
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 30 March 2023
Sec. Brain Disease Mechanisms
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1121944
 Shu-Hao Miao1†
Shu-Hao Miao1† Sheng-Qing Gao2†
Sheng-Qing Gao2† Hui-Xin Li3
Hui-Xin Li3 Yun-Song Zhuang1Xue Wang2
Yun-Song Zhuang1Xue Wang2 Tao Li1
Tao Li1 Chao-Chao Gao1
Chao-Chao Gao1 Yan-Ling Han2Jia-Yin Qiu2
Yan-Ling Han2Jia-Yin Qiu2 Meng-Liang Zhou1*
Meng-Liang Zhou1*Introduction: Endothelial nitric oxide synthase (eNOS) uncoupling plays a significant role in acute vasoconstriction during early brain injury (EBI) after subarachnoid hemorrhage (SAH). Astrocytes in the neurovascular unit extend their foot processes around endothelia. In our study, we tested the hypothesis that increased nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) expression in astrocytes after SAH leads to eNOS uncoupling.
Methods: We utilized laser speckle contrast imaging for monitoring cortical blood flow changes in mice, nitric oxide (NO) kits to measure the level of NO, and a co-culture system to study the effect of astrocytes on endothelial cells. Moreover, the protein levels were assessed by Western blot and immunofluorescence staining. We used CCK-8 to measure the viability of astrocytes and endothelial cells, and we used the H2O2 kit to measure the H2O2 released from astrocytes. We used GSK2795039 as an inhibitor of NOX2, whereas lentivirus and adeno-associated virus were used for dihydrofolate reductase (DHFR) knockdown in vivo and in vitro.
Results: The expression of NOX2 and the release of H2O2 in astrocytes are increased, which was accompanied by a decrease in endothelial DHFR 12 h after SAH. Moreover, the eNOS monomer/dimer ratio increased, leading to a decrease in NO and acute cerebral ischemia. All of the above were significantly alleviated after the administration of GSK2795039. However, after knocking down DHFR both in vivo and in vitro, the protective effect of GSK2795039 was greatly reversed.
Discussion: The increased level of NOX2 in astrocytes contributes to decreased DHFR in endothelial cells, thus aggravating eNOS uncoupling, which is an essential mechanism underlying acute vasoconstriction after SAH.
Subarachnoid hemorrhage (SAH), primarily caused by ruptured aneurysm, is a common neurologically critical disease with high morbidity and mortality (Macdonald and Schweizer, 2017). As more than half of the patients died within 48 h of SAH and reversal of large-arterial spasm does not improve outcomes, early brain injury (EBI) that occurs within 72 h after SAH seems to be the main target of future research (Mayberg et al., 1994; Hop et al., 1997; Wang et al., 2012). One of the main features of EBI after SAH is a severe reduction in cerebral blood flow (CBF) (Adams et al., 1981; Schubert et al., 2009). Acute vasoconstriction, which has been observed in both experimental animals and patients after SAH, is a putative mechanism responsible for the decreased CBF in EBI (Bederson et al., 1995; Schubert et al., 2009; Yang et al., 2018). However, the mechanisms underlying the formation of acute cerebral vasoconstriction have not yet been fully elucidated, and effective treatments for SAH are still being explored (Ayer and Zhang, 2008).
Evidence suggests that oxidative stress is a factor leading to vasoconstriction after hemorrhage, but the specific mechanism by which oxidative stress regulates cerebral vasoconstriction still requires further study (Macdonald and Weir, 1994; Ayer and Zhang, 2008). As one of the main sources of reactive oxygen species (ROS) after SAH, nicotinamide adenine dinucleotide phosphate oxidase (NOX) has received extensive attention. To date, seven NOX isoforms (NOX1-5 and DUOX1-2) have been discovered (Cheng et al., 2001; Bedard and Krause, 2007). Among them, nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) is highly expressed in cells throughout the central nervous system, including astrocytes (Cheng et al., 2001; Zhang et al., 2017). Several studies have found that increased NOX expression in the cerebrovascular system is associated with ROS-mediated vasospasm after SAH, while inhibition of NOX can reduce arterial constriction and improve CBF (Shin et al., 2002; Paravicini and Sobey, 2003; Miller et al., 2006). However, no previous study has determined the specific mechanism of acute vasoconstriction after SAH caused by increased NOX2 expression.
Reactive oxygen species, such as superoxide anion (O2–), and hydrogen peroxide (H2O2), are widely involved in cell signaling (Thannickal and Fanburg, 2000; Thannickal et al., 2000; Waghray et al., 2005). H2O2 is generally less reactive, longer-lived, and more stable than other ROS such as O2– in most biological environments (Waghray et al., 2005). Normally, NOX2 produces O2–, which is converted to stable H2O2 by abundant, and ubiquitous extracellular and extracellular superoxide dismutase (Marklund, 1984; Jung et al., 2003). In addition, H2O2 is lipid soluble and can cross the plasma membrane to access targets in the cytoplasm to drive intracellular signaling cascades. Aquaporin (AQP) is a membrane protein that bidirectionally transports H2O and H2O2 across the membrane (Winterbourn, 2018). AQP is expressed in the plasma membrane and organelle membrane of cells throughout the animal kingdom (Lindskog et al., 2016; Nauseef, 2019). It is reasonable to guess that H2O2 released by astrocytes can enter endothelial cells.
It has been demonstrated that ROS generated by increased NOX2 expression contributes to brain endothelial dysfunction in angiotensin II-dependent hypertension, aging, and hypercholesterolemia (Iadecola et al., 2009; Miller et al., 2010; Chrissobolis et al., 2012). Nitric oxide (NO) is an important mediator in maintaining normal cerebrovascular function. Both clinical and experimental studies have shown a decline in NO after SAH (Schwartz et al., 2000; Sehba et al., 2000; Terpolilli et al., 2015, 2016). This early NO reduction is associated with endothelial dysfunction and is an important mechanism for early vasoconstriction in EBI (Sehba et al., 1999; Terpolilli et al., 2016). Endothelial nitric oxide synthase (eNOS) is a well-known member of the NOS enzyme family, which is highly expressed in endothelial cells, and eNOS is the main source of NO produced by endothelial cells (Förstermann and Sessa, 2012; Bendall et al., 2014). eNOS is a homodimer that can be converted from the dimer producing NO to the monomer producing superoxide anion in a process known as eNOS uncoupling (Förstermann and Sessa, 2012). Although the mechanism of eNOS uncoupling has not been fully elucidated, oxidation of the eNOS cofactor tetrahydrobiopterin (BH4) appears to be the main cause (Xia et al., 1998; Laursen et al., 2001). Dihydrofolate reductase (DHFR) catalyzes the regenerated BH4 from dihydrobiopterin (BH2), the oxidized form of BH4, in several cell types (Thöny et al., 2000). Moreover, studies have shown that DHFR deficiency leads to eNOS uncoupling by decreasing BH4 levels, thereby contributing to the development of vascular disease (Oak and Cai, 2007; Gao et al., 2009). Therefore, we hypothesized that this pathway may be involved in SAH leading to acute vasoconstriction.
Neurons, astrocytes, and endothelial cells are the main cells in the neurovascular unit, and they interact to ensure adequate levels of CBF in all areas of the brain (Abbott et al., 2006); this is the anatomical basis for the hypothesis that NOX2 in astrocytes may induce endothelial dysfunction through eNOS uncoupling after SAH, which was explored in this study.
All animal procedures were approved by the Jinling Hospital Animal Care and Use Committee and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication Nos. 80–23). Male C57/BL6 mice (6–8 w, 22–26 g) were purchased from Nanjing University (Nanjing, China). These mice were kept in an air-conditioned room with air humidity (50 ± 10%) and constant temperature (26 ± 2°C), under a 12 h light/dark cycle. All animals were provided with adequate food and water.
Human astrocytes (HA) cells were purchased from BeNa Culture Collection (Kunshan, China) and were cultured in a medium containing 90% high glucose DMEM, 10% fetal bovine serum (purchased from Gibco BRL, Grand Island, NY, USA). Human cerebral microvascular endothelial cells (hCMEC/D3) were purchased from ABM technology (Zhenjiang, China) and were cultured in a medium containing 89% Prigrow I Medium (TM001), 10% fetal bovine serum, and 1% penicillin-streptomycin. We created an in vitro SAH model by adding 20 μmol/L of oxyhemoglobin (OxyHb) (H2625, Sigma-Aldrich, St. Louis, MO, USA) to the above medium. The OxyHb we purchased from sigma is a qualified product derived from bovine blood. The hCMEC/D3 cell line is a stable, extensively characterized, and well-differentiated human brain endothelial cell line and it is suitable for research on blood–brain barrier function and the response of brain vascular endothelium to inflammation (Weksler et al., 2013).
An experimental SAH model was established by injecting autologous arterial blood into the prechiasmatic cistern of the optic chiasm as described previously, with minor modifications (Deng et al., 2021). The mice were anesthetized using 100% O2 containing 2% isoflurane and fixed on a stereotaxic apparatus while maintaining anesthesia with 1% isoflurane. Under sterile conditions, we made an incision in the midline of the mouse scalp to expose the skull and subsequently drilled a 0.9 mm depth hole which located 4 mm in front of the bregma. A 27-gauge needle was passed through the burr hole at a 40° angle to the base of the skull. For the SAH group, 50 μl non-heparinized autologous femoral arterial blood was slowly injected into the prechiasmatic cisterna of mice using a syringe pump over 40 s. For the sham group, we injected an equal volume of normal saline. The 27-gauge needle was gently withdrawn 2 min after the blood/saline injections to prevent the backflow of blood and the leakage of cerebrospinal fluid. After sealing the hole using bone wax, the incision was sutured immediately. After injection, we monitored the mice for 40 min for recovery and then returned to their cages.
The schematic diagram of experimental design is demonstrated in Supplementary Figure 1. Our research is containing in vivo and in vitro parts. For the in vivo experiments, we first used laser speckle contrast imaging (LSCI) to compare the CBF of the sham and the 12 h groups after SAH (n = 6 each group). The mice in sham group were euthanized 12 h after operation (n = 6). We measured the time change of NOX2 expression in the temporal lobe of mice, and the change of NO and DHFR levels in endothelial cells extracted from the temporal lobe (12 h, 24 h, and 3 days; n = 6 each group). According to the results of the above experiments, we chose 12 h for further experiments. Then, 12 mice were randomly divided into sham group (n = 6), and 12 h group (n = 6), and collected their brain samples for immunofluorescence staining (Supplementary Figure 1A). To verify the hypothesized pathway, the mice were randomly divided into the following six groups: SAH, Vehicle + SAH, GSK + SAH(1), GSK + SAH(2), AAV-shDHFR + GSK + SAH, and AAV-shCtrl + GSK + SAH groups. For GSK/Vehicle + SAH group, two hours after the injection of GSK/Vehicle into the lateral ventricle of mice, we began to induce experimental SAH in vivo. For AAV-shCtrl/AAV-shDHFR + GSK + SAH group, AAV-shCtrl/AAV-shDHFR were administrated via tail vein injection 14 days before GSK injection. Before euthanasia of the mice in the above experiment, Neurological Score (n = 6 each group) and LSCI (n = 6 each group) were performed. As previously mentioned, their temporal lobe samples were gathered for NO content measurement (n = 6 each group), Western blot (n = 6 each group) and immunofluorescence staining (n = 6 each group) (Supplementary Figure 1B). For in vitro experiments, HA cells contained five groups: Control, OxyHb(1), OxyHb(2), Vehicle + OxyHb, and GSK + OxyHb groups. hCMEC/D3 cells contained the Control group and the OxyHb group. HA cells were co-cultured with hCMEC/D3 cells to explore the effect of astrocytes on endothelial cells, and the co-cultured cells were divided into six groups: AS + OxyHb, AS + Vehicle + OxyHb, AS + GSK + OxyHb(1), AS + GSK + OxyHb(2), LV-shCtrl + AS + GSK + OxyHb, and LV-shDHFR + AS + GSK + OxyHb groups. In the AS + Vehicle/GSK + OxyHb group, after using GSK/Vehicle alone on astrocytes for 2 h, we co-cultured astrocytes with endothelial cells and replaced the medium containing GSK/Vehicle with the medium containing OxyHb without GSK/Vehicle. We used DMSO as the vehicle in the in vitro experiments, and the dose of GSK2795039 was 20 μmol/ml. For LV-shCtrl/LV-shDHFR + AS + GSK + OxyHb groups, 48 h before co-culture, LV-shCtrl/LV-shDHFR were added to hCMEC/D3 cell culture medium. Vehicle/GSK were added to HA cell culture medium 2 h before co-culture. When HA cells were co-cultured with hCMEC/D3 cells, the culture medium containing OxyHb without GSK/Vehicle was replaced. The 6-well plates cultured cells were collected for Western blot (WB, n = 6 each group) while their corresponding culture medium of hCMEC/D3 cells was collected to evaluate the NO level (n = 6 each group). We also collected the medium of astrocytes to measure the amount of H2O2 released by astrocytes (n = 6 each group). In addition, we did immunofluorescence staining (IF)in 12-well plates cultured cells (n = 6 each group). We used 96-well plates to measure the viability of astrocytes and endothelial cells (n = 6 each group) (Supplementary Figure 1C).
Laser speckle contrast imaging is based on blurring the interference pattern of scattered laser light through the flow of blood cells to instantaneously visualize blood perfusion in the circulation. We have been measured CBF and other experimental operations on mice on a constant temperature pad at 37°C, and we used air conditioning to maintain the room temperature at 28°C. Briefly, mice were anesthetized with gas anesthesia (2% isoflurane in 100% O2), their heads were fixed in a stereotaxic frame, and they were subjected to continuous inhalation of 1% isoflurane throughout the experiment. We made an incision in the midline of the mouse scalp. The blood flow was detected by a CCD camera, and image acquisition was performed using custom software. The same regions of interest were selected for different cortical samples and analyzed by two independent pathological researchers who were blinded to the groupings.
We isolated cerebral blood vessels in the temporal lobe of brain, as previously published, with minor modifications (Bales et al., 2016). After cerebellum and olfactory bulb were removed, we homogenized the remaining brain tissue in 3 ml 4°C sucrose buffer (5 mM HEPES, 0.32 M sucrose, pH 7.4) with homogenizer. Then the homogenate of brain samples was centrifuged at 1,000 g for 12 min, after which the supernatant mainly containing neurons and the dense white myelin sheath on the top of the granules were discarded. Then we resuspended the particles in 3 ml 4°C sucrose solution and centrifuged them at 1,000 g for 12 min. The final pellet, which contained numerous cerebral blood vessels, was lysed in radioimmunoprecipitation assay which contained 1% phenylmethanesulfonyl fluoride (PMSF) for WB or NO content test.
Nitric oxide content of the extracted cortical blood vessels or culture medium was detected using a total nitric oxide assay kit (S0024, Beyotime). In brief, 80 μl of the extracted sample was added in a 96-well plate at room temperature, protected from light. Then, 7.5 μl of 2 mM nicotinamide adenine dinucleotide phosphate (NADPH), 15 μl of flavin adenine dinucleotide (FAD), and 7.5 μl of nitrate reductase were added to the sample sequentially, mixed, and incubated at 37°C for 30 min. Subsequently, 15 μl of lactate dehydrogenase (LDH) buffer and 15 μl of LDH were added, mixed, and incubated at 37°C for an additional 30 min. Finally, 50 μl of Griess reagent I and 50 μl of Griess reagent II were added, mixed, and incubated for 10 min at room temperature. The absorbance of each well was measured at 540 nm using a microplate reader. We quantified the protein content of the different samples and compared the NO levels at the same protein concentration.
For Western blot, cells, and tissues were lysed in radioimmunoprecipitation assay buffer containing 1% PMSF and 1% phosphatase inhibitor cocktail (P5726, Sigma-Aldrich). The protein content was quantified using the Bicinchoninic acid protein assay (BCA) kit (P0011, Beyotime). The molecular weight of NOX2 is 60 kDa, the molecular weight of DHFR is 20 kDa, the molecular weight of eNOS monomer is 140 kDa, and the molecular weight of eNOS dimer is 280 kDa. The samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. After the membranes were blocked in 5% skim milk at room temperature for 2 h, they were incubated with the following primary antibodies at 4°C overnight: anti-β-actin (1:5,000, 81115-1-RR, Proteintech, Chicago, IL, USA), anti-eNOS (1:500, 9572, Cell Signaling Technology, Danvers, MA, USA), anti-NOX2 (1:1,000, ab129068, Abcam) and anti-DHFR (1:2,000, ab124814, Abcam, Cambridge, MA, USA). The membranes were incubated using the appropriate secondary antibody (1:10,000, 7074, Cell Signaling Technology) for 1 h. The membrane is divided into distinct bands containing β-actin and other proteins of interest. Finally, the immunoreactive bands were detected using the enhanced chemiluminescence system. ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to analyze the relative densities of at least six independent experiments.
The mouse brain sections (6 μm) were incubated with primary antibodies against NOX2 (1:200, ab133303, Abcam), DHFR (1:200, ab124814, Abcam), GFAP (1:200, 2389127, Invitrogen, Carlsbad, CA, USA), and CD31 (1:200, ab24590, Abcam) at 4°C overnight. GFAP is considered a standard marker of mature astrocytes and it is often used to label astrocytes in IF. CD31 is also commonly used to label endothelial cells in IF. So, we used CD31 as a marker to distinguish endothelial cells and GFAP as a marker to distinguish astrocytes. The brain slices were incubated using the corresponding goat anti-rabbit IgG antibodies (1:250, SA00009-2, Proteintech) or goat anti-mouse IgG (1:250, SA00013-1, Proteintech) for 1.5 h at room temperature. Subsequently, the brain sections were sealed with anti-fluorescence solution after incubated DAPI (C1005, Beyotime) for 6 min. Finally, the sections were observed and captured under the fluorescence microscope. Immunofluorescence staining of hCMEC/D3 and HA was similar to brain sections after fixing cells with 4% paraformaldehyde.
The co-culture system consisted of lower and upper chambers separated by a selectively permeable membrane (Corning, Transwell 3450) with 0.4 μm-diameter pores. HA cells were plated in the upper chamber, hCMEC/D3 cells were plated in the lower chamber, and both were co-cultured to mimic neurovascular units.
We measure cell viability using the Cell Counting Kit 8 (CCK-8). HA was seeded in 96-well plates at a density of 5,000 cells/well, and then treated as described in the experimental design for different groups. Because 96-well plates cannot be used for co-cultivation, we diluted different groups of hCMEC/D3 to the same multiple after treatment and seeded them in 96-well plates. After the cells were grown in the incubator for 24 h, they were washed 3 times with medium. Cells were subsequently incubated with CCK-8 solution and culture medium (diluted 1:10) at 37°C for 2 h, and then the absorbance was measured at 450 nm with a microplate reader.
Hydrogen peroxide was measured in vitro using a hydrogen peroxide detection kit (S0038, Beyotime) and 96-well plates as previously described (Gao et al., 2022). Briefly, the media of astrocytes after different groups of treatments were collected. We added 50 μl of sample to each well, followed by 100 μl of detection reagent, mixed them and incubated them at room temperature for 30 min. The absorbance of each well was measured at 560 nm using a microplate reader, and then the hydrogen peroxide level in the medium was calculated according to the standard concentration curve.
The mice were anesthetized using 100% O2 containing 2% isoflurane and their heads were fixed on a stereotaxic apparatus while maintaining anesthesia with 1% isoflurane. Under sterile conditions, an incision was made in the middle of the mouse scalp to expose the skull, and drills were used to drill 1.0 mm posterior to the bregma and 2.0 mm lateral to the bregma until the dura was penetrated. The vehicle-diluted GSK2795039 (HY-18950, MedChemExpress, NJ, USA) was injected vertically into the lateral ventricle through the hole at a flow rate of 2 μl/min using a syringe pump. For the vehicle group mice, an equal volume of vehicle was injected into the lateral ventricle. In the in vivo experiment, we mixed 10% DMSO, 40% PEG300, 5% Tween-80, and 45% saline as the vehicle, and the dose of GSK2795039 was 3 mg/kg. We kept the vehicle and GSK2795039 in a 37°C water bath before intraventricular injection. The needle was left in place after injection for 5 additional min before immediately sealing the drilled hole using bone wax. We observed the mice for 1 h during recovery before proceeding to the next experimental operation.
Lentiviral vectors which contained shRNA to knock down DHFR (the targeted sequence was CCUCUUCAGUAGAAGGUAATT; Lv-shDHFR) were obtained from Hanbio Biotechnology (Shanghai, China) and 30% proliferating fused hCMEC/D3 was incubated with the Lv-shDHFR mixture for 48 h before the next experiment.
Neurological function was assessed in mice using a modified Garcia Score by two investigators who were blinded to the grouping, using the following six separate tests: locomotion, symmetry of limb movements, forepaw extension, climbing, body proprioception, and response to touch (Gao et al., 2020). After calculating the total of the individual scores of all tests, the lowest neurobehavioral score was 3 points, which represented severely impaired neurological function, and the highest score was 18 points, which represented normal neurological function.
Statistical analysis was performed using Prism 8.02 (GraphPad Software, La Jolla, CA, USA). For each group, the values are expressed as the mean ± standard deviation (mean ± SD). Statistical differences were determined using one-way analysis of variance (ANOVA) when more than two groups were present. When ANOVA showed a significant difference, Tukey’s post-hoc test for multiple comparisons was applied to evaluate that difference. When only two groups were compared, student t-test was utilized. Statistical significance was inferred when the p-value was <0.05.
We applied the prechiasmatic cistern injection model in vivo. We found that the bottom of the brain of mice in the 12 h group was filled with blood compared with the Sham group, demonstrating that the injection model well mimics the situation after SAH in humans (Figure 1A). We performed laser speckle examination on the mice in the Sham and 12 h groups (Figure 1B). The analysis results showed that compared to the sham group, the CBF of the mice in the 12 h group decreased significantly (Figure 1C). We examined the expression of DHFR, eNOS monomer, eNOS dimer, and NO content in cerebral vessels isolated from the temporal lobe. The results showed that compared to the Sham group, the endothelial NO content in the mice in the 12 h groups decreased significantly (Figure 1D). The results of Western blot showed that the eNOS monomer/dimer ratio was significantly increased at 12 h after SAH, which indicated that the uncoupling of eNOS was significantly increased 12 h after SAH (Figures 1E, F). The results of Western blot showed that NOX2 was significantly increased 12 h after SAH (Figures 1G, H). Therefore, we chose to conduct follow-up experiments 12 h after SAH. Likewise, we chose 12 h after SAH as the time point for Immunofluorescence staining analysis. Immunofluorescence staining showed the expression of NOX2 in astrocytes, which was notably increased 12 h after SAH (Figure 1K). The results of Western blot showed that compared to the sham group, DHFR decreased significantly at 12 h, 24 h, and 3 days after SAH, and the decrease was more obvious at 12 h (Figures 1I, J). Furthermore, Immunofluorescence staining showed that DHFR was mainly located in endothelial cells, and the expression of DHFR was significantly decreased 12 h after SAH (Figure 1L).
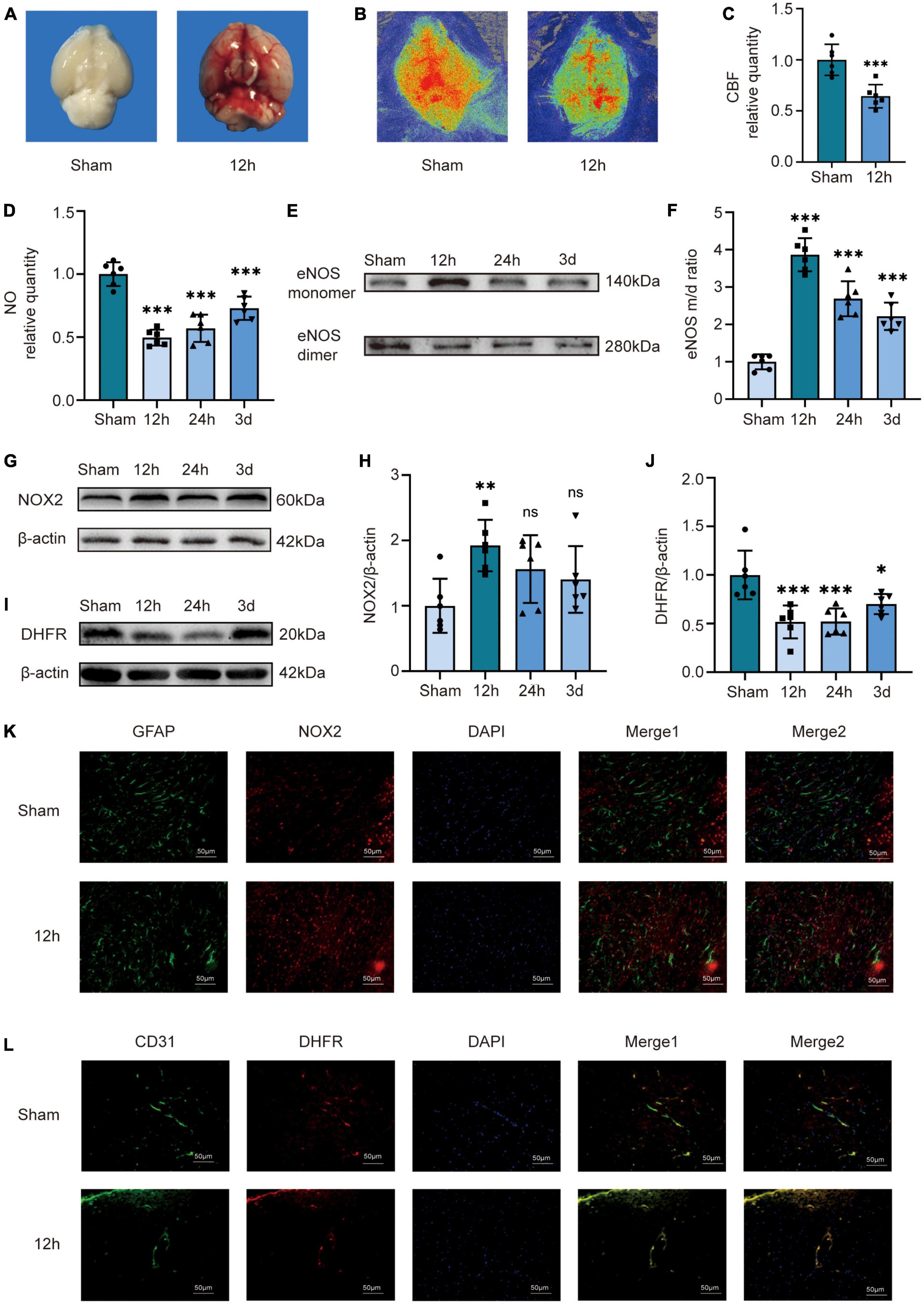
Figure 1. Cerebral blood flow, NO content, NOX2, DHFR, and the ratio of eNOS monomer/dimer in the temporal lobes of mice at different time courses after SAH in mice. (A) Compared to the Sham group, there was a larger amount of blood at the bottom of the brains of the 12 h group mice. (B) Representative LSCI images in these two groups. (C) Quantitative analysis of CBF in these two groups. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001). (D) NO content in extracted endothelial cells of the Sham group and different time course groups after SAH. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001). (E,F) Representative Western blot images and quantitative analysis of eNOS monomer and eNOS dimer expression in the Sham group and different time course groups after SAH by ImageJ. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001). (G,H) Representative Western blot images and quantitative analysis of NOX2 expression in the temporal lobes of the Sham group and different time-course groups after SAH by ImageJ. Data are shown as mean ± standard deviation (n = 6 mice each group, **p < 0.01, ns: no statistically significant difference). (I,J) Representative Western blot images and quantitative analysis for endothelial DHFR expression in the temporal lobe of mice in the Sham group and different time course groups after SAH by ImageJ. Data are shown as mean ± SD (n = 6 mice each group, *p < 0.05, ***p < 0.001, ns: no statistically significant difference). (K) Immunofluorescence images showed GFAP and NOX2 colocalization. All scale bars, 50 μm. (L) Immunofluorescence images showed CD31 and DHFR colocalization. All scale bars, 50 μm. CBF, cerebral blood flow; eNOS, endothelial nitric oxide synthase; NOX2, nicotinamide adenine dinucleotide phosphate oxidase 2; GFAP, glial fibrillary acidic protein; DHFR, dihydrofolate reductase.
Subsequently, we used the SAH in vitro model to verify the above results. To explore the effect of astrocytes on endothelial cells, we co-cultured endothelial cells with astrocytes (Figure 2A). The data showed that the NO release from endothelial cells in the lower chamber medium in the OxyHb group was significantly lower than that in the control group and the NO in the medium of AS + OxyHb group was lower than that in the OxyHb group (Figure 2B). According to the results of Western blot, the OxyHb group showed a significant increase in the ratio of eNOS monomer/dimer in endothelial cells compared to the control group (Figures 2C, D). More importantly, the eNOS monomer/dimer ratio was significantly increased in endothelial cells in the AS + OxyHb group when compared to the OxyHb group (Figures 2C, D). The expression of DHFR also showed opposite trends to those of the eNOS monomer/dimer ratios (Figures 2E, F). Consistent with the results of Western blot, Immunofluorescence staining indicated that DHFR expression was significantly decreased in endothelial cells 12 h after SAH (Figure 2G). Taken together, these findings indicate that the presence of astrocytes increases endothelial cell eNOS uncoupling after SAH. Simultaneously, we also found that the expression of NOX2 in astrocytes increased significantly 12 h after adding OxyHb (Figures 2H, I). Moreover, in line with the results of Western blot, Immunofluorescence staining showed similar changes in NOX2 expression in astrocytes from diverse groups (Figure 2J).

Figure 2. Alterations in NO release, NOX2 and DHFR expression, and eNOS monomer/dimer ratio in endothelial cells after SAH. (A) Diagram of co-culture of HA and hCMEC/D3 cells. (B) NO released by endothelial cells among different experimental groups. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001, ns: no statistically significant difference). (C,D) Representative Western blot images and quantitative analysis of eNOS monomer and eNOS dimer expression by ImageJ. Data are shown as mean ± SD (n = 6 each group, **p < 0.01, ***p < 0.001). (E,F) Representative Western blot images and quantitative analysis of endothelial DHFR expression by ImageJ. Data are shown as mean ± SD (n = 6 each group, **p < 0.01). (H,I) Representative Western blot images and quantitative analysis of astrocytes NOX2 expression by ImageJ. Data are shown as mean ± SD (n = 6 each group, **p < 0.01). (G) Immunofluorescence images showed the co-localization of CD31 and DHFR. All scale bars, 50μm. (J) Immunofluorescence images showed GFAP and NOX2 colocalization. All scale bars, 50 μm.
Because the above in vivo and in vitro results were similar, we set out to validate our pathway using NOX2 inhibitors and Lv-shDHFR. To further verify the effect of NOX2 in astrocytes on endothelial cells, we used GSK2795039 to inhibit NOX2 in astrocytes and co-cultured astrocytes with endothelial cells. After adding OxyHb for 12 h, we found no significant difference in NO content, eNOS monomer/dimer ratio, and DHFR expression in the AS + OxyHb and AS + vehicle + OxyHb groups but surprisingly found that compared to the AS + OxyHb group, the NO content was significantly increased in the AS + GSK + OxyHb group (Figure 3A). The results of Western blot showed that compared to the AS + OxyHb group, the eNOS monomer/dimer ratio in the AS + GSK + OxyHb group was significantly decreased (Figures 3B, C) and the expression of DHFR was significantly increased (Figures 3D, E). Moreover, compared to the AS + OxyHb group, the DHFR expression was significantly increased in endothelial cells in the AS + GSK + OxyHb group (Figure 3F). We found that there was no significant difference in the viability of astrocytes in the OxyHb group, Vehicle + OxyHb group, and GSK + OxyHb group (Supplementary Figure 2A). However, we found that the viability of endothelial cells in the AS + GSK + OxyHb group was significantly increased compared with the AS + OxyHb group, while there was no significant difference in the viability of endothelial cells between the AS + OxyHb group and the AS + Vehicle + OxyHb group (Supplementary Figure 2B). We found that compared with the Control group, the H2O2 released from astrocytes in the medium in the OxyHb group was significantly increased, while compared with the OxyHb group, the H2O2 in the GSK + OxyHb group was significantly decreased (Supplementary Figure 3). This proves that a large part of H2O2 released by astrocytes after SAH is mediated by NOX2. These findings demonstrate that eNOS and DHFR are downstream effectors of NOX2 in astrocytes.
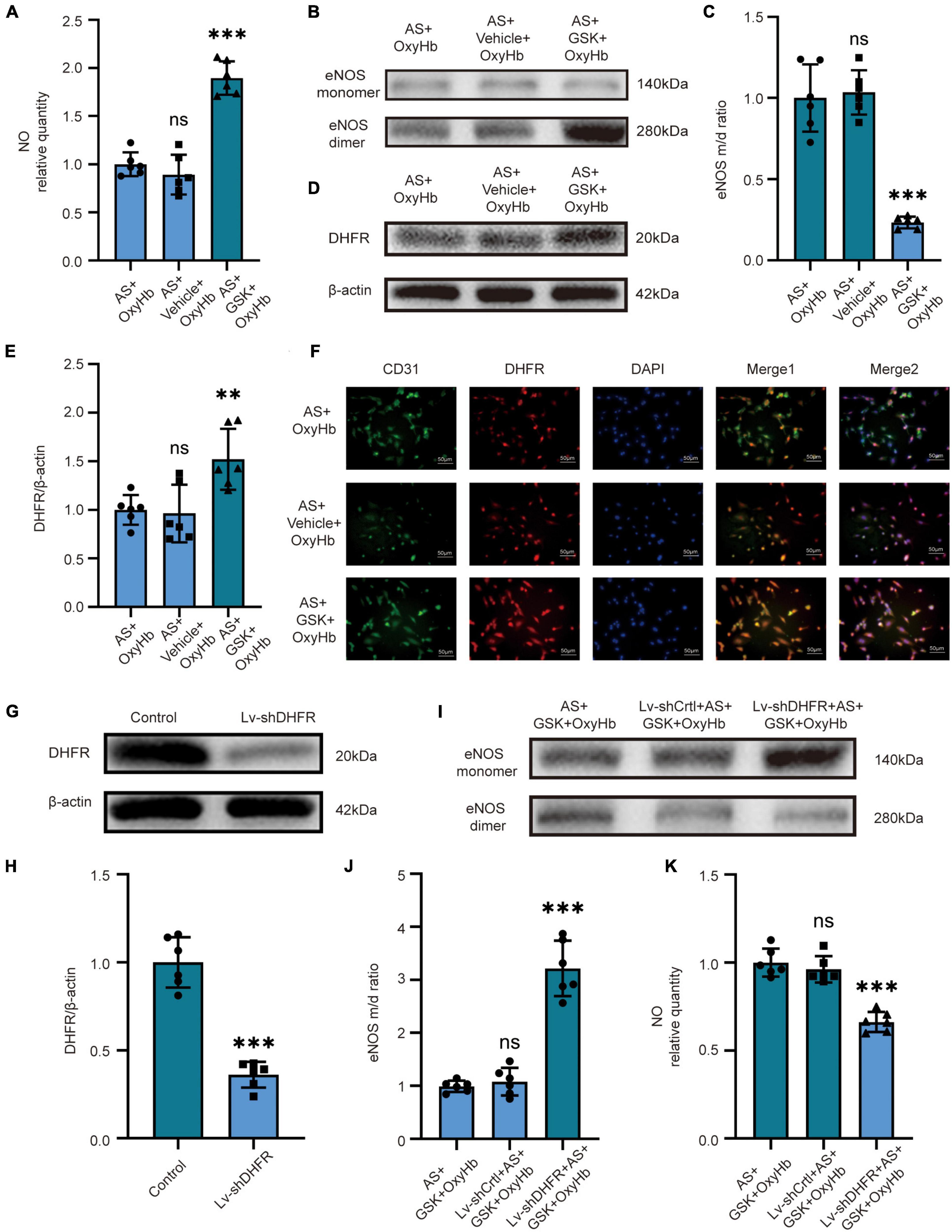
Figure 3. In vitro, inhibition of NOX2 in astrocytes after SAH recouples eNOS by increasing the expression of DHFR in endothelial cells. In vitro, LV-shDHFR largely attenuated the protective effect of NOX2 inhibitors after SAH. (A) NO released by cells among different experimental groups. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001, ns: no significant difference). (B,C) Representative Western blot images and quantitative analysis of eNOS monomer and eNOS dimer expression by ImageJ. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001, ns: no statistically significant difference). (D,E) Representative Western blot images and quantitative analysis of endothelial DHFR expression by ImageJ. Data are shown as mean ± SD (n = 6 each group, **p < 0.01, ns: no statistically significant difference). (F) Immunofluorescence images showed CD31 and DHFR colocalization. All scale bars, 50μm. (G,H) Representative Western blot images and quantitative analysis of endothelial DHFR expression by ImageJ. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001). (I,J) Representative Western blot images and quantitative analysis of eNOS monomer and eNOS dimer expression by ImageJ. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001). (K) NO released by cells among different experimental groups. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001, ns: no statistically significant difference).
We demonstrated the knockdown efficacy of Lv-shDHFR in endothelial cells by Western blot (Figures 3G, H). The results showed no significant difference in NO content or eNOS monomer/dimer ratio in the AS + GSK + OxyHb and Lv-shCtrl + AS + GSK + OxyHb groups. The Western blot results showed that compared to the AS + GSK + OxyHb group, the eNOS monomer/dimer ratio was significantly increased in the Lv-shDHFR + AS + GSK + OxyHb group (Figures 3I, J). Compared to the AS + GSK + OxyHb group, the amount of NO released by endothelial cells was significantly decreased in the Lv-shDHFR + AS + GSK + OxyHb group (Figure 3K). These results demonstrate that knockdown of DHFR reduces the protective effect of GSK2795039. These results also confirmed the existence of our conjectured pathway.
We then set out to validate our pathway in vivo. For in vivo experiments, we injected GSK2795039 into the ventricles of mice to inhibit NOX2. The laser speckle diagram showed that there was no significant difference in CBF between the SAH group and the vehicle + SAH group, however, compared to the SAH group, mice in the GSK + SAH group had increased CBF (Figures 4A, B). Simultaneously, we found no significant difference in endothelial NO content, eNOS monomer/dimer ratio, and DHFR expression in the SAH and vehicle + SAH groups. However, compared to the SAH group, the endothelial NO content was significantly increased in the GSK + SAH group (Figure 4C). Moreover, as shown in the cell experiments, compared to the vehicle + SAH group, the ratio of eNOS monomer/dimer and the expression of DHFR were increased significantly in the GSK + SAH group (Figures 4D–G). After injection of GSK2795039, the deteriorating neurological function was also significantly relieved (Figure 4H). Moreover, the results of Immunofluorescence staining showed that compared to the vehicle group, the NOX2 inhibitor significantly increased the expression of DHFR in endothelial cells (Figure 4I). Similar to the results of cell experiments, these findings also demonstrate that eNOS and DHFR are downstream effectors of NOX2.
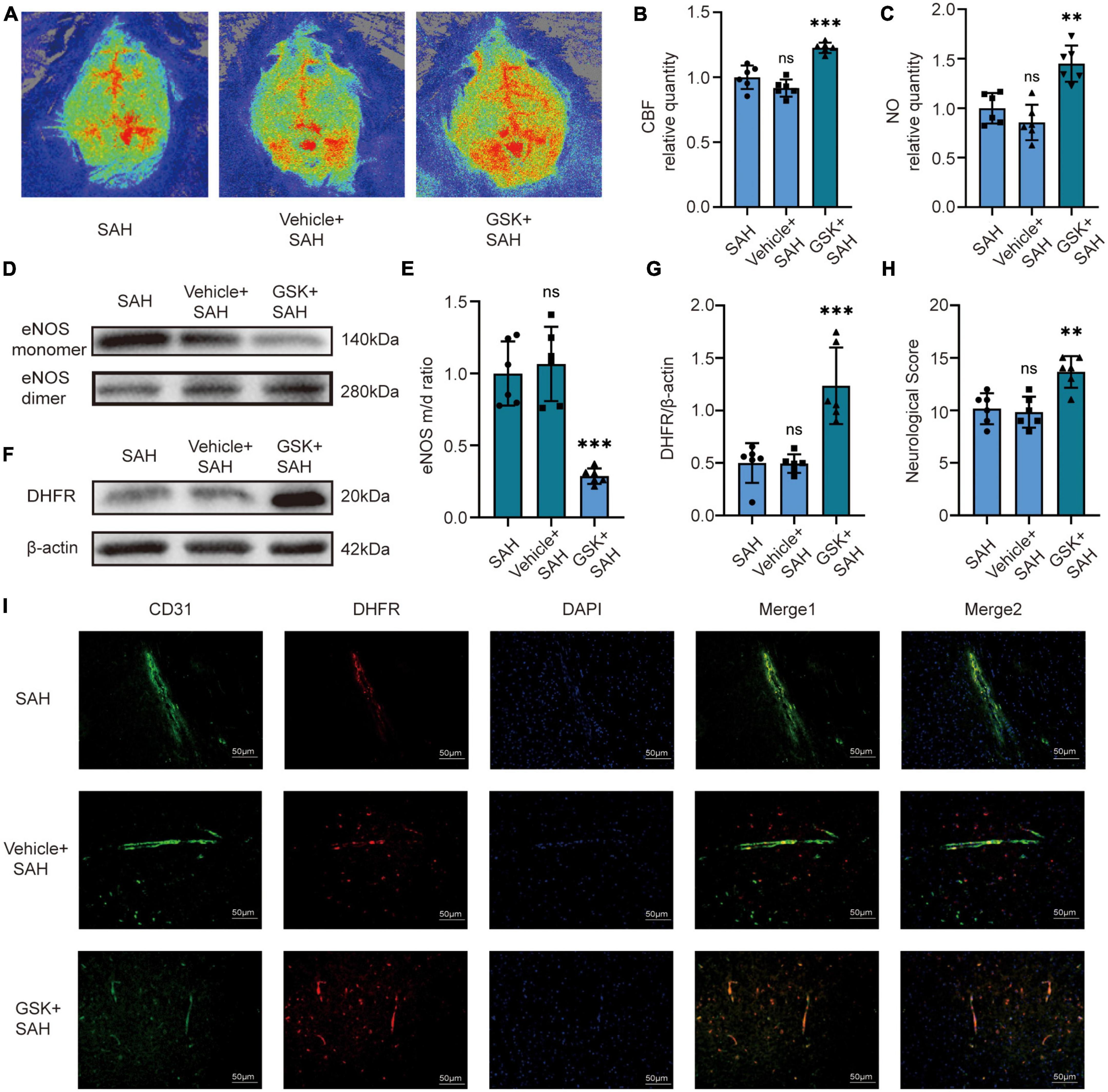
Figure 4. In vivo, NOX2 inhibitors increased CBF in mice by recoupling eNOS after SAH. (A) Representative LSCI images among different experimental groups. (B) Quantitative analysis of CBF among different experimental groups. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001, ns, no statistically significant difference). (C) NO content in extracted endothelial cells among different experimental groups. Data are shown as mean ± SD (n = 6 mice each group, **p < 0.01, ns: no statistically significant difference). (D,E) Representative Western blot images and quantitative analysis of eNOS monomer and eNOS dimer expression by ImageJ. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001). (F,G) Representative Western blot images and quantitative analysis of endothelial DHFR expression by ImageJ. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001). (H) Modified Garcia Score among different experimental groups. Data are shown as mean ± SD (n = 6 mice each group, **p < 0.01, ns: no statistically significant difference). (I) Immunofluorescence images showed CD31 and DHFR colocalization. All scale bars, 50μm.
We first knocked down DHFR by injecting an adeno-associated virus (AAV) into the tail vein of mice, and the knockdown efficacy of AAV-shDHFR was demonstrated by Western blot (Figures 5A, B). The laser speckle diagram demonstrates no significant difference in CBF between the GSK + SAH and AAV-shCtrl + GSK + SAH groups, however, compared to the GSK + SAH group, mice in the AAV-shDHFR + GSK + SAH group had increased CBF (Figures 5C, D). We also found no significant difference in endothelial NO content and eNOS monomer/dimer ratio between the GSK + SAH and AAV-shCtrl + GSK + SAH groups. The data showed that compared to the GSK + SAH group, the endothelial NO content was significantly decreased in the GSK + AAV-shDHFR + SAH group (Figure 5E). The Western blot results showed that the eNOS monomer/dimer ratio in the GSK + AAV-shDHFR + SAH group was significantly increased compared to that in the GSK + SAH group (Figures 5F, G). The protective effect of GSK2795039 in attenuating neurological deficits was also abolished by DHFR knockdown in endothelial cells (Figure 5H). This result proves that DHFR is a downstream factor of NOX2, and its knockdown counteracts the partial alleviation of acute cerebral ischemia in SAH by NOX2 inhibitors.

Figure 5. In vivo, knockdown of DHFR partially counteracted the effect of NOX2 inhibitors on relieving acute cerebral ischemia in SAH. (A,B) Representative Western blot images and quantitative analysis of endothelial DHFR expression by ImageJ. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001). (C) Representative LSCI images among different experimental groups. (D) Quantitative analysis of CBF among different experimental groups. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001, ns: no statistically significant difference). (E) NO content in extracted endothelial cells among different experimental groups. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001, ns: no statistically significant difference). (F,G) Representative Western blot images and quantitative analysis of eNOS monomer and eNOS dimer expression by ImageJ. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001). (H) Modified Garcia Score among different experimental groups. Data are shown as mean ± SD (n = 6 mice each group, ***p < 0.001, ns: no statistically significant difference).
In the present study, we found that NOX2 of astrocytes was increased after experimental SAH, contributing to the decreased expression of endothelial DHFR and aggravated eNOS uncoupling, which was an essential mechanism underlying the acute cerebral ischemia after SAH. The mechanism diagram is shown in Figure 6.

Figure 6. Schematic representation of the molecular mechanism of eNOS uncoupling in acute vasoconstriction after subarachnoid hemorrhage (SAH). In SAH, the increased expression of NOX2 in astrocytes causes the cells to release excess H2O2, and H2O2 enters the endothelial cells to reduce the expression of DHFR, which leads to the decrease of BH4, a key cofactor of eNOS. The decrease of BH4 increases the uncoupling of eNOS and reduces its production of NO. This leads to acute cerebral ischemia after SAH, most likely by mediating acute vasoconstriction.
We applied the prechiasmatic cistern injection model in vivo by infusing quantitative amounts of fresh autologous blood into the subarachnoid space under appropriate pressure. This closely mimics the situation in humans with SAH where blood fills the subarachnoid space and surrounds the main conducting arteries (Sabri et al., 2009). The intravascular perforation model has the disadvantages of difficulty to control bleeding volume and high mortality (up to 50%) (Bederson et al., 1995). While the injection model has a lower mortality rate, the mortality rate of each group of mice in our in vivo experiments is listed in Supplementary Table 1. Moreover, using the injection model, we can ensure a constant infusion pressure as well as a quantitative blood volume (Matz et al., 2000; Hansen-Schwartz et al., 2003). It ensures that the bleeding intensity and the damage caused by each experiment are repeatable, which is very important for the accuracy of our measurement of CBF and other experimental results (Sehba et al., 2012).
Therapeutic strategies that inhibit ROS-generating enzymes or scavenge ROS alleviate vasoconstriction in animal models of SAH (Horky et al., 1998; McGirt et al., 2002b). Among various ROS sources, such as NOX, cyclooxygenase, and xanthine synthase, NOX is considered as the main source of ROS in the brain (Ayer and Zhang, 2008). In experimental models of cerebral ischemia, NOX2 knockout mice have smaller cerebral infarct size and less blood-brain barrier disruption compared to wild-type mice (DiNapoli et al., 2008; Cairns et al., 2012). Furthermore, hyperbaric oxygen induces neuroprotection after SAH by downregulating NOX2 and subsequently reducing oxidative stress (Ostrowski et al., 2006b,c). We demonstrated that NOX2 in astrocytes was elevated 12 h after SAH, which was similarly demonstrated by Zhang et al. (2017) who demonstrated that silencing NOX2 inhibited neuronal apoptosis after SAH. We showed that elevated NOX2 expression in astrocytes contributed to acute cerebral ischemia after SAH. A previous study demonstrated that the application of the NOX inhibitor diphenyleneiodonium in the cisterna magna can partially attenuate cerebral vasospasm (Shin et al., 2002). Our experiments also demonstrated that the application of the NOX2 inhibitor, GSK2795039, effectively alleviates acute cerebral ischemia.
In the brain, astrocytes produce and release various molecular mediators, such as prostaglandins and arachidonic acid, that regulate vessel diameter (Iadecola and Nedergaard, 2007; Gordon et al., 2011; Liu and Chopp, 2016). It has been shown in the literature that NADPH oxidase autocrine and paracrine H2O2 play a signaling role in various biological environments (Lambeth and Neish, 2014; Breitenbach et al., 2018). Similarly, it has been documented that the ROS produced by NADPH oxidase in the adventitia enters the inner side of the blood vessel and causes intimal hypertrophy, and other experiments have shown that the diffusion of H2O2 produced by the adventitia causes contraction of the abdominal aorta in mice (Wang et al., 1998; Di Wang et al., 1999; Rey et al., 2002). We also demonstrated that astrocytes can release H2O2 into the medium, which may enter endothelial cells through diffusion and AQP. Our results and these findings above suggest that astrocytes can release H2O2 into endothelial cells to mediate eNOS uncoupling.
There is evidence that persistent oxidative stress leads to the uncoupling of eNOS, reducing its production of NO, while increasing evidence suggests that NOX plays a key role in the phenomenon of eNOS uncoupling (Landmesser et al., 2003; Förstermann and Münzel, 2006; Förstermann, 2008, 2010). It has been demonstrated that NOX activation occurs upstream of eNOS uncoupling in endothelial cells in diabetic mice and ischemia/reperfusion-injured hearts (Chalupsky and Cai, 2005; Oak and Cai, 2007). We also demonstrated that inhibition of NOX2 after SAH resulted in reduced uncoupling of eNOS.
Numerous experiments have shown that endothelial DHFR deficiency leads to decreased BH4 and eNOS uncoupling in many cardiovascular diseases, including hypertension, aortic aneurysm, and diabetic vascular complications (Gao et al., 2012; Youn et al., 2012; Li et al., 2019). Another previous experiment showed that restoring DHFR can recouple eNOS to lower blood pressure (Li et al., 2015). We demonstrate that, after SAH, there is a decrease in DHFR expression in endothelial cells that leads to eNOS uncoupling, which subsequently leads to acute cerebral ischemia.
Both our research and that of several other studies have demonstrated an increase in eNOS uncoupling after SAH by a decrease in NO and an increase in the eNOS monomer/dimer ratio (Sabri et al., 2011a,b). It has been shown that 3 h after SAH, eNOS-deficient mice have less microvascular perfusion and increased microvascular spasm compared to wild-type mice (Lenz et al., 2021). However, SAH treatment with simvastatin, a compound that increases eNOS expression in cerebral blood vessels, alleviates cerebral vasospasm in a mouse model of SAH (McGirt et al., 2002a). An article from our laboratory and our study showed that DHFR was significantly decreased at 12 h, 24 h, and 3 days after SAH while eNOS uncoupling was significantly increased at 12 h, 24 h, and 3 days after SAH. This may indicate that eNOS uncoupling occurs before 12 h after SAH, which may be a direction worthy of further investigation (Gao et al., 2022). Our study demonstrated that inhibition of NOX2 decreased eNOS uncoupling after SAH, resulting in elevated NO and reduced acute cerebral ischemia in mice. Taken together, these findings provide evidence that eNOS coupling plays an important role in maintaining proper vascular function after SAH.
Cell interactions in neurovascular units are complex, with one study showing that SAH increases the amplitude of astrocyte terminal Ca2+ oscillations and induces parenchymal arteriole constriction (Pappas et al., 2015). We used co-culture techniques to explore the role of astrocytes in endothelial cells and to link oxidative stress and endothelial dysfunction after SAH. Our study is the first to demonstrate that in SAH, elevated NOX2 expression in astrocytes leads to decreased DHFR expression and subsequent eNOS uncoupling in endothelial cells.
At present, there are two methods for measuring cerebral perfusion: (1) The more intuitive methods for evaluating cerebral perfusion include traditional measurement of Intracranial pressure (ICP)/cerebral perfusion pressure (CPP), optical methods including LSCI, transcranial Doppler (TCD), CT perfusion imaging (PCT), For example, PCT mainly conducts qualitative and quantitative analysis through hemodynamic parameters such as cerebral blood volume and mean transit time. (2) Indirect assessment of CBF adequacy by monitoring global or regional oxygenation and metabolism (jugular venous oxygen saturation, regional cerebral oxygen saturation, and microdialysis) (Artru et al., 2004; Rao and Durga, 2011; Mir et al., 2014; Deffieux et al., 2018). LSCI provides high-resolution images with a large field of view, so we chose LSCI to directly measure CBF in mice (Dunn, 2012). ICP rises with the release of blood in the brain after SAH, rising to the level of mean arterial pressure within 1 min after SAH. The ICP then drops above but close to its physiological level within minutes (Grote and Hassler, 1988). Concurrently, CPP decreases, and CBF decreases, which leads to severe cerebral ischemia (Cahill et al., 2006; Ostrowski et al., 2006a). CPP is the difference between mean arterial pressure and ICP. Both ICP and CPP are important indicators related to CBF. CBF is determined by CPP and cerebrovascular resistance (CVR). CVR is determined by the diameter and length of blood vessels and the viscosity of blood (Fan et al., 2022). It has been widely documented that the presence of acute vasoconstriction in SAH can reduce CBF by reducing the diameter of blood vessels.
The temporal order in which early microvascular constriction occurs has been roughly elucidated in experimental studies using histological techniques and in vivo imaging (Plesnila, 2013). As early as 5 min after SAH, the small blood vessels at the base of the brain begin to constrict. Subsequently, intraparenchymal and pial microvessels contract sharply within 24 h of SAH (Bederson et al., 1998; Sun et al., 2009; Friedrich et al., 2012). In studies observing the cerebral vessels in patients and experimental animals within hours after SAH, they showed that arteriolar constriction reduced CBF by 60–80% (Uhl et al., 2003; Pennings et al., 2004). Our experiments also demonstrated a sharp decrease in CBF at 12 h after SAH by measuring CBF in mice using LSCI.
In EBI after SAH, not only early vasoconstriction but also microvascular thrombus existed. Both mechanisms lead to an early decline in CBF, but studies have shown a significant correlation between vasoconstriction and microvascular thrombosis (Clarke et al., 2020). Microvascular thrombosis almost only occurs in small arteries with significant constriction. Dynamic imaging of arteriolar constriction then demonstrates vessel constriction within seconds after SAH, leading to secondary thrombus formation (Friedrich et al., 2012). Another study showed that no microvascular thrombosis was found in unconstricted blood vessels (Sabri et al., 2012). These results may indicate that vasoconstriction leads to thrombus formation, rather than thrombus occlusion of microvessels causing their secondary constriction. Further, studies have pointed out that 70% of arterioles in the brain after SAH constricted and the proportion of blood vessel diameter decreased by more than 30% (Friedrich et al., 2012). A 30% decrease in the diameter of blood vessels leads to an approximately 80% decrease in CBF, according to the Hagen–Poiseuille law. This is enough to cause cerebral ischemia. At the same time, because half of the observed small arteries have a stronger degree of constriction, and the constriction of small arteries in blood vessels has the greatest impact on blood flow, the number and degree of constriction of small arteries can indicate that a large part of the decline in CBF is due to vasoconstriction (Friedrich et al., 2012). The current study shows that reduction of endothelial DHFR mainly results in reduction of NO, a well-known factor promoting vasodilation, in several diseases including SAH (Gao et al., 2009, 2022). So, we can speculate that a large part of the decrease in blood flow caused by the decrease in NO in our experiment is mediated by vasoconstriction.
However, our study has some limitations. The types of astrocytes we used were mixed. We did not repeat the validation using fibrous or protoplasmic astrocytes or different cell lines. It has been reported that the NOX2 protein level of astrocytes in the perihematomal brain tissue of SAH patients is increased, and it has been proved that the increase of NOX2 will produce excessive ROS in many types of cells, and our experiments and supplementary experiments also demonstrated the increased expression of NOX2 and the increased release of ROS in astrocytes (Marchetto et al., 2008; Zhang et al., 2017; Nauseef, 2019). At the same time, both fibrous and protoplasmic astrocytes have been shown to wrap around cerebral blood vessels, so they both may secrete ROS into endothelial cells (Bozic et al., 2021). Many articles have proved that the increase of ROS in endothelial cells will lead to the decrease of DHFR and the uncoupling of eNOS (Li et al., 2019; Gao et al., 2022). So even though the degree of DHFR reduction may vary with increasing ROS content released by different cell lines or astrocyte types, this would not rebut our conclusions. We will use different types of astrocytes to verify the conclusion in future work.
Both NOX2 from astrocytes and uncoupled eNOS from endothelial cells can release H2O2, and limited by technical conditions, we could not distinguish the specific source of H2O2 in the medium of co-cultivation after SAH. Meanwhile, the measurement of ROS in vivo remains a challenge, which may require confocal imaging techniques and ESR analysis. However, the low levels and relatively short lifetime of ROS make measurement difficult, and several processes and side effects associated with laser exposure (such as photobleaching or opposite effects of photoactivation) can make data difficult to analyze and interpret (Fuloria et al., 2021). These existing technical problems still need to be solved, and we will study how to solve these problems in future research.
In conclusion, our study revealed a novel mechanism by which oxidative stress leads to acute cerebral ischemia after SAH, that is, elevated NOX2 expression in astrocytes leads to eNOS uncoupling through endothelial DHFR downregulation. This signaling pathway leads to a decrease in NO, which contributes to acute cerebral ischemia after SAH most likely by causing acute vasoconstriction. This signaling cascade may represent a pervasive mechanism of eNOS uncoupling under pathophysiological conditions associated with oxidative stress. These findings are also highly translational in facilitating the development of drugs for treating SAH.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Jinling Hospital Animal Care and Use Committee.
M-LZ, TL, Y-LH, and S-HM contributed to conception and design of the study. S-HM, S-QG, H-XL, Y-SZ, XW, and J-YQ conducted the experiments. M-LZ, C-CG, and TL performed the statistical analysis. S-HM wrote the first draft of the manuscript. S-QG and H-XL wrote the sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by the Natural Science Foundation of Jiangsu Province (SBK2022023139), the Scientific Research Project of Jiangsu Provincial Health Commission (M2021008), and the National Natural Science Foundation of China (Nos. 81771292 and 81571162).
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1121944/full#supplementary-material
Supplementary Figure 1 | Experimental schematic in vivo and in vitro. (A) Experimental design diagram of the Sham group and the 12 h, 24 h, and 3 days groups after SAH. (B) Experimental design diagram to explore signaling pathways in vivo. (C) Experimental design diagram to explore signaling pathways in vitro. SAH, subarachnoid hemorrhage; OxyHb, oxyhemoglobin; AS, astrocyte; WB, Western blot; IF, immunofluorescence staining; H2O2, hydrogen peroxide; NO, nitric oxide; LSCI, laser speckle contrast imaging; GSK, GSK2795039; LV, lentivirus; AAV, adeno-associated virus.
Supplementary Figure 2 | The viability of different groups of astrocytes and endothelial cells. (A) Astrocyte viability among different experimental groups. Data are shown as mean ± SD (n = 6 each group, ns: no statistically significant difference). (B) Endothelial cell viability among different experimental groups. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001, ns: no statistically significant difference).
Supplementary Figure 3 | The content of H2O2 released by astrocytes in the medium. (A) H2O2 released by astrocytes among different experimental groups. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001). (B) H2O2 released by astrocytes among different experimental groups. Data are shown as mean ± SD (n = 6 each group, ***p < 0.001, ns: no statistically significant difference).
SAH, subarachnoid hemorrhage; EBI, early brain injury; CBF, cerebral blood flow; ROS, reactive oxygen species; H2O2, hydrogen peroxide; NOX, nicotinamide adenine dinucleotide phosphate oxidase; NOX2, nicotinamide adenine dinucleotide phosphate oxidase 2; NO, nitric oxide; eNOS, endothelial nitric oxide synthase; BH4, tetrahydrobiopterin; DHFR, dihydrofolate reductase; BH2, dihydrobiopterin; OxyHb, oxyhemoglobin; FAD, flavin adenine dinucleotide; LDH, lactate dehydrogenase; TBS, Tris-buffered saline; PBS, phosphate buffered saline; DAPI, 4 ′,6-diamidino-2-phenylindole; GFAP, glial fibrillary acidic protein; GSK, GSK2795039; LV-shDHFR, lentivirus targeting DHFR; LV-shCtrl, lentivirus containing scramble RNA; AAV-shDHFR, adeno-associated virus targeting DHFR; AAV-shCtrl, adeno-associated virus containing scramble RNA.
Abbott, N. J., Rönnbäck, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi: 10.1038/nrn1824
Adams, H. P. Jr., Kassell, N. F., Torner, J. C., Nibbelink, D. W., and Sahs, A. L. (1981). Early management of aneurysmal subarachnoid hemorrhage. A report of the Cooperative Aneurysm Study. J. Neurosurg. 54, 141–145. doi: 10.3171/jns.1981.54.2.0141
Artru, F., Dailler, F., Burel, E., Bodonian, C., Grousson, S., Convert, J., et al. (2004). Assessment of jugular blood oxygen and lactate indices for detection of cerebral ischemia and prognosis. J. Neurosurg. Anesthesiol. 16, 226–231. doi: 10.1097/00008506-200407000-00007
Ayer, R. E., and Zhang, J. H. (2008). Oxidative stress in subarachnoid haemorrhage: Significance in acute brain injury and vasospasm. Acta Neurochir. Suppl. 104, 33–41. doi: 10.1007/978-3-211-75718-5_7
Bales, K. R., O’neill, S. M., Pozdnyakov, N., Pan, F., Caouette, D., Pi, Y., et al. (2016). Passive immunotherapy targeting amyloid-β reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain 139, 563–577. doi: 10.1093/brain/awv313
Bedard, K., and Krause, K. H. (2007). The Nox family of Ros-generating Nadph oxidases: Physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Bederson, J. B., Germano, I. M., and Guarino, L. (1995). Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke 26, 1086–1091; discussion 1091–1092. doi: 10.1161/01.STR.26.6.1086
Bederson, J. B., Levy, A. L., Ding, W. H., Kahn, R., Diperna, C. A., and Jenkins, A. L. III, et al. (1998). Acute vasoconstriction after subarachnoid hemorrhage. Neurosurgery 42, 352–360; discussion 360–362. doi: 10.1097/00006123-199802000-00091
Bendall, J. K., Douglas, G., Mcneill, E., Channon, K. M., and Crabtree, M. J. (2014). Tetrahydrobiopterin in cardiovascular health and disease. Antioxid. Redox Signal. 20, 3040–3077. doi: 10.1089/ars.2013.5566
Bozic, I., Savic, D., and Lavrnja, I. (2021). Astrocyte phenotypes: Emphasis on potential markers in neuroinflammation. Histol. Histopathol. 36, 267–290.
Breitenbach, M., Rinnerthaler, M., Weber, M., Breitenbach-Koller, H., Karl, T., Cullen, P., et al. (2018). The defense and signaling role of NADPH oxidases in eukaryotic cells : Review. Wien. Med. Wochenschr. 168, 286–299. doi: 10.1007/s10354-018-0640-4
Cahill, J., Calvert, J. W., and Zhang, J. H. (2006). Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 26, 1341–1353. doi: 10.1038/sj.jcbfm.9600283
Cairns, B., Kim, J. Y., Tang, X. N., and Yenari, M. A. (2012). NOX inhibitors as a therapeutic strategy for stroke and neurodegenerative disease. Curr. Drug Targets 13, 199–206. doi: 10.2174/138945012799201676
Chalupsky, K., and Cai, H. (2005). Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 102, 9056–9061. doi: 10.1073/pnas.0409594102
Cheng, G., Cao, Z., Xu, X., Van Meir, E. G., and Lambeth, J. D. (2001). Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269, 131–140. doi: 10.1016/S0378-1119(01)00449-8
Chrissobolis, S., Banfi, B., Sobey, C. G., and Faraci, F. M. (2012). Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J. Appl. Physiol. 113, 184–191. doi: 10.1152/japplphysiol.00455.2012
Clarke, J. V., Suggs, J. M., Diwan, D., Lee, J. V., Lipsey, K., Vellimana, A. K., et al. (2020). Microvascular platelet aggregation and thrombosis after subarachnoid hemorrhage: A review and synthesis. J. Cereb. Blood Flow Metab. 40, 1565–1575. doi: 10.1177/0271678X20921974
Deffieux, T., Demene, C., Pernot, M., and Tanter, M. (2018). Functional ultrasound neuroimaging: A review of the preclinical and clinical state of the art. Curr. Opin. Neurobiol. 50, 128–135. doi: 10.1016/j.conb.2018.02.001
Deng, H. J., Deji, Q., Zhaba, W., Liu, J. Q., Gao, S. Q., Han, Y. L., et al. (2021). A20 establishes negative feedback with TRAF6/NF-κB and attenuates early brain injury after experimental subarachnoid hemorrhage. Front. Immunol. 12:623256. doi: 10.3389/fimmu.2021.623256
Di Wang, H., Hope, S., Du, Y., Quinn, M. T., Cayatte, A., Pagano, P. J., et al. (1999). Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension 33, 1225–1232. doi: 10.1161/01.HYP.33.5.1225
DiNapoli, V. A., Huber, J. D., Houser, K., Li, X., and Rosen, C. L. (2008). Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol. Aging 29, 753–764. doi: 10.1016/j.neurobiolaging.2006.12.007
Dunn, A. K. (2012). Laser speckle contrast imaging of cerebral blood flow. Ann. Biomed. Eng. 40, 367–377. doi: 10.1007/s10439-011-0469-0
Fan, J. L., Nogueira, R. C., Brassard, P., Rickards, C. A., Page, M., Nasr, N., et al. (2022). Integrative physiological assessment of cerebral hemodynamics and metabolism in acute ischemic stroke. J. Cereb. Blood Flow Metab. 42, 454–470. doi: 10.1177/0271678X211033732
Förstermann, U. (2008). Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 5, 338–349. doi: 10.1038/ncpcardio1211
Förstermann, U. (2010). Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 459, 923–939. doi: 10.1007/s00424-010-0808-2
Förstermann, U., and Münzel, T. (2006). Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 113, 1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532
Förstermann, U., and Sessa, W. C. (2012). Nitric oxide synthases: Regulation and function. Eur. Heart J. 33, 29–37, 837a–837d. doi: 10.1093/eurheartj/ehr304
Friedrich, B., Müller, F., Feiler, S., Schöller, K., and Plesnila, N. (2012). Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: An in-vivo microscopy study. J. Cereb. Blood Flow Metab. 32, 447–455. doi: 10.1038/jcbfm.2011.154
Fuloria, S., Subramaniyan, V., Karupiah, S., Kumari, U., Sathasivam, K., Meenakshi, D. U., et al. (2021). Comprehensive review of methodology to detect reactive oxygen species (ROS) in mammalian species and establish its relationship with antioxidants and cancer. Antioxidants 10:128. doi: 10.3390/antiox10010128
Gao, L., Chalupsky, K., Stefani, E., and Cai, H. (2009). Mechanistic insights into folic acid-dependent vascular protection: Dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: A novel HPLC-based fluorescent assay for DHFR activity. J. Mol. Cell Cardiol. 47, 752–760. doi: 10.1016/j.yjmcc.2009.07.025
Gao, L., Siu, K. L., Chalupsky, K., Nguyen, A., Chen, P., Weintraub, N. L., et al. (2012). Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: Treatment with folic acid. Hypertension 59, 158–166. doi: 10.1161/HYPERTENSIONAHA.111.181644
Gao, S. Q., Liu, J. Q., Han, Y. L., Deji, Q. Z., Zhaba, W. D., Deng, H. J., et al. (2020). Neuroprotective role of glutathione peroxidase 4 in experimental subarachnoid hemorrhage models. Life Sci. 257:118050. doi: 10.1016/j.lfs.2020.118050
Gao, S. Q., Shi, J. J., Xue, W., Miao, S. H., Li, T., Gao, C. C., et al. (2022). Endothelial NOX4 aggravates eNOS uncoupling by decreasing dihydrofolate reductase after subarachnoid hemorrhage. Free Radic. Biol. Med. 193, 499–510. doi: 10.1016/j.freeradbiomed.2022.10.318
Gordon, G. R., Howarth, C., and Macvicar, B. A. (2011). Bidirectional control of arteriole diameter by astrocytes. Exp. Physiol. 96, 393–399. doi: 10.1113/expphysiol.2010.053132
Grote, E., and Hassler, W. (1988). The critical first minutes after subarachnoid hemorrhage. Neurosurgery 22, 654–661. doi: 10.1227/00006123-198804000-00006
Hansen-Schwartz, J., Hoel, N. L., Zhou, M., Xu, C. B., Svendgaard, N. A., and Edvinsson, L. (2003). Subarachnoid hemorrhage enhances endothelin receptor expression and function in rat cerebral arteries. Neurosurgery 52, 1188–1194; 1194–1195. doi: 10.1227/01.NEU.0000058467.82442.64
Hop, J. W., Rinkel, G. J., Algra, A., and Van Gijn, J. (1997). Case-fatality rates and functional outcome after subarachnoid hemorrhage: A systematic review. Stroke 28, 660–664. doi: 10.1161/01.STR.28.3.660
Horky, L. L., Pluta, R. M., Boock, R. J., and Oldfield, E. H. (1998). Role of ferrous iron chelator 2,2’-dipyridyl in preventing delayed vasospasm in a primate model of subarachnoid hemorrhage. J. Neurosurg. 88, 298–303. doi: 10.3171/jns.1998.88.2.0298
Iadecola, C., and Nedergaard, M. (2007). Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376. doi: 10.1038/nn2003
Iadecola, C., Park, L., and Capone, C. (2009). Threats to the mind: Aging, amyloid, and hypertension. Stroke 40, S40–S44. doi: 10.1161/STROKEAHA.108.533638
Jung, O., Marklund, S. L., Geiger, H., Pedrazzini, T., Busse, R., and Brandes, R. P. (2003). Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: In vivo and ex vivo evidence from ecSOD-deficient mice. Circ. Res. 93, 622–629. doi: 10.1161/01.RES.0000092140.81594.A8
Lambeth, J. D., and Neish, A. S. (2014). Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 9, 119–145. doi: 10.1146/annurev-pathol-012513-104651
Landmesser, U., Dikalov, S., Price, S. R., Mccann, L., Fukai, T., Holland, S. M., et al. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111, 1201–1209. doi: 10.1172/JCI200314172
Laursen, J. B., Somers, M., Kurz, S., Mccann, L., Warnholtz, A., Freeman, B. A., et al. (2001). Endothelial regulation of vasomotion in apoE-deficient mice: Implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103, 1282–1288. doi: 10.1161/01.CIR.103.9.1282
Lenz, I. J., Plesnila, N., and Terpolilli, N. A. (2021). Role of endothelial nitric oxide synthase for early brain injury after subarachnoid hemorrhage in mice. J. Cereb. Blood Flow Metab. 41, 1669–1681. doi: 10.1177/0271678X20973787
Li, H., Li, Q., Zhang, Y., Liu, W., Gu, B., Narumi, T., et al. (2019). Novel treatment of hypertension by specifically targeting E2F for restoration of endothelial dihydrofolate reductase and eNOS function under oxidative stress. Hypertension 73, 179–189. doi: 10.1161/HYPERTENSIONAHA.118.11643
Li, Q., Youn, J. Y., and Cai, H. (2015). Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 33, 1128–1136. doi: 10.1097/HJH.0000000000000587
Lindskog, C., Asplund, A., Catrina, A., Nielsen, S., and Rützler, M. (2016). A systematic characterization of aquaporin-9 expression in human normal and pathological tissues. J. Histochem. Cytochem. 64, 287–300. doi: 10.1369/0022155416641028
Liu, Z., and Chopp, M. (2016). Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 144, 103–120. doi: 10.1016/j.pneurobio.2015.09.008
Macdonald, R. L., and Schweizer, T. A. (2017). Spontaneous subarachnoid haemorrhage. Lancet 389, 655–666. doi: 10.1016/S0140-6736(16)30668-7
Macdonald, R. L., and Weir, B. K. (1994). Cerebral vasospasm and free radicals. Free Radic. Biol. Med. 16, 633–643. doi: 10.1016/0891-5849(94)90064-7
Marchetto, M. C., Muotri, A. R., Mu, Y., Smith, A. M., Cezar, G. G., and Gage, F. H. (2008). Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3, 649–657. doi: 10.1016/j.stem.2008.10.001
Marklund, S. L. (1984). Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochem. J. 222, 649–655. doi: 10.1042/bj2220649
Matz, P. G., Copin, J. C., and Chan, P. H. (2000). Cell death after exposure to subarachnoid hemolysate correlates inversely with expression of CuZn-superoxide dismutase. Stroke 31, 2450–2459. doi: 10.1161/01.STR.31.10.2450
Mayberg, M. R., Batjer, H. H., Dacey, R., Diringer, M., Haley, E. C., Heros, R. C., et al. (1994). Guidelines for the management of aneurysmal subarachnoid hemorrhage. A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Circulation 90, 2592–2605. doi: 10.1161/01.CIR.90.5.2592
McGirt, M. J., Parra, A., Sheng, H., Higuchi, Y., Oury, T. D., Laskowitz, D. T., et al. (2002b). Attenuation of cerebral vasospasm after subarachnoid hemorrhage in mice overexpressing extracellular superoxide dismutase. Stroke 33, 2317–2323. doi: 10.1161/01.STR.0000027207.67639.1E
McGirt, M. J., Lynch, J. R., Parra, A., Sheng, H., Pearlstein, R. D., Laskowitz, D. T., et al. (2002a). Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke 33, 2950–2956. doi: 10.1161/01.STR.0000038986.68044.39
Miller, A. A., De Silva, T. M., Judkins, C. P., Diep, H., Drummond, G. R., and Sobey, C. G. (2010). Augmented superoxide production by Nox2-containing NADPH oxidase causes cerebral artery dysfunction during hypercholesterolemia. Stroke 41, 784–789. doi: 10.1161/STROKEAHA.109.575365
Miller, A. A., Drummond, G. R., and Sobey, C. G. (2006). Novel isoforms of Nadph-oxidase in cerebral vascular control. Pharmacol. Ther. 111, 928–948. doi: 10.1016/j.pharmthera.2006.02.005
Mir, D. I., Gupta, A., Dunning, A., Puchi, L., Robinson, C. L., Epstein, H. A., et al. (2014). CT perfusion for detection of delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. AJNR Am. J. Neuroradiol. 35, 866–871. doi: 10.3174/ajnr.A3787
Nauseef, W. M. (2019). The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 60, 130–140. doi: 10.1016/j.coi.2019.05.006
Oak, J. H., and Cai, H. (2007). Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 56, 118–126. doi: 10.2337/db06-0288
Ostrowski, R. P., Colohan, A. R., and Zhang, J. H. (2006b). Neuroprotective effect of hyperbaric oxygen in a rat model of subarachnoid hemorrhage. Acta Neurochir. Suppl. 96, 188–193. doi: 10.1007/3-211-30714-1_41
Ostrowski, R. P., Tang, J., and Zhang, J. H. (2006c). Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke 37, 1314–1318. doi: 10.1161/01.STR.0000217310.88450.c3
Ostrowski, R. P., Colohan, A. R., and Zhang, J. H. (2006a). Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol. Res. 28, 399–414. doi: 10.1179/016164106X115008
Pappas, A. C., Koide, M., and Wellman, G. C. (2015). Astrocyte Ca2+ signaling drives inversion of neurovascular coupling after subarachnoid hemorrhage. J. Neurosci. 35, 13375–13384. doi: 10.1523/JNEUROSCI.1551-15.2015
Paravicini, T. M., and Sobey, C. G. (2003). Cerebral vascular effects of reactive oxygen species: Recent evidence for a role of NADPH-oxidase. Clin. Exp. Pharmacol. Physiol. 30, 855–859. doi: 10.1046/j.1440-1681.2003.03920.x
Pennings, F. A., Bouma, G. J., and Ince, C. (2004). Direct observation of the human cerebral microcirculation during aneurysm surgery reveals increased arteriolar contractility. Stroke 35, 1284–1288. doi: 10.1161/01.STR.0000126039.91400.cb
Plesnila, N. (2013). Pathophysiological role of global cerebral ischemia following subarachnoid hemorrhage: The current experimental evidence. Stroke Res. Treat. 2013:651958. doi: 10.1155/2013/651958
Rao, G. S., and Durga, P. (2011). Changing trends in monitoring brain ischemia: From intracranial pressure to cerebral oximetry. Curr. Opin. Anaesthesiol. 24, 487–494. doi: 10.1097/ACO.0b013e32834a8965
Rey, F. E., Li, X. C., Carretero, O. A., Garvin, J. L., and Pagano, P. J. (2002). Perivascular superoxide anion contributes to impairment of endothelium-dependent relaxation: Role of gp91(phox). Circulation 106, 2497–2502. doi: 10.1161/01.CIR.0000038108.71560.70
Sabri, M., Ai, J., Knight, B., Tariq, A., Jeon, H., Shang, X., et al. (2011a). Uncoupling of endothelial nitric oxide synthase after experimental subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 31, 190–199. doi: 10.1038/jcbfm.2010.76
Sabri, M., Ai, J., Marsden, P. A., and Macdonald, R. L. (2011b). Simvastatin re-couples dysfunctional endothelial nitric oxide synthase in experimental subarachnoid hemorrhage. PLoS One 6:e17062. doi: 10.1371/journal.pone.0017062
Sabri, M., Ai, J., Lakovic, K., D’abbondanza, J., Ilodigwe, D., and Macdonald, R. L. (2012). Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience 224, 26–37. doi: 10.1016/j.neuroscience.2012.08.002
Sabri, M., Jeon, H., Ai, J., Tariq, A., Shang, X., Chen, G., et al. (2009). Anterior circulation mouse model of subarachnoid hemorrhage. Brain Res. 1295, 179–185. doi: 10.1016/j.brainres.2009.08.021
Schubert, G. A., Seiz, M., Hegewald, A. A., Manville, J., and Thomé, C. (2009). Acute hypoperfusion immediately after subarachnoid hemorrhage: A xenon contrast-enhanced CT study. J. Neurotrauma 26, 2225–2231. doi: 10.1089/neu.2009.0924
Schwartz, A. Y., Sehba, F. A., and Bederson, J. B. (2000). Decreased nitric oxide availability contributes to acute cerebral ischemia after subarachnoid hemorrhage. Neurosurgery 47, 208–214; discussion 214–215. doi: 10.1227/00006123-200007000-00042
Sehba, F. A., Ding, W. H., Chereshnev, I., and Bederson, J. B. (1999). Effects of S-nitrosoglutathione on acute vasoconstriction and glutamate release after subarachnoid hemorrhage. Stroke 30, 1955–1961. doi: 10.1161/01.STR.30.9.1955
Sehba, F. A., Hou, J., Pluta, R. M., and Zhang, J. H. (2012). The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 97, 14–37. doi: 10.1016/j.pneurobio.2012.02.003
Sehba, F. A., Schwartz, A. Y., Chereshnev, I., and Bederson, J. B. (2000). Acute decrease in cerebral nitric oxide levels after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 20, 604–611. doi: 10.1097/00004647-200003000-00018
Shin, H. K., Lee, J. H., Kim, K. Y., Kim, C. D., Lee, W. S., Rhim, B. Y., et al. (2002). Impairment of autoregulatory vasodilation by NAD(P)H oxidase-dependent superoxide generation during acute stage of subarachnoid hemorrhage in rat pial artery. J. Cereb. Blood Flow Metab. 22, 869–877. doi: 10.1097/00004647-200207000-00012
Sun, B. L., Zheng, C. B., Yang, M. F., Yuan, H., Zhang, S. M., and Wang, L. X. (2009). Dynamic alterations of cerebral pial microcirculation during experimental subarachnoid hemorrhage. Cell. Mol. Neurobiol. 29, 235–241. doi: 10.1007/s10571-008-9316-8
Terpolilli, N. A., Brem, C., Bühler, D., and Plesnila, N. (2015). Are we barking up the wrong vessels? Cerebral microcirculation after subarachnoid hemorrhage. Stroke 46, 3014–3019. doi: 10.1161/STROKEAHA.115.006353
Terpolilli, N. A., Feiler, S., Dienel, A., Müller, F., Heumos, N., Friedrich, B., et al. (2016). Nitric oxide inhalation reduces brain damage, prevents mortality, and improves neurological outcome after subarachnoid hemorrhage by resolving early pial microvasospasms. J. Cereb. Blood Flow Metab. 36, 2096–2107. doi: 10.1177/0271678X15605848
Thannickal, V. J., and Fanburg, B. L. (2000). Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L1005-28. doi: 10.1152/ajplung.2000.279.6.L1005
Thannickal, V. J., Day, R. M., Klinz, S. G., Bastien, M. C., Larios, J. M., and Fanburg, B. L. (2000). Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and Tgf-beta1. FASEB J. 14, 1741–1748. doi: 10.1096/fj.99-0878com
Thöny, B., Auerbach, G., and Blau, N. (2000). Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 347(Pt 1), 1–16. doi: 10.1042/bj3470001
Uhl, E., Lehmberg, J., Steiger, H. J., and Messmer, K. (2003). Intraoperative detection of early microvasospasm in patients with subarachnoid hemorrhage by using orthogonal polarization spectral imaging. Neurosurgery 52, 1307–1315; discussion 1315–1317. doi: 10.1227/01.NEU.0000065154.04824.9E
Waghray, M., Cui, Z., Horowitz, J. C., Subramanian, I. M., Martinez, F. J., Toews, G. B., et al. (2005). Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 19, 854–856. doi: 10.1096/fj.04-2882fje
Wang, H. D., Pagano, P. J., Du, Y., Cayatte, A. J., Quinn, M. T., Brecher, P., et al. (1998). Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circ. Res. 82, 810–818. doi: 10.1161/01.RES.82.7.810
Wang, X., Li, Y. M., Li, W. Q., Huang, C. G., Lu, Y. C., and Hou, L. J. (2012). Effect of clazosentan in patients with aneurysmal subarachnoid hemorrhage: A meta-analysis of randomized controlled trials. PLoS One 7:e47778. doi: 10.1371/journal.pone.0047778
Weksler, B., Romero, I. A., and Couraud, P. O. (2013). The hcmec/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 10:16. doi: 10.1186/2045-8118-10-16
Winterbourn, C. C. (2018). Biological production, detection, and fate of hydrogen peroxide. Antioxid. Redox Signal. 29, 541–551. doi: 10.1089/ars.2017.7425
Xia, Y., Tsai, A. L., Berka, V., and Zweier, J. L. (1998). Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem. 273, 25804–25808. doi: 10.1074/jbc.273.40.25804
Yang, X. M., Chen, X. H., Lu, J. F., Zhou, C. M., Han, J. Y., and Chen, C. H. (2018). In vivo observation of cerebral microcirculation after experimental subarachnoid hemorrhage in mice. Neural Regen. Res. 13, 456–462. doi: 10.4103/1673-5374.228728
Youn, J. Y., Gao, L., and Cai, H. (2012). The p47phox- and Nadph oxidase organiser 1 (NOXO1)-dependent activation of Nadph oxidase 1 (Nox1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 55, 2069–2079. doi: 10.1007/s00125-012-2557-6
Keywords: subarachnoid hemorrhage, acute vasoconstriction, endothelial nitric oxide synthase uncoupling, nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2), dihydrofolate reductase, neurovascular unit, acute cerebral ischemia
Citation: Miao S-H, Gao S-Q, Li H-X, Zhuang Y-S, Wang X, Li T, Gao C-C, Han Y-L, Qiu J-Y and Zhou M-L (2023) Increased NOX2 expression in astrocytes leads to eNOS uncoupling through dihydrofolate reductase in endothelial cells after subarachnoid hemorrhage. Front. Mol. Neurosci. 16:1121944. doi: 10.3389/fnmol.2023.1121944
Received: 12 December 2022; Accepted: 13 March 2023;
Published: 30 March 2023.
Edited by:
Mukesh K. Pandey, Mayo Clinic, United StatesReviewed by:
Brian Burkett, Mayo Clinic, United StatesCopyright © 2023 Miao, Gao, Li, Zhuang, Wang, Li, Gao, Han, Qiu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng-Liang Zhou, emhvdW1lbmdsaWFuZ0BuanUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.