762. doi: 10.1002/glia.23338
Cerpa, W., Ramos-Fernandez, E., and Inestrosa, N. C. (2016). Modulation of the NMDA receptor through secreted soluble factors. Mol. Neurobiol. 53, 299–309. doi: 10.1007/s12035-014-9009-x
Chan, C. C., Tuo, J., Bojanowski, C. M., Csaky, K. G., and Green, W. R. (2005). Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol. Histopathol. 20, 857–863. doi: 10.14670/HH-20.857
Chang, B., Hawes, N. L., Hurd, R. E., Davisson, M. T., Nusinowitz, S., and Heckenlively, J. R. (2002). Retinal degeneration mutants in the mouse. Vis. Res. 42, 517–525. doi: 10.1016/s0042-6989(01)00146-8
Chapman, H. A. Jr., Reilly, J. J. Jr., Yee, R., and Grubb, A. (1990). Identification of cystatin C, a cysteine proteinase inhibitor, as a major secretory product of human alveolar macrophages in vitro. Am. Rev. Respir. Dis. 141, 698–705. doi: 10.1164/ajrccm/141.3.698
Chen, S., Luo, D., Streit, W. J., and Harrison, J. K. (2002). TGF-beta1 upregulates CX3CR1 expression and inhibits fractalkine-stimulated signaling in rat microglia. J. Neuroimmunol. 133, 46–55. doi: 10.1016/s0165-5728(02)00354-5
Chen, C. D., Tung, T. Y., Liang, J., Zeldich, E., Tucker Zhou, T. B., Turk, B. E., et al. (2014). Identification of cleavage sites leading to the shed form of the anti-aging protein klotho. Biochemistry 53, 5579–5587. doi: 10.1021/bi500409n
Cho, S. H., Sun, B., Zhou, Y., Kauppinen, T. M., Halabisky, B., Wes, P., et al. (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 286, 32713–32722. doi: 10.1074/jbc.M111.254268
Clark, A. K., Yip, P. K., Grist, J., Gentry, C., Staniland, A. A., Marchand, F., et al. (2007). Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc. Natl. Acad. Sci. U. S. A. 104, 10655–10660. doi: 10.1073/pnas.0610811104
Clark, A. K., Yip, P. K., and Malcangio, M. (2009). The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J. Neurosci. 29, 6945–6954. doi: 10.1523/JNEUROSCI.0828-09.2009
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Munch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U. S. A. 115, E1896–E1905. doi: 10.1073/pnas.1800165115
Cohen, J., and Torres, C. (2019). Astrocyte senescence: evidence and significance. Aging Cell 18:e12937. doi: 10.1111/acel.12937
Coleman, P. D., and Flood, D. G. (1987). Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiol. Aging 8, 521–545. doi: 10.1016/0197-4580(87)90127-8
Combadiere, C., Feumi, C., Raoul, W., Keller, N., Rodero, M., Pezard, A., et al. (2007). CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Invest. 117, 2920–2928. doi: 10.1172/JCI31692
Conboy, M. J., Conboy, I. M., and Rando, T. A. (2013). Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging Cell 12, 525–530. doi: 10.1111/acel.12065
Conboy, I. M., Conboy, M. J., Wagers, A. J., Girma, E. R., Weissman, I. L., and Rando, T. A. (2005). Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764. doi: 10.1038/nature03260
Cornejo, F., and von Bernhardi, R. (2013). Role of scavenger receptors in glia-mediated neuroinflammatory response associated with Alzheimer's disease. Mediat. Inflamm. 2013:895651. doi: 10.1155/2013/895651
Cornejo, F., Vruwink, M., Metz, C., Munoz, P., Salgado, N., Poblete, J., et al. (2018). Scavenger receptor-a deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer's disease pathophysiology. Brain Behav. Immun. 69, 336–350. doi: 10.1016/j.bbi.2017.12.007
Dawson, T. M., Ko, H. S., and Dawson, V. L. (2010). Genetic animal models of Parkinson's disease. Neuron 66, 646–661. doi: 10.1016/j.neuron.2010.04.034
Dawson, M. R., Polito, A., Levine, J. M., and Reynolds, R. (2003). NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 24, 476–488. doi: 10.1016/s1044-7431(03)00210-0
de la Fuente, A. G., Queiroz, R. M. L., Ghosh, T., McMurran, C. E., Cubillos, J. F., Bergles, D. E., et al. (2020). Changes in the oligodendrocyte progenitor cell proteome with ageing. Mol. Cell. Proteomics 19, 1281–1302. doi: 10.1074/mcp.RA120.002102
de Lau, L. M., and Breteler, M. M. (2006). Epidemiology of Parkinson's disease. Lancet Neurol. 5, 525–535. doi: 10.1016/S1474-4422(06)70471-9
Deiva, K., Geeraerts, T., Salim, H., Leclerc, P., Hery, C., Hugel, B., et al. (2004). Fractalkine reduces N-methyl-d-aspartate-induced calcium flux and apoptosis in human neurons through extracellular signal-regulated kinase activation. Eur. J. Neurosci. 20, 3222–3232. doi: 10.1111/j.1460-9568.2004.03800.x
Denes, A., Ferenczi, S., Halasz, J., Kornyei, Z., and Kovacs, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721. doi: 10.1038/jcbfm.2008.64
Di Micco, R., Krizhanovsky, V., Baker, D., and d'Adda di Fagagna, F. (2021). Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95. doi: 10.1038/s41580-020-00314-w
Dickstein, D. L., Weaver, C. M., Luebke, J. I., and Hof, P. R. (2013). Dendritic spine changes associated with normal aging. Neuroscience 251, 21–32. doi: 10.1016/j.neuroscience.2012.09.077
Dimou, L., Simon, C., Kirchhoff, F., Takebayashi, H., and Gotz, M. (2008). Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci. 28, 10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008
Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 92, 9363–9367. doi: 10.1073/pnas.92.20.9363
Dobson, R., and Giovannoni, G. (2019). Multiple sclerosis – a review. Eur. J. Neurol. 26, 27–40. doi: 10.1111/ene.13819
Dong, X., Luo, M., Huang, G., Zhang, J., Tong, F., Cheng, Y., et al. (2015). Relationship between irradiation-induced neuro-inflammatory environments and impaired cognitive function in the developing brain of mice. Int. J. Radiat. Biol. 91, 224–239. doi: 10.3109/09553002.2014.988895
Doonan, F., O'Driscoll, C., Kenna, P., and Cotter, T. G. (2011). Enhancing survival of photoreceptor cells in vivo using the synthetic progestin Norgestrel. J. Neurochem. 118, 915–927. doi: 10.1111/j.1471-4159.2011.07354.x
Dos-Santos-Pereira, M., Acuna, L., Hamadat, S., Rocca, J., Gonzalez-Lizarraga, F., Chehin, R., et al. (2018). Microglial glutamate release evoked by alpha-synuclein aggregates is prevented by dopamine. Glia 66, 2353–2365. doi: 10.1002/glia.23472
Dworzak, J., Renvoise, B., Habchi, J., Yates, E. V., Combadiere, C., Knowles, T. P., et al. (2015). Neuronal Cx3cr1 deficiency protects against amyloid beta-induced neurotoxicity. PLoS One 10:e0127730. doi: 10.1371/journal.pone.0127730
Edler, M. K., Mhatre-Winters, I., and Richardson, J. R. (2021). Microglia in aging and Alzheimer's disease: a comparative species review. Cells 10:1138. doi: 10.3390/cells10051138
Ekstrom, U., Wallin, H., Lorenzo, J., Holmqvist, B., Abrahamson, M., and Aviles, F. X. (2008). Internalization of cystatin C in human cell lines. FEBS J. 275, 4571–4582. doi: 10.1111/j.1742-4658.2008.06600.x
Escartin, C., Galea, E., Lakatos, A., O'Callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325. doi: 10.1038/s41593-020-00783-4
Eugenin, J., Vecchiola, A., Murgas, P., Arroyo, P., Cornejo, F., and von Bernhardi, R. (2016). Expression pattern of scavenger receptors and amyloid-beta phagocytosis of astrocytes and microglia in culture are modified by acidosis: implications for Alzheimer's disease. J. Alzheimers Dis. 53, 857–873. doi: 10.3233/JAD-160083
Eugenin von Bernhardi, J., and Dimou, L. (2022). Oligodendrogenesis is a key process for cognitive performance improvement induced by voluntary physical activity. Glia 70, 1052–1067. doi: 10.1002/glia.24155
Fan, Q., Gayen, M., Singh, N., Gao, F., He, W., Hu, X., et al. (2019). The intracellular domain of CX3CL1 regulates adult neurogenesis and Alzheimer's amyloid pathology. J. Exp. Med. 216, 1891–1903. doi: 10.1084/jem.20182238
Faust, T. E., Gunner, G., and Schafer, D. P. (2021). Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nat. Rev. Neurosci. 22, 657–673. doi: 10.1038/s41583-021-00507-y
Finneran, D., Li, Q., Subbarayan, M. S., Joly-Amado, A., Kamath, S., Dengler, D. G., et al. (2023). Concentration and proteolysis of CX3CL1 may regulate the microglial response to CX3CL1. Glia 71, 245–258. doi: 10.1002/glia.24269
Finneran, D. J., Morgan, D., Gordon, M. N., and Nash, K. R. (2019). CNS-wide over expression of Fractalkine improves cognitive functioning in a Tauopathy model. J. Neuroimmune Pharmacol. 14, 312–325. doi: 10.1007/s11481-018-9822-5
Floden, A. M., and Combs, C. K. (2011). Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J. Alzheimers Dis. 25, 279–293. doi: 10.3233/JAD-2011-101014
Flores, B., and von Bernhardi, R. (2012). Transforming growth factor beta1 modulates amyloid beta-induced glial activation through the Smad3-dependent induction of MAPK phosphatase-1. J. Alzheimers Dis. 32, 417–429. doi: 10.3233/JAD-2012-120721
Fonovic, U. P., Jevnikar, Z., and Kos, J. (2013). Cathepsin S generates soluble CX3CL1 (fractalkine) in vascular smooth muscle cells. Biol. Chem. 394, 1349–1352. doi: 10.1515/hsz-2013-0189
Franceschi, C. (2007). Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr. Rev. 65, S173–S176. doi: 10.1111/j.1753-4887.2007.tb00358.x
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Franceschi, C., Capri, M., Monti, D., Giunta, S., Olivieri, F., Sevini, F., et al. (2007). Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 128, 92–105. doi: 10.1016/j.mad.2006.11.016
Franklin, R. J. M., and Ffrench-Constant, C. (2017). Regenerating CNS myelin – from mechanisms to experimental medicines. Nat. Rev. Neurosci. 18, 753–769. doi: 10.1038/nrn.2017.136
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Galan-Ganga, M., Garcia-Yague, A. J., and Lastres-Becker, I. (2019). Role of MSK1 in the induction of NF-kappaB by the chemokine CX3CL1 in microglial cells. Cell. Mol. Neurobiol. 39, 331–340. doi: 10.1007/s10571-019-00664-w
Gao, H. M., and Hong, J. S. (2008). Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365. doi: 10.1016/j.it.2008.05.002
Garton, K. J., Gough, P. J., Blobel, C. P., Murphy, G., Greaves, D. R., Dempsey, P. J., et al. (2001). Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 276, 37993–38001. doi: 10.1074/jbc.M106434200
Gaspar-Silva, F., Trigo, D., and Magalhaes, J. (2023). Ageing in the brain: mechanisms and rejuvenating strategies. Cell. Mol. Life Sci. 80:190. doi: 10.1007/s00018-023-04832-6
Ge, Y., Grossman, R. I., Babb, J. S., Rabin, M. L., Mannon, L. J., and Kolson, D. L. (2002). Age-related total gray matter and white matter changes in normal adult brain. Part I: volumetric MR imaging analysis. AJNR Am. J. Neuroradiol. 23, 1327–1333.
Ghidoni, R., Paterlini, A., Albertini, V., Glionna, M., Monti, E., Schiaffonati, L., et al. (2011). Cystatin C is released in association with exosomes: a new tool of neuronal communication which is unbalanced in Alzheimer's disease. Neurobiol. Aging 32, 1435–1442. doi: 10.1016/j.neurobiolaging.2009.08.013
Ghys, L. F., Paepe, D., Duchateau, L., Taffin, E. R., Marynissen, S., Delanghe, J., et al. (2015). Biological validation of feline serum cystatin C: the effect of breed, age and sex and establishment of a reference interval. Vet. J. 204, 168–173. doi: 10.1016/j.tvjl.2015.02.018
Giovannoni, F., and Quintana, F. J. (2020). The role of astrocytes in CNS inflammation. Trends Immunol. 41, 805–819. doi: 10.1016/j.it.2020.07.007
Godoy, B., Murgas, P., Tichauer, J., and Von Bernhardi, R. (2012). Scavenger receptor class a ligands induce secretion of IL1beta and exert a modulatory effect on the inflammatory activation of astrocytes in culture. J. Neuroimmunol. 251, 6–13. doi: 10.1016/j.jneuroim.2012.06.004
Gomez-Arboledas, A., Acharya, M. M., and Tenner, A. J. (2021). The role of complement in synaptic pruning and neurodegeneration. Immunotargets Ther 10, 373–386. doi: 10.2147/ITT.S305420
Gomez-Gonzalo, M., Martin-Fernandez, M., Martinez-Murillo, R., Mederos, S., Hernandez-Vivanco, A., Jamison, S., et al. (2017). Neuron-astrocyte signaling is preserved in the aging brain. Glia 65, 569–580. doi: 10.1002/glia.23112
Gonzales, M. M., Garbarino, V. R., Pollet, E., Palavicini, J. P., Kellogg, D. L. Jr., Kraig, E., et al. (2022). Biological aging processes underlying cognitive decline and neurodegenerative disease. J. Clin. Invest. 132:8453. doi: 10.1172/JCI158453
Gu, H. J., Zuo, S., Liu, H. Y., Gu, L. L., Yang, X. W., Liao, J., et al. (2019). CX3CR1 participates in pulmonary angiogenesis in experimental hepatopulmonary syndrome mice through inhibiting AKT/ERK signaling pathway and regulating NO/NOS release. Eur. Rev. Med. Pharmacol. Sci. 23, 6645–6656. doi: 10.26355/eurrev_201908_18555
Gudi, V., Gingele, S., Skripuletz, T., and Stangel, M. (2014). Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front. Cell. Neurosci. 8:73. doi: 10.3389/fncel.2014.00073
Guerreiro, R., and Bras, J. (2015). The age factor in Alzheimer's disease. Genome Med. 7:106. doi: 10.1186/s13073-015-0232-5
Halassa, M. M., and Haydon, P. G. (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 72, 335–355. doi: 10.1146/annurev-physiol-021909-135843
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U. S. A. 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Hart, A. D., Wyttenbach, A., Perry, V. H., and Teeling, J. L. (2012). Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav. Immun. 26, 754–765. doi: 10.1016/j.bbi.2011.11.006
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368, 1795–1809. doi: 10.1016/S0140-6736(06)69740-7
Hayashi, Y., Yoshida, M., Yamato, M., Ide, T., Wu, Z., Ochi-Shindou, M., et al. (2008). Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J. Neurosci. 28, 8624–8634. doi: 10.1523/JNEUROSCI.1957-08.2008
Hedman, A. M., van Haren, N. E., Schnack, H. G., Kahn, R. S., and Hulshoff Pol, H. E. (2012). Human brain changes across the life span: a review of 56 longitudinal magnetic resonance imaging studies. Hum. Brain Mapp. 33, 1987–2002. doi: 10.1002/hbm.21334
Hermand, P., Pincet, F., Carvalho, S., Ansanay, H., Trinquet, E., Daoudi, M., et al. (2008). Functional adhesiveness of the CX3CL1 chemokine requires its aggregation. Role of the transmembrane domain. J. Biol. Chem. 283, 30225–30234. doi: 10.1074/jbc.M802638200
Herrera-Molina, R., Flores, B., Orellana, J. A., and von Bernhardi, R. (2012). Modulation of interferon-gamma-induced glial cell activation by transforming growth factor beta1: a role for STAT1 and MAPK pathways. J. Neurochem. 123, 113–123. doi: 10.1111/j.1471-4159.2012.07887.x
Hickman, S. E., Allison, E. K., Coleman, U., Kingery-Gallagher, N. D., and El Khoury, J. (2019). Heterozygous CX3CR1 deficiency in microglia restores neuronal beta-amyloid clearance pathways and slows progression of Alzheimer's like-disease in PS1-APP mice. Front. Immunol. 10:2780. doi: 10.3389/fimmu.2019.02780
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Hill, R. A., Li, A. M., and Grutzendler, J. (2018). Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 21, 683–695. doi: 10.1038/s41593-018-0120-6
Hiremath, M. M., Saito, Y., Knapp, G. W., Ting, J. P., Suzuki, K., and Matsushima, G. K. (1998). Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J. Neuroimmunol. 92, 38–49. doi: 10.1016/s0165-5728(98)00168-4
Hoek, R. M., Ruuls, S. R., Murphy, C. A., Wright, G. J., Goddard, R., Zurawski, S. M., et al. (2000). Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290, 1768–1771. doi: 10.1126/science.290.5497.1768
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N., and Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Hughes, E. G., Kang, S. H., Fukaya, M., and Bergles, D. E. (2013). Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 16, 668–676. doi: 10.1038/nn.3390
Hundhausen, C., Misztela, D., Berkhout, T. A., Broadway, N., Saftig, P., Reiss, K., et al. (2003). The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102, 1186–1195. doi: 10.1182/blood-2002-12-3775
Hundhausen, C., Schulte, A., Schulz, B., Andrzejewski, M. G., Schwarz, N., von Hundelshausen, P., et al. (2007). Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J. Immunol. 178, 8064–8072. doi: 10.4049/jimmunol.178.12.8064
Imai, T., Hieshima, K., Haskell, C., Baba, M., Nagira, M., Nishimura, M., et al. (1997). Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cells 91, 521–530. doi: 10.1016/s0092-8674(00)80438-9
Imura, Y., Morizawa, Y., Komatsu, R., Shibata, K., Shinozaki, Y., Kasai, H., et al. (2013). Microglia release ATP by exocytosis. Glia 61, 1320–1330. doi: 10.1002/glia.22517
Inoue, K., Morimoto, H., Ohgidani, M., and Ueki, T. (2021). Modulation of inflammatory responses by fractalkine signaling in microglia. PLoS One 16:e0252118. doi: 10.1371/journal.pone.0252118
Iwasa, N., Takashima, S., Iwasa, T., Kumazawa, R., Nomura, S., Asami, S., et al. (2022). Effect of age, sex, and breed on serum cystatin C and creatinine concentrations in dogs. Vet. Res. Commun. 46, 183–188. doi: 10.1007/s11259-021-09844-w
Jankovic, J. (2008). Parkinson's disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. doi: 10.1136/jnnp.2007.131045
Javanmehr, N., Saleki, K., Alijanizadeh, P., and Rezaei, N. (2022). Microglia dynamics in aging-related neurobehavioral and neuroinflammatory diseases. J. Neuroinflammation 19:273. doi: 10.1186/s12974-022-02637-1
Jellinger, K. A. (2020). Neuropathology of the Alzheimer's continuum: an update. Free Neuropathol 1:3050. doi: 10.17879/freeneuropathology-2020-3050
Johnson-Venkatesh, E. M., and Umemori, H. (2010). Secreted factors as synaptic organizers. Eur. J. Neurosci. 32, 181–190. doi: 10.1111/j.1460-9568.2010.07338.x
Jones, B. A., Beamer, M., and Ahmed, S. (2010). Fractalkine/CX3CL1: a potential new target for inflammatory diseases. Mol. Interv. 10, 263–270. doi: 10.1124/mi.10.5.3
Kansra, V., Groves, C., Gutierrez-Ramos, J. C., and Polakiewicz, R. D. (2001). Phosphatidylinositol 3-kinase-dependent extracellular calcium influx is essential for CX(3)CR1-mediated activation of the mitogen-activated protein kinase cascade. J. Biol. Chem. 276, 31831–31838. doi: 10.1074/jbc.M009374200
Karussis, D. (2014). The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J. Autoimmun. 48-49, 134–142. doi: 10.1016/j.jaut.2014.01.022
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cells 169, 1276–1290 e1217. doi: 10.1016/j.cell.2017.05.018
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kigerl, K. A., Gensel, J. C., Ankeny, D. P., Alexander, J. K., Donnelly, D. J., and Popovich, P. G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009
Kolodziejczyk, R., Michalska, K., Hernandez-Santoyo, A., Wahlbom, M., Grubb, A., and Jaskolski, M. (2010). Crystal structure of human cystatin C stabilized against amyloid formation. FEBS J. 277, 1726–1737. doi: 10.1111/j.1742-4658.2010.07596.x
Kotter, M. R., Stadelmann, C., and Hartung, H. P. (2011). Enhancing remyelination in disease--can we wrap it up? Brain 134, 1882–1900. doi: 10.1093/brain/awr014
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581 e569. doi: 10.1016/j.immuni.2017.08.008
Kuhn, S., Gritti, L., Crooks, D., and Dombrowski, Y. (2019). Oligodendrocytes in development, myelin generation and beyond. Cells 8:1424. doi: 10.3390/cells8111424
Lampron, A., Larochelle, A., Laflamme, N., Prefontaine, P., Plante, M. M., Sanchez, M. G., et al. (2015). Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 212, 481–495. doi: 10.1084/jem.20141656
Lasiene, J., Matsui, A., Sawa, Y., Wong, F., and Horner, P. J. (2009). Age-related myelin dynamics revealed by increased oligodendrogenesis and short internodes. Aging Cell 8, 201–213. doi: 10.1111/j.1474-9726.2009.00462.x
Lastres-Becker, I., Innamorato, N. G., Jaworski, T., Rabano, A., Kugler, S., Van Leuven, F., et al. (2014). Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 137, 78–91. doi: 10.1093/brain/awt323
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Lee, S. J., Namkoong, S., Kim, Y. M., Kim, C. K., Lee, H., Ha, K. S., et al. (2006). Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am. J. Physiol. Heart Circ. Physiol. 291, H2836–H2846. doi: 10.1152/ajpheart.00113.2006
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am. J. Pathol. 177, 2549–2562. doi: 10.2353/ajpath.2010.100265
Lee, S., Xu, G., Jay, T. R., Bhatta, S., Kim, K. W., Jung, S., et al. (2014). Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J. Neurosci. 34, 12538–12546. doi: 10.1523/JNEUROSCI.0853-14.2014
Lenarcic, B., Krasovec, M., Ritonja, A., Olafsson, I., and Turk, V. (1991). Inactivation of human cystatin C and kininogen by human cathepsin D. FEBS Lett. 280, 211–215. doi: 10.1016/0014-5793(91)80295-e
Li, R., Jia, Z., and Zhu, H. (2019). Regulation of Nrf2 Signaling. React Oxyg Species (Apex) 8, 312–322. doi: 10.20455/ros.2019.865
Li, L., Lu, J., Tay, S. S., Moochhala, S. M., and He, B. P. (2007). The function of microglia, either neuroprotection or neurotoxicity, is determined by the equilibrium among factors released from activated microglia in vitro. Brain Res. 1159, 8–17. doi: 10.1016/j.brainres.2007.04.066
Li Puma, D. D., Piacentini, R., and Grassi, C. (2020). Does impairment of adult neurogenesis contribute to pathophysiology of Alzheimer's disease? A still open question. Front. Mol. Neurosci. 13:578211. doi: 10.3389/fnmol.2020.578211
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Liguori, I., Russo, G., Curcio, F., Bulli, G., Aran, L., Della-Morte, D., et al. (2018). Oxidative stress, aging, and diseases. Clin. Interv. Aging 13, 757–772. doi: 10.2147/CIA.S158513
Liu, C., Hong, K., Chen, H., Niu, Y., Duan, W., Liu, Y., et al. (2019). Evidence for a protective role of the CX3CL1/CX3CR1 axis in a model of amyotrophic lateral sclerosis. Biol. Chem. 400, 651–661. doi: 10.1515/hsz-2018-0204
Liu, Q., Zhang, J., Tran, H., Verbeek, M. M., Reiss, K., Estus, S., et al. (2009). LRP1 shedding in human brain: roles of ADAM10 and ADAM17. Mol. Neurodegener. 4:17. doi: 10.1186/1750-1326-4-17
Liuzzo, J. P., Petanceska, S. S., and Devi, L. A. (1999a). Neurotrophic factors regulate cathepsin S in macrophages and microglia: a role in the degradation of myelin basic protein and amyloid beta peptide. Mol. Med. 5, 334–343. doi: 10.1007/BF03402069
Liuzzo, J. P., Petanceska, S. S., Moscatelli, D., and Devi, L. A. (1999b). Inflammatory mediators regulate cathepsin S in macrophages and microglia: a role in attenuating heparan sulfate interactions. Mol. Med. 5, 320–333.
Lopez-Lopez, A., Gamez, J., Syriani, E., Morales, M., Salvado, M., Rodriguez, M. J., et al. (2014). CX3CR1 is a modifying gene of survival and progression in amyotrophic lateral sclerosis. PLoS One 9:e96528. doi: 10.1371/journal.pone.0096528
Lopez-Teros, M., Alarcon-Aguilar, A., Lopez-Diazguerrero, N. E., Luna-Lopez, A., and Konigsberg, M. (2022). Contribution of senescent and reactive astrocytes on central nervous system inflammaging. Biogerontology 23, 21–33. doi: 10.1007/s10522-022-09952-3
Ludwig, A., and Mentlein, R. (2008). Glial cross-talk by transmembrane chemokines CX3CL1 and CXCL16. J. Neuroimmunol. 198, 92–97. doi: 10.1016/j.jneuroim.2008.04.024
Lynch, A. M., Murphy, K. J., Deighan, B. F., O'Reilly, J. A., Gun'ko, Y. K., Cowley, T. R., et al. (2010). The impact of glial activation in the aging brain. Aging Dis. 1, 262–278.
Lynch, J., and Smith, G. D. (2005). A life course approach to chronic disease epidemiology. Annu. Rev. Public Health 26, 1–35. doi: 10.1146/annurev.publhealth.26.021304.144505
Lyons, A., Lynch, A. M., Downer, E. J., Hanley, R., O'Sullivan, J. B., Smith, A., et al. (2009). Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 110, 1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x
Ma, C. L., Ma, X. T., Wang, J. J., Liu, H., Chen, Y. F., and Yang, Y. (2017). Physical exercise induces hippocampal neurogenesis and prevents cognitive decline. Behav. Brain Res. 317, 332–339. doi: 10.1016/j.bbr.2016.09.067
Maggi, L., Scianni, M., Branchi, I., D'Andrea, I., Lauro, C., and Limatola, C. (2011). CX(3)CR1 deficiency alters hippocampal-dependent plasticity phenomena blunting the effects of enriched environment. Front. Cell. Neurosci. 5:22. doi: 10.3389/fncel.2011.00022
Maggi, L., Trettel, F., Scianni, M., Bertollini, C., Eusebi, F., Fredholm, B. B., et al. (2009). LTP impairment by fractalkine/CX3CL1 in mouse hippocampus is mediated through the activity of adenosine receptor type 3 (A3R). J. Neuroimmunol. 215, 36–42. doi: 10.1016/j.jneuroim.2009.07.016
Makabe, K., Sugita, S., Mandai, M., Futatsugi, Y., and Takahashi, M. (2020). Microglia dynamics in retinitis pigmentosa model: formation of fundus whitening and autofluorescence as an indicator of activity of retinal degeneration. Sci. Rep. 10:14700. doi: 10.1038/s41598-020-71626-2
Manich, G., Recasens, M., Valente, T., Almolda, B., Gonzalez, B., and Castellano, B. (2019). Role of the CD200-CD200R Axis during homeostasis and Neuroinflammation. Neuroscience 405, 118–136. doi: 10.1016/j.neuroscience.2018.10.030
Mathews, P. M., and Levy, E. (2016). Cystatin C in aging and in Alzheimer's disease. Ageing Res. Rev. 32, 38–50. doi: 10.1016/j.arr.2016.06.003
Mecca, C., Giambanco, I., Donato, R., and Arcuri, C. (2018). Microglia and aging: the role of the TREM2-DAP12 and CX3CL1-CX3CR1 axes. Int. J. Mol. Sci. 19:318. doi: 10.3390/ijms19010318
Mendiola, A. S., Church, K. A., Cardona, S. M., Vanegas, D., Garcia, S. A., Macklin, W., et al. (2022). Defective fractalkine-CX3CR1 signaling aggravates neuroinflammation and affects recovery from cuprizone-induced demyelination. J. Neurochem. 162, 430–443. doi: 10.1111/jnc.15616
Merz, G. S., Benedikz, E., Schwenk, V., Johansen, T. E., Vogel, L. K., Rushbrook, J. I., et al. (1997). Human cystatin C forms an inactive dimer during intracellular trafficking in transfected CHO cells. J. Cell. Physiol. 173, 423–432. doi: 10.1002/(SICI)1097-4652(199712)173:3<423::AID-JCP15>3.0.CO;2-C
Meucci, O., Fatatis, A., Simen, A. A., and Miller, R. J. (2000). Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U. S. A. 97, 8075–8080. doi: 10.1073/pnas.090017497
Milligan, E. D., Zapata, V., Chacur, M., Schoeniger, D., Biedenkapp, J., O'Connor, K. A., et al. (2004). Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur. J. Neurosci. 20, 2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x
Miron, V. E., Boyd, A., Zhao, J. W., Yuen, T. J., Ruckh, J. M., Shadrach, J. L., et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218. doi: 10.1038/nn.3469
Miyake, T., Gahara, Y., Nakayama, M., Yamada, H., Uwabe, K., and Kitamura, T. (1996). Up-regulation of cystatin C by microglia in the rat facial nucleus following axotomy. Brain Res. Mol. Brain Res. 37, 273–282. doi: 10.1016/0169-328x(95)00337-r
Mizuno, T., Kawanokuchi, J., Numata, K., and Suzumura, A. (2003). Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 979, 65–70. doi: 10.1016/s0006-8993(03)02867-1
Morganti, J. M., Nash, K. R., Grimmig, B. A., Ranjit, S., Small, B., Bickford, P. C., et al. (2012). The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson's disease. J. Neurosci. 32, 14592–14601. doi: 10.1523/JNEUROSCI.0539-12.2012
Munoz-Castro, C., Noori, A., Magdamo, C. G., Li, Z., Marks, J. D., Frosch, M. P., et al. (2022). Cyclic multiplex fluorescent immunohistochemistry and machine learning reveal distinct states of astrocytes and microglia in normal aging and Alzheimer's disease. J. Neuroinflammation 19:30. doi: 10.1186/s12974-022-02383-4
Murgas, P., Cornejo, F. A., Merino, G., and von Bernhardi, R. (2014). SR-A regulates the inflammatory activation of astrocytes. Neurotox. Res. 25, 68–80. doi: 10.1007/s12640-013-9432-1
Murgas, P., Godoy, B., and von Bernhardi, R. (2012). Abeta potentiates inflammatory activation of glial cells induced by scavenger receptor ligands and inflammatory mediators in culture. Neurotox. Res. 22, 69–78. doi: 10.1007/s12640-011-9306-3
Murro, V., Banfi, S., Testa, F., Iarossi, G., Falsini, B., Sodi, A., et al. (2023). A multidisciplinary approach to inherited retinal dystrophies from diagnosis to initial care: a narrative review with inputs from clinical practice. Orphanet J. Rare Dis. 18:223. doi: 10.1186/s13023-023-02798-z
Nakajima, K., Tohyama, Y., Maeda, S., Kohsaka, S., and Kurihara, T. (2007). Neuronal regulation by which microglia enhance the production of neurotrophic factors for GABAergic, catecholaminergic, and cholinergic neurons. Neurochem. Int. 50, 807–820. doi: 10.1016/j.neuint.2007.02.006
Nakanishi, H. (2003). Neuronal and microglial cathepsins in aging and age-related diseases. Ageing Res. Rev. 2, 367–381. doi: 10.1016/s1568-1637(03)00027-8
Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., et al. (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cells 113, 703–716. doi: 10.1016/s0092-8674(03)00401-x
Nash, K. R., Lee, D. C., Hunt, J. B. Jr., Morganti, J. M., Selenica, M. L., Moran, P., et al. (2013). Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 34, 1540–1548. doi: 10.1016/j.neurobiolaging.2012.12.011
Nash, K. R., Moran, P., Finneran, D. J., Hudson, C., Robinson, J., Morgan, D., et al. (2015). Fractalkine over expression suppresses alpha-synuclein-mediated neurodegeneration. Mol. Ther. 23, 17–23. doi: 10.1038/mt.2014.175
Neumann, B., Baror, R., Zhao, C., Segel, M., Dietmann, S., Rawji, K. S., et al. (2019). Metformin restores CNS Remyelination capacity by rejuvenating aged stem cells. Cell Stem Cell 25, 473–485.e478. doi: 10.1016/j.stem.2019.08.015
Nguyen, M. D., Julien, J. P., and Rivest, S. (2002). Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 3, 216–227. doi: 10.1038/nrn752
Nichols, N. R., Day, J. R., Laping, N. J., Johnson, S. A., and Finch, C. E. (1993). GFAP mRNA increases with age in rat and human brain. Neurobiol. Aging 14, 421–429.
Oh, J., Vidal-Jordana, A., and Montalban, X. (2018). Multiple sclerosis: clinical aspects. Curr. Opin. Neurol. 31, 752–759. doi: 10.1097/WCO.0000000000000622
Ohara, G., Miyazaki, K., Kurishima, K., Kagohashi, K., Ishikawa, H., Satoh, H., et al. (2012). Serum levels of cystatin C in elderly lung cancer patients. Oncol. Lett. 3, 303–306. doi: 10.3892/ol.2011.377
Orellana, J. A., Montero, T. D., and von Bernhardi, R. (2013). Astrocytes inhibit nitric oxide-dependent ca(2+) dynamics in activated microglia: involvement of ATP released via pannexin 1 channels. Glia 61, 2023–2037. doi: 10.1002/glia.22573
Ostuni, M. A., Guellec, J., Hermand, P., Durand, P., Combadiere, C., Pincet, F., et al. (2014). CX3CL1, a chemokine finely tuned to adhesion: critical roles of the stalk glycosylation and the membrane domain. Biol Open 3, 1173–1182. doi: 10.1242/bio.20149845
Ostuni, M. A., Hermand, P., Saindoy, E., Guillou, N., Guellec, J., Coens, A., et al. (2020). CX3CL1 homo-oligomerization drives cell-to-cell adherence. Sci. Rep. 10:9069. doi: 10.1038/s41598-020-65988-w
Pabon, M. M., Bachstetter, A. D., Hudson, C. E., Gemma, C., and Bickford, P. C. (2011). CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson's disease. J. Neuroinflammation 8:9. doi: 10.1186/1742-2094-8-9
Palm, D. E., Knuckey, N. W., Primiano, M. J., Spangenberger, A. G., and Johanson, C. E. (1995). Cystatin C, a protease inhibitor, in degenerating rat hippocampal neurons following transient forebrain ischemia. Brain Res. 691, 1–8. doi: 10.1016/0006-8993(95)00520-z
Pannese, E. (2011). Morphological changes in nerve cells during normal aging. Brain Struct. Funct. 216, 85–89. doi: 10.1007/s00429-011-0308-y
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Paolicelli, R. C., Sierra, A., Stevens, B., Tremblay, M. E., Aguzzi, A., Ajami, B., et al. (2022). Microglia states and nomenclature: a field at its crossroads. Neuron 110, 3458–3483. doi: 10.1016/j.neuron.2022.10.020
Paraoan, L., White, M. R., Spiller, D. G., Grierson, I., and Maden, B. E. (2001). Precursor cystatin C in cultured retinal pigment epithelium cells: evidence for processing through the secretory pathway. Mol. Membr. Biol. 18, 229–236. doi: 10.1080/09687680110075101
Park, S. K., Kim, K., Page, G. P., Allison, D. B., Weindruch, R., and Prolla, T. A. (2009). Gene expression profiling of aging in multiple mouse strains: identification of aging biomarkers and impact of dietary antioxidants. Aging Cell 8, 484–495. doi: 10.1111/j.1474-9726.2009.00496.x
Partridge, L., Deelen, J., and Slagboom, P. E. (2018). Facing up to the global challenges of ageing. Nature 561, 45–56. doi: 10.1038/s41586-018-0457-8
Pawate, S., Shen, Q., Fan, F., and Bhat, N. R. (2004). Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J. Neurosci. Res. 77, 540–551. doi: 10.1002/jnr.20180
Peschon, J. J., Slack, J. L., Reddy, P., Stocking, K. L., Sunnarborg, S. W., Lee, D. C., et al. (1998). An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284. doi: 10.1126/science.282.5392.1281
Petanceska, S., Canoll, P., and Devi, L. A. (1996). Expression of rat cathepsin S in phagocytic cells. J. Biol. Chem. 271, 4403–4409. doi: 10.1074/jbc.271.8.4403
Peters, A. (2009). The effects of normal aging on myelinated nerve fibers in monkey central nervous system. Front. Neuroanat. 3:11. doi: 10.3389/neuro.05.011.2009
Peters, A., and Sethares, C. (2003). Is there remyelination during aging of the primate central nervous system? J. Comp. Neurol. 460, 238–254. doi: 10.1002/cne.10639
Pierre, P., and Mellman, I. (1998). Developmental regulation of invariant chain proteolysis controls MHC class II trafficking in mouse dendritic cells. Cells 93, 1135–1145. doi: 10.1016/s0092-8674(00)81458-0
Porchet, R., Probst, A., Bouras, C., Draberova, E., Draber, P., and Riederer, B. M. (2003). Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer's disease. Proteomics 3, 1476–1485. doi: 10.1002/pmic.200300456
Postina, R. (2012). Activation of alpha-secretase cleavage. J. Neurochem. 120, 46–54. doi: 10.1111/j.1471-4159.2011.07459.x
Qin, L., Li, G., Qian, X., Liu, Y., Wu, X., Liu, B., et al. (2005). Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 52, 78–84. doi: 10.1002/glia.20225
Ragozzino, D., Di Angelantonio, S., Trettel, F., Bertollini, C., Maggi, L., Gross, C., et al. (2006). Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J. Neurosci. 26, 10488–10498. doi: 10.1523/JNEUROSCI.3192-06.2006
Ramirez, G., Rey, S., and von Bernhardi, R. (2008). Proinflammatory stimuli are needed for induction of microglial cell-mediated AbetaPP_{244-C} and Abeta-neurotoxicity in hippocampal cultures. J. Alzheimers Dis. 15, 45–59. doi: 10.3233/jad-2008-15104
Rawji, K. S., Neumann, B., and Franklin, R. J. M. (2023). Glial aging and its impact on central nervous system myelin regeneration. Ann. N. Y. Acad. Sci. 1519, 34–45. doi: 10.1111/nyas.14933
Raz, N., Gunning, F. M., Head, D., Dupuis, J. H., McQuain, J., Briggs, S. D., et al. (1997). Selective aging of the human cerebral cortex observed in vivo: differential vulnerability of the prefrontal gray matter. Cereb. Cortex 7, 268–282. doi: 10.1093/cercor/7.3.268
Reinhard, C., Hebert, S. S., and De Strooper, B. (2005). The amyloid-beta precursor protein: integrating structure with biological function. EMBO J. 24, 3996–4006. doi: 10.1038/sj.emboj.7600860
Rivas-Fuentes, S., Salgado-Aguayo, A., Arratia-Quijada, J., and Gorocica-Rosete, P. (2021). Regulation and biological functions of the CX3CL1-CX3CR1 axis and its relevance in solid cancer: a mini-review. J. Cancer 12, 571–583. doi: 10.7150/jca.47022
Rivest, S. (2009). Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439. doi: 10.1038/nri2565
Rivest, S. (2015). CX3CR1 in multiple sclerosis. Oncotarget 6, 19946–19947. doi: 10.18632/oncotarget.4650
Roche, S. L., Wyse-Jackson, A. C., Gomez-Vicente, V., Lax, P., Ruiz-Lopez, A. M., Byrne, A. M., et al. (2016). Progesterone attenuates microglial-driven retinal degeneration and stimulates protective Fractalkine-CX3CR1 Signaling. PLoS One 11:e0165197. doi: 10.1371/journal.pone.0165197
Roche, S. L., Wyse-Jackson, A. C., Ruiz-Lopez, A. M., Byrne, A. M., and Cotter, T. G. (2017). Fractalkine-CX3CR1 signaling is critical for progesterone-mediated neuroprotection in the retina. Sci. Rep. 7:43067. doi: 10.1038/srep43067
Rodier, F., and Campisi, J. (2011). Four faces of cellular senescence. J. Cell Biol. 192, 547–556. doi: 10.1083/jcb.201009094
Rodriguez, J. J., Yeh, C. Y., Terzieva, S., Olabarria, M., Kulijewicz-Nawrot, M., and Verkhratsky, A. (2014). Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 35, 15–23. doi: 10.1016/j.neurobiolaging.2013.07.002
Rogers, J. T., Morganti, J. M., Bachstetter, A. D., Hudson, C. E., Peters, M. M., Grimmig, B. A., et al. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011
Sahu, M. R., Rani, L., Subba, R., and Mondal, A. C. (2022). Cellular senescence in the aging brain: a promising target for neurodegenerative diseases. Mech. Ageing Dev. 204:111675. doi: 10.1016/j.mad.2022.111675
Saito, T., and Saido, T. C. (2018). Neuroinflammation in mouse models of Alzheimer's disease. Clin Exp Neuroimmunol 9, 211–218. doi: 10.1111/cen3.12475
Salat, D. H., Buckner, R. L., Snyder, A. Z., Greve, D. N., Desikan, R. S., Busa, E., et al. (2004). Thinning of the cerebral cortex in aging. Cereb. Cortex 14, 721–730. doi: 10.1093/cercor/bhh032
Sastre, M., Calero, M., Pawlik, M., Mathews, P. M., Kumar, A., Danilov, V., et al. (2004). Binding of cystatin C to Alzheimer's amyloid beta inhibits in vitro amyloid fibril formation. Neurobiol. Aging 25, 1033–1043. doi: 10.1016/j.neurobiolaging.2003.11.006
Schafer, D. P., Lehrman, E. K., and Stevens, B. (2013). The "quad-partite" synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61, 24–36. doi: 10.1002/glia.22389
Segel, M., Neumann, B., Hill, M. F. E., Weber, I. P., Viscomi, C., Zhao, C., et al. (2019). Niche stiffness underlies the ageing of central nervous system progenitor cells. Nature 573, 130–134. doi: 10.1038/s41586-019-1484-9
Sessler, K., Blechschmidt, V., Hoheisel, U., Mense, S., Schirmer, L., and Treede, R. D. (2021). Spinal cord fractalkine (CX3CL1) signaling is critical for neuronal sensitization in experimental nonspecific, myofascial low back pain. J. Neurophysiol. 125, 1598–1611. doi: 10.1152/jn.00348.2020
Shan, S., Hong-Min, T., Yi, F., Jun-Peng, G., Yue, F., Yan-Hong, T., et al. (2011). New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson's disease. Neurobiol. Aging 32, 443–458. doi: 10.1016/j.neurobiolaging.2009.03.004
Sharma, L., Sharma, A., Kumar, D., Asthana, M. K., Lalhlenmawia, H., Kumar, A., et al. (2022). Promising protein biomarkers in the early diagnosis of Alzheimer's disease. Metab. Brain Dis. 37, 1727–1744. doi: 10.1007/s11011-021-00847-9
Sheridan, G. K., Wdowicz, A., Pickering, M., Watters, O., Halley, P., O'Sullivan, N. C., et al. (2014). CX3CL1 is up-regulated in the rat hippocampus during memory-associated synaptic plasticity. Front. Cell. Neurosci. 8:233. doi: 10.3389/fncel.2014.00233
Shi, L., Li, S., Maurer, K., Zhang, Z., Petri, M., and Sullivan, K. E. (2018). Enhancer RNA and NFkappaB-dependent P300 regulation of ADAMDEC1. Mol. Immunol. 103, 312–321. doi: 10.1016/j.molimm.2018.09.019
Shimi, T., Butin-Israeli, V., Adam, S. A., Hamanaka, R. B., Goldman, A. E., Lucas, C. A., et al. (2011). The role of nuclear Lamin B1 in cell proliferation and senescence. Genes Dev. 25, 2579–2593. doi: 10.1101/gad.179515.111
Sierra, A., Gottfried-Blackmore, A. C., McEwen, B. S., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi: 10.1002/glia.20468
Sikora, E., Bielak-Zmijewska, A., Dudkowska, M., Krzystyniak, A., Mosieniak, G., Wesierska, M., et al. (2021). Cellular senescence in brain aging. Front. Aging Neurosci. 13:646924. doi: 10.3389/fnagi.2021.646924
Sim, F. J., Zhao, C., Penderis, J., and Franklin, R. J. (2002). The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 22, 2451–2459. doi: 10.1523/JNEUROSCI.22-07-02451.2002
Sobue, A., Komine, O., and Yamanaka, K. (2023). Neuroinflammation in Alzheimer's disease: microglial signature and their relevance to disease. Inflamm Regen 43:26. doi: 10.1186/s41232-023-00277-3
Soreq, L., UK Brain Expression Consortium, North American Brain Expression Consortium, Rose, J., Soreq, E., Hardy, J., et al. (2017). Major shifts in glial regional identity are a transcriptional Hallmark of human brain aging. Cell Rep. 18, 557–570. doi: 10.1016/j.celrep.2016.12.011
Spittau, B. (2017). Aging microglia-phenotypes, functions and implications for age-related neurodegenerative diseases. Front. Aging Neurosci. 9:194. doi: 10.3389/fnagi.2017.00194
Stephan, A. H., Madison, D. V., Mateos, J. M., Fraser, D. A., Lovelett, E. A., Coutellier, L., et al. (2013). A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013
Stojkovic, L., Djuric, T., Stankovic, A., Dincic, E., Stancic, O., Veljkovic, N., et al. (2012). The association of V249I and T280M fractalkine receptor haplotypes with disease course of multiple sclerosis. J. Neuroimmunol. 245, 87–92. doi: 10.1016/j.jneuroim.2011.12.028
St-Pierre, M. K., Carrier, M., Gonzalez Ibanez, F., Simoncicova, E., Wallman, M. J., Vallieres, L., et al. (2022). Ultrastructural characterization of dark microglia during aging in a mouse model of Alzheimer's disease pathology and in human post-mortem brain samples. J. Neuroinflammation 19:235. doi: 10.1186/s12974-022-02595-8
Streit, W. J., Khoshbouei, H., and Bechmann, I. (2021). The role of microglia in sporadic Alzheimer's disease. J. Alzheimers Dis. 79, 961–968. doi: 10.3233/JAD-201248
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. doi: 10.1002/glia.10319
Sun, J. L., Xiao, C., Lu, B., Zhang, J., Yuan, X. Z., Chen, W., et al. (2013). CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J. Neurosci. Res. 91, 545–553. doi: 10.1002/jnr.23168
Switon, K., Kotulska, K., Janusz-Kaminska, A., Zmorzynska, J., and Jaworski, J. (2017). Molecular neurobiology of mTOR. Neuroscience 341, 112–153. doi: 10.1016/j.neuroscience.2016.11.017
Szepesi, Z., Manouchehrian, O., Bachiller, S., and Deierborg, T. (2018). Bidirectional microglia-neuron communication in health and disease. Front. Cell. Neurosci. 12:323. doi: 10.3389/fncel.2018.00323
Tarkowski, E., Issa, R., Sjogren, M., Wallin, A., Blennow, K., Tarkowski, A., et al. (2002). Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer's disease and vascular dementia. Neurobiol. Aging 23, 237–243. doi: 10.1016/s0197-4580(01)00285-8
Taubert, M., Roggenhofer, E., Melie-Garcia, L., Muller, S., Lehmann, N., Preisig, M., et al. (2020). Converging patterns of aging-associated brain volume loss and tissue microstructure differences. Neurobiol. Aging 88, 108–118. doi: 10.1016/j.neurobiolaging.2020.01.006
Tavera, C., Leung-Tack, J., Prevot, D., Gensac, M. C., Martinez, J., Fulcrand, P., et al. (1992). Cystatin C secretion by rat glomerular mesangial cells: autocrine loop for in vitro growth-promoting activity. Biochem. Biophys. Res. Commun. 182, 1082–1088. doi: 10.1016/0006-291x(92)91842-e
Theodore, S., Cao, S., McLean, P. J., and Standaert, D. G. (2008). Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 67, 1149–1158. doi: 10.1097/NEN.0b013e31818e5e99
Tichauer, J. E., Flores, B., Soler, B., Eugenin-von Bernhardi, L., Ramirez, G., and von Bernhardi, R. (2014). Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 37, 187–196. doi: 10.1016/j.bbi.2013.12.018
Tichauer, J., Saud, K., and von Bernhardi, R. (2007). Modulation by astrocytes of microglial cell-mediated neuroinflammation: effect on the activation of microglial signaling pathways. Neuroimmunomodulation 14, 168–174. doi: 10.1159/000110642
Tichauer, J. E., and von Bernhardi, R. (2012). Transforming growth factor-beta stimulates beta amyloid uptake by microglia through Smad3-dependent mechanisms. J. Neurosci. Res. 90, 1970–1980. doi: 10.1002/jnr.23082
Tischer, J., Krueger, M., Mueller, W., Staszewski, O., Prinz, M., Streit, W. J., et al. (2016). Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer's disease. Glia 64, 1562–1572. doi: 10.1002/glia.23024
Tremblay, M. E., Stevens, B., Sierra, A., Wake, H., Bessis, A., and Nimmerjahn, A. (2011). The role of microglia in the healthy brain. J. Neurosci. 31, 16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011
Tristao, F. S., Lazzarini, M., Martin, S., Amar, M., Stuhmer, W., Kirchhoff, F., et al. (2016). CX3CR1 disruption differentially influences dopaminergic neuron degeneration in parkinsonian mice depending on the neurotoxin and route of administration. Neurotox. Res. 29, 364–380. doi: 10.1007/s12640-015-9557-5
Trougakos, I. P. (2019). Nrf2, stress and aging. Aging (Albany NY) 11, 5289–5291. doi: 10.18632/aging.102143
Tse, K. H., and Herrup, K. (2017). Re-imagining Alzheimer's disease – the diminishing importance of amyloid and a glimpse of what lies ahead. J. Neurochem. 143, 432–444. doi: 10.1111/jnc.14079
Tsou, C. L., Haskell, C. A., and Charo, I. F. (2001). Tumor necrosis factor-alpha-converting enzyme mediates the inducible cleavage of fractalkine. J. Biol. Chem. 276, 44622–44626. doi: 10.1074/jbc.M107327200
Tuo, J., Smith, B. C., Bojanowski, C. M., Meleth, A. D., Gery, I., Csaky, K. G., et al. (2004). The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 18, 1297–1299. doi: 10.1096/fj.04-1862fje
Turk, V., and Bode, W. (1991). The cystatins: protein inhibitors of cysteine proteinases. FEBS Lett. 285, 213–219. doi: 10.1016/0014-5793(91)80804-c
Turk, V., Stoka, V., and Turk, D. (2008). Cystatins: biochemical and structural properties, and medical relevance. Front. Biosci. 13, 5406–5420. doi: 10.2741/3089
Turk, B., Turk, V., and Turk, D. (1997). Structural and functional aspects of papain-like cysteine proteinases and their protein inhibitors. Biol. Chem. 378, 141–150.
Turk, B., Turk, D., and Turk, V. (2000). Lysosomal cysteine proteases: more than scavengers. Biochim. Biophys. Acta 1477, 98–111. doi: 10.1016/s0167-4838(99)00263-0
Turner, P. R., O'Connor, K., Tate, W. P., and Abraham, W. C. (2003). Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 70, 1–32. doi: 10.1016/s0301-0082(03)00089-3
Ueno, M., Fujita, Y., Tanaka, T., Nakamura, Y., Kikuta, J., Ishii, M., et al. (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16, 543–551. doi: 10.1038/nn.3358
Uribe-San Martin, R., Herrera-Molina, R., Olavarria, L., Ramirez, G., and von Bernhardi, R. (2009). Reduction of beta-amyloid-induced neurotoxicity on hippocampal cell cultures by moderate acidosis is mediated by transforming growth factor beta. Neuroscience 158, 1338–1347. doi: 10.1016/j.neuroscience.2008.11.002
Villeda, S. A., Plambeck, K. E., Middeldorp, J., Castellano, J. M., Mosher, K. I., Luo, J., et al. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663. doi: 10.1038/nm.3569
Volin, M. V., Huynh, N., Klosowska, K., Chong, K. K., and Woods, J. M. (2007). Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and Akt. Arthritis Rheum. 56, 2512–2522. doi: 10.1002/art.22806
von Bernhardi, R. (2007). Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotox. Res. 12, 215–232. doi: 10.1007/BF03033906
von Bernhardi, R., and Eugenin, J. (2004). Microglial reactivity to beta-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res. 1025, 186–193. doi: 10.1016/j.brainres.2004.07.084
von Bernhardi, R., and Eugenin, J. (2012). Alzheimer's disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid. Redox Signal. 16, 974–1031. doi: 10.1089/ars.2011.4082
von Bernhardi, R., Eugenin-von Bernhardi, L., and Eugenin, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
von Bernhardi, R., Ponce, E., Muñoz, P., Rodríguez, V., Zuñiga-Traslaviña, C., Beltrán-Castillo, S., et al. (2019). Age-related changes in Smad- and non-Smad TGFβ signaling in Neuroinflammation. 10th IBRO world congress of neuroscience Daegu, Korea. September 21–25.
von Bernhardi, R., Tichauer, J. E., and Eugenin, J. (2010). Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 112, 1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x
von Bernhardi, R., Tichauer, J., and Eugenin-von Bernhardi, L. (2011). Proliferating culture of aged microglia for the study of neurodegenerative diseases. J. Neurosci. Methods 202, 65–69. doi: 10.1016/j.jneumeth.2011.08.027
Wallin, H., Abrahamson, M., and Ekstrom, U. (2013). Cystatin C properties crucial for uptake and inhibition of intracellular target enzymes. J. Biol. Chem. 288, 17019–17029. doi: 10.1074/jbc.M113.453449
Wang, S. K., and Cepko, C. L. (2022). Targeting microglia to treat degenerative eye diseases. Front. Immunol. 13:843558. doi: 10.3389/fimmu.2022.843558
Wang, J., Gan, Y., Han, P., Yin, J., Liu, Q., Ghanian, S., et al. (2018). Ischemia-induced neuronal cell death is mediated by chemokine receptor CX3CR1. Sci. Rep. 8:556. doi: 10.1038/s41598-017-18774-0
Wang, J., Pan, H., Lin, Z., Xiong, C., Wei, C., Li, H., et al. (2021). Neuroprotective effect of Fractalkine on radiation-induced brain injury through promoting the M2 polarization of microglia. Mol. Neurobiol. 58, 1074–1087. doi: 10.1007/s12035-020-02138-3
Welser-Alves, J. V., and Milner, R. (2013). Microglia are the major source of TNF-alpha and TGF-beta1 in postnatal glial cultures; regulation by cytokines, lipopolysaccharide, and vitronectin. Neurochem. Int. 63, 47–53. doi: 10.1016/j.neuint.2013.04.007
Wendimu, M. Y., and Hooks, S. B. (2022). Microglia phenotypes in aging and neurodegenerative diseases. Cells 11:2091. doi: 10.3390/cells11132091
Wendt, W., Lubbert, H., and Stichel, C. C. (2008). Upregulation of cathepsin S in the aging and pathological nervous system of mice. Brain Res. 1232, 7–20. doi: 10.1016/j.brainres.2008.07.067
Williams, J. L., Holman, D. W., and Klein, R. S. (2014). Chemokines in the balance: maintenance of homeostasis and protection at CNS barriers. Front. Cell. Neurosci. 8:154. doi: 10.3389/fncel.2014.00154
Winter, A. N., Subbarayan, M. S., Grimmig, B., Weesner, J. A., Moss, L., Peters, M., et al. (2020). Two forms of CX3CL1 display differential activity and rescue cognitive deficits in CX3CL1 knockout mice. J. Neuroinflammation 17:157. doi: 10.1186/s12974-020-01828-y
Wu, Y., Dissing-Olesen, L., MacVicar, B. A., and Stevens, B. (2015). Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 36, 605–613. doi: 10.1016/j.it.2015.08.008
Wynne, A. M., Henry, C. J., Huang, Y., Cleland, A., and Godbout, J. P. (2010). Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 24, 1190–1201. doi: 10.1016/j.bbi.2010.05.011
Ximerakis, M., Lipnick, S. L., Innes, B. T., Simmons, S. K., Adiconis, X., Dionne, D., et al. (2019). Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 22, 1696–1708. doi: 10.1038/s41593-019-0491-3
Xu, Y. J., Au, N. P. B., and Ma, C. H. E. (2022). Functional and phenotypic diversity of microglia: implication for microglia-based therapies for Alzheimer's disease. Front. Aging Neurosci. 14:896852. doi: 10.3389/fnagi.2022.896852
Xue, J., Dong, J. H., Huang, G. D., Qu, X. F., Wu, G., and Dong, X. R. (2014). NF-kappaB signaling modulates radiation-induced microglial activation. Oncol. Rep. 31, 2555–2560. doi: 10.3892/or.2014.3144
Yang, X. P., Mattagajasingh, S., Su, S., Chen, G., Cai, Z., Fox-Talbot, K., et al. (2007). Fractalkine upregulates intercellular adhesion molecule-1 in endothelial cells through CX3CR1 and the Jak Stat5 pathway. Circ. Res. 101, 1001–1008. doi: 10.1161/CIRCRESAHA.107.160812
Yasuda, Y., Shimoda, T., Uno, K., Tateishi, N., Furuya, S., Yagi, K., et al. (2008). The effects of MPTP on the activation of microglia/astrocytes and cytokine/chemokine levels in different mice strains. J. Neuroimmunol. 204, 43–51. doi: 10.1016/j.jneuroim.2008.08.003
Yasuhara, O., Hanai, K., Ohkubo, I., Sasaki, M., McGeer, P. L., and Kimura, H. (1993). Expression of cystatin C in rat, monkey and human brains. Brain Res. 628, 85–92. doi: 10.1016/0006-8993(93)90941-f
Ye, S. M., and Johnson, R. W. (1999). Increased interleukin-6 expression by microglia from brain of aged mice. J. Neuroimmunol. 93, 139–148. doi: 10.1016/s0165-5728(98)00217-3
Yin, J. A., Liu, X. J., Yuan, J., Jiang, J., and Cai, S. Q. (2014). Longevity manipulations differentially affect serotonin/dopamine level and behavioral deterioration in aging Caenorhabditis elegans. J. Neurosci. 34, 3947–3958. doi: 10.1523/JNEUROSCI.4013-13.2014
Yin, X., Qiu, Y., Zhao, C., Zhou, Z., Bao, J., and Qian, W. (2021). The role of amyloid-Beta and tau in the early pathogenesis of Alzheimer's disease. Med. Sci. Monit. 27:e933084. doi: 10.12659/MSM.933084
Yoshida, H., Imaizumi, T., Fujimoto, K., Matsuo, N., Kimura, K., Cui, X., et al. (2001). Synergistic stimulation, by tumor necrosis factor-alpha and interferon-gamma, of fractalkine expression in human astrocytes. Neurosci. Lett. 303, 132–136. doi: 10.1016/s0304-3940(01)01699-8
Young, K. M., Psachoulia, K., Tripathi, R. B., Dunn, S. J., Cossell, L., Attwell, D., et al. (2013). Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–885. doi: 10.1016/j.neuron.2013.01.006
Zabel, M. K., Zhao, L., Zhang, Y., Gonzalez, S. R., Ma, W., Wang, X., et al. (2016). Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 64, 1479–1491. doi: 10.1002/glia.23016
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zhang, L., Xu, J., Gao, J., Wu, Y., Yin, M., and Zhao, W. (2018). CD200-, CX3CL1-, and TREM2-mediated neuron-microglia interactions and their involvements in Alzheimer's disease. Rev. Neurosci. 29, 837–848. doi: 10.1515/revneuro-2017-0084
Zheng, H., and Koo, E. H. (2006). The amyloid precursor protein: beyond amyloid. Mol. Neurodegener. 1:5. doi: 10.1186/1750-1326-1-5
Zhu, W., Acosta, C., MacNeil, B., Cortes, C., Intrater, H., Gong, Y., et al. (2013). Elevated expression of fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) in the dorsal root ganglia and spinal cord in experimental autoimmune encephalomyelitis: implications in multiple sclerosis-induced neuropathic pain. Biomed. Res. Int. 2013:480702. doi: 10.1155/2013/480702
Zieger, M., Ahnelt, P. K., and Uhrin, P. (2014). CX3CL1 (fractalkine) protein expression in normal and degenerating mouse retina: in vivo studies. PLoS One 9:e106562. doi: 10.1371/journal.pone.0106562
Zucker-Franklin, D., Warfel, A., Grusky, G., Frangione, B., and Teitel, D. (1987). Novel monocyte-like properties of microglial/astroglial cells. Constitutive secretion of lysozyme and cystatin-C. Lab. Investig. 57, 176–185.
Zujovic, V., Benavides, J., Vige, X., Carter, C., and Taupin, V. (2000). Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia 29, 305–315.
Zujovic, V., Schussler, N., Jourdain, D., Duverger, D., and Taupin, V. (2001). In vivo neutralization of endogenous brain fractalkine increases hippocampal TNFalpha and 8-isoprostane production induced by intracerebroventricular injection of LPS. J. Neuroimmunol. 115, 135–143. doi: 10.1016/s0165-5728(01)00259-4
Keywords: aging, Alzheimer’s disease, CX3CL1, CX3CR1, metalloproteases, neurodegenerative disease, neuroinflammation, Parkinson’s disease
Citation: Eugenín J, Eugenín-von Bernhardi L and von Bernhardi R (2023) Age-dependent changes on fractalkine forms and their contribution to neurodegenerative diseases. Front. Mol. Neurosci. 16:1249320. doi: 10.3389/fnmol.2023.1249320
Edited by:
Björn Spittau, Bielefeld University, GermanyReviewed by:
Angela Gomez-Arboledas, University of California, Irvine, United StatesPeter Wieghofer, Leipzig University, Germany
Copyright © 2023 Eugenín, Eugenín-von Bernhardi and von Bernhardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaime Eugenín, amFpbWUuZXVnZW5pbkB1c2FjaC5jbA==; Rommy von Bernhardi, cm9tbXkudm9uYmVybmhhcmRpQHVzcy5jbA==