 Anna Maria Caroleo1
Anna Maria Caroleo1 Silvia Rotulo2
Silvia Rotulo2 Emanuele Agolini3
Emanuele Agolini3 Marina Macchiaiolo4
Marina Macchiaiolo4 Luigi Boccuto5
Luigi Boccuto5 Manila Antonelli6
Manila Antonelli6 Giovanna Stefania Colafati7
Giovanna Stefania Colafati7 Antonella Cacchione1
Antonella Cacchione1 Giacomina Megaro1
Giacomina Megaro1 Andrea Carai8
Andrea Carai8 Maria Antonietta De Ioris1
Maria Antonietta De Ioris1 Mariachiara Lodi1
Mariachiara Lodi1 Assunta Tornesello9
Assunta Tornesello9 Valeria Simone9Filippo Torroni10Giuseppe Cinalli11
Valeria Simone9Filippo Torroni10Giuseppe Cinalli11 Angela Mastronuzzi1*
Angela Mastronuzzi1*- 1Department of Onco-Hematology, Cell Therapy, Gene Therapy and Hemopoietic Transplant, Bambino Gesù Children’s Hospital (IRCCS), Rome, Italy
- 2Department of Pediatrics, Sapienza University of Rome, Rome, Italy
- 3Laboratory of Medical Genetics, Translational Cytogenomics Research Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 4Rare Diseases and Medical Genetics Unit, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
- 5School of Nursing, College of Behavioral, Social and Health Sciences Healthcare Genetics Interdisciplinary Doctoral Program, Clemson University, Clemson, SC, United States
- 6Faculty of Medicine and Dentistry, Department of Radiological, Oncological, and Pathological Anatomy Sciences, Sapienza University of Rome, Rome, Italy
- 7Neuroradiology Unit, Department of Imaging, Bambino Gesù Children’s Hospital (IRCCS), Rome, Italy
- 8Neurosurgery Unit, Department of Neurosciences, Bambino Gesù Children’s Hospital (IRCCS), Rome, Italy
- 9Pediatric Oncology Unit, Ospedale Vito Fazzi, Lecce, Italy
- 10Digestive Endoscopy and Surgery Unit, Bambino Gesù Children Hospital, IRCCS, Rome, Italy
- 11Pediatric Neurosurgery Unit, Department of Neuroscience, Santobono-Pausilipon Children’s Hospital, Naples, Italy
Phosphatase and tensin homolog (PTEN) hamartoma tumor syndrome (PHTS) is a cancer predisposition syndrome characterized by an increased risk of developing benign and malignant tumors, caused by germline pathogenic variants of the PTEN tumour suppressor gene. PTEN gene variants often present in childhood with macrocephaly, developmental delay, and/or autism spectrum disorder while tumors and intestinal polyps are commonly detected in adults. PHTS is rarely associated with childhood brain tumors with only two reported cases of medulloblastoma (MB). We report the exceptional case of an infant carrying a germline and somatic pathogenic variant of PTEN and a germline and somatic pathogenic variant of CHEK2 who developed a MB SHH in addition to intestinal polyposis.
1. Introduction
Phosphatase and tensin homolog (PTEN) hamartoma tumor syndrome (PHTS) is a rare neurocutaneous syndrome caused by germline pathogenic variants of the PTEN tumor suppressor gene (Hendricks et al., 2020; Isik et al., 2020; Kim et al., 2020) that cause an increased risk of developing benign and malignant tumors of the thyroid, breast, endometrium, skin, and brain. In addition to cancer susceptibility, PHTS features include macrocephaly, autism spectrum disorder, atypical neurodevelopment, benign thyroid lesions, and dermatologic findings (trichilemmomas, papillomas). PHTS may be considered a non-classical brain tumor polyposis syndrome, as central nervous system (CNS) manifestations are a rare component of the patient’s clinical burden (Kim et al., 2020). It is a rare disease with an estimated prevalence of 1/200.000, but it is probably underestimated because most patients are not recognized as such (Nelen et al., 1999; Ngeow and Eng, 2015; Karczewski et al., 2020). Approximately 50% of PHTS cases are inherited in an autosomal dominant manner, with the remainder of cases having a de novo mutation; in approximately 80% of case mutations of the PTEN gene affects the germline (Kim et al., 2020). All types of pathogenic variants (loss-of-function, deletions, missense, and promoter abnormalities) have been reported with no clear genotype–phenotype correlation (Smith et al., 2019). PTEN gene mutations show age-related penetrance (Lachlan et al., 2007): in childhood, they are often associated with macrocephaly, developmental delay (DD), and/or autism spectrum disorder, less commonly with thyroid lesions, while the development of tumors and intestinal polyps are rare, being more frequently detected in adult individuals (Heald et al., 2010; Hansen-Kiss et al., 2017; Ciaccio et al., 2019; Macken et al., 2019).
Medulloblastoma (MB) is a heterogeneous tumor that represents about 10% of CNS malignancies in children between 0 and 14 years of age (Millard and De Braganca, 2016). There are four MB subgroups (Sonic Hedgehog or SHH, WNT, group 3, and group 4), which are associated with specific transcriptional, epigenetic, and clinical characteristics (Vladoiu et al., 2019). However, the molecular details of each subgroup are not fully understood to date (14). Recently, it has been shown that the SHH subgroup is most frequently (approximately 20–40%) associated with germline mutations (BRCA2, PALB2, PTCH1, SUFU, and TP53; Waszak et al., 2018; Garcia-Lopez et al., 2021). Currently, MB cases are rarely described in individuals with Cowden syndrome (Waszak et al., 2018; Tolonen et al., 2020), a condition included in the PHTS spectrum. Instead, cases associated with other CNS tumors such as dysplastic gangliocytoma, meningioma, pineal tumor, oligodendroglioma, and glioblastoma have been reported in patients with this condition (Kim et al., 2020).
Susceptibility to develop intestinal polyps is one of the most distinctive features of PHTS, involving up to 95% of PHTS patients throughout life (Heald et al., 2010). Bowel polyps may be found from the stomach to the colon, and histology may include hamartomatous polyps (most common), ganglioneuromas, adenomas, and inflammatory polyps (Heald et al., 2010). This clinical manifestation is similar to Juvenile Polyposis Syndrome (JPS): hamartomatous polyps are indistinguishable (Schreibman et al., 2005) but tend to occur in adulthood (Lachlan et al., 2007; Heald et al., 2010). While the increased risk for the development of breast and thyroid cancers is well documented, the development of hamartomatous polyps does not lead to an increased risk of colorectal cancer. Heald et al. documented that colorectal cancer occurred in 7.1% of cases of their series (Heald et al., 2010).
Here we report the first pediatric case of PHTS with both a germline and somatic variant in PTEN and in CHEK2, who presented a significantly early onset of MB SHH (15 months), in addition to a remarkably early picture of hamartomatous intestinal polyposis.
2. Materials and methods
The patient and his legal guardians conferred informed consent for the study. A centralized review of histological characterization was performed. Molecular genetics studies were performed on genomic DNA extracted from peripheral blood using a next-generation sequencing (NGS) panel including medulloblastoma and cancer predisposition genes (APC, BRCA2, PALB2, PTCH1, PTCH2, SUFU, PTEN, TP53, CHEK2, and GPR161), according to the manufacturer’s protocol (Twist Bioscience, CA, USA). The presence of deletions and duplications in PTCH1 and SUFU genes on peripheral blood was also excluded by multiplex ligation-dependent probe amplification (MLPA) according to the manufacturer’s protocol (MRC Holland, Amsterdam, Netherlands).
3. Case report
The patient is a Caucasian male, referred to the Bambino Gesù Children’s Hospital at 15 months of age after the removal of a cerebellar mass, histologically compatible with MB at another center. He is the firstborn child to unrelated parents. His family history is free of neurocognitive developmental alterations, his father has intestinal polyposis, his paternal grandfather and uncle died of intestinal cancer; his paternal grandmother died of pancreatic cancer. He was born at 39 weeks of gestational age after an uneventful pregnancy. His birth weight (3,700 gr, 60 percentile, +0.79 SD) and height (50 cm, 20 percentile, −0.9 SD) were normal, while his head circumference was above normal (38 cm, 98 percentile, +3.0 SD). On arrival to the hospital, at the age of 15 months, he presented with macrocephaly (+3.0 SD) and psychomotor delay with major weaknesses related to language skills as detected by Griffiths Developmental Scales.
The surgical removal was fraught with difficulty, despite neuroimaging suggested a superficial, almost extra-axial lesion. The tumor was in fact very hard and bled profusely, to the point of reminding more of a hemangioblastoma, with a complex pattern of intratumoral vessels, than of an MB, which was moreover completely isodense at the pre-operative computed tomography (CT) scan. Complete resection was confirmed by postoperative magnetic resonance imaging (MRI) (Figure 1). Cerebrospinal fluid was free of neoplastic cells.
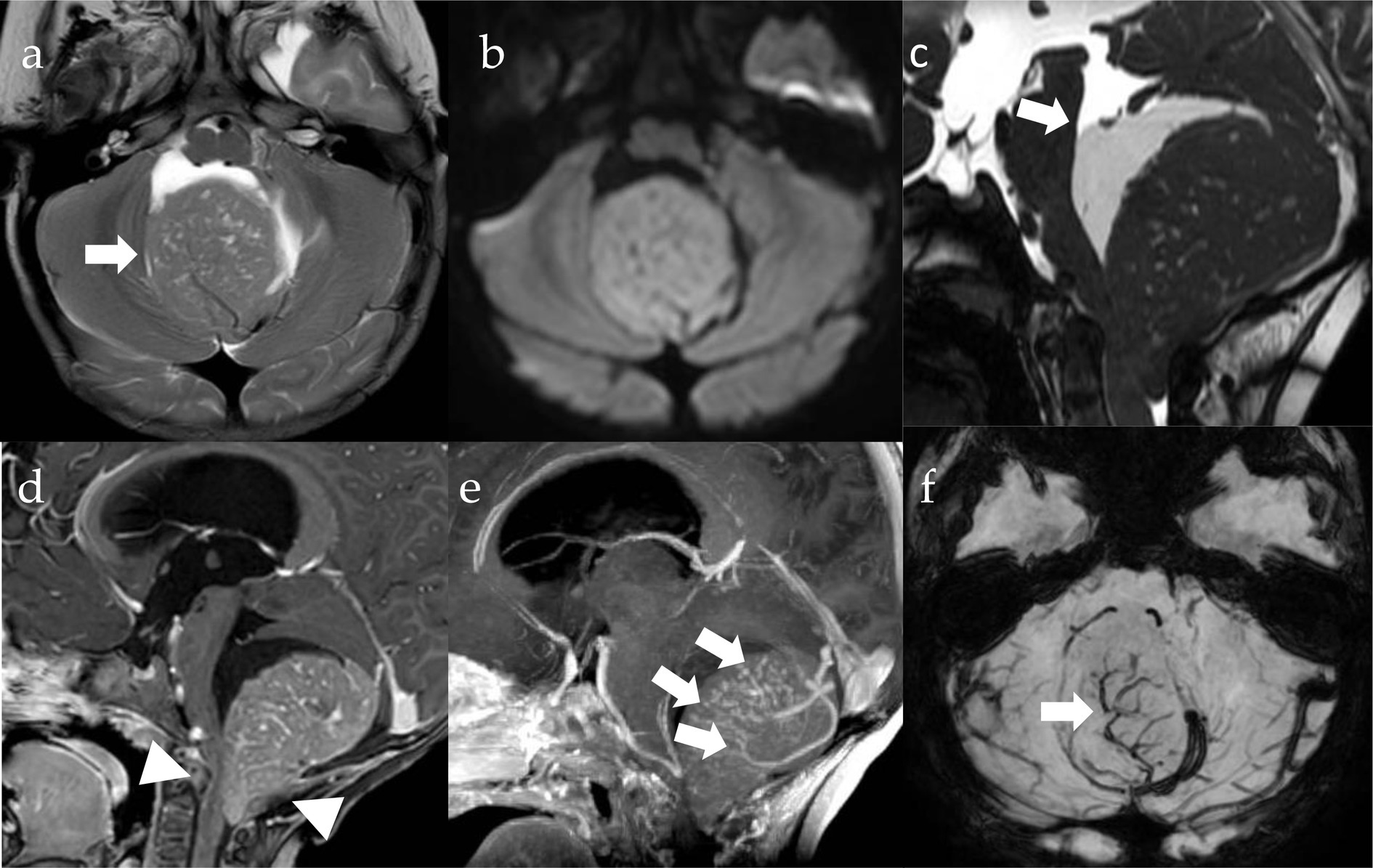
Figure 1. MRI imaging. Pre-operative axial T2w (A), DWI (diffusion weighted imaging, B) and SWI (susceptibility weighted imaging, F) images, sagittal CISS (three-dimensional constructive interference in steady state, C) Gd T1w (D) and MIP (maximum intensity projection, E) images. There is a well-circumscribed lesion in the posterior fossa (A, arrow), which is centered on the cisterna magna and pushing the vermis cranially, with growth into the fourth ventricle and extension through the foramen of Magendie onto the posterior aspect of the upper cervical cord (D, arrowheads). The tumor is isointense to the cerebellar cortex on T2 (A) and shows restriction of diffusivity (B) due to high cell density along with high nuclear-to-cytoplasmic ratio (B). There is significant contrast-enhancement (D) and intralesional vessels (E,F, arrows). Cystic components are appreciable and appear larger in the cranial portions of the lesion (arrow, D).
The histological examination revealed an embryonic neoplasm characterized by the presence of nodular and internodular areas. The nodular areas showed elongated aspects and consisted of neurocytic-type cells immersed in a fibrillar stroma. In the internodular areas, the cells were markedly hyperchromic with frequent mitosis. The immunohistochemical investigation showed a pattern coherent with MB SHH. The cells were positive for synaptophysin in the nodular areas; positivity was observed for GAB1, YAP1, and Filamin A. The proliferation index evaluated with Ki67 was high in the internodular areas (about 30%).
Molecular genetic characterization by NGS was performed on genomic DNA extracted from circulating leukocytes of the patient and unaffected parents to check for the presence of germline variants in high-risk cancer-predisposition genes. None of the genes typically associated with MB (APC, BRCA2, PALB2, PTCH1, SUFU, and TP53) (Waszak et al., 2018) were found to be mutated. Sequence analysis showed a germline heterozygous variant c.79 T > A in the PTEN gene (NM_000314.6) determining the missense change p.Tyr27Asn (rs746128825), previously reported in association with PHTS (Ngeow et al., 2014). This single-base substitution affects the last nucleotide position of the exon 1 and could be a splicing variant. However, further RNA studies are needed to test this hypothesis but are not feasible at present due to sample unavailability. This variant can be classified as pathogenic according to the ACMG criteria (PP3, PP2, PM2, PM1, PM5 and PS2).
Segregation analysis performed on the parents confirmed the de novo nature of the variant.
Genetic analysis also revealed the presence of a germline heterozygous variant c.507delT (p.Phe169LeufsTer2, rs587780183) in the CHEK2 gene (NM_007194.3). The analysis of this gene was recently included in the panel of genes studied in patients with MB at our center, as an association between CHEK2 and MB is reported in the literature, although not well established (Shah and Walter, 2018). The variant was inherited from the patient’s father and has previously been reported as likely pathogenetic, associated with a hereditary cancer-predisposing syndrome (Manoukian et al., 2011).
NGS was also performed on genomic DNA extracted from the tumor sample. Sequencing analysis revealed a somatic variant (allele burden 40%) in PTEN, c.388C > G (p.Arg130Gly) in addition to the germline change p.Tyr27Asn (allele burden 48%). This variant has been reported in the literature in individuals with clinical features characteristic of a PTEN-related disorder and identified as somatic variant in multiple malignancies (Fusco et al., 2020). The p.Arg130Gly variant affects PTEN function abolishing the phosphatase activities (Han et al., 2000). The variant in CHEK2 was also found in the tumor sample with an allele burden of 46%.
Post-surgical chemotherapy was performed according to the Italian Association of Pediatric Hematology and Oncology MB infant reccomandations. It consisted in three courses of induction chemotherapy (methotrexate 8 g/m2 plus vincristine 1.5 mg/m2 week 0; etoposide 2.4 g/m2 week 1; cyclophosphamide 4 g/m2 plus vincristine 1.5 mg/m2 week 4) and two courses of high-dose thiotepa (300 mg/m2 for 3 days, week 7 and 12) followed by autologous hematopoietic stem cell transplantation (Massimino et al., 2013). Four years after diagnosis, the child is currently in remission from MB.
At 3 years of age, the patient presented with blood and mucus in stools, inappetence, recurrent abdominal pain and weight loss. For these reasons a colonoscopy was performed, and colic polyposis was found (>50 sessile lesions, others pedunculated). Some skin lesions compatible with PHTS (Tan et al., 2011) were also found: punctate keratosis of the palm-plantar region, hyperkeratotic papular lesions on the back of the hands and feet (trichilemmomas), numerous papular lesions on the back of the feet, periungual and axillary (acrochordon), papillomatous lesions of the oral cavity, and a melanocytic lesions in the abdominal region (compound melanocytic nevus). The child underwent the removal of 30 polyps (diameter 2 cm), during two endoscopic sessions; all polyps were hamartomatous (Figure 2).

Figure 2. Endoscopic picture (A): colon macro-polyp. Capsular picture (B): middle ileum micro-polyp.
At 4 years of age, an additional brain lesion (a right frontal with dural implant amartoma) was diagnosed and removed. The following year (at 5 years of age) severe hypoglycaemia was found and the patient needed to positionate a sensor and initially started feeding with the nasogastric tube. Severe hypoglycemia, although not always present in PTHS, is described in the literature as part of the clinical picture [always linked to PTEN regulation of the PI3K-AKT/mTOR pathway (Maines et al., 2021)].
4. Discussion
We present the first known case of a child carrying a germline and somatic pathogenic variant of PTEN associated with a germline and somatic variant of CHEK2, with a phenotype characterized by macrocephaly, DD, skin lesions, and very early onset of MB SHH and intestinal polyps.
There are only a few other cases with this type of MB-associated mutation that have been described: two pediatric cases, both with MB SHH (Table 1), and two cases of young adults (19 and 23 years) with MB SHH (Gröbner et al., 2018). It should be noted that the latter two patients are part of the series of Gröbner et al. (2018), which also includes a 4 year-old MB G3 patient with a PTEN low allele frequencies, in whom genetic analysis was performed only at the somatic level. Several studies have shown that PTEN variants are associated in 5% of cases with the development of CNS tumors (Liaw et al., 1997; Lynch et al., 1997; Staal et al., 2002; Sturm et al., 2014; Yakubov et al., 2016; Gröbner et al., 2018; Waszak et al., 2018; Kim et al., 2020), in particular glioblastoma, meningioma, dysplastic gangliocytoma, pineal tumor, and oligodendroglioma. The data currently available in the literature, although scarce, would suggest a possible association with MB as well. In contrast, it is not surprising that all patients with PHTS-associated MB belonged to the SHH subgroup, as more than 40% of pediatric SHH MBs have damaging germline mutations (Garcia-Lopez et al., 2021). The peculiarity of our case lies in the early onset of the brain tumor [presented before the first peak incidence of 3–4 years according to Millard and De Braganca (2016)], and the very early onset of the gastrointestinal manifestations, which usually occur in adulthood. Even cases of juvenile intestinal polyps in patients younger than 12 years with PHTS are rarely reported (Table 2). In fact, to our knowledge, this is the sixth described case of intestinal polyps in PHTS before the age of 12 years, and our patient is the youngest case reported so far. Due to the rarity of pediatric age intestinal manifestations, considering that even the National Comprehensive Cancer Network (NCCN) guidelines recommend “starting at 35 years old, unless symptomatic or close relative with colon cancer under age 40 years” a follow up with colonoscopy was not initially set (Daly et al., 2020).

Table 1. Known pediatric cases of MB with germline variants of PTEN.

Table 2. Known cases of affected by PHTS with bowel polyps’ onset before age of 12 years.
Nearly 90% of patients with PHTS develop clinical manifestations before 20 years of age, although they may not be diagnosed until 30 years (Kim et al., 2020). There is an increased risk of developing breast or endometrial cancer for women, and thyroid cancer for both men and women. Colorectal cancer is also seen in 9–13% of cases, while polyps are found in 40–60% of cases (Heald et al., 2010). Other cancers were detected in the kidney and skin (Tan et al., 2011; Ngeow and Eng, 2015).
The PTEN protein acts as a potent suppressor of oncogenesis by inhibiting the PI3K-AKT/mTOR pathway and regulating cell proliferation and survival (Mester and Eng, 2013). Reinforcing the hypothesis that inhibition of this trail plays a crucial role in tumor pathogenesis. A study recently reported a reduction in hamartomas in patients with PHTS after rapamycin treatment, suggesting that patients with disorders in the PTEN hamartoma tumor syndrome spectrum might respond to therapies designed to inhibit the PI3-K/mTOR pathway (Marsh D. J. et al., 2008).
The SHH and PI3K pathways converge to promote the proliferation of granule cell progenitors in the outer granular layer of the cerebellum in vitro (Kenney et al., 2004). It has been observed in a mouse model that inactivation of the PTEN gene creates an abnormal perivascular proliferative niche in the cerebellum, persistent in adult animals, characterized by undifferentiated cells but without the tendency for malignancy, and in the absence of TP53 or PTCH1 codeletion (Zhu et al., 2017). Alterations in PTEN could therefore create a predisposing substrate for the development of MB, especially the SHH subgroup. A genomic analysis of medulloblastoma tumors showed that of 13 SHH subgroup patients, 2 had loss-of-function somatic mutations in PTEN (Robinson et al., 2012). Of 66 patients profiled from the other subgroups, none had loss of PTEN. Another study found a number of PTEN mutations in medulloblastoma tumors, one of which co-occurred with a homozygous PTCH mutation (Parsons et al., 2011). In addition, epigenetic inactivation of PTEN has been reported to occur at a high frequency in medulloblastoma samples (Hartmann et al., 2006). In our patient, sequencing analysis on tumor revealed the well-characterized loss of function somatic variant of PTEN p.Arg130Gly, that together with the germline missense change p.Tyr27Asn likely determines the complete loss of phosphatase activities of the protein, providing a strong evidence that the MB in our patient is associated with PHTS. On the contrary, we did not observe a loss of heterozygosity or the presence of a second deleterious somatic variant in CHEK2, suggesting this gene could have a marginal role in the tumorigenesis in our patient.
The increased susceptibility to develop hamartomatous polyps in the gastrointestinal tract is also related to uncontrolled cell growth in patients with PTEN mutation, especially subjects with heterozygous PTEN deletions developing intestinal epithelial dysplasia with subsequent invasion of the lamina propria, as described in adenoma-carcinoma progression (Marsh V. et al., 2008). The occurrence of bowel polyps has been described especially in patients with overlapping phenotypes between JPS and PTHS. BMPR1A, the gene associated with JPS, shares the same chromosomal region as PTEN (10q23.2): if large deletions encompass these genes the phenotypic expression can include features of both PHTS and JPS, most typically with juvenile polyposis of infancy (JPI), an aggressive subtype of JPS characterized by severe gastrointestinal symptoms, including diarrhea, intestinal bleeding, rectal prolapse, protein-losing enteropathy with a high risk of intussusception and consequently high infant mortality (Jelsig et al., 2014). The severity of this condition was hypothesized to be due to the loss of these two tumor suppressors, which function in two different but cooperative pathways (Delnatte et al., 2006; Salviati et al., 2006; Menko et al., 2008; Hiljadnikova Bajro et al., 2013). Our case did not present overlapping mutations between these genes, but rather a variant in the CHEK2 gene.
The frameshift variant in the CHEK2 gene, related to the TP53 pathway, has been previously described in an Italian family with hereditary breast/ovarian cancer (HBOC) and is considered to be likely pathogenetic for cancer predisposition syndromes. However, the role of this variant is not yet fully understood, and it might be speculated that it elicits its effect in a context of polygenic inheritance, contributing to cancer risk in association with other susceptibility alleles and increasing the oncological recurrence risk in the family (Manoukian et al., 2011; Teodorczyk et al., 2013).
5. Conclusion
Although the association is rare, the panel of genes to be tested in the presence of an MB SHH could be extended to PTEN. The role of CHEK2, instead, remains uncertain at this time. The discovery of a PTEN germline mutation, even if in childhood, should induce the clinician to promptly provide genetic counseling in order to assess and monitor the occurrence of other PHTS clinical features and set up careful surveillance.
Author contributions
AMC, AM, EA, and LB: conceptualization. AMC and SR: investigation and writing—original draft preparation. AMC, SR, LB, EA, MM, and AntC: data curation. AM, LB, EA, MM, FT, GSC, and AT: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
The authors thank Megan Eckley for assisting with the final English version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ciaccio, C., Saletti, V., D’Arrigo, S., Esposito, S., Alfei, E., Moroni, I., et al. (2019). Clinical spectrum of PTEN mutation in pediatric patients. A bicenter experience. Eur. J. Med. Genet. 62:103596. doi: 10.1016/j.ejmg.2018.12.001
Daly, M. B., Pilarski, R., Yurgelun, M. B., Berry, M. P., Buys, S. S., Dickson, P., et al. (2020). NCCN guidelines insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 1.2020. J. Natl. Compr. Canc. Netw. 18, 380–391. doi: 10.6004/jnccn.2020.0017
Delnatte, C., Sanlaville, D., Mougenot, J. F., Vermeesch, J. R., Houdayer, C., Blois, M. C., et al. (2006). Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am. J. Hum. Genet. 78, 1066–1074. doi: 10.1086/504301
Fusco, N., Sajjadi, E., Venetis, K., Gaudioso, G., Lopez, G., Corti, C., et al. (2020). PTEN alterations and their role in cancer management: are we making headway on precision medicine? Genes (Basel) 11:719. doi: 10.3390/genes11070719
Garcia-Lopez, J., Kumar, R., Smith, K. S., and Northcott, P. A. (2021). Deconstructing sonic hedgehog Medulloblastoma: molecular subtypes, drivers, and beyond. Trends Genet. 37, 235–250. doi: 10.1016/j.tig.2020.11.001
Gorensek, M., Matko, I., Skralovnik, A., Rode, M., Satler, J., and Jutersek, A. (1984). Disseminated hereditary gastrointestinal polyposis with orocutaneous hamartomatosis (Cowden’s disease). Endoscopy 16, 59–63. doi: 10.1055/s-2007-1018534
Gröbner, S. N., Worst, B. C., Weischenfeldt, J., Buchhalter, I., Kleinheinz, K., Rudneva, V. A., et al. (2018). The landscape of genomic alterations across childhood cancers. Nature 555, 321–327. doi: 10.1038/nature25480
Han, S. Y., Kato, H., Kato, S., Suzuki, T., Shibata, H., Ishii, S., et al. (2000). Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 60, 3147–3151.
Hansen-Kiss, E., Beinkampen, S., Adler, B., Frazier, T., Prior, T., Erdman, S., et al. (2017). A retrospective chart review of the features of PTEN hamartoma tumour syndrome in children. J. Med. Genet. 54, 471–478. doi: 10.1136/jmedgenet-2016-104484
Hartmann, W., Digon-Söntgerath, B., Koch, A., Waha, A., Endl, E., Dani, I., et al. (2006). Phosphatidylinositol 3′-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin. Cancer Res. 12, 3019–3027. doi: 10.1158/1078-0432.CCR-05-2187
Heald, B., Mester, J., Rybicki, L., Orloff, M. S., Burke, C. A., and Eng, C. (2010). Frequent gastrointestinal polyps and colorectal adenocarcinomas in prospective series of PTEN mutation carriers. Gastroenterology 139, 1927–1933. doi: 10.1053/j.gastro.2010.06.061
Hendricks, L. A. J., Hoogerbrugge, N., Schuurs-Hoeijmakers, J. H. M., and Vos, J. R. (2020). A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin Genet 99, 219–225. doi: 10.1111/cge.13875
Hiljadnikova Bajro, M., Sukarova-Angelovska, E., Adélaïde, J., Chaffanet, M., and Dimovski, A. J. (2013). A new case with 10q23 interstitial deletion encompassing both PTEN and BMPR1A narrows the genetic region deleted in juvenile polyposis syndrome. J. Appl. Genet. 54, 43–47. doi: 10.1007/s13353-012-0115-z
Isik, E., Simsir, O. S., Solmaz, A. E., Onay, H., Atik, T., Aykut, A., et al. (2020). Clinical and molecular aspects of PTEN mutations in 10 pediatric patients. Ann. Hum. Genet. 84, 324–330. doi: 10.1111/ahg.12380
Jelsig, A. M., Qvist, N., Brusgaard, K., Nielsen, C. B., Hansen, T. P., and Ousager, L. B. (2014). Hamartomatous polyposis syndromes: a review. Orphanet. J. Rare Dis. 9:101. doi: 10.1186/1750-1172-9-101
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kenney, A. M., Widlund, H. R., and Rowitch, D. H. (2004). Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 131, 217–228. doi: 10.1242/dev.00891
Kim, B., Tabori, U., and Hawkins, C. (2020). An update on the CNS manifestations of brain tumor polyposis syndromes. Acta. Neuropathol. 139, 703–715. doi: 10.1007/s00401-020-02124-y
Lachlan, K. L., Lucassen, A. M., Bunyan, D., and Temple, I. K. (2007). Cowden syndrome and Bannayan Riley Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance: results of a clinical study of PTEN mutation carriers. J. Med. Genet. 44, 579–585. doi: 10.1136/jmg.2007.049981
Liaw, D., Marsh, D. J., Li, J., Dahia, P. L., Wang, S. I., Zheng, Z., et al. (1997). Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 16, 64–67. doi: 10.1038/ng0597-64
Lynch, E. D., Ostermeyer, E. A., Lee, M. K., Arena, J. F., Ji, H., Dann, J., et al. (1997). Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am. J. Hum. Genet. 61, 1254–1260. doi: 10.1086/301639
Macken, W. L., Tischkowitz, M., and Lachlan, K. L. (2019). PTEN hamartoma tumor syndrome in childhood: a review of the clinical literature. Am. J. Med. Genet. C. Semin. Med. Genet. 181, 591–610. doi: 10.1002/ajmg.c.31743
Maines, E., Franceschi, R., Martinelli, D., Soli, F., Lepri, F. R., Piccoli, G., et al. (2021). Hypoglycemia due to PI3K/AKT/mTOR signaling pathway defects: two novel cases and review of the literature. Hormones (Athens) 20, 623–640. doi: 10.1007/s42000-021-00287-1
Manoukian, S., Peissel, B., Frigerio, S., Lecis, D., Bartkova, J., Roversi, G., et al. (2011). Two new CHEK2 germ-line variants detected in breast cancer/sarcoma families negative for BRCA1, BRCA2, and TP53 gene mutations. Breast Cancer Res. Treat. 130, 207–215. doi: 10.1007/s10549-011-1548-5
Marsh, D. J., Trahair, T. N., Martin, J. L., Chee, W. Y., Walker, J., Kirk, E. P., et al. (2008). Rapamycin treatment for a child with germline PTEN mutation. Nat. Clin. Pract. Oncol. 5, 357–361. doi: 10.1038/ncponc1112
Marsh, V., Winton, D. J., Williams, G. T., Dubois, N., Trumpp, A., Sansom, O. J., et al. (2008). Epithelial Pten is dispensable for intestinal homeostasis but suppresses adenoma development and progression after Apc mutation. Nat. Genet. 40, 1436–1444. doi: 10.1038/ng.256
Massimino, M., Gandola, L., Biassoni, V., Spreafico, F., Schiavello, E., Poggi, G., et al. (2013). Evolving of therapeutic strategies for CNS-PNET. Pediatr. Blood Cancer 60, 2031–2035. doi: 10.1002/pbc.24540
Menko, F. H., Kneepkens, C. M. F., Leeuw, N. D., Peeters, E. A. J., Maldergem, L. V., Kamsteeg, E. J., et al. (2008). Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clin. Genet. 74, 145–154. doi: 10.1111/j.1399-0004.2008.01026.x
Mester, J., and Eng, C. (2013). When overgrowth bumps into cancer: the PTEN-Opathies. Am. J. Med. Genet. C: Semin. Med. Genet. 163, 114–121. doi: 10.1002/ajmg.c.31364
Millard, N. E., and De Braganca, K. C. (2016). Medulloblastoma. J. Child Neurol. 31, 1341–1353. doi: 10.1177/0883073815600866
Nelen, M. R., Kremer, H., Konings, I. B., Schoute, F., van Essen, A. J., Koch, R., et al. (1999). Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur. J. Hum. Genet. 7, 267–273. doi: 10.1038/sj.ejhg.5200289
Ngeow, J., and Eng, C. (2015). PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol. Methods 77-78, 11–19. doi: 10.1016/j.ymeth.2014.10.011
Ngeow, J., Stanuch, K., Mester, J. L., Barnholtz-Sloan, J. S., and Eng, C. (2014). Second malignant neoplasms in patients with Cowden syndrome with underlying germline PTEN mutations. J. Clin. Oncol. 32, 1818–1824. doi: 10.1200/JCO.2013.53.6656
Parsons, D. W., Li, M., Zhang, X., Jones, S., Leary, R. J., Lin, J. C. H., et al. (2011). The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439. doi: 10.1126/science.1198056
Robinson, G., Parker, M., Kranenburg, T. A., Lu, C., Chen, X., Ding, L., et al. (2012). Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48. doi: 10.1038/nature11213
Salviati, L., Patricelli, M., Guariso, G., Sturniolo, G. C., Alaggio, R., Bernardi, F., et al. (2006). Deletion of PTEN and BMPR1A on chromosome 10q23 is not always associated with juvenile polyposis of infancy. Am. J. Hum. Genet. 79, 593–596. doi: 10.1086/507151
Schreibman, I. R., Baker, M., Amos, C., and McGarrity, T. J. (2005). The hamartomatous polyposis syndromes: a clinical and molecular review. Am. J. Gastroenterol. 100, 476–490. doi: 10.1111/j.1572-0241.2005.40237.x
Shah, N., and Walter, A. (2018). MBCL-18.CHEK2 mutation in high-risk MEDULLOBLASTOMA. Neuro-Oncology 20:i120. doi: 10.1093/neuonc/noy059.415
Smith, I. N., Thacker, S., Seyfi, M., Cheng, F., and Eng, C. (2019). Conformational dynamics and allosteric regulation landscapes of germline PTEN mutations associated with autism compared to those associated with cancer. Am J Hum Genet 104, 861–878. doi: 10.1016/j.ajhg.2019.03.009
Staal, F. J. T., van der Luijt, R. B., Baert, M. R. M., van Drunen, J., van Bakel, H., Peters, E., et al. (2002). A novel germline mutation of PTEN associated with brain tumours of multiple lineages. Br. J. Cancer 86, 1586–1591. doi: 10.1038/sj.bjc.6600206
Sturm, D., Bender, S., Jones, D. T. W., Lichter, P., Grill, J., Becher, O., et al. (2014). Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 14, 92–107. doi: 10.1038/nrc3655
Tan, M. H., Mester, J., Peterson, C., Yang, Y., Chen, J. L., Rybicki, L. A., et al. (2011). A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 88, 42–56. doi: 10.1016/j.ajhg.2010.11.013
Teodorczyk, U., Cybulski, C., Wokołorczyk, D., Jakubowska, A., Starzyńska, T., Lawniczak, M., et al. (2013). The risk of gastric cancer in carriers of CHEK2 mutations. Fam. Cancer 12, 473–478. doi: 10.1007/s10689-012-9599-2
Tolonen, J. P., Hekkala, A., Kuismin, O., Tuominen, H., Suo-Palosaari, M., Tynninen, O., et al. (2020). Medulloblastoma, macrocephaly, and a pathogenic germline PTEN variant: cause or coincidence? Mol. Genet. Genomic Med. 8:e1302. doi: 10.1002/mgg3.1302
Tsuchiya, K. D., Wiesner, G., Cassidy, S. B., Limwongse, C., Boyle, J. T., and Schwartz, S. (1998). Deletion 10q23.2-q23.33 in a patient with gastrointestinal juvenile polyposis and other features of a Cowden-like syndrome. Genes Chromosomes Cancer 21, 113–118. doi: 10.1002/(SICI)1098-2264(199802)21:2<113::AID-GCC6>3.0.CO;2-3
Vladoiu, M. C., El-Hamamy, I., Donovan, L. K., Farooq, H., Holgado, B. L., Sundaravadanam, Y., et al. (2019). Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73. doi: 10.1038/s41586-019-1158-7
Waszak, S. M., Northcott, P. A., Buchhalter, I., Robinson, G. W., Sutter, C., Groebner, S., et al. (2018). Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 19, 785–798. doi: 10.1016/S1470-2045(18)30242-0
Yakubov, E., Ghoochani, A., Buslei, R., Buchfelder, M., Eyüpoglu, I. Y., and Savaskan, N. (2016). Hidden association of Cowden syndrome, PTEN mutation and meningioma frequency. Onco. Targets. Ther. 3, 149–155. doi: 10.18632/oncoscience.305
Keywords: cancer predisposition syndrome (CPS), pediatric, PTHS, medulloblastoma (MB), intestinal polyp, PTEN hamartoma tumor syndrome
Citation: Caroleo AM, Rotulo S, Agolini E, Macchiaiolo M, Boccuto L, Antonelli M, Colafati GS, Cacchione A, Megaro G, Carai A, De Ioris MA, Lodi M, Tornesello A, Simone V, Torroni F, Cinalli G and Mastronuzzi A (2023) SHH medulloblastoma and very early onset of bowel polyps in a child with PTEN hamartoma tumor syndrome. Front. Mol. Neurosci. 16:1228389. doi: 10.3389/fnmol.2023.1228389
Edited by:
Shilpa S. Dhar, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Moatasem El-Ayadi, Cairo University, EgyptCopyright © 2023 Caroleo, Rotulo, Agolini, Macchiaiolo, Boccuto, Antonelli, Colafati, Cacchione, Megaro, Carai, De Ioris, Lodi, Tornesello, Simone, Torroni, Cinalli and Mastronuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Mastronuzzi, YW5nZWxhLm1hc3Ryb251enppQG9wYmcubmV0;