 Viviana Brembati
Viviana Brembati Gaia Faustini
Gaia Faustini Francesca Longhena
Francesca Longhena Arianna Bellucci
Arianna Bellucci- Division of Pharmacology, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
Parkinson’s disease (PD) is the most common neurodegenerative disorder with motor symptoms. The neuropathological alterations characterizing the brain of patients with PD include the loss of dopaminergic neurons of the nigrostriatal system and the presence of Lewy bodies (LB), intraneuronal inclusions that are mainly composed of alpha-synuclein (α-Syn) fibrils. The accumulation of α-Syn in insoluble aggregates is a main neuropathological feature in PD and in other neurodegenerative diseases, including LB dementia (LBD) and multiple system atrophy (MSA), which are therefore defined as synucleinopathies. Compelling evidence supports that α-Syn post translational modifications (PTMs) such as phosphorylation, nitration, acetylation, O-GlcNAcylation, glycation, SUMOylation, ubiquitination and C-terminal cleavage, play important roles in the modulation α-Syn aggregation, solubility, turnover and membrane binding. In particular, PTMs can impact on α-Syn conformational state, thus supporting that their modulation can in turn affect α-Syn aggregation and its ability to seed further soluble α-Syn fibrillation. This review focuses on the importance of α-Syn PTMs in PD pathophysiology but also aims at highlighting their general relevance as possible biomarkers and, more importantly, as innovative therapeutic targets for synucleinopathies. In addition, we call attention to the multiple challenges that we still need to face to enable the development of novel therapeutic approaches modulating α-Syn PTMs.
1. Introduction
Parkinson’s disease (PD) is the second most common movement disorder, affecting 2% of the world population over 65 years of age (Baker and Graham, 2004).
Motor symptoms mainly arise from the loss of dopaminergic nigrostriatal neurons, that alters the homeostasis of basal ganglia networks (Hornykiewicz, 2001). Beyond motor manifestations, PD patients may also exhibit a wide range of non-motor and psychiatric symptoms, which are caused by functional changes in central nervous system (CNS) and peripheral network system (PNS) circuits (Pfeiffer, 2016; Engelender and Isacson, 2017; Takamatsu et al., 2018; Kulkarni et al., 2022).
Key neuropathological hallmark of PD is the deposition of insoluble proteinaceous inclusions in cell bodies and neurites (Gibb, 1986), which are called Lewy bodies (LB) and Lewy neurites (LN), respectively. In 1997, these were found to be mainly composed of alpha synuclein (α-Syn) insoluble fibrils (Spillantini et al., 1997). In the last decades, it has been shown that α-Syn is particularly enriched at synaptic terminals, where it regulates synaptic function (Spillantini et al., 1997; Burre et al., 2010; Longhena et al., 2019). Since then, other disorders such as LB dementia (LBD), multiple system atrophy (MSA), Alzheimer’s disease (AD) LB variant or neurodegeneration with brain iron accumulation (NBIA), have been found to be characterized by brain accumulation of insoluble α-Syn deposits, and have been defined as synucleinopathies (Spillantini et al., 1998; Spillantini, 1999; Spillantini and Goedert, 2016; Goedert et al., 2017).
Interestingly, α-Syn deposits have been observed also in the PNS innervating the gastrointestinal tract, blood, salivary glands, olfactory mucosa, skin, retina, adrenal gland, heart and muscles (Qualman et al., 1984; Fumimura et al., 2007; Beach et al., 2010; Gelpi et al., 2014; Zange et al., 2015; Stoessl, 2016; Rey et al., 2016a,b, 2018; Wakabayashi, 2020). This peripheral α-Syn pathology is thought to contribute to the onset of PD non-motor manifestations in the prodromal and symptomatic phase (Abbott et al., 2007).
Remarkably, numerous studies in experimental models of synucleinopathy, post-mortem PD brains and neuroimaging evidences support that α-Syn pathological aggregation can severely impair synaptic function, thus consequently perturbing neuronal network dynamics and inducing neurodegeneration (Bellucci et al., 2016, 2017; Longhena et al., 2017, 2019; Kulkarni et al., 2022). This notwithstanding, we still ignore the mechanisms that drive pathological α-Syn aggregation in neuronal cells, and this has hampered the development of innovative effective therapies that block α-Syn pathological deposition as disease modifying approaches for PD and other synucleinopathies (Fields et al., 2019; Lashuel, 2021; Oliveira et al., 2021; Engelender et al., 2022). Indeed, current α-Syn-targeting strategies mainly include immunotherapy-based removal of extracellular α-Syn fibrils, gene therapy-based reduction of α-Syn, general and non-selective small molecule inhibitors of protein aggregation and protein degradation enhancers, but we still miss a cutting edge approach interfering with the culprit of α-Syn aggregate formation.
Interestingly, several post translational modifications (PTMs) of α-Syn have been found to differently modulate α-Syn aggregation either by predisposing or interfering with it (Zhang et al., 2019; Table 1). Indeed, they can affect α-Syn aggregation propensity, solubility and turnover, membrane binding and interaction with other proteins and metals (Oueslati et al., 2010; Zhang et al., 2017, 2019; Bell and Vendruscolo, 2021; Bell et al., 2022a,b). Moreover, α-Syn PTMs can serve as markers for environmental changes, may play a role in gene expression by impinging on cellular responses to stimuli and are also under study as possible disease biomarkers for synucleinopathies (Vicente Miranda et al., 2017a; Fayyad et al., 2019; Vivacqua et al., 2019; Petricca et al., 2022; Sonustun et al., 2022).
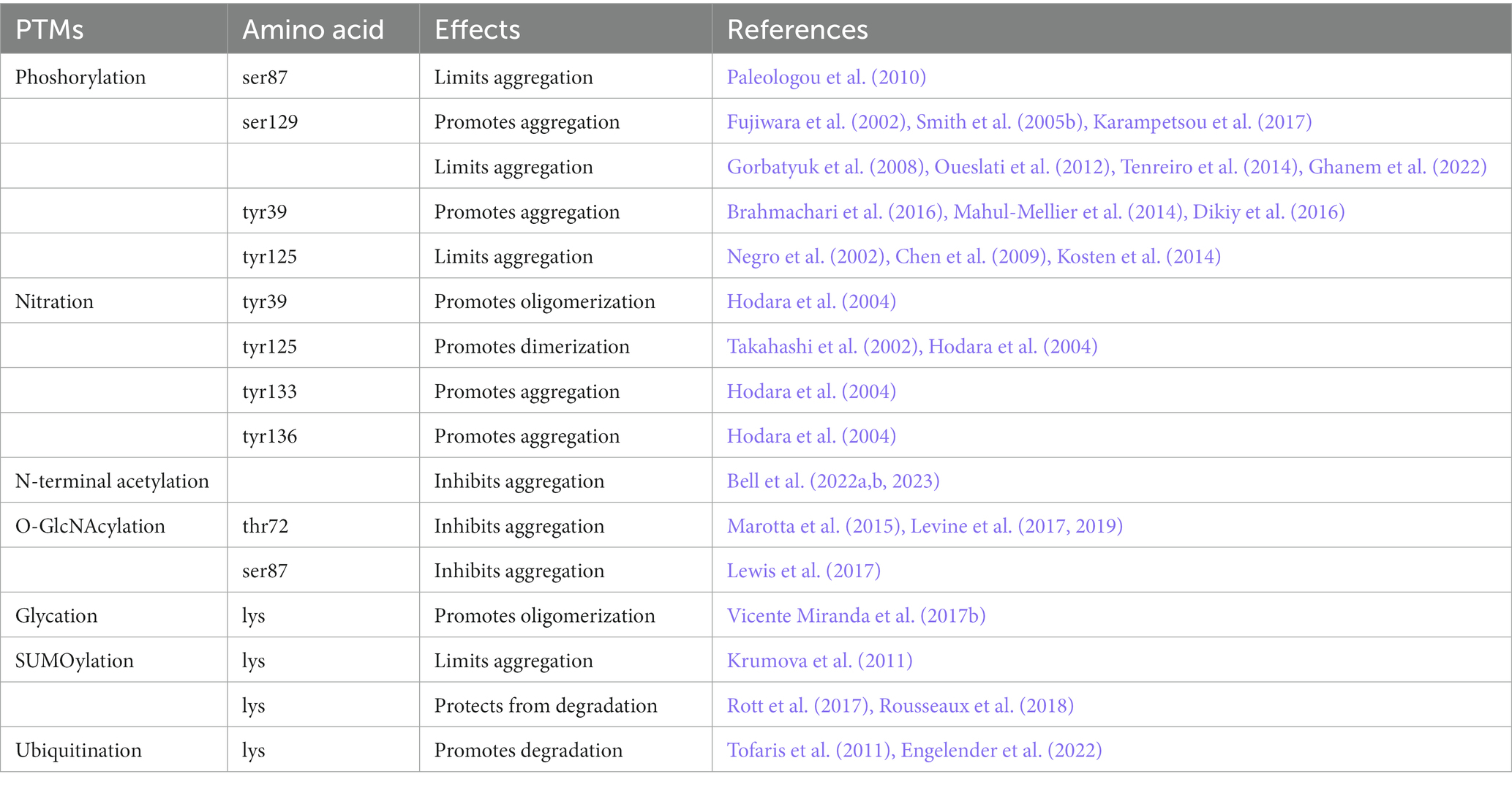
Table 1. Functional consequences of the majorly studied α-Syn PTMs.
In this review, we summarize and discuss the main findings on α-Syn PTMs, in order to define a route to decipher whether these modifications can be rationally considered as achievable druggable targets for synucleinopathies or effective biomarkers monitoring the progression or enabling patient stratification in these neurodegenerative disorders.
2. α-Syn and its post-translational modifications
α-Syn is a member of synuclein family, which also includes β-and γ-synuclein (Clayton and George, 1998). In humans, α-Syn is encoded by the SNCA gene located on chromosome 4q21 (Shibasaki et al., 1995; Lavedan, 1998).
Although the physiological role of α-Syn has not been fully elucidated yet, numerous studies demonstrated its involvement in the control of synaptic release. Indeed, it regulates synaptic vesicle clustering, the coupling and fusion of vesicles participating in SNARE complex assembly, the extent of phasic and tonic neurotransmitter release as well as neurotransmitter reuptake (Choi et al., 2013; Ghiglieri et al., 2018; Longhena et al., 2019). Moreover, α-Syn regulates mitochondrial function, fusion as well as mitochondria and endoplasmic reticulum interaction at mitochondria-associated membranes (MAM; Dauer et al., 2002; Ellis et al., 2005; Di Maio et al., 2016; Ludtmann et al., 2016; Menges et al., 2017; Faustini et al., 2019; Risiglione et al., 2021; Thorne and Tumbarello, 2022) and is involved in neuronal plasticity (Liu et al., 2004b, 2007; Watson et al., 2009; Ullman et al., 2011; Leite et al., 2022; Calabresi et al., 2023).
α-Syn is composed of 140 amino acids and its molecular weight is 14 kDa. α-Syn structure encompasses 3 domains: (1) the N-terminal region (residues 1–60), is positively charged and contains imperfect repeats with a highly conserved hexameric motif (KTKEGV), typically involved in the formation of amphipathic α-helices which mediate membrane binding (Clayton and George, 1998; George, 2002; Vamvaca et al., 2009); (2) the central hydrophobic region (residues 61–95), also known as non-amyloid component (NAC) portion, is prone to intermolecular interactions and is crucial for aggregation and fibril formation (Giasson et al., 2001; Ma et al., 2003); (3) the C-terminal region (residues 96–140) is highly enriched in acidic proline residues (Bellucci et al., 2012). This part of the protein reduces the NAC propensity for aggregation, mediates the majority of α-Syn interactions with proteins, metal ions and other ligands, including dopamine and polyamines, and harbors the majority of PTMs sites (Jensen et al., 1999; Paik et al., 1999; Giasson et al., 2003; Fernandez et al., 2004; Hoyer et al., 2004; Brown, 2007).
α-Syn does not present a defined structure in aqueous solutions and for this reason is defined “natively unfolded” (Stefanis, 2012), but it can shift to α-helix structure in association with membrane phospholipids, suggesting that it acquires different roles in different subcellular compartments based on its dynamic structure (Ahn et al., 2002). Indeed, in function of its capacity to acquire different conformations, α-Syn can interact with lipid membranes, enzymes, chaperones, synaptic and cytoskeletal proteins. Some studies also suggested a physiological α-helical structure forming dimers that counteract synaptic vesicle fission or tetramers that resist aggregation (Bartels et al., 2011; Wang et al., 2011; Medeiros et al., 2017).
Compelling evidence supports that PTMs play an important role in promoting conformational changes that make α-Syn more or less prone to aggregation (Table 1). Indeed, several PTMs such as phosphorylation, nitration, acetylation, glycation, truncation, ubiquitination, SUMOylation and O-GlcNAcylation can affect α-Syn structure. In particular, PTMs can either promote or inhibit α-Syn oligomerization, fibrillization and degradation (Feany and Bender, 2000; Fujiwara et al., 2002; Hodara et al., 2004; Smith et al., 2005a; Kasai et al., 2008; Lee et al., 2008; Rott et al., 2008, 2017; Tetzlaff et al., 2008; Danielson et al., 2009; Oueslati et al., 2010, 2013; Levine et al., 2017; Lewis et al., 2017; Zhang et al., 2019). Moreover, it has been described that LB contain phosphorylated, nitrated, ubiquitinated, SUMOylated and C-terminally truncated α-Syn, further supporting the role of PTMs in the modulation of α-Syn aggregation (Baba et al., 1998; Crowther et al., 1998; Giasson et al., 2000; Gomez-Tortosa et al., 2000; Campbell et al., 2001; Hasegawa et al., 2002; Anderson et al., 2006; Paleologou et al., 2010; Rott et al., 2017).
3. α-Syn post-translational modifications as possible biomarkers for PD and other synucleinopathies
Of note, α-Syn and post translational modified α-Syn in peripheral and accessible tissues have been investigated as possible biomarkers for the diagnosis of PD and other synucleinopathies. Nevertheless, since none of them has been validated across different cohorts so far, we still miss a clear cut evidence supporting their factual clinical significance (Witt et al., 2009; Pouclet et al., 2012; Shannon et al., 2012; Donadio et al., 2014, 2018; Sprenger et al., 2015; Zange et al., 2015; Stokholm et al., 2016; Vilas et al., 2016; Fereshtehnejad et al., 2017).
Biomarkers are defined as cellular, biochemical or molecular alterations that are measurable in biological samples such as human tissues, cells, or fluids (Hulka, 1990). The definition has been extended in order to define biomarkers as biological characteristics that can be objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention (Naylor, 2003). In particular, biomarkers include tools and technologies that can help disease prediction, cause, diagnosis, progression, regression, or the outcome of treatments (Mayeux, 2004). The importance of biomarkers is particularly relevant in the context of diseases affecting CNS, where it is impossible to have the direct access to the unhealthy tissue. CNS biomarkers detection can be pursued by positron emission tomography (PET) or magnetic resonance imaging (MRI) as well as by biological fluids [blood, cerebrospinal fluid (CSF), saliva], skin and gastrointestinal system biopsies or nasal mucosa analysis.
The fact that α-Syn can be found in different forms (monomeric, oligomeric, aggregated or post translational modified) in accessible and peripheral tissues such as CSF, blood, saliva, tears, colon, esophagus and skin (Tokuda et al., 2010; Devic et al., 2011; Foulds et al., 2011, 2012; Mollenhauer et al., 2013; Abd-Elhadi et al., 2015; Koehler et al., 2015; Chung et al., 2016; Cariulo et al., 2019; Fenyi et al., 2019; Hamm-Alvarez et al., 2019; Vivacqua et al., 2019; Maass et al., 2020; Wang et al., 2020b; Tanei et al., 2021; Bakhit et al., 2022), opened up the possibility to evaluate whether these different proteoforms may be useful for the diagnosis of PD and or other synucleinopathies (Witt et al., 2009; Pouclet et al., 2012; Shannon et al., 2012; Donadio et al., 2014, 2018; Sprenger et al., 2015; Zange et al., 2015; Stokholm et al., 2016; Vilas et al., 2016; Fereshtehnejad et al., 2017; Fayyad et al., 2019; Parnetti et al., 2019; Vivacqua et al., 2019, 2023; Wang et al., 2020b; Ganguly et al., 2021).
Several studies demonstrated that the levels of α-Syn phosphorylated at serine 129 (p-ser129), a PTM that is considered a marker of mature α-Syn aggregates (Ghanem et al., 2022), are elevated in the CSF and plasma of PD patients (Foulds et al., 2011, 2012; Wang et al., 2014; Landeck et al., 2016; Majbour et al., 2016a,b), while total α-Syn levels are decreased (Vivacqua et al., 2019, 2023). Remarkably, the levels of p-ser129 α-Syn were also found to significantly correlate with symptom severity in PD patients, suggesting that p-ser129 may serve as a biomarker for disease progression (Wang et al., 2014; Stewart et al., 2015).
In a recent study, increased levels of total and aggregated α-Syn in the membrane fraction of erythrocytes and high levels of p-ser129 α-Syn in cytosolic fractions were detected in PD cases versus healthy controls (Tian et al., 2019). Another report that analyzed oxidized and p-ser129 α-Syn demonstrated that higher levels of total and proteinase K resistant α-Syn and p-ser129 α-Syn can be detected in PD patients with motor symptoms (without dementia) with a high degree of accuracy (Abd Elhadi et al., 2019). Interestingly, p-ser129 α-Syn can be detected in skin nerve fibers biopsies and saliva (Vivacqua et al., 2019, 2023; Bougea et al., 2019a; Infante et al., 2020; Wang et al., 2020a; De Bartolo et al., 2023). Interestingly, α-Syn isolated from the skin and saliva has aggregation seeding activity and could serve as a biomarker for PD and as a differential biomarker to distinguish synucleoinopathies from tauopathies (Wang et al., 2020b).
p-ser129 α-Syn has also been detected in the lysate of red blood cells in synucleinopathies (Tian et al., 2019; Li et al., 2020, 2021). Higher levels of both Tyrosine (tyr) 125-phosphorylated α-Syn (p-tyr125) and p-ser129 α-Syn can be also detected in the blood of PD patients (Foulds et al., 2011, 2013; Vicente Miranda et al., 2017a).
Two recent meta-analysis showed that patients with PD have higher blood oxidative stress (OS) markers such as malondialdehyde (MDA), 8-Oxo-2′-deoxyguanosine lipid hydro-peroxide, nitrate and ferritine and lower antioxidant activity of superoxide dismutase (SOD), glucose 6 phosphate dehydrogenase, catalase, and glutathione peroxidase (GPx) compared with healthy control (Khan and Ali, 2018). Nitration of tyr and tryptophan residues as a consequence of the formation of peroxynitrite byproducts easily occurs at OS sites, i.e., in inflamed tissue, and can alter the structure and function of proteins. Nitric oxide (NO) and superoxide react to form peroxynitrite which promotes the nitrification of tyr residues in proteins. Specifically, the nitro group (−NO2) is added to replace a hydrogen atom in the 3′ position of the tyr phenolic ring to form 3-nitrotyrosine (Chavarria and Souza, 2013). Several studies reported the presence of nitrated α-Syn in in vivo and in vitro experimental models of PD and also in LB (Giasson et al., 2000; Yu et al., 2010; He et al., 2019; Manzanza et al., 2021; Simon et al., 2021; Magalhaes and Lashuel, 2022). Of note, Fernandez et al. (2013) reported the presence of tyr125/136 nitrated α-Syn in the CSF and serum of early PD patients, while a more recent study showed increased levels of nitrated α-Syn at tyr39 (n-tyr39) in the red blood cells of PD patients (Vicente Miranda et al., 2017a). In the same study, Vicente Miranda et al. (2017a) showed also reduced levels of SUMOylated α-Syn and increased levels of glycated α-Syn in PD patients erythrocytes with respect to controls. Since SUMOylation can increase α-Syn solubility and reduce aggregation (Krumova et al., 2011) and glycation can potentiate neuronal loss and motor impairment (Vicente Miranda et al., 2017b), the observed results may reflect brain α-Syn pathological alterations and toxicity (Vicente Miranda et al., 2017a,b).
These findings suggest that α-Syn PTMs, and in particular α-Syn nitration or phosphorylation, can be valuable biomarkers for synucleinopathies. This notwithstanding, we miss large cross-sectional and follow-up studies that will be pivotal for the implementation of post-translationally-modified α-Syn as a biomarker and we need to standardize the most reliable detection methods and several technical issues dealing with the detection or quantification of α-Syn have to be solved (Schmid et al., 2013; Mollenhauer et al., 2017; Magalhaes and Lashuel, 2022; Petricca et al., 2022). Indeed, the assay developed in the different studies exhibited different sensitivity and specificity and also led to conflicting results (Malek et al., 2014; Vivacqua et al., 2019, 2023; Bougea et al., 2019a,b; De Bartolo et al., 2023). For instance, Lin et al. (2019) recently reported a marked increase in total and phosphorylated α-Syn levels as well as in their ratio in the plasma of PD patients vs. healthy controls with assays exhibiting elevated specificity (AUC of ROC curves: 0.94, 0.91 and 0.74, respectively). This is in contrast to the findings of a previous study (Foulds et al., 2012) describing a reduction of total α-Syn and a parallel increase in phosphorylated α-Syn levels detected in the plasma of PD patients with a phosphorylated α-Syn assay exhibiting a ROC AUC = 0.68. Consistently, other reports showed that levels of phosphorylated α-Syn are increased in spite of the decrease of total α-Syn levels in plasma of PD patients (Hong et al., 2010; Gorostidi et al., 2012; Cariulo et al., 2019). When considering that because of sensitivity and specificity issues even CSF or plasma α-Syn cannot be considered as valuable markers of PD yet, it is clear that, as the reliable detection of post-translationally modified α-Syn is even more problematic, much work is warranted for achieving the exhaustive clinical translation of these kind of assay. This notwithstanding, the integrated measurement of α-Syn PTM may offer the possibility to single out patient-specific signatures that in the future could be of great help to settle precision-medicine-based approaches if disease-modifying therapies targeting α-Syn pathology will be developed.
4. Phosphorylation
Among α-Syn PTMs, phosphorylation is the most studied. The primary cause of this interest is mainly due to the fact that in normal brains only 4% of α-Syn is phosphorylated, whereas in LB extracted from PD brains 90% of α-Syn is phosphorylated at ser87 (p-ser87) and at ser129 (Anderson et al., 2006; Paleologou et al., 2010). Other sites of phosphorylation have been found on tyr residues at position 39, 125, 133, and 136.
Phosphorylation is the chemical addition of a phosphoryl group (PO3−) to an organic molecule. Phosphorylation and dephosphorylation (the removal of a phosphoryl group) are carried out by enzymes (e.g., kinases, phosphatases) and the processes orchestrate a plethora of cellular functions in response to external stimuli. In vitro and cell culture-based studies have identified a number of kinases, which phosphorylate α-Syn at ser129 and/or ser87, including casein kinase I (CKI; ser87 and ser129), casein kinase II (CKII; ser129; Okochi et al., 2000), G protein-coupled receptor kinases (GRKs 1, 2, 5 and 6; ser129; Pronin et al., 2000), leucine-rich repeat kinase 2 (LRRK2; ser129; Qing et al., 2009b), polo-like kinase (PLK; ser129; Inglis et al., 2009, Mbefo et al., 2010) protein kinase C-related kinase (PKR; ser129; Reimer et al., 2018) and LK6/Mnk2a (ser129; Zhang et al., 2015).
α-Syn phosphorylation at tyr125 can be mediated by the proto-oncogene tyrosine-protein kinase Fyn (Nakamura et al., 2001) and SRC proto-oncogene non-receptor (Src) tyr kinases such as spleen associated tyrosine kinase (Syk), the non-receptor tyrosine-protein kinase Lyn, the protein tyrosine kinase expressed by the protooncogene c-fgr (Ellis et al., 2001; Negro et al., 2002). Syk also phosphorylates α-Syn at try133 and tyr136.
Although the contribution of α-Syn pathology to LRRK2-associated PD is debated (Schneider and Alcalay, 2017) and the relevance of LRRK2-mediated α-Syn phosphorylation in PD is still to be determined, several studies reported that LRRK2 co-localizes with α-Syn in the lower brainstem of PD and LBD patients at early stages (Alegre-Abarrategui et al., 2008; Qing et al., 2009b; Zimprich et al., 2011). Still, in vitro studies hint that G2019S-mutant LRRK2 exhibit an improved ability to phosphorylate α-Syn on ser129 when compared to wt LRRK2 (Qing et al., 2009a).
On the other hand, the phosphatases involved in the dephosphorylation are phosphoprotein phosphatase 2A and 2C (PP2A and PP2C).
Increased ser129 α-Syn phosphorylation has been detected in PD, LBD and MSA (Kahle et al., 2000; Okochi et al., 2000; Fujiwara et al., 2002; Takahashi et al., 2003; Anderson et al., 2006). A recent study analyzing post-mortem tissue from PD and MSA patients at different disease stages reported that ser129 α-Syn phosphorylation is the dominant and earliest PTMs, while lower amounts of p-ser87 α-Syn appeared later along PD progression (Sonustun et al., 2022).
Almost all phosphorylation sites cluster at the C-terminal region of α-Syn (residues 120–140), which is involved in protein–protein, protein-ligand and protein-metal interactions, suggesting a possible role of the modification in the regulation of these functions. Only ser87 lies in the hydrophobic NAC region of α-Syn, which is essential for α-Syn aggregation and fibrillogenesis (El-Agnaf et al., 1998b).
Ser129 is the most studied phosphorylation site because it was linked with increased cytotoxicity and neuronal death (Zhang et al., 2015; Karampetsou et al., 2017; Zhong et al., 2017; Reimer et al., 2018). Furthermore, it has been described that p-ser129 enhances intracellular aggregate formation in SH-SY5Y cells (Smith et al., 2005b) and mediates cell death through activation of the unfolded protein response (UPR) pathway (Sugeno et al., 2008). Still, Karampetsou et al. (2017) observed that mice who received intrastriatal injection of p-ser129 α-Syn exhibited enhanced α-Syn pathology deposition and neurodegeneration in the substantia nigra (SN) compared to the mice injected with wild type (wt) α-Syn.
However, other studies in cellular and animal models claimed that phosphorylated α-Syn exherts a neuroprotective role (Gorbatyuk et al., 2008; Oueslati et al., 2012; Tenreiro et al., 2014; Ghanem et al., 2022). In particular, it has been demonstrated that p-ser129 phosphorylation occurs secondarily to α-Syn accumulation, reducing cytotoxicity and aggregation propensity of α-Syn (Ghanem et al., 2022). Interestingly, p-tyr125 α-Syn can also prevent α-Syn neurotoxicity and aggregation and is pivotal for ser129 phosphorylation (Kosten et al., 2014).
The role of p-ser87 is also controversial as this PTM falls in the NAC region of α-Syn, which is crucial for α-Syn aggregation and fibrillogenesis in vitro (Ueda et al., 1993; El-Agnaf et al., 1998a,b; Giasson et al., 2001). In addition, though p-ser87 phosphorylation is increased in the membrane fractions of post mortem brains of patients affected by LBD, MSA and AD and healthy controls and of rats overexpressing wt α-Syn, p-ser87 was found to reduce α-Syn membrane binding (Paleologou et al., 2010), supporting that this phosphorylation may be crucial for modulating the physiological effect of α-Syn on synaptic vesicle mobility. Moreover, the unilateral p-ser87 α-Syn overexpression in the nigrostriatal system of rats results in reduced formation of aggregates and does not exert toxicity for nigral dopaminergic neurons in contrast to what has been observed following wt α-Syn overexpression (Decressac et al., 2012; Lundblad et al., 2012; Oueslati et al., 2012; Faustini et al., 2018).
Differently, p-tyr125 was reported to decrease with aging and in PD brains, in Drosophila melanogaster and mice (Chen et al., 2009). As this phosphorylation has been found to reduce α-Syn oligomerization, it has been hypothesized that it may play a protective role against aggregate formation (Chen et al., 2009). On this line, Negro et al. (2002) showed that the kinase Syk phosphorylates the C-terminal tyr125 of α-Syn to block α-Syn fibrillation. Moreover, p-tyr125 facilitates the deposition of p-ser129 under physiological conditions (Kosten et al., 2014).
PLK2 has been found to phosphorylate α-Syn, but not β-or γ-syn, at ser129 in HEK293T cells and in primary neurons (Arawaka et al., 2006; Inglis et al., 2009; Mbefo et al., 2010). In particular, PLKs can phosphorylate both monomeric or fibrillary α-Syn (Waxman and Giasson, 2011) and overexpression of PLK2 enhances α-Syn turnover via the autophagic degradation pathway, thus suppressing its toxicity in vivo (Oueslati et al., 2013). Despite the role of PLK2 in centriole duplication and cell cycle regulation, PLK2 inhibitors do not appear to cause cytotoxicity nor genotoxicity in vitro or in vivo at doses and exposures that engage the target in rat (Fitzgerald et al., 2013), but clinical trials on PLK2 inhibitors have shown difficulties in targeting specifically PLK2 in order to avoid off-target-related side effects (Vancraenenbroeck et al., 2011).
c-Abelson tyrosine kinase (c-Abl) is a 120 kDa protein majorly known in relation to human leukemias. c-Abl is distributed in the nucleus and cytosol and is involved in a wide range of functions, including apoptosis and development of the CNS in which it affects neurogenesis, neurite outgrowth, and neuronal plasticity. Moreover, it is involved in several neurodegenerative diseases including PD (Tremblay et al., 2010; Imam et al., 2011). For instance, c-Abl is elevated in postmortem nigrostriatal region of PD patients (Ko et al., 2010; Imam et al., 2011) where it is majorly phosphorylated at tyr412 (Mehdi et al., 2016). c-Abl was found to phosphorylate parkin thus impairing its E3 ligase activity and leading to the loss of dopaminergic neurons in the SN (Ko et al., 2010). It has been described that c-Abl aberrant activation induced a progressive accumulation of α-Syn in the human A53T mutant α-Syn tg mouse model of genetic PD (Brahmachari et al., 2016) through the phosphorylation at tyr39 (Mahul-Mellier et al., 2014; Brahmachari et al., 2016; Dikiy et al., 2016), thus contributing to neurodegeneration. Furthermore, c-Abl is activated by OS (Brasher and Van Etten, 2000; Sun et al., 2000; Gonfloni et al., 2012), and in turn it disrupts antioxidant defense mechanisms driving oxidative injury (Li et al., 2004). It may thus be inferred that c-Abl inhibitors may impact on α-Syn pathology by affecting the phosphorylation and nitration state of the protein.
Consistently, Hebron et al. (2013) showed that c-Abl activation promotes α-Syn accumulation and that the treatment with nilotinib, a brain-permeable second-generation c-Abl inhibitor, developed from the first generation anticancer agent, named imatinib, favored the clearance of α-Syn, improved motor performances (Hebron et al., 2013), restored the levels of dopamine transporter (DAT) and dopamine production in the striatum as well as the expression of tyrosine hydroxylase (TH) in the SN (Hebron et al., 2013, Karuppagounder et al., 2014; Table 2).

Table 2. Kinase-inhibitors tested in preclinical models of PD and in clinical trials.
Of note, results from nilotinib clinical trials showed that the drug could reduce oligomeric α-Syn (only at 150 mg dose) as well as phosphorylated tau. Nilotinib treatment also improved dopamine metabolism in patients with PD. In particular, it increased the levels of homovanillic acid (HVA) and 3,4-Dihydroxyphenylacetic acid (DOPAC) in the CSF (Pagan et al., 2016, 2020) but without improving motor and nonmotor outcomes.
Simuni et al. (2021) run a double-blind, placebo-controlled trial on 173 PD patients. The results about safety, tolerability, adverse effects and lack of the symptomatic effect of nilotinib were in line with the study by Pagan et al. (2020). However, they could not observe changes in biomarkers. Although these evidences support that nilotinib is not suitable for further testing the collected data did not exclude the importance of c-Abl modulation in PD therapeutic strategy (Simuni et al., 2021).
The fact that no clinically meaningful benefit in PD patients in two double-blind studies was reported, is discouraging, but this can find an explanation by the fact that nilotinib does not accumulate in the brain at concentrations sufficient to inhibit c-Abl. As a competitive inhibitor of c-Abl with an IC50 of ≈48 nM it would require a sustained concentration of 150 nM to exert the adequate functions (Pagan et al., 2019). Other c-Abl inhibitors such as IkT-148,009 and vodobatinib (Table 2), are currently under development. The chronic oral treatment with IkT-148,009 was found to significantly reduce p-tyr39 and p-ser129 α-Syn levels thus preventing neurodegeneration in the brain of human A53T mutated α-Syn transgenic (tg) mice and of mice who received striatal injections of mouse recombinant α-Syn pre-formed fibrils (PFF; Karuppagounder et al., 2023). IkT-148009 is a derivative of the commercial anticancer imatinib and it has an IC50 of 33 nM for c-Abl, an improvement in potency of more than 20-fold over imatinib (Werner and Olanow, 2022). The randomized phase I/Ib study in older adult or elderly healthy volunteer was then extended to PD patients to identify the safety, tolerability, maximum tolerated dose and the pharmacokinetic profile of the molecule in single doses up to 325 mg and multiple doses up to 100 mg (Clinical trial identifier: NCT04350177). A randomized, double-blind study in non-treated PD patients is also ongoing (Clinical trial identifier: NCT05424276).
Vodobatinib, also known as K0706 or as SCC-138 is a chemical mixture of other two commercial anticancer agents (Dasatinib and Ponatinib) and it has a reported IC50 for wt c-Abl of 0.9 nM (Antelope et al., 2019). In preclinical models of PD it has been shown that it inhibited preferentially, with a sub-nanomolar potency, the protein kinase activity of c-Abl. Moreover, it increased autophagic flux, it had appreciable BBB penetration in vivo and protected both 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mice and rats overexpressing α-Syn from nigrostriatal neuron loss (Mandhane et al., 2019).
A phase I clinical trial showed that vodobatinib was well-tolerated and allowed the selection of two doses that are likely to produce therapeutic effects (Clinical trial identifier: NCT03316820). A new double-blind, placebo-controlled phase II study is now recruiting for evaluating the safety and effectiveness of the two selected K0706 doses in people with early PD who are not receiving dopaminergic therapy (Clinical trial identifier: NCT03655236). The primary endpoints focus on changes from baseline in the sum of Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Parts 2 and 3, but among other outcome measures there will be the evaluation of CSF and blood K0706 levels and dopamine transporter single-photon emission computed tomography (DAT-SPECT).
Collectively, the above summarized studies on c-Abl inhibitors support that the use of protein kinase modulators in PD may be beneficial. Nevertheless, we need to achieve a deeper understanding of the role of the α-Syn phosphorylation and, more generally, on protein kinase and phosphatases activity in synucleinopathies, before to conclude that strategies modulating this PTM may constitute a possible therapeutic approach for this class of neurodegenerative disorders.
5. Nitration
Post-mortem PD brains are rich in lipid peroxidation products such as 4-hydroxyl-2-nonenal (HNE) as well as DNA and RNA oxidation products (Alam et al., 1997; Floor and Wetzel, 1998; Zhang et al., 1999). Moreover, several lines of evidence support that OS is involved in the degeneration of dopaminergic neurons in PD (Jenner and Olanow, 2006; Schapira and Tolosa, 2010).
OS is the result of a disequilibrium between the production of reactive oxygen species (ROS) or reactive nitrogen species (RNS) and the system for the detoxification leading to the production of free radicals byproducts that damage proteins, lipids, nucleic acids and organelles (Ryan et al., 2014). Although the brain represents only 2% of the body weight, it consumes 20% of the total body oxygen (Quastel and Wheatley, 1932; Magistretti and Pellerin, 1996), which is majorly converted in ROS. To defend against oxidative injuries, cells own a series of enzyme-based antioxidant mechanisms, such as glutathione (GSH), SOD and DJ-1. However, these systems are feeble in preventing the damage. In particular, nigral dopaminergic neurons are particularly sensitive to oxidative injuries as they own long, highly branched axons with a huge number of release sites that renders these cells bioenergentically demanding and at risk of developing mitochondrial OS (Pissadaki and Bolam, 2013). Nigral dopaminergic neurons also own a pacemaking activity characterized by broad and slow action potentials in the absence of synaptic input (Grace and Bunney, 1983). This activity engages continuously L-type Ca2+ channel, creating a basal mitochondrial OS in SN dopaminergic neurons (Guzman et al., 2010) and elevating intracellular Ca2+ levels (Wilson and Callaway, 2000; Chan et al., 2007). In light of the fact that cytoplasmic Ca2+ controls a huge number of pathways within a cell, its presence inside a neuron must be strictly controlled, and it is rapidly sequestered or pumped back in an ATP-dependent manner, thus resulting highly energy demanding (Wilson and Callaway, 2000). Still, dopamine turnover by monoamine oxidases (MAO) is involved in the production of cytotoxic free radicals, causing the death of dopaminergic neurons (Greenamyre and Hastings, 2004). Among them, the MAO-derived dopamine catabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL) exhibits an enhanced reactivity towards proteins especially at synaptic terminals (Rees et al., 2009) and has been recently found to contribute to the initiation of α-Syn-dependent impaired proteostasis and degeneration of neuronal projections in different experimental models of PD (Masato et al., 2023).
Consistently, it is well established that mitochondria dysfunction is crucially involved in the pathogenesis of PD. This is also supported by the fact that several gene mutations implicated in familial and idiopathic forms of PD are located on loci encoding for mitochondria-linked proteins (Moore et al., 2005; Abou-Sleiman et al., 2006; Schapira, 2008). Moreover, postmortem studies on the SN of sporadic PD patients reported a decreased activity of mitochondrial complex I and higher levels of iron in the SN (Mann et al., 1994; Keeney et al., 2006). Free iron is toxic since it can donate or accept an electron from neighboring molecules and cause damage to cellular components and it can create ROS through the Fenton and Haber-Weiss reaction, in which ferric iron (Fe3+) and ferrous iron (Fe2+) react with superoxide and hydrogen peroxide to form hydroxyl radical (Beard and Connor, 2003; Jomova and Valko, 2011; Eid et al., 2017). Neuromelanin, the dark colored granular pigment present in the dopaminergic neurons of the SN, has the ability to chelate metals, in particular the ferric Fe3+ form (Gerlach et al., 2003), thus blocking the Fenton reaction and protecting the cells from hydroxyl radical production. The huge increase of iron found in SN of PD brains might saturate the iron-chelating site of neuromelanin, increasing the production of free radical species. Finally, neuroinflammation can also contribute to OS in the PD brain (Mosley et al., 2006; Picca et al., 2020; Teleanu et al., 2022).
The interplay between α-Syn and OS is still not fully elucidated. In vitro and in vivo studies support that increased OS in the brain may promote α-Syn aggregation (Paxinou et al., 2001), but α-Syn itself can increase ROS production (Junn and Mouradian, 2002; Winklhofer and Haass, 2010) or it can bind to mitochondrial complex I causing mitochondrial dysfunction in turn favoring OS (Chinta et al., 2010; Winklhofer and Haass, 2010; Wilkaniec et al., 2013).
Nitrated α-Syn can be easily formed under OS conditions. α-Syn has four tyr residues, placed in positions 39 (at the N-terminal region), 125, 133, and 136 (at the C-terminal region). The positions of the nitration sites suggest a possible modulation of membrane binding ability (Hodara et al., 2004) and protein–protein and protein-metal interactions. α-Syn is sensitive to the presence of nitrating agents and the presence of peroxynitrite not only induces the deposition of 3-nitrotyrosines but also the formation of 3,3-dityrosine via the oxidation of tyr residues, which results in α-Syn dimers and oligomer formation (Souza et al., 2000). Danielson et al. (2009) demonstrated a selective 9-fold increase in nitration on tyr39 of α-Syn in oxidative cellular model of PD. In addition, nitration of tyr39 induces high rate of oligomerization (Hodara et al., 2004) similarly to n-tyr125 that contributes to α-Syn dimer formation upon the exposure of recombinant α-Syn to nitrating agents (Takahashi et al., 2002).
Interestingly, a recent study analyzing post-mortem tissue from PD and MSA patients at different disease stages reported that ser129 α-Syn phosphorylation is the dominant and earliest PTMs, followed by tyr39 nitration, while lower amounts of p-ser87 α-Syn appeared later along PD progression (Sonustun et al., 2022). However, in the MSA brain glial cytoplasmatic inclusions, neuronal inclusions and small threads are mainly positive for tyr39 nitrated while ser129 α-Syn can be mainly detected in Schwan cell and neuronal inclusions (Sonustun et al., 2022; Wakabayashi et al., 2022).
Nitrated α-Syn monomers and dimers have been shown to accelerate fibril formation while nitrated α-Syn oligomers inhibit this process (Hodara et al., 2004). This supports that improving the amount of nitrated α-Syn oligomers may delay the formation of mature fibrils. This notwithstanding, as we still ignore whether fibrils or oligomers are the major neurotoxic species in PD, it is hard to predict whether this may be beneficial or detrimental.
Nevertheless, it may be feasible that antioxidant supplementation may be used to reduce α-Syn nitration. In this framework, some antioxidant schemes have been attempted, such as the supplementation of vitamin C, E and β-carotene as well as an adequate diet (Percario et al., 2020). Vitamin A and its precursor β-carotene, have been involved in the destabilization of fibrillary α-Syn in vitro (Ono et al., 2004; Ono and Yamada, 2007). Vitamin E (i.e., α-tocopherol) and Vitamin C (i.e., ascorbic acid) are antioxidants that are thought to have a protective effect by either reducing or preventing oxidative damage, preventing or interacting directly with free radicals, respectively. A lot of studies tried to investigate the relation between the intake of vitamins and the protection from PD, but they generated only conflicting results (Kieburtz et al., 1994; Hellenbrand et al., 1996; Morens et al., 1996; de Rijk et al., 1997; Scheider et al., 1997; Etminan et al., 2005; Miyake et al., 2011; Hughes et al., 2016; Schirinzi et al., 2019; Zhao et al., 2019). It has been demonstrated that NXP031, a new compound composed of aptamin C and vitamin C, blocks α-Syn aggregation in the hippocampus of AAV-human α-Syn-injected mice (Song et al., 2022). Similarly, also vitamin B12 was found to inhibit α-Syn fibrillogenesis in in vitro models (Jia et al., 2019).
Recent studies on MPTP in vivo and in vitro models support that γ-and δ-tocotrienol reduces dopaminergic neuron toxicity and improves motor performances through estrogen receptor/PI3K/Akt signaling pathway activation, hence in an antioxidant-independent way (Matsura, 2019). The supplementation of α-and δ-tocotrienol significantly ameliorates motor behavior and prevents the loss of nigra dopaminergic neurons and striatal fibers and neuroinflammation in 6-Hydroxydopamine (6-OHDA)-injected rats (Kumari et al., 2021). The vitamin E family compound tocotrienol is currently under study as a potential agent to delay motor symptoms in PD patients at Hoehn & Yahr stage 2 in a phase II clinical trial (Clinical trial identifier: NCT04491383; Table 3).
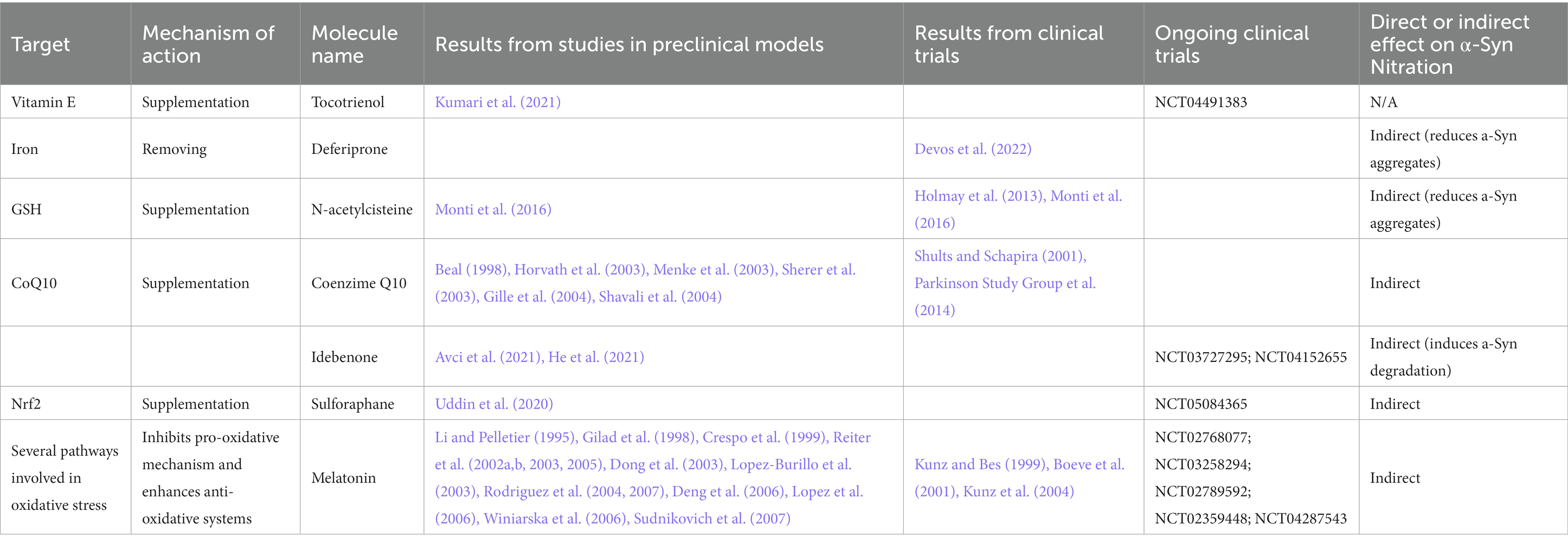
Table 3. List of OS modulators tested in preclinical models of PD and in clinical trials.
This notwithstanding, a multicenter, phase II, randomized, double-blind trial in early drug-naïve PD patients evaluating the efficacy of the iron chelator deferiprone (Table 3) on disease progression indicate that 36 weeks of therapy with deferiprone could remove specifically, safely and gradually the iron content in the nigrostriatal system of PD patients but it worsened the progression of symptoms (Devos et al., 2022). Studies on the efficacy of deferiprone in experimental in vivo models of synucleinopathies led to conflicting results. Indeed, human A57T α-Syn tg mice showed improvement in behavioral performances upon deferiprone treatment but without reduction of α-Syn aggregation (Carboni et al., 2017), while deferiprone treated mouse model of MSA exhibited rescued motor performance, higher neuronal survival and reduced density of α-Syn aggregates in SN (Shukla et al., 2021).
Another possible strategy to counteract OS is based on GSH rebalancing. In particular, since GSH is neither able to pass the blood brain barrier (BBB) nor the cellular membrane of neurons, the dietary supplementation of this enzyme is not possible. However, cysteine, which is rate-limiting in the GSH synthesis pathway, crosses both the BBB and most cell membranes. Therefore, cysteine and its derivative N-acetylcysteine have been investigated as a possible dietary supplementation to implement GSH amount, with several clinical trials ongoing (Table 3). Intravenous N-acetylcysteine injection increased blood GSH redox ratios in PD and healthy subjects and magnetic resonance spectroscopy (MRS) showed higher brain GSH concentrations in all subjects. This supports that it is possible to directly monitor GSH levels that could help during clinical trial to determine the activities and the doses of this antioxidant therapy (Holmay et al., 2013).
Another study aimed at assessing the effect of N-acetylcysteine on human embryonic stem cells-derived midbrain dopaminergic neurons treated with rotenone and on PD patients and showed that N-acetylcysteine exposure significantly improved the survival of midbrain dopaminergic neurons treated with rotenone (Monti et al., 2016). Furthermore, Dopamine Transporter scan (DaTscan) analysis on patients treated for 3 months with N-acetylcysteine resulted in increased DAT binding in the caudate and putamen (Monti et al., 2016). These results support a potential direct effect of N-acetylcysteine (Table 3) on the dopamine system in PD patients, but we still ignore whether this compound affects α-Syn nitration state though N-acetylcysteine has shown protective effects against the damage in dopaminergic terminals concomitant with a reduction in α-Syn levels in transgenic mice (Clark et al., 2010).
Coenzime Q10 (CoQ10) is a key component of the electron transport chain that leads to decreased free radical generation, and, in its reduced form, acts as a powerful antioxidant (Shults, 2005). CoQ10 levels were altered in PD cases (Matsubara, 1991; Shults et al., 1997; Molina et al., 2002) with a significant increase in the percentage of oxidized CoQ10 in affected patients (Sohmiya et al., 2004). Numerous studies in in vitro and in vivo models of PD demonstrated that CoQ10 protects neurons against MPTP and rotenone toxicity (Beal, 1998; Horvath et al., 2003; Menke et al., 2003; Sherer et al., 2003; Gille et al., 2004), and 1-Benzyl-1,2,3,4-tetrahydroisoquinol (Shavali et al., 2004; Table 3).
A randomized, double-blind, placebo-controlled, multicenter phase II study in early PD examined the effects of 300, 600, and 1,200 mg per day of CoQ10 vs. placebo. CoQ10 supplementation decreased functional decline in participants and increased platelet mitochondrial complex I and II/III activities. These results suggested a possible disease-modifying effect (Shults and Schapira, 2001). Based on these results, in a phase III study, the group tested whether high doses (1,200 and 2,400 mg/d) of CoQ10 could slow functional decline in early PD. The results showed that CoQ10 could be safely administered to patients with early PD, however no therapeutic efficacy was demonstrated (Parkinson Study Group et al., 2014).
The hydrophilic analogue of CoQ10, idebenone, is well-known antioxidant compound with better pharmacological properties. Clinical safety of idebenone was well described, and the molecule is currently used to treat Freidrich’s ataxia and AD (Orsucci et al., 2011; Montenegro et al., 2018). Two clinical trials assessing the efficacy and safety of idebenone in PD are currently ongoing (Clinical trial identifier: NCT03727295; NCT04152655) and results obtained on PD models are encouraging (Table 3). Indeed, idebenone improved motor coordination and locomotor activity while decreasing TH-positive neurons damage, lipid peroxidation, ferroptosis and other OS markers in rotenone-induced PD models (Avci et al., 2021). Moreover, idebenone activated autophagy and promoted α-Syn degradation by suppressing the AKT/mTOR pathway in SH-SY5Y overexpressing the A53T mutant form of α-Syn (He et al., 2021). This mechanism appears unusual for this compound, but recently idebenone has been demonstrated to act as cytoprotective molecule activating fundamental pathways rather than by functioning as a direct antioxidant agent (Gueven et al., 2021; He et al., 2021).
A new interesting agent for OS modulation is sulforaphane, a phytocompound belonging to the isothiocyanate family and owning lipophilic nature and a molecular size that makes it highly bioavailable (Schepici et al., 2020; Uddin et al., 2020). Its molecular target is nuclear factor erythroid 2 related factor 2 (Nrf2), which is a crucial controller of enzymes involved in antioxidation and detoxification of xenobiotics (Eggler et al., 2008; Zhang et al., 2013; Stefanson and Bakovic, 2014; Sajja et al., 2017). In vitro studies on cellular models of PD treated with sulforaphane showed reduced OS, cell damage and death (Uddin et al., 2020; Table 3). In line with the in vitro studies, in vivo experiments demonstrated that in C57BL/6 mice sulforaphane administration improved motor deficits and counteracted nigrostriatal dopaminergic neurons degeneration and apoptosis attenuating OS and neuroinflammation (Uddin et al., 2020). A phase II clinical trial is currently ongoing to evaluate the efficacy and safety of sulforaphane in PD patients (Clinical trial identifier: NCT05084365).
An interesting molecule to counteract OS is melatonin, a hormone produced endogenously by pineal gland and other tissues. It regulates circadian cycle and also plays a relevant role in neuroprotection, anti-inflammation and anti-oxidation. For all these reasons, it has been considered as a candidate for PD therapy (Table 3). Melatonin is an indoleamine and it can yield electron easily, hence it is a potent reducer agent. It acts as a scavenger for oxygen-and nitrogen-based reactive molecules (Reiter et al., 2002a,b, 2003; Lopez-Burillo et al., 2003; Sudnikovich et al., 2007) and it works as an inhibitor of inducible NO synthase (iNOS; Gilad et al., 1998; Crespo et al., 1999; Dong et al., 2003; Rodriguez et al., 2004, 2007; Lopez et al., 2006). The ability to interact with iNOS and peroxinitrite is the one that makes melatonin a special candidate for the treatment of OS as none of the previous mentioned antioxidant is able to exert this action. It has been demonstrated that melatonin also helps antioxidant enzymes, including SOD and GPx, stimulating the production of GSH (Rodriguez et al., 2004; Reiter et al., 2005; Winiarska et al., 2006). In addition, melatonin has been found to inhibit cyclooxygenase-2 reducing the severity of inflammation (Deng et al., 2006). In particular, it ameliorates inflammation blocking tumor necrosis factor-α (TNF-α; Li and Pelletier, 1995; Reiter et al., 2003) and it impacts on mitochondrial respiration, protecting both proteins of electron transport chain and mitochondrial DNA from oxidative damage (Reiter et al., 2008). Interestingly, melatonin has been found to reduce α-Syn secretion in rat adipose-derived mesenchymal stem cells (Ibrahim et al., 2022). Several phase II and III clinical trials are evaluating the effect of melatonin on sleep disturbances in PD patients (Clinical trial identifiers: NCT02768077; NCT03258294; NCT02789592; NCT02359448; NCT04287543; Table 3). Interestingly, trial NCT04287543 aimed at following the activity of mitochondrial complex I, the levels of MDA and 4-hydroxyalkene and the production of NO among the secondary outcome measures, but it was withdrawn because of COVID-19 pandemic. Other studies on exogenous melatonin investigated the effect of the molecule on rapid eye movement (REM) sleep behavior disorder (RBD), which is a prodromal sign for PD. Among them, the study by Kunz et al. (2004) demonstrated that medical melatonin increased REM sleep percentage to normal levels in patients with reduced REM sleep duration and re-organized REM sleep episode length during night-time sleep. The effect lasted for several weeks after the discontinuation of the therapy. Other studies reported a resolution of clinical RBD symptoms lasting for up to 3 years after discontinuation of melatonin treatment (Kunz and Bes, 1999; Boeve et al., 2001; Kunz et al., 2004).
It is worth considering that unfortunately the limitations offered by OS targeting therapeutic strategies are challenging. Moreover, despite OS is common to several diseases, it rarely constitutes the primary cause of a disease, supporting that the use of an antioxidant may have mild impact on pathology progression. Moreover, in vitro and in vivo evidences demonstrated that endogenous antioxidants support the progression of different types of tumors (Singh et al., 2008; DeNicola et al., 2011; Sayin et al., 2014; George and Abrahamse, 2020; Harris and DeNicola, 2020). This effect is even greater in older people, where the activation of Nrf2 pathway, which usually is chemopreventing, can be deleterious and it could predispose for tumor progression (Forman and Zhang, 2021). Still, all classical antioxidants, excluding melatonin, are potential electron donors and they exhibit both reduced and oxidized forms. In general, these oxidized molecules should be regenerated to the reduce form through a process of recycling that consumes GSH to be exploited or through a redox reaction that, eventually, oxidizes other molecules. This means that the classical antioxidant may act as prooxidant molecules, causing other damages. However, the toxic concentrations of most of these prooxidant regenerated compounds are extremely high and their toxic potential appears negligible.
Another issue is related to the discrepancy that exists in the ratio of in vitro vs. in vivo exogenous agents. In general, in in vitro studies free radicals are produced at much greater rates than what would be observed in real physiological or pathological conditions (Forman et al., 2014). In addition, antioxidant defenses may not be able to reach effective concentrations in vivo. Therefore, it is hard to think that antioxidant approaches may significantly impact on PD progression though we cannot exclude that they may contribute in reducing α-Syn nitration.
6. Acetylation
Protein acetylation is one of the major PTM found in eukaryotes, in which the acetyl group from acetyl coenzyme A is transferred to a specific site on a polypeptide chain. Acetylation is mostly known for the role on gene transcription regulation, indeed through the reversible accumulation of acetylation on the lysines (ac-lys) of the histones, the transcription is activated.
In humans, 80–90% of all proteins become co-translationally acetylated at their N-terminal (Nt) of the nascent polypeptide chains (Arnesen, 2009; Aksnes et al., 2015) in an irreversible way. Nt-acetylation is a general mechanism for stabilizing α-helical structures in both proteins and peptides (Chakrabartty et al., 1993), and makes α-Syn resistant for amyloid aggregation enhancing both protein–protein and protein-membrane interaction (Bartels et al., 2014). Indeed, recent findings indicate that all the in vivo detectable α-Syn is post-translationally modified by an acetyl group attached to the amino group of the first N-terminal amino acid (Anderson et al., 2006; Bartels et al., 2011; Ohrfelt et al., 2011). This modification alters the charge and structure of α-Syn molecules affecting their interaction with lipid membranes, as well as their aggregation process (Bell et al., 2022a,b, 2023). It has been found that ac-lys impacts on α-Syn aggregation (Fauvet et al., 2012; Kang et al., 2012; Gruschus et al., 2013; Bu et al., 2017; de Oliveira et al., 2017) and that acetylated α-Syn and α-tubulin inhibit oligomers formation (Kazantsev and Kolchinsky, 2008). Interestingly, studies demonstrated that increases in histone acetylation are disease-dependently associated with PD progression (Park et al., 2016; Harrison et al., 2018; Toker et al., 2021) and histone-3 or-4 hyperacetylation is a key epigenetic change in dopaminergic neurons exposed to other PD-related neurotoxins. Conversely, the deacetylation of histones operated by histone deacetylase (HDAC) is implicated in the control of α-Syn toxicity. The activity of HDAC6 has been linked with PD pathogenesis (Lemos and Stefanova, 2020) and HDAC6 is highly expressed in LB in PD patients’ brain sections, indicating that HDAC6 may play a key role in the clearance of those misfolded and aggregated protein (Kawaguchi et al., 2003; Du et al., 2010; Richter-Landsberg and Leyk, 2013). Indeed, HDAC6 decreased activity is an essential factor for impaired autophagic flux in PD pathophysiology (Wang et al., 2019). Several studies demonstrated that the inhibitors of HDAC worsen the motor abilities of mice and exacerbate cell death in primary neuron cells (Du et al., 2014), while other demonstrated that HDAC inhibitors restore axonal transport and motor behavior (Godena et al., 2014; Pinho et al., 2016), reduce ROS production, and alleviate dopaminergic neurotoxicity (Jian et al., 2017). Other studies demonstrated the protective effect of pan-HDAC inhibitors such as valproic acid, sodium butyrate, phenylbutyrate, suberoylanilide hydroxamic acid and trichostatin A in in vitro and in vivo models of PD acting through different mechanism listed in Table 4 (Gardian et al., 2004; Chen et al., 2007, 2012; Wu and Guo, 2008; Kidd and Schneider, 2010, 2011; Zhou et al., 2011, 2014; Rane et al., 2012; St Laurent et al., 2013; Harrison et al., 2015; Suo et al., 2015; Sharma et al., 2015a; Kim et al., 2019; Getachew et al., 2020; Hsu et al., 2020). The specific inhibitors of HDAC1, 2 and 3, RGFP109, K560, K-856, MS-275, MC-1568, and LMK235 also showed neuroprotection against α-Syn toxicity (Table 4; Johnston et al., 2013, Formisano et al., 2015, Choong et al., 2016, Hirata et al., 2018, Mazzocchi et al., 2021).
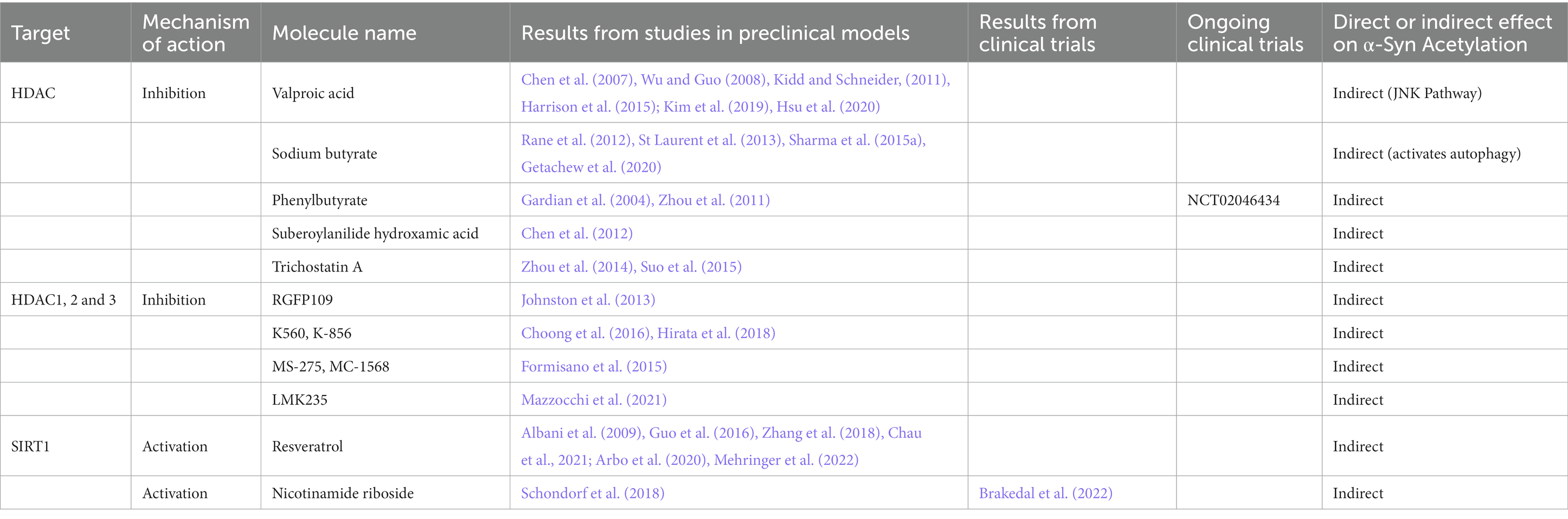
Table 4. HDAC-modulators tested in preclinical models of PD and in clinical trials.
On this line, a recent phase I clinical trial investigated whether phenylbutyrate (Table 4) can increase the removal of α-Syn from the brain into the bloodstream (Clinical trial identifier: NCT02046434), but results are not available yet.
Sirtuins (SIRT) are nicotinamide adenine dinucleotide (NAD+)-dependent HDAC, proteins implied in neurodegenerative disorders (Satoh and Imai, 2014). In mammals, there are seven members of the SIRT family: SIRT1-SIRT7. SIRT2 is the most abundant SIRT in the brain and its levels increase with aging (Maxwell et al., 2011). De Oliveira et al. (2017) recently described that SIRT2 interacts with and removes acetyl groups from α-Syn. They also demonstrated both in vitro and in vivo that the inhibition of SIRT2 decreased α-Syn toxicity (Outeiro et al., 2007; de Oliveira et al., 2017).
On the other hand, SIRT1 increases lifespan in mammals (Cohen et al., 2004), promotes mitochondrial biogenesis (Wenz, 2013), protects against neurodegeneration (Kim et al., 2007) and mitigates α-Syn pathology through the induction of the chaperone heat shock protein 70, which prevents the misfolding or clear the aggregates by degradation (Donmez et al., 2012). By reducing signs of aging, the SIRT1-activating drugs, such as resveratrol may have a role in the counteract of neurodegenerative diseases (Barger et al., 2008; Pearson et al., 2008). Indeed, resveratrol and its derivatives are able to alleviate motor and cognitive deficits and neuropathology in different mouse model of PD (Table 4; Guo et al., 2016, Zhang et al., 2018) and to reduce α-Syn toxicity and OS in in vitro models of the pathology (Albani et al., 2009; Arbo et al., 2020; Chau et al., 2021). Interestingly, though the bioavailability and brain penetration of resveratrol are problematic, some modified forms of this molecule have been developed to overcome these issues (Intagliata et al., 2019) and it has been demonstrated that one of the more bioavailable forms of resveratrol acts as a protein aggregation suppressor in vitro and in vivo (Mehringer et al., 2022).
The upstream regulation of SIRT through a replenishment of NAD within the brain has been attempted through the nicotinamide riboside supplementation. Brakedal et al. (2022) summarized the double-blinded, randomized, placebo-controlled phase I study of nicotinamide riboside in which they demonstrated a mild improvement in motor ability and a neuroprotective effect that was previously shown in murine, Drosophila melanogaster and induced pluripotent stem cells-based experimental models of noise induced hearing loss, amyotrophic lateral sclerosis, depression and PD (Table 4; Brown et al., 2014, Sorrentino et al., 2017, Schondorf et al., 2018, Han et al., 2020, Harlan et al., 2020, Xie et al., 2020). Nicotinamide riboside may target multiple processes implicated in the pathophysiology of the disease by upregulating the expression of genes involved in mitochondrial respiration, oxidative damage response, lysosomal and proteasomal function as well as by downregulating inflammatory cytokines in the central nervous system (Canto et al., 2012; Gong et al., 2013; Mehmel et al., 2020; Brakedal et al., 2022). In addition, it is possible that nicotinamide riboside may mitigate epigenomic dysregulation in PD by regulating histone acetylation. Increasing neuronal NAD levels would boost the activity of the NAD-dependent histone deacetylases of the SIRT family, potentially ameliorating histone hyperacetylation in PD.
7. O-GlcNAcylation
O-linked N-acetylglucosamine (O-GlcNAc) is a form of protein glycosylation in which N-acetylglucosamine (GlcNAc) residues are O-linked to ser and threonine (thr) hydroxyl groups of proteins (Butkinaree et al., 2010). The enzymes which control the levels of GlcNAc are O-GlcNAc transferase (OGT) which attaches O-GlcNAc and O-GlcNAcase (OGA), which instead removes the O-GlcNAc (Bond and Hanover, 2013).
O-GlcNAcylation reduces the aggregation propensity and the toxicity of amyloidogenic proteins including and α-Syn (Marotta et al., 2015; Levine et al., 2017; Lewis et al., 2017). α-Syn has several O-GlcNAcylation sites (Cole and Hart, 2001), especially located in the NAC region of the protein (Marotta et al., 2015; Levine et al., 2017, 2019; Lewis et al., 2017). The O-GlcNAcylation at thr72 of α-Syn decreases aggregation propensity and toxicity in cultured cells (Marotta et al., 2015). Moreover, O-GlcNAcylation hampers the cleavage of α-Syn by calpain (Levine et al., 2017), a process involved in the formation of aggregates, and is implicated in the modulation of endocytic and autophagic pathways (Dufty et al., 2007). In addition, it has been demonstrated that pharmacological inhibition or the knockdown of OGA hampers α-Syn pre-formed fibrils internalization (Tavassoly et al., 2021).
Selective inhibitors of OGA are of interest for their potential to reduce the aggregation of the amyloidogenic proteins within brain (Selnick et al., 2019). In this context, thiamet G, a brain permeable molecule, has been shown to increase cerebral O-GlcNAc levels to hamper neurodegeneration and reduce phosphorylation and aggregation of tau (Liu et al., 2004a; Yuzwa et al., 2008; Gong et al., 2012). Moreover, thiamet G improves behavioral features in preclinical models of tauopathies (Yuzwa et al., 2008, 2012, 2014a,b; Yu et al., 2012; Borghgraef et al., 2013; Graham et al., 2014; Hastings et al., 2017). A novel, highly potent and selective OGA inhibitor, MK-8719, has been developed and showing promising results in in vitro and in vivo tauopathies model. The OGA inhibitor ASN120290, that has been recently assigned the Orphan Drug Designation for the treatment of progressive supranuclear palsy (PSP) by the Food and Drug Administration has granted to ASN120290 reduced neurofibrillary tangles in mouse model of tauopathy. Permanne et al. (2022) demonstrated that the administration of ASN120290 enhance α-Syn O-GlcNAcylation and slows the progression of motor impairment in a α-Syn tg mouse model of PD (Table 5). In June 2021, a phase I first-in-human trial assessing the diffusion of ASN121151 to the CNS and the safety and pharmacokinetic profile in elderly healthy and AD subjects has been started (Clinical trial identifier: NCT04759365). Furthermore, a multiple ascending doses PET study is currently ongoing to investigate the brain occupancy of OGA and the pharmacodynamic response in peripheral blood mononuclear cells after repeated doses of ASN121151 to healthy subjects (Clinical trial identifier: NCT05725005; Table 5).
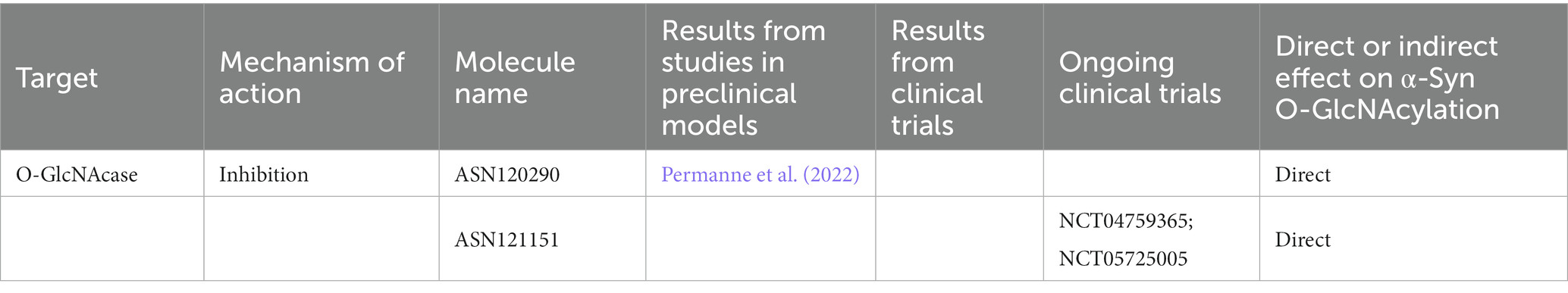
Table 5. Inhibitors of OGA tested in preclinical models of PD or in clinical trials.
8. Glycation
In the context of sugar-based modifications we can find glycation. Glycation is a non-enzymatic reaction that proceeds under hyperglycemia and during aging. Through the Maillard reaction the reduced carbohydrates and amino compounds form the intermediate Amadori products which in turn break down, thus creating a variety of different carbonyl and dicarbonyl intermediate products, including glyoxal and methylglyoxal (MGO) that are able to bound to the proteins (Hodge, 1955). Lastly, higher molecular weight species or advanced glycation end products (AGEs) can be formed from these lower molecular weight species (Henning and Glomb, 2016). These reactions are generally rather slow and their end products are very stable (Henning and Glomb, 2016). Therefore, short lived proteins are usually not involved in this process, however long-lived proteins, such as α-Syn can be modified in AGEs (Ahmed, 2005; Vicente Miranda and Outeiro, 2010). AGEs colocalize with α-Syn in LB in the SN (Munch et al., 2000) and glycated α-Syn has been identified in brain tissue from PD patients (Vicente Miranda et al., 2017b). MGO reacts with α-Syn to form oligomers, increasing the toxicity (Vicente Miranda et al., 2017b). In addition, diabetes is associated with the accumulation of AGEs (Kopytek et al., 2020) and patients with type 2 diabetes mellitus experience an increased risk to develop PD (Yang et al., 2017; Vaccari et al., 2021), indicating a possible insulin-modulating role in this latter condition. Both diabetes and PD are characterized by altered homeostasis of sugar metabolism (Dunn et al., 2014; Shamsaldeen et al., 2016; Trezzi et al., 2017). Interestingly, antidiabetic drugs have been suggested to exert a neuroprotective role both in PD models and in patients (Konig et al., 2018; Iravanpour et al., 2021). For instance, insulin modulates α-Syn expression and aggregation (Sharma et al., 2015b,c), regulates vesicular monoamine transporter 2 (VMAT2; Kong et al., 2020) and intranasal administration of insulin ameliorated mitochondrial function, motor impairment and dopaminergic neuron death in a rat model of PD (Iravanpour et al., 2021).
Glucagon-like peptide-1 (GLP1) is secreted in response to ingestion and absorption, preferably of carbohydrates and fats (Drucker and Nauck, 2006; Wu et al., 2015; Nauck and Meier, 2018). The binding of GLP1 to its receptor (GLP1R) induces the glucose-dependent pancreatic insulin secretion (Flock et al., 2007; Holst, 2007). It has been demonstrated that agonists (GLP1RA) such as exendin-4 (Ex-4) can regulate several functions related to neurodegeneration, OS and neurogenesis (Kim et al., 2017). Consistently, Ex-4 and derivatives showed beneficial effects in PD animal models (Bertilsson et al., 2008, Rampersaud et al., 2012, Liu et al., 2015, Palleria et al., 2017, Chen et al., 2018, Elbassuoni and Ahmed, 2019, Zhang et al., 2021; Table 6). Indeed, it has been demonstrated that GLP1RA ameliorates MPTP-induced neurotoxicity acting on mitophagy flux, OS and α-Syn aggregation in both the MPTP-mouse model of PD (Lin et al., 2021) and in α-Syn transgenic mice (Yun et al., 2018).
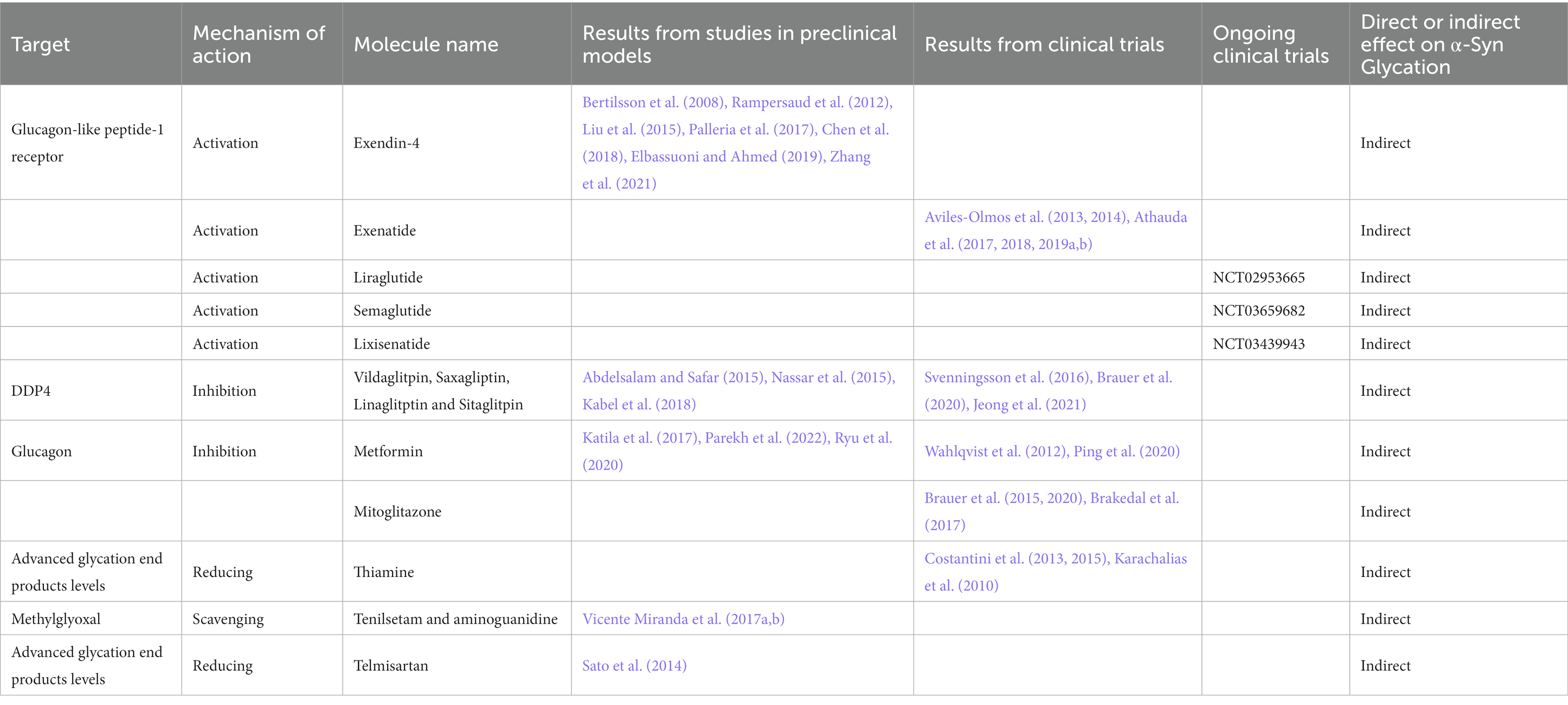
Table 6. Glycation-modifying agents tested in preclinical models of PD and in clinical trials.
Phase II clinical trials assessing the effect of 12 or 24 months treatments with exenatide, a synthetic Ex-4 derivative, showed cognitive and motor benefits which persisted for 12 months after drug washout in moderate PD patients (Aviles-Olmos et al., 2013, 2014; Table 6). In a next randomized, placebo-controlled, double-blind trial the authors analyzed the improvements of exenatide treated PD patients regarding motor abilities (Athauda et al., 2017), mood and cognition (Athauda et al., 2018). A post hoc analysis showed that younger patients with lower MDS-UPDRS-2 scores and tremor-dominant phenotype had the best response to exenatide (Athauda et al., 2019b). Moreover, there was a positive trend in obese patients or those with insulin resistance (Athauda et al., 2019a). Several other trials are evaluating other GLP1RA such as liraglutide, semaglutide or lixisenatide (Clinical trial identifier: NCT02953665; NCT03659682; NCT03439943; Table 6).
Dipeptidyl peptidase 4 (DDP4) inhibitors such as Vildaglitpin, Saxagliptin, Linaglitptin and Sitaglitpin have also been tested in animals as blockers of peripheral GLP1 degradation (Abdelsalam and Safar, 2015; Nassar et al., 2015; Kabel et al., 2018). In humans DDP4 inhibitors administration showed decrease in PD incidence (Svenningsson et al., 2016; Brauer et al., 2020) and beneficial effect in diabetic PD patients (Jeong et al., 2021; Table 6).
The most common treatment for type 2 diabetes, metformin, showed promising results in MPTP animal models (Katila et al., 2017; Table 6). Moreover, it reduced mitochondrial respiration dysfunction, activating AMP-activated protein kinase (AMPK), which has pro-survival functions and increases α-Syn clearance in animal models of PD (Parekh et al., 2022). Recently, it has been demonstrated that metformin is able to control microglial and astrocyte activation, eventually leading to neuroprotection and controlling dyskinesia development (Ryu et al., 2020). So far, metformin treatments in humans gave rise to conflicting results (Wahlqvist et al., 2012; Ping et al., 2020).
Mitoglitazone, an antidiabetic molecule which was found to protect against MPTP toxicity in cells, rodents and nematodes, reduced the incidence of PD in diabetic patients (Brauer et al., 2015, 2020; Table 6) exerting a better effect when compared to metformin (Brakedal et al., 2017).
Furthermore, high doses of thiamine improved motor function in PD patients by acting on AGE levels (Karachalias et al., 2010; Costantini et al., 2013, 2015; Table 6).
Other molecules showed promising results in preclinical models such as, MGO-scavengers tenilsetam and aminoguanidine that reduced α-Syn aggregation while improving its clearance and motor behavior in a PD models (Vicente Miranda et al., 2017b; Table 6). Telmisartan an anti-hypertension molecule, which was shown to reduce AGEs levels in rodents, demonstrated a protective role in MPTP models (Sato et al., 2014; Table 6).
9. SUMOylation
The covalent addition of a small ubiquitin like modifiers (SUMO) is one of the PTM which characterizes α-Syn. SUMO is a 12 kDa protein attached covalently to the lys-residues of a protein and it is essential for normal cellular processes including cell cycle regulation, nuclear-cytosolic transport, gene transcription, protein stability, response to stress, apoptosis and many others functions (Matunis et al., 1996; Hershko and Ciechanover, 1998).
SUMOylation is mediated by a three-step reaction that involves SUMO activating enzyme (SAE1), Ubc9 conjugating enzyme and SUMO-E3 ligase (Muller et al., 2001; Wilkinson and Henley, 2010). SUMO peptides can be recycled through a process of deSUMOylation by the SUMO proteases from the Ulp/SENP family.
SUMOylation machinery and protein SUMOylation dramatically increase in response to cellular stresses, and so in PD (Zhou et al., 2004; Enserink, 2015). Furthermore, rotenone-injected mice exhibit increased α-Syn and SUMO levels (Weetman et al., 2013). SUMOylation participates in several pathways connected to PD such as regulation of DJ-1 activity, modulation of transcription factors involved in mitochondrial and lysosomal biogenesis, and regulation of mitochondrial fission machinery (Harder et al., 2004; Ariga et al., 2013; Savyon and Engelender, 2020).
SUMO has been shown to enhance the solubility of aggregation-prone proteins like α-Syn, and impaired SUMOylation increased α-Syn aggregation and toxicity in HEK293 cells and a PD rat models (Krumova et al., 2011). On the other hand, SUMOylation competes with ubiquitination on the same lys residue, protecting the protein from degradation (Rott et al., 2017; Rousseaux et al., 2018). The discrepancies seen on α-Syn aggregation may be related to the different SUMO isoforms and SUMO-ligases that may be involved in the processes (Tatham et al., 2001; Bohren et al., 2004; Wilkinson and Henley, 2010).
The only tested molecule for the interference with E1-SUMO complex formation in PD like model, is ginkgolic acid (Fukuda et al., 2009; Table 7), which decreases the levels of SUMOylation stimulating the macroautophagic clearance of α-Syn aggregates (Vijayakumaran et al., 2019).

Table 7. SUMOylation inhibitors tested in preclinical models of PD.
So far, SUMOylation targeting has been achieved especially in oncology, indeed spectomycin B1 had been proposed as therapeutic agent to cure breast cancer through the blocking of SUMOylation preventing the formation of the Ubc9-SUMO (Hirohama et al., 2013). In addition, the potent SAE inhibitor ML-792 impairs SUMO conjugation but also induces significant loss of viability in multiple cancer cell lines (He et al., 2017). On the other hand, global cellular SUMOylation is enhanced in response to interferons (Maroui et al., 2018).
10. Ubiquitination
The ubiquitin–proteasome system (UPS) mediates the degradation of proteins in mammalian cells (Ross and Pickart, 2004). The addition of multiple molecules of ubiquitin, a conserved 8.5-kDa polypeptide, constitute the signal for proteasome-mediated degradation. Ubiquitin–substrate ligation is mediated by different enzymatic steps which are mainly mediated by E3 ligases. These latter recognize specific substrate-based signals in a manner that is frequently regulated by covalent modification (Weissman, 2001), in which the first ubiquitin is covalently joined to proteins through an isopeptide bond between the C-terminus of ubiquitin and a lys residue, and must be proteolytically processed by ubiquitin C-terminal hydrolases (UCHs) before it can acquire activity (Weissman, 2001). Additional ubiquitins are then linked to the first one to form a polyubiquitin chain that is a potent attractive signal for the regulatory complex of the proteasome. The UPS is vitally important for protecting cells against the toxic effects of misfolded proteins (Engelender et al., 2022). The 26S proteasome consists of more than 60 subunits. It is composed by: (1) a central, barrel-shaped catalytic (20S) complex carrying multiple active sites, which are sequestered in an interior chamber that is only accessible through a narrow axial pore; (2) two distally positioned regulatory (19S) complexes which unfold the substrate polypeptide chain and translocate it through this pore and into the active-site chamber, using integral chaperone subunits placed immediately adjacent to the axial pore of the 20S complex (Ross and Pickart, 2004). Of note, studies in the post-mortem brains of sporadic PD patients showed that LB contain ubiquitinated α-Syn that is not associated with UPS impairment (Tofaris et al., 2003). However, even non-ubiquitinated α-Syn appears to be degraded by the 20S proteasome (Tofaris et al., 2001), supporting the occurrence of ubiquitin-independent mechanism of UPS-mediated α-Syn degradation in synucleinopathies.
Studies in cell models or purified systems led to conflicting results either supporting that both 20S and 26S proteasomes degrade α-Syn or failing to detect α-Syn accumulation upon UPS inhibition (Bennett et al., 1999; Tofaris et al., 2001, 2011; Webb et al., 2003; Emmanouilidou et al., 2010; Shabek et al., 2012) hinting that the UPS may play a relevant role in degrading a fraction of α-Syn, whose relative abundance may vary between cell types and experimental conditions (Stefanis et al., 2019).
Promoting the activity of the UPS can thus be considered as a possible therapeutic strategy for combating α-Syn accumulation (Engelender et al., 2022; Table 8). For instance, following evidence that p38 mitogen-activated protein kinase (MAPK) negatively regulates proteasome activity, the p38 MAPK inhibitor PD169316 has been identified as a proteasome activator that decreases α-Syn toxicity in cells (Braun et al., 2021; Engelender et al., 2022). Several p38 MAPK inhibitors tested in clinical trials for chronic inflammatory diseases and cancer may also be considered as possible UPS stimulators, though their neuroprotective effects may not be solely ascribed to UPS induction. Indeed, studies in experimental models of synucleinopathies and of other neurodegenerative diseases such as AD have shown that p38 MAPK plays a relevant role in mediating other key processes involved in neurodegeneration, neuroinflammation and disease protein-mediated brain damage (Giovannini et al., 2002, 2008; Cuenda and Rousseau, 2007).
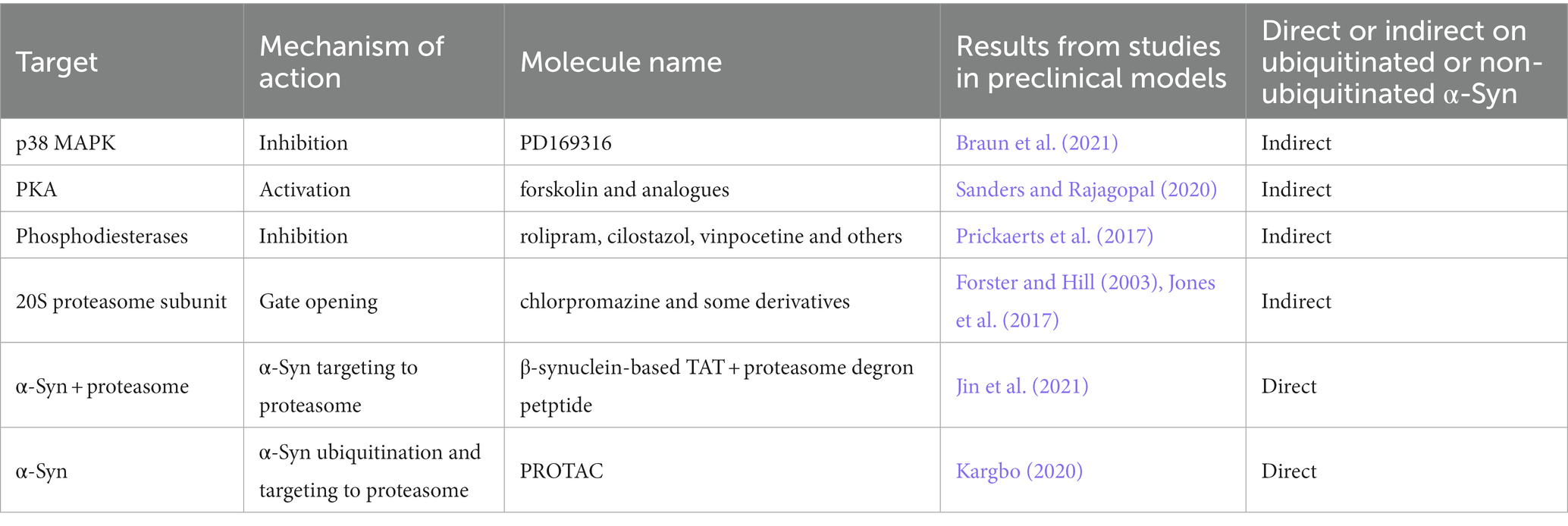
Table 8. UPS modulators tested in preclinical models of PD.
Alternatively, compounds that work as gate-openers of the 20S proteasome by preventing the barrel closing may also promote α-Syn clearing (Forster and Hill, 2003; Jones et al., 2017). For instance, chlorpromazine and some derivatives devoid of dopamine receptors D2 binding were shown to promote the degradation of α-Syn by interacting with the 20S subunits and preventing its closure (Jones et al., 2017).
Another strategy to increase proteasomal activity is to modulate the phosphorylation status of its subunits that are influenced by several protein kinases (Kors et al., 2019). In particular, cAMP-dependent protein kinase A (PKA) phosphorylates the 19S subunits Rpt6 and Rpn6, leading to activation of 20S proteolytic activities in a process that may involve changes in proteasomal conformation (Zhang et al., 2007; Lokireddy et al., 2015). Despite the benefits of PKA activators, no positive outcome on improving cognition has been observed in clinical trials with forskolin analogs (Sanders and Rajagopal, 2020). On the other hand, several clinical trials assessing the efficacy of phosphodiesterase inhibitors are currently under way, including rolipram, cilostazol and vinpocetine (Prickaerts et al., 2017) and may hold promise for treating synucleinopathies.
A more recent approach to promote the proteasomal degradation of disease proteins is cell-penetrating peptides that specifically interact with the target protein and the proteasome. One promising peptide consists of a portion of β-synuclein peptide that interacts with α-Syn, which was fused to the cell-penetrating peptide TAT and a proteasomal degron and significantly decreased the neuronal levels of α-Syn via proteasome as well as neurotoxicity in mice (Jin et al., 2021).
Finally, the proteasomal degradation of disease proteins can also be improved Proteolysis Targeting Chimeric (PROTAC) compounds (Sakamoto et al., 2001). The technology relies on the fusion of a ligand for the target protein to a ligand for an E3 ubiquitin-ligase, such as cereblon and Van Hippel-Landau (VHL; Au et al., 2020). α-Syn-targeting PROTAC are currently in preclinical development (Kargbo, 2020).
11. Discussion
The evidence summarized in this review highlights the relevance of α-Syn PTMs in PD pathophysiology. In the last few years, α-Syn PTMs have been investigated as biomarker for the diagnosis and progression of PD and other synucleinopathies. Moreover, studies supporting that PTMs control structural changes in α-Syn thus influencing its aggregation propensity, have blossomed great interest for the development of innovative therapeutic strategies, that by modulating α-Syn PTM, could reduce its pathological aggregation or spreading. Interestingly, some novel therapeutic strategies modulating α-Syn PTMs are already under investigation in clinical trials. This notwithstanding, further studies are warranted to better clarify the role of PTMs on α-Syn pathophysiology, to confirm the translational potential of PTMs-modifying drugs in synucleinopathies as well as to disclose whether the evaluation of α-Syn PTMs in peripheral tissues can be a valuable readout to monitor the effect of such approaches.
Author contributions
FL, GF, VB, and AB conceived the manuscript. VB and AB collected references, wrote the main text, and prepared illustrations and tables. AB revised manuscript text and tables. All authors contributed to the article and approved the submitted version.
Funding
We are grateful to the Michael J. Fox Foundation for Parkinson’s Research, NY, USA (grant ID: MJFF-021179), the Multiple system atrophy coalition, USA, and the MIUR PRIN 2017-1065.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbott, R. D., Ross, G. W., Petrovitch, H., Tanner, C. M., Davis, D. G., Masaki, K. H., et al. (2007). Bowel movement frequency in late-life and incidental Lewy bodies. Mov. Disord. 22, 1581–1586. doi: 10.1002/mds.21560
Abd-Elhadi, S., Honig, A., Simhi-Haham, D., Schechter, M., Linetsky, E., Ben-Hur, T., et al. (2015). Total and proteinase K-resistant alpha-synuclein levels in erythrocytes, determined by their ability to bind phospholipids, associate with Parkinson's disease. Sci. Rep. 5:11120. doi: 10.1038/srep11120
Abd Elhadi, S., Grigoletto, J., Poli, M., Arosio, P., Arkadir, D., and Sharon, R. (2019). Alpha-synuclein in blood cells differentiates Parkinson's disease from healthy controls. Ann. Clin. Transl. Neurol. 6, 2426–2436. doi: 10.1002/acn3.50944
Abdelsalam, R. M., and Safar, M. M. (2015). Neuroprotective effects of vildagliptin in rat rotenone Parkinson's disease model: role of RAGE-NFkappaB and Nrf2-antioxidant signaling pathways. J. Neurochem. 133, 700–707. doi: 10.1111/jnc.13087
Abou-Sleiman, P. M., Muqit, M. M., and Wood, N. W. (2006). Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 7, 207–219. doi: 10.1038/nrn1868
Ahmed, N. (2005). Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 67, 3–21. doi: 10.1016/j.diabres.2004.09.004
Ahn, B. H., Rhim, H., Kim, S. Y., Sung, Y. M., Lee, M. Y., Choi, J. Y., et al. (2002). Alpha-synuclein interacts with phospholipase D isozymes and inhibits pervanadate-induced phospholipase D activation in human embryonic kidney-293 cells. J. Biol. Chem. 277, 12334–12342. doi: 10.1074/jbc.M110414200
Aksnes, H., Hole, K., and Arnesen, T. (2015). Molecular, cellular, and physiological significance of N-terminal acetylation. Int. Rev. Cell Mol. Biol. 316, 267–305. doi: 10.1016/bs.ircmb.2015.01.001
Alam, Z. I., Jenner, A., Daniel, S. E., Lees, A. J., Cairns, N., Marsden, C. D., et al. (1997). Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 69, 1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x
Albani, D., Polito, L., Batelli, S., De Mauro, S., Fracasso, C., Martelli, G., et al. (2009). The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J. Neurochem. 110, 1445–1456. doi: 10.1111/j.1471-4159.2009.06228.x
Alegre-Abarrategui, J., Ansorge, O., Esiri, M., and Wade-Martins, R. (2008). LRRK2 is a component of granular alpha-synuclein pathology in the brainstem of Parkinson's disease. Neuropathol. Appl. Neurobiol. 34, 272–283. doi: 10.1111/j.1365-2990.2007.00888.x
Anderson, J. P., Walker, D. E., Goldstein, J. M., De Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Antelope, O., Vellore, N. A., Pomicter, A. D., Patel, A. B., Van Scoyk, A., Clair, P. M., et al. (2019). BCR-ABL1 tyrosine kinase inhibitor K0706 exhibits preclinical activity in Philadelphia chromosome-positive leukemia. Exp. Hematol. 77, 36–40.e2. doi: 10.1016/j.exphem.2019.08.007
Arawaka, S., Wada, M., Goto, S., Karube, H., Sakamoto, M., Ren, C. H., et al. (2006). The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J. Neurosci. 26, 9227–9238. doi: 10.1523/JNEUROSCI.0341-06.2006
Arbo, B. D., Andre-Miral, C., Nasre-Nasser, R. G., Schimith, L. E., Santos, M. G., Costa-Silva, D., et al. (2020). Resveratrol derivatives as potential treatments for Alzheimer's and Parkinson's disease. Front. Aging Neurosci. 12:103. doi: 10.3389/fnagi.2020.00103
Ariga, H., Takahashi-Niki, K., Kato, I., Maita, H., Niki, T., and Iguchi-Ariga, S. M. (2013). Neuroprotective function of DJ-1 in Parkinson's disease. Oxidative Med. Cell. Longev. 2013:683920. doi: 10.1155/2013/683920
Arnesen, T. (2009). Protein N-terminal acetylation: NAT 2007-2008 Symposia. BMC Proc. 3:S1. doi: 10.1186/1753-6561-3-S6-S1
Athauda, D., Gulyani, S., Karnati, H. K., Li, Y., Tweedie, D., Mustapic, M., et al. (2019a). Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: a secondary analysis of the exenatide-PD trial. JAMA Neurol. 76, 420–429. doi: 10.1001/jamaneurol.2018.4304
Athauda, D., Maclagan, K., Budnik, N., Zampedri, L., Hibbert, S., Aviles-Olmos, I., et al. (2019b). Post hoc analysis of the exenatide-PD trial-factors that predict response. Eur. J. Neurosci. 49, 410–421. doi: 10.1111/ejn.14096
Athauda, D., Maclagan, K., Budnik, N., Zampedri, L., Hibbert, S., Skene, S. S., et al. (2018). What effects might exenatide have on non-motor symptoms in Parkinson's disease: a post hoc analysis. J. Parkinsons Dis. 8, 247–258. doi: 10.3233/JPD-181329
Athauda, D., Maclagan, K., Skene, S. S., Bajwa-Joseph, M., Letchford, D., Chowdhury, K., et al. (2017). Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675. doi: 10.1016/S0140-6736(17)31585-4
Au, Y. Z., Wang, T., Sigua, L. H., and Qi, J. (2020). Peptide-based PROTAC: the predator of pathological proteins. Cell Chem. Biol. 27, 637–639. doi: 10.1016/j.chembiol.2020.06.002
Avci, B., Gunaydin, C., Guvenc, T., Yavuz, C. K., Kuruca, N., and Bilge, S. S. (2021). Idebenone ameliorates rotenone-induced Parkinson's disease in rats through decreasing lipid peroxidation. Neurochem. Res. 46, 513–522. doi: 10.1007/s11064-020-03186-w
Aviles-Olmos, I., Dickson, J., Kefalopoulou, Z., Djamshidian, A., Ell, P., Soderlund, T., et al. (2013). Exenatide and the treatment of patients with Parkinson's disease. J. Clin. Invest. 123, 2730–2736. doi: 10.1172/JCI68295
Aviles-Olmos, I., Dickson, J., Kefalopoulou, Z., Djamshidian, A., Kahan, J., Ell, P., et al. (2014). Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson's disease. J. Parkinsons Dis. 4, 337–344. doi: 10.3233/JPD-140364
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Baker, M. G., and Graham, L. (2004). The journey: Parkinson's disease. BMJ 329, 611–614. doi: 10.1136/bmj.329.7466.611
Bakhit, Y., Schmitt, I., Hamed, A., Ibrahim, E. A. A., Mohamed, I. N., El-Sadig, S. M., et al. (2022). Methylation of alpha-synuclein in a Sudanese cohort. Parkinsonism Relat. Disord. 101, 6–8. doi: 10.1016/j.parkreldis.2022.05.009
Barger, J. L., Kayo, T., Vann, J. M., Arias, E. B., Wang, J., Hacker, T. A., et al. (2008). A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS One 3:e2264. doi: 10.1371/annotation/c54ef754-1962-4125-bf19-76d3ec6f19e5
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bartels, T., Kim, N. C., Luth, E. S., and Selkoe, D. J. (2014). N-alpha-acetylation of alpha-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS One 9:e103727. doi: 10.1371/journal.pone.0103727
Beach, T. G., Adler, C. H., Sue, L. I., Vedders, L., Lue, L., White Iii, C. L., et al. (2010). Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 119, 689–702. doi: 10.1007/s00401-010-0664-3
Beal, M. F. (1998). Excitotoxicity and nitric oxide in Parkinson's disease pathogenesis. Ann. Neurol. 44, S110–S114. doi: 10.1002/ana.410440716
Beard, J. L., and Connor, J. R. (2003). Iron status and neural functioning. Annu. Rev. Nutr. 23, 41–58. doi: 10.1146/annurev.nutr.23.020102.075739
Bell, R., Castellana-Cruz, M., Nene, A., Thrush, R. J., Xu, C. K., Kumita, J. R., et al. (2022a). Effects of N-terminal acetylation on the aggregation of disease-related alpha-synuclein variants. J. Mol. Biol. 167825
Bell, R., Castellana-Cruz, M., Nene, A., Thrush, R. J., Xu, C. K., Kumita, J. R., et al. (2023). Effects of N-terminal acetylation on the aggregation of disease-related alpha-synuclein variants. J. Mol. Biol. 435:167825. doi: 10.1016/j.jmb.2022.167825
Bell, R., Thrush, R. J., Castellana-Cruz, M., Oeller, M., Staats, R., Nene, A., et al. (2022b). N-terminal acetylation of alpha-synuclein slows down its aggregation process and alters the morphology of the resulting aggregates. Biochemistry 61, 1743–1756. doi: 10.1021/acs.biochem.2c00104
Bell, R., and Vendruscolo, M. (2021). Modulation of the interactions between alpha-synuclein and lipid membranes by Post-translational modifications. Front. Neurol. 12:661117. doi: 10.3389/fneur.2021.661117
Bellucci, A., Antonini, A., Pizzi, M., and Spano, P. (2017). The end is the beginning: Parkinson's disease in the light of brain imaging. Front. Aging Neurosci. 9:330. doi: 10.3389/fnagi.2017.00330
Bellucci, A., Mercuri, N. B., Venneri, A., Faustini, G., Longhena, F., Pizzi, M., et al. (2016). Review: Parkinson's disease: from synaptic loss to connectome dysfunction. Neuropathol. Appl. Neurobiol. 42, 77–94. doi: 10.1111/nan.12297
Bellucci, A., Navarria, L., Zaltieri, M., Missale, C., and Spano, P. (2012). Alpha-synuclein synaptic pathology and its implications in the development of novel therapeutic approaches to cure Parkinson's disease. Brain Res. 1432, 95–113. doi: 10.1016/j.brainres.2011.11.031
Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N., and Mouradian, M. M. (1999). Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858. doi: 10.1074/jbc.274.48.33855
Bertilsson, G., Patrone, C., Zachrisson, O., Andersson, A., Dannaeus, K., Heidrich, J., et al. (2008). Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson's disease. J. Neurosci. Res. 86, 326–338. doi: 10.1002/jnr.21483
Boeve, B. F., Silber, M. H., Ferman, T. J., Lucas, J. A., and Parisi, J. E. (2001). Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov. Disord. 16, 622–630. doi: 10.1002/mds.1120
Bohren, K. M., Nadkarni, V., Song, J. H., Gabbay, K. H., and Owerbach, D. (2004). A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 279, 27233–27238. doi: 10.1074/jbc.M402273200
Bond, M. R., and Hanover, J. A. (2013). O-GlcNAc cycling: a link between metabolism and chronic disease. Annu. Rev. Nutr. 33, 205–229. doi: 10.1146/annurev-nutr-071812-161240
Borghgraef, P., Menuet, C., Theunis, C., Louis, J. V., Devijver, H., Maurin, H., et al. (2013). Increasing brain protein O-GlcNAc-ylation mitigates breathing defects and mortality of Tau.P301L mice. PLoS One 8:e84442. doi: 10.1371/journal.pone.0084442
Bougea, A., Koros, C., and Stefanis, L. (2019a). Salivary alpha-synuclein as a biomarker for Parkinson's disease: a systematic review. J. Neural Transm. (Vienna) 126, 1373–1382. doi: 10.1007/s00702-019-02062-4
Bougea, A., Stefanis, L., Paraskevas, G. P., Emmanouilidou, E., Vekrelis, K., and Kapaki, E. (2019b). Plasma alpha-synuclein levels in patients with Parkinson's disease: a systematic review and meta-analysis. Neurol. Sci. 40, 929–938. doi: 10.1007/s10072-019-03738-1
Brahmachari, S., Ge, P., Lee, S. H., Kim, D., Karuppagounder, S. S., Kumar, M., et al. (2016). Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. J. Clin. Invest. 126, 2970–2988. doi: 10.1172/JCI85456
Brakedal, B., Dolle, C., Riemer, F., Ma, Y., Nido, G. S., Skeie, G. O., et al. (2022). The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson's disease. Cell Metab. 34:e6. doi: 10.1016/j.cmet.2022.02.001
Brakedal, B., Flones, I., Reiter, S. F., Torkildsen, O., Dolle, C., Assmus, J., et al. (2017). Glitazone use associated with reduced risk of Parkinson's disease. Mov. Disord. 32, 1594–1599. doi: 10.1002/mds.27128
Brasher, B. B., and Van Etten, R. A. (2000). c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 275, 35631–35637. doi: 10.1074/jbc.M005401200
Brauer, R., Bhaskaran, K., Chaturvedi, N., Dexter, D. T., Smeeth, L., and Douglas, I. (2015). Glitazone treatment and incidence of Parkinson's disease among people with diabetes: a retrospective cohort study. PLoS Med. 12:e1001854. doi: 10.1371/journal.pmed.1001854
Brauer, R., Wei, L., Ma, T., Athauda, D., Girges, C., Vijiaratnam, N., et al. (2020). Diabetes medications and risk of Parkinson's disease: a cohort study of patients with diabetes. Brain 143, 3067–3076. doi: 10.1093/brain/awaa262
Braun, A. R., Liao, E. E., Horvath, M., Kalra, P., Acosta, K., Young, M. C., et al. (2021). Potent inhibitors of toxic alpha-synuclein identified via cellular time-resolved FRET biosensors. NPJ Parkinsons Dis. 7:52. doi: 10.1038/s41531-021-00195-6
Brown, D. R. (2007). Interactions between metals and alpha-synuclein--function or artefact? FEBS J. 274, 3766–3774. doi: 10.1111/j.1742-4658.2007.05917.x
Brown, K. D., Maqsood, S., Huang, J. Y., Pan, Y., Harkcom, W., Li, W., et al. (2014). Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 20, 1059–1068. doi: 10.1016/j.cmet.2014.11.003
Bu, B., Tong, X., Li, D., Hu, Y., He, W., Zhao, C., et al. (2017). N-terminal acetylation preserves alpha-Synuclein from Oligomerization by blocking intermolecular hydrogen bonds. ACS Chem. Neurosci. 8, 2145–2151. doi: 10.1021/acschemneuro.7b00250
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Butkinaree, C., Park, K., and Hart, G. W. (2010). O-linked beta-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta 1800, 96–106. doi: 10.1016/j.bbagen.2009.07.018
Calabresi, P., Mechelli, A., Natale, G., Volpicelli-Daley, L., Di Lazzaro, G., and Ghiglieri, V. (2023). Alpha-synuclein in Parkinson's disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 14:176. doi: 10.1038/s41419-023-05672-9
Campbell, B. C., Mclean, C. A., Culvenor, J. G., Gai, W. P., Blumbergs, P. C., Jakala, P., et al. (2001). The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease. J. Neurochem. 76, 87–96. doi: 10.1046/j.1471-4159.2001.00021.x
Canto, C., Houtkooper, R. H., Pirinen, E., Youn, D. Y., Oosterveer, M. H., Cen, Y., et al. (2012). The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15, 838–847. doi: 10.1016/j.cmet.2012.04.022
Carboni, E., Tatenhorst, L., Tonges, L., Barski, E., Dambeck, V., Bahr, M., et al. (2017). Deferiprone rescues behavioral deficits induced by mild Iron exposure in a mouse model of alpha-synuclein aggregation. NeuroMolecular Med. 19, 309–321. doi: 10.1007/s12017-017-8447-9
Cariulo, C., Martufi, P., Verani, M., Azzollini, L., Bruni, G., Weiss, A., et al. (2019). Phospho-S129 alpha-synuclein is present in human plasma but not in cerebrospinal fluid as determined by an ultrasensitive immunoassay. Front. Neurosci. 13:889. doi: 10.3389/fnins.2019.00889
Chakrabartty, A., Doig, A. J., and Baldwin, R. L. (1993). Helix capping propensities in peptides parallel those in proteins. Proc. Natl. Acad. Sci. U. S. A. 90, 11332–11336. doi: 10.1073/pnas.90.23.11332
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Chau, E., Kim, H., Shin, J., Martinez, A., and Kim, J. R. (2021). Inhibition of alpha-synuclein aggregation by AM17, a synthetic resveratrol derivative. Biochem. Biophys. Res. Commun. 574, 85–90. doi: 10.1016/j.bbrc.2021.08.049
Chavarria, C., and Souza, J. M. (2013). Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch. Biochem. Biophys. 533, 25–32. doi: 10.1016/j.abb.2013.02.009
Chen, L., Periquet, M., Wang, X., Negro, A., Mclean, P. J., Hyman, B. T., et al. (2009). Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Investig. 119, 3257–3265. doi: 10.1172/JCI39088
Chen, P. S., Wang, C. C., Bortner, C. D., Peng, G. S., Wu, X., Pang, H., et al. (2007). Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 149, 203–212. doi: 10.1016/j.neuroscience.2007.06.053
Chen, S., Yu, S. J., Li, Y., Lecca, D., Glotfelty, E., Kim, H. K., et al. (2018). Author correction: post-treatment with PT302, a long-acting Exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-Hydroxydopamine rat model of Parkinson's disease. Sci. Rep. 8:13953. doi: 10.1038/s41598-018-31455-w
Chen, S. H., Wu, H. M., Ossola, B., Schendzielorz, N., Wilson, B. C., Chu, C. H., et al. (2012). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Br. J. Pharmacol. 165, 494–505. doi: 10.1111/j.1476-5381.2011.01575.x
Chinta, S. J., Mallajosyula, J. K., Rane, A., and Andersen, J. K. (2010). Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 486, 235–239. doi: 10.1016/j.neulet.2010.09.061
Choi, B. K., Choi, M. G., Kim, J. Y., Yang, Y., Lai, Y., Kweon, D. H., et al. (2013). Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. U. S. A. 110, 4087–4092. doi: 10.1073/pnas.1218424110
Choong, C. J., Sasaki, T., Hayakawa, H., Yasuda, T., Baba, K., Hirata, Y., et al. (2016). A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson's disease. Neurobiol. Aging 37, 103–116. doi: 10.1016/j.neurobiolaging.2015.10.001
Chung, S. J., Kim, J., Lee, H. J., Ryu, H. S., Kim, K., Lee, J. H., et al. (2016). Alpha-synuclein in gastric and colonic mucosa in Parkinson's disease: limited role as a biomarker. Mov. Disord. 31, 241–249. doi: 10.1002/mds.26473
Clark, J., Clore, E. L., Zheng, K., Adame, A., Masliah, E., and Simon, D. K. (2010). Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS One 5:e12333. doi: 10.1371/journal.pone.0012333
Clayton, D. F., and George, J. M. (1998). The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254. doi: 10.1016/S0166-2236(97)01213-7
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., et al. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392. doi: 10.1021/acschembio.8b00466
Cole, R. N., and Hart, G. W. (2001). Cytosolic O-glycosylation is abundant in nerve terminals. J. Neurochem. 79, 1080–1089. doi: 10.1046/j.1471-4159.2001.00655.x
Costantini, A., Pala, M. I., Compagnoni, L., and Colangeli, M. (2013). High-dose thiamine as initial treatment for Parkinson's disease. BMJ Case Rep. 2013:bcr2013009289. doi: 10.1136/bcr-2013-009289
Costantini, A., Pala, M. I., Grossi, E., Mondonico, S., Cardelli, L. E., Jenner, C., et al. (2015). Long-term treatment with high-dose thiamine in Parkinson disease: an open-label pilot study. J. Altern. Complement. Med. 21, 740–747. doi: 10.1089/acm.2014.0353
Crespo, E., Macias, M., Pozo, D., Escames, G., Martin, M., Vives, F., et al. (1999). Melatonin inhibits expression of the inducible NO synthase II in liver and lung and prevents endotoxemia in lipopolysaccharide-induced multiple organ dysfunction syndrome in rats. FASEB J. 13, 1537–1546. doi: 10.1096/fasebj.13.12.1537
Crowther, R. A., Jakes, R., Spillantini, M. G., and Goedert, M. (1998). Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 436, 309–312. doi: 10.1016/S0014-5793(98)01146-6
Cuenda, A., and Rousseau, S. (2007). p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 1773, 1358–1375. doi: 10.1016/j.bbamcr.2007.03.010
Danielson, S. R., Held, J. M., Schilling, B., Oo, M., Gibson, B. W., and Andersen, J. K. (2009). Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson's disease. Anal. Chem. 81, 7823–7828. doi: 10.1021/ac901176t
Dauer, W., Kholodilov, N., Vila, M., Trillat, A. C., Goodchild, R., Larsen, K. E., et al. (2002). Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U. S. A. 99, 14524–14529. doi: 10.1073/pnas.172514599
De Bartolo, M. I., Vivacqua, G., Belvisi, D., Mancinelli, R., Fabbrini, A., Manzo, N., et al. (2023). A combined panel of salivary biomarkers in de novo Parkinson's disease. Ann. Neurol. 93, 446–459. doi: 10.1002/ana.26550
De Oliveira, R. M., Vicente Miranda, H., Francelle, L., Pinho, R., Szego, E. M., Martinho, R., et al. (2017). Correction: the mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 15:e1002601. doi: 10.1371/journal.pbio.1002601
De Rijk, M. C., Breteler, M. M., Den Breeijen, J. H., Launer, L. J., Grobbee, D. E., Van Der Meche, F. G., et al. (1997). Dietary antioxidants and Parkinson disease. The rotterdam study. Arch. Neurol. 54, 762–765. doi: 10.1001/archneur.1997.00550180070015
Decressac, M., Mattsson, B., Lundblad, M., Weikop, P., and Bjorklund, A. (2012). Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol. Dis. 45, 939–953. doi: 10.1016/j.nbd.2011.12.013
Deng, W. G., Tang, S. T., Tseng, H. P., and Wu, K. K. (2006). Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 108, 518–524. doi: 10.1182/blood-2005-09-3691
Denicola, G. M., Karreth, F. A., Humpton, T. J., Gopinathan, A., Wei, C., Frese, K., et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. doi: 10.1038/nature10189
Devic, I., Hwang, H., Edgar, J. S., Izutsu, K., Presland, R., Pan, C., et al. (2011). Salivary alpha-synuclein and DJ-1: potential biomarkers for Parkinson's disease. Brain 134:e178. doi: 10.1093/brain/awr015
Devos, D., Labreuche, J., Rascol, O., Corvol, J. C., Duhamel, A., Guyon Delannoy, P., et al. (2022). Trial of Deferiprone in Parkinson's Disease. N. Engl. J. Med. 387, 2045–2055. doi: 10.1056/NEJMoa2209254
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). Alpha-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci. Transl. Med. 8:342ra78. doi: 10.1126/scitranslmed.aaf3634
Dikiy, I., Fauvet, B., Jovicic, A., Mahul-Mellier, A. L., Desobry, C., El-Turk, F., et al. (2016). Semisynthetic and in vitro phosphorylation of alpha-synuclein at Y39 promotes functional partly helical membrane-bound states resembling those induced by PD mutations. ACS Chem. Biol. 11, 2428–2437. doi: 10.1021/acschembio.6b00539
Donadio, V., Incensi, A., El-Agnaf, O., Rizzo, G., Vaikath, N., Del Sorbo, F., et al. (2018). Skin alpha-synuclein deposits differ in clinical variants of synucleinopathy: an in vivo study. Sci. Rep. 8:14246. doi: 10.1038/s41598-018-32588-8
Donadio, V., Incensi, A., Leta, V., Giannoccaro, M. P., Scaglione, C., Martinelli, P., et al. (2014). Skin nerve alpha-synuclein deposits: a biomarker for idiopathic Parkinson disease. Neurology 82, 1362–1369. doi: 10.1212/WNL.0000000000000316
Dong, W. G., Mei, Q., Yu, J. P., Xu, J. M., Xiang, L., and Xu, Y. (2003). Effects of melatonin on the expression of iNOS and COX-2 in rat models of colitis. World J. Gastroenterol. 9, 1307–1311. doi: 10.3748/wjg.v9.i6.1307
Donmez, G., Arun, A., Chung, C. Y., Mclean, P. J., Lindquist, S., and Guarente, L. (2012). SIRT1 protects against alpha-synuclein aggregation by activating molecular chaperones. J. Neurosci. 32, 124–132. doi: 10.1523/JNEUROSCI.3442-11.2012
Drucker, D. J., and Nauck, M. A. (2006). The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368, 1696–1705. doi: 10.1016/S0140-6736(06)69705-5
Du, G., Liu, X., Chen, X., Song, M., Yan, Y., Jiao, R., et al. (2010). Drosophila histone deacetylase 6 protects dopaminergic neurons against alpha-synuclein toxicity by promoting inclusion formation. Mol. Biol. Cell 21, 2128–2137. doi: 10.1091/mbc.e10-03-0200
Du, Y., Wang, F., Zou, J., Le, W., Dong, Q., Wang, Z., et al. (2014). Histone deacetylase 6 regulates cytotoxic alpha-synuclein accumulation through induction of the heat shock response. Neurobiol. Aging 35, 2316–2328. doi: 10.1016/j.neurobiolaging.2014.04.029
Dufty, B. M., Warner, L. R., Hou, S. T., Jiang, S. X., Gomez-Isla, T., Leenhouts, K. M., et al. (2007). Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 170, 1725–1738. doi: 10.2353/ajpath.2007.061232
Dunn, L., Allen, G. F., Mamais, A., Ling, H., Li, A., Duberley, K. E., et al. (2014). Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol. Aging 35, 1111–1115. doi: 10.1016/j.neurobiolaging.2013.11.001
Eggler, A. L., Gay, K. A., and Mesecar, A. D. (2008). Molecular mechanisms of natural products in chemoprevention: induction of cytoprotective enzymes by Nrf2. Mol. Nutr. Food Res. 52, S84–S94. doi: 10.1002/mnfr.200700249
Eid, R., Arab, N. T., and Greenwood, M. T. (2017). Iron mediated toxicity and programmed cell death: a review and a re-examination of existing paradigms. Biochim. Biophys. Acta, Mol. Cell Res. 1864, 399–430. doi: 10.1016/j.bbamcr.2016.12.002
El-Agnaf, O. M., Bodles, A. M., Guthrie, D. J., Harriott, P., and Irvine, G. B. (1998a). The N-terminal region of non-a beta component of Alzheimer's disease amyloid is responsible for its tendency to assume beta-sheet and aggregate to form fibrils. Eur. J. Biochem. 258, 157–163.
El-Agnaf, O. M., Jakes, R., Curran, M. D., Middleton, D., Ingenito, R., Bianchi, E., et al. (1998b). Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett. 440, 71–75.
Elbassuoni, E. A., and Ahmed, R. F. (2019). Mechanism of the neuroprotective effect of GLP-1 in a rat model of Parkinson's with pre-existing diabetes. Neurochem. Int. 131:104583. doi: 10.1016/j.neuint.2019.104583
Ellis, C. E., Murphy, E. J., Mitchell, D. C., Golovko, M. Y., Scaglia, F., Barcelo-Coblijn, G. C., et al. (2005). Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell. Biol. 25, 10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005
Ellis, C. E., Schwartzberg, P. L., Grider, T. L., Fink, D. W., and Nussbaum, R. L. (2001). Alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J. Biol. Chem. 276, 3879–3884. doi: 10.1074/jbc.M010316200
Emmanouilidou, E., Stefanis, L., and Vekrellis, K. (2010). Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 31, 953–968. doi: 10.1016/j.neurobiolaging.2008.07.008
Engelender, S., and Isacson, O. (2017). The threshold theory for Parkinson's Disease. Trends Neurosci. 40, 4–14. doi: 10.1016/j.tins.2016.10.008
Engelender, S., Stefanis, L., Oddo, S., and Bellucci, A. (2022). Can we treat neurodegenerative Proteinopathies by enhancing protein degradation? Mov. Disord. 37, 1346–1359. doi: 10.1002/mds.29058
Enserink, J. M. (2015). Sumo and the cellular stress response. Cell Div 10:4. doi: 10.1186/s13008-015-0010-1
Etminan, M., Gill, S. S., and Samii, A. (2005). Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson's disease: a meta-analysis. Lancet Neurol. 4, 362–365. doi: 10.1016/S1474-4422(05)70097-1
Faustini, G., Longhena, F., Varanita, T., Bubacco, L., Pizzi, M., Missale, C., et al. (2018). Synapsin III deficiency hampers alpha-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson's disease. Acta Neuropathol. 136, 621–639. doi: 10.1007/s00401-018-1892-1
Faustini, G., Marchesan, E., Zonta, L., Bono, F., Bottani, E., Longhena, F., et al. (2019). Alpha-Synuclein preserves mitochondrial fusion and function in neuronal cells. Oxidative Med. Cell. Longev. 2019:4246350. doi: 10.1155/2019/4246350
Fauvet, B., Fares, M. B., Samuel, F., Dikiy, I., Tandon, A., Eliezer, D., et al. (2012). Characterization of semisynthetic and naturally Nalpha-acetylated alpha-synuclein in vitro and in intact cells: implications for aggregation and cellular properties of alpha-synuclein. J. Biol. Chem. 287, 28243–28262. doi: 10.1074/jbc.M112.383711
Fayyad, M., Salim, S., Majbour, N., Erskine, D., Stoops, E., Mollenhauer, B., et al. (2019). Parkinson's disease biomarkers based on alpha-synuclein. J. Neurochem. 150, 626–636. doi: 10.1111/jnc.14809
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson's disease. Nature 404, 394–398. doi: 10.1038/35006074
Fenyi, A., Leclair-Visonneau, L., Clairembault, T., Coron, E., Neunlist, M., Melki, R., et al. (2019). Detection of alpha-synuclein aggregates in gastrointestinal biopsies by protein misfolding cyclic amplification. Neurobiol. Dis. 129, 38–43. doi: 10.1016/j.nbd.2019.05.002
Fereshtehnejad, S. M., Zeighami, Y., Dagher, A., and Postuma, R. B. (2017). Clinical criteria for subtyping Parkinson's disease: biomarkers and longitudinal progression. Brain 140, 1959–1976. doi: 10.1093/brain/awx118
Fernandez, C. O., Hoyer, W., Zweckstetter, M., Jares-Erijman, E. A., Subramaniam, V., Griesinger, C., et al. (2004). NMR of alpha-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 23, 2039–2046. doi: 10.1038/sj.emboj.7600211
Fernandez, E., Garcia-Moreno, J. M., Martin De Pablos, A., and Chacon, J. (2013). May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosine proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum alpha-synuclein serve for diagnosis of sporadic Parkinson's disease? Antioxid. Redox Signal. 19, 912–918. doi: 10.1089/ars.2013.5250
Fields, C. R., Bengoa-Vergniory, N., and Wade-Martins, R. (2019). Targeting alpha-Synuclein as a therapy for Parkinson's Disease. Front. Mol. Neurosci. 12:299. doi: 10.3389/fnmol.2019.00299
Fitzgerald, K., Bergeron, M., Willits, C., Bowers, S., Aubele, D. L., Goldbach, E., et al. (2013). Pharmacological inhibition of polo like kinase 2 (PLK2) does not cause chromosomal damage or result in the formation of micronuclei. Toxicol. Appl. Pharmacol. 269, 1–7. doi: 10.1016/j.taap.2013.02.012
Flock, G., Baggio, L. L., Longuet, C., and Drucker, D. J. (2007). Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes 56, 3006–3013. doi: 10.2337/db07-0697
Floor, E., and Wetzel, M. G. (1998). Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J. Neurochem. 70, 268–275. doi: 10.1046/j.1471-4159.1998.70010268.x
Forman, H. J., Davies, K. J., and Ursini, F. (2014). How do nutritional antioxidants really work: nucleophilic tone and Para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 66, 24–35. doi: 10.1016/j.freeradbiomed.2013.05.045
Forman, H. J., and Zhang, H. (2021). Author correction: targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20:652. doi: 10.1038/s41573-021-00267-5
Formisano, L., Guida, N., Laudati, G., Mascolo, L., Di Renzo, G., and Canzoniero, L. M. (2015). MS-275 inhibits aroclor 1254-induced SH-SY5Y neuronal cell toxicity by preventing the formation of the HDAC3/REST complex on the synapsin-1 promoter. J. Pharmacol. Exp. Ther. 352, 236–243. doi: 10.1124/jpet.114.219345
Forster, A., and Hill, C. P. (2003). Proteasome degradation: enter the substrate. Trends Cell Biol. 13, 550–553. doi: 10.1016/j.tcb.2003.09.001
Foulds, P., Mann, D. M., and Allsop, D. (2012). Phosphorylated alpha-synuclein as a potential biomarker for Parkinson's disease and related disorders. Expert. Rev. Mol. Diagn. 12, 115–117. doi: 10.1586/erm.12.5
Foulds, P. G., Diggle, P., Mitchell, J. D., Parker, A., Hasegawa, M., Masuda-Suzukake, M., et al. (2013). A longitudinal study on alpha-synuclein in blood plasma as a biomarker for Parkinson's disease. Sci. Rep. 3:2540. doi: 10.1038/srep02540
Foulds, P. G., Mitchell, J. D., Parker, A., Turner, R., Green, G., Diggle, P., et al. (2011). Phosphorylated alpha-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson's disease. FASEB J. 25, 4127–4137. doi: 10.1096/fj.10-179192
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Fukuda, I., Ito, A., Hirai, G., Nishimura, S., Kawasaki, H., Saitoh, H., et al. (2009). Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 16, 133–140. doi: 10.1016/j.chembiol.2009.01.009
Fumimura, Y., Ikemura, M., Saito, Y., Sengoku, R., Kanemaru, K., Sawabe, M., et al. (2007). Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J. Neuropathol. Exp. Neurol. 66, 354–362. doi: 10.1097/nen.0b013e3180517454
Ganguly, U., Singh, S., Pal, S., Prasad, S., Agrawal, B. K., Saini, R. V., et al. (2021). Alpha-synuclein as a biomarker of Parkinson's disease: good, but not good enough. Front. Aging Neurosci. 13:702639. doi: 10.3389/fnagi.2021.702639
Gardian, G., Yang, L., Cleren, C., Calingasan, N. Y., Klivenyi, P., and Beal, M. F. (2004). Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. NeuroMolecular Med. 5, 235–241. doi: 10.1385/NMM:5:3:235
Gelpi, E., Navarro-Otano, J., Tolosa, E., Gaig, C., Compta, Y., Rey, M. J., et al. (2014). Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 29, 1010–1018. doi: 10.1002/mds.25776
George, J. M. (2002). The Synucleins. Genome Biol. 3:Reviews3002. doi: 10.1186/gb-2001-3-1-reviews3002
George, S., and Abrahamse, H. (2020). Redox potential of antioxidants in cancer progression and prevention. Antioxidants 9:1156. doi: 10.3390/antiox9111156
Gerlach, M., Double, K. L., Ben-Shachar, D., Zecca, L., Youdim, M. B., and Riederer, P. (2003). Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson's disease. Neurotox. Res. 5, 35–44. doi: 10.1007/BF03033371
Getachew, B., Csoka, A. B., Bhatti, A., Copeland, R. L., and Tizabi, Y. (2020). Butyrate protects against salsolinol-induced toxicity in SH-SY5Y cells: implication for Parkinson's disease. Neurotox. Res. 38, 596–602. doi: 10.1007/s12640-020-00238-5
Ghanem, S. S., Majbour, N. K., Vaikath, N. N., Ardah, M. T., Erskine, D., Jensen, N. M., et al. (2022). Alpha-synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. U. S. A. 119:e2109617119. doi: 10.1073/pnas.2109617119
Ghiglieri, V., Calabrese, V., and Calabresi, P. (2018). Alpha-Synuclein: from early synaptic dysfunction to Neurodegeneration. Front. Neurol. 9:295. doi: 10.3389/fneur.2018.00295
Giasson, B. I., Duda, J. E., Forman, M. S., Lee, V. M., and Trojanowski, J. Q. (2001). Prominent perikaryal expression of alpha-and beta-synuclein in neurons of dorsal root ganglion and in medullary neurons. Exp. Neurol. 172, 354–362. doi: 10.1006/exnr.2001.7805
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Giasson, B. I., Forman, M. S., Higuchi, M., Golbe, L. I., Graves, C. L., Kotzbauer, P. T., et al. (2003). Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–640. doi: 10.1126/science.1082324
Gibb, W. R. (1986). Idiopathic Parkinson's disease and the Lewy body disorders. Neuropathol. Appl. Neurobiol. 12, 223–234. doi: 10.1111/j.1365-2990.1986.tb00136.x
Gilad, E., Wong, H. R., Zingarelli, B., Virag, L., O'connor, M., Salzman, A. L., et al. (1998). Melatonin inhibits expression of the inducible isoform of nitric oxide synthase in murine macrophages: role of inhibition of NFkappaB activation. FASEB J. 12, 685–693. doi: 10.1096/fasebj.12.9.685
Gille, G., Hung, S. T., Reichmann, H., and Rausch, W. D. (2004). Oxidative stress to dopaminergic neurons as models of Parkinson's disease. Ann. N. Y. Acad. Sci. 1018, 533–540. doi: 10.1196/annals.1296.066
GioVannini, M. G., Cerbai, F., Bellucci, A., Melani, C., Grossi, C., Bartolozzi, C., et al. (2008). Differential activation of mitogen-activated protein kinase signalling pathways in the hippocampus of CRND8 transgenic mouse, a model of Alzheimer's disease. Neuroscience 153, 618–633. doi: 10.1016/j.neuroscience.2008.02.061
Giovannini, M. G., Scali, C., Prosperi, C., Bellucci, A., Vannucchi, M. G., Rosi, S., et al. (2002). Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol. Dis. 11, 257–274. doi: 10.1006/nbdi.2002.0538
Godena, V. K., Brookes-Hocking, N., Moller, A., Shaw, G., Oswald, M., Sancho, R. M., et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 5:5245. doi: 10.1038/ncomms6245
Goedert, M., Jakes, R., and Spillantini, M. G. (2017). The synucleinopathies: twenty years on. J. Parkinsons Dis. 7, S51–S69. doi: 10.3233/JPD-179005
Gomez-Tortosa, E., Newell, K., Irizarry, M. C., Sanders, J. L., and Hyman, B. T. (2000). Alpha-synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 99, 352–357. doi: 10.1007/s004010051135
Gonfloni, S., Maiani, E., Di Bartolomeo, C., Diederich, M., and Cesareni, G. (2012). Oxidative stress, DNA damage, and c-Abl signaling: at the crossroad in neurodegenerative diseases? Int. J. Cell. Biol. 2012:683097. doi: 10.1155/2012/683097
Gong, B., Pan, Y., Vempati, P., Zhao, W., Knable, L., Ho, L., et al. (2013). Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol. Aging 34, 1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005
Gong, C. X., Liu, F., and Iqbal, K. (2012). O-GlcNAc cycling modulates neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 109, 17319–17320. doi: 10.1073/pnas.1215395109
Gorbatyuk, O. S., Li, S., Sullivan, L. F., Chen, W., Kondrikova, G., Manfredsson, F. P., et al. (2008). The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. U. S. A. 105, 763–768. doi: 10.1073/pnas.0711053105
Gorostidi, A., Bergareche, A., Ruiz-Martinez, J., Marti-Masso, J. F., Cruz, M., Varghese, S., et al. (2012). Alphalpha-synuclein levels in blood plasma from LRRK2 mutation carriers. PLoS One 7:e52312. doi: 10.1371/journal.pone.0052312
Grace, A. A., and Bunney, B. S. (1983). Intracellular and extracellular electrophysiology of nigral dopaminergic neurons—3. Evidence for electrotonic coupling. Neuroscience 10, 333–348. doi: 10.1016/0306-4522(83)90137-9
Graham, D. L., Gray, A. J., Joyce, J. A., Yu, D., Omoore, J., Carlson, G. A., et al. (2014). Increased O-GlcNAcylation reduces pathological tau without affecting its normal phosphorylation in a mouse model of tauopathy. Neuropharmacology 79, 307–313. doi: 10.1016/j.neuropharm.2013.11.025
Greenamyre, J. T., and Hastings, T. G. (2004). Biomedicine. Parkinson's--divergent causes, convergent mechanisms. Science 304, 1120–1122. doi: 10.1126/science.1098966
Gruschus, J. M., Yap, T. L., Pistolesi, S., Maltsev, A. S., and Lee, J. C. (2013). NMR structure of calmodulin complexed to an N-terminally acetylated alpha-synuclein peptide. Biochemistry 52, 3436–3445. doi: 10.1021/bi400199p
Gueven, N., Ravishankar, P., Eri, R., and Rybalka, E. (2021). Idebenone: when an antioxidant is not an antioxidant. Redox Biol. 38:101812. doi: 10.1016/j.redox.2020.101812
Guo, Y. J., Dong, S. Y., Cui, X. X., Feng, Y., Liu, T., Yin, M., et al. (2016). Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of alpha-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 60, 2161–2175. doi: 10.1002/mnfr.201600111
Guzman, E., Taylor, G., Charleston, B., and Ellis, S. A. (2010). Induction of a cross-reactive CD8(+) T cell response following foot-and-mouth disease virus vaccination. J. Virol. 84, 12375–12384. doi: 10.1128/JVI.01545-10
Hamm-Alvarez, S. F., Okamoto, C. T., Janga, S. R., Feigenbaum, D., Edman, M. C., Freire, D., et al. (2019). Oligomeric alpha-synuclein is increased in basal tears of Parkinson's patients. Biomark. Med 13, 941–952. doi: 10.2217/bmm-2019-0167
Han, S., Du, Z., Liu, K., and Gong, S. (2020). Nicotinamide riboside protects noise-induced hearing loss by recovering the hair cell ribbon synapses. Neurosci. Lett. 725:134910. doi: 10.1016/j.neulet.2020.134910
Harder, Z., Zunino, R., and Mcbride, H. (2004). Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 14, 340–345. doi: 10.1016/j.cub.2004.02.004
Harlan, B. A., Killoy, K. M., Pehar, M., Liu, L., Auwerx, J., and Vargas, M. R. (2020). Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1-linked ALS mouse models. Exp. Neurol. 327:113219. doi: 10.1016/j.expneurol.2020.113219
Harris, I. S., and Denicola, G. M. (2020). The complex interplay between antioxidants and ROS in Cancer. Trends Cell Biol. 30, 440–451. doi: 10.1016/j.tcb.2020.03.002
Harrison, I. F., Crum, W. R., Vernon, A. C., and Dexter, D. T. (2015). Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson's is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 172, 4200–4215. doi: 10.1111/bph.13208
Harrison, I. F., Smith, A. D., and Dexter, D. T. (2018). Pathological histone acetylation in Parkinson's disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 666, 48–57. doi: 10.1016/j.neulet.2017.12.037
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M., et al. (2002). Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. doi: 10.1074/jbc.M208046200
Hastings, N. B., Wang, X., Song, L., Butts, B. D., Grotz, D., Hargreaves, R., et al. (2017). Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol. Neurodegener. 12:39. doi: 10.1186/s13024-017-0181-0
He, P. K., Gao, Y. Y., Lyu, F. J., Chen, J. N., Zhang, Y. H., Nie, K., et al. (2021). Idebenone-activating Autophagic degradation of alpha-synuclein via inhibition of AKT-mTOR pathway in a SH-SY5Y-A53T model of Parkinson's disease: a network pharmacological approach. Evid. Based Complement. Alternat. Med. 2021:8548380. doi: 10.1155/2021/8548380
He, X., Riceberg, J., Soucy, T., Koenig, E., Minissale, J., Gallery, M., et al. (2017). Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 13, 1164–1171. doi: 10.1038/nchembio.2463
He, Y., Yu, Z., and Chen, S. (2019). Alpha-synuclein nitration and its implications in Parkinson's disease. ACS Chem. Neurosci. 10, 777–782. doi: 10.1021/acschemneuro.8b00288
Hebron, M. L., Lonskaya, I., and Moussa, C. E. (2013). Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum. Mol. Genet. 22, 3315–3328. doi: 10.1093/hmg/ddt192
Hellenbrand, W., Boeing, H., Robra, B. P., Seidler, A., Vieregge, P., Nischan, P., et al. (1996). Diet and Parkinson's disease. II: a possible role for the past intake of specific nutrients. Results from a self-administered food-frequency questionnaire in a case-control study. Neurology 47, 644–650. doi: 10.1212/WNL.47.3.644
Henning, C., and Glomb, M. A. (2016). Pathways of the Maillard reaction under physiological conditions. Glycoconj. J. 33, 499–512. doi: 10.1007/s10719-016-9694-y
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hirata, Y., Sasaki, T., Kanki, H., Choong, C. J., Nishiyama, K., Kubo, G., et al. (2018). New 5-aryl-substituted 2-aminobenzamide-type HDAC inhibitors with a diketopiperazine group and their ameliorating effects on ischemia-induced neuronal cell death. Sci. Rep. 8:1400. doi: 10.1038/s41598-018-19664-9
HIrohama, M., Kumar, A., Fukuda, I., Matsuoka, S., Igarashi, Y., Saitoh, H., et al. (2013). Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem. Biol. 8, 2635–2642. doi: 10.1021/cb400630z
Hodara, R., Norris, E. H., Giasson, B. I., Mishizen-Eberz, A. J., Lynch, D. R., Lee, V. M., et al. (2004). Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 279, 47746–47753. doi: 10.1074/jbc.M408906200
Hodge, J. E. (1955). The Amadori rearrangement. Adv. Carbohydr. Chem. 10, 169–205. doi: 10.1016/s0096-5332(08)60392-6
Holmay, M. J., Terpstra, M., Coles, L. D., Mishra, U., Ahlskog, M., Oz, G., et al. (2013). N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 36, 103–106. doi: 10.1097/WNF.0b013e31829ae713
Holst, J. J. (2007). The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439. doi: 10.1152/physrev.00034.2006
Hong, Z., Shi, M., Chung, K. A., Quinn, J. F., Peskind, E. R., Galasko, D., et al. (2010). DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain 133, 713–726. doi: 10.1093/brain/awq008
Hornykiewicz, O. (2001). Chemical neuroanatomy of the basal ganglia--normal and in Parkinson's disease. J. Chem. Neuroanat. 22, 3–12. doi: 10.1016/S0891-0618(01)00100-4
Horvath, T. L., Diano, S., Leranth, C., Garcia-Segura, L. M., Cowley, M. A., Shanabrough, M., et al. (2003). Coenzyme Q induces nigral mitochondrial uncoupling and prevents dopamine cell loss in a primate model of Parkinson's disease. Endocrinology 144, 2757–2760. doi: 10.1210/en.2003-0163
Hoyer, W., Cherny, D., Subramaniam, V., and Jovin, T. M. (2004). Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 43, 16233–16242. doi: 10.1021/bi048453u
Hsu, S. W., Hsu, P. C., Chang, W. S., Yu, C. C., Wang, Y. C., Yang, J. S., et al. (2020). Protective effects of valproic acid on 6-hydroxydopamine-induced neuroinjury. Environ. Toxicol. 35, 840–848. doi: 10.1002/tox.22920
Hughes, K. C., Gao, X., Kim, I. Y., Rimm, E. B., Wang, M., Weisskopf, M. G., et al. (2016). Intake of antioxidant vitamins and risk of Parkinson's disease. Mov. Disord. 31, 1909–1914. doi: 10.1002/mds.26819
Hulka, B. S. (1990). Principles of bladder cancer screening in an intervention trial. J. Occup. Med. 32, 812–816. doi: 10.1097/00043764-199009000-00011
Ibrahim, H. A. M., Hussein, A. M., Gabr, M., El-Saeed, R. A., Ammar, O. A., Mosa, A. A. H., et al. (2022). Effect of melatonin on alpha synuclein and autophagy in dopaminergic neuronal differentiation of adipose mesenchymal stem cells. Res. Sq. doi: 10.21203/rs.3.rs-1746786/v
Imam, S. Z., Zhou, Q., Yamamoto, A., Valente, A. J., Ali, S. F., Bains, M., et al. (2011). Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J. Neurosci. 31, 157–163. doi: 10.1523/JNEUROSCI.1833-10.2011
Infante, R., Scaglione, C., Incensi, A., Rizzo, G., Liguori, R., and Donadio, V. (2020). A longitudinal skin biopsy study of phosphorylated alpha-synuclein in a patient with Parkinson disease and orthostatic hypotension. J. Neuropathol. Exp. Neurol. 79, 813–816. doi: 10.1093/jnen/nlaa048
Inglis, K. J., Chereau, D., Brigham, E. F., Chiou, S. S., Schobel, S., Frigon, N. L., et al. (2009). Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602. doi: 10.1074/jbc.C800206200
Intagliata, S., Modica, M. N., Santagati, L. M., and Montenegro, L. (2019). Strategies to improve resveratrol systemic and topical bioavailability: an update. Antioxidants 8:244. doi: 10.3390/antiox8080244
Iravanpour, F., Dargahi, L., Rezaei, M., Haghani, M., Heidari, R., Valian, N., et al. (2021). Intranasal insulin improves mitochondrial function and attenuates motor deficits in a rat 6-OHDA model of Parkinson's disease. CNS Neurosci. Ther. 27, 308–319. doi: 10.1111/cns.13609
Jenner, P., and Olanow, C. W. (2006). The pathogenesis of cell death in Parkinson's disease. Neurology 66, S24–S36. doi: 10.1212/WNL.66.10_suppl_4.S24
Jensen, P. H., Hager, H., Nielsen, M. S., Hojrup, P., Gliemann, J., and Jakes, R. (1999). Alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 274, 25481–25489. doi: 10.1074/jbc.274.36.25481
Jeong, S. H., Chung, S. J., Yoo, H. S., Hong, N., Jung, J. H., Baik, K., et al. (2021). Beneficial effects of dipeptidyl peptidase-4 inhibitors in diabetic Parkinson's disease. Brain 144, 1127–1137. doi: 10.1093/brain/awab015
Jia, L., Wang, Y., Wei, W., Zhao, W., Lu, F., and Liu, F. (2019). Vitamin B12 inhibits alpha-synuclein fibrillogenesis and protects against amyloid-induced cytotoxicity. Food Funct. 10, 2861–2870. doi: 10.1039/C8FO02471E
Jian, W., Wei, X., Chen, L., Wang, Z., Sun, Y., Zhu, S., et al. (2017). Inhibition of HDAC6 increases acetylation of peroxiredoxin1/2 and ameliorates 6-OHDA induced dopaminergic injury. Neurosci. Lett. 658, 114–120. doi: 10.1016/j.neulet.2017.08.029
Jin, J. W., Fan, X., Del Cid-Pellitero, E., Liu, X. X., Zhou, L., Dai, C., et al. (2021). Development of an alpha-synuclein knockdown peptide and evaluation of its efficacy in Parkinson's disease models. Commun. Biol. 4:232. doi: 10.1038/s42003-021-01746-6
Johnston, T. H., Huot, P., Damude, S., Fox, S. H., Jones, S. W., Rusche, J. R., et al. (2013). RGFP109, a histone deacetylase inhibitor attenuates L-DOPA-induced dyskinesia in the MPTP-lesioned marmoset: a proof-of-concept study. Parkinsonism Relat. Disord. 19, 260–264. doi: 10.1016/j.parkreldis.2012.07.001
Jomova, K., and Valko, M. (2011). Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr. Pharm. Des. 17, 3460–3473. doi: 10.2174/138161211798072463
Jones, C. L., Njomen, E., Sjogren, B., Dexheimer, T. S., and Tepe, J. J. (2017). Small molecule enhancement of 20S proteasome activity targets intrinsically disordered proteins. ACS Chem. Biol. 12, 2240–2247. doi: 10.1021/acschembio.7b00489
Junn, E., and Mouradian, M. M. (2002). Human alpha-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 320, 146–150. doi: 10.1016/S0304-3940(02)00016-2
Kabel, A. M., Omar, M. S., Alhadhrami, A., Alharthi, S. S., and Alrobaian, M. M. (2018). Linagliptin potentiates the effect of l-dopa on the behavioural, biochemical and immunohistochemical changes in experimentally-induced Parkinsonism: role of toll-like receptor 4, TGF-beta1, NF-kappaB and glucagon-like peptide 1. Physiol. Behav. 188, 108–118. doi: 10.1016/j.physbeh.2018.01.028
Kahle, P. J., Neumann, M., Ozmen, L., and Haass, C. (2000). Physiology and pathophysiology of alpha-synuclein. Cell culture and transgenic animal models based on a Parkinson's disease-associated protein. Ann. N. Y. Acad. Sci. 920, 33–41. doi: 10.1111/j.1749-6632.2000.tb06902.x
Kang, L., Moriarty, G. M., Woods, L. A., Ashcroft, A. E., Radford, S. E., and Baum, J. (2012). N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 21, 911–917. doi: 10.1002/pro.2088
Karachalias, N., Babaei-Jadidi, R., Rabbani, N., and Thornalley, P. J. (2010). Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia 53, 1506–1516. doi: 10.1007/s00125-010-1722-z
Karampetsou, M., Ardah, M. T., Semitekolou, M., Polissidis, A., Samiotaki, M., Kalomoiri, M., et al. (2017). Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 7:16533. doi: 10.1038/s41598-017-15813-8
Kargbo, R. B. (2020). PROTAC compounds targeting alpha-synuclein protein for treating neurogenerative disorders: Alzheimer's and Parkinson's diseases. ACS Med. Chem. Lett. 11, 1086–1087. doi: 10.1021/acsmedchemlett.0c00192
Karuppagounder, S. S., Brahmachari, S., Lee, Y., Dawson, V. L., Dawson, T. M., and Ko, H. S. (2014). The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson's disease. Sci. Rep. 4:4874. doi: 10.1038/srep04874
Karuppagounder, S. S., Wang, H., Kelly, T., Rush, R., Nguyen, R., Bisen, S., et al. (2023). The c-Abl inhibitor IkT-148009 suppresses neurodegeneration in mouse models of heritable and sporadic Parkinson's disease. Sci. Transl. Med. 15:eabp9352.
Kasai, T., Tokuda, T., Yamaguchi, N., Watanabe, Y., Kametani, F., Nakagawa, M., et al. (2008). Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci. Lett. 436, 52–56. doi: 10.1016/j.neulet.2008.02.057
Katila, N., Bhurtel, S., Shadfar, S., Srivastav, S., Neupane, S., Ojha, U., et al. (2017). Metformin lowers alpha-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson's disease. Neuropharmacology 125, 396–407. doi: 10.1016/j.neuropharm.2017.08.015
Kawaguchi, Y., Kovacs, J. J., Mclaurin, A., Vance, J. M., Ito, A., and Yao, T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cells 115, 727–738. doi: 10.1016/S0092-8674(03)00939-5
Kazantsev, A. G., and Kolchinsky, A. M. (2008). Central role of alpha-synuclein oligomers in neurodegeneration in Parkinson disease. Arch. Neurol. 65, 1577–1581. doi: 10.1001/archneur.65.12.1577
Keeney, P. M., Xie, J., Capaldi, R. A., and Bennett, J. P. Jr. (2006). Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 26, 5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006
Khan, Z., and Ali, S. A. (2018). Oxidative stress-related biomarkers in Parkinson's disease: a systematic review and meta-analysis. Iran. J. Neurol. 17, 137–144.
Kidd, S. K., and Schneider, J. S. (2010). Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res. 1354, 172–178. doi: 10.1016/j.brainres.2010.07.041
Kidd, S. K., and Schneider, J. S. (2011). Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Neuroscience 194, 189–194. doi: 10.1016/j.neuroscience.2011.08.010
Kieburtz, K., Mcdermott, M., Como, P., Growdon, J., Brady, J., Carter, J., et al. (1994). The effect of deprenyl and tocopherol on cognitive performance in early untreated Parkinson's disease. Parkinson study group. Neurology 44, 1756–1759. doi: 10.1212/WNL.44.9.1756
Kim, D., Nguyen, M. D., Dobbin, M. M., Fischer, A., Sananbenesi, F., Rodgers, J. T., et al. (2007). SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179. doi: 10.1038/sj.emboj.7601758
Kim, D. S., Choi, H. I., Wang, Y., Luo, Y., Hoffer, B. J., and Greig, N. H. (2017). A new treatment strategy for Parkinson's disease through the gut-brain axis: the glucagon-like peptide-1 receptor pathway. Cell Transplant. 26, 1560–1571. doi: 10.1177/0963689717721234
Kim, T., Song, S., Park, Y., Kang, S., and Seo, H. (2019). HDAC inhibition by valproic acid induces neuroprotection and improvement of PD-like behaviors in LRRK2 R1441G transgenic mice. Exp. Neurobiol. 28, 504–515. doi: 10.5607/en.2019.28.4.504
Ko, H. S., Lee, Y., Shin, J. H., Karuppagounder, S. S., Gadad, B. S., Koleske, A. J., et al. (2010). Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc. Natl. Acad. Sci. U. S. A. 107, 16691–16696. doi: 10.1073/pnas.1006083107
Koehler, N. K., Stransky, E., Meyer, M., Gaertner, S., Shing, M., Schnaidt, M., et al. (2015). Alpha-synuclein levels in blood plasma decline with healthy aging. PLoS One 10:e0123444. doi: 10.1371/journal.pone.0123444
Kong, Y., Zhou, H., Feng, H., Zhuang, J., Wen, T., Zhang, C., et al. (2020). Elucidating the relationship between diabetes mellitus and Parkinson's disease using (18) F-FP-(+)-DTBZ, a positron-emission tomography probe for vesicular monoamine transporter 2. Front. Neurosci. 14:682.
Konig, A., Vicente Miranda, H., and Outeiro, T. F. (2018). Alpha-synuclein glycation and the action of anti-diabetic agents in Parkinson's disease. J. Parkinsons Dis. 8, 33–43. doi: 10.3233/JPD-171285
Kopytek, M., Zabczyk, M., Mazur, P., Undas, A., and Natorska, J. (2020). Accumulation of advanced glycation end products (AGEs) is associated with the severity of aortic stenosis in patients with concomitant type 2 diabetes. Cardiovasc. Diabetol. 19:92. doi: 10.3389/fnins.2020.00682
Kors, S., Geijtenbeek, K., Reits, E., and Schipper-Krom, S. (2019). Regulation of proteasome activity by (Post-)transcriptional mechanisms. Front. Mol. Biosci. 6:48. doi: 10.3389/fmolb.2019.00048
Kosten, J., Binolfi, A., Stuiver, M., Verzini, S., Theillet, F. X., Bekei, B., et al. (2014). Efficient modification of alpha-synuclein serine 129 by protein kinase CK1 requires phosphorylation of tyrosine 125 as a priming event. ACS Chem. Neurosci. 5, 1203–1208. doi: 10.1021/cn5002254
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H. H., Bossis, G., et al. (2011). Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60. doi: 10.1083/jcb.201010117
Kulkarni, A. S., Burns, M. R., Brundin, P., and Wesson, D. W. (2022). Linking alpha-synuclein-induced synaptopathy and neural network dysfunction in early Parkinson's disease. Brain Commun. 4:fcac165. doi: 10.1093/braincomms/fcac165
Kumari, P., Ghosh, D., Vanas, A., Fleischmann, Y., Wiegand, T., Jeschke, G., et al. (2021). Structural insights into alpha-synuclein monomer-fibril interactions. Proc. Natl. Acad. Sci. U. S. A. 118:e2012171118. doi: 10.1073/pnas.2012171118
Kunz, D., and Bes, F. (1999). Melatonin as a therapy in REM sleep behavior disorder patients: an open-labeled pilot study on the possible influence of melatonin on REM-sleep regulation. Mov. Disord. 14, 507–511. doi: 10.1002/1531-8257(199905)14:3<507::AID-MDS1021>3.0.CO;2-8
Kunz, D., Mahlberg, R., Muller, C., Tilmann, A., and Bes, F. (2004). Melatonin in patients with reduced REM sleep duration: two randomized controlled trials. J. Clin. Endocrinol. Metab. 89, 128–134. doi: 10.1210/jc.2002-021057
Landeck, N., Hall, H., Ardah, M. T., Majbour, N. K., El-Agnaf, O. M., Halliday, G., et al. (2016). A novel multiplex assay for simultaneous quantification of total and S129 phosphorylated human alpha-synuclein. Mol. Neurodegener. 11:61. doi: 10.1186/s13024-016-0125-0
Lashuel, H. A. (2021). Rethinking protein aggregation and drug discovery in neurodegenerative diseases: why we need to embrace complexity? Curr. Opin. Chem. Biol. 64, 67–75. doi: 10.1016/j.cbpa.2021.05.006
Lee, J. T., Wheeler, T. C., Li, L., and Chin, L. S. (2008). Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 17, 906–917. doi: 10.1093/hmg/ddm363
Leite, K., Garg, P., Spitzner, F. P., Guerin Darvas, S., Bahr, M., Priesemann, V., et al. (2022). Alpha-synuclein impacts on intrinsic neuronal network activity through reduced levels of cyclic AMP and diminished numbers of active presynaptic terminals. Front. Mol. Neurosci. 15:868790. doi: 10.3389/fnmol.2022.868790
Lemos, M., and Stefanova, N. (2020). Histone deacetylase 6 and the disease mechanisms of alpha-synucleinopathies. Front. Synaptic. Neurosci. 12:586453. doi: 10.3389/fnsyn.2020.586453
Levine, P. M., De Leon, C. A., Galesic, A., Balana, A., Marotta, N. P., Lewis, Y. E., et al. (2017). O-GlcNAc modification inhibits the calpain-mediated cleavage of alpha-synuclein. Bioorg. Med. Chem. 25, 4977–4982. doi: 10.1016/j.bmc.2017.04.038
Levine, P. M., Galesic, A., Balana, A. T., Mahul-Mellier, A. L., Navarro, M. X., De Leon, C. A., et al. (2019). Alpha-synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 116, 1511–1519. doi: 10.1073/pnas.1808845116
Lewis, Y. E., Galesic, A., Levine, P. M., De Leon, C. A., Lamiri, N., Brennan, C. K., et al. (2017). O-GlcNAcylation of alpha-synuclein at serine 87 reduces aggregation without affecting membrane binding. ACS Chem. Biol. 12, 1020–1027. doi: 10.1021/acschembio.7b00113
Li, B., Wang, X., Rasheed, N., Hu, Y., Boast, S., Ishii, T., et al. (2004). Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev. 18, 1824–1837. doi: 10.1101/gad.1223504
Li, S., and Pelletier, G. (1995). Effects of pinealectomy and melatonin on gonadotropin-releasing hormone (GnRH) gene expression in the male rat brain. Endocrine 3, 533–536. doi: 10.1007/BF02738829
Li, X. Y., Li, W., Li, X., Li, X. R., Sun, L., Yang, W., et al. (2021). Alterations of erythrocytic phosphorylated alpha-synuclein in different subtypes and stages of Parkinson's disease. Front. Aging Neurosci. 13:623977. doi: 10.3389/fnagi.2021.623977
Li, X. Y., Yang, W., Li, X., Li, X. R., Li, W., Song, Q., et al. (2020). Phosphorylated alpha-synuclein in red blood cells as a potential diagnostic biomarker for multiple system atrophy: a pilot study. Parkinsons Dis. 2020:8740419. doi: 10.1155/2020/8740419
Lin, T. K., Lin, K. J., Lin, H. Y., Lin, K. L., Lan, M. Y., Wang, P. W., et al. (2021). Glucagon-Like Peptide-1 Receptor Agonist Ameliorates 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Neurotoxicity Through Enhancing Mitophagy Flux and Reducing alpha-Synuclein and Oxidative Stress. Front. Mol. Neurosci. 14:697440. doi: 10.3389/fnmol.2021.697440
Lin, C. H., Liu, H. C., Yang, S. Y., Yang, K. C., Wu, C. C., and Chiu, M. J. (2019). Plasma pS129-alpha-synuclein is a surrogate biofluid marker of motor severity and progression in Parkinson's disease. J. Clin. Med. 8:1601. doi: 10.3390/jcm8101601
Liu, F., Iqbal, K., Grundke-Iqbal, I., Hart, G. W., and Gong, C. X. (2004a). O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 101, 10804–10809. doi: 10.1073/pnas.0400348101
Liu, S., Fa, M., Ninan, I., Trinchese, F., Dauer, W., and Arancio, O. (2007). Alpha-synuclein involvement in hippocampal synaptic plasticity: role of NO, cGMP, cGK and CaMKII. Eur. J. Neurosci. 25, 3583–3596. doi: 10.1111/j.1460-9568.2007.05569.x
Liu, S., Ninan, I., Antonova, I., Battaglia, F., Trinchese, F., Narasanna, A., et al. (2004b). Alpha-synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 23, 4506–4516. doi: 10.1038/sj.emboj.7600451
Liu, W., Jalewa, J., Sharma, M., Li, G., Li, L., and Holscher, C. (2015). Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Neuroscience 303, 42–50. doi: 10.1016/j.neuroscience.2015.06.054
Lokireddy, S., Kukushkin, N. V., and Goldberg, A. L. (2015). cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. U. S. A. 112, E7176–E7185. doi: 10.1073/pnas.1522332112
Longhena, F., Faustini, G., Missale, C., Pizzi, M., Spano, P., and Bellucci, A. (2017). The contribution of alpha-Synuclein spreading to Parkinson's Disease Synaptopathy. Neural Plast. 2017:5012129. doi: 10.1155/2017/5012129
Longhena, F., Faustini, G., Spillantini, M. G., and Bellucci, A. (2019). Living in promiscuity: the multiple Partners of alpha-synuclein at the synapse in physiology and pathology. Int. J. Mol. Sci. 20:141. doi: 10.3390/ijms20010141
Lopez-Burillo, S., Tan, D. X., Mayo, J. C., Sainz, R. M., Manchester, L. C., and Reiter, R. J. (2003). Melatonin, xanthurenic acid, resveratrol, EGCG, vitamin C and alpha-lipoic acid differentially reduce oxidative DNA damage induced by Fenton reagents: a study of their individual and synergistic actions. J. Pineal Res. 34, 269–277. doi: 10.1034/j.1600-079X.2003.00041.x
Lopez, L. C., Escames, G., Tapias, V., Utrilla, P., Leon, J., and Acuna-Castroviejo, D. (2006). Identification of an inducible nitric oxide synthase in diaphragm mitochondria from septic mice: its relation with mitochondrial dysfunction and prevention by melatonin. Int. J. Biochem. Cell Biol. 38, 267–278. doi: 10.1016/j.biocel.2005.09.008
Ludtmann, M. H., Angelova, P. R., Ninkina, N. N., Gandhi, S., Buchman, V. L., and Abramov, A. Y. (2016). Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J. Neurosci. 36, 10510–10521. doi: 10.1523/JNEUROSCI.1659-16.2016
Lundblad, M., Decressac, M., Mattsson, B., and Bjorklund, A. (2012). Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. U. S. A. 109, 3213–3219. doi: 10.1073/pnas.1200575109
Ma, Q. L., Chan, P., Yoshii, M., and Ueda, K. (2003). Alpha-synuclein aggregation and neurodegenerative diseases. J. Alzheimers Dis. 5, 139–148. doi: 10.3233/JAD-2003-5208
Maass, F., Rikker, S., Dambeck, V., Warth, C., Tatenhorst, L., Csoti, I., et al. (2020). Increased alpha-synuclein tear fluid levels in patients with Parkinson's disease. Sci. Rep. 10:8507. doi: 10.1038/s41598-020-65503-1
Magalhaes, P., and Lashuel, H. A. (2022). Opportunities and challenges of alpha-synuclein as a potential biomarker for Parkinson's disease and other synucleinopathies. NPJ Parkinsons Dis. 8:93. doi: 10.1038/s41531-022-00357-0
Magistretti, P. J., and Pellerin, L. (1996). Cellular mechanisms of brain energy metabolism. Relevance to functional brain imaging and to neurodegenerative disorders. Ann. N. Y. Acad. Sci. 777, 380–387.
Mahul-Mellier, A. L., Fauvet, B., Gysbers, A., Dikiy, I., Oueslati, A., Georgeon, S., et al. (2014). C-Abl phosphorylates alpha-synuclein and regulates its degradation: implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson's disease. Hum. Mol. Genet. 23, 2858–2879. doi: 10.1093/hmg/ddt674
Majbour, N. K., Vaikath, N. N., Eusebi, P., Chiasserini, D., Ardah, M., Varghese, S., et al. (2016a). Longitudinal changes in CSF alpha-synuclein species reflect Parkinson's disease progression. Mov. Disord. 31, 1535–1542. doi: 10.1002/mds.26754
Majbour, N. K., Vaikath, N. N., Van Dijk, K. D., Ardah, M. T., Varghese, S., Vesterager, L. B., et al. (2016b). Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson's disease. Mol. Neurodegener. 11:7. doi: 10.1186/s13024-016-0072-9
Malek, N., Swallow, D., Grosset, K. A., Anichtchik, O., Spillantini, M., and Grosset, D. G. (2014). Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson's disease - a systematic review. Acta Neurol. Scand. 130, 59–72. doi: 10.1111/ane.12247
Mandhane, S., Soni, D., Jani, K., Sengupta, P., Patel, A., Bambal, R., et al. (2019). K0706, a potent orally bioavailable brain-penetrating selective inhibitor of cABL protein tyrosine kinase, exhibits neuroprotective activity in preclinical models of Parkinson’s disease [online] Available at: https://www.mdsabstracts.org/abstract/k0706-a-potent-orally-bioavailable-brain-penetrating-selective-inhibitor-of-cabl-protein-tyrosine-kinase-exhibits-neuroprotective-activity-in-preclinical-models-of-parkinsons-disease/ [Accessed May 3, 2023].
Mann, V. M., Cooper, J. M., Daniel, S. E., Srai, K., Jenner, P., Marsden, C. D., et al. (1994). Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann. Neurol. 36, 876–881. doi: 10.1002/ana.410360612
Manzanza, N. O., Sedlackova, L., and Kalaria, R. N. (2021). Alpha-synuclein post-translational modifications: implications for pathogenesis of Lewy body disorders. Front. Aging Neurosci. 13:690293. doi: 10.3389/fnagi.2021.690293
Marotta, N. P., Lin, Y. H., Lewis, Y. E., Ambroso, M. R., Zaro, B. W., Roth, M. T., et al. (2015). O-GlcNAc modification blocks the aggregation and toxicity of the protein alpha-synuclein associated with Parkinson's disease. Nat. Chem. 7, 913–920. doi: 10.1038/nchem.2361
Maroui, M. A., Maarifi, G., Mcmanus, F. P., Lamoliatte, F., Thibault, P., and Chelbi-Alix, M. K. (2018). Promyelocytic leukemia protein (PML) requirement for interferon-induced global cellular SUMOylation. Mol. Cell. Proteomics 17, 1196–1208. doi: 10.1074/mcp.RA117.000447
Masato, A., Plotegher, N., Terrin, F., Sandre, M., Faustini, G., Thor, A., et al. (2023). DOPAL initiates alphaSynuclein-dependent impaired proteostasis and degeneration of neuronal projections in Parkinson's disease. NPJ Parkinsons Dis. 9:42. doi: 10.1038/s41531-023-00485-1
Matsubara, T. (1991). Interleukin 6 activities and tumor necrosis factor-alpha levels in serum of patients with Kawasaki disease. Arerugi 40, 147–154.
Matsura, T. (2019). Protective effect of tocotrienol on in vitro and in vivo models of Parkinson's disease. J. Nutr. Sci. Vitaminol. (Tokyo) 65, S51–S53. doi: 10.3177/jnsv.65.S51
Matunis, M. J., Coutavas, E., and Blobel, G. (1996). A novel ubiquitin-like modification modulates the partitioning of the ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 135, 1457–1470. doi: 10.1083/jcb.135.6.1457
Maxwell, M. M., Tomkinson, E. M., Nobles, J., Wizeman, J. W., Amore, A. M., Quinti, L., et al. (2011). The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 20, 3986–3996. doi: 10.1093/hmg/ddr326
Mayeux, R. (2004). Biomarkers: potential uses and limitations. NeuroRx 1, 182–188. doi: 10.1602/neurorx.1.2.182
Mazzocchi, M., Goulding, S. R., Wyatt, S. L., Collins, L. M., Sullivan, A. M., and O'keeffe, G. W. (2021). LMK235, a small molecule inhibitor of HDAC4/5, protects dopaminergic neurons against neurotoxin-and alpha-synuclein-induced degeneration in cellular models of Parkinson's disease. Mol. Cell. Neurosci. 115:103642. doi: 10.1016/j.mcn.2021.103642
Mbefo, M. K., Paleologou, K. E., Boucharaba, A., Oueslati, A., Schell, H., Fournier, M., et al. (2010). Phosphorylation of synucleins by members of the polo-like kinase family. J. Biol. Chem. 285, 2807–2822. doi: 10.1074/jbc.M109.081950
Medeiros, A. T., Soll, L. G., Tessari, I., Bubacco, L., and Morgan, J. R. (2017). Alpha-synuclein dimers impair vesicle fission during clathrin-mediated synaptic vesicle recycling. Front. Cell. Neurosci. 11:388. doi: 10.3389/fncel.2017.00388
Mehdi, S. J., Rosas-Hernandez, H., Cuevas, E., Lantz, S. M., Barger, S. W., Sarkar, S., et al. (2016). Protein kinases and Parkinson's disease. Int. J. Mol. Sci. 17:1585. doi: 10.3390/ijms17091585
Mehmel, M., Jovanovic, N., and Spitz, U. (2020). Nicotinamide Riboside-the current state of research and therapeutic uses. Nutrients 12:1616. doi: 10.3390/nu12061616
Mehringer, J., Navarro, J. A., Touraud, D., Schneuwly, S., and Kunz, W. (2022). Phosphorylated resveratrol as a protein aggregation suppressor in vitro and in vivo. RSC Chem. Biol. 3, 250–260. doi: 10.1039/D1CB00220A
Menges, S., Minakaki, G., Schaefer, P. M., Meixner, H., Prots, I., Schlotzer-Schrehardt, U., et al. (2017). Alpha-synuclein prevents the formation of spherical mitochondria and apoptosis under oxidative stress. Sci. Rep. 7:42942. doi: 10.1038/srep42942
Menke, T., Gille, G., Reber, F., Janetzky, B., Andler, W., Funk, R. H., et al. (2003). Coenzyme Q10 reduces the toxicity of rotenone in neuronal cultures by preserving the mitochondrial membrane potential. Biofactors 18, 65–72. doi: 10.1002/biof.5520180208
Miyake, Y., Fukushima, W., Tanaka, K., Sasaki, S., Kiyohara, C., Tsuboi, Y., et al. (2011). Dietary intake of antioxidant vitamins and risk of Parkinson's disease: a case-control study in Japan. Eur. J. Neurol. 18, 106–113. doi: 10.1111/j.1468-1331.2010.03088.x
Molina, J. A., De Bustos, F., Ortiz, S., Del Ser, T., Seijo, M., Benito-Leon, J., et al. (2002). Serum levels of coenzyme Q in patients with Lewy body disease. J. Neural Transm. (Vienna) 109, 1195–1201. doi: 10.1007/s00702-001-0761-5
Mollenhauer, B., Batrla, R., El-Agnaf, O., Galasko, D. R., Lashuel, H. A., Merchant, K. M., et al. (2017). A user's guide for alpha-synuclein biomarker studies in biological fluids: Perianalytical considerations. Mov. Disord. 32, 1117–1130. doi: 10.1002/mds.27090
Mollenhauer, B., Trautmann, E., Taylor, P., Manninger, P., Sixel-Doring, F., Ebentheuer, J., et al. (2013). Total CSF alpha-synuclein is lower in de novo Parkinson patients than in healthy subjects. Neurosci. Lett. 532, 44–48. doi: 10.1016/j.neulet.2012.11.004
Montenegro, L., Turnaturi, R., Parenti, C., and Pasquinucci, L. (2018). Idebenone: novel strategies to improve its systemic and local efficacy. Nanomaterials 8:87. doi: 10.3390/nano8020087
Monti, D. A., Zabrecky, G., Kremens, D., Liang, T. W., Wintering, N. A., Cai, J., et al. (2016). N-acetyl cysteine may support dopamine neurons in Parkinson's disease: preliminary clinical and cell line data. PLoS One 11:e0157602. doi: 10.1371/journal.pone.0157602
Moore, D. J., West, A. B., Dawson, V. L., and Dawson, T. M. (2005). Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 28, 57–87. doi: 10.1146/annurev.neuro.28.061604.135718
Morens, D. M., Grandinetti, A., Waslien, C. I., Park, C. B., Ross, G. W., and White, L. R. (1996). Case-control study of idiopathic Parkinson's disease and dietary vitamin E intake. Neurology 46, 1270–1274. doi: 10.1212/WNL.46.5.1270
Mosley, R. L., Benner, E. J., Kadiu, I., Thomas, M., Boska, M. D., Hasan, K., et al. (2006). Neuroinflammation, oxidative stress and the pathogenesis of Parkinson's Disease. Clin. Neurosci. Res. 6, 261–281. doi: 10.1016/j.cnr.2006.09.006
Muller, S., Hoege, C., Pyrowolakis, G., and Jentsch, S. (2001). SUMO, ubiquitin's mysterious cousin. Nat. Rev. Mol. Cell Biol. 2, 202–210. doi: 10.1038/35056591
Munch, G., Luth, H. J., Wong, A., Arendt, T., Hirsch, E., Ravid, R., et al. (2000). Crosslinking of alpha-synuclein by advanced glycation endproducts--an early pathophysiological step in Lewy body formation? J. Chem. Neuroanat. 20, 253–257. doi: 10.1016/S0891-0618(00)00096-X
Nakamura, T., Yamashita, H., Takahashi, T., and Nakamura, S. (2001). Activated Fyn phosphorylates alpha-synuclein at tyrosine residue 125. Biochem. Biophys. Res. Commun. 280, 1085–1092. doi: 10.1006/bbrc.2000.4253
Nassar, N. N., Al-Shorbagy, M. Y., Arab, H. H., and Abdallah, D. M. (2015). Saxagliptin: a novel antiparkinsonian approach. Neuropharmacology 89, 308–317. doi: 10.1016/j.neuropharm.2014.10.007
Nauck, M. A., and Meier, J. J. (2018). Incretin hormones: their role in health and disease. Diabetes Obes. Metab. 20, 5–21. doi: 10.1111/dom.13129
Naylor, S. (2003). Biomarkers: current perspectives and future prospects. Expert. Rev. Mol. Diagn. 3, 525–529. doi: 10.1586/14737159.3.5.525
Negro, A., Brunati, A. M., Donella-Deana, A., Massimino, M. L., and Pinna, L. A. (2002). Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB J. 16, 210–212. doi: 10.1096/fj.01-0517fje
Ohrfelt, A., Zetterberg, H., Andersson, K., Persson, R., Secic, D., Brinkmalm, G., et al. (2011). Identification of novel alpha-synuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochem. Res. 36, 2029–2042. doi: 10.1007/s11064-011-0527-x
Okochi, M., Walter, J., Koyama, A., Nakajo, S., Baba, M., Iwatsubo, T., et al. (2000). Constitutive phosphorylation of the Parkinson's disease associated alpha-synuclein. J. Biol. Chem. 275, 390–397. doi: 10.1074/jbc.275.1.390
Oliveira, L. M. A., Gasser, T., Edwards, R., Zweckstetter, M., Melki, R., Stefanis, L., et al. (2021). Alpha-synuclein research: defining strategic moves in the battle against Parkinson's disease. NPJ Parkinsons Dis. 7:65. doi: 10.1038/s41531-021-00203-9
Ono, K., and Yamada, M. (2007). Vitamin a potently destabilizes preformed alpha-synuclein fibrils in vitro: implications for Lewy body diseases. Neurobiol. Dis. 25, 446–454. doi: 10.1016/j.nbd.2006.10.010
Ono, K., Yoshiike, Y., Takashima, A., Hasegawa, K., Naiki, H., and Yamada, M. (2004). Vitamin a exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp. Neurol. 189, 380–392. doi: 10.1016/j.expneurol.2004.05.035
Orsucci, D., Mancuso, M., Ienco, E. C., Logerfo, A., and Siciliano, G. (2011). Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Curr. Med. Chem. 18, 4053–4064. doi: 10.2174/092986711796957257
Oueslati, A., Fournier, M., and Lashuel, H. A. (2010). Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145. doi: 10.1016/S0079-6123(10)83007-9
Oueslati, A., Paleologou, K. E., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2012). Mimicking phosphorylation at serine 87 inhibits the aggregation of human alpha-synuclein and protects against its toxicity in a rat model of Parkinson's disease. J. Neurosci. 32, 1536–1544. doi: 10.1523/JNEUROSCI.3784-11.2012
Oueslati, A., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2013). Polo-like kinase 2 regulates selective autophagic alpha-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. U. S. A. 110, E3945–E3954. doi: 10.1073/pnas.1309991110
Outeiro, T. F., Kontopoulos, E., Altmann, S. M., Kufareva, I., Strathearn, K. E., Amore, A. M., et al. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science 317, 516–519. doi: 10.1126/science.1143780
Pagan, F., Hebron, M., Valadez, E. H., Torres-Yaghi, Y., Huang, X., Mills, R. R., et al. (2016). Nilotinib effects in Parkinson's disease and dementia with Lewy bodies. J. Parkinsons Dis. 6, 503–517. doi: 10.3233/JPD-160867
Pagan, F. L., Hebron, M. L., Wilmarth, B., Torres-Yaghi, Y., Lawler, A., Mundel, E. E., et al. (2020). Nilotinib effects on safety, tolerability, and potential biomarkers in Parkinson Disease: a phase 2 randomized clinical trial. JAMA Neurol. 77, 309–317. doi: 10.1001/jamaneurol.2019.4200
Pagan, F. L., Hebron, M. L., Wilmarth, B., Torres-Yaghi, Y., Lawler, A., Mundel, E. E., et al. (2019). Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson's disease. Pharmacol. Res. Perspect. 7:e00470. doi: 10.1002/prp2.470
Paik, S. R., Shin, H. J., Lee, J. H., Chang, C. S., and Kim, J. (1999). Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem. J. 340, 821–828.
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., et al. (2010). Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 30, 3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010
Palleria, C., Leo, A., Andreozzi, F., Citraro, R., Iannone, M., Spiga, R., et al. (2017). Liraglutide prevents cognitive decline in a rat model of streptozotocin-induced diabetes independently from its peripheral metabolic effects. Behav. Brain Res. 321, 157–169. doi: 10.1016/j.bbr.2017.01.004
Parekh, P., Sharma, N., Sharma, M., Gadepalli, A., Sayyed, A. A., Chatterjee, S., et al. (2022). AMPK-dependent autophagy activation and alpha-synuclein clearance: a putative mechanism behind alpha-mangostin's neuroprotection in a rotenone-induced mouse model of Parkinson's disease. Metab. Brain Dis. 37, 2853–2870. doi: 10.1007/s11011-022-01087-1
Park, G., Tan, J., Garcia, G., Kang, Y., Salvesen, G., and Zhang, Z. (2016). Regulation of histone acetylation by autophagy in Parkinson disease. J. Biol. Chem. 291, 3531–3540. doi: 10.1074/jbc.M115.675488
Parkinson Study GroupBeal, M. F., Oakes, D., Shoulson, I., Henchcliffe, C., Galpern, W. R., et al. (2014). A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 71, 543–552. doi: 10.1001/jamaneurol.2014.131
Parnetti, L., Gaetani, L., Eusebi, P., Paciotti, S., Hansson, O., El-Agnaf, O., et al. (2019). CSF and blood biomarkers for Parkinson's disease. Lancet Neurol. 18, 573–586. doi: 10.1016/S1474-4422(19)30024-9
Paxinou, E., Chen, Q., Weisse, M., Giasson, B. I., Norris, E. H., Rueter, S. M., et al. (2001). Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 21, 8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001
Pearson, K. J., Baur, J. A., Lewis, K. N., Peshkin, L., Price, N. L., Labinskyy, N., et al. (2008). Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 8, 157–168. doi: 10.1016/j.cmet.2008.06.011
Percario, S., Da Silva Barbosa, A., Varela, E. L. P., Gomes, A. R. Q., Ferreira, M. E. S., Moreira, D. N. A., et al. (2020). Oxidative stress in Parkinson's Disease: potential benefits of antioxidant supplementation. Oxidative Med. Cell. Longev. 2020:2360872. doi: 10.1155/2020/2360872
Permanne, B., Sand, A., Ousson, S., Neny, M., Hantson, J., Schubert, R., et al. (2022). O-GlcNAcase inhibitor ASN90 is a multimodal drug candidate for Tau and alpha-synuclein proteinopathies. ACS Chem. Neurosci. 13, 1296–1314. doi: 10.1021/acschemneuro.2c00057
Petricca, L., Chiki, N., Hanna-El-Daher, L., Aeschbach, L., Burai, R., Stoops, E., et al. (2022). Comparative analysis of Total alpha-synuclein (alphaSYN) immunoassays reveals that they do not capture the diversity of modified alphaSYN proteoforms. J. Parkinsons Dis. 12, 1449–1462. doi: 10.3233/JPD-223285
Pfeiffer, R. F. (2016). Non-motor symptoms in Parkinson's disease. Parkinsonism Relat. Disord. 22, S119–S122. doi: 10.1016/j.parkreldis.2015.09.004
Picca, A., Guerra, F., Calvani, R., Marini, F., Biancolillo, A., Landi, G., et al. (2020). Mitochondrial signatures in circulating extracellular vesicles of older adults with Parkinson's disease: results from the EXosomes in PArkiNson's disease (EXPAND) study. J. Clin. Med. 9:504. doi: 10.3390/jcm9020504
Ping, F., Jiang, N., and Li, Y. (2020). Association between metformin and neurodegenerative diseases of observational studies: systematic review and meta-analysis. BMJ Open Diabetes Res. Care 8:e001370. doi: 10.1136/bmjdrc-2020-001370
Pinho, B. R., Reis, S. D., Guedes-Dias, P., Leitao-Rocha, A., Quintas, C., Valentao, P., et al. (2016). Pharmacological modulation of HDAC1 and HDAC6 in vivo in a zebrafish model: therapeutic implications for Parkinson's disease. Pharmacol. Res. 103, 328–339. doi: 10.1016/j.phrs.2015.11.024
Pissadaki, E. K., and Bolam, J. P. (2013). The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Front. Comput. Neurosci. 7:13. doi: 10.3389/fncom.2013.00013
Pouclet, H., Lebouvier, T., Coron, E., Rouaud, T., Flamant, M., Toulgoat, F., et al. (2012). Analysis of colonic alpha-synuclein pathology in multiple system atrophy. Parkinsonism Relat. Disord. 18, 893–895. doi: 10.1016/j.parkreldis.2012.04.020
Prickaerts, J., Heckman, P. R. A., and Blokland, A. (2017). Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer's disease. Expert Opin. Investig. Drugs 26, 1033–1048. doi: 10.1080/13543784.2017.1364360
Pronin, A. N., Morris, A. J., Surguchov, A., and Benovic, J. L. (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275, 26515–26522. doi: 10.1074/jbc.M003542200
Qing, H., Wong, W., Mcgeer, E. G., and Mcgeer, P. L. (2009a). Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem. Biophys. Res. Commun. 387, 149–152. doi: 10.1016/j.bbrc.2009.06.142
Qing, H., Zhang, Y., Deng, Y., Mcgeer, E. G., and Mcgeer, P. L. (2009b). Lrrk2 interaction with alpha-synuclein in diffuse Lewy body disease. Biochem. Biophys. Res. Commun. 390, 1229–1234. doi: 10.1016/j.bbrc.2009.10.126
Qualman, S. J., Haupt, H. M., Yang, P., and Hamilton, S. R. (1984). Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson's disease. Gastroenterology 87, 848–856. doi: 10.1016/0016-5085(84)90079-9
Quastel, J. H., and Wheatley, A. H. (1932). Oxidations by the brain. Biochem. J. 26, 725–744. doi: 10.1042/bj0260725
Rampersaud, N., Harkavyi, A., Giordano, G., Lever, R., WhittON, J., and WHitton, P. (2012). Exendin-4 reverts behavioural and neurochemical dysfunction in a pre-motor rodent model of Parkinson's disease with noradrenergic deficit. Br. J. Pharmacol. 167, 1467–1479. doi: 10.1111/j.1476-5381.2012.02100.x
Rane, P., Shields, J., Heffernan, M., Guo, Y., Akbarian, S., and King, J. A. (2012). The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 62, 2409–2412. doi: 10.1016/j.neuropharm.2012.01.026
Rees, J. N., Florang, V. R., Eckert, L. L., and Doorn, J. A. (2009). Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem. Res. Toxicol. 22, 1256–1263. doi: 10.1021/tx9000557
Reimer, L., Vesterager, L. B., Betzer, C., Zheng, J., Nielsen, L. D., Kofoed, R. H., et al. (2018). Inflammation kinase PKR phosphorylates alpha-synuclein and causes alpha-synuclein-dependent cell death. Neurobiol. Dis. 115, 17–28. doi: 10.1016/j.nbd.2018.03.001
Reiter, R. J., Paredes, S. D., Korkmaz, A., Jou, M. J., and Tan, D. X. (2008). Melatonin combats molecular terrorism at the mitochondrial level. Interdiscip. Toxicol. 1, 137–149. doi: 10.2478/v10102-010-0030-2
Reiter, R. J., Sainz, R. M., Lopez-Burillo, S., Mayo, J. C., Manchester, L. C., and Tan, D. X. (2003). Melatonin ameliorates neurologic damage and neurophysiologic deficits in experimental models of stroke. Ann. N. Y. Acad. Sci. 993, 35–47. discussion 48-53. doi: 10.1111/j.1749-6632.2003.tb07509.x
Reiter, R. J., Tan, D. X., and Allegra, M. (2002a). Melatonin: reducing molecular pathology and dysfunction due to free radicals and associated reactants. Neuro Endocrinol. Lett. 23, 3–8.
Reiter, R. J., Tan, D. X., and Burkhardt, S. (2002b). Reactive oxygen and nitrogen species and cellular and organismal decline: amelioration with melatonin. Mech. Ageing Dev. 123, 1007–1019. doi: 10.1016/s0047-6374(01)00384-0
Reiter, R. J., Tan, D. X., Leon, J., Kilic, U., and Kilic, E. (2005). When melatonin gets on your nerves: its beneficial actions in experimental models of stroke. Exp. Biol. Med. 230, 104–117. doi: 10.1177/153537020523000205
Rey, N. L., George, S., and Brundin, P. (2016a). Review: spreading the word: precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol. Appl. Neurobiol. 42, 51–76. doi: 10.1111/nan.12299
Rey, N. L., George, S., Steiner, J. A., Madaj, Z., Luk, K. C., Trojanowski, J. Q., et al. (2018). Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 135, 65–83. doi: 10.1007/s00401-017-1792-9
Rey, N. L., Steiner, J. A., Maroof, N., Luk, K. C., Madaj, Z., Trojanowski, J. Q., et al. (2016b). Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. J. Exp. Med. 213, 1759–1778. doi: 10.1084/jem.20160368
Richter-Landsberg, C., and Leyk, J. (2013). Inclusion body formation, macroautophagy, and the role of HDAC6 in neurodegeneration. Acta Neuropathol. 126, 793–807. doi: 10.1007/s00401-013-1158-x
Risiglione, P., Zinghirino, F., Di Rosa, M. C., Magri, A., and Messina, A. (2021). Alpha-Synuclein and mitochondrial dysfunction in Parkinson's disease: the emerging role of VDAC. Biomol. Ther. 11:718. doi: 10.3390/biom11050718
Rodriguez, C., Mayo, J. C., Sainz, R. M., Antolin, I., Herrera, F., Martin, V., et al. (2004). Regulation of antioxidant enzymes: a significant role for melatonin. J. Pineal Res. 36, 1–9. doi: 10.1046/j.1600-079X.2003.00092.x
Rodriguez, M. I., Escames, G., Lopez, L. C., Lopez, A., Garcia, J. A., Ortiz, F., et al. (2007). Chronic melatonin treatment reduces the age-dependent inflammatory process in senescence-accelerated mice. J. Pineal Res. 42, 272–279. doi: 10.1111/j.1600-079X.2006.00416.x
Ross, C. A., and Pickart, C. M. (2004). The ubiquitin-proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol. 14, 703–711. doi: 10.1016/j.tcb.2004.10.006
Rott, R., Szargel, R., Haskin, J., Shani, V., Shainskaya, A., Manov, I., et al. (2008). Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328. doi: 10.1074/jbc.M704809200
Rott, R., Szargel, R., Shani, V., Hamza, H., Savyon, M., Abd Elghani, F., et al. (2017). SUMOylation and ubiquitination reciprocally regulate alpha-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. U. S. A. 114, 13176–13181. doi: 10.1073/pnas.1704351114
Rousseaux, M. W., Revelli, J. P., Vazquez-Velez, G. E., Kim, J. Y., Craigen, E., Gonzales, K., et al. (2018). Depleting Trim28 in adult mice is well tolerated and reduces levels of alpha-synuclein and tau. elife 7:e36768. doi: 10.7554/eLife.36768
Ryan, B. J., Lourenco-Venda, L. L., Crabtree, M. J., Hale, A. B., Channon, K. M., and Wade-Martins, R. (2014). Alpha-synuclein and mitochondrial bioenergetics regulate tetrahydrobiopterin levels in a human dopaminergic model of Parkinson disease. Free Radic. Biol. Med. 67, 58–68. doi: 10.1016/j.freeradbiomed.2013.10.008
Ryu, Y. K., Go, J., Park, H. Y., Choi, Y. K., Seo, Y. J., Choi, J. H., et al. (2020). Metformin regulates astrocyte reactivity in Parkinson's disease and normal aging. Neuropharmacology 175:108173. doi: 10.1016/j.neuropharm.2020.108173
Sajja, R. K., Prasad, S., Tang, S., Kaisar, M. A., and Cucullo, L. (2017). Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: role of ABCB10? Neurosci. Lett. 653, 152–158. doi: 10.1016/j.neulet.2017.05.059
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 98, 8554–8559. doi: 10.1073/pnas.141230798
Sanders, O., and Rajagopal, L. (2020). Phosphodiesterase inhibitors for Alzheimer's disease: a systematic review of clinical trials and epidemiology with a mechanistic rationale. J. Alzheimers Dis. Rep. 4, 185–215. doi: 10.3233/ADR-200191
Sato, K., Yamashita, T., Kurata, T., Lukic, V., Fukui, Y., Hishikawa, N., et al. (2014). Telmisartan reduces progressive oxidative stress and phosphorylated alpha-synuclein accumulation in stroke-resistant spontaneously hypertensive rats after transient middle cerebral artery occlusion. J. Stroke Cerebrovasc. Dis. 23, 1554–1563. doi: 10.1016/j.jstrokecerebrovasdis.2013.12.051
Satoh, A., and Imai, S. (2014). Systemic regulation of mammalian ageing and longevity by brain sirtuins. Nat. Commun. 5:4211. doi: 10.1038/ncomms5211
Savyon, M., and Engelender, S. (2020). SUMOylation in alpha-Synuclein homeostasis and pathology. Front. Aging Neurosci. 12:167. doi: 10.3389/fnagi.2020.00167
Sayin, V. I., Ibrahim, M. X., Larsson, E., Nilsson, J. A., Lindahl, P., and Bergo, M. O. (2014). Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 6:221ra15. doi: 10.1126/scitranslmed.3007653
Schapira, A. H. (2008). Mitochondrial dysfunction in neurodegenerative diseases. Neurochem. Res. 33, 2502–2509. doi: 10.1007/s11064-008-9855-x
Schapira, A. H., and Tolosa, E. (2010). Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat. Rev. Neurol. 6, 309–317. doi: 10.1038/nrneurol.2010.52
Scheider, W. L., Hershey, L. A., Vena, J. E., Holmlund, T., and Marshall, J. R., Freudenheim (1997). Dietary antioxidants and other dietary factors in the etiology of Parkinson's disease. Mov. Disord. 12, 190–196. doi: 10.1002/mds.870120209
Schepici, G., Bramanti, P., and Mazzon, E. (2020). Efficacy of sulforaphane in neurodegenerative diseases. Int. J. Mol. Sci. 21:8637. doi: 10.3390/ijms21228637
Schirinzi, T., Martella, G., Imbriani, P., Di Lazzaro, G., Franco, D., Colona, V. L., et al. (2019). Dietary vitamin E as a protective factor for Parkinson's disease: clinical and experimental evidence. Front. Neurol. 10:148. doi: 10.3389/fneur.2019.00148
Schmid, A. W., Fauvet, B., Moniatte, M., and Lashuel, H. A. (2013). Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol. Cell. Proteomics 12, 3543–3558. doi: 10.1074/mcp.R113.032730
Schneider, S. A., and Alcalay, R. N. (2017). Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov. Disord. 32, 1504–1523. doi: 10.1002/mds.27193
Schondorf, D. C., Ivanyuk, D., Baden, P., Sanchez-Martinez, A., De Cicco, S., Yu, C., et al. (2018). The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and Fly models of Parkinson's disease. Cell Rep. 23, 2976–2988. doi: 10.1016/j.celrep.2018.05.009
Selnick, H. G., Hess, J. F., Tang, C., Liu, K., Schachter, J. B., Ballard, J. E., et al. (2019). Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for Tauopathies. J. Med. Chem. 62, 10062–10097. doi: 10.1021/acs.jmedchem.9b01090
Shabek, N., Herman-Bachinsky, Y., Buchsbaum, S., Lewinson, O., Haj-Yahya, M., Hejjaoui, M., et al. (2012). The size of the proteasomal substrate determines whether its degradation will be mediated by mono-or polyubiquitylation. Mol. Cell 48, 87–97. doi: 10.1016/j.molcel.2012.07.011
Shamsaldeen, Y. A., Mackenzie, L. S., Lione, L. A., and Benham, C. D. (2016). Methylglyoxal, a metabolite increased in diabetes is associated with insulin resistance, vascular dysfunction and neuropathies. Curr. Drug Metab. 17, 359–367. doi: 10.2174/1389200217666151222155216
Shannon, K. M., Keshavarzian, A., Mutlu, E., Dodiya, H. B., Daian, D., Jaglin, J. A., et al. (2012). Alpha-synuclein in colonic submucosa in early untreated Parkinson's disease. Mov. Disord. 27, 709–715. doi: 10.1002/mds.23838
Sharma, S., Taliyan, R., and Ramagiri, S. (2015a). Histone deacetylase inhibitor, trichostatin A, improves learning and memory in high-fat diet-induced cognitive deficits in mice. J. Mol. Neurosci. 56, 1–11. doi: 10.1007/s12031-014-0461-x
Sharma, S. K., Chorell, E., Steneberg, P., Vernersson-Lindahl, E., Edlund, H., and Wittung-Stafshede, P. (2015b). Insulin-degrading enzyme prevents alpha-synuclein fibril formation in a nonproteolytical manner. Sci. Rep. 5:12531. doi: 10.1038/srep12531
Sharma, S. K., Chorell, E., and Wittung-Stafshede, P. (2015c). Insulin-degrading enzyme is activated by the C-terminus of alpha-synuclein. Biochem. Biophys. Res. Commun. 466, 192–195. doi: 10.1016/j.bbrc.2015.09.002
Shavali, S., Carlson, E. C., Swinscoe, J. C., and Ebadi, M. (2004). 1-Benzyl-1,2,3,4-tetrahydroisoquinoline, a Parkinsonism-inducing endogenous toxin, increases alpha-synuclein expression and causes nuclear damage in human dopaminergic cells. J. Neurosci. Res. 76, 563–571. doi: 10.1002/jnr.20082
Sherer, T. B., Betarbet, R., Testa, C. M., Seo, B. B., Richardson, J. R., Kim, J. H., et al. (2003). Mechanism of toxicity in rotenone models of Parkinson's disease. J. Neurosci. 23, 10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003
Shibasaki, Y., Baillie, D. A., St Clair, D., and Brookes, A. J. (1995). High-resolution mapping of SNCA encoding alpha-synuclein, the non-A beta component of Alzheimer's disease amyloid precursor, to human chromosome 4q21.3-->q22 by fluorescence in situ hybridization. Cytogenet. Cell Genet. 71, 54–55.
Shukla, J. J., Stefanova, N., Bush, A. I., Mccoll, G., Finkelstein, D. I., and Mcallum, E. J. (2021). Therapeutic potential of iron modulating drugs in a mouse model of multiple system atrophy. Neurobiol. Dis. 159:105509. doi: 10.1016/j.nbd.2021.105509
Shults, C. W. (2005). Therapeutic role of coenzyme Q(10) in Parkinson's disease. Pharmacol. Ther. 107, 120–130. doi: 10.1016/j.pharmthera.2005.02.002
Shults, C. W., Haas, R. H., Passov, D., and Beal, M. F. (1997). Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann. Neurol. 42, 261–264. doi: 10.1002/ana.410420221
Shults, C. W., and Schapira, A. H. (2001). A cue to queue for CoQ? Neurology 57, 375–376. doi: 10.1212/WNL.57.3.375
Simon, C., Soga, T., Okano, H. J., and Parhar, I. (2021). Alpha-synuclein-mediated neurodegeneration in dementia with Lewy bodies: the pathobiology of a paradox. Cell Biosci. 11:196. doi: 10.1186/s13578-021-00709-y
Simuni, T., Fiske, B., Merchant, K., Coffey, C. S., Klingner, E., Caspell-Garcia, C., et al. (2021). Efficacy of Nilotinib in patients with moderately advanced Parkinson disease: a randomized clinical trial. JAMA Neurol. 78, 312–320. doi: 10.1001/jamaneurol.2020.4725
Singh, A., Boldin-Adamsky, S., Thimmulappa, R. K., Rath, S. K., Ashush, H., Coulter, J., et al. (2008). RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 68, 7975–7984. doi: 10.1158/0008-5472.CAN-08-1401
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., et al. (2005a). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 14, 3801–3811. doi: 10.1093/hmg/ddi396
Smith, W. W., Margolis, R. L., Li, X., Troncoso, J. C., Lee, M. K., Dawson, V. L., et al. (2005b). Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 25, 5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005
Sohmiya, M., Tanaka, M., Tak, N. W., Yanagisawa, M., Tanino, Y., Suzuki, Y., et al. (2004). Redox status of plasma coenzyme Q10 indicates elevated systemic oxidative stress in Parkinson's disease. J. Neurol. Sci. 223, 161–166. doi: 10.1016/j.jns.2004.05.007
Song, M. K., Adams, L., Lee, J. H., and Kim, Y. S. (2022). NXP031 prevents dopaminergic neuronal loss and oxidative damage in the AAV-WT-alpha-synuclein mouse model of Parkinson's disease. PLoS One 17:e0272085. doi: 10.1371/journal.pone.0272085
Sonustun, B., Altay, M. F., Strand, C., Ebanks, K., Hondhamuni, G., Warner, T. T., et al. (2022). Pathological relevance of Post-Translationally modified alpha-synuclein (pSer87, pSer129, nTyr39) in idiopathic Parkinson's Disease and multiple system atrophy. Cells 11:906. doi: 10.3390/cells11050906
Sorrentino, L., Cossu, F., Milani, M., Aliverti, A., and Mastrangelo, E. (2017). Structural bases of the altered catalytic properties of a pathogenic variant of apoptosis inducing factor. Biochem. Biophys. Res. Commun. 490, 1011–1017. doi: 10.1016/j.bbrc.2017.06.156
Souza, J. M., Giasson, B. I., Chen, Q., Lee, V. M., and Ischiropoulos, H. (2000). Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 275, 18344–18349. doi: 10.1074/jbc.M000206200
Spillantini, M. G. (1999). Parkinson's disease, dementia with Lewy bodies and multiple system atrophy are alpha-synucleinopathies. Parkinsonism Relat. Disord. 5, 157–162. doi: 10.1016/S1353-8020(99)00031-0
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U. S. A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spillantini, M. G., and Goedert, M. (2016). Synucleinopathies: past, present and future. Neuropathol. Appl. Neurobiol. 42, 3–5. doi: 10.1111/nan.12311
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Sprenger, F. S., Stefanova, N., Gelpi, E., Seppi, K., Navarro-Otano, J., Offner, F., et al. (2015). Enteric nervous system alpha-synuclein immunoreactivity in idiopathic REM sleep behavior disorder. Neurology 85, 1761–1768. doi: 10.1212/WNL.0000000000002126
St Laurent, R., Obrien, L. M., and Ahmad, S. T. (2013). Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson's disease. Neuroscience 246, 382–390. doi: 10.1016/j.neuroscience.2013.04.037
Stefanis, L. (2012). Alpha-synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Stefanis, L., Emmanouilidou, E., Pantazopoulou, M., Kirik, D., Vekrellis, K., and Tofaris, G. K. (2019). How is alpha-synuclein cleared from the cell? J. Neurochem. 150, 577–590. doi: 10.1111/jnc.14704
Stefanson, A. L., and Bakovic, M. (2014). Dietary regulation of Keap1/Nrf2/ARE pathway: focus on plant-derived compounds and trace minerals. Nutrients 6, 3777–3801. doi: 10.3390/nu6093777
Stewart, T., Sossi, V., Aasly, J. O., Wszolek, Z. K., Uitti, R. J., Hasegawa, K., et al. (2015). Phosphorylated alpha-synuclein in Parkinson's disease: correlation depends on disease severity. Acta Neuropathol. Commun. 3:7. doi: 10.1186/s40478-015-0185-3
Stoessl, A. J. (2016). Salivary gland biopsy for diagnosis of Parkinson's disease? Lancet Neurol. 15, 654–656. doi: 10.1016/S1474-4422(16)30031-X
Stokholm, M. G., Danielsen, E. H., Hamilton-Dutoit, S. J., and Borghammer, P. (2016). Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 79, 940–949. doi: 10.1002/ana.24648
Sudnikovich, E. J., Maksimchik, Y. Z., Zabrodskaya, S. V., Kubyshin, V. L., Lapshina, E. A., Bryszewska, M., et al. (2007). Melatonin attenuates metabolic disorders due to streptozotocin-induced diabetes in rats. Eur. J. Pharmacol. 569, 180–187. doi: 10.1016/j.ejphar.2007.05.018
Sugeno, N., Takeda, A., Hasegawa, T., Kobayashi, M., Kikuchi, A., Mori, F., et al. (2008). Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 283, 23179–23188. doi: 10.1074/jbc.M802223200
Sun, X., Majumder, P., Shioya, H., Wu, F., Kumar, S., Weichselbaum, R., et al. (2000). Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J. Biol. Chem. 275, 17237–17240. doi: 10.1074/jbc.C000099200
Suo, H., Wang, P., Tong, J., Cai, L., Liu, J., Huang, D., et al. (2015). NRSF is an essential mediator for the neuroprotection of trichostatin a in the MPTP mouse model of Parkinson's disease. Neuropharmacology 99, 67–78. doi: 10.1016/j.neuropharm.2015.07.015
Svenningsson, P., Wirdefeldt, K., Yin, L., Fang, F., Markaki, I., Efendic, S., et al. (2016). Reduced incidence of Parkinson's disease after dipeptidyl peptidase-4 inhibitors-a nationwide case-control study. Mov. Disord. 31, 1422–1423. doi: 10.1002/mds.26734
Takahashi, M., Kanuka, H., Fujiwara, H., Koyama, A., Hasegawa, M., Miura, M., et al. (2003). Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 336, 155–158. doi: 10.1016/S0304-3940(02)01258-2
Takahashi, T., Yamashita, H., Nakamura, T., Nagano, Y., and Nakamura, S. (2002). Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 938, 73–80. doi: 10.1016/S0006-8993(02)02498-8
Takamatsu, Y., Fujita, M., Ho, G. J., Wada, R., Sugama, S., Takenouchi, T., et al. (2018). Motor and nonmotor symptoms of Parkinson's disease: antagonistic pleiotropy phenomena derived from alpha-synuclein evolvability? Parkinsons Dis. 2018:5789424. doi: 10.1155/2018/5789424
Tanei, Z. I., Saito, Y., Ito, S., Matsubara, T., Motoda, A., Yamazaki, M., et al. (2021). Lewy pathology of the esophagus correlates with the progression of Lewy body disease: a Japanese cohort study of autopsy cases. Acta Neuropathol. 141, 25–37. doi: 10.1007/s00401-020-02233-8
Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., et al. (2001). Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276, 35368–35374. doi: 10.1074/jbc.M104214200
Tavassoly, O., Yue, J., and Vocadlo, D. J. (2021). Pharmacological inhibition and knockdown of O-GlcNAcase reduces cellular internalization of alpha-synuclein preformed fibrils. FEBS J. 288, 452–470. doi: 10.1111/febs.15349
Teleanu, D. M., Niculescu, A. G., Lungu, I., Radu, C. I., Vladacenco, O., Roza, E., et al. (2022). An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int. J. Mol. Sci. 23:5938. doi: 10.3390/ijms23115938
Tenreiro, S., Reimao-Pinto, M. M., Antas, P., Rino, J., Wawrzycka, D., Macedo, D., et al. (2014). Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson's disease. PLoS Genet. 10:e1004302. doi: 10.1371/journal.pgen.1004302
Tetzlaff, J. E., Putcha, P., Outeiro, T. F., Ivanov, A., Berezovska, O., Hyman, B. T., et al. (2008). CHIP targets toxic alpha-Synuclein oligomers for degradation. J. Biol. Chem. 283, 17962–17968. doi: 10.1074/jbc.M802283200
Thorne, N. J., and Tumbarello, D. A. (2022). The relationship of alpha-synuclein to mitochondrial dynamics and quality control. Front. Mol. Neurosci. 15:947191. doi: 10.3389/fnmol.2022.947191
Tian, C., Liu, G., Gao, L., Soltys, D., Pan, C., Stewart, T., et al. (2019). Erythrocytic alpha-Synuclein as a potential biomarker for Parkinson's disease. Transl. Neurodegener. 8:15. doi: 10.1186/s40035-019-0155-y
Tofaris, G. K., Kim, H. T., Hourez, R., Jung, J. W., Kim, K. P., and Goldberg, A. L. (2011). Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. U. S. A. 108, 17004–17009. doi: 10.1073/pnas.1109356108
Tofaris, G. K., Layfield, R., and Spillantini, M. G. (2001). Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 509, 22–26. doi: 10.1016/S0014-5793(01)03115-5
Tofaris, G. K., Razzaq, A., Ghetti, B., Lilley, K. S., and Spillantini, M. G. (2003). Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 278, 44405–44411. doi: 10.1074/jbc.M308041200
Toker, L., Tran, G. T., Sundaresan, J., Tysnes, O. B., Alves, G., Haugarvoll, K., et al. (2021). Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson's disease brain. Mol. Neurodegener. 16:31. doi: 10.1186/s13024-021-00450-7
Tokuda, T., Qureshi, M. M., Ardah, M. T., Varghese, S., Shehab, S. A., Kasai, T., et al. (2010). Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75, 1766–1772. doi: 10.1212/WNL.0b013e3181fd613b
Tremblay, M. A., Acker, C. M., and Davies, P. (2010). Tau phosphorylated at tyrosine 394 is found in Alzheimer's disease tangles and can be a product of the Abl-related kinase, Arg. J. Alzheimers Dis. 19, 721–733. doi: 10.3233/JAD-2010-1271
Trezzi, J. P., Galozzi, S., Jaeger, C., Barkovits, K., Brockmann, K., Maetzler, W., et al. (2017). Distinct metabolomic signature in cerebrospinal fluid in early parkinson's disease. Mov. Disord. 32, 1401–1408. doi: 10.1002/mds.27132
Uddin, M. S., Mamun, A. A., Jakaria, M., Thangapandiyan, S., Ahmad, J., Rahman, M. A., et al. (2020). Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 707:135624. doi: 10.1016/j.scitotenv.2019.135624
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 90, 11282–11286. doi: 10.1073/pnas.90.23.11282
Ullman, O., Fisher, C. K., and Stultz, C. M. (2011). Explaining the structural plasticity of alpha-synuclein. J. Am. Chem. Soc. 133, 19536–19546. doi: 10.1021/ja208657z
Vaccari, C., Grotto, D., Pereira, T. D. V., De Camargo, J. L. V., and Lopes, L. C. (2021). GLP-1 and GIP receptor agonists in the treatment of Parkinson's disease: translational systematic review and meta-analysis protocol of clinical and preclinical studies. PLoS One 16:e0255726. doi: 10.1371/journal.pone.0255726
Vamvaca, K., Volles, M. J., and Lansbury, P. T. Jr. (2009). The first N-terminal amino acids of alpha-synuclein are essential for alpha-helical structure formation in vitro and membrane binding in yeast. J. Mol. Biol. 389, 413–424. doi: 10.1016/j.jmb.2009.03.021
Vancraenenbroeck, R., Lobbestael, E., Maeyer, M. D., Baekelandt, V., and Taymans, J. M. (2011). Kinases as targets for Parkinson's disease: from genetics to therapy. CNS Neurol. Disord. Drug Targets 10, 724–740. doi: 10.2174/187152711797247858
Vicente Miranda, H., Cassio, R., Correia-Guedes, L., Gomes, M. A., Chegao, A., Miranda, E., et al. (2017a). Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson's disease. Sci. Rep. 7:13713. doi: 10.1038/s41598-017-14175-5
Vicente Miranda, H., and Outeiro, T. F. (2010). The sour side of neurodegenerative disorders: the effects of protein glycation. J. Pathol. 221, 13–25. doi: 10.1002/path.2682
Vicente Miranda, H., Szego, E. M., Oliveira, L. M. A., Breda, C., Darendelioglu, E., De Oliveira, R. M., et al. (2017b). Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 140, 1399–1419. doi: 10.1093/brain/awx056
Vijayakumaran, S., Nakamura, Y., Henley, J. M., and Pountney, D. L. (2019). Ginkgolic acid promotes autophagy-dependent clearance of intracellular alpha-synuclein aggregates. Mol. Cell. Neurosci. 101:103416. doi: 10.1016/j.mcn.2019.103416
Vilas, D., Iranzo, A., Tolosa, E., Aldecoa, I., Berenguer, J., Vilaseca, I., et al. (2016). Assessment of alpha-synuclein in submandibular glands of patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol. 15, 708–718. doi: 10.1016/S1474-4422(16)00080-6
Vivacqua, G., Mason, M., De Bartolo, M. I., Wegrzynowicz, M., Calo, L., Belvisi, D., et al. (2023). Salivary alpha-synuclein RT-QuIC correlates with disease severity in de novo Parkinson's disease. Mov. Disord. 38, 153–155. doi: 10.1002/mds.29246
Vivacqua, G., Suppa, A., Mancinelli, R., Belvisi, D., Fabbrini, A., Costanzo, M., et al. (2019). Salivary alpha-synuclein in the diagnosis of Parkinson's disease and progressive supranuclear palsy. Parkinsonism Relat. Disord. 63, 143–148. doi: 10.1016/j.parkreldis.2019.02.014
Wahlqvist, M. L., Lee, M. S., Hsu, C. C., Chuang, S. Y., Lee, J. T., and Tsai, H. N. (2012). Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson's disease occurring with type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 18, 753–758. doi: 10.1016/j.parkreldis.2012.03.010
Wakabayashi, K. (2020). Where and how alpha-synuclein pathology spreads in Parkinson's disease. Neuropathology 40, 415–425. doi: 10.1111/neup.12691
Wakabayashi, K., Miki, Y., Tanji, K., and Mori, F. (2022). Neuropathology of multiple system atrophy, a glioneuronal degenerative disease. Cerebellum, doi: 10.1007/s12311-022-01407-2 [Epub ahead of print].
Wang, D. B., Kinoshita, C., Kinoshita, Y., Sopher, B. L., Uo, T., Lee, R. J., et al. (2019). Neuronal susceptibility to beta-amyloid toxicity and ischemic injury involves histone deacetylase-2 regulation of endophilin-B1. Brain Pathol. 29, 164–175. doi: 10.1111/bpa.12647
Wang, N., Garcia, J., Freeman, R., and Gibbons, C. H. (2020a). Phosphorylated alpha-synuclein within cutaneous autonomic nerves of patients with Parkinson's disease: the implications of sample thickness on results. J. Histochem. Cytochem. 68, 669–678. doi: 10.1369/0022155420960250
Wang, W., Perovic, I., Chittuluru, J., Kaganovich, A., Nguyen, L. T., Liao, J., et al. (2011). A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U. S. A. 108, 17797–17802. doi: 10.1073/pnas.1113260108
Wang, X. Y., Kang, W. Y., Yang, Q., Zhang, L. Y., Chen, S. D., and Liu, J. (2014). Using gastrocnemius sEMG and plasma alpha-synuclein for the prediction of freezing of gait in Parkinson's disease patients. PLoS One 9:e89353. doi: 10.1371/journal.pone.0116382
Wang, Z., Becker, K., Donadio, V., Siedlak, S., Yuan, J., Rezaee, M., et al. (2020b). Skin alpha-synuclein aggregation seeding activity as a novel biomarker for Parkinson disease. JAMA Neurol. 78, 1–11. doi: 10.1001/jamaneurol.2020.3311
Watson, J. B., Hatami, A., David, H., Masliah, E., Roberts, K., Evans, C. E., et al. (2009). Alterations in corticostriatal synaptic plasticity in mice overexpressing human alpha-synuclein. Neuroscience 159, 501–513. doi: 10.1016/j.neuroscience.2009.01.021
Waxman, E. A., and Giasson, B. I. (2011). Characterization of kinases involved in the phosphorylation of aggregated alpha-synuclein. J. Neurosci. Res. 89, 231–247. doi: 10.1002/jnr.22537
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.M300227200
Weetman, J., Wong, M. B., Sharry, S., Rcom-H'cheo-Gauthier, A., Gai, W. P., Meedeniya, A., et al. (2013). Increased SUMO-1 expression in the unilateral rotenone-lesioned mouse model of Parkinson's disease. Neurosci. Lett. 544, 119–124. doi: 10.1016/j.neulet.2013.03.057
Weissman, A. M. (2001). Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2, 169–178. doi: 10.1038/35056563
Wenz, T. (2013). Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion 13, 134–142. doi: 10.1016/j.mito.2013.01.006
Werner, M. H., and Olanow, C. W. (2022). Parkinson's disease modification through Abl kinase inhibition: an opportunity. Mov. Disord. 37, 6–15. doi: 10.1002/mds.28858
Wilkaniec, A., Strosznajder, J. B., and Adamczyk, A. (2013). Toxicity of extracellular secreted alpha-synuclein: its role in nitrosative stress and neurodegeneration. Neurochem. Int. 62, 776–783. doi: 10.1016/j.neuint.2013.02.004
Wilkinson, K. A., and Henley, J. M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145. doi: 10.1042/BJ20100158
Wilson, C. J., and Callaway, J. C. (2000). Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J. Neurophysiol. 83, 3084–3100. doi: 10.1152/jn.2000.83.5.3084
Winiarska, K., Fraczyk, T., Malinska, D., Drozak, J., and Bryla, J. (2006). Melatonin attenuates diabetes-induced oxidative stress in rabbits. J. Pineal Res. 40, 168–176. doi: 10.1111/j.1600-079X.2005.00295.x
Winklhofer, K. F., and Haass, C. (2010). Mitochondrial dysfunction in Parkinson's disease. Biochim. Biophys. Acta 1802, 29–44. doi: 10.1016/j.bbadis.2009.08.013
Witt, M., Bormann, K., Gudziol, V., Pehlke, K., Barth, K., Minovi, A., et al. (2009). Biopsies of olfactory epithelium in patients with Parkinson's disease. Mov. Disord. 24, 906–914. doi: 10.1002/mds.22464
Wu, T., Thazhath, S. S., Marathe, C. S., Bound, M. J., Jones, K. L., Horowitz, M., et al. (2015). Comparative effect of intraduodenal and intrajejunal glucose infusion on the gut-incretin axis response in healthy males. Nutr. Diabetes 5:e156. doi: 10.1038/nutd.2015.6
Wu, Y., and Guo, S. W. (2008). Histone deacetylase inhibitors trichostatin A and valproic acid induce cell cycle arrest and p21 expression in immortalized human endometrial stromal cells. Eur. J. Obstet. Gynecol. Reprod. Biol. 137, 198–203. doi: 10.1016/j.ejogrb.2007.02.014
Xie, X., Yu, C., Zhou, J., Xiao, Q., Shen, Q., Xiong, Z., et al. (2020). Nicotinamide mononucleotide ameliorates the depression-like behaviors and is associated with attenuating the disruption of mitochondrial bioenergetics in depressed mice. J. Affect. Disord. 263, 166–174. doi: 10.1016/j.jad.2019.11.147
Yang, Y. W., Hsieh, T. F., Li, C. I., Liu, C. S., Lin, W. Y., Chiang, J. H., et al. (2017). Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Medicine (Baltimore) 96:e5921. doi: 10.1097/MD.0000000000009419
Yu, Y., Zhang, L., Li, X., Run, X., Liang, Z., Li, Y., et al. (2012). Differential effects of an O-GlcNAcase inhibitor on tau phosphorylation. PLoS One 7:e35277. doi: 10.1371/journal.pone.0051967
Yu, Z., Xu, X., Xiang, Z., Zhou, J., Zhang, Z., Hu, C., et al. (2010). Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS One 5:e9956. doi: 10.1371/journal.pone.0015623
Yun, S. P.., Kim, D., Kim, S., Kim, S., Karuppagounder, S. S., Kwon, S. H., et al. (2018). Biopsiesalpha-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol Neurodegener. 13:1. doi: 10.1186/s13024-017-0233-5
Yuzwa, S. A., Cheung, A. H., Okon, M., Mcintosh, L. P., and Vocadlo, D. J. (2014a). O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J. Mol. Biol. 426, 1736–1752. doi: 10.1016/j.jmb.2014.01.004
Yuzwa, S. A., Macauley, M. S., Heinonen, J. E., Shan, X., Dennis, R. J., He, Y., et al. (2008). A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 4, 483–490. doi: 10.1038/nchembio.96
Yuzwa, S. A., Shan, X., Jones, B. A., Zhao, G., Woodward, M. L., Li, X., et al. (2014b). Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol. Neurodegener. 9:42. doi: 10.1186/1750-1326-9-42
Yuzwa, S. A., Shan, X., Macauley, M. S., Clark, T., Skorobogatko, Y., Vosseller, K., et al. (2012). Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 8, 393–399. doi: 10.1038/nchembio.797
Zange, L., Noack, C., Hahn, K., Stenzel, W., and Lipp, A. (2015). Phosphorylated alpha-synuclein in skin nerve fibres differentiates Parkinson's disease from multiple system atrophy. Brain 138, 2310–2321. doi: 10.1093/brain/awv138
Zhang, F., Hu, Y., Huang, P., Toleman, C. A., Paterson, A. J., and Kudlow, J. E. (2007). Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J. Biol. Chem. 282, 22460–22471. doi: 10.1074/jbc.M702439200
Zhang, J., Lei, H., Chen, Y., Ma, Y. T., Jiang, F., Tan, J., et al. (2017). Enzymatic O-GlcNAcylation of alpha-synuclein reduces aggregation and increases SDS-resistant soluble oligomers. Neurosci. Lett. 655, 90–94. doi: 10.1016/j.neulet.2017.06.034
Zhang, J., Li, X., and Li, J. D. (2019). The roles of post-translational modifications on alpha-synuclein in the pathogenesis of Parkinson's diseases. Front. Neurosci. 13:381. doi: 10.3389/fnins.2019.00381
Zhang, J., Perry, G., Smith, M. A., Robertson, D., Olson, S. J., Graham, D. G., et al. (1999). Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 154, 1423–1429. doi: 10.1016/S0002-9440(10)65396-5
Zhang, L. F., Yu, X. L., Ji, M., Liu, S. Y., Wu, X. L., Wang, Y. J., et al. (2018). Resveratrol alleviates motor and cognitive deficits and neuropathology in the A53T alpha-synuclein mouse model of Parkinson's disease. Food Funct. 9, 6414–6426. doi: 10.1039/C8FO00964C
Zhang, L. Y., Jin, Q. Q., Holscher, C., and Li, L. (2021). Glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist DA-CH5 is superior to exendin-4 in protecting neurons in the 6-hydroxydopamine rat Parkinson model. Neural Regen. Res. 16, 1660–1670. doi: 10.4103/1673-5374.303045
Zhang, S., Xie, J., Xia, Y., Yu, S., Gu, Z., Feng, R., et al. (2015). LK6/Mnk2a is a new kinase of alpha synuclein phosphorylation mediating neurodegeneration. Sci. Rep. 5:12564. doi: 10.1038/srep12564
Zhang, Y. C., Gan, F. F., Shelar, S. B., Ng, K. Y., and Chew, E. H. (2013). Antioxidant and Nrf2 inducing activities of luteolin, a flavonoid constituent in Ixeris sonchifolia Hance, provide neuroprotective effects against ischemia-induced cellular injury. Food Chem. Toxicol. 59, 272–280. doi: 10.1016/j.fct.2013.05.058
Zhao, X., Zhang, M., Li, C., Jiang, X., Su, Y., and Zhang, Y. (2019). Benefits of vitamins in the treatment of Parkinson's Disease. Oxidative Med. Cell. Longev. 2019:9426867. doi: 10.1155/2019/9426867
Zhong, C. B., Chen, Q. Q., Haikal, C., Li, W., Svanbergsson, A., Diepenbroek, M., et al. (2017). Age-dependent alpha-synuclein accumulation and phosphorylation in the enteric nervous system in a transgenic mouse model of Parkinson's disease. Neurosci. Bull. 33, 483–492. doi: 10.1007/s12264-017-0179-1
Zhou, C., Ji, J., Shi, M., Yang, L., Yu, Y., Liu, B., et al. (2014). Suberoylanilide hydroxamic acid enhances the antitumor activity of oxaliplatin by reversing the oxaliplatin-induced Src activation in gastric cancer cells. Mol. Med. Rep. 10, 2729–2735. doi: 10.3892/mmr.2014.2548
Zhou, W., Bercury, K., Cummiskey, J., Luong, N., Lebin, J., and Freed, C. R. (2011). Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 286, 14941–14951. doi: 10.1074/jbc.M110.211029
Zhou, W., Ryan, J. J., and Zhou, H. (2004). Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein sumoylation by cellular stresses. J. Biol. Chem. 279, 32262–32268. doi: 10.1074/jbc.M404173200
Keywords: alpha synuclein, post translational modifications, Parkinson’s disease, synucleinopathies, therapeutic targets, biomarkers
Citation: Brembati V, Faustini G, Longhena F and Bellucci A (2023) Alpha synuclein post translational modifications: potential targets for Parkinson’s disease therapy? Front. Mol. Neurosci. 16:1197853. doi: 10.3389/fnmol.2023.1197853
Edited by:
Laura Musazzi, University of Milano Bicocca, ItalyReviewed by:
Ayse Ulusoy, Helmholtz Association of German Research Centers (HZ), GermanyGeorge K. Tofaris, University of Oxford, United Kingdom
Copyright © 2023 Brembati, Faustini, Longhena and Bellucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arianna Bellucci, YXJpYW5uYS5iZWxsdWNjaUB1bmlicy5pdA==