
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 01 February 2023
Sec. Brain Disease Mechanisms
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1052288
This article is part of the Research Topic The Role of GABA-Shift in Neurodevelopment and Psychiatric Disorders View all 13 articles
 Junhui Huang1†
Junhui Huang1† Fei Xu1†
Fei Xu1† Liping Yang2Lina Tuolihong3
Liping Yang2Lina Tuolihong3 Xiaoyu Wang4Zibo Du4Yiqi Zhang4Xuanlin Yin3
Xiaoyu Wang4Zibo Du4Yiqi Zhang4Xuanlin Yin3 Yingjun Li5
Yingjun Li5 Kangrong Lu6*Wanshan Wang7*
Kangrong Lu6*Wanshan Wang7*The neurobiological mechanism of post-traumatic stress disorder (PTSD) is poorly understood. The inhibition of GABA neurons, especially in the amygdala, is crucial for the precise regulation of the consolidation, expression, and extinction of fear conditioning. The GABAergic system is involved in the pathophysiological process of PTSD, with several studies demonstrating that the function of the GABAergic system decreases in PTSD patients. This paper reviews the preclinical and clinical studies, neuroimaging techniques, and pharmacological studies of the GABAergic system in PTSD and summarizes the role of the GABAergic system in PTSD. Understanding the role of the GABAergic system in PTSD and searching for new drug targets will be helpful in the treatment of PTSD.
Post-traumatic stress disorder (PTSD) refers to a delayed and persistent mental disorder that is caused by an individual’s experience of a sudden and traumatic event, such as war, earthquake, car accident, sexual assault, or exposure to extreme stress (Battle, 2013). More than 70% of adults worldwide have experienced at least one traumatic event in their lifetime, and 31% have experienced four or more (Benjet et al., 2016). The lifetime prevalence of PTSD varies by social background and country of residence, ranging from 1.3 to 12.2%, and the 1-year prevalence ranges from 0.2 to 3.8% (Karam et al., 2014). As PTSD was originally thought to be a physiological disorder rather than a mental one, some early studies used physiological methods to explore the physical abnormalities of patients, such as heart rate, skin conductance, and facial electromyography (EMG). These measures have been widely used in PTSD studies and have strongly demonstrated high emotional responses to trauma-related cues and excessive startle responses (Vasterling and Brewin, 2005). One of the earliest and most common findings of PTSD studies is the autonomic reactivity of patients to traumatic stimuli (e.g., heart rate and skin conductance) and facial EMG. Studies have demonstrated that the response to trauma-related cues is related to the severity of the disease (Wilson and Keane, 1997; Wolfe et al., 2000; Suendermann et al., 2010). In addition, the exaggerated startle response of PTSD patients has been documented in numerous blinks and electromyogram measurements (Orr et al., 1997, 2003; Pole, 2007; Pole et al., 2009). In mammalian studies (Koch and Schnitzler, 1997), the acoustic startle response may be a valuable model for studying the general principles of sensorimotor-motivational information processing at the behavioral and neurophysiological levels. Besides, studies of acoustic startle responses in rodents have shown that phasic fear is mediated by the amygdala, which sends outputs to the hypothalamus and brainstem to produce fear symptoms (Davis et al., 2010). However, it is still not clear whether this represents an increased neurological sensitivity to situational threats in people with PTSD. According to the latest Diagnostic and Statistical Manual of Mental Disorders by the American Psychiatric Association (DSM-5, 5th edition) (Battle, 2013) and the International Classification of Diseases, 11th Edition (ICD-11) published by the World Health Organization (Almeida et al., 2020), the core features of PTSD include intrusive symptoms, avoidance symptoms, and excessive alertness. According to ICD-11, these three symptom groups are also part of the complex post-traumatic stress disorder (CPTSD) diagnosis. Additionally, In CPTSD three other symptom groups can be summarized as disturbances in self-organization: Emotion regulation difficulties, relationship difficulties, and negative self-concept (Maercker, 2021). The DSM-5 also emphasizes cognitive and emotional changes, and patients may experience cognitive decline, depression, loss of interest, indifference, and other manifestations. The clinical diagnosis of PTSD is established when these symptoms persist for more than 1 month.
At present, the neurobiological mechanism of PTSD has not been confirmed, and the research directions mainly include four aspects: (1) Genes involved with monoamine and the hypothalamic-pituitary-adrenal (HPA) axis function have been examined extensively in epigenetic and genetic studies of PTSD risk and separately in studies of disease risk and response to treatments for mood disorders (Kato and Serretti, 2010; Domschke et al., 2014; Zannas et al., 2015; Smoller, 2016). Two of the most commonly characterized genes in this regard are the serotonin transporter (SLC6A4) and FK506 binding protein 5 (FKBP5) (Bishop et al., 2021); (2) neuroendocrine dysfunction, such as the increased secretion of catecholamines (Olson et al., 2011) and decreased secretion of 5-hydroxytryptamine (5-HT) hormones (Liu et al., 2018) and corticosterone (Geracioti et al., 2008); (3) changes in the neural structure and circuitry. Basic and clinical studies have demonstrated that structural and functional abnormalities in the hippocampus, prefrontal cortex (PFC), amygdala, and other brain areas were observed in both animal models and individuals with PTSD (Rauch et al., 2006; Hayes et al., 2012; Disner et al., 2018).
Gamma-aminobutyric acid (GABA) is an important inhibitory neurotransmitter in the central nervous system (CNS), which can reduce neuronal activity, prevent nerve cells from overheating, and calm nerves. Besides, GABA is also an active amino acid that plays an important role in the process of energy metabolism in the human brain. For example, GABA participates in the tricarboxylic acid cycle in the brain and promotes the metabolism of brain cells. At the same time, GABA can also improve the activity of glucose phosphatase during glucose metabolism, increase the generation of acetylcholine, expand blood vessels to increase blood flow, and reduce blood ammonia to promote brain metabolism and restore the function of brain cells (Petroff, 2002). As one of the inhibitory neurotransmitters of the CNS, GABA plays an essential role in regulating the stress response, emotion, and registration and encoding of fear memory (Corcoran and Maren, 2001). Dysfunction of the GABAergic system has been proven to be one of the mechanisms of PTSD, with several studies demonstrating that PTSD can reduce the levels of GABA and its receptors in some brain regions. Positron emission tomography (PET) was used to identify post-war PTSD patients, and it was observed that the distribution of benzodiazepine-GABA receptors decreased in the PFC, as well as the entire cortex, hippocampus, and thalamus (Geuze et al., 2008). Other studies have shown that the levels of GABA in the occipital and temporal lobes of PTSD patients were significantly decreased compared to controls, resulting in sleep disorders (Meyerhoff et al., 2014). Studies on animals have shown that the anxiety and fear behaviors of mouse models of PTSD improved after the administration of exogenous tetrahydroprogesteroneal, which may be induced by the enhancement of GABA function mediated by allopregnanolone (Evans et al., 2012).
Recent preclinical and clinical data indicate that GABA, which is a major inhibitory neurotransmitter involved in the pathophysiology of PTSD, plays an important role in stress. Changes in the GABA system are related to the pathogenesis of PTSD. Understanding the systemic changes in GABA in PTSD will not only contribute to the diagnosis of PTSD but also reveal new targets for pharmacological intervention.
In this review, we review the role of the GABAergic system from the phenomenon to the mechanism, and then to the clinical guidance in PTSD. We discuss fear memory, an important component of PTSD, and the role of GABA receptors in fear memory formation and extinction. By reviewing the changes in the GABA system in different brain regions in PTSD, we state the ubiquitous and heterogeneous nature of GABA in the brain. Furthermore, the role of GABA in PTSD and its mechanism were further discussed, which involved how the GABA system interacts with other systems, including the HPA axis and the endocannabinoid system (ECS), as well as the role of glutamate (Glu) and GABA signal imbalance in the brain in PTSD. Finally, we bring together the clinical, preclinical and neuroimaging evidence of changes in the GABA system in PTSD, as well as GABA mechanisms of several clinical drugs. We treat the “GABAergic system” as a single unified neurotransmitter system. It is useful to highlight large-scale, non-specific changes in GABA signaling to establish the importance of dysregulation of GABA function in PTSD.
The GABAergic system plays a certain role in the onset of depression, anxiety, and other mental disorders. Clinical and preclinical studies have shown that the inhibitory effect of the GABAergic system in anxiety patients is reduced (Domschke and Zwanzger, 2008). The receptor of GABA refers to the part of the postsynaptic membrane that can recognize and bind GABA. When it binds to GABA, it can cause changes in membrane ion permeability. The expression or dysfunction of GABA receptors is associated with mental illness. Three main subtypes of GABA receptors, namely, GABA-A, GABA-B, and GABA-C, have been identified so far. Among them, GABA-A and GABA-C receptors are ligand-gated ion channels. The transmembrane receptor GABA-B binds with the G protein to activate the second messenger system (Chebib and Johnston, 1999). GABA-A receptors are widely distributed throughout the nervous system and peripheral tissues. GABA-B receptors are found in the olfactory bulb, neocortex, hippocampus, thalamus and cerebellum of mammals (Lujan and Ciruela, 2012). GABA-C receptors are found mainly in the retina, which is also distributed in the spinal cord, thalamus, pituitary gland, and intestine of mammals (Zhang et al., 2001). The rapid inhibition of the neurotransmitter GABA is mediated by GABA-A receptors. Furthermore, various subtypes of the GABA-A receptor have been identified, including α1–6, β1–3, γ1–3, δ, ε1–3, θ, and π (Jacob et al., 2008). The main receptors mediating neural inhibition in the brain are GABA-A receptors, and changes in the expression or function of these receptors in patients are increasingly related to the etiology of anxiety and depression (Merali et al., 2004; Sanacora et al., 2004; Bhagwagar et al., 2008; Poulter et al., 2008; Klempan et al., 2009; Sequeira et al., 2009; Craddock et al., 2010; Levinson et al., 2010; Figure 1). In particular, the changes in GABA-A receptor subunits play an important role in the amygdala and PFC mediating fear memory, as detailed in section “2.1.2. Effects of the GABAergic system on fear memory in specific brain regions.” In addition, decreased levels of GABA are associated with stress responses (Dolfen et al., 2021), and GABA transmitters mediate stress and fear responses mainly by binding to their receptors.
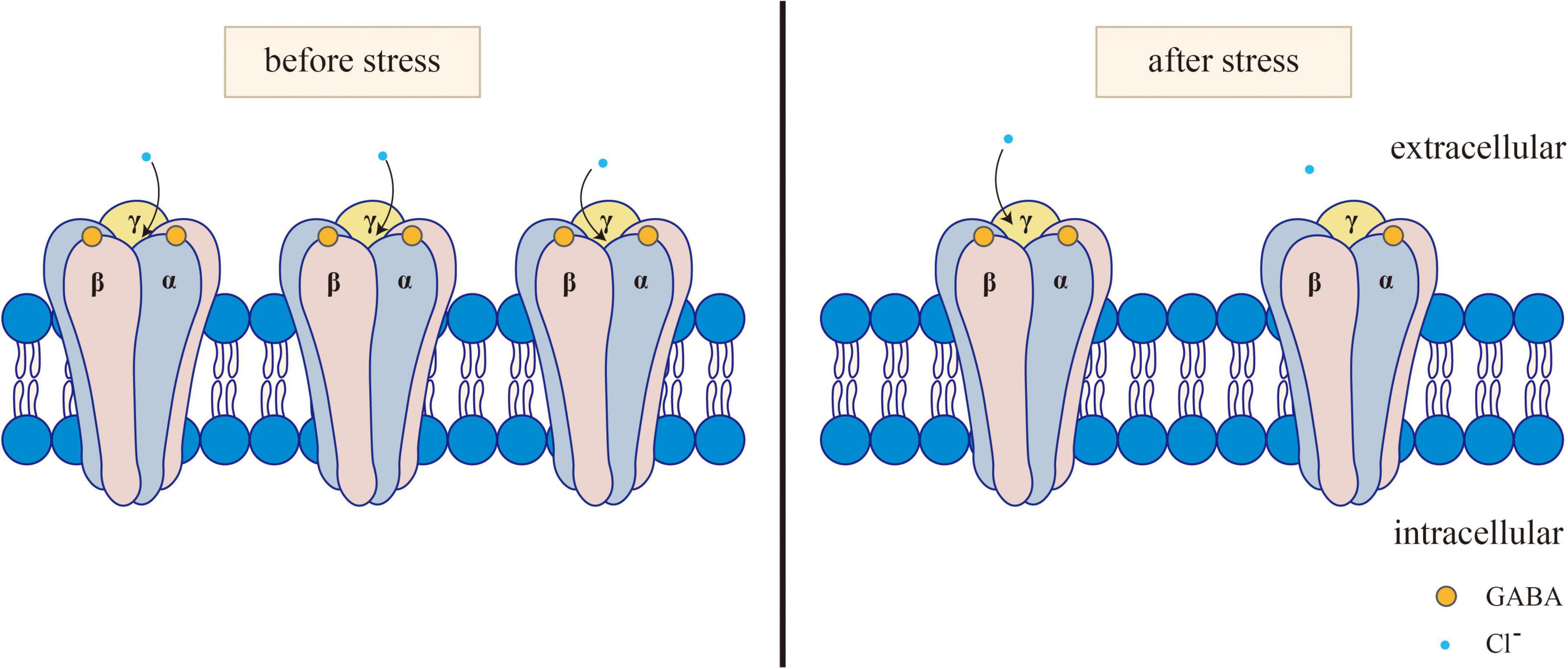
Figure 1. Changes of GABAA receptor before and after stress. Normally, GABA activates the opening of GABAA receptors. After the GABAA receptor is activated, it can selectively let Cl– through, causing the hyperpolarization of neurons. After stress, GABA levels and GABA-A receptors decrease, and GABA binding to GABA-A receptors subsequently decreases, resulting in reduced Cl- influx.
Generally, PTSD is regarded as a high fear response to a threat. Thus, fear is an important target in the neurobiology of PTSD. The process of fear memory is divided into several stages, including fear acquisition, fear consolidation, fear destabilization/reconsolidation and fear extinction (Liberzon and Abelson, 2016; Figure 2).

Figure 2. The process of fear memory. When a neutral cue or context [a sound, light, or conditioned stimulus (CS)] occurs with a highly aversive signal or unconditioned stimuli (US) and leads to fear behaviors or conditioned fear responses (CRs), fear acquisition arises. Fear memory will be consolidated within a few hours to a day of fear acquisition. When CS occurs alone, the animal will retrieve the fear memory and lead to CR. After that, if CS has been presented alone without US, fear memory becomes unstable and two different processes could occur. One is called fear extinction, which occurs after CS appears alone repeatedly many times. As a result, fear strength will decrease. The other is reconsolidation, where the fear the memory can be updated or left intact.
Classical (or Pavlovian) fear conditioning is an effective behavioral paradigm for studying the mechanisms of associative fear learning and memory processes. In rodents, fear indicators are typically assessed by freezing behaviors (Bouton and Bolles, 1980), which is characterized by immobility and the absence of any movement except for breathing. Freezing is a potent conditioned fear response in rats and mice. And it has a great advantage over many other fear measures because freezing is not a typical response of rats or mice to ordinary new stimuli (Anagnostaras et al., 2010). A computerized method based on latency between photobeam interruption measures is used as a reliable scoring criterion in mice, which can reduce bias or inconsistencies (Valentinuzzi et al., 1998). It is important to note that when testing different strains of mice, it would be essential to validate the testing or scoring procedure (Valentinuzzi et al., 1998). To avoid artificially inflated lighting sources and thus ensure freezing behavior is induced in a well-controlled manner, it is suggested to report Lux and control the lighting properly (Neuwirth et al., 2022). When a neutral cue or context [a sound, light, or conditioned stimulus (CS)] occurs with a highly aversive signal or unconditioned stimuli (US) and leads to fear behaviors or conditioned fear responses (CRs), which include freezing, a systemic response to behavior besides breathing, fear acquisition arises [47]. The training box used in the Pavlov paradigm itself can be used as an environmental situational stimulus (CS), which is matched with an aversive plantar shock (US) multiple times. Animals learn the connection between the CS and the US and obtain the fear response (CR) to the environment. This process is called contextual fear conditioning (CFC). If a cue [usually an auditory stimulus (CS)] is matched with a foot shock (US) multiple times during the training, the auditory stimulus (CS) will be separated from the environment and associated with the foot shock (US) to form the CR to the auditory stimulus (CS). This process is called cued fear conditioning (Maren et al., 2001). The exposure of an individual to CS stimulation is sufficient to trigger a fear response (CR). This process is called retrieval of fear memory (Pape and Pare, 2010). Following retrieval, previously formed memory is destabilized, which is similar to the unstable state when the memory has just been acquired, and requires new protein synthesis to restabilize, a process referred to as reconsolidation. Reconsolidation acts to stabilize, update, or integrate new information into long-term memories (Izquierdo et al., 2016). Memory-dependent reconsolidation modification may serve as a therapeutic target to modulate the enhanced fear response commonly associated with debilitating mental disorders. People with PTSD are unable to modify or weaken memories through the reconsolidation process (Ferrara et al., 2019). Once the animals have acquired a CR, repetition of the CS alone without the US usually reduces the CR: this is called extinction. Fear extinction indicates the formation of a new competing memory, which is similar to fear conditioning, rather than the elimination of the original fear memory. In this process, the organism is enabled to acquire an association between CS and no-US, which competes with the conditioned fear memory (Bouton and Bolles, 1980). It is known that extinction generates a new memory engram, so it can be regarded as a process of active learning (Maddox et al., 2019).
Exposure to trauma can not only damage the body’s physiological and psychological adaptation to stress (Lissek and van Meurs, 2015; Hill et al., 2018) but also alter to the formation and consolidation of associative fear memories (Parsons and Ressler, 2013; Elms et al., 2019). Frequent exposure to fear-related cues can impact the initial consolidation and subsequent retrieval of memory (Maddox et al., 2019), causing anxiety and trauma-related ailments such as PTSD. The persistent presence of negative cognition will impair the inhibition of invasive memory (Meiser-Stedman et al., 2009; Catarino et al., 2015). Thus, memories related to traumatic events will be repeated, while frequent flashbacks and avoidance behaviors will, in turn, worsen negative cognition, resulting in anxiety and depressive symptoms. Thus, a vicious cycle is formed.
Fear conditioning is a highly conservative form of emotional learning that occurs when an environmental stimulus predicts aversive events. This type of learning allows a previously neutral stimulus to trigger a fear response that prepares the animal for the threat and helps it escape (LeDoux et al., 1988; Blanchard and Blanchard, 1989; Fendt and Fanselow, 1999; Babaev et al., 2018). Neural circuits and cellular mechanisms that mediate fear conditioning have been extensively described, among which the inhibitory regulation of GABA neurons is crucial for the precise regulation of the consolidation, expression, and extinction of fear conditioning (Fendt and Fanselow, 1999; Zhang and Cranney, 2008; Makkar et al., 2010). The antagonists of GABA such as bicuculline (Castellano and McGaugh, 1990) have been demonstrated to enhance memory consolidation, while GABA agonists such as muscimol (Akirav et al., 2006) inhibit memory consolidation. These data suggest the involvement of GABA receptors in memory acquisition and consolidation. The role and mechanism of GABA in PTSD and fear memory extinction are still unclear. A study indicated that GABA signals promoted extinction (Berlau and McGaugh, 2006), while another study found that the activation of GABA signals prevented extinction (Singewald et al., 2015). The divergent role of GABA in extinction may be related to the different distribution of GABAergic neurons in the brain, but this still needs further explanation.
Most commonly, studies involved in fear memory include the PFC, amygdala and hippocampus. A summary of the changes and effects of the GABAergic system in specific brain regions can be seen in Figure 3. Under normal conditions, the PFC exerts inhibitory control over the amygdala. When the inhibitory effect of the PFC on the amygdala is weakened, it leads to excessive activation of the amygdala. This reduction in top-down control leads to impaired fear extinction. Michels et al. (2014) found increased GABA in the dorsolateral PFC of PTSD patients, indicating an overall shift toward inhibitory tone. Schneider et al. (2016) also showed similar changes in PFC inhibitory tone. GABA was increased in the PFC, but Glu was not changed. This change in GABA may indicate reduced excitatory activity of the PFC. A temporary increase of GABA in PFC can initiate long-term plasticity of the PFC. This change in PFC may affect the activity of the network, transmit through connections to the midline thalamus or entorhinal cortex, and ultimately affect the excitability of the hippocampus (Kyd and Bilkey, 2005). The increased excitatory tone in the hippocampus may lead to impaired fear extinction. The ventral hippocampus (vHPC) can directly project to the PFC and amygdala, affecting their activity (Ishikawa and Nakamura, 2006; Sierra-Mercado et al., 2011). The vHPC projections to the PFC can activate GABAergic neurons in the dorsal PFC and anterior cingulate.
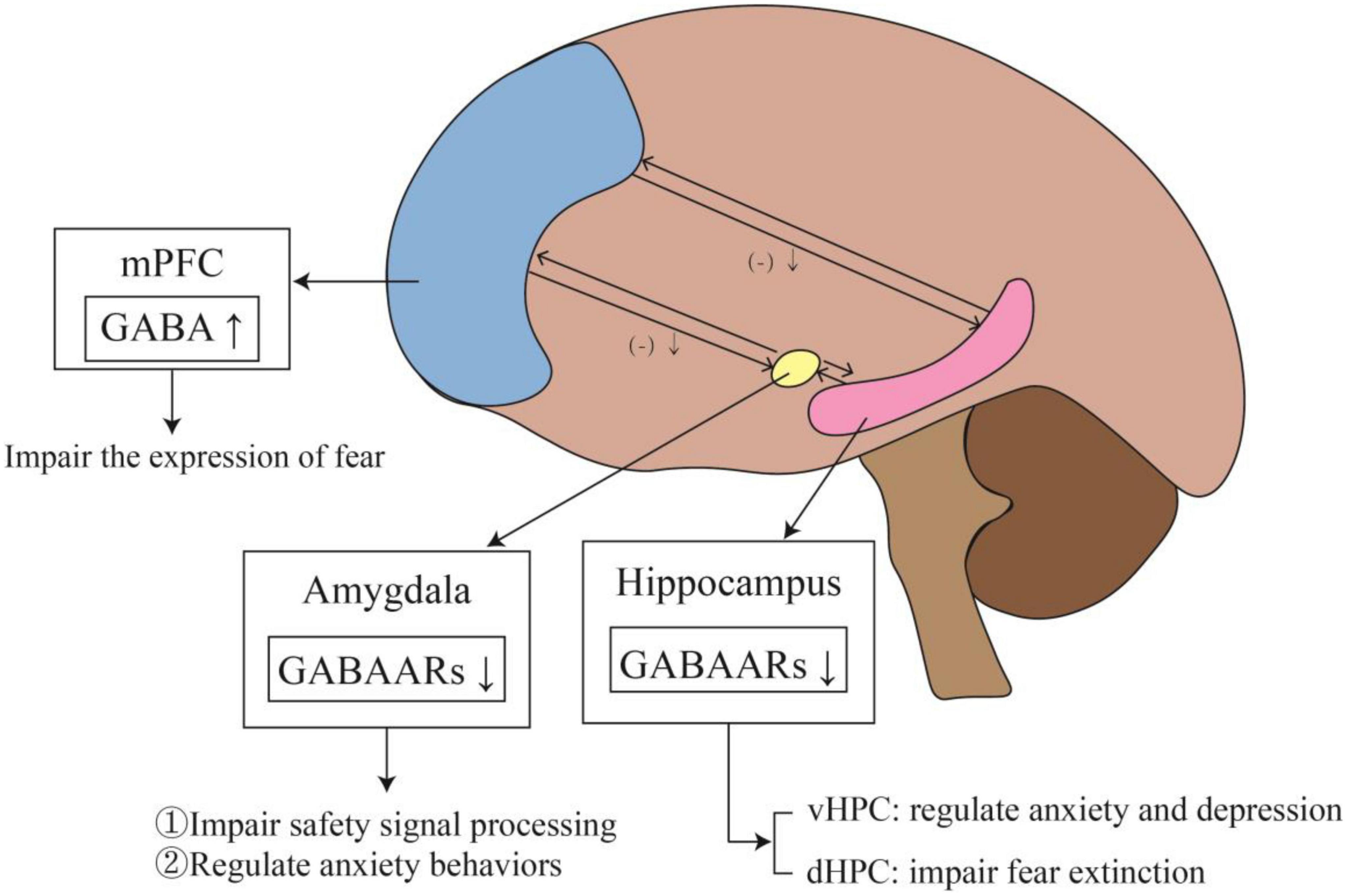
Figure 3. The changes and effects of the GABAergic system in specific brain regions. The outputs of mPFC to the amygdala and hippocampus are inhibitory. When PTSD develops, the level of GABA in mPFC rises, which weakens the inhibition of mPFC to amygdala and hippocampus, affecting fear memory. Meanwhile, elevated GABA level in mPFC impairs fear expression. In addition, the reduction of GABAARs in the amygdala impairs safety signal processing and regulates anxiety behaviors. The expression of GABAARs also decreases in the hippocampus. In vHPC, it leads to anxiety and depression, while in dHPC affected fear extinction.
Previous studies have used specific agonists of GABA receptors to inhibit the function of specific brain regions, aiming to clarify the role of each brain region in behavior. The amygdala is crucial for fear conditioning and is a key brain structure for the acquisition and expression of conditioned fear responses (LeDoux, 2000; Sah et al., 2003; Chen et al., 2006). The amygdala consists of the basolateral amygdala (BLA) and the central amygdala (CeA). The BLA can be subdivided into the lateral amygdala (LA), basal amygdala (BA), and basal medial amygdala (BMA), while CeA can be further subdivided into lateral CEA (CEl) and medial CEA (CEm) (Tovote et al., 2015). The BLA and the CeA are the main research objects of fear memory, and both receive input from the cortex. The BLA consists of glutamatergic spiny projection neurons (approximately 80%) and GABAergic interneurons (McDonald, 1982; McDonald and Augustine, 1993). In contrast, the CeA is composed of GABAergic medium spiny neurons (Sah et al., 2003). In addition, there is a class of GABAergic neuronuclear nuclei between the BLA and CeA that provide feedforward inhibition signals for the BLA and CeA (Tovote et al., 2015). When the amygdala was inactivated with the GABA-A agonist muscimol, the inactivation of the amygdala resulted in deficits in situational and delayed conditioning (Raybuck and Lattal, 2011). Conversely, hyperactivity of the amygdala may be associated with impaired safety signal processing in PTSD (Christianson et al., 2012). In the classical fear conditioning circuit model, the LA is the main part of the formation and storage association of CS and US (LeDoux, 2000; Maren and Quirk, 2004; Chen et al., 2006; Sigurdsson et al., 2007; Sah et al., 2008). Besides, CeA is involved not only in fear expression but also in the learning and consolidation of Pavlovian fear conditioning (Wilensky et al., 2006). Conditioned fear can activate the inhibitory network in the CeA, inhibit the CeA-periaqueduct gray pathway, and finally promote fear (Ball et al., 2017). Conditioned fear was observed to reduce the frequency of miniature inhibitory postsynaptic current (mIPSC), as well as the expression levels of GABA type-A (GABAAR) γ2 and β2 receptors of the amygdala (Lin et al., 2011). Moreover, α2 receptors in the amygdala are involved in the regulation of anxiety behaviors (Lehner et al., 2010).
The hippocampus plays a key role in the encoding, storage, and retrieval of fear memories (Anagnostaras et al., 1999; Bast et al., 2001). The vHPC inputting directly into prelimbic cortex enables to form contextual fear memory, which is necessary for the subsequent rapid expression of a fear response (Twining et al., 2020). Contextual fear conditioning was impaired after unilateral microinjection of GABAA agonist muscimol into the vHPC (Gilmartin et al., 2012). Another study (Herbst et al., 2022) showed that shock encoding in the vHPC modulated the expression of learned fear in a sex-specific manner, i.e., preventing hippocampal shock-evoked firing led to a more prolonged expression of CS freezing across test trials, an effect observed in males but not in females. Moreover, Almada et al. (2013) reported that the injection of the GABAA agonist muscimol into the dorsal hippocampus (dHPC) of mice impaired the expression of situation-associated fear memory. Similarly, another experiment revealed that the injection of muscimol into the dHPC inhibited the retrieval of short-term fear memory but not long-term fear memory (Raybuck and Lattal, 2014). This may be because the enhanced transmission of GABAergic signals can inhibit excitatory pyramidal neurons and prevent them from participating in the encoding and storage of fear memories. For example, the inhibition of hippocampal CA1 pyramidal neurons can impair the expression of fear memories associated with recent situations (Sakaguchi et al., 2015). In addition, one study showed that after stress exposure, the expression of the α1 and α2 subunits of GABA-A receptors in the vHPC is increased, contributing to stress recovery in rats (Ardi et al., 2016).
Stress and stress hormones such as corticosterone affect long-term potentiation (LTP) differently in the ventral and dorsal hippocampus. Maggio’s study (Maggio and Segal, 2007) found that the magnitude of LTP in the vHPC was significantly smaller than that elicited in the dHPC. However, stress actually enhanced LTP in the vHPC, which is unlike the case in the dHPC. This different property is controlled by a metabolic Glu receptor (Sakaguchi et al., 2015). Glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) antagonists blocked the stress on dHPC and vHPC, respectively. The vHPC and dHPC are distinguished not only by differences in anatomical structure but also by differences in function and gene expression. The vHPC is more involved in anxiety and emotional processing, while the dHPC is more involved in learning and memory (Fanselow and Dong, 2010). People with PTSD may suffer from anxiety and depression, and the neurobiological mechanism of PTSD involves fear learning and fear memory. Therefore, it may be difficult to distinguish the role between the vHPC and dHPC in PTSD, and further research is needed in the future.
Early studies on the connection between the medial prefrontal cortex (mPFC) and fear have been conducted through damage experiments. Morgan and LeDoux (1995) demonstrated that the overall damage to the mPFC, including the anterior cingulate cortex (ACC), prelimbic cortex (PL), and infralimbic cortex (IL), impaired fear acquisition rather than fear extinction. Subsequent studies have shown that damage to PL alone (mainly the dorsomedial region) enhances the acquisition of fear memories and prevents fear extinction (Davidson et al., 2000). The expression of fear was also impaired by the injection of muscimol to reduce the activity of PL neurons before extinction training (Marquis et al., 2007). Another study revealed that specific damage to the ventromedial prefrontal cortex (vmPFC) is necessary for recalling a previously learned extinction memory, rather than learning extinction per se (Quirk et al., 2000). These studies suggest that PL is involved in the expression of extinction learning, whereas the vmPFC is in extinction consolidation. Additionally, different subregions of the mPFC play different roles in fear expression, which may be related to differences in the distribution of GABA receptor subtypes.
Moreover, the PFC controls the stress response and participates in the regulation of emotion through inhibitory GABAergic projection to the amygdala (Davidson et al., 2000). The findings of Lu et al. (2017) indicate that early traumatic stress can increase the expression of the α2 subunit in the PFC. Deficiency of GABAAR in the PFC will weaken its control over downstream neuroanatomical regions and increase the expression of the γ2 subunit in the amygdala, leading to defective GABAergic neurotransmission. This fails to erase fear-related memories (Koenigs and Grafman, 2009; Milad et al., 2009) and impairs the control of fear responses in PTSD (Aupperle et al., 2012), thereby increasing susceptibility to PTSD in adulthood.
Exposure to war or other traumatic events is associated with a higher risk of mental health problems, including PTSD. The clinical symptoms of these stress-related anxiety disorders are associated with the long-term dysfunction of inhibitory and excitatory neurotransmission and potential neuronal damage (Hageman et al., 2001; Charney, 2004). Both GABA and Glu are important neurotransmitters that are associated with general neurological function, memory registration, and encoding of emotional and fearful memories in anxiety disorders, especially anxiety (Pham et al., 2009). Chronic stress reduces the activity of the neurotransmitter GABA in the hippocampus and PFC, as well as the inhibition of the HPA axis, resulting in anxiety and depression (Mikkelsen et al., 2008). In previous neuroimaging studies, concentrations of these amino acids (in addition to the density and activity of neurotransmitter receptors and transporters) have been associated with mood disorders and memory dysfunction (Krystal et al., 2002). It is suggested that the normal excitation-inhibition balance in the CNS is broken between excitatory glutamatergic neurons and inhibitory GABAergic neurons due to the weakened function of GABAergic neurons, which may lead to the onset of mental disorders. The decrease in plasma GABA levels after trauma can predict the development of PTSD (Vaiva et al., 2004), suggesting that the GABAergic system is involved in the pathophysiology of PTSD. Various studies, including preclinical and clinical studies, neuroimaging techniques, and pharmacological studies, have reported the involvement of the GABAergic system in PTSD.
Preclinical and clinical data indicate that GABA plays an important role in stress and that changes in the GABAergic system are related to the pathogenesis of PTSD, as it is the main inhibitory neurotransmitter involved in the pathophysiology of PTSD (Shin and Liberzon, 2010; Averill et al., 2017; Ghosal et al., 2017). Increasing evidence has suggested that the overactivation of the amygdala in PTSD patients is associated with a decreased inhibitory function of GABA (Pitman et al., 2012). Decreased inhibitory function of GABA thus plays an important role in the expression of stress-induced fear memory and the production of neuronal LTP. Reduced binding of GABA-A receptors in the cortex, hippocampus, and thalamus has been reported in veterans with PTSD, and the overall inhibitory function of the brain is deficient (Geuze et al., 2008). In victims of traffic accidents that meet the criteria of trauma exposure, a lower level of post-traumatic plasma GABA predicts the development and more chronic course of PTSD (Vaiva et al., 2006). In addition, PET studies have demonstrated that the binding of hippocampal GABAARs to benzodiazepine (BDZ) in PTSD patients is reduced (Geuze et al., 2008; Möller et al., 2016). These studies suggest that the increased expression of GABA-A receptors under stress may contribute to stress adaptation, while decreased expression of GABA-A receptors may contribute to PTSD susceptibility. Consistent with this view, a recent study in the ventral tegmental region reported that the mRNA levels of GABAergic genes were down-regulated in stress-prone mice but not in stress-adapted mice (Sun et al., 2018). Other clinical studies have reported that PTSD patients have impaired recognition of environmental cues, suggesting that impaired hippocampal function may play an important role in the development of PTSD. Exposure to unavoidable foot shock results in reduced GABA-A receptor function, as well as cortical and hippocampal binding (Lippa et al., 1978; Medina et al., 1983; Drugan et al., 1986). In the stress-restress paradigm, an animal model exhibits a similar pattern as PTSD, wherein stress causes a sustained decline in hippocampal GABA levels (Harvey et al., 2004). Animal experiments have demonstrated that chronic stress can reduce the number of GABA neurons in the hippocampal CA1 and CA3 regions (Megahed et al., 2015). Chronic exposure to stress levels of adrenal corticosteroids leads to the abnormal expression of GABA and receptor subunits in the hippocampal CA1 and CA3 regions, although the changes in the expression of different subunits follow different patterns in the CA1 and CA3 regions (Orchinik et al., 1995). Moreover, the expression of the GABAA α2 and γ2 receptors in the PFC of adult rats decreased after shock stress in childhood, indicating that traumatic stress may reduce the activity of GABA in PFC, thus reducing the inhibition of amygdala (Lu et al., 2017). Rodent models are usually used to study social behavior disorders because they can simulate human social behavior and provide guiding information for the treatment of human social-emotional disorders. However, current rodent models of PTSD are still unable to simulate real PTSD patients well and can only simulate some features of PTSD. Therefore, more suitable animal models of PTSD still need to be explored.
In addition, several other studies have linked the GABAergic system to PTSD. Spivak et al. (2000) reported significantly elevated levels of dehydroepiandrosterone and dehydroepiandrosterone sulfate in response to GABAARs in the blood of patients with war-related chronic PTSD. These results strongly suggested that the increase in steroids and the inhibition of GABAARs are closely related to the delayed recovery of PTSD patients. A molecular genetic study reported that the polymorphism of the microsatellite sequence at the third end of the GABA β3 subunit is correlated with the incidence of PTSD (Feusner et al., 2001).
Currently, intracranial GABA levels can be measured using proton magnetic resonance spectroscopy. Neuroimaging studies of BDZ-GABAA receptors in PTSD are rare. The GABAAR complex includes the GABAergic postsynaptic membrane, GABA receptor and chloride ion channel. When BDZ binds to BDZ receptors on the GABAAR complex, chloride channels open, and GABA binds to GABAAR to increase the frequency of chloride channel opening, which promotes chloride influx in a dual way to enhance central inhibition. Bremner et al. (2000) examined the distribution of benzodiazepine receptor recognition sites (a recognition site on the GABAAR complex) in the PFC of PTSD patients and observed that the density was significantly lower than that of normal controls. It has been suggested that changes in the function of GABAAR in this region may underlie several symptoms of PTSD. However, one study (Randall et al., 1995) showed no increase in anxiety and PTSD symptoms in PTSD patients treated with the GABAAR antagonist flumazenil. Flumazenil can reverse the central sedative effects of BDZ, but the results of this study suggest that PTSD and panic disorder (a type of anxiety disorder) differ in BDZ/GABAA system function. This may indicate the complexity of the pathological mechanism of PTSD. Changes in GABA have also been reported in Magnetic resonance spectroscopy studies on the in vivo neurochemistry of PTSD. Rosso et al. (2014) reported a 30% decrease in the insular GABA/creatine ratio in 13 adults with PTSD compared to healthy controls. Male veterans and civilians exhibited lower levels of GABA in the parietal occipital cortex (POC) and the proximal temporal lobe than trauma-exposed controls, while POC-GABA levels were inversely correlated with the severity of sleep symptoms (Meyerhoff et al., 2014). Sheth et al. (2019) observed that the exposure of veterans to trauma might be related to lower GABA/H2O and glutamine (Gln)/H2O levels in the dorsal ACC (dACC), indicating the interruption of the GABA-Gln-Glu cycle. Moreover, changes in Glu/GABA in the dACC in the PTSD group may indicate an excitation-inhibition (E-I) imbalance (Sheth et al., 2019). The E-I balance can represent the ratio of excitatory and inhibitory inputs to a neuron, and the E-I balance of a single neuron is viewed as a combination of two reaction forces projected on the neuron: excitatory and inhibitory. That is, if the excitatory input level increases, the firing rate of the neuron will increase, and if the inhibitory input level increases, the firing rate of the neuron will decrease accordingly. Furthermore, reduced GABA/H2O in the ACC was associated with poor sleep quality in the PTSD group (Meyerhoff et al., 2014). Treatments that restore the GABAergic balance may be particularly effective in reducing sleep symptoms of PTSD. Reduction in GABAergic tone may also disturb the E/I balance, which disrupts not only regional neural activity but also functional connections in large-scale brain networks (Gu et al., 2019). These studies collectively suggest that reduced GABAergic activity may underlie the symptoms of PTSD.
Pharmacological studies also support the involvement of the GABAergic system in PTSD. Potential drugs targeting the GABAergic system can be seen in Table 1.
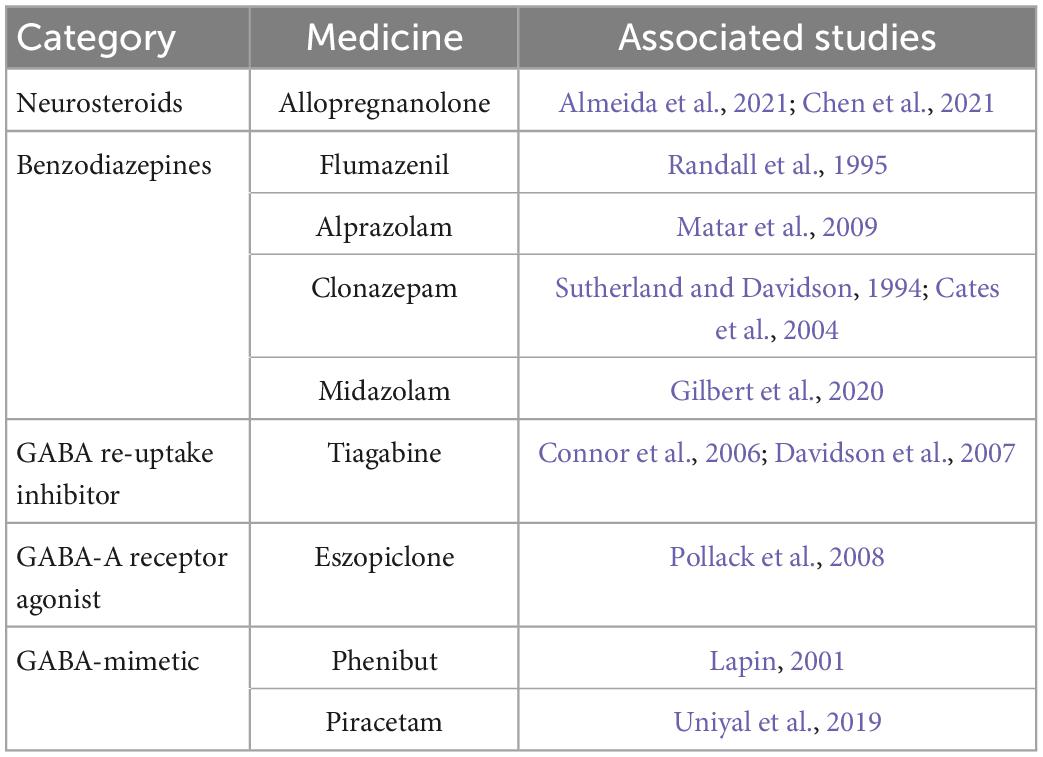
Table 1. Potential drugs targeting GABAergic system.
Neurosteroids, including 5-dihydro-progesterone (5-DHP), allopregnanolone (ALLO), and their stereoisomers, such as progesterone, are synthesized directly by brain neurons (glutamatergic and GABAergic long-projection neurons) in the CNS (Baulieu and Robel, 1990; Agís-Balboa et al., 2006, 2007). Neurosteroids not only act on classical steroid hormone receptors (which regulate gene expression and have long-term effects) but also rapidly regulate neuronal excitability by binding membrane receptors to ion channels. They effectively modulate GABAARs (Belelli and Lambert, 2005). Neurosteroid levels and the expression of GABAARs are affected by normal physiological changes, such as pregnancy and the ovarian cycle (Concas et al., 1998; Maguire et al., 2005), as well as by pathological conditions caused by delayed or traumatic stress, such as anxiety, depression, and PTSD (Romeo et al., 1998; Uzunova et al., 1998; Rasmusson et al., 2006). In a physiological state, the mRNA expression level of the GABAAR γ2 L subunit in the cerebral cortex and hippocampus decreased during pregnancy and recovered to the control level 2 days after delivery. Subchronic administration of finasteride (a 5α-reductase inhibitor) in pregnant rats reduced ALLO in the brain to a greater extent than in plasma and prevented the observed decline in γ2S mRNA during pregnancy (Belelli and Lambert, 2005). Pollack et al. (2008) demonstrated that periodic changes in the specific GABAAR subunit during the estrous cycle in mice lead to periodic changes in the tonic inhibition of hippocampal neurons. In late diabetes (the high progesterone phase), tonic inhibition was increased by increased expression of deltaGABA(A)Rs, and decreased neuronal excitability was reflected in decreased susceptibility to epilepsy and anxiety. Elimination of the circulation of deltaGABA(A)Rs by antisense RNA treatment or gene knockout may prevent decreased excitability in diabetes. In a pathological state, Szabó et al. (2014) found that low levels of ALLO in the cerebrospinal fluid of premenopausal women with PTSD may lead to an imbalance between inhibitory and excitatory neurotransmitters, leading to PTSD re-experience and increased depressive symptoms. Romeo et al. (1998) found that GABA receptor positive modulators 3α,5α-tetrahydrogestrone (3α,5α-THP) and 3α,5β-THP concentrations decreased significantly during depression, while 3β,5α-THP levels increased. This imbalance of neuroactive steroids can be corrected with different antidepressant medications. Moreover, ALLO (3α5α and 3α5β isomers) has a high affinity for GABAAR and promotes the action of GABA at these receptors. In one study (Kamprath et al., 2006), SSRIs treatment induced a normal increase in ALLO levels in the cerebrospinal fluid of depressed patients. Normalization of ALLO levels in the cerebrospinal fluid of depressed patients appears to be sufficient to mediate fluoxetine or fluvoxamine to modulate the antianxiety effects of positive allosteric modulation of GABAAR. Progesterone or its neuroactive metabolite tetrahydrogestrinone can produce anti-anxiety, sedation, anesthesia, analgesic, and anticonvulsant effects in rodents and humans by effectively increasing the flux of Cl– by binding GABA to GABAARs (Belelli et al., 2009). Allopregnanolone regulates neuroendocrine changes induced by the stress response, especially in combination with the GABAAR-mediated feedback mechanism that regulates the HPA axis (Almeida et al., 2021). Studies have demonstrated that low levels of allopregnanolone can improve PTSD symptoms by regulating the activity of GABAARs (Schüle et al., 2014).
Generally, BDZ is widely used to treat anxiety disorders, including PTSD, because of its quick-relief properties. In animal studies, BDZ regulates the function of GABAARs and inhibits the “startle” response caused by the threat from predators (Adamec et al., 2006). The role of GABA in PTSD is also supported by studies on treatment using other GABAergic compounds, such as the selective GABA re-uptake inhibitor tiagabine (Taylor, 2003; Connor et al., 2006). In addition, pharmacological studies have found that the combination of piracetam (a GABA derivative drug) and risperidone can enhance the therapeutic of PTSD (Uniyal et al., 2019). Insomnia is among the main features of PTSD, and pharmacological drugs that regulate GABA effectively alleviate insomnia (Randall et al., 1995; Krystal et al., 2014). Although BDZ effectively reduces anxiety in PTSD patients, it do not affect the core symptoms of the disorder, such as intrusive thinking, numbness, and hyperarousal (Gelpin et al., 1996; Viola et al., 1997; Davidson, 2004). This suggests that drugs targeting GABAA receptors may not be effective in the treatment of PTSD, so future studies could explore drugs targeting other targets of the GABAergic system, such as GABA reuptake inhibitors.
Stress responses mediated by the HPA axis are most closely related to psychiatric disorders, such as PTSD and depression (Almeida et al., 2021). There are intricate afferent and efferent connections between the hypothalamus and other brain regions of CNS. Neuroendocrine cells in the hypophysiotrophic area of the hypothalamus have endocrine functions and can produce peptide neurohormones to be transported to the adenohypophysis to regulate the secretion of corresponding hormones (Clarke, 2015). Activation of the HPA axis originates from the release of corticotropin-releasing hormone (CRH) from the paraventricular nucleus (PVN) of the hypothalamus, then CRH induces the release of adrenocorticotropic hormone (ACTH), which stimulates the adrenal gland to release glucocorticoid (de Kloet et al., 2005). Glucocorticoid inhibits the release of CRH in the hypothalamus by activating MR and GR with negative feedback (de Kloet et al., 2005). During acute stress, the HPA axis is activated, which plays an important role in mobilizing energy and maintaining homeostasis. However, exposure to a single but extremely intense traumatic event, combined with chronic stress conditions, can lead to anxiety spectrum disorders or induce PTSD. Patients with PTSD showed abnormal regulation of the HPA axis, such as altered cortisol levels or failure to suppress cortisol release during dexamethasone suppression tests (Almeida et al., 2021). Preclinical and clinical studies have demonstrated that continuous activation of the HPA axis leads to elevated levels of corticosterone, which is strongly associated with stress-related psychiatric disorders (Ouellet-Morin et al., 2011; Rasmusson and Pineles, 2018). In addition, the HPA axis and GABAergic system have mutual regulatory effects.
First, changes in GABA signaling cause HPA axis dysfunction in fear memory. GABAergic connects the brain regions involved in the HPA axis and the PVN, as well as its surrounding regions, including the subventricular area, the anterior hypothalamic area, the dorsomedial nucleus of the hypothalamus, the medial preoptic area, the lateral hypothalamic area, and some subnuclei of the bed nucleus of the stria terminalis. Thus, CRH neurons receive several GABAergic inputs (Cullinan et al., 2008). The fear response is regulated by GABAergic signaling through the HPA axis, mainly through the activation of GABAARs (Brickley and Mody, 2012). Previous studies have demonstrated that CRF plays a part in fear extinction learning. Ouellet-Morin et al. (2011) reported that the absence of either GABAAα1 or NMDAR1 gene expression in CRFergic neurons results in significant and long-term deficiency of fear extinction rather than acquisition or retention.
Besides, the HPA axis regulates stress by regulating GABAergic signaling (Mikkelsen et al., 2008). Studies have reported that the injection of metyrapone, an inhibitor of corticosterone synthesis, can prevent the enhancement of intrinsic excitability in BLA→vHPC pyramidal neurons caused by foot shock. Moreover, the overexpression of GR and MR in amygdala neurons provides the basis for corticosterone regulation of BLA neurons (Kohda et al., 2007; Deppermann et al., 2014). Furthermore, changes in the intrinsic excitability of neurons are regulated by several factors, among which the weakening of inhibitory signals by the GABAAR antagonist bicuculline is an important factor (Song et al., 2017). Long-term corticosterone feeding has been observed to decrease GABAAR-mediated tonic inhibition in amygdala neurons. Moreover, corticosterone can alter the expression and function of GABAAR and drive the GABAAR-mediated chloride current (Liu et al., 2017). Liu et al. (2017) found that the recruitment of GABAA(δ)R by researchers through pharmacological and physiological methods leads to the weakening of GABAergic transmission in LA projection neurons, which indicates the disinhibition of GABAA(δ)R. Activation of GABAA(δ)R reduces the input resistance of local interneurons and inhibits the activation of local interneurons. Deletion of the GABAA(δ)R gene weakened its inhibitory effect on LA interneurons. Therefore, corticosterone may enhance the intrinsic excitability of BLA→vHPC pyramidal neurons by reducing their inhibitory signals. In addition, studies have found that (Verkuyl and Joëls, 2003), parvocellular neurons in the PVN receive hormonal inputs mediated by corticosterone as well as GABAergic inhibitory projection. It has been proven that increases in glucocorticoid levels due to stress can inhibit GABAergic tone on parvocellular hypothalamic neurons (Verkuyl et al., 2005). Interestingly, some current studies (Hartmann et al., 2017) suggest that the regulation of fear and stress-related behaviors by corticosterone and glucocorticoids in the forebrain is mediated more by glutamatergic neurons than by GABA. Therefore, there may be two different explanations for the enhancement of neuronal excitability caused by the HPA axis in different brain regions, and more studies are needed to show which mechanism plays a dominant role in other brain regions in the future.
The endocannabinoid system (ECS) plays an important role in regulating behaviors such as fear and anxiety (Lafenêtre et al., 2007; Ruehle et al., 2012; Gunduz-Cinar et al., 2013; McLaughlin et al., 2014). Moreover, the abnormal function of this system can cause a series of neuropsychiatric disorders, such as depression and anxiety (Hillard et al., 2012; Mechoulam and Parker, 2013). Studies in the past decade have reported the relationship between The ECS and traumatic stress disorder. Piomelli (2003) and Neumeister et al. (2013) found abnormal levels of endocannabinoids in the blood and their receptors in PTSD patients, suggesting possible changes in the function of the ECS in PTSD patients.
Endocannabinoids mainly include N-arachidonoylethanolamine (AEA) and 2-arachidonoylglycerol (2-AG), which are mainly synthesized at the postsynaptic and serve as retrograde first messengers. It regulates the release of neurotransmitters by activating cannabinoid receptors located in the presynaptic membrane (Piomelli, 2003). Endocannabinoids primarily regulate the transport of GABA and Glu neurotransmitters. In addition, they also regulate the release of acetylcholine, biogenic amines noradrenaline and serotonin, and the neuropeptide CCK-8 (Piomelli, 2003).
There are two main types of cannabinoid receptors in the human body, the CB1 receptor and CB2 receptor, both of which are G-protein-coupled receptors that can activate intracellular signal transduction pathways and regulate the release of neurotransmitters and synaptic function (Adolphs et al., 2002). Animal studies have observed that the knockdown of CB1 receptors or the inhibition of CB1 receptor signaling with drugs can cause anxiety symptoms (Haller et al., 2002, 2004). The CB1 receptor-knockout mice could not eliminate fear memory after fear extinction training, indicating that the CB1 receptor plays an important role in the process of fear extinction (Marsicano et al., 2002; Cannich et al., 2004; Dubreucq et al., 2012). The agonists of CB1 such as nabilone and △9-THC can relieve some of the clinical symptoms of PTSD, such as over-stress, forgetfulness, nightmares, and pain (Bremner et al., 1996; Fraser, 2009; Passie et al., 2012). These findings suggest that the CB1 receptor is involved in regulating the transmission of neural information in the PTSD brain and plays an important role in the pathogenesis of PTSD.
Recent studies have reported that endocannabinoids regulate the processing of fear memory (Marsicano et al., 2002; Barad et al., 2006; Mahan and Ressler, 2012; Drumond et al., 2017). They are involved in the acquisition, consolidation, retrieval, and extinction of fear memories, as well as in the regulation of emotional states (Ruehle et al., 2012; Trezza and Campolongo, 2013; Xu and Südhof, 2013). In the hippocampus and amygdala, the activation of GABAergic presynaptic CB1 receptors decreases the release of GABA transmitters, thereby modulating the processing of fear memories (Morales et al., 2004; Szabó et al., 2014). The CB1 receptors are located in the presynaptic membranes of glutamatergic and GABAergic neurons, and their activation inhibits the release of Glu (Kamprath et al., 2006; Hoffman et al., 2010) and GABA (Laaris et al., 2010). Several researchers have suggested that there exists a dynamic balance system that is composed of Glu and GABA in the brain, which jointly maintains a balance between the inhibition and excitation of nerve cells. Once this dynamic balance is broken, anxiety, depression, and other emotional-related behaviors may occur (Martin et al., 2002; Haller et al., 2004), and the ECB system is an important part of the regulation of this balance. Studies have demonstrated that activation of the CB1 receptor in the dorsolateral periaqueductal gray can induce anxiety-like effects in the elevated plus maze, while the CB1 receptor antagonist AM251 prevents anxiety-like behaviors resulting from the Vogel conflict test (Lisboa et al., 2008). The activation of CB1 receptors on GABAergic neurons leads to the reduced release of GABA transmitters, resulting in anxiety-like responses (Roohbakhsh et al., 2009).
The main inhibitory neurotransmitter in the brain is GABA, acting along with Glu to control the balance of excitation and inhibition in several brain circuits (Petroff, 2002). The neurotoxicity of Glu not only leads to the apoptosis and necrosis of brain neurons in patients with ischemic cerebrovascular disease (Sun et al., 2015) but also affects synaptic plasticity in the brains of patients with PTSD (Stachowicz, 2018). The GABAergic system is an important inhibitory system in the CNS that can protect the normal regulatory function of the nervous system by inhibiting the hyper-excitatory effect of Glu. During the metabolic switch between Glu and GABA, two pathways eliminate excess Glu in the synaptic cleft or mitigate its neurotoxicity (Gao et al., 2014).
The production of GABA from Glu promotes the release of more Glu, which is mediated by glutamic acid decarboxylase (GAD). The GAD is the key rate-limiting enzyme in GABA synthesis. There are two kinds of isoenzymes of GAD, GAD65 and GAD67. Abnormal function of GAD65/67 can also lead to decreased GABA levels. Generally, GAD65 is primarily responsible for the synthesis of GABA that is released at synapses, while GAD67 is the 67 kDa isoform of GAD (GABA synthase), which is responsible for the synthesis of GABA in the metabolic pool of neurons. The distribution of GAD is consistent with that of GABAergic neurons, which makes GAD an excellent marker enzyme for GABAergic neurons, with the functional status of GABAergic neurons in the CNS being often reflected by measuring the activity of GAD in brain tissue. It was reported that chronic adverse stimulation further decreased the expression level of GAD67 and the number of GAD67-positive neurons, suggesting that stress impaired the conversion of Glu to GABA by reducing the level of GAD, thereby limiting GABA synthesis (Fogaça and Duman, 2019). The levels of GAD67 in the hippocampus, amygdala, and PFC of PTSD rats were decreased, suggesting that the reduction in GABA content could weaken the central inhibitory function and disrupt the balance between GABA and Glu. The results of autopsies of patients with major depressive disorder showed significant reductions in the activity of GAD65 in the PFC brain region (Karolewicz et al., 2010), as well as the density of GAD65/67 in the PVN of the hypothalamus (Gao et al., 2013). Similarly, the expression of GAD67 was downregulated in the PFC (Pochwat et al., 2016) in both mouse and rat models of depression. The GAD65 knockout mice showed fear generalization and fear resolution disorders in preclinical trials (Sangha et al., 2009), suggesting an important role for GAD65 in preventing anxiety-like behaviors, fear memory extinction, and resilience to the development of contextual fear generalization.
The GABA transporter (GAT) may play an important role in mental disorders such as anxiety and depression by affecting GABAergic transmission. However, evidence for this hypothesis is scarce, with studies reporting that patients with anxiety disorders exhibit positive clinical responses to the selective GAT-1 inhibitor tiagabine (Schwartz and Nihalani, 2006). In the study of Liu et al. (2007) the researchers used the forced swimming test and the tail suspension test to model depression and anxiety in mice and measured resilience through the open field test, the dark light exploration test, the emergence test and the elevated plus maze (EPM) behavior test. The GAT-1-deficient mice exhibit increased resilience to acute stress in response to behavioral paradigms such as antidepressant- and antianxiety-like activities (Liu et al., 2007; Gong et al., 2015), which substantiates the role of GAT-1 in mood disorders. However, the role of GAT in PTSD needs investigation. The concentration of GABA in the extra-synaptic space is regulated in several ways. In addition to GAT-1, the GABA transporters GAT-2 and betaine/GABA transporter 1 (BGT-1) are located on astrocytes that recycle GABA. Additionally, GABA in astrocytes can enter the intercellular space through the bestrophin channel as a non-vesicular release (Brickley and Mody, 2012), and these regulatory mechanisms maintain the concentration of GABA at a stable level outside synapses. However, there are very few studies linking GABA- astrocytes to PTSD, which gives us an indication that future studies may focus on this aspect.
Psychological stress can cause an imbalance between excitatory Glu and inhibitory GABA neurotransmitters, which can lead to a series of neurochemical changes. First, the increased expression of apoptotic markers in the muscle, thymus, and nervous system is related to stress. Moreover, the survival and apoptosis of neurons are related to the mechanism of the balance between Glu and GABA (Engelbrecht et al., 2010). The glutamate/gamma-aminobutyric acid-glutamine metabolic circuit in the CNS is the main pathway that regulates the metabolism of Glu and GABA, which is important to maintain the dynamic balance between excitatory and inhibitory systems in the brain (Jiang et al., 2022). During the metabolic conversion of Glu to GABA, excess Glu in the synaptic cleft can be eliminated or reduced through the excitatory amino acid transporters (EAATs) and GAD pathways. In other words, when the conversion level of Glu to GABA is insufficient, resulting in too much Glu and too little GABA in the CNS, an imbalance of excitation and inhibition can occur. The elevation or accumulation of Glu can induce apoptosis in sensitive neural cells. When GABA is catalyzed by Glu and directed by GAD, it has positive feedback on its own release. Such imbalance further leads to dysfunctional excitatory spasms. The findings of Fogaça and Duman (2019) suggest that the imbalance between the excitatory (Glu) and inhibitory (GABA) transmitters may promote apoptosis in the hippocampal neurons and play an important role in the pathogenesis of PTSD. However, this finding is inconsistent with the brain imaging results of patients, which may indicate that humans and rodents have different responses to PTSD-like stress. Therefore, the specific reasons for this need further investigation.
Several types of drugs, mainly antidepressants, antipsychotics, and anticonvulsants, target PTSD. However, anti-stress drugs are not widely used in PTSD, although they can regulate the GABAergic system, stimulate GABA receptors, inhibit GABA transporters, and restore the function of the GABAergic system in the brain.
As anti-anxiety agents, BDZ is widely used clinically. For instance, BDZ such as diazepam, flurazepam, clorazepate, oxazepam, and triazolam, is also used in the treatment of PTSD symptoms, but prolonged use of these agents can cause dizziness, drowsiness, fatigue, and other adverse effects. In addition, BDZ promotes GABA-mediated neural transmission in the CNS (Mao et al., 1975). Such drugs do not directly excite GABA recognition sites and accelerate the release of GABA but increase the affinity of low-affinity GABAAR or the number of GABAARs through allosteric regulation to enhance the inhibitory function of GABA. Promoting the E-I balance caused by stress produces anti-anxiety and sedative effects. Studies on the GABAAR family reported that the α2 and α3 subunits are closely related to anxiety behaviors under the effect of BDZ (Dias et al., 2005). The α5 subunit is involved in hippocampus-associated associative memory (Rudolph et al., 1999; Rudolph and Möhler, 2004). The γ2 subunit is extensively regulated by BDZ in the brain (Günther et al., 1995).
Topiramate is an anticonvulsant, which is also among the supplementary drugs in the clinical use of PTSD (Khan and Liberzon, 2004). Topiramate enhances the function of the GABA receptor and induces the influx of chloride ions. It is also involved in the intervention of PTSD symptoms by regulating and restoring the content of GABA in the brain and maintaining the excitation-inhibitory stability of Glu and GABA in the CNS (Berlant, 2001). In clinical studies on PTSD, it was observed that the rate of relief from PTSD symptoms in the topiramate-treatment group was significantly higher than that in the control group. Topiramate has been proven to relieve the symptoms of recurrent traumatic experiences, nightmares, and other PTSD symptoms, such as high arousal, although these drugs also have significant side effects, including dizziness and lethargy.
Studies have also found that neurosteroids have an anti-anxiety effect by acting on the GABAergic system, as they can directly act on the GABAAR, prolong the opening time and increase the opening frequency of its chloride channel, thus increasing the inhibitory effect of GABA (Lambert et al., 1995).
The GABAergic system is significant in the regulation of PTSD. After traumatic events, the formation of fear memory and synaptic plasticity are affected by the GABA inhibition system, and the balance in excitation-inhibition homeostasis between neural networks is destroyed, resulting in complex changes in the thoughts, feelings, and behaviors of the subject. Anti-stress drugs that regulate the GABAergic system restore GABA function in the brain by stimulating GABA receptors or inhibiting GABA transporters. Although such drugs can relieve PTSD symptoms, they can also have side effects such as dizziness, drowsiness, and fatigue. Future research is needed to further explore the common mechanisms underlying the core symptoms of PTSD to guide the discovery of ideal targets for the treatment of PTSD. In summary, the consequence of PTSD may be related to the downregulation of GABAergic system. However, although patients with PTSD have been found reduced GABAARs, the GABAergic system has weaker pharmacodynamics at the receptor level.
JH and FX conceived and designed the study and wrote the draft. LY and LT collected the literature and organized the structure. XW, ZD, and YZ contributed to the supervision. XY and YL used software to do the drawing. KL and WW reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adamec, R., Strasser, K., Blundell, J., Burton, P., and McKay, D. W. (2006). Protein synthesis and the mechanisms of lasting change in anxiety induced by severe stress. Behav. Brain Res. 167, 270–286. doi: 10.1016/j.bbr.2005.09.019
Adolphs, R., Baron-Cohen, S., and Tranel, D. (2002). Impaired recognition of social emotions following amygdala damage. J. Cogn. Neurosci. 14, 1264–1274. doi: 10.1162/089892902760807258
Agís-Balboa, R. C., Pinna, G., Pibiri, F., Kadriu, B., Costa, E., and Guidotti, A. (2007). Down-regulation of neurosteroid biosynthesis in corticolimbic circuits mediates social isolation-induced behavior in mice. Proc. Natl. Acad. Sci. U.S.A. 104, 18736–18741. doi: 10.1073/pnas.0709419104
Agís-Balboa, R. C., Pinna, G., Zhubi, A., Maloku, E., Veldic, M., Costa, E., et al. (2006). Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 103, 14602–14607. doi: 10.1073/pnas.0606544103
Akirav, I., Raizel, H., and Maroun, M. (2006). Enhancement of conditioned fear extinction by infusion of the GABA(A) agonist muscimol into the rat prefrontal cortex and amygdala. Eur. J. Neurosci. 23, 758–764. doi: 10.1111/j.1460-9568.2006.04603.x
Almada, R. C., Albrechet-Souza, L., and Brandão, M. L. (2013). Further evidence for involvement of the dorsal hippocampus serotonergic and γ-aminobutyric acid (GABA)ergic pathways in the expression of contextual fear conditioning in rats. J. Psychopharmacol. (Oxford, England) 27, 1160–1168. doi: 10.1177/0269881113482840
Almeida, F. B., Pinna, G., and Barros, H. M. T. (2021). The role of HPA axis and allopregnanolone on the neurobiology of major depressive disorders and PTSD. Int. J. Mol. Sci. 22:5495. doi: 10.3390/ijms22115495
Almeida, M., Sousa Filho, L. F., Rabello, P. M., and Santiago, B. M. (2020). International classification of diseases - 11th revision: From design to implementation. Rev. Saude Publica 54:104. doi: 10.11606/s1518-8787.2020054002120
Anagnostaras, S. G., Maren, S., and Fanselow, M. S. (1999). Temporally graded retrograde amnesia of contextual fear after hippocampal damage in rats: Within-subjects examination. J. Neurosci. 19, 1106–1114. doi: 10.1523/JNEUROSCI.19-03-01106.1999
Anagnostaras, S. G., Wood, S. C., Shuman, T., Cai, D. J., Leduc, A. D., Zurn, K. R., et al. (2010). Automated assessment of pavlovian conditioned freezing and shock reactivity in mice using the video freeze system. Front. Behav. Neurosci. 4:158. doi: 10.3389/fnbeh.2010.00158
Ardi, Z., Albrecht, A., Richter-Levin, A., Saha, R., and Richter-Levin, G. (2016). Behavioral profiling as a translational approach in an animal model of posttraumatic stress disorder. Neurobiol. Dis. 88, 139–147. doi: 10.1016/j.nbd.2016.01.012
Aupperle, R. L., Melrose, A. J., Stein, M. B., and Paulus, M. P. (2012). Executive function and PTSD: Disengaging from trauma. Neuropharmacology 62, 686–694. doi: 10.1016/j.neuropharm.2011.02.008
Averill, L. A., Purohit, P., Averill, C. L., Boesl, M. A., Krystal, J. H., and Abdallah, C. G. (2017). Glutamate dysregulation and glutamatergic therapeutics for PTSD: Evidence from human studies. Neurosci. Lett. 649, 147–155. doi: 10.1016/j.neulet.2016.11.064
Babaev, O., Piletti Chatain, C., and Krueger-Burg, D. (2018). Inhibition in the amygdala anxiety circuitry. Exp. Mol. Med. 50, 1–16. doi: 10.1038/s12276-018-0063-8
Ball, T. M., Knapp, S. E., Paulus, M. P., and Stein, M. B. (2017). Brain activation during fear extinction predicts exposure success. Depress. Anxiety 34, 257–266. doi: 10.1002/da.22583
Barad, M., Gean, P. W., and Lutz, B. (2006). The role of the amygdala in the extinction of conditioned fear. Biol. Psychiatry 60, 322–328. doi: 10.1016/j.biopsych.2006.05.029
Bast, T., Zhang, W. N., and Feldon, J. (2001). The ventral hippocampus and fear conditioning in rats. Different anterograde amnesias of fear after tetrodotoxin inactivation and infusion of the GABA(A) agonist muscimol. Exp. Brain Res. 139, 39–52. doi: 10.1007/s002210100746
Battle, D. E. (2013). Diagnostic and statistical manual of mental disorders (DSM). CoDAS 25, 191–192. doi: 10.1590/s2317-17822013000200017
Baulieu, E. E., and Robel, P. (1990). Neurosteroids: A new brain function? J. Steroid Biochem. Mol. Biol. 37, 395–403. doi: 10.1016/0960-0760(90)90490-c
Belelli, D., and Lambert, J. J. (2005). Neurosteroids: Endogenous regulators of the GABA(A) receptor. Nat. Rev. Neurosci. 6, 565–575. doi: 10.1038/nrn1703
Belelli, D., Harrison, N. L., Maguire, J., Macdonald, R. L., Walker, M. C., and Cope, D. W. (2009). Extrasynaptic GABAA receptors: Form, pharmacology, and function. J. Neurosci. 29, 12757–12763. doi: 10.1523/JNEUROSCI.3340-09.2009
Benjet, C., Bromet, E., Karam, E. G., Kessler, R. C., McLaughlin, K. A., Ruscio, A. M., et al. (2016). The epidemiology of traumatic event exposure worldwide: results from the World Mental Health Survey Consortium. Psychol. Med. 46, 327–343. doi: 10.1017/S0033291715001981
Berlant, J. L. (2001). Topiramate in posttraumatic stress disorder: Preliminary clinical observations. J. Clin. Psychiatry 62 Suppl 17, 60–63.
Berlau, D. J., and McGaugh, J. L. (2006). Enhancement of extinction memory consolidation: The role of the noradrenergic and GABAergic systems within the basolateral amygdala. Neurobiol. Learn. Mem. 86, 123–132. doi: 10.1016/j.nlm.2005.12.008
Bhagwagar, Z., Wylezinska, M., Jezzard, P., Evans, J., Boorman, E., Matthews, P., et al. (2008). Low GABA concentrations in occipital cortex and anterior cingulate cortex in medication-free, recovered depressed patients. Int. J. Neuropsychopharmacol. 11, 255–260. doi: 10.1017/S1461145707007924
Bishop, J. R., Lee, A. M., Mills, L. J., Thuras, P. D., Eum, S., Clancy, D., et al. (2021). Corrigendum: Methylation of FKBP5 and SLC6A4 in relation to treatment response to mindfulness based stress reduction for posttraumatic stress disorder. Front. Psychiatry 12:642245. doi: 10.3389/fpsyt.2021.642245
Blanchard, R. J., and Blanchard, D. C. (1989). Attack and defense in rodents as ethoexperimental models for the study of emotion. Prog. Neuro Psychopharmacol. Biol. Psychiatry 13 Suppl, S3–S14. doi: 10.1016/0278-5846(89)90105-x
Bouton, M., and Bolles, R. (1980). Conditioned fear assessed by freezing and by the suppression of three different baselines. Anim. Learn. Behav. 8, 429–434.
Bremner, J. D., Innis, R. B., Southwick, S. M., Staib, L., Zoghbi, S., and Charney, D. S. (2000). Decreased benzodiazepine receptor binding in prefrontal cortex in combat-related posttraumatic stress disorder. Am. J. Psychiatry 157, 1120–1126. doi: 10.1176/appi.ajp.157.7.1120
Bremner, J. D., Southwick, S. M., Darnell, A., and Charney, D. S. (1996). Chronic PTSD in Vietnam combat veterans: Course of illness and substance abuse. Am. J. Psychiatry 153, 369–375. doi: 10.1176/ajp.153.3.369
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABA(A) receptors: Their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Cannich, A., Wotjak, C. T., Kamprath, K., Hermann, H., Lutz, B., and Marsicano, G. (2004). CB1 cannabinoid receptors modulate kinase and phosphatase activity during extinction of conditioned fear in mice. Learn. Mem. (Cold Spring Harbor, N.Y.) 11, 625–632. doi: 10.1101/lm.77904
Castellano, C., and McGaugh, J. L. (1990). Effects of post-training bicuculline and muscimol on retention: Lack of state dependency. Behav. Neural Biol. 54, 156–164. doi: 10.1016/0163-1047(90)91352-c
Catarino, A., Küpper, C. S., Werner-Seidler, A., Dalgleish, T., and Anderson, M. C. (2015). Failing to forget: Inhibitory-control deficits compromise memory suppression in posttraumatic stress disorder. Psychol. Sci. 26, 604–616. doi: 10.1177/0956797615569889Meiser-Stedman
Cates, M. E., Bishop, M. H., Davis, L. L., Lowe, J. S., and Woolley, T. W. (2004). Clonazepam for treatment of sleep disturbances associated with combat-related posttraumatic stress disorder. Ann. Pharmacother. 38, 1395–1399. doi: 10.1345/aph.1E043
Charney, D. S. (2004). Psychobiological mechanisms of resilience and vulnerability: Implications for successful adaptation to extreme stress. Am. J. Psychiatry 161, 195–216. doi: 10.1176/appi.ajp.161.2.195
Chebib, M., and Johnston, G. A. (1999). The ‘ABC’ of GABA receptors: A brief review. Clin. Exp. Pharmacol. Physiol. 26, 937–940. doi: 10.1046/j.1440-1681.1999.03151.x
Chen, S. W., Shemyakin, A., and Wiedenmayer, C. P. (2006). The role of the amygdala and olfaction in unconditioned fear in developing rats. J. Neurosci. 26, 233–240. doi: 10.1523/JNEUROSCI.2890-05.2006
Chen, S., Gao, L., Li, X., and Ye, Y. (2021). Allopregnanolone in mood disorders: Mechanism and therapeutic development. Pharmacol. Res. 169:105682. doi: 10.1016/j.phrs.2021.105682
Christianson, J., Fernando, A., Kazama, A., Jovanovic, T., Ostroff, L., and Sangha, S. (2012). Inhibition of fear by learned safety signals: A mini-symposium review. J. Neurosci. 32, 14118–14124. doi: 10.1523/JNEUROSCI.3340-12.2012
Clarke, I. J. (2015). Hypothalamus as an endocrine organ. Compr. Physiol. 5, 217–253. doi: 10.1002/cphy.c140019
Concas, A., Mostallino, M. C., Porcu, P., Follesa, P., Barbaccia, M. L., Trabucchi, M., et al. (1998). Role of brain allopregnanolone in the plasticity of gamma-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc. Natl. Acad. Sci. U.S.A. 95, 13284–13289. doi: 10.1073/pnas.95.22.13284
Connor, K. M., Davidson, J. R., Weisler, R. H., Zhang, W., and Abraham, K. (2006). Tiagabine for posttraumatic stress disorder: Effects of open-label and double-blind discontinuation treatment. Psychopharmacology 184, 21–25. doi: 10.1007/s00213-005-0265-3
Corcoran, K. A., and Maren, S. (2001). Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J. Neurosci. 21, 1720–1726. doi: 10.1523/JNEUROSCI.21-05-01720.2001
Craddock, N., Jones, L., Jones, I. R., Kirov, G., Green, E. K., Grozeva, D., et al. (2010). Strong genetic evidence for a selective influence of GABAA receptors on a component of the bipolar disorder phenotype. Mol. Psychiatry 15, 146–153. doi: 10.1038/mp.2008.66
Cullinan, W. E., Ziegler, D. R., and Herman, J. P. (2008). Functional role of local GABAergic influences on the HPA axis. Brain Struct. Funct. 213, 63–72. doi: 10.1007/s00429-008-0192-2
Davidson, J. R. (2004). Use of benzodiazepines in social anxiety disorder, generalized anxiety disorder, and posttraumatic stress disorder. J. Clin. Psychiatry 65 Suppl 5, 29–33.
Davidson, J. R., Brady, K., Mellman, T. A., Stein, M. B., and Pollack, M. H. (2007). The efficacy and tolerability of tiagabine in adult patients with post-traumatic stress disorder. J. Clin. Psychopharmacol. 27, 85–88. doi: 10.1097/JCP.0b013e31802e5115
Davidson, R. J., Putnam, K. M., and Larson, C. L. (2000). Dysfunction in the neural circuitry of emotion regulation–A possible prelude to violence. Science (New York, N.Y.) 289, 591–594. doi: 10.1126/science.289.5479.591
Davis, M., Walker, D. L., Miles, L., and Grillon, C. (2010). Phasic vs sustained fear in rats and humans: Role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology 35, 105–135. doi: 10.1038/npp.2009.109
de Kloet, E. R., Joëls, M., and Holsboer, F. (2005). Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 6, 463–475. doi: 10.1038/nrn1683
Deppermann, S., Storchak, H., Fallgatter, A. J., and Ehlis, A. C. (2014). Stress-induced neuroplasticity: (Mal)adaptation to adverse life events in patients with PTSD–a critical overview. Neuroscience 283, 166–177. doi: 10.1016/j.neuroscience.2014.08.037
Dias, R., Sheppard, W. F., Fradley, R. L., Garrett, E. M., Stanley, J. L., Tye, S. J., et al. (2005). Evidence for a significant role of alpha 3-containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J. Neurosci. 25, 10682–10688. doi: 10.1523/JNEUROSCI.1166-05.2005
Disner, S. G., Marquardt, C. A., Mueller, B. A., Burton, P. C., and Sponheim, S. R. (2018). Spontaneous neural activity differences in posttraumatic stress disorder: A quantitative resting-state meta-analysis and fMRI validation. Hum. Brain Mapp. 39, 837–850. doi: 10.1002/hbm.23886
Dolfen, N., Veldman, M. P., Gann, M. A., von Leupoldt, A., Puts, N. A. J., Edden, R. A. E., et al. (2021). A role for GABA in the modulation of striatal and hippocampal systems under stress. Commun. Biol. 4:1033. doi: 10.1038/s42003-021-02535-x
Domschke, K., and Zwanzger, P. (2008). GABAergic and endocannabinoid dysfunction in anxiety – Future therapeutic targets? Curr. Pharm. Design 14, 3508–3517. doi: 10.2174/138161208786848784
Domschke, K., Tidow, N., Schwarte, K., Deckert, J., Lesch, K., Arolt, V., et al. (2014). Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int. J. Neuropsychopharmacol. 17, 1167–1176. doi: 10.1017/S146114571400039X
Drugan, R. C., Basile, A. S., Crawley, J. N., Paul, S. M., and Skolnick, P. (1986). Inescapable shock reduces [3H]Ro 5-4864 binding to “peripheral-type” benzodiazepine receptors in the rat. Pharmacol. Biochem. Behav. 24, 1673–1677. doi: 10.1016/0091-3057(86)90504-6
Drumond, A., Madeira, N., and Fonseca, R. (2017). Endocannabinoid signaling and memory dynamics: A synaptic perspective. Neurobiol. Learn. Mem. 138, 62–77. doi: 10.1016/j.nlm.2016.07.031
Dubreucq, S., Matias, I., Cardinal, P., Häring, M., Lutz, B., Marsicano, G., et al. (2012). Genetic dissection of the role of cannabinoid type-1 receptors in the emotional consequences of repeated social stress in mice. Neuropsychopharmacology 37, 1885–1900. doi: 10.1038/npp.2012.36
Elms, L., Shannon, S., Hughes, S., and Lewis, N. (2019). Cannabidiol in the treatment of post-traumatic stress disorder: A case series. J. Altern. Complement. Med. 25, 392–397.
Engelbrecht, A. M., Smith, C., Neethling, I., Thomas, M., Ellis, B., Mattheyse, M., et al. (2010). Daily brief restraint stress alters signaling pathways and induces atrophy and apoptosis in rat skeletal muscle. Stress (Amsterdam, Netherlands) 13, 132–141. doi: 10.3109/10253890903089834
Evans, J., Sun, Y., McGregor, A., and Connor, B. (2012). Allopregnanolone regulates neurogenesis and depressive/anxiety-like behaviour in a social isolation rodent model of chronic stress. Neuropharmacology 63, 1315–1326. doi: 10.1016/j.neuropharm.2012.08.012
Fanselow, M. S., and Dong, H. W. (2010). Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7–19. doi: 10.1016/j.neuron.2009.11.031
Fendt, M., and Fanselow, M. S. (1999). The neuroanatomical and neurochemical basis of conditioned fear. Neurosci. Biobehav. Rev. 23, 743–760. doi: 10.1016/s0149-7634(99)00016-0
Ferrara, N. C., Jarome, T. J., Cullen, P. K., Orsi, S. A., Kwapis, J. L., Trask, S., et al. (2019). GluR2 endocytosis-dependent protein degradation in the amygdala mediates memory updating. Sci. Rep. 9:5180. doi: 10.1038/s41598-019-41526-1
Feusner, J., Ritchie, T., Lawford, B., Young, R. M., Kann, B., and Noble, E. P. (2001). GABA(A) receptor beta 3 subunit gene and psychiatric morbidity in a post-traumatic stress disorder population. Psychiatry Res. 104, 109–117. doi: 10.1016/s0165-1781(01)00296-7
Fogaça, M. V., and Duman, R. S. (2019). Cortical GABAergic dysfunction in stress and depression: New insights for therapeutic interventions. Front. cell. Neurosci. 13:87. doi: 10.3389/fncel.2019.00087
Fraser, G. A. (2009). The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci. Ther. 15, 84–88. doi: 10.1111/j.1755-5949.2008.00071.x
Gao, J., Wang, H., Liu, Y., Li, Y. Y., Chen, C., Liu, L. M., et al. (2014). Glutamate and GABA imbalance promotes neuronal apoptosis in hippocampus after stress. Med. Sci. Monit. 20, 499–512. doi: 10.12659/MSM.890589
Gao, S. F., Klomp, A., Wu, J. L., Swaab, D. F., and Bao, A. M. (2013). Reduced GAD(65/67) immunoreactivity in the hypothalamic paraventricular nucleus in depression: A postmortem study. J. Affect. Disord. 149, 422–425. doi: 10.1016/j.jad.2012.12.003
Gelpin, E., Bonne, O., Peri, T., Brandes, D., and Shalev, A. Y. (1996). Treatment of recent trauma survivors with benzodiazepines: A prospective study. J. Clin. Psychiatry 57, 390–394.
Geracioti, T. D. Jr., Baker, D. G., Kasckow, J. W., Strawn, J. R., Jeffrey Mulchahey, J., Dashevsky, B. A., et al. (2008). Effects of trauma-related audiovisual stimulation on cerebrospinal fluid norepinephrine and corticotropin-releasing hormone concentrations in post-traumatic stress disorder. Psychoneuroendocrinology 33, 416–424. doi: 10.1016/j.psyneuen.2007.12.012
Geuze, E., van Berckel, B. N., Lammertsma, A. A., Boellaard, R., de Kloet, C. S., Vermetten, E., et al. (2008). Reduced GABAA benzodiazepine receptor binding in veterans with post-traumatic stress disorder. Mol. Psychiatry 13, 74–83, 3. doi: 10.1038/sj.mp.4002054
Ghosal, S., Hare, B., and Duman, R. S. (2017). Prefrontal cortex GABAergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Curr. Opin. Behav. Sci. 14, 1–8. doi: 10.1016/j.cobeha.2016.09.012
Gilbert, M., Dinh La, A., Romulo Delapaz, N., Kenneth Hor, W., Fan, P., Qi, X., et al. (2020). An emulation of randomized trials of administrating benzodiazepines in PTSD patients for outcomes of suicide-related events. J. Clin. Med. 9:3492. doi: 10.3390/jcm9113492
Gilmartin, M. R., Kwapis, J. L., and Helmstetter, F. J. (2012). Trace and contextual fear conditioning are impaired following unilateral microinjection of muscimol in the ventral hippocampus or amygdala, but not the medial prefrontal cortex. Neurobiol. Learn. Mem. 97, 452–464. doi: 10.1016/j.nlm.2012.03.009
Gong, X., Shao, Y., Li, B., Chen, L., Wang, C., and Chen, Y. (2015). γ-aminobutyric acid transporter-1 is involved in anxiety-like behaviors and cognitive function in knockout mice. Exp. Ther. Med. 10, 653–658. doi: 10.3892/etm.2015.2577
Gu, H., Hu, Y., Chen, X., He, Y., and Yang, Y. (2019). Regional excitation-inhibition balance predicts default-mode network deactivation via functional connectivity. NeuroImage 185, 388–397. doi: 10.1016/j.neuroimage.2018.10.055
Gunduz-Cinar, O., Hill, M. N., McEwen, B. S., and Holmes, A. (2013). Amygdala FAAH and anandamide: Mediating protection and recovery from stress. Trends Pharmacol. Sci. 34, 637–644. doi: 10.1016/j.tips.2013.08.008
Günther, U., Benson, J., Benke, D., Fritschy, J. M., Reyes, G., Knoflach, F., et al. (1995). Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 92, 7749–7753. doi: 10.1073/pnas.92.17.7749
Hageman, I., Andersen, H. S., and Jørgensen, M. B. (2001). Post-traumatic stress disorder: A review of psychobiology and pharmacotherapy. Acta Psychiatr. Scand. 104, 411–422. doi: 10.1034/j.1600-0447.2001.00237.x
Haller, J., Bakos, N., Szirmay, M., Ledent, C., and Freund, T. F. (2002). The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur. J. Neurosci. 16, 1395–1398. doi: 10.1046/j.1460-9568.2002.02192.x
Haller, J., Varga, B., Ledent, C., and Freund, T. F. (2004). CB1 cannabinoid receptors mediate anxiolytic effects: Convergent genetic and pharmacological evidence with CB1-specific agents. Behav. Pharmacol. 15, 299–304. doi: 10.1097/01.fbp.0000135704.56422.40
Hartmann, J., Dedic, N., Pöhlmann, M. L., Häusl, A., Karst, H., Engelhardt, C., et al. (2017). Forebrain glutamatergic, but not GABAergic, neurons mediate anxiogenic effects of the glucocorticoid receptor. Mol. Psychiatry 22, 466–475. doi: 10.1038/mp.2016.87
Harvey, B. H., Oosthuizen, F., Brand, L., Wegener, G., and Stein, D. J. (2004). Stress-restress evokes sustained iNOS activity and altered GABA levels and NMDA receptors in rat hippocampus. Psychopharmacology 175, 494–502. doi: 10.1007/s00213-004-1836-4
Hayes, J. P., Hayes, S. M., and Mikedis, A. M. (2012). Quantitative meta-analysis of neural activity in posttraumatic stress disorder. Biol. Mood Anxiety Disord. 2:9. doi: 10.1186/2045-5380-2-9
Herbst, M. R., Twining, R. C., and Gilmartin, M. R. (2022). Ventral hippocampal shock encoding modulates the expression of trace cued fear. Neurobiol. Learn. Mem. 190:107610. doi: 10.1016/j.nlm.2022.107610
Hill, M. N., Campolongo, P., Yehuda, R., and Patel, S. (2018). Integrating endocannabinoid signaling and cannabinoids into the biology and treatment of posttraumatic stress disorder. Neuropsychopharmacology 43, 80–102. doi: 10.1038/npp.2017.162
Hillard, C. J., Weinlander, K. M., and Stuhr, K. L. (2012). Contributions of endocannabinoid signaling to psychiatric disorders in humans: Genetic and biochemical evidence. Neuroscience 204, 207–229. doi: 10.1016/j.neuroscience.2011.11.020
Hoffman, A. F., Laaris, N., Kawamura, M., Masino, S. A., and Lupica, C. R. (2010). Control of cannabinoid CB1 receptor function on glutamate axon terminals by endogenous adenosine acting at A1 receptors. J. Neurosci. 30, 545–555. doi: 10.1523/JNEUROSCI.4920-09.2010
Ishikawa, A., and Nakamura, S. (2006). Ventral hippocampal neurons project axons simultaneously to the medial prefrontal cortex and amygdala in the rat. J. Neurophysiol. 96, 2134–2138. doi: 10.1152/jn.00069.2006
Izquierdo, I., Furini, C. R., and Myskiw, J. C. (2016). Fear memory. Physiol. Rev. 96, 695–750. doi: 10.1152/physrev.00018.2015
Jacob, T. C., Moss, S. J., and Jurd, R. (2008). GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343. doi: 10.1038/nrn2370
Jiang, C., Wang, H., Qi, J., Li, J., He, Q., Wang, C., et al. (2022). Antidepressant effects of cherry leaf decoction on a chronic unpredictable mild stress rat model based on the Glu/GABA-Gln metabolic loop. Metab. Brain Dis. 37, 2883–2901. doi: 10.1007/s11011-022-01081-7
Kamprath, K., Marsicano, G., Tang, J., Monory, K., Bisogno, T., Di Marzo, V., et al. (2006). Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J. Neurosci. 26, 6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006
Karam, E. G., Friedman, M. J., Hill, E. D., Kessler, R. C., McLaughlin, K. A., Petukhova, M., et al. (2014). Cumulative traumas and risk thresholds: 12-month PTSD in the World Mental Health (WMH) surveys. Depress. Anxiety 31, 130–142. doi: 10.1002/da.22169
Karolewicz, B., Maciag, D., O’Dwyer, G., Stockmeier, C. A., Feyissa, A. M., and Rajkowska, G. (2010). Reduced level of glutamic acid decarboxylase-67 kDa in the prefrontal cortex in major depression. Int. J. Neuropsychopharmacol. 13, 411–420. doi: 10.1017/S1461145709990587
Kato, M., and Serretti, A. (2010). Review and meta-analysis of antidepressant pharmacogenetic fifindings in major depressive disorder. Mol. Psychiatry 15, 473–500. doi: 10.1038/mp.2008.116
Khan, S., and Liberzon, I. (2004). Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology 172, 225–229. doi: 10.1007/s00213-003-1634-4
Klempan, T. A., Sequeira, A., Canetti, L., Lalovic, A., Ernst, C., ffrench-Mullen, J., et al. (2009). Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol. Psychiatry 14, 175–189. doi: 10.1038/sj.mp.4002110
Koch, M., and Schnitzler, H. U. (1997). The acoustic startle response in rats–circuits mediating evocation, inhibition and potentiation. Behav. Brain Res. 89, 35–49. doi: 10.1016/s0166-4328(97)02296-1
Koenigs, M., and Grafman, J. (2009). Posttraumatic stress disorder: The role of medial prefrontal cortex and amygdala. Neuroscientist 15, 540–548.
Kohda, K., Harada, K., Kato, K., Hoshino, A., Motohashi, J., Yamaji, T., et al. (2007). Glucocorticoid receptor activation is involved in producing abnormal phenotypes of single-prolonged stress rats: A putative post-traumatic stress disorder model. Neuroscience 148, 22–33. doi: 10.1016/j.neuroscience.2007.05.041
Krystal, A. D., Zhang, W., Davidson, J. R., and Connor, K. M. (2014). The sleep effects of tiagabine on the first night of treatment predict post-traumatic stress disorder response at three weeks. J. Psychopharmacol. (Oxford, England) 28, 457–465. doi: 10.1177/0269881113509903
Krystal, J. H., Sanacora, G., Blumberg, H., Anand, A., Charney, D. S., Marek, G., et al. (2002). Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol. Psychiatry 7 Suppl 1, S71–S80. doi: 10.1038/sj.mp.4001021
Kyd, R. J., and Bilkey, D. K. (2005). Hippocampal place cells show increased sensitivity to changes in the local environment following prefrontal cortex lesions. Cereb. Cortex (New York, N.Y. : 1991) 15, 720–731. doi: 10.1093/cercor/bhh173
Laaris, N., Good, C. H., and Lupica, C. R. (2010). Delta9-tetrahydrocannabinol is a full agonist at CB1 receptors on GABA neuron axon terminals in the hippocampus. Neuropharmacology 59, 121–127. doi: 10.1016/j.neuropharm.2010.04.013
Lafenêtre, P., Chaouloff, F., and Marsicano, G. (2007). The endocannabinoid system in the processing of anxiety and fear and how CB1 receptors may modulate fear extinction. Pharmacol. Res. 56, 367–381. doi: 10.1016/j.phrs.2007.09.006
Lambert, J. J., Belelli, D., Hill-Venning, C., and Peters, J. A. (1995). Neurosteroids and GABAA receptor function. Trends Pharmacol. Sci. 16, 295–303. doi: 10.1016/s0165-6147(00)89058-6
Lapin, I. (2001). Phenibut (beta-phenyl-GABA): A tranquilizer and nootropic drug. CNS Drug Rev. 7, 471–481. doi: 10.1111/j.1527-3458.2001.tb00211.x
LeDoux, J. E. (2000). Emotion circuits in the brain. Annu. Rev. Neurosci. 23, 155–184. doi: 10.1146/annurev.neuro.23.1.155
LeDoux, J. E., Iwata, J., Cicchetti, P., and Reis, D. J. (1988). Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J. Neurosci. 8, 2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988
Lehner, M., Wisłowska-Stanek, A., Skórzewska, A., Maciejak, P., Szyndler, J., Turzyńska, D., et al. (2010). Differences in the density of GABA-A receptor alpha-2 subunits and gephyrin in brain structures of rats selected for low and high anxiety in basal and fear-stimulated conditions, in a model of contextual fear conditioning. Neurobiol. Learn. Mem. 94, 499–508. doi: 10.1016/j.nlm.2010.09.001
Levinson, A. J., Fitzgerald, P. B., Favalli, G., Blumberger, D. M., Daigle, M., and Daskalakis, Z. J. (2010). Evidence of cortical inhibitory deficits in major depressive disorder. Biol. Psychiatry 67, 458–464. doi: 10.1016/j.biopsych.2009.09.025
Liberzon, I., and Abelson, J. L. (2016). Context processing and the neurobiology of post-traumatic stress disorder. Neuron 92, 14–30. doi: 10.1016/j.neuron.2016.09.039
Lin, H. C., Tseng, Y. C., Mao, S. C., Chen, P. S., and Gean, P. W. (2011). GABAA receptor endocytosis in the basolateral amygdala is critical to the reinstatement of fear memory measured by fear-potentiated startle. J. Neurosci. 31, 8851–8861. doi: 10.1523/JNEUROSCI.0979-11.2011
Lippa, A. S., Klepner, C. A., Yunger, L., Sano, M. C., Smith, W. V., and Beer, B. (1978). Relationship between benzodiazepine receptors and experimental anxiety in rats. Pharmacol. Biochem. Behav. 9, 853–856. doi: 10.1016/0091-3057(78)90368-4
Lisboa, S. F., Resstel, L. B., Aguiar, D. C., and Guimarães, F. S. (2008). Activation of cannabinoid CB1 receptors in the dorsolateral periaqueductal gray induces anxiolytic effects in rats submitted to the Vogel conflict test. Eur. J. Pharmacol. 593, 73–78. doi: 10.1016/j.ejphar.2008.07.032
Lissek, S., and van Meurs, B. (2015). Learning models of PTSD: Theoretical accounts and psychobiological evidence. Int. J. Psychophysiol. 98(3 Pt 2), 594–605. doi: 10.1016/j.ijpsycho.2014.11.006
Liu, G. X., Cai, G. Q., Cai, Y. Q., Sheng, Z. J., Jiang, J., Mei, Z., et al. (2007). Reduced anxiety and depression-like behaviors in mice lacking GABA transporter subtype 1. Neuropsychopharmacology 32, 1531–1539. doi: 10.1038/sj.npp.1301281
Liu, L., Wang, L., Cao, C., Cao, X., Zhu, Y., Liu, P., et al. (2018). Serotonin transporter 5-HTTLPR genotype is associated with intrusion and avoidance symptoms of DSM-5 posttraumatic stress disorder (PTSD) in Chinese earthquake survivors. Anxiety Stress Coping 31, 318–327. doi: 10.1080/10615806.2017.1420174
Liu, Z. P., He, Q. H., Pan, H. Q., Xu, X. B., Chen, W. B., He, Y., et al. (2017). Delta subunit-containing gamma-aminobutyric acid a receptor disinhibits lateral amygdala and facilitates fear expression in mice. Biol. Psychiatry 81, 990–1002. doi: 10.1016/j.biopsych.2016.06.022
Lu, C. Y., Liu, X., Jiang, H., Pan, F., Ho, C. S., and Ho, R. C. (2017). Effects of traumatic stress induced in the juvenile period on the expression of gamma-aminobutyric acid receptor type a subunits in adult rat brain. Neural Plast. 2017:5715816. doi: 10.1155/2017/5715816
Lujan, R., and Ciruela, F. (2012). GABAB receptors-associated proteins: Potential drug targets in neurological disorders? Curr. Drug Targets 13, 129–144. doi: 10.2174/138945012798868425
Maddox, S. A., Hartmann, J., Ross, R. A., and Ressler, K. J. (2019). Deconstructing the gestalt: Mechanisms of fear, threat, and trauma memory encoding. Neuron 102, 60–74. doi: 10.1016/j.neuron.2019.03.017
Maercker, A. (2021). Development of the new CPTSD diagnosis for ICD-11. Borderline Pers. Disord. Emot. Dysregul. 8:7. doi: 10.1186/s40479-021-00148-8
Maggio, N., and Segal, M. (2007). Striking variations in corticosteroid modulation of long-term potentiation along the septotemporal axis of the hippocampus. J. Neurosci. 27, 5757–5765. doi: 10.1523/JNEUROSCI.0155-07.2007
Maguire, J. L., Stell, B. M., Rafizadeh, M., and Mody, I. (2005). Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat. Neurosci. 8, 797–804. doi: 10.1038/nn1469
Mahan, A. L., and Ressler, K. J. (2012). Fear conditioning, synaptic plasticity and the amygdala: Implications for posttraumatic stress disorder. Trends Neurosci. 35, 24–35. doi: 10.1016/j.tins.2011.06.007
Makkar, S. R., Zhang, S. Q., and Cranney, J. (2010). Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology 35, 1625–1652. doi: 10.1038/npp.2010.53
Mao, C. C., Guidotti, A., and Costa, E. (1975). Evidence for an involvement of GABA in the mediation of the cerebellar cGMP decrease and the anticonvulsant action diazepam. Naunyn Schmiedebergs Arch. Pharmacol. 289, 369–378. doi: 10.1007/BF00508411
Maren, S., and Quirk, G. J. (2004). Neuronal signalling of fear memory. Nat. Rev. Neurosci. 5, 844–852. doi: 10.1038/nrn1535
Maren, S., Yap, S. A., and Goosens, K. A. (2001). The amygdala is essential for the development of neuronal plasticity in the medial geniculate nucleus during auditory fear conditioning in rats. J. Neurosci. 21:RC135. doi: 10.1523/JNEUROSCI.21-06-j0001.2001
Marquis, J. P., Killcross, S., and Haddon, J. E. (2007). Inactivation of the prelimbic, but not infralimbic, prefrontal cortex impairs the contextual control of response conflict in rats. Eur. J. Neurosci. 25, 559–566. doi: 10.1111/j.1460-9568.2006.05295.x
Marsicano, G., Wotjak, C. T., Azad, S. C., Bisogno, T., Rammes, G., Cascio, M. G., et al. (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534. doi: 10.1038/nature00839
Martin, M., Ledent, C., Parmentier, M., Maldonado, R., and Valverde, O. (2002). Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology 159, 379–387. doi: 10.1007/s00213-001-0946-5
Matar, M. A., Zohar, J., Kaplan, Z., and Cohen, H. (2009). Alprazolam treatment immediately after stress exposure interferes with the normal HPA-stress response and increases vulnerability to subsequent stress in an animal model of PTSD. Eur. Neuropsychopharmacol. 19, 283–295. doi: 10.1016/j.euroneuro.2008.12.004
McDonald, A. J. (1982). Neurons of the lateral and basolateral amygdaloid nuclei: A Golgi study in the rat. J. Comp. Neurol. 212, 293–312. doi: 10.1002/cne.902120307
McDonald, A. J., and Augustine, J. R. (1993). Localization of GABA-like immunoreactivity in the monkey amygdala. Neuroscience 52, 281–294. doi: 10.1016/0306-4522(93)90156-a
McLaughlin, R. J., Hill, M. N., and Gorzalka, B. B. (2014). A critical role for prefrontocortical endocannabinoid signaling in the regulation of stress and emotional behavior. Neurosci. Biobehav. Rev. 42, 116–131. doi: 10.1016/j.neubiorev.2014.02.006
Mechoulam, R., and Parker, L. A. (2013). The endocannabinoid system and the brain. Annu. Rev. Psychol. 64, 21–47. doi: 10.1146/annurev-psych-113011-143739
Medina, J. H., Novas, M. L., Wolfman, C. N., Levi de Stein, M., and De Robertis, E. (1983). Benzodiazepine receptors in rat cerebral cortex and hippocampus undergo rapid and reversible changes after acute stress. Neuroscience 9, 331–335. doi: 10.1016/0306-4522(83)90298-1
Megahed, T., Hattiangady, B., Shuai, B., and Shetty, A. K. (2015). Parvalbumin and neuropeptide Y expressing hippocampal GABA-ergic inhibitory interneuron numbers decline in a model of Gulf War illness. Front. Cell. Neurosci. 8:447. doi: 10.3389/fncel.2014.00447
Meiser-Stedman, R., Dalgleish, T., Glucksman, E., Yule, W., and Smith, P. (2009). Maladaptive cognitive appraisals mediate the evolution of posttraumatic stress reactions: A 6-month follow-up of child and adolescent assault and motor vehicle accident survivors. J. Abnorm. Psychol. 118, 778–787. doi: 10.1037/a0016945
Merali, Z., Du, L., Hrdina, P., Palkovits, M., Faludi, G., Poulter, M. O., et al. (2004). Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J. Neurosci. 24, 1478–1485. doi: 10.1523/JNEUROSCI.4734-03.2004
Meyerhoff, D. J., Mon, A., Metzler, T., and Neylan, T. C. (2014). Cortical gamma-aminobutyric acid and glutamate in posttraumatic stress disorder and their relationships to self-reported sleep quality. Sleep 37, 893–900. doi: 10.5665/sleep.3654
Michels, L., Schulte-Vels, T., Schick, M., O’Gorman, R. L., Zeffiro, T., Hasler, G., et al. (2014). Prefrontal GABA and glutathione imbalance in posttraumatic stress disorder: Preliminary findings. Psychiatry Res. 224, 288–295. doi: 10.1016/j.pscychresns.2014.09.007
Mikkelsen, J. D., Bundzikova, J., Larsen, M. H., Hansen, H. H., and Kiss, A. (2008). GABA regulates the rat hypothalamic-pituitary-adrenocortical axis via different GABA-A receptor alpha-subtypes. Ann. N. Y. Acad. Sci. 1148, 384–392. doi: 10.1196/annals.1410.044
Milad, M. R., Pitman, R. K., Ellis, C. B., Gold, A. L., Shin, L. M., Lasko, N. B., et al. (2009). Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol. Psychiatry 66, 1075–1082. doi: 10.1016/j.biopsych.2009.06.026
Möller, A. T., Bäckström, T., Nyberg, S., Söndergaard, H. P., and Helström, L. (2016). Women with PTSD have a changed sensitivity to GABA-A receptor active substances. Psychopharmacology 233, 2025–2033. doi: 10.1007/s00213-014-3776-y
Morales, M., Wang, S. D., Diaz-Ruiz, O., and Jho, D. H. (2004). Cannabinoid CB1 receptor and serotonin 3 receptor subunit A (5-HT3A) are co-expressed in GABA neurons in the rat telencephalon. J. Comp. Neurol. 468, 205–216. doi: 10.1002/cne.10968
Morgan, M. A., and LeDoux, J. E. (1995). Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav. Neurosci. 109, 681–688. doi: 10.1037//0735-7044.109.4.681
Neumeister, A., Normandin, M. D., Pietrzak, R. H., Piomelli, D., Zheng, M. Q., Gujarro-Anton, A., et al. (2013). Elevated brain cannabinoid CB1 receptor availability in post-traumatic stress disorder: a positron emission tomography study. Mol. Psychiatry 18, 1034–1040. doi: 10.1038/mp.2013.61
Neuwirth, L. S., Verrengia, M. T., Harikinish-Murrary, Z. I., Orens, J. E., and Lopez, O. E. (2022). Under or absent reporting of light stimuli in testing of anxiety-like behaviors in rodents: the need for standardization. Front. Mol. Neurosci. 15:912146. doi: 10.3389/fnmol.2022.912146
Olson, V. G., Rockett, H. R., Reh, R. K., Redila, V. A., Tran, P. M., Venkov, H. A., et al. (2011). The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol. Psychiatry 70, 441–448. doi: 10.1016/j.biopsych.2010.11.029
Orchinik, M., Weiland, N. G., and McEwen, B. S. (1995). Chronic exposure to stress levels of corticosterone alters GABAA receptor subunit mRNA levels in rat hippocampus. Brain Res. Mol. Brain Res. 34, 29–37. doi: 10.1016/0169-328x(95)00118-c
Orr, S. P., Lasko, N. B., Metzger, L. J., and Pitman, R. K. (1997). Physiologic responses to non-startling tones in Vietnam veterans with post-traumatic stress disorder. Psychiatry Res. 73, 103–107. doi: 10.1016/s0165-1781(97)00110-8
Orr, S. P., Metzger, L. J., Lasko, N. B., Macklin, M. L., Hu, F. B., Shalev, A. Y., et al. (2003). Physiologic responses to sudden, loud tones in monozygotic twins discordant for combat exposure: Association with posttraumatic stress disorder. Arch. Gene. Psychiatry 60, 283–288. doi: 10.1001/archpsyc.60.3.283
Ouellet-Morin, I., Odgers, C. L., Danese, A., Bowes, L., Shakoor, S., Papadopoulos, A. S., et al. (2011). Blunted cortisol responses to stress signal social and behavioral problems among maltreated/bullied 12-year-old children. Biol. Psychiatry 70, 1016–1023. doi: 10.1016/j.biopsych.2011.06.017
Pape, H. C., and Pare, D. (2010). Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol. Rev. 90, 419–463. doi: 10.1152/physrev.00037.2009
Parsons, R., and Ressler, K. (2013). Implications of memory modulation for post-traumatic stress and fear disorders. Nat. Neurosci. 16, 146–153. doi: 10.1038/nn.3296
Passie, T., Emrich, H. M., Karst, M., Brandt, S. D., and Halpern, J. H. (2012). Mitigation of post-traumatic stress symptoms by Cannabis resin: A review of the clinical and neurobiological evidence. Drug Test. Anal. 4, 649–659. doi: 10.1002/dta.1377
Petroff, O. A. (2002). GABA and glutamate in the human brain. Neuroscientist 8, 562–573. doi: 10.1177/1073858402238515
Pham, X., Sun, C., Chen, X., van den Oord, E. J., Neale, M. C., Kendler, K. S., et al. (2009). Association study between GABA receptor genes and anxiety spectrum disorders. Depression Anxiety 26, 998–1003. doi: 10.1002/da.20628
Piomelli, D. (2003). The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4, 873–884. doi: 10.1038/nrn1247
Pitman, R. K., Rasmusson, A. M., Koenen, K. C., Shin, L. M., Orr, S. P., Gilbertson, M. W., et al. (2012). Biological studies of post-traumatic stress disorder. Nat. Rev. Neurosci. 13, 769–787. doi: 10.1038/nrn3339
Pochwat, B., Nowak, G., and Szewczyk, B. (2016). Brain glutamic acid decarboxylase-67kDa alterations induced by magnesium treatment in olfactory bulbectomy and chronic mild stress models in rats. Pharmacol. Rep. 68, 881–885. doi: 10.1016/j.pharep.2016.04.011
Pole, N. (2007). The psychophysiology of posttraumatic stress disorder: A meta-analysis. Psychol. Bull. 133, 725–746. doi: 10.1037/0033-2909.133.5.725
Pole, N., Neylan, T. C., Otte, C., Henn-Hasse, C., Metzler, T. J., and Marmar, C. R. (2009). Prospective prediction of posttraumatic stress disorder symptoms using fear potentiated auditory startle responses. Biol. Psychiatry 65, 235–240. doi: 10.1016/j.biopsych.2008.07.015
Pollack, M. H., Jensen, J. E., Simon, N. M., Kaufman, R. E., and Renshaw, P. F. (2008). High-field MRS study of GABA, glutamate and glutamine in social anxiety disorder: Response to treatment with levetiracetam. Prog. Neuro Psychopharmacol. Biol. Psychiatry 32, 739–743. doi: 10.1016/j.pnpbp.2007.11.023
Poulter, M. O., Du, L., Weaver, I., Palkovits, M., Faludi, G., Merali, Z., et al. (2008). GABAA receptor promoter hypermethylation in suicide brain: Implications for the involvement of epigenetic processes. Biol. Psychiatry 64, 645–652. doi: 10.1016/j.biopsych.2008.05.028
Quirk, G. J., Russo, G. K., Barron, J. L., and Lebron, K. (2000). The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J. Neurosci. 20, 6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000
Randall, P. K., Bremner, J. D., Krystal, J. H., Nagy, L. M., Heninger, G. R., Nicolaou, A. L., et al. (1995). Effects of the benzodiazepine antagonist flumazenil in PTSD. Biol. Psychiatry 38, 319–324. doi: 10.1016/0006-3223(94)00306-n
Rasmusson, A. M., and Pineles, S. L. (2018). Neurotransmitter, peptide, and steroid hormone abnormalities in PTSD: Biological endophenotypes relevant to treatment. Curr. Psychiatry Rep. 20:52. doi: 10.1007/s11920-018-0908-9
Rasmusson, A. M., Pinna, G., Paliwal, P., Weisman, D., Gottschalk, C., Charney, D., et al. (2006). Decreased cerebrospinal fluid allopregnanolone levels in women with posttraumatic stress disorder. Biol. Psychiatry 60, 704–713. doi: 10.1016/j.biopsych.2006.03.026
Rauch, S. L., Shin, L. M., and Phelps, E. A. (2006). Neurocircuitry models of posttraumatic stress disorder and extinction: Human neuroimaging research–Past, present, and future. Biol. Psychiatry 60, 376–382. doi: 10.1016/j.biopsych.2006.06.004
Raybuck, J. D., and Lattal, K. M. (2011). Double dissociation of amygdala and hippocampal contributions to trace and delay fear conditioning. PLoS One 6:e15982. doi: 10.1371/journal.pone.0015982
Raybuck, J. D., and Lattal, K. M. (2014). Differential effects of dorsal hippocampal inactivation on expression of recent and remote drug and fear memory. Neurosci. Lett. 569, 1–5. doi: 10.1016/j.neulet.2014.02.063
Romeo, E., Ströhle, A., Spalletta, G., di Michele, F., Hermann, B., Holsboer, F., et al. (1998). Effects of antidepressant treatment on neuroactive steroids in major depression. Am. J. Psychiatry 155, 910–913. doi: 10.1176/ajp.155.7.910
Roohbakhsh, A., Keshavarz, S., Hasanein, P., Rezvani, M. E., and Moghaddam, A. H. (2009). Role of endocannabinoid system in the ventral hippocampus of rats in the modulation of anxiety-like behaviours. Basic Clin. Pharmacol. Toxicol. 105, 333–338. doi: 10.1111/j.1742-7843.2009.00449.x
Rosso, I. M., Weiner, M. R., Crowley, D. J., Silveri, M. M., Rauch, S. L., and Jensen, J. E. (2014). Insula and anterior cingulate GABA levels in posttraumatic stress disorder: Preliminary findings using magnetic resonance spectroscopy. Depression Anxiety 31, 115–123. doi: 10.1002/da.22155
Rudolph, U., and Möhler, H. (2004). Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu. Rev. Pharmacol. Toxicol. 44, 475–498. doi: 10.1146/annurev.pharmtox.44.101802.121429
Rudolph, U., Crestani, F., Benke, D., Brünig, I., Benson, J. A., Fritschy, J. M., et al. (1999). Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 401, 796–800. doi: 10.1038/44579
Ruehle, S., Rey, A. A., Remmers, F., and Lutz, B. (2012). The endocannabinoid system in anxiety, fear memory and habituation. J. Psychopharmacol. (Oxford, England) 26, 23–39. doi: 10.1177/0269881111408958
Sah, P., Faber, E. S., Lopez De Armentia, M., and Power, J. (2003). The amygdaloid complex: Anatomy and physiology. Physiol. Rev. 83, 803–834. doi: 10.1152/physrev.00002.2003
Sah, P., Westbrook, R. F., and Lüthi, A. (2008). Fear conditioning and long-term potentiation in the amygdala: What really is the connection? Ann. N. Y. Acad. Sci. 1129, 88–95. doi: 10.1196/annals.1417.020
Sakaguchi, M., Kim, K., Yu, L. M., Hashikawa, Y., Sekine, Y., Okumura, Y., et al. (2015). Inhibiting the activity of CA1 hippocampal neurons prevents the recall of contextual fear memory in inducible ArchT transgenic mice. PLoS One 10:e0130163. doi: 10.1371/journal.pone.0130163
Sanacora, G., Gueorguieva, R., Epperson, C. N., Wu, Y. T., Appel, M., Rothman, D. L., et al. (2004). Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch. Gen. Psychiatry 61, 705–713. doi: 10.1001/archpsyc.61.7.705
Sangha, S., Narayanan, R. T., Bergado-Acosta, J. R., Stork, O., Seidenbecher, T., and Pape, H. C. (2009). Deficiency of the 65 kDa isoform of glutamic acid decarboxylase impairs extinction of cued but not contextual fear memory. J. Neurosci. 29, 15713–15720. doi: 10.1523/JNEUROSCI.2620-09.2009
Schneider, B. L., Ghoddoussi, F., Charlton, J. L., Kohler, R. J., Galloway, M. P., Perrine, S. A., et al. (2016). Increased cortical gamma-aminobutyric acid precedes incomplete extinction of conditioned fear and increased hippocampal excitatory tone in a mouse model of mild traumatic brain injury. J. Neurotrauma 33, 1614–1624. doi: 10.1089/neu.2015.4190
Schüle, C., Nothdurfter, C., and Rupprecht, R. (2014). The role of allopregnanolone in depression and anxiety. Prog. Neurobiol. 113, 79–87. doi: 10.1016/j.pneurobio.2013.09.003
Schwartz, T. L., and Nihalani, N. (2006). Tiagabine in anxiety disorders. Expert Opin. Pharmacother. 7, 1977–1987. doi: 10.1517/14656566.7.14.1977
Sequeira, A., Mamdani, F., Ernst, C., Vawter, M. P., Bunney, W. E., Lebel, V., et al. (2009). Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS One 4:e6585. doi: 10.1371/journal.pone.0006585
Sheth, C., Prescot, A. P., Legarreta, M., Renshaw, P. F., McGlade, E., and Yurgelun-Todd, D. (2019). Reduced gamma-amino butyric acid (GABA) and glutamine in the anterior cingulate cortex (ACC) of veterans exposed to trauma. J. Affect. Disord. 248, 166–174. doi: 10.1016/j.jad.2019.01.037
Shin, L. M., and Liberzon, I. (2010). The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology 35, 169–191. doi: 10.1038/npp.2009.83
Sierra-Mercado, D., Padilla-Coreano, N., and Quirk, G. J. (2011). Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36, 529–538. doi: 10.1038/npp.2010.184
Sigurdsson, T., Doyère, V., Cain, C. K., and LeDoux, J. E. (2007). Long-term potentiation in the amygdala: A cellular mechanism of fear learning and memory. Neuropharmacology 52, 215–227. doi: 10.1016/j.neuropharm.2006.06.022
Singewald, N., Schmuckermair, C., Whittle, N., Holmes, A., and Ressler, K. J. (2015). Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol. Ther. 149, 150–190. doi: 10.1016/j.pharmthera.2014.12.004
Smoller, J. (2016). The genetics of stress-related disorders: PTSD, depression, and anxiety disorders. Neuropsychopharmacology 41, 297–319. doi: 10.1038/npp.2015.266
Song, C., Zhang, W. H., Wang, X. H., Zhang, J. Y., Tian, X. L., Yin, X. P., et al. (2017). Acute stress enhances the glutamatergic transmission onto basoamygdala neurons embedded in distinct microcircuits. Mol. Brain 10:3. doi: 10.1186/s13041-016-0283-6
Spivak, B., Maayan, R., Kotler, M., Mester, R., Gil-Ad, I., Shtaif, B., et al. (2000). Elevated circulatory level of GABA(A)–antagonistic neurosteroids in patients with combat-related post-traumatic stress disorder. Psychol. Med. 30, 1227–1231. doi: 10.1017/s0033291799002731
Stachowicz, K. (2018). The role of DSCAM in the regulation of synaptic plasticity: Possible involvement in neuropsychiatric disorders. Acta Neurobiol. Exp. 78, 210–219.
Suendermann, O., Ehlers, A., Boellinghaus, I., Gamer, M., and Glucksman, E. (2010). Early heart rate responses to standardized trauma-related pictures predict posttraumatic stress disorder: A prospective study. Psychosom. Med. 72, 301–308. doi: 10.1097/PSY.0b013e3181d07db8
Sun, K., Fan, J., and Han, J. (2015). Ameliorating effects of traditional Chinese medicine preparation, Chinese materia medica and active compounds on ischemia/reperfusion-induced cerebral microcirculatory disturbances and neuron damage. Acta Pharm. Sin. B 5, 8–24. doi: 10.1016/j.apsb.2014.11.002
Sun, X., Song, Z., Si, Y., and Wang, J. H. (2018). microRNA and mRNA profiles in ventral tegmental area relevant to stress-induced depression and resilience. Prog. Neuro Psychopharmacol. Biol. Psychiatry 86, 150–165. doi: 10.1016/j.pnpbp.2018.05.023
Sutherland, S. M., and Davidson, J. R. (1994). Pharmacotherapy for post-traumatic stress disorder. Psychiatric Clin. North Am. 17, 409–423.
Szabó, G. G., Lenkey, N., Holderith, N., Andrási, T., Nusser, Z., and Hájos, N. (2014). Presynaptic calcium channel inhibition underlies CB1 cannabinoid receptor-mediated suppression of GABA release. J. Neurosci. 34, 7958–7963. doi: 10.1523/JNEUROSCI.0247-14.2014
Taylor, F. B. (2003). Tiagabine for posttraumatic stress disorder: A case series of 7 women. J. Clin. Psychiatry 64, 1421–1425. doi: 10.4088/jcp.v64n1204
Tovote, P., Fadok, J. P., and Lüthi, A. (2015). Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 16, 317–331. doi: 10.1038/nrn3945
Trezza, V., and Campolongo, P. (2013). The endocannabinoid system as a possible target to treat both the cognitive and emotional features of post-traumatic stress disorder (PTSD). Front. Behav. Neurosci. 7:100. doi: 10.3389/fnbeh.2013.00100
Twining, R. C., Lepak, K., Kirry, A. J., and Gilmartin, M. R. (2020). Ventral hippocampal input to the prelimbic cortex dissociates the context from the cue association in trace fear memory. J. Neurosci. 40, 3217–3230. doi: 10.1523/JNEUROSCI.1453-19.2020
Uniyal, A., Singh, R., Akhtar, A., Bansal, Y., Kuhad, A., and Sah, S. P. (2019). Co-treatment of piracetam with risperidone rescued extinction deficits in experimental paradigms of post-traumatic stress disorder by restoring the physiological alterations in cortex and hippocampus. Pharmacol. Biochem. Behav. 185:172763. doi: 10.1016/j.pbb.2019.172763
Uzunova, V., Sheline, Y., Davis, J. M., Rasmusson, A., Uzunov, D. P., Costa, E., et al. (1998). Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc. Natl. Acad. Sci. U.S.A. 95, 3239–3244. doi: 10.1073/pnas.95.6.3239
Vaiva, G., Boss, V., Ducrocq, F., Fontaine, M., Devos, P., Brunet, A., et al. (2006). Relationship between posttrauma GABA plasma levels and PTSD at 1-year follow-up. Am. J. Psychiatry 163, 1446–1448. doi: 10.1176/ajp.2006.163.8.1446
Vaiva, G., Thomas, P., Ducrocq, F., Fontaine, M., Boss, V., Devos, P., et al. (2004). Low posttrauma GABA plasma levels as a predictive factor in the development of acute posttraumatic stress disorder. Biol. Psychiatry 55, 250–254. doi: 10.1016/j.biopsych.2003.08.009
Valentinuzzi, V. S., Kolker, D. E., Vitaterna, M. H., Shimomura, K., Whiteley, A., Low-Zeddies, S., et al. (1998). Automated measurement of mouse freezing behavior and its use for quantitative trait locus analysis of contextual fear conditioning in (BALB/cJ x C57BL/6J)F2 mice. Learn. Mem. (Cold Spring Harbor, N.Y.) 5, 391–403.
Vasterling, J., and Brewin, C. R. (2005). Neuropsychology of PTSD : Biological, cognitive, and clinical perspectives. New York, NY: Guilford Press.
Verkuyl, J. M., and Joëls, M. (2003). Effect of adrenalectomy on miniature inhibitory postsynaptic currents in the paraventricular nucleus of the hypothalamus. J. Neurophysiol. 89, 237–245. doi: 10.1152/jn.00401.2002
Verkuyl, J. M., Karst, H., and Joëls, M. (2005). GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur. J. Neurosci. 21, 113–121. doi: 10.1111/j.1460-9568.2004.03846.x
Viola, J., Ditzler, T., Batzer, W., Harazin, J., Adams, D., Lettich, L., et al. (1997). Pharmacological management of post-traumatic stress disorder: clinical summary of a five-year retrospective study, 1990-1995. Military Med. 162, 616–619.
Wilensky, A. E., Schafe, G. E., Kristensen, M. P., and LeDoux, J. E. (2006). Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J. Neurosci. 26, 12387–12396. doi: 10.1523/JNEUROSCI.4316-06.2006
Wilson, J., and Keane, T. M. (1997). Assessing psychological trauma and PTSD. New York, NY: Guilford Press.
Wolfe, J., Chrestman, K. R., Ouimette, P. C., Kaloupek, D., Harley, R. M., and Bucsela, M. (2000). Trauma-related psychophysiological reactivity in women exposed to war-zone stress. J. Clin. Psychol. 56, 1371–1379. doi: 10.1002/1097-4679(200010)56:10<1371::AID-JCLP8<3.0.CO;2-X
Xu, W., and Südhof, T. C. (2013). A neural circuit for memory specificity and generalization. Science (New York, N.Y.) 339, 1290–1295. doi: 10.1126/science.1229534
Zannas, A., Provençal, N., and Binder, E. (2015). Epigenetics of posttraumatic stress disorder: Current evidence, challenges, and future directions. Biol. Psychiatry 78, 327–335. doi: 10.1016/j.biopsych.2015.04.003
Zhang, D., Pan, Z. H., Awobuluyi, M., and Lipton, S. A. (2001). Structure and function of GABA(C) receptors: A comparison of native versus recombinant receptors. Trends Pharmacol. Sci. 22, 121–132. doi: 10.1016/s0165-6147(00)01625-4
Keywords: post-traumatic stress disorder, GABA, fear memory, amygdala, neurotransmitter
Citation: Huang J, Xu F, Yang L, Tuolihong L, Wang X, Du Z, Zhang Y, Yin X, Li Y, Lu K and Wang W (2023) Involvement of the GABAergic system in PTSD and its therapeutic significance. Front. Mol. Neurosci. 16:1052288. doi: 10.3389/fnmol.2023.1052288
Received: 23 September 2022; Accepted: 16 January 2023;
Published: 01 February 2023.
Edited by:
Lorenz S. Neuwirth, State University of New York at Old Westbury, United StatesReviewed by:
Nicole Ferrara, Rosalind Franklin University of Medicine and Science, United StatesCopyright © 2023 Huang, Xu, Yang, Tuolihong, Wang, Du, Zhang, Yin, Li, Lu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kangrong Lu,  MjYxMjY3NDQzQHFxLmNvbQ==; Wanshan Wang, d3dzQHNtdS5lZHUuY24=
MjYxMjY3NDQzQHFxLmNvbQ==; Wanshan Wang, d3dzQHNtdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.