
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 20 April 2023
Sec. Pain Mechanisms and Modulators
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1153870
This article is part of the Research Topic Insights into Molecular and Cellular Mechanisms of Chronic Pain and Neuroinflammation View all 15 articles
 Cheng Wu1,2,3,4†
Cheng Wu1,2,3,4† Hui-Fang Shang3,4†
Hui-Fang Shang3,4† Yong-Jie Wang3,4†
Yong-Jie Wang3,4† Jing-Hua Wang3,4Zhen-Xing Zuo5
Jing-Hua Wang3,4Zhen-Xing Zuo5 Yan-Na Lian3,4
Yan-Na Lian3,4 Li Liu6
Li Liu6 Chen Zhang7
Chen Zhang7 Xiang-Yao Li1,2,3,4*
Xiang-Yao Li1,2,3,4*The deficit of fragile X messenger ribonucleoprotein (FMRP) leads to intellectual disability in human and animal models, which also leads to desensitization of pain after nerve injury. Recently, it was shown that the protein arginine methyltransferases 1 (PRMT1) regulates the phase separation of FMRP. However, the role of PRMT1 in pain regulation has been less investigated. Here we showed that the downregulation of PRMT1 in the anterior cingulate cortex (ACC) contributes to the development of peripheral pain hypersensitivity. We observed that the peripheral nerve injury decreased the expression of PRMT1 in the ACC; knockdown of the PRMT1 via shRNA in the ACC decreased the paw withdrawal thresholds (PWTs) of naïve mice. Moreover, the deficits of FMRP abolished the effects of PRMT1 on pain sensation. Furthermore, overexpression of PRMT1 in the ACC increased the PWTs of mice with nerve injury. These observations indicate that the downregulation of cingulate PRMT1 was necessary and sufficient to develop peripheral hypersensitivity after nerve injury. Thus, we provided evidence that PRMT1 is vital in regulating peripheral pain hypersensitivity after nerve injury via the FMRP.
• Peripheral nerve injury decreased the expression of PRMT1 in the ACC.
• The downregulation of cingulate PRMT1 was sufficient to induce peripheral hypersensitivity.
• FMRP was necessary for PRMT1’s regulation of peripheral hypersensitivity.
• The downregulation of cingulate PRMT1 was necessary for peripheral hypersensitivity.
The absence of the RNA-binding protein Fragile X messenger ribonucleoprotein (FMRP) results in fragile X syndrome. This condition causes intellectual disabilities and social deficits in affected individuals (Hagerman et al., 2018). The FMRP regulates the synaptic plasticity in multiple brain regions, including the anterior cingulate cortex (ACC) (Zhao et al., 2005) and hippocampus (Martinez and Tejada-Simon, 2018; Asiminas et al., 2019; Prieto et al., 2021). ACC and hippocampus share common molecular mechanisms regulating synaptic transmission, and ACC is a vital target for regulating neuropathic pain (Bliss et al., 2016). It has been extensively reported that ACC could be activated by neuropathic pain and enhanced excitatory synaptic transmissions (Xu et al., 2008; Bliss et al., 2016; Wang et al., 2017). Furthermore, previous studies have detected sensory abnormalities in the Fragile X messenger ribonucleoprotein 1 (Fmr1) knock-out (KO) mice (Wang et al., 2016; He et al., 2017) and FXS patients (Juczewski et al., 2016), who have more robust and more frequent responses and reduced habituation to sensory stimulations, including olfactory, auditory, visual, tactile, and vestibular stimuli, as measured by electrodermal responses (Miller et al., 1999; Kazdoba et al., 2014; Kozono et al., 2020). More specifically, Fmr1 KO mice exhibit hyporesponsiveness to inflammatory and neuropathic pain. Also, patients with FXS display altered sensory processing of nocifensive response, including sensory hyperresponsiveness and hyporesponsiveness (Wang et al., 2016). Furthermore, previous studies have detected sensory abnormalities in the Fragile X messenger ribonucleoprotein 1 (Fmr1) KO mice (Wang et al., 2016; He et al., 2017) and FXS patients (Juczewski et al., 2016). Consistently, the deficit of FMRP is desensitized to pain after nerve injury (Price et al., 2007). These studies indicate that FMRP has a critical role in regulating somatic sensation.
The liquid–liquid phase separation of the FMRP-RNA complex is critical to delivering the bonded RNA to the target locations (Kim et al., 2019; Tsang et al., 2019); this process is vital for RNA transporting and initiating protein translation (Tsang et al., 2019). PRMT1 was the first identified mammalian methyltransferase (Bedford and Clarke, 2009). It catalyzes the formation of ω-NG-monomethyl arginine (MMA) and asymmetric di-methylarginine (ADMA) (McBride and Silver, 2001) and regulates ~85% of total protein arginine methylation observed in cultured rat fibroblasts and mouse liver (Blanc and Richard, 2017). Previous studies showed that the activities of PRMT1 determined the methylated level of FMRP (Stetler et al., 2006; Blackwell et al., 2010), which regulates the phase separation of the FMRP-RNA complex (Tsang et al., 2019), and determines the binding ability of FMRP (Blackwell et al., 2010). These studies indicate that PRMT1 has a critical role in regulating the functions of FMRP (Stetler et al., 2006; Blackwell et al., 2010), especially in the central nervous system (CNS). However, this point has not been evaluated. By employing the pain evaluation assay, we evaluated the involvement of PRMT1-FMRP in ACC’s pathological changes of pain. Our data suggest that PRMT1 may be a new target for chronic pain treatments in the future.
Adult (8–10 weeks old) male C57BL/6 J mice were purchased from Shanghai SLAC. Fmr1 knock-out (Fmr1 KO, FVB;129P-Fmr1tm1Cgr/J) mice with an FVB genetic background were gifted from Prof. Chen Zhang (Capital Medical University, Beijing, China). Mice were bred and maintained in the experimental animal center of Zhejiang University. Fmr1 KO mice were generated by crossing heterozygous female Fmr1+/− mice with male Fmr1+/y mice. The offspring were genotyped by PCR using mouse tail-tip DNA and standard primers (5′-CTTCTGGCACCT CCAGCTT-3′), WT primers (5′-TGTGATAGAATATGCAGCATGTGA-3′) for 131 bp and mutant allele-specific primers (5′-CACGAGACTAGTGAGACGTG-3′) for 400 bp. The PCR products were visualized with ethidium bromide staining. Animals were maintained on a 12 h/12 h light/dark cycle. Food and water were provided ad libitum. The animal care and use committee of Zhejiang University approved all mice protocols.
For the CPN ligation mouse model, in brief, mice were anesthetized with isoflurane (1% ~ 3%, as needed, RWD, Shenzhen, China, R510-22). The left CPN between the anterior and posterior groups of muscles was slowly ligated with a chromic gut suture 5–0 (Jinhuan, Shanghai, China, F503) until the digits began to twitch. The skin was sutured using a 5–0 silk suture and cleaned with povidone-iodine (Caoshanhu, Nanchang, China). Sham surgery was conducted in the same manner, but the nerve was not ligated. All animals were kept in a standard living chamber after surgery. The mice could recover for at least 7 days before the behavioral test.
To induce inflammatory pain, 10 μl of 50% CFA (Sigma, St. Louis, MO, F5881) was injected subcutaneously into the dorsal surface of one hindpaw. As a Sham group, saline was injected.
The von Frey behavioral test was performed according to the up-down algorithm described by Chaplan et al. (1994). To determine speedy withdrawal or paw flinching evoked by mechanical stimuli, animals were placed on a raised mesh grid and covered with a clear plastic box for containment. Calibrated von Frey filaments were applied to the middle of the plantar surface of each paw until the filament bent. Brisk withdrawal or paw flinching was considered a positive response. Lifting of the paw due to normal locomotor behavior was ignored. In the absence of a response, the filament of the following greater force was applied. Following a response, the filament of the next lower force was applied. The tactile stimulus producing a 50% likelihood of a withdrawal response was calculated and treated as the paw withdrawal threshold (PWT).
For the cannula surgery and microinjection, mice were anesthetized with isoflurane (1–3%, as needed, RWD, Shenzhen, China, R510-22) inhalation of 100% oxygen with a flow of 0.5 L/min delivered by facemask. The scalp was shaved and then cleaned with iodine (Caoshanhu, Nanchang, China) and alcohol. The head of each mouse was fixed into a stereotaxic adapter mounted on a stereotaxic frame (RWD, Shenzhen, China, 68,025), and eye ointment (Cisen, Jining, China) was applied to the eyes. An incision was made over the skull, and the surface was exposed. Two small holes were drilled above the ACC, and the dura was gently reflected. Guide cannulas were placed 0.7 mm anterior to the bregma, 0.3 mm lateral to the midline, and 1.75 mm ventral to the surface of the skull. For the microinjection, the mice were restrained in a plastic cone (RWD, Shenzhen, China, 68,025), and a small hole was cut in the plastic overlying the microinjection guides. The dummy cannulas were removed, and the microinjection cannula was inserted into the guide. A 30-gage injection cannula was placed 0.7 mm lower than the guide. AMI-1 (0.5 μL, 0.1 ng/μL, Sigma-Aldrich, Saint Louis, MO 63103, United States, A9232) was bilaterally delivered at 0.5 μL/min using a syringe driven by an infusion pump (Alcott, Shanghai, China, ALC-IP600). The volume delivered was confirmed by watching the movement of the meniscus down a length of calibrated polyethylene (PE10) tubing. After delivery to each side of the brain, the injection cannula was left in place for 1 min to prevent any solution from flowing back up the guide. The cannula was then retracted and inserted into the opposite side of the brain. Ten min after microinjection, the mechanical allodynia test was administered.
The adeno-associated viruse 8 (AAV8) was constructed for overexpressing FMRP (NM_008031.3) and PRMT1 (NM_019830.3). The lentiviruses were constructed for the knockdown of FMRP (5’-GCTGTTGGTGGTTAGCTAAAG-3′) and PRMT1 (5′-GCATTAAAGACGTGGCCATCA-3′). All the viruses were packaged by OBiO Technology (Shanghai) Corp., Ltd. (Shanghai, China). For viral injection, mice were anesthetized with ketamine (100 mg/kg of body weight) and xylazine (8 mg/kg) by intraperitoneal (IP) injection and placed in a stereotactic frame. Mice were injected bilaterally with purified and concentrated AAV8 or lentivirus into the ACC (1 μl, coordinates from Bregma: −0.7 mm anterior/posterior, ±0.3 mm medial/lateral, −1.75 mm dorsal/ventral) using glass microelectrodes at a slow rate (100 nL/min). The injection microelectrode was slowly withdrawn 10 min after the virus infusion. Experiments were started from at least 21 days for AAV and 10 days for lentivirus after the injection. The injection sites were examined at the end of all the behavioral tests, and only data from animals with correct injections were included. Brain slices of ACC were directly examined under a fluorescent microscope. The PWT was examined in a blind manner.
Primers were designed by referring to the sequences of β-actin and Prmt1 in GenBank. The specific sequence information is shown in Table 1 below. Hangzhou Youkang Biological Co., LTD synthesized the designed primers.

Table 1. Primer sequences for real-time quantitative fluorescence PCR.
The mice were lightly anesthetized with isoflurane and then decapitated. ACC was dissected after 7 days of CPN ligation. According to the TransZol Up Plus RNA kit (TransGen Biotech, Beijing, China, ER501-01), RNA was extracted from the ACC, and its purity and concentration were measured. A total RNA of 1.0 μg was taken from each group, and reverse transcription was performed by using the reverse transcription reagent HiScript®II Q RT SuperMix (Vazyme, Nanjing, China, R223). The transcribed cDNA was used as the real-time quantitative PCR reaction template, as shown in Table 2. The real-time PCR experiment obtained the Ct value, amplification curve, and dissolution curve. According to the formula ΔCt = CTTarget -CTactin, the average Ct value of 3 duplicates of each sample was taken to calculate the ΔCt value of each target gene relative to the reference gene β-actin, using 2-ΔΔCt (Livak and Schmittgen, 2001) method, the relative expression level of each gene was calculated.
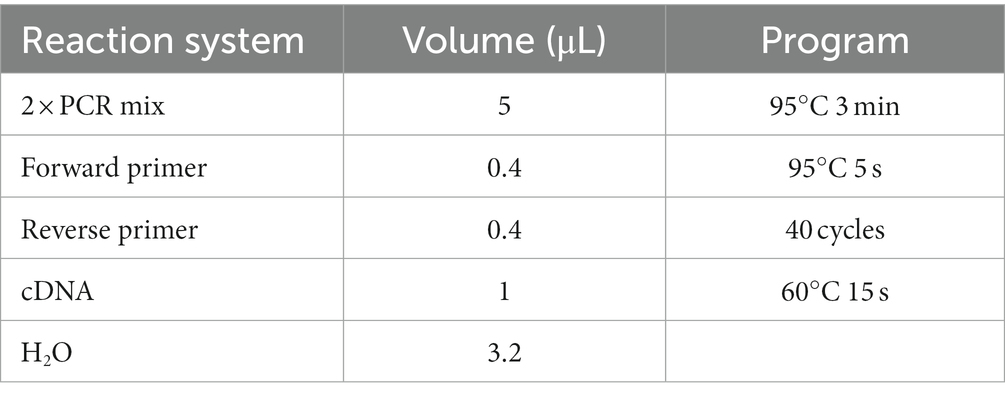
Table 2. Real-time quantitative PCR reaction system and procedure.
The mice were lightly anesthetized with isoflurane and then decapitated. The regions needed were dissected after 7 days of CPN ligation and then homogenized in a RIPA buffer (50 mM pH 7.6 Tris-Cl, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 1 mM DTT, 0.5% sodium deoxycholate) containing a protease inhibitor cocktail. After centrifugation, the supernatants were used for protein quantification by the Bradford assay. Electrophoresis of equal amounts of total protein was performed on SDS-polyacrylamide gels. The separated proteins were transferred onto polyvinylidene membranes at 4°C. The membranes were blocked for 2 h with 5% milk in TBST (Tris-buffered saline with Tween-20, room temperature) and incubated with a primary antibody (PRMT1, 1:1000, Abcam, Cambridge, UK, ab73246; PRMT1, 1:1000, Proteintech, Wuhan, China, 11,279-1-AP) at 4°C for overnight. After being washed, the membranes were incubated for 1 h with the appropriate HRP-coupled secondary antibody (Jackson immune research, goat anti-rabbit, 111–035-144) diluted 1:3000, followed by enhanced chemiluminescence detection of the proteins with Western Lightning Chemiluminescence Reagent Plus (Life Technologies, California, United States, 1863097), according to the manufacturer’s instructions. To verify equal loading, we probed the membranes with antibodies against tubulin (1:10000, Sigma, T5201) or actin (1:5000, Sigma, A3853). The density of the immunoblots was measured with the NIH ImageJ program.
Mice were anesthetized with 1% pentobarbital sodium (50 mg/kg, Sigma-Aldrich, Saint Louis, MO 63103, United States, P3761) and subjected to cardiac perfusion with 0.01 M phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. Brains were post-fixed overnight in PFA at 4°C, then transferred to 15% sucrose in PBS, followed by 30% sucrose until saturated. The brain was embedded in Tissue-Tek O.C.T. compound, frozen in liquid nitrogen, and stored at-80°C before being sliced to 25 μm coronal cryostat sections at-20°C (Leica CM3050S). Free-floating sections containing ACC were washed with PBS. For immunostaining, sections were incubated with blocking buffer (5% normal goat serum and 0.3% Triton X-100 in PBS) for 1 h at room temperature and incubated with primary antibodies (FMRP, 1:150, Proteintech, Wuhan, China, 66,548-1-Ig; PRMT1, 1:150, Abcam, Cambridge, UK, ab73246) overnight at 4°C. Sections were washed in PBS and then added appropriate secondary antibodies (goat anti-mouse 594, Life Technologies, California, United States, A-11005; goat anti-rabbit 488, Life Technologies, California, United States, A-11034) for 2 h at room temperature. Sections were rewashed for 3 × 10 min in PBS. Following washing, the stained coverslips were mounted using Fluoroshield Mounting Medium with DAPI (Abcam, Cambridge, UK, ab104139) for image collection.
White-colored plastic boxes were used as the open field chambers (dimension: 45 × 45 × 45 cm3). Mice were individually placed into the center of the chambers and allowed to explore for 5 min freely. The locomotion and exploratory behaviors of mice were recorded with Anymaze software (Stoelting, UK). The total traveling distance was used to evaluate locomotor activity.
Human embryonic kidney cells (HEK-293 T) were cultured in DMEM (Corning Cellgro, Virginia, United States, 10-017-CVA) containing 5% fetal bovine serum (Excel. Bio, Hefei, China, FCS500). The medium also contained 5% F12 (Gibco™, California, USA, 11765–054) and penicillin/streptomycin (Gibco™, California, USA, 15140122). 0.25% trypsin (Gibco™, California, USA, 25200056) digestion was performed every 2 ~ 3 days. All the cultures were placed in a 3.5 cm NEST dish at 37°C and 5.0% CO2. FMRP-GFP plasmid was transfected into HEK-293 T using Lipofectamine™ 3,000 transfection reagent (Invitrogen™, California, United States, L3000075). After transfection for 12–14 h, the fluorescence of the fluorescent protein could be seen under a fluorescence microscope (Olympus, Tokyo, Japan, CKX53). AMI-1 (Sigma-Aldrich, Saint Louis, United States, A9232) was added to the medium at the final concentration of 100 μM after transfection for 24 h and incubated for 1 h continuously.
Kill three or four postnatal rat pups using an approved method of euthanasia; the head was cut off quickly, the hippocampus was taken out, and the tissue was chopped up on an ice plate and put into 1 ml 0.25% trypsin for digestion at 37°C for 12 min. After complete digestion, the tissue was blown 12 times with a rounded tip three times to obtain the supernatant. Centrifugation followed, in which the cell suspension and debris were stratified, and the cell suspension was carefully sucked out. Plate 1 × 106 cells per 3.5 cm dish. Change half of the culture medium with fresh medium, including Neurobasal Medium (Invitrogen™, California, United States, 21103–049), GlutaMax-1 (Invitrogen™, California, USA, 35050–061), and B27 supplement (Invitrogen™, California, United States, 17504–044), every 3 days. Ten days after plating, neurons were transfected with AAV8 carried with GFP or Fmr1. 48 h after transfection, AMI-1 was added to the medium at the final concentration of 100 μM and incubated for 1 h continuously.
Cells were initially fixed with 4% PFA for 10 min for cluster quantification after AMI-1 incubation. DAPI was used for nuclear counter-staining. Images were taken using Olympus IX83-FV3000. To quantify the percentage of cells with FMRP granule clusters, more than 50 cells were counted for each sample.
GraphPad 8.0 was used to plot and fit the data. Statistical comparisons were made using the paired or unpaired t-test, one-way or two-way ANOVA (Student–Newman–Keuls test was used for post hoc comparison), and chi-square test. All data were presented as the mean ± SEM. In all cases, p < 0.05 is considered statistically significant.
To investigate the involvement of PRMT1 in the development of peripheral pain hypersensitivity, we first examined the expression of PRMT1 in the ACC and found that PRMT1, respectively, expressed in layer 1 (PRMT1/DAPI%; 33.95%), layer 2/3 (80.4%), layer 5 (73.41%) and layer 6 (73.55%) (Figures 1A,B). After that, we performed ligation of the common peroneal nerve (CPN) (Vadakkan et al., 2005) on naïve mice (Figure 1C), which decreased paw withdrawal thresholds (PWTs) on day seven after the operation (Figure 1D). Interestingly, the lower levels of Prmt1 mRNA and PRMT1 protein were, respectively, observed in the ACC (Figures 1E,F). In order to examine whether the down-regulation of PRMT1 selectively occurred in ACC after CPN ligation, we also detected the PRMT1 expression in the auditory cortex (AuD) and lateral amygdaloid nucleus (La). The down-regulation of PRMT1 was also observed in the AuD but not in the La (Figures 1G,H). Cumulatively, these results demonstrate that PRMT1 in the ACC is downregulated after peripheral nerve injury.
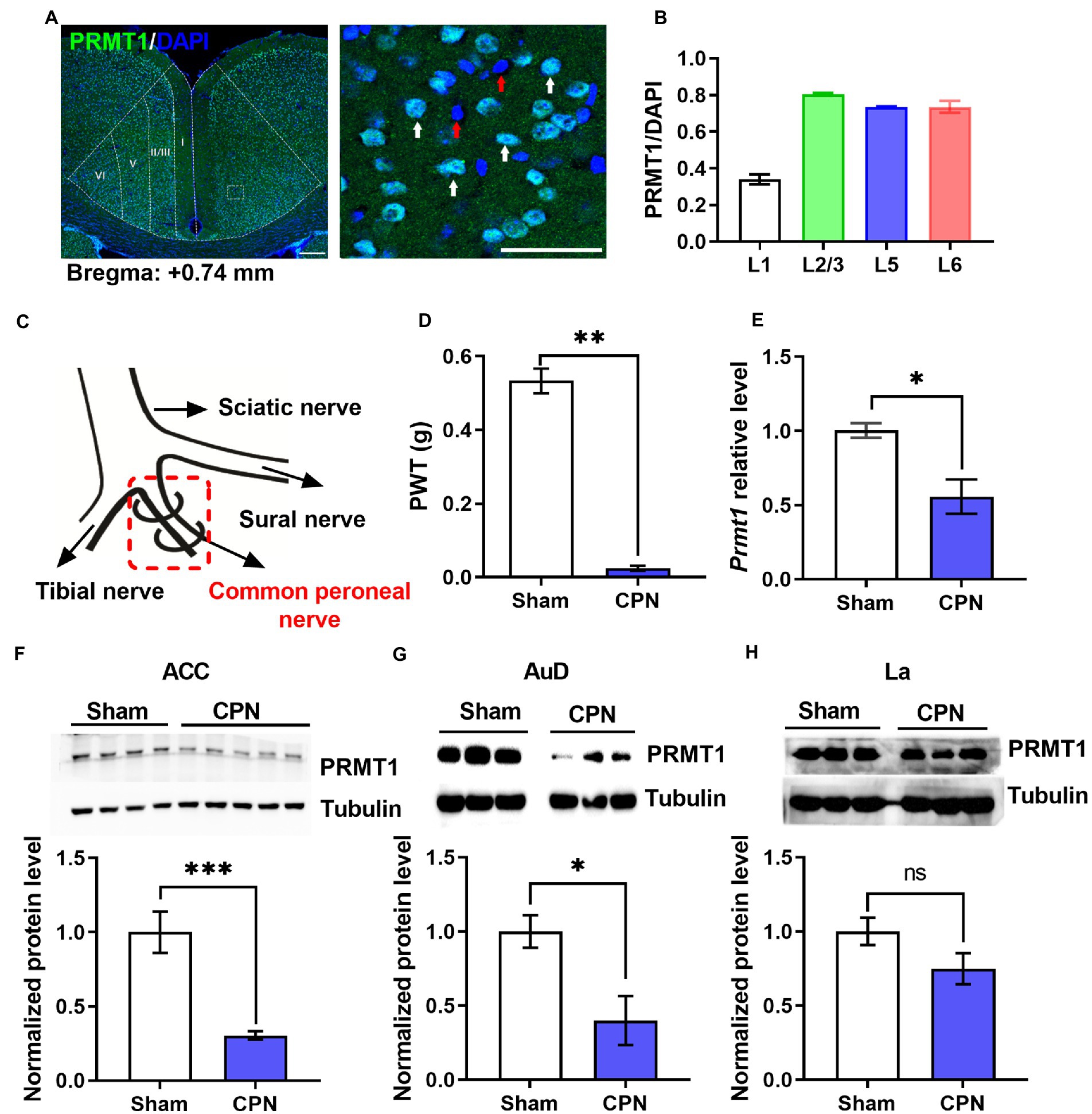
Figure 1. Peripheral nerve injury decreased PRMT1. (A) Left: representative photomicrograph of PRMT1 (green) in the anterior cingulate cortex (ACC). Right: zoomed-in view of the boxed region in the left panel. Blue: DAPI; green: PRMT1; red arrow: DAPI; white arrow: PRMT1 and DAPI; scale bars, 200 μm (left), 50 μm (right). (B) Quantification of PRMT1 positive cells in different layers of the ACC. (C) Schematic diagram of the ligation of the common peroneal nerve (CPN) in mouse. (D) Paw withdrawal thresholds (PWTs) assessed by von Frey behavioral test after common peroneal nerve (CPN) ligation (Sham and CPN, n = 6/group, unpaired t-test, p < 0.01; **p < 0.01). (E) Prmt1 mRNA level in the ACC of Sham and CPN mice (Sham: n = 3, CPN: n = 3; unpaired t-test, p = 0.0237; *p < 0.05). (F) PRMT1 protein level in the ACC of Sham and CPN mice. Top: representative immunoblot images; bottom: statistical analysis (Sham: n = 4, CPN: n = 5; unpaired t-test, p = 0.0009; ***p < 0.001). (G) PRMT1 protein level in the auditory cortex (AuD) of Sham and CPN mice. Top: representative immunoblot images; bottom: statistical analysis (Sham: n = 3, CPN: n = 3; unpaired t-test, p = 0.039; *p < 0.01). (H) PRMT1 protein level in the lateral amygdaloid nucleus (La) of Sham and CPN mice. Top: representative immunoblot images; bottom: statistical analysis (Sham: n = 3, CPN: n = 3; unpaired t-test, p = 0.1453). Data are represented as mean ± SEM; ns, not significant.
We next want to know whether the downregulation of cingulate PRMT1 contributes to peripheral hypersensitivity after a nerve injury. In order to answer this question, we first examined the causality of the PRMT1 downregulation, expressing the Prmt1 shRNA in the ACC (Figures 2A,B), to peripheral pain hypersensitivity. Behaviorally, we observed significantly lower PWTs (Figure 2C) after shRNA expression but not the control (Ctrl) virus. The PRMT1 downregulation did not affect the motor function because there were no changes in the center time (Figure 2D) and travel distance (Figure 2E) in the open field. Further examinations confirmed the downregulation of PRMT1 (Figure 2F).
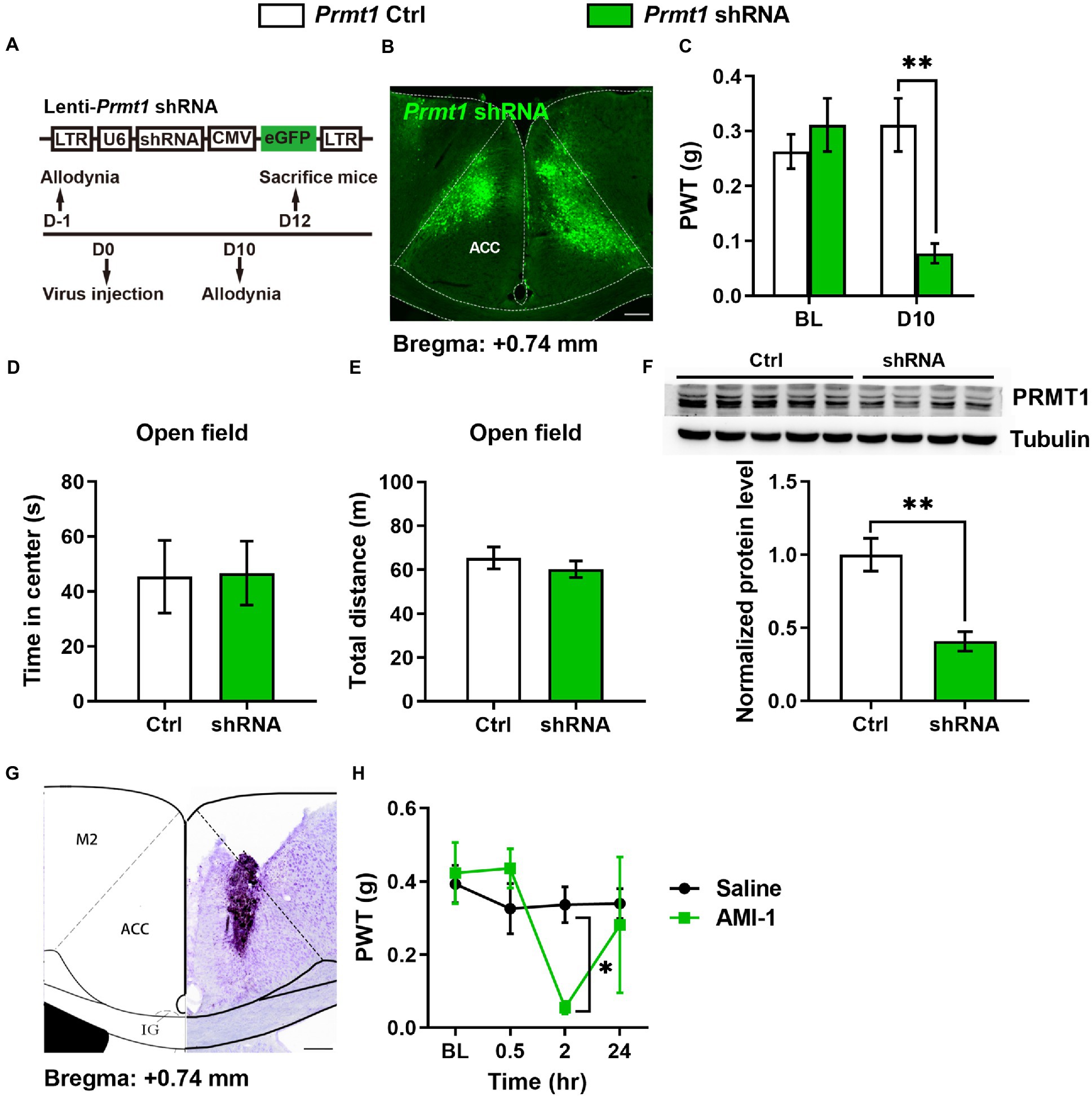
Figure 2. Downregulation of cingulate PRMT1 was sufficient and necessary for the development of peripheral hypersensitivity (A) Up: schematics of lentivirus vector engineered shRNA to knock down Prmt1 and eGFP control (Ctrl). Bottom: the experimental paradigm for behavioral testing. scale bar, 200 μm. (B) Illustration of bilateral expression of lenti-Prmt1 in C57BL/6 J mouse ACC. (C) PWT tests after Prmt1 shRNA expressed in the ACC (two-way RM ANOVA, interaction: F(2;36) = 23.95, p < 0.01, Time: F(2;36) = 16.95, p < 0.01, Ctrl vs. shRNA: F(1;36) = 152.63, p < 0.01, Ctrl: n = 10, shRNA: n = 10; “**” indicates p < 0.01; BL: baseline). (D) Statistics for the center time in the open-field test (Ctrl: n = 5, shRNA: n = 4, unpaired t-test, p > 0.05). (E) Statistics for the total distance in the open-field test (Ctrl: n = 5, shRNA: n = 4, unpaired t-test, p > 0.05). (F) Western blots and quantification showing efficient knockdown of PRMT1 by shRNA in the ACC (Ctrl: n = 5, shRNA: n = 4, unpaired t-test, p = 0.0039; “**” indicates p < 0.01). (G) Example injection site of cannula in the ACC of C57BL/6 J mice. (H) PWT tests after micro infusion of PRMT1 inhibitor, AMI-1, into ACC (Two-way RM ANOVA, interaction: F (1;9) = 10.19, p < 0.05, Time: F (1;9) = 13.51, p < 0.05, saline vs. AMI-1: (F) (1;9) = 7.24, p < 0.05, Ctrl: n = 6, AMI-1: n = 5; “*” indicates p < 0.05). Data are represented as mean ± SEM; ns, not significant.
We also evaluated the effects of blocking the activity of PRMT1 with AMI-1, a selective and potent PRMT1 inhibitor. When we micro-infused AMI-1 into the ACC of naïve mice (Figure 2G), the PWTs markedly decreased (Figure 2H). Thus, by combining genetic and pharmacological approaches, we showed that the downregulation of cingulate PRMT1 is sufficient to induce peripheral hypersensitivity.
Since the phase separation of FMRP is essential to its combining with RNA, which is regulated by PRMT1 (Tsang et al., 2019), we confirmed the regulation of PRMT1 on the phase separation of FMRP. We expressed the FMRP conjected with GFP in the HEK-293 T cell and observed cluster-like fluorescence signals in the FMRP-GFP group 24 h later. We then applied AMI-1 to inhibit the activities of PRMT1 (Figure 3A) and observed a larger size (Figure 3B) and higher fluorescence intensity (Figure 3C) of FMRP in the AMI-1 group than in the Ctrl group. This result suggests that PRMT1 regulates the condensation of FMRP in the HEK-293 T system.
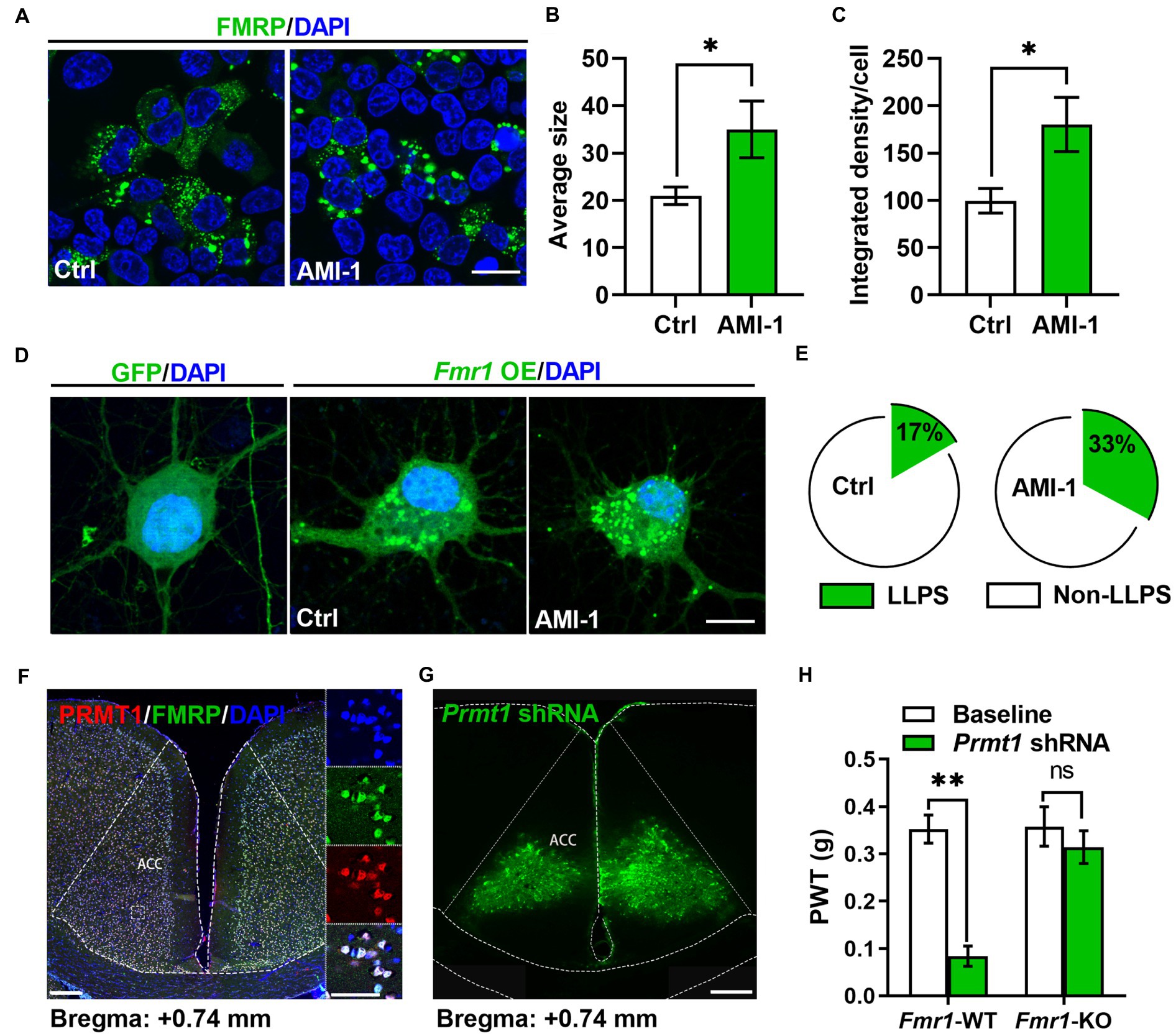
Figure 3. FMRP was involved in the development of peripheral hypersensitivity induced by PRMT1 downregulation. (A) Representative examples of control (Ctrl, left) and the application of AMI-1 (right) of FMRP-GFP in HEK-293 T cells. Scale bar, 20 μm. (B) The average size of the condensed FMRP-GFP clusters in Ctrl and AMI-1 groups (Ctrl: n = 8, AMI-1: n = 7; unpaired t-test, p = 0.0338; *p < 0.05). (C) The integrated density of the condensed FMRP-GFP clusters in Ctrl and AMI-1 groups (Ctrl: n = 8, AMI-1: n = 7; unpaired t-test, p = 0.0185; *p < 0.05). (D) Representative examples of GFP (left), Ctrl (middle, transfected FMRP-GFP) and the application of AMI-1 (right, transfected FMRP-GFP) of FMRP-GFP in the cultured neurons. Scale bar, 10 μm. (E) The percentage of condensed FMRP in the FMRP-GFP clusters after applying solvent (Ctrl group, left) or AMI-1 (right). LLPS: liquid-liquid phase separation; Non-LLPS: no liquid-liquid phase separation (Chi-square test, Ctrl vs. AMI-1, *p < 0.05). (F) Confocal images of FMRP-and PRMT1-expressing cells in ACC; zoomed-in view of the boxed region in the left panel (blue, DAPI; red, PRMT1; green, FMRP; scale bars, 200 μm (left), 50 μm (right)). (G) Illustration of bilateral expression of lenti-Prmt1 shRNA in ACC of Fmr1 wild-type (WT)/knock-out (KO) mice (scale bar, 200 μm). (H) PWT tests after Prmt1 shRNA expressed in ACC of Fmr1 WT and KO mice (Two-way RM ANOVA, interaction: F(2;32) = 10.34, p < 0.01, Time: F(2;32) = 14.15, p < 0.01, WT vs. KO: F(1;32) = 47.84, p < 0.01; Baseline: WT vs. KO, p > 0.05; WT: n = 8, KO: n = 10; **p < 0.01). Data are represented as mean ± SEM; ns, not significant.
We further expressed the FMRP-conjected GFP in the cultured neurons via the AAV virus. Consistently, we observed both dispersed and condensed GFP signals after virus transfection (Figure 3D), while, in the Ctrl group, we only observed dispersed GFP signals (Figure 3D). We then applied AMI-1 to inhibit the activities of PRMT1. Unlike the observations from HEK-293 T, we did not observe any differences in the average size and fluorescence intensity of FMRP between the two groups. However, a higher proportion of neurons had condensed FMRP (Figure 3E). Therefore, inhibiting the activities of PRMT1 in the cultured neurons also increased the condensation of FMRP, which mirrors that the activities of PRMT1 determined the methylated level of the FMRP-RNA complex.
Since PRMT1 regulates the liquid–liquid phase separation of the FMRP-RNA complex, this observation prompts us to explore further how PRMT1 regulates peripheral hypersensitivity and raises an interesting question of whether the FMRP is involved in regulating peripheral hypersensitivity induced by PRMT1 downregulation. Firstly, we investigated whether PRMT1 has the same distribution patterns as FMRP in the ACC. As shown in Figure 3F, about 90% of cells expressed both PRMT1 and FMRP. Next, we wanted to know whether the PRMT1-FMRP was involved in the development of peripheral hypersensitivity. When we delivered a Prmt1 shRNA into the ACC of both Fmr1 wild-type (WT) and knock-out (KO) mice (Figure 3G), we recorded decreased PWTs in the WT but not in the KO mice (Figure 3H). These data suggest that FMRP is critical for the development of hypersensitivity induced by PRMT1 downregulation.
Since deficits of FMRP is extensive, the absence of FMRP in other brain areas may mask the effects of Prmt1 shRNA on PWTs. If this is true, manipulation of the expression of cingulate FMRP will not affect the development of peripheral hypersensitivity in the Fmr1 KO mice. We performed CPN ligation on Fmr1 WT and KO mice to test the above deduction. We recorded lower PWTs in the WT but not in the KO mice (Figure 4A). Next, we designed an Fmr1-specific shRNA (Figure 4B) and delivered it to the ACC of naïve mice using lentivirus (Figure 4C). There were not any noticeable changes in PWTs on day five after virus injection (Figure 4D). Moreover, CPN ligation decreased the PWTs in mice with Fmr1 Ctrl virus, but not in the mice expressing Fmr1 shRNA (Figure 4D). Above-mentioned results suggest that knockdown cingulate FMRP prevents the development of hypersensitivity after nerve injury.
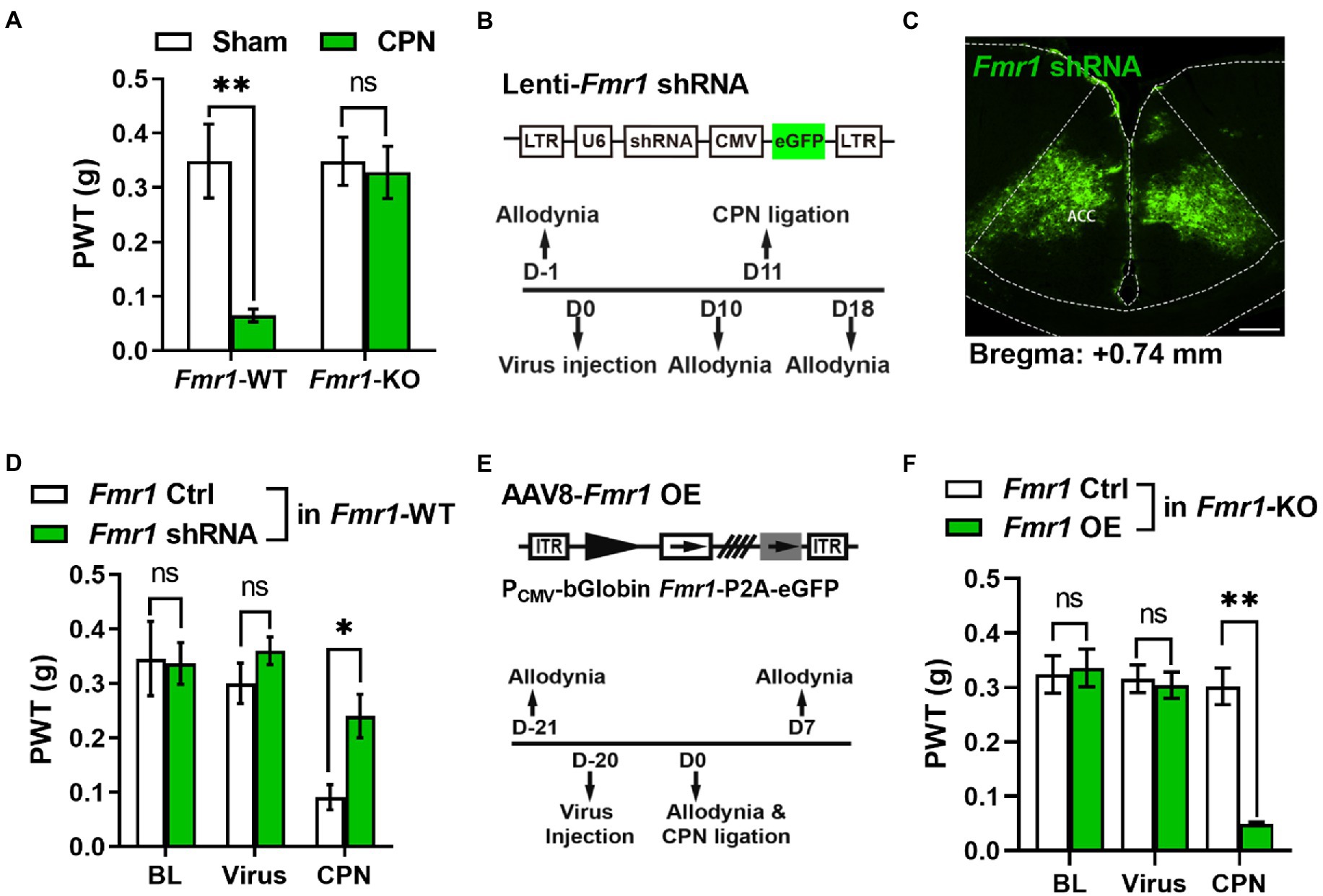
Figure 4. Cingulate FMRP is necessary for the development of peripheral hypersensitivity after nerve injury (A) PWT tests after CPN ligation in Fmr1 WT and KO mice (Two-way RM ANOVA, interaction: F(1;11) = 4.90, p < 0.05, Time: F(1;11) = 6.50, p < 0.05, WT vs. KO: F(1;11) = 10.37, p < 0.01; Sham: WT vs. KO, p > 0.05; WT: n = 8, KO: n = 5; “**” indicates p < 0.01). (B) Upper schematics of lentivirus vectors engineered shRNA to knock down Fmr1 and eGFP Ctrl. Lower experimental paradigm for behavioral testing after injection of Fmr1-shRNA virus into ACC. (C) Illustration of bilateral expression of lenti-Fmr1 shRNA virus in ACC of Fmr1 WT mice (scale bar, 200 μm). (D) PWT tests in Fmr1 WT mice conducted at BL, after virus injection of either Fmr1 Ctrl or shRNA in ACC, further after CPN ligation (Two-way RM ANOVA, interaction: F(3;30) = 1.43, p > 0.05, Time: F(3;30) = 19.10, p < 0.01, Ctrl vs. shRNA: F(1;30) = 8.24, p < 0.05, n = 6/group; “*” indicates p < 0.05). (E) Upper schematics of AAV8 vector engineered to overexpress Fmr1 and an eGFP Ctrl. Lower experimental paradigm for behavioral testing. (F) PWTs in Fmr1 KO mice detected at BL, after virus injection of either Fmr1 Ctrl or OE in ACC, further after CPN ligation (Two-way RM ANOVA, interaction: F(4;72) = 12.36, p < 0.01, Time: F(4;72) = 16.11, p < 0.01, Ctrl vs. OE: F(1;72) = 102.31, p < 0.01, n = 10/group; “**” indicates p < 0.01). Data are represented as mean ± SEM; ns, not significant.
We next re-expressed FMRP in the ACC of Fmr1 KO mice via Fmr1 overexpression (OE) virus (AAV8-PCMV-bGlobin-Fmr1-P2A-eGFP) (Figure 4E) and did not find any pronounced differences in PWTs between the Fmr1 Ctrl and OE virus groups (Figure 4F). Interestingly, in Fmr1 KO mice, compared with the Fmr1 Ctrl virus group, further CPN ligation significantly decreased the PWTs in Fmr1 OE group (Figure 4F). Therefore, the functions of cingulate FMRP in the regulation of hypersensitivity were not masked by the absence of FMRP in other areas. Together, these results reveal that FMRP is involved in pain hypersensitivity after the PRMT1 downregulation.
We next investigated the necessity of PRMT1 downregulation in developing peripheral pain hypersensitivity. We overexpressed PRMT1 in the ACC using AAV8-PCMV-bGlobin-Prmt1-P2A-eGFP (Figures 5A,B) and did not detect any remarkable changes in PWTs (Figure 5C) on day 21 after virus expression. We further ligated CPN and observed that, unlike mice with the Prmt1 Ctrl virus, CPN ligation did not decrease the PWTs (Figure 5C) in mice with the Prmt1 OE virus. Furthermore, the CFA injection did not change the PWTs (Figure 5D) in mice with the Prmt1 OE virus. The overexpression of PRMT1 did not notably affect motor function because neither the center time nor travel distance in the open-field test exhibited apparent change (data not shown). Taken together, these results suggest that overexpression of PRMT1 in the ACC prevents the development of peripheral hypersensitivity induced by peripheral inflammation or nerve injury.
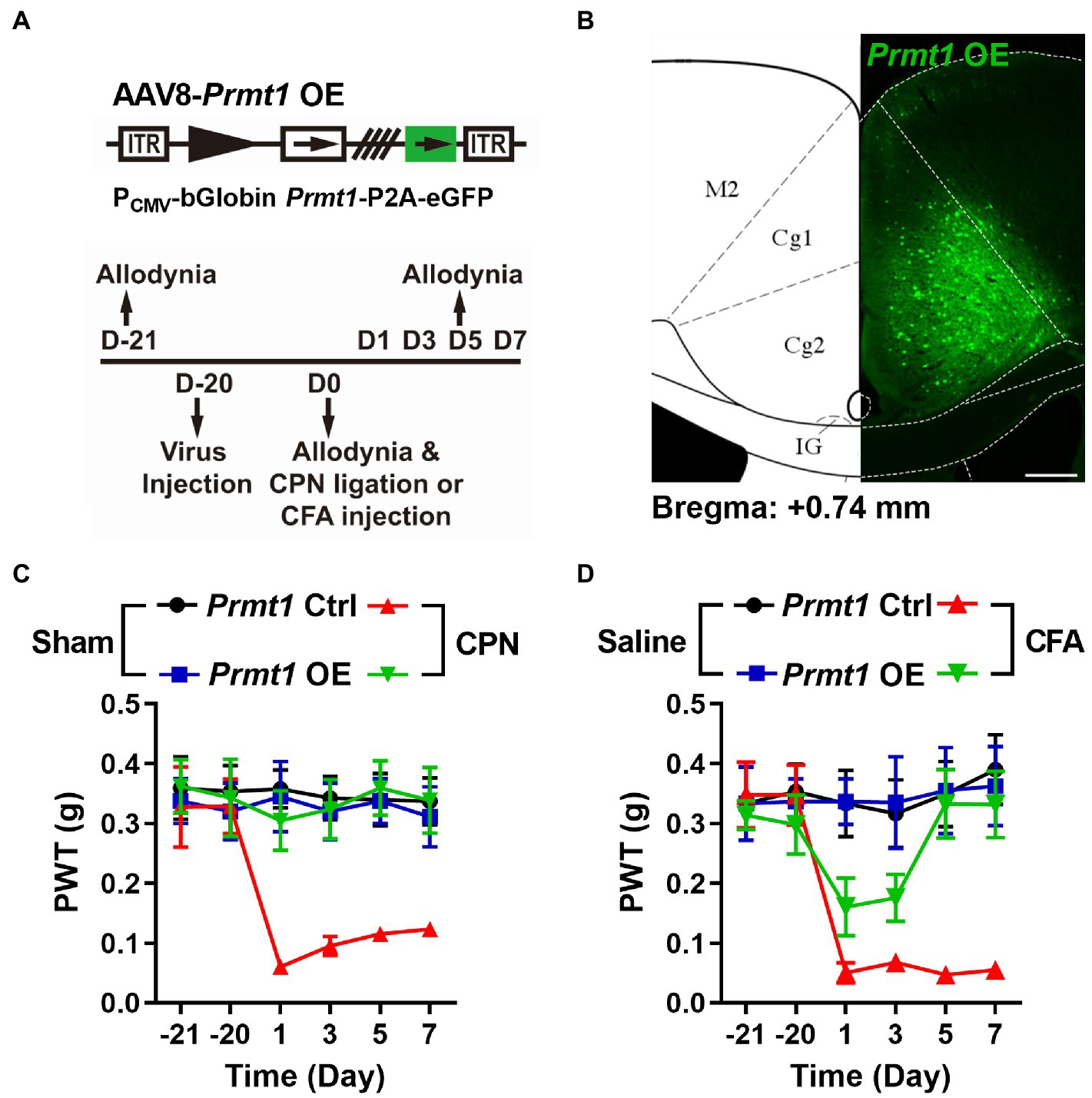
Figure 5. Downregulation of cingulate PRMT1 is necessary to develop peripheral hypersensitivity. (A) Upper schematics of AAV8 vectors engineered to overexpress PRMT1 and an eGFP Ctrl. Lower experimental paradigm for behavioral testing. (B) Illustration of bilateral AAV8-Prmt1 OE virus in the mouse ACC. (C) PWT tests conducted in both sham and CPN-ligated mice injected either Prmt1 Ctrl or OE virus in ACC (Sham Prmt1 Ctrl, n = 9; Sham Prmt1 OE, n = 8; CPN Prmt1 Ctrl, n = 9; CPN Prmt1 OE, n = 8, Two-way RM ANOVA, interaction: F(15;150) = 2.0, p < 0.05, Time: F(5;150) = 2.70, p < 0.05, Prmt1 Ctrl vs. Prmt1 OE: F(3;150) = 15.60, p < 0.01). (D) PWTs measured in both saline-and CFA-injected mice either with Prmt1 Ctrl or OE virus in ACC (Saline Prmt1 Ctrl, n = 6; Saline Prmt1 OE, n = 6; CFA Prmt1 Ctrl, n = 8; CFA Prmt1 OE, n = 10, Two-way RM ANOVA, interaction: F(15;130) = 3.40, p < 0.01, Time: F(5;130) = 4.72, p < 0.01, Prmt1 Ctrl vs. Prmt1 OE: F(3;130) = 11.41, p < 0.01). Data are represented as mean ± SEM; ns, not significant.
In the current study, we examined the possible role of PRMT1 in the development of peripheral pain hypersensitivity. We observed the downregulation of PRMT1 after peripheral nerve injury. Behaviorally, the downregulation of PRMT1 in the ACC was sufficient and necessary to develop peripheral pain hypersensitivity. However, in the Fmr1 KO mice, the role of the downregulation of PRMT1 on pain perception was abolished. Thus, to our knowledge, this is the first study to show that the downregulation of PRMT1 is essential for peripheral hypersensitivity.
In this work, we observed the downregulation of PRMT1 in the ACC of a kind of neuropathic pain model with CPN ligation. Also, arginine methylation is an essential part of protein post-translational modification, and PRMT1 is the predominant protein arginine methyltransferases in mammalian cells (Blanc and Richard, 2017). Mo and co-workers reported that the expression of coactivator-associated arginine methyltransferase 1 in the dorsal root ganglia was upregulated after nerve injury, contributing to peripheral hypersensitivity via epigenetic regulation (Mo et al., 2018). Unlike Mo′s study, we showed that PRMT1 is involved in pain regulation via the activities on FMRP.
Plus, we observed significant pain hypersensitivity through the ethological tests after either PRMT1 knockdown or PRMT1 activity inhibiting. We employed the genetic approach, downregulated the PRMT1 via shRNA in the ACC, and observed the apparent peripheral pain hypersensitivity. Thus, we provided solid evidence to show that the activities-dependent downregulation of PRMT1 was sufficient to develop peripheral pain hypersensitivity. Furthermore, overexpression of PRMT1 in ACC rescued mechanical allodynia of mice with peripheral nerve injury; this suggests that the downregulation of PRMT1 was necessary for hypersensitivity. Therefore, we provided new evidence for the protein arginine methylation’s involvement in the CNS’s functions.
FMRP is a crucial factor for the hypersensitivity induced by PRMT1 downregulation. Downregulation of cingulate PRMT1 failed to induce allodynia when FMRP was absent. The FMRP is broadly distributed in the CNS, and the lack of FMRP in other areas may mask PRMT1 downregulation in the ACC. Here we found that knockdown cingulate FMRP prevented the decrease of PWTs induced by nerve injury. Furthermore, nerve injury decreased the PWTs of Fmr1 KO mice with WT FMRP re-expression in the ACC. These data suggest that the cingulate FMRP is necessary for developing hypersensitivity after nerve injury. Furthermore, deficits of FMRP did not change the expression of PRMT1. Taking together, we concluded that in the ACC, the downregulation of PRMT1 was involved in developing hypersensitivity via changing the level of FMRP.
Arginine methylation on the C terminal is crucial to regulating FMRP functionally. Arginine methylation of FMRP was first observed by HeLa cell extracts (Liu and Dreyfuss, 1995). Later, it was reported that a synthetic peptide with RGG sequence presented in human FMRP could be methylated by rat brain extract (Ai et al., 1999). The exact methylated sites were identified in 2006 (Stetler et al., 2006). By mutating arginine to lysine or blocking methylation with adenosine-2′, 3′-dialdehyde (AdOx), it was found that the methylation of the RGG box modulated the quantity of bound mRNA by FMRP (Blackwell et al., 2010). Given that the arginine methylation regulates the condensation of FMRP with the binding RNA (Tsang et al., 2019), we also observed a higher ratio of condensed FMRP in the cultured neurons with the AMI-1 application; the downregulation of PRMT1 may modulate the condensation of FMRP, therefore changing the expression of excitatory synaptic transmission in the ACC.
Meanwhile, our study also provides promising practical pain management to administer drugs to ACC directly. Combined with our previous study (Wang et al., 2017), cannular microinjection delivered an analgesic to ACC, and the PWTs of mice were markedly increased. Therefore, precisely applying analgesics to ACC is a potential option for treating neuropathic pain.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the animal care and use committee of Zhejiang University approved all mice protocols.
CW, H-FS, and Y-JW performed western blot, immunostaining, mechanical allodynia testing, data analysis, and approved the draft. J-HW, Z-XZ, Y-NL, CW, and LL performed virus injection and allodynia testing, and analyzed the data. CZ provided the Fmr1 KO mice and analyzed data. X-YL designed the experiments, approved the draft, and wrote the paper. All authors contributed to the article and approved the submitted version.
This study was supported by the Hangzhou Science and Technology Plan Project (20190101A10), the National Natural Science Foundation of China (31871062, 82071181, and 32271042), the Fundamental Research Funds for the Central Universities, the MOE Frontier Science Center for Brain Science & Brain-Machine Integration, Zhejiang University.
We thank Shu-Min Duan and Hai-Lan Hu for their helpful suggestions on the manuscript. We thank Jing-Wei Zhao and Jian-Hong Luo for their suggestions on experiments. We thank Melissa Lepp. from the University of Toronto for the help with text editing. We are grateful to San-Hua Fang at the Core Facilities of Zhejiang University Institute of Neuroscience for technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ai, L. S., Lin, C. H., Hsieh, M., and Li, C. (1999). “Arginine methylation of a glycine and arginine rich peptide derived from sequences of human FMRP and fibrillarin,” Proceedings of the National Science Council, Republic of China. Vol. 23. Part B, Life sciences, 175–180.
Asiminas, A., Jackson, A. D., Louros, S. R., Till, S. M., Spano, T., Dando, O., et al. (2019). Sustained correction of associative learning deficits after brief, early treatment in a rat model of fragile X syndrome. Sci. Transl. Med. 11:eaao0498. doi: 10.1126/scitranslmed.aao0498
Bedford, M. T., and Clarke, S. G. (2009). Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 33, 1–13. doi: 10.1016/j.molcel.2008.12.013
Blackwell, E., Zhang, X., and Ceman, S. (2010). Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA. Hum. Mol. Genet. 19, 1314–1323. doi: 10.1093/hmg/ddq007
Blanc, R. S., and Richard, S. (2017). Arginine methylation: the coming of age. Mol. Cell. 65, 8–24. doi: 10.1016/j.molcel.2016.11.003
Bliss, T., Collingridge, G. L., Kaang, B. K., and Zhuo, M. (2016). Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci. 17. doi: 10.1038/nrn.2016.68
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M., and Yaksh, T. L. (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. doi: 10.1016/0165-0270(94)90144-9
Hagerman, R. J., Protic, D., Rajaratnam, A., Salcedo-Arellano, M. J., Aydin, E. Y., and Schneider, A. (2018). Fragile X-associated neuropsychiatric disorders (FXAND). Front. Psych. 9:564. doi: 10.3389/fpsyt.2018.00564
He, C. X., Cantu, D. A., Mantri, S. S., Zeiger, W. A., Goel, A., and Portera-Cailliau, C. (2017). Tactile defensiveness and impaired adaptation of neuronal activity in the Fmr1 knock-out mouse model of autism. J. Neurosci. 37, 6475–6487. doi: 10.1523/JNEUROSCI.0651-17.2017
Juczewski, K., von Richthofen, H., Bagni, C., Celikel, T., Fisone, G., and Krieger, P. (2016). Somatosensory map expansion and altered processing of tactile inputs in a mouse model of fragile X syndrome. Neurobiol. Dis. 96, 201–215. doi: 10.1016/j.nbd.2016.09.007
Kazdoba, T. M., Leach, P. T., Silverman, J. L., and Crawley, J. N. (2014). Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare. Dis. Res. 3, 118–133. doi: 10.5582/irdr.2014.01024
Kim, T. H., Tsang, B., Vernon, R. M., Sonenberg, N., Kay, L. E., and Forman-Kay, J. D. (2019). Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 365, 825–829. doi: 10.1126/science.aax4240
Kozono, N., Okamura, A., Honda, S., Matsumoto, M., and Mihara, T. (2020). Gamma power abnormalities in a Fmr1-targeted transgenic rat model of fragile X syndrome. Sci. Rep. 10:18799. doi: 10.1038/s41598-020-75893-x
Livak, K. J., and Schmittgen, T. D. (2001). ‘Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.’ Methods (San Diego, Calif.) 25, 402–408. doi: 10.1006/meth.2001.1262
Liu, Q., and Dreyfuss, G. (1995). In vivo and in vitro arginine methylation of RNA-binding proteins. Mol. Cell. Biol. 15, 2800–2808. doi: 10.1128/mcb.15.5.2800
Martinez, L. A., and Tejada-Simon, M. V. (2018). Pharmacological Rescue of Hippocampal Fear Learning Deficits in fragile X syndrome. Mol. Neurobiol. 55, 5951–5961. doi: 10.1007/s12035-017-0819-5
Miller, L. J., McIntosh, D. N., McGrath, J., Shyu, V., Lampe, M, Taylor, A. K., et al. (1999). Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am. J. Med. Genet. 83, 268–279.
McBride, A. E., and Silver, P. A. (2001). State of the Arg: protein methylation at arginine comes of age. Cell 106, 5–8. doi: 10.1016/S0092-8674(01)00423-8
Mo, K., Xu, H., Gong, H., Lei, H., Wang, Y., Guo, W., et al. (2018). Dorsal root ganglia Coactivator-associated arginine Methyltransferase 1 contributes to peripheral nerve injury-induced pain hypersensitivities. Neuroscience 394, 232–242. doi: 10.1016/j.neuroscience.2018.10.038
Price, T. J., Rashid, M. H., Millecamps, M., Sanoja, R., Entrena, J. M., and Cervero, F. (2007). Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J. Neurosci. 27, 13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007
Prieto, M., Folci, A., Poupon, G., Schiavi, S., Buzzelli, V., Pronot, M., et al. (2021). Missense mutation of Fmr1 results in impaired AMPAR-mediated plasticity and socio-cognitive deficits in mice. Nat. Commun. 12:1557. doi: 10.1038/s41467-021-21820-1
Stetler, A., Winograd, C., Sayegh, J., Cheever, A., Patton, E., Zhang, X., et al. (2006). Identification and characterization of the methyl arginines in the fragile X mental retardation protein Fmrp. Hum. Mol. Genet. 15, 87–96. doi: 10.1093/hmg/ddi429
Tsang, B., Arsenault, J., Vernon, R. M., Lin, H., Sonenberg, N., Wang, L. Y., et al. (2019). Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl. Acad. Sci. U. S. A. 116, 4218–4227. doi: 10.1073/pnas.1814385116
Vadakkan, K. I., Jia, Y. H., and Zhuo, M. (2005). A behavioral model of neuropathic pain induced by ligation of the common Peroneal nerve in mice. J. Pain 6, 747–756. doi: 10.1016/j.jpain.2005.07.005
Wang, L., Almeida, L. E. F., Nettleton, M., Khaibullina, A., Albani, S., Kamimura, S., et al. (2016). Altered nocifensive behavior in animal models of autism spectrum disorder: the role of the nicotinic cholinergic system. Neuropharmacology 111, 323–334. doi: 10.1016/j.neuropharm.2016.09.013
Wang, Y. J., Zuo, Z. X., Wu, C., Liu, L., Feng, Z. H., and Li, Y. X. (2017). Cingulate alpha-2A adrenoceptors mediate the effects of clonidine on spontaneous pain induced by peripheral nerve injury. Front. Mol. Neurosci. doi: 10.3389/fnmol.2017.00289
Xu, H., Wu, L. J., Wang, H., Zhang, X., Vadakkan, K. I., Kim, S. S., et al. (2008). Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci: the official journal of the Society for Neuroscience 28, 7445–7453. doi: 10.1523/JNEUROSCI.1812-08.2008
Keywords: protein arginine methyltransferases 1, peripheral hypersensitivity, cingulate cortex, peripheral nerve injury, fragile X messenger ribonucleoprotein
Citation: Wu C, Shang H-F, Wang Y-J, Wang J-H, Zuo Z-X, Lian Y-N, Liu L, Zhang C and Li X-Y (2023) Cingulate protein arginine methyltransferases 1 regulates peripheral hypersensitivity via fragile X messenger ribonucleoprotein. Front. Mol. Neurosci. 16:1153870. doi: 10.3389/fnmol.2023.1153870
Edited by:
Silke Neumann, University of Otago, New ZealandReviewed by:
Takatoshi Iijima, Tokai University, JapanCopyright © 2023 Wu, Shang, Wang, Wang, Zuo, Lian, Liu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang-Yao Li, TGl4aWFuZ3lAemp1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.