 Xi Zhang1†
Xi Zhang1† Neng Xiao2†Yang Cao3Ying Peng4Aojie Lian4,5Yuanlu Chen6Pengchao Wang7Weiyue Gu7Bo Xiao1Jing Yu8*
Neng Xiao2†Yang Cao3Ying Peng4Aojie Lian4,5Yuanlu Chen6Pengchao Wang7Weiyue Gu7Bo Xiao1Jing Yu8* Hua Wang9*Li Shu10,11*
Hua Wang9*Li Shu10,11*- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2Department of Pediatric Neurology, Chenzhou First People’s Hospital, Chenzhou, China
- 3Department of Radiology, Chenzhou First People’s Hospital, Chenzhou, China
- 4National Health Commission Key Laboratory for Birth Defect Research and Prevention, Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
- 5Clinical Research Center for Placental Medicine in Hunan Province, Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
- 6Department of Pharmacy, Chenzhou First People’s Hospital, Chenzhou, China
- 7Chigene (Beijing) Translational Medical Research Center Co., Ltd., Beijing, China
- 8Department of Neurology, Children’s Hospital of Xinjiang Uygur Autonomous Region, Ürümqi, China
- 9Department of Medical Genetics, Hunan Children’s Hospital, Changsha, China
- 10Department of Biochemistry, Molecular Biology and Medical Genetics, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 11Alberta Children’s Hospital Research Institute, University of Calgary, Calgary, AB, Canada
Objective: This study aims to prove that the de novo variants in MAST4 gene are associated with neurodevelopmental disorders (NDD) with developmental delay (DD) and infantile spasm (IS) and to determine the genotype-phenotype correlations.
Methods: Trio-based exome sequencing (ES) was performed on the four families enrolled in this study. We collected and systematically reviewed the four probands’ clinical data, magnetic resonance images (MRI), and electroencephalography (EEG). We also carried out bioinformatics analysis by integrating published exome/genome sequencing data and human brain transcriptomic data.
Results: We described four patients whose median age of seizure onset was 5 months. The primary manifestation was infantile spasms with typical hypsarrhythmia on EEG. Developmental delays or intellectual disabilities varied among the four individuals. Three de novo missense variants in MAST4 gene were identified from four families, including chr5:66438324 (c.2693T > C: p.Ile898Thr) z, chr5:66459419 (c.4412C > T: p.Thr1471Ile), and chr5:66462662 (c.7655C > G:p.Ser2552Trp). The missense variant p.Ile898Thr is mapped to the AGC-kinase C-terminal with phosphatase activity. The other variant p.Ser2552Trp is located in a phosphoserine-modified residue which may affect cell membrane stability and signal transduction. Besides, the variant p.Thr1471Ile is a recurrent site screened out in two unrelated patients. Compared to private mutations (found only in a single family or a small population) of MAST4 in the gnomAD non-neuro subset, all de novo variants were predicted to be damaging or probably damaging through different bioinformatic analyses. Significantly higher CADD scores of the variant p.Thr1471Ile indicate more deleteriousness of the recurrent site. And the affected amino acids are highly conserved across multiple species. According to the Brainspan Atlas database, MAST4 is expressed primarily in the mediodorsal nucleus of the thalamus and medial prefrontal cortex during the prenatal period, potentially contributing to embryonic brain development.
Conclusion: Our results revealed that the variants of MAST4 gene might lead to neurodevelopmental disorders with developmental delay and infantile spasm. Thus, MAST4 variants should be considered the potential candidate gene in patients with neurodevelopmental disorders clinically marked by infantile spasms.
Introduction
Neurodevelopmental disorders (NDDs) are a group of conditions with a high level of clinical and etiologic heterogeneity, such as attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), intellectual disability, and learning and communication disorders (Thapar et al., 2017). More than 3% of the population suffers from NDDs, ranging from mild impairments living a fairly normal life to severe disorders requiring lifelong care (Bitta et al., 2017; Mitani et al., 2021). To date, de novo variants currently account for only about 25% of unsolved NDD cases (Deciphering Developmental Disorders Study, 2017), indicating more NDD-associated genes remain to be discovered.
MAST family members (MAST1–4, and MAST-like) sharing approximately 49–64% sequence homology contain four distinct domains: The DUF 1,908 domain, the serine/threonine kinase domain, an AGC-kinase C-terminal domain and the PDZ domain (Garland et al., 2008). The MAST family plays a critical role in regulating microtubule system and normal cell division in mammals and fruit flies (Chudinova et al., 2017). So far, only MAST1 and MAST3 associated with the neurodevelopmental disease have been reported. MAST1 is highly expressed in the post-mitotic neurons of the cerebellum, cerebral cortex, and hippocampus (Garland et al., 2008; Tripathy et al., 2018). Latterly, de novo variants in MAST1 gene have been linked to mega-corpus-callosum syndrome and intellectual disability (Tripathy et al., 2018; Ben-Mahmoud et al., 2020; Hecher et al., 2020; Rodríguez-García et al., 2020). As a member of the MAST family, MAST3 has recently drawn growing attention as a novel pathogenic gene in neurodevelopmental disorders. More recently, patients with de novo variants in MAST3 are related to neurodevelopmental disabilities with or without epileptic seizure (Iwama et al., 2019; Shu et al., 2021; Spinelli et al., 2021). However, there have been no MAST2/MAST-L-related neurological diseases reported. MAST2 was reported to interact with the PTEN protein, which functions as a negative regulator of cell survival pathways and neuronal survival (Terrien et al., 2012). MAST-L is predominantly expressed in only heart and testis rather than in the brain, which is involved in the modulation of the microtubule system (Chudinova et al., 2017).
However, there are only a few reports on the association between MAST4 and neurological diseases, and the genotype-phenotype correlation needs to be clarified. Genome-wide association studies related MAST4 variations to myoclonic epilepsy and abnormalities in hippocampal anatomy (EPICURE Consortium et al., 2012; Hibar et al., 2017). Other studies have also reported the association of MAST4 with other systemic conditions. Furthermore, Mast4(−/−) mice exhibited excessive cartilage synthesis and osteopetrosis phenotype (Kim et al., 2022). In addition, Lee et al. (2021) revealed that MAST4 is associated with the fibroblast growth factor 2 (FGF2)/ERM pathway and is essential for spermatogenesis in mice.
Given that many other genes of the MAST family are highly correlated with neurological disorders, we hypothesized that MAST4 is also associated with it. In this study, we firstly reported three de novo missense variants in MAST4 gene in four unrelated individuals manifesting NDD with DD and epileptic spasms using trio-based ES. The four affected individuals’ clinical data and imaging investigation were collected and systematically reviewed. Together with the bioinformatic analysis, our results support the possibility that MAST4 relates to neurodevelopmental disorders with infantile spasms as a promising novel pathogenic gene. In addition, we further prove that the MAST family members have a critical role in nervous system development.
Materials and methods
Study participants
The patients were recruited from an NDD cohort of more than 1,000 at Hunan Provincial Maternal and Child Health Care Hospital. Patients diagnosed with NDD without any identifiable risk factors were enrolled in the study. This study was approved by the Ethics Committee (2020-S003) of Hunan Maternal and Child Health Hospital. The clinical data, laboratory findings, brain MRI and EEG of the four affected individuals were collected and systematically reviewed by experienced neurophysiologists and neuroradiologists. Written informed consent was obtained from all participants in the study.
Trio-exome sequencing and quality control
The peripheral blood was collected from the probands, their parents and other available family members to determine the origin of the identified genetic variants. Genomic DNA (>3 μg) was extracted using the DNA mini kit (Aidlab Biotech, China). The kinship of four trios’ samples have been checked, and the data is shown in Supplementary Table 1 and novel variants identified in this study were submitted to ClinVar (SCV002817420- SCV002817422). Sequencing reads were aligned to the reference human genome (GRCh37/hg19) using the BWA2 (v0.7.15). Picard was applied to sort by chromosome coordinates and mark duplicates1. Single nucleotide variants and insertions/deletions of samples were called and the Variant Call Format (VCF) file was generated (format v4.1) by HaplotypeCaller v4.0 from Genome Analysis Toolkit (GATK) (Van der Auwera et al., 2013). BCFtools (Narasimhan et al., 2016), VCFtools (Danecek et al., 2011) and GATK (v4.1.8.1) were applied to left-align, normalize, extract and retain rare events allele frequency <0.1% in gnomAD v2.1.1 non-neuro subset exomes (Karczewski et al., 2020) in the VCF files. We removed variants within mean mapping quality of mapped reads (MQ) <35, quality by depth (QD) ≤2, Fisher Strand (FS) ≤60, Strand Odds Ratio (SOR) >3, MQ Rank Sum −12.5 and Read Pos Rank Sum ≤−8. Genotype quality (GQ), read-depth (RD) and allele balance (AB) were used for genotype-level quality controls. Variants within 0.25 < AB < 0.75, DP > 10, and GQ > 25 were reserved. Finally, we chose loss of function mutations and missense variants with CADD > 20. Pubvar variant annotation engine and VEP6 (release 88) were used to annotate variants2. Relatedness was calculated between each pair of samples using King v2.2.7 (Manichaikul et al., 2010). The pathogenicity of variants was interpreted according to American College of Medical Genetics (ACMG)/AMP guidelines and modified based on ClinGen recommendations. Validation of the identified variants was performed using sanger sequencing.
Bioinformatic analysis
In silico variants analysis
To evaluate the conservation of the affected amino acid, we performed multiple sequence alignments across different species (Madeira et al., 2022). The conservation and pathogenicity of missense variations were evaluated through an integrated and accessible genetic database containing multiple prediction tools (i.e., SIFT, REVEL, CADD, and VEST3 et al.) (Li et al., 2018; Kopanos et al., 2019). ANNOVAR was used to annotate all MAST4 variants (Wang et al., 2010). Published MAST4 de novo mutations were obtained from Gene4Denovo3. Protein domain structures were visualized using DOG 2.0 (Ren et al., 2009).
Co-expression analysis
A gene with RPKM >0.5 in 50% of all developing cortex tissues was defined as a cortex-expressed gene. A total of 37 well-established IS-associated genes were considered a known IS gene set (Pavone et al., 2020). A total of 43 known genes associated with dominant epilepsy disorders, 33 genes related to neurodevelopmental disorders with epilepsy and 50 known genes associated with dominant EE syndromes were defined as a known EE gene set (Epi4K consortium and Epilepsy Phenome/Genome Project, 2017; Heyne et al., 2018). A total of 789 mono-allelic DD genes within definitive or strong evidence were from The Developmental Disorders Genotype-to-Phenotype database4 (Supplementary Table 2). Three gene sets were all applied to calculate the Spearman’s correlation coefficient with all cortex-expressed genes. Next, the mean of Spearman’s correlation coefficient of each cortex-expressed gene with known IS/EE/DD gene sets was computed. Percentiles of the average correlation coefficient between MAST4 and known IS/EE/DD sets were acquired.
MAST4 expression pattern in development brain
RNA-seq data at different developmental stages (from 8 postconceptional weeks to 40 years) for multiple brain areas were obtained from Brainspan5. RNA expression was normalized to reads per kilobase million (RPKM). Univariate linear regression for analyzing MAST4 expression mode in CC and FC was achieved by lm() function in R6.
Statistical methods
Comparison for CADD was performed using Wilcoxon test. Comparison for SIFT and Poly then was carried out using the Fisher test.
Results
Four individuals presented typical neurodevelopmental disorders with infantile spasm
Firstly, we did not find Pathogenic and Like Pathogenic variation sites of genes related to NDD and epilepsy based on the trios’ exome sequencing. Then, we chose our candidate variant based on variant pathogenicity, conservation and hereditary mode. For the possible candidate variants, we further look into the gene functions which were related to neurodevelopment. Through all these methods to identify candidate variant, we finally chose MAST4 variants as candidates for our patients. Three de novo variants were identified in four unrelated patients (Figure 1). Clinical features of the individual with MAST4 variants were summarized in Table 1.
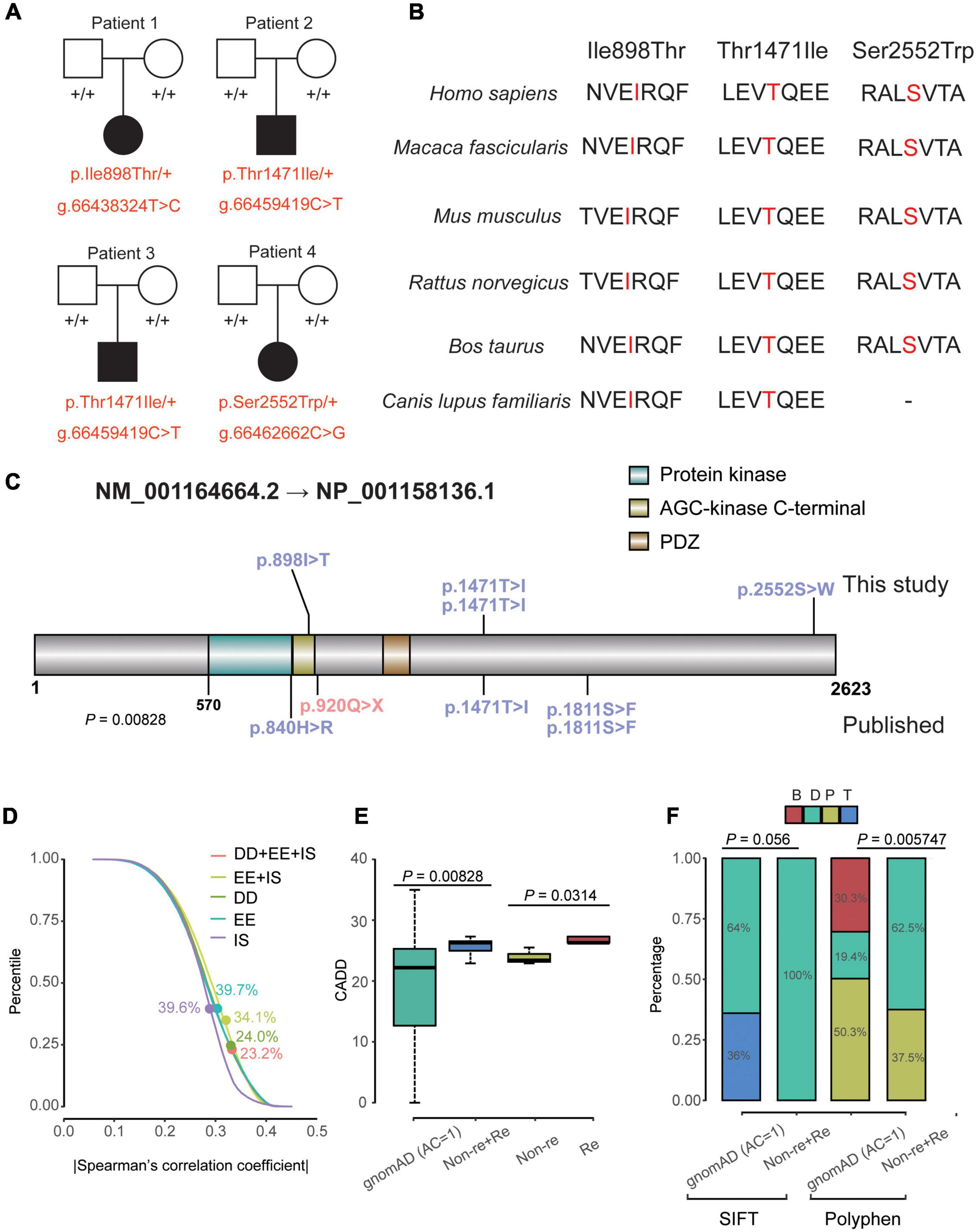
Figure 1. Clinical data and genetics of variants in MAST4. (A) Pedigree plot of the four unrelated families in this research. All affected probands (patient 1–4) are children of unaffected parents. Solid squares and solid circles represent affected males and females, respectively. Hollow squares and hollow circles represent unaffected males and females, respectively, +/+ represents a wild type and –/+ (red) represents a heterozygous genotype. (B) Comparison of conservative property of three de novo MAST4 sites among multiple species. (C) Location of the variants in MAST4. Variants in blue and red represent missense and LGD events, respectively. Mutations on and under the MAST4 belong to this study and published researches, respectively. (D) Percentile of |Spearman’s correlation coefficient| of the co-expression for all non-DD + EE + ISs genes vs. DD + EE + ISs genes, all non-DD vs. DD genes, all non- EE genes vs. EE genes and all non- ISs genes vs. ISs genes. Dots represent the percentile of MAST4. (E,F) Comparison of the CADD scores, SIFT bin and Polyphen bin between de novo missense variants (recurrent and non-recurrent) identified in Table 1 and the private missense variants identified in the non-neuro subset of gnomAD. AC, allele count; B, benign; CADD, Combined Annotation Dependent Depletion; D, deleterious; DD, developmental disorder; EE, epileptic encephalopathy; gnomAD, genome aggregation database; IS, infantile spasm; P, possibly damaging; SIFT, sortig intolerant from tolerant; T, Tolerant.
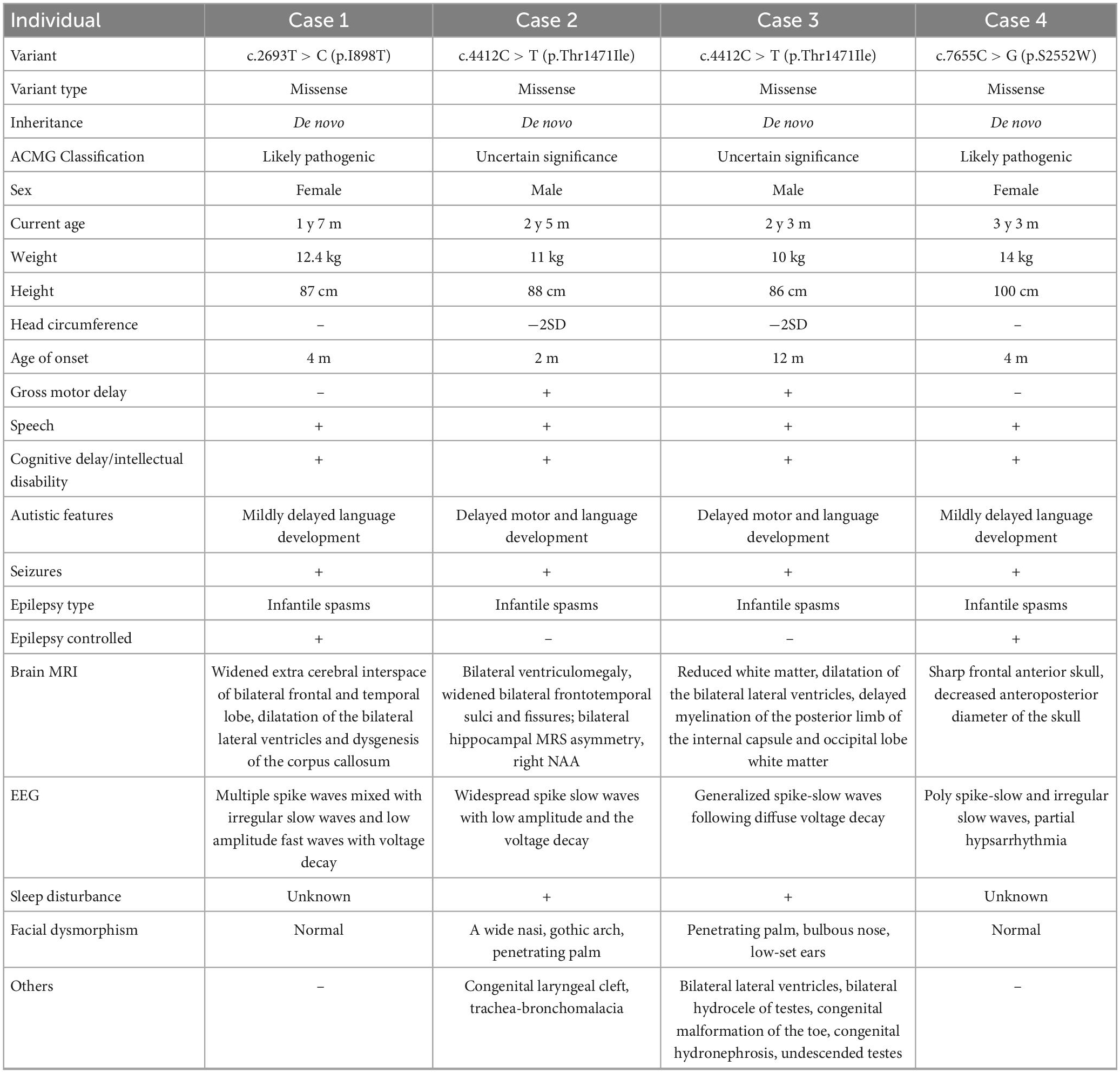
Table 1. The clinical features of the affected individual with the MAST4 variants.
Patient 1 is a 1 year and 8 months old girl born to a healthy, non-consanguineous couple. At 2 months of age, the girl could not hold her head up steadily and lacked eye contact. Two months later, she started to present recurrent seizures, characterized by crying with abduction motions in bilateral upper limbs, flexion motions in lower limbs, body writhing toward right and both eyes gazing up and left. Cluster episodes of seizures occurred tens of seconds every time and two to three times a day. Convulsions of limbs occurred mainly at midnight after wake. At 5-month-old, the proband cannot lift her head steadily and presented hypertonia of the extremities. Her EEG mainly showed intermittent partial hypsarrhythmia at 5-month-old (Figure 2A) and clusters of spastic seizures: multiple spike waves mixed with irregular slow waves and low amplitude fast waves with voltage declining for 3–5 s (Figure 2B). Brain MRI showed widened extracerebral interspace of bilateral frontal and temporal lobe, dilatation of the bilateral lateral ventricles and dysgenesis of the corpus callosum (Figures 3A–C). At present, the 20-month-old girl had no cluster episodes of convulsions after the treatment with adrenocorticotropic hormone (ACTH) and her motor developmental milestone nearly reached normal.
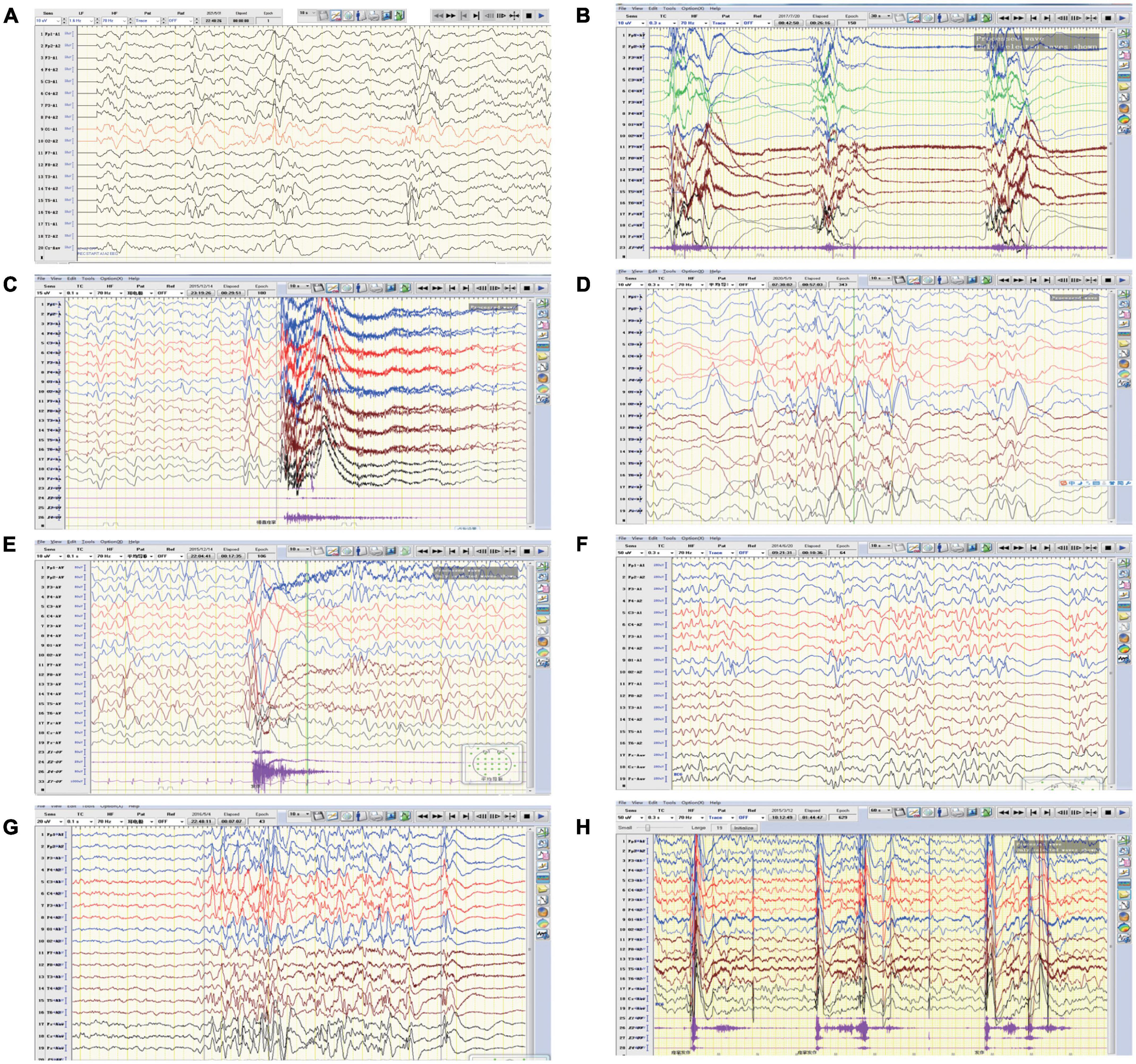
Figure 2. Representative EEGs of the affected individuals. (A) EEG of patient one revealed intermittent partial hypsarrhythmia (obtained at the age of 5 months). (B) EEG of patient one showed clusters of spastic seizures (at the age of 10 months). (C,D) EEG of patient two revealed convulsive episode and widespread spike slow waves with low amplitude at 4-month-old and 12-month-old, respectively. (E) EEG of patient three showed generalized spike-slow waves following diffuse voltage decay (at 15 months old). (F–H) EEG of patient four demonstrated poly spike-slow and irregular slow waves were more prominent on left, accompany with partial hypsarrhythmia and cluster of spasms (obtained at the age of 4 months and 12 months, respectively).
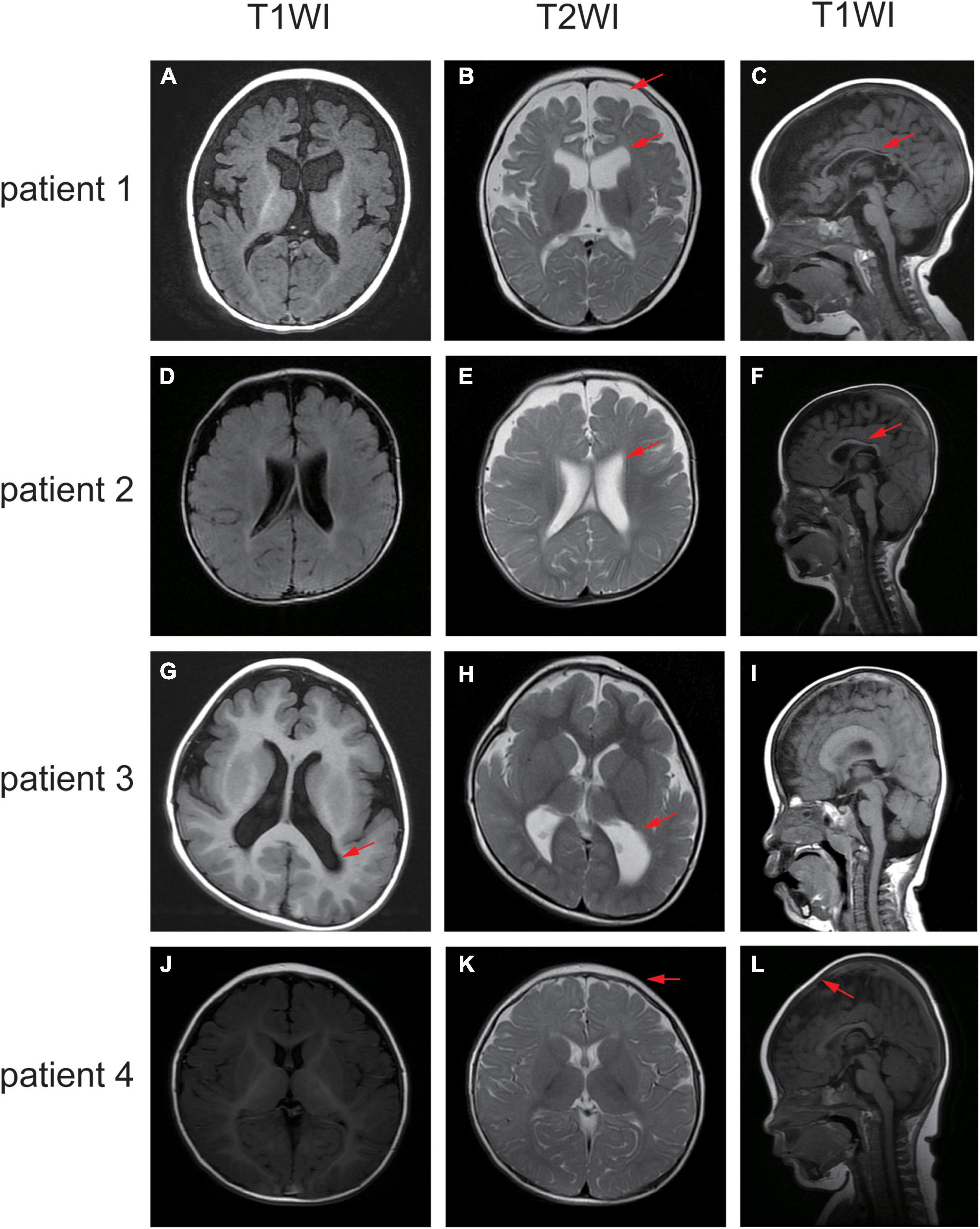
Figure 3. Different sequence of the brain MRI of the affected individuals. (A–C) MRI of patient one demonstrated widened extracerebral interspace, dilatation of the bilateral lateral ventricles and dysgenesis of the corpus callosum. (D–F) MRI of patient two showed bilateral ventriculomegaly and thin corpus callosum. (G–I) MRI of patient three revealed the posterior horn of bilateral ventricle and the trigone were enlarged bilaterally. (J–L) MRI of patient four elucidated sharp frontal anterior skull and decreased anteroposterior diameter of the skull. The red arrows indicate the positions of the lesions.
Patient 2 is a boy, aged of 2 years and 6 months, who was born at 38 weeks via vaginal delivery due to oligohydramnios. At 2 months old, the infant was admitted to hospital because of intermittent convulsions and fever and the physical examination suggested microcephaly with a head circumference of 35 cm (<−2SD). His echocardiogram revealed patent foramen ovale (PFO) and tricuspid regurgitation. Brain MRI showed bilateral ventriculomegaly and widened bilateral frontotemporal sulci and fissures, suggesting brain developmental delay, bilateral hippocampal MRS asymmetry, and right low NAA indicating neuronal damage or loss in the hippocampal (Figures 3D–F). The onset EEG of the boy showed widespread spike slow waves with low amplitude and the voltage declined about 4.5 s (Figure 2C). Diffuse slow wave, spike-slow and slow wave were still seen after half a year (Figure 2D). Two months later, he was re-admitted due to recurrent convulsions manifested as facial flushing, cyanosis of lips, closed eyes, head pitched back, stiffness and shaking of limbs and foaming at the mouth. He was diagnosed with NDD, infantile spasm, trachea-bronchomalacia, cortical corpus callosum dysgenesis, and congenital laryngeal cleft. Seizures were poorly controlled with levetiracetam and topiramate. EEG at 1 year of age was still abnormal. At present, the patient was presented with delayed cognitive and language development.
Patient 3 is a 2-year-old male born after an uneventful delivery. The color ultrasound at 34 weeks of gestation showed widened bilateral ventricle. Shortly after birth, the child was diagnosed with dilatation of the bilateral lateral ventricles, tricuspid regurgitation, and bilateral hydrocele of the testes. The patient was hospitalized at 5 months of age because of head-up instability with a head circumference of 38.5 cm (<−2SD) and suboptimal limb activity. His developmental milestones were also severely delayed. He couldn’t raise his head until 7 months old, turn over until 10 months and still couldn’t crawl until now. Since the age of 3 months, he has been undergoing a continuous rehabilitation program including physical, speech and neuropsychology therapy. Brain MRI at 9 months old revealed reduced white matter, dilatation of the bilateral lateral ventricles, delayed myelination of the posterior limb of the internal capsule and occipital lobe white matter (Figures 3G–I). The boy had episodes of seizures at 1 year of age manifested as upturned eyes, trismus, and flexed twitching limbs. The onset EEG mainly showed generalized spike-slow waves following diffuse voltage decay suggesting infantile spasm (Figure 2E).
Patient 4 is a 3-year-old female who was born at full term from a normal pregnancy. She couldn’t make sounds and head up unsteadily until 4 months of age. Shortly after, the girl developed seizures characterized by dozen times of nodding and body flexing, 5–6 min in each episode. The EEG showed poly spike-slow and irregular slow waves were more prominent on the left, accompany by partial hypsarrhythmia at 4 months old (Figures 2F, G) and a cluster of spasms at 1 year old (Figure 2H). Brain MRI at 11 months old showed a sharp frontal anterior skull and decreased anteroposterior diameter of the skull (Figures 3J–L). Then the girl was diagnosed with infantile spasm and her seizures were well controlled with topiramate monotherapy. At present, the girl had reached a mildly delayed motor development combined with mildly delayed behavioral development.
In silico parameters prompt de novo MAST4 variants are pathogenic especially its recurrent sites
Although clinical evidence has been presented to implicate MAST4 an EE/DD risk gene, its pathogenicity needs further elucidation based on the characteristics of MAST4 and variants on its protein.
For gene, constraint’s parameters of MAST4 (Missense Z-score = 1.93 and RVIS% = 8.35) suggested that MAST4 is moderately intolerant for missense variants. Statistical analyses of publically available data on co-expression revealed MAST4 ranks higher concerning the correlations with known DD genes (Percentile = 24.0%) compared with well-established IS/EE genes (IS: Percentile = 39.6%; EE: Percentile = 39.7%) (Figure 1D and Supplementary Table 3).
For variants on it, all three variants are not found in human population databases gnomAD, ExAC, and 1,000 genomes (non-neuro subset) (Table 2). These three heterozygote missense variants were identified in four unrelated cases as follows: chr5:66438324 (c.2693T > C: p.Ile898Thr), chr5:66459419 (c.4412C > T: p.Thr1471Ile) and chr5:66462662 (c.7655C > G:p.Ser2552Trp). All of them are de novo variants, where in bidirectional Sanger sequencing confirmed such findings (Figures 1A, 4). All three variants are predicted to be damaging or probably damaging through multiple different bioinformatics software (Table 2) and the affected amino acid is highly conserved across multiple species (Figure 1B). The missense variant p.Ile898Thr is mapped to the AGC-kinase C-terminal, which can modify other proteins by adding a phosphate group (Figure 1C). And the other variant p.Ser2552Trp is located in a phosphoserine-modified residue which may affect cell membrane stability and signal transduction (Uniport, O15021). The last variant p.Thr1471Ile was screened out in two unrelated patients and is located in the domain with the function unclear yet.
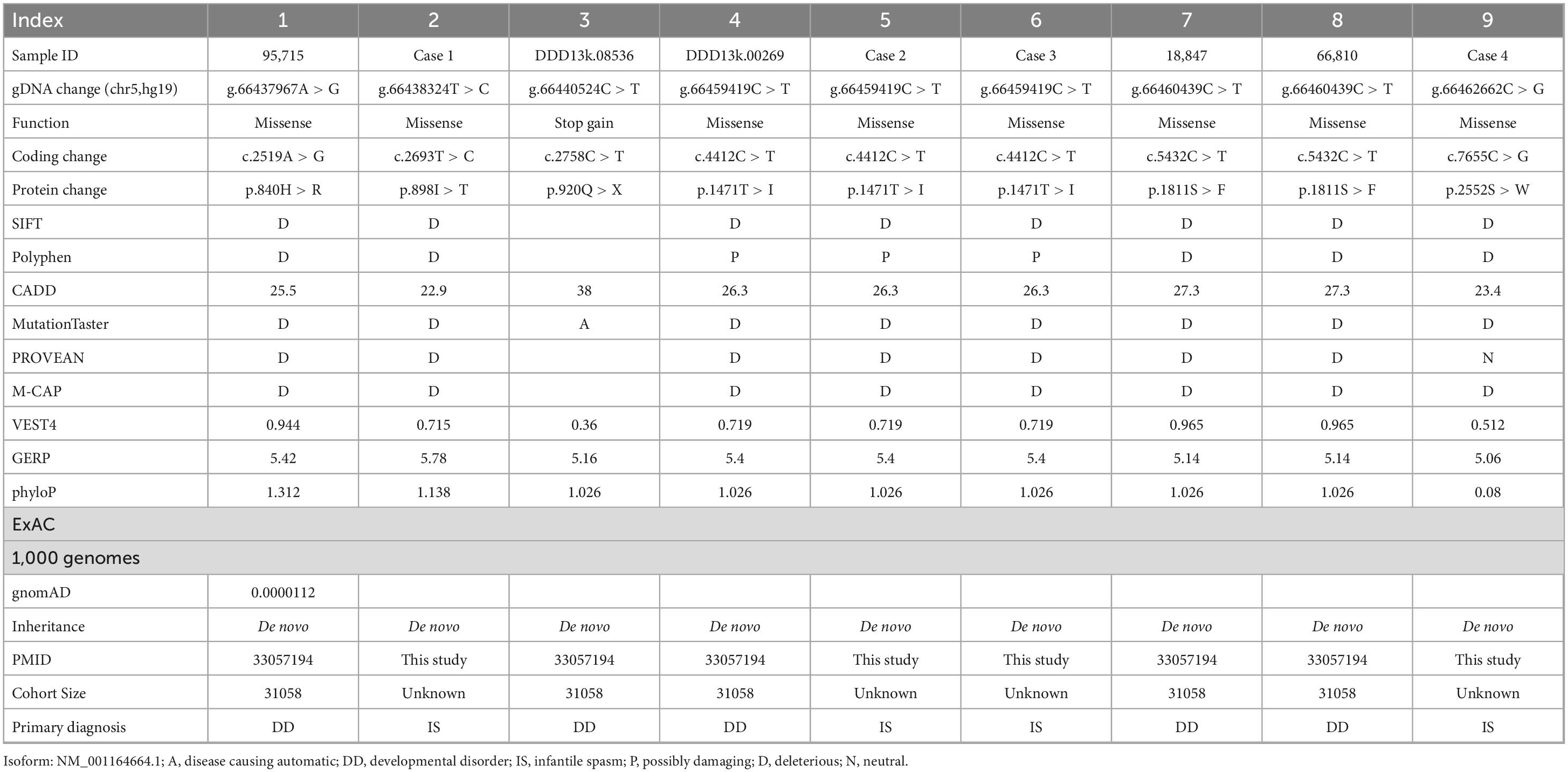
Table 2. Summary of MAST4 de novo missense variants identified in neurodevelopmental disorders.
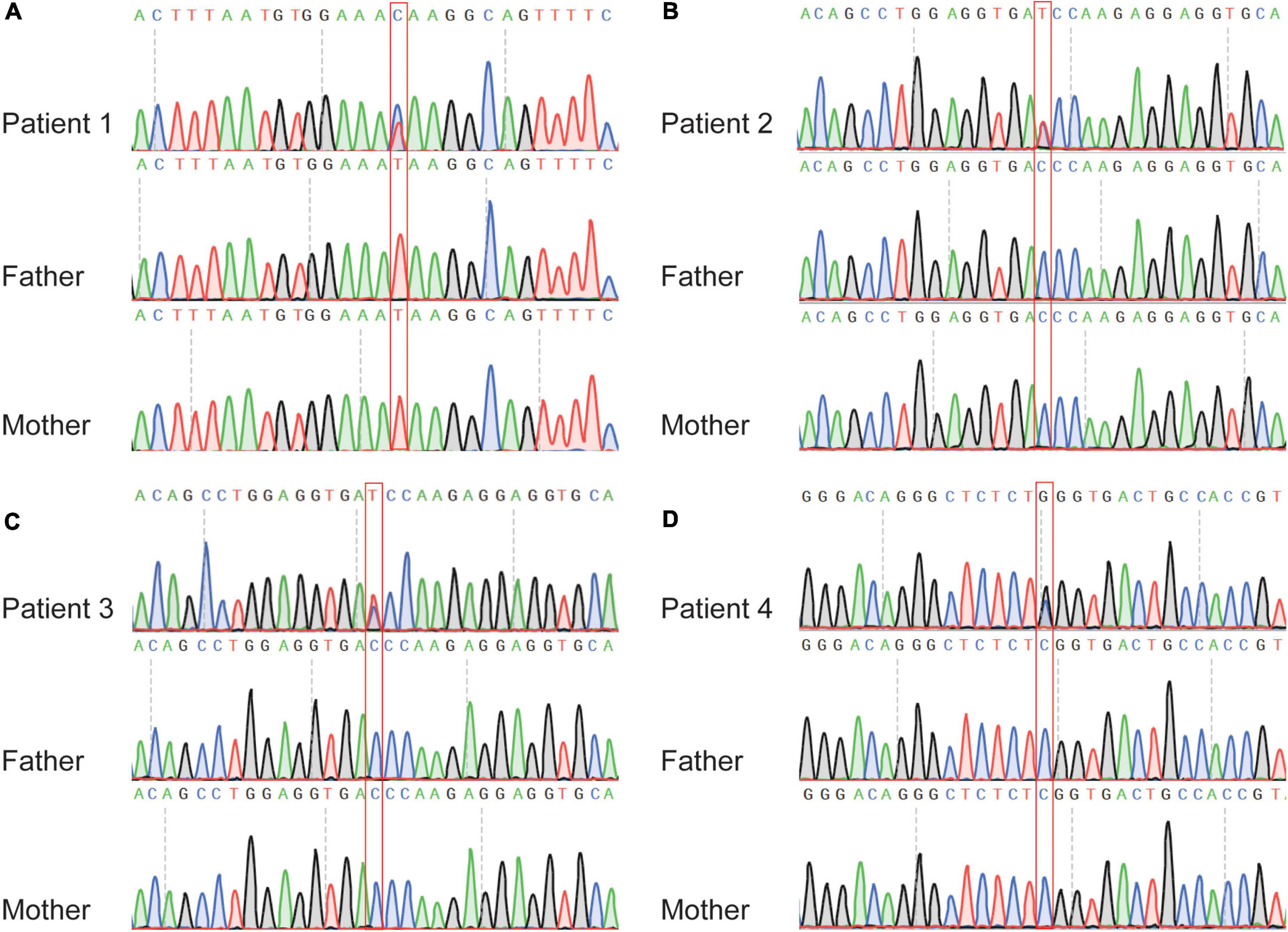
Figure 4. Sanger sequencing of the affected individuals with MAST4 variants identified in this study. (A) c.2693T > C in patient one trio. (B) c.4412C > T in patient two trio. (C) c.4412C > T in patient three trio. (D) c.7655C > G in patient four trio. The red boxes denote the positions of the variants.
After we integrated IS/EE/DD studies with large GS/ES cohorts from Gene4Denovo, eight de novo missense variants and one nonsense mutation were obtained (Table 2 and Figure 1C). Probands carrying MAST4 variants from published research had DD symptoms, which implied that de novo MAST4 variants may mainly contribute to DD phenotypes combined with previous results in Figure 1D and Supplementary Table 3. Furthermore, we chose three representative constraint’s metrics of missense variants for follow-up analysis, Combined Annotation Dependent Depletion (CADD), Sorting Intolerant From Tolerant (SIFT) and Polyphen. Compared to private MAST4 missense mutations in gnomAD non-neuro subset, higher CADD scores were found in de novo missense variants of integrated IS/EE/DD cohorts (P = 0.00828) (Figure 1E). Similarly, integrated de novo missense events had more pathogenic components predicted by SIFT and Polyphen in contrast to private missense variants in gnomAD non-neuro subset (SITF: P = 0.056; Polyphen: P = 0.005747), even if P-value in SIFT was close to significance (Figure 1F). Strikingly, two recurrent sites, p.Thr1471Ile and p.Ser1811Glu, were carried by three (two from our study and one from published research) and two (all from published research) unrelated probands, separately (Table 2 and Figure 1C). We noticed higher CADD scores from two recurrent events than those in non-recurrent mutations from integrated IS/EE/DD cohorts (P = 0.0314), which indicated more deleteriousness of recurrent variants in MAST4 (Figure 1E).
Spatio-temporal modes of MAST4
In order to clarify the important role of MAST4 in brain development, the RNA sequencing data from BrainSpan was applied to find out the spatiotemporal expression pattern of MAST4. We found a time-dependent up-regulation during 50–150th days in utero for MAST4 (Figure 5A). Differently, most brain regions retained the high expression of MAST4 during whole post-birth periods (Figure 5A). To reveal a dynamic change of MAST4 in IS/EE/DD relevant brain regions during periods of brain development, like cerebral cortex (CC) and prefrontal cortex (FC) (Treiman, 2001; Hsu and Jaeggi, 2014; Severino et al., 2020; Galovic et al., 2021), a univariate linear regression analysis was performed. In CC and FC, expression of MAST4 was more significantly up-regulated during the period of embryonic development (CC: R2 = 0.4203 and P = 2.2 × 10–16. FC: R2 = 0.4115 and P = 1.97 × 10–10) than that during the postnatal period (CC: R2 = 0.1237 and P = 1.951 × 10–7. FC: R2 = 0.1309 and P = 0.0003386) (Figures 5B, C). These data implicated a potential role of MAST4 during embryonic developing periods.
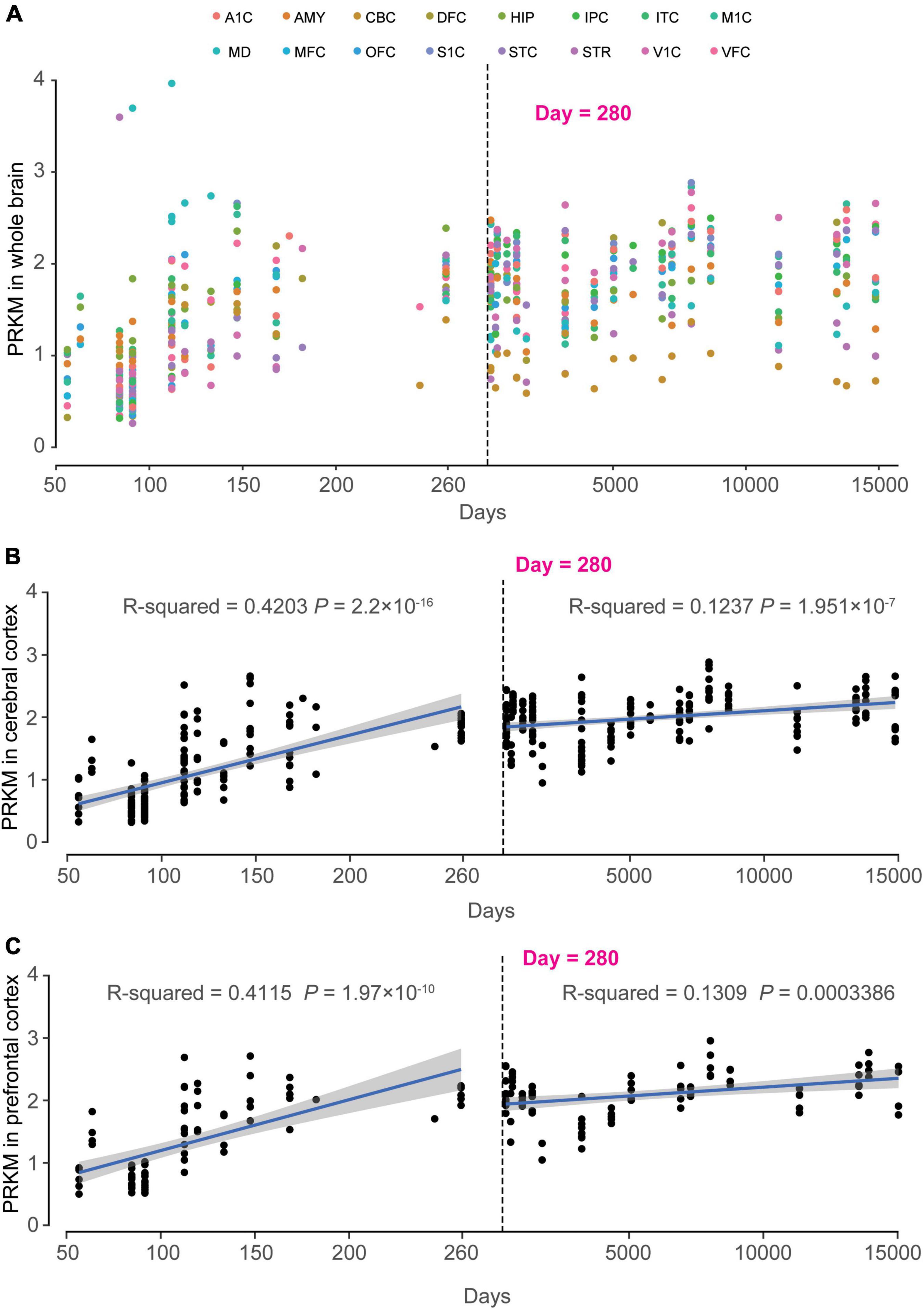
Figure 5. Dynamic expression pattern of MAST4 in human development brains. The x-axis is the age of samples in days and y-axis is the RPKM of MAST4. The dashed line indicates the birth day. (A) Expression of MAST4 during the whole life in different brain regions. A1C, primary auditory cortex; AMY, amygdaloid complex; CBC, cerebellar cortex; DFC, dorsolateral prefrontal cortex; HIP, hippocampus; IPC, inferior parietal cortex; ITC, inferolateral temporal cortex; M1C, primary motor cortex; MD, mediodorsal nucleus of thalamus; MFC, medial prefrontal cortex; OFC, orbital frontal cortex; S1C, primary somatosensory cortex; STC, superior temporal cortex; STR, striatum; V1C, primary visual cortex; VFC, ventrolateral prefrontal cortex. (B,C) Expression of MAST4 in the cerebral cortex and prefrontal cortex regions of human brain, respectively. Univariate regression analysis was applied in prenatal brains (left) and postnatal brains (right), separately. Blue line represents the regression line and the gray region indicates 95% confidence interval. The text shows the R2 and P-value of regression.
Discussion
In the present study, we described three de novo heterozygous missense MAST4 variants in four unrelated patients with IS and varying levels of developmental delays. The detail variations were identified as follows: 5:66438324-T-C (p.Ile898Thr), 5:66459419-C-T (p.Thr1471Ile) and 5:66462662-C-G (p.Ser2552Trp). The variant p.Ile898Thr located in the AGC-kinase C-terminal domain and p.Ser2552Trp was right in a phosphoserine-modified residue which may affect cell membrane stability and signal transduction. The variant p.Thr1471Ile was identified in two unrelated individuals with severe developmental retardation, indicating a greater probability of function-altering and pathogenic hotspot variants. According to a previous study reanalyzing large-scale whole exome sequencing data, MAST4 was identified as a novel gene significantly associated with developmental disorders (Kaplanis et al., 2020). Intriguingly, coupled with our findings, two recurrent missense variants, 5-66460439-C-T and 5-66459419-C-T were identified, which greatly improved the possibility of pathogenicity. The results of the bioinformatic analysis tend to elucidate that de novo MAST4 variants may contribute to DD phenotypes compared to IS combined with previous report. However, this might because MAST4 gene variations are rarely reported and the patients may also have seizures in the cohort with DD. Our study presented evidence that MAST4 could be a candidate NDD with IS gene and therefore expanded the genetic and phenotypic spectrum.
MAST4 belongs to the microtubule-associated serine/threonine kinase (MAST) family and is widely expressed in the nervous system (Garland et al., 2008). MAST4 protein distribution in the central nervous system is predominant in cerebellar Purkinje cells, hippocampus and white matter-containing regions (Garland et al., 2008) and its expression was increased in the mouse hippocampus following seizure activity (French et al., 2001). At present, there have been few basic studies on MAST4 in the nervous system. These results suggest that MAST4 variants disrupt cells’ self-renewal, proliferation, and differentiation. Gongol et al. (2017) constructed a missense mutant plasmid (E682A) and the detection of kinase activity in vitro suggested that the missense variant disrupts kinase activity. We also constructed the MAST4 wild type and three corresponding mutant plasmids. However, the plasmids were too long to be transfected into cells, and neither was the lentivirus.
Recent studies have shown close relationships between NDD and variants in the MAST family members. MAST1 was the first family member found to be linked with neurological conditions (McMichael et al., 2015). And it appeared to play a vital role in interacting with the dystrophin/utrophin-associated complex located in the neuromuscular junction and postsynaptic region of the central synapses (Lumeng et al., 1999). Previous studies suggested that MAST1 was a novel candidate gene associated with multiple neurological diseases, including mega-corpus-callosum syndrome, cerebral palsy, developmental delay and intellectual disability (McMichael et al., 2015; Tripathy et al., 2018; Ben-Mahmoud et al., 2020; Rodríguez-García et al., 2020). These observations suggest that MAST1 is crucial in neuronal development and may be a new potential biomarker for neuronal developmental disorders. Mast 3, as well as Mast 1, another protein of this family, has been previously reported to be involved in neurological disease. Iwama et al. (2019) firstly showed that de novo variants in the STK domain of the MAST3 gene resulted in Rett syndrome-like phenotype. Subsequently, two studies illustrated the role of MAST3 variants in neurodevelopmental delay and developmental epileptic encephalopathy, respectively (Shu et al., 2021; Spinelli et al., 2021). Morange et al. (2021) proposed that MAST2 variants may cause venous thromboembolism by interfering with the hemostatic balance of endothelial cells. Taken together, our analysis firstly reported the variations in MAST4 associated with NDD and IS and took a step further indicating the MAST family members play an essential role in neurodevelopment.
Combining the findings of genetic and bioinformatic analysis, our findings may confirm MAST4 as a novel gene for NDD with IS. However, there are some limitations in this study to be mentioned. The variants were mainly identified in in-person visiting patients with seizures and the lack of an observable phenotype in other systems does not rule out the possibility that other undiscovered biological and clinical phenotypes exist. Furthermore, more cases and functional investigations are necessary to demonstrate robust genotype-phenotype correlations and the underlying mechanisms.
Conclusion
Here, we identified three heterozygous missense MAST4 variants in four unrelated patients with a spectrum of IS and varying developmental delays. Our findings evaluate the genotype-phenotype correlations of MAST4 gene in NDDs and IS, which expands the genetic landscape and future genetic counseling of NDDs with IS.
Data availability statement
The datasets that support this article are available on request from the corresponding authors. The data are not publicly available due to concerns regarding privacy and participant/patient anonymity.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Maternal and Child Health Hospital of Hunan Province (2020-S003). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
JY and HW: conceptualization and methodology. XZ and NX: data curation and writing—original draft preparation. YC, YLC, PW, and WG: collection and evaluation of the clinical and genetic evidence. AL and YP: data analysis. BX and LS: writing—reviewing and editing. All authors read and approval of the final manuscript.
Funding
This research was supported by the National Key Research and Development Program of China (grant number: 2021YFC1005300), the Natural Science Foundation of Hunan Province, China (grant numbers: 2021JJ40280 and 2022JJ40206), the Technical Innovation Guidance Project of Chenzhou (grant number: lcyl2021042), and the Science and Technology Development Plan of Chenzhou Science and Technology Bureau (grant number: ZDYF2020147).
Acknowledgments
We thank the patients and their families for their cooperation and participation in our study. We also thank the coordinators from the Chigene (Beijing) Translational Medical Research Center Co., Ltd., for sequencing technology support and matching the cases from Chigene genotype-phenotype database.
Conflict of interest
PW and WG were employed by Chigene (Beijing) Translational Medical Research Center Co., Ltd., Beijing.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1097553/full#supplementary-material
Supplementary Table 1 | The relationships of family 1–4 in this study. Kinship: Estimated kinship coefficient from the SNP data. >0.354, (0.177, 0.354), (0.0884, 0.177) and (0.0442, 0.0884) corresponds to duplicate/MZ twin, 1st-degree, 2nd-degree, and 3rd-degree relationships respectively.
Supplementary Table 2 | Well-established known IS/EE/DD gene sets.
Supplementary Table 3 | Percentile of Spearman’s correlation coefficient| of the co-expression for all non-DD + EE + ISs genes vs. DD + EE + ISs genes, all non-DD vs. DD genes, all non- EE genes vs. EE genes and all non- ISs genes vs. ISs genes.
Footnotes
- ^ http://broadinstitute.github.io/picard/
- ^ www.pubvar.com
- ^ http://www.genemed.tech/gene4denovo/home
- ^ www.deciphergenomics.org/ddd/ddgenes
- ^ http://www.brainspan.org/
- ^ www.r-project.org/
References
Ben-Mahmoud, A., Al-Shamsi, A. M., Ali, B. R., and Al-Gazali, L. (2020). Evaluating the role of MAST1 as an intellectual disability disease gene: Identification of a novel de novo variant in a patient with developmental disabilities. J. Mol. Neurosci. 70, 320–327. doi: 10.1007/s12031-019-01415-8
Bitta, M., Kariuki, S. M., Abubakar, A., and Newton, C. R. J. C. (2017). Burden of neurodevelopmental disorders in low and middle-income countries: A systematic review and meta-analysis. Wellcome Open Res. 2:121. doi: 10.12688/wellcomeopenres.13540.3
Chudinova, E. M., Karpov, P. A., Fokin, A. I., Yemets, A. I., Lytvyn, D. I., Nadezhdina, E. S., et al. (2017). MAST-like protein kinase IREH1 from Arabidopsis thaliana co-localizes with the centrosome when expressed in animal cells. Planta 246, 959–969. doi: 10.1007/s00425-017-2742-4
EPICURE Consortium, EMINet Consortium, Steffens, M., Leu, C., Ruppert, A.-K., Zara, F., et al. (2012). Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum. Mol. Genet. 21, 5359–5372. doi: 10.1093/hmg/dds373
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Epi4K consortium and Epilepsy Phenome/Genome Project (2017). Ultra-rare genetic variation in common epilepsies: A case-control sequencing study. Lancet Neurol. 16, 135–143. doi: 10.1016/S1474-4422(16)30359-3
French, P. J., O’Connor, V., Jones, M. W., Davis, S., Errington, M. L., Voss, K., et al. (2001). Subfield-specific immediate early gene expression associated with hippocampal long-term potentiation in vivo. Eur. J. Neurosci. 13, 968–976. doi: 10.1046/j.0953-816x.2001.01467.x
Galovic, M., Ferreira-Atuesta, C., Abraira, L., Döhler, N., Sinka, L., Brigo, F., et al. (2021). Seizures and epilepsy after stroke: Epidemiology, biomarkers and management. Drugs Aging 38, 285–299. doi: 10.1007/s40266-021-00837-7
Garland, P., Quraishe, S., French, P., and O’Connor, V. (2008). Expression of the MAST family of serine/threonine kinases. Brain Res. 1195, 12–19. doi: 10.1016/j.brainres.2007.12.027
Gongol, B., Marin, T. L., Jeppson, J. D., Mayagoitia, K., Shin, S., Sanchez, N., et al. (2017). Cellular hormetic response to 27-hydroxycholesterol promotes neuroprotection through AICD induction of MAST4 abundance and kinase activity. Sci. Rep. 7:13898. doi: 10.1038/s41598-017-13933-9
Hecher, L., Johannsen, J., Bierhals, T., Buhk, J.-H., Hempel, M., and Denecke, J. (2020). The clinical picture of a bilateral perisylvian syndrome as the initial symptom of mega-corpus-callosum syndrome due to a MAST1-gene mutation. Neuropediatrics 51, 435–439. doi: 10.1055/s-0040-1710588
Heyne, H. O., Singh, T., Stamberger, H., Abou Jamra, R., Caglayan, H., Craiu, D., et al. (2018). De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 50, 1048–1053. doi: 10.1038/s41588-018-0143-7
Hibar, D. P., Adams, H. H. H., Jahanshad, N., Chauhan, G., Stein, J. L., Hofer, E., et al. (2017). Novel genetic loci associated with hippocampal volume. Nat. Commun. 8:13624. doi: 10.1038/ncomms13624
Hsu, N. S., and Jaeggi, S. M. (2014). The emergence of cognitive control abilities in childhood. Curr. Top. Behav. Neurosci. 16, 149–166. doi: 10.1007/7854_2013_241
Iwama, K., Mizuguchi, T., Takeshita, E., Nakagawa, E., Okazaki, T., Nomura, Y., et al. (2019). Genetic landscape of rett syndrome-like phenotypes revealed by whole exome sequencing. J. Med. Genet. 56, 396–407. doi: 10.1136/jmedgenet-2018-105775
Kaplanis, J., Samocha, K. E., Wiel, L., Zhang, Z., Arvai, K. J., Eberhardt, R. Y., et al. (2020). Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762. doi: 10.1038/s41586-020-2832-5
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kim, P., Park, J., Lee, D.-J., Mizuno, S., Shinohara, M., Hong, C. P., et al. (2022). Mast4 determines the cell fate of MSCs for bone and cartilage development. Nat. Commun. 13:3960. doi: 10.1038/s41467-022-31697-3
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: The human genomic variant search engine. Bioinformatics 35, 1978–1980. doi: 10.1093/bioinformatics/bty897
Lee, S.-J., Park, J., Lee, D.-J., Otsu, K., Kim, P., Mizuno, S., et al. (2021). Mast4 knockout shows the regulation of spermatogonial stem cell self-renewal via the FGF2/ERM pathway. Cell Death Differ. 28, 1441–1454. doi: 10.1038/s41418-020-00670-2
Li, J., Shi, L., Zhang, K., Zhang, Y., Hu, S., Zhao, T., et al. (2018). VarCards: An integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46, D1039–D1048. doi: 10.1093/nar/gkx1039
Lumeng, C., Phelps, S., Crawford, G. E., Walden, P. D., Barald, K., and Chamberlain, J. S. (1999). Interactions between beta 2-syntrophin and a family of microtubule-associated serine/threonine kinases. Nat. Neurosci. 2, 611–617. doi: 10.1038/10165
Madeira, F., Pearce, M., Tivey, A. R. N., Basutkar, P., Lee, J., Edbali, O., et al. (2022). Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279. doi: 10.1093/nar/gkac240
Manichaikul, A., Mychaleckyj, J. C., Rich, S. S., Daly, K., Sale, M., and Chen, W.-M. (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. doi: 10.1093/bioinformatics/btq559
McMichael, G., Bainbridge, M. N., Haan, E., Corbett, M., Gardner, A., Thompson, S., et al. (2015). Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 20, 176–182. doi: 10.1038/mp.2014.189
Mitani, T., Isikay, S., Gezdirici, A., Gulec, E. Y., Punetha, J., Fatih, J. M., et al. (2021). High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am. J. Hum. Genet. 108, 1981–2005. doi: 10.1016/j.ajhg.2021.08.009
Morange, P.-E., Peiretti, F., Gourhant, L., Proust, C., Soukarieh, O., Pulcrano-Nicolas, A.-S., et al. (2021). A rare coding mutation in the MAST2 gene causes venous thrombosis in a French family with unexplained thrombophilia: The Breizh MAST2 Arg89Gln variant. PLoS Genet. 17:e1009284. doi: 10.1371/journal.pgen.1009284
Narasimhan, V., Danecek, P., Scally, A., Xue, Y., Tyler-Smith, C., and Durbin, R. (2016). BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751. doi: 10.1093/bioinformatics/btw044
Pavone, P., Polizzi, A., Marino, S. D., Corsello, G., Falsaperla, R., Marino, S., et al. (2020). West syndrome: A comprehensive review. Neurol. Sci. 41, 3547–3562. doi: 10.1007/s10072-020-04600-5
Ren, J., Wen, L., Gao, X., Jin, C., Xue, Y., and Yao, X. (2009). DOG 1.0: Illustrator of protein domain structures. Cell Res. 19, 271–273. doi: 10.1038/cr.2009.6
Rodríguez-García, M. E., Cotrina-Vinagre, F. J., Gómez-Cano, M., Martínez de Aragón, A., Martín-Hernández, E., and Martínez-Azorín, F. (2020). MAST1 variant causes mega-corpus-callosum syndrome with cortical malformations but without cerebellar hypoplasia. Am. J. Med. Genet. A 182, 1483–1490. doi: 10.1002/ajmg.a.61560
Severino, M., Geraldo, A. F., Utz, N., Tortora, D., Pogledic, I., Klonowski, W., et al. (2020). Definitions and classification of malformations of cortical development: Practical guidelines. Brain 143, 2874–2894. doi: 10.1093/brain/awaa174
Shu, L., Xiao, N., Qin, J., Tian, Q., Zhang, Y., Li, H., et al. (2021). The role of microtubule associated serine/threonine kinase 3 variants in neurodevelopmental diseases: Genotype-phenotype association. Front. Mol. Neurosci. 14:775479. doi: 10.3389/fnmol.2021.775479
Spinelli, E., Christensen, K. R., Bryant, E., Schneider, A., Rakotomamonjy, J., Muir, A. M., et al. (2021). Pathogenic MAST3 variants in the STK domain are associated with epilepsy. Ann. Neurol. 90, 274–284. doi: 10.1002/ana.26147
Terrien, E., Chaffotte, A., Lafage, M., Khan, Z., Préhaud, C., Cordier, F., et al. (2012). Interference with the PTEN-MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci. Signal. 5:ra58. doi: 10.1126/scisignal.2002941
Thapar, A., Cooper, M., and Rutter, M. (2017). Neurodevelopmental disorders. Lancet Psychiatry 4, 339–346. doi: 10.1016/S2215-0366(16)30376-5
Treiman, D. M. (2001). GABAergic mechanisms in epilepsy. Epilepsia 42, 8–12. doi: 10.1046/j.1528-1157.2001.042suppl.3008.x
Tripathy, R., Leca, I., van Dijk, T., Weiss, J., van Bon, B. W., Sergaki, M. C., et al. (2018). Mutations in MAST1 cause mega-corpus-callosum syndrome with cerebellar hypoplasia and cortical malformations. Neuron 100, 1354–1368.e5. doi: 10.1016/j.neuron.2018.10.044
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
Keywords: MAST4, neurodevelopmental delay, infantile spasms, genetic, epilepsy
Citation: Zhang X, Xiao N, Cao Y, Peng Y, Lian A, Chen Y, Wang P, Gu W, Xiao B, Yu J, Wang H and Shu L (2023) De novo variants in MAST4 related to neurodevelopmental disorders with developmental delay and infantile spasms: Genotype-phenotype association. Front. Mol. Neurosci. 16:1097553. doi: 10.3389/fnmol.2023.1097553
Received: 14 November 2022; Accepted: 06 February 2023;
Published: 22 February 2023.
Edited by:
Clive R. Bramham, University of Bergen, NorwayReviewed by:
Pierre Szepetowski, INSERM U901 Institut de Neurobiologie de la Méditerranée, FranceLin Zhao, Duke University, United States
Copyright © 2023 Zhang, Xiao, Cao, Peng, Lian, Chen, Wang, Gu, Xiao, Yu, Wang and Shu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Shu,  c2h1bGliYm1AZ21haWwuY29t; Hua Wang, d2FuZ2h1YV8yMTNAaG90bWFpbC5jb20=; Jing Yu, NTY4NDc1OTY4QHFxLmNvbQ==
c2h1bGliYm1AZ21haWwuY29t; Hua Wang, d2FuZ2h1YV8yMTNAaG90bWFpbC5jb20=; Jing Yu, NTY4NDc1OTY4QHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship