 Aditi Naskar
Aditi Naskar Asima Nayak
Asima Nayak Muthu Raj Salaikumaran
Muthu Raj Salaikumaran Sonali S. Vishal
Sonali S. Vishal Pallavi P. Gopal
Pallavi P. Gopal- 1Department of Pathology, Yale School of Medicine, New Haven, CT, United States
- 2Program in Cellular Neuroscience, Neurodegeneration, and Repair, Yale School of Medicine, New Haven, CT, United States
Liquid–liquid phase separation results in the formation of dynamic biomolecular condensates, also known as membrane-less organelles, that allow for the assembly of functional compartments and higher order structures within cells. Multivalent, reversible interactions between RNA-binding proteins (RBPs), including FUS, TDP-43, and hnRNPA1, and/or RNA (e.g., RBP-RBP, RBP-RNA, RNA-RNA), result in the formation of ribonucleoprotein (RNP) condensates, which are critical for RNA processing, mRNA transport, stability, stress granule assembly, and translation. Stress granules, neuronal transport granules, and processing bodies are examples of cytoplasmic RNP condensates, while the nucleolus and Cajal bodies are representative nuclear RNP condensates. In neurons, RNP condensates promote long-range mRNA transport and local translation in the dendrites and axon, and are essential for spatiotemporal regulation of gene expression, axonal integrity and synaptic function. Mutations of RBPs and/or pathologic mislocalization and aggregation of RBPs are hallmarks of several neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Alzheimer’s disease. ALS/FTD-linked mutations of RBPs alter the strength and reversibility of multivalent interactions with other RBPs and RNAs, resulting in aberrant phase transitions. These aberrant RNP condensates have detrimental functional consequences on mRNA stability, localization, and translation, and ultimately lead to compromised axonal integrity and synaptic function in disease. Pathogenic protein aggregation is dependent on various factors, and aberrant dynamically arrested RNP condensates may serve as an initial nucleation step for pathologic aggregate formation. Recent studies have focused on identifying mechanisms by which neurons resolve phase transitioned condensates to prevent the formation of pathogenic inclusions/aggregates. The present review focuses on the phase separation of neurodegenerative disease-linked RBPs, physiological functions of RNP condensates, and the pathologic role of aberrant phase transitions in neurodegenerative disease, particularly ALS/FTD. We also examine cellular mechanisms that contribute to the resolution of aberrant condensates in neurons, and potential therapeutic approaches to resolve aberrantly phase transitioned condensates at a molecular level.
Introduction
Neurons are large, highly polarized cells that require precise spatial and temporal organization of gene expression, particularly in the axon and dendrites, in order to regulate synaptic structure and function. A growing body of evidence suggests the physical process of liquid–liquid phase separation (LLPS) allows for biological macromolecules, such as proteins and nucleic acids, to self-assemble into higher order structures within cells, including in neurons. Post-transcriptional control of RNA is accomplished by ribonucleoprotein (RNP) granules, dynamic membrane-less cellular compartments enriched in RNA and RNA-binding proteins (RBPs), which often form through LLPS (Banani et al., 2017; Yang et al., 2020). These phase separated RNP condensates include nucleoli, Cajal bodies, processing bodies, stress granules, and neuronal RNP transport granules, and each regulates distinct aspects of the RNA lifecycle, from ribonucleoprotein biogenesis, to RNA splicing, mRNA stability, transport, and translation. Strikingly, many of the RBPs involved in LLPS have been implicated in the pathogenesis of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), fatal neurodegenerative diseases with distinct but overlapping clinical, genetic and pathologic features (Ling et al., 2013).
ALS, also known as Lou Gehrig’s disease, is characterized by the selective loss of motor neurons in the brain and spinal cord and progressive muscle atrophy (Hardiman et al., 2017). In contrast, FTD is defined by neurocognitive decline and changes in social behavior, personality and language due to degeneration of the frontal and temporal cortex (Van Mossevelde et al., 2018). As many as 50% of ALS patients also develop cognitive impairment, and motor deficits are relatively common in FTD patients, underscoring the fact that these disorders exist on a clinical disease continuum (Lomen-Hoerth et al., 2002; Baizabal-Carvallo and Jankovic, 2016). While ~90% of ALS/FTD cases are “sporadic” and present without a clear family history, the remaining 5–10% of cases are associated with familial disease. Inherited and rare sporadic cases of ALS/FTD have been linked to mutations in ~40 genes, many of which encode RBPs, including transactive DNA response binding protein of 43 kDa (TDP-43) and fused in sarcoma (FUS) (Sreedharan et al., 2008; Kwiatkowski et al., 2009; Vance et al., 2009; Kim G. et al., 2020). The vast majority (>95%) of ALS and about half of FTD patients also share a common molecular histopathology, characterized by the mislocalization and aggregation of TDP-43 in the affected cells (Neumann et al., 2006; Lee et al., 2011).
In this review, we specifically focus on the role of LLPS in cellular organization into liquid-like compartments, which offer advantages to cells and drive several aspects of neuronal cell biology, but also likely contribute to disease. We outline evidence that neurodegenerative disease-linked mutations of RBPs lead to aberrant phase separation, altered condensate dynamics and biophysical properties, and disruption of critical cellular and molecular functions (Figure 1). Although we largely concentrate on aberrant LLPS in ALS/FTD, we also discuss data that suggest aberrant phase separation plays a role in pathological protein aggregation in Alzheimer’s disease (AD) and Parkinson’s disease (PD). Finally, we discuss cellular mechanisms to combat aberrant LLPS as well as potential therapeutic approaches.
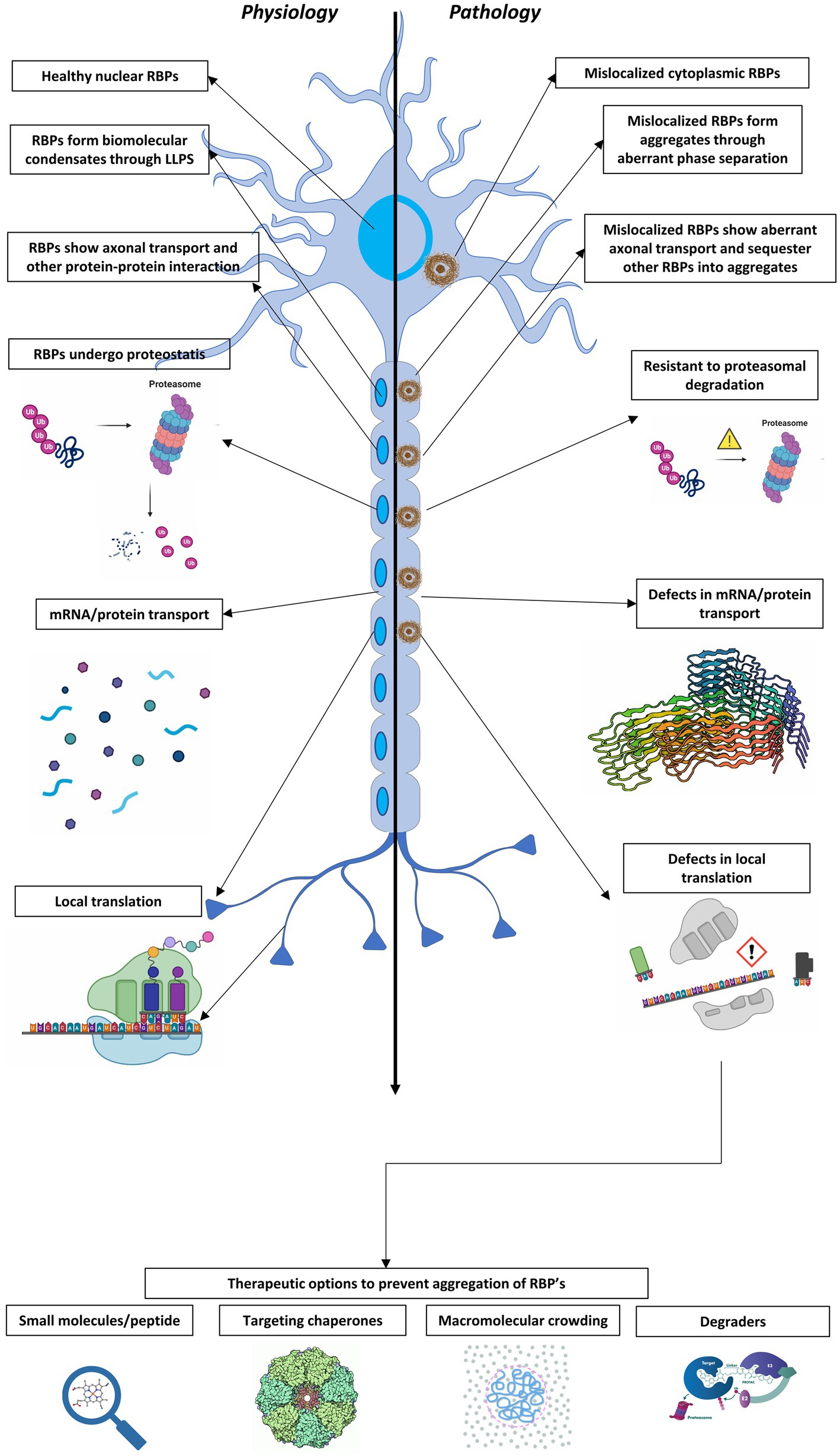
Figure 1. A schematic representation depicting a healthy neuron (left) and an ALS/FTD-affected neuron (right) along with the major differences in RBPs and RNP condensates under physiologic and pathologic conditions. 1. Physiological RBPs are found predominantly in the nucleus whereas RBPs are mislocalized to cytoplasm in disease. 2. Resistance to proteasomal degradation: Aggregates composed of RBPs in affected neurons show increased resistance to degradation by the cellular proteasome system. 3. Defects in mRNA/protein transport: There is impaired stability and transport of mRNA. 4. Defects in local translation: Affected neurons show deficiencies in local translation. The bottom of the figure represents several therapeutic options to target neurodegenerative diseases aimed to resolve aberrant RNP condensates and prevent the aggregation of RBPs in the affected neurons.
Liquid–liquid phase separation: a mechanism for the assembly of biomolecular condensates
Phase separation is a fundamental thermodynamic process in which two (or more) macromolecules preferentially interact with one another and demix from a homogeneous solution to form distinct phases (Banani et al., 2017). In cells, multivalent low-affinity interactions between biomolecules present at high local concentrations (e.g., RNA–RNA, RNA-protein, protein–protein) drive the condensation of proteins, RNA, and membrane-bound organelles into liquid-like foci, allowing for the formation of biomolecular condensates, also known as membrane-less organelles (MLOs) (Banani et al., 2017; Ma and Mayr, 2018). In neurons, LLPS facilitates the assembly of specialized biomolecular condensates including RNP condensates (e.g., neuronal RNP transport granules, discussed below), membrane-associated higher-order assemblies at the synapse (e.g., pre-synaptic active zone) and synaptic vesicle condensates (Milovanovic et al., 2018; Liao et al., 2019; Wu et al., 2019; McDonald et al., 2020). The phenomenon of LLPS is governed by the balance between interaction energies of biomolecules (i.e., enthalpy) and entropy of the system, which can be modulated by temperature, molecular crowding, solvent conditions, macromolecule concentrations and composition (Hyman et al., 2014; Nott et al., 2016). These multivalent and low-affinity interactions promote a dynamic exchange of components within the biomolecular condensate and the surrounding environment (Banani et al., 2017).
Biomolecular condensates are often composed of proteins with modular interaction domains or intrinsically disordered regions (IDRs) (Li et al., 2012; Lin et al., 2015). IDRs lack stable tertiary structures, can adopt multiple conformations and frequently contain repetitive low-complexity sequences (Banani et al., 2017). For example, the IDRs of many RBPs are referred to as low complexity domains (LCDs) because they are enriched in polar (e.g., glycine, serine, glutamine, asparagine), aromatic (e.g., tryptophan and tyrosine), and/or charged residues (Kato et al., 2012; Mittag and Parker, 2018; Figure 2). Several studies suggest these repeated sequence elements drive multivalent, low-affinity electrostatic, dipolar or hydrophobic interactions between aromatic, polar and/or charged residues in the IDRs of RBPs, and thus promote LLPS in vitro and condensate formation in cells (Gilks et al., 2004; Decker et al., 2007; Kato et al., 2012; Brangwynne et al., 2015; Nott et al., 2015; Lin et al., 2017; Wang et al., 2018). In addition, purified RBPs with IDRs, including many disease associated RBPs such as FUS (Han et al., 2012; Patel et al., 2015), hnRNPA1 (Kim et al., 2013; Molliex et al., 2015), TDP-43 (Conicella et al., 2016; Li et al., 2018; McGurk et al., 2018), TIA-1 (Mackenzie et al., 2017), and FMRP (Kim et al., 2019; Tsang et al., 2019), have been shown to undergo LLPS in vitro. Furthermore, multivalent interactions between aromatic and/or charged residues in the IDRs and RNA-binding domains of RBPs as well as RNA–RNA and RBP-RNA interactions also play an important role in driving phase separation (Elbaum-Garfinkle et al., 2015; Lin et al., 2015; Molliex et al., 2015; Zhang H. et al., 2015; Wang et al., 2018; Grese et al., 2021). In cells, the formation of RNP condensates integrates both specific protein-RNA and protein–protein interactions along with the relatively weak non-specific IDR interactions that drive LLPS in vitro (Mittag and Parker, 2018). However, the relative contributions of these different types of interactions for RNP condensate formation in cells is not well understood but is likely context-specific and involves post-translational modifications, for example, that alter weak multivalent interactions between IDRs (Sanders et al., 2020; Yang et al., 2020). Additional research is required to elucidate the complex network of interactions driving RNP condensate assembly and regulation in neurons and other disease-relevant cells.
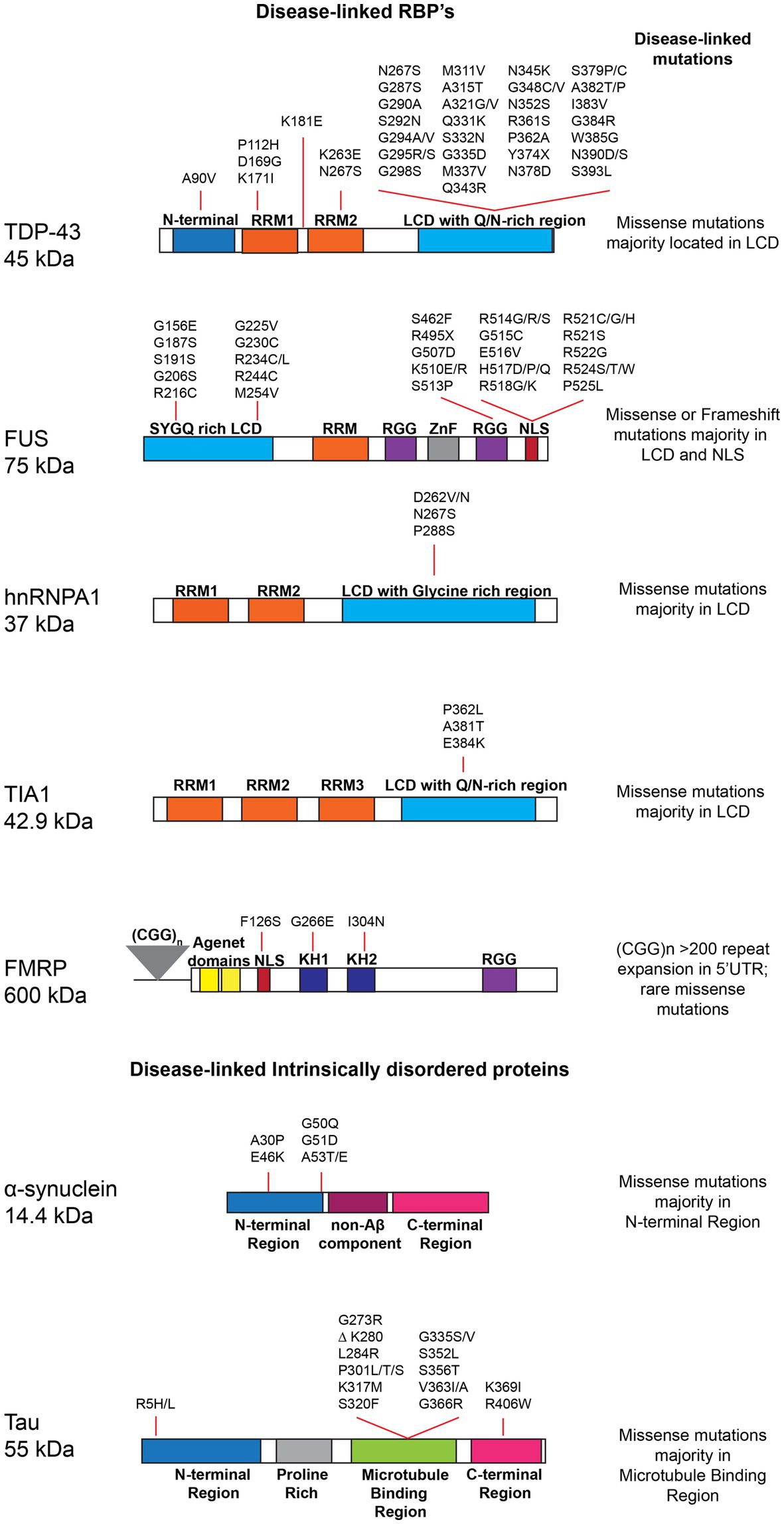
Figure 2. Schematic highlighting RNA-binding proteins (RBPs) and intrinsically disordered proteins (IDPs), domain structure and disease-linked mutations: TAR DNA-binding protein 43 (TDP-43 - Uniprot Q13148), Fused in sarcoma (FUS - Uniprot P35637), Heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1 Uniprot P09651), T-cell intracellular antigen 1 (TIA1 - Uniprot P31483), Fragile X mental retardation protein (FMRP – Uniprot Q06787), α-Synuclein (Alpha-synuclein – Uniprot P37840), and Microtubule-associated protein tau (Tau - Uniprot P10636-5). The major domains for each protein and disease-linked mutations retrieved from Uniprot, are depicted. RNA recognition motif (RRM); Low complexity domain (LCD); Zinc finger (ZnF); Tyrosine- and glycine-rich region (YG-box); Frameshift (FS); Loss of function (LOF); Nuclear localization signal (NLS); hnRNP K protein homology (KH); arginine-glycine–glycine box (RGG); Non-Amyloid-beta component (NAC) region, Microtubule binding region (MTBR).
Functions of physiological RNP condensates in neurons
In cells, biomolecular condensates serve to accelerate biochemical reactions by increasing local enzyme and substrate concentrations, or alternatively, may modulate reactions by sequestering or buffering condensate components from the cytoplasm (Strulson et al., 2012; Banjade and Rosen, 2014; Tatomer et al., 2016). RNP condensates in particular serve as temporary reservoirs for specific mRNAs and RBPs, where they store, stabilize and/or transport translationally repressed mRNA, ultimately contributing to the function of synapses, as well as the architecture and organization of neurons (Kim N. Y. et al., 2020; Nedelsky and Taylor, 2022).
Neuronal transport granules, stress granules, and processing (P)-bodies are distinct but related cytoplasmic RNP condensates that have vital functions in the post-transcriptional processing of RNA, as well as mRNA subcellular localization, translation, storage, and turnover (Kiebler and Bassell, 2006; Buchan and Parker, 2009; Donlin-Asp et al., 2017). Stress granules (SGs) are transient RNP condensates that contain translationally stalled mRNAs, pre-initiation factors, ribosomal components and specific RBPs (Buchan and Parker, 2009; Kedersha et al., 2013; Jain et al., 2016). SGs emerge in response to various forms of cellular stress such as viral infections, heat shock, and oxidative stress (Anderson and Kedersha, 2009). SGs prevent the degradation of mRNAs that have undergone translation arrest, by storing them alongside the necessary translation initiation factors and RBPs. During stress, SGs also act as signaling hubs and work to suppress the production of new proteins that might be unnecessary (those involved with cellular growth) or detrimental (those associated with apoptosis or cytotoxicity) to promote cell survival (Kedersha et al., 2013; Wippich et al., 2013; Kanda et al., 2021). Independent of stress, depletion of SG scaffolding protein G3BP1 alters intracellular calcium homeostasis and results in abnormal synaptic plasticity in neurons (Martin et al., 2013; Yang et al., 2020). Recent research also suggests that cellular aging impacts SG assembly and dynamics of primary motor neurons, though the relevance of these findings for function in vivo remains to be elucidated (Khalfallah et al., 2018).
P-bodies are involved in mRNA breakdown, storage, and translational repression (Adeli, 2011). They contain a wide variety of enzymes involved in mRNA degradation, including decapping enzymes, exonucleases, and RISC (RNA-induced silencing complex) components (Parker and Sheth, 2007). P-bodies may regulate synaptic plasticity, neuronal development, and stress response, but further research is needed to completely understand the functions of P-bodies in neurons (Nonhoff et al., 2007). Dysregulation of P-bodies has been linked to a number of neurodegenerative disorders, including AD and ALS (Wootz et al., 2013; Wen et al., 2014; Barmada et al., 2015).
Neuronal transport granules facilitate the long-distance transport of mRNAs from the cell body to the axon and dendrites, enabling local translation and activity-dependent regulation of protein synthesis in distal neuronal compartments and at the synapse (Ainger et al., 1993; Krichevsky and Kosik, 2001; Kanai et al., 2004; Willis et al., 2005). Maintaining local translation is crucial for synaptic function, regulating dendritic spine morphology, and modulating long-term potentiation (LTP) and long-term depression (LTD) (Sutton and Schuman, 2006; Holt and Schuman, 2013). In neurons, mRNA transport and local translation are tightly regulated by RBPs. In response to certain cues, the mRNAs are released from the RNP granules and translated by local ribosomes, resulting in the synthesis of proteins necessary for synaptic function, plasticity, and neuronal homeostasis (Steward and Schuman, 2003; Bramham and Wells, 2007; Buxbaum et al., 2014). For instance FMRP is believed to regulate the local storage and translation of mRNAs at synapses, where it binds to many mRNAs in neurons (Siomi et al., 1993; Brown et al., 2001; Hagerman et al., 2001; Huber et al., 2002; Antar et al., 2004; Muddashetty et al., 2007; Darnell et al., 2011; Nelson et al., 2013; Alpatov et al., 2014; Pasciuto and Bagni, 2014; Hu et al., 2015; Myrick et al., 2015; D'Souza et al., 2018; Kute et al., 2019) forming RNP complexes that are then transported along microtubules by kinesin and dynein motor proteins (Antar et al., 2005, 2006; Dictenberg et al., 2008). Similar to FMRP, ALS/FTD-linked RBPs such as FUS and TDP-43, are components of neuronal transport granules (Kanai et al., 2004; Elvira et al., 2006; Fallini et al., 2012; Liu-Yesucevitz et al., 2014). In addition to their critical roles in the nucleus, TDP-43 and FUS also regulate the trafficking of specific mRNAs in the dendrites and axon, particularly transcripts encoding cytoskeletal-related proteins (Fujii et al., 2005; Fujii and Takumi, 2005; Strong et al., 2007; Alami et al., 2014; Coyne et al., 2014; Chu et al., 2019). The pool of mRNAs stored in neuronal transport granules must be readily available for translation in response to signals from the cell as reported in the case of Staufen proteins (STAU1 and STAU2) (Duchaîne et al., 2002; Graber et al., 2013). Thus, neuronal transport granules play a critical role in the storage and regulated translation of mRNA, as well as its localization and stability (Buxbaum et al., 2014, 2015).
Aberrant phase separation in neurodegenerative disease
Defects in RNA metabolism characterize a large proportion of neurodevelopmental and neurodegenerative disorders (Prashad and Gopal, 2021). In fact, there is strong human genetic evidence that mutations of numerous RBPs are linked to ALS and FTD (Ling et al., 2013). Notably, many of these ALS/FTD-linked mutations cluster in the low complexity domains of RBPs and/or disrupt their nuclear localization (Figure 2), raising the possibility that these mutations disturb the delicate balance of low affinity interactions driving physiological phase separation, alter subcellular localization of RBPs, and/or increase the propensity for pathological aggregation (Buratti, 2015; Nedelsky and Taylor, 2022). Indeed, several in vitro studies of purified ALS/FTD-linked mutant forms of TDP-43, FUS, hnRNPA1, and TIA-1 show that they exhibit aberrant phase separation, accelerate the formation of solid or gel-like structures and lead to thioflavin positive fibrillar aggregates (Molliex et al., 2015; Patel et al., 2015; Conicella et al., 2016; Mackenzie et al., 2017; McGurk et al., 2018). Moreover, ALS-linked mutations of RBPs, have been shown to perturb post-transcriptional RNA processing, mRNA trafficking and local translation, stress granule formation/dissolution kinetics, and/or mRNA stability in addition to altering RNP condensate behavior in vivo and in cellular disease models (Kim et al., 2013; Coyne et al., 2015; Murakami et al., 2015; Gopal et al., 2017; Fratta et al., 2018; Wolozin and Ivanov, 2019; Hallegger et al., 2021).
While it is difficult to distinguish functional defects induced by the RBP mutation itself vs. compromised function due to aberrantly phase separated RNP condensates, it has been suggested that aberrant phase separation may offer a unifying explanation for the plethora of functional defects in RNA regulation observed with pathological mutations of RBPs (Nedelsky and Taylor, 2022). In addition, examples of aberrant condensates associated with pathologic post-translational modifications of wild type RBPs or other disordered proteins such as tau, suggest that aberrant phase separation may also be relevant in the setting of sporadic neurodegenerative disease (Cohen et al., 2015; Wegmann et al., 2018; Yu et al., 2021; Lu et al., 2022). Here we consider potential mechanisms of how aberrant phase separation may be detrimental to function and evaluate the data indicating that aberrant LLPS contributes to the formation of pathological aggregates in neurodegenerative disease (Patel et al., 2015; Alberti et al., 2019). Table 1 provides a summary of notable RBPs and intrinsically disordered proteins linked to aberrant phase separation in ALS/FTD and other neurodegenerative diseases.

Table 1. Neurodegenerative disease-linked RBPs and Intrinsically disordered proteins.
Loss of RNP condensate liquid-like properties impairs mRNA transport
A growing body of evidence supports the view that aberrantly phase separated RNP condensates composed of mutant RBPs result in loss of dynamic liquid-like biophysical properties, which compromises their RNA regulatory functions in neurons. For example, ALS-linked mutations of FUS and TDP-43 promote hardening of neuronal RNP condensates (Murakami et al., 2015; Gopal et al., 2017). Neuronal RNP transport granules containing mutant TDP-43 display impaired transport in dendrites (Liu-Yesucevitz et al., 2014) and along the axon of primary cortical neurons or ALS patient iPSC-derived neurons (Alami et al., 2014; Gopal et al., 2017) as well as increased viscosity and loss of liquid-like properties (Gopal et al., 2017; Vishal et al., 2022). Furthermore, ALS-linked mutations of TDP-43 disrupt axonal transport and/or stability of Nefl and futsch/MAP1B mRNA (Alami et al., 2014; Coyne et al., 2014). Similarly, FUS mutations disrupt axonal transport of Dynll2 and Kif5b mRNA in primary hippocampal neurons and localization of Ddr2 mRNA to the cell periphery (Yasuda et al., 2017). Thus, defects in neuronal RNP transport granule dynamics impair mRNA trafficking and localization of critical transcripts to the axon, dendrites, and neuromuscular junction (NMJ), which may in turn compromise synaptic and NMJ homeostasis and trigger axonal degeneration.
Sequestration of mRNAs and RBPs dysregulate local and global translation
Several studies suggest that another key consequence of dynamically arrested, aberrant RNP condensates and is the sequestration of mRNA and RBPs resulting in defects in multiple cellular functions, most prominently translation (Kim N. Y. et al., 2020). Missense mutations of arginine residues in FUS (i.e., R244C, R216G, R521G) sequester wild type (WT) FUS and disrupt WT FUS-RNA droplet fluidity (Rhine et al., 2020). Moreover, ALS-linked mutant FUS mislocalizes to the cytoplasm, sequesters FMRP into FUS-FMRP condensates in an RNA-dependent manner, and suppresses translation in motor neurons (Birsa et al., 2021). Additional support for translation dysregulation associated with aberrant FUS condensates comes from a proteomic analysis of ALS-mutant FUS cytoplasmic inclusions, which reveals an enrichment of stalled ribosomal complexes and factors promoting nonsense mediated decay, compared to wild type FUS (Kamelgarn et al., 2018). Spinal cords of FUS mutant [R521C and R521H] transgenic mice show depletion of mRNAs encoding ion channels, transporters and ribosomal proteins, and furthermore, these FUS mutations inhibit local translation in axons by inducing stress mediated phosphorylation of eIF2α (Lopez-Erauskin et al., 2018). In other instances, mutant FUS binds the 3’UTR of transcripts, such as the ELAVL4 mRNA, and increases its translation, but also sequesters ELAVL4 (HuD) and other RBPs within stiff, gel-like condensates (De Santis et al., 2019).
There is also evidence that pathologic C-terminal fragments of wild type TDP-43 and mislocalized hyper-phosphorylated wild type TDP-43 are associated with aberrant RNP condensates and aggregate formation (Igaz et al., 2009; Altman et al., 2021). For example, pathologic C-terminal fragments of wild type TDP-43 sequester RBPs such as TIA-1, PABPC1, hnRNPA2/B1 in a RNA dependent manner (Jiang et al., 2022). Moreover, aberrant phosphorylated TDP-43 condensates in the axon of motor neurons sequester G3BP and nuclear encoded mitochondrial mRNA Cox4i1 and ATP5A1, and impair local protein synthesis of nuclear encoded mitochondrial genes (Altman et al., 2021). Aside from inhibiting translation, aberrant condensates are also likely to sequester key proteins in the proteosome and autophagy pathways, compromising proteostasis and organelle quality control mechanisms (discussed below) (Wakabayashi et al., 2002; Kuusisto et al., 2003; Shao et al., 2022).
Aberrant condensates and transition to pathological fibrillar aggregates
In addition to functional defects in RNA regulation associated with aberrantly phase separated condensates, evidence suggests that in some cases, these dynamically arrested condensates can give rise to fibrillar protein aggregates that are the pathologic hallmarks of ALS/FTD and other neurodegenerative diseases (Ramaswami et al., 2013). For example, aspartate to valine mutations in the prion-like domain of hnRNPA1 family of proteins accelerate the formation of fibrils and of cytoplasmic inclusions leading to multisystem proteinopathy and ALS (Kim et al., 2013). Similarly, ALS-linked mutations of FUS promote the molecular aging of liquid FUS condensates and conversion to fibrillar aggregates, leading to formation of cytoplasmic inclusions (Patel et al., 2015; Birsa et al., 2021). PD-linked mutations of α-Synuclein, which is a synaptic protein normally found at axon terminals, promote LLPS and maturation of α-Synuclein condensates, resulting in the formation of perinuclear cytoplasmic α-Synuclein aggregates (Ray et al., 2020). On the other hand, mutations in the alpha-helical region of TDP-43’s low complexity domain disrupt LLPS but also promote aggregation (Buratti, 2015; Che et al., 2015; Conicella et al., 2016). In addition, under stress conditions in cells, cytoplasmic TDP-43 aggregates can form independently and are distinct from stress granules (Gasset-Rosa et al., 2019).
Apart from aberrant condensates associated with neurodegenerative disease-linked mutations, post-translational modifications of wild type RBPs or other IDPs such as tau, may also result in aberrant condensates or aggregates. Pathologic TDP-43 aggregates found in the majority of sporadic ALS cases display numerous post-translational modifications of wild type TDP-43, including phosphorylation, ubiquitination, cysteine oxidation, acetylation, and truncation (Neumann et al., 2006; Buratti, 2015; Cohen et al., 2015; Arseni et al., 2022). Acetylation of TDP-43 in particular promotes the formation of nuclear TDP-43 condensates but also alters TDP-43 solubility and promotes aggregation (Cohen et al., 2015). A similar observation is seen in the case of phosphorylated wild type Tau protein, which forms phase separated phospho-tau droplets that go on to initiate Tau aggregates. Furthermore, high molecular weight Tau isolated from human AD brain tissue also undergoes LLPS and aggregation (Wegmann et al., 2018).
Role of persistent SGs in neurodegenerative disease
Due to their complex structure and post-mitotic nature, neurons experience various forms of acute and chronic stress over their long life-span. To overcome oxidative or proteo-stress conditions for instance, neurons depend on an efficiently regulated SG response (Wolozin and Ivanov, 2019). Dysregulation of SG formation/dissolution kinetics and persistence of SGs has been suggested as the first step toward aberrant phase separation and formation of insoluble protein aggregates in ALS/FTD (Li et al., 2013; Mackenzie et al., 2017). Indeed, a subset of TDP-43 positive aggregates and phosphorylated-tau in ALS/FTD and AD patient brain tissue, respectively, contain SG markers (Liu-Yesucevitz et al., 2010; Bentmann et al., 2012; Maziuk et al., 2018; Wolozin and Ivanov, 2019). For example, TDP-43 aggregates in the spinal cord of ALS patients are positive for TIA-1, PABP-1, or other canonical SG markers (Liu-Yesucevitz et al., 2010; Bentmann et al., 2012). However, in the hippocampus of FTD patients with TDP-43 pathology, SG RBPs show more inconsistent overlap with aggregates (Bentmann et al., 2012; Aulas and Vande Velde, 2015), and mature neurofibrillary tangles in AD patient tissue do not colocalize with SG markers (Maziuk et al., 2018). These findings may reflect potential differences in SG biology of motor neurons compared to hippocampal and cortical neurons, and/or perhaps highlight differences in the role of persistent SGs in ALS vs. FTD and AD. Furthermore, as mentioned above, there is also evidence that under cellular stress paradigms, cytoplasmic TDP-43 aggregates which form are distinct from SGs, suggesting that there are both SG-dependent and independent mechanisms driving TDP-43 pathology (Aulas and Vande Velde, 2015; Gasset-Rosa et al., 2019). Additional in vivo evidence that demonstrates the transformation of persistent SGs to mature pathologic aggregates is needed in longitudinal ALS/FTD and AD disease models in order to elucidate the mechanistic role of SGs in these diseases.
Taken together, the literature suggests that while pathologic aggregation of RBPs and other intrinsically disordered proteins in neurodegenerative disease may occur via aberrant phase separation and persistence of SGs in some instances, protein misfolding and aggregation in disease also occurs independent of LLPS. Regardless of the mechanism leading to pathologic aggregates in disease, cells have evolved several mechanisms to prevent aggregation and/or resolve aberrantly phase separated condensates.
Cellular mechanisms to overcome aberrant phase separation
For the majority of patients with ALS/FTD or other neurodegenerative disease who do not have highly penetrant mutations in RBPs or other IDPs such as α-synuclein, it has been suggested that aging and cellular stress, induce changes in protein conformation or stability that may promote aberrant phase separation and/or aggregation. Indeed, human genetics data strongly implicate defects in lysosomal trafficking, autophagy, and protein quality control mechanisms in ALS/FTD, AD and PD, indicating that mutations in these pathways may also trigger neurodegenerative disease (Rogaeva et al., 2007; Caglayan et al., 2014; Bras et al., 2015; Guerreiro et al., 2015). Under stress conditions, proteins may adopt altered conformations that expose hydrophobic regions, which can promote the formation of solid-gel like aggregates (Gasset-Rosa et al., 2019). Therefore, to combat aberrantly phase separated condensates, cells have evolved mechanisms to regulate LLPS as well as quality control mechanisms (Prasad et al., 2010). Post-translational modifications, such as phosphorylation, ubiquitination, and acetylation, can also modulate the phase behavior of proteins by altering their charge or conformation, ultimately affecting their ability to undergo aberrant phase separation (Hofweber et al., 2018; Maziuk et al., 2018). In this section we describe our current understanding of these homeostatic mechanisms and their neuroprotective functions.
Role of lysosomes
Biomolecular condensates are known to interact with and recruit membrane-bound organelles like endoplasmic reticulum and lysosomes (Ma and Mayr, 2018; Liao et al., 2019; Zhou, 2022). For example, Liao and colleagues showed that Annexin A11 (ANXA11) serves as an adaptor protein that tethers RNP granules and lysosomes, allowing RNP condensates to hitchhike on lysosomes (Liao et al., 2019). Whether membrane-bound organelles like lysosomes are recruited by cells to clear protein aggregates is poorly understood. One of the hallmark features of ALS is the aggregation of FUS upon cellular stress. FUS aggregates are reported to recruit lysosomes (Trnka et al., 2021). This observation suggests that lysosomes may be able to sense aberrantly phase separated condensates and amass more near solid-gel like aggregates rather than fluid condensates. Additional studies are needed to determine whether this is a potential mechanism to clear aggregates and aberrantly phase separated condensates.
Role of ubiquitin proteasome system and autophagy
The ubiquitin proteasome system (UPS) and autophagy are the two major proteolytic systems in the eukaryotic cells responsible for the degradation of proteins and protein aggregates. Aggregation prone proteins such as poly (Q) expansion repeats, mutant (SOD1), α-synuclein and tau can be degraded by both (UPS) and autophagy (Zheng et al., 2009). The UPS involves ubiquitination of proteins with the help of a cascade of enzymes (E1-ubiquitin activating enzyme, E2-ubiquitin conjugase, E3- ubiquitin ligase) consequently targeting them to the proteasomal machinery (Ciechanover and Schwartz, 1998). Multiple studies have shown the clearance of soluble TDP-43 (van Eersel et al., 2011) and mutant TDP-43 aggregates by the UPS (Scotter et al., 2014). Autophagy pathway is activated when the UPS cannot degrade aggregated proteins because of their failure to enter the proteasomal barrel (Verhoef et al., 2002).
Autophagy is a cellular degradation process that involves engulfing and enclosing the cytoplasmic contents, mainly the damaged cellular components, in an autophagosome preceding lysosomal degradation (Nixon, 2013). An autophagosome engulfs parts of the cytosol and fuses with a lysosome leading to autophagolysosome where its contents are broken down (Deter et al., 1967). The autophagosomes play a key role in degrading the stress granules that undergo a decelerated degradation process or that fail to disassemble (Buchan et al., 2013). According to a study, C9orf72 repeat expansions hoard arginine methylated proteins and incorporate autophagy receptor P62 to promote elimination of stress granules by autophagy (Chitiprolu et al., 2018). Kinases like TANK-binding kinase (TBK-1) helps in autophagy mediated degradation of protein aggregates by phosphorylating autophagy adaptor p62 (Shao et al., 2022). A recent study showed C9orf72 expansion sequesters TBK-1 into poly GA inclusions thereby diminishing TBK1’s function which in turn impedes endosomal maturation (Shao et al., 2022). Hence, kinase activity of TBK1 is important to target the clearance of aggresomes with the help of the endolysosomal system.
Role of chaperones and nuclear import receptors (NIRs)
Aberrantly phase transitioned stress granules carrying misfolded proteins are disassembled by chaperones upon subsidence of stress. Heat shock proteins (HSPs) act as chaperones in both prokaryotes and eukaryotes, thereby promoting proteostasis and protecting from cellular stress (Lindquist and Craig, 1988). Although few HSPs are constitutively expressed while the remaining are upregulated in stressful conditions (Miller and Fort, 2018). Studies in yeast and Drosophila showed that the chaperones of the HSP70, HSP104 families help disaggregate (Cherkasov et al., 2013; Kroschwald et al., 2015) the misfolded proteins from SGs. In mammalian cells, chaperone HSP72 is critical for SG dissolution (Mazroui et al., 2007). Other studies have shown that HSP70, BAG3 (nucleotide-exchange factor), HSPB8 (small heat shock protein) promote SG quality control and favor the disassembly of stress granules (Minoia et al., 2014; Crippa et al., 2016; Ganassi et al., 2016). Further, ALS-associated variants of SOD-1, susceptible to misfolding, aggregate into SGs thereby interfering with the dynamic liquid like property of SG and turning them into an aberrant condensate (Das et al., 2023). After the removal of stress, chaperones like HSP27, HSP70 can avert aberrant SG formation and promote SG disassembly in SOD1 positive SGs (Mateju et al., 2017).
Physiological stress granules, if not resolved, can transform into aberrantly phase transitioned condensates. The intrinsically disordered region of FUS is prone to form aggregates and accumulates in SGs and promotes their maturation (Li et al., 2013; Patel et al., 2015). HSPB8 can decelerate FUS induced SG maturation (Boczek et al., 2021). HSP family protein variants of HSP104 are known to suppress toxicity and aggregation caused by TDP-43, FUS and α-synuclein in yeast models (Jackrel et al., 2014; Jackrel and Shorter, 2014). Moreover, synthetic HSP104 variants are capable of dissolving cytoplasmic ALS-linked FUS aggregates in mammalian cells (Yasuda et al., 2017). Mammalian HSPB1, an ATP dependent chaperone in association with other chaperones of HSP-70 family, BAG2, BAG3 and HSP40, help in disassembly of de-mixed droplets of TDP-43 in ALS upon removal of stress thereby preventing the formation of TDP-43 gel like condensates (Lu et al., 2022).
Not only chaperones, but kinases are also involved in stress granule disassembly. For example, kinases like Casein kinase 2 (CK2) cause SG disassembly by phosphorylating the SG nucleating protein G3BP1 (Reineke et al., 2017). DYRK3 (dual specificity tyrosine phosphorylation regulated kinase 3), another kinase can regulate liquid–liquid unmixing in cells. This kinase is capable of dissolving SGs by promoting phosphorylation of several RNA binding proteins (Wippich et al., 2013), as well as other MLOs, such as splicing speckles, centrosomes (Rai et al., 2018).
Various nuclear RNA-binding proteins (RBPs) like TDP-43, FUS, hnRNPA1, and hnRNPA2, upon mutation or stress can mislocalize to cytoplasm and form aggregates leading to several neurodegenerative disorders. Guo et al. (2018) demonstrated that nuclear import receptors (NIRs) can recognize nuclear localization signals (NLS) of wild type and disease linked RBPs with prion like domains, including TDP-43, FUS, and hnRNPA1/A2, and disaggregate them (Guo et al., 2018). Karyopherin-β2 acts as a molecular chaperone for RBPs with NLS containing proline and tyrosine residues (PY-NLS) like FUS, hnRNPA1and reverses their aberrant phase transitions (Fare et al., 2023) whereas Importin-α, in consort with Karyopherin-β1, reverses TDP-43 fibrillization (Guo et al., 2018).
To summarize, autophagosomes, endolysosomal systems, chaperones, NIRs and kinases are critical cellular defense mechanisms to combat aberrant phase separation. Further research is required to exploit these mechanisms to generate therapeutic treatments modalities for neurodegenerative diseases.
Molecular therapeutic approaches targeting aberrantly phase separated condensates and aggregation of intrinsically disordered proteins in neurodegenerative disease
As discussed above, in disease states and/or during aging, aberrant phase transitions of RNP condensates may lead to the accumulation of misfolded proteins and/or the formation of pathologic aggregates over time (Tauber et al., 2020). The increasing number of examples of aberrant condensate behavior associated with cellular and molecular dysfunction in neurodegenerative disease states highlight a need to develop new therapeutic approaches for modulating biomolecular condensates as well as preventing protein aggregation.
However, because disease-linked RBPs are enriched in IDRs and lack stable, well-defined binding pockets, they have largely been considered undruggable. Furthermore, the specific structural aberrations observed in RBPs under pathological conditions can vary depending on the proteins involved, the cytoplasmic milieu, and the disease context (Molliex et al., 2015; Nedelsky and Taylor, 2022). For example, the same protein may exhibit different structural forms, such as amyloid fibrils, liquid droplets with gel like properties, amorphous aggregates and membrane associated structures, depending on the stage of disease progression and/or the presence of additional factors (Hamad et al., 2021). Elucidating the detailed structures and mechanisms of aberrant phase transitions in RBPs is an active area of research to better understand the underlying molecular processes and to develop potential therapeutic strategies. Despite these challenges, several approaches to modify biomolecular condensate composition, rates of formation or clearance, and biophysical properties are under intense investigation, not only for ALS/FTD-linked RBPs but also targeting intrinsically disordered proteins in Parkinson’s disease (PD), Alzheimer’s disease (AD) and related dementias (Biesaga et al., 2021; Mitrea et al., 2022). In this section, we discuss recent advances in designing small molecules, peptides, and degrader compounds and other molecular approaches to potentially modulate aberrant RNP condensates and inhibit disease-associated protein aggregation (Table 2). Additionally, we explore the challenges and future prospects of utilizing these small molecules and peptides as therapeutic interventions for neurodegenerative diseases associated with RBP aggregation.

Table 2. Therapeutic approaches targeting protein aggregation.
Based on our current understanding of biomolecular condensate formation and regulation, an emerging strategy is to design or screen for therapeutics that modulate the biophysical properties, macromolecular composition, dynamics and/or free energy landscape of condensate assembly/disassembly (Mitrea et al., 2022). Given the difficulties with rational drug design targeting IDRs, a recent study employed a phenotypic high-content screen to identify small molecules that robustly modulate SG assembly and/or promote disassembly. Among the many hits were ~ 20 SG-modulating compounds that contained extended planar moieties with nucleic acid intercalating properties. One of the hits, mitoxantrone, was able to reduce persistent TDP-43 cytoplasmic puncta in iPSC-derived motor neurons, though the exact mechanism of action remains to be clarified (Fang et al., 2019). Additional small molecules have been tested for modulation of TDP-43 LLPS and/or inhibiting its aggregation. For example, AIM4 ([4,5-bis{(N-carboxy methyl imidazolium)methyl}acridine] dibromide), refers to a class of compounds known as imidazolium-tagged acridines. These compounds have demonstrated the ability to impede the formation of pre-formed TDP-43 aggregates in in vitro studies (Prasad et al., 2016). Furthermore, follow-up studies suggest that AIM4 can directly bind to TDP-43 (aa: 288–319) and inhibits LLPS and amyloid-like aggregation of mutant TDP-43 A315T (Girdhar et al., 2020). Another study identified bis-ANS, a small molecule that modulates TDP-43 phase separation in a biphasic manner in vitro; however, this compound requires further optimization since it was unable to de-condense SGs in cells (Babinchak et al., 2020).
Additional potential approaches include macromolecular crowding and post-translational/chemical modification, to prevent formation of aberrant condensates or protein aggregates. Molecular crowding refers to the phenomenon where high concentrations of macromolecules in a cellular environment can reduce the propensity of proteins to aggregate. The process of molecular crowding can enhance the protein stability, prevent protein misfolding, and inhibit protein aggregation by increasing the thermodynamic stability of proteins and reducing their conformational entropy (Schreck et al., 2020). Several crowding agent molecules have shown promising results in reducing aggregation propensity of Aβ peptides (Munishkina et al., 2004; Siddiqui and Naeem, 2023). Recently, post translational modifications such as lysine acetylation mimic molecules such as K_ac-mimic molecules have been observed to limit the size and growth of Aβ fibrils (Adhikari et al., 2020).
Modulation of protein aggregation with small molecules, peptides, and chaperones
Another potential strategy is to identify compounds that can modulate or prevent various steps of fibril formation, regardless of whether fibrillation occurs directly or via an aberrant condensate state (Vendruscolo and Fuxreiter, 2022). Numerous therapeutic approaches that aim to disrupt the aggregation process and mitigate the detrimental effects associated with aggregate formation have been investigated for the treatment of PD (Troncoso-Escudero et al., 2020). The recognition of α-synuclein accumulation as a hallmark of PD has accelerated the development of pharmacological strategies that specifically address misfolded α-synuclein to prevent its aggregation. NPT200-11, a small molecule is designed to specifically target crucial regions of the α-synuclein protein involved in aggregate formation. Through its unique molecular structure, NPT200-11 utilizes its influence on the aggregation process, potentially preventing the formation of toxic oligomers (Wrasidlo et al., 2016). In the transgenic mouse model of PD, oral administration of NPT200-11 demonstrated its capability to penetrate the blood–brain barrier and exert beneficial effects on neuropathological and behavioral outcomes by reducing α-synuclein aggregation (Price et al., 2018). However, additional validation is necessary to establish the safety and effectiveness of NPT200-11. Notably, UCB0599, which is the R-enantiomer of NPT200-11, has demonstrated a favorable safety and tolerability profile in PD patients (Smit et al., 2022). Similarly, several protein structure based small molecules have been discovered to inhibit the formation of Aβ fibrils in the context of AD. For example, the small molecule (10074-G5) stabilizes the soluble state of Aβ and inhibits nucleation steps in the aggregation process (Heller et al., 2020). Tricyclic pyrone molecules (CP2 and TP3) (Hong et al., 2009), beta sheet breaker peptide (Jani et al., 2021), D-enantiomeric peptide (Rösener et al., 2018) and others (Seidler et al., 2022; Murray et al., 2023), also have been highlighted as therapeutic options to prevent the formation of Aβ fibrils.
Likewise, chaperone proteins could be employed to bind unfolded or misfolded proteins and facilitate their proper folding and/or to stabilize native protein conformations, in turn preventing protein aggregation. Additionally, chaperones may bind to early-stage aggregates and inhibit their maturation into fibrils. One well-known chaperone protein is HSP70, which can recognize and bind to hydrophobic regions of proteins, preventing them from forming toxic aggregates in major neurodegenerative diseases such as AD and PD (Auluck et al., 2002; Gifondorwa et al., 2007; Lindstedt et al., 2019).
Degraders: clearance of protein aggregates
Targeted protein degradation (TPD) has emerged as a promising area of research for modulating proteins that are difficult to target with traditional small molecule inhibitors, particularly those implicated in neurodegenerative diseases. The hotspot regions of RBPs often have broad, shallow active sites or smooth surfaces with few binding sites for small molecules. Therefore, small molecules lose their efficiency as a potential therapeutic option. To address these challenges, Crews and Deshaies developed PROteolysis TArgeting Chimeras (PROTACs) in 2001. PROTACs are hetero bi-functional compounds that consist of a ligand that binds to the target protein and a ubiquitin E3 ligase recruiting group that brings the target protein and the ubiquitin E3 ligase closer together. This proximity facilitates effective degradation of the target protein by the proteasomal machinery (Lai and Crews, 2017). PROTACs have received significant interest in neurodegenerative diseases as a potential therapeutic strategy due to their ability to cross the blood–brain barrier and their ease of administration via various routes, including oral, intravenous, subcutaneous, and intrathecal routes. Arvinas Biopharmaceuticals, Pfizer, and Genentech have all investigated the application of PROTACs in neurodegenerative diseases, particularly in targeting pathological proteins such as tau and α-synuclein in AD and PD. Pioneering research in the use of PROTACs for AD demonstrated the activity of TH006 against the elimination of Tau by recruiting von Hippel–Lindau (VHL) E3 ligase (Ramsay and Di Giovanni, 2017). Since then, different groups have developed Tau-Keap1-CPP, QC-01–175, and C004019 PROTACs employing Keap1, CRBN, and VHL E3 ligases, respectively, for the targeted degradation of Tau in AD models (Cummings, 2021; Karlawish and Grill, 2021; Wang et al., 2021). Small molecule PROTACs have also been shown to induce positive cross-interaction of neuroprotective proteins with toxic proteins, such as transthyretin (a transport protein) with toxic β-amyloids, preventing their oligomerization and subsequent aggregation. Furthermore, small molecule PROTACs that target both Tau and/or α-Synuclein were recently described. This strategy employs Cereblon (CRBN) and VHL E3 ligases with different linkage lengths to design PROTAC compounds against the Tau protein (Kargbo, 2019, 2020). These developments demonstrate the potential of PROTAC technology for targeting “undruggable” targets in neurodegenerative diseases.
An alternative approach for targeting Tau involves the utilization of passively transferred antibodies that bind and degrade tau assemblies (Mukadam et al., 2023). Notably, Mukadam and colleagues showed that Tau-antibody complexes were internalized into the cytosol of neurons, leading to engagement of the cytosolic antibody receptor and E3 ligase TRIM21 and degradation via the ubiquitin-proteasome system. This TRIM21-dependent mechanism was remarkably effective in protecting against seeded aggregation of tau. These findings underscore the significance of the cytosolic compartment as a potential site for immunotherapeutic protection, which holds promise for the development of antibody-based therapies in the treatment of neurodegenerative diseases.
Concluding remarks
Concentrated efforts by many investigators have highlighted LLPS as an important mechanism for organization of cellular compartments, formation of RNP condensates enriched in RNA and RBPs, and higher order assemblies of condensates with membrane-bound organelles. While a large body of evidence suggests ALS/FTD-linked mutations and associated aberrant RNP condensates have detrimental functional consequences for mRNA stability, transport, and translation in neurons, it is not clear that pathologic aggregation of TDP-43 and other RBPs in ALS/FTD necessarily requires aberrant condensate formation. Rather, it is likely that aberrant LLPS as well as several “hits’” to cellular protein and organelle quality control mechanisms lead to multiple, non-mutually exclusive aggregation pathways. Future work and challenges lie in elucidating effective therapeutic targets shared across familial and sporadic forms of ALS/FTD.
Author contributions
PG provided the scope and template of the work, and reviewed, edited and wrote sections of the manuscript. AdN and SV wrote the manuscript and reviewed each section. AsN wrote the manuscript, and collected and prepared information for the tables. MS wrote the manuscript, collected and prepared information for the tables, and prepared the schematic figure. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Institute of Neurological Disorders and Stroke/NIH under Awards R01NS122907 (to PG).
Acknowledgments
We thank past and present members of the Gopal lab for thoughtful discussion and comments on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adeli, K. (2011). Translational control mechanisms in metabolic regulation: critical role of RNA binding proteins, microRNAs, and cytoplasmic RNA granules. Am. J. Physiol. Endocrinol. Metab. 301, E1051–E1064. doi: 10.1152/ajpendo.00399.2011
Adhikari, R., Yang, M., Saikia, N., Dutta, C., Alharbi, W. F. A., Shan, Z., et al. (2020). Acetylation of Aβ42 at lysine 16 disrupts amyloid formation. ACS Chem. Neurosci. 11, 1178–1191. doi: 10.1021/acschemneuro.0c00069
Ainger, K., Avossa, D., Morgan, F., Hill, S. J., Barry, C., Barbarese, E., et al. (1993). Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. J. Cell Biol. 123, 431–441. doi: 10.1083/jcb.123.2.431
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S. W., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Alberti, S., Gladfelter, A., and Mittag, T. (2019). Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cells 176, 419–434. doi: 10.1016/j.cell.2018.12.035
Alpatov, R., Lesch, B. J., Nakamoto-Kinoshita, M., Blanco, A., Chen, S., Stützer, A., et al. (2014). A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cells 157, 869–881. doi: 10.1016/j.cell.2014.03.040
Altman, T., Ionescu, A., Ibraheem, A., Priesmann, D., Gradus-Pery, T., Farberov, L., et al. (2021). Axonal TDP-43 condensates drive neuromuscular junction disruption through inhibition of local synthesis of nuclear encoded mitochondrial proteins. Nat. Commun. 12:6914. doi: 10.1038/s41467-021-27221-8
Anderson, P., and Kedersha, N. (2009). Stress granules. Curr. Biol. 19, R397–R398. doi: 10.1016/j.cub.2009.03.013
Antar, L. N., Afroz, R., Dictenberg, J. B., Carroll, R. C., and Bassell, G. J. (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci. 24, 2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004
Antar, L. N., Dictenberg, J. B., Plociniak, M., Afroz, R., and Bassell, G. J. (2005). Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 4, 350–359. doi: 10.1111/j.1601-183X.2005.00128.x
Antar, L. N., Li, C., Zhang, H., Carroll, R. C., and Bassell, G. J. (2006). Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 32, 37–48. doi: 10.1016/j.mcn.2006.02.001
Arseni, D., Hasegawa, M., Murzin, A. G., Kametani, F., Arai, M., Yoshida, M., et al. (2022). Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 601, 139–143. doi: 10.1038/s41586-021-04199-3
Aulas, A., and Vande Velde, C. (2015). Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front. Cell. Neurosci. 9:423. doi: 10.3389/fncel.2015.00423
Auluck, P. K., Chan, H. Y. E., Trojanowski, J. Q., Lee, V. M. Y., and Bonini, N. M. (2002). Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295, 865–868. doi: 10.1126/science.1067389
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. doi: 10.1152/physrev.00024.2003
Ayala, Y. M., De Conti, L., Avendaño-Vázquez, S. E., Dhir, A., Romano, M., D'Ambrogio, A., et al. (2011). TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288. doi: 10.1038/emboj.2010.310
Babinchak, W. M., Dumm, B. K., Venus, S., Boyko, S., Putnam, A. A., Jankowsky, E., et al. (2020). Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat. Commun. 11:5574. doi: 10.1038/s41467-020-19211-z
Baizabal-Carvallo, J. F., and Jankovic, J. (2016). Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 12, 175–185. doi: 10.1038/nrneurol.2016.14
Banani, S. F., Lee, H. O., Hyman, A. A., and Rosen, M. K. (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298. doi: 10.1038/nrm.2017.7
Banjade, S., and Rosen, M. K. (2014). Phase transitions of multivalent proteins can promote clustering of membrane receptors. elife 3:e04123. doi: 10.7554/eLife.04123
Barmada, S. J., Ju, S., Arjun, A., Batarse, A., Archbold, H. C., Peisach, D., et al. (2015). Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc. Natl. Acad. Sci. U. S. A. 112, 7821–7826. doi: 10.1073/pnas.1509744112
Beijer, D., Kim, H. J., Guo, L., O'Donovan, K., Mademan, I., Deconinck, T., et al. (2021). Characterization of HNRNPA1 mutations defines diversity in pathogenic mechanisms and clinical presentation. JCI Insight 6:e148363. doi: 10.1172/jci.insight.148363
Bentmann, E., Neumann, M., Tahirovic, S., Rodde, R., Dormann, D., and Haass, C. (2012). Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094. doi: 10.1074/jbc.M111.328757
Biesaga, M., Frigole-Vivas, M., and Salvatella, X. (2021). Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr. Opin. Chem. Biol. 62, 90–100. doi: 10.1016/j.cbpa.2021.02.009
Birsa, N., Ule, A. M., Garone, M. G., Tsang, B., Mattedi, F., Chong, P. A., et al. (2021). FUS-ALS mutants alter FMRP phase separation equilibrium and impair protein translation. Sci. Adv. 7:eabf8660. doi: 10.1126/sciadv.abf8660
Boczek, E. E., Fürsch, J., Niedermeier, M. L., Jawerth, L., Jahnel, M., Ruer-Gruß, M., et al. (2021). HspB8 prevents aberrant phase transitions of FUS by chaperoning its folded RNA-binding domain. elife 10:e69377. doi: 10.7554/eLife.69377
Boehringer, A., Garcia-Mansfield, K., Singh, G., Bakkar, N., Pirrotte, P., and Bowser, R. (2017). ALS associated mutations in matrin 3 alter protein-protein interactions and impede mRNA nuclear export. Sci. Rep. 7:14529. doi: 10.1038/s41598-017-14924-6
Bramham, C. R., and Wells, D. G. (2007). Dendritic mRNA: transport, translation and function. Nat. Rev. Neurosci. 8, 776–789. doi: 10.1038/nrn2150
Brangwynne, C. P., Tompa, P., and Pappu, R. V. (2015). Polymer physics of intracellular phase transitions. Nat. Phys. 11, 899–904. doi: 10.1038/Nphys3532
Bras, J., Guerreiro, R., and Hardy, J. (2015). SnapShot: genetics of Parkinson's disease. Cells 160, 570–570 e571. doi: 10.1016/j.cell.2015.01.019
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O'Donnell, W. T., Tenenbaum, S. A., et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cells 107, 477–487. doi: 10.1016/s0092-8674(01)00568-2
Buchan, J. R., Kolaitis, R.-M., Taylor, J. P., and Parker, R. (2013). Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cells 153, 1461–1474. doi: 10.1016/j.cell.2013.05.037
Buchan, J. R., and Parker, R. (2009). Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941. doi: 10.1016/j.molcel.2009.11.020
Buratti, E. (2015). Functional significance of TDP-43 mutations in disease. Adv. Genet. 91, 1–53. doi: 10.1016/bs.adgen.2015.07.001
Buxbaum, A. R., Haimovich, G., and Singer, R. H. (2015). In the right place at the right time: visualizing and understanding mRNA localization. Nat. Rev. Mol. Cell Biol. 16, 95–109. doi: 10.1038/nrm3918
Buxbaum, A. R., Wu, B., and Singer, R. H. (2014). Single beta-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science 343, 419–422. doi: 10.1126/science.1242939
Caglayan, S., Takagi-Niidome, S., Liao, F., Carlo, A. S., Schmidt, V., Burgert, T., et al. (2014). Lysosomal sorting of amyloid-beta by the SORLA receptor is impaired by a familial Alzheimer's disease mutation. Sci. Transl. Med. 6:223ra220. doi: 10.1126/scitranslmed.3007747
Che, M. X., Jiang, L. L., Li, H. Y., Jiang, Y. J., and Hu, H. Y. (2015). TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett. 589, 1920–1928. doi: 10.1016/j.febslet.2015.06.009
Chen, L., Jin, J., Davis, J., Zhou, Y., Wang, Y., Liu, J., et al. (2007). Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem. Biophys. Res. Commun. 356, 548–553. doi: 10.1016/j.bbrc.2007.02.163
Chen, Z. X., Wallis, K., Fell, S. M., Sobrado, V. R., Hemmer, M. C., Ramsköld, D., et al. (2014). RNA helicase A is a downstream mediator of KIF1Bβ tumor-suppressor function in neuroblastoma. Cancer Discov. 4, 434–451. doi: 10.1158/2159-8290.CD-13-0362
Chen, R. H. C., Wislet-Gendebien, S., Samuel, F., Visanji, N. P., Zhang, G., Marsilio, D., et al. (2013). α-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 288, 7438–7449. doi: 10.1074/jbc.M112.439497
Cherkasov, V., Hofmann, S., Druffel-Augustin, S., Mogk, A., Tyedmers, J., Stoecklin, G., et al. (2013). Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 23, 2452–2462. doi: 10.1016/j.cub.2013.09.058
Chitiprolu, M., Jagow, C., Tremblay, V., Bondy-Chorney, E., Paris, G., Savard, A., et al. (2018). A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 9:2794. doi: 10.1038/s41467-018-05273-7
Choi, Y. H., Lim, J.-K., Jeong, M.-W., and Kim, K.-T. (2012). HnRNP A1 phosphorylated by VRK1 stimulates telomerase and its binding to telomeric DNA sequence. Nucleic Acids Res. 40, 8499–8518. doi: 10.1093/nar/gks634
Chornenkyy, Y., Fardo, D. W., and Nelson, P. T. (2019). Tau and TDP-43 proteinopathies: kindred pathologic cascades and genetic pleiotropy. Lab. Investig. 99, 993–1007. doi: 10.1038/s41374-019-0196-y
Chu, J. F., Majumder, P., Chatterjee, B., Huang, S. L., and Shen, C. J. (2019). TDP-43 regulates coupled dendritic mRNA transport-translation processes in co-operation with FMRP and Staufen1. Cell Rep. 29, 3118–3133 e3116. doi: 10.1016/j.celrep.2019.10.061
Ciechanover, A., and Schwartz, A. L. (1998). The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. U. S. A. 95, 2727–2730. doi: 10.1073/pnas.95.6.2727
Cohen, T. J., Hwang, A. W., Restrepo, C. R., Yuan, C. X., Trojanowski, J. Q., and Lee, V. M. (2015). An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 6:5845. doi: 10.1038/ncomms6845
Conicella, A. E., Zerze, G. H., Mittal, J., and Fawzi, N. L. (2016). ALS mutations disrupt phase separation mediated by alpha-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549. doi: 10.1016/j.str.2016.07.007
Coyne, A. N., Siddegowda, B. B., Estes, P. S., Johannesmeyer, J., Kovalik, T., Daniel, S. G., et al. (2014). Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 34, 15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014
Coyne, A. N., Yamada, S. B., Siddegowda, B. B., Estes, P. S., Zaepfel, B. L., Johannesmeyer, J. S., et al. (2015). Fragile X protein mitigates TDP-43 toxicity by remodeling RNA granules and restoring translation. Hum. Mol. Genet. 24, 6886–6898. doi: 10.1093/hmg/ddv389
Crippa, V., Cicardi, M. E., Ramesh, N., Seguin, S. J., Ganassi, M., Bigi, I., et al. (2016). The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum. Mol. Genet. 25, 3908–3924. doi: 10.1093/hmg/ddw232
Cummings, J. (2021). Why aducanumab is important. Nat. Med. 27:1498. doi: 10.1038/s41591-021-01478-4
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y. S., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cells 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Das, B., Roychowdhury, S., Mohanty, P., Rizuan, A., Chakraborty, J., Mittal, J., et al. (2023). A Zn-dependent structural transition of SOD1 modulates its ability to undergo phase separation. EMBO J. 42:e111185. doi: 10.15252/embj.2022111185
De Santis, R., Alfano, V., de Turris, V., Colantoni, A., Santini, L., Garone, M. G., et al. (2019). Mutant FUS and ELAVL4 (HuD) aberrant crosstalk in amyotrophic lateral sclerosis. Cell Rep. 27, 3818–3831 e3815. doi: 10.1016/j.celrep.2019.05.085
Decker, C. J., Teixeira, D., and Parker, R. (2007). Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol. 179, 437–449. doi: 10.1083/jcb.200704147
Deng, H.-X., Chen, W., Hong, S.-T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Deng, Q., Holler, C. J., Taylor, G., Hudson, K. F., Watkins, W., Gearing, M., et al. (2014). FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 34, 7802–7813. doi: 10.1523/JNEUROSCI.0172-14.2014
Deter, R. L., Baudhuin, P., and De Duve, C. (1967). Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 35, C11–C16. doi: 10.1083/jcb.35.2.c11
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939. doi: 10.1016/j.devcel.2008.04.003
Donev, R., Newall, A., Thome, J., and Sheer, D. (2007). A role for SC35 and hnRNPA1 in the determination of amyloid precursor protein isoforms. Mol. Psychiatry 12, 681–690. doi: 10.1038/sj.mp.4001971
Donlin-Asp, P. G., Rossoll, W., and Bassell, G. J. (2017). Spatially and temporally regulating translation via mRNA-binding proteins in cellular and neuronal function. FEBS Lett. 591, 1508–1525. doi: 10.1002/1873-3468.12621
D'Souza, M. N., Gowda, N. K. C., Tiwari, V., Babu, R. O., Anand, P., Dastidar, S. G., et al. (2018). FMRP interacts with C/D box snoRNA in the nucleus and regulates ribosomal RNA methylation. iScience 9, 399–411. doi: 10.1016/j.isci.2018.11.007
Duchaîne, T. F., Hemraj, I., Furic, L., Deitinghoff, A., Kiebler, M. A., and DesGroseillers, L. (2002). Staufen2 isoforms localize to the somatodendritic domain of neurons and interact with different organelles. J. Cell Sci. 115, 3285–3295. doi: 10.1242/jcs.115.16.3285
Ehrnhoefer, D. E., Bieschke, J., Boeddrich, A., Herbst, M., Masino, L., Lurz, R., et al. (2008). EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 15, 558–566. doi: 10.1038/nsmb.1437
Elbaum-Garfinkle, S., Kim, Y., Szczepaniak, K., Chen, C. C., Eckmann, C. R., Myong, S., et al. (2015). The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. U. S. A. 112, 7189–7194. doi: 10.1073/pnas.1504822112
Elvira, G., Wasiak, S., Blandford, V., Tong, X. K., Serrano, A., Fan, X., et al. (2006). Characterization of an RNA granule from developing brain. Mol. Cell. Proteomics 5, 635–651. doi: 10.1074/mcp.M500255-MCP200
Fallini, C., Bassell, G. J., and Rossoll, W. (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 21, 3703–3718. doi: 10.1093/hmg/dds205
Fang, M. Y., Markmiller, S., Vu, A. Q., Javaherian, A., Dowdle, W. E., Jolivet, P., et al. (2019). Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103, 802–819 e811. doi: 10.1016/j.neuron.2019.05.048
Fare, C. M., Rhine, K., Lam, A., Myong, S., and Shorter, J. (2023). A minimal construct of nuclear-import receptor Karyopherin-beta2 defines the regions critical for chaperone and disaggregation activity. J. Biol. Chem. 299:102806. doi: 10.1016/j.jbc.2022.102806
Fernandes, L., Cardim-Pires, T. R., Foguel, D., and Palhano, F. L. (2021). Green tea polyphenol epigallocatechin-Gallate in amyloid aggregation and neurodegenerative diseases. Front. Neurosci. 15:718188. doi: 10.3389/fnins.2021.718188
Fratta, P., Sivakumar, P., Humphrey, J., Lo, K., Ricketts, T., Oliveira, H., et al. (2018). Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 37:e98684. doi: 10.15252/embj.201798684
Fritzsching, K. J., Yang, Y., Pogue, E. M., Rayman, J. B., Kandel, E. R., and McDermott, A. E. (2020). Micellar TIA1 with folded RNA binding domains as a model for reversible stress granule formation. Proc. Natl. Acad. Sci. U. S. A. 117, 31832–31837. doi: 10.1073/pnas.2007423117
Fujii, R., Okabe, S., Urushido, T., Inoue, K., Yoshimura, A., Tachibana, T., et al. (2005). The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 15, 587–593. doi: 10.1016/j.cub.2005.01.058
Fujii, R., and Takumi, T. (2005). TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 118, 5755–5765. doi: 10.1242/jcs.02692
Ganassi, M., Mateju, D., Bigi, I., Mediani, L., Poser, I., Lee, H. O., et al. (2016). A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell 63, 796–810. doi: 10.1016/j.molcel.2016.07.021
Gao, N., Chu, T.-T., Li, Q.-Q., Lim, Y.-J., Qiu, T., Ma, M.-R., et al. (2017). Hydrophobic tagging-mediated degradation of Alzheimer's disease related Tau. RSC Adv. 7, 40362–40366. doi: 10.1039/C7RA05347A
Gao, Y. L., Wang, N., Sun, F. R., Cao, X. P., Zhang, W., and Yu, J. T. (2018). Tau in neurodegenerative disease. Ann. Transl. Med. 6:175. doi: 10.21037/atm.2018.04.23
Gasset-Rosa, F., Lu, S., Yu, H., Chen, C., Melamed, Z., Guo, L., et al. (2019). Cytoplasmic TDP-43 De-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102, 339–357 e337. doi: 10.1016/j.neuron.2019.02.038
Ge, Y., Masoura, A., Yang, J., and Aprile, F. A. (2022). A chemical mutagenesis approach to insert post-translational modifications in aggregation-prone proteins. ACS Chem. Neurosci. 13, 1714–1718. doi: 10.1021/acschemneuro.2c00077
Gifondorwa, D. J., Robinson, M. B., Hayes, C. D., Taylor, A. R., Prevette, D. M., Oppenheim, R. W., et al. (2007). Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 27, 13173–13180. doi: 10.1523/JNEUROSCI.4057-07.2007
Gilks, N., Kedersha, N., Ayodele, M., Shen, L., Stoecklin, G., Dember, L. M., et al. (2004). Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398. doi: 10.1091/mbc.e04-08-0715
Girdhar, A., Bharathi, V., Tiwari, V. R., Abhishek, S., Deeksha, W., Mahawar, U. S., et al. (2020). Computational insights into mechanism of AIM4-mediated inhibition of aggregation of TDP-43 protein implicated in ALS and evidence for in vitro inhibition of liquid-liquid phase separation (LLPS) of TDP-432C-A315T by AIM4. Int. J. Biol. Macromol. 147, 117–130. doi: 10.1016/j.ijbiomac.2020.01.032
Gopal, P. P., Nirschl, J. J., Klinman, E., and Holzbaur, E. L. (2017). Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. U. S. A. 114, E2466–E2475. doi: 10.1073/pnas.1614462114
Gorbatyuk, O. S., Li, S., Nguyen, F. N., Manfredsson, F. P., Kondrikova, G., Sullivan, L. F., et al. (2010). α-Synuclein expression in rat substantia nigra suppresses phospholipase D2 toxicity and nigral neurodegeneration. Mol. Ther. 18, 1758–1768. doi: 10.1038/mt.2010.137
Graber, T. E., Hebert-Seropian, S., Khoutorsky, A., David, A., Yewdell, J. W., Lacaille, J. C., et al. (2013). Reactivation of stalled polyribosomes in synaptic plasticity. Proc. Natl. Acad. Sci. U. S. A. 110, 16205–16210. doi: 10.1073/pnas.1307747110
Grese, Z. R., Bastos, A. C., Mamede, L. D., French, R. L., Miller, T. M., and Ayala, Y. M. (2021). Specific RNA interactions promote TDP-43 multivalent phase separation and maintain liquid properties. EMBO Rep. 22:e53632. doi: 10.15252/embr.202153632
Guerreiro, R., Bras, J., and Hardy, J. (2015). SnapShot: genetics of ALS and FTD. Cells 160, 798–798 e791. doi: 10.1016/j.cell.2015.01.052
Guo, L., Kim, H. J., Wang, H., Monaghan, J., Freyermuth, F., Sung, J. C., et al. (2018). Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cells 173, 677–692.e620. doi: 10.1016/j.cell.2018.03.002
Hagerman, R. J., Leehey, M., Heinrichs, W., Tassone, F., Wilson, R., Hills, J., et al. (2001). Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57, 127–130. doi: 10.1212/wnl.57.1.127
Hallegger, M., Chakrabarti, A. M., Lee, F. C. Y., Lee, B. L., Amalietti, A. G., Odeh, H. M., et al. (2021). TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cells 184, 4680–4696 e4622. doi: 10.1016/j.cell.2021.07.018
Hamad, N., Yoneda, R., So, M., Kurokawa, R., Nagata, T., and Katahira, M. (2021). Non-coding RNA suppresses FUS aggregation caused by mechanistic shear stress on pipetting in a sequence-dependent manner. Sci. Rep. 11:9523. doi: 10.1038/s41598-021-89075-w
Han, T. W., Kato, M., Xie, S., Wu, L. C., Mirzaei, H., Pei, J., et al. (2012). Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cells 149, 768–779. doi: 10.1016/j.cell.2012.04.016
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers. 3:17071. doi: 10.1038/nrdp.2017.71
Heller, G. T., Aprile, F. A., Michaels, T. C. T., Limbocker, R., Perni, M., Ruggeri, F. S., et al. (2020). Small-molecule sequestration of amyloid-β as a drug discovery strategy for Alzheimer's disease. Sci. Adv. 6:eabb5924. doi: 10.1126/sciadv.abb5924
Hofweber, M., Hutten, S., Bourgeois, B., Spreitzer, E., Niedner-Boblenz, A., Schifferer, M., et al. (2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cells 173, 706–719.e713. doi: 10.1016/j.cell.2018.03.004
Holt, C. E., and Schuman, E. M. (2013). The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80, 648–657. doi: 10.1016/j.neuron.2013.10.036
Honda, H., Hamasaki, H., Wakamiya, T., Koyama, S., Suzuki, S. O., Fujii, N., et al. (2015). Loss of hnRNPA1 in ALS spinal cord motor neurons with TDP-43-positive inclusions. Neuropathology 35, 37–43. doi: 10.1111/neup.12153
Hong, H.-S., Rana, S., Barrigan, L., Shi, A., Zhang, Y., Zhou, F., et al. (2009). Inhibition of Alzheimer's amyloid toxicity with a tricyclic pyrone molecule in vitro and in vivo. J. Neurochem. 108, 1097–1108. doi: 10.1111/j.1471-4159.2008.05866.x
Hu, Y., Chen, Z., Fu, Y., He, Q., Jiang, L., Zheng, J., et al. (2015). The amino-terminal structure of human fragile X mental retardation protein obtained using precipitant-immobilized imprinted polymers. Nat. Commun. 6:6634. doi: 10.1038/ncomms7634
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U. S. A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Hyman, A. A., Weber, C. A., and Jülicher, F. (2014). Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 30, 39–58. doi: 10.1146/annurev-cellbio-100913-013325
Igaz, L. M., Kwong, L. K., Chen-Plotkin, A., Winton, M. J., Unger, T. L., Xu, Y., et al. (2009). Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 Proteinopathies. J. Biol. Chem. 284, 8516–8524. doi: 10.1074/jbc.M809462200
Ishigaki, S., Masuda, A., Fujioka, Y., Iguchi, Y., Katsuno, M., Shibata, A., et al. (2012). Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2:529. doi: 10.1038/srep00529
Jackrel, M. E., DeSantis, M. E., Martinez, B. A., Castellano, L. M., Stewart, R. M., Caldwell, K. A., et al. (2014). Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cells 156:170. doi: 10.1016/j.cell.2013.11.047
Jackrel, M. E., and Shorter, J. (2014). Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis. Model. Mech. 7, 1175–1184. doi: 10.1242/dmm.016113
Jain, S., Wheeler, J. R., Walters, R. W., Agrawal, A., Barsic, A., and Parker, R. (2016). ATPase-modulated stress granules contain a diverse proteome and substructure. Cells 164, 487–498. doi: 10.1016/j.cell.2015.12.038
Jani, V., Sonavane, U., and Joshi, R. (2021). Destabilization potential of beta sheet breaker peptides on Abeta fibril structure: an insight from molecular dynamics simulation study. RSC Adv. 11, 23557–23573. doi: 10.1039/d1ra03609b
Jiang, L., Ash, P. E. A., Maziuk, B. F., Ballance, H. I., Boudeau, S., Abdullatif, A. A., et al. (2019). TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 137, 259–277. doi: 10.1007/s00401-018-1937-5
Jiang, L. L., Guan, W. L., Wang, J. Y., Zhang, S. X., and Hu, H. Y. (2022). RNA-assisted sequestration of RNA-binding proteins by cytoplasmic inclusions of the C-terminal 35-kDa fragment of TDP-43. J. Cell Sci. 135:jcs259380. doi: 10.1242/jcs.259380
Jinwal, U. K., Trotter, J. H., Abisambra, J. F., Koren, J. 3rd, Lawson, L. Y., Vestal, G. D., et al. (2011). The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J. Biol. Chem. 286, 16976–16983. doi: 10.1074/jbc.M110.182493
Ju, S., Tardiff, D. F., Han, H., Divya, K., Zhong, Q., Maquat, L. E., et al. (2011). A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 9:e1001052. doi: 10.1371/journal.pbio.1001052
Kamelgarn, M., Chen, J., Kuang, L., Jin, H., Kasarskis, E. J., and Zhu, H. (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. U. S. A. 115, E11904–E11913. doi: 10.1073/pnas.1810413115
Kanai, Y., Dohmae, N., and Hirokawa, N. (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. doi: 10.1016/j.neuron.2004.07.022
Kanda, Y., Satoh, R., Takasaki, T., Tomimoto, N., Tsuchiya, K., Tsai, C. A., et al. (2021). Sequestration of the PKC ortholog Pck2 in stress granules as a feedback mechanism of MAPK signaling in fission yeast. J. Cell Sci. 134:jcs250191. doi: 10.1242/jcs.250191
Kargbo, R. B. (2019). Treatment of Alzheimer's by PROTAC-tau protein degradation. ACS Med. Chem. Lett. 10, 699–700. doi: 10.1021/acsmedchemlett.9b00083
Kargbo, R. B. (2020). PROTAC compounds targeting α-Synuclein protein for treating Neurogenerative disorders: Alzheimer's and Parkinson's diseases. ACS Med. Chem. Lett. 11, 1086–1087. doi: 10.1021/acsmedchemlett.0c00192
Karlawish, J., and Grill, J. D. (2021). The approval of Aduhelm risks eroding public trust in Alzheimer research and the FDA. Nat. Rev. Neurol. 17, 523–524. doi: 10.1038/s41582-021-00540-6
Kato, M., Han, T. W., Xie, S., Shi, K., Du, X., Wu, L. C., et al. (2012). Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cells 149, 753–767. doi: 10.1016/j.cell.2012.04.017
Kawahara, K., Hashimoto, M., Bar-On, P., Ho, G. J., Crews, L., Mizuno, H., et al. (2008). Alpha-Synuclein aggregates interfere with Parkin solubility and distribution: role in the pathogenesis of Parkinson disease. J. Biol. Chem. 283, 6979–6987. doi: 10.1074/jbc.M710418200
Kedersha, N., and Anderson, P. (2002). Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 30, 963–969. doi: 10.1042/bst0300963
Kedersha, N., Ivanov, P., and Anderson, P. (2013). Stress granules and cell signaling: more than just a passing phase? Trends Biochem. Sci. 38, 494–506. doi: 10.1016/j.tibs.2013.07.004
Khalfallah, Y., Kuta, R., Grasmuck, C., Prat, A., Durham, H. D., and Vande Velde, C. (2018). TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 8:7551. doi: 10.1038/s41598-018-25767-0
Kiebler, M. A., and Bassell, G. J. (2006). Neuronal RNA granules: movers and makers. Neuron 51, 685–690. doi: 10.1016/j.neuron.2006.08.021
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R., and Gitler, A. D. (2020). ALS genetics: gains, losses, and implications for future therapies. Neuron 108, 822–842. doi: 10.1016/j.neuron.2020.08.022
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Kim, N. Y., Lee, S., Yu, J., Kim, N., Won, S. S., Park, H., et al. (2020). Optogenetic control of mRNA localization and translation in live cells. Nat. Cell Biol. 22, 341–352. doi: 10.1038/s41556-020-0468-1
Kim, T. H., Tsang, B., Vernon, R. M., Sonenberg, N., Kay, L. E., and Forman-Kay, J. D. (2019). Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 365, 825–829. doi: 10.1126/science.aax4240
Kitchell, B. B., Rauckman, E. J., and Rosen, G. M. (1978). The effect of temperature on mixed function amine oxidase intrinsic fluorescence and oxidative activity. Mol. Pharmacol. 14, 1092–1098.
Krichevsky, A. M., and Kosik, K. S. (2001). Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron 32, 683–696. doi: 10.1016/s0896-6273(01)00508-6
Krone, M. G., Baumketner, A., Bernstein, S. L., Wyttenbach, T., Lazo, N. D., Teplow, D. B., et al. (2008). Effects of familial Alzheimer’s disease mutations on the folding nucleation of the amyloid beta-protein. J. Mol. Biol. 381, 221–228. doi: 10.1016/j.jmb.2008.05.069
Kroschwald, S., Maharana, S., Mateju, D., Malinovska, L., Nuske, E., Poser, I., et al. (2015). Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. elife 4:e06807. doi: 10.7554/eLife.06807
Kute, P. M., Ramakrishna, S., Neelagandan, N., Chattarji, S., and Muddashetty, R. S. (2019). NMDAR mediated translation at the synapse is regulated by MOV10 and FMRP. Mol. Brain 12:65. doi: 10.1186/s13041-019-0473-0
Kuusisto, E., Parkkinen, L., and Alafuzoff, I. (2003). Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 62, 1241–1253. doi: 10.1093/jnen/62.12.1241
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lagier-Tourenne, C., Polymenidou, M., and Cleveland, D. W. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64. doi: 10.1093/hmg/ddq137
Lai, A. C., and Crews, C. M. (2017). Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114. doi: 10.1038/nrd.2016.211
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Lee, E. B., Lee, V. M., and Trojanowski, J. Q. (2011). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50. doi: 10.1038/nrn3121
Lee, F. J., Liu, F., Pristupa, Z. B., and Niznik, H. B. (2001). Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 15, 916–926. doi: 10.1096/fj.00-0334com
Lewis-Smith, D. J., Duff, J., Pyle, A., Griffin, H., Polvikoski, T., Birchall, D., et al. (2016). Novel HSPB1 mutation causes both motor neuronopathy and distal myopathy. Neurol. Genet. 2:e110. doi: 10.1212/NXG.0000000000000110
Li, P., Banjade, S., Cheng, H. C., Kim, S., Chen, B., Guo, L., et al. (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340. doi: 10.1038/nature10879
Li, H. R., Chiang, W. C., Chou, P. C., Wang, W. J., and Huang, J. R. (2018). TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 293, 6090–6098. doi: 10.1074/jbc.AC117.001037
Li, Y. R., King, O. D., Shorter, J., and Gitler, A. D. (2013). Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372. doi: 10.1083/jcb.201302044
Liao, Y. C., Fernandopulle, M. S., Wang, G., Choi, H., Hao, L., Drerup, C. M., et al. (2019). RNA granules hitchhike on lysosomes for long-distance transport, using Annexin A11 as a molecular tether. Cells 179, 147–164 e120. doi: 10.1016/j.cell.2019.08.050
Lin, Y., Currie, S. L., and Rosen, M. K. (2017). Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 292, 19110–19120. doi: 10.1074/jbc.M117.800466
Lin, Y., Protter, D. S., Rosen, M. K., and Parker, R. (2015). Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219. doi: 10.1016/j.molcel.2015.08.018
Lindquist, S., and Craig, E. A. (1988). The heat-shock proteins. Annu. Rev. Genet. 22, 631–677. doi: 10.1146/annurev.ge.22.120188.003215
Lindstedt, P. R., Aprile, F. A., Matos, M. J., Perni, M., Bertoldo, J. B., Bernardim, B., et al. (2019). Enhancement of the anti-aggregation activity of a molecular chaperone using a rationally designed post-translational modification. ACS Cent Sci. 5, 1417–1424. doi: 10.1021/acscentsci.9b00467
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderweyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. doi: 10.1371/journal.pone.0013250
Liu-Yesucevitz, L., Lin, A. Y., Ebata, A., Boon, J. Y., Reid, W., Xu, Y. F., et al. (2014). ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J. Neurosci. 34, 4167–4174. doi: 10.1523/JNEUROSCI.2350-13.2014
Lomen-Hoerth, C., Anderson, T., and Miller, B. (2002). The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. doi: 10.1212/wnl.59.7.1077
Lopez-Erauskin, J., Tadokoro, T., Baughn, M. W., Myers, B., McAlonis-Downes, M., Chillon-Marinas, C., et al. (2018). ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100, 816–830 e817. doi: 10.1016/j.neuron.2018.09.044
Loughlin, F. E., West, D. L., Gunzburg, M. J., Waris, S., Crawford, S. A., Wilce, M. C. J., et al. (2021). Tandem RNA binding sites induce self-association of the stress granule marker protein TIA-1. Nucleic Acids Res. 49, 2403–2417. doi: 10.1093/nar/gkab080
Lu, S., Hu, J., Arogundade, O. A., Goginashvili, A., Vazquez-Sanchez, S., Diedrich, J. K., et al. (2022). Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat. Cell Biol. 24, 1378–1393. doi: 10.1038/s41556-022-00988-8
Ma, W., and Mayr, C. (2018). A Membraneless organelle associated with the endoplasmic reticulum enables 3'UTR-mediated protein-protein interactions. Cells 175, 1492–1506 e1419. doi: 10.1016/j.cell.2018.10.007
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and Alter stress granule dynamics. Neuron 95, 808–816 e809. doi: 10.1016/j.neuron.2017.07.025
Malik, A. M., and Barmada, S. J. (2021). Matrin 3 in neuromuscular disease: physiology and pathophysiology. JCI. Insight 6:e143948. doi: 10.1172/jci.insight.143948
Markmiller, S., Soltanieh, S., Server, K. L., Mak, R., Jin, W., Fang, M. Y., et al. (2018). Context-dependent and disease-specific diversity in protein interactions within stress granules. Cells 172, 590–604.e513. doi: 10.1016/j.cell.2017.12.032
Martin, S., Zekri, L., Metz, A., Maurice, T., Chebli, K., Vignes, M., et al. (2013). Deficiency of G3BP1, the stress granules assembly factor, results in abnormal synaptic plasticity and calcium homeostasis in neurons. J. Neurochem. 125, 175–184. doi: 10.1111/jnc.12189
Mateju, D., Franzmann, T. M., Patel, A., Kopach, A., Boczek, E. E., Maharana, S., et al. (2017). An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 36, 1669–1687. doi: 10.15252/embj.201695957
Maziuk, B. F., Apicco, D. J., Cruz, A. L., Jiang, L., Ash, P. E. A., da Rocha, E. L., et al. (2018). RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 6:71. doi: 10.1186/s40478-018-0574-5
Mazroui, R., Di Marco, S., Kaufman, R. J., and Gallouzi, I.-E. (2007). Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 18, 2603–2618. doi: 10.1091/mbc.e06-12-1079
McDonald, N. A., Fetter, R. D., and Shen, K. (2020). Assembly of synaptic active zones requires phase separation of scaffold molecules. Nature 588, 454–458. doi: 10.1038/s41586-020-2942-0
McGurk, L., Gomes, E., Guo, L., Mojsilovic-Petrovic, J., Tran, V., Kalb, R. G., et al. (2018). Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol. Cell 71:703-+. doi: 10.1016/j.molcel.2018.07.002
McLean, P. J., Kawamata, H., Ribich, S., and Hyman, B. T. (2000). Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson's disease-linked mutations. J. Biol. Chem. 275, 8812–8816. doi: 10.1074/jbc.275.12.8812
Miller, D. J., and Fort, P. E. (2018). Heat shock proteins regulatory role in neurodevelopment. Front. Neurosci. 12:821. doi: 10.3389/fnins.2018.00821
Milovanovic, D., Wu, Y., Bian, X., and De Camilli, P. (2018). A liquid phase of synapsin and lipid vesicles. Science 361, 604–607. doi: 10.1126/science.aat5671
Minoia, M., Boncoraglio, A., Vinet, J., Morelli, F. F., Brunsting, J. F., Poletti, A., et al. (2014). BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: implications for a proteasome-to-autophagy switch. Autophagy 10, 1603–1621. doi: 10.4161/auto.29409
Mitrea, D. M., Mittasch, M., Gomes, B. F., Klein, I. A., and Murcko, M. A. (2022). Modulating biomolecular condensates: a novel approach to drug discovery. Nat. Rev. Drug Discov. 21, 841–862. doi: 10.1038/s41573-022-00505-4
Mittag, T., and Parker, R. (2018). Multiple modes of protein-protein interactions promote RNP granule assembly. J. Mol. Biol. 430, 4636–4649. doi: 10.1016/j.jmb.2018.08.005
Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., et al. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cells 163, 123–133. doi: 10.1016/j.cell.2015.09.015
Monahan, Z., Shewmaker, F., and Pandey, U. B. (2016). Stress granules at the intersection of autophagy and ALS. Brain Res. 1649, 189–200. doi: 10.1016/j.brainres.2016.05.022
Muddashetty, R. S., Kelić, S., Gross, C., Xu, M., and Bassell, G. J. (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 27, 5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007
Mukadam, A. S., Miller, L. V. C., Smith, A. E., Vaysburd, M., Sakya, S. A., Sanford, S., et al. (2023). Cytosolic antibody receptor TRIM21 is required for effective tau immunotherapy in mouse models. Science 379, 1336–1341. doi: 10.1126/science.abn1366
Munishkina, L. A., Cooper, E. M., Uversky, V. N., and Fink, A. L. (2004). The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 17, 456–464. doi: 10.1002/jmr.699
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Murray, K. A., Hu, C. J., Pan, H., Lu, J., Abskharon, R., Bowler, J. T., et al. (2023). Small molecules disaggregate alpha-synuclein and prevent seeding from patient brain-derived fibrils. Proc Natl Acad Sci U S A. 120:e2217835120. doi: 10.1073/pnas.2217835120
Myrick, L. K., Hashimoto, H., Cheng, X., and Warren, S. T. (2015). Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum. Mol. Genet. 24, 1733–1740. doi: 10.1093/hmg/ddu586
Nedelsky, N. B., and Taylor, J. P. (2022). Pathological phase transitions in ALS-FTD impair dynamic RNA-protein granules. RNA 28, 97–113. doi: 10.1261/rna.079001.121
Nelson, D. L., Orr, H. T., and Warren, S. T. (2013). The unstable repeats--three evolving faces of neurological disease. Neuron 77, 825–843. doi: 10.1016/j.neuron.2013.02.022
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Nonhoff, U., Ralser, M., Welzel, F., Piccini, I., Balzereit, D., Yaspo, M. L., et al. (2007). Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol. Biol. Cell 18, 1385–1396. doi: 10.1091/mbc.E06-12-1120
Nott, T. J., Craggs, T. D., and Baldwin, A. J. (2016). Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat. Chem. 8, 569–575. doi: 10.1038/nchem.2519
Nott, T. J., Petsalaki, E., Farber, P., Jervis, D., Fussner, E., Plochowietz, A., et al. (2015). Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 57, 936–947. doi: 10.1016/j.molcel.2015.01.013
Ostrerova, N., Petrucelli, L., Farrer, M., Mehta, N., Choi, P., Hardy, J., et al. (1999). Alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J. Neurosci. 19, 5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999
Parker, R., and Sheth, U. (2007). P bodies and the control of mRNA translation and degradation. Mol. Cell 25, 635–646. doi: 10.1016/j.molcel.2007.02.011
Pasciuto, E., and Bagni, C. (2014). SnapShot: FMRP interacting proteins. Cells 159, 218–218.e211. doi: 10.1016/j.cell.2014.08.036
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cells 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Paterno, G., Bell, B. M., Gorion, K. M., Prokop, S., and Giasson, B. I. (2022). Reassessment of neuronal tau distribution in adult human brain and implications for tau pathobiology. Acta Neuropathol. Commun. 10:94. doi: 10.1186/s40478-022-01394-9
Prasad, R., Kawaguchi, S., and Ng, D. T. W. (2010). A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell 21, 2117–2127. doi: 10.1091/mbc.e10-02-0111
Prasad, A., Raju, G., Sivalingam, V., Girdhar, A., Verma, M., Vats, A., et al. (2016). An acridine derivative, [4,5-bis{(N-carboxy methyl imidazolium)methyl}acridine] dibromide, shows anti-TDP-43 aggregation effect in ALS disease models. Sci. Rep. 6:39490. doi: 10.1038/srep39490
Prashad, S., and Gopal, P. P. (2021). RNA-binding proteins in neurological development and disease. RNA Biol. 18, 972–987. doi: 10.1080/15476286.2020.1809186
Price, D. L., Koike, M. A., Khan, A., Wrasidlo, W., Rockenstein, E., Masliah, E., et al. (2018). The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson's disease. Sci. Rep. 8:16165. doi: 10.1038/s41598-018-34490-9
Rai, A. K., Chen, J.-X., Selbach, M., and Pelkmans, L. (2018). Kinase-controlled phase transition of membraneless organelles in mitosis. Nature 559, 211–216. doi: 10.1038/s41586-018-0279-8
Ramaswami, M., Taylor, J. P., and Parker, R. (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cells 154, 727–736. doi: 10.1016/j.cell.2013.07.038
Ramesh, N., Daley, E. L., Gleixner, A. M., Mann, J. R., Kour, S., Mawrie, D., et al. (2020). RNA dependent suppression of C9orf72 ALS/FTD associated neurodegeneration by Matrin-3. Acta Neuropathol. Commun. 8:177. doi: 10.1186/s40478-020-01060-y
Ramsay, R. R., and Di Giovanni, G. (2017). Structure-based drug design for diagnosis and treatment of neurological diseases. Front. Pharmacol. 8:13. doi: 10.3389/fphar.2017.00013
Ray, S., Singh, N., Kumar, R., Patel, K., Pandey, S., Datta, D., et al. (2020). Alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 12, 705–716. doi: 10.1038/s41557-020-0465-9
Reineke, L. C., Tsai, W.-C., Jain, A., Kaelber, J. T., Jung, S. Y., and Lloyd, R. E. (2017). Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol. 37:e00596-16. doi: 10.1128/MCB.00596-16
Rhine, K., Makurath, M. A., Liu, J., Skanchy, S., Lopez, C., Catalan, K. F., et al. (2020). ALS/FTLD-linked mutations in FUS Glycine residues cause accelerated gelation and reduced interactions with wild-type FUS. Mol. Cell 80:1139. doi: 10.1016/j.molcel.2020.11.031
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T., Zou, F., et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177. doi: 10.1038/ng1943
Rösener, N. S., Gremer, L., Reinartz, E., König, A., Brener, O., Heise, H., et al. (2018). A d-enantiomeric peptide interferes with heteroassociation of amyloid-β oligomers and prion protein. J. Biol. Chem. 293, 15748–15764. doi: 10.1074/jbc.RA118.003116
Salton, M., Elkon, R., Borodina, T., Davydov, A., Yaspo, M.-L., Halperin, E., et al. (2011). Matrin 3 binds and stabilizes mRNA. PLoS One 6:e23882. doi: 10.1371/journal.pone.0023882
Sanders, D. W., Kedersha, N., Lee, D. S. W., Strom, A. R., Drake, V., Riback, J. A., et al. (2020). Competing protein-RNA interaction networks control multiphase intracellular organization. Cells 181, 306–324 e328. doi: 10.1016/j.cell.2020.03.050
Schmidt, H. B., Barreau, A., and Rohatgi, R. (2019). Phase separation-deficient TDP43 remains functional in splicing. Nat. Commun. 10:4890. doi: 10.1038/s41467-019-12740-2
Schreck, J. S., Bridstrup, J., and Yuan, J.-M. (2020). Investigating the effects of molecular crowding on the kinetics of protein aggregation. J. Phys. Chem. B 124, 9829–9839. doi: 10.1021/acs.jpcb.0c07175
Scotter, E. L., Vance, C., Nishimura, A. L., Lee, Y.-B., Chen, H.-J., Urwin, H., et al. (2014). Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 127, 1263–1278. doi: 10.1242/jcs.140087
Seidler, P. M., Murray, K. A., Boyer, D. R., Ge, P., Sawaya, M. R., Hu, C. J., et al. (2022). Structure-based discovery of small molecules that disaggregate Alzheimer's disease tissue derived tau fibrils in vitro. Nat. Commun. 13:5451. doi: 10.1038/s41467-022-32951-4
Shaltiel-Karyo, R., Frenkel-Pinter, M., Egoz-Matia, N., Frydman-Marom, A., Shalev, D. E., Segal, D., et al. (2010). Inhibiting α-synuclein oligomerization by stable cell-penetrating β-synuclein fragments recovers phenotype of Parkinson's disease model flies. PLoS One 5:e13863. doi: 10.1371/journal.pone.0013863
Shao, W., Todd, T. W., Wu, Y., Jones, C. Y., Tong, J., Jansen-West, K., et al. (2022). Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration. Science 378, 94–99. doi: 10.1126/science.abq7860
Siddiqui, G. A., and Naeem, A. (2023). Connecting the dots: macromolecular crowding and protein aggregation. J. Fluoresc. 33, 1–11. doi: 10.1007/s10895-022-03082-2
Siomi, H., Siomi, M. C., Nussbaum, R. L., and Dreyfuss, G. (1993). The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cells 74, 291–298. doi: 10.1016/0092-8674(93)90420-u
Smit, J. W., Basile, P., Prato, M. K., Detalle, L., Mathy, F. X., Schmidt, A., et al. (2022). Phase 1/1b studies of UCB0599, an oral inhibitor of α-synuclein misfolding, including a randomized study in Parkinson's disease. Mov. Disord. 37, 2056–2045.
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Steward, O., and Schuman, E. M. (2003). Compartmentalized synthesis and degradation of proteins in neurons. Neuron 40, 347–359. doi: 10.1016/s0896-6273(03)00635-4
Strong, M. J., Volkening, K., Hammond, R., Yang, W., Strong, W., Leystra-Lantz, C., et al. (2007). TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 35, 320–327. doi: 10.1016/j.mcn.2007.03.007
Strulson, C. A., Molden, R. C., Keating, C. D., and Bevilacqua, P. C. (2012). RNA catalysis through compartmentalization. Nat. Chem. 4, 941–946. doi: 10.1038/nchem.1466
Sutton, M. A., and Schuman, E. M. (2006). Dendritic protein synthesis, synaptic plasticity, and memory. Cells 127, 49–58. doi: 10.1016/j.cell.2006.09.014
Tatomer, D. C., Terzo, E., Curry, K. P., Salzler, H., Sabath, I., Zapotoczny, G., et al. (2016). Concentrating pre-mRNA processing factors in the histone locus body facilitates efficient histone mRNA biogenesis. J. Cell Biol. 213, 557–570. doi: 10.1083/jcb.201504043
Tauber, D., Tauber, G., and Parker, R. (2020). Mechanisms and regulation of RNA condensation in RNP granule formation. Trends Biochem. Sci. 45, 764–778. doi: 10.1016/j.tibs.2020.05.002
Trnka, F., Hoffmann, C., Wang, H., Sansevrino, R., Rankovic, B., Rost, B. R., et al. (2021). Aberrant phase separation of FUS leads to lysosome sequestering and acidification. Front. Cell Dev. Biol. 9:716919. doi: 10.3389/fcell.2021.716919
Troncoso-Escudero, P., Sepulveda, D., Pérez-Arancibia, R., Parra, A. V., Arcos, J., Grunenwald, F., et al. (2020). On the right track to treat movement disorders: promising therapeutic approaches for Parkinson's and Huntington's disease. Front. Aging Neurosci. 12:571185. doi: 10.3389/fnagi.2020.571185
Tsang, B., Arsenault, J., Vernon, R. M., Lin, H., Sonenberg, N., Wang, L. Y., et al. (2019). Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl. Acad. Sci. U. S. A. 116, 4218–4227. doi: 10.1073/pnas.1814385116
van Eersel, J., Ke, Y. D., Gladbach, A., Bi, M., Götz, J., Kril, J. J., et al. (2011). Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS One 6:e22850. doi: 10.1371/journal.pone.0022850
Van Mossevelde, S., Engelborghs, S., van der Zee, J., and Van Broeckhoven, C. (2018). Genotype-phenotype links in frontotemporal lobar degeneration. Nat. Rev. Neurol. 14, 363–378. doi: 10.1038/s41582-018-0009-8
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vanderweyde, T., Apicco, D. J., Youmans-Kidder, K., Ash, P. E. A., Cook, C., Lummertz da Rocha, E., et al. (2016). Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 15, 1455–1466. doi: 10.1016/j.celrep.2016.04.045
Venda, L. L., Cragg, S. J., Buchman, V. L., and Wade-Martins, R. (2010). α-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci. 33, 559–568. doi: 10.1016/j.tins.2010.09.004
Vendruscolo, M., and Fuxreiter, M. (2022). Protein condensation diseases: therapeutic opportunities. Nat. Commun. 13:5550. doi: 10.1038/s41467-022-32940-7
Verhoef, L. G. G. C., Lindsten, K., Masucci, M. G., and Dantuma, N. P. (2002). Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 11, 2689–2700. doi: 10.1093/hmg/11.22.2689
Vishal, S. S., Wijegunawardana, D., Salaikumaran, M. R., and Gopal, P. P. (2022). Sequence determinants of TDP-43 ribonucleoprotein condensate formation and axonal transport in neurons. Front. Cell Dev. Biol. 10:876893. doi: 10.3389/fcell.2022.876893
Wakabayashi, K., Engelender, S., Tanaka, Y., Yoshimoto, M., Mori, F., Tsuji, S., et al. (2002). Immunocytochemical localization of synphilin-1, an alpha-synuclein-associated protein, in neurodegenerative disorders. Acta Neuropathol. 103, 209–214. doi: 10.1007/s004010100451
Wang, J., Choi, J. M., Holehouse, A. S., Lee, H. O., Zhang, X. J., Jahnel, M., et al. (2018). A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cells 174:688-+. doi: 10.1016/j.cell.2018.06.006
Wang, W., Zhou, Q., Jiang, T., Li, S., Ye, J., Zheng, J., et al. (2021). A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics 11, 5279–5295. doi: 10.7150/thno.55680
Wegmann, S., Eftekharzadeh, B., Tepper, K., Zoltowska, K. M., Bennett, R. E., Dujardin, S., et al. (2018). Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 37:e98049. doi: 10.15252/embj.201798049
Wen, X., Tan, W., Westergard, T., Krishnamurthy, K., Markandaiah, S. S., Shi, Y., et al. (2014). Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. doi: 10.1016/j.neuron.2014.12.010
Willis, D., Li, K. W., Zheng, J.-Q., Chang, J. H., Smit, A. B., Kelly, T., et al. (2005). Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J. Neurosci. 25, 778–791. doi: 10.1523/JNEUROSCI.4235-04.2005
Wippich, F., Bodenmiller, B., Trajkovska, M. G., Wanka, S., Aebersold, R., and Pelkmans, L. (2013). Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cells 152, 791–805. doi: 10.1016/j.cell.2013.01.033
Wolozin, B., and Ivanov, P. (2019). Stress granules and neurodegeneration. Nat. Rev. Neurosci. 20, 649–666. doi: 10.1038/s41583-019-0222-5
Wootz, H., Fitzsimons-Kantamneni, E., Larhammar, M., Rotterman, T. M., Enjin, A., Patra, K., et al. (2013). Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J. Comp. Neurol. 521, 1449–1469. doi: 10.1002/cne.23266
Wrasidlo, W., Tsigelny, I. F., Price, D. L., Dutta, G., Rockenstein, E., Schwarz, T. C., et al. (2016). A de novo compound targeting α-synuclein improves deficits in models of Parkinson's disease. Brain 139, 3217–3236. doi: 10.1093/brain/aww238
Wu, X., Cai, Q., Shen, Z., Chen, X., Zeng, M., Du, S., et al. (2019). RIM and RIM-BP form presynaptic active-zone-like condensates via phase separation. Mol. Cell 73, 971–984 e975. doi: 10.1016/j.molcel.2018.12.007
Xue, Y. C., Ng, C. S., Xiang, P., Liu, H., Zhang, K., Mohamud, Y., et al. (2020). Dysregulation of RNA-binding proteins in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 13:78. doi: 10.3389/fnmol.2020.00078
Yang, P., Mathieu, C., Kolaitis, R. M., Zhang, P., Messing, J., Yurtsever, U., et al. (2020). G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cells 181, 325–345 e328. doi: 10.1016/j.cell.2020.03.046
Yasuda, K., Clatterbuck-Soper, S. F., Jackrel, M. E., Shorter, J., and Mili, S. (2017). FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol. 216, 1015–1034. doi: 10.1083/jcb.201608022
Yu, S., Li, X., Liu, G., Han, J., Zhang, C., Li, Y., et al. (2007). Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience 145, 539–555. doi: 10.1016/j.neuroscience.2006.12.028
Yu, H., Lu, S., Gasior, K., Singh, D., Vazquez-Sanchez, S., Tapia, O., et al. (2021). HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 371:eabb4309. doi: 10.1126/science.abb4309
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, H., Elbaum-Garfinkle, S., Langdon, E. M., Taylor, N., Occhipinti, P., Bridges, A. A., et al. (2015). RNA controls PolyQ protein phase transitions. Mol. Cell 60, 220–230. doi: 10.1016/j.molcel.2015.09.017
Zheng, Q., Li, J., and Wang, X. (2009). Interplay between the ubiquitin-proteasome system and autophagy in proteinopathies. Int. J. Physiol. Pathophysiol. Pharmacol. 1, 127–142.
Zhou, C. (2022). The molecular and functional interaction between membrane-bound organelles and membrane-less condensates. Front. Cell Dev. Biol. 10:896305. doi: 10.3389/fcell.2022.896305
Keywords: neurodegenerative disease, RNA-binding proteins, aggregation, phase separation, biomolecular condensates
Citation: Naskar A, Nayak A, Salaikumaran MR, Vishal SS and Gopal PP (2023) Phase separation and pathologic transitions of RNP condensates in neurons: implications for amyotrophic lateral sclerosis, frontotemporal dementia and other neurodegenerative disorders. Front. Mol. Neurosci. 16:1242925. doi: 10.3389/fnmol.2023.1242925
Edited by:
Anca Flavia Savulescu, University of Cape Town, South AfricaReviewed by:
Julie Qiaojin Lin, University of Cambridge, United KingdomShu Yang, Macquarie University, Australia
Copyright © 2023 Naskar, Nayak, Salaikumaran, Vishal and Gopal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pallavi P. Gopal, cGFsbGF2aS5nb3BhbEB5YWxlLmVkdQ==
†These authors have contributed equally to this work and share first authorship