
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci., 24 July 2023
Sec. Brain Disease Mechanisms
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1222321
This article is part of the Research TopicFocus on Ion Channels as a Potential Therapeutic Target for Neurodegenerative DiseasesView all 5 articles
 Miriam Kessi1,2,3
Miriam Kessi1,2,3 Baiyu Chen1,2,3
Baiyu Chen1,2,3 Nan Pang1,2,3
Nan Pang1,2,3 Lifen Yang1,2,3
Lifen Yang1,2,3 Jing Peng1,2,3*Fang He1,2,3*
Jing Peng1,2,3*Fang He1,2,3* Fei Yin1,2,3*
Fei Yin1,2,3*Background: Genotype–phenotype correlations of the CACNA1A-related neurodevelopmental disorders such as global developmental delay (GDD)/intellectual disability (ID), epileptic encephalopathy (EE), and autism spectrum disorder (ASD) are unknown. We aimed to summarize genotype–phenotype correlations and potential treatment for CACNA1A-related neurodevelopmental disorders.
Methods: Six children diagnosed with CACNA1A-related neurodevelopmental disorders at Xiangya Hospital, Central South University from April 2018 to July 2021 were enrolled. The PubMed database was systematically searched for all reported patients with CACNA1A-related neurodevelopmental disorders until February 2023. Thereafter, we divided patients into several groups for comparison.
Results: Six patients were recruited from our hospital. Three cases presented with epilepsy, five with GDD/ID, five with ataxia, and two with ASD. The variants included p.G701R, p.R279C, p.D1644N, p.Y62C, p.L1422Sfs*8, and p. R1664Q [two gain-of-function (GOF) and four loss-of-function (LOF) variants]. About 187 individuals with GDD/ID harboring 123 variants were found (case series plus data from literature). Of those 123 variants, p.A713T and p.R1664* were recurrent, 37 were LOF, and 7 were GOF. GOF variants were linked with severe-profound GDD/ID while LOF variants were associated with mild–moderate GDD/ID (p = 0.001). The p.A713T variant correlated with severe-profound GDD/ID (p = 0.003). A total of 130 epileptic patients harboring 83 variants were identified. The epileptic manifestations included status epilepticus (n = 64), provoked seizures (n = 49), focal seizures (n = 37), EE (n = 29), absence seizures (n = 26), and myoclonic seizures (n = 10). About 49 (42.20%) patients had controlled seizures while 67 (57.80%) individuals remained with refractory seizures. Status epilepticus correlated with variants located on S4, S5, and S6 (p = 0.000). Among the 83 epilepsy-related variants, 23 were recurrent, 32 were LOF, and 11 were GOF. Status epilepticus was linked with GOF variants (p = 0.000). LOF variants were associated with absence seizures (p = 0.000). Six patients died at an early age (3 months to ≤5 years). We found 18 children with ASD. Thirteen variants including recurrent ones were identified in those 18 cases. GOF changes were more linked to ASD.
Conclusion: The p.A713T variant is linked with severe-profound GDD/ID. More than half of CACNA1A-related epilepsy is refractory. The most common epileptic manifestation is status epilepticus, which correlates with variants located on S4, S5, and S6.
CACNA1A (calcium voltage-gated channel subunit alpha1 A) is a bicistronic gene that encodes both the alpha-1 (α1A) subunit of a calcium channel, also called Cav2.1 (Bourinet et al., 1999), and α1ACT (a transcription factor). Cav2.1 channels are highly expressed in the cerebral cortex, hippocampus, and cerebellum (Kessi et al., 2021). They are found in the pre-synaptic area, where they stimulate the secretion of neurotransmitters which hasten synaptic transmission as well as synapse plasticity (Martínez-Monseny et al., 2021). CACNA1A variants are related to a broad spectrum of neurological phenotypes. The classical phenotypes include episodic ataxia 2 (EA2) which is mostly caused by nonsense mutations (Strupp et al., 2007), familial hemiplegic migraine type 1 (FHM1) which is frequently caused by missense mutations (mainly gain-of-function (GOF) variants) (Ducros et al., 2001), and spinocerebellar ataxia type 6 (SCA6) which usually occurs because of the expanded CAG repeats (Klockgether, 2008). However, with advanced next-generation sequencing techniques, CACNA1A variants have been linked to more wider phenotypic spectrum including global developmental delay (GDD)/intellectual disability (ID), epileptic encephalopathy (EE), and autism spectrum disorder (ASD) (Kessi et al., 2021; Indelicato and Boesch, 2023). Currently, genotype–phenotype correlations of the CACNA1A – related neurodevelopmental disorders such as GDD/ID, ASD, and epilepsy are unknown.
Thus, we aimed to investigate genotype–phenotype correlations, underlying mechanisms, and potential therapies for the for CACNA1A-related neurodevelopmental disorders. Knowledge about genotype–phenotype correlations will aid in knowing disease prognosis which might help in the preparation of future research.
The institutional ethics committee of Xiangya Hospital, Central South University reviewed and approved this study according to the World Medical Association on ethical principles of human research for medical purposes agreement. Written informed consent was acquired from the parents or guardians of the subjects.
Children diagnosed with CACNA1A-related neurodevelopmental disorders in the pediatric neurology department, Xiangya Hospital, Central South University from April 2018 to July 2021 were recruited. In addition, we collected information of all reported patients with CACNA1A-related neurodevelopmental disorders systematically from the PubMed database. The keywords used for searching articles were the combination of the CACNA1A OR Cav2.1 channel AND (epilepsy OR intellectual disability OR global developmental delay OR autism spectrum disorder OR neurological disease OR neurological disorder OR treatment OR animal model OR mechanisms) for all years to date February 2023. This review included only clinical papers published in English, however, the discussion was supplemented by all animal or mechanisms studies available in the literature. It excluded abstracts, patents, reviews, book chapters, and conference papers. The final reference list was produced according to the originality and relevance to the broad scope of this study. Overall, 1024 papers were found at the first search which were filtered to get the desired information. About 90 papers that met the inclusion criteria were included in the final analysis (Supplementary Flow Chart 1). The systematic review was carried out based on the PRISMA 2020 statement: an updated guideline for reporting systematic reviews (Page et al., 2021).
The cross-sectional retrospective cohort study was conducted. This was accompanied by a comprehensive literature review of all CACNA1A reported cases in the literature. Our patients along with those reported in the literature were divided into groups according to the major phenotypes; epilepsy, GDD/ID, and ASD. We further categorized patients into other smaller groups for comparison, particularly for the epilepsy and GDD/ID groups. For ID/GDD patients, we created groups of patients with GOF and loss of function (LOF) variants, individuals with missense and nonsense variants as well as patients with mild–moderate and severe-profound ID/GDD. For the epileptic patients, we formed groups of patients with GOF and LOF variants, individuals with missense and nonsense variants, patients with status epilepticus, absence, and myoclonic seizures as well as patients with controlled seizures (including those who achieved seizure freedom), and refractory epilepsy. The sample size depended on the available CACNA1A-related disordered patients from our hospital and in the literature.
For the case series, epilepsy was diagnosed according to International League against Epilepsy (ILAE) standards. Whereas, GDD/ID and ASD were diagnosed according to diagnostic criteria of the DSM-5 for Intellectual disabilities (Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, American Psychiatric Association 2013). Clinical interviews, observations, and standardized age-related rating scales were used for assessment of the adaptive functioning. The GDD diagnosis was often initially formulated based on clinical judgment rather than on formal standardized assessments, especially for young patients (Shevell, 2008). We used standardized age-related rating scales such as Gesell Developmental Schedules for patients younger than 2–4 years of age, Wechsler Preschool and Primary Scale of Intelligence Fourth Edition (WPPSI-IV) for patients between 4 and 6 years, and Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV) for patients who were 6 years old or older than 6 years. The GDD/ID severity was graded as described in our previously published articles (Kessi et al., 2018, 2023). For the cases from the literature, there was no specific definition for GDD/ID. Some of the authors defined GDD/ID according to DSM-5 and provided information in regards to severity while other authors did not provide the severity grades. The information of all GDD/ID patients was collected, however, we only analyzed those with information related to GDD/ID severity.
Data was collected by senior neurologists and medical geneticists. The expert opinions of radiologists and electroencephalogram technicians were considered. Clinical characteristics of the patients such as demographic data, prenatal, natal, post-natal, family and behavioral histories, seizure characteristics, growth and development assessment results, and neurological examinations were collected. Auxiliary examination findings including blood and urine test results, brain magnetic resonance imaging (MRI) or computerized tomography (CT) results, cardiac and kidney imaging findings, electroencephalography (EEG) findings, metabolic findings, and genetic test results along with their functional experimental results were collected.
After informing and receiving the signed consent forms from the patients, guardians, or parents, blood samples of the patients and their biological parents were collected. Genomic DNA was extracted, prepared, and analyzed according to the previous protocols. Whole exome sequencing (WES) was performed on all six cases. Sanger sequencing confirmed the parenteral origin of the variants. The genetic results were interpreted as per American College of Medical Genetics (ACMG) guidelines (Richards et al., 2015; Riggs et al., 2020).
Statistical analysis was performed by using the Statistical Package for Social Science (IBM, SPSS Statistics Version 25). Categorical data were summarized in the form of frequencies and proportions, and analyzed with the Chi-square test. Mann–Whitney test was used to compare non-parametric continuous data. The p ≤ 0.05 indicated significant differences between groups.
A total of six cases were recruited from our hospital: three boys and three girls. The onset age ranged from 1 year and 20 days to 10 years. Three cases presented with seizures (three focal and one absence seizures, and one had status epilepticus). Two cases had a previous history of febrile seizures. Five cases had GDD/ID, five cases had ataxia, and two had ASD. Two cases had profound GDD/ID, one severe and two mild. Three cases had cerebellar atrophy (Supplementary Table 1). We identified six CACNA1A variants of which four were de novo and two inherited. The variants included p.G701R, p.R279C, p.D1644N, p.Y62C, p.L1422Sfs*8, and p. R1664Q (2 GOF and 4 LOF variants) (Table 1). These variants can be found in ClinVar (SCV003930352 – SCV003930359).
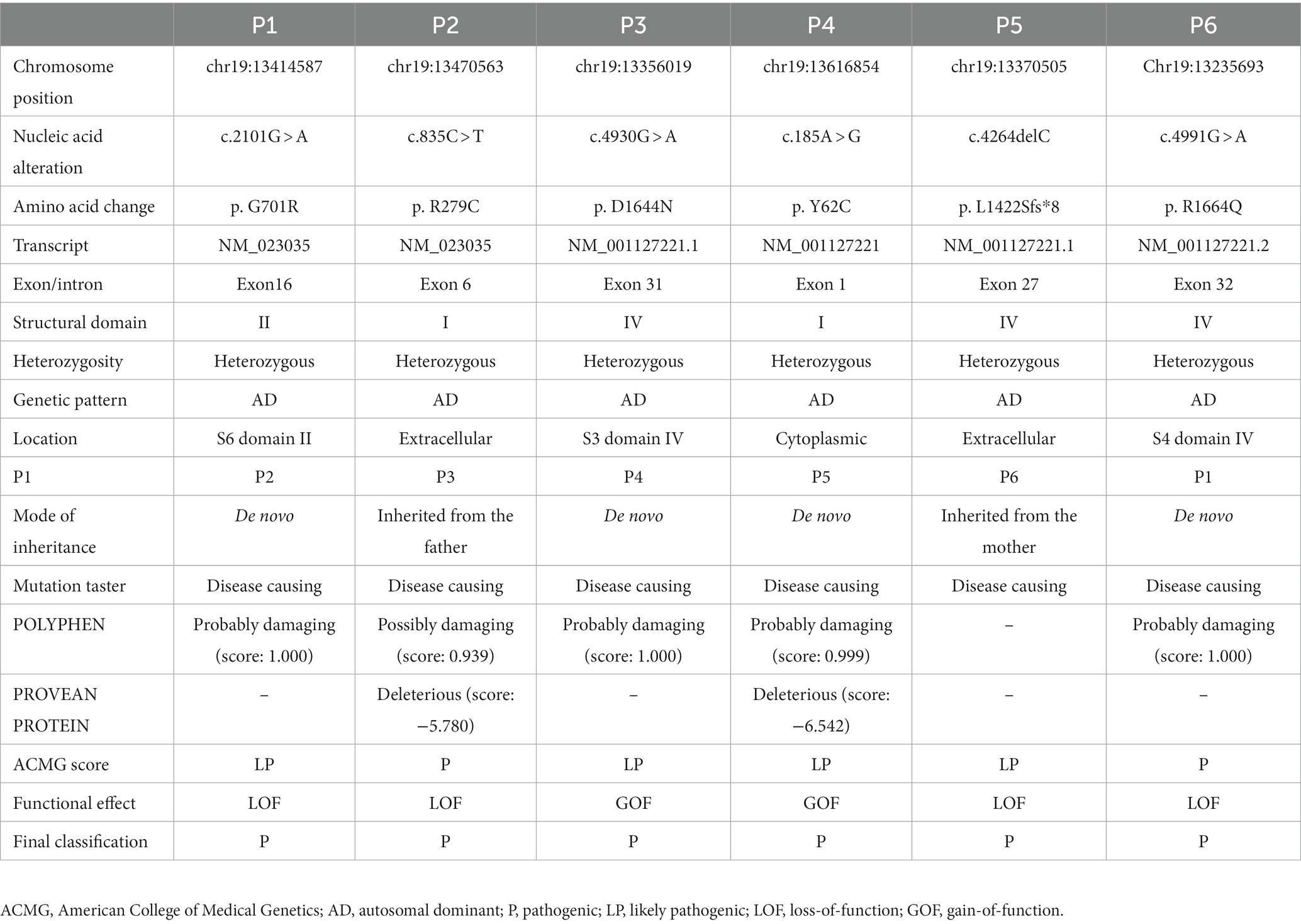
Table 1. Genetic summary of 6 patients with CACNA1A-associated neurodevelopmental disorders from our hospital.
We collected information on patients with epilepsy from our hospital and combined them with those reported in the literature. We found 130 patients carrying 83 pathogenic or likely pathogenic CACNA1A variants. Of the 83 variants, 23 were recurrent.
Ninety-two individuals had information related to sex, of whom 45 were males and 47 were females. The mean age of seizure onset was 59 months, the median age was 25 months ± 107.5SD, and the age range was 0 days – 50 years. Of the 130 patients, there were 42 (32.30%) from China, 27 (20.76%) from the USA, 18 (13.84%) from France and Belgium, 18 (13.85%) from Korea, 6 (4.62%) from the UK, 6 (4.62%) from the Netherlands, 4 (3.078%) from ‘North America’, 4 (3.08%) from Italy, 3 (2.31%) from Iran, 3 (2.31%) from Canada, 3 (2.31%) from Japan, 3 (2.31%) from Estonia, 2 (1.54%) from Germany, 1 (0.77%) from Poland, 1 (0.77%) from India, 1 (0.77%) from Australia, and it was unclear which country 5 (3.85%) were from (from the Epilepsy Phenome/Genome Project). Of the 116 patients with information about the treatment outcome, 67 (57.80%) individuals remained with refractory seizures while 49 (42.20%) achieved seizure control. Six cases died at a very young age (from the age of 3 months – ≤ 5 years) (Supplementary Table 2). The epileptic phenotypes were very diverse and had some provoking factors for the 49 cases. The epileptic manifestations included status epilepticus (n = 64), provoked seizures (n = 49), focal seizures (n = 37), epileptic encephalopathy (EE, n = 29), absence seizures (n = 26), and myoclonic seizures (n = 10) (Supplementary Figure 1). Seizure-provoking factors included fever for most of the cases, infection, mild head trauma, stress, excitation, bathing, climatic changes, excitation, agitation, and traveling.
There were 83 variants reported to relate to epilepsy (Imbrici et al., 2004; Kors et al., 2004; Rajakulendran et al., 2010; Zangaladze et al., 2010; Yamazaki et al., 2011; Damaj et al., 2015; Byers et al., 2016; Reinson et al., 2016; Du et al., 2017; Lv et al., 2017; Epperson et al., 2018; Hayashida et al., 2018; Kothur et al., 2018; Lee et al., 2018; Angelini et al., 2019; Jiang et al., 2019; Yamamoto et al., 2019; Gauquelin et al., 2020; Gudenkauf et al., 2020; Mitta et al., 2020; Stendel et al., 2020; Zhang et al., 2020; Ho et al., 2021; Le Roux et al., 2021; Stubberud et al., 2021; Verriello et al., 2021; Alehabib et al., 2022; Bolte et al., 2022; Hommersom et al., 2022; Krygier et al., 2022; Li et al., 2022; Lipman et al., 2022; Niu X. et al., 2022; Niu Y. et al., 2022) as shown in Supplementary Table 2. The majority of the variants were missense (77.20%) followed by nonsense (8.80%) as shown in Figure 1A. For the 83 variants with functional results, 32 (38.55%) were LOF and 11 (13.25%) were GOF (Figure 1B). Of the 83 variants, 21 were recurrent, including p.A713T (n = 10), p.V1392M (n = 8), p.V1393M (n = 7), p.A710T (n = 3), p.L226W (n = 3), p.R583* (n = 3), p.R279C (n = 3), p.E101Q (n = 3), p.Y62C (n = 3), p.R1352Q (n = 3), and so on (Supplementary Figure 2).

Figure 1. (A) The distribution of the variants related to epilepsy. (B) The distribution of the 83 epilepsy-related variants according to the electrophysiological changes.
Some recurrent variants showed correlations with status epilepticus, absence seizures, and myoclonic seizures as summarized in Supplementary Figure 3. Status epilepticus showed an association with GOF variants (72.1% Vs. 15.8%, p = 0.000). In contrast, LOF variants revealed an association with absence seizures (2.4% Vs. 47.4%, p = 0.000) (Supplementary Table 3). We attempted to investigate whether the location of the variants could influence epileptic manifestations. We found that status epilepticus had a correlation with variants located on S4, S5, and S6 (p = 0.000) as well as missense mutations (p = 0.000). The hotspots for the status epilepticus included p.A710T, p.I711M, p.A712T, p.A713T, p.V1392M, p.V1393M, p.R1348Q, p.R1349Q, and p.Y62C. There was no correlation between the variant location with absence nor myoclonic seizures (Supplementary Table 4).
Epilepsy treatment outcome was not influenced by either electrophysiological changes (GOF or LOF) or type of mutations (missense or nonsense). Nevertheless, patients with refractory epilepsy were more likely to receive topiramate (TPM) (p = 0.001), phenobarbital (PB) (p = 0.001), and levetiracetam (LEV) (p = 0.000) (Supplementary Table 5). Figure 2 summarizes the distribution of the therapies. None of the anti-seizure medications (ASMs) could control seizures among patients in the GOF group. Notably, patients within the refractory seizure group were more likely to receive LEV (p = 0.032) (Supplementary Table 6). Supplementary Figure 4 summarizes the distribution of the common ASMs used for the GOF variants and their outcome. None of the ASMs could control seizures among the patients in the LOF group (Supplementary Table 7). Supplementary Figure 5 summarizes the distribution of the common ASMs used for the LOF variants and their outcome.
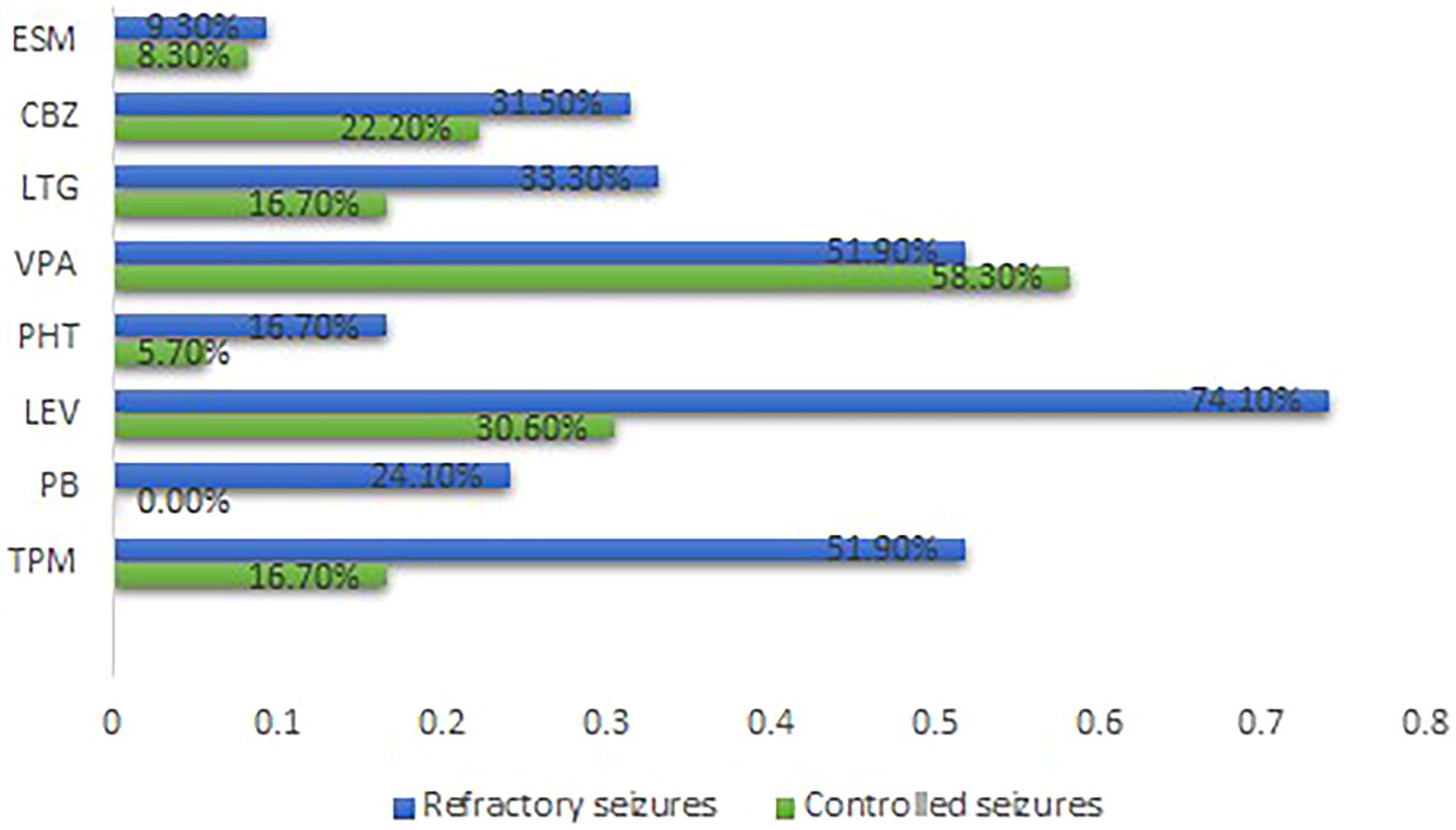
Figure 2. Distribution of the common ASMs used and outcome for all cases Carbamazepine. ESM, Ethosuximide; GOF, Gain-of-function; LEV, Levetiracetam; LTG, Lamotrigine; LOF, Loss-of-function; PB, Phenobarbital; PHT, phenytoin; TPM, Topiramate; VPA, Sodium Valproate.
We found 5 patients from our hospital and 182 individuals from the literature reported to have GDD/ID, and harbor CACNA1A pathogenic or likely pathogenic variants. The mean onset age was 25 months, the median was 11 months ±37.1744 SD, the range was 0–144 months. Of the 187 cases, there were 35 (18.72%) from China, 20 (10.70%) from Israel, 19 (10.16%) from France, 17 (9.09%) from France and Belgium, 11 (5.88%) from the USA, 10 (5.35%) from Canada, 10 (5.35%) from Austria, 9 (4.81%) from Spain, 9 (4.81%) from the Netherlands, 8 (4.28%) from Australia, 7 (3.74%) from the UK, 6 (3.21%) from Italy, 6 (3.21%) from Germany, 6 (3.21%) from Japan, 4 (2.14%) from ‘North America’, 2 (1.07%) from Estonia, 1 (0.53%) from Switzerland, 1 (0.53%) from Minnesota, 1 (0.53%) from India, and 5 (2.67%) were from unknown countries (from the Epilepsy Phenome/Genome Project). Of the 99 cases with information about sex, males comprised 41 (41.4%) and females 58 (58.6%) (Supplementary Table 8).
The GDD/ID severity distribution is summarized in Figure 3. Progressive cerebellar atrophy (congenital/early onset/isolated) was observed in 46 patients, progressive cerebral atrophy was found in 15 individuals, and hypomyelination/delayed myelination was identified in 4 patients. In addition, progressive optic nerve atrophy was reported in 2 patients, shoulder girdle atrophy in one individual, normal brain imaging findings in 44 cases, and 52 individuals had unknown brain changes (Supplementary Table 8).
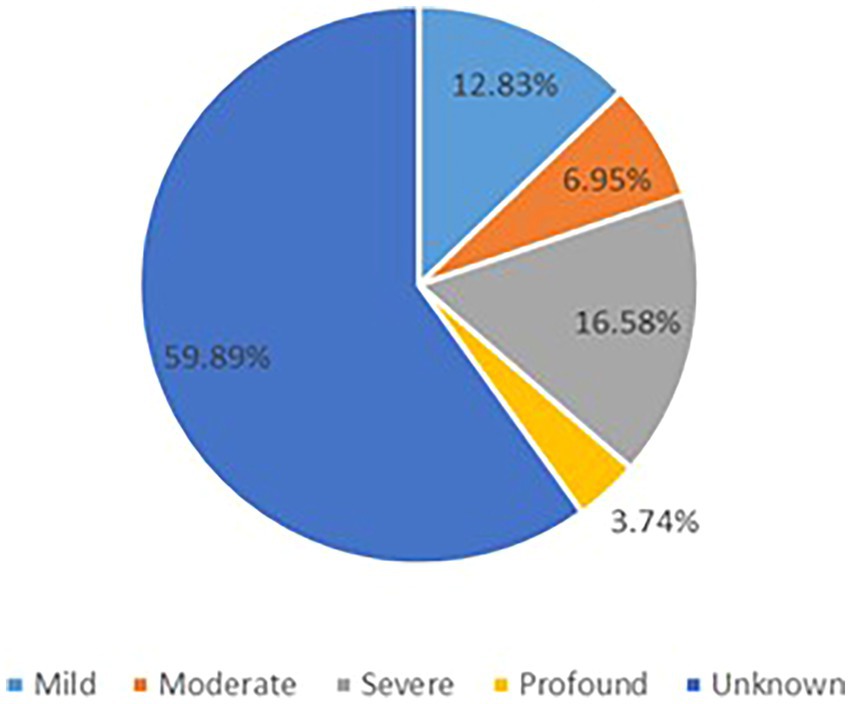
Figure 3. The GDD/ID severity distribution.
A total of 123 variants were identified in 187 cases (Fitzsimons and Wolfenden, 1985; Vahedi et al., 2000; Kors et al., 2001; Wada et al., 2002; De Vries et al., 2008; Guerin et al., 2008; Bertholon et al., 2009; Blumkin et al., 2010, 2015; Jung et al., 2010; Mantuano et al., 2010; Rajakulendran et al., 2010; Riant et al., 2010; Carreño et al., 2011; Freilinger et al., 2011; Naik et al., 2011; Yamazaki et al., 2011; Epi4K Consortium et al., 2013; Ohba et al., 2013; Bürk et al., 2014; García Segarra et al., 2014; Bahamonde et al., 2015; Damaj et al., 2015; Epi4K Consortium, 2016; Reinson et al., 2016; Tantsis et al., 2016; Weyhrauch et al., 2016; Balck et al., 2017; Luo et al., 2017; Epperson et al., 2018; Hayashida et al., 2018; Humbertclaude et al., 2018, 2020; Kothur et al., 2018; Angelini et al., 2019; Indelicato et al., 2019; Jiang et al., 2019; Kashimada et al., 2019; Tyagi et al., 2019; Yamamoto et al., 2019; Gauquelin et al., 2020; Gudenkauf et al., 2020; Meloche et al., 2020; Zhang et al., 2020, 2021; Arteche-López et al., 2021; Gandini et al., 2021; Gur-Hartman et al., 2021; Ho et al., 2021; Le Roux et al., 2021; Martínez-Monseny et al., 2021; Saathoff et al., 2021; Stubberud et al., 2021; Alehabib et al., 2022; Bolte et al., 2022; Gajam et al., 2022; Grosso et al., 2022; Hommersom et al., 2022; Li et al., 2022; Mellone et al., 2022; Niu X. et al., 2022; Niu Y. et al., 2022; Wong-Spracklen et al., 2022; Yuan et al., 2022) as shown in Supplementary Table 8. There were 25 recurrent variants which have been summarized in Figure 4. The type of mutations have been summarized in Figure 5A. The distribution of the functional changes of the variants has been summarized in Figure 5B.
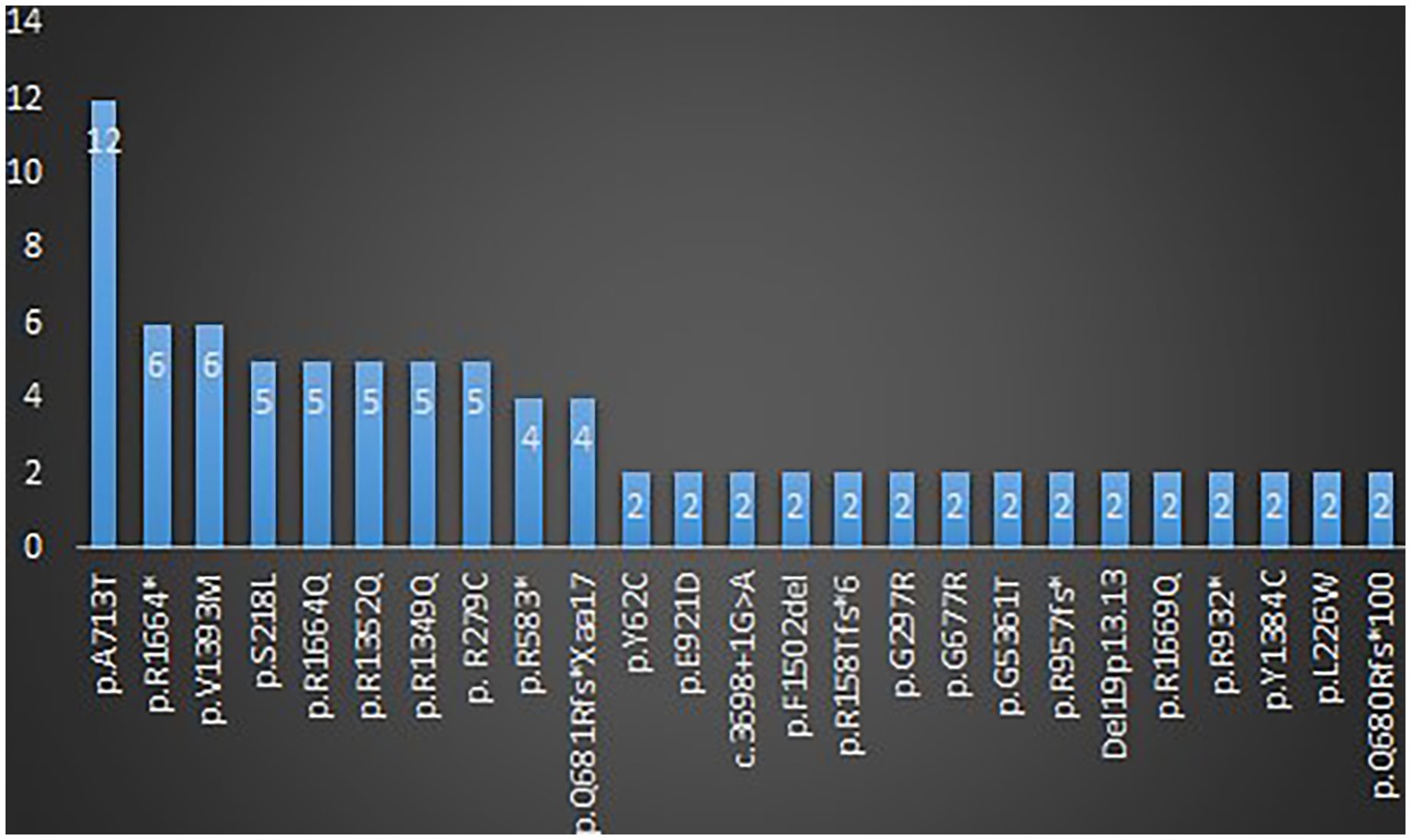
Figure 4. Recurrent variants related to GDD/ID.
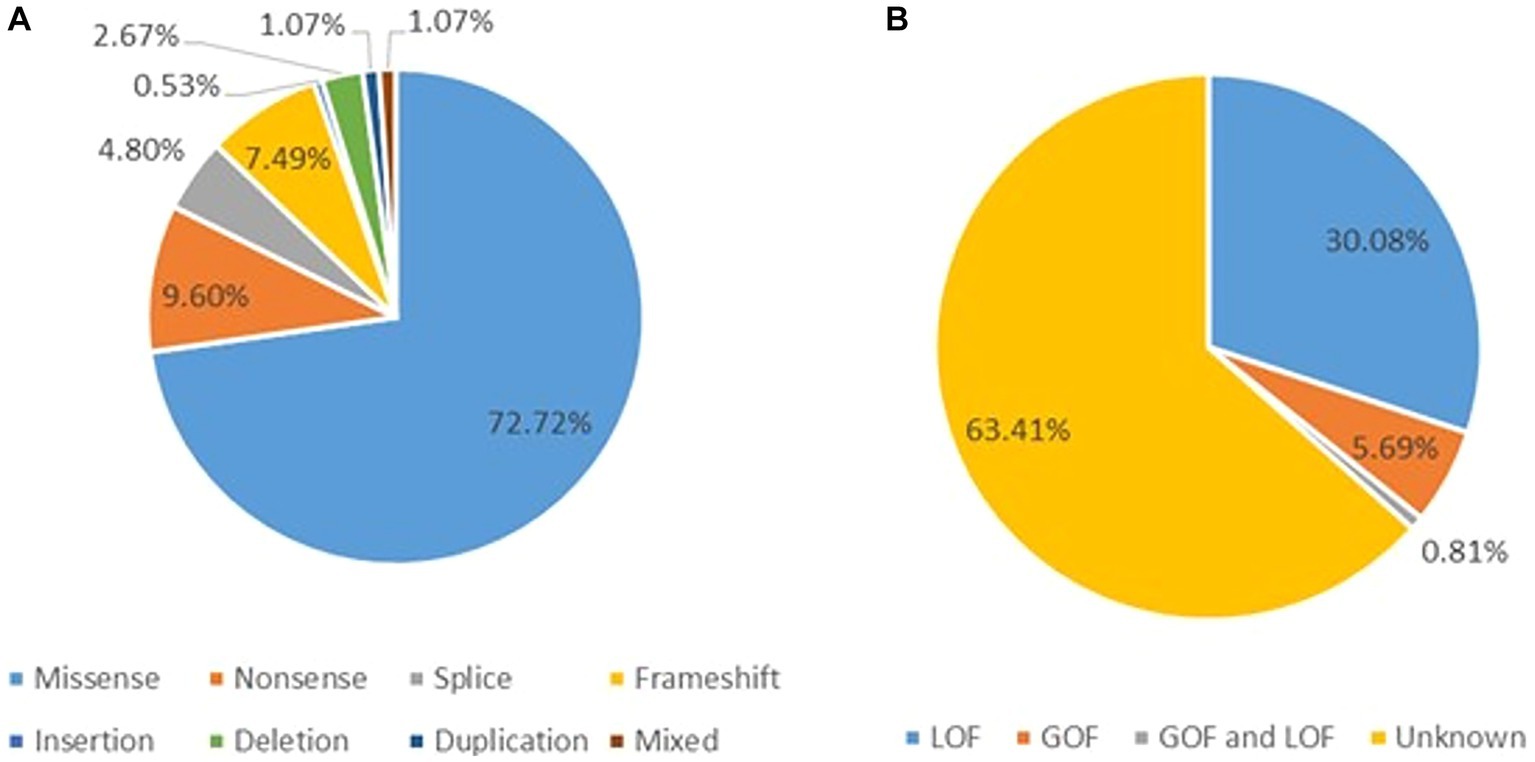
Figure 5. (A) Types of the GDD/ID-related mutations. (B) The distribution of the GDD/ID-related variants according to the electrophysiological changes.
GOF variants showed a substantial relationship with severe – profound GDD/ID, in contrast, LOF variants displayed a correlation with mild–moderate GDD/ID (p = 0.001). Cerebellar atrophy was observed more in patients with severe – profound GDD/ID although the result was not statistically significant (p = 0.061). Missense mutations were related to severe–profound GDD/ID, however, results were not significant (p = 0.072). Supplementary Table 9 and Figure 6 below summarize this information. The p.A713T variant located on S6 with a GOF effect showed a correlation with severe – profound GDD/ID (p = 0.003). Supplementary Table 10 and Figure 7 summarize this information.
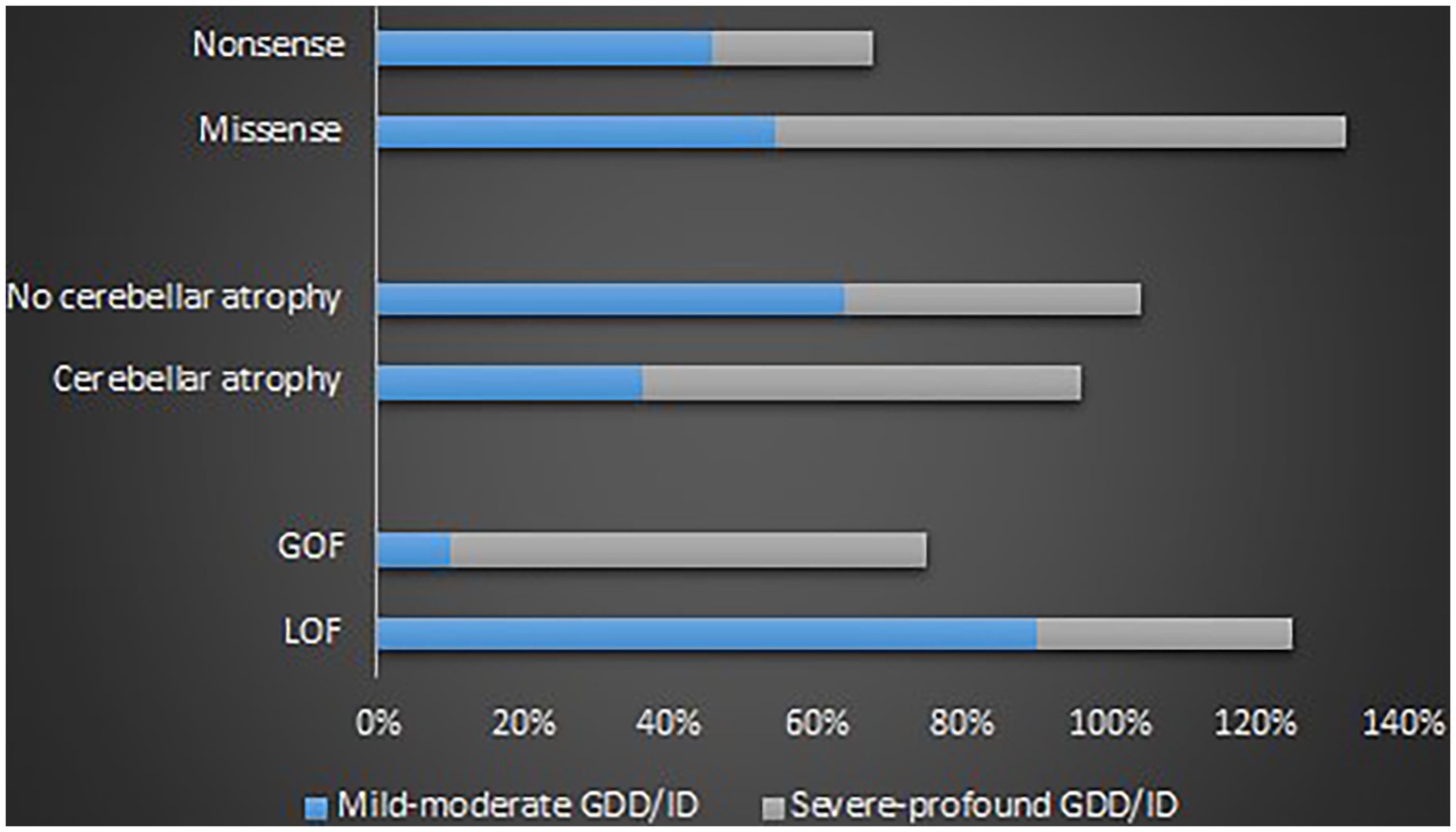
Figure 6. Proportions of the determinants of the GDD/ID severity.
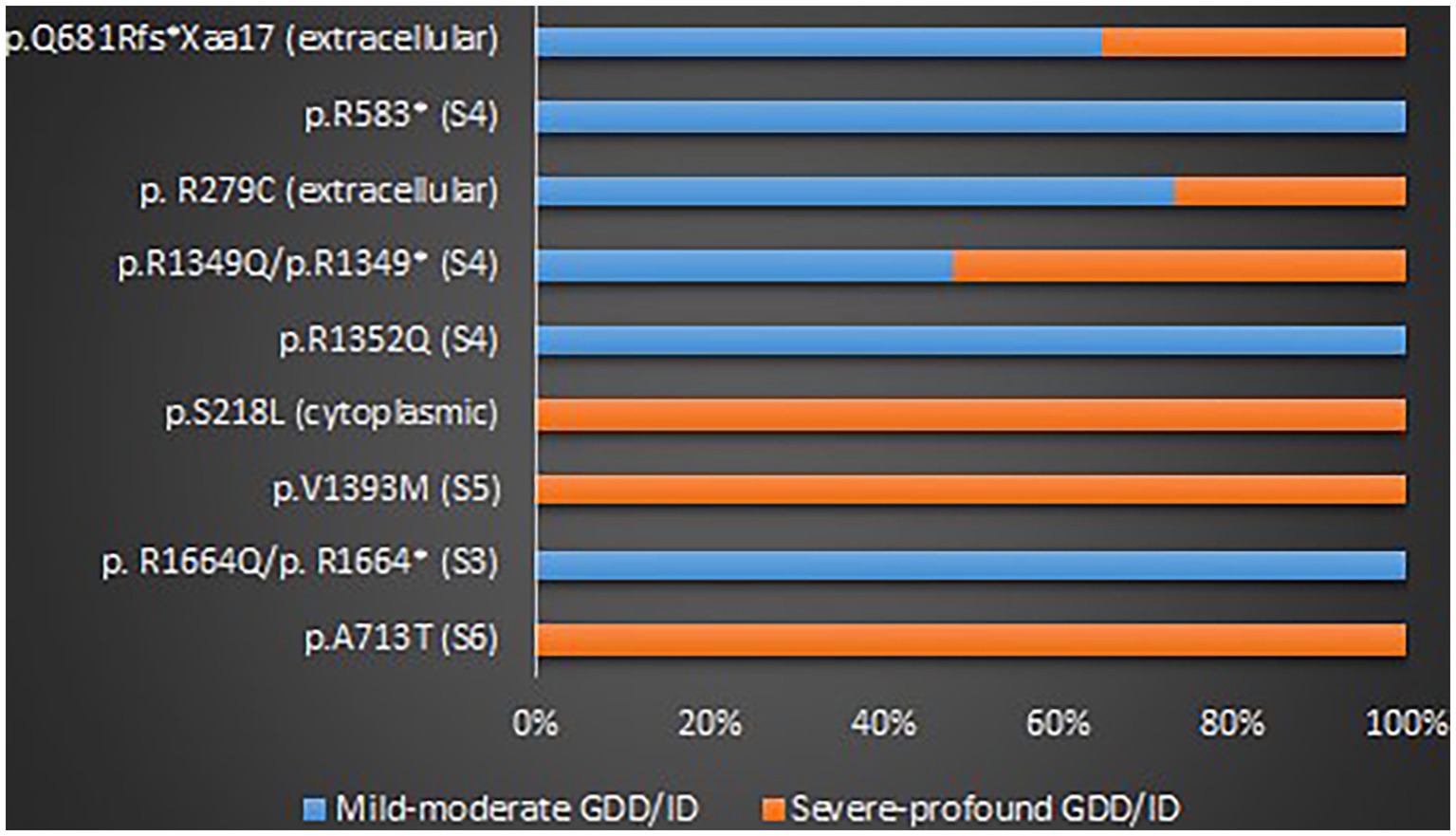
Figure 7. The proportions of the recurrent variants in different GDD/ID severity.
We found a total of 18 cases that presented with ASD. Ten were males, 5 were females, and 3 had unknown sex. The onset age was unclear for all cases, but mostly during childhood according to their recent ages.
In addition to ASD, 16 had epilepsy, 13 cases had GDD/ID, 9 were hypotonic, 8 had non-episodic ataxia, 3 had ADHD, and 2 had episodic ataxia (Supplementary Table 11).
Thirteen variants were identified in those 18 cases including p.V1392M (n = 3), p.A712T (n = 2), p.E533K (n = 2), p.R1278Ter (n = 2), p. G701R (n = 1), p.Y62C (n = 1), p.R279C (n = 1), p.R1348Q (n = 1), p.I711M (n = 1), p.G700E (n = 1), p.S1798L (n = 1), p.I1707T (n = 1), and p.P2312_ Q2313ins (n = 1) (Vila-Pueyo et al., 2014; Damaj et al., 2015; Meloche et al., 2020; Gur-Hartman et al., 2021; Martínez-Monseny et al., 2021). Supplementary Table 11 summarizes this information. The most common type of mutation was missense (83%) as shown in Figure 8. Of the 13 variants, 4 were recurrent; p.V1392M (n = 3), p.A712T (n = 2), p.E533K (n = 2), and p.R1278Ter (n = 2) as shown in Supplementary Figure 6. Most of the variants were GOF (53.84%), followed by LOF (30.76%), and 15.38% were unknown (Supplementary Figure 7). Variants were located on S6 (n = 6), S5 (n = 4), and S2 (n = 4).
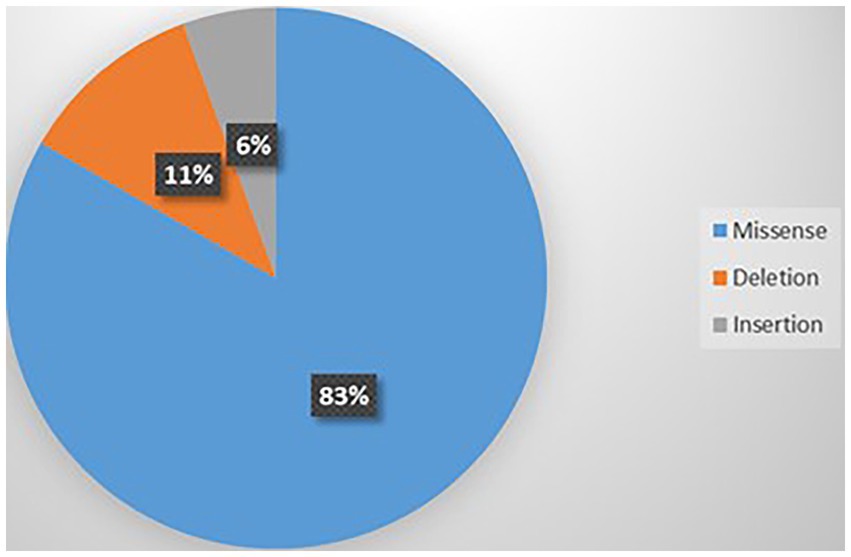
Figure 8. Types of mutations related to ASD.
Some of the variants have been reported to associate with epilepsy, GDD/ID, and ASD. The variants include p.A713T, p.R1664*, p.P2312_Q2313ins, p.V1393M, p.Y62C, p.V1392M, p.A712T, p.R1348Q, p.I711M, p.G700E, p.S1798L, p.I1707T, and p.R1278Ter. There is a cluster of six critical residues in the S6 domain II. Most of the variants are located on S4 and S6 as shown in Figure 9.
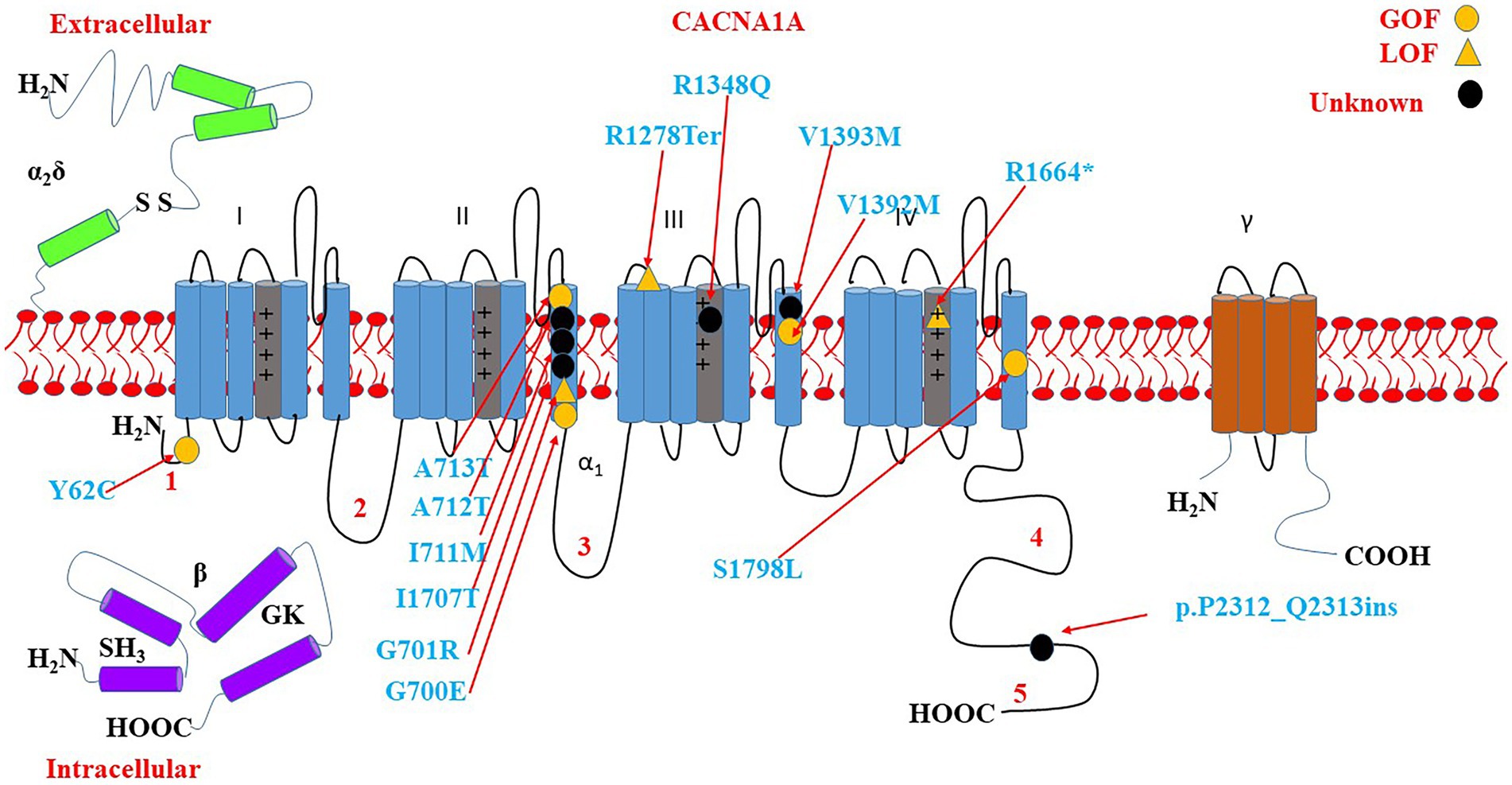
Figure 9. Location of the identified CACNA1A amino acid substitutions related to GDD/ID, epilepsy, and ASD. There is a cluster of 5 critical residues in the S6 domain II. Round yellow dots represent gain-of-function variants. Triangular yellow dots represent loss-of-function variants. Round black dots represent unknown functional change variants.
CACNA1A is related to a wide range of phenotypes including GDD/ID, epilepsy, and ASD. Different forms of epilepsy including status epilepticus, absence seizures, juvenile myoclonic seizures, and EE have been reported. More than a third of the cases had provoking factors for the seizures/status epilepticus. Status epilepticus correlated with GOF variants whereas, LOF variants were associated with absence seizures. Status epilepticus also showed correlations with missense mutations. We could find some hotspots for the status epilepticus, and variants on S4, S5, and S6 showed a statistically substantial correlation with status epilepticus. More than half of the cases remained with refractory epilepsy. Refractory cases in both GOF and LOF groups were more likely to receive TPM, PB, and LEV. For the GOF patients, individuals within the refractory seizure group were more likely to receive LEV. CACNA1A variants caused early deaths in six cases. Most of the reported cases originated from China, the USA, France and/or Belgium, and Korea, suggesting the possibility of the presence of racial disparity. Nevertheless, this finding could also signify that these countries perform more genetic tests compared to other countries in the world.
Diverse forms of epilepsy including status epilepticus, absence seizures, juvenile myoclonic seizures, and EE were related to both GOF, LOF, and mixed GOF and LOF variants. Epilepsy could present a few hours after delivery, approximately half of the patients presented with status epilepticus, and 37.7% had provoking factors. We could observe genotype–phenotype correlations. Status epilepticus and myoclonus showed correlations with GOF variants whereas, LOF variants were linked with absence seizures. The status epilepticus was often focal which could be generalized later. Of interest, patients carrying mutations located on S4, S5, and S6 were more likely to have status epilepticus which corresponds with the specific function of these transmembrane helices. The S4 helix is positively charged, therefore, it is in charge of controlling voltage-dependent activation. The loop between S5 and S6 has negatively charged residues that produce the selectivity filter (Simms and Zamponi, 2014). The hotspots for the status epilepticus include p.A710T, p.I711M, p.A712T, p.A713T, p.V1392M, p.V1393M, p.R1348Q, p.R1349Q, and p.Y62C. It is known that an excessive calcium ions influx through NMDARs can lead to glutamate excitotoxicity as observed in neurodegenerative disorders (Stanika et al., 2012). Moreover, excessive build-up of calcium ions in the neuronal mitochondria can lead to excessive firing like in epileptic seizure, and subsequently resulting in neuronal death (Kramer and Bressan, 2018). A patient diagnosed with intractable epilepsy as well as early stroke, and yet carrying p.L1692Q was found to have mitochondrial depletion (according to muscle biopsy), and an electron transport chain study showed a reduction in several respiratory chain complex activities (Gudenkauf et al., 2020). Noteworthily, CACNA1A-related epilepsy has some provoking factors including fever for most of the cases, infection, mild head trauma, stress, excitation, bathing, climatic changes, excitation, agitation, and traveling which somehow present like mitochondrial diseases.
The p.R1667P mutation with mixed GOF and LOF effects has been observed in two children (Grosso et al., 2022). The first patient presented with focal seizures, hypotonia, severe congenital ataxia, dysarthria, and severe cerebral edema which led to death at 5 years of age. The clinical manifestations of the first patient mimicked the autoimmune encephalitis. Post-mortem brain biopsy revealed atrophic cerebellar vermis, loss of Purkinje cells, and hemorrhagic lesions involving the cerebral cortex (Gauquelin et al., 2020). The second patient presented with GDD, microcephaly, pontocerebellar hypoplasia, and progressive cerebellar ataxia (Grosso et al., 2022). The two cases with the same mutations and yet diverse clinical severity or prognosis suggest the complexity of the balance of GOF and LOF changes on the Cav2.1 channel as suggested before (Grosso et al., 2022).
More than half of the cases remained with refractory epilepsy. We have observed that refractory cases for both GOF and LOF were more likely to receive TPM, PB, and LEV. Besides, LEV showed an association with refractory seizures within the GOF group. Whether LEV can worsen seizures is not clear now as many ASMs were prescribed, and it is hard to tell which of them improved or worsened seizures. LEV has been shown to inhibit N-type calcium channels, ryanodine receptor (RyR), and IP3 receptors (IP3R) – activated calcium-induced calcium release (CICR) in hippocampal neurons (Nagarkatti et al., 2008). LEV can also minimize seizures by inhibiting synaptic vesicle glycoprotein 2A (SV2A) (Lynch et al., 2004; Gillard et al., 2006). Notably, LEV has been reported to be effective for generalized tonic–clonic seizures, focal seizures, and generalized myoclonic seizures (Abou-Khalil, 2019). The inability of the LEV to control seizures might be explained by the fact that it has less effect on the Cav2.1 channel. The ability of LEV to worsen seizures might be due to its capacity to interfere with other channels or receptors needed for the regulation of the calcium ions homeostasis. PB works by binding the GABA-A receptor and thus prolongs the opening of the associated chloride channel leading to seizure inhibition. It has been reported that PB is effective against focal seizures and generalized tonic–clonic seizures but is not effective against absence seizures (Abou-Khalil, 2019). Besides, the parenteral solution seems to be effective for status epilepticus (Abou-Khalil, 2019). LTG has been proposed to work on the Cav2.1 channel due to its observed ability to control seizures in very few cases that received several ASMs (Byers et al., 2016; Le Roux et al., 2021). However, we could not find any link between LTG and seizure control in this study. Therefore, the efficacy of LTG on a few CACNA1A patients need to be interpreted cautiously because several ASMs were prescribed. The regulation of the calcium ions in the cell is very complex and depends on several channels, transporters, and pumps within organelles and does not depend only on VGCCs.
Cacna1a missense mutation mouse model of absence seizures revealed that the cerebellar neurons are powerful regulators of the pathological oscillations in the thalamocortical system (Kros et al., 2015). Although the Cav 2.1 channel deletion lead to epilepsy and ataxia in human, the Cav 2.1 channel deletion in GABAergic interneurons from the medial ganglionic eminence affects cortical inhibition leading to generalized seizures in Nkx2.1Cre; Cacna1ac/c mice. Whereas, the Cav 2.1 channel deletion in somatostatin-expressing interneurons does not cause seizures, signifying a vital role of parvalbumin-expressing interneurons (Danzer, 2019). The deletion of the Cacna1a in adult mice reproduced the absence epilepsy phenotype by different thalamic bursting mechanisms suggesting that Cav2.1 channels are important for preserving standard thalamocortical oscillations as well as motor regulation in the adult brain (Miao et al., 2020). Tamoxifen prompted adult onset ablation of the Cacna1a in layer VI pyramidal neurons of mice that exhibited spontaneous absence seizure phenotype similar to human (Bomben et al., 2016). There is a downregulation of CACNA1A and GABRD proteins expression in the cortical region of the rat brain during epileptogenesis suggesting that the impaired learning and memory of animals might be linked with the dysregulation of both proteins (Kumar and Sharma, 2020). About 90% knockdown of Cacna1a in Larval Zebrafish (absence seizure model) on the protein level induced epileptiform-like discharges in the optic tectum of larval zebrafish brains, and the incubation with ASMs (VPA, ethosuximide, LTG, and TPM) significantly decreased the number and duration of epileptiform-like discharges (Gawel et al., 2020). The Cav2.1 mutation impairs GABAergic inhibition, resulting in abnormal discharges in the hippocampi of epileptic mice without tg (Nakao et al., 2015).
Sudden unexpected death in epilepsy (SUDEP) is a lethal epilepsy complication. A total of six patients died at a young age (from 3 months – 5 years). CACNA1A gene compound mutations (p.T1439R, and p.A158Tfs) were identified in two siblings, and they led to the death of one patient at the age of 5 years (Reinson et al., 2016). Both children developed daily recurrent seizures from the age of 4 months which were accompanied by very severe hypotonia, hypokinesia, and GDD. The patients had small corpus callosum, diffuse hypomyelination, as well as progressive cerebral, cerebellar, and optic nerve atrophy. At the age of 5 years, both siblings were blind and bedbound with profound GDD. The elder sister developed significant muscular atrophy and rigidity, and she died at 5 years of age. A nonsense p.R932* was identified in four newborn siblings who died between 3 and 6 months of life (Arteche-López et al., 2021). All four newborns presented with severe hypotonia, encephalopathy, seizures, and mild dysmorphic features. A de novo heterozygous pathogenic variant in the CACNA1A gene p.R1667P (mixed GOF and LOF variant) was also found in a patient who presented with very severe congenital ataxia and died at the age of 5 years as described in detail in the previous paragraph (Gauquelin et al., 2020). The p.A951V and p.P2421V variants in CACNA1A have been reported as risk factors for SUDEP (Coll et al., 2016). Transgenic mice unveiled the main role of the cortical neuronal downregulation and brainstem spreading depolarization in the occurrence of the SUDEP (Loonen et al., 2019). The optogenetic stimulation of the colliculi in Cacna1a mice mimicked the SUDEP process (Cain et al., 2022).
The majority of the reported GDD/ID cases originated from China, France and/or Belgium, Israel, the USA, Canada, and Austria. Similar to the geographical distribution of CACNA1A-related epilepsy, there could be a racial disparity in the distribution of CACNA1A-related GDD/ID. However, it is important to consider the possibility that these phenomena could be due to genetic testing bias. Interestingly, GOF, LOF, and mixed GOF and LOF variants of this gene were associated with mild to severe GDD/ID, and more than half of the cases presented with moderate to profound GDD/ID. We have identified some hotspots for the ID/GDD including p.A713T, p.S218L, p.R1664Q, p.V1393M, p.R279C, p.R1352Q, p.R1349Q, p.V1396M, and p.L226W. GOF variants were associated with severe – profound GDD/ID, in contrast, LOF variants showed an association with mild to moderate GDD/ID. The possible explanation for this finding is, GOF variants might induce neuronal death. Cerebellar atrophy was linked with severe – profound GDD/ID. The p.A713T variant located on S6 with a GOF effect showed a correlation with severe to profound GDD/ID. As we know, S6 forms a pore that allows calcium ions influx through the neurons, therefore, GOF variants might have led to calcium ions overload leading to neuronal overexcitability and death. Therefore, there is a probability that excessive calcium ions influx led to neuronal cell death leading to severe GDD/ID.
The calcium ion signaling process is crucial for synaptic plasticity, learning, and memory. In the Cav2.1 model of schizophrenia (Drosophila), memory impairment was accompanied by a reduction in calcium ions transients at the presynaptic terminals suggesting that loss of the Cav2.1 channel function can lead to cognitive and behavioral deficits (Hidalgo et al., 2021). The CAG repetitions in the CACNA1A gene are related to cognitive function in old age plus brain volume changes (Gardiner et al., 2019). Homozygous tottering (tg) mice with a Cacna1a mutation with LOF effect exhibited impaired learning in Pavlovian eyeblink conditioning implying that calcium ion homeostasis in neurons is very crucial for learning and memory formation (de Oude et al., 2021). The Cacna1a mutant mice tottering (tg) exhibited impaired hippocampus-related memory and synaptic plasticity, similar to the human phenotype (Nakao et al., 2022). Neonatal lesions in the cerebellar system of the Cacna1a Rolling mouse Nagoya mice exhibited sensorimotor disturbances similar to children with cerebellar lesions signifying the importance of the cerebellum and its connections in the regulations and development of motor functions (Lalonde and Strazielle, 2015). Moreover, it has been hypothesized that the cerebellum has a role to play in cognition (Shipman and Green, 2019).
Calcium channel blockers (verapamil) showed partial efficacy in the treatment of hemiplegic migraine in individuals with GOF variants, however, there was no comment given regarding its effects on ID (Tantsis et al., 2016). A conditional (forebrain specific) Cav2.1 knock-out mouse exhibited an impairment of synaptic transmission in hippocampal synapses as well as spatial learning and memory deficits (Mallmann et al., 2013). Knock-in mice expressing p.A192G (GOF variant) which is associated with migraine and ID, revealed enhanced hippocampal excitatory transmission and long-term potentiation, however, learning and memory were impaired (Dilekoz et al., 2015). This study demonstrated how unexpected changes in plasticity can affect learning and how heightened neuronal excitability may lead to ID (Dilekoz et al., 2015). A Cav2.1-channel mutant, the heterozygous leaner mouse [tg(la)/+] demonstrated cognitive and motor deficits (Alonso et al., 2008). Ifenprodil (a selective blocker of NMDAR) can block Cav2.1 channels, leading to the reduction of presynaptic excitatory synaptic transmission (Delaney et al., 2012). The blockage of Cav2.1 channels by D1-like dopamine receptors led to low glutamate release into the cholinergic basal forebrain neurons of immature mice (Momiyama and Fukazawa, 2007; Momiyama, 2010).
ASD is also a common phenotype that can be observed in CACNA1A mutations. The commonest causative variants were those with the GOF effect, however, how GOF variants lead to ASD is not clear at present. CACNA1A rs7249246 and rs12609735 have been linked with ASD in the Chinese Han population (Li et al., 2015). CACNA1A single nucleotide polymorphisms are among the top causes of ASD in all VGCCs (Liao and Li, 2020). Homozygous Groggy dams carrying Cacna1a missense mutation have no concern for their babies leading to their deaths, which somehow present like ASD (Kawakami et al., 2020). Auts2 knockout mice have small and malformed cerebella with reduced expression of Cacna1a protein, and impaired motor learning and communication (Yamashiro et al., 2020). Since Cacna1a is a regulator of the synapse development in Purkinje cells, it was suggested that AUTS2 is needed for the typical development of Purkinje cells synapses, therefore, AUTS2 impairment can lead to cerebellar dysfunction linked to ASD (Yamashiro et al., 2020).
We have observed an overlap of the genotypes and phenotypes. Notably, most of the variants are located on S4 and S6; the critical regions that regulate the channel gating and calcium ions influx to the cells (Kessi et al., 2021). This suggests that epilepsy, GDD/ID, and ASD might have common underlying mechanisms.
CACNA1A variants are related to a wide range of neurodevelopmental disorder phenotypes including epilepsy, GDD/ID, and ASD. CACNA1A-related GDD/ID range from mild to profound, and more than half of the patients present with moderate to profound ID/GDD. The p.A713T variant located on S6 with a GOF effect showed a correlation with severe to profound GDD/ID. More than half of CACNA1A-related epilepsy is refractory. The most common epileptic manifestation is status epilepticus which correlates with variants located on S4, S5, and S6. Epilepsy treatment outcome is not influenced by either the electrophysiological changes or mutation types.
There is no prospective study that assessed the potential treatment for epilepsy, therefore, it is difficult to provide a proper conclusion. Future prospective studies are needed to know more about the natural history of CACNA1A-related disorders and potential therapies. The literature review is prone to selection and publication bias. In addition, we might have missed some data due to literature retrieval methodology.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The studies involving human participants were reviewed and approved by the study including all methods adhered to the tenets of the Declaration of Helsinki and received approval from the Institutional Review Board and Research Ethics Committee of Xiangya Hospital, Central South University, Changsha, Hunan. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
MK conceptualized, designed the study, performed literature review, collected and analyzed data, and drafted and revised the manuscript. BC and NP aided in data collection and analysis. LF participated in data collection and revised the manuscript. JP, FH, and FY critically reviewed the manuscript for important intellectual content. All authors reviewed the manuscript and approved the submitted version.
This work was supported by the Hunan Natural Science Foundation (2021JJQNJJ1515) and the Natural Science Foundation of Changsha City (kq2208384).
We thank the participating patients and their families.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1222321/full#supplementary-material
Abou-Khalil, B. W. (2019). Update on antiepileptic drugs 2019. Continuum 25, 508–536. doi: 10.1212/CON.0000000000000715
Alehabib, E., Kokotović, T., Ranji-Burachaloo, S., Tafakhori, A., Ramshe, S. M., Esmaeilizadeh, Z., et al. (2022). Leu226trp Cacna1a variant associated with juvenile myoclonic epilepsy with and without intellectual disability. Clin. Neurol. Neurosurg. 213:107108. doi: 10.1016/j.clineuro.2021.107108
Alonso, I., Marques, J. M., Sousa, N., Sequeiros, J., Olsson, I. A. S., and Silveira, I. (2008). Motor and cognitive deficits in the heterozygous leaner mouse, a Cav2.1 voltage-gated Ca2+ channel mutant. Neurobiol. Aging 29, 1733–1743. doi: 10.1016/j.neurobiolaging.2007.04.005
Angelini, C., Van Gils, J., Bigourdan, A., Jouk, P.-S., Lacombe, D., Menegon, P., et al. (2019). Major intra-familial phenotypic heterogeneity and incomplete penetrance due to a Cacna1a pathogenic variant. Eur. J. Med. Genet. 62:103530. doi: 10.1016/j.ejmg.2018.08.011
Arteche-López, A., Álvarez-Mora, M. I., Sánchez Calvin, M. T., Lezana Rosales, J. M., Palma Milla, C., Gómez Rodríguez, M. J., et al. (2021). Biallelic variants in genes previously associated with dominant inheritance: Cacna1a, ret and Slc20a2. Eur. J. Hum. Genet. 29, 1520–1526. doi: 10.1038/s41431-021-00919-5
Bahamonde, M. I., Serra, S. A., Drechsel, O., Rahman, R., Marcé-Grau, A., Prieto, M., et al. (2015). A single amino acid deletion (Δf1502) in the S6 segment of Cav2.1 domain iii associated with congenital Ataxia increases channel activity and promotes Ca2+ influx. PLoS One 10:E0146035:e0146035. doi: 10.1371/journal.pone.0146035
Balck, A., Hanssen, H., Hellenbroich, Y., Lohmann, K., and Münchau, A. (2017). Adult-onset Ataxia or developmental disorder with seizures: two sides of missense changes in Cacna1a. J. Neurol. 264, 1520–1522. doi: 10.1007/s00415-017-8494-z
Bertholon, P., Chabrier, S., Riant, F., Tournier-Lasserve, E., and Peyron, R. (2009). Episodic Ataxia type 2: unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J. Neurol. Neurosurg. Psychiatry 80, 1289–1292. doi: 10.1136/jnnp.2008.159103
Blumkin, L., Leshinsky-Silver, E., Michelson, M., Zerem, A., Kivity, S., Lev, D., et al. (2015). Paroxysmal tonic upward gaze as a presentation of De-novo mutations in Cacna1a. Eur. J. Paediatr. Neurol. 19, 292–297. doi: 10.1016/j.ejpn.2014.12.018
Blumkin, L., Michelson, M., Leshinsky-Silver, E., Kivity, S., Lev, D., and Lerman-Sagie, T. (2010). Congenital Ataxia, mental retardation, and dyskinesia associated with a novel Cacna1a mutation. J. Child Neurol. 25, 892–897. doi: 10.1177/0883073809351316
Bolte, K. N., Assaf, M., Zach, T., and Peche, S. (2022). Two children with early-onset strokes and intractable epilepsy, both with Cacna1a mutations. Child Neurol. Open 9:2329048X2210949. doi: 10.1177/2329048X221094977
Bomben, V. C., Aiba, I., Qian, J., Mark, M. D., Herlitze, S., and Noebels, J. L. (2016). Isolated P/Q Calcium Channel deletion in layer vi Corticothalamic neurons generates absence epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 36, 405–418. doi: 10.1523/JNEUROSCI.2555-15.2016
Bourinet, E., Soong, T. W., Sutton, K., Slaymaker, S., Mathews, E., Monteil, A., et al. (1999). Splicing of alpha 1a subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat. Neurosci. 2, 407–415. doi: 10.1038/8070
Bürk, K., Kaiser, F. J., Tennstedt, S., Schöls, L., Kreuz, F. R., Wieland, T., et al. (2014). A novel missense mutation in Cacna1a evaluated by in silico protein modeling is associated with non-episodic spinocerebellar Ataxia with slow progression. Eur. J. Med. Genet. 57, 207–211. doi: 10.1016/j.ejmg.2014.01.005
Byers, H. M., Beatty, C. W., Hahn, S. H., and Gospe, S. M. J. (2016). Dramatic response after lamotrigine in a patient with epileptic encephalopathy and a De Novocacna1a variant. Pediatr. Neurol. 60, 79–82. doi: 10.1016/j.pediatrneurol.2016.03.012
Cain, S. M., Bernier, L.-P., Zhang, Y., Yung, A. C., Kass, J., Bohnet, B., et al. (2022). Hyperexcitable superior colliculus and fatal brainstem spreading depolarization in a model of sudden unexpected death in epilepsy. Brain Commun. 4:Fcac006. doi: 10.1093/braincomms/fcac006
Carreño, O., García-Silva, M. T., García-Campos, Ó., Martínez-De Aragón, A., Cormand, B., and Macaya, A. (2011). Acute striatal necrosis in hemiplegic migraine with De novo Cacna1a mutation. Headache 51, 1542–1546. doi: 10.1111/j.1526-4610.2011.02014.x
Coll, M., Allegue, C., Partemi, S., Mates, J., Del Olmo, B., Campuzano, O., et al. (2016). Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int. J. Legal Med. 130, 331–339. doi: 10.1007/s00414-015-1269-0
Damaj, L., Lupien-Meilleur, A., Lortie, A., Riou, É., Ospina, L. H., Gagnon, L., et al. (2015). Cacna1a Haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 23, 1505–1512. doi: 10.1038/ejhg.2015.21
De Oude, N. L., Hoebeek, F. E., Ten Brinke, M. M., De Zeeuw, C. I., and Boele, H.-J. (2021). Pavlovian Eyeblink conditioning is severely impaired in tottering mice. J. Neurophysiol. 125, 398–407. doi: 10.1152/jn.00578.2020
De Vries, B., Stam, A. H., Beker, F., Van Den Maagdenberg, A. M., Vanmolkot, K. R., Laan, L., et al. (2008). Cacna1a mutation linking hemiplegic migraine and alternating hemiplegia of childhood. Cephalalgia 28, 887–891. doi: 10.1111/j.1468-2982.2008.01596.x
Delaney, A. J., Power, J. M., and Sah, P. (2012). Ifenprodil reduces excitatory synaptic transmission by blocking presynaptic P/Q type calcium channels. J. Neurophysiol. 107, 1571–1575. doi: 10.1152/jn.01066.2011
Dilekoz, E., Houben, T., Eikermann-Haerter, K., Balkaya, M., Lenselink, A. M., Whalen, M. J., et al. (2015). Migraine mutations impair hippocampal learning despite enhanced long-term potentiation. J. Neurosci. Off. J. Soc. Neurosci. 35, 3397–3402. doi: 10.1523/JNEUROSCI.2630-14.2015
Du, X., Chen, Y., Zhao, Y., Luo, W., Cen, Z., and Hao, W. (2017). Dramatic response to pyridoxine in a girl with absence epilepsy with Ataxia caused by a De novo Cacna1a mutation. Seizure 45, 189–191. doi: 10.1016/j.seizure.2016.12.020
Ducros, A., Denier, C., Joutel, A., Cecillon, M., Lescoat, C., Vahedi, K., et al. (2001). The clinical Spectrum of familial hemiplegic migraine associated with mutations in a neuronal Calcium Channel. N. Engl. J. Med. 345, 17–24. doi: 10.1056/NEJM200107053450103
Epi4K Consortium (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Epi4K Consortium (2016). De novo mutations in Slc1a2 and Cacna1a are important causes of epileptic encephalopathies. Am. J. Hum. Genet. 99, 287–298. doi: 10.1016/j.ajhg.2016.06.003
Epperson, M. V., Haws, M. E., Standridge, S. M., and Gilbert, D. L. (2018). An atypical Rett syndrome phenotype due to a novel missense mutation in Cacna1a. J. Child Neurol. 33, 286–289. doi: 10.1177/0883073818754987
Fitzsimons, R. B., and Wolfenden, W. H. (1985). Migraine coma. Meningitic migraine with cerebral Oedema associated with a new form of autosomal dominant cerebellar Ataxia. Brain J. Neurol. 108, 555–577. doi: 10.1093/brain/108.3.555-a
Freilinger, T., Ackl, N., Ebert, A., Schmidt, C., Rautenstrauss, B., Dichgans, M., et al. (2011). A novel mutation in Cacna1a associated with hemiplegic migraine, cerebellar dysfunction and late-onset cognitive decline. J. Neurol. Sci. 300, 160–163. doi: 10.1016/j.jns.2010.09.032
Gajam, S., Peterson, R. R., Mathew, A. A., and Thomas, A. (2022). Sporadic hemiplegic migraine with Cacna1a mutation masquerading as acute meningoencephalitis. Ann. Indian Acad. Neurol. 25, 528–529. doi: 10.4103/aian.aian_908_21
Gandini, M. A., Souza, I. A., Ferron, L., Innes, A. M., and Zamponi, G. W. (2021). The De novo Cacna1a pathogenic variant Y1384c associated with hemiplegic migraine, early onset cerebellar atrophy and developmental delay leads to a loss of Cav2.1 channel function. Mol. Brain 14:27. doi: 10.1186/s13041-021-00745-2
García Segarra, N., Gautschi, I., Mittaz-Crettol, L., Kallay Zetchi, C., Al-Qusairi, L., Van Bemmelen, M. X., et al. (2014). Congenital Ataxia and hemiplegic migraine with cerebral edema associated with a novel gain of function mutation in the Calcium Channel Cacna1a. J. Neurol. Sci. 342, 69–78. doi: 10.1016/j.jns.2014.04.027
Gardiner, S. L., Trompet, S., Sabayan, B., Boogaard, M. W., Jukema, J. W., Slagboom, P. E., et al. (2019). Repeat variations in Polyglutamine disease-associated genes and cognitive function in old age. Neurobiol. Aging 84, 236.E17–236.E28. doi: 10.1016/j.neurobiolaging.2019.08.002
Gauquelin, L., Hawkins, C., Tam, E. W. Y., Miller, S. P., and Yoon, G. (2020). Pearls & oy-Sters: fatal brain edema is a rare complication of severe Cacna1a-related disorder. Neurology 94, 631–634. doi: 10.1212/WNL.0000000000009223
Gawel, K., Turski, W. A., Van Der Ent, W., Mathai, B. J., Kirstein-Smardzewska, K. J., Simonsen, A., et al. (2020). Phenotypic characterization of larval zebrafish (Danio Rerio) with partial knockdown of the Cacna1a gene. Mol. Neurobiol. 57, 1904–1916. doi: 10.1007/s12035-019-01860-x
Gillard, M., Chatelain, P., and Fuks, B. (2006). Binding characteristics of Levetiracetam to synaptic vesicle protein 2a (Sv2a) in human brain and in Cho cells expressing the human recombinant protein. Eur. J. Pharmacol. 536, 102–108. doi: 10.1016/j.ejphar.2006.02.022
Grosso, B. J., Kramer, A. A., Tyagi, S., Bennett, D. F., Tifft, C. J., D'souza, P., et al. (2022). Complex effects on ca(V)2.1 channel gating caused by a Cacna1a variant associated with a severe neurodevelopmental disorder. Sci. Rep. 12:9186. doi: 10.1038/s41598-022-12789-y
Gudenkauf, F. J., Azamian, M. S., Hunter, J. V., Nayak, A., and Lalani, S. R. (2020). A novel Cacna1a variant in a child with early stroke and intractable epilepsy. Mol Genet Genomic Med 8:E1383. doi: 10.1002/mgg3.1383
Guerin, A. A., Feigenbaum, A., Donner, E. J., and Yoon, G. (2008). Stepwise developmental regression associated with novel Cacna1a mutation. Pediatr. Neurol. 39, 363–364. doi: 10.1016/j.pediatrneurol.2008.07.030
Gur-Hartman, T., Berkowitz, O., Yosovich, K., Roubertie, A., Zanni, G., Macaya, A., et al. (2021). Clinical phenotypes of infantile onset Cacna1a-related disorder. Eur. J. Paediatr. Neurol. 30, 144–154. doi: 10.1016/j.ejpn.2020.10.004
Hayashida, T., Saito, Y., Ishii, A., Yamada, H., Itakura, A., Minato, T., et al. (2018). Cacna1a-related early-onset encephalopathy with myoclonic epilepsy: a case report. Brain Dev. 40, 130–133. doi: 10.1016/j.braindev.2017.08.006
Hidalgo, S., Campusano, J. M., and Hodge, J. J. L. (2021). The Drosophila Ortholog of the schizophrenia-associated Cacna1a and Cacna1b voltage-gated calcium channels regulate memory, sleep and circadian rhythms. Neurobiol. Dis. 155:105394. doi: 10.1016/j.nbd.2021.105394
Ho, C. Y., Love, H. L., Sokol, D. K., and Walsh, L. E. (2021). Longitudinal Mri brain findings in the R1349q pathogenic variant of Cacna1a. Radiol Case Rep 16, 1276–1279. doi: 10.1016/j.radcr.2021.02.052
Hommersom, M. P., Van Prooije, T. H., Pennings, M., Schouten, M. I., Van Bokhoven, H., Kamsteeg, E. J., et al. (2022). The complexities of Cacna1a in clinical neurogenetics. J. Neurol. 269, 3094–3108. doi: 10.1007/s00415-021-10897-9
Humbertclaude, V., Krams, B., Nogue, E., Nagot, N., Annequin, D., Tourniaire, B., et al. (2018). Benign paroxysmal torticollis, benign paroxysmal Vertigo, and benign tonic upward gaze are not benign disorders. Dev. Med. Child Neurol. 60, 1256–1263. doi: 10.1111/dmcn.13935
Humbertclaude, V., Riant, F., Krams, B., Zimmermann, V., Nagot, N., Annequin, D., et al. (2020). Cognitive impairment in children with Cacna1a mutations. Dev. Med. Child Neurol. 62, 330–337. doi: 10.1111/dmcn.14261
Imbrici, P., Jaffe, S. L., Eunson, L. H., Davies, N. P., Herd, C., Robertson, R., et al. (2004). Dysfunction of the brain Calcium Channel Cav2.1 in absence epilepsy and episodic Ataxia. Brain 127, 2682–2692. doi: 10.1093/brain/awh301
Indelicato, E., and Boesch, S. (2023). Cacna1a-related channelopathies: clinical manifestations and treatment options. Handb. Exp. Pharmacol. doi: 10.1007/164_2022_625
Indelicato, E., Nachbauer, W., Karner, E., Eigentler, A., Wagner, M., Unterberger, I., et al. (2019). The neuropsychiatric phenotype in Cacna1a mutations: a retrospective single center study and review of the literature. Eur. J. Neurol. 26:66-E7. doi: 10.1111/ene.13765
Jiang, X., Raju, P. K., D'avanzo, N., Lachance, M., Pepin, J., Dubeau, F., et al. (2019). Both gain-of-function and loss-of-function De novo Cacna1a mutations cause severe developmental epileptic encephalopathies in the Spectrum of Lennox-Gastaut syndrome. Epilepsia 60, 1881–1894. doi: 10.1111/epi.16316
Jung, J., Testard, H., Tournier-Lasserve, E., Riant, F., Vallet, A.-E., Berroir, S., et al. (2010). Phenotypic variability of episodic Ataxia type 2 mutations: a family study. Eur. Neurol. 64, 114–116. doi: 10.1159/000315145
Kashimada, A., Hasegawa, S., Nomura, T., Shiraku, H., Moriyama, K., Suzuki, T., et al. (2019). Genetic analysis of undiagnosed Ataxia-telangiectasia-like disorders. Brain Dev. 41, 150–157. doi: 10.1016/j.braindev.2018.09.007
Kawakami, N., Kobayashi, K., Nishimura, A., and Ohmori, I. (2020). Poor mother-offspring relationships in rats with Cacna1a mutation. Exp. Anim. 69, 153–160. doi: 10.1538/expanim.19-0086
Kessi, M., Chen, B., Peng, J., Yan, F., Yang, L., and Yin, F. (2021). Calcium Channelopathies and intellectual disability: a systematic review. Orphanet J. Rare Dis. 16:219. doi: 10.1186/s13023-021-01850-0
Kessi, M., Chen, B., Shan, L. D., Wang, Y., Yang, L., Yin, F., et al. (2023). Genotype-phenotype correlations of Stxbp1 pathogenic variants and the treatment choices for Stxbp1-related disorders in China. BMC Med. Genet. 16:46. doi: 10.1186/s12920-023-01474-2
Kessi, M., Xiong, J., Wu, L., Yang, L., He, F., Chen, C., et al. (2018). Rare copy number variations and predictors in children with intellectual disability and epilepsy. Front. Neurol. 9:947. doi: 10.3389/fneur.2018.00947
Klockgether, T. (2008). The clinical diagnosis of autosomal dominant spinocerebellar ataxias. Cerebellum 7, 101–105. doi: 10.1007/s12311-008-0023-2
Kors, E. E., Melberg, A., Vanmolkot, K. R. J., Kumlien, E., Haan, J., Raininko, R., et al. (2004). Childhood epilepsy, familial hemiplegic migraine, cerebellar Ataxia, and a new Cacna1a mutation. Neurology 63, 1136–1137. doi: 10.1212/01.WNL.0000138571.48593.FC
Kors, E. E., Terwindt, G. M., Vermeulen, F. L., Fitzsimons, R. B., Jardine, P. E., Heywood, P., et al. (2001). Delayed cerebral edema and fatal coma after minor head trauma: role of the Cacna1a Calcium Channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 49, 753–760. doi: 10.1002/ana.1031
Kothur, K., Holman, K., Farnsworth, E., Ho, G., Lorentzos, M., Troedson, C., et al. (2018). Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 59, 132–140. doi: 10.1016/j.seizure.2018.05.005
Kramer, P., and Bressan, P. (2018). Our (Mother's) mitochondria and our mind. Perspect. Psychol. Sci. 13, 88–100. doi: 10.1177/1745691617718356
Kros, L., Eelkman Rooda, O. H. J., Spanke, J. K., Alva, P., Van Dongen, M. N., Karapatis, A., et al. (2015). Cerebellar output controls generalized spike-and-wave discharge occurrence. Ann. Neurol. 77, 1027–1049. doi: 10.1002/ana.24399
Krygier, M., Zawadzka, M., Sawicka, A., and Mazurkiewicz-Bełdzińska, M. (2022). Reflex seizures in rare monogenic epilepsies. Seizure 97, 32–34. doi: 10.1016/j.seizure.2022.03.004
Kumar, P., and Sharma, D. (2020). Ameliorative effect of curcumin on altered expression of Cacna1a and Gabrd in the pathogenesis of Fecl(3)-induced epilepsy. Mol. Biol. Rep. 47, 5699–5710. doi: 10.1007/s11033-020-05538-9
Lalonde, R., and Strazielle, C. (2015). Behavioral effects of neonatal lesions on the cerebellar system. Int. J. Dev. Neurosci. 43, 58–65. doi: 10.1016/j.ijdevneu.2015.04.007
Le Roux, M., Barth, M., Gueden, S., Desbordes De Cepoy, P., Aeby, A., Vilain, C., et al. (2021). Cacna1a-associated epilepsy: electroclinical findings and treatment response on seizures in 18 patients. Eur. J. Paediatr. Neurol. 33, 75–85. doi: 10.1016/j.ejpn.2021.05.010
Lee, C. G., Lee, J., and Lee, M. (2018). Multi-gene panel testing in Korean patients with common genetic generalized epilepsy syndromes. PLoS One 13:E0199321. doi: 10.1371/journal.pone.0199321
Li, X.-L., Li, Z.-J., Liang, X.-Y., Liu, D.-T., Jiang, M., Gao, L.-D., et al. (2022). Cacna1a mutations associated with epilepsies and their molecular sub-regional implications. Front. Mol. Neurosci. 15:860662. doi: 10.3389/fnmol.2022.860662
Li, J., You, Y., Yue, W., Jia, M., Yu, H., Lu, T., et al. (2015). Genetic evidence for possible involvement of the Calcium Channel gene Cacna1a in autism pathogenesis in Chinese Han population. PLoS One 10:E0142887. doi: 10.1371/journal.pone.0142887
Liao, X., and Li, Y. (2020). Genetic associations between voltage-gated calcium channels and autism Spectrum disorder: a systematic review. Mol. Brain 13:96. doi: 10.1186/s13041-020-00634-0
Lipman, A. R., Fan, X., Shen, Y., and Chung, W. K. (2022). Clinical and genetic characterization of Cacna1a-related disease. Clin. Genet. 102, 288–295. doi: 10.1111/cge.14180
Loonen, I. C. M., Jansen, N. A., Cain, S. M., Schenke, M., Voskuyl, R. A., Yung, A. C., et al. (2019). Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218l mice. Brain J. Neurol. 142, 412–425. doi: 10.1093/brain/awy325
Luo, X., Rosenfeld, J. A., Yamamoto, S., Harel, T., Zuo, Z., Hall, M., et al. (2017). Clinically severe Cacna1a alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 13:E1006905. doi: 10.1371/journal.pgen.1006905
Lv, Y., Wang, Z., Liu, C., and Cui, L. (2017). Identification of a novel Cacna1a mutation in a Chinese family with autosomal recessive progressive myoclonic epilepsy. Neuropsychiatr. Dis. Treat. 13, 2631–2636. doi: 10.2147/NDT.S145774
Lynch, B. A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S. M., Matagne, A., et al. (2004). The synaptic vesicle protein Sv2a is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. U. S. A. 101, 9861–9866. doi: 10.1073/pnas.0308208101
Mallmann, R. T., Elgueta, C., Sleman, F., Castonguay, J., Wilmes, T., Van Den Maagdenberg, A., et al. (2013). Ablation of CaV2.1 voltage-gated Ca2+ channels in mouse forebrain generates multiple cognitive impairments. PLoS One 8:E78598. doi: 10.1371/journal.pone.0078598
Mantuano, E., Romano, S., Veneziano, L., Gellera, C., Castellotti, B., Caimi, S., et al. (2010). Identification of novel and recurrent Cacna1a gene mutations in fifteen patients with episodic Ataxia type 2. J. Neurol. Sci. 291, 30–36. doi: 10.1016/j.jns.2010.01.010
Martínez-Monseny, A. F., Edo, A., Casas-Alba, D., Izquierdo-Serra, M., Bolasell, M., Conejo, D., et al. (2021). Cacna1a mutations causing early onset Ataxia: profiling clinical, dysmorphic and structural-functional findings. Int. J. Mol. Sci. 22:5180. doi: 10.3390/ijms22105180
Mellone, S., Puricelli, C., Vurchio, D., Ronzani, S., Favini, S., Maruzzi, A., et al. (2022). The usefulness of a targeted next generation sequencing gene panel in providing molecular diagnosis to patients with a broad Spectrum of neurodevelopmental disorders. Front. Genet. 13:875182. doi: 10.3389/fgene.2022.875182
Meloche, J., Brunet, V., Gagnon, P.-A., Lavoie, M.-E., Bouchard, J.-B., Nadaf, J., et al. (2020). Exome sequencing study of partial agenesis of the Corpus callosum in men with developmental delay, epilepsy, and microcephaly. Mol. Genet. Genomic Med. 8:e992. doi: 10.1002/mgg3.992
Miao, Q.-L., Herlitze, S., Mark, M. D., and Noebels, J. L. (2020). Adult loss of Cacna1a in mice recapitulates childhood absence epilepsy by distinct thalamic bursting mechanisms. Brain J. Neurol. 143, 161–174. doi: 10.1093/brain/awz365
Mitta, N., Menon, R. N., Mctague, A., Radhakrishnan, A., Sundaram, S., Cherian, A., et al. (2020). Genotype-phenotype correlates of infantile-onset developmental & epileptic encephalopathy syndromes in South India: a single centre experience. Epilepsy Res. 166:106398. doi: 10.1016/j.eplepsyres.2020.106398
Momiyama, T. (2010). Developmental increase in D1-like dopamine receptor-mediated inhibition of glutamatergic transmission through P/Q-Type Channel regulation in the basal forebrain of rats. Eur. J. Neurosci. 32, 579–590. doi: 10.1111/j.1460-9568.2010.07306.x
Momiyama, T., and Fukazawa, Y. (2007). D1-like dopamine receptors selectively block P/Q-type calcium channels to reduce glutamate release onto cholinergic basal forebrain neurones of immature rats. J. Physiol. 580, 103–117. doi: 10.1113/jphysiol.2006.125724
Nagarkatti, N., Deshpande, L. S., and Delorenzo, R. J. (2008). Levetiracetam inhibits both ryanodine and Ip3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci. Lett. 436, 289–293. doi: 10.1016/j.neulet.2008.02.076
Naik, S., Pohl, K., Malik, M., Siddiqui, A., and Josifova, D. (2011). Early-onset cerebellar atrophy associated with mutation in the Cacna1a gene. Pediatr. Neurol. 45, 328–330. doi: 10.1016/j.pediatrneurol.2011.08.002
Nakao, A., Hayashida, K., Ogura, H., Mori, Y., and Imoto, K. (2022). Hippocampus-related cognitive disorders develop in the absence of epilepsy and Ataxia in the heterozygous Cacna1a mutant mice tottering. Channels 16, 113–126. doi: 10.1080/19336950.2022.2072449
Nakao, A., Miki, T., Shimono, K., Oka, H., Numata, T., Kiyonaka, S., et al. (2015). Compromised maturation of Gabaergic inhibition underlies abnormal network activity in the Hippocampus of epileptic Ca2+ channel mutant mice, tottering. Pflugers Arch. 467, 737–752. doi: 10.1007/s00424-014-1555-6
Niu, Y., Gong, P., Jiao, X., Xu, Z., Zhang, Y., and Yang, Z. (2022). Genetic and phenotypic Spectrum of Chinese patients with epilepsy and photosensitivity. Front. Neurol. 13:907228. doi: 10.3389/fneur.2022.907228
Niu, X., Yang, Y., Chen, Y., Cheng, M., Liu, M., Ding, C., et al. (2022). Genotype-phenotype correlation of Cacna1a variants in children with epilepsy. Dev. Med. Child Neurol. 64, 105–111. doi: 10.1111/dmcn.14985
Ohba, C., Osaka, H., Iai, M., Yamashita, S., Suzuki, Y., Aida, N., et al. (2013). Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 14, 225–232. doi: 10.1007/s10048-013-0375-8
Page, M. J., Mckenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). The Prisma 2020 statement: an updated guideline for reporting systematic reviews. BMJ 372:N71. doi: 10.1136/bmj.n71
Rajakulendran, S., Graves, T. D., Labrum, R. W., Kotzadimitriou, D., Eunson, L., Davis, M. B., et al. (2010). Genetic and functional characterisation of the P/Q Calcium Channel in episodic Ataxia with epilepsy. J. Physiol. 588, 1905–1913. doi: 10.1113/jphysiol.2009.186437
Reinson, K., Õiglane-Shlik, E., Talvik, I., Vaher, U., Õunapuu, A., Ennok, M., et al. (2016). Biallelic Cacna1a mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am. J. Med. Genet. A 170, 2173–2176. doi: 10.1002/ajmg.a.37678
Riant, F., Ducros, A., Ploton, C., Barbance, C., Depienne, C., and Tournier-Lasserve, E. (2010). De novo mutations in Atp1a2 and Cacna1a are frequent in early-onset sporadic hemiplegic migraine. Neurology 75, 967–972. doi: 10.1212/WNL.0b013e3181f25e8f
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American college of medical genetics and genomics (Acmg) and the clinical genome resource (Clingen). Genet. Med. 22, 245–257. doi: 10.1038/s41436-019-0686-8
Saathoff, Y., Biskup, S., Funke, C., and Roth, C. (2021). New nonsense variant C.2983g>T; P.Glu995* in the Cacna1a gene causes progressive autosomal dominant Ataxia. J. Mov. Disord. 14, 70–74. doi: 10.14802/jmd.20082
Shevell, M. (2008). Global developmental delay and mental retardation or intellectual disability: conceptualization, evaluation, and etiology. Pediatr. Clin. N. Am. 55:Xi, 1071–1084. doi: 10.1016/j.pcl.2008.07.010
Shipman, M. L., and Green, J. T. (2019). Cerebellum and cognition: does the rodent cerebellum participate in cognitive functions? Neurobiol. Learn. Mem. 170:106996. doi: 10.1016/j.nlm.2019.02.006
Simms, B. A., and Zamponi, G. W. (2014). Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82, 24–45. doi: 10.1016/j.neuron.2014.03.016
Stanika, R. I., Villanueva, I., Kazanina, G., Andrews, S. B., and Pivovarova, N. B. (2012). Comparative impact of voltage-gated calcium channels and Nmda receptors on mitochondria-mediated neuronal injury. J. Neurosci. 32, 6642–6650. doi: 10.1523/JNEUROSCI.6008-11.2012
Stendel, C., D'adamo, M. C., Wiessner, M., Dusl, M., Cenciarini, M., Belia, S., et al. (2020). Association of a novel splice site mutation in P/Q-type calcium channels with childhood epilepsy and late-onset slowly progressive non-episodic cerebellar Ataxia. Int. J. Mol. Sci. 21:3810. doi: 10.3390/ijms21113810
Strupp, M., Zwergal, A., and Brandt, T. (2007). Episodic ataxia type 2. Neurotherapeutics 4, 267–273. doi: 10.1016/j.nurt.2007.01.014
Stubberud, A., O'connor, E., Tronvik, E., Houlden, H., and Matharu, M. (2021). R1352q Cacna1a variant in a patient with sporadic hemiplegic migraine, ataxia, seizures and cerebral oedema: a case report. Case Rep. Neurol. 13, 123–130. doi: 10.1159/000512275
Tantsis, E. M., Gill, D., Griffiths, L., Gupta, S., Lawson, J., Maksemous, N., et al. (2016). Eye movement disorders are an early manifestation of Cacna1a mutations in children. Dev. Med. Child Neurol. 58, 639–644. doi: 10.1111/dmcn.13033
Tyagi, S., Bendrick, T. R., Filipova, D., Papadopoulos, S., and Bannister, R. A. (2019). A mutation in ca(V)2.1 linked to a severe neurodevelopmental disorder Impairs Channel gating. J. Gen. Physiol. 151, 850–859. doi: 10.1085/jgp.201812237
Vahedi, K., Denier, C., Ducros, A., Bousson, V., Levy, C., Chabriat, H., et al. (2000). Cacna1a gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology 55, 1040–1042. doi: 10.1212/WNL.55.7.1040
Verriello, L., Pauletto, G., Nilo, A., Lonigro, I., Betto, E., Valente, M., et al. (2021). Epilepsy and episodic ataxia type 2: family study and review of the literature. J. Neurol. 268, 4296–4302. doi: 10.1007/s00415-021-10555-0
Vila-Pueyo, M., Gené, G. G., Flotats-Bastardes, M., Elorza, X., Sintas, C., Valverde, M. A., et al. (2014). A loss-of-function Cacna1a mutation causing benign paroxysmal torticollis of infancy. Eur. J. Paediatr. Neurol. 18, 430–433. doi: 10.1016/j.ejpn.2013.12.011
Wada, T., Kobayashi, N., Takahashi, Y., Aoki, T., Watanabe, T., and Saitoh, S. (2002). Wide clinical variability in a family with a Cacna1a T666m mutation: hemiplegic migraine, coma, and progressive Ataxia. Pediatr. Neurol. 26, 47–50. doi: 10.1016/S0887-8994(01)00371-X
Weyhrauch, D. L., Ye, D., Boczek, N. J., Tester, D. J., Gavrilova, R. H., Patterson, M. C., et al. (2016). Whole exome sequencing and heterologous cellular electrophysiology studies elucidate a novel loss-of-function mutation in the Cacna1a-encoded neuronal P/Q-type Calcium Channel in a child with congenital Hypotonia and developmental delay. Pediatr. Neurol. 55, 46–51. doi: 10.1016/j.pediatrneurol.2015.10.014
Wong-Spracklen, V. M. Y., Kolesnik, A., Eck, J., Sabanathan, S., Spasic-Boskovic, O., Maw, A., et al. (2022). Biallelic Cacna1a variants: review of literature and report of a child with drug-resistant epilepsy and developmental delay. Am. J. Med. Genet. A 188, 3306–3311. doi: 10.1002/ajmg.a.62960
Yamamoto, T., Imaizumi, T., Yamamoto-Shimojima, K., Lu, Y., Yanagishita, T., Shimada, S., et al. (2019). Genomic backgrounds of Japanese patients with undiagnosed neurodevelopmental disorders. Brain Dev. 41, 776–782. doi: 10.1016/j.braindev.2019.05.007
Yamashiro, K., Hori, K., Lai, E. S. K., Aoki, R., Shimaoka, K., Arimura, N., et al. (2020). Auts2 governs cerebellar development, Purkinje cell maturation, motor function and social communication. Iscience 23:101820. doi: 10.1016/j.isci.2020.101820
Yamazaki, S., Ikeno, K., Abe, T., Tohyama, J., and Adachi, Y. (2011). Hemiconvulsion-hemiplegia-epilepsy syndrome associated with Cacna1a S218l mutation. Pediatr. Neurol. 45, 193–196. doi: 10.1016/j.pediatrneurol.2011.04.010
Yuan, X., Zheng, Y., Gao, F., Sun, W., Wang, Z., and Zhao, G. (2022). Case report: a novel Cacna1a mutation caused Flunarizine-responsive type 2 episodic Ataxia and hemiplegic migraine with abnormal Mri of cerebral white matter. Front. Neurol. 13:899813. doi: 10.3389/fneur.2022.899813
Zangaladze, A., Asadi-Pooya, A. A., Ashkenazi, A., and Sperling, M. R. (2010). Sporadic hemiplegic migraine and epilepsy associated with Cacna1a gene mutation. Epilepsy Behav. 17, 293–295. doi: 10.1016/j.yebeh.2009.12.017
Zhang, L.-P., Jia, Y., and Wang, Y.-P. (2021). Identification of two De novo variants of Cacna1a in pediatric Chinese patients with paroxysmal tonic upgaze. Front. Pediatr. 9:722105. doi: 10.3389/fped.2021.722105
Keywords: CACNA1A , genotype–phenotype correlations, global developmental delay, intellectual disability, epilepsy, autism spectrum disorder
Citation: Kessi M, Chen B, Pang N, Yang L, Peng J, He F and Yin F (2023) The genotype–phenotype correlations of the CACNA1A-related neurodevelopmental disorders: a small case series and literature reviews. Front. Mol. Neurosci. 16:1222321. doi: 10.3389/fnmol.2023.1222321
Edited by:
Daniel Souza Monteiro De Araújo, University of Florence, ItalyReviewed by:
Alessandro Capuano, Azienda Sanitaria Locale di Viterbo, ItalyCopyright © 2023 Kessi, Chen, Pang, Yang, Peng, He and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Peng, cGVuZ2ppbmc0MzQ2QDE2My5jb20=; Fang He, YnViYmx5X2hvQDE2My5jb20=; Fei Yin, eWYyMzIzQGhvdG1haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.