 Honglu Yu
Honglu Yu Min Xiong1
Min Xiong1 Zhentao Zhang
Zhentao Zhang- 1Department of Neurology, Renmin Hospital of Wuhan University, Wuhan, China
- 2TaiKang Center for Life and Medical Sciences, Wuhan University, Wuhan, China
Neurodegenerative diseases (NDDs) pose an increasingly prevalent threat to the well-being and survival of elderly individuals worldwide. NDDs include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and so on. They are characterized by progressive loss or dysfunction of neurons in the central or peripheral nervous system and share several cellular and molecular mechanisms, including protein aggregation, mitochondrial dysfunction, gene mutations, and chronic neuroinflammation. Glycogen synthase kinase-3 beta (GSK-3β) is a serine/threonine kinase that is believed to play a pivotal role in the pathogenesis of NDDs. Here we summarize the structure and physiological functions of GSK3β and explore its involvement in NDDs. We also discussed its potential as a therapeutic target.
1. Introduction
Neurodegenerative diseases (NDDs) are characterized by the progressive loss of neurons in the nervous system. These diseases typically cause damage to the brain and spinal cord, which in turn affects various physiological functions such as cognition, movement, perception, the autonomic nervous system, and even breathing and circulation (Dugger and Dickson, 2017). NDDs include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and Amyotrophic lateral sclerosis (ALS), among others (Boland et al., 2018). Symptoms of these conditions worsen over time. Treatment focuses on improving patients’ quality of life by alleviating symptoms, but there is currently no effective cure. Increasing evidence suggests that changes occur in the activity and levels of GSK-3β in NDDs, and the GSK-3β signaling pathway is now recognized as a key signaling pathway promoting neurodegeneration. Inhibition of GSK-3β has emerged as a promising therapeutic approach for NDDs (Rippin and Eldar-Finkelman, 2021).
2. Structure and biological function of GSK3β
Glycogen synthase kinase-3 (GSK-3) was initially discovered for its ability to phosphorylate glycogen synthase and regulate glucose metabolism in response to insulin. Subsequent studies have shown that it is a widely expressed serine/threonine kinase that can phosphorylate over 100 protein substrates and is located at the intersection of many signaling pathways (Beurel et al., 2015). Encoded by two different genes located on chromosomes 19 and 3, GSK-3 has two unique isoforms: GSK-3α (51 kDa) and GSK-3β (47 kDa). These isoforms share a striking 97% amino acid sequence identity within the catalytic domain and 84% overall amino acid sequence similarity, suggesting potential common biological functions. For example, they regulate cell signaling, participate in biological metabolism, cell growth, proliferation, and differentiation, and are critical regulatory factors in many neurodevelopmental processes (Marosi et al., 2022). GSK-3β exhibits high levels of expression in the central nervous system (CNS; Yao et al., 2002). The kinase activity of GSK-3β is regulated by phosphorylation. Within the cell, serine kinases such as protein kinase B (PKB/AKT) and AMP-activated protein kinase (AMPK) target the 9th serine residue (Ser9) of GSK3β, resulting in its phosphorylation (pGSK-3β-Ser9) and subsequent inactivation. In contrast, tyrosine kinases can phosphorylate the 216th tyrosine residue (Tyr216) of GSK-3β (resulting in pGSK-3β-Tyr216), leading to a fivefold increase in its enzymatic activity. GSK-3β interacts with numerous molecules intricately associated with NDDs, including hippocampal cell proliferation, neuronal development and regeneration, cell cycle regulation, and neuronal polarization (Pardo et al., 2016; Demuro et al., 2021; see Figure 1).
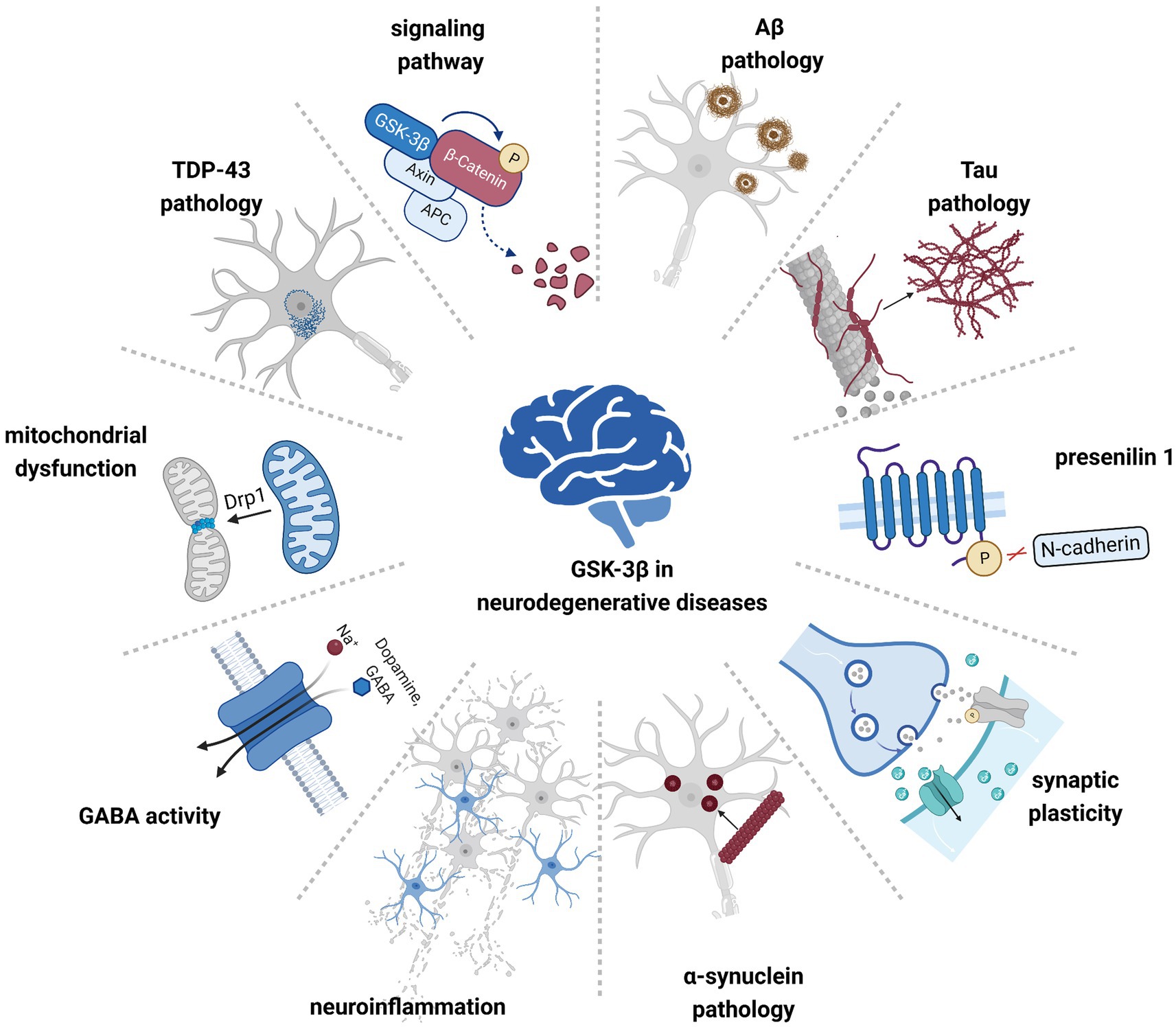
Figure 1. GSK-3β is involved in the pathogenesis of multiple neurodegenerative diseases. Hyperactivated GSK-3β contributes to pathological protein aggregation, neuroinflammation, and mitochondrial dysfunction, affecting cellular signaling pathways and neuronal synaptic plasticity.
3. GSK-3β in AD
AD is the most common neurodegenerative disease, encompassing 60–80% of global dementia cases (Gauthier et al., 2022). Cognitive and non-cognitive symptoms are prominent clinical features. Early stages manifest with challenges in recalling recent discussions, names, events, emotional responses, or even depressive symptoms, which evolved into communication impairments, confusion, compromised decision-making, and behavioral alterations. As the disease advances, walking difficulties, speech impairments, and swallowing problems become prevalent (Breijyeh and Karaman, 2020). Regarding genetic characteristics, Alzheimer’s disease (AD) can be categorized into familial and sporadic forms. Familial AD is associated with mutations in three genes: APP, PSEN1, and PSEN2. Sporadic AD is the most prevalent form (Uddin et al., 2021). It has prompted various hypotheses elucidating its onset, encompassing the cholinergic, amyloid, Tau protein proliferation, mitochondrial cascade, inflammation, and neurovascular hypotheses. The pathological features include the formation of senile plaques composed of amyloid-β (Aβ) and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated Tau (Penke et al., 2019; Kent et al., 2020). This aggregation of Aβ and Tau detrimentally affects synaptic plasticity and triggers neuronal cell demise (Scheltens et al., 2021). Elevated GSK-3β activity has been observed in the brains of AD patients and various AD mouse models. Upregulation of GSK-3β has been shown to instigate AD pathology, cognitive decline, and glial cell proliferation (Albeely et al., 2022; Chauhan et al., 2022).
3.1. GSK-3β regulates Aβ pathology
In the context of Alzheimer’s disease (AD), the accumulation of Aβ protein precedes the formation of paired helical filaments (PHFs), which constitute the neurofibrillary tangles (NFTs). The principal events underlying AD involve the aberrant metabolism of amyloid precursor protein (APP) and subsequent Aβ deposition. The processing of APP occurs through either the non-amyloidogenic or the amyloidogenic pathway. Sequential cleavage of APP by α-secretase and γ-secretase generates three fragments: the secreted C-terminal fragment (sAPPα), p3, and the APP intracellular domain (AICD) (Aguzzi and O'Connor, 2010). The alternative pathway includes cleavage by β-secretase, also known as β-site APP cleaving enzyme (BACE-1), followed by cleavage by γ-secretase, which produces Aβ (Dimitrov et al., 2013). Normally, APP cleavage predominantly follows the non-amyloidogenic pathway. Cleavage of APP via the amyloidogenic pathway leads to the production of Aβ. β-Secretase (BACE1) is the rate-limiting enzyme that generates Aβ in the amyloidogenic pathway (Bahn et al., 2019). GSK-3β intensifies the activity of β-secretase (BACE1) and mediates the toxicity of Aβ aggregates. Inhibition of GSK-3β diminishes BACE1-mediated APP cleavage, thus reducing Aβ generation (Ly et al., 2013). Aβ aggregates activate microglia and trigger inflammation, leading to neurodegeneration and cell death (Wang et al., 2023).
3.2. GSK-3β regulates tau pathology
Tau is predominantly expressed in the axons of neurons and functions as a microtubule-associated protein. It possesses 45 phosphorylation sites and 79 serine and threonine residues available for phosphorylation (Duan et al., 2017). Abnormal phosphorylation of Tau protein leads to the formation of aggregates known as neurofibrillary tangles (NFTs; Sayas and Ávila, 2021). One of the primary kinases responsible for Tau phosphorylation is GSK-3β, which phosphorylates numerous serine or threonine residues on Tau. The aggregation of hyperphosphorylated Tau represents an additional pathological hallmark of AD (Muralidar et al., 2020). This aggregation of Tau is directly linked to the upregulation of GSK-3β, further intensifying Tau pathology in murine brains. Conversely, deletion of GSK-3β attenuates Tau phosphorylation, hippocampal degeneration, and learning defects in mouse models of AD. Inhibition of GSK-3β reduces Tau phosphorylation and neurodegeneration (Hurtado et al., 2012; Sayas and Ávila, 2021).
3.3. GSK-3β regulates the function of presenilin 1
Presenilin 1 (PS1), a product of the PSEN1 gene, has been verified as an important causative factor for familial AD (FAD). PS1 is a transmembrane protein with nine domains linked by hydrophilic loops in the extracellular area or cytosol. It plays a role in cleaving APP and also affects other processes like Notch signaling, β-cadherin processing, and calcium metabolism (Giau et al., 2019). PS1 functions in the C-terminal transmembrane region of APP, directing the generation of amyloid-β peptide (Aβ42) through γ-secretase after α- and β-secretase cleavage (Bagaria et al., 2022). GSK-3β phosphorylates PS1 at serine residues (Ser353 and Ser357), restraining APP cleavage and Aβ production while modulating the Aβ 42/40 ratio, thereby exacerbating AD (Maesako et al., 2011). Moreover, PS1 forms a synaptic trimeric complex with N-cadherin/β-catenin, facilitated by its ring structure domain. GSK-3β-mediated PS1 phosphorylation also affects its interaction with N-cadherin, disrupting their binding and leading to synaptic and neuronal activity deficits (Uemura et al., 2007). This intricate interplay among PS1, GSK-3β, and their impact on Aβ metabolism and synaptic function contributes significantly to the pathogenesis of Alzheimer’s disease.
3.4. GSK-3β regulates synaptic plasticity
Synaptic dysfunction is an early sign of AD (Tönnies and Trushina, 2017). Long-term potentiation (LTP) and long-term depression (LTD) are crucial for regulating synaptic connections between neurons (Warpechowski et al., 2023). LTP refers to the long-lasting increase in synaptic strength, while LTD is defined as the opposing process. In LTD, N-methyl-D-aspartate (NMDA) receptor channels and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor channels on the postsynaptic membrane play a critical role. AMPA is a sodium-potassium cation channel that helps activate neurons, while NMDA is a calcium ion channel. High-frequency stimulation leads to a high influx of calcium ions, which then bind with calmodulin (CaM) to form Ca2+/CaM complexes. These complexes activate calcium/calmodulin-dependent protein kinase II (CaMKII), thereby inducing LTP. LTP is involved in synaptic formation, plasticity, learning, memory, as well as excitotoxicity (Vyklicky et al., 2014). Low-frequency electrical stimulation causes a minor influx of calcium ions, leading to AMPA phosphorylation and LTD (Collingridge and Monaghan, 2022). GSK-3β is widely expressed in the hippocampus, a key brain region involved in learning and memory, where it plays a crucial regulatory role in balancing LTP and LTD. Inducing LTP can prevent the occurrence of LTD, and the induction of LTD is associated with a decrease in the phosphorylation of GSK-3β at Ser9. Inhibiting the active form of GSK-3β is beneficial for the induction of LTP (Peineau et al., 2007). Transgenic mice overexpressing the active form of GSK-3β display reduced LTP and abnormalities in hippocampus-dependent spatial and fear memory formation. Defective LTP can be alleviated or rescued by the long-term administration of the GSK-3β inhibitor lithium (Hooper et al., 2007). Therefore, GSK-3β plays a critical role in initiating NMDA-induced LTD in neurons.
4. GSK-3β in PD
Parkinson’s disease is a progressive neurodegenerative disorder with significant morbidity and mortality (Halli-Tierney et al., 2020). The pathological hallmark of PD is the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the accumulation of misfolded α-synuclein (Armstrong and Okun, 2020). Cardinal motor symptoms such as tremors, rigidity, bradykinesia, and postural instability are typically observed. PD can also present with non-motor symptoms (NMSs), including sleep disorders, constipation, urinary dysfunction, orthostatic hypotension, cognitive decline, depression, and anxiety (Schapira et al., 2017). Multiple pathways and mechanisms are involved in the molecular pathogenesis of PD, including α-synuclein deposition, oxidative stress, mitochondrial function, calcium homeostasis, axonal transport, and neuroinflammation (Balestrino and Schapira, 2020). Both environmental and genetic factors have been found to contribute to the development of PD (Yuan et al., 2022). Notably, the activity of GSK-3β is elevated in the striatum of PD patients compared to controls, indicating the involvement of GSK-3β in the pathogenesis of PD.
4.1. GSK-3β regulates α-synuclein pathology
α-Synuclein is a neuronal protein primarily located at the presynaptic terminal, and it is the major proteinaceous component of Lewy bodies (Lashuel, 2020). α-Synuclein plays a role in regulating synaptic activity and neurotransmitter release (Burré, 2015). During the onset of PD, soluble monomers of α-synuclein form oligomers, which then aggregate to form insoluble fibrils. Mutations or overexpression of α-synuclein promote its aggregation (Burré et al., 2018). Increasing evidence suggests that GSK-3β may play a crucial role in the expression and aggregation of α-synuclein. Both active and inactive forms of GSK-3β colocalize with pathologic α-synuclein in Lewy bodies in postmortem examinations of PD patient (Nagao and Hayashi, 2009). Transgenic mice overexpressing α-synuclein demonstrate heightened GSK-3β activity (Duka et al., 2009). Inhibition of GSK-3β can reduce the expression of α-synuclein and prevent cell death in cellular models of PD, suggesting that inhibiting GSK-3β may have neuroprotective effects on dopaminergic neurons by reducing the toxicity of α-synuclein overexpression. These findings emphasize the pivotal role of GSK-3β in α-synuclein pathology.
4.2. GSK-3β regulates dopamine receptor signaling
Dopamine is a pivotal neurotransmitter within the central nervous system, intricately regulating various functions such as motor control, cognition, emotion, and reward. It plays a critical role in the pathogenesis of PD (Latif et al., 2021). The PI3K/Akt pathway serves as the upstream regulatory factor of GSK-3β. The PI3K/Akt pathway is integral to dopamine receptor signaling, playing a pivotal role in various cellular processes, including apoptosis, transcription, and cell proliferation. Dysregulation of this pathway is associated with PD pathology (Timmons et al., 2009). In the dopaminergic neurons of individuals with PD, there is a reduction in the phospho-Akt/total Akt ratio (Malagelada et al., 2008). Specific PD medications activate the PI3K/Akt pathway, protecting dopaminergic neurons in the substantia nigra (Lim et al., 2008; Zhang et al., 2011). The Akt/GSK-3 cascade also intersects with dopamine D2 receptor effect (Gomez-Sintes et al., 2014). Dopamine D2 receptor activation triggers a signaling complex (PP2A, β-arrestin-2, and Akt) (Beaulieu et al., 2007). This complex leads to Akt dephosphorylation and subsequent activation of GSK-3β. When dopamine is present in excess, the activation of striatal GSK-3β depends on D2 receptor activity. This interaction underscores the involvement of GSK-3β signaling in dopamine-mediated locomotor activity. In PD models, inhibition of GSK-3β safeguards dopaminergic neurons from stress-induced damage, subsequently reducing dopamine-driven locomotor deficit (Beaulieu et al., 2004; Wang et al., 2007). These findings underscore the crucial role of the Akt/GSK-3β cascade as a significant signaling pathway influenced by dopamine dysregulation.
5. GSK-3β in HD
HD is a rare autosomal dominant genetic disorder resulting from the abnormal expansion of CAG trinucleotide repeats in the Huntingtin gene on the chromosome (Tabrizi et al., 2020). This expansion triggers the toxic form of the Huntingtin protein, leading to synaptic impairment, mitochondrial dysfunction, and disrupted axonal transport, ultimately contributing to motor, cognitive, and psychiatric impairments (Baake et al., 2017; Ghosh and Tabrizi, 2018). Emerging research also suggests that HD may be classified as a secondary tauopathy, with tau insoluble aggregates observed in late-stage HD (Salem and Cicchetti, 2023). Furthermore, there is increased activity of pGSK-3β-Tyr216 in the hippocampus of HD patients and mice, underscoring the potential significance of abnormal GSK-3β signaling in HD progression (L'Episcopo et al., 2016).
5.1. GSK-3β regulates tau pathology and neuronal degeneration in HD
Recent findings indicate that Tau undergoes pathological changes in HD, encompassing insoluble aggregate formation, altered levels, mis-splicing, hyperphosphorylation, and truncation within the brain (Gratuze et al., 2016; St-Amour et al., 2018; Masnata et al., 2020; Petry et al., 2023). Hyperphosphorylated Tau aggregates are observed in postmortem HD brain neurons within cortical, striatal, and hippocampal regions, coinciding with hippocampal neuronal loss (Fernández-Nogales et al., 2014; Vuono et al., 2015). Significantly, Tau knock-down has been demonstrated to attenuate motor abnormalities in an HD mouse model (Fernández-Nogales et al., 2014). This suggests the potential involvement of hyperphosphorylated Tau in HD’s behavioral and pathological aspects. The association between the mutation in the huntingtin gene and Tau dysregulation in HD remains unclear. Several hypotheses propose that the mutant huntingtin does not directly interact with Tau (Fernández-Nogales et al., 2014). Instead, it interacts with Tau kinases, phosphatases, and proteins engaged in Tau alternative splicing. Notably, GSK-3β and its active form (pGSK-3β-Tyr216) are elevated in the hippocampus of postmortem HD brain (L'Episcopo et al., 2016). Researchers have examined different Tau kinases and phosphatases in HD mouse models to unveil mechanisms underlying Tau hyperphosphorylation (Blum et al., 2015; Gratuze et al., 2015). They observed heightened levels of GSK-3β and its active form (pGSK-3β-Tyr216) in neurons and astrocytes of HD brains. Meanwhile, the localization of GSK-3β and its active form shifted from the cytoplasm to the nucleus in both neurons and astrocytes (L'Episcopo et al., 2016). This shift in localization from the cytoplasm to the nucleus suggests that GSK-3β may not only be heightened at the protein level but also upregulated in gene transcription in HD, ultimately contributing to excessive Tau phosphorylation (L'Episcopo et al., 2016). Furthermore, as HD progresses, levels of pGSK-3-Tyr216 and pTau steadily increase in the hippocampus, highlighting GSK-3β’s role in facilitating Tau hyperphosphorylation and pathological accumulation in HD.
5.2. GSK-3β regulates GABA activity
Both neurons and astrocytes are capable of synthesizing and releasing gamma-aminobutyric acid (GABA). The balance between excitatory glutamatergic and inhibitory GABAergic systems is crucial for motor and behavioral control (Koch and Raymond, 2019). One of the mechanisms underlying the development of HD is the disruption of GABAergic neurotransmission (Garret et al., 2018). Dysfunction in this circuitry may lead to the development of HD symptoms. Studies on postmortem brain tissue from HD patients have shown that GABA content is lower in the tail of the caudate nucleus, putamen, and cortex compared to non-HD controls. GABA typically suppresses dopamine release by activating GABA receptors on the soma and terminals of nigrostriatal neuron (Tyagarajan et al., 2011). Increased initial dopamine release or elevated extracellular glutamate levels may induce excitotoxicity, loss of cortical and striatal neurons, and loss of dopaminergic terminals, leading to motor disorder (Koch and Raymond, 2019). There is a unique phosphorylation site, Ser270, on the GABAergic postsynaptic scaffold protein Gephyrin, and phosphorylation of Gephyrin by GSK-3β reduces GABAergic transmission (Tyagarajan et al., 2011). Blocking Ser270 phosphorylation increases the density of Gephyrin clusters and the frequency of GABAergic postsynaptic currents in cultured hippocampal neurons (Rippin et al., 2021).
6. GSK-3β in ALS
ALS, also known as Lou Gehrig’s disease, is a neurodegenerative disorder that primarily affects motor neurons in the spinal cord, brainstem, and motor cortex, resulting in the gradual weakening of the muscles and eventually respiratory failure. The cause of ALS is unknown, with suggested involvement of signaling pathway alterations and factors such as protein misfolding and misassembly, mitochondrial dysfunction, oxidative stress, and neuroinflammation (Sever et al., 2022). ALS is divided into two types, with approximately 90–95% being sporadic cases (sALS) and the remaining 5–10% being familial cases (fALS) (Prasad et al., 2019). Research has found an increase in the levels of GSK-3β and phosphorylated GSK-3β at Tyr216 in the frontal cortex and hippocampus of ALS patients (Yang et al., 2008). GSK-3β is believed to affect the superoxide dismutase SOD1 gene and TAR DNA binding protein 43 (TDP-43), thus promoting the progression of ALS.
6.1. GSK-3β regulates TDP-43 pathology
TDP-43 is a 43 kDa protein encoded by the TARDBP gene that binds to DNA and RNA. It plays a crucial role in regulating RNA metabolism, axonal transport, vesicular trafficking, and stress response mechanisms (Palomo et al., 2019). Hyperphosphorylated and ubiquitinated TDP-43-positive neuronal cytoplasmic inclusions are identified in the brain and spinal cord in most cases of ALS. While TDP-43 is predominantly expressed in the nucleus, it can shuttle between the nucleus and cytoplasm and undergo various posttranslational modifications, such as phosphorylation and ubiquitination, in the cytoplasm (Tziortzouda et al., 2021). TDP-43 dysfunctions and cytoplasmic aggregation seem to be the central pathogenicity in ALS, which can result in neuronal and glial cell damage (Tamaki and Urushitani, 2022). Increased expression and activity of GSK-3β in the spinal cord, frontal, and temporal cortices have been linked to TDP-43 phosphorylation and cytoplasmic accumulation in ALS patient (Yang et al., 2008). There is a correlation between the level of GSK-3β and dysfunction in ALS individuals, indicating a potential biomarker for disease progression (Sreedharan et al., 2015). Studies have shown that treatment with a non-ATP competitive GSK-3β inhibitor, tideglusib, in TDP-43 (A315T) transgenic mice not only reduces the levels of phosphorylated TDP-43 in the mouse spinal cord but also delays symptom onset, improves motor function, and slows disease progression, further supporting the role of GSK-3β in the pathogenesis of ALS (Martínez-González et al., 2021).
6.2. GSK-3β regulates the Wnt signaling pathway
The Wnt signaling pathway is crucial in both physiological and pathophysiological processes of the CNS (Chen et al., 2012; Yu et al., 2013). There are two Wnt signaling pathways: the β-catenin-dependent pathway (WNT/β-catenin pathway) and the β-catenin-independent pathway, which were formerly referred to as the canonical and noncanonical Wnt signaling pathways, respectively (Akoumianakis et al., 2022). The WNT/β-catenin pathway drives neural progenitor cell differentiation into neurons, regulates hippocampal neurogenesis, enhances neural stem cell proliferation, and promotes synaptic stability and plasticity (Hayat et al., 2022). GSK-3β is a component of the complex that disrupts the WNT/β-catenin complex, phosphorylates β-catenin, and targets it for ubiquitin-proteasome degradation (Aberle et al., 1997; Duda et al., 2020). Inhibiting GSK-3β promotes the nuclear translocation of β-catenin in the lateral ventricles of postnatal mice, stimulating the proliferation of oligodendrocyte progenitor cells (Azim and Butt, 2011). Oligodendrocyte progenitor cells support myelin formation and neuronal metabolism. Therefore, inhibiting GSK-3β is considered necessary for the normal physiological function of the CNS (Lee et al., 2012). These findings support the hypothesis that GSK-3β is a crucial modulator in the pathway.
7. GSK-3β’s collective role in neurodegenerative diseases
Neuroinflammation is considered a contributing factor in NDDs (Gianferrara et al., 2022; Samim Khan et al., 2023). GSK-3β is a nexus for various signaling pathways, recognized as a pivotal inflammation regulator. Microglia activation and heightened proinflammatory cytokines define brain inflammation. Activated microglia release proinflammatory and neurotoxic agents, intensifying neuron harm through oxidative stress and cytokine toxicity (Kwon and Koh, 2020; Bennett and Sloan, 2021). GSK-3β activation stimulates microglia, amplifying inflammatory cytokine production, culminating in neuronal demise (Beurel, 2011; Cao et al., 2017). This perpetuates an ongoing inflammatory response, indicating GSK-3β’s vital role in the harmful feedback loop with microglia and impaired neurons. Inhibiting GSK-3β enhances tolerance to inflammation, curbing repeated cytokine release, and ameliorating symptoms in inflammatory conditions (Wang et al., 2013; Beurel et al., 2015). These findings underscore GSK-3β’s involvement in microglia-mediated neuroinflammation and neuronal death.
Mitochondria play a crucial role in the development and progression of NDD (Johnson et al., 2021). Mitochondria can synthesize ATP and regulate cellular apoptosis, ferroptosis, and inflammasome activation. Deficits in mitochondrial biogenesis may contribute to dysfunction in various neurodegenerative conditions. Drp1 is key in maintaining healthy mitochondrial dynamics by inducing fission through contraction and GTPase activity (Kleele et al., 2021). GSK-3β triggers GTPase activity by phosphorylating Drp1 at Ser40 and Ser44, resulting in fragmented mitochondria (Yan et al., 2015). Furthermore, mitochondrial dysfunction-induced hydrogen peroxide further activates GSK3β, exacerbating disease progression. Inhibition of GSK3β-induced phosphorylation can protect mitochondria, maintain energy metabolism, and rescue memory impairment in transgenic mice (Baek et al., 2017). In summary, overactive GSK-3β disrupts mitochondrial function and energy production, leading to neurodegeneration.
8. GSK-3β in other neurological diseases
Recent studies have also suggested potential links between GSK-3β dysregulation and other neuropsychiatric disorders, such as schizophrenia, depression, and anxiety (Cole, 2013; Tamura et al., 2016). Disrupted-in-schizophrenia 1 (DISC1) is associated with schizophrenia, and its deficiency causes behavioral abnormalities in mice (Johnstone et al., 2011). DISC1 also interacts with Translin-associated protein X (TRAXd), and GSK-3β regulates the function of DISC1/TRAXd (Weng et al., 2018). Inhibiting GSK-3β can protect DNA and restore neuronal function, thereby preserving neuron (Chien et al., 2018). Dysregulation of brain serotonin (5-HT) function is one of the pathogenic mechanisms of depression and anxiety (Carhart-Harris and Nutt, 2017). Increasing evidence has shown that GSK-3β acts as a modulator in the serotonin neurotransmission system, including direct interaction with serotonin 1B (5-HT1B) receptors in a highly selective manner and a prominent modulating effect on 5-HT1B receptor activity. Inactivation of GSK-3β in the brain by either pharmacological or genetic methods leads to amelioration of the abnormal behavior resulting from 5-HT deficiency (Beaulieu et al., 2008; Zhou et al., 2012). Therefore, targeting GSK-3β and related signaling pathways may provide therapeutic advantages for treating certain DISC1- and 5-HT-related neuropsychiatric disorders.
9. GSK-3β as a therapeutic target
GSK-3β plays a crucial role in many neurological disorders. GSK-3β may serve as a promising therapeutic target for treating diseases, promoting recovery, and maintaining organism stability (Rippin and Eldar-Finkelman, 2021). Several categories of GSK-3β inhibitors have been developed (Wadhwa et al., 2020), including (1) magnesium-competitive inhibitors; (2) ATP-competitive inhibitors; (3) substrate-competitive inhibitors; and (4) regulators of GSK-3β Ser9 phosphorylation.
The currently used drugs for AD, including cholinesterase inhibitors and NMDA receptor antagonists, only mildly alleviate the symptoms but do not halt or delay the progression of the disease (Birks and Harvey, 2018; McShane et al., 2019). Lithium salts are one of the earliest drugs used for AD treatment and can inhibit GSK-3β and reduce Tau phosphorylation and amyloid production but they have poor efficacy for mild AD patient (Hampel et al., 2009). The selective inhibitors SB216763 and FLZs can reduce Tau phosphorylation and protect neurons from the toxicity of Aβ oligomers, improving spatial memory (Bao et al., 2013; Deng et al., 2014). Noncompetitive irreversible Adenosine Triphosphate (ATP) inhibitor derivatives of 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD), such as Tideglusib and TDZD-8, can stimulate GSK-3β Ser9 phosphorylation, thereby reducing Tau and amyloid deposition (Domínguez et al., 2012). Although there are many potential inhibitors available for AD treatment, few have succeeded in clinical trials (Chauhan et al., 2022).
GSK-3β inhibitors, including SB216763, lithium chloride, and TDZD-8, have demonstrated significant effects in the treatment of PD. These inhibitors efficiently inhibit the generation of proinflammatory cytokines such as TNF-α and IL-12 while promoting the production of anti-inflammatory cytokines such as IL-10 (Coant et al., 2011), which helps protect dopaminergic neurons from neurotoxicity, preserve mitochondrial function, and eliminate oxidative damage in PD model (Chiu et al., 2014). Additionally, GSK-3β inhibitors can promote the proliferation of neural progenitor cells and stimulate neurogenesis, playing a role in neural regeneration and repair (Eom and Jope, 2009). Therefore, GSK-3β inhibitors have broad application prospects in the treatment of PD.
GSK-3β inhibition has emerged as a potential therapeutic approach for HD (D'Mello, 2021). Studies have revealed GSK-3β’s association with mHtt aggregation in HD mice and neurons (Valencia et al., 2010). Elevated GSK-3β expression and activity in the hippocampus of HD patients and mouse models correlate with increased tau phosphorylation (L'Episcopo et al., 2016). In cellular and mouse HD models, GSK-3β silencing and inhibition reduce mutant huntingtin aggregates and neuronal death, while selective GSK-3 inhibition improves motor function and neuroprotection (Rippin et al., 2021). Lithium, a GSK-3β inhibitor, exhibits beneficial effects in preclinical HD models, enhancing motor function and mitigating striatal deficits (Snitow et al., 2021). Co-treatment with lithium and valproate further alleviates HD-associated deficits (Chiu et al., 2011). These findings underscore GSK-3β’s relevance in HD pathogenesis, driving ongoing exploration of its therapeutic potential.
GSK-3β inhibitors have been studied as potential therapies for ALS in vivo. It has been shown that lithium treatment can prevent apoptosis, alleviate motor function defects, and slow disease progression in SOD1-G93A mice (Shin et al., 2007). Another potential therapeutic option for ALS is valproic acid (VPA), which has been shown to slow disease progression and increase lifespan in SOD1-G93A mice (Sugai et al., 2004). Combined therapy with lithium and VPA shows greater rescue effects on motor dysfunction and disease progression in the SOD1-G93A mouse model (Jiang et al., 2016). These findings indicate that GSK-3β holds great potential as a therapeutic target for ALS.
10. Conclusion
NDDs such as AD, HD, PD, and ALS are increasingly recognized as major contributors to death and disability worldwide (Carroll, 2019). Although these four common NDDs exhibit different clinical profiles, they share common molecular pathogenic mechanisms. These mechanisms encompass proteostasis, cellular signaling pathways, neuroinflammation, and mitochondrial deficits, suggesting converging pathways of neurodegeneration (Durrenberger et al., 2015; Noori et al., 2021). GSK-3β plays a pivotal role in multiple NDDs. In AD, GSK-3β influences the processing of APP and hyperphosphorylation of Tau protein, contributing to Aβ plaque formation and neurofibrillary tangle development. In PD, GSK-3β affects α-synuclein aggregation and dopamine receptor signaling. In HD, GSK-3β contributes to Tau pathology and GABAergic dysfunction. In ALS, GSK-3β impacts TDP-43 pathology and the Wnt signaling pathway. The intricate involvement of GSK-3β in the pathogenesis of various NDDs underscores its significance as a potential therapeutic target. GSK-3β inhibitors have the potential to protect neurons in various disease models. In conclusion, by unraveling its diverse functions and their interactions, we can advance our understanding of disease mechanisms and design targeted strategies to counteract neurodegeneration.
11. Discussion and future directions
The diverse functions of GSK-3β in controlling protein aggregation, mitochondrial dysfunction, neuroinflammation, and cellular signaling contribute to a comprehensive understanding of disease mechanisms. Although GSK-3β’s involvements in AD, PD, HD, and ALS are established, its potential impact on other neurodegenerative and neuropsychiatric disorders presents an intriguing field for further investigation (Lin et al., 2020).
The interactions of GSK-3β with the Wnt pathway, dopamine receptor signaling, and various kinases are intricately complex. Delving into these interactions deepens our understanding of potential molecular mechanisms and may unveil novel treatment strategies. Selectively modulating the activity of specific cellular pools of GSK-3β or its specific downstream or upstream partners may offer effective approaches for combating neurodegenerative diseases. Thus, GSK3β and its signaling pathway partners hold great promise as therapeutic targets for a multitude of neurological disorders.
A global endeavor is currently in progress to discover more effective methods for clinically managing NDDs, aiming to delay their onset and hinder their progression. Inhibition of GSK-3β was considered a promising therapeutic approach. However, most GSK-3β inhibitors that were developed function as ATP competitive inhibitors, with typical limitations in specificity, safety, and drug-induced resistance. Clinical and preclinical trials with GSK-3β inhibitors have shown limited effectiveness (Llorens-Martín et al., 2014; Xia et al., 2021). Thus, more selective and potent GSK-3β inhibitors should be developed, which can effectively target pathological processes and spare essential physiological functions. Additionally, considering the complex nature of NDDs, it appears that single-target drugs might fall short of achieving satisfactory therapeutic outcomes. Multi-target design strategy may potentially enhance both therapeutic safety and efficacy (Zhang et al., 2019; Gontijo et al., 2020).
Author contributions
HY wrote the draft. MX revised the manuscript. ZZ conceived the manuscript and made revisions. All authors contributed to the article and approved the submission.
Funding
This work was supported by grants from the Innovative Research Groups of Hubei Province (2022CFA026) and the National Natural Science Foundation of China (Nos. 82271447 and 81901090).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aberle, H., Bauer, A., Stappert, J., Kispert, A., and Kemler, R. (1997). beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804. doi: 10.1093/emboj/16.13.3797
Aguzzi, A., and O'Connor, T. (2010). Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 9, 237–248. doi: 10.1038/nrd3050
Akoumianakis, I., Polkinghorne, M., and Antoniades, C. (2022). Non-canonical WNT signalling in cardiovascular disease: mechanisms and therapeutic implications. Nat. Rev. Cardiol. 19, 783–797. doi: 10.1038/s41569-022-00718-5
Albeely, A. M., Williams, O. O. F., and Perreault, M. L. (2022). GSK-3β disrupts neuronal oscillatory function to inhibit learning and memory in male rats. Cell. Mol. Neurobiol. 42, 1341–1353. doi: 10.1007/s10571-020-01020-z
Armstrong, M. J., and Okun, M. S. (2020). Diagnosis and treatment of Parkinson disease: a review. JAMA 323, 548–560. doi: 10.1001/jama.2019.22360
Azim, K., and Butt, A. M. (2011). GSK3β negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 59, 540–553. doi: 10.1002/glia.21122
Baake, V., Reijntjes, R., Dumas, E. M., Thompson, J. C., and Roos, R. A. C. (2017). Cognitive decline in Huntington's disease expansion gene carriers. Cortex 95, 51–62.
Baek, S. H., Park, S. J., Jeong, J. I., Kim, S. H., Han, J., Kyung, J. W., et al. (2017). Inhibition of Drp 1 ameliorates synaptic depression, Aβ deposition, and cognitive impairment in an Alzheimer's disease model. J. Neurosci. 37, 5099–5110. doi: 10.1523/JNEUROSCI.2385-16.2017
Bagaria, J., Bagyinszky, E., and An, S. S. A. (2022). Genetics, functions, and clinical impact of Presenilin-1 (PSEN1) gene. Int. J. Mol. Sci. 23:10970. doi: 10.3390/ijms231810970
Bahn, G., Park, J. S., Yun, U. J., Lee, Y. J., Choi, Y., Park, J. S., et al. (2019). NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer's models. Proc. Natl. Acad. Sci. U. S. A. 116, 12516–12523. doi: 10.1073/pnas.1819541116
Balestrino, R., and Schapira, A. H. V. (2020). Parkinson disease. Eur. J. Neurol. 27, 27–42. doi: 10.1111/ene.14108
Bao, X. Q., Li, N., Wang, T., Kong, X. C., Tai, W. J., Sun, H., et al. (2013). FLZ alleviates the memory deficits in transgenic mouse model of Alzheimer's disease via decreasing beta-amyloid production and tau hyperphosphorylation. PLoS One 8:e78033. doi: 10.1371/journal.pone.0078033
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2007). The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 28, 166–172. doi: 10.1016/j.tips.2007.02.006
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., et al. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U. S. A. 101, 5099–5104. doi: 10.1073/pnas.0307921101
Beaulieu, J. M., Zhang, X., Rodriguiz, R. M., Sotnikova, T. D., Cools, M. J., Wetsel, W. C., et al. (2008). Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U. S. A. 105, 1333–1338. doi: 10.1073/pnas.0711496105
Bennett, F. C., and Sloan, S. A. (2021). Glia in neurodegeneration. Neurobiol. Dis. 151:105260. doi: 10.1016/j.nbd.2021.105260
Beurel, E. (2011). Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front. Mol. Neurosci. 4:18. doi: 10.3389/fnmol.2011.00018
Beurel, E., Grieco, S. F., and Jope, R. S. (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131. doi: 10.1016/j.pharmthera.2014.11.016
Birks, J. S., and Harvey, R. J. (2018). Donepezil for dementia due to Alzheimer's disease. Cochrane Database Syst. Rev. 6:Cd001190. doi: 10.1002/14651858.CD001190.pub3
Blum, D., Herrera, F., Francelle, L., Mendes, T., Basquin, M., et al. (2015). Mutant huntingtin alters tau phosphorylation and subcellular distribution. Hum. Mol. Genet. 24, 76–85. doi: 10.1093/hmg/ddu421
Boland, B., Yu, W. H., Corti, O., Mollereau, B., Henriques, A., Bezard, E., et al. (2018). Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 17, 660–688. doi: 10.1038/nrd.2018.109
Breijyeh, Z., and Karaman, R. (2020). Comprehensive review on Alzheimer's disease: causes and treatment. Molecules 25:5789. doi: 10.3390/molecules25245789
Burré, J. (2015). The synaptic function of α-Synuclein. J. Parkinsons Dis. 5, 699–713. doi: 10.3233/JPD-150642
Burré, J., Sharma, M., and Südhof, T. C. (2018). Cell biology and pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 8:a024091. doi: 10.1101/cshperspect.a024091
Cao, Q., Karthikeyan, A., Dheen, S. T., Kaur, C., and Ling, E. A. (2017). Production of proinflammatory mediators in activated microglia is synergistically regulated by Notch-1, glycogen synthase kinase (GSK-3β) and NF-κB/p 65 signalling. PLoS One 12:e0186764. doi: 10.1371/journal.pone.0186764
Carhart-Harris, R. L., and Nutt, D. J. (2017). Serotonin and brain function: a tale of two receptors. J. Psychopharmacol. 31, 1091–1120. doi: 10.1177/0269881117725915
Carroll, W. M. (2019). The global burden of neurological disorders. Lancet Neurol. 18, 418–419. doi: 10.1016/S1474-4422(19)30029-8
Chauhan, N., Paliwal, S., Jain, S., Verma, K., Paliwal, S., and Sharma, S. (2022). GSK-3β and its inhibitors in Alzheimer's disease: a recent update. Mini Rev. Med. Chem. 22, 2881–2895. doi: 10.2174/1389557522666220420094317
Chen, Y., Guan, Y., Liu, H., Wu, X., Yu, L., et al. (2012). Activation of the Wnt/β-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochem. Biophys. Res. Commun. 420, 397–403. doi: 10.1016/j.bbrc.2012.03.006
Chien, T., Weng, Y. T., Chang, S. Y., Lai, H. L., Chiu, F. L., Kuo, H. C., et al. (2018). GSK3β negatively regulates TRAX, a scaffold protein implicated in mental disorders, for NHEJ-mediated DNA repair in neurons. Mol. Psychiatry 23, 2375–2390. doi: 10.1038/s41380-017-0007-z
Chiu, C. T., Liu, G., Leeds, P., and Chuang, D. M. (2011). Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington's disease. Neuropsychopharmacology 36, 2406–2421. doi: 10.1038/npp.2011.128
Chiu, C. T., Scheuing, L., Liu, G., Liao, H. M., Linares, G. R., Lin, D., et al. (2014). The mood stabilizer lithium potentiates the antidepressant-like effects and ameliorates oxidative stress induced by acute ketamine in a mouse model of stress. Int. J. Neuropsychopharmacol. 18:pyu102. doi: 10.1093/ijnp/pyu102
Coant, N., Simon-Rudler, M., Gustot, T., Fasseu, M., Gandoura, S., Ragot, K., et al. (2011). Glycogen synthase kinase 3 involvement in the excessive proinflammatory response to LPS in patients with decompensated cirrhosis. J. Hepatol. 55, 784–793. doi: 10.1016/j.jhep.2010.12.039
Cole, A. R. (2013). Glycogen synthase kinase 3 substrates in mood disorders and schizophrenia. FEBS J. 280, 5213–5227. doi: 10.1111/febs.12407
Collingridge, G. L., and Monaghan, D. T. (2022). The continually evolving role of NMDA receptors in neurobiology and disease. Neuropharmacology 210:109042. doi: 10.1016/j.neuropharm.2022.109042
Demuro, S., Di Martino, R. M. C., Ortega, J. A., and Cavalli, A. (2021). GSK-3β, FYN, and DYRK1A: master regulators in neurodegenerative pathways. Int. J. Mol. Sci. 22:9098. doi: 10.3390/ijms22169098
Deng, Y., Xiong, Z., Chen, P., Wei, J., Chen, S., and Yan, Z. (2014). β-Amyloid impairs the regulation of N-methyl-D-aspartate receptors by glycogen synthase kinase 3. Neurobiol. Aging 35, 449–459. doi: 10.1016/j.neurobiolaging.2013.08.031
Dimitrov, M., Alattia, J. R., Lemmin, T., Lehal, R., Fligier, A., Houacine, J., et al. (2013). Alzheimer's disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat. Commun. 4:2246. doi: 10.1038/ncomms3246
D'Mello, S. R. (2021). When good kinases go rogue: GSK3, p 38 MAPK and CDKs as therapeutic targets for Alzheimer's and Huntington's disease. Int. J. Mol. Sci. 22:5911. doi: 10.3390/ijms22115911
Domínguez, J. M., Fuertes, A., Orozco, L., del Monte-Millán, M., Delgado, E., and Medina, M. (2012). Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J. Biol. Chem. 287, 893–904. doi: 10.1074/jbc.M111.306472
Duan, A. R., Jonasson, E. M., Alberico, E. O., Li, C., Scripture, J. P., Miller, R. A., et al. (2017). Interactions between tau and different conformations of tubulin: implications for tau function and mechanism. J. Mol. Biol. 429, 1424–1438. doi: 10.1016/j.jmb.2017.03.018
Duda, P., Akula, S. M., Abrams, S. L., Steelman, L. S., Martelli, A. M., Cocco, L., et al. (2020). Targeting GSK3 and associated signaling pathways involved in Cancer. Cells 9:1110. doi: 10.3390/cells9051110
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Duka, T., Duka, V., Joyce, J. N., and Sidhu, A. (2009). Alpha-Synuclein contributes to GSK-3beta-catalyzed tau phosphorylation in Parkinson's disease models. FASEB J. 23, 2820–2830. doi: 10.1096/fj.08-120410
Durrenberger, P. F., Fernando, F. S., Kashefi, S. N., Bonnert, T. P., Seilhean, D., Nait-Oumesmar, B., et al. (2015). Common mechanisms in neurodegeneration and neuroinflammation: a brain net Europe gene expression microarray study. J. Neural Transm. (Vienna) 122, 1055–1068. doi: 10.1007/s00702-014-1293-0
Eom, T. Y., and Jope, R. S. (2009). Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol. Psychiatry 66, 494–502. doi: 10.1016/j.biopsych.2009.04.015
Fernández-Nogales, M., Cabrera, J. R., Santos-Galindo, M., et al. (2014). Huntington's disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 20, 881–885. doi: 10.1038/nm.3617
Garret, M., Du, Z., Chazalon, M., Cho, Y. H., and Baufreton, J. (2018). Alteration of GABAergic neurotransmission in Huntington's disease. CNS Neurosci. Ther. 24, 292–300. doi: 10.1111/cns.12826
Gauthier, S. W. C., Servaes, S., and Morais, J. A., Rosa-Neto P World Alzheimer Report (2022).Life after diagnosis: navigating treatment, care and support. Available at: www.alzint.org/u/World-Alzheimer-Report-2022.pdf.
Ghosh, R., and Tabrizi, S. J. (2018). Huntington disease. Handb. Clin. Neurol. 147, 255–278. doi: 10.1016/B978-0-444-63233-3.00017-8
Gianferrara, T., Cescon, E., Grieco, I., Spalluto, G., and Federico, S. (2022). Glycogen synthase kinase 3β involvement in Neuroinflammation and neurodegenerative diseases. Curr. Med. Chem. 29, 4631–4697. doi: 10.2174/0929867329666220216113517
Giau, V. V., Bagyinszky, E., Youn, Y. C., An, S. S. A., and Kim, S. (2019). APP, PSEN1, and PSEN2 mutations in Asian patients with early-onset Alzheimer disease. Int. J. Mol. Sci. 20:4757. doi: 10.3390/ijms20194757
Gomez-Sintes, R., Bortolozzi, A., Artigas, F., and Lucas, J. J. (2014). Reduced striatal dopamine DA D2 receptor function in dominant-negative GSK-3 transgenic mice. Eur. Neuropsychopharmacol. 24, 1524–1533. doi: 10.1016/j.euroneuro.2014.07.004
Gontijo, V. S., Viegas, F. P. D., Ortiz, C. J. C., de Freitas Silva, M., Damasio, C. M., Rosa, M. C., et al. (2020). Molecular hybridization as a tool in the Design of Multi-target Directed Drug Candidates for neurodegenerative diseases. Curr. Neuropharmacol. 18, 348–407. doi: 10.2174/1385272823666191021124443
Gratuze, M., Cisbani, G., Cicchetti, F., and Planel, E. (2016). Is Huntington's disease a tauopathy? Brain 139, 1014–1025. doi: 10.1093/brain/aww021
Gratuze, M., Noël, A., Julien, C., Cisbani, G., Milot-Rousseau, P., Morin, F., et al. (2015). Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington's disease. Hum. Mol. Genet. 24, 86–99. doi: 10.1093/hmg/ddu456
Halli-Tierney, A. D., Luker, J., and Carroll, D. G. (2020). Parkinson disease. Am. Fam. Physician 102, 679–691.
Hampel, H., Ewers, M., Bürger, K., Annas, P., Mörtberg, A., et al. (2009). Lithium trial in Alzheimer's disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931. doi: 10.4088/JCP.08m04606
Hayat, R., Manzoor, M., and Hussain, A. (2022). Wnt signaling pathway: a comprehensive review. Cell Biol. Int. 46, 863–877. doi: 10.1002/cbin.11797
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., et al. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86. doi: 10.1111/j.1460-9568.2006.05245.x
Hurtado, D. E., Molina-Porcel, L., Carroll, J. C., Macdonald, C., Aboagye, A. K., Trojanowski, J. Q., et al. (2012). Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer's disease. J. Neurosci. 32, 7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012
Jiang, H. Z., Wang, S. Y., Yin, X., Jiang, H. Q., Wang, X. D., Wang, J., et al. (2016). Downregulation of Homer 1b/c in SOD1 G93A models of ALS: a novel mechanism of neuroprotective effect of Lithium and Valproic acid. Int. J. Mol. Sci. 17:2129. doi: 10.3390/ijms17122129
Johnson, J., Mercado-Ayon, E., Mercado-Ayon, Y., Dong, Y. N., Halawani, S., Ngaba, L., et al. (2021). Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 702:108698. doi: 10.1016/j.abb.2020.108698
Johnstone, M., Thomson, P. A., Hall, J., McIntosh, A. M., Lawrie, S. M., and Porteous, D. J. (2011). DISC1 in schizophrenia: genetic mouse models and human genomic imaging. Schizophr. Bull. 37, 14–20. doi: 10.1093/schbul/sbq135
Kent, S. A., Spires-Jones, T. L., and Durrant, C. S. (2020). The physiological roles of tau and Aβ: implications for Alzheimer's disease pathology and therapeutics. Acta Neuropathol. 140, 417–447. doi: 10.1007/s00401-020-02196-w
Kleele, T., Rey, T., Winter, J., Zaganelli, S., Mahecic, D., Perreten Lambert, H., et al. (2021). Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439. doi: 10.1038/s41586-021-03510-6
Koch, E. T., and Raymond, L. A. (2019). Dysfunctional striatal dopamine signaling in Huntington's disease. J. Neurosci. Res. 97, 1636–1654. doi: 10.1002/jnr.24495
Kwon, H. S., and Koh, S. H. (2020). Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9:42. doi: 10.1186/s40035-020-00221-2
Lashuel, H. A. (2020). Do Lewy bodies contain alpha-synuclein fibrils? And does it matter? A brief history and critical analysis of recent reports. Neurobiol. Dis. 141:104876.doi: 10.1016/j.nbd.2020.104876
Latif, S., Jahangeer, M., Maknoon Razia, D., Ashiq, M., Ghaffar, A., Akram, M., et al. (2021). Dopamine in Parkinson's disease. Clin. Chim. Acta 522, 114–126. doi: 10.1016/j.cca.2021.08.009
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
L'Episcopo, F., Drouin-Ouellet, J., Tirolo, C., Pulvirenti, A., Giugno, R., Testa, N., et al. (2016). GSK-3β-induced tau pathology drives hippocampal neuronal cell death in Huntington's disease: involvement of astrocyte-neuron interactions. Cell Death Dis. 7:e2206. doi: 10.1038/cddis.2016.104
Lim, J. H., Kim, K. M., Kim, S. W., Hwang, O., and Choi, H. J. (2008). Bromocriptine activates NQO1 via Nrf2-PI3K/Akt signaling: novel cytoprotective mechanism against oxidative damage. Pharmacol. Res. 57, 325–331. doi: 10.1016/j.phrs.2008.03.004
Lin, R., Jones, N. C., and Kwan, P. (2020). Unravelling the role of glycogen synthase Kinase-3 in Alzheimer's disease-related epileptic seizures. Int. J. Mol. Sci. 21:3676. doi: 10.3390/ijms21103676
Llorens-Martín, M., Jurado, J., Hernández, F., and Avila, J. (2014). GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 7:46. doi: 10.3389/fnmol.2014.00046
Ly, P. T., Wu, Y., Zou, H., Wang, R., Zhou, W., Kinoshita, A., et al. (2013). Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 123, 224–235. doi: 10.1172/JCI64516
Maesako, M., Uemura, K., Kubota, M., Hiyoshi, K., Ando, K., Kuzuya, A., et al. (2011). Effect of glycogen synthase kinase 3 β-mediated presenilin 1 phosphorylation on amyloid β production is negatively regulated by insulin receptor cleavage. Neuroscience 177, 298–307. doi: 10.1016/j.neuroscience.2010.12.017
Malagelada, C., Jin, Z. H., and Greene, L. A. (2008). RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 28, 14363–14371. doi: 10.1523/JNEUROSCI.3928-08.2008
Marosi, M., Arman, P., Aceto, G., D’Ascenzo, M., and Laezza, F. (2022). Glycogen synthase kinase 3: ion channels, plasticity, and diseases. Int. J. Mol. Sci. 23:4413. doi: 10.3390/ijms23084413
Martínez-González, L., Gonzalo-Consuegra, C., Gómez-Almería, M., Porras, G., de Lago, E., Martín-Requero, Á., et al. (2021). Tideglusib, a non-ATP competitive inhibitor of GSK-3β as a drug candidate for the treatment of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 22:8975. doi: 10.3390/ijms22168975
Masnata, M., Salem, S., de Rus Jacquet, A., Anwer, M., and Cicchetti, F. (2020). Targeting tau to treat clinical features of Huntington's disease. Front. Neurol. 11:580732
McShane, R., Westby, M. J., Roberts, E., Minakaran, N., Schneider, L., Farrimond, L. E., et al. (2019). Memantine for dementia. Cochrane Database Syst. Rev. 3:Cd003154. doi: 10.1002/14651858.CD003154.pub6
Muralidar, S., Ambi, S. V., Sekaran, S., Thirumalai, D., and Palaniappan, B. (2020). Role of tau protein in Alzheimer's disease: the prime pathological player. Int. J. Biol. Macromol. 163, 1599–1617. doi: 10.1016/j.ijbiomac.2020.07.327
Nagao, M., and Hayashi, H. (2009). Glycogen synthase kinase-3beta is associated with Parkinson's disease. Neurosci. Lett. 449, 103–107. doi: 10.1016/j.neulet.2008.10.104
Noori, A., Mezlini, A. M., Hyman, B. T., Serrano-Pozo, A., and Das, S. (2021). Systematic review and meta-analysis of human transcriptomics reveals neuroinflammation, deficient energy metabolism, and proteostasis failure across neurodegeneration. Neurobiol. Dis. 149:105225. doi: 10.1016/j.nbd.2020.105225
Palomo, V., Tosat-Bitrian, C., Nozal, V., Nagaraj, S., Martin-Requero, A., and Martinez, A. (2019). TDP-43: a key therapeutic target beyond amyotrophic lateral sclerosis. ACS Chem. Neurosci. 10, 1183–1196. doi: 10.1021/acschemneuro.9b00026
Pardo, M., Abrial, E., Jope, R. S., and Beurel, E. (2016). GSK3β isoform-selective regulation of depression, memory and hippocampal cell proliferation. Genes Brain Behav. 15, 348–355. doi: 10.1111/gbb.12283
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., et al. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Penke, B., Bogár, F., Paragi, G., Gera, J., and Fülöp, L. (2019). Key peptides and proteins in Alzheimer's disease. Curr. Protein Pept. Sci. 20, 577–599. doi: 10.2174/1389203720666190103123434
Petry, S., Nateghi, B., Keraudren, R., Sergeant, N., Planel, E., Hébert, S. S., et al. (2023). Differential regulation of tau exon 2 and 10 isoforms in Huntington's disease brain. Neuroscience 518, 54–63. doi: 10.1016/j.neuroscience.2022.07.014
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 Misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Rippin, I., Bonder, K., Joseph, S., Sarsor, A., Vaks, L., and Eldar-Finkelman, H. (2021). Inhibition of GSK-3 ameliorates the pathogenesis of Huntington's disease. Neurobiol. Dis. 154:105336. doi: 10.1016/j.nbd.2021.105336
Rippin, I., and Eldar-Finkelman, H. (2021). Mechanisms and therapeutic implications of GSK-3 in treating neurodegeneration. Cells 10:262. doi: 10.3390/cells10020262
Salem, S., and Cicchetti, F. (2023). Untangling the role of tau in Huntington's disease pathology. J. Hunting. Dis. 12, 15–29. doi: 10.3233/JHD-220557
Samim Khan, S., Janrao, S., Srivastava, S., Bala Singh, S., Vora, L., and Kumar Khatri, D. (2023). GSK-3β: An exuberating neuroinflammatory mediator in Parkinson's disease. Biochem. Pharmacol. 210:115496. doi: 10.1016/j.bcp.2023.115496
Sayas, C. L., and Ávila, J. (2021). GSK-3 and tau: a key duet in Alzheimer's disease. Cells 10:721. doi: 10.3390/cells10040721
Schapira, A. H. V., Chaudhuri, K. R., and Jenner, P. (2017). Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18, 435–450. doi: 10.1038/nrn.2017.62
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chételat, G., Teunissen, C. E., et al. (2021). Alzheimer's disease. Lancet 397, 1577–1590. doi: 10.1016/S0140-6736(20)32205-4
Sever, B., Ciftci, H., DeMirci, H., Sever, H., Ocak, F., Yulug, B., et al. (2022). Comprehensive research on past and future therapeutic strategies devoted to treatment of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 23:2400. doi: 10.3390/ijms23052400
Shin, J. H., Cho, S. I., Lim, H. R., Lee, J. K., Lee, Y. A., Noh, J. S., et al. (2007). Concurrent administration of Neu 2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol. Pharmacol. 71, 965–975. doi: 10.1124/mol.106.030676
Snitow, M. E., Bhansali, R. S., and Klein, P. S. (2021). Lithium and therapeutic targeting of GSK-3. Cells 10:255. doi: 10.3390/cells10020255
Sreedharan, J., Neukomm, L. J., Brown, R. H., and Freeman, M. R. (2015). Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr. Biol. 25, 2130–2136. doi: 10.1016/j.cub.2015.06.045
St-Amour, I., Turgeon, A., Goupil, C., Planel, E., and Hébert, S. S. (2018). Co-occurrence of mixed proteinopathies in late-stage Huntington's disease. Acta Neuropathol. 135, 249–265. doi: 10.1007/s00401-017-1786-7
Sugai, F., Yamamoto, Y., Miyaguchi, K., Zhou, Z., Sumi, H., Hamasaki, T., et al. (2004). Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur. J. Neurosci. 20, 3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x
Tabrizi, S. J., Flower, M. D., Ross, C. A., and Wild, E. J. (2020). Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 16, 529–546. doi: 10.1038/s41582-020-0389-4
Tamaki, Y., and Urushitani, M. (2022). Molecular dissection of TDP-43 as a leading cause of ALS/FTLD. Int. J. Mol. Sci. 23:12508. doi: 10.3390/ijms232012508
Tamura, M., Mukai, J., Gordon, J. A., and Gogos, J. A. (2016). Developmental inhibition of Gsk 3 rescues behavioral and neurophysiological deficits in a mouse model of schizophrenia predisposition. Neuron 89, 1100–1109. doi: 10.1016/j.neuron.2016.01.025
Timmons, S., Coakley, M. F., and Moloney, A. M. (2009). Akt signal transduction dysfunction in Parkinson's disease. Neurosci. Lett. 467, 30–35. doi: 10.1016/j.neulet.2009.09.055
Tönnies, E., and Trushina, E. (2017). Oxidative stress, synaptic dysfunction, and Alzheimer's disease. J. Alzheimers Dis. 57, 1105–1121. doi: 10.3233/JAD-161088
Tyagarajan, S. K., Ghosh, H., Yévenes, G. E., Nikonenko, I., Ebeling, C., Schwerdel, C., et al. (2011). Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. U. S. A. 108, 379–384. doi: 10.1073/pnas.1011824108
Tziortzouda, P., Van Den Bosch, L., and Hirth, F. (2021). Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat. Rev. Neurosci. 22, 197–208. doi: 10.1038/s41583-021-00431-1
Uddin, M. S., Hasana, S., Hossain, M. F., Islam, M. S., Behl, T., Perveen, A., et al. (2021). Molecular genetics of early- and late-onset Alzheimer's disease. Curr. Gene Ther. 21, 43–52. doi: 10.2174/1566523220666201123112822
Uemura, K., Kuzuya, A., Shimozono, Y., Aoyagi, N., Ando, K., Shimohama, S., et al. (2007). GSK3beta activity modifies the localization and function of presenilin 1. J. Biol. Chem. 282, 15823–15832. doi: 10.1074/jbc.M610708200
Valencia, A., Reeves, P. B., Sapp, E., Li, X., Alexander, J., Kegel, K. B., et al. (2010). Mutant huntingtin and glycogen synthase kinase 3-beta accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington's disease. J. Neurosci. Res. 88, 179–190. doi: 10.1002/jnr.22184
Vuono, R., Winder-Rhodes, S., de Silva, R., Cisbani, G., Drouin-Ouellet, J., Spillantini, M. G., et al. (2015). The role of tau in the pathological process and clinical expression of Huntington's disease. Brain 138, 1907–1918. doi: 10.1093/brain/awv107
Vyklicky, V., Korinek, M., Smejkalova, T., Balik, A., Krausova, B., Kaniakova, M., et al. (2014). Structure, function, and pharmacology of NMDA receptor channels. Physiol. Res. 63, S191–S203. doi: 10.33549/physiolres.932678
Wadhwa, P., Jain, P., and Jadhav, H. R. (2020). Glycogen synthase kinase 3 (GSK3): its role and inhibitors. Curr. Top. Med. Chem. 20, 1522–1534. doi: 10.2174/1568026620666200516153136
Wang, W., Yang, Y., Ying, C., Li, W., Ruan, H., Zhu, X., et al. (2007). Inhibition of glycogen synthase kinase-3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology 52, 1678–1684. doi: 10.1016/j.neuropharm.2007.03.017
Wang, H. M., Zhang, T., Li, Q., Huang, J. K., Chen, R. F., and Sun, X. J. (2013). Inhibition of glycogen synthase kinase-3β by lithium chloride suppresses 6-hydroxydopamine-induced inflammatory response in primary cultured astrocytes. Neurochem. Int. 63, 345–353. doi: 10.1016/j.neuint.2013.07.003
Wang, C., Zong, S., Cui, X., Wang, X., Wu, S., Wang, L., et al. (2023). The effects of microglia-associated neuroinflammation on Alzheimer's disease. Front. Immunol. 14:1117172. doi: 10.3389/fimmu.2023.1117172
Warpechowski, M., Warpechowski, J., Kulczyńska-Przybik, A., and Mroczko, B. (2023). Biomarkers of activity-dependent plasticity and persistent enhancement of synaptic transmission in Alzheimer disease: a review of the current status. Med. Sci. Monit. 29:e938826. doi: 10.12659/MSM.938826
Weng, Y. T., Chien, T., Kuan, I. I., and Chern, Y. (2018). The TRAX, DISC1, and GSK3 complex in mental disorders and therapeutic interventions. J. Biomed. Sci. 25:71. doi: 10.1186/s12929-018-0473-x
Xia, Y., Prokop, S., and Giasson, B. I. (2021). "Don't Phos over tau": recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer's disease and other tauopathies. Mol. Neurodegener. 16:37. doi: 10.1186/s13024-021-00460-5
Yan, J., Liu, X. H., Han, M. Z., Wang, Y. M., Sun, X. L., Yu, N., et al. (2015). Blockage of GSK3β-mediated Drp 1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer's disease. Neurobiol. Aging 36, 211–227. doi: 10.1016/j.neurobiolaging.2014.08.005
Yang, W., Leystra-Lantz, C., and Strong, M. J. (2008). Upregulation of GSK3beta expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci). Brain Res. 1196, 131–139. doi: 10.1016/j.brainres.2007.12.031
Yao, H. B., Shaw, P. C., Wong, C. C., and Wan, D. C. (2002). Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 23, 291–297. doi: 10.1016/S0891-0618(02)00014-5
Yu, L., Guan, Y., Wu, X., Chen, Y., Liu, Z., Du, H., et al. (2013). Wnt signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem. Res. 38, 1904–1913. doi: 10.1007/s11064-013-1096-y
Yuan, X., Tian, Y., Liu, C., and Zhang, Z. (2022). Environmental factors in Parkinson's disease: new insights into the molecular mechanisms. Toxicol. Lett. 356, 1–10. doi: 10.1016/j.toxlet.2021.12.003
Zhang, H., Mak, S., Cui, W., Li, W., Han, R., Hu, S., et al. (2011). Tacrine (2)-ferulic acid, a novel multifunctional dimer, attenuates 6-hydroxydopamine-induced apoptosis in PC12 cells by activating Akt pathway. Neurochem. Int. 59, 981–988. doi: 10.1016/j.neuint.2011.09.001
Zhang, P., Xu, S., Zhu, Z., and Xu, J. (2019). Multi-target design strategies for the improved treatment of Alzheimer's disease. Eur. J. Med. Chem. 176, 228–247. doi: 10.1016/j.ejmech.2019.05.020
Keywords: protein aggregation, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis
Citation: Yu H, Xiong M and Zhang Z (2023) The role of glycogen synthase kinase 3 beta in neurodegenerative diseases. Front. Mol. Neurosci. 16:1209703. doi: 10.3389/fnmol.2023.1209703
Edited by:
Jonasz Jeremiasz Weber, Ruhr University Bochum, GermanyReviewed by:
Victoria Campos-Peña, Manuel Velasco Suárez National Institute of Neurology and Neurosurgery, MexicoRenuka Khatik, Washington University in St. Louis, United States
Copyright © 2023 Yu, Xiong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhentao Zhang, emhlbnRhb3poYW5nQHdodS5lZHUuY24=