 Oualid Sbai
Oualid Sbai Veronica Bazzani
Veronica Bazzani Shreya Tapaswi3
Shreya Tapaswi3 Joshua McHale
Joshua McHale Carlo Vascotto
Carlo Vascotto Lorena Perrone
Lorena Perrone- 1Institut Pasteur de Tunis, LR11IPT02, Laboratory of Transmission, Control and Immunobiology of Infections (LTCII), Tunis, Tunisia
- 2Department of Medicine, University of Udine, Udine, Italy
- 3IMol Polish Academy of Sciences, Warsaw, Poland
- 4Department of Advanced Medical and Surgical Sciences, University of Campania Luigi Vanvitelli, Naples, Italy
Recent advances highlight that inflammation is critical to Alzheimer Disease (AD) pathogenesis. Indeed, several diseases characterized by inflammation are considered risk factors for AD, such as type 2 diabetes, obesity, hypertension, and traumatic brain injury. Moreover, allelic variations in genes involved in the inflammatory cascade are risk factors for AD. AD is also characterized by mitochondrial dysfunction, which affects the energy homeostasis of the brain. The role of mitochondrial dysfunction has been characterized mostly in neuronal cells. However, recent data are demonstrating that mitochondrial dysfunction occurs also in inflammatory cells, promoting inflammation and the secretion of pro-inflammatory cytokines, which in turn induce neurodegeneration. In this review, we summarize the recent finding supporting the hypothesis of the inflammatory-amyloid cascade in AD. Moreover, we describe the recent data that demonstrate the link between altered mitochondrial dysfunction and the inflammatory cascade. We focus in summarizing the role of Drp1, which is involved in mitochondrial fission, showing that altered Drp1 activation affects the mitochondrial homeostasis and leads to the activation of the NLRP3 inflammasome, promoting the inflammatory cascade, which in turn aggravates Amyloid beta (Ab) deposition and tau-induced neurodegeneration, showing the relevance of this pro-inflammatory pathway as an early event in AD.
1. Introduction
Alzheimer’s disease (AD) is the most common type of dementia, with prevalence rates of 11% in those of 65 years and older (Hebert et al., 2013) and 68% in memory disorder clinics (Paulino Ramirez Diaz et al., 2005). With the progression of the disease, macroscopic atrophy affects the entorhinal area and hippocampus, amygdala, and associative regions of the neocortex. AD is characterized by white matter loss and myelin degeneration due to death of oligodendrocytes occurring in the early phase of AD. Hallmark signs of AD are the formation of amyloid plaques and neurofibrillary tangles (NFT) in the hippocampal and entorhinal regions (Perrone et al., 2012). Amyloid plaques are constituted by accumulation of the beta amyloid peptides (Aβ), which aggregate both intracellularly and extracellularly and is produced by the processing of the amyloid precursor protein (APP; Hardy and Selkoe, 2002). NFT are formed in neurons by intracellular aggregation of hyperphosphorylated tau. About 90–95% of AD cases are sporadic and only 5–10% are familiar, showing mutations in APP gene or the genes encoding the proteins involved in APP cleavage and Aβ production (presenilin 1-PSEN1-and presenilin 2 PSEN2). The presence of mutations in APP or PSEN1/2 in familiar AD initially supported the hypothesis of the “Amyloid cascade” as a central player in AD onset and progression, indicating the over-production of Aβ as the causative event responsible for the pathophysiological process leading to AD progression. However, recent studies underline that Aβ deposition and NFT are not sufficient to clarify AD onset and progression, opening the way to the amyloid-inflammatory cascade (Wang and Mandelkow, 2016; Long and Holtzman, 2019). In agreement, aging and several diseases that are risks for AD, such as type 2 diabetes, obesity, hypertension, and metabolic syndrome, are characterized by chronic inflammation (Matrone et al., 2015). It has been hypothesized a central role of microglia activation as initial step initiating the pathological cascade leading to AD. Indeed, several AD risk genes play a central role in the innate immunity (Scheltens et al., 2021). Microglia are central for the brain homeostasis, ensuring effective synapse pruning and plasticity, as well as supporting myelin stability. Several microglia phenotypes have been described, showing different morphology, molecular and metabolic characteristics that correspond to the different functions of the microglia. Recent data underline that dysfunctional microglia affect the synaptic plasticity and alter the cognitive function, contributing to AD pathophysiology. Interestingly, mitochondrial fission promotes inflammatory activation, showing a link between mitochondrial dynamics and microglia activation (Lawrence et al., 2022). NOD-like receptor family, pyrin domain containing 3 (NLRP3) plays a central role in inflammation, by producing pro-inflammatory cytokines. On the other hands, Dynamin Related Protein 1 (Drp1) is essential for the homeostasis of mitochondria dynamics and is implicated in mitochondria fission. We will summarize below the link between Drp1 and NLRP3 in promoting inflammation and participating in AD pathophysiology.
2. AD and inflammation
In the last decades, increasing evidence has demonstrated that a sustained immune response can be classified as an essential factor involved in AD pathophysiology as well as Aβ aggregation and tau hyperphosphorylation (Matrone et al., 2015; Kinney et al., 2018). In normal conditions, acute inflammation is a response counteracting injury to the brain. This well-established mechanism serves as protection and may be activated upon different stimuli. However, the disruption of the equilibrium between pro-inflammatory and anti-inflammatory signaling can results in chronic neuroinflammation, where the sustained immune response becomes a central feature of neurodegenerative disorders (Matrone et al., 2015). Recent studies underline that inflammation plays a central role in AD pathophysiology and is implicated in the development of the pathological changes (Aβ aggregation and NFT) observed in AD. In agreement, various inflammatory molecules have been proposed as AD biomarkers (Novoa et al., 2022). In addition, diseases characterized by systemic inflammation, such as obesity, type 2 diabetes, and cerebrovascular diseases, are considered risk factors for AD (Novoa et al., 2022). In neurodegenerative diseases, inflammation can be produced in two ways. In one mechanism, peripheral inflammation produces cytokines, which alters and cross the Blood Brain Barrier (BBB), further inducing the release of pro-inflammatory factors by the brain endothelial cells and by glial cells associated to the BBB. This process enhances the BBB permeability to peripheral immune cells, leading to the entry of leucocytes into the brain (Varatharaj and Galea, 2017). These events promote the activation of astrocytes and microglia, leading to further production of cytokines into the brain, ultimately promoting neuronal dysfunction. This mechanism is defined as neuroinflammation (Carson et al., 2006). In a second mechanism, the innate immune system of the brain (such as the microglia) promotes a cascade leading to neuroinflammation. This mechanism can be induced by neuronal lesions or aggregated proteins, such as Aβ, which activates the astro-glial cells, leading to cytokine production and ultimately promoting synaptic dysfunction and neurodegeneration (Lull and Block, 2010).
As with most of the other mechanisms investigated in AD, it is not yet definitively understood whether inflammation is the cause, contribution, or secondary phenomenon of this disorder. In the next paragraphs, we will delineate a comprehensive report of the studies performed to elucidate the role of inflammation and the new frontiers aimed to target inflammation in AD.
2.1. Role of inflammation in AD: clinical data
The majority age related diseases, such as diabetes, and obesity-that are risk factors for AD-and AD are characterized by chronic inflammation (Chung et al., 2009). In the 1980s, it has been demonstrated the presence of inflammatory proteins and immune-related cells in the proximity of Aβ plaques (Rogers et al., 1988; Griffin et al., 1989). Since the 1990s, researchers have demonstrated a significant presence of sustained inflammation in patients with AD (Aisen and Davis, 1994), confirmed by post-mortem tissues analysis (Gomez-Nicola and Boche, 2015). Epidemiological studies of large-scale cohorts have shown that people showing enhanced pro-inflammatory proteins in the blood in mid-life are at higher risk of cognitive decline over the decades compared to subjects maintaining a low presence of pro-inflammatory factors in the blood (Leung et al., 2013; Huang et al., 2022). In addition, in their later life, these individuals are characterized by lower volume of brain with abnormal microstructure of white matter, increased myelin loss and the inability of the oligodendrocytes, the cells responsible for the production and maintenance of myelin, to repair myelin damages (Nasrabady et al., 2018; Collij et al., 2021). These observations were among the first to support the idea that systemic inflammation occurs one or two decades before the appearance of dementia symptoms, suggesting its active role in promoting the progression of cognitive decline and neurodegeneration. In parallel, genome-wide association studies (GWAS) reported that more than 60% of the genes linked to late-onset sporadic AD are inflammation-related (Efthymiou and Goate, 2017; Misra et al., 2018; Carpanini et al., 2021). In this scenario, preliminary studies aimed to evaluate the potential beneficial effect of long-term administration of anti-inflammatory drugs. Indeed, people who regularly took anti-inflammatory drugs over a long period of time showed reduced risk of developing AD later in life. Unfortunately, the search for anti-inflammatory drugs effective in preventing AD was less straight forward than it could seem looking at the plethora of data supporting the pivotal role of inflammation. Indeed, the effect of anti-inflammatory drugs remains under debate. Chronic inflammation exacerbates amyloid β deposition and tau hyperphosphorylation and participate to the pathogenesis of AD (Matrone et al., 2015). Interestingly, chronic administration of non-steroidal anti-inflammatory drugs (NSAIDs) appears to be beneficial only in the very early stages of the AD process along with initial Aβ deposition, early microglia activation and subsequent release of pro-inflammatory mediators (Imbimbo et al., 2010). On the other side, over time the beneficial effect of NSAIDs is no more significant, especially when older groups of patients are studied. In 2009, Breitner and colleagues suggested that the differences seen in these observational studies may be due to the fact that NSAIDs may not simply reduce the risk of AD, but delay AD onset in later ages (Breitner et al., 2009), suggesting that NSAIDs may be beneficial in delaying AD only when administered in the young age. Since AD is more common in old subjects and occurs as a chronic effect after decades from the initial pathological alteration, NSAID is beneficial in preventing AD when administered to young subject, while NSAID is less effective when administered in the old age, explaining the variance observed among different patient groups. Other factors may also cause the inconsistencies reported in several publications. The selection of the most effective NSAIDs for a study and the presence of other factors as concomitant pathologies to AD can affect the epidemiological results, producing discrepancies among different studies. To overcome these limits, in recent years, more homogeneous approaches have been taken in clinical trials to clarify the effects of NSAIDs without confounding variables. Jack Rivers-Auty and colleagues used logistic regression and an innovative approach of negative binomial generalized linear mixed modeling to investigate both prevalence and cognitive decline in the AD Neuroimaging dataset for commonly used NSAIDs and paracetamol (Rivers-Auty et al., 2020). They demonstrated that most NSAIDs can reduce the prevalence of AD, but not cognitive decline. Interestingly, paracetamol also had a similar effect, which lead the authors to hypothesize that the prevalence of AD is independent of inflammation. Finally, they also analyzed the use of diclofenac (a non-steroidal anti-inflammatory drug), finding a significant association between diclofenac intake and the reduction in AD incidence and similarly to slower cognitive decline, suggesting a possible therapeutic effect of this compound in AD. Recent genetic data strongly support the key role of inflammation and immune-related genes in AD pathogenesis by demonstrating that a mutation in the Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) confers a very high likelihood to develop AD (Basha et al., 2023). The R47H allelic variant of TREM2 confers a 2–4,5-fold increased risk of developing AD (Basha et al., 2023). TREM2 variants are the second genetic risk factor for AD, behind apolipoprotein E4 (ApoE4), demonstrating the key role of the innate immunity in AD pathogenesis (Basha et al., 2023). As we have seen, clinical studies present limits that are difficult to overcome: they require large cohort of individuals, long periods of observations, they can be affected by the presence of co-morbidities, environmental factors, and habits that can alter the outcome of a study. Moreover, it is almost impossible to standardize the protocols adopted among different studies in order to compare their results. Finally, the investigations of the molecular mechanisms of inflammation involved in the pathogenesis of AD is pivotal to develop drugs blocking the chronic mechanism that support AD development. For these reasons, pre-clinical studies are crucial and mouse transgenic models (expressing mutant amyloid precursor protein and presenilin mutants, resulting in increased Aβ deposition, or human tau mutant leading to tau hyperphosphorylation) have helped the identification of the focal pathways dysregulated in neuroinflammatory diseases, as described in the next paragraph.
2.2. Role of inflammation in AD: pre-clinical data
Initially the observed presence of inflammation in AD patients was considered the consequence of neuronal damage, which in turn activate the immune system promoting the inflammatory response. However, recent studies demonstrate that chronic inflammation in AD enhances both Aβ- and NFT-induced pathology. Notably, recent studies underline that the amyloid cascade hypothesis is not sufficient to explain the development of NFT and suggest that inflammation may represent the link between the initial Aβ-induced dysfunction and the subsequent development of NFT. In agreement, recent investigations demonstrate that inflammation exacerbates both Aβ- and NFT-induced pathology leading to AD (Kinney et al., 2018).
Studies carried out in different animal models are essential to clarify the role of inflammation in AD. The role of pro-inflammatory factors in promoting neurodegeneration has been summarized by Chen and colleagues (Chen et al., 2016). Some researchers investigated the effect of anti-inflammatory compounds in AD. NSAIDs treatment in AD mice models ameliorates AD pathophysiology (McGeer and McGeer, 2007). Studies have also been carried out in not transgenic models of AD, further supporting the role of ani-inflammatory compounds in ameliorating AD. Indeed, Lindsay and colleagues demonstrate that andrographolide—a natural compound-ameliorates not only inflammation but also oxidative stress in Octodon degus, which is a rodent that develops AD spontaneously (Lindsay et al., 2020). Moreover, the effect of an anti-inflammatory compound (GsRb1) has been analyzed in a rat model of AD, where Aβ was injected intraventricularly. GsRb1 treatment ameliorates the inflammatory reaction and restored the learning capability in this AD model, further supporting the role of inflammation in AD pathology (Novoa et al., 2022). To further elucidate the pathological role of neuroinflammation in AD, different strategies using immune challenge-based models and neurotoxin-induced AD models have also been employed (Nazem et al., 2015). According to the endotoxin hypothesis, the endotoxin molecules cause or contribute to the neurodegenerative process (Brown, 2019). LPS-induced rodent models are used for studying neuroinflammation and inflammation-induced amyloidosis. These rodents develop memory impairment by affecting the consolidation of memory process. Acute treatment with LPS before training blocks contextual-cue fear conditioning, which is a hippocampal-dependent learning paradigm (Zakaria et al., 2017). These data further support the role of inflammation in promoting neuronal dysfunction (Brown, 2019). Since LPS treatment in rodents induces both amyloidosis and cognitive dysfunction, it has been proposed as AD model (Zakaria et al., 2017). However, when an LPS-induced memory impairment AD model is designed, the following factors must be taken into consideration: route of administration (mainly intraperitoneal and intracerebroventricular), duration of exposure, age, and sex of the animal. LPS administration causes many behavioral effects, namely, fever, hypersomnia, activation of hypothalamus-pituitary–adrenal (HPA) axis-causing sympathetic activation, reduction in exploration, social interaction, consumption, and activity. According to studies based on LPS injection, researchers have showed that peripheral inflammation induces neuroendocrine alterations, astrocyte and microglia activation, as well as cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS) and pro-inflammatory cytokine expression in the brain (Brown, 2019). Interestingly, gut microbiome derived LPS accumulates in AD affected brain, further aggravating the pro-inflammatory environment of the brain (Zhao et al., 2021). In addition, intracellular accumulation of APP, Aβ peptide, and hyperphosphorylated tau as well as exacerbation of memory deficits were observed in LPS-treated APP transgenic mice (Sheng et al., 2003; Lee et al., 2008). However, this approach has limitations, as seen when evaluating the number of injections and the route of administrations used (directly into the central nervous system vs. systemic), which can lead to different pathological effects and contradictory results (Valero et al., 2014). The data obtained by injecting LPS in wild type mice and rats are important to demonstrate the role of inflammation in promoting neuronal dysfunction and an AD-like phenotype. More detailed analysis of the molecular pathways involved have been carried out in transgenic AD mice models, unveiling specific inflammatory pathways that are central in AD pathophysiology.
The App NL-G-F knock in (KI) model carries a combination of Swedish, Arctic, and Iberian APP mutations. This KI model more closely represents human amyloidogenic pathways than other APP models. Another KI model used to unveil the early impact of neuroinflammation in AD is the App NL-F. Both KI models show an Aβ plaque composition comparable to that of AD patients. Interestingly, in these model microglia is associated with diffuse plaques and mushroom spine loss, underlying the crucial function of microglia-mediated synapse loss and supporting that the expression of neuroinflammation-related genes is an AD risk factor (Saito et al., 2014).
The role of TREM2 in AD pathophysiology is well characterized in the APP/PS1 and 5xFAD AD mice models. Both heterozygous and homozygous Knock out (KO) of TREM2 in these AD models strongly reduce the presence of macrophages associated to Aβ plaques, reducing Aβ plaques load in 4 months old AD mice, while Aβ load is increased in 8 months old AD mice lacking TREM2 (Basha et al., 2023). TREM2 KO in AD mice leads also to reduced production of pro-inflammatory cytokines and ameliorates astrocytosis, as observed by decreased expression of glial fibrillary acidic protein (GFAP; Basha et al., 2023). However, microglia and macrophages lacking TREM2 show a decreased capability to phagocytose Aβ and apoptotic cells and TREM KO in AD mice finally lead to enhanced neurodegeneration (Kinney et al., 2018). TREM2 is also involved in tau-mediated pathology. Indeed, the KO of TREM2 in mice expressing human tau aggravates tau pathology (Kinney et al., 2018). Studies in AD and tau mice models suggest that TREM2 plays a dual role in Aβ and tau pathology, appearing to being beneficial in the initial phase of the disease by contributing in altered protein phagocytosis, whereas it exerts a pathological role in the later phase of the disease by enhancing inflammation and neurodegeneration (Kinney et al., 2018). This dual role of TREM2 is currently explained by three distinct mechanisms related to inflammation that are relevant in AD: (i) the function of phagocytosis of damaged and misfolded proteins; (ii) the survival and proliferation of cells involved in the inflammatory response; (iii) the regulation of the whole inflammatory process (Kinney et al., 2018).
Further supporting the role of inflammation in AD, several studies carried out in AD mice models reveal the relevance of the Receptor for Advanced Glycation Endproducts (RAGE) in AD pathophysiology (Perrone et al., 2012). RAGE is a multi-ligand receptor whose activity is also triggered by Aβ. Silencing of RAGE specifically in microglia ameliorates neuronal dysfunction in an AD contest (Origlia et al., 2010). Interestingly, High Mobility Group Box 1 (HMGB1)—another ligand of RAGE-seems to be involved in AD progression (Mo et al., 2023). HMGB1 is a ubiquitous non-histone DNA binding protein, which may exert various functions depending on its subcellular localization. Inside the nucleus, HMGB1 acts as a structural chromatin protein, regulating DNA repair and gene expression (Mo et al., 2023). In the cytoplasm, HMGB1 modulates autophagy and is implicated in the removal of damaged mitochondria (Mo et al., 2023). HMGB1 can also be secreted by inflammatory cells or released by necrotic cells. Extracellular HMGB1 is considered as an alarm protein or a damage associated molecular pattern (DAMP) protein and induces inflammation through interaction with various receptors, such as RAGE, Toll-like receptor 4 (TLR 4), CD24, and CXCR4 (Mo et al., 2023). Inhibition of HMGB1 is beneficial in AD mice (Paudel et al., 2020), further supporting the key role of inflammation in AD onset and progression.
3. Cell types promoting inflammation in AD
3.1. Microglia
Microglia are known resident immune cells within the central nervous system (CNS) and are among the main responsible for the surveillance of the surrounding neurons health. Among the various microglia phenotypes described, three of them are more relevant for the understanding of AD pathophysiology: the steady state, the activated and the primed phenotype (Bivona et al., 2023). Steady state microglia exert a neuroprotective function and present a basic and low level of cytokine production, with an anti-inflammatory function. Activated microglia show a protective function against injuries, enhances the production of cytokines, both pro-inflammatory (IL-1β, IL-18, and TNF) and anti-inflammatory cytokines in order to counteract an excessive inflammation and inhibit a subsequent neuronal damage. Activated microglia can revert its phenotype to steady state, when the activating stimulation is removed. Primed microglia show a more aggressive and pro-inflammatory activity and maintain the memory of the specific stimulation with a Toll-like receptor 4 (TLR4)-mediated mechanism. Primed microglia cannot revert his phenotype and turn again on the steady state phenotype (Bivona et al., 2023).
Microglial activation is highly regulated. In normal conditions, the microglia are maintained inactive by healthy neurons through the continuous release of inhibitors such as the chemokine CX3CL1, whose receptor CX3CR1 is expressed uniquely on microglia surface (Arnoux and Audinat, 2015). In addition, the CD200 protein expressed on the surface of neurons, astrocytes and oligodendrocytes interacts with its receptor CD200R, which is expressed only by macrophages and microglia. The interaction between CD200 and CD200R induces microglia inactivation and maintains microglia in a resting state (Biber et al., 2007). Microglia in AD are initially activated by Aβ formation, through the ability of the pattern recognition receptors (PRRs) to recognize misfolded and aggregated proteins with a consequent trigger of the innate immune response (Tu et al., 2015), leading to migration of the microglia close to the plaques and subsequent phagocytosis of Aβ. When the pro-inflammatory stimulus is chronic, microglia efficacy to bind and phagocyte Aβ decreases and the overall clearance becomes compromised while the immune activation continues (Hickman et al., 2008). The accumulation of Aβ together with chronic release of pro-inflammatory cytokines drives neuronal damage by reducing trophic factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor (IGF; Sheng et al., 1998; Hickman et al., 2008).
The colony stimulating factor 1 (Csf 1) and IL-34 are also important for microglia proliferation during the neurodegenerative process occurring in AD (Gómez-Nicola et al., 2013).
The mechanisms involved in microglia activation induced by Tau are not yet fully elucidated. Recent data show that microglia phagocytose of aggregated Tau, which is targeted to lysosomes, leading to NLRP3 inflammasome activation (Stancu et al., 2019). Moreover, tau interacts with polyglutamine binding protein 1 (PQBP1), which in turn induces the cGAS-STING pathway, leading to microglia activation (Jin et al., 2021).
The microglial myeloid differentiation primary response 88 (MyD88) and the p38 mitogen activated protein kinase (MAPK) signaling pathways are also involved and drive the release of neurotoxins, in a process initiated by the pro-inflammatory cytokine TNF, contributing to the damage of neurons (Michaud et al., 2011; Wang et al., 2015; Schnöder et al., 2016). Moreover, in these conditions, microglia are characterized by a phenotypic change: the retraction of their processes that correlates with an impaired ability to remodel synapses, a phenomenon that contributes to impaired synaptic plasticity observed in AD (Nimmerjahn et al., 2005). All these events contribute to the microgliosis that leads to neurodegeneration (Zhang J. et al., 2021).
Microglia activation leads to the secretion of IL-1β and IL-18, which plays a role in AD progression, as we will describe in more detail in the paragraphs below.
3.2. Astrocytes
Astrocytes provide trophic support to neurons and form a protective barrier that isolates neurons from amyloid deposits. Astrocytes are not only essential for the maintenance of neuronal health, but also are an important component implicated in synaptic transmission, they modulate brain energetics and cerebrovascular function (Delekate et al., 2014). In the healthy brain, astrocytes are assembled into dynamic networks. Connexins control this network and their expression is reshaped in AD, leading to a perturbation of the astrocytic network as occurs when astrocytes are activated leading to reactive astrogliosis (Delekate et al., 2014). Besides microglia, astrocytes contribute to the clearance of Aβ but this role is affected in the presence of chronic stress and inflammation. Accumulation of astrocytes, indeed, has been detected in proximity of the Aβ deposits in AD patients (Kuchibhotla et al., 2009). Recently, it has been shown that the functional connectivity of astrocytes is altered early in AD (Shah et al., 2022). Indeed, it is observed a decreased calcium signaling in astrocytes of AD mice before the appearance of Aβ plaques (Shah et al., 2022). Moreover, disruption of astrocyte network in AD affects the cortical neuronal activity, promoting cognitive decline (Lines et al., 2022). The prolonged response of astrocytes to Aβ accumulation promotes neuroinflammation, contributing to nitric oxide (NO) mediated neurotoxicity. Activated astrocytes can participate to the formation of Aβ, as suggested by Rossner et al. in a study on the overexpression of β-secretase (BACE1) in astrocytes affected by chronic stress (Rossner et al., 2005). Moreover, the end-feet of these cells form a lacework of fine lamellae closely linked to the outer surface of the BBB endothelium and basement membrane. Inflammation induces astrocytes proliferation and activation, followed by astrocytes loss and changes in the end-feet structures, affecting the integrity of the BBB (Escartin et al., 2021). Finally, the ability of astrocytes to produce pro-inflammatory prostaglandins and cytokines, such as IL-1β and IL-18, in large quantities during prolonged inflammation negatively alter the BBB protective function, as better reported in the next paragraph (Sofroniew, 2015).
3.3. Blood brain barrier
The blood brain barrier (BBB) is one of the main brain barriers that impedes free diffusion between brain and blood and regulates the transport of essential nutrients, ions, and metabolic waste products. It is formed by endothelial cells which are tightly linked by tight junctions (TJs) and it is surrounded by pericytes and astrocytes, which provide support and regulatory functions.
Systemic inflammation has disruptive effects on the BBB integrity, leading to the diffusion of peripheral inflammatory factors into the brain. These factors induce alterations into the brain that in turn may participate to neuronal dysfunction and cognitive decline in AD patients. Among the known factors, the most important are prostanoids and NO. The mediators involved in their release, the Matrix Metalloproteinases (MMPs) and the Reactive Oxygen Species (ROS), activate pathways such as the MAPK, and induce mitochondrial dysfunctions with a subsequent destruction of the BBB integrity (Ralay Ranaivo et al., 2012; Takata et al., 2021). Some miRNAs have a role in the BBB structure and function integrity maintenance. The expression of miR-155 in microvessels is strongly and rapidly upregulated by inflammatory cytokines and alters BBB function by affecting expression of TJs and adhesion components (Lopez-Ramirez et al., 2014). Next to direct morphological changes in the BBB, systemic inflammation can also cause non-disruptive changes that affect BBB functionality (Perrone et al., 2012). Chronic inflammation downregulates the multi-functional efflux transporter permeability glycoprotein (P-gp) and upregulates the expression of the influx carriers responsible for TNF translocation (Roberts, 2008). Systemic inflammation is also responsible for the reduced bulk flow of cerebrospinal and interstitial fluids, resulting in impaired Aβ clearance (Tarasoff-Conway et al., 2015; Mogensen et al., 2021). In AD, the elevated production of TNFα and IL-1β by microglia and astrocytes promotes BBB dysfunction by affecting the TJs, leading to BBB hyperpermeability. In addition, TNFα and IL-1β reduce the expression of the low-density lipoprotein receptor-related 1 (LRP1), which drives the efflux of Aβ from the brain to the blood, resulting in a reduced Aβ clearance (Versele et al., 2022).
4. Role of the inflammasome in AD
4.1. The NLRP3 complex
We underlined above the crucial role of cytokines and chemokines in the inflammation process. However, the molecular pathways involved are not yet fully characterized. Although it is well documented that IL-1β, IL-6, TNF-α, IL-8, and TGF-β and macrophage inflammatory protein-1a (MIP-1a) are upregulated in AD patients (Domingues et al., 2017), it is not yet fully clarified their role in the onset or progression of AD. Pro-inflammatory cytokines can promote Aβ formation by upregulating BACE1 and APP levels. Further studies are necessary to elucidate the mechanisms induced by pro-inflammatory molecules and their effect in AD progression. Recently, several publications described the role of nucleotide-binding domain, leucine-rich-repeat containing family, pyrin domain-containing 3 (NLRP3) inflammasome in AD (Milner et al., 2021; Barczuk et al., 2022; Sharma et al., 2022). The NLRP3 inflammasome is a multiprotein complex, activated upon infection or stress. The inflammasome is formed by NLRP3 protein and the apoptosis associated speck-like protein containing a caspase recruitment domain (ASC). This macromolecular complex responds to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs; Kelley et al., 2019).
NOD-like receptor family, pyrin domain containing 3 contains an N-terminal pyrin domain (PYD), which clusters upon stimulation, allowing the interaction with ASC and the procaspase-1. Procaspase-1 clustering drives its autocleavage and the formation of the active caspase-1, which induces the cleavage of inactive cytokines pro-IL-1β and pro-IL-18 into their active forms, IL-1β and IL-18, respectively. Notably, enhanced levels of IL-1β induces the production of other cytokines, including IL-6, which in turn promotes the activation of the kinase CDK5 that hyper-phosphorylates tau, further promoting AD progression (Rauf et al., 2022). Furthermore, chronic production of IL-1β enhances brain acetylcholinesterase activity and microglia activation, producing a vitious circle of dysfunction that promotes AD progression by reducing the acetylcholine function and promoting inflammation (Rauf et al., 2022). Chronic IL-1β secretion leads also to astrocytes activation and subsequent production of the pro-inflammatory S100β, further exacerbating neuroinflammation (Rauf et al., 2022). In addition to caspase-1-dependent cytokine production, caspase-1 activates Gasdermin D (GSDMD) by cleaving it, producing a fragment that oligomerizes generating a pore-forming complex that translocates to the plasma membrane leading to release of pro-inflammatory cytokines and pyroptosis, a lytic form of cell death (Yang et al., 2019). Inflammasome activation exacerbates amyloidogenesis, as shown by the study of Venegas et al. (2017), which shows that ASC specks can cross seed Aβ in AD and consequently perpetuate the vicious cycle of brain inflammageing. Intriguingly, NLRP3 inflammasome activation is associated with mitochondrial function, as described in the next paragraph.
4.2. Mitochondria alterations in AD
Mitochondria are dynamic double-membrane organelle which undertake several roles from energy production to apoptosis regulation, Ca2+ signaling, lipid, and amino acid synthesis. Mitochondrial ATP, produced by aerobic oxidative phosphorylation (OXPHOS), plays an important role for the cell functions. Indeed, enzymatic complexes, present in the inner membrane of the mitochondria, synthesize ATP from ADP and phosphate. However, OXPHOS is a major source of endogenous toxic free radicals, including hydrogen peroxide (H2O2), hydroxyl (HO), and superoxide (O2−) radicals that are products of normal cellular respiration (Moreira et al., 2010). Data demonstrate an impairment of the mitochondrial enzymatic complex in AD brain (Castellani et al., 2002). Neurons in AD show striking and significant increase of mitochondrial DNA (mtDNA) localized in the cytoplasm, demonstrating mitochondrial damage (Castellani et al., 2002). ROS overproduction in AD impairs mitochondrial function and reduces the supply of the ATP. Several research demonstrate that oxidative damage occurs before Aβ plaque formation (Nunomura et al., 2001), suggesting that mitochondrial dysfunction plays an early role in AD pathophysiology. Mitochondrial abnormalities appear to be key features during the maturation of AD-like pathology in YAC and C57B6/SJL transgenic mice. In the absence of energy source, abnormal mitochondria produce an excess of free radicals, which can reduce the supply of ATP and contribute to mitochondrial dysfunction in AD (Reed et al., 2008). AD mice show APP mitochondrial localization in cortical neurons, overexpression of oxidative stress markers, deletions in mtDNA, and impaired energy metabolism (Devi et al., 2006). In particular, mitochondrial APP localization inhibits the translocation of COX subunits IV from the cytoplasm to mitochondria, reducing COX activity and increasing H2O2 levels (Devi et al., 2006). Intra-cerebroventricular injection of Aβ in mice induces H2O2 production in neocortex mitochondria, leading to neurotoxicity (Morais Cardoso et al., 2002). Aβ also affects the mitochondrial membrane potential (Sbai et al., 2022), affects the mitochondrial fission/fusion balance and mitochondrial distribution (Wang et al., 2020).
Neuronal function is also modulated by the Ca2+ concentration and mitochondria are high-capacity Ca2+ sinks that control the cytosolic Ca2+ loads. Aβ in the presence of Ca2+ promotes mitochondria dysfunction by inducing a complete uncoupling of respiration, altering the morphology of mitochondria, decreasing the mitochondrial membrane potential, and affecting the mitochondria capacity to accumulate Ca2+ (Moreira et al., 2001). The excessive Ca2+ uptake into mitochondria enhances ROS production, which inhibits ATP synthesis and increases the mitochondrial permeability transition pore (PTP). Increased PTP levels promote the release from the mitochondria of various proteins, such as cytochrome c and apoptosis-inducing factor (AIF), promoting the initiation of apoptosis by activating the caspase cascade (Hengartner, 2000). Moreover, pro-apoptotic proteins released by mitochondria can translocate into the nucleus, leading to DNA fragmentation, which in turn promote cell death.
4.3. Role of mitochondria in activating NLRP3 inflammasome
Activators of NLRP3 induce both NLRP3 deubiquitination and the destabilization of mitochondria membrane potential, its permeabilization, and permeability transition that results in the externalization and release of mitochondria derived molecules (i.e., mitochondrial DNA, ATP, etc.; Liu et al., 2018). These molecules bind NLRP3 and cause its translocation to mitochondria surface, with consequent inflammasome activation, which occurs in the cytoplasm.
A two-step model has been proposed to describe the activation of the NLRP3 inflammasome: a first signal, in response to proinflammatory receptors, induces NF-κB-dependent expression of both proIL-1β and NLRP3 (transcriptional priming) and NLRP3 deubiquitination (non-transcriptional priming); later, a second mechanism triggers the assembly and activation of the inflammasome (Sutterwala et al., 2014). Exception to this general mechanism has been described: the model is not always applicable, with signal I or II missing depending on the molecule activating the NLRP3 inflammasome assembly (Gaidt et al., 2016).
Reactive Oxygen Species have a crucial role in the NLRP3 activation and assembly. Many pathogens and endogenous signals promote ROS generation (Martinon, 2010). Several mitochondrial processes lead to ROS production, which can activate signaling cascades involved in proliferation, apoptosis, and senescence. Antioxidant enzymes such as superoxide dismutase (SOD) and glutathione peroxidase control ROS homeostasis, protecting the cells from the dangerous effects caused by an imbalance in ROS production. When the anti-oxidant systems fail in maintaining the ROS homeostasis, the cells are subjected to oxidative stress and cytotoxicity. Oxidative stress, indeed, activates inflammatory pathways and promotes the formation of pro-mutagenic DNA adducts, creating genetic instability that participate in the progression of various diseases (Sosa et al., 2013; Huang et al., 2016). Considering that NLRP3 and ASC localize within mitochondria upon NLRP3 activators stimulation, studies suggested that NLRP3 activators could induce mitochondrial ROS generation which in turns activate the NLRP3 inflammasome (Zhong et al., 2013; Pang et al., 2021). Studies investigating the effect of ROS scavengers and ROS inhibitors support this hypothesis (Bauernfeind et al., 2011; Alfonso-Loeches et al., 2014; Dai et al., 2017; Liu et al., 2017). Nevertheless, ROS seems to function in the priming step, but not in the activation one (Bauernfeind et al., 2011). Further studies are needed to elucidate the link between ROS and NLRP3 activation (Abais et al., 2015).
4.4. Role of NLRP3 in AD
Recent studies demonstrate that NLRP3 inflammasome activation contributes to the pathogenesis of chronic inflammatory or metabolic disorders.
In the AD context, Aβ has been described as priming stimulus to NLRP3 transcription (Nakanishi et al., 2018), however, the molecular mechanisms involved are not yet fully elucidated. Aβ phagocytosis by microglia represents a key step for lysosomal destabilization and consequent release of lysosome content. The assembly of the inflammasome is later promoted by Cathepsin B, a lysosomal proteolytic enzyme, by a still unknown mechanism (Campden and Zhang, 2019). Moreover, Aβ induction of NLRP3 activation leads to GSDMD cleavage and production of a Gasdermin D (GSMD) fragment that oligomerizes at the plasma membrane, resulting in pyroptosis (Han et al., 2020). Another fascinating hypothesis suggests that a negative regulator of NLRP3, NLRP10 (Eisenbarth et al., 2012), reduces NLRP3 assembly. NLRP10 is able to bind ASC, decreasing its availability to interact with NLRP3, reducing the assembly of the inflammasome. In stressful conditions, NLRP10 is degraded by the cathepsin released upon Aβ phagocytosis, leaving ASC free to bind NLRP3 and form a functional inflammasome (Murphy et al., 2014).
Moreover, the release of ATP from dying neurons modulates the activation of NLRP3 inflammasome by activating the P2X7 receptors on microglia, causing K+ efflux, ROS generation, and inducing the recruitment of pore-forming pannexin-1. The formation of this channel by pannexin-1 promotes the entry of extracellular molecules, altering the homeostasis of NLRP3 activation (Gombault et al., 2012).
Although several studies investigate the relationship between Aβ and NLRP3, there are only few reports analyzing the link between tau and NLRP3 inflammasome. In 3xTg AD mice model, the inhibition of IL-1β receptor by injection of blocking antibodies results in reduced tau kinase activity and ameliorates the cognitive impairment, implying that NLRP3 inflammasome activation exacerbates tau phosphorylation and subsequent neuronal damage (Kitazawa et al., 2011). Ising and colleagues demonstrate that loss of function of NLRP3 reduces tau hyperphosphorylation and aggregation (Ising et al., 2019). Another study shows that tau activates the NLRP3 inflammasome after being phagocytosed by microglia, further enhancing tau-mediated damage (Stancu et al., 2019). Furthermore, in tau transgenic mice, ASC deletion or NLRP3 inhibition blocks tau-induced pathology (Liang et al., 2022). These data indicate that activation of NLRP3 exacerbate tau pathology through a positive feedback loop, which contributes to AD pathology.
While the role of NLRP3 in microglia is well characterized, only recently researchers focused on NLRP3 role in astrocyte. Studies carried out analyzing astrocytic cell lines stimulated with LPS indicate that NLRP3, ASC, and IL-1β are not expressed in astrocytes (Gustin et al., 2015). This observation is supported by data obtained by employing neurosphere-derived astrocytes (Tarassishin et al., 2014). On the other side, Tg2576 transgenic mice, which develops Aβ plaques, show a strong expression of IL-1β in the reactive astrocytes surrounding the Aβ deposits (Apelt and Schliebs, 2001). In agreement, other studies carried out in animal models presenting lesions of the central nervous system demonstrate that astrocytes express NLRP3, ASC, and caspase-1 (Kawana et al., 2013). The mechanism of activation of NLRP3 in astrocytes is still controversial. Recent investigations provide data indicating that GSMD is cleaved by a noncanonical caspase 4 pathway in astrocytes in AD (Moonen et al., 2023). The NLRP3 inflammasome is active also in the BBB, leading to BBB dysfunction (Liu et al., 2022). Knock out of NLRP3 in AD mice (APP/PS1/NLRP3−/− mice) ameliorates memory deficit as well as decreases Aβ production and deposition. These mice show an increment in anti-inflammatory microglia, promoting the phagocytosis and clearance of Aβ (Heneka et al., 2013). NLRP3 inflammasome inhibitor Mcc950 exerts a beneficial effect in a rat model of AD by ameliorating the synaptic plasticity (Qi et al., 2018). Indeed, in this rat model of AD the long-term potentiation (LTP) is impaired and treatment with Mcc950 ameliorates the LTP impairment (Qi et al., 2018). Enhanced NLRP3 activation promotes a chronic inflammatory state leading to tau hyperphosphorylation, NFT formation, and synaptic dysfunction (Heneka, 2017). Knock out of NLRP3 reduces tau hyperphosphorylation and ameliorates spatial memory impairment in Tau22 mice (Ising et al., 2019). In early phase AD patients, the levels of IL-1β and caspase 1 activity are enhanced, confirming the over activation of NLRP3 as early event in AD (Venegas et al., 2017). Peripheral blood mononuclear cells (PBMCs) derived from AD and amnestic mild cognitive impairment (aMCI) patients shows elevated levels of NLRP3, IL-1β and caspase 1, showing the presence of systemic NLRP3 activation and inflammation in aMCI and AD patients (Rui et al., 2021; Figure 1).
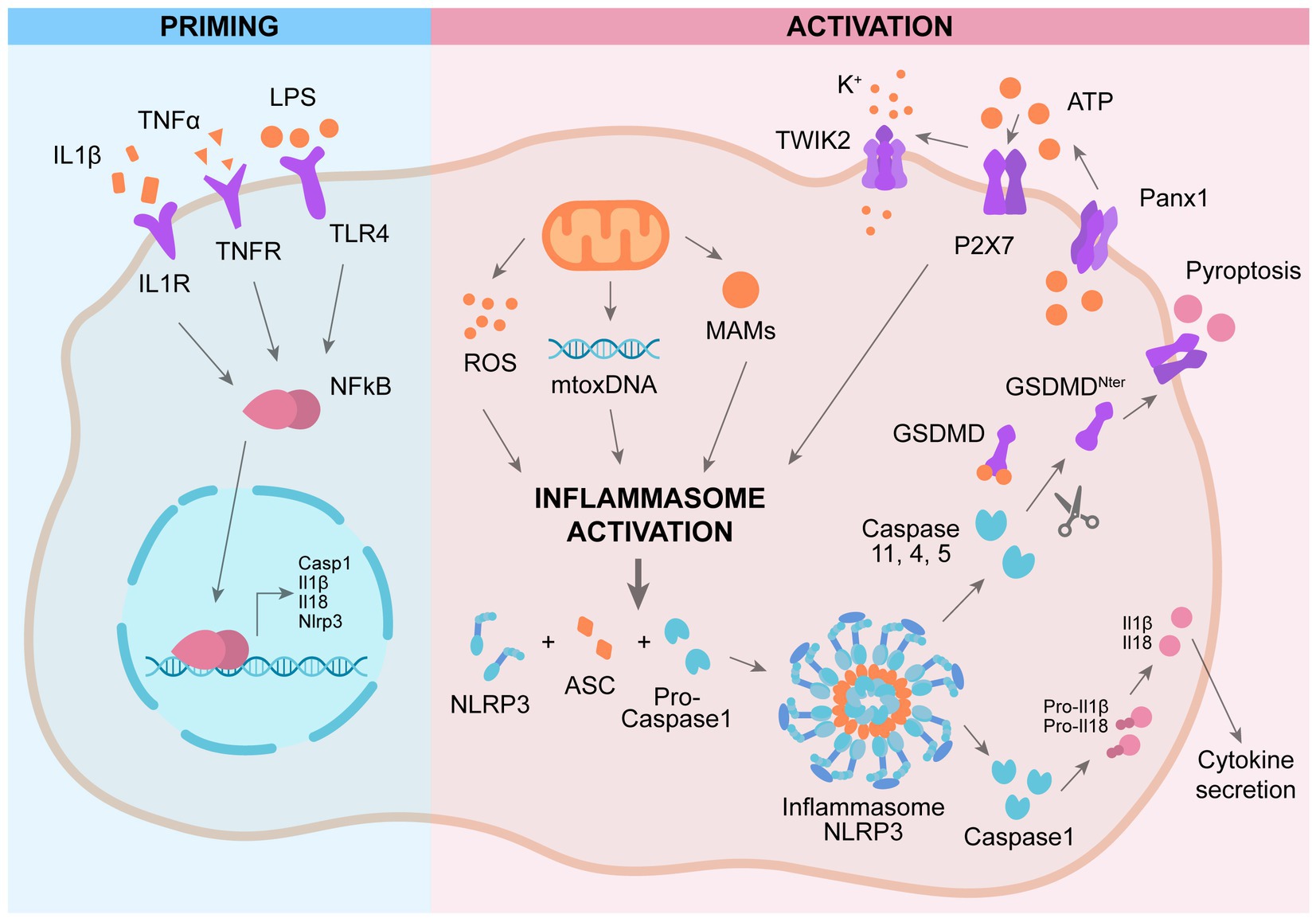
Figure 1. Mechanism of activation of NLRP3 inflammasome. Priming: activation of plasma-membrane receptors induces NFkB activation and subsequent expression of genes involved in NLRP3 cascade. Activation: a second activation of different plasma-membrane receptors promotes the assembly of NLRP3 complex, leading to activation of caspase 1 and subsequent cleavage pf pro-IL1β and GSMD, promoting IL1β secretion and pyroptosis.
4.5. Other inflammasomes and their role in AD
There exist 22 human NLR proteins which can bind various caspases involved in the production of inflammatory cytokines, more frequently caspase-1. NLRs can bind the caspases either directly or via the ASC adaptor, an apoptosis-associated speck-like protein containing CARD and PYD domains. The NLR type of inflammasomes can be subdivided on the basis of their structures and assembly model into the NLRA, NLRB, NLRC (containing, e.g., NOD1, NLRC4, NLRC5, and NLRX), and NLRP (e.g., NLRP1 and NLRP3) subfamilies. The NLRP3 inflammasome is considered a predominant element in the inflammatory process, but the activation of other inflammasomes is also induced by mitochondria dysfunction. Indeed, mitochondrial DNA released in the cytoplasm activates the NLRC4 (Jabir et al., 2015), the NLRP3 and the AIM2 inflammasomes (Korhonen et al., 2021).
Recent findings indicate that NLR family CARD Domain Containing 4 (NLRC4), AIM2 (Interferon-inducible protein AIM2), and NLR Family Pyrin Domain Containing 1 (NLRP1) are also linked to mitochondrial dysfunction and appear to play a key role in neuronal diseases (Patergnani et al., 2021). In a rat model of AD, obtained by intracerebral injection of streptozotocin (STZ), NLRC4 expression is increased as well as IL-1β production, while the expression of NLRP1, NLRP3, and AIM2 is unvaried compared to control rats, suggesting that NLRC4 promotes inflammation in this AD rat model (Saadi et al., 2020). NLRP1 levels are significantly increased in the brain of AD patients and NLRP1 genetic variants are associated with increased risk of AD (Pontillo et al., 2012). NLRP1 is expressed in neurons, where it activates caspase 1, which in turn activates caspase 6, leading to IL-1b production (Kaushal et al., 2015). In the APP/PS1 AD mice model, NLRP1 is upregulated and NLRP1 silencing in this AD model reduces neuronal pyroptosis and cognitive impairment (Tan et al., 2014). An in vitro model of AD is obtained by treating astrocytes with palmitate, which activates a pro-inflammatory response and IL-1β production, which in turn leads to enhanced Aβ production in neurons (Liu and Chan, 2014). In this AD model, palmitate induces the activation of ice protease-activating factor (IPAF)-which interact with ASC, leading to IL-1β production. Silencing of IPAF in astrocytes decreases IL-1β secretion and diminishes Aβ production in neurons (Liu and Chan, 2014).
5. Role of Drp1 in AD inflammation
5.1. Structure and function of Drp1
The Dynamin-related protein 1 (Drp1), is a dynamin-like GTPase protein required to modulate the dynamics of fusion and fission of mitochondria, in particular it is required for the fission of mitochondria (Kandimalla and Reddy, 2016). Drp1 shows various subcellular localizations: cytoplasm, mitochondria, Golgi and peroxisomes. It is encoded by the Dnm1 gene, which is located on 12p11.21 chromosome in mice and on 11q23 in human (Oliver and Reddy, 2019). The RNA transcribed from the Dnm1 gene can be subjected to alternative splicing, which give rise to various isoforms of Drp1 (Oliver and Reddy, 2019). The largest isoform (variant 1) is constituted by 736 amino acids and its calculated molecular mass is 81.6 kDa. Exon 15 is spliced out in variant 2 isoform, producing a protein with 710 amino acids; both exons 15 and 16 are spliced out in variant 3, resulting in a protein with 699 amino acids; variant 4 has 725 amino acids; variant 5 is constituted by 710 amino acids and variant 6-which is present only in neurons-is formed by 749 amino acids (Reddy et al., 2011).
Dynamin Related Protein 1 is characterized by the presence of highly conserved domains. It contains four GTPase domains: the N-terminal GTPase domain, the middle domain, a variable or B domain, and the C-terminal GTPase effector domain. Drp1 has two major phosphorylation sites: CDK phosphorylates in S579, a PKA site is located at S600 in Drp1 isoform 3 (Kandimalla and Reddy, 2016; Oliver and Reddy, 2019).
The mitochondrial fusion and fission processes are controlled during cellular activation by several proteins and not yet fully elucidated mechanisms (Sharma et al., 2019). Current research has demonstrated that Drp1, Fis1, Miro, Opa1, Mfn1, Mfn2, Mid49, Mid51, and Mff are associated with mitochondrial dynamics, morphology, distribution, and function (Kandimalla and Reddy, 2016; Oliver and Reddy, 2019). Drp1 is a key regulator of mitochondrial fission. Drp1 activation induces its translocation from cytoplasm to the mitochondria outer membrane, where it interacts with the fission protein 1 (Fis1), promoting a contraction and a split of the mitochondria. Ramonett et al. (2022), based on proteomic interactome analysis, reported that Drp1 interacts with GAIP/RGS19-interacting protein (GIPC) through its atypical C-terminal PDZ-binding motif. Next, GIPC mediates the actin-based retrograde transport of Drp1 toward the perinuclear mitochondria to enhance their fission. GIPC (Ramonett et al., 2022).
Notably, the balance between mitochondrial fusion and fission is crucial to maintaining a physiological dynamic regulation of mitochondrial function. Downregulation of Drp1 promotes fusion. Loss of Drp1 triggers genome instability, cell cycle arrest and initiates the DNA damage response by disrupting the mitochondrial dynamics and distribution (Qian et al., 2012). Drp1 is enriched at neuronal terminals and involved in synapse formation and synaptic sprouting. Drp1 can be phosphorylated at different sites, which play opposite functions, leading to either increased fragmentation or enhanced fusion of mitochondria (Oliver and Reddy, 2019).
Dynamin Related Protein 1 is involved in the mechanisms underlying various diseases, such as myocardial ischemia/reperfusion injury, heart failure, and cancer. Indeed, tumor progression can be affected by Drp1 mediated regulation of mitochondrial metabolism. In agreement, in vitro studies indicate that phosphorylation of Drp1 promoting mitochondrial fission inhibits mitochondrial oxidative phosphorylation and enhances aerobic glycolysis, which promotes growth and metastasis in cancer cells. Drp1 silencing causes mitochondrial elongation and significantly suppresses the metastatic abilities of breast cancer cells (Rodrigues and Ferraz, 2020).
Moreover, Drp1 modulates the production of the cytokine IL-1β, IL-6, and IFN-β in response to immune stimulation and infection (Tiku et al., 2020). Indeed, swine influenza virus (SIV) infection leads to Drp1 phosphorylation at serine 579 and subsequent mitochondrial fission and IL-1β production in alveolar macrophages (Park et al., 2018). In particular, SIV infection induces ROS formation as well as the activation of the receptor-interacting protein kinase 1 (RIPK1), which in turn phosphorylates Drp1. RIPK1-dependent Drp1 phosphorylation is necessary for mitochondrial fission and ROS release, which in turn activate the NLRP3 inflammasome and subsequent IL-1β production (Park et al., 2018).
In agreement, it is emerging a role of Drp1 in modulating NLRP3 activation (Park et al., 2015). Parkin et al. provide a molecular insight into the relevance of mitochondrial dynamics in potentiating NLRP3 inflammasome activation, leading to aberrant inflammation. Knockdown of dynamin-related protein 1 (Drp1) induces aberrant mitochondrial elongation, promoting a marked increase in NLRP3-dependent caspase-1 activation and IL-1β secretion in mouse bone marrow-derived macrophages (Park et al., 2015). Several studies highlight the importance of Drp1 for mitochondrial balance between fission and fusion, which is modulated in response to infection to enhance macrophage effector function. Indeed, infection with bacteria commonly results in mitochondrial fragmentation, whereas viral infection often leads to mitochondrial fusion in macrophages (Tiku et al., 2020). Surprisingly, both enhanced fission and fusion can promote NLRP3 activation through different pathways. It seems that mitochondrial fission is implicated in IL-1β production following bacterial infection. Indeed, mitochondrial fission results in an increased cytosolic level of mitochondrial DAMPs, such as mitoDNA or mitoROS, inducing NLRP3 inflammasome activation (Park et al., 2015). On the other hands, the cellular response to cytosolic viral RNA promotes mitochondrial fusion and the activation of the mitochondrial antiviral-signaling (MAVS) protein, which in turn activates the NLRP3 inflammasome (Park et al., 2013; Figure 2).
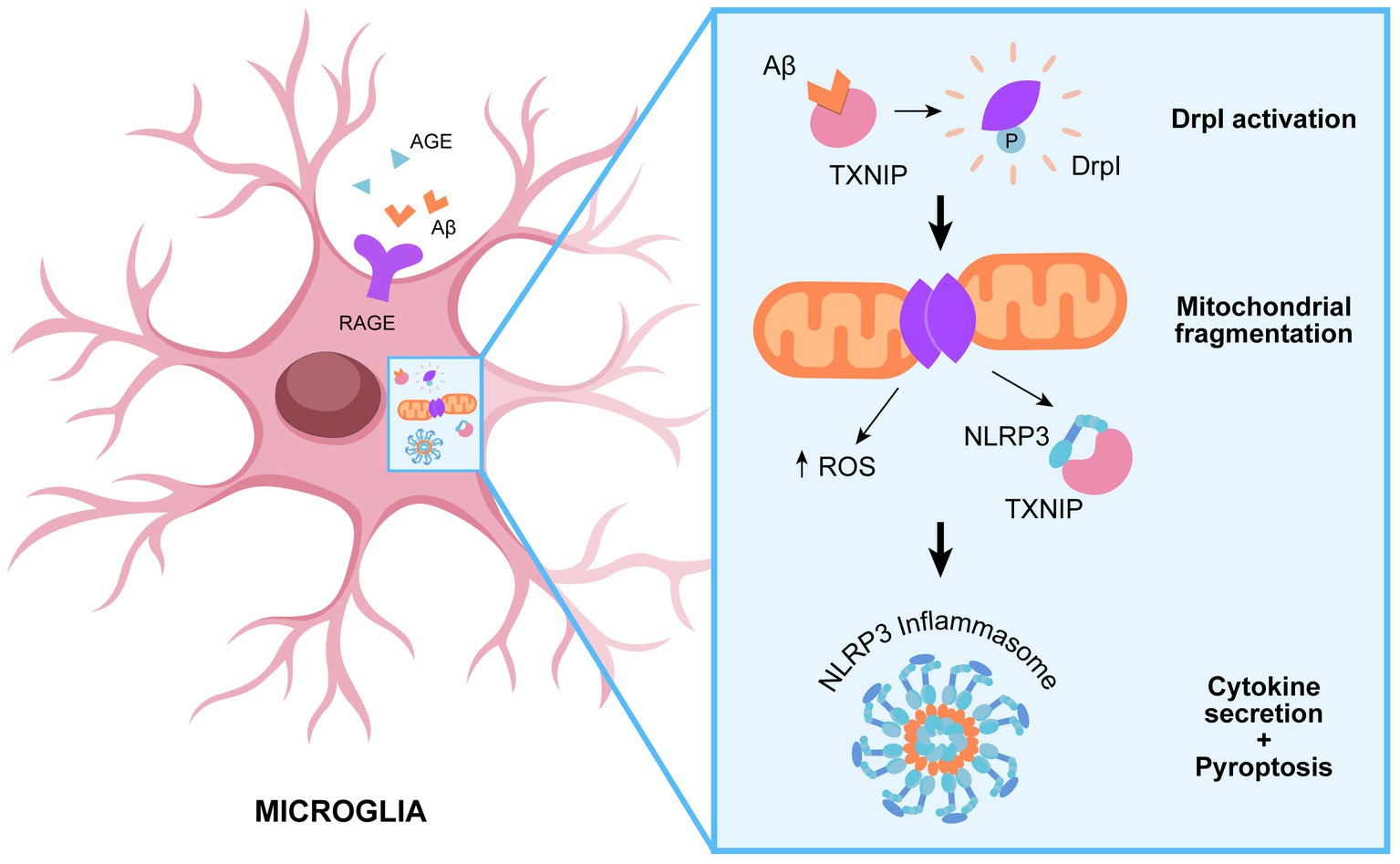
Figure 2. Role of Drp1 in promoting NLRP3 assembly. TXNIP-mediated recruitment of DRp1 to mitochondrial results in aberrant mitochondrial fission and ROS production, which in turn promote NLRP3 assembly and subsequent inflammatory cascade.
5.2. Role of Drp1 in AD
Alzheimer Disease is characterized by a diminished expression of genes involved in mitochondrial biogenesis, affecting the mitochondrial homeostasis and producing defective mitochondria biogenesis (Kandimalla et al., 2018; Manczak et al., 2018). Recent data show that Drp1 expression is significantly enhanced in the brain of AD patients and AD mice compared to healthy human controls and wild type mice, as well as Drp1 expression increases in neurons exposed to Aβ peptides in vitro (Medala et al., 2021; Bera et al., 2022). Moreover, a study carried out in an in vitro AD model revealed an enhanced expression of genes involved in mitochondrial fission (such as Drp1 and Fis1) and a decreased expression of those related to mitochondrial fusion (such as Mfn1 and Mfn2), altering the mitochondria dynamics and affecting the synaptic function (see below). These observations are corroborated by the analysis of post-mortem brains of AD patients, where the expression levels of Opa-1, Mfn1, and Mfn2 are decreased, while Fis1 is significantly increased. Notably, Aβ induces the S-nitrosylation of Drp1, leading to its hyperactivation and subsequent excessive mitochondrial fragmentation, which in turn generates ROS, leading to synaptic damage (Nakamura et al., 2010). Notably, AD is characterized by enhanced mitochondrial fission, suggesting that Drp1 plays a role in AD (Baek et al., 2017). In agreement, treatment of hemizygous APP/PS1 mice with mitochondrial division inhibitor 1 (Mdivi-1), an indirect inhibitor of Drp1, ameliorates anterograde mitochondrial transport, oxidative stress, and synaptic damage (Baek et al., 2017). Mdivi-1 ameliorates mitochondria fragmentation, distribution, and function also in CRND8 mice (APP strain; Wang et al., 2017). Double transgenic mice obtained crossing heterozygote Drp1 (+/−) mice with AD mice (Tg2576 strain) reveals that partial reduction of Drp1 is beneficial by ameliorating mitochondrial dysfunction, reducing Aβ production, increasing mitochondrial biogenesis and enhancing synaptic activity (Manczak et al., 2016). Reduction of Drp1 is protective also against mutant Tau-induced neuronal dysfunction in AD. A double transgenic mice obtained by crossing Drp1 (+/−) mice with Tau mice (P301L strain) show reduced mitochondrial dysfunction, decreased Tau phosphorylation, enhanced mitochondrial biogenesis and synaptic activity (Kandimalla et al., 2016). Drp1 interacts directly with Aβ in neurons of AD patients, in AD transgenic mice, and also in vitro in neurons derived from AD transgenic mice, leading to mitochondrial dysfunction and synaptic damage (Manczak et al., 2011). Tau plays a critical role in the assembly, stabilization, and modulation of microtubules, which are important for the normal function of neurons and the brain. Drp1 interacts directly also with phosphorylated Tau, further inducing neuronal dysfunction (Manczak and Reddy, 2012).
Many researchers have demonstrated that mitochondrial dysfunction can promote energy impairment in AD. Thus, Drp1-induced excessive mitochondrial fragmentation and defective transport of mitochondria to synapses, leads also to reduced synaptic ATP production and subsequent synaptic dysfunction (Pradeepkiran and Reddy, 2020). Aβ accumulation affects mitochondrial dynamics also in astrocytes, inducing a shift in the metabolic pathways (Zyśk et al., 2023). In human induced pluripotent cell (hiPSC)-derived astrocytes treated with Aβ it is observed an increased Drp1 phosphorylation, which localizes in lipid droplets and is secreted in extracellular vesicles (EV) and transported in the surrounding astrocytes through tunneling nanotubes (TNTs), enhancing the mitochondrial OXPHOX and increasing the glycolysis, switching toward fatty acid β oxidation for energy production (Zyśk et al., 2023). Astrocytic EVs (called astrosomes) derived from AD mice (5xFAD) contain Aβ (Elsherbini et al., 2020). 5xFAD-derived astrosomes are transported to mitochondria when added to neuronal cells in vitro, leading to enhanced Drp1 expression, altering the mitochondria dynamics and promoting caspase 3 activation, which in turn induces neuronal cell death (Elsherbini et al., 2020).
Oligodendrocytes are required for the myelination process, which is strongly dependent on glycolysis, which requires an efficient mitochondrial function. Thus, hyperactivated Drp1 can affect myelination by disrupting the mitochondrial activity. Notably, mature oligodendrocytes both in human AD patients and AD mice show inflammatory injuries associated with the NLRP3 inflammasome (Zhang et al., 2020). NLRP3 can be activated by components of glycolytic pathway and ROS, suggesting the role of Drp1 also in promoting the inflammatory process in AD. In agreement, excessive activation of Drp1 causes a glycolytic dysfunction in AD mice, which in turn activates NLRP3 inflammasome and subsequent pyroptosis. Knock down of Dp1 in oligodendrocytes abolishes NLRP3-induced inflammation and ameliorates myelin loss (Zhang et al., 2020). These data demonstrate that Drp1-promoted mitochondrial dysfunction leads to inflammation, which plays a key role in myelin loss and neuronal dysfunction.
Reactive Oxygen Species is the major factor responsible of imbalance in mitochondria function. Several recent findings report that advanced glycation end products (AGEs) participate in inflammation and neurodegenerative diseases (Perrone et al., 2012). AGE and its receptor RAGE are over expressed in AD brain samples, induce ROS production and mitochondria dysfunction. AGEs can influence mitochondrial dynamics by impairing fusion-fission balance, with a significant increment of mitochondrial fission in AD (Pradeepkiran and Reddy, 2020). AGEs contribute significantly to mitochondria dysfunction, by upregulating the expression of fission proteins Drp1 and Fis1 and down-regulating fusion proteins Mfn1, Mfn2, and Opa1. AGEs are also involved in abnormal APP processing and Aβ production. Indeed, AGEs activate RAGE, which in turn enhances the expression of BACE 1, a key enzyme in APP processing that initiates Aβ production (Perrone et al., 2012). Both AGE and Aβ trigger RAGE activation, which induces redox sensitive pathways by activating the transcription factor nuclear factor kappa B (NFkB), which promotes the expression of pro-inflammatory genes (Perrone et al., 2012). These pathological pathways promote an inflammatory cascade, which lower glucose consumption, decreases ATP levels, and downregulates mitochondrial activity, ultimately promoting neuronal death (Sivitz and Yorek, 2010). Oxidative stress caused by RAGE triggering promotes neuroinflammation and enhances Aβ levels into the brain by enhancing the rate of Aβ influx, leading to a vicious circle of RAGE activation following interaction with Aβ and further promoting neurodegeneration (Perrone et al., 2012). RAGE promotes inflammation also by inducing the expression of Thioredoxin-Interacting Protein (TXNIP), which is an β-arrestin-containing protein that can bind to and inhibit the antioxidant protein thioredoxin (TXN), promoting oxidative stress (Perrone et al., 2009; Sbai et al., 2010). TXNIP is over expressed in the brain of AD patients and in several AD mice models and recent data suggest that it participates in AD pathophysiology (Melone et al., 2018; Nasoohi et al., 2018a,b; Wang et al., 2019; Tsubaki et al., 2020; Perrone and Valente, 2021; Zhang M. et al., 2021). TXNIP plays a key role in modulating the cell and body glucose homeostasis (Perrone and Valente, 2021). Notably, TXNIP plays a key role in activating the NLRP3 inflammasome (Schroder et al., 2010) and its association with NLRP3 has been shown also in AD (Li et al., 2019; Sbai et al., 2022). Sbai et al. demonstrate that TXNIP drives the transport of Aβ to mitochondria in microglia both in vivo AD mice (5xFAD strain) and in vitro primary microglia, leading to Drp1 translocation to mitochondria, leading to mitochondria dysfunction, ROS production and NLRP3 activation and subsequent production of cytokines and pyroptosis of microglia, showing that it is an early even in AD, occurring in the early phases of AD when the first signs of cognitive alterations occur (Sbai et al., 2022). Drp1 appears to being transported to mitochondria in a complex together with TXNIP and Aβ, resulting in Drp1 activation. Silencing of TXNIP or blockade of RAGE reduces Aβ transport from the cellular surface to mitochondria in microglia, restores mitochondrial functionality, mitigates Aβ toxicity, inhibits NLRP3-induced inflammation and blocks pyroptosis in vivo AD mice and in vitro primary microglia (Sbai et al., 2022).
Up until now it has been demonstrated that there is a link between enhanced Drp1 activation and NLRP3 activation. We cannot exclude that Drp1 may modulate the assembly/function of other inflammasome complexes other than NLRP3. However, the data published so far in AD and other pathologies, such as cancer, report a link only between Drp1 and NLRP3 inflammasome. Further studies are needed in order to unveil the role of Drp1 in altering the mitochondrial function and activating inflammasome complexes other than NLRP3. However, the studies described above clearly show the relevance of Drp1 in linking the mitochondrial function to the induction of inflammation.
6. Conclusion
Several data are indicating a key role of Drp1 in promoting neurodegeneration by altering mitochondrial function in neurons. However, recent data are underline the role of Drp1 in promoting the activation of NRP3 in not neuronal cells, leading to inflammatory process that in turn leads to neuronal dysfunction. These data support the relevance of Drp1 in promoting mitochondrial dysfunction as an early even in AD. In addition, Drp1 links mitochondrial dysfunction to NLRP3 activation and the subsequent inflammatory cascade. In addition, these data demonstrate the pathological effect of inflammation as early even in AD. Indeed, inflammation occurs before the detection of neuronal dysfunction in all AD mice models. We described publications demonstrating that Drp1 promotes early inflammatory alterations in oligodendrocytes and microglia, further promoting neurodegeneration. These data are essential for the development of innovative strategies aimed to prevent and cure AD at the early stages.
Author contributions
OS, VB, ST, and LP wrote the manuscript. OS, VB, ST, JM, and CV corrected the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 956070 and by the European Union’s Horzion Europe research and innovation program under the Marie Skłodowska-Curie grant agreement No. 101072515 to CV.
Conflict of interest
OS is founder and CEO of Caminnov sas. LP plays a consulting role for Caminnov sas.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abais, J. M., Xia, M., Zhang, Y., Boini, K. M., and Li, P. L. (2015). Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 22, 1111–1129. doi: 10.1089/ars.2014.5994
Aisen, P. S., and Davis, K. L. (1994). Inflammatory mechanisms in Alzheimer's disease: implications for therapy. Am. J. Psychiatry 151, 1105–1113. doi: 10.1176/ajp.151.8.1105
Alfonso-Loeches, S., Ureña-Peralta, J. R., Morillo-Bargues, M. J., Oliver-De La Cruz, J., and Guerri, C. (2014). Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell. Neurosci. 8:216. doi: 10.3389/fncel.2014.00216
Apelt, J., and Schliebs, R. (2001). Beta-amyloid-induced glial expression of both pro-and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 894, 21–30. doi: 10.1016/S0006-8993(00)03176-0
Arnoux, I., and Audinat, E. (2015). Fractalkine signaling and microglia functions in the developing brain. Neural Plast. 2015:689404. doi: 10.1155/2015/689404
Baek, S. H., Park, S. J., Jeong, J. I., Kim, S. H., Han, J., Kyung, J. W., et al. (2017). Inhibition of Drp 1 ameliorates synaptic depression, Aβ deposition, and cognitive impairment in an Alzheimer's disease model. J. Neurosci. 37, 5099–5110. doi: 10.1523/JNEUROSCI.2385-16.2017
Barczuk, J., Siwecka, N., Lusa, W., Rozpędek-Kamińska, W., Kucharska, E., and Majsterek, I. (2022). Targeting NLRP3-mediated Neuroinflammation in Alzheimer’s disease treatment. Int. J. Mol. Sci. 23:8979. doi: 10.3390/ijms23168979
Basha, SC, Ramaiah, MJ, and Kosagisharaf, JR. Untangling the role of TREM2 in conjugation with microglia in neuronal dysfunction: a hypothesis on a novel pathway in the pathophysiology of Alzheimer's disease. J. Alzheimers Dis. (2023). doi: 10.3233/JAD-221070 (Epub ahead of print).
Bauernfeind, F., Bartok, E., Rieger, A., Franchi, L., Núñez, G., and Hornung, V. (2011). Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617. doi: 10.4049/jimmunol.1100613
Bera, A., Lavanya, G., Reshmi, R., Dev, K., and Kumar, R. (2022). Mechanistic and therapeutic role of Drp 1 in the pathogenesis of Alzheimer's disease. Eur. J. Neurosci. 56, 5516–5531. doi: 10.1111/ejn.15611
Biber, K., Neumann, H., Inoue, K., and Boddeke, H. W. G. M. (2007). Neuronal “on” and “off” signals control microglia. Trends Neurosci. 30, 596–602. doi: 10.1016/j.tins.2007.08.007
Bivona, G., Iemmolo, M., Agnello, L., Lo Sasso, B., Gambino, C. M., Giglio, R. V., et al. (2023). Microglial activation and priming in Alzheimer's disease: state of the art and future perspectives. Int. J. Mol. Sci. 24:884. doi: 10.3390/ijms24010884
Breitner, J. C. S., Haneuse, S. J. P. A., Walker, R., Dublin, S., Crane, P. K., Gray, S. L., et al. (2009). Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 72, 1899–1905. doi: 10.1212/WNL.0b013e3181a18691
Brown, G. C. (2019). The endotoxin hypothesis of neurodegeneration. J. Neuroinflammation 16:180. doi: 10.1186/s12974-019-1564-7
Campden, R. I., and Zhang, Y. (2019). The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 670, 32–42. doi: 10.1016/j.abb.2019.02.015
Carpanini, S. M., Harwood, J. C., Baker, E., Torvell, M., Sims, R., Williams, J., et al. (2021). The impact of complement genes on the risk of late-onset Alzheimer’s disease. Gene 12:443. doi: 10.3390/genes12030443
Carson, M. J., Thrash, J. C., and Walter, B. (2006). The cellular response in neuroinflammation: the role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 6, 237–245. doi: 10.1016/j.cnr.2006.09.004
Castellani, R. K., Hirai, K. G., Aliev, G., Drew, K. L., Nunomura, A., Takeda, A., et al. (2002). Role of mitochondrial dysfunction in Alzheimer's disease. J. Neurosci. Res. 70, 357–360. doi: 10.1002/jnr.10389
Chen, W. W., Zhang, X., and Huang, W. J. (2016). Role of neuroinflammation in neurodegenerative diseases. Mol. Med. Rep. 13, 3391–3396. doi: 10.3892/mmr.2016.4948
Chung, H. Y., Cesari, M., Anton, S., Marzetti, E., Giovannini, S., Seo, A. Y., et al. (2009). Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res. Rev. 8, 18–30. doi: 10.1016/j.arr.2008.07.002
Collij, L. E., Ingala, S., Top, H., Wottschel, V., Stickney, K. E., Tomassen, J., et al. (2021). White matter microstructure disruption in early stage amyloid pathology. Alzheimers Dement. Diagn. Assess. Dis. Monit. 13:e12124. doi: 10.1002/dad2.12124
Dai, J., Zhang, X., Wang, Y., Chen, H., and Chai, Y. (2017). ROS-activated NLRP3 inflammasome initiates inflammation in delayed wound healing in diabetic rats. Int. J. Clin. Exp. Pathol. 10, 9902–9909.
Delekate, A., Füchtemeier, M., Schumacher, T., Ulbrich, C., Foddis, M., and Petzold, G. C. (2014). Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 5:5422.
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Domingues, C., da Cruz, E., Silva, O. A. B., and Henriques, A. G. (2017). Impact of cytokines and chemokines on Alzheimer’s disease neuropathological hallmarks. Curr. Alzheimer Res. 14, 870–882. doi: 10.2174/1567205014666170317113606
Efthymiou, A. G., and Goate, A. M. (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12:43. doi: 10.1186/s13024-017-0184-x
Eisenbarth, S. C., Williams, A., Colegio, O. R., Meng, H., Strowig, T., Rongvaux, A., et al. (2012). NLRP10 is a NOD-like receptor essential to initiate adaptive immunity by dendritic cells. Nature 484, 510–513. doi: 10.1038/nature11012
Elsherbini, A., Kirov, A. S., Dinkins, M. B., Wang, G., Qin, H., Zhu, Z., et al. (2020). Association of Aβ with ceramide-enriched astrosomes mediates Aβ neurotoxicity. Acta Neuropathol. Commun. 8:60. doi: 10.1186/s40478-020-00931-8
Escartin, C., Galea, E., Lakatos, A., O’Callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325. doi: 10.1038/s41593-020-00783-4
Gaidt, M. M., Ebert, T. S., Chauhan, D., Schmidt, T., Schmid-Burgk, J. L., Rapino, F., et al. (2016). Human monocytes engage an alternative inflammasome pathway. Immunity 44, 833–846. doi: 10.1016/j.immuni.2016.01.012
Gombault, A., Baron, L., and Couillin, I. (2012). ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 3:414. doi: 10.3389/fimmu.2012.00414
Gomez-Nicola, D., and Boche, D. (2015). Post-mortem analysis of neuroinflammatory changes in human Alzheimer's disease. Alzheimers Res. Ther. 7:42. doi: 10.1186/s13195-015-0126-1
Gómez-Nicola, D., Fransen, N. L., Suzzi, S., and Perry, V. H. (2013). Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 33, 2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013
Griffin, W. S., Stanley, L. C., Ling, C., White, L., Mac Leod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Gustin, A., Kirchmeyer, M., Koncina, E., Felten, P., Losciuto, S., Heurtaux, T., et al. (2015). NLRP3 Inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10:e0130624. doi: 10.1371/journal.pone.0130624
Han, C., Yang, Y., Guan, Q., Zhang, X., Shen, H., Sheng, Y., et al. (2020). New mechanism of nerve injury in Alzheimer’s disease: β-amyloid-induced neuronal pyroptosis. J. Cell. Mol. Med. 24, 8078–8090. doi: 10.1111/jcmm.15439
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hebert, L. E., Weuve, J., Scherr, P. A., and Evans, D. A. (2013). Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783. doi: 10.1212/WNL.0b013e31828726f5
Heneka, M. T. (2017). Inflammasome activation and innate immunity in Alzheimer's disease. Brain Pathol. 27, 220–222. doi: 10.1111/bpa.12483
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Huang, J., Tao, Q., Ang, T. F. A., Farrell, J., Zhu, C., Wang, Y., et al. (2022). The impact of increasing levels of blood C-reactive protein on the inflammatory loci SPI1 and CD33 in Alzheimer’s disease. Transl. Psychiatry 12, 1–11. doi: 10.1038/s41398-022-02281-6
Huang, W. J., Zhang, X., and Chen, W. W. (2016). Role of oxidative stress in Alzheimer’s disease. Biomed Rep. 4, 519–522. doi: 10.3892/br.2016.630
Imbimbo, B. P., Solfrizzi, V., and Panza, F. (2010). Are NSAIDs useful to treat Alzheimer's disease or mild cognitive impairment? Front. Aging Neurosci. 2:19. doi: 10.3389/fnagi.2010.00019
Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S. V., Vieira-Saecker, A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi: 10.1038/s41586-019-1769-z
Jabir, M. S., Hopkins, L., Ritchie, N. D., Ullah, I., Bayes, H. K., Li, D., et al. (2015). Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11, 166–182. doi: 10.4161/15548627.2014.981915
Jin, M., Shiwaku, H., Tanaka, H., Obita, T., Ohuchi, S., Yoshioka, Y., et al. (2021). Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 12:6565. doi: 10.1038/s41467-021-26851-2
Kandimalla, R., Manczak, M., Fry, D., Suneetha, Y., Sesaki, H., and Reddy, P. H. (2016). Reduced dynamin-related protein 1 protects against phosphorylated tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum. Mol. Genet. 25, 4881–4897. doi: 10.1093/hmg/ddw312
Kandimalla, R., Manczak, M., Yin, X., Wang, R., and Reddy, P. H. (2018). Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 27, 30–40. doi: 10.1093/hmg/ddx381
Kandimalla, R., and Reddy, P. H. (2016). Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta 1862, 814–828. doi: 10.1016/j.bbadis.2015.12.018
Kaushal, V., Dye, R., Pakavathkumar, P., Foveau, B., Flores, J., Hyman, B., et al. (2015). Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 22, 1676–1686. doi: 10.1038/cdd.2015.16
Kawana, N., Yamamoto, Y., Ishida, T., Saito, Y., Konno, H., Arima, K., et al. (2013). Reactive astrocytes and perivascular macrophages express NLRP3 inflammasome in active demyelinating lesions of multiple sclerosis and necrotic lesions of neuromyelitis optica and cerebral infarction. Clin. Exp. Neuroimmunol. 4, 296–304. doi: 10.1111/cen3.12068
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 Inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20:3328. doi: 10.3390/ijms20133328
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. 4, 575–590. doi: 10.1016/j.trci.2018.06.014
Kitazawa, M., Cheng, D., Tsukamoto, M. R., Koike, M. A., Wes, P. D., Vasilevko, V., et al. (2011). Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 187, 6539–6549. doi: 10.4049/jimmunol.1100620
Korhonen, E., Hytti, M., Piippo, N., Kaarniranta, K., and Kauppinen, A. (2021). Antimycin A-induced mitochondrial dysfunction regulates inflammasome signaling in human retinal pigment epithelial cells. Exp. Eye Res. 209:108687. doi: 10.1016/j.exer.2021.108687
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., and Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215. doi: 10.1126/science.1169096
Lawrence, G. M. E. P., Holley, C. L., and Schroder, K. (2022). Parkinson's disease: connecting mitochondria to inflammasomes. Trends Immunol. 43, 877–885. doi: 10.1016/j.it.2022.09.010
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 5:37. doi: 10.1186/1742-2094-5-37
Leung, R., Proitsi, P., Simmons, A., Lunnon, K., Güntert, A., Kronenberg, D., et al. (2013). Inflammatory proteins in plasma are associated with severity of Alzheimer’s disease. PLoS One 8:e64971. doi: 10.1371/journal.pone.0064971
Li, L., Ismael, S., Nasoohi, S., Sakata, K., Liao, F. F., McDonald, M. P., et al. (2019). Thioredoxin-interacting protein (TXNIP) associated NLRP3 inflammasome activation in human Alzheimer's disease brain. J. Alzheimers Dis. 68, 255–265. doi: 10.3233/JAD-180814
Liang, T., Zhang, Y., Wu, S., Chen, Q., and Wang, L. (2022). The role of NLRP3 inflammasome in Alzheimer’s disease and potential therapeutic targets. Front. Pharmacol. 13:845185. doi: 10.3389/fphar.2022.845185
Lindsay, C. B., Zolezzi, J. M., Rivera, D. S., Cisternas, P., Bozinovic, F., and Inestrosa, N. C. (2020). Andrographolide reduces Neuroinflammation and oxidative stress in aged Octodon degus. Mol. Neurobiol. 57, 1131–1145. doi: 10.1007/s12035-019-01784-6
Lines, J., Baraibar, A. M., Fang, C., Martin, E. D., Aguilar, J., Lee, M. K., et al. (2022). Astrocyte-neuronal network interplay is disrupted in Alzheimer’s disease mice. GLIA 70, 368–378. doi: 10.1002/glia.24112
Liu, L., and Chan, C. (2014). IPAF inflammasome is involved in interleukin-1β production from astrocytes, induced by palmitate; implications for Alzheimer's disease. Neurobiol. Aging 35, 309–321. doi: 10.1016/j.neurobiolaging.2013.08.016
Liu, L., Wang, N., Kalionis, B., Xia, S., and He, Q. (2022). HMGB1 plays an important role in pyroptosis induced blood brain barrier breakdown in diabetes-associated cognitive decline. J. Neuroimmunol. 362:577763. doi: 10.1016/j.jneuroim.2021.577763
Liu, X., Zhang, X., Ding, Y., Zhou, W., Tao, L., Lu, P., et al. (2017). Nuclear factor E2-related Factor-2 negatively regulates NLRP3 Inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxid. Redox Signal. 26, 28–43. doi: 10.1089/ars.2015.6615
Liu, Q., Zhang, D., Hu, D., Zhou, X., and Zhou, Y. (2018). The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 103, 115–124. doi: 10.1016/j.molimm.2018.09.010
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cells 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Lopez-Ramirez, M. A., Wu, D., Pryce, G., Simpson, J. E., Reijerkerk, A., King-Robson, J., et al. (2014). Micro RNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J. 28, 2551–2565. doi: 10.1096/fj.13-248880
Lull, M. E., and Block, M. L. (2010). Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. doi: 10.1016/j.nurt.2010.05.014
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp 1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Manczak, M., Kandimalla, R., Fry, D., Sesaki, H., and Reddy, P. H. (2016). Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum. Mol. Genet. 25, 5148–5166. doi: 10.1093/hmg/ddw330
Manczak, M., Kandimalla, R., Yin, X., and Reddy, P. H. (2018). Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 27, 1332–1342. doi: 10.1093/hmg/ddy042
Manczak, M., and Reddy, P. H. (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547. doi: 10.1093/hmg/dds072
Martinon, F. (2010). Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 40, 616–619. doi: 10.1002/eji.200940168
Matrone, C., Djelloul, M., Taglialatela, G., and Perrone, L. (2015). Inflammatory risk factors and pathologies promoting Alzheimer’s disease progression: is RAGE the key? Histol. Histopathol. 30, 125–139. doi: 10.14670/HH-30.125
McGeer, P. L., and McGeer, E. G. (2007). NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol. Aging 28, 639–647. doi: 10.1016/j.neurobiolaging.2006.03.013
Medala, V. K., Gollapelli, B., Dewanjee, S., Ogunmokun, G., Kandimalla, R., and Vallamkondu, J. (2021). Mitochondrial dysfunction, mitophagy, and role of dynamin-related protein 1 in Alzheimer's disease. J. Neurosci. Res. 99, 1120–1135. doi: 10.1002/jnr.24781
Melone, M. A. B., Dato, C., Paladino, S., Coppola, C., Trebini, C., Giordana, M., et al. (2018). Verapamil inhibits Ser202/Thr205 phosphorylation of tau by blocking TXNIP/ROS/p38 MAPK pathway. Pharm. Res. 35:44. doi: 10.1007/s11095-017-2276-2
Michaud, J. P., Richard, K. L., and Rivest, S. (2011). MyD88-adaptor protein acts as a preventive mechanism for memory deficits in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 6:5. doi: 10.1186/1750-1326-6-5
Milner, M. T., Maddugoda, M., Götz, J., Burgener, S. S., and Schroder, K. (2021). The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr. Opin. Immunol. 68, 116–124. doi: 10.1016/j.coi.2020.10.011
Misra, A., Chakrabarti, S. S., and Gambhir, I. S. (2018). New genetic players in late-onset Alzheimer’s disease: findings of genome-wide association studies. Indian J. Med. Res. 148, 135–144. doi: 10.4103/ijmr.IJMR_473_17
Mo, J., Hu, J., and Cheng, X. (2023). The role of high mobility group box 1 in neuroinflammatory related diseases. Biomed. Pharmacother. 161:114541. doi: 10.1016/j.biopha.2023.114541
Mogensen, F. L. H., Delle, C., and Nedergaard, M. (2021). The Glymphatic system (En) during inflammation. Int. J. Mol. Sci. 22:7491. doi: 10.3390/ijms22147491
Moonen, S., Koper, M. J., Van Schoor, E., Schaeverbeke, J. M., Vandenberghe, R., von Arnim, C. A. F., et al. (2023). Pyroptosis in Alzheimer's disease: cell type-specific activation in microglia, astrocytes and neurons. Acta Neuropathol. 23, 175–195.
Morais Cardoso, S., Swerdlow, R. H., and Oliveira, C. R. (2002). Induction of cytochrome c-mediated apoptosis by amyloid beta 25–35 requires functional mitochondria. Brain Res. 931, 117–125. doi: 10.1016/S0006-8993(02)02256-4
Moreira, P. I., Carvalho, C., Zhu, X., Smith, M. A., and Perry, G. (2010). Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim. Biophys. Acta 1802, 2–10. doi: 10.1016/j.bbadis.2009.10.006
Moreira, P. I., Santos, M. S., Moreno, A., and Oliveira, C. (2001). Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci. Rep. 21, 789–800. doi: 10.1023/A:1015536808304
Murphy, N., Grehan, B., and Lynch, M. A. (2014). Glial uptake of amyloid beta induces NLRP3 inflammasome formation via cathepsin-dependent degradation of NLRP10. NeuroMolecular Med. 16, 205–215. doi: 10.1007/s12017-013-8274-6
Nakamura, T., Cieplak, P., Cho, D. H., Godzik, A., and Lipton, S. A. (2010). S-nitrosylation of Drp 1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10, 573–578. doi: 10.1016/j.mito.2010.04.007
Nakanishi, A., Kaneko, N., Takeda, H., Sawasaki, T., Morikawa, S., Zhou, W., et al. (2018). Amyloid β directly interacts with NLRP3 to initiate inflammasome activation: identification of an intrinsic NLRP3 ligand in a cell-free system. Inflamm. Regen. 38:27. doi: 10.1186/s41232-018-0085-6
Nasoohi, S., Ismael, S., and Ishrat, T. (2018a). Thioredoxin-interacting protein (TXNIP) in cerebrovascular and neurodegenerative diseases: regulation and implication. Mol. Neurobiol. 55, 7900–7920. doi: 10.1007/s12035-018-0917-z
Nasoohi, S., Parveen, K., and Ishrat, T. (2018b). Metabolic syndrome, brain insulin resistance, and Alzheimer's disease: Thioredoxin interacting protein (TXNIP) and Inflammasome as Core amplifiers. J. Alzheimers Dis. 66, 857–885. doi: 10.3233/JAD-180735
Nasrabady, S. E., Rizvi, B., Goldman, J. E., and Brickman, A. M. (2018). White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6:22. doi: 10.1186/s40478-018-0515-3
Nazem, A., Sankowski, R., Bacher, M., and Al-Abed, Y. (2015). Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflammation 12:74. doi: 10.1186/s12974-015-0291-y
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Novoa, C., Salazar, P., Cisternas, P., Gherardelli, C., Vera-Salazar, R., Zolezzi, J. M., et al. (2022). Inflammation context in Alzheimer's disease, a relationship intricate to define. Biol. Res. 55:39. doi: 10.1186/s40659-022-00404-3
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
Oliver, D., and Reddy, P. H. (2019). Dynamics of dynamin-related protein 1 in Alzheimer's disease and other neurodegenerative diseases. Cells 8:961. doi: 10.3390/cells8090961
Origlia, N., Bonadonna, C., Rosellini, A., Leznik, E., Arancio, O., Yan, S. S., et al. (2010). Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. 30, 11414–11425. doi: 10.1523/JNEUROSCI.2127-10.2010
Pang, Y., Wu, D., Ma, Y., Cao, Y., Liu, Q., Tang, M., et al. (2021). Reactive oxygen species trigger NF-κB-mediated NLRP3 inflammasome activation involvement in low-dose CdTe QDs exposure-induced hepatotoxicity. Redox Biol. 47:102157. doi: 10.1016/j.redox.2021.102157
Park, S., Juliana, C., Hong, S., Datta, P., Hwang, I., Fernandes-Alnemri, T., et al. (2013). The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366. doi: 10.4049/jimmunol.1301170
Park, H. S., Liu, G., Liu, Q., and Zhou, Y. (2018). Swine influenza virus induces RIPK1/DRP1-mediated Interleukin-1 Beta production. Viruses 10:419. doi: 10.3390/v10080419
Park, S., Won, J. H., Hwang, I., Hong, S., Lee, H. K., and Yu, J. W. (2015). Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 5:15489. doi: 10.1038/srep15489
Patergnani, S., Bouhamida, E., Leo, S., Pinton, P., and Rimessi, A. (2021). Mitochondrial oxidative stress and "Mito-inflammation": actors in the diseases. Biomedicine 9:216. doi: 10.3390/biomedicines9020216
Paudel, Y. N., Angelopoulou, E., Semple, B., Piperi, C., Othman, I., and Shaikh, M. F. (2020). Potential neuroprotective effect of the HMGB1 inhibitor glycyrrhizin in neurological disorders. ACS Chem. Neurosci. 11, 485–500. doi: 10.1021/acschemneuro.9b00640
Paulino Ramirez Diaz, S., Gil Gregório, P., Manuel Ribera Casado, J., Reynish, E., Jean Ousset, P., Vellas, B., et al. (2005). The need for a consensus in the use of assessment tools for Alzheimer's disease: the feasibility study (assessment tools for dementia in Alzheimer Centres across Europe), a European Alzheimer's disease Consortium's (EADC) survey. Int. J. Geriatr. Psychiatry 20, 744–748. doi: 10.1002/gps.1355
Perrone, L., Devi, T. S., Hosoya, K. C., Terasaki, T., and Singh, L. P. (2009). Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 221, 262–272. doi: 10.1002/jcp.21852
Perrone, L., Sbai, O., Nawroth, P. P., and Bierhaus, A. (2012). The complexity of sporadic Alzheimer's disease pathogenesis: the role of RAGE as therapeutic target to promote neuroprotection by inhibiting neurovascular dysfunction. Int. J. Alzheimers Dis. 2012, 1–13. doi: 10.1155/2012/734956
Perrone, L., and Valente, M. (2021). The emerging role of metabolism in brain-heart axis: new challenge for the therapy and prevention of Alzheimer Disease. May Thioredoxin interacting protein (TXNIP) play a role? Biomol. Ther. 11:1652. doi: 10.3390/biom11111652
Pontillo, A., Catamo, E., Arosio, B., Mari, D., and Crovella, S. (2012). NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 26, 277–281. doi: 10.1097/WAD.0b013e318231a8ac
Pradeepkiran, J. A., and Reddy, P. H. (2020). Defective mitophagy in Alzheimer's disease. Ageing Res. Rev. 64:101191. doi: 10.1016/j.arr.2020.101191
Qi, Y., Klyubin, I., Cuello, A. C., and Rowan, M. J. (2018). NLRP3-dependent synaptic plasticity deficit in an Alzheimer's disease amyloidosis model in vivo. Neurobiol. Dis. 114, 24–30. doi: 10.1016/j.nbd.2018.02.016
Qian, W., Choi, S., Gibson, G. A., Watkins, S. C., Bakkenist, C. J., and Van Houten, B. (2012). Mitochondrial hyperfusion induced by loss of the fission protein Drp 1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 125, 5745–5757. doi: 10.1242/jcs.109769
Ralay Ranaivo, H., Hodge, J. N., Choi, N., and Wainwright, M. S. (2012). Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J. Neuroinflammation 9:68. doi: 10.1186/1742-2094-9-68
Ramonett, A., Kwak, E. A., Ahmed, T., Flores, P. C., Ortiz, H. R., Lee, Y. S., et al. (2022). Regulation of mitochondrial fission by GIPC-mediated Drp 1 retrograde transport. Mol. Biol. Cell 33:ar4. doi: 10.1091/mbc.E21-06-0286
Rauf, A., Badoni, H., Abu-Izneid, T., Olatunde, A., Rahman, M. M., Painuli, S., et al. (2022). Neuroinflammatory markers: key indicators in the pathology of neurodegenerative diseases. Molecules 27:3194. doi: 10.3390/molecules27103194
Reddy, P. H., Reddy, T. P., Manczak, M., Calkins, M. J., Shirendeb, U., and Mao, P. (2011). Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 67, 103–118. doi: 10.1016/j.brainresrev.2010.11.004
Reed, T., Perluigi, M., Sultana, R., Pierce, W. M., Klein, J. B., Turner, D. M., et al. (2008). Redox proteomic identification of 4–hydroxy–2–nonenal–modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 30, 107–120. doi: 10.1016/j.nbd.2007.12.007
Rivers-Auty, J., Mather, A. E., Peters, R., Lawrence, C. B., and Brough, D. (2020). Anti-inflammatories in Alzheimer’s disease-potential therapy or spurious correlate? Brain Commun. 2:fcaa109. doi: 10.1093/braincomms/fcaa109
Roberts, D. J. (2008). Goralski KBA critical overview of the influence of inflammation and infection on P-glycoprotein expression and activity in the brain. Expert Opin. Drug Metab. Toxicol. 4, 1245–1264. doi: 10.1517/17425255.4.10.1245
Rodrigues, T., and Ferraz, L. S. (2020). Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharmacol. 182:114282. doi: 10.1016/j.bcp.2020.114282
Rogers, J., Luber-Narod, J., Styren, S. D., and Civin, W. H. (1988). Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol. Aging 9, 339–349. doi: 10.1016/S0197-4580(88)80079-4
Rossner, S., Lange-Dohna, C., Zeitschel, U., and Perez-Polo, J. (2005). Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J. Neurochem. 92, 226–234. doi: 10.1111/j.1471-4159.2004.02857.x
Rui, W., Xiao, H., Fan, Y., Ma, Z., Xiao, M., Li, S., et al. (2021). Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer's disease. J. Neuroinflammation 18:280. doi: 10.1186/s12974-021-02329-2
Saadi, M., Karkhah, A., Pourabdolhossein, F., Ataie, A., Monif, M., and Nouri, H. R. (2020). Involvement of NLRC4 inflammasome through caspase-1 and IL-1β augments neuroinflammation and contributes to memory impairment in an experimental model of Alzheimer's like disease. Brain Res. Bull. 154, 81–90. doi: 10.1016/j.brainresbull.2019.10.010
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single app knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 17, 661–663. doi: 10.1038/nn.3697
Sbai, O., Devi, T. S., Melone, M. A., Feron, F., Khrestchatisky, M., Singh, L. P., et al. (2010). RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J. Cell Sci. 123, 4332–4339. doi: 10.1242/jcs.074674
Sbai, O., Djelloul, M., Auletta, A., Ieraci, A., Vascotto, C., and Perrone, L. (2022). RAGE-TXNIP axis drives inflammation in Alzheimer’s by targeting Aβ to mitochondria in microglia. Cell Death Dis. 13:302. doi: 10.1038/s41419-022-04758-0
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, M., Chételat, G., Teunissen, C. E., et al. (2021). Alzheimer’s disease. Lancet 397, 1577–1590. doi: 10.1016/S0140-6736(20)32205-4
Schnöder, L., Hao, W., Qin, Y., Liu, S., Tomic, I., Liu, X., et al. (2016). Deficiency of neuronal p38α MAPK attenuates amyloid pathology in Alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J. Biol. Chem. 291, 2067–2079. doi: 10.1074/jbc.M115.695916
Schroder, K., Zhou, R., and Tschopp, J. (2010). The NLRP3 inflammasome: a sensor for metabolic danger? Science 327, 296–300. doi: 10.1126/science.1184003
Shah, D., Gsell, W., Wahis, J., Luckett, E. S., Jamoulle, T., Vermaercke, B., et al. (2022). Astrocyte calcium dysfunction causes early network hyperactivity in Alzheimer’s disease. Cell Rep. 40:111280. doi: 10.1016/j.celrep.2022.111280
Sharma, B., Satija, G., Madan, A., Garg, M., Alam, M. M., Shaquiquzzaman, M., et al. (2022). Role of NLRP3 Inflammasome and its inhibitors as emerging therapeutic drug candidate for Alzheimer’s disease: a review of mechanism of activation, regulation, and inhibition. Inflammation 2022, 1–32. doi: 10.1007/s10753-022-01730-0
Sharma, A., Smith, H. J., Yao, P., and Mair, W. B. (2019). Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 20:e48395. doi: 10.15252/embr.201948395
Sheng, J. G., Bora, S. H., Xu, G., Borchelt, D. R., Price, D. L., and Koliatsos, V. E. (2003). Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 14, 133–145. doi: 10.1016/S0969-9961(03)00069-X
Sheng, J. G., Zhou, X. Q., Mrak, R. E., and Griffin, W. S. (1998). Progressive neuronal injury associated with amyloid plaque formation in Alzheimer disease. J. Neuropathol. Exp. Neurol. 57, 714–717. doi: 10.1097/00005072-199807000-00008
Sivitz, W. I., and Yorek, M. A. (2010). Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 12, 537–577. doi: 10.1089/ars.2009.2531
Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263. doi: 10.1038/nrn3898
Sosa, V., Moliné, T., Somoza, R., Paciucci, R., Kondoh, H., and Leonart, M. E. (2013). Oxidative stress and cancer: an overview. Ageing Res. Rev. 12, 376–390. doi: 10.1016/j.arr.2012.10.004
Stancu, I. C., Cremers, N., Vanrusselt, H., Couturier, J., Vanoosthuyse, A., Kessels, S., et al. (2019). Aggregated tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded tau pathology in vivo. Acta Neuropathol. 137, 599–617. doi: 10.1007/s00401-018-01957-y
Sutterwala, F. S., Haasken, S., and Cassel, S. L. (2014). Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 1319, 82–95. doi: 10.1111/nyas.12458
Takata, F., Nakagawa, S., Matsumoto, J., and Dohgu, S. (2021). Blood-brain barrier dysfunction amplifies the development of Neuroinflammation: understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction. Front. Cell. Neurosci. 15:661838. doi: 10.3389/fncel.2021.661838
Tan, M. S., Tan, L., Jiang, T., Zhu, X. C., Wang, H. F., Jia, C. D., et al. (2014). Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis. 5:e1382. doi: 10.1038/cddis.2014.348
Tarasoff-Conway, J. M., Carare, R. O., Osorio, R. S., Glodzik, L., Butler, T., Fieremans, E., et al. (2015). Clearance systems in the brain—implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470. doi: 10.1038/nrneurol.2015.119
Tarassishin, L., Casper, D., and Lee, S. C. (2014). Aberrant expression of interleukin-1β and Inflammasome activation in human malignant gliomas. PLoS One 9:e103432. doi: 10.1371/journal.pone.0103432
Tiku, V., Tan, M. W., and Dikic, I. (2020). Mitochondrial functions in infection and immunity. Trends Cell Biol. 30, 263–275. doi: 10.1016/j.tcb.2020.01.006
Tsubaki, H., Tooyama, I., and Walker, D. G. (2020). Thioredoxin-interacting protein (TXNIP) with focus on brain and neurodegenerative diseases. Int. J. Mol. Sci. 21:9357. doi: 10.3390/ijms21249357
Tu, J., Chen, B., Yang, L., Qi, K., Lu, J., and Zhao, D. (2015). Amyloid-β activates microglia and regulates protein expression in a manner similar to prions. J. Mol. Neurosci. 56, 509–518. doi: 10.1007/s12031-015-0553-2
Valero, J., Mastrella, G., Neiva, I., Sánchez, S., and Malva, J. O. (2014). Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front. Neurosci. 8:83. doi: 10.3389/fnins.2014.00083
Varatharaj, A., and Galea, I. (2017). The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12. doi: 10.1016/j.bbi.2016.03.010
Venegas, C., Kumar, S., Franklin, B. S., Dierkes, T., Brinkschulte, R., Tejera, D., et al. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361. doi: 10.1038/nature25158
Versele, R., Sevin, E., Gosselet, F., Fenart, L., and Candela, P. (2022). TNF-α and IL-1β modulate blood-brain barrier permeability and decrease amyloid-β peptide efflux in a human blood-brain barrier model. Int. J. Mol. Sci. 23:10235. doi: 10.3390/ijms231810235
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. doi: 10.1038/nrn.2015.1
Wang, W. Y., Tan, M. S., Yu, J. T., and Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
Wang, Y., Wang, Y., Bharti, V., Zhou, H., Hoi, V., Tan, H., et al. (2019). Upregulation of Thioredoxin-interacting protein in brain of amyloid-β protein precursor/Presenilin 1 transgenic mice and amyloid-β treated neuronal cells. J. Alzheimers Dis. 72, 139–150. doi: 10.3233/JAD-190223
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of mitochondrial fragmentation protects against Alzheimer's disease in rodent model. Hum. Mol. Genet. 26, 4118–4131. doi: 10.1093/hmg/ddx299
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15:30. doi: 10.1186/s13024-020-00376-6
Yang, Y., Wang, H., Kouadir, M., Song, H., and Shi, F. (2019). Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 10:128. doi: 10.1038/s41419-019-1413-8
Zakaria, R., Wan Yaacob, W. M., Othman, Z., Long, I., Ahmad, A. H., and Al-Rahbi, B. (2017). Lipopolysaccharide-induced memory impairment in rats: a model of Alzheimer's disease. Physiol. Res. 66, 553–565. doi: 10.33549/physiolres.933480
Zhang, M., Hu, G., Shao, N., Qin, Y., Chen, Q., Wang, Y., et al. (2021). Thioredoxin-interacting protein (TXNIP) as a target for Alzheimer's disease: flavonoids and phenols. Inflammopharmacology 29, 1317–1329. doi: 10.1007/s10787-021-00861-4
Zhang, X., Wang, R., Hu, D., Sun, X., Fujioka, H., Lundberg, K., et al. (2020). Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer's disease. Sci. Adv. 6:eabb8680. doi: 10.1126/sciadv.abb8680
Zhang, J., Wu, N., Wang, S., Yao, Z., Xiao, F., Lu, J., et al. (2021). Neuronal loss and microgliosis are restricted to the core of Aβ deposits in mouse models of Alzheimer's disease. Aging Cell 20:e13380. doi: 10.1111/acel.13380
Zhao, Y., Jaber, V., and Lukiw, W. J. (2021). Gastrointestinal tract microbiome-derived pro-inflammatory neurotoxins in Alzheimer’s disease. J. Aging Sci. 9:002.
Zhong, Z., Zhai, Y., Liang, S., Mori, Y., Han, R., Sutterwala, F. S., et al. (2013). TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 4:1611. doi: 10.1038/ncomms2608
Keywords: Alzheimer inflammation, mitochondria, DRP1, NLRP3, TXNIP
Citation: Sbai O, Bazzani V, Tapaswi S, McHale J, Vascotto C and Perrone L (2023) Is Drp1 a link between mitochondrial dysfunction and inflammation in Alzheimer’s disease? Front. Mol. Neurosci. 16:1166879. doi: 10.3389/fnmol.2023.1166879
Edited by:
Florian Plattner, Neuro-Research Strategies, United StatesReviewed by:
Manikandan Samidurai, SENS Research Foundation, United StatesDerek Costello, University College Dublin, Ireland
Copyright © 2023 Sbai, Bazzani, Tapaswi, McHale, Vascotto and Perrone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorena Perrone, cGVycm9uZWxvcmVuYTFAZ21haWwuY29t