 Bin Dong
Bin Dong Yang Yue
Yang Yue Han Dong
Han Dong Yuehui Wang1*
Yuehui Wang1*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 15 June 2023
Sec. Brain Disease Mechanisms
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1174738
This article is part of the Research Topic Molecular Mechanisms of Glutamatergic Synapse Function and Dysfunction - Volume II View all 5 articles
N-methyl-D-aspartate receptors (NMDA) are glutamate-gated ion channels critical for synaptic transmission and plasticity. A slight variation of NMDAR expression and function can result in devastating consequences, and both hyperactivation and hypoactivation of NMDARs are detrimental to neural function. Compared to NMDAR hyperfunction, NMDAR hypofunction is widely implicated in many neurological disorders, such as intellectual disability, autism, schizophrenia, and age-related cognitive decline. Additionally, NMDAR hypofunction is associated with the progression and manifestation of these diseases. Here, we review the underlying mechanisms of NMDAR hypofunction in the progression of these neurological disorders and highlight that targeting NMDAR hypofunction is a promising therapeutic intervention in some neurological disorders.
N-methyl-D-aspartate receptors (NMDARs) are glutamate-gated ion channels permeable to Ca2+ found throughout the brain. Glutamate is the principal excitatory neurotransmitter by binding to postsynaptic NMDARs, contributing to NMDAR excitatory postsynaptic currents (EPSCs), and participating in almost all brain physiological functions. In addition, NMDAR has been implicated in synaptic plasticity, which is the key substrate for learning and memory processes (Traynelis et al., 2010; Paoletti et al., 2013). The NMDA receptor is a heterotetramer composed of four subunits, forming ion channel pores in the middle (Hansen et al., 2018). Functional NMDA receptors consist of two GluN1 subunits and two GluN2 subunits, or two GluN1 subunits, one GluN2 subunit and one GluN3 subunit (Traynelis et al., 2010). While the GluN1 subunit is encoded by GRIN1, which has eight splice variants (GluN1-1a, GluN1-1b, GluN1-2a, GluN1-2b, GluN1-3a, GluN1-3b, GluN1-4a, and GluN1-4b), the GluN2 subunit has four different isoforms (GluN2A–GluN2D) encoded by four different genes (GRIN2A–GRIN2D), and GluN3 subunit has two distinct isoforms, GluN3A and GluN3B, encoded by GRIN3A and GRIN3B. Each subunit has four similar architectural domains (Figure 1): an extracellular N-terminal domain (NTD), an extracellular ligand-binding domain (LBD) binding glutamate in GluN2 subunits, and the co-agonist glycine in GluN1 and GluN3 subunits; a pore-forming transmembrane domain (TMD); and a cytoplasmic carboxyl-terminal domain (CTD). Distinct selective features are associated with each of these different subunit domains (Swanger et al., 2016; Ogden et al., 2017; Li et al., 2019), so different isoforms of subunits combine to produce multiple NMDA receptor isoforms with unique functions. In order to activate NMDAR, it is necessary to bind both glutamate and glycine co-agonists, remove voltage-dependent ion channel blockage by Mg2+, and then open the receptor-gated ion channel, resulting in the influx of Ca2+ from the extracellular space into neurons (Hansen et al., 2018). NMDA receptor-gated Ca2+ influx regulates several intracellular enzymes and signaling processes critical to synaptic plasticity (Dingledine et al., 1999; Lynch, 2004; Traynelis et al., 2010; Huganir and Nicoll, 2013).
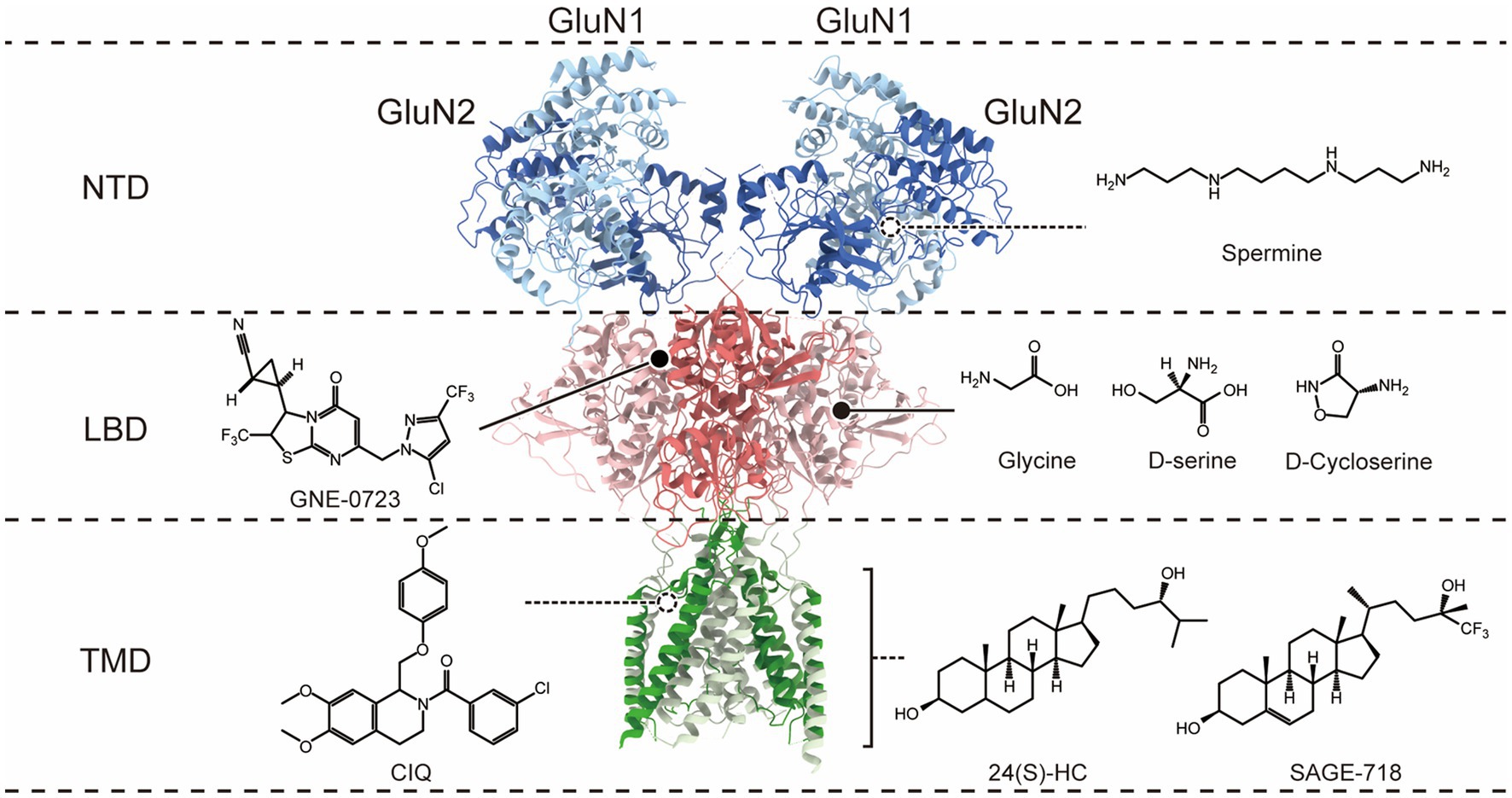
Figure 1. Co-agonist and allosteric modulatory binding sites on the N-methyl-D-aspartate (NMDA) receptor. The molecular structure of NMDARs is depicted, with only a dimer of GluN1 and GluN2 subunits shown for clarity. The receptor exhibits a layered architecture, with the N-terminal domains (NTDs) positioned at the top, the ligand-binding domains (LBDs) that bind glycine (Gly; or D-serine) in GluN1 and glutamate (Glu) in GluN2, the transmembrane domain (TMD), and the intracellular carboxyl-terminal domain is not present in the structure. The different domains corresponding to the binding sites of co-agonists and PAMs on the receptor are distinguished on the right.
N-methyl-D-aspartate receptor dysfunction is implicated in the pathophysiology of many neurologic and psychiatric disorders. Hyperactivation of NMDARs, as observed in cerebral ischemia, brain injury, epilepsy, Huntington’s disease, and Parkinson’s disease (Mony et al., 2009; Parsons and Raymond, 2014; Wang et al., 2020), involves a process of excessive calcium influx and causing excitotoxicity (Rothman and Olney, 1995; Traynelis et al., 2010; Paoletti et al., 2013). Accordingly, most recent studies focus on NMDAR hyperfunction-associated diseases and NMDAR antagonists. However, the hypofunction of NMDARs can also have detrimental effects, such as in the pathophysiology of schizophrenia (SZ), where reduced NMDAR function is critically important. The deleterious effects of NMDAR hypofunction were confirmed by observations that the administration of noncompetitive NMDAR antagonists, phencyclidine (PCP), MK-801 and ketamine (Javitt and Zukin, 1991; Krystal et al., 1994; Coyle et al., 2003), by healthy volunteers could induce all three symptoms similar to SZ (positive, negative, and cognitive; Olney and Farber, 1995; Olney et al., 1999). SZ patients suffer worse symptoms when exposed to these compounds (Javitt and Zukin, 1991; Beck et al., 2020). Moreover, in vertebrate models, psychotic symptoms can be provoked by administrating NMDAR antagonists or genetic decline in NMDAR function (Eyer and Oghaddam, 2002; Jones et al., 2011). NMDAR hypofunction is also identified in GRIN-related diseases, especially loss-of-function genetic mutations, which make up a large part of the genetic mutation in the GRIN genes. The-loss-of function GRIN variants cause NMDAR hypofunction by various mechanisms, including reduced glutamate/glycine potency, accelerated deactivation time course, decreased surface expression, reduced channel open probability, or both. Ultimately, these variants are associated with many neurodevelopmental disorders, including epilepsy, developmental delay, autism spectrum disorder (ASD), intellectual disability (ID), and SZ (Yuan et al., 2015; XiangWei et al., 2018; Myers et al., 2019; García-Recio et al., 2021). Additionally, anti-NMDAR encephalitis is an NMDAR hypofunction-related disease that can cause several neurological symptoms, such as psychosis, seizures, abnormal behaviors, and cognitive manifestations (Lynch et al., 2018; Dalmau et al., 2019). Finally, age-related changes in cognitive performance are associated with NMDAR hypofunction (Clayton et al., 2002; Foster, 2007; Mostany et al., 2013; Kumar and Foster, 2019).
N-methyl-D-aspartate receptor hypofunction is a convergence point of pathological processes in many diseases; therefore, enhancing NMDAR function is highly therapeutically relevant. Herein, we discuss the recent research progress of NMDAR hypofunction-related diseases and pharmacological agents, hoping to provide more ideas for the research of NMDAR hypofunction-related neurological disorders.
Schizophrenia is a progressive neurodevelopmental disorder that affects the brain before its clinical symptoms appear. Despite different neurochemical pathways and mechanisms that contribute to SZ, such as glutamatergic, GABAergic, and dopaminergic systems, the precise etiology of SZ remains unknown. Over the past few decades, the dopamine (DA) model dominated the pathophysiology hypothesis of SZ. Almost all current medications for SZ primarily work by blocking dopamine D2 receptors in the nigrostriatal pathway (Gray and Roth, 2007). Conversely, current therapeutics targeting the dopamine system have had limited success, especially in managing negative and cognitive symptoms of SZ (Uno et al., 2019; McCutcheon et al., 2020). Furthermore, most functional and structural changes of SZ patients present in the early phases of postnatal development, particularly during adolescence and juvenile years, in contrast to the emergence of dopamine dysfunction during late adolescence (Snyder and Gao, 2013; Selemon and Zecevic, 2015; Tagliabue et al., 2017; Chini and Hanganu-Opatz, 2021). Consequently, on its own, dopaminergic dysfunction cannot fully explain the wide range of symptoms and neurological deficits in SZ. The paradigm of studying schizophrenia has beyond the DA hypothesis. A major shift in the understanding of the etiology of schizophrenia proposes that multiple genetic and environmental risk factors converge on the glutamatergic system mediated by NMDARs, leading to NMDAR hypofunction in the cortical circuitry during neurodevelopment. NMDARs are widely considered critical for synaptic plasticity and circuit formation during early and late phases of neural development (Monyer et al., 1994; Sheng et al., 1994; Quinlan et al., 1999; Wang et al., 2008; Roberts et al., 2009; Wang and Gao, 2009; Snyder et al., 2013; Monaco et al., 2015). Many studies indicate that the maturation of brain circuits usually coincides with NMDAR subtype switching (e.g., from GluN2B to GluN2A and GluN3A to GluN3B), which occurs at the beginning of critical developmental periods (Monyer et al., 1994; Sheng et al., 1994; Quinlan et al., 1999; Wang et al., 2008; Roberts et al., 2009; Snyder et al., 2013). Therefore, NMDAR subtype switching renders NMDARs vulnerable to genetic and environmental risk factors (Spear, 2000). A glutamatergic model mediated by NMDARs can provide an alternative way of conceptualizing schizophrenia-associated brain abnormalities (Harrison and Weinberger, 2005; Lewis and Moghaddam, 2006; Lisman et al., 2008).
N-methyl-D-aspartate receptor hypofunction is a convergence point for the progression and symptoms of SZ, especially cognitive deficits (Moghaddam, 2003; Coyle, 2006; Lisman et al., 2008; Marek, 2010; Snyder et al., 2013; Cohen et al., 2015; Nakazawa et al., 2017). Hypofunction of NMDAR induces progressive symptoms of SZ, such as cognitive impairment and psychosis, by intervening in local deficits of gamma-aminobutyric acid (GABA; Snyder and Gao, 2013; Cohen et al., 2015; Nakazawa et al., 2017; Datta and Arnsten, 2018) and long-range disconnections between regions of the brain (Howes and Kapur, 2009; Grace, 2016). The NMDAR hypofunction hypothesis of SZ is based on observations that non-competitive NMDAR antagonists [phencyclidine (PCP), MK-801, and ketamine; Javitt and Zukin, 1991; Krystal et al., 1994; Coyle et al., 2003] can induce all three SZ-like symptoms (positive, negative, and cognitive) in healthy individuals (Olney and Farber, 1995; Olney et al., 1999). Patients with SZ patients suffer worse symptoms when exposed to these compounds (Javitt and Zukin, 1991; Beck et al., 2020). Most non-competitive antagonists work as open channel blockers of NMDAR channels (Lodge and Mercier, 2015) and directly block NMDAR channels during the depolarization phase. Therefore, NMDAR antagonists inhibit the reactivation of NMDA receptors, and synaptic transmission is disrupted concomitantly. Accordingly, the hypofunction of NMDAR is regarded to be associated with SZ-related neurocognitive symptoms.
The γ-aminobutyric acid-ergic (GABAergic) interneurons are the major inhibitory system in the brain and play a critical role in the pathological process of SZ (Lisman et al., 2008; Nakazawa et al., 2012). NMDAR antagonists mainly target constantly depolarizing neurons, especially parvalbumin fast-spiking GABAergic interneurons (Homayoun and Moghaddam, 2007). Administering antagonists to NMDA receptors causes a reduction in excitatory glutamatergic inputs to fast-spiking GABAergic interneurons, resulting in the disinhibition of pyramidal neurons and hyperexcitability in pyramidal neurons (Olney et al., 1991; Moghaddam et al., 1997; Jentsch et al., 1999; Benes, 2000; Homayoun and Moghaddam, 2007; Lisman et al., 2008). Consequently, the process evokes SZ-related phenotypes and neurochemical changes in animal models (Lindsley et al., 2006; Rujescu et al., 2006; Carpenter and Koenig, 2008; Gunduz-Bruce, 2009). For example, MK-801 displays higher affinity and specificity for NMDAR than other noncompetitive antagonists (Koek et al., 1988; Kovacic and Somanathan, 2010). MK-801 could induce SZ-like symptoms in adult rodents, including increased locomotor activity and reduced prepulse inhibition (Zhou et al., 2018). The GluN2A subunit of NMDA receptor is a faster target of MK-801 than other NMDA receptor subunits (Gielen et al., 2009), and GluN2A subunits are highly distributed in fast-spiking GABAergic interneurons (Kinney et al., 2006; Xi et al., 2009). MK-801 exhibits cell type-specific effects, blocking presynaptic NMDA receptors on axon terminals of fast-spiking neurons rather than pyramidal neurons. After 5 consecutive days of MK-801 administration in adolescent rats, blocking presynaptic NMDA receptors on excitatory inputs to fast-spiking neurons significantly reduces the amplitude of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated miniature excitatory postsynaptic currents (mEPSCs), while the frequency and amplitude of AMPAR-mEPSCs in pyramidal neurons significantly increase (Wang and Gao, 2012). The excitation-inhibition balance is changed due to reduced excitatory drive in fast-spiking interneurons and increased excitation in pyramidal neurons, resulting in excessive glutamate release, excitotoxicity, and overexcitation of pyramidal neurons (Olney and Farber, 1995; Lisman et al., 2008). Consequently, NMDAR hypofunction may cause dysfunction of the GABAergic circuitry and interruption of input and output connections between neurons, which may contribute to the major clinical characteristics of SZ.
Moreover, transgenic mice with NMDAR hypofunction displayed SZ-related phenotypes (Mohn et al., 1999). According to recent genetic studies, the mutation of the NMDAR subunit has been implicated in various neurological disorders, including SZ. However, 25 out of 370 Japanese SZ patients with rare mutations in six GRIN subunits (except GRIN2B) were identified through exome sequencing, indicating an estimated 7% of patients with idiopathic SZ carry mutations in the gene for the GRIN subunit (Yu et al., 2018). Accordingly, GRIN gene variants are relatively rare and unlikely to explain most SZ cases. Rather, there remain several high-risk genes that contribute to the hypofunction of NMDAR to increase the susceptibility to SZ, including neuregulin 1 (NRG1; Roy et al., 2007; Mei and Xiong, 2008; Kato et al., 2011), disrupted in schizophrenia-1 (DISC-1; Lipina et al., 2010; Niwa et al., 2010), and dystrobrevin-binding protein 1 (dysbindin-1; Ji et al., 2009; Papaleo and Weinberger, 2011; Papaleo et al., 2012). Neuregulin (NRG) is an SZ risk gene that signals via the receptor tyrosine kinase ErbB4 (Hahn et al., 2006). NRG1-ErbB4 signaling may reduce NMDAR-mediated currents in interneurons (Kotzadimitriou et al., 2018), contributing to the pathophysiology of SZ. NRG2 promotes ErbB4 association with GluN2B-containing NMDA receptors, followed by rapid internalization of surface receptors and potent downregulation of NMDA but not AMPA receptor currents (Vullhorst et al., 2015). Disrupted-In-Schizophrenia 1 (DISC1) is a genetic risk factor implicated in major mental disorders and plays an important role in synaptic and dendritic development in neurons (Brandon et al., 2009). DISC1 binds to and stabilizes serine racemase (SR) to regulate D-serine (an endogenous NMDAR co-agonist) production (Ma et al., 2013) and contributes to NMDAR activation. Cellular knockdown of DISC1 significantly increased NMDAR currents and NMDAR subunit expression through a phosphodiesterase 4 (PDE4)/protein kinase A (PKA)/cAMP response element-binding protein (CREB)-dependent mechanism (Wei et al., 2014), providing a potential molecular basis of DISC1 in influencing NMDAR-dependent cognitive and emotional processes. Furthermore, dysbindin-1 is highly expressed in the axon terminals of glutamatergic pyramidal neurons (Talbot et al., 2004), and it is likely to play a role in vesicular protein trafficking and synaptic transmission to influence glutamatergic function (Chen et al., 2008; Jentsch et al., 2009). The reduction of dysbindin reduces glutamate release, resulting in the dysfunction of NMDARs. Dysbindin-induced degradation of GluN1 is associated with impairments in spatial working memory (Numakawa et al., 2004; Chen et al., 2008; Jentsch et al., 2009; Karlsgodt et al., 2011). Furthermore, deficiencies in dysbindin-1 result in significant reductions in NMDAR-mediated synaptic potentiation and impaired synaptic plasticity and play a role in the cognitive impairments of SZ (Glen et al., 2014). Consequently, there is considerable evidence that SZ susceptibility genes are linked to glutamate receptors, specifically NMDARs.
In vivo, studies using PET imaging and single-photon emission tomography (SPECT) have revealed that the distribution volume ratio of NMDAR in the hippocampus of SZ patients was significantly lower than that of healthy controls (Pilowsky et al., 2006; Beck et al., 2021). Additionally, postmortem studies have shown that SZ patients had decreased NMDAR density and lower GluN1 mRNA and protein levels in their hippocampus than healthy controls (Vrajová et al., 2010; Rubio et al., 2012). Furthermore, results from postmortem brain studies measuring the expression of NMDAR subunits showed that in 49–73% of the GABAergic neurons in the prefrontal cortex (Bitanihirwe et al., 2009) and anterior cingulate cortex (Woo et al., 2004) of SZ patients, GluN2A mRNA expression was decreased to a level that it can no longer be detected experimentally. Moreover, a meta-analysis reported a reduction in GluN1 mRNA and protein levels in the prefrontal cortex in SZ people compared to controls (Catts et al., 2016).
Based on these observations, the hypothesis of hypofunction of NMDAR makes sense as a plausible theory for SZ (Stahl, 2007), and many researchers accept the dysfunction of NMDAR as a convergence point for SZ (Kantrowitz and Javitt, 2010).
N-methyl-D-aspartate receptor hypofunction also is identified in GRIN-related diseases, mainly focusing on loss-of-function variants. As next-generation sequencing becomes more prevalent, numerous genetic studies have reported genetic associations between GRIN gene variants and neurodevelopmental disorders, especially rare pediatric encephalopathies. GRIN disease-associated variants affect NMDAR function by various mechanisms involving glutamate/glycine potency, course of deactivation time, surface expression, and/or open probability, ultimately resulting in neurodevelopmental disorders, including epilepsy, developmental delay, ASD, ID, and SZ (XiangWei et al., 2018; García-Recio et al., 2021). Many of these are loss-of-function variants, which could reduce overall charge transfer associated with NMDAR activation and drive some initial aspects of clinical phenotypes.
The NMDA receptor is a heterotetramer composed of two indispensable GluN1 subunits and two GluN2 and/or GluN3 subunits (Traynelis et al., 2010). Unlike GluN2 and GluN3 subunits, the presence of the GluN1 subunit is invariable and exerts a pivotal role in the normal function of NMDAR (Forrest et al., 1994; Li et al., 1994). Concomitantly, mutations affecting the GluN1 subunit could impair glutamatergic circuitry and cause severe neurodevelopmental alterations. With pathogenic GRIN1 variants first diagnosed in two patients with intellectual disability in 2011 (Hamdan et al., 2011), pathogenic GRIN1 variants have been identified in many neurological disorders. For example, loss-of-function variants of GRIN1 have been found in many patients with SZ (Begni et al., 2003; Galehdari et al., 2009; Yuan et al., 2015; Yu et al., 2018), and most epilepsy-related GRIN 1 variants are loss-of-function (D552E, Q556*, S560dup, Y647S, G815R, and G827R; Lemke et al., 2016; Xu and Luo, 2018). Meanwhile, mutation scattered along the M4-CTD region and the ATD induces a dramatic decline in NMDAR surface expression, generating a loss-of-function effect and clinical symptoms (Santos-Gómez et al., 2021b). A vitro study using GRIN 1−/− knockdown mice (GluN1KD) found that loss-of-function GRIN 1 mutations reduce the volume of dopaminergic structures in the early stages of development. In contrast, delayed changes in the structure of limbic and white matter of the brain are more evident after puberty (Intson et al., 2019). Furthermore, smaller brain volume has been reported in patients with GRIN1 variants with loss of function related to encephalopathy and SZ (Brown et al., 1986; Cahn et al., 2002; van Haren et al., 2008; Lemke et al., 2016; Rossi et al., 2017; Fry et al., 2018).
The GluN2 subunit genes, especially GRIN2A and GRIN2B (which encode GluN2A and GluN2B subunits, respectively), are highly intolerant to genetic variations, and the incidence of de novo genetic variations is high (Higley and Sabatini, 2012; Otsu et al., 2019). Mutations in these genes were generally correlated with NMDAR-related neurodevelopment disorders, with 43 and 35% of NMDAR mutations, respectively (Hansen et al., 2021).
An in-depth investigation of GRIN2A-related phenotypes, encompassing 248 affected individuals with pathogenic or probable pathogenic variants in GRIN2A, suggested that missense variants, like C436R, G483R, V685G, P699S, M705V, P716T, A727T, and D731N, within amino-terminational or ligand-binding domains exclusively caused NMDAR loss-of-function (Lemke et al., 2013; Lesca et al., 2013; Swanger et al., 2016; Addis et al., 2017; Strehlow et al., 2019). Most rare variants of GluN2A were associated with epilepsy and ID. The GluN2A(D731N) variant, which resides in a section of the LBD, could decrease glutamate potency, channel open probability, charge transfer, and receptor surface expression (Gao et al., 2017). The GluN2A (C436R) variant, which also resides in LBD, impairs the folding and stability of protein structure by disrupting the disulfide bond formation (Elmasri et al., 2022). Additionally, GRIN2A and GRIN2B pathogenic protein truncating variants lead to the reduction or total loss of NMDARs function, as variant products are insufficient to access the cell surface, resulting in a significant reduction of NMDAR-mediated currents (Santos-Gómez et al., 2021a; De Bernardi et al., 2022). Each of these GRIN2 mutations results in a loss-of-function effect on NMDARs.
While homozygous mice with GRIN 2B deletion showed a long-term depression impairment in the hippocampal area and died with a deficiency of suckling response, heterozygous mice survived with decreased NMDAR surface expression (Kutsuwada et al., 1996). It has been identified that loss-of-function mutations in GRIN2B are associated with ID and ASD (Endele et al., 2010; O'Roak et al., 2011). Some missense genetic variants of GRIN2B can damage GluN2B function and cause loss of function. For example, GluN2B (E413G and C461F), located in the agonist-binding domain, reduces Glu potency, current density, and deactivation time (Adams et al., 2014; Swanger et al., 2016; Bell et al., 2018; Fedele et al., 2018; Wells et al., 2018). GluN2B S541R, located in linker regions, reduces Glu potency (Platzer et al., 2017). GluN2B (E413G, C436R, and C456Y) revealed decreased surface expression (O'Roak et al., 2012; Adams et al., 2014; Swanger et al., 2016; Platzer et al., 2017; Bell et al., 2018; Fedele et al., 2018; Wells et al., 2018). Additionally, analyses of GRIN2B protein truncation variants revealed decreased NMDAR-mediated currents and surface expression, eventually leading to NMDAR functional haploinsufficiency (Sceniak et al., 2019; Santos-Gómez et al., 2021a). Interestingly, using clonal models of loss of function mutations and electrophysiology and calcium imaging method, Bell et al. (2018) demonstrated that loss of GRIN2B function might be in a more proliferative-like state, altering differentiation and, presumably, how neurons integrate into developing circuits.
Anti-NMDAR encephalitis is a severe neuropsychiatric disorder, considered a NMDAR hypofunction-related disease. It is responsible for various neurological symptoms such as psychosis, seizures, abnormal behaviors, and cognitive manifestations (Lynch et al., 2018; Dalmau et al., 2019). NMDAR internalization causes NMDAR hypofunction in anti-NMDAR encephalitis, a process where antibodies crosslink NMDARs on the surface of neurons, resulting in a reversible reduction of NMDARs on the surface (Hughes et al., 2010; Mikasova et al., 2012; Moscato et al., 2014; Jézéquel et al., 2018). Molecular studies about the antibody binding sites identified that antibodies recognize the epitope on NTD of GluN1 in anti-NMDAR encephalitis, which contains N368/G369 (Dalmau et al., 2008; Gleichman et al., 2012; Kreye et al., 2016; Sharma et al., 2018; Jones et al., 2019). Antibody-binding epitopes overlap or be adjacent to the GluN1-EphB2-interacting sites on GluN1 (Mikasova et al., 2012), and the NMDAR-EphB2 interaction contributes to the stability of NMDARs in synapses (Dalva et al., 2000). Therefore, NMDAR antibodies could disrupt the interaction between NMDARs and EphB2 receptors, and then alter the localization of NMDARs accompanied by internalization (Mikasova et al., 2012). Although studies with super-resolution and single molecule localization microscopy have shown that treatment with NMDAR antibodies in cultured neurons induces a reduction of NMDARs on the surface and diffusion of remaining receptors (Ladépêche et al., 2018), it does not affect the ionotropic function of NMDAR channels before receptor internalization. Treatment with autoantibody-containing cerebrospinal fluid (CSF) in patients does not reduce NMDAR-mediated currents within 30 min (Mikasova et al., 2012; Moscato et al., 2014), significantly reduces them after 24 h due to receptor internalization/lateral diffusion (Hughes et al., 2010).
Research utilizing passive transfer models has provided positive evidence that the chronic transfer of patient antibodies leads to a decrease in endogenous NMDARs in synapses, a reduction in NMDAR-mediated currents, impaired NMDAR-dependent LTP, compromised spatial memory and novel object recognition, depression-like behaviors, and seizures. Mice treated with autoantibody-containing CSF of patients exhibited impaired LTP in Schaffer collateral-CA1 synapses and decreased NMDAR densities and EphB2 expression (Planagumà et al., 2016). Meanwhile, hippocampal CA1 and CA3 also observed the impairment of NMDAR-dependent LTP in female Wistar rats with the infusion of autoantibody-containing CSF of patients (Blome et al., 2018; Kersten et al., 2019). Additionally, various studies have reported that patient’s NMDAR antibodies consistently decrease the novel object discrimination index in mice and rats after the passive transfer of patient antibodies, indicating a decline in short-term/working memory (Planagumà et al., 2015, 2016; Malviya et al., 2017; Kersten et al., 2019; Carceles-Cordon et al., 2020). Cortical inhibitory neurons—GABAergic interneurons are the main target of hNR1 antibodies (a class of human monoclonal antibodies targeting the GluN1 subunit; Kreye et al., 2016; Andrzejak et al., 2022). With the internalization and hypofunction of NMDARs in GABAergic interneurons, the inhibitory drive onto excitatory neurons dramatically decreases (Nakazawa et al., 2017), which may explain the psychosis symptom of anti-NMDAR encephalitis. Furthermore, Wright et al. (2021) found that the decrease in excitatory neurotransmitter transport caused by NMDAR antibodies may be the cause of seizures in anti-NMDAR encephalitis patients.
Ovarian teratomas and herpes simplex virus (HSE) infections may be the main causes of anti-NMDAR encephalitis. The clinical manifestations of anti-NMDAR encephalitis are consistent with the results of in vitro and animal experiments. It is widely accepted that NMDAR autoimmune antibodies are responsible for encephalitis, as they crosslink NMDARs, disrupt NMDAR-EphB2 interactions, and cause NMDAR internalization. This internalization leads to NMDAR hypofunction, which is believed to be the cause of impaired long-term potentiation, memory, and behavioral deficits, and increased susceptibility to seizures. Once diagnosed, immunotherapy should be given actively, and antibodies can be neutralized and degraded indirectly. In addition, tumor removal should be performed as soon as possible for patients with associated tumors. The above treatments can accelerate symptom recovery and reduce the risk of recurrence.
Normal aging, a natural process characterized by structural and functional changes in the brain, leads to cognitive decline, mainly in learning and memory performance. Age-related cognitive performance is associated with hypofunction of NMDAR (Clayton et al., 2002; Foster, 2007; Mostany et al., 2013; Kumar and Foster, 2019). The NMDAR plays a crucial role in cognitive performance, particularly in learning and memory performance (Kuehl-Kovarik et al., 2000; Paoletti and Neyton, 2007; Forsyth et al., 2015). The expression of NMDA receptor-associated signaling molecules attenuates with increasing age (Gonzales et al., 1991; Pittaluga et al., 1993).
Cognitive declines are related to changes in the specific subunits in NMDARs with advancing age. The GluN2B subunit plays a major role in forming and maintaining learning and memory (Sakimura et al., 1995; Malenka, 2003; Zhuo, 2008). The expression of GluN2B subunits decreased significantly than the other subunits in the forebrain of aged rodents, especially in the cerebral cortex and hippocampus (Magnusson, 2001; Ontl et al., 2004; Magnusson et al., 2006; Zhao et al., 2009). This reduction induced a decrease in the transmission of GluN2B-dependent signaling molecules that contribute to long-term potentiation and synaptic plasticity (Hardingham and Bading, 2010; Lussier et al., 2015), followed by decreased NMDAR function and NMDAR-related cognitive decline (Clayton and Browning, 2001; Magnusson et al., 2007; Zhao et al., 2009). Further analyses of human temporal cortical tissue obtained during neurosurgery declared a progressive reduction in GluN2B subunits in the aging human brain (Pegasiou et al., 2020). This indicates that NMDARs containing GluN2B subunits exert a critical role in synaptic responses in the adult human brain, but their influence is diminished in the elderly. NMDAR hypofunction is also implicated in Alzheimer’s disease (AD). Significant reductions in the expression of the GluN2B subunit have also been seen in the hippocampus and the entorhinal cortex of brains with AD (Sze et al., 2001), suggesting a relationship between GluN2B-related NMDAR hypofunction and AD. Furthermore, upregulating GluN2B expression in the brain by dephosphorylating (Plattner et al., 2014), protein kinase inhibitors (Hawasli et al., 2007), or genetic overexpression (Wong et al., 2002; Cao et al., 2007; Yin et al., 2011, 2012) resulted in significant changes in learning and memory in aged rodents. These data suggest that the GluN2B subunit is an ideal target for improving age-related cognitive decline.
Although many previous works supported the viewpoint that the expression of the GluN2B subunit decreased with age and contributed to the decline of cognitive function in the elderly, there are still some studies that proposed different views. Proximity ligation assay (PLA) analyses revealed disrupted GluN2B-containing NMDAR trafficking in aged mice, resulting in synaptic accumulation of these receptors in the apical dendrites of dCA1 (Zamzow et al., 2013; Al Abed et al., 2020). In the hippocampal region, GluN2B association with membrane scaffolding proteins, including PSD95, augments with age (Zamzow et al., 2013). These all resulted in hypofunction of NMDARs, followed by age-related memory impairment. Interestingly, some researchers examined the role of the GluN2A and GluN2B subunits in the hypofunction of the NMDAR synaptic due to aging, indicating that the age-related decrease in response to NMDAR is not due to a change in the ratio of diheteromeric GluN2A/GluN2B subunits in the synapse. The meaningful result is that age-related oxidative stress disrupts synaptic plasticity and suppresses NMDAR function (Kumar et al., 2019).
Meanwhile, compared to the GluN2B subunit, there are conflicting results about the GluN1 expression by aging, with meaningful declines in some studies but not in others (Magnusson et al., 2005; Das and Magnusson, 2011). Age-related lower expression of the GluN1 subunit shows a relationship to the hypofunction of the NMDAR, impairing spatial reference memory performance in old rats and mice (Adams et al., 2001; Magnusson et al., 2007; Das and Magnusson, 2008). Nonetheless, a study by Zhao et al. (2009) found no significant alteration in GluN1 protein expression in the hippocampus of aged mice, while another study by Gramuntell et al. (2021) reported no variation in the density of GluN1 protein in the oriens-lacunosum molecular cells in the hippocampal CA1 region with aging.
Other factors, including hormones (Lichtenwalner et al., 2001; Li et al., 2004), changes of neuropeptides, and synaptic loss (Burke and Barnes, 2006), oxidative stress, inflammatory response, also play a critical role in age-related cognitive decline. The damage to NMDAR structure and function can be caused by these factors, which may converge to cause NMDAR dysfunction, making it a potential convergence point in the pathological process of age-related cognitive impairment. While the exact mechanisms and treatments of NMDAR dysfunction remain unclear, investigating this area is highly significant for comprehending the pathological processes of cognitive disorders and discovering novel approaches to treatment.
Regarding therapeutic potential, NMDARs are highly interesting targets. Overactivation of NMDARs may have a deleterious effect, which limits the therapeutic window for enhancing NMDAR activity. This may lead to the difficulty of improving NMDAR function through direct activation via glutamate binding sites located in the LBD. Therefore, most of the current studies aimed at improving NMDAR hypofunction are based on endogenous co-agonists and positive allosteric modulators (PAMs).
Clinical interventions for NMDAR hypofunction mainly rely on co-agonists to increase the occupancy of glycine co-agonist binding sites (Figure 1), such as glycine, D-serine, and D-cycloserine. However, the application of high concentrations of glycine may lead to the internalization of NMDAR in vitro (Nong et al., 2003), so the application of glycine may ultimately reduce rather than increase NMDAR activity. D-serine is an endogenous NMDAR co-agonist that functions in a glycine environment at physiological concentrations (Harsing and Matyus, 2013). In the aging brain, the level of D-serine in the brain decreases, leading to NMDAR hypofunction. Studies have shown that supplementing D-serine in rat experiments reversed age-related cognitive flexibility and dendritic spine density decline and partially restored large-scale functional connections (Nava-Gómez et al., 2022). Another clinical study showed that administering a combined metabolic activator containing 61.75% L-serine (the endogenous D-serine precursor) orally to AD patients improved cognitive function by 29% after 84 days (Yulug et al., 2023). Furthermore, L-serine has been administered as a dietary supplement to a 5-year and 10-month-old patient diagnosed with GRIN2B-related loss of function mutation and suffering from severe encephalopathy. Encouragingly, the patient showed significant improvements in motor and cognitive function after 11 and 17 months of L-serine dietary supplementation (Soto et al., 2019), suggesting that L-serine may have a therapeutic role in treating NMDAR hypofunction. D-cycloserine (DCS), another partial agonist located at the NMDAR glycine binding site (Monahan et al., 1989), enhances receptor activation in the presence of glutamate. In vitro, studies have shown that DCS promoted NMDAR-dependent synaptic potentials (Billard and Rouaud, 2007), and intrahippocampal infusion of DCS enhanced the expression of the NMDAR subunit GluN2B in the hippocampus of young rats (Ren et al., 2013), which may contribute to enhancing long-term potentiation (LTP) and memory. In the treatment of schizophrenia, the role of co-agonists (including glycine, D-serine, and DCS) has also been extensively studied, but most of the results are contradictory or, overall, do not significantly improve negative and cognitive symptoms.
Over the past decade, NMDAR-positive allosteric modulators have gained substantial interest in counteracting diseases associated with the hypofunction of NMDARs. Allosteric modulators target allosteric sites that have high freedom and low conservatism (Figure 1), which allows for higher selectivity and avoids competition with ligands. Some allosteric modulators are relatively safe, even at high doses. The binding of allosteric modulators to specific structural domains can lead to long-range structural rearrangements that affect receptor activity. For example, conformational changes in NTDs can induce concomitant motions of the LBD and TMD, providing allosteric control of channel activity (Zhu and Paoletti, 2015; Hansen et al., 2018; Esmenjaud et al., 2019). Some of these PAMs have shown therapeutic potential in animal models or humans with NMDAR hypofunction-related diseases.
Polyamines at micromolar concentrations can selectively enhance the GluN2B subtype of NMDARs (Williams, 1994; Mony et al., 2009). Polyamine spermine binding to the lower lobe interface of GluN1 and GluN2B NTDs stabilizes this interface in a tight conformation, possibly by reducing the repellency between negative charges in the lower lobe of the NTD, which may result in a higher probability of channel opening (Mony et al., 2011; Sirrieh et al., 2015; Esmenjaud et al., 2019). The effects of polyamines have been studied in various cognitive and behavioral paradigms in animal experiments, and spermine administration has been shown to rescue learning and memory deficits in neuroinflammatory animal models and Huntington’s disease models, both of which are associated with neurodegeneration and cognitive decline (Velloso et al., 2008; Frühauf et al., 2015). However, polyamines have shown harmful effects in Alzheimer’s disease models (Guerra et al., 2016). GNE compounds are specific PAMs for GluN2A-NMDARs discovered through high-throughput screening. GNE PAMs may bind at the D1-D1 interface between GluN1 and GluN2A LBDs (Hackos et al., 2016) and enhance NMDAR activity by stabilizing the dimer interfaces between LBDs and preventing the receptor from desensitization. Chronic administration of an oral bioavailable derivative of GNE, GNE-0723, has been shown to improve learning and memory retention in AD mice in the Morris water maze task and in Dravet Syndrome mice in the contextual fear conditioning task, and the research also revealed the therapeutic potential of GNEs in conditions characterized by epileptic discharges and network overactivity (Hanson et al., 2020). CIQ is a selective PAM for GluN2C/2D-NMDARs, with no activity on other NMDAR, AMPA, kainate, GABA, or glycine receptors. The CIQ binding site is located in the outer transmembrane region and involves the pre-M1 and M3 regions of GluN2, as well as the neighboring pre-M4/M4 region of GluN1, forming the “gating triad” (Perszyk et al., 2020), and acts by disrupting the closed state of the receptor (Ogden and Traynelis, 2013; McDaniel et al., 2020). CIQ administered systemically partially alleviated abnormal sensory-motor gating, hyperactivity, and stereotypy in animals treated with MK-801 and methamphetamine, as well as improved working memory in mice treated with MK-801 (Suryavanshi et al., 2014). 24(S)-hydroxycholesterol (24(S)-HC), an endogenous compound, is the most prevalent metabolite of cholesterol in the brain. The compound has a high selectivity toward NMDARs, with little or no effect on AMPAR or GABAAR (Paul et al., 2013). 24(S)-HC exerts its activity through the TMD (Wilding et al., 2016), increases channel open probability, and enhances NMDARs function at saturating concentrations of agonist. Reduced levels of 24(S)-HC in the brain have been implicated in several neurodegenerative diseases, such as aging, AD, or Parkinson’s disease. Mice deficient in cholesterol 24-hydroxylase, an enzyme responsible for converting cholesterol to 24(S)-hydroxycholesterol, exhibited severe learning deficits and impaired CA3-CA1 synaptic LTP (Kotti et al., 2006; Russell et al., 2009). Studies have shown that 24(S)-HC or its derivatives SGE-201 and SGE-301 can increase STP and LTP in brain slices of aged rats, partially reversed behavioral deficits in rats caused by MK801 or PCP injections (a pharmacological model of SZ; Paul et al., 2013), and reversed memory loss in mice in the anti-NMDAR encephalitis model after chronic administration of SGE-301 (Mannara et al., 2020). Additionally, a 24(S)-HC derivative SAGE-718 has shown promising results in a phase 2 clinical trial (LUMINARY) for the treatment of Alzheimer’s disease (AD), improving executive function, learning, and memory in patients with mild cognitive impairment (MCI) and mild dementia, with good tolerability (Koenig et al., 2022). Although 24(S)-hydroxycholesterol (24(S)-HC) has shown great therapeutic potential in various neurological disorders, it is important to note that high concentrations of 24(S)-HC may trigger or aggravate NMDAR-induced neurotoxicity (Sun et al., 2017).
Herein, we provide an overview of the research progress on the neurobiological effects of NMDAR hypofunction, which is associated with many neurological and psychiatric disorders. We also discuss the research progress on pharmacological agents that directly target the gating mechanisms of NMDAR to enhance its function. We are pleased to see that there is currently a lot of research on co-agonists and PAMs that aim to improve NMDAR hypofunction, and encouraging progress has been made in various NMDAR hypofunction-related neurological diseases. In particular, PAMs have great therapeutic potential. However, there are still many issues that need to be addressed in the research on co-agonists and PAMs.
N-methyl-D-aspartate receptor PAMs can act in various ways, including increasing the maximum open channel probability of the receptor (e.g., glycine and GNE −6901), increasing the sensitivity of the receptor to glycine (or polyamines), and increasing the potency of glutamate (e.g., GNE -8304 or GNE -0723; Hackos and Hanson, 2017). Several NMDAR PAMs can effectively alleviate some behavioral and cognitive symptoms in animal models of NMDAR hypofunction. However, the physiological effects and therapeutic potential of any given NMDAR PAM usually depend on multiple factors, so it seems very difficult to predict the physiological outcomes of NMDAR PAMs based solely on the characteristics of their recombinant receptors. For example, two compounds, GNE-6901 and GNE-8314, which have the same GluN2A subunit selectivity and binding site, may have completely opposite effects on synaptic physiology (LTP) because GNE-8314 may act preferentially on interneurons (Hackos et al., 2016; Yao et al., 2018). Furthermore, due to the low conservation of allosteric sites, there are huge differences in allosteric regulation mechanisms between different species. Therefore, it is difficult to evaluate or predict the results of clinical trials of allosteric modulators based on animal experiments, and even contradictory results may be obtained. In elderly animals and humans, NMDAR expression levels and NMDAR-mediated EPSCs are all decreased, especially the expression of the GluN2B subunit. Similarly, in patients with anti-NMDAR encephalitis and mice induced with anti-NMDAR IgGs, NMDAR expression is reduced by 50–80% (depending on the titer; Mannara et al., 2020). However, when the remaining NMDARs are too few, it may be difficult to achieve significant cognitive and behavioral rescue by just enhancing the remaining NMDARs. In neurological diseases caused by NMDAR gene mutations, gene mutations may cause structural changes in allosteric binding sites, preventing allosteric modulators from binding, or may affect the signaling pathways of allosteric regulation, resulting in unknown effects. However, it is worth noting that a child with the loss-of-function mutation GluN2B-P553T showed improvement in behavioral and cognitive symptoms after treatment with L-serine (Soto et al., 2019). This clinical study serves as a compelling example of how enhancing the overall activity of NMDAR may be beneficial in rescuing the hypofunction of a specific NMDAR subtype.
The allosteric modulation mechanism may transform non-druggable targets into druggable targets, with corresponding allosteric modulators having higher selectivity and safety. This brings new opportunities for the study of NMDAR structure and function and the treatment of some NMDAR hypofunction-related neurological diseases. Furthermore, studies have proposed an unconventional NMDAR signaling (Dore et al., 2017), non-ionotropic NMDAR signaling, which is believed to arise from conformational changes in the receptor induced by agonists, independent of channel opening. This type of signaling is more likely to occur when there is a decrease in Ca2+ influx through the NMDAR, resulting in synaptic depression and reduction of spines. These changes could be a contributing factor to the reduced number of dendritic spines and cognitive impairments observed in individuals with schizophrenia. These findings may potentially pave the way for novel pharmacological interventions aimed at targeting distinct synaptic signaling pathways in neurological disorders such as schizophrenia. Due to the highly complex nature of NMDAR, the relationship between its structure and function, as well as the molecular basis of its diverse functions, warrant further research and exploration in the future.
BD: conceptualization, writing—original draft. YY and HD wrote the sections of the manuscript. YW: conceptualization, resources, supervision, and writing—review and editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adams, M. M., Smith, T. D., Moga, D., Gallagher, M., Wang, Y., Wolfe, B. B., et al. (2001). Hippocampal dependent learning ability correlates with N-methyl-D-aspartate (NMDA) receptor levels in CA3 neurons of young and aged rats. J. Comp. Neurol. 432, 230–243. doi: 10.1002/cne.1099
Adams, D. R., Yuan, H., Holyoak, T., Arajs, K. H., Hakimi, P., Markello, T. C., et al. (2014). Three rare diseases in one sib pair: RAI1, PCK1, GRIN2B mutations associated with Smith-Magenis syndrome, cytosolic PEPCK deficiency and NMDA receptor glutamate insensitivity. Mol. Genet. Metab. 113, 161–170. doi: 10.1016/j.ymgme.2014.04.001
Addis, L., Virdee, J. K., Vidler, L. R., Collier, D. A., Pal, D. K., and Ursu, D. (2017). Epilepsy-associated GRIN2A mutations reduce NMDA receptor trafficking and agonist potency—molecular profiling and functional rescue. Sci. Rep. 7:66. doi: 10.1038/s41598-017-00115-w
Al Abed, A. S., Sellami, A., Potier, M., Ducourneau, E. G., Gerbeaud-Lassau, P., Brayda-Bruno, L., et al. (2020). Age-related impairment of declarative memory: linking memorization of temporal associations to GluN2B redistribution in dorsal CA1. Aging Cell 19:e13243. doi: 10.1111/acel.13243
Andrzejak, E., Rabinovitch, E., Kreye, J., Prüss, H., Rosenmund, C., Ziv, N. E., et al. (2022). Patient-derived anti-NMDAR antibody disinhibits cortical neuronal networks through dysfunction of inhibitory neuron output. J. Neurosci. 42, 3253–3270. doi: 10.1523/JNEUROSCI.1689-21.2022
Beck, K., Arumuham, A., Veronese, M., Santangelo, B., McGinnity, C. J., Dunn, J., et al. (2021). N-methyl-D-aspartate receptor availability in first-episode psychosis: a PET-MR brain imaging study. Transl. Psychiatry 11:425. doi: 10.1038/s41398-021-01540-2
Beck, K., Hindley, G., Borgan, F., Ginestet, C., McCutcheon, R., Brugger, S., et al. (2020). Association of Ketamine with Psychiatric Symptoms and Implications for its therapeutic use and for understanding schizophrenia: a systematic review and Meta-analysis. JAMA Netw. Open 3:e204693. doi: 10.1001/jamanetworkopen.2020.4693
Begni, S., Moraschi, S., Bignotti, S., Fumagalli, F., Rillosi, L., Perez, J., et al. (2003). Association between the G1001C polymorphism in the GRIN1 gene promoter region and schizophrenia. Biol. Psychiatry 53, 617–619. doi: 10.1016/S0006-3223(02)01783-3
Bell, S., Maussion, G., Jefri, M., Peng, H., Theroux, J. F., Silveira, H., et al. (2018). Disruption of GRIN2B impairs differentiation in human neurons. Stem Cell Rep. 11, 183–196. doi: 10.1016/j.stemcr.2018.05.018
Benes, F. M. J. B. R. R. (2000). Emerging principles of altered neural circuitry in schizophrenia. Brain Res. Brain Res. Rev. 31, 251–269. doi: 10.1016/s0165-0173(99)00041-7
Billard, J. M., and Rouaud, E. (2007). Deficit of NMDA receptor activation in CA1 hippocampal area of aged rats is rescued by D-cycloserine. Eur. J. Neurosci. 25, 2260–2268. doi: 10.1111/j.1460-9568.2007.05488.x
Bitanihirwe, B. K., Lim, M. P., Kelley, J. F., Kaneko, T., and Woo, T. U. (2009). Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 9:71. doi: 10.1186/1471-244x-9-71
Blome, R., Bach, W., Guli, X., Porath, K., Sellmann, T., Bien, C. G., et al. (2018). Differentially altered NMDAR dependent and independent long-term potentiation in the CA3 subfield in a model of anti-NMDAR encephalitis. Front. Synap. Neurosci. 10:26. doi: 10.3389/fnsyn.2018.00026
Brandon, N. J., Millar, J. K., Korth, C., Sive, H., Singh, K. K., and Sawa, A. (2009). Understanding the role of DISC1 in psychiatric disease and during normal development. J. Neurosci. 29, 12768–12775. doi: 10.1523/JNEUROSCI.3355-09.2009
Brown, R., Colter, N., Corsellis, J. A., Crow, T. J., Frith, C. D., Jagoe, R., et al. (1986). Postmortem evidence of structural brain changes in schizophrenia. Differences in brain weight, temporal horn area, and parahippocampal gyrus compared with affective disorder. Arch. Gen. Psychiatry 43, 36–42. doi: 10.1001/archpsyc.1986.01800010038005
Burke, S. N., and Barnes, C. A. (2006). Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 7, 30–40. doi: 10.1038/nrn1809
Cahn, W., Hulshoff Pol, H. E., Lems, E. B., van Haren, N. E., Schnack, H. G., van der Linden, J. A., et al. (2002). Brain volume changes in first-episode schizophrenia: a 1-year follow-up study. Arch. Gen. Psychiatry 59, 1002–1010. doi: 10.1001/archpsyc.59.11.1002
Cao, X., Cui, Z., Feng, R., Tang, Y. P., Qin, Z., Mei, B., et al. (2007). Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur. J. Neurosci. 25, 1815–1822. doi: 10.1111/j.1460-9568.2007.05431.x
Carceles-Cordon, M., Mannara, F., Aguilar, E., Castellanos, A., Planagumà, J., and Dalmau, J. (2020). NMDAR antibodies Alter dopamine receptors and cause psychotic behavior in mice. Ann. Neurol. 88, 603–613. doi: 10.1002/ana.25829
Carpenter, W. T., and Koenig, J. I. (2008). The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology 33, 2061–2079. doi: 10.1038/sj.npp.1301639
Catts, V. S., Lai, Y. L., Weickert, C. S., Weickert, T. W., and Catts, S. V. (2016). A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: how can we link molecular abnormalities to mismatch negativity deficits? Biol. Psychol. 116, 57–67. doi: 10.1016/j.biopsycho.2015.10.013
Chen, X. W., Feng, Y. Q., Hao, C. J., Guo, X. L., He, X., Zhou, Z. Y., et al. (2008). DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J. Cell Biol. 181, 791–801. doi: 10.1083/jcb.200711021
Chini, M., and Hanganu-Opatz, I. L. (2021). Prefrontal cortex development in health and disease: lessons from rodents and humans. Trends Neurosci. 44, 227–240. doi: 10.1016/j.tins.2020.10.017
Clayton, D. A., and Browning, M. D. (2001). Deficits in the expression of the NR2B subunit in the hippocampus of aged fisher 344 rats. Neurobiol. Aging 22, 165–168. doi: 10.1016/S0197-4580(00)00196-2
Clayton, D. A., Grosshans, D. R., and Browning, M. D. (2002). Aging and surface expression of hippocampal NMDA receptors. J. Biol. Chem. 277, 14367–14369. doi: 10.1074/jbc.C200074200
Cohen, S. M., Tsien, R. W., Goff, D. C., and Halassa, M. M. (2015). The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr. Res. 167, 98–107. doi: 10.1016/j.schres.2014.12.026
Coyle, J. T. (2006). Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 26, 365–384. doi: 10.1007/s10571-006-9062-8
Coyle, J. T., Tsai, G., and Goff, D. (2003). Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann. N. Y. Acad. Sci. 1003, 318–327. doi: 10.1196/annals.1300.020
Dalmau, J., Armangué, T., Planagumà, J., Radosevic, M., Mannara, F., Leypoldt, F., et al. (2019). An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 18, 1045–1057. doi: 10.1016/S1474-4422(19)30244-3
Dalmau, J., Gleichman, A. J., Hughes, E. G., Rossi, J. E., Peng, X., Lai, M., et al. (2008). Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 7, 1091–1098. doi: 10.1016/S1474-4422(08)70224-2
Dalva, M. B., Takasu, M. A., Lin, M. Z., Shamah, S. M., Hu, L., Gale, N. W., et al. (2000). EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cells 103, 945–956. doi: 10.1016/S0092-8674(00)00197-5
Das, S. R., and Magnusson, K. R. (2008). Relationship between mRNA expression of splice forms of the zeta1 subunit of the N-methyl-D-aspartate receptor and spatial memory in aged mice. Brain Res. 1207, 142–154. doi: 10.1016/j.brainres.2008.02.046
Das, S. R., and Magnusson, K. R. (2011). Changes in expression of splice cassettes of NMDA receptor GluN1 subunits within the frontal lobe and memory in mice during aging. Behav. Brain Res. 222, 122–133. doi: 10.1016/j.bbr.2011.03.045
Datta, D., and Arnsten, A. F. T. (2018). Unique molecular regulation of higher-order prefrontal cortical circuits: insights into the neurobiology of schizophrenia. ACS Chem. Neurosci. 9, 2127–2145. doi: 10.1021/acschemneuro.7b00505
De Bernardi, M. L., Di Stazio, A., Romano, A., Minardi, R., Bisulli, F., Licchetta, L., et al. (2022). Mild neurological phenotype in a family carrying a novel N-terminal null GRIN2A variant. Eur. J. Med. Genet. 65:104500. doi: 10.1016/j.ejmg.2022.104500
Dingledine, R., Borges, K., Bowie, D., and Traynelis, S. F. (1999). The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61.
Dore, K., Stein, I. S., Brock, J. A., Castillo, P. E., Zito, K., and Sjöström, P. J. (2017). Unconventional NMDA receptor signaling. J. Neurosci. 37, 10800–10807. doi: 10.1523/JNEUROSCI.1825-17.2017
Elmasri, M., Hunter, D. W., Winchester, G., Bates, E. E., Aziz, W., Van Der Does, D. M., et al. (2022). Common synaptic phenotypes arising from diverse mutations in the human NMDA receptor subunit GluN2A. Commun. Biol. 5:174. doi: 10.1038/s42003-022-03115-3
Endele, S., Rosenberger, G., Geider, K., Popp, B., Tamer, C., Stefanova, I., et al. (2010). Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 42, 1021–1026. doi: 10.1038/ng.677
Esmenjaud, J. B., Stroebel, D., Chan, K., Grand, T., David, M., Wollmuth, L. P., et al. (2019). An inter-dimer allosteric switch controls NMDA receptor activity. EMBO J. 38, 1–16. doi: 10.15252/embj.201899894
Fedele, L., Newcombe, J., Topf, M., Gibb, A., Harvey, R. J., and Smart, T. G. (2018). Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat. Commun. 9:957. doi: 10.1038/s41467-018-02927-4
Forrest, D., Yuzaki, M., Soares, H. D., Ng, L., Luk, D. C., Sheng, M., et al. (1994). Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13, 325–338. doi: 10.1016/0896-6273(94)90350-6
Forsyth, J. K., Bachman, P., Mathalon, D. H., Roach, B. J., and Asarnow, R. F. (2015). Augmenting NMDA receptor signaling boosts experience-dependent neuroplasticity in the adult human brain. Proc. Natl. Acad. Sci. U. S. A. 112, 15331–15336. doi: 10.1073/pnas.1509262112
Foster, T. C. (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 6, 319–325. doi: 10.1111/j.1474-9726.2007.00283.x
Frühauf, P. K., Ineu, R. P., Tomazi, L., Duarte, T., Mello, C. F., and Rubin, M. A. (2015). Spermine reverses lipopolysaccharide-induced memory deficit in mice. J. Neuroinflammation 12:3. doi: 10.1186/s12974-014-0220-5
Fry, A. E., Fawcett, K. A., Zelnik, N., Yuan, H., Thompson, B. A. N., Shemer-Meiri, L., et al. (2018). De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain 141, 698–712. doi: 10.1093/brain/awx358
Galehdari, H., Pooryasin, A., Foroughmand, A., Daneshmand, S., and Saadat, M. (2009). Association between the G1001C polymorphism in the GRIN1 gene promoter and schizophrenia in the Iranian population. J. Mol. Neurosci. 38, 178–181. doi: 10.1007/s12031-008-9148-5
Gao, K., Tankovic, A., Zhang, Y., Kusumoto, H., Zhang, J., Chen, W., et al. (2017). A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS One 12:e0170818. doi: 10.1371/journal.pone.0170818
García-Recio, A., Santos-Gómez, A., Soto, D., Julia-Palacios, N., García-Cazorla, À., Altafaj, X., et al. (2021). GRIN database: a unified and manually curated repertoire of GRIN variants. Hum. Mutat. 42, 8–18. doi: 10.1002/humu.24141
Gielen, M., Siegler Retchless, B., Mony, L., Johnson, J. W., and Paoletti, P. (2009). Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459, 703–707. doi: 10.1038/nature07993
Gleichman, A. J., Spruce, L. A., Dalmau, J., Seeholzer, S. H., and Lynch, D. R. (2012). Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J. Neurosci. 32, 11082–11094. doi: 10.1523/JNEUROSCI.0064-12.2012
Glen, W. B. Jr., Horowitz, B., Carlson, G. C., Cannon, T. D., Talbot, K., Jentsch, J. D., et al. (2014). Dysbindin-1 loss compromises NMDAR-dependent synaptic plasticity and contextual fear conditioning. Hippocampus 24, 204–213. doi: 10.1002/hipo.22215
Gonzales, R. A., Brown, L. M., Jones, T. W., Trent, R. D., Westbrook, S. L., and Leslie, S. W. (1991). N-methyl-D-aspartate mediated responses decrease with age in Fischer 344 rat brain. Neurobiol. Aging 12, 219–225. doi: 10.1016/0197-4580(91)90100-X
Grace, A. A. (2016). Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 17, 524–532. doi: 10.1038/nrn.2016.57
Gramuntell, Y., Klimczak, P., Coviello, S., Perez-Rando, M., and Nacher, J. (2021). Effects of aging on the structure and expression of NMDA receptors of Somatostatin expressing neurons in the mouse Hippocampus. Front. Aging Neurosci. 13:782737. doi: 10.3389/fnagi.2021.782737
Gray, J. A., and Roth, B. L. (2007). The pipeline and future of drug development in schizophrenia. Mol. Psychiatry 12, 904–922. doi: 10.1038/sj.mp.4002062
Guerra, G. P., Rubin, M. A., and Mello, C. F. (2016). Modulation of learning and memory by natural polyamines. Pharmacol. Res. 112, 99–118. doi: 10.1016/j.phrs.2016.03.023
Gunduz-Bruce, H. (2009). The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res. Rev. 60, 279–286. doi: 10.1016/j.brainresrev.2008.07.006
Hackos, D. H., and Hanson, J. E. (2017). Diverse modes of NMDA receptor positive allosteric modulation: mechanisms and consequences. Neuropharmacology 112, 34–45. doi: 10.1016/j.neuropharm.2016.07.037
Hackos, D. H., Lupardus, P. J., Grand, T., Chen, Y., Wang, T. M., Reynen, P., et al. (2016). Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron 89, 983–999. doi: 10.1016/j.neuron.2016.01.016
Hahn, C. G., Wang, H. Y., Cho, D. S., Talbot, K., Gur, R. E., Berrettini, W. H., et al. (2006). Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 12, 824–828. doi: 10.1038/nm1418
Hamdan, F. F., Gauthier, J., Araki, Y., Lin, D. T., Yoshizawa, Y., Higashi, K., et al. (2011). Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 88, 306–316. doi: 10.1016/j.ajhg.2011.02.001
Hansen, K. B., Wollmuth, L. P., Bowie, D., Furukawa, H., Menniti, F. S., Sobolevsky, A. I., et al. (2021). Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 73, 298–487. doi: 10.1124/pharmrev.120.000131
Hansen, K. B., Yi, F., Perszyk, R. E., Furukawa, H., Wollmuth, L. P., Gibb, A. J., et al. (2018). Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 150, 1081–1105. doi: 10.1085/jgp.201812032
Hanson, J. E., Ma, K., Elstrott, J., Weber, M., Saillet, S., Khan, A. S., et al. (2020). GluN2A NMDA receptor enhancement improves brain oscillations, synchrony, and cognitive functions in Dravet syndrome and Alzheimer's disease models. Cell Rep. 30, 381–396.e384. doi: 10.1016/j.celrep.2019.12.030
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Harrison, P. J., and Weinberger, D. R. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68. doi: 10.1038/sj.mp.4001558
Harsing, L. G. Jr., and Matyus, P. (2013). Mechanisms of glycine release, which build up synaptic and extrasynaptic glycine levels: the role of synaptic and non-synaptic glycine transporters. Brain Res. Bull. 93, 110–119. doi: 10.1016/j.brainresbull.2012.12.002
Hawasli, A. H., Benavides, D. R., Nguyen, C., Kansy, J. W., Hayashi, K., Chambon, P., et al. (2007). Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat. Neurosci. 10, 880–886. doi: 10.1038/nn1914
Higley, M. J., and Sabatini, B. L. (2012). Calcium signaling in dendritic spines. Cold Spring Harb. Perspect. Biol. 4:a005686. doi: 10.1101/cshperspect.a005686
Homayoun, H., and Moghaddam, B. (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 27, 11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007
Howes, O. D., and Kapur, S. (2009). The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr. Bull. 35, 549–562. doi: 10.1093/schbul/sbp006
Huganir, R. L., and Nicoll, R. A. (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. doi: 10.1016/j.neuron.2013.10.025
Hughes, E. G., Peng, X., Gleichman, A. J., Lai, M., Zhou, L., Tsou, R., et al. (2010). Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 30, 5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010
Intson, K., van Eede, M. C., Islam, R., Milenkovic, M., Yan, Y., Salahpour, A., et al. (2019). Progressive neuroanatomical changes caused by Grin1 loss-of-function mutation. Neurobiol. Dis. 132:104527. doi: 10.1016/j.nbd.2019.104527
Javitt, D. C., and Zukin, S. R. (1991). Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308. doi: 10.1176/ajp.148.10.1301
Jentsch, J. D., Taylor, J. R., Elsworth, J. D., Redmond, D. E. Jr., and Roth, R. H. (1999). Altered frontal cortical dopaminergic transmission in monkeys after subchronic phencyclidine exposure: Involvement in frontostriatal cognitive deficits. Neuroscience 90, 823–832. doi: 10.1016/s0306-4522(98)00481-3
Jentsch, J. D., Trantham-Davidson, H., Jairl, C., Tinsley, M., Cannon, T. D., and Lavin, A. (2009). Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology 34, 2601–2608. doi: 10.1038/npp.2009.90
Jézéquel, J., Johansson, E. M., Leboyer, M., and Groc, L. (2018). Pathogenicity of antibodies against NMDA receptor: molecular insights into autoimmune psychosis. Trends Neurosci. 41, 502–511. doi: 10.1016/j.tins.2018.05.002
Ji, Y., Yang, F., Papaleo, F., Wang, H. X., Gao, W. J., Weinberger, D. R., et al. (2009). Role of dysbindin in dopamine receptor trafficking and cortical GABA function. Proc. Natl. Acad. Sci. U. S. A. 106, 19593–19598. doi: 10.1073/pnas.0904289106
Jones, B. E., Tovar, K. R., Goehring, A., Jalali-Yazdi, F., Okada, N. J., Gouaux, E., et al. (2019). Autoimmune receptor encephalitis in mice induced by active immunization with conformationally stabilized holoreceptors. Sci. Transl. Med. 11, 1–28. doi: 10.1126/scitranslmed.aaw0044
Jones, C. A., Watson, D. J., and Fone, K. C. (2011). Animal models of schizophrenia. Br. J. Pharmacol. 164, 1162–1194. doi: 10.1111/j.1476-5381.2011.01386.x
Kantrowitz, J. T., and Javitt, D. C. (2010). N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res. Bull. 83, 108–121. doi: 10.1016/j.brainresbull.2010.04.006
Karlsgodt, K. H., Robleto, K., Trantham-Davidson, H., Jairl, C., Cannon, T. D., Lavin, A., et al. (2011). Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biol. Psychiatry 69, 28–34. doi: 10.1016/j.biopsych.2010.09.012
Kato, T., Abe, Y., Sotoyama, H., Kakita, A., Kominami, R., Hirokawa, S., et al. (2011). Transient exposure of neonatal mice to neuregulin-1 results in hyperdopaminergic states in adulthood: implication in neurodevelopmental hypothesis for schizophrenia. Mol. Psychiatry 16, 307–320. doi: 10.1038/mp.2010.10
Kersten, M., Rabbe, T., Blome, R., Porath, K., Sellmann, T., Bien, C. G., et al. (2019). Novel object recognition in rats with NMDAR dysfunction in CA1 after stereotactic injection of anti-NMDAR encephalitis cerebrospinal fluid. Front. Neurol. 10:586. doi: 10.3389/fneur.2019.00586
Kinney, J. W., Davis, C. N., Tabarean, I., Conti, B., Bartfai, T., and Behrens, M. M. (2006). A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J. Neurosci. 26, 1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006
Koek, W., Woods, J. H., and Winger, G. D. (1988). MK-801, a proposed noncompetitive antagonist of excitatory amino acid neurotransmission, produces phencyclidine-like behavioral effects in pigeons, rats and rhesus monkeys. J. Pharmacol. Exp. Ther. 245, 969–974.
Koenig, A., Malhotra, S., Wald, J., Petrillo, J., Paumier, K., Johannensen, J., et al. (2022). SAGE-718 in patients with mild cognitive impairment or mild dementia due to Alzheimer’s disease: results from the phase 2 LUMINARY study. Drug Dev. 18:e063896. doi: 10.1002/alz.063896
Kotti, T. J., Ramirez, D. M., Pfeiffer, B. E., Huber, K. M., and Russell, D. W. (2006). Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. U. S. A. 103, 3869–3874. doi: 10.1073/pnas.0600316103
Kotzadimitriou, D., Nissen, W., Paizs, M., Newton, K., Harrison, P. J., Paulsen, O., et al. (2018). Neuregulin 1 type I overexpression is associated with reduced NMDA receptor-mediated synaptic signaling in hippocampal interneurons expressing PV or CCK. eNeuro 5, 1–18. doi: 10.1523/ENEURO.0418-17.2018
Kovacic, P., and Somanathan, R. (2010). Clinical physiology and mechanism of dizocilpine (MK-801): electron transfer, radicals, redox metabolites and bioactivity. Oxidative Med. Cell. Longev. 3, 13–22. doi: 10.4161/oxim.3.1.10028
Kreye, J., Wenke, N. K., Chayka, M., Leubner, J., Murugan, R., Maier, N., et al. (2016). Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 139, 2641–2652. doi: 10.1093/brain/aww208
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., et al. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 51, 199–214. doi: 10.1001/archpsyc.1994.03950030035004
Kuehl-Kovarik, M. C., Magnusson, K. R., Premkumar, L. S., and Partin, K. M. (2000). Electrophysiological analysis of NMDA receptor subunit changes in the aging mouse cortex. Mech. Ageing Dev. 115, 39–59. doi: 10.1016/S0047-6374(00)00104-4
Kumar, A., and Foster, T. C. (2019). Alteration in NMDA receptor mediated Glutamatergic neurotransmission in the Hippocampus during senescence. Neurochem. Res. 44, 38–48. doi: 10.1007/s11064-018-2634-4
Kumar, A., Thinschmidt, J. S., and Foster, T. C. (2019). Subunit contribution to NMDA receptor hypofunction and redox sensitivity of hippocampal synaptic transmission during aging. Aging (Albany NY) 11, 5140–5157. doi: 10.18632/aging.102108
Kutsuwada, T., Sakimura, K., Manabe, T., Takayama, C., Katakura, N., Kushiya, E., et al. (1996). Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16, 333–344. doi: 10.1016/S0896-6273(00)80051-3
Ladépêche, L., Planagumà, J., Thakur, S., Suárez, I., Hara, M., Borbely, J. S., et al. (2018). NMDA receptor autoantibodies in autoimmune encephalitis cause a subunit-specific Nanoscale redistribution of NMDA receptors. Cell Rep. 23, 3759–3768. doi: 10.1016/j.celrep.2018.05.096
Lemke, J. R., Geider, K., Helbig, K. L., Heyne, H. O., Schütz, H., Hentschel, J., et al. (2016). Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 86, 2171–2178. doi: 10.1212/WNL.0000000000002740
Lemke, J. R., Lal, D., Reinthaler, E. M., Steiner, I., Nothnagel, M., Alber, M., et al. (2013). Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 45, 1067–1072. doi: 10.1038/ng.2728
Lesca, G., Rudolf, G., Bruneau, N., Lozovaya, N., Labalme, A., Boutry-Kryza, N., et al. (2013). GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat. Genet. 45, 1061–1066. doi: 10.1038/ng.2726
Lewis, D. A., and Moghaddam, B. (2006). Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 63, 1372–1376. doi: 10.1001/archneur.63.10.1372
Li, C., Brake, W. G., Romeo, R. D., Dunlop, J. C., Gordon, M., Buzescu, R., et al. (2004). Estrogen alters hippocampal dendritic spine shape and enhances synaptic protein immunoreactivity and spatial memory in female mice. Proc. Natl. Acad. Sci. U. S. A. 101, 2185–2190. doi: 10.1073/pnas.0307313101
Li, Y., Erzurumlu, R. S., Chen, C., Jhaveri, S., and Tonegawa, S. (1994). Whisker-related neuronal patterns fail to develop in the trigeminal brainstem nuclei of NMDAR1 knockout mice. Cells 76, 427–437. doi: 10.1016/0092-8674(94)90108-2
Li, J., Zhang, J., Tang, W., Mizu, R. K., Kusumoto, H., XiangWei, W., et al. (2019). De novo GRIN variants in NMDA receptor M2 channel pore-forming loop are associated with neurological diseases. Hum. Mutat. 40, 2393–2413. doi: 10.1002/humu.23895
Lichtenwalner, R. J., Forbes, M. E., Bennett, S. A., Lynch, C. D., Sonntag, W. E., and Riddle, D. R. (2001). Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience 107, 603–613. doi: 10.1016/S0306-4522(01)00378-5
Lindsley, C. W., Shipe, W. D., Wolkenberg, S. E., Theberge, C. R., Williams, D. L. Jr., Sur, C., et al. (2006). Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr. Top. Med. Chem. 6, 771–785. doi: 10.2174/156802606777057599
Lipina, T. V., Niwa, M., Jaaro-Peled, H., Fletcher, P. J., Seeman, P., Sawa, A., et al. (2010). Enhanced dopamine function in DISC1-L100P mutant mice: implications for schizophrenia. Genes Brain Behav. 9, 777–789. doi: 10.1111/j.1601-183X.2010.00615.x
Lisman, J. E., Coyle, J. T., Green, R. W., Javitt, D. C., Benes, F. M., Heckers, S., et al. (2008). Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 31, 234–242. doi: 10.1016/j.tins.2008.02.005
Lodge, D., and Mercier, M. S. (2015). Ketamine and phencyclidine: the good, the bad and the unexpected. Br. J. Pharmacol. 172, 4254–4276. doi: 10.1111/bph.13222
Lussier, M. P., Sanz-Clemente, A., and Roche, K. W. (2015). Dynamic regulation of N-methyl-d-aspartate (NMDA) and α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J. Biol. Chem. 290, 28596–28603. doi: 10.1074/jbc.R115.652750
Lynch, M. A. (2004). Long-term potentiation and memory. Physiol. Rev. 84, 87–136. doi: 10.1152/physrev.00014.2003
Lynch, D. R., Rattelle, A., Dong, Y. N., Roslin, K., Gleichman, A. J., and Panzer, J. A. (2018). Anti-NMDA receptor encephalitis: clinical features and basic mechanisms. Adv. Pharmacol. 82, 235–260. doi: 10.1016/bs.apha.2017.08.005
Ma, T. M., Abazyan, S., Abazyan, B., Nomura, J., Yang, C., Seshadri, S., et al. (2013). Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via D-serine depletion. Mol. Psychiatry 18, 557–567. doi: 10.1038/mp.2012.97
Magnusson, K. R. (2001). Influence of diet restriction on NMDA receptor subunits and learning during aging. Neurobiol. Aging 22, 613–627. doi: 10.1016/S0197-4580(00)00258-X
Magnusson, K. R., Bai, L., and Zhao, X. (2005). The effects of aging on different C-terminal splice forms of the zeta1(NR1) subunit of the N-methyl-d-aspartate receptor in mice. Brain Res. Mol. Brain Res. 135, 141–149. doi: 10.1016/j.molbrainres.2004.12.012
Magnusson, K. R., Kresge, D., and Supon, J. (2006). Differential effects of aging on NMDA receptors in the intermediate versus the dorsal hippocampus. Neurobiol. Aging 27, 324–333. doi: 10.1016/j.neurobiolaging.2005.01.012
Magnusson, K. R., Scruggs, B., Zhao, X., and Hammersmark, R. (2007). Age-related declines in a two-day reference memory task are associated with changes in NMDA receptor subunits in mice. BMC Neurosci. 8:43. doi: 10.1186/1471-2202-8-43
Malenka, R. C. (2003). Synaptic plasticity and AMPA receptor trafficking. Ann. N. Y. Acad. Sci. 1003, 1–11. doi: 10.1196/annals.1300.001
Malviya, M., Barman, S., Golombeck, K. S., Planagumà, J., Mannara, F., Strutz-Seebohm, N., et al. (2017). NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann. Clin. Transl. Neurol. 4, 768–783. doi: 10.1002/acn3.444
Mannara, F., Radosevic, M., Planagumà, J., Soto, D., Aguilar, E., García-Serra, A., et al. (2020). Allosteric modulation of NMDA receptors prevents the antibody effects of patients with anti-NMDAR encephalitis. Brain 143, 2709–2720. doi: 10.1093/brain/awaa195
Marek, G. J. (2010). Metabotropic glutamate2/3 (mGlu2/3) receptors, schizophrenia and cognition. Eur. J. Pharmacol. 639, 81–90. doi: 10.1016/j.ejphar.2010.02.058
McCutcheon, R. A., Krystal, J. H., and Howes, O. D. (2020). Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry 19, 15–33. doi: 10.1002/wps.20693
McDaniel, M. J., Ogden, K. K., Kell, S. A., Burger, P. B., Liotta, D. C., and Traynelis, S. F. (2020). NMDA receptor channel gating control by the pre-M1 helix. J. Gen. Physiol. 152, 1–16. doi: 10.1085/jgp.201912362
Mei, L., and Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452. doi: 10.1038/nrn2392
Mikasova, L., De Rossi, P., Bouchet, D., Georges, F., Rogemond, V., Didelot, A., et al. (2012). Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 135, 1606–1621. doi: 10.1093/brain/aws092
Moghaddam, B. (2003). Bringing order to the glutamate chaos in schizophrenia. Neuron 40, 881–884. doi: 10.1016/S0896-6273(03)00757-8
Moghaddam, B., Adams, B., Verma, A., and Daly, D. (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 17, 2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997
Mohn, A. R., Gainetdinov, R. R., Caron, M. G., and Koller, B. H. (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cells 98, 427–436. doi: 10.1016/S0092-8674(00)81972-8
Monaco, S. A., Gulchina, Y., and Gao, W. J. (2015). NR2B subunit in the prefrontal cortex: a double-edged sword for working memory function and psychiatric disorders. Neurosci. Biobehav. Rev. 56, 127–138. doi: 10.1016/j.neubiorev.2015.06.022
Monahan, J. B., Handelmann, G. E., Hood, W. F., and Cordi, A. A. (1989). D-cycloserine, a positive modulator of the N-methyl-D-aspartate receptor, enhances performance of learning tasks in rats. Pharmacol. Biochem. Behav. 34, 649–653. doi: 10.1016/0091-3057(89)90571-6
Mony, L., Kew, J. N., Gunthorpe, M. J., and Paoletti, P. (2009). Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 157, 1301–1317. doi: 10.1111/j.1476-5381.2009.00304.x
Mony, L., Zhu, S., Carvalho, S., and Paoletti, P. (2011). Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J. 30, 3134–3146. doi: 10.1038/emboj.2011.203
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. doi: 10.1016/0896-6273(94)90210-0
Moscato, E. H., Peng, X., Jain, A., Parsons, T. D., Dalmau, J., and Balice-Gordon, R. J. (2014). Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 76, 108–119. doi: 10.1002/ana.24195
Mostany, R., Anstey, J. E., Crump, K. L., Maco, B., Knott, G., and Portera-Cailliau, C. (2013). Altered synaptic dynamics during normal brain aging. J. Neurosci. 33, 4094–4104. doi: 10.1523/JNEUROSCI.4825-12.2013
Myers, S. J., Yuan, H., Kang, J. Q., Tan, F. C. K., Traynelis, S. F., and Low, C. M. (2019). Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Res 8, 1–14. doi: 10.12688/f1000research.18949.1
Nakazawa, K., Jeevakumar, V., and Nakao, K. (2017). Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr. 3:7. doi: 10.1038/s41537-016-0003-3
Nakazawa, K., Zsiros, V., Jiang, Z., Nakao, K., Kolata, S., Zhang, S., et al. (2012). GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62, 1574–1583. doi: 10.1016/j.neuropharm.2011.01.022
Nava-Gómez, L., Calero-Vargas, I., Higinio-Rodríguez, F., Vázquez-Prieto, B., Olivares-Moreno, R., Ortiz-Retana, J., et al. (2022). Aging-associated cognitive decline is reversed by D-serine supplementation. eNeuro 9, 1–22. doi: 10.1523/ENEURO.0176-22.2022
Niwa, M., Kamiya, A., Murai, R., Kubo, K., Gruber, A. J., Tomita, K., et al. (2010). Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron 65, 480–489. doi: 10.1016/j.neuron.2010.01.019
Nong, Y., Huang, Y. Q., Ju, W., Kalia, L. V., Ahmadian, G., Wang, Y. T., et al. (2003). Glycine binding primes NMDA receptor internalization. Nature 422, 302–307. doi: 10.1038/nature01497
Numakawa, T., Yagasaki, Y., Ishimoto, T., Okada, T., Suzuki, T., Iwata, N., et al. (2004). Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum. Mol. Genet. 13, 2699–2708. doi: 10.1093/hmg/ddh280
Ogden, K. K., Chen, W., Swanger, S. A., McDaniel, M. J., Fan, L. Z., Hu, C., et al. (2017). Molecular mechanism of disease-associated mutations in the pre-M1 Helix of NMDA receptors and potential rescue pharmacology. PLoS Genet. 13:e1006536. doi: 10.1371/journal.pgen.1006536
Ogden, K. K., and Traynelis, S. F. (2013). Contribution of the M1 transmembrane helix and pre-M1 region to positive allosteric modulation and gating of N-methyl-D-aspartate receptors. Mol. Pharmacol. 83, 1045–1056. doi: 10.1124/mol.113.085209
Olney, J. W., and Farber, N. B. (1995). Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 52, 998–1007. doi: 10.1001/archpsyc.1995.03950240016004
Olney, J. W., Labruyere, J., Wang, G., Wozniak, D. F., Price, M. T., and Sesma, M. A. (1991). NMDA antagonist neurotoxicity: mechanism and prevention. Science 254, 1515–1518. doi: 10.1126/science.1835799
Olney, J. W., Newcomer, J. W., and Farber, N. B. (1999). NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 33, 523–533. doi: 10.1016/S0022-3956(99)00029-1
Ontl, T., Xing, Y., Bai, L., Kennedy, E., Nelson, S., Wakeman, M., et al. (2004). Development and aging of N-methyl-D-aspartate receptor expression in the prefrontal/frontal cortex of mice. Neuroscience 123, 467–479. doi: 10.1016/j.neuroscience.2003.09.006
O'Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/ng.835
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
Otsu, Y., Darcq, E., Pietrajtis, K., Mátyás, F., Schwartz, E., Bessaih, T., et al. (2019). Control of aversion by glycine-gated GluN1/GluN3A NMDA receptors in the adult medial habenula. Science 366, 250–254. doi: 10.1126/science.aax1522
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi: 10.1038/nrn3504
Paoletti, P., and Neyton, J. (2007). NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 7, 39–47. doi: 10.1016/j.coph.2006.08.011
Papaleo, F., Lipska, B. K., and Weinberger, D. R. (2012). Mouse models of genetic effects on cognition: relevance to schizophrenia. Neuropharmacology 62, 1204–1220. doi: 10.1016/j.neuropharm.2011.04.025
Papaleo, F., and Weinberger, D. R. (2011). Dysbindin and schizophrenia: it's dopamine and glutamate all over again. Biol. Psychiatry 69, 2–4. doi: 10.1016/j.biopsych.2010.10.028
Parsons, M. P., and Raymond, L. A. (2014). Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–293. doi: 10.1016/j.neuron.2014.03.030
Paul, S. M., Doherty, J. J., Robichaud, A. J., Belfort, G. M., Chow, B. Y., Hammond, R. S., et al. (2013). The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J. Neurosci. 33, 17290–17300. doi: 10.1523/JNEUROSCI.2619-13.2013
Pegasiou, C. M., Zolnourian, A., Gomez-Nicola, D., Deinhardt, K., Nicoll, J. A. R., Ahmed, A. I., et al. (2020). Age-dependent changes in synaptic NMDA receptor composition in adult human cortical neurons. Cereb. Cortex 30, 4246–4256. doi: 10.1093/cercor/bhaa052
Perszyk, R. E., Myers, S. J., Yuan, H., Gibb, A. J., Furukawa, H., Sobolevsky, A. I., et al. (2020). Hodgkin-Huxley-Katz prize lecture: genetic and pharmacological control of glutamate receptor channel through a highly conserved gating motif. J. Physiol. 598, 3071–3083. doi: 10.1113/JP278086
Pilowsky, L. S., Bressan, R. A., Stone, J. M., Erlandsson, K., Mulligan, R. S., Krystal, J. H., et al. (2006). First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry 11, 118–119. doi: 10.1038/sj.mp.4001751
Pittaluga, A., Fedele, E., Risiglione, C., and Raiteri, M. (1993). Age-related decrease of the NMDA receptor-mediated noradrenaline release in rat hippocampus and partial restoration by D-cycloserine. Eur. J. Pharmacol. 231, 129–134. doi: 10.1016/0014-2999(93)90693-C
Planagumà, J., Haselmann, H., Mannara, F., Petit-Pedrol, M., Grünewald, B., Aguilar, E., et al. (2016). Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann. Neurol. 80, 388–400. doi: 10.1002/ana.24721
Planagumà, J., Leypoldt, F., Mannara, F., Gutiérrez-Cuesta, J., Martín-García, E., Aguilar, E., et al. (2015). Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 138, 94–109. doi: 10.1093/brain/awu310
Plattner, F., Hernández, A., Kistler, T. M., Pozo, K., Zhong, P., Yuen, E. Y., et al. (2014). Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron 81, 1070–1083. doi: 10.1016/j.neuron.2014.01.022
Platzer, K., Yuan, H., Schütz, H., Winschel, A., Chen, W., Hu, C., et al. (2017). GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 54, 460–470. doi: 10.1136/jmedgenet-2016-104509
Quinlan, E. M., Olstein, D. H., and Bear, M. F. (1999). Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc. Natl. Acad. Sci. U. S. A. 96, 12876–12880. doi: 10.1073/pnas.96.22.12876
Ren, J., Li, X., Zhang, X., Li, M., Wang, Y., and Ma, Y. (2013). The effects of intra-hippocampal microinfusion of D-cycloserine on fear extinction, and the expression of NMDA receptor subunit NR2B and neurogenesis in the hippocampus in rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 44, 257–264. doi: 10.1016/j.pnpbp.2013.02.017
Roberts, A. C., Díez-García, J., Rodriguiz, R. M., López, I. P., Luján, R., Martínez-Turrillas, R., et al. (2009). Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron 63, 342–356. doi: 10.1016/j.neuron.2009.06.016
Rossi, M., Chatron, N., Labalme, A., Ville, D., Carneiro, M., Edery, P., et al. (2017). Novel homozygous missense variant of GRIN1 in two sibs with intellectual disability and autistic features without epilepsy. Eur. J. Hum. Genet. 25, 376–380. doi: 10.1038/ejhg.2016.163
Rothman, S. M., and Olney, J. W. (1995). Excitotoxicity and the NMDA receptor--still lethal after eight years. Trends Neurosci. 18, 57–58. doi: 10.1016/0166-2236(95)93869-y
Roy, K., Murtie, J. C., El-Khodor, B. F., Edgar, N., Sardi, S. P., Hooks, B. M., et al. (2007). Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. U. S. A. 104, 8131–8136. doi: 10.1073/pnas.0702157104
Rubio, M. D., Drummond, J. B., and Meador-Woodruff, J. H. (2012). Glutamate receptor abnormalities in schizophrenia: implications for innovative treatments. Biomol. Ther. 20, 1–18. doi: 10.4062/biomolther.2012.20.1.001
Rujescu, D., Bender, A., Keck, M., Hartmann, A. M., Ohl, F., Raeder, H., et al. (2006). A pharmacological model for psychosis based on N-methyl-D-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol. Psychiatry 59, 721–729. doi: 10.1016/j.biopsych.2005.08.029
Russell, D. W., Halford, R. W., Ramirez, D. M., Shah, R., and Kotti, T. (2009). Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 78, 1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859
Sakimura, K., Kutsuwada, T., Ito, I., Manabe, T., Takayama, C., Kushiya, E., et al. (1995). Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 373, 151–155. doi: 10.1038/373151a0
Santos-Gómez, A., Miguez-Cabello, F., García-Recio, A., Locubiche-Serra, S., García-Díaz, R., Soto-Insuga, V., et al. (2021a). Disease-associated GRIN protein truncating variants trigger NMDA receptor loss-of-function. Hum. Mol. Genet. 29, 3859–3871. doi: 10.1093/hmg/ddaa220
Santos-Gómez, A., Miguez-Cabello, F., Juliá-Palacios, N., García-Navas, D., Soto-Insuga, V., García-Peñas, J. J., et al. (2021b). Paradigmatic De novo GRIN1 variants recapitulate pathophysiological mechanisms underlying GRIN1-related disorder clinical Spectrum. Int. J. Mol. Sci. 22, 1–19. doi: 10.3390/ijms222312656
Sceniak, M. P., Fedder, K. N., Wang, Q., Droubi, S., Babcock, K., Patwardhan, S., et al. (2019). An autism-associated mutation in GluN2B prevents NMDA receptor trafficking and interferes with dendrite growth. J. Cell Sci. 132, 1–14. doi: 10.1242/jcs.232892
Selemon, L. D., and Zecevic, N. (2015). Schizophrenia: a tale of two critical periods for prefrontal cortical development. Transl. Psychiatry 5:e623. doi: 10.1038/tp.2015.115
Sharma, R., Al-Saleem, F. H., Panzer, J., Lee, J., Puligedda, R. D., Felicori, L. F., et al. (2018). Monoclonal antibodies from a patient with anti-NMDA receptor encephalitis. Ann. Clin. Transl. Neurol. 5, 935–951. doi: 10.1002/acn3.592
Sheng, M., Cummings, J., Roldan, L. A., Jan, Y. N., and Jan, L. Y. (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. doi: 10.1038/368144a0
Sirrieh, R. E., MacLean, D. M., and Jayaraman, V. (2015). Subtype-dependent N-methyl-D-aspartate receptor amino-terminal domain conformations and modulation by spermine. J. Biol. Chem. 290, 12812–12820. doi: 10.1074/jbc.M115.649723
Snyder, M. A., Adelman, A. E., and Gao, W. J. (2013). Gestational methylazoxymethanol exposure leads to NMDAR dysfunction in hippocampus during early development and lasting deficits in learning. Neuropsychopharmacology 38, 328–340. doi: 10.1038/npp.2012.180
Snyder, M. A., and Gao, W. J. (2013). NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia. Front. Cell. Neurosci. 7:31. doi: 10.3389/fncel.2013.00031
Soto, D., Olivella, M., Grau, C., Armstrong, J., Alcon, C., Gasull, X., et al. (2019). L-serine dietary supplementation is associated with clinical improvement of loss-of-function GRIN2B-related pediatric encephalopathy. Sci. Signal. 12, 1–15. doi: 10.1126/scisignal.aaw0936
Spear, L. P. (2000). The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev. 24, 417–463. doi: 10.1016/S0149-7634(00)00014-2
Stahl, S. M. (2007). Beyond the dopamine hypothesis to the NMDA glutamate receptor hypofunction hypothesis of schizophrenia. CNS Spectr. 12, 265–268. doi: 10.1017/S1092852900021015
Strehlow, V., Heyne, H. O., Vlaskamp, D. R. M., Marwick, K. F. M., Rudolf, G., de Bellescize, J., et al. (2019). GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain 142, 80–92. doi: 10.1093/brain/awy304
Sun, M. Y., Taylor, A., Zorumski, C. F., and Mennerick, S. (2017). 24S-hydroxycholesterol and 25-hydroxycholesterol differentially impact hippocampal neuronal survival following oxygen-glucose deprivation. PLoS One 12:e0174416. doi: 10.1371/journal.pone.0174416
Suryavanshi, P. S., Ugale, R. R., Yilmazer-Hanke, D., Stairs, D. J., and Dravid, S. M. (2014). GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br. J. Pharmacol. 171, 799–809. doi: 10.1111/bph.12518
Swanger, S. A., Chen, W., Wells, G., Burger, P. B., Tankovic, A., Bhattacharya, S., et al. (2016). Mechanistic insight into NMDA receptor Dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am. J. Hum. Genet. 99, 1261–1280. doi: 10.1016/j.ajhg.2016.10.002
Sze, C., Bi, H., Kleinschmidt-DeMasters, B. K., Filley, C. M., and Martin, L. J. (2001). N-methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J. Neurol. Sci. 182, 151–159. doi: 10.1016/s0022-510x(00)00467-6
Tagliabue, E., Pouvreau, T., Eybrard, S., Meyer, F., and Louilot, A. (2017). Dopaminergic responses in the core part of the nucleus accumbens to subcutaneous MK801 administration are increased following postnatal transient blockade of the prefrontal cortex. Behav. Brain Res. 335, 191–198. doi: 10.1016/j.bbr.2017.08.021
Talbot, K., Eidem, W. L., Tinsley, C. L., Benson, M. A., Thompson, E. W., Smith, R. J., et al. (2004). Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J. Clin. Invest. 113, 1353–1363. doi: 10.1172/jci20425
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Uno, Y., Coyle, J. T. J. P., and Neurosciences, C. (2019). Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 73, 204–215. doi: 10.1111/pcn.12823
van Haren, N. E., Hulshoff Pol, H. E., Schnack, H. G., Cahn, W., Brans, R., Carati, I., et al. (2008). Progressive brain volume loss in schizophrenia over the course of the illness: evidence of maturational abnormalities in early adulthood. Biol. Psychiatry 63, 106–113. doi: 10.1016/j.biopsych.2007.01.004
Velloso, N. A., Dalmolin, G. D., Fonini, G., Gindri Sinhorin, V. D., Ferreira da Silveira, A., Rubin, M. A., et al. (2008). Spermine attenuates behavioral and biochemical alterations induced by quinolinic acid in the striatum of rats. Brain Res. 1198, 107–114. doi: 10.1016/j.brainres.2007.12.056
Vrajová, M., Stastný, F., Horácek, J., Lochman, J., Serý, O., Peková, S., et al. (2010). Expression of the hippocampal NMDA receptor GluN1 subunit and its splicing isoforms in schizophrenia: postmortem study. Neurochem. Res. 35, 994–1002. doi: 10.1007/s11064-010-0145-z
Vullhorst, D., Mitchell, R. M., Keating, C., Roychowdhury, S., Karavanova, I., Tao-Cheng, J. H., et al. (2015). A negative feedback loop controls NMDA receptor function in cortical interneurons via neuregulin 2/ErbB4 signalling. Nat. Commun. 6:7222. doi: 10.1038/ncomms8222
Wang, H. X., and Gao, W. J. (2009). Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology 34, 2028–2040. doi: 10.1038/npp.2009.20
Wang, H. X., and Gao, W. J. (2012). Prolonged exposure to NMDAR antagonist induces cell-type specific changes of glutamatergic receptors in rat prefrontal cortex. Neuropharmacology 62, 1808–1822. doi: 10.1016/j.neuropharm.2011.11.024
Wang, H., Stradtman, G. G. 3rd, Wang, X. J., and Gao, W. J. (2008). A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc. Natl. Acad. Sci. U. S. A. 105, 16791–16796. doi: 10.1073/pnas.0804318105
Wang, J., Wang, F., Mai, D., and Qu, S. (2020). Molecular mechanisms of glutamate toxicity in Parkinson's disease. Front. Neurosci. 14:585584. doi: 10.3389/fnins.2020.585584
Wei, J., Graziane, N. M., Wang, H., Zhong, P., Wang, Q., Liu, W., et al. (2014). Regulation of N-methyl-D-aspartate receptors by disrupted-in-schizophrenia-1. Biol. Psychiatry 75, 414–424. doi: 10.1016/j.biopsych.2013.06.009
Wells, G., Yuan, H., McDaniel, M. J., Kusumoto, H., Snyder, J. P., Liotta, D. C., et al. (2018). The GluN2B-Glu413Gly NMDA receptor variant arising from a de novo GRIN2B mutation promotes ligand-unbinding and domain opening. Proteins 86, 1265–1276. doi: 10.1002/prot.25595
Wilding, T. J., Lopez, M. N., and Huettner, J. E. (2016). Chimeric glutamate receptor subunits reveal the Transmembrane domain is sufficient for NMDA receptor pore properties but some positive allosteric modulators require additional domains. J. Neurosci. 36, 8815–8825. doi: 10.1523/JNEUROSCI.0345-16.2016
Williams, K. (1994). Mechanisms influencing stimulatory effects of spermine at recombinant N-methyl-D-aspartate receptors. Mol. Pharmacol. 46, 161–168.
Wong, R. W., Setou, M., Teng, J., Takei, Y., and Hirokawa, N. (2002). Overexpression of motor protein KIF17 enhances spatial and working memory in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 99, 14500–14505. doi: 10.1073/pnas.222371099
Woo, T. U., Walsh, J. P., and Benes, F. M. (2004). Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch. Gen. Psychiatry 61, 649–657. doi: 10.1001/archpsyc.61.7.649
Wright, S. K., Rosch, R. E., Wilson, M. A., Upadhya, M. A., Dhangar, D. R., Clarke-Bland, C., et al. (2021). Multimodal electrophysiological analyses reveal that reduced synaptic excitatory neurotransmission underlies seizures in a model of NMDAR antibody-mediated encephalitis. Commun. Biol. 4:1106. doi: 10.1038/s42003-021-02635-8
Xi, D., Keeler, B., Zhang, W., Houle, J. D., and Gao, W. J. (2009). NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J. Neurosci. Methods 176, 172–181. doi: 10.1016/j.jneumeth.2008.09.013
XiangWei, W., Jiang, Y., and Yuan, H. (2018). De novo mutations and rare variants occurring in NMDA receptors. Curr. Opin. Physiol. 2, 27–35. doi: 10.1016/j.cophys.2017.12.013
Xu, X. X., and Luo, J. H. (2018). Mutations of N-methyl-D-aspartate receptor subunits in epilepsy. Neurosci. Bull. 34, 549–565. doi: 10.1007/s12264-017-0191-5
Yao, L., Grand, T., Hanson, J. E., Paoletti, P., and Zhou, Q. (2018). Higher ambient synaptic glutamate at inhibitory versus excitatory neurons differentially impacts NMDA receptor activity. Nat. Commun. 9:4000. doi: 10.1038/s41467-018-06512-7
Yin, X., Feng, X., Takei, Y., and Hirokawa, N. (2012). Regulation of NMDA receptor transport: a KIF17-cargo binding/releasing underlies synaptic plasticity and memory in vivo. J. Neurosci. 32, 5486–5499. doi: 10.1523/JNEUROSCI.0718-12.2012
Yin, X., Takei, Y., Kido, M. A., and Hirokawa, N. (2011). Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron 70, 310–325. doi: 10.1016/j.neuron.2011.02.049
Yu, Y., Lin, Y., Takasaki, Y., Wang, C., Kimura, H., Xing, J., et al. (2018). Rare loss of function mutations in N-methyl-D-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl. Psychiatry 8:12. doi: 10.1038/s41398-017-0061-y
Yuan, H., Low, C. M., Moody, O. A., Jenkins, A., and Traynelis, S. F. (2015). Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol. Pharmacol. 88, 203–217. doi: 10.1124/mol.115.097998
Yulug, B., Altay, O., Li, X., Hanoglu, L., Cankaya, S., Lam, S., et al. (2023). Combined metabolic activators improve cognitive functions in Alzheimer's disease patients: a randomised, double-blinded, placebo-controlled phase-II trial. Transl. Neurodegener. 12:4. doi: 10.1186/s40035-023-00336-2
Zamzow, D. R., Elias, V., Shumaker, M., Larson, C., and Magnusson, K. R. (2013). An increase in the association of GluN2B containing NMDA receptors with membrane scaffolding proteins was related to memory declines during aging. J. Neurosci. 33, 12300–12305. doi: 10.1523/JNEUROSCI.0312-13.2013
Zhao, X., Rosenke, R., Kronemann, D., Brim, B., Das, S. R., Dunah, A. W., et al. (2009). The effects of aging on N-methyl-D-aspartate receptor subunits in the synaptic membrane and relationships to long-term spatial memory. Neuroscience 162, 933–945. doi: 10.1016/j.neuroscience.2009.05.018
Zhou, D., Lv, D., Wang, Z., Zhang, Y., Chen, Z., and Wang, C. (2018). GLYX-13 ameliorates schizophrenia-like phenotype induced by MK-801 in mice: role of hippocampal NR2B and DISC1. Front. Mol. Neurosci. 11:121. doi: 10.3389/fnmol.2018.00121
Zhu, S., and Paoletti, P. (2015). Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr. Opin. Pharmacol. 20, 14–23. doi: 10.1016/j.coph.2014.10.009
Keywords: N-methyl-D-aspartate (NMDA) receptors, schizophrenia, GRIN related disease, NMDAR antibody encephalitis, age-related cognitive decline
Citation: Dong B, Yue Y, Dong H and Wang Y (2023) N-methyl-D-aspartate receptor hypofunction as a potential contributor to the progression and manifestation of many neurological disorders. Front. Mol. Neurosci. 16:1174738. doi: 10.3389/fnmol.2023.1174738
Edited by:
Laura Musazzi, University of Milano Bicocca, ItalyReviewed by:
Bodo Laube, Darmstadt University of Technology, GermanyCopyright © 2023 Dong, Yue, Dong and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuehui Wang, eXVlaHVpd2FuZzMwMEBqbHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.