
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci., 06 October 2022
Sec. Neuroplasticity and Development
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.979061
This article is part of the Research TopicMolecular Advances and Applications of Machine Learning in Understanding Autism and Comorbid Psychiatric DisordersView all 16 articles
 Afif Ben-Mahmoud1†
Afif Ben-Mahmoud1† Kyung Ran Jun2†
Kyung Ran Jun2† Vijay Gupta1
Vijay Gupta1 Pinang Shastri3
Pinang Shastri3 Alberto de la Fuente4
Alberto de la Fuente4 Yongsoo Park1
Yongsoo Park1 Kyung Chul Shin1
Kyung Chul Shin1 Chong Ae Kim5
Chong Ae Kim5 Aparecido Divino da Cruz6,7
Aparecido Divino da Cruz6,7 Irene Plaza Pinto6,7
Irene Plaza Pinto6,7 Lysa Bernardes Minasi6,7
Lysa Bernardes Minasi6,7 Alex Silva da Cruz6,7
Alex Silva da Cruz6,7 Laurence Faivre8,9Patrick Callier10
Laurence Faivre8,9Patrick Callier10 Caroline Racine10Lawrence C. Layman11,12
Caroline Racine10Lawrence C. Layman11,12 Il-Keun Kong13
Il-Keun Kong13 Cheol-Hee Kim14
Cheol-Hee Kim14 Woo-Yang Kim15
Woo-Yang Kim15 Hyung-Goo Kim1*
Hyung-Goo Kim1*Genome-wide chromosomal microarray is extensively used to detect copy number variations (CNVs), which can diagnose microdeletion and microduplication syndromes. These small unbalanced chromosomal structural rearrangements ranging from 1 kb to 10 Mb comprise up to 15% of human mutations leading to monogenic or contiguous genomic disorders. Albeit rare, CNVs at 1p13.3 cause a variety of neurodevelopmental disorders (NDDs) including development delay (DD), intellectual disability (ID), autism, epilepsy, and craniofacial anomalies (CFA). Most of the 1p13.3 CNV cases reported in the pre-microarray era encompassed a large number of genes and lacked the demarcating genomic coordinates, hampering the discovery of positional candidate genes within the boundaries. In this study, we present four subjects with 1p13.3 microdeletions displaying DD, ID, autism, epilepsy, and CFA. In silico comparative genomic mapping with three previously reported subjects with CNVs and 22 unreported DECIPHER CNV cases has resulted in the identification of four different sub-genomic loci harboring five positional candidate genes for DD, ID, and CFA at 1p13.3. Most of these genes have pathogenic variants reported, and their interacting genes are involved in NDDs. RT-qPCR in various human tissues revealed a high expression pattern in the brain and fetal brain, supporting their functional roles in NDDs. Interrogation of variant databases and interacting protein partners led to the identification of another set of 11 potential candidate genes, which might have been dysregulated by the position effect of these CNVs at 1p13.3. Our studies define 1p13.3 as a genomic region harboring 16 NDD candidate genes and underscore the critical roles of small CNVs in in silico comparative genomic mapping for disease gene discovery. Our candidate genes will help accelerate the isolation of pathogenic heterozygous variants from exome/genome sequencing (ES/GS) databases.
Chromosomal deletions and duplications, called copy number variations (CNVs), are unbalanced chromosomal structural abnormalities that may result in genomic disorders, the manifestations of which are frequently syndromic neurodevelopmental disorders (NDDs) (Lee et al., 2015). With its genomic length of 249 Mb, chromosome 1 is the largest human chromosome and contains about 2,000 human genes comprising about 9% of the total human genes (Murphy et al., 2003). A large number of genes on this chromosome are involved in mental and physical development, and thus, large heterozygous deletions or duplications can cause congenital abnormalities or pregnancy loss (Chen et al., 2017).
Subjects with CNVs within the proximal short arm of chromosome 1, specifically in the 1p13.3 region, are rare, and only seven cases have been reported so far (Tabata et al., 1991; Mattia et al., 1992; Lo et al., 1998; Utkus et al., 1999; Bisgaard et al., 2007; van Kuilenburg et al., 2009; Piccione et al., 2010). Importantly, these seven subjects share an overlapping phenotype of developmental delay (DD), intellectual disability (ID), epilepsy, and craniofacial anomalies (CFA) (Cases 1–7), suggesting the presence of syndromic NDD disease genes within 1p13.3. Four of them (Cases 4–7) are from the pre-comparative genomic hybridization (CGH) era without demarcating coordinates and thus not very informative (Table 1).
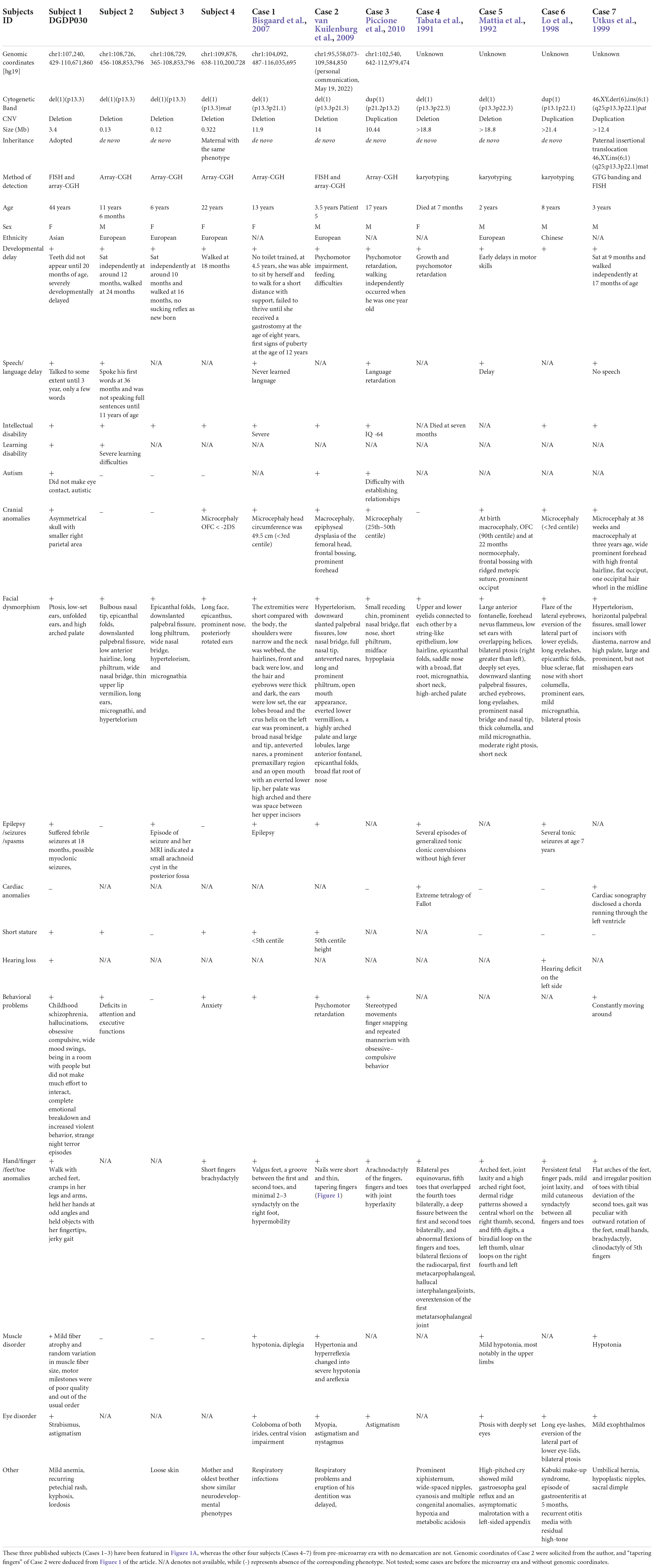
Table 1. Clinical features of eleven CNV subjects with 1p13.3 microdeletion/microduplication. Our four CNV carriers 1–4 are indicated here as Subjects 1–4.
In this study, we describe the clinical phenotype of four subjects having microdeletions of different sizes at 1p13.3 with an overlapping phenotype of DD, ID, autism, epilepsy, and CFA. In silico comparative genomic mapping of these four subjects (Subjects 1–4 on Figure 1A and Table 1) with three reported demarcated deletions and duplication (Cases 1-3 on Table 1 and Figure 1A) alongside twenty-two informative DECIPHER cases1, version 11.14) (Firth et al., 2009; Figure 1A and Supplementary Table 3) has allowed us to narrow down the 1p13.3 genomic region harboring approximately 59 genes to four sub-genomic segments that encompass five positional candidate genes for syndromic NDDs at 1p13.3 (Figure 1A and Table 2). Upon analyzing various human disease databases including Human Gene Mutation Database (HGMD, 2 version Professional 2022.2), MGI (Mouse Genome Informatics3, version 6.21), BioGrid4 (version 4.4.212), and STRING5 (version 11.5), we found that these five candidate genes have either NDD-associated nucleotide variants reported in them or their interactors, and a behavioral phenotype was shown in KO mice (Table 2). A high RNA expression pattern of these five positional candidate genes in human adult and fetal brain further supported this postulate (Figures 2, 3). Individual interrogation of the 54 remaining genes identified another set of 11 candidate genes. Their candidacy was substantiated by sporadic variants reported in them and their interactors, and their physical interaction with known NDD genes based on BioGrid and STRING, as well as KO mice phenotype. (Supplementary Table 1). Disease Gene Network Analysis (DisGeNET6, version 7.0) and HPO (Human Phenotype Ontology7, version 1.7.16) provided additional substantiation of the candidacy of our candidate genes in NDDs.
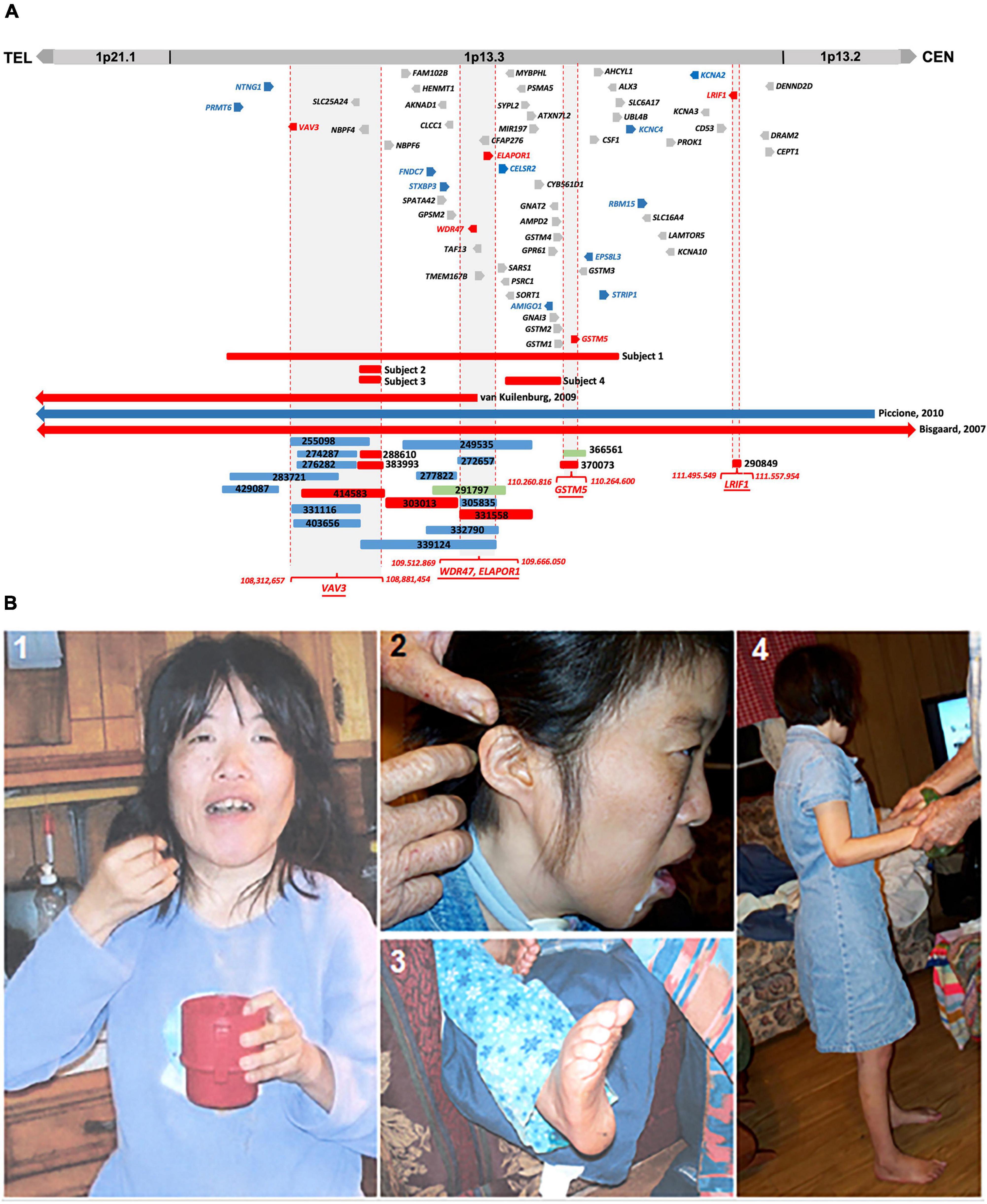
Figure 1. (A) Comparative CNV mapping of subjects with CNVs at 1p13.3. Deletions are represented by red bars and duplications are represented by blue bars. CNV cases from the DECIPHER database are denoted by a six-digit reference number. The sizes of the deleted and duplicated regions from Bisgaard et al. (2007) (Case 1), van Kuilenburg et al. (2009) (Case 2), Piccione et al. (2010) (Case 3), and 22 DECIPHER cases are presented relative to our four subjects. Vertical dotted red lines flanking gray background represent the refined four candidate gene loci; 59 genes located within the 4.6 Mb genomic region at 1p13.3 are depicted either in red for positional candidate genes, blue for functional candidate genes, or gray for the remaining genes. (B) Phenotypic features of Subject 1. (1) Picture shows facial dysmorphism, asymmetrical head, and small hands; (2) Low set, small, and unfolded ears; (3) arched feet; and (4) full body picture showing short stature.

Table 2. Five positional candidate genes for syndromic NDDs identified by in silico CNV mapping at 1p13.3.
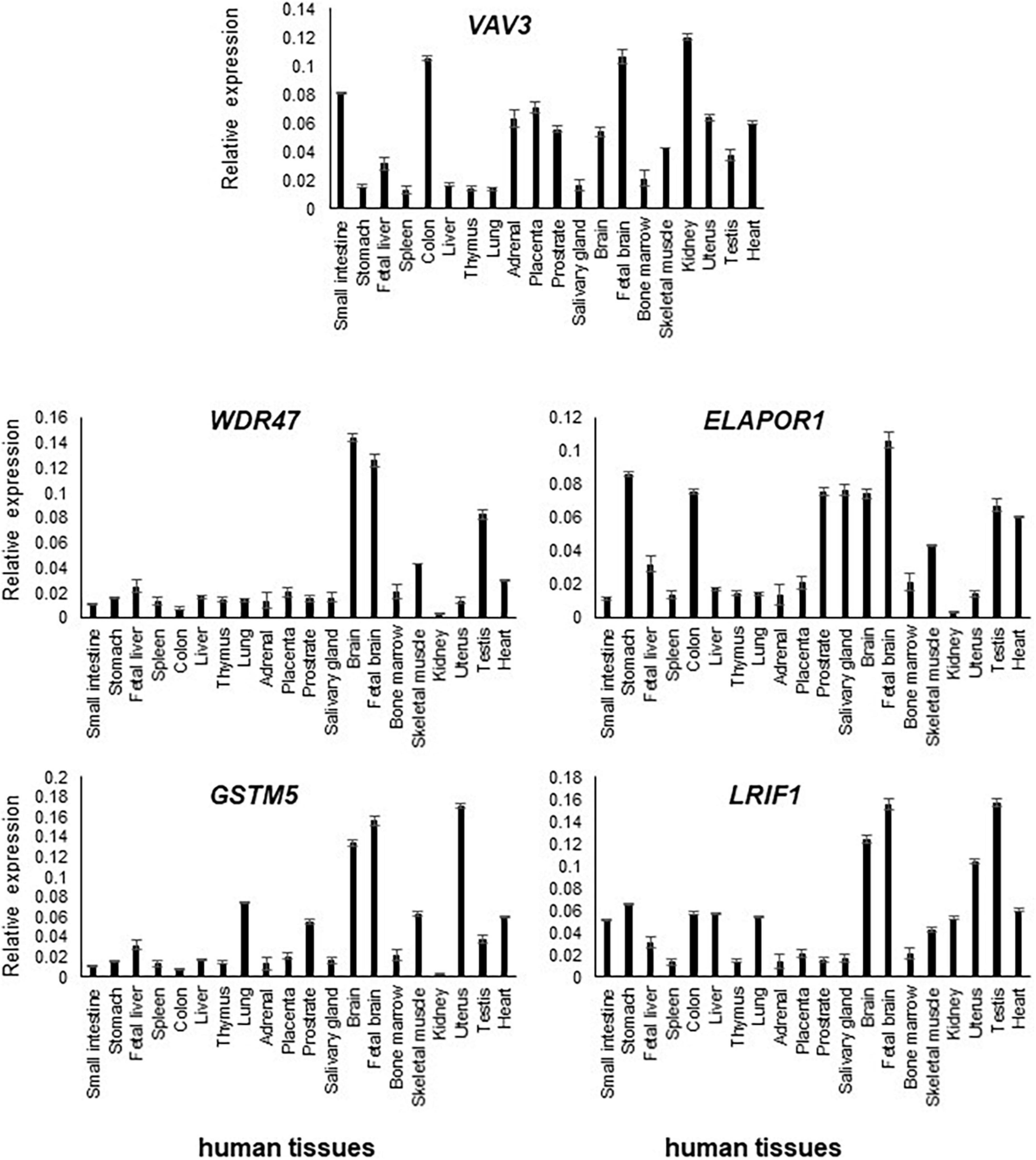
Figure 2. Transcript levels of VAV3, ELAPOR1, WDR47, GSTM5, and LRIF1 in various human tissues were determined by RT-qPCR. Relative high expression of them was detected in the adult and fetal brain compared to other tissues.
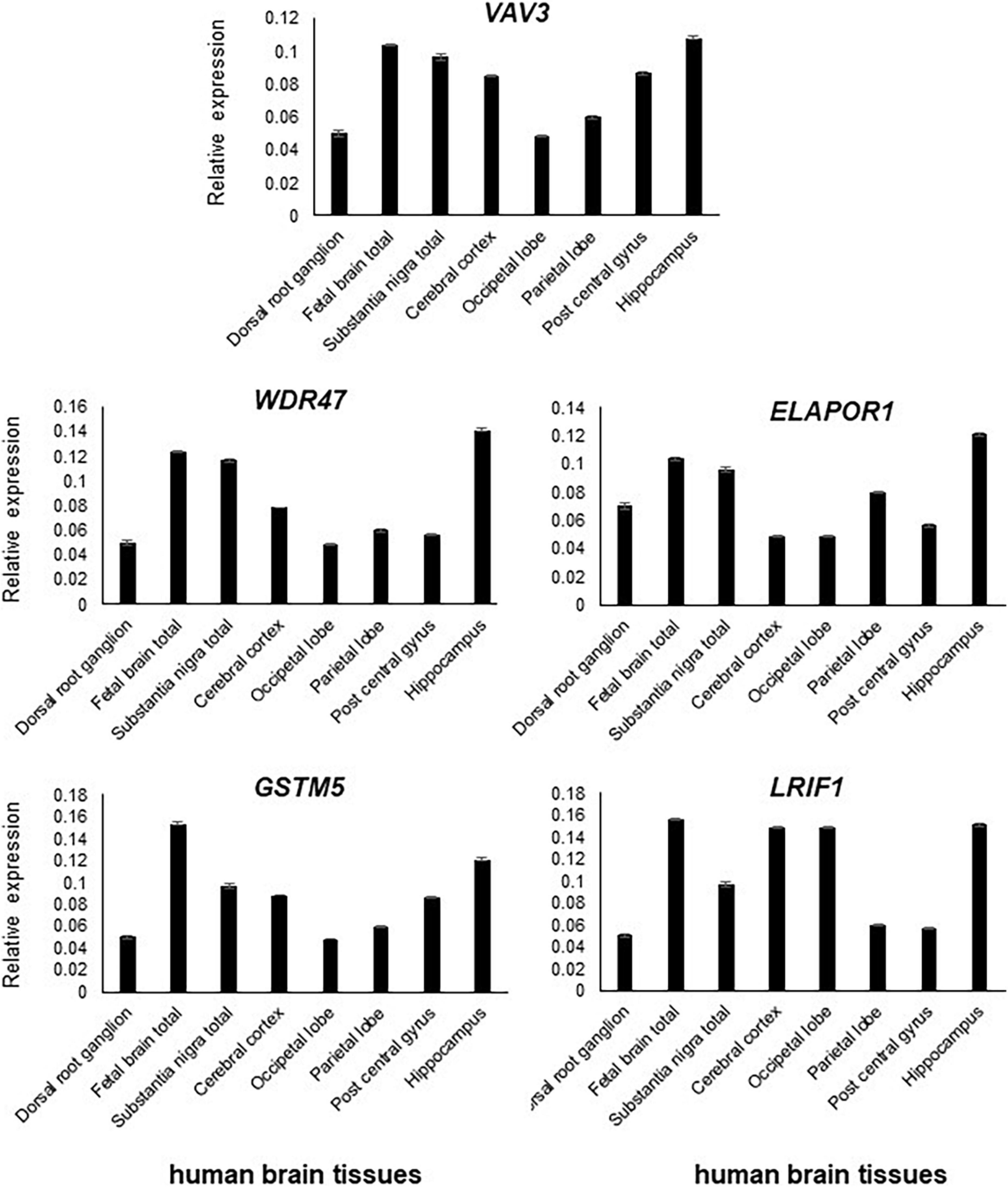
Figure 3. All five genes, namely, VAV3, WDR47, ELAPOR1, GSTM5, and LRIF1, are expressed in the various parts of the brain with varying levels.
Microarray analysis carried out on genomic DNA from Subject 1 revealed a 3.4 Mb heterozygous deletion at 1p13.3 (chr1: 107,240,429-110,671,860, [hg19]) and its inheritance was not determined. Additional microarray performed on Subject 2, Subject 3, and Subject 4 revealed a 130 kb del(1)(p13.3) arr[hg19](chr1: 108,726,456-108,853,796)x1dn, 120 kb del(1)(p13.3) arr[hg19](chr1; 108,729,365-108,853,796)x1dn, and 322 kb del(1)(p13.3) arr[hg19](chr1: 109,878,638-110,200,728)x1mat, respectively (Table 1 and Figure 1A). Subject 4 has inherited the deletion from her mother with the same phenotype.
We compared the clinical phenotypes and flanking genomic boundaries displayed in our four subjects (Subjects 1–4) with three previously reported CNV cases encompassing 1p13.3 (Cases 1–3) (Bisgaard et al., 2007; van Kuilenburg et al., 2009; Piccione et al., 2010) (Table 1 and Figure 1A) as well as 22 CNVs with NDDs from a total of 82 cases in the DECIPHER database, based on the criteria of less than 1 Mb in size (Supplementary Table 3). In silico comparative CNV mapping resulted in four refined candidate gene loci, where the maximal number of CNVs are overlapped (Figure 1A), and identified five positional candidate genes (Table 2). Loci one, three, and four have a single candidate gene, VAV3, GSTM5, and LRIF1, respectively, whereas locus two has two positional candidate genes, WDR47 and ELAPOR1.
VAV3 (VAV guanine nucleotide exchange factor 3, MIM 605541) from locus 1 is a member of the VAV family of guanine nucleotide exchange factors for Rho and Rac GTPases. The VAV family is a group of signal transduction molecules that are regulated by tyrosine phosphorylation and positioned downstream of protein tyrosine kinases. In the ventral medulla, VAV3 is involved in cerebellum development and axon wiring processes. In the cerebellum, it is involved in Purkinje cell dendritogenesis and the migration and survival of granule cells (Quevedo et al., 2010). In the early postnatal stages of KO mice, the loss of VAV3 causes motor coordination defects and abnormal gait (Quevedo et al., 2010). Haploinsufficiency of VAV3 and other four genes (NTNG1, LPPR4, GPSM2, and COL11A1) was postulated to be contributing factors toward the psychomotor impairment and abnormal craniofacial features reported in a subject with a 14 Mb interstitial deletion encompassing 1p13.3 (van Kuilenburg et al., 2009). Interestingly, three de novo variants in VAV3 were found in individuals with autism spectrum disorder (ASD) (Neale et al., 2012; Iossifov et al., 2014; Li et al., 2017; Lim et al., 2017; Turner et al., 2019), one with neurodevelopmental disorder (Turner et al., 2019), and one with schizophrenia (Aleksic et al., 2013; Table 2).
SLC25A24 (solute carrier family 25 member 24, MIM 608744) is within candidate locus one, which also includes NBPF4 (Figure 1A). Two recurrent missense mutations in SLC25A24 have been reported in Gorlin-Chaudhry-Moss syndrome (GCMS) (Ehmke et al., 2017) and Fontaine syndrome (Writzl et al., 2017), which are progeroid syndromes associated with accelerated aging. Due to the extreme aging facial phenotype caused by reported heterozygous mutations, the low level of expression in the human brain, and the absence of sporadic variants in the subjects with DD or ASD from the literature, this gene was excluded from the positional candidate gene list.
NBPF4 (neuroblastoma breakpoint family, member 4, MIM 613994) is a member of the neuroblastoma breakpoint family (NBPF) and contains the DUF1220 protein domain of approximately 65 amino acids in length. This domain has undergone unusually rapid and extensive duplications due to its repetitive structure during recent primate evolution. Chromosome 1 has 20 NBPF genes containing highly conserved DUF1220 domains and 13 of them are dispersed at 1q21 (Vandepoele et al., 2005; Diskin et al., 2009). Although DUF1220 sequences appear to exhibit a significant direct correlation with brain-size phenotypes in humans (Dumas et al., 2012), it was excluded due to no expression in the human brain and the absence of any reported genetic variants in NDDs.
Among the five genes, namely, WDR47, TAF13, TMEM167B, CFAP276, and ELAPOR1, located in the second locus, we found two positional candidate genes.
WDR47 (WD repeat-containing protein 47, MIM 615734), sharing structural homology with lissencephaly gene LIS1 (PAFAH1B1), participates in core microtubule-mediated processes, including neural stem cell proliferation, radial migration, and growth cone dynamics. KO mice lacking Wdr47 exhibited partial lethality, extensive fiber defects, microcephaly, thinner cortices, and abnormal sensory motor gait (Kannan et al., 2017). Kannan et al. speculated that mutations in this gene, particularly, truncating ones, would cause an embryonic lethal phenotype in humans, which would explain why no human mutations were identified. However, 2 years later, one frameshift variant in this gene was reported in a patient with ASD (Callaghan et al., 2019).
TAF13 (TATA-box-binding-protein-associated factor 13, MIM 600774), associated with ID combined with microcephaly, was excluded due to its autosomal recessive pattern (Tawamie et al., 2017).
TMEM167B (transmembrane protein 167B, aka C1orf119) was not considered as a candidate because no genetic variants in this gene were reported in NDD subjects and it is not expressed at high levels in the human brain.
CFAP276 (cilia and flagella-associated protein 276, aka C1orf194, MIM 618682) was excluded because two missense mutations cause dominant Charcott-Marie-Tooth disease (Sun et al., 2019); it is expressed at very low levels in the human brain, and no genetic variant was reported in NDD subjects.
Four genetic variants in ELAPOR1 (endosome-lysosome associated apoptosis and autophagy regulator 1, aka KIAA1324, MIM 611298) in DD and ASD have been reported (Iossifov et al., 2014; Turner et al., 2019), and at least six of its interacting genes at the protein levels are strongly involved in diverse NDDs. Moreover, KO mice exhibit decreased exploration in a new environment (Table 2), suggesting ELAPOR1 is likely a candidate gene (Figure 1A and Table 2).
The third candidate locus refined to 86 kb as depicted in Figure 1A implies GSTM5 (glutathione S-transferase mu 5, MIM 138385), a member of the glutathione S-transferase (GST) family possibly involved in NDDs, because of variants found in interacting genes at the protein levels and KO mice showing decreased exploration in a new environment (Figure 1A and Table 2). Its paralog GSTM1 null genotype in human carriers is more likely to develop ASD (Mandic-Maravic et al., 2021). GSTs are a group of enzymes that play a major role in the antioxidant defense mechanism by performing the inactivation of a large number of endogenous oxidative stress products. So far, a number of studies have suggested a link between oxidative stress and ASD (Mandic-Maravic et al., 2021). GSTM5 has at least five interacting genes at the protein level, variants of which were reported in various NDD subjects (Table 2).
Finally, the fifth positional candidate gene LRIF1 (ligand-dependent nuclear receptor-interacting factor 1, MIM 615354) is aligned near the centromeric region of 1p13.3. It overlaps with a 62 kb DECIPHER microdeletion case 290849 who presented with ID and behavioral abnormalities (Figure 1A and Supplementary Table 3). This variant was inherited from the father, whose phenotype is unknown. Importantly, however, LRIF1 interacts with two genes among others at the protein level, namely, PQBP1 (polyglutamine-binding protein 1, MIM 300463) and CHD4 (chromodomain helicase DNA-binding protein 4, MIM 603277). Mutations in PQBP1 are the cause of X-linked ID (Kalscheuer et al., 2003), while missense mutations in CHD4 are linked to ID syndrome with distinctive dysmorphisms (Weiss et al., 2016). Furthermore, another interactor ADHC1 (AT-hook DNA-binding motif-containing protein 1, MIM 615790) is involved in Xia-Gibbs syndrome with symptoms of ID, speech/motor delay, and facial dysmorphism (Xia et al., 2014), whereas interactor CHD3 is associated with Snijders Blok-Campeau syndrome, an NDD exhibiting macrocephaly and speech/language delay (Snijders Blok et al., 2018). Missense variants of the interactor CDC42 cause a diverse neurodevelopmental phenotype resembling Noonan syndrome (Martinelli et al., 2018), and the interactor SETD1B (aka KMT2G) is associated with the syndromic ID (Hiraide et al., 2018; Roston et al., 2021; Weerts et al., 2021; Table 2). We, therefore, suggest that LRIF1 could be considered a strong candidate for NDD, and this single DECIPHER case underscores the possibility of finding a neurodevelopmental disease gene (Figure 1A).
We also checked the interacting proteins of these five positional candidate genes using STRING (Functional Protein Association Networks, string-db.org, version 11.5) and BIOGRID (Database of Protein, Chemical, and Genetic Interactions, thebiogrid.org, version 4.4) protein–protein interaction databases and found that all of them interact with proteins already known to be associated with various NDDs. This outlines the potential role played by the proteins encoded from these five positional candidate genes and their interacting protein partners (Table 2).
We have interrogated the remaining 54 genes at 1p13.3, some of which might be dysregulated by position effect even if they are not contained in the four candidate gene loci. The criteria for selecting additional candidate genes are 3-fold: (1) sporadic de novo genetic variants in them were reported in subjects with NDDs, (2) animal models substantiated their pathological roles in NDDs, and (3) sporadic de novo genetic variants in NDD subjects were reported in their interacting genes at the protein level. This approach has identified additional 11 candidate genes which might be dysregulated by position effect (Supplementary Table 1).
Transcript levels of the five positional candidate genes were determined by RT-qPCR in various human tissues to assess their functional importance in phenotype-relevant tissues. The spatio-temporal regulation of gene expression leads to disparate expression patterns dependent on a variety of factors including the detection methods, which may not properly define the expression patterns. Differential expression patterns in multiple publicly available resources including GTEx Portal8 (version 8) and NCBI prompted us to use commercially available human RNA samples to measure the expression patterns through RT-qPCR experiment so that we would have a reference of expression of the genes of interest.
In comparison to other tissues, VAV3, WDR47, ELAPOR1, GSTM5, and LRIF1 expressions were found relatively high in the adult brain and/or fetal brain (Figure 2). The high expression of these five genes in the human fetal brain suggests that they might be involved in an early neurodevelopmental phenotype such as DD when mutated. The variable expression pattern in tissues at different developmental stages underscores the significance of analyzing the fetal brain along with the adult brain.
All five genes (VAV3, WDR47, ELAPOR1, GSTM5, and LRIF1) were highly expressed in various subsections of the brain with differing levels (Figure 3).
To obtain additional evidence for our candidate genes being functionally involved in the clinical phenotype observed in our subjects, we consulted DisGeNET, one of the largest publicly available collections of genes and variants associated with human diseases (Pinero et al., 2020). After combining the lists of the five positional candidates (Table 2) and the 11 functional candidates (Supplementary Table 1), we focused on two specific disorders relevant to the phenotype studied here: “mental disorders” and “nervous system diseases.” Out of our 16 candidates, FNDC7 is not present in DisGeNET, and seven are connected to at least one mental disorder confirming their involvement in the phenotype studied here (Figure 4). Among the 11 functional candidate genes, STXBP3 and KCNA2 seem to be particularly strong candidates since they are connected to disease terms like “impaired cognition” and “mental deterioration” among other relevant terms. The network for nervous system diseases is given in Supplementary Figure 1.
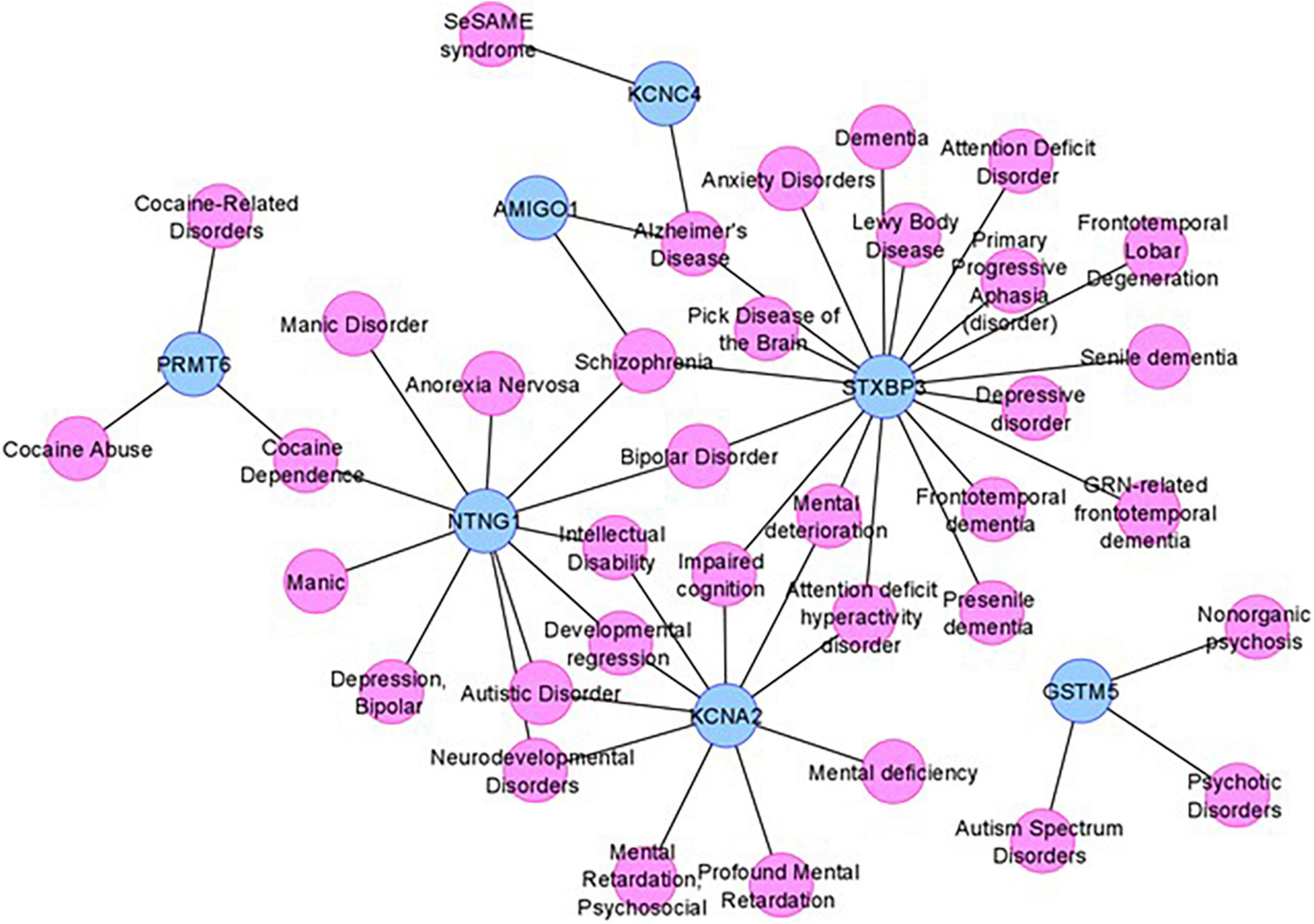
Figure 4. The mental disorder network. The candidate genes are depicted by blue nodes, and the disease terms are depicted by pink nodes. Edges correspond to disease associations.
As DisGeNET was last updated in June 2020, we also consulted Human Phenotype Ontology (HPO), another resource for gene–disease relationships, which was last updated in June 2022. As in the results from DisGeNET, KCNA2 is again linked to relevant terms like HP:0001268 (mental deterioration), HP:0001249 (intellectual disability), as well as HP:0000750 (delayed speech and language development), HP:0001263 (global developmental delay), and HP:0002317 (unsteady gait), additional relevant terms to the phenotype described in this article. KCNA4 is also linked to HP:0000750 (delayed speech and language development) and HP:0002064 (spastic gait). Unfortunately, STXBP3 could not be found in the HPO database nor any other genes given in Figure 4.
The CNVs represent a rich source of structural chromosomal variations that can result in severe phenotypic consequences as a result of gene dosage alteration, disruptions in coding sequences, and long-range gene regulators (Stranger et al., 2007). With the advancement of detection technology, it has become apparent that the collective involvement of CNVs underlies neurodevelopmental syndromes (Lee et al., 2015). Clinical features of seven heterozygous CNVs located at 1p13.3 have been described through karyotype–phenotype correlations (Tabata et al., 1991; Mattia et al., 1992; Lo et al., 1998; Utkus et al., 1999; Bisgaard et al., 2007; van Kuilenburg et al., 2009; Piccione et al., 2010) (Cases 1–7 in Table 1). Four of these published cases (Cases 4–7 in Table 1) are from the pre-microarray era, so the exact sizes, as well as the distal and proximal genomic breakpoints, of these large 1p13.3 deletions (9–21 Mb) are unknown.
Causative disease gene identification has been aided by refinement of critical regions through comparison of patients with overlapping large deletions like in Kallmann syndrome (Dode et al., 2003) and CHARGE syndrome (Vissers et al., 2004). However, understanding the genetic etiology of large pathogenic CNVs still remains a major challenge, because they frequently contain multiple genes (Rice and McLysaght, 2017).
By contrast, small CNVs containing a couple of genes provide a great potential in the identification of disease genes through comparative genomic mapping by aligning the genomic boundaries of CNVs with varying sizes and overlapping phenotypes. This was evidenced by an NDD candidate gene SETD1B identified from a critical 445 kb genomic region encompassing seven genes at 12q24.31 (Labonne et al., 2016). In the ensuing years, a large number of mutations in this gene were identified from next-generation sequencing (NGS) databases, verifying it as an NDD disease gene (Hiraide et al., 2018; Roston et al., 2021; Weerts et al., 2021).
Recently, we have recruited four new subjects having heterozygous microdeletions at 1p13.3 with common features of NDDs. To find out the candidate genes responsible for disease phenotypes, we aligned our cases with the aforementioned three published CNVs along with 22 informative DECIPHER cases on 1p13.3 as described in the “Results” section.
DECIPHER cases at 1p13.3 comprise 13 duplications, seven deletions, and two triplications (Supplementary Table 3). Nonetheless, most cases displayed neurodevelopmental phenotypes with various comorbidities. NDDs are linked to dosage alterations of several genes in both deletions and duplications (Rice and McLysaght, 2017). In addition, the majority of CNV cases are inherited from one parent with an unknown phenotype, while the inheritance of the remainders is unknown. This suggests that some CNVs at 1p13.3 might be inherited from an affected parent, have varying penetrance, or are imprinted epigenetically.
Our four subjects and seven patients reported in the literature share an overlapping phenotype of DD, ID, and facial dysmorphism (Table 1). This offered an opportunity for the identification of NDD genes that are responsible for a common set of phenotypes and features among all subjects with 1p13.3 interstitial deletions and duplications, regardless of whether they are inherited or de novo.
Based on our comparative in silico genomic mapping, we narrowed down the 4.6 Mb genomic region of 1p13.3 to four individual candidate gene loci and identified five positional candidate genes (VAV3, WDR47, ELAPOR1, GSTM5, and LRIF1) out of 59 genes in total (Figure 1A and Table 2). It is likely that the neurodevelopmental features in the majority of our CNV cases encompassing one or more of the five potential candidate genes are caused either by a single gene or by a combination of multiple genes among them.
Each gene has been interrogated in order to identify sporadic de novo variants in them or their interacting genes at the protein level in NDD subjects from the literature. Furthermore, their KO mice phenotype has been investigated to obtain supporting evidence (Table 2 and Supplementary Table 1), which has substantiated the candidacy of these positional candidate genes. We also confirmed the high expression of these genes in the brain and subsequently specific brain regions, suggesting their pathogenicity in NDDs (Figures 2, 3). The collective effort of in silico genome mapping informed by various disease gene databases, the role of interacting protein partners, and confirmation of gene expression in brain tissues strongly indicate the functional candidacy of these five positional genes in causing various NDDs.
Among the candidate genes we found, VAV3 plays an important role in bone mass and remodeling, axon wiring in the ventrolateral medulla, and cerebellar development (Faccio et al., 2005). The cerebellum is known to be involved in motor learning and coordination. Consequentially, impairment of this region leads to dysfunctions in movement (Mapelli et al., 2022). This suggests a reason for the short stature, bone anomalies, and jerky gait pertaining to Subject 1 in this study (Table 1). VAV proteins have key signaling roles in the immune, cardiovascular, and nervous systems (Bustelo, 2014).
WDR47 belongs to the family of WD40-repeat (WDR) proteins. So far, mutations in 27 genes (9.4%) among 286 WDR genes have been linked to brain disorders, most notably ID associated with corpus callosum defects (Kannan et al., 2017). A smaller corpus callosum has been linked to an increased risk of autism (Paul et al., 2007), bipolar disorder (Li et al., 2014), and schizophrenia (Balevich et al., 2015). WDR47 is necessary for proper radial migration of projection neurons, and Wdr47 depletion compromises growth cone morphology and microtubule. The neuroanatomical defects found in Wdr47 KO result in hyperactivity and sensory motor gating abnormalities both in male and female mice (Kannan et al., 2017), suggesting a link between WDR47 and neurological disorder.
LRIF1 is required for accurate chromosome segregation in mitosis (Akram et al., 2018). A growing number of genes that regulate mitotic division are involved in neurodevelopmental diseases. Mitotic gene mutations give rise to insufficient cell proliferation and/or failure to replenish the neuronal stem cells in early development. The reduced number of neurons results in the underdevelopment of the central nervous system (CNS) and causes microcephaly and neuronal migration disorders (Degrassi et al., 2019). LRIF1 also interacts with many known genes involved in NDDs, which justifies its status as an NDD candidate gene (Table 2).
Another observation in the subjects and cases was that the severity of the phenotypes increases with the CNV size (Table 1 and Figure 1A). Three of the published cases (Cases 1–3 in Table 1) and Subject 1 with CNVs ranging from 3 to 14 Mb displayed severe cranial anomalies including microcephaly, macrocephaly, and asymmetry of the skull. However, among the remaining subjects with small CNVs, only Subject 4 displayed cranial anomalies that were considerably less pronounced than larger CNVs. Although Case 4 with the largest deletion did not show any apparent cranial anomalies, it remains to be seen whether she will develop secondary cranial anomalies after her infancy (Table 1). Larger deletions (Subject 1, Case 1, Case 2, Case 5, and Case 7) exhibited a distinct muscle disorder (mild fiber atrophy, diplegia, hypertonia, areflexia, and hypotonia) in comparison to Subjects 2–4 with small deletions, who did not show any muscle disorders (Table 1). Additionally, in contrast to Subjects 2–4 with small deletions, epilepsy was more pronounced in published cases with larger CNVs, including Subject 1, Case 4, and Case 6 (Table 1 and Figure 1A). Similarly, multiple hand, finger, feet, and toe anomalies were observed in all the published Cases 1–7 and Subject 1 that had significant large CNVs.
Furthermore, we also propose 11 additional genes, not encompassed within the demarcated loci, yet, pose as NDD candidate genes due to genetic variants in NDD subjects and KO mice with neurological phenotype. To support their candidacy, we also analyzed and compiled a list of interacting protein partners known to play a role in NDDs (Supplementary Table 1). These genes included in large CNVs (Subject 1 and Cases 1–3, Figure 1A and Table 1) were not contained in small CNVs, which were aligned to refine the candidate gene loci in in silico comparative genomic mapping. Hence, the clinical symptoms seen in these small CNVs might be caused by the position effect leading to altered expression of these 11 genes adjacent to them (Redon et al., 2005). Or these 11 genes might contribute to the phenotypes in the individuals with larger deletions and be irrelevant for cases with smaller CNVs.
Being the largest chromosome (249 Mb and representing 9% of the total human genome) containing a maximum number of 2,000 genes, chromosome 1 harbors the highest number of disease genes (Murphy et al., 2003).
We hypothesized that a total of 16 novel autosomal dominant candidate genes identified at 1p13.3 in this study are dosage sensitive in either direction and thus might not tolerate copy number change. These candidate genes will help zero in on NDD disease genes from NGS databases containing a large number of loss-of-function VUSs (variants of unknown significance) and autosomal dominant GUSs (genes of unknown significance).
Subject 1 (DGDP030) is a 44-year-old Asian woman, exhibiting a severe clinical phenotype as a result of a 3.4 Mb interstitial heterozygous deletion at 1p13.3. Her records indicate a normal birth, and her examination was within normal limits. She was adopted at 4 months of age; healthy appearing and quiet. She did not make eye contact or smile much; she would only cry when hungry and was unable to lie on her stomach. Yet, she was able to roam around the room on her back and she had a forceful grip. Her teeth did not appear until 20 months of age, after which, all grew within 3 months. Her mouth was too small for all her teeth and five had to be extracted. She then started to display mild hypotonia and suffered febrile seizures at 18 months of age.
At 3 years of age, she was diagnosed with the following symptoms: asymmetrical skull, mild anemia, febrile seizures, possible myoclonic seizures, hypotonia, hearing loss, and a recurring petechial rash. She was having seizures once a month. She also had strange night terror episodes and childhood schizophrenia. She was obsessive–compulsive and had wide mood swings that lasted 2–3 weeks each. The biopsy of her skeletal muscle from both gluteus and maximus at 18 years of age showed mild fiber atrophy and random variation in muscle fiber size. Her motor milestones were of poor quality and out of the usual order. She had a learning disability but could be directed to play; however, she preferred not to. She liked being in a room with people but did not make much of an effort to interact.
At the age of 31 years, she remained severely developmentally delayed and autistic. She had a jerky gait and would walk with arched feet. She constantly suffered from cramps in her legs and arms. She had an asymmetrical head with a smaller right parietal area, ptosis, astigmatism, low-set ears, unfolded ears, kyphosis, lordosis, and a high arched palate (Figure 1B). She held her hands at odd angles and held objects with her fingertips. At the age of 31 years, she had a complete emotional breakdown and increased violent behavior. At the same time, her tegretol levels were in the toxic range, and her thyroid levels were in the hypothyroid range.
At this age, she has had several bouts of myoglobinuria, unexplained fevers, recurrent infections, paradoxic and toxic reactions to medications, allergies to insect bites, hypothyroidism likely related to her seizure medication, thrombocytopenia due to medication, malignant hyperthermia, easily developed bruises, petechial rash, anemia, intermittent diarrhea, severe constipation, and both external canals intermittently plugged with cerumen. An auditory brainstem response evaluation was suggestive of a moderate peripheral deficit, potentially sensorineural, more so in the right ear than in the left ear. She was usually remarkably healthy with a normal echocardiogram and normal eye examination.
Subject 2 is an 11-year 6-month-old Caucasian boy with a history of global developmental delay, ID, and dysmorphic features. The child was born full term at 38 weeks of pregnancy and delivered by elective cesarean section from non-consanguineous parents. At birth, he weighed 2.650 kg (10th percentile) and his crown-heel length was 51 cm (50th percentile). Global developmental delay was observed at an early age. Up until the age of 11 years, no morphological phenotypic data were available for the child until specific genetic physicals revealed a bulbous nasal tip, epicanthal folds, downslanted palpebral fissure, low anterior hairline, long philtrum, wide nasal bridge, thin upper lip vermilion, long ears, micrognathia, and hypertelorism. At the age of 11 years, he weighed 32.5 kg (25th percentile) and measured 139 cm in height (25th percentile). He sat independently at around 12 months, walked at 24 months, spoke his first words at 36 months, and was not communicating in full sentences until 11 years of age. Now, at the age of 18 years, his speech has improved but he is not yet able to speak fluently. The MRI of the head showed no remarkable findings and remained likewise subsequently. Despite being recognized as an inattentive patient, psychological assessments revealed no attention deficit hyperactivity disorder (ADHD). However, he has shown severe learning difficulties, requiring individualized educational assistance to date. He has no significant anxiety disorder and shows continued progress; nevertheless, his deficits in attention and executive functions persist. Additional clinical laboratory tests have been carried out for this patient, including an extensive metabolic workup, chromosome analysis, and chromosomal microarray analysis. Except for the microarray result, all of the tests were uneventful. However, microarray indicated a de novo 130 kb heterozygous genomic loss at 1p13.3.
Subject 3 is a 6-year-old Caucasian girl with a history of neurodevelopmental delay, ID, epilepsy, and dysmorphic features. The child was born full term at 36 weeks of pregnancy and delivered by elective cesarean section. The biological parents stated that they could not rule out consanguinity; nonetheless, the child’s chromosomal microarray analysis did not show long continuous stretches of homozygosity. At birth, the newborn weighed 2.415 kg (5th percentile), and her crown-heel length was 50 cm (50th percentile). The Apgar scores were 07/09. As a newborn, the child showed no sucking reflex, requiring assistance to nursing. Moreover, at the age of 3 days old, she displayed jaundice. Up until the age of 6 years, no morphological phenotypic data were available for the child, when specific genetic physicals revealed epicanthal folds, downslanted palpebral fissure, long philtrum, wide nasal bridge, hypertelorism, and micrognathia. At the age of 6 years, the child had one seizure episode, and her MRI indicated a small arachnoid cyst in the posterior fossa. Global developmental delay was observed at an early age. She sat independently at around 10 months and walked at 16 months. Since birth, the child has shown loose skin. Additional clinical laboratory tests were performed including an extensive metabolic workup, chromosome analysis, and microarray analysis, which indicated a de novo 120 kb heterozygous genomic loss at 1p13.3.
Subject 4 is the first of four children of an unrelated Caucasian couple. The mother with the same microdeletion presents with global DD, behavioral disorders, and ID. Otherwise, the medical history of the father’s family was non-contributory. Vaginal delivery occurred at 39 weeks. At birth, growth parameters were within the normal range except for OFC < –2DS (birth weight 20th percentile/birth height 30th percentile). She walked at 18 months of age. She suffered from anxiety and intellectual disability, the reason why she was referred to a medical geneticist at the age of 15 years. On examination, she had a long face, epicanthus, prominent nose, posteriorly rotated ears, and short fingers. Her microcephaly was persistent. She had an array-CGH that revealed a maternally inherited 322 kb heterozygous deletion at 1p13.3 arr[hg19](chr1:109,878,638-110,200,728). Her oldest brother presents with global developmental delay, speech delay, and intellectual disability. Unfortunately, this family was lost to follow-up, and the segregation including the brother within the family could not get done. We were not able to get updated information regarding her development now that she is 22 years.
For high-resolution karyotypes, peripheral blood lymphocytes from subjects were cultured with phytohematoagglutinin and harvested for cytogenetic analysis using standard techniques. Chromosome analysis was performed on GTL-banded chromosomes at an approximately 550 band level.
For Subjects 2 and 3, conventional cell cultures, harvesting, and GTG banding with a > 550 band resolution were performed following standard procedures. Chromosome analyses were done using Zeiss Axioscope® (Göttingen, Germany) and the software IKAROS® (Metasystems Corporation, Altlussheim, Germany).
For Subject 4, array-comparative genomic hybridization analysis (CGH) was performed using 180K Agilent microarray (Agilent Technologies, Santa Clara, CA, USA). Image analysis, normalization, and annotation were done with Feature Extraction 10.5.1.1 (Agilent, Santa Clara, USA) using the default settings. Data visualization and further analysis were performed with Cytogenomics 2.7.6.0 (Agilent, Santa Clara, USA). The array-CGH also detected a duplication of 17p11.2 of 194 kb (chr17:21307889-21502083), considered as benign.
The extraction of genomic DNA from Subject 1 was carried out using a standard phenol-chloroform protocol. To analyze submicroscopic copy number alterations in subjects, DNA samples from the subject’s blood and pooled normal controls (PROMEGA) were differentially labeled and cohybridized using a 44K whole-genome oligonucleotide array employing the protocols for array CGH provided by the manufacturer (Agilent, Santa Clara, USA). No deletion or duplication of other chromosomes was observed using the laboratory standard cutoff values (0.0Log2 ratio at a resolution of 0.3 Mb). Image analysis, normalization, and annotation were done with Feature Extraction 10.5.1.1 (Agilent, Santa Clara, USA) using the default settings. Data visualization and further analysis were performed with GenomeCAT9. CNVs were determined by circular binary segmentation.
Genomic DNAs from Subjects 2 and 3 as well as their parents were isolated from whole blood samples using the Illustra Blood Genomic Prep Mini Spin Kit (GE Healthcare Life Sciences, USA), following the manufacturer’s instructions. Chromosomal microarray analysis (CMA) was carried out on probands and their biological parents using the GeneChip® CytoScanHD™ (ThermoFisher, USA) with 2.7 million polymorphic and non-polymorphic markers. Array analyses were done using the Chromosome Analysis Suite (ChAS®) software. The CNVs found in probands were analyzed in comparison with public databases, including the Database of Genomic Variants10 (DGV, version 107), the Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHER), and the CytoScan™ HD (High Density) Array Database.
For Subject 1, BACs for probing genomic regions of interest were selected from the RPCI-11 (Roswell Park Cancer Institute) library via the UCSC genome browser11. Human genomic DNA inserts were extracted, fluorescently labeled using nick translation, and hybridized to a metaphase spread of the subject’s lymphocytes using standard procedures (Lichter et al., 1990).
The phenotypes and genomic coordinates from our four subjects (Subjects 1–4) were compared with three previously reported literature cases (Cases 1–3) (Bisgaard et al., 2007; van Kuilenburg et al., 2009; Piccione et al., 2010) and 22 unpublished DECIPHER CNVs with NDDs and less than 1 Mb in size (Figure 1A, Table 1, and Supplementary Table 3). Genomic coordinates from Case 1 were converted from hg17 to hg19 before the comparison was carried out. Furthermore, for the case from van Kuilenburg et al. (2009), the flanking distal and proximal genomic boundaries were between TLCD4 and WDR47 with the approximate genomic coordinates 95,558,073-109,584,850 (A.B.P. van Kuilenburg, personal communication, 19 May 2022) as reported in Figure 5 of their study.
RT-qPCR was performed from the total RNA of the human brain and other tissues (Human Total RNA Master Panel II, Cat# 636643, Clontech). The catalog numbers of eight brain tissues from Clontech were as follows: dorsal root ganglion-636150, fetal brain total-636526, substantia nigra total-636560, cerebral cortex-636561, occipital lobe-636570, parietal lobe-636571, postcentral gyrus-636573, and hippocampus-636565. The cDNA synthesis was performed using 1–2 μg of total RNA using high-capacity cDNA reverse transcription kit and analyzed by RT-PCR on QuantStudio 6 Flex system using SYBR Green (ThermoFisher, Waltham, MA). We used ΔCt method to calculate the relative expression of each gene. In summary, relative gene expression was calculated by the difference between the Ct value (ΔCt) of the gene of interest and reference gene, GAPDH. After determining ΔCt, the fold change (2–ΔCt) was measured, and the relative expression was plotted as excel graphs.
We performed disease-gene network analysis with DisGeNET (v7.0) (1) which contains 1,134,942 gene-disease associations between 21,671 genes and 30,170 diseases, disorders, traits, and clinical or abnormal human phenotypes. We used the DisGeNET Cytoscape app (Pinero et al., 2021) to browse and visualize the networks around the candidate genes. We used all sources, any association type and evidence level, and two specific disease classes of interest, namely, “mental disorders” and “nervous system diseases.”
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Augusta University, Georgia, USA. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
AB-M and KJ contributed to the manuscript preparation including figure design and phenotypic data analysis. VG and PS performed the RT-qPCR and clinical examination of the patient data, respectively. ADLF performed the bioinformatics analysis. YP and KS collected and analyzed the data. CK, ADDC, ASDC, IP, LM, AS, LF, PC, and CR recruited the subjects and/or performed the clinical follow-up. LL, I-KK, C-HK, and W-YK edited the subsequent manuscript drafts and analyzed the data. H-GK conceived and designed the study, analyzed the data, and drafted the first manuscript. All authors read and approved the final manuscript.
This work was supported by an internal grant QB17 of Qatar Biomedical Research Institute (H-GK), a grant from the National Research Foundation of Korea (2018M3A9B8021980 to C-HK), an NICHD grant (R01HD092505 to LL), and a grant from the National Research Foundation of Korea (2020R1A2C2006614 to I-KK). We also thank the support of funding provided by Caroline Jones-Carrick and Collin Carrick.
We thank Jill A Rosenfeld, Jade Deborah D Mello, and Shahad Sabaawi Ibrahim AlHassan for their assistance in proofreading the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.979061/full#supplementary-material
Supplementary Figure 1 | The nervous system diseases network. The candidate genes are depicted by blue nodes and the disease terms by pink nodes. Edges correspond to disease associations. Out of our 16 candidates, 10 are connected to at least one nervous system disease providing more evidence for their involvement in the phenotype studied here.
CNV, copy number variation; NDDs, neurodevelopmental disorders; DD, development delay; ASD, autism spectrum disorder; ID, intellectual disability; CFA, craniofacial anomalies; ADHD, attention deficit hyperactivity disorder; NGS, next-generation sequencing.
Abdel-Salam, G. M. H., Miyake, N., Abdel-Hamid, M. S., Sayed, I. S. M., Gadelhak, M. I., Ismail, S. I., et al. (2018). Phenotypic and molecular insights into PQBP1-related intellectual disability. Am. J. Med. Genet. A 176, 2446–2450. doi: 10.1002/ajmg.a.40479
Akram, S., Yang, F., Li, J., Adams, G., Liu, Y., Zhuang, X., et al. (2018). LRIF1 interacts with HP1alpha to coordinate accurate chromosome segregation during mitosis. J. Mol. Cell Biol. 10, 527–538. doi: 10.1093/jmcb/mjy040
Aleksic, B., Kushima, I., Hashimoto, R., Ohi, K., Ikeda, M., Yoshimi, A., et al. (2013). Analysis of the VAV3 as candidate gene for schizophrenia: Evidences from voxel-based morphometry and mutation screening. Schizophr. Bull. 39, 720–728. doi: 10.1093/schbul/sbs038
Al-Mubarak, B., Abouelhoda, M., Omar, A., Aldhalaan, H., Aldosari, M., Nester, M., et al. (2017). Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: A trio study from Saudi families. Sci. Rep. 7:5679.
Al-Nabhani, M., Al-Rashdi, S., Al-Murshedi, F., Al-Kindi, A., Al-Thihli, K., Al-Saegh, A., et al. (2018). Reanalysis of exome sequencing data of intellectual disability samples: Yields and benefits. Clin. Genet. 94, 495–501. doi: 10.1111/cge.13438
Amiel, J., Rio, M., De Pontual, L., Redon, R., Malan, V., Boddaert, N., et al. (2007). Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am. J. Hum. Genet. 80, 988–993. doi: 10.1086/515582
An, J. Y., Cristino, A. S., Zhao, Q., Edson, J., Williams, S. M., Ravine, D., et al. (2014). Towards a molecular characterization of autism spectrum disorders: An exome sequencing and systems approach. Transl. Psychiatry 4:e394.
Anazi, S., Maddirevula, S., Salpietro, V., Asi, Y. T., Alsahli, S., Alhashem, A., et al. (2017). Expanding the genetic heterogeneity of intellectual disability. Hum. Genet. 136, 1419–1429.
Ando, Y., Sawada, M., Kawakami, T., Morita, M., and Aoki, Y. (2021). A patient with noonan syndrome with a KRAS mutation who presented severe nerve root hypertrophy. Case Rep. Neurol. 13, 108–118. doi: 10.1159/000512265
Balevich, E. C., Haznedar, M. M., Wang, E., Newmark, R. E., Bloom, R., Schneiderman, J. S., et al. (2015). Corpus callosum size and diffusion tensor anisotropy in adolescents and adults with schizophrenia. Psychiatry Res. 231, 244–251. doi: 10.1016/j.pscychresns.2014.12.005
Bisgaard, A. M., Rasmussen, L. N., Moller, H. U., Kirchhoff, M., and Bryndorf, T. (2007). Interstitial deletion of the short arm of chromosome 1 (1p13.1p21.1) in a girl with mental retardation, short stature and colobomata. Clin. Dysmorphol. 16, 109–112. doi: 10.1097/01.mcd.0000228425.89660.bf
Bowling, K. M., Thompson, M. L., Amaral, M. D., Finnila, C. R., Hiatt, S. M., Engel, K. L., et al. (2017). Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 9:43.
Brunet, T., Jech, R., Brugger, M., Kovacs, R., Alhaddad, B., Leszinski, G., et al. (2021). De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin. Genet. 100, 14–28. doi: 10.1111/cge.13946
Bustelo, X. R. (2014). Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 5:9. doi: 10.4161/21541248.2014.973757
Buysse, K., Delle Chiaie, B., Van Coster, R., Loeys, B., De Paepe, A., Mortier, G., et al. (2009). Challenges for CNV interpretation in clinical molecular karyotyping: Lessons learned from a 1001 sample experience. Eur. J. Med. Genet. 52, 398–403. doi: 10.1016/j.ejmg.2009.09.002
Callaghan, D. B., Rogic, S., Tan, P. P. C., Calli, K., Qiao, Y., Baldwin, R., et al. (2019). Whole genome sequencing and variant discovery in the ASPIRE autism spectrum disorder cohort. Clin. Genet. 96, 199–206. doi: 10.1111/cge.13556
Cappi, C., Oliphant, M. E., Peter, Z., Zai, G., Conceicao Do Rosario, M., Sullivan, C. A. W., et al. (2020). De novo damaging DNA coding mutations are associated with obsessive-compulsive disorder and overlap with Tourette’s disorder and autism. Biol. Psychiatry 87, 1035–1044. doi: 10.1016/j.biopsych.2019.09.029
Carroll, R., Kumar, R., Shaw, M., Slee, J., Kalscheuer, V. M., Corbett, M. A., et al. (2017). Variant in the X-chromosome spliceosomal gene GPKOW causes male-lethal microcephaly with intrauterine growth restriction. Eur. J. Hum. Genet. 25, 1078–1082. doi: 10.1038/ejhg.2017.97
Chen, Y., Bartanus, J., Liang, D., Zhu, H., Breman, A. M., Smith, J. L., et al. (2017). Characterization of chromosomal abnormalities in pregnancy losses reveals critical genes and loci for human early development. Hum. Mutat. 38, 669–677. doi: 10.1002/humu.23207
D’Amore, A., Tessa, A., Casali, C., Dotti, M. T., Filla, A., Silvestri, G., et al. (2018). Next generation molecular diagnosis of hereditary spastic paraplegias: An Italian Cross-Sectional Study. Front. Neurol. 9:981. doi: 10.3389/fneur.2018.00981
Deciphering Developmental Disorders, S. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438.
Degrassi, F., Damizia, M., and Lavia, P. (2019). The mitotic apparatus and kinetochores in microcephaly and neurodevelopmental diseases. Cells 9:49. doi: 10.3390/cells9010049
Diskin, S. J., Hou, C., Glessner, J. T., Attiyeh, E. F., Laudenslager, M., Bosse, K., et al. (2009). Copy number variation at 1q21.1 associated with neuroblastoma. Nature 459, 987–991. doi: 10.1038/nature08035
Dode, C., Levilliers, J., Dupont, J. M., De Paepe, A., Le Du, N., Soussi-Yanicostas, N., et al. (2003). Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 33, 463–465. doi: 10.1038/ng1122
Dumas, L. J., O’Bleness, M. S., Davis, J. M., Dickens, C. M., Anderson, N., Keeney, J. G., et al. (2012). DUF1220-domain copy number implicated in human brain-size pathology and evolution. Am. J. Hum. Genet. 91, 444–454. doi: 10.1016/j.ajhg.2012.07.016
Eaton, D. L., and Bammler, T. K. (1999). Concise review of the glutathione S-transferases and their significance to toxicology. Toxicol. Sci. 49, 156–164.
Ehmke, N., Graul-Neumann, L., Smorag, L., Koenig, R., Segebrecht, L., Magoulas, P., et al. (2017). De novo mutations in SLC25A24 cause a craniosynostosis syndrome with hypertrichosis, progeroid appearance, and mitochondrial dysfunction. Am. J. Hum. Genet. 101, 833–843. doi: 10.1016/j.ajhg.2017.09.016
Erdem-Eraslan, L., Gao, Y., Kloosterhof, N. K., Atlasi, Y., Demmers, J., Sacchetti, A., et al. (2015). Mutation specific functions of EGFR result in a mutation-specific downstream pathway activation. Eur. J. Cancer 51, 893–903. doi: 10.1016/j.ejca.2015.02.006
Faccio, R., Teitelbaum, S. L., Fujikawa, K., Chappel, J., Zallone, A., Tybulewicz, V. L., et al. (2005). Vav3 regulates osteoclast function and bone mass. Nat. Med. 11, 284–290. doi: 10.1038/nm1194
Faergeman, S. L., Bojesen, A. B., Rasmussen, M., Becher, N., Andreasen, L., Andersen, B. N., et al. (2021). Phenotypic heterogeneity and mosaicism in Xia-Gibbs syndrome: Five Danish patients with novel variants in AHDC1. Eur. J. Med. Genet. 64:104280. doi: 10.1016/j.ejmg.2021.104280
Faundes, V., Newman, W. G., Bernardini, L., Canham, N., Clayton-Smith, J., Dallapiccola, B., et al. (2018). Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am. J. Hum. Genet. 102, 175–187. doi: 10.1016/j.ajhg.2017.11.013
Firth, H. V., Richards, S. M., Bevan, A. P., Clayton, S., Corpas, M., Rajan, D., et al. (2009). DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 84, 524–533. doi: 10.1016/j.ajhg.2009.03.010
Garcia-Acero, M., and Acosta, J. (2017). Whole-exome sequencing identifies a de novo AHDC1 mutation in a colombian patient with Xia-Gibbs Syndrome. Mol. Syndromol. 8, 308–312. doi: 10.1159/000479357
Gilks, W. P., Hill, M., Gill, M., Donohoe, G., Corvin, A. P., and Morris, D. W. (2012). Functional investigation of a schizophrenia GWAS signal at the CDC42 gene. World J. Biol. Psychiatry 13, 550–554. doi: 10.3109/15622975.2012.666359
Girard, S. L., Gauthier, J., Noreau, A., Xiong, L., Zhou, S., Jouan, L., et al. (2011). Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863. doi: 10.1038/ng.886
Goehler, H., Lalowski, M., Stelzl, U., Waelter, S., Stroedicke, M., Worm, U., et al. (2004). A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol. Cell 15, 853–865. doi: 10.1016/j.molcel.2004.09.016
Grozeva, D., Carss, K., Spasic-Boskovic, O., Tejada, M. I., Gecz, J., Shaw, M., et al. (2015). Targeted next-generation sequencing analysis of 1,000 individuals with intellectual disability. Hum. Mutat. 36, 1197–1204.
Guard, S. E., Poss, Z. C., Ebmeier, C. C., Pagratis, M., Simpson, H., Taatjes, D. J., et al. (2019). The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Sci. Rep. 9:6539. doi: 10.1038/s41598-019-42990-5
Gulsuner, S., Walsh, T., Watts, A. C., Lee, M. K., Thornton, A. M., Casadei, S., et al. (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529. doi: 10.1016/j.cell.2013.06.049
Guo, H., Wang, T., Wu, H., Long, M., Coe, B. P., Li, H., et al. (2018). Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 9:64. doi: 10.1186/s13229-018-0247-z
Gupta, H. V., Vengoechea, J., Sahaya, K., and Virmani, T. (2015). A splice site mutation in ATP6AP2 causes X-linked intellectual disability, epilepsy, and parkinsonism. Parkinsonism Relat. Disord. 21, 1473–1475. doi: 10.1016/j.parkreldis.2015.10.001
Hashimoto, R., Nakazawa, T., Tsurusaki, Y., Yasuda, Y., Nagayasu, K., Matsumura, K., et al. (2016). Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J. Hum. Genet. 61, 199–206. doi: 10.1038/jhg.2015.141
Helbig, K. L., Farwell Hagman, K. D., Shinde, D. N., Mroske, C., Powis, Z., Li, S., et al. (2016). Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet. Med. 18, 898–905.
Hiraide, T., Hattori, A., Ieda, D., Hori, I., Saitoh, S., Nakashima, M., et al. (2019). De novo variants in SETD1B cause intellectual disability, autism spectrum disorder, and epilepsy with myoclonic absences. Epilepsia Open 4, 476–481. doi: 10.1002/epi4.12339
Hiraide, T., Nakashima, M., Yamoto, K., Fukuda, T., Kato, M., Ikeda, H., et al. (2018). De novo variants in SETD1B are associated with intellectual disability, epilepsy and autism. Hum. Genet. 137, 95–104.
Hiraide, T., Yamoto, K., Masunaga, Y., Asahina, M., Endoh, Y., Ohkubo, Y., et al. (2021). Genetic and phenotypic analysis of 101 patients with developmental delay or intellectual disability using whole-exome sequencing. Clin. Genet. 100, 40–50.
Hu, H., Haas, S. A., Chelly, J., Van Esch, H., Raynaud, M., De Brouwer, A. P., et al. (2016). X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 21, 133–148. doi: 10.1038/mp.2014.193
Hu, H., Kahrizi, K., Musante, L., Fattahi, Z., Herwig, R., Hosseini, M., et al. (2019). Genetics of intellectual disability in consanguineous families. Mol. Psychiatry 24, 1027–1039.
Huttlin, E. L., Bruckner, R. J., Navarrete-Perea, J., Cannon, J. R., Baltier, K., Gebreab, F., et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040 e3028. doi: 10.1016/j.cell.2021.04.011
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299.
Jensen, L. R., Chen, W., Moser, B., Lipkowitz, B., Schroeder, C., Musante, L., et al. (2011). Hybridisation-based resequencing of 17 X-linked intellectual disability genes in 135 patients reveals novel mutations in ATRX, SLC6A8 and PQBP1. Eur. J. Hum. Genet. 19, 717–720. doi: 10.1038/ejhg.2010.244
Ji, J., Lee, H., Argiropoulos, B., Dorrani, N., Mann, J., Martinez-Agosto, J. A., et al. (2015). DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet. 23, 1473–1481. doi: 10.1038/ejhg.2015.71
Jiang, Y., Wangler, M. F., Mcguire, A. L., Lupski, J. R., Posey, J. E., Khayat, M. M., et al. (2018). The phenotypic spectrum of Xia-Gibbs syndrome. Am. J. Med. Genet. A 176, 1315–1326.
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., Mcconkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574.
Kahrizi, K., Hu, H., Hosseini, M., Kalscheuer, V. M., Fattahi, Z., Beheshtian, M., et al. (2019). Effect of inbreeding on intellectual disability revisited by trio sequencing. Clin. Genet. 95, 151–159. doi: 10.1111/cge.13463
Kalscheuer, V. M., Freude, K., Musante, L., Jensen, L. R., Yntema, H. G., Gecz, J., et al. (2003). Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 35, 313–315. doi: 10.1038/ng1264
Kannan, M., Bayam, E., Wagner, C., Rinaldi, B., Kretz, P. F., Tilly, P., et al. (2017). WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc. Natl. Acad. Sci. U.S.A. 114, E9308–E9317. doi: 10.1073/pnas.1713625114
Kosmicki, J. A., Samocha, K. E., Howrigan, D. P., Sanders, S. J., Slowikowski, K., Lek, M., et al. (2017). Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510. doi: 10.1038/ng.3789
Labonne, J. D., Lee, K. H., Iwase, S., Kong, I. K., Diamond, M. P., Layman, L. C., et al. (2016). An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum. Genet. 135, 757–771. doi: 10.1007/s00439-016-1668-4
Le, V. S., Tran, K. T., Bui, H. T. P., Le, H. T. T., Nguyen, C. D., Do, D. H., et al. (2019). A Vietnamese human genetic variation database. Hum. Mutat. 40, 1664–1675.
Lee, C. T., Freed, W. J., and Mash, D. C. (2015). CNVs in neurodevelopmental disorders. Oncotarget 6, 18238–18239. doi: 10.18632/oncotarget.4853
Lelieveld, S. H., Reijnders, M. R., Pfundt, R., Yntema, H. G., Kamsteeg, E. J., De Vries, P., et al. (2016). Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19, 1194–1196. doi: 10.1038/nn.4352
Lenski, C., Abidi, F., Meindl, A., Gibson, A., Platzer, M., Frank Kooy, R., et al. (2004). Novel truncating mutations in the polyglutamine tract binding protein 1 gene (PQBP1) cause Renpenning syndrome and X-linked mental retardation in another family with microcephaly. Am. J. Hum. Genet. 74, 777–780. doi: 10.1086/383205
Leventopoulos, G., Denayer, E., Makrythanasis, P., Papapolychroniou, C., and Fryssira, H. (2010). Noonan syndrome and systemic lupus erythematosus in a patient with a novel KRAS mutation. Clin. Exp. Rheumatol. 28, 556–557.
Li, H. J., Haque, Z. K., Chen, A., and Mendelsohn, M. (2007). RIF-1, a novel nuclear receptor corepressor that associates with the nuclear matrix. J. Cell Biochem. 102, 1021–1035. doi: 10.1002/jcb.21340
Li, J., Cai, T., Jiang, Y., Chen, H., He, X., Chen, C., et al. (2016). Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol. Psychiatry 21:298.
Li, J., Kale Edmiston, E., Chen, K., Tang, Y., Ouyang, X., Jiang, Y., et al. (2014). A comparative diffusion tensor imaging study of corpus callosum subregion integrity in bipolar disorder and schizophrenia. Psychiatry Res. 221, 58–62. doi: 10.1016/j.pscychresns.2013.10.007
Li, J., Wang, L., Yu, P., Shi, L., Zhang, K., Sun, Z. S., et al. (2017). Vitamin D-related genes are subjected to significant de novo mutation burdens in autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 568–577. doi: 10.1002/ajmg.b.32543
Li, X., Wang, W., Wang, J., Malovannaya, A., Xi, Y., Li, W., et al. (2015). Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 11:775. doi: 10.15252/msb.20145504
Lichter, P., Tang, C. J., Call, K., Hermanson, G., Evans, G. A., Housman, D., et al. (1990). High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science 247, 64–69. doi: 10.1126/science.2294592
Lim, E. T., Uddin, M., De Rubeis, S., Chan, Y., Kamumbu, A. S., Zhang, X., et al. (2017). Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 20, 1217–1224. doi: 10.1038/nn.4598
Lin, W., Zhang, J., Liu, Y., Wu, R., Yang, H., Hu, X., et al. (2017). Studies on diagnostic biomarkers and therapeutic mechanism of Alzheimer’s disease through metabolomics and hippocampal proteomics. Eur. J. Pharm. Sci. 105, 119–126. doi: 10.1016/j.ejps.2017.05.003
Liu, Y. F., Sowell, S. M., Luo, Y., Chaubey, A., Cameron, R. S., Kim, H. G., et al. (2015). Autism and intellectual disability-associated KIRREL3 interacts with neuronal proteins MAP1B and MYO16 with potential roles in neurodevelopment. PLoS One 10:e0123106. doi: 10.1371/journal.pone.0123106
Lo, I. F., Cheung, L. Y., Ng, A. Y., and Lam, S. T. (1998). Interstitial Dup(1p) with findings of Kabuki make-up syndrome. Am. J. Med. Genet. 78, 55–57.
Luck, K., Kim, D. K., Lambourne, L., Spirohn, K., Begg, B. E., Bian, W., et al. (2020). A reference map of the human binary protein interactome. Nature 580, 402–408.
Mandic-Maravic, V., Mitkovic-Voncina, M., Pljesa-Ercegovac, M., Savic-Radojevic, A., Djordjevic, M., Ercegovac, M., et al. (2021). Glutathione S-transferase polymorphisms and clinical characteristics in autism spectrum disorders. Front. Psychiatry 12:672389. doi: 10.3389/fpsyt.2021.672389
Mapelli, L., Soda, T., D’Angelo, E., and Prestori, F. (2022). The Cerebellar involvement in autism spectrum disorders: From the social brain to mouse models. Int. J. Mol. Sci. 23:3894. doi: 10.3390/ijms23073894
Marcon, E., Ni, Z., Pu, S., Turinsky, A. L., Trimble, S. S., Olsen, J. B., et al. (2014). Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation. Cell Rep. 8, 297–310. doi: 10.1016/j.celrep.2014.05.050
Martin, T. D., Cook, D. R., Choi, M. Y., Li, M. Z., Haigis, K. M., and Elledge, S. J. (2017). A role for mitochondrial translation in promotion of viability in K-Ras mutant cells. Cell Rep. 20, 427–438. doi: 10.1016/j.celrep.2017.06.061
Martinelli, S., Krumbach, O. H. F., Pantaleoni, F., Coppola, S., Amin, E., Pannone, L., et al. (2018). Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am. J. Hum. Genet. 102, 309–320. doi: 10.1016/j.ajhg.2017.12.015
Mary, L., Piton, A., Schaefer, E., Mattioli, F., Nourisson, E., Feger, C., et al. (2018). Disease-causing variants in TCF4 are a frequent cause of intellectual disability: Lessons from large-scale sequencing approaches in diagnosis. Eur. J. Hum. Genet. 26, 996–1006. doi: 10.1038/s41431-018-0096-4
Matsunami, N., Hensel, C. H., Baird, L., Stevens, J., Otterud, B., Leppert, T., et al. (2014). Identification of rare DNA sequence variants in high-risk autism families and their prevalence in a large case/control population. Mol. Autism 5:5. doi: 10.1186/2040-2392-5-5
Mattia, F. R., Wardinsky, T. D., Tuttle, D. J., Grix, A. Jr., Smith, K. A., and Walling, P. (1992). Interstitial deletion of the short arm of chromosome 1 (46XY, del(1)(p13p22.3)). Am. J. Med. Genet. 44, 551–554. doi: 10.1002/ajmg.1320440503
Monroe, G. R., Frederix, G. W., Savelberg, S. M., De Vries, T. I., Duran, K. J., Van Der Smagt, J. J., et al. (2016). Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet. Med. 18, 949–956. doi: 10.1038/gim.2015.200
Morais, S., Raymond, L., Mairey, M., Coutinho, P., Brandao, E., Ribeiro, P., et al. (2017). Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. Eur. J. Hum. Genet. 25, 1217–1228.
Movilla, N., and Bustelo, X. R. (1999). Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell Biol. 19, 7870–7885. doi: 10.1128/MCB.19.11.7870
Muller, P. M., Rademacher, J., Bagshaw, R. D., Wortmann, C., Barth, C., Van Unen, J., et al. (2020). Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions. Nat. Cell Biol. 22, 498–511. doi: 10.1038/s41556-020-0488-x
Murphy, W. J., Fronicke, L., O’Brien, S. J., and Stanyon, R. (2003). The origin of human chromosome 1 and its homologs in placental mammals. Genome Res. 13, 1880–1888. doi: 10.1101/gr.1022303
Najmabadi, H., Hu, H., Garshasbi, M., Zemojtel, T., Abedini, S. S., Chen, W., et al. (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478, 57–63. doi: 10.1038/nature10423
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Palumbo, O., Palumbo, P., Delvecchio, M., Palladino, T., Stallone, R., Crisetti, M., et al. (2015). Microdeletion of 12q24.31: Report of a girl with intellectual disability, stereotypies, seizures and facial dysmorphisms. Am. J. Med. Genet. A 167A, 438–444. doi: 10.1002/ajmg.a.36872
Park, J. M., Lee, B., Kim, J. H., Park, S. Y., Yu, J., Kim, U. K., et al. (2020). An autosomal dominant ERLIN2 mutation leads to a pure HSP phenotype distinct from the autosomal recessive ERLIN2 mutations (SPG18). Sci. Rep. 10:3295. doi: 10.1038/s41598-020-60374-y
Paul, L. K., Brown, W. S., Adolphs, R., Tyszka, J. M., Richards, L. J., Mukherjee, P., et al. (2007). Agenesis of the corpus callosum: Genetic, developmental and functional aspects of connectivity. Nat. Rev. Neurosci. 8, 287–299. doi: 10.1038/nrn2107
Pekeles, H., Accogli, A., Boudrahem-Addour, N., Russell, L., Parente, F., and Srour, M. (2019). Diagnostic yield of intellectual disability gene panels. Pediatr. Neurol. 92, 32–36.
Piccione, M., Antona, V., Antona, R., Gambino, G., Pierluigi, M., Malacarne, M., et al. (2010). Array-CGH defined chromosome 1p duplication in a patient with autism spectrum disorder, mild mental deficiency, and minor dysmorphic features. Am. J. Med. Genet. A 152A, 486–489. doi: 10.1002/ajmg.a.33212
Pinero, J., Ramirez-Anguita, J. M., Sauch-Pitarch, J., Ronzano, F., Centeno, E., Sanz, F., et al. (2020). The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 48, D845–D855. doi: 10.1093/nar/gkz1021
Pinero, J., Sauch, J., Sanz, F., and Furlong, L. I. (2021). The DisGeNET cytoscape app: Exploring and visualizing disease genomics data. Comput. Struct. Biotechnol. J. 19, 2960–2967. doi: 10.1016/j.csbj.2021.05.015
Popp, B., Ekici, A. B., Thiel, C. T., Hoyer, J., Wiesener, A., Kraus, C., et al. (2017). Exome Pool-Seq in neurodevelopmental disorders. Eur. J. Hum. Genet. 25, 1364–1376. doi: 10.1038/s41431-017-0022-1
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Quevedo, C., Sauzeau, V., Menacho-Marquez, M., Castro-Castro, A., and Bustelo, X. R. (2010). Vav3-deficient mice exhibit a transient delay in cerebellar development. Mol. Biol. Cell 21, 1125–1139. doi: 10.1091/mbc.E09-04-0292
Rahman, S. K., Okazawa, H., and Chen, Y. W. (2019). Frameshift PQBP-1 mutants K192S(fs*7) and R153S(fs*41) implicated in X-linked intellectual disability form stable dimers. J. Struct. Biol. 206, 305–313. doi: 10.1016/j.jsb.2019.04.003
Ramser, J., Abidi, F. E., Burckle, C. A., Lenski, C., Toriello, H., Wen, G., et al. (2005). A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum. Mol. Genet. 14, 1019–1027. doi: 10.1093/hmg/ddi094
Redin, C., Gerard, B., Lauer, J., Herenger, Y., Muller, J., Quartier, A., et al. (2014). Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 51, 724–736.
Redon, R., Rio, M., Gregory, S. G., Cooper, R. A., Fiegler, H., Sanlaville, D., et al. (2005). Tiling path resolution mapping of constitutional 1p36 deletions by array-CGH: Contiguous gene deletion or “deletion with positional effect” syndrome? J. Med. Genet. 42, 166–171. doi: 10.1136/jmg.2004.023861
Reijnders, M. R. F., Ansor, N. M., Kousi, M., Yue, W. W., Tan, P. L., Clarkson, K., et al. (2017). RAC1 missense mutations in developmental disorders with diverse phenotypes. Am. J. Hum. Genet. 101, 466–477. doi: 10.1016/j.ajhg.2017.08.007
Rice, A. M., and McLysaght, A. (2017). Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 8:14366. doi: 10.1038/ncomms14366
Rodriguez-Fdez, S., Lorenzo-Martin, L. F., Fabbiano, S., Menacho-Marquez, M., Sauzeau, V., Dosil, M., et al. (2021). New functions of vav family proteins in cardiovascular biology, skeletal muscle, and the nervous system. Biology 10:857. doi: 10.3390/biology10090857
Roohi, J., Tegay, D. H., Pomeroy, J. C., Burkett, S., Stone, G., Stanyon, R., et al. (2008). A de novo apparently balanced translocation [46,XY,t(2;9)(p13;p24)] interrupting RAB11FIP5 identifies a potential candidate gene for autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 411–417.
Roston, A., Evans, D., Gill, H., McKinnon, M., Isidor, B., Cogne, B., et al. (2021). SETD1B-associated neurodevelopmental disorder. J. Med. Genet. 58, 196–204. doi: 10.1136/jmedgenet-2019-106756
Rydning, S. L., Dudesek, A., Rimmele, F., Funke, C., Kruger, S., Biskup, S., et al. (2018). A novel heterozygous variant in ERLIN2 causes autosomal dominant pure hereditary spastic paraplegia. Eur. J. Neurol. 25, 943–e971. doi: 10.1111/ene.13625
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of Autism. Cell 180, 568–584 e523. doi: 10.1016/j.cell.2019.12.036
Sauzeau, V., Horta-Junior, J. A., Riolobos, A. S., Fernandez, G., Sevilla, M. A., Lopez, D. E., et al. (2010). Vav3 is involved in GABAergic axon guidance events important for the proper function of brainstem neurons controlling cardiovascular, respiratory, and renal parameters. Mol. Biol. Cell 21, 4251–4263. doi: 10.1091/mbc.E10-07-0639
Schubbert, S., Zenker, M., Rowe, S. L., Boll, S., Klein, C., Bollag, G., et al. (2006). Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 38, 331–336.
Searles Nielsen, S., Bammler, T. K., Gallagher, L. G., Farin, F. M., Longstreth, W. Jr., Franklin, G. M., et al. (2013). Genotype and age at Parkinson disease diagnosis. Int. J. Mol. Epidemiol. Genet. 4, 61–69.
Shaheen, R., Maddirevula, S., Ewida, N., Alsahli, S., Abdel-Salam, G. M. H., Zaki, M. S., et al. (2019). Genomic and phenotypic delineation of congenital microcephaly. Genet. Med. 21, 545–552.
Shami Shah, A., Batrouni, A. G., Kim, D., Punyala, A., Cao, W., Han, C., et al. (2019). PLEKHA4/kramer attenuates dishevelled ubiquitination to modulate Wnt and planar cell polarity signaling. Cell Rep. 27, 2157–2170 e2158. doi: 10.1016/j.celrep.2019.04.060
Silva, L. E., Souza, R. C., Kitano, E. S., Monteiro, L. F., Iwai, L. K., and Forti, F. L. (2019). Proteomic and interactome approaches reveal PAK4, PHB-2, and 14-3-3eta as targets of overactivated Cdc42 in cellular responses to genomic instability. J. Proteome Res. 18, 3597–3614. doi: 10.1021/acs.jproteome.9b00260
Snijders Blok, L., Rousseau, J., Twist, J., Ehresmann, S., Takaku, M., Venselaar, H., et al. (2018). CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat. Commun. 9:4619. doi: 10.1038/s41467-018-06014-6
Snijders Blok, L., Rousseau, J., Twist, J., Ehresmann, S., Takaku, M., Venselaar, H., et al. (2019). Author correction: CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat. Commun. 10:2079.
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672.
Stelzl, U., Worm, U., Lalowski, M., Haenig, C., Brembeck, F. H., Goehler, H., et al. (2005). A human protein-protein interaction network: A resource for annotating the proteome. Cell 122, 957–968.
Stranger, B. E., Forrest, M. S., Dunning, M., Ingle, C. E., Beazley, C., Thorne, N., et al. (2007). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853. doi: 10.1126/science.1136678
Sun, S. C., Ma, D., Li, M. Y., Zhang, R. X., Huang, C., Huang, H. J., et al. (2019). Mutations in C1orf194, encoding a calcium regulator, cause dominant Charcot-Marie-Tooth disease. Brain 142, 2215–2229. doi: 10.1093/brain/awz151
Szczawinska-Poplonyk, A., Ploski, R., Bernatowska, E., and Pac, M. (2020). A Novel CDC42 mutation in an 11-year old child manifesting as syndromic immunodeficiency, autoinflammation, hemophagocytic lymphohistiocytosis, and malignancy: A case report. Front. Immunol. 11:318. doi: 10.3389/fimmu.2020.00318
Tabata, H., Sone, K., Kobayashi, T., Yanagisawa, T., Tamura, T., Shimizu, N., et al. (1991). Short arm deletion of chromosome 1: Del(1)(p13.3 p22.3) in a female infant with an extreme tetralogy of Fallot. Clin. Genet. 39, 132–135. doi: 10.1111/j.1399-0004.1991.tb02999.x
Takata, A., Nakashima, M., Saitsu, H., Mizuguchi, T., Mitsuhashi, S., Takahashi, Y., et al. (2019). Comprehensive analysis of coding variants highlights genetic complexity in developmental and epileptic encephalopathy. Nat. Commun. 10:2506. doi: 10.1038/s41467-019-10482-9
Tawamie, H., Martianov, I., Wohlfahrt, N., Buchert, R., Mengus, G., Uebe, S., et al. (2017). Hypomorphic pathogenic variants in TAF13 are associated with autosomal-recessive intellectual disability and microcephaly. Am. J. Hum. Genet. 100, 555–561. doi: 10.1016/j.ajhg.2017.01.032
Toma, C., Shaw, A. D., Overs, B. J., Mitchell, P. B., Schofield, P. R., Cooper, A. A., et al. (2020). De novo gene variants and familial bipolar disorder. JAMA Netw. Open 3:e203382.
Tran Mau-Them, F., Moutton, S., Racine, C., Vitobello, A., Bruel, A. L., Nambot, S., et al. (2020). Second-tier trio exome sequencing after negative solo clinical exome sequencing: An efficient strategy to increase diagnostic yield and decipher molecular bases in undiagnosed developmental disorders. Hum. Genet. 139, 1381–1390. doi: 10.1007/s00439-020-02178-8
Travaglini, L., Aiello, C., Stregapede, F., D’amico, A., Alesi, V., Ciolfi, A., et al. (2018). The impact of next-generation sequencing on the diagnosis of pediatric-onset hereditary spastic paraplegias: New genotype-phenotype correlations for rare HSP-related genes. Neurogenetics 19, 111–121. doi: 10.1007/s10048-018-0545-9
Trinh, J., Kandaswamy, K. K., Werber, M., Weiss, M. E. R., Oprea, G., Kishore, S., et al. (2019). Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. J. Neurodev. Disord. 11:11.
Turner, T. N., Coe, B. P., Dickel, D. E., Hoekzema, K., Nelson, B. J., Zody, M. C., et al. (2017). Genomic patterns of de novo mutation in simplex autism. Cell 171, 710–722.e712.
Turner, T. N., Wilfert, A. B., Bakken, T. E., Bernier, R. A., Pepper, M. R., Zhang, Z., et al. (2019). Sex-based analysis of de novo variants in neurodevelopmental disorders. Am. J. Hum. Genet. 105, 1274–1285. doi: 10.1016/j.ajhg.2019.11.003
Turro, E., Astle, W. J., Megy, K., Graf, S., Greene, D., Shamardina, O., et al. (2020). Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583, 96–102. doi: 10.1038/s41586-020-2434-2
Uddin, M., Tammimies, K., Pellecchia, G., Alipanahi, B., Hu, P., Wang, Z., et al. (2014). Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat. Genet. 46, 742–747. doi: 10.1038/ng.2980
Uehara, D. T., Hayashi, S., Okamoto, N., Mizuno, S., Chinen, Y., Kosaki, R., et al. (2016). SNP array screening of cryptic genomic imbalances in 450 Japanese subjects with intellectual disability and multiple congenital anomalies previously negative for large rearrangements. J. Hum. Genet. 61, 335–343. doi: 10.1038/jhg.2015.154
Utkus, A., Sorokina, I., Kucinskas, V., Rothlisberger, B., Balmer, D., Brecevic, L., et al. (1999). Duplication of segment 1p21 following paternal insertional translocation, ins(6;1)(q25;p13.3p22.1). J. Med. Genet. 36, 73–76.
van Kuilenburg, A. B., Meijer, J., Mul, A. N., Hennekam, R. C., Hoovers, J. M., de Die-Smulders, C. E., et al. (2009). Analysis of severely affected patients with dihydropyrimidine dehydrogenase deficiency reveals large intragenic rearrangements of DPYD and a de novo interstitial deletion del(1)(p13.3p21.3). Hum. Genet. 125, 581–590. doi: 10.1007/s00439-009-0653-6
Vandepoele, K., Van Roy, N., Staes, K., Speleman, F., and van Roy, F. (2005). A novel gene family NBPF: Intricate structure generated by gene duplications during primate evolution. Mol. Biol. Evol. 22, 2265–2274. doi: 10.1093/molbev/msi222
Vergult, S., Van Binsbergen, E., Sante, T., Nowak, S., Vanakker, O., Claes, K., et al. (2014). Mate pair sequencing for the detection of chromosomal aberrations in patients with intellectual disability and congenital malformations. Eur. J. Hum. Genet. 22, 652–659. doi: 10.1038/ejhg.2013.220
Vissers, L. E., van Ravenswaaij, C. M., Admiraal, R., Hurst, J. A., de Vries, B. B., Janssen, I. M., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957. doi: 10.1038/ng1407
Wang, Q., Li, M., Yang, Z., Hu, X., Wu, H. M., Ni, P., et al. (2015). Increased co-expression of genes harboring the damaging de novo mutations in Chinese schizophrenic patients during prenatal development. Sci. Rep. 5:18209. doi: 10.1038/srep18209
Wang, T., Guo, H., Xiong, B., Stessman, H. A., Wu, H., Coe, B. P., et al. (2016). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 7:13316. doi: 10.1038/ncomms13316
Wang, T., Hoekzema, K., Vecchio, D., Wu, H., Sulovari, A., Coe, B. P., et al. (2020). Author correction: Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun. 11:. doi: 10.1038/s41467-020-19289-5
Weerts, M. J. A., Lanko, K., Guzman-Vega, F. J., Jackson, A., Ramakrishnan, R., Cardona-Londono, K. J., et al. (2021). Delineating the molecular and phenotypic spectrum of the SETD1B-related syndrome. Genet. Med. 23, 2122–2137. doi: 10.1038/s41436-021-01246-2
Weiss, K., Lazar, H. P., Kurolap, A., Martinez, A. F., Paperna, T., Cohen, L., et al. (2020). Correction: The CHD4-related syndrome: A comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis. Genet. Med. 22:669. doi: 10.1038/s41436-019-0727-3
Weiss, K., Terhal, P. A., Cohen, L., Bruccoleri, M., Irving, M., Martinez, A. F., et al. (2016). De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am. J. Hum. Genet. 99, 934–941. doi: 10.1016/j.ajhg.2016.08.001
Wright, F. A., Bonzerato, C. G., Sliter, D. A., and Wojcikiewicz, R. J. H. (2018). The erlin2 T65I mutation inhibits erlin1/2 complex-mediated inositol 1,4,5-trisphosphate receptor ubiquitination and phosphatidylinositol 3-phosphate binding. J. Biol. Chem. 293, 15706–15714. doi: 10.1074/jbc.RA118.004547
Writzl, K., Maver, A., Kovacic, L., Martinez-Valero, P., Contreras, L., Satrustegui, J., et al. (2017). De novo mutations in slc25a24 cause a disorder characterized by early aging, bone dysplasia, characteristic face, and early demise. Am. J. Hum. Genet. 101, 844–855. doi: 10.1016/j.ajhg.2017.09.017
Wu, S. F., Xia, L., Shi, X. D., Dai, Y. J., Zhang, W. N., Zhao, J. M., et al. (2020). RIG-I regulates myeloid differentiation by promoting TRIM25-mediated ISGylation. Proc. Natl. Acad. Sci. U.S.A. 117, 14395–14404. doi: 10.1073/pnas.1918596117
Xia, F., Bainbridge, M. N., Tan, T. Y., Wangler, M. F., Scheuerle, A. E., Zackai, E. H., et al. (2014). De novo truncating mutations in AHDC1 in individuals with syndromic expressive language delay, hypotonia, and sleep apnea. Am. J. Hum. Genet. 94, 784–789. doi: 10.1016/j.ajhg.2014.04.006
Yang, H., Douglas, G., Monaghan, K. G., Retterer, K., Cho, M. T., Escobar, L. F., et al. (2015). De novo truncating variants in the AHDC1 gene encoding the AT-hook DNA-binding motif-containing protein 1 are associated with intellectual disability and developmental delay. Cold Spring Harb. Mol. Case Stud. 1: a000562.
Yildirim, Y., Orhan, E.K., Iseri, S.A., Serdaroglu-Oflazer, P., Kara, B., Solakoglu, S. et al. (2011). A frameshift mutation of ERLIN2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum Mol Genet 20, 1886-1892
Yuen, R.K., Merico, D., Cao, H., Pellecchia, G., Alipanahi, B., Thiruvahindrapuram, B. et al. (2016). Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med 1:, 160271–1602710.
Zeng, L., Sachdev, P., Yan, L., Chan, J.L., Trenkle, T., Mcclelland, M. et al. (2000). Vav3 mediates receptor protein tyrosine kinase signaling, regulates GTPase activity, modulates cell morphology, and induces cell transformation. Mol Cell Biol 20:, 9212-9224.
Zenker, M., Lehmann, K., Schulz, A.L., Barth, H., Hansmann, D., Koenig, R. et al. (2007). Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet 44:, 131-135.
Zhang, Y., Wang, T., Wang, Y., Xia, K., Li, J., and Sun, Z. (2021). Targeted sequencing and integrative analysis to prioritize candidate genes in neurodevelopmental disorders. Mol Neurobiol 58, 3863-3873.
Keywords: 1p13.3 deletion, intellectual disability, autism, VAV3, WDR47, ELAPOR1, GSTM5, LRIF1
Citation: Ben-Mahmoud A, Jun KR, Gupta V, Shastri P, de la Fuente A, Park Y, Shin KC, Kim CA, da Cruz AD, Pinto IP, Minasi LB, Silva da Cruz A, Faivre L, Callier P, Racine C, Layman LC, Kong I-K, Kim C-H, Kim W-Y and Kim H-G (2022) A rigorous in silico genomic interrogation at 1p13.3 reveals 16 autosomal dominant candidate genes in syndromic neurodevelopmental disorders. Front. Mol. Neurosci. 15:979061. doi: 10.3389/fnmol.2022.979061
Received: 27 June 2022; Accepted: 30 August 2022;
Published: 06 October 2022.
Edited by:
Javier Corral, University of Murcia, SpainReviewed by:
Ferdinando Di Cunto, University of Turin, ItalyCopyright © 2022 Ben-Mahmoud, Jun, Gupta, Shastri, de la Fuente, Park, Shin, Kim, da Cruz, Pinto, Minasi, Silva da Cruz, Faivre, Callier, Racine, Layman, Kong, Kim, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyung-Goo Kim, aGtpbUBoYmt1LmVkdS5xYQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.