 Liang Wang1†
Liang Wang1† Ziyun Yang1,2,3,4†
Ziyun Yang1,2,3,4† Xiumei He2,3,4
Xiumei He2,3,4 Shiming Pu2,3,4
Shiming Pu2,3,4 Cheng Yang2,3,4
Cheng Yang2,3,4 Qiong Wu2,3,4
Qiong Wu2,3,4 Zuping Zhou3,4
Zuping Zhou3,4 Xiaobo Cen1
Xiaobo Cen1 Hongxia Zhao2,3,4,5*
Hongxia Zhao2,3,4,5*- 1National Chengdu Center for Safety Evaluation of Drugs, State Key Laboratory of Biotherapy/Collaborative Innovation Center for Biotherapy, West China Hospital of Sichuan University, Chengdu, China
- 2School of Life Sciences, Guangxi Normal University, Guilin, China
- 3Guangxi Universities, Key Laboratory of Stem Cell and Biopharmaceutical Technology, Guangxi Normal University, Guilin, China
- 4Research Center for Biomedical Sciences, Guangxi Normal University, Guilin, China
- 5Faculty of Biological and Environmental Sciences, University of Helsinki, Helsinki, Finland
Mitochondria are essential organelles for neuronal function and cell survival. Besides the well-known bioenergetics, additional mitochondrial roles in calcium signaling, lipid biogenesis, regulation of reactive oxygen species, and apoptosis are pivotal in diverse cellular processes. The mitochondrial proteome encompasses about 1,500 proteins encoded by both the nuclear DNA and the maternally inherited mitochondrial DNA. Mutations in the nuclear or mitochondrial genome, or combinations of both, can result in mitochondrial protein deficiencies and mitochondrial malfunction. Therefore, mitochondrial quality control by proteins involved in various surveillance mechanisms is critical for neuronal integrity and viability. Abnormal proteins involved in mitochondrial bioenergetics, dynamics, mitophagy, import machinery, ion channels, and mitochondrial DNA maintenance have been linked to the pathogenesis of a number of neurological diseases. The goal of this review is to give an overview of these pathways and to summarize the interconnections between mitochondrial protein dysfunction and neurological diseases.
Introduction
The central nervous system (CNS) is a highly specialized organ and consists of distinct cells, such as neurons, astrocytes, microglia, oligodendrocytes, and ependymal cells, all of which work independently but collectively to perform a variety of neuronal functions (Caruso Bavisotto et al., 2019). As one of the most metabolically active organs in the body, the CNS accounts for only ~2% of total body weight, yet consumes at least 20% of the body’s energy (Sokoloff, 1973). It is thus not surprising that the CNS is densely populated with mitochondria (Magistretti and Allaman, 2015). Growing evidence indicates that mitochondria are important for neural development and activities, including neuronal sprouting and plasticity, synaptic transmission and connection, neural oscillations, and cognition (Misgeld and Schwarz, 2017; Nortley and Attwell, 2017; Fang et al., 2019; Filiou and Sandi, 2019). Therefore, mitochondrial dysfunction has been demonstrated as a key player in the development and progression of many neurological disorders.
Mitochondria perform critical roles beyond simply serving as a powerhouse for producing adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS). They also play vital roles in calcium (Ca2+) buffering, biogenesis of lipids and iron-sulfur [Fe-S] clusters, production of reactive oxygen species (ROS), and apoptosis (Osellame et al., 2012; Rangaraju et al., 2019). To fulfill these functions, mitochondria form highly dynamic and motile networks that undergo constant morphology and positioning changes through fusion and fission (El-Hattab et al., 2018). Mitochondria in neurons are extremely dynamic and move quickly along microtubule tracks to facilitate transport to match energy supply and demand, ensure their distribution to the neuronal periphery, allow the exchange of components between mitochondria, and mediate the removal of damaged mitochondria via mitophagy (Seager et al., 2020).
A large variety of mitochondrial functions, as well as their morphogenesis and dynamics, have been assigned to mitochondrial proteins and protein complexes (Pfanner et al., 2019). In humans, it is estimated that mitochondria contain at least 1,500 different proteins (Taylor et al., 2003; Lefort et al., 2009). These proteins are involved in mitochondrial bioenergetics, dynamics, mitophagy, import machinery, ion channels, and mitochondrial DNA maintenance (Figure 1). As semi-autonomous organelles, mitochondria encode and synthesize 13 proteins, all of which are core subunits of the respiratory chain. The remaining nearly 99% of mitochondrial proteins are encoded by the nuclear genome, synthesized in the cytosol, and imported into the mitochondria by import, sorting, and assembly machinery (Schmidt et al., 2010). Protein functional analysis shows that approximately 15% of the proteins are directly involved in OXPHOS, 10% of proteins participate in the metabolism of iron, lipids, and amino acids, 20%–25% of proteins maintain and regulate the mitochondrial genome, and about 25% of proteins are involved in a variety of cell signaling, redox, protein and metabolite transport, mitochondrial morphology, and dynamics. Additionally, the cellular roles of almost 20% of mitochondrial proteins still remain unknown (Fox, 2012; Pfanner et al., 2019). The central importance of mitochondrial proteins has been widely highlighted by the association of aberrant mitochondrial protein expression (Lin et al., 2004; Ekstrand et al., 2007; Tysnes and Storstein, 2017), localization (Gandhi, 2006; Lu and Guo, 2020), folding, and function (Castro et al., 2018; Franco-Iborra et al., 2018) with neurological diseases (Figure 1).
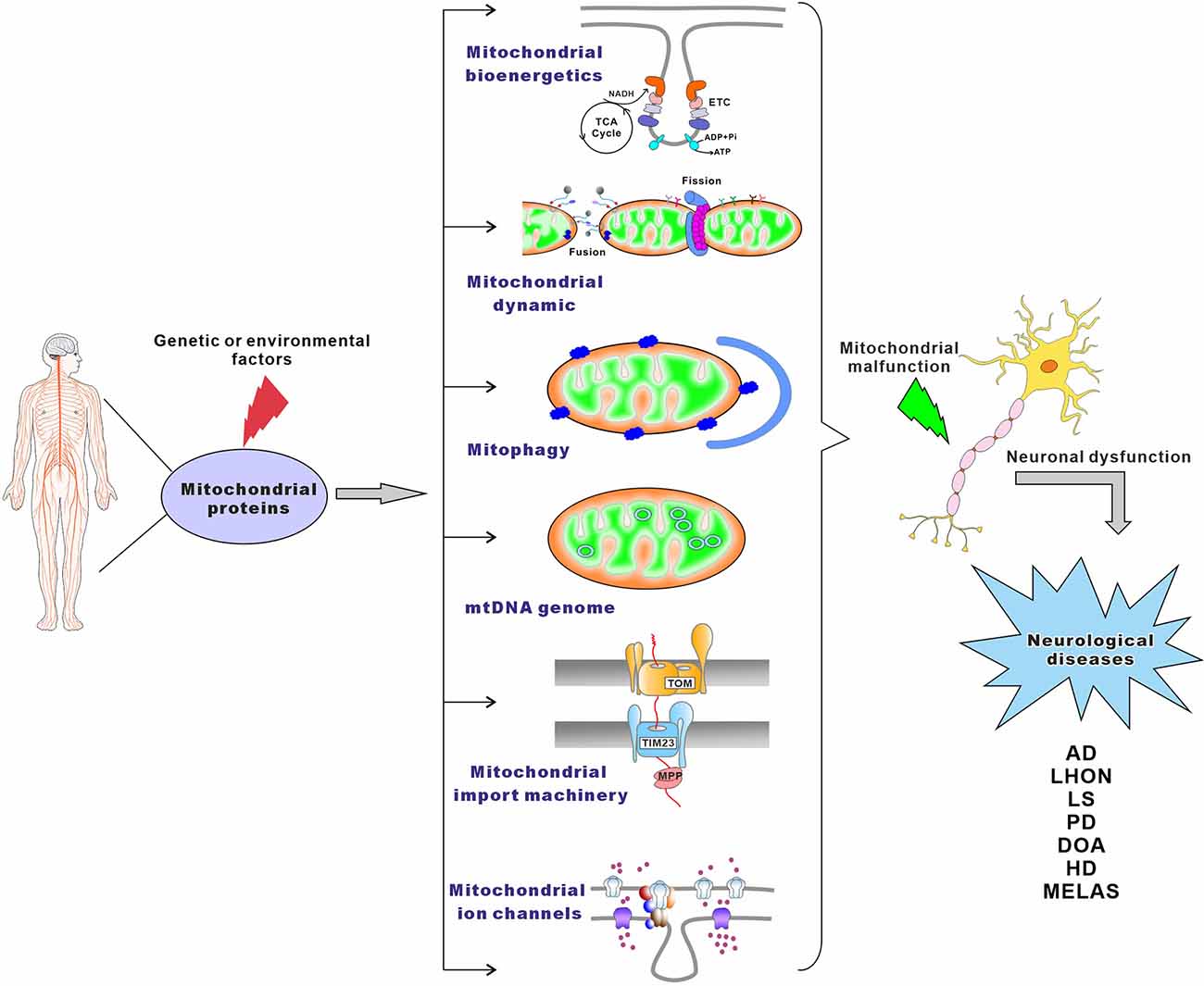
Figure 1. The main pathways leading to mitochondrial-associated neurological disorders. Genetic or environmental factors can cause mutations of mitochondrial proteins, which lead to mitochondrial malfunction. Neurological disorders may result from damaged mitochondrial bioenergetics, imbalanced mitochondrial dynamics, defective mitophagy, impaired maintenance of mitochondrial genome, compromised mitochondrial importing machinery, altered mitochondrial ion channels, or combinations thereof.
Neurological diseases constitute a group of disorders characterized by a progressive deterioration of the brain and spinal cord functions (Farooqui, 2019). Common neurodegenerative diseases include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (Bredesen et al., 2006; Fu et al., 2018). Neuropsychiatric diseases mainly refer to the abnormalities in the cerebral cortex and limbic system, including both neurodevelopmental disorders and behavioral or psychological difficulties associated with some neurological disorders (Farooqui, 2019). Common neuropsychiatric diseases are autism, depression, schizophrenia, and some forms of bipolar disorders, such as depression and anxiety (Lyalina et al., 2013).
In this review, we focus on the association of mitochondrial protein dysfunction with the pathogenesis of neurological diseases, such as AD, PD, and ALS, as well as psychiatric disorders like depression, anxiety, and schizophrenia. Based on the functional classification of mitochondrial proteins, we begin by introducing their key roles in mitochondria. We then incorporate new evidence demonstrating that abnormal mitochondrial protein causes dysregulation of mitochondrial morphology, dynamics, and function in neurological disorders. The literature reviewed here is not exhaustive, but it highlights major findings that characterize the current status of the field in terms of protein-linked mitochondrial quality control in the CNS.
Dysregulation of Mitochondrial Bioenergetics
The human brain is particularly reliant on a steady supply of energy, almost entirely in the form of glucose, to meet its high metabolic demands (Harris et al., 2012; Nortley and Attwell, 2017). Among all brain cell types, neurons are the principal consumers, using 80%–90% of the total brain energy (Howarth et al., 2012; Zimmer et al., 2017). However, under basal conditions, neurons only uptake almost an equal amount of glucose as astrocytes, and their uptake is significantly lower in the functional activation state (Magistretti and Allaman, 2018). Unlike neurons, which synthesize ATP through OXPHOS, the most effective ATP-producing pathway, astrocytes predominantly metabolize glucose through aerobic glycolysis, resulting in a huge amount of lactate and pyruvate generation from glucose. The produced lactate can be transported to neurons by monocarboxylate transporters (MCTs) and hydrocarboxylic acid receptor 1 (HCAR1), where it serves as an energy substrate for OXPHOS (Morland et al., 2015; Sotelo-Hitschfeld et al., 2015; Díaz-García et al., 2017; Magistretti and Allaman, 2018).
OXPHOS occurs via the electron transport chain (ETC), which transfers electrons from donors like NADH and FADH2 to the oxygen (MITCHELL, 1961; Cogliati et al., 2021). ETC consists of four enzyme complexes that all reside in the inner mitochondrial membrane (IMM): complexes I (NADH ubiquinone oxidoreductase), II (succinate ubiquinone oxidoreductase), III (ubiquinone cytochrome c oxidoreductase or cytochrome bc1 complex), and IV (cytochrome c oxidase, COX; Breuer et al., 2013). During electron transfer, three of these complexes (complexes I, III, and IV) pump protons from the matrix to the mitochondrial intermembrane space (IMS), thereby generating an electrochemical proton gradient across the IMM that the F1Fo-ATP Synthase (also known as ATPase Synthase or complex V) exploits to drive ATP synthesis from adenosine diphosphate (ADP; Breuer et al., 2013; Guo R. et al., 2018; Signes and Fernandez-Vizarra, 2018; Nolfi-Donegan et al., 2020). The fundamental role of ETC in mitochondrial energy production is essential for neuronal survival and activity (Li et al., 2021a). However, both complexes I and III can also transfer single electrons to oxygen, resulting in the formation of oxygen superoxide in the mitochondrial matrix. Consequently, mitochondrial DNA (mtDNA), lipids, and proteins including subunits of the OXPHOS complexes exposed to this region experience oxidative damage from the superoxide radical (Dröse and Brandt, 2012; Lenaz, 2012). Thus, dysfunctions in the ETC not only reduce energy production but also result in the excessive accumulation of ROS, which are involved in the development of neurological illnesses, such as PD, AD, ALS, Leigh Syndrome (LS), and HD (Angelova and Abramov, 2018; Pepperberg, 2019; Bakare et al., 2021). Mounting evidence demonstrates that primary mitochondrial disorder-mediated human diseases often arise from mutations in the OXPHOS subunits or the proteins and RNAs required for their expression (Craven et al., 2017; Figure 2 and Table 1).
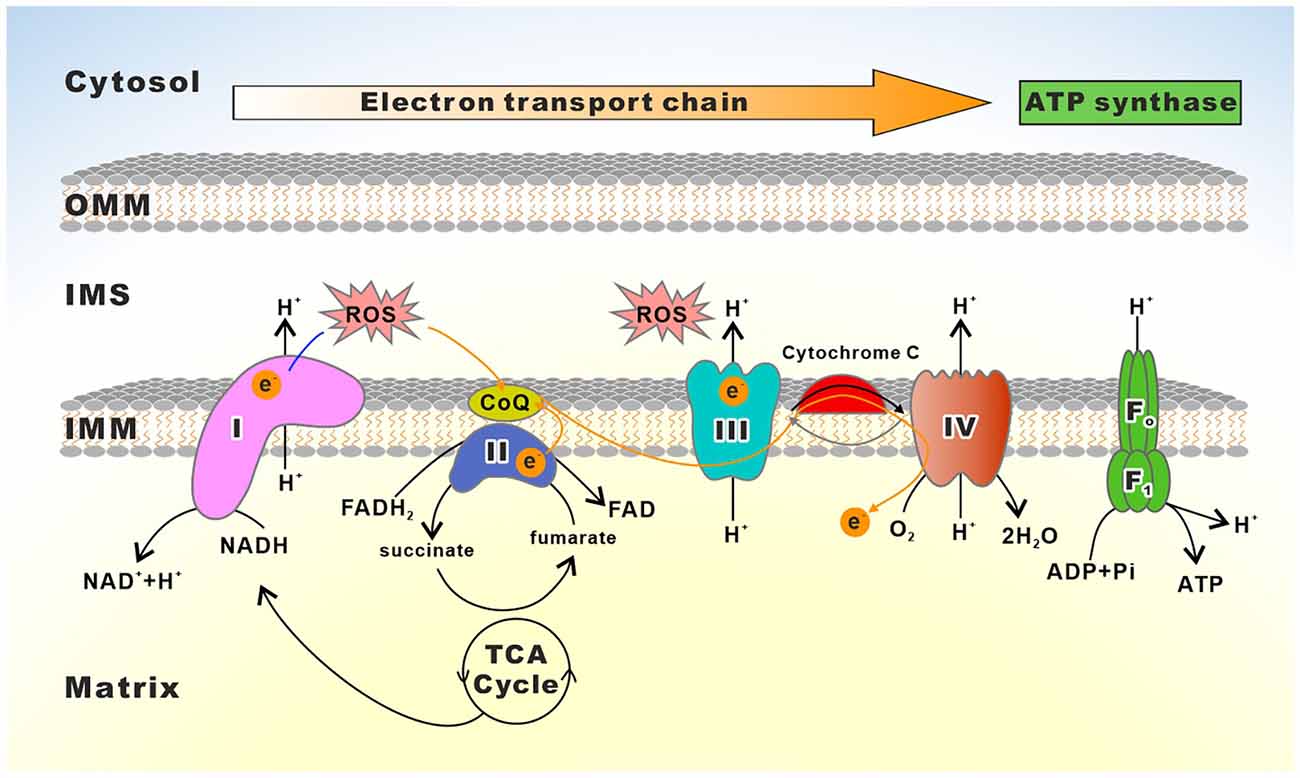
Figure 2. Schematic depiction of the mitochondrial electron transport chain. The electron transport chain (ETC) consists of complexes I to IV as well as two free electron carriers, CoQ and Cyt c. Reducing equivalents (NADH, FADH2) provide electrons flowing through complexes I, the ubiquinone cycle (Q/QH2), III, IV, and to the final acceptor O2. The electron flows through complexes resulting in the pumping of protons to the IMS, which creates a membrane potential used by the F1Fo-ATP synthase to drive the production of ATP.
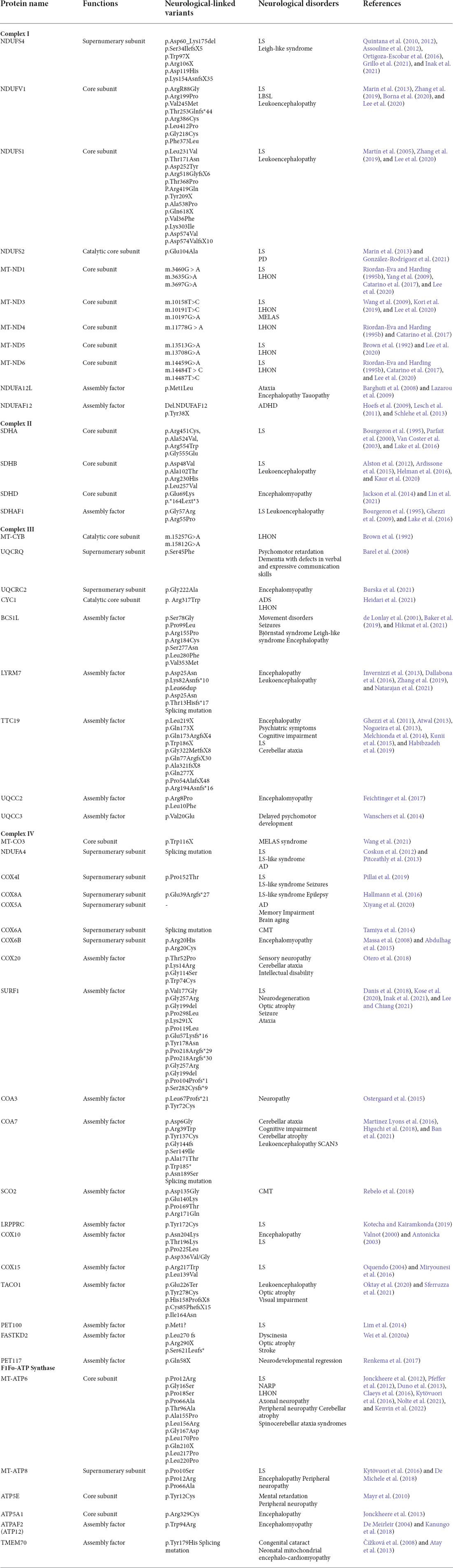
Table 1. Neurological diseases caused by protein variants involved in mitochondrial bioenergetics.
Complex I
Complex I (CI) is the first enzyme of ETC, pumping four protons into the IMS upon each NADH oxidation in the matrix and transferring two electrons to ubiquinone in the IMM via flavin mononucleotide and a series of iron-sulfur centers (Walker, 1992). Being the largest component, CI is comprised of more than 45 subunits, with seven subunits (MT-ND1- MT-ND6 and MT-ND4L) encoded by mtDNA (Sazanov, 2015). Fourteen CI subunits, which are highly conserved from prokaryotes to humans, constitute the minimal working core of the enzyme and harbor all the redox and proton-motive activity. These supernumerary subunits are thought to increase genetic complexity, susceptibility, and phenotypic diversity to some extent (Carroll et al., 2006; Papa et al., 2012; Sánchez-Caballero et al., 2016). As the point of entry in the mitochondrial ETC for NADH reducing equivalents, CI acts as a regulatable pacemaker for respiratory ATP synthesis in mammalian cells. Given the importance of CI in energy provision, any mutation that disrupts its structure or function seriously perturbs the ETC, resulting in a reduction of ATP synthesis and an accumulation of ROS and other reactive species (Fiedorczuk and Sazanov, 2018; Yin et al., 2021). Lots of studies have discovered that a large proportion of mitochondrial-related diseases are caused by mutations or chemical inhibition of the mitochondrial CI (Papa and De Rasmo, 2013; Rodenburg, 2016; Abramov and Angelova, 2019; Holper et al., 2019; González-Rodríguez et al., 2021).
NDUFS4, an accessory subunit of CI, plays an essential role in the assembly and stability of the CI (Scacco et al., 2003; Kahlhöfer et al., 2017). At the clinical level, mutations in NDUFS4 severely affect the brainstem and basal ganglia, which are typically associated with hypotonia, abnormal ocular movements, visual impairment, psychomotor arrest/regression, and episodes of respiratory failure. Children with NDUFS4 mutations (such as p.97Ter) show a homogeneous early-onset LS and a severe, lethal course of the disease (Ortigoza-Escobar et al., 2016). Additionally, the NDUFS4 knockout mouse (ndufs4−/−) is one of the most widely used mouse models for investigating the pathomechanism of LS, a rare, complicated, and incurable early-onset pediatric disorder with both phenotypic and genetic heterogeneity featuring brain-specific anomalies (Quintana et al., 2010, 2012; Grillo et al., 2021). Except for NDUFS4, the mutations in other components of CI, such as NDUFV1 and NDUFS1, have also been linked to LS (Martín et al., 2005; Marin et al., 2013; Borna et al., 2020). One recent study on 37 children with cavitating leukoencephalopathies (a brain disorder that predominantly damages the cerebral white matter) was found caused by mutations in NDUFV1 (2/37) and NDUFS1 (5/37; Zhang et al., 2019). Besides LS, one infant with a biallelic loss-of-function mutation in NDUFV1 is also compatible with the phenotype of LBSL (Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation), a rare condition characterized by progressive pyramidal, cerebellar, and dorsal column dysfunction (Borna et al., 2020). Moreover, partially disrupted CI function in dopaminergic neurons by deleting its NDUFS2, an essential subunit for the catalytic core of CI, caused progressive, human-like parkinsonism in which the loss of nigral dopamine release makes a critical contribution to motor dysfunction (González-Rodríguez et al., 2021).
Besides the nuclear DNA (nDNA) encoded subunits, aberrant mtDNA-encoded components, MT-ND1, MT-ND3, MT-ND4, MT-ND5, and MT-ND6, caused the dysfunction of CI that is also involved in the pathomechanism of neurological disorders, such as LS and Leber hereditary optic neuropathy (LHON; Catarino et al., 2017; Lee et al., 2020; Bakare et al., 2021). For instance, the MT-ND3 mutation disrupted the active/inactive transition of CI, and the MT-ND5 mutation inhibited proton translocation, both of which were found in patients with LS and LHON (Wang et al., 2009; Vodopivec et al., 2016; Lee et al., 2020). Additionally, an m.10158T>C mutation in MT-ND3 has also been linked to mitochondrial encephalomyopathy with lactic acid and stroke-like episodes (MELAS) syndrome (Kori et al., 2019). Three primary point mutations in mtDNA, m.3460G>A, m.11778G>A, and m.14484T>C, have been shown to be responsible for more than 95% of LHON patients (Riordan-Eva and Harding, 1995a; Catarino et al., 2017).
Observations from the mammalian CI suggest an “L” shaped structure with a hydrophilic arm protruding into the matrix and a hydrophobic arm embedding within the IMM (Wirth et al., 2016; Zhu et al., 2016; Parey et al., 2020). This highly complicated structure with over 45 subunits requires at least 15 assembly factors for its complete maturation (Andrews et al., 2013; Formosa et al., 2018; Parey et al., 2020). Hence, mutations in the genes encoding assembly factors lead to a reduced amount of fully assembled functional CI by affecting the rate of CI stability (Bakare et al., 2021). For example, Barghuti et al. (2008) discovered that one CI assembly factor NDUFA12L is mutated in two patients who presented in late infancy with hypotonia, nystagmus, and ataxia interspersed with acute episodes of encephalopathy (Barghuti et al., 2008). Additionally, mutations in NDUFA12L may also have a functional impact on the etiology of tauopathies, suggesting that NDUFA12L could be a crucial factor in the development of neurodegenerative tauopathy, a possible contributing factor in the development of PD (Salama and Mohamed, 2015). In addition, NDUFS4 deletion also results in the nearly complete absence of another accessory subunit NDUFAF12 (Adjobo-Hermans et al., 2020). In NDUFAF12-mutated patient cells, the level of assembled and active CI is reduced, eventually leading to mitochondrial deterioration by increasing mitochondrial ROS and damaging mtDNA and lipids (Schlehe et al., 2013). A heterozygous NDUFAF12 deletion has been observed in a patient with attention-deficit/hyperactivity disorder (ADHD), a common, highly heritable neurodevelopmental syndrome characterized by hyperactivity, inattention, and increased impulsivity (Lesch et al., 2011).
Complex II
Complex II (CII), also known as succinate dehydrogenase (SDH), is the smallest complex of the ETC. This complex is composed of only four subunits SDHA, SDHB, SDHC, and SDHD, which are all encoded by the nuclear genome (Sun et al., 2005; Bezawork-Geleta et al., 2017). Moreover, four assembly factors are required for the assembly and maturation of CII (Bezawork-Geleta et al., 2017). As part of ETC, CII transfers electrons from succinate to ubiquinone (UbQ) via its [Fe-S] clusters. SDHA, the largest catalytic subunit of CII, oxidizes succinate and couples this to the reduction of its flavine cofactor, FAD; whereas the other catalytic subunit SDHB shuttles electrons to UbQ in a coordinated manner (Cecchini, 2003; Miyadera et al., 2003). In addition to its role in ETC, CII also participates in the tricarboxylic acid cycle (TCA), forming a functional link between these two vital processes (Cecchini, 2003; Cardaci et al., 2015; Lussey-Lepoutre et al., 2015).
Although fewer neurological disorders are reported to be linked to the mutations in CII, multiple types of mutations in SDHA, and SDHB, as well as one assembly factor SDHAF1, have been reported to cause LS or LS-like symptoms (Bourgeron et al., 1995; Birch-Machin et al., 2000; Pagnamenta et al., 2006; Jain-Ghai et al., 2013; Bezawork-Geleta et al., 2017; Kaur et al., 2020). Mutations in the SDHA gene are the most commonly reported cause of isolated complex II deficiency (Fullerton et al., 2020). People who carry one of the SDHA mutations, p.R451C, p.A524V, p.R554W, or p.G555E mutations, are diagnosed with LS (Bourgeron et al., 1995; Parfait et al., 2000; Van Coster et al., 2003). Except for LS, multiple point mutations in SDHB are reported in patients with leukoencephalopathy (Alston et al., 2012; Ardissone et al., 2015; Helman et al., 2016; Kaur et al., 2020). In patients with early progressive encephalomyopathy, a homozygous or compound heterozygous mutations in SDHD p.E69K and p.*164Lext*3 (a mutation that extends the SDHD C-terminus by the three extra amino acids Leu, Pro, and Phe) was identified (Jackson et al., 2014; Lin et al., 2021). Moreover, although CI and CIII are the main sources of mitochondrial ROS, growing studies indeed reveal that CII also serves as a source and modulator of ROS (Yankovskaya et al., 2003; Hadrava Vanova et al., 2020). Both functional loss of CII and its pharmacological inhibition lead to excessive ROS accumulation, which has a relevant impact on the development of pathophysiological conditions such as neurodegenerative disorders (Lin and Beal, 2006; Iverson et al., 2012).
Complex III
Complex III (CIII) catalyzes the transfer of electrons from ubiquinol (CoQH2) to cytochrome c (Cyt c), as well as couples this electron transfer to the vectorial translocation of two protons from the matrix, releasing four into the IMS (Zhang et al., 1998; Meunier et al., 2013). CIII forms a homodimeric complex consisting of 10–11 subunits per monomer, and the biogenesis of CIII requires approximately eight assembly factors (Ndi et al., 2018; Vercellino and Sazanov, 2022). Except for MT-CYB encoded by mtDNA, the remaining subunits are encoded by nDNA. The enzyme has two distinct quinone-binding sites (Qo or QP, quinol oxidation site, and Qi or QN, quinone reduction site), which are located on opposite sides of the IMM and linked by a transmembrane electron-transfer pathway. Each monomer of the CIII contains three catalytic subunits that are highly conserved from prokaryotes to eukaryotes: MT-CYB, cytochrome c1 (also known as CYC1), and the Rieske iron-sulfur protein (UQCRFS1; Xia et al., 2013). MT-CYB provides both the Qo and Qi pockets and the transmembrane electron pathway (via hemes bL and bH; Xia et al., 1997; Iwata et al., 1998).
Mutations in subunits of CIII (MT-CYB, UQCRB, UQCRQ, UQCRC2, CYC1, and UQCRFS1) and its assembly factors (BCS1L, LYRM7, TTC19, UQCC2, UQCC3) lead to inherited disorders in humans (Fernández-Vizarra and Zeviani, 2015; Feichtinger et al., 2017). A homozygous missense mutation of CYC1 (p.R317Y) caused CIII deficiency, resulting in mitochondrial respiratory chain abnormalities and eventually leading to acute demyelinating syndrome or LHON (Heidari et al., 2021). Another homozygous missense (p.S45F) mutation in UQCRQ, one of the CIII subunits that joins and stabilizes the mature MT-CYB by forming a stable intermediate complex together with UQCRB (Gruschke et al., 2012; Vercellino and Sazanov, 2022), caused the reduced activity of CIII and CI. This defect in OXPHOS, especially for CIII, causes severe neurological disorders characterized by severe psychomotor retardation and global dementia with defects in verbal and expressive communication skills (Barel et al., 2008). Another homozygous missense mutation (p.G222A) in one core subunit UQCRC2, which plays an important role in CIII dimerization during the early period of the CIII assembly (Stephan and Ott, 2020), also causes neurological disorders, presenting with severe encephalomyopathy, paleocerebellar symptomatology, delay in motoric and cognitive functions (Burska et al., 2021). Additionally, one girl with two homozygous missense variations in UQCC2 leads to a severe reduction of UQCC2 protein, a key assembly factor that regulates MT-CYB expression and subsequent complex III assembly (Tucker et al., 2013). The reduced UQCC2 inhibits the activity of CI and CIII, leading to the progression of epileptic seizures (Feichtinger et al., 2017). Furthermore, patients with homozygous mutations in another assembly factor TTC19 (tetratricopeptide repeat domain 19), such as p.L219*, p.E173*, are found to develop a series of neurodegenerative disorders, including severe psychiatric symptoms, cerebellar ataxia, cognitive impairment, LS and so on (Ghezzi et al., 2011; Atwal, 2013; Malek et al., 2018; Habibzadeh et al., 2019).
Complex IV
Complex IV (CIV) is the terminal enzyme of the ETC, catalyzing the transfer of electrons from reduced Cyt c to molecular oxygen. It is comprised of 14 subunits, three (MT-COX1, MT-COX2, and MT-COX3) of which are encoded by the mtDNA. These three mtDNA-encoded subunits form the catalytic fraction of CIV, and the remaining ten nDNA-encoded subunits contribute to the assembly and biogenesis of the CIV (Antonicka, 2003; Böhm et al., 2006). Although the number of unique subunits between CIII and CIV is similar, CIV requires far more assembly factors for biogenesis than CIII, with almost 50 assembly factors necessary for CIV assembly (Zong et al., 2018; Brzezinski et al., 2021; Vercellino and Sazanov, 2022).
CIV deficiency is the second most common ETC complex deficiency, occurring in 19%–27% of all mitochondrial disease patients (Renkema et al., 2017). The majority of recorded occurrences present a severe, often fatal, infantile disease (Balsa et al., 2012; Pitceathly et al., 2013). Multiple homozygous mutations of CIV subunits, such as NDUFA4 (c.42 + 1G>C, aberrant splicing), COX4I (c.454C>A, p.P152T), and COX8A (c.115-1G>C, p.E39Rfs*27), have been discovered in patients with LS or LS-like syndrome. These mutations cause a frameshift with premature stop-codon formation, which results in truncated proteins or even loss of proteins, leading to the reduced quantity or activity of CIV (Pitceathly et al., 2013; Hallmann et al., 2016; Pillai et al., 2019; Čunátová et al., 2020). Mutation in mtDNA encoded core subunit MT-CO3 (m.9553G>A, p.W116X) was founded in a female patient with the MELAS syndrome (Wang et al., 2021). Except for spontaneous mutations in its subunits, some dysregulated proteins, like β-amyloid (Aβ) peptides, can specifically inhibit the activity of CIV by binding to the heme of CIV and forming an Aβ-heme complex, resulting in cognitive deficits and neuronal morphological abnormalities, which are implicated in the development of AD and PD (Atamna and Boyle, 2006; Coskun et al., 2012). Furthermore, the profound changes in two CIV subunits, COX5A and NDUFA4, also suggest a link between CIV abnormalities and mitochondrial dysfunction in the etiology of AD (Shim et al., 2017). COX6A mutations at various loci have been associated with various forms of neuropathy. For example, mutations in COX6B1 (p.R20H/C) and a 5 bp deletion (c.247-10_247-6delCACTC, aberrant splicing) in a splicing element of COX6A1 cause severe infantile encephalomyopathy and an axonal form of Charcot-Marie Tooth (CMT) syndrome, respectively (Massa et al., 2008; Tamiya et al., 2014; Abdulhag et al., 2015).
The current knowledge of neurological disorders linked to CIV deficits reveals that mutations in assembly factors are far more common than mutations in the structural subunits of the CIV (Renkema et al., 2017). Bi-allelic loss of function variants in the COX20 (c.41A>G or c.157+3G>C) gene perturbs the assembly of complex IV, which leads to mitochondrial bioenergetic failure, resulting in ataxia and autosomal recessive sensory neuronopathies (Otero et al., 2018; Dong et al., 2021). Except for COX20, defects or mutations in other CIV assembly proteins such as SURF1, COA3, COA7, and SCO2 may impair axonal transport or mitochondrial copper homeostasis, as well as increase ROS damage, resulting in peripheral neuropathies (Echaniz-Laguna et al., 2013; Ostergaard et al., 2015; Higuchi et al., 2018; Rebelo et al., 2018; Finsterer and Winklehner, 2021). Mutations in these assembly factors, which are involved in the biogenesis of CIV, LRPPRC (p.Y172C), COX10, SURF1 (834G→A or 820-824dupTACAT), COX15, TACO1, and PET100 (c.3G>C), have been identified in patients with LS (Bugiani, 2005; Tay et al., 2005; Coenen et al., 2006; Weraarpachai et al., 2009; Lim et al., 2014; Kotecha and Kairamkonda, 2019). Furthermore, a mutation in the FASTKD2 (fas activated serine-threonine kinase domain 2) gene induces the occurrence of epilepsy in infancy (Ghezzi et al., 2008). Patients with bradykinesia and cognitive impairment have a homozygous mutation in PET117, a potential complex IV assembly component (Renkema et al., 2017; Finsterer and Winklehner, 2021).
F1Fo-ATP Synthase
F1Fo-ATP synthase is the terminal complex of the OXPHOS and thus is also referred to complex V (CV). F1Fo-ATP synthase is composed of 19 structural subunits, two of which (MT-ATP6 and MT-ATP8) are encoded by mtDNA and the other 17 by nDNA. Five assembly factors are involved in the biogenesis of this multi-subunit enzyme (Genov et al., 2020; Pinke et al., 2020; Vercellino and Sazanov, 2022). Based on the structural and functional differences, F1Fo-ATP synthase is divided into two domains: a hydrophobic domain embedded in the IMM (Fo) is responsible for proton translocation by forming a rotatory proton channel, and a hydrophilic ATPase domain (F1) exposed to the matrix synthases ATP by phosphorylating ADP to ATP (MITCHELL, 1961; Zhou and Sazanov, 2019; Spikes et al., 2020). Mutations in either of the nuclear or mitochondrial encoded subunits may damage the activity of F1Fo-ATP Synthase, resulting in reduced energy production (Kucharczyk et al., 2009; Ebanks et al., 2020; Mnatsakanyan and Jonas, 2020; Patro et al., 2021).
ATP synthase deficiency-caused illnesses are often severe encephalopathies or cardiomyopathies and appear soon after birth (Dautant et al., 2018). The better-characterized aberrant ATP synthase-caused diseases are attributed to mutations in the mtDNA-encoded MT-ATP6 and MT-ATP8 genes, which have overlapped 46 nucleotides at the 5’ part of MT-ATP6 with the open reading frame of MT-ATP8 (Taanman, 1999). As a result, alterations in this region probably cause double mutations (Ware et al., 2009; Imai et al., 2016; Fragaki et al., 2019; Galber et al., 2021). For instance, the heteroplasmic m.8561C>G or m.8561C>T mutations in the overlapping region of MT-ATP6 and MT-ATP8 in adult and childhood caused severe neurological signs, presenting with cerebellar ataxia, psychomotor delay, peripheral neuropathy, and microcephaly (Kytövuori et al., 2016; Fragaki et al., 2019). Compared to MT-ATP8, mutations in MT-ATP6-caused defects of ATP synthase occur more frequently in humans. Mutations in MT-ATP6, like the most common mutations (m.8993T>G/C and m.9176T>G/C), as well as less frequent mutations (m.9035T>C, m.9185T>C, m.9191T>C, m.8914C>T, m.8701A>G), cause different clinical phenotypes of neuropathy, varying from NARP (Neuropathy, Ataxia, Retinitis Pigmentosa) to, encephalomyopathy and MILS (Maternally Inherited Leigh’s Syndrome; Guo Y. et al., 2018; Ichikawa et al., 2019; Capiau et al., 2022; Na and Lee, 2022). Besides mutations in the mtDNA encoded subunits, ATP synthase deficits induced by nDNA-encoded subunits, such as ATP5E and ATP5A1, as well as the assembly factors ATPAF2 (ATP12) and TMEM70, have also been observed in patients with neurological disorders (De Meirleir, 2004; Čížková et al., 2008; Mayr et al., 2010; Jonckheere et al., 2013). For example, exome sequencing of the two siblings with severe neonatal encephalopathy reveals a heterozygous mutation in CV subunit ATP5A1, resulting in a disrupted inter-subunit interaction and thereby compromising CV stability (Jonckheere et al., 2013).
Dysregulation of Mitochondrial Dynamics
Mitochondria are dynamic organelles that divide and fuse rapidly, which determine the mitochondrial architecture and bioenergetics (El-Hattab et al., 2018). Both the fission and fusion processes are predominately performed by several canonical dynamin-like guanosine triphosphatases (GTPase) enzymes (Trevisan et al., 2018; Sinha and Manoj, 2019). Mitochondrial fusion is primarily accomplished by two outer mitochondrial membrane (OMM) proteins, mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2), and one IMM protein, optic dominant atrophy (OPA1). Mitochondrial fission is mediated by dynamin-related GTPase protein 1 (Drp1) and dynamin 2 (Dnm2), which are responsible for membrane constriction and scission, respectively. In addition, the trans-localization of Drp1 relies on multiple OMM-localized transmembrane receptors like MiD51, MiD49, Fis1, and Mff (Giacomello et al., 2020; Yang et al., 2022). Furthermore, mitochondria actively move along microtubes in both anterograde and retrograde directions with the help of ATP-dependent “motor” proteins (Pozo Devoto and Falzone, 2017). Mitochondrial anterograde transport from the soma to synapses is mediated by kinesins, while retrograde transport from the synapse to the soma is carried out by dynein motor proteins. Mitochondria do not bind directly to the motor proteins but, instead, bind to adaptor proteins, TRAK and Miro, that link the mitochondrial membrane to the motor protein (Maday et al., 2014; Zheng et al., 2019).
Although mitochondrial biogenesis can occur in the axon, the majority of new mitochondria are produced in the soma and transported to the peripheral area of the distal synapse (Amiri and Hollenbeck, 2008; Sheng and Cai, 2012) As a result, mitochondrial anterograde transport can deliver young mitochondria to distal regions (Farías et al., 2017; Zheng et al., 2019). In addition, aged or damaged mitochondria in the distal region are transported back to soma for complete degradation via mitophagy (Zheng et al., 2019; López-Doménech et al., 2021). Neurons are particularly vulnerable to both autophagic impairment and mitochondrial malfunction. Defects in mitochondrial fusion, division, or transportation result in the accumulation of dysfunctional mitochondria, which promotes neurological disease pathogenesis (Martinez-Vicente, 2017; Han et al., 2020; Doxaki and Palikaras, 2021; Figure 3 and Table 2).
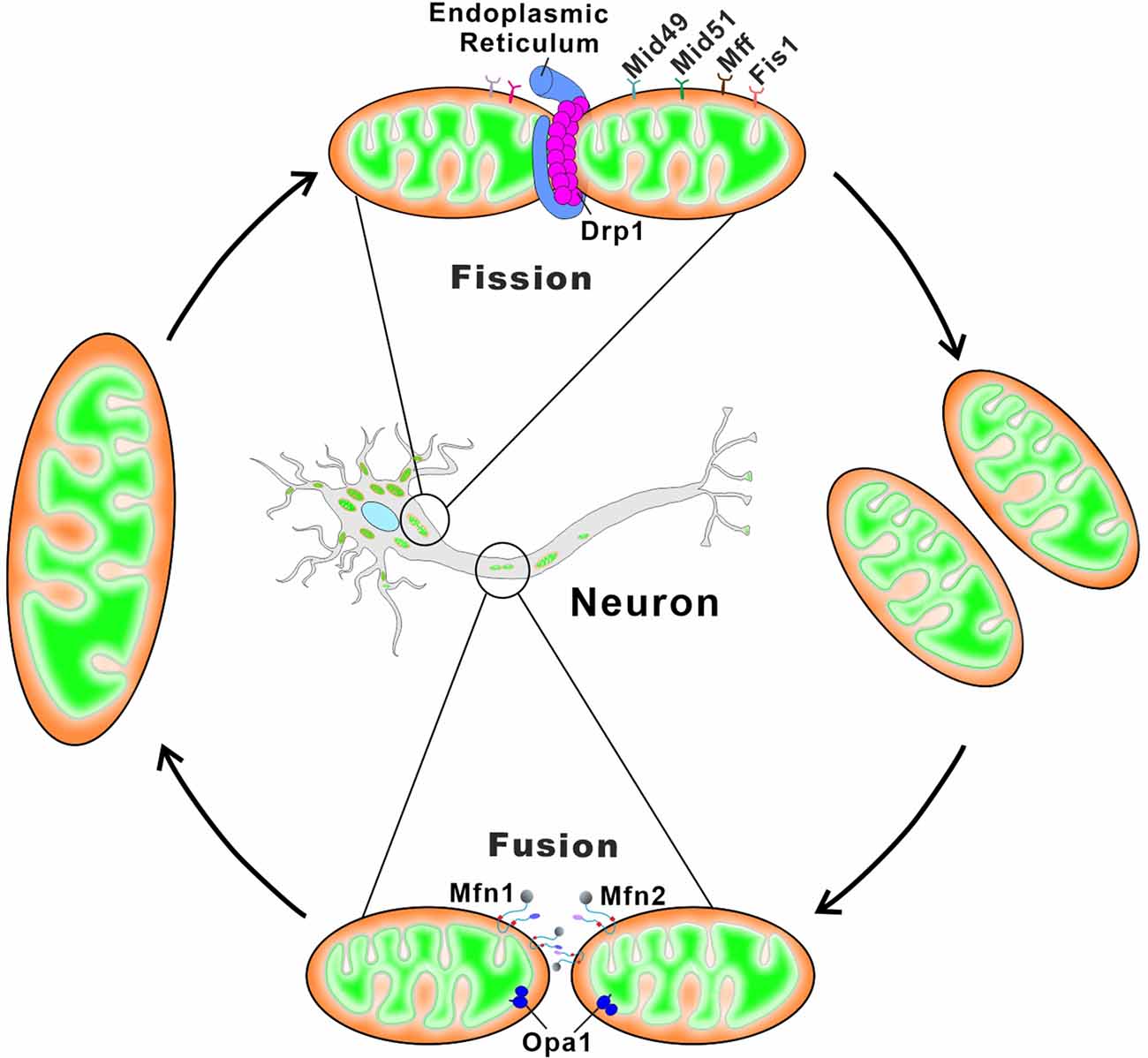
Figure 3. Schematic depiction of mitochondrial fusion and fission in neurons. Mitochondrial fusion relies on membrane proteins Mfn1/2 and OPA1 residing in the OMM and the IMM, respectively. Mfn1 and Mfn2 form homo-oligomeric (Mfn1-Mfn1 or Mfn2-Mfn2) and hetero-oligomeric (Mfn1-Mfn2) complexes in trans between the opposing mitochondria to induce the OMM fusion. The long and short forms of OPA1 synergistically catalyze the tethering and fusion of the IMM. Mitochondrial fission begins with the ER contacts with the OMM at the ER-mitochondria contact sites, and ER wraps tightly around the mitochondria to form constrictions. The cytosolic Drp1 is subsequently recruited to the OMM via multiple membrane adaptors MiD51, MiD49, Fis1, and Mff. Drp1 oligomerizes at the ER-marked pre-constriction sites, forming a ring-like structure around the mitochondria for further membrane constriction.
Table 2. Neurological diseases caused by protein variants involved in mitochondrial dynamics.
Mitochondrial fusion
Mfn1 and Mfn2
The integral mitochondrial outer membrane protein mitofusins Mfn1 and Mfn2 can form both homo-oligomeric (Mfn1-Mfn1 or Mfn2-Mfn2) and hetero-oligomeric (Mfn1-Mfn2) complexes in trans between the opposing mitochondria to induce the OMM fusion (Chen et al., 2003; Griffin and Chan, 2006; Qa et al., 2016). The primary protein sequence, as well as the structure of Mfn1/2, are quite similar, with an N-terminal GTPase domain, two transmembrane domains spanning the OMM, and two regions essential for protein-protein interactions (Santel and Fuller, 2001; Legros et al., 2002; de Brito and Scorrano, 2008; Yang et al., 2022). Despite both proteins being essential for mitochondrial fusion, Mfn1 has considerably higher GTPase activity than Mfn2 (Ishihara et al., 2004). In addition to modulating mitochondrial morphology, Mfn2 also participates in the bridging of mitochondria to the endoplasmic reticulum (ER) as well as multiple signaling pathways, regulating mitochondrial metabolism, apoptosis, and even the cell cycle (de Brito and Scorrano, 2008; Muñoz et al., 2013).
Although the loss of either Mfn1 or Mfn2 causes lethality in mice due to overtly fragmented mitochondria, mutations in human Mfn2 but not Mfn1 lead to Charcot-Marie-Tooth disease type 2A (CMT2A), peripheral neuropathy of long motor and sensory neurons (Züchner et al., 2004). Although no Mfn1 mutation is found in CMT2A patients, Mfn1 but not Mfn2 functionally complements the CMT2A mutant Mfn2R94Q to induce mitochondrial fusion (Detmer and Chan, 2007). Mfn2 ablation mediated aberrant mitochondrial dynamics causes abnormal mitochondrial distribution, ultrastructure, and ETC activity in Purkinje cells, which impairs dendritic outgrowth and spine formation, resulting in cell death and animal movement defects (Chen et al., 2007). Another in vivo study found that loss of Mfn2 results in severe, age-dependent motor deficits characterized by reduced activity and rearing, which are preceded by the loss of dopaminergic terminals in the striatum. These nigrostriatal abnormalities may be a risk factor for neurodegeneration in PD (Pham et al., 2012). In addition, the expression of the Mfn2 mutant in the juvenile stage of a mouse results in fatality. However, Mfn2 mutant expression in adulthood does not cause abnormalities until 150 days. The progressive neurodegeneration, irregular behaviors, and learning and memory deficiencies start to emerge after this silent 150-day period, which is similar to those seen in human neurodegenerative diseases (Ishikawa et al., 2019). These findings indicate that defects in Mfn2 cause irregular neuronal mitochondrial dynamics, which not only seriously affect survival during early life stages but also significantly damage brain function and even maturation.
OPA1 and OMA1
In 2000, OPA1 mutations, primarily frameshift and missense variants, were first identified as the cause of dominant optic atrophy (DOA), a blinding disease characterized by retinal ganglion cell degeneration leading to optic neuropathy (Delettre et al., 2000). In mammalian mitochondria, nuclear-encoded OPA1 ubiquitously exists in eight different isoforms, ranging from 924 to 1,014 amino acids (Olichon et al., 2007; Del Dotto et al., 2018). After import of the precursor OPA1 protein through the OMM and IMM translocases, the N-terminal mitochondrial targeting sequence (MTS) is cleaved by the mitochondrial processing peptidase (MPP), generating the membrane-anchored OPA1 long forms (L-OPA1). The L-OPA1 may be further proteolytically processed at the N terminus by two IMM peptidases, OMA1, and i-AAA protease YME1L, to produce the short forms of OPA1 (S-OPA1) that are soluble in the IMS (MacVicar and Langer, 2016). According to Anand et al., L-OPA1 is sufficient to promote mitochondrial fusion, whereas S-OPA1 is found to function in mitochondrial fission. Under normal metabolic control, YME1L is activated for L-OPA1 processing, whereas OMA1 is inactivated or undergoes an autocatalytic self-degradation (Baker et al., 2014). However, OMA1 is activated upon various stress insults and mitochondrial dysfunction, for example, dissipation of mitochondrial membrane potential (ΔΨm), which results in the complete degradation of L-OPA1 and an increase in S-OPA1, inducing mitochondrial fragmentation (Baker et al., 2014; Zhang et al., 2014).
Eight OPA1 isoforms with diverse roles are variably expressed in different tissues, with the highest levels observed in the retina, brain, testis, heart, and muscle. According to current research, the most affected tissues from the OPA1 mutation and depletion are the retina and brain, leading to a variety of neurodegenerative diseases, such as DOA, chronic progressive external ophthalmoplegia (CPEO), PD, and dementia (Ferré et al., 2005; Olichon et al., 2007; Zanna et al., 2008; Williams et al., 2012; Kushnareva et al., 2013; Carelli et al., 2015a; Santarelli et al., 2015; Zorzano and Claret, 2015). The OPA1 mutation caused DOA is the most prevalent inherited form of optic neuropathy, characterized by the gradual degeneration of retinal ganglion cells (RGCs) and the optic nerve. Over a hundred distinct OPA1 variants have been identified in patients with DOA so far (Ferré et al., 2005; Weisschuh et al., 2021). The mechanistic investigation discovered that the OPA1-involved dysregulated mitochondrial fusion, crista morphology, OXPHOS, and Ca2+ buffering are the primary sources of RGCs and optic nerve injury (Zanna et al., 2008; Kushnareva et al., 2013). OPA1 mutations can be substitutions, deletions, or insertions, which are spread throughout the coding region of the gene, with the majority of them clustered around the GTPase and GED domains. In DOA patients of European heritage, the c.2708delTTAG frameshift-inducing microdeletion in the GED domain is the most frequent OPA1 mutation (Zanna et al., 2008).
OMA1-mediated OPA1 processing often occurs during stress, and a low ratio of L-OPA1 to S-OPA1 disrupts mitochondrial dynamics, which is the cause of neuronal degeneration. Hence, OMA1 is a critical regulator of neuronal survival, particularly under stressful situations (Merkwirth et al., 2012; Korwitz et al., 2016). For example, ischemia-reperfusion (I/R) injury induces a dramatic increase in L-OPA1 cleavage, which results in L-OPA1 loss, fragmented mitochondrial morphology, and retinal neuron death. Overexpression of the OMA1 resistant cleavage OPA1 mutant (Opa1-ΔS1) prevented retinal neuronal I/R injury by restoring ATP production and mitochondrial networks, thus attenuating necroptosis and neuronal damage (Sun Y. et al., 2016). Another study also reports that stress-induced OPA1 processing by OMA1 promotes neuronal death and neuroinflammatory responses, whereas OMA1 ablation delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria (Korwitz et al., 2016).
SLC25A46
The 46th isoform of superfamily A of the solute carrier (SLC) family 25, termed SLC25A46, is an integral mitochondrial outer membrane protein that interacts physically with Mfn2, OPA1, and the mitochondrial contact site and cristae organizing system (MICOS) complex (Abrams et al., 2015; Li et al., 2019). SLC25A46 plays important roles in the regulation of mitochondrial dynamics, cristae architecture, as well as mitochondrial distribution (Abrams et al., 2015; Janer et al., 2016). SLC25A46 deficiency not only leads to hyper fused mitochondrial morphology but also abnormal crista architecture, whereas SLC25A46 overexpression induces fragmented mitochondria (Abrams et al., 2015; Janer et al., 2016), indicating that SLC25A46 acts as an independent pro-fission factor. Abnormal crista architecture and enlarged mitochondria with swollen cristae after depletion of SLC25A46 confirmed its role in crista morphogenesis (Wan et al., 2016). In addition, SLC25A46 interacts with components of the MICOS complex, which is critical for the maintenance of mitochondrial crista junctions (Janer et al., 2016). In SLC25A46-mutated Purkinje cells, mitochondria had an aberrant distribution and mobility (Abrams et al., 2015; Terzenidou et al., 2017). Moreover, SLC25A46 serves as a regulator for Mfn1/2 oligomerization and facilitates lipid transfer between the ER and mitochondria (Li et al., 2017, 2019).
SLC25A46 mutations have been linked to a wide spectrum of neurological diseases with recessive inheritance, including LS, optic atrophy, progressive ataxia, peripheral neuropathy, and lethal congenital pontocerebellar hypoplasia (Janer et al., 2016; Li et al., 2017, 2019). One clinical study reported that eight patients from four unrelated families encompassed autosomal dominant optic atrophy (ADOA) like optic atrophy, CMT-like axonal peripheral neuropathy, and cerebellar atrophy. The whole-exome sequencing of those patients found the recessive mutations in SLC25A46 (Abrams et al., 2015). The pathogenic mechanism investigation using SLC25A46 knockout mice discovered that animals lacking SLC25A46 have severe ataxia, which is mostly caused by Purkinje cell degeneration. Investigation of the underlying cellular basis found increased numbers of small, unmyelinated, and degenerated optic nerves as well as loss of RGCs, indicating optic atrophy. Cerebellar neurons having SLC25A46 mutation have large mitochondria with aberrant distribution and transport (Li et al., 2017).
Mitochondrial fission
Drp1
Drp1 is encoded by the nuclear gene DNML1 and serves as a central molecular player in mitochondrial fission (Smirnova et al., 2001). During the mitochondrial fission process, Drp1 is recruited from a cytosolic pool onto the mitochondrial surface where ER marks the constriction sites (Pagliuso et al., 2018). Four mitochondrial outer membrane-localized adaptors, Mff, Fis1, MiD49, and MiD51, serve as receptors and independently recruit Drp1 onto the mitochondrial outer membrane. By self-assembling into a spiral superstructure, Drp1 wraps around and constricts mitochondrial tubules to promote the scission event (Jimah and Hinshaw, 2019). Because Drp1 is unable to complete the mitochondrial tubule scission, another GTPase Dnm2 is recruited to the mitochondrial fission site. By assembling into a collar-like structure around the constricting lipid “necks” of the budding membrane, Dnm2 mediates final membrane scission, creating individual mitochondria (Ferguson and De Camilli, 2012; Tilokani et al., 2018). Depletion of any of these fission-related proteins disrupts the mitochondrial dynamics with a reduction of mitochondrial fission, resulting in the elongated mitochondrial morphology (Ferguson and De Camilli, 2012; Losón et al., 2013).
Drp1 deletion leads to extensive mitochondria networks, and the mouse embryos fail to undergo developmentally regulated apoptosis during neural tube formation, which is lethal for the embryos. Additionally, brain-specific Drp1 ablation caused giant mitochondria in Purkinje cells and may be linked to cerebellum developmental abnormalities (Wakabayashi et al., 2009; Yamada et al., 2016). Besides protein-loss-caused neuronal degeneration, mutations of Drp1 are also able to impair self-assembly at the fission site and act in a dominant-negative manner (Chan, 2020). In neurological diseases, the Drp1 mutation appears to be de novo, heterozygous and dominantly acting. One clinical case reports that a single point mutation in Drp1 A395D causes elongated mitochondrial morphology, which may be associated with multi-system damage, including microcephaly, abnormal brain development, optic atrophy, and hypoplasia, resulting in neonatal lethality (Waterham et al., 2007). Another mutation in Drp1 (R403C) impairs mitochondrial fission via reducing Drp1 recruitment to mitochondria and Drp1 oligomerization at the fission site. Unlike the severe lethality of the Drp1 A395D mutation, the impairment of the missense mutation in Drp1 R403C is compatible with normal conditions for several years but develops refractory focal status epilepticus and subsequent rapid neurological degeneration later on (Fahrner et al., 2016).
The dynamic recruitment of Drp1 onto the OMM surface, assembly, and its GTPase activity are frequently regulated by several post-translational modifications like phosphorylation (Chang and Blackstone, 2010), ubiquitination (Wang et al., 2011), SUMOylation (Wasiak et al., 2007), and S-nitrosylation (Cho et al., 2009). Among them, phosphorylation of Drp1 at S600 or S579 by multiple kinases including PKA, Cdk1, ERK1/2, or Cdk5 has gotten the greatest interest (Taguchi et al., 2007; Valera-Alberni et al., 2021). Through phosphorylation of Drp1 at Ser579, Cdk5 promotes Aβ1-42-induced mitochondrial fission and mitophagy, whereas blocking Drp1 phosphorylation at Ser579 protects neurons from Aβ1-42-induced neuron degeneration (Han et al., 2017; Xu et al., 2021). In 1-methyl-4-phenylpyridinium ion (MPP+) treated cells, the overproduced nitric oxide (NO) triggers the phosphorylation of Drp1 Ser616 and leads to elevated mitochondrial fission. These events create a death-prone environment, which contributes to the loss of dopaminergic (DA) neurons and may explain the pathogenesis of PD (Zhang et al., 2016). Moreover, in human postmortem brains of HD patients, the elevated S-nitrosylation of Drp1 (SNO-Drp1) profoundly up-regulates the GTPase activity of Drp1, which leads to mitochondrial fragmentation, thus impairing bioenergetics and inducing synaptic damage and neuronal loss (Haun et al., 2013).
Mff
Mitochondrial fission factor, termed Mff, is a tail-anchored mitochondrial outer membrane protein (Gandre-Babbe and van der Bliek, 2008). Mff recruits cytosolic Drp1 to the mitochondrial surface and promotes mitochondrial fission by forming a complex with Drp1 (Otera et al., 2010; Dikov and Reichert, 2011). Despite the fact that four canonical receptors are involved in Drp1 recruitment, Mff depletion without disturbance of the other three receptors still results in a reduced number of Drp1-positive foci at the OMM, accompanied by impairment of mitochondrial fission, showing severely elongated and interconnected mitochondria (Losón et al., 2013). In contrast, Mff overexpression produces the opposite effects (Otera et al., 2010), demonstrating that Mff can function independently in Drp1 recruitment.
The loss of Mff in neurons reduces mitochondrial fission, which restricts the entrance of mitochondria into the axon and along the axonal shaft. Although Mff deletion has only a minimal impact on mitochondrial trafficking along the axon, presynaptic location, membrane potential, or their ability to generate ATP, it significantly boosts their Ca2+ buffering capacity. The size-dependent increase in mitochondrial Ca2+ uptake reduces presynaptic cytoplasmic Ca2+ levels, resulting in decreased neurotransmitter release and terminal axon branching during evoked activity (Lewis et al., 2018). In humans, a truncating mutation in Mff caused loss of function in mitochondrial fission is highlighted in those patients with early-onset Leigh-like basal ganglia disease, encephalopathy, seizures, developmental delay, and acquired microcephaly appearing in the first year of life, followed by dysphagia, spasticity, optic neuropathy, and peripheral neuropathy in subsequent years (Shamseldin et al., 2012; Koch et al., 2016; Panda et al., 2020).
Dysregulation of Mitophagy
Mitophagy is an autophagic mechanism that specifically targets excess or damaged mitochondria and delivers them to lysosomes for degradation (Lemasters, 2005; Palikaras et al., 2015; Sun N. et al., 2016; Pickles et al., 2018; Onishi et al., 2021). Mitophagy is a set of processes that begins with the isolation of excess or damaged mitochondria and ends with the complete degradation of damaged mitochondria. In brief, the entire process can be completed in the following steps: (1) The isolation of excess or damaged mitochondria by fission in response to intracellular or external stimuli; (2) The recruitment and/or activation of mitophagy receptors or ubiquitin-autophagy adaptors on the mitochondrial surface, conferring selectivity for degradation; (3) The recognition of selected mitochondria by core autophagy-related proteins, mediating the isolation membrane/phagophore enclosing mitochondria; (4) The formation of spherical double-membrane structure “autophagosome”; (5) Transportation of autophagosomes to lysosomes for fusion; and (6) Complete degradation of selected mitochondria in lysosomes for recycling of contents (Ding and Yin, 2012; Wang Y. et al., 2019; Onishi et al., 2021).
Mitophagy-mediated elimination of impaired mitochondria plays critical roles in a variety of processes, including early embryonic development, cell differentiation, inflammation, and apoptosis (Onishi et al., 2021). Mitochondrial damage is particularly harmful to the nervous system. As a result, poor mitophagy could be detrimental to neuronal health. Defects in mitophagy have been linked to aging and the pathogenesis of age-associated neurodegenerative disorders, such as PD (Mouton-Liger et al., 2017; Tanaka, 2020; Malpartida et al., 2021), AD (Fang et al., 2019; Reddy and Oliver, 2019; Pradeepkiran and Reddy, 2020), ALS (Harding et al., 2021; Madruga et al., 2021), frontotemporal dementia (FTD; Ruffoli et al., 2015) and HD (Franco-Iborra et al., 2021; Šonský et al., 2021).
Mitophagy can be robustly induced in response to a variety of pathological stimuli (Lou et al., 2020; Pradeepkiran and Reddy, 2020). There are a number of mitophagy pathways that have been identified at present, including: (1) PTEN-induced putative kinase protein 1 (PINK1)-Parkin-mediated mitophagy; (2) Aβ and p-tau-induced mitophagy; (3) stress-induced mitophagy; (4) Parkin-independent ubiquitin-mediated mitophagy; and (5) basal mitophagy. Among all, PINK1-Parkin-mediated mitophagy is the most heavily studied and best-understood mitophagy pathway (Gautier et al., 2008; Narendra et al., 2008; Palikaras et al., 2018; Chu, 2019; Cai and Jeong, 2020). In brief, the mitochondrial imported serine-threonine protein kinase PINK1 is constitutively proteolyzed by mitochondrial proteases at the IMM under healthy conditions. Whereas, following the dissipation of mitochondrial ΔΨm, the injured mitochondria are unable to degrade PINK1, resulting in translocation of unproteolyzed PINK1 onto the OMM surface. Through phosphorylation of ubiquitin, the accumulated PINK1 recruits and activates Parkin, an E3 ubiquitin ligase (Kawajiri et al., 2010; Matsuda et al., 2010; Narendra et al., 2010; Shiba-Fukushima et al., 2012; Kane et al., 2014; Koyano et al., 2014). The activated Parkin then ubiquitinates a variety of OMM substrates, triggering the ubiquitin-proteasome system (UPS) to degrade these ubiquitinated OMM proteins. These events eventually induce the autophagy machinery, allowing the engulfment and degradation of damaged mitochondria through the above mitophagy process (Poole et al., 2010; Chan et al., 2011; Yoshii et al., 2011). In addition to mitophagy, PINK1 and Parkin regulate mitochondrial motility, which appears to be particularly important for neuronal function (Scarffe et al., 2014; Arano and Imai, 2015). These two proteins are the most well-known monogenic PD-associated genes involved in mitochondrial quality control. Therefore, loss-of-function mutations in PINK1 and Parkin are the most common known causes of autosomal recessive and early-onset PD (before the age of 45; Kitada et al., 1998; Valente et al., 2004; Thomas et al., 2007; Houlden and Singleton, 2012; Giannoccaro et al., 2017; Figure 4).
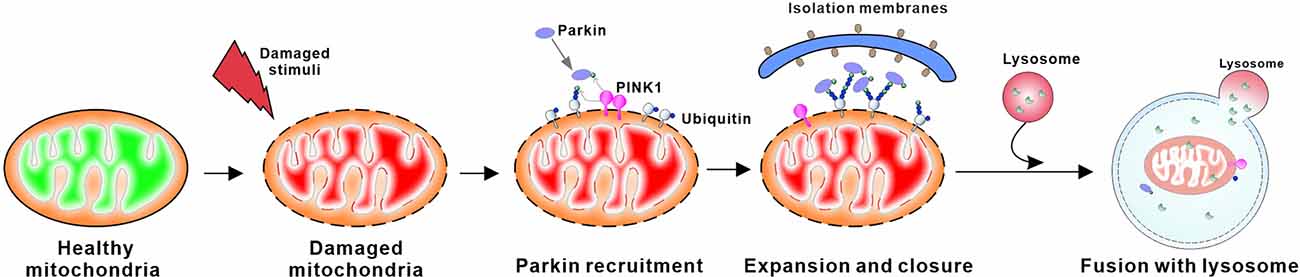
Figure 4. Schematic overview of PINK1/Parkin mediated mitophagy. PINK1 accumulates at the surface of damaged mitochondria when the constitutive degradation of PINK in the mitochondrial matrix is inhibited. Subsequent homodimerization of PINK1 on the OMM leads to autophosphorylation, which promotes its activation. Activated PINK1 phosphorylates ubiquitin to recruit parkin, an E3 ubiquitin ligase, to the mitochondrial membrane. PINK1 regulates the localization and activity of parkin through phosphorylation of both ubiquitin and the ubiquitin-like domain of parkin. This process leads to ubiquitination of mitochondrial proteins on the OMM that can then be bound by autophagic proteins, ultimately triggering the formation of autophagosomes that deliver the damaged mitochondria to lysosomes for degradation.
Parkin
Parkin is an E3 ubiquitin ligase that ubiquitinates diverse substrates (Martin et al., 2011). Protein structural analysis shows that Parkin protein consists of a ubiquitin-like (Ubl) domain at the amino terminus and four RING domains: RING0, RING1, IBR, and RING2 (Shimura et al., 2000; Hristova et al., 2009; Trempe et al., 2013). This RING-in-between-RING protein structure leads to an autoinhibited state of protein activity, resulting in a very low basal activity. Mutations in protein, on the other hand, can disrupt these autoinhibitory interactions of protein domains, leading to the activation of Parkin (Trempe et al., 2013). Parkin deficiency is neurotoxic, whereas the rise of its level is neuroprotective (Hatano et al., 2009; Martin et al., 2011). More than 120 mutations in Parkin have been shown to cause autosomal recessive PD, with point mutations found in every domain of the protein (Kitada et al., 1998; Lücking et al., 2000; Mata, 2004).
The involvement of Parkin in PD was highlighted in 1997 (Matsumine et al., 1997). In a genetic linkage analysis of autosomal recessive juvenile parkinsonism (AR-JP), PRKN, the encoded gene of Parkin (also known as PARK2), was found to have a deletion at exon 4 or a large-scale deletion between exons 3 and 7 in all analyzed AR-JP patients (Matsumine et al., 1997; Kitada et al., 1998). Moreover, Parkin mutations with varied deletions or point mutations that mediate Parkin protein loss of function were soon identified in other patients of various ethnicities with early-onset PD (Hattori et al., 1998a, b; Leroy et al., 1998; Lücking et al., 1998).
PINK1
PINK1 (also known as PARK6), the upstream protein kinase of Parkin, is one of the most diverse human protein kinases (Manning et al., 2002). PINK1 can phosphorylate both ubiquitin and the Ubl domain of Parkin on structurally protected Ser65 residue, triggering mitophagy (Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Wauer et al., 2015). The PINK1 gene has eight exons that encode a protein kinase with 581 amino acids in total. It contains an N-terminal mitochondrial importing sequence and a transmembrane segment, an unconserved region, a kinase domain with three insertions in the N lobe, and a conserved C-terminal region (CTR) of unclear function and structure (Valente et al., 2004; Sim et al., 2006; Schubert et al., 2017). Under healthy conditions, the PINK1 precursor interacts with the translocase of the outer membrane (TOM) complex and is imported through the translocase of the inner membrane (TIM) complex to the IMM, where it is initially cleaved by the MPP like the importing of all traditional MTS-containing proteins (Greene et al., 2012). PINK1 is subsequently digested in its hydrophobic domain spanning the IMM by the rhomboid family protease PARL (Lazarou et al., 2012; Kato et al., 2013). The cleaved PINK1 is reversely released into the cytosol, where it is subjected to degradation by the ubiquitin ligases UBR1, UBR2, and UBR4 (Yamano and Youle, 2013). This continuous import, release, and degradation cycle yields very low to undetectable levels of PINK1 in healthy mitochondria.
However, under the damaged mitochondrial conditions, including the depolarized mitochondrial membrane potential, mutagenic or environmental stresses, OXPHOS suppression, and proteotoxicity, PINK1 import into the IMM where MPP and PARL reside is blocked, resulting in the prevention of PINK1 processing. Instead, the uncleaved PINK1 accumulates on the OMM, flagging them for elimination. Homodimerization of PINK1 on the OMM causes autophosphorylation events, which promote its kinase activation and facilitate binding to the substrates Parkin and ubiquitin (Lazarou et al., 2012; Pickrell and Youle, 2015). Due to its propensity to rapidly accumulate and activate in response to mitochondrial stress, PINK1 acts as a mitochondrial damage sensor. The mutation of the PARK6 locus on the short arm of chromosome 1 was discovered in a large Italian family with autosomal recessive early-onset parkinsonism in 2001, and the PINK1 gene is suggested to be one of the causal genes that can result in parkinsonism (Valente et al., 2001). A number of PINK1 mutations, including point mutations, frameshift, and truncating mutations, have been further reported in patients with PD (Hatano et al., 2004; Rogaeva et al., 2004; Rohé et al., 2004).
Dysregulation of Mitochondrial Genome Maintenance
Mitochondria possess their own circular genomic DNA (mtDNA), which encodes 13 core protein subunits of complexes I, III, IV, and V, as well as 22 transfer RNAs (mt-tRNAs) and two ribosomal RNAs (mt-rRNAs; mt-rRNAs; Clayton, 2000; Park and Larsson, 2011; Chocron et al., 2019). Due to multiple copies of mtDNA (approximately ~1,000–10,000 copies per cell), the absence of complex chromatin organization, limited mtDNA repair activities, or proximity to the ROS as a result of being close to the mitochondrial ETC, thus mutations in mtDNA occur far more frequently in mtDNA than in nDNA (~10–17-fold higher mutation rate compared to nDNA). Both deletions and point mutations can occur in the mtDNA (Marcelino and Thilly, 1999; Su et al., 2010; Tuppen et al., 2010). In addition, mtDNA mutagenesis in humans becomes more common as people get older (Bender et al., 2006). In neurons, aberrant mtDNA including primary mutations in the mtDNA itself as well as reduced mtDNA copy number contributes to bioenergetic impairments, reduced synaptic function, and an increased risk of degeneration (Keeney and Bennett, 2010).
Unlike the nuclear-encoded genome DNA, mtDNA does not encode any genes involved in its DNA maintenance or repair. Therefore, the integrity of mtDNA is entirely dependent on the nuclear-encoded proteins (Zinovkina, 2018; Allkanjari and Baldock, 2021). Any defects in mtDNA maintenance or repair machinery, including not only the deficiencies in the related proteins themselves but also the abnormal mitochondrial importing machinery, typically result in secondary multiple deletions, duplications, or depletion of mtDNA. These changes cause subsequent poor mitochondrial respiration and dysfunction, which are linked to a broad spectrum of mitochondrial and age-related diseases (Larsson, 2010; Nunnari and Suomalainen, 2012; Figure 5 and Table 3).
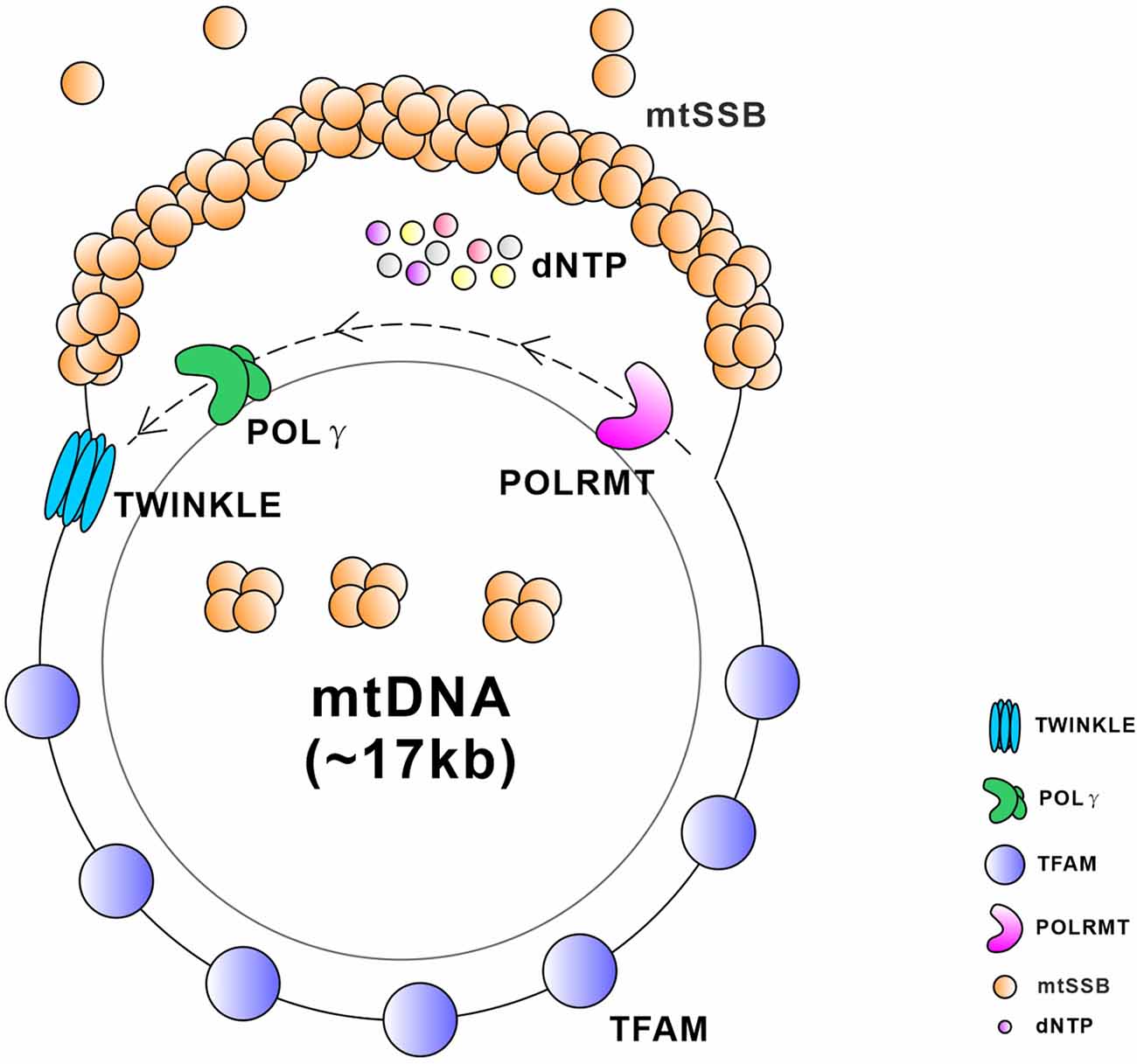
Figure 5. Schematic diagram of critical proteins required for the replication and transcription of mtDNA. TWINKLE is an mtDNA helicase that catalyzes the unwinding of duplex DNA using energy from NTP hydrolysis. Mitochondrial single stranded DNA-binding protein (mtSSB) exists as a stable tetramer and binds single-stranded DNA intermediates that form transiently during genome maintenance. DNA polymerase gamma (POLγ) is a DNA polymerase that synthesizes a nascent strand using one of the unwound parental template strands at the replication fork. Mitochondrial transcription factor A (TFAM) is a master regulator of mitochondrial transcription initiation and mtDNA replication that binds the promoter region of mtDNA in a sequence-specific manner and thereby initiates promoter-specific transcription or replication from the light-strand promoter. Mitochondrial RNA polymerase (POLRMT) is an RNA polymerase that executes the transcription of the mtDNA.
Table 3. Neurological diseases caused by protein variants involved in mitochondrial genome maintenance.
TWINKLE
TWINKLE helicase is the main helicase in mitochondria encoded by the TWNK gene, serving as the only replicative helicase required for mtDNA replication (Peter and Falkenberg, 2020). Helicases are a collection of motor proteins that catalyze the unwinding of duplex DNA/RNA using energy from nucleotide triphosphate (NTP) hydrolysis. They function in almost all nucleic acid transactions, including DNA replication, transcription, translation, recombination, and DNA repair (Donmez and Patel, 2006; Singleton et al., 2007; Lohman et al., 2008; Pyle, 2008). Based on structure and sequence similarities, helicases are classified into six superfamilies (SF1–6; Singleton et al., 2007; Fairman-Williams et al., 2010), and TWINKLE is a member of the SF4 helicase superfamily, which shares five conserved helicase motifs and forms a ring-shaped higher-order structure to unwind mtDNA (Spelbrink et al., 2001). TWINKLE is hexameric and each monomer is comprised of an N-terminal domain (NTD) and a C-terminal domain (CTD), which are joined by a flexible linker helix. The five SF4 helicase motifs are located in the highly conserved CTD domain (Singleton et al., 2007). The activity of TWINKLE helicase is stimulated by interacting with mitochondrial single-stranded DNA (ssDNA)-binding protein (mtSSB; Korhonen et al., 2003; Oliveira and Kaguni, 2011). In order to efficiently initiate DNA unwinding, TWINKLE requires a fork structure comprising a single-stranded 5’ DNA end and a short 3’ tail (Patel and Picha, 2000; Korhonen et al., 2003).
TWINKLE mutations were initially detected in autosomal dominant progressive external ophthalmoplegia (PEO), a neurodegenerative disease that largely affects the muscles controlling eye movement (Spelbrink et al., 2001; Iakovenko et al., 2020). Up to 40 point mutations in the TWNK gene have been identified in patients with PEO, and these mutations can occur at both the primase and helicase domains as well as the linker region of the TWINKLE protein (Peter and Falkenberg, 2020). These mutations impair the helicase activity of TWINKLE, which is likely to impede mtDNA replication and lead to the gradual accumulation of large mtDNA deletions over time (Zeviani et al., 1989; Peter and Falkenberg, 2020). For example, TWINKLE mutations in the linker region (p.A359T, p.I367T, p.S369P, p.R374Q, and p.L381P) disrupt the ring-shaped structure, causing ATP hydrolysis to be reduced and DNA helicase function to be lost (Korhonen et al., 2008; Peter et al., 2019).
Apart from PEO, mutations in the TWNK gene have been discovered in a variety of mitochondrial illnesses, including mtDNA depletion syndromes (MDSs), Perrault syndrome, infantile-onset spinocerebellar ataxia (IOSCA), and other ataxia neuropathies (Hudson et al., 2005; Hakonen et al., 2008; Lonnqvist et al., 2009; Van Hove et al., 2009; Fratter et al., 2010). For example, several TWNK mutations like p.A318T, p.T457I, or p.Y508C, are supposed to disrupt the NTP binding/hydrolysis and/or oligomerization, which leads to severe mtDNA depletion and impaired OXPHOS activity, manifesting clinically as seizures, developmental delay, and peripheral neuropathy (Hakonen et al., 2008; Lonnqvist et al., 2009).
Polymerase-γ (POLγ)
POLγ is the most important DNA polymerase executing mtDNA replication and consists of two subunits, a catalytic subunit POLγA, and a homodimeric accessory subunit POLγB (Ropp and Copeland, 1996). POLγA, which is encoded by the POLG gene, contains an amino-terminal exonuclease domain coupled to the carboxy-terminal polymerase domain by a linker region. The POLG2-encoded POLγB provides high processivity of the POLγ complex by accelerating its DNA binding affinity (Johnson et al., 2000; Young et al., 2015). B-stimulated TWINKLE helicase activity is thus required to unwind the mtDNA duplex at the replication fork, where POLγ synthesizes the nascent strand using one of the unwound parental template strands (Clayton, 1982; Bogenhagen and Clayton, 2003; Bowmaker et al., 2003).
POLG mutations are the most common cause of mitochondrial disease, particularly mitochondrial epilepsy, polyneuropathy, ataxia, and PEO (Rahman and Copeland, 2019). More than 300 pathogenic mutations have been mapped to the POLG gene based on the Human DNA Polymerase Gamma Mutation Database, and both homozygous and heterozygous POLγA mutations have been found in individuals ranging from newborns with myocerebrohepatopathy spectrum (MCHS) to the elderly with Parkinsonism (Tzoulis et al., 2013; Hikmat et al., 2017; Rahman and Copeland, 2019). Multiple mtDNA deletions and depletion are two of the most common consequences of POLG mutations (Rahman and Copeland, 2019). The same POLG mutation can often lead to deletions and depletion of mtDNA, or both, which may result in different diseases. For example, the homozygous mutation in POLγA p.A467T has been observed in patients with AHS (Alpers-Huttenlocher syndrome), MELAS, MEMSA (Myoclonic Epilepsy, Myopathy, and Sensory Ataxia), and SANDO (sensory ataxia neuropathy dysarthria and ophthalmoplegia), accompanied by a profound mtDNA depletion or deletions (Neeve et al., 2012; Rajakulendran et al., 2016). Therefore, it is difficult to predict the possible phenotype on the basis of POLG mutations. Despite the wide-ranging mitochondrial-related sickness, there are still common clinical features. For example, POLγA mutations in the spacer domain, such as p.W748S, have been found to be a significant cause of inherited neurodegenerative phenotypes, like ataxia, parkinsonism, and seizures (Van Goethem et al., 2003; Luoma et al., 2004; Hakonen et al., 2005).
In comparison to the POLγA-related neurological diseases, a relatively less neuronal pathology is linked to the mutation in POLγB. However, one research reported that one patient with childhood-onset and progressive neuro-ophthalmic manifestation with optic atrophy, mixed polyneuropathy, spinal and cerebellar ataxia, and generalized chorea, is caused by a homozygous missense mutation in POLG2 p.D433Y, which is associated with mtDNA depletion (Dosekova et al., 2020).
Mitochondrial transcription factor A (TFAM)
TFAM is a high-mobility group-box (HMG) protein encoded by the nuclear genome, which binds to mtDNA in a sequence-specific or non-specific manner. It functions as a master regulator of mitochondrial transcription initiation and mtDNA copy number (Ekstrand, 2004; Campbell et al., 2012; Kang et al., 2018). TFAM binds to the promoter region of mtDNA in a sequence-specific manner and thereby initiates promoter-specific transcription or replication from the light-strand promoter (Gustafsson et al., 2016; Ramachandran et al., 2017). In a non-sequence-specific manner, TFAM binds non-specifically to all sequences of mtDNA, which plays a role as an mtDNA packaging factor that can bind, wrap, and bend the mitochondrial genome into nucleoid-like structures (Kaufman et al., 2007; Kukat et al., 2015).
TFAM protein levels in the CNS were shown to be reduced by ~43% in some AD, PD, and HD patients, demonstrating the association of TFAM abnormalities with a variety of neurodegenerative diseases (Sheng et al., 2012; Kang et al., 2018). For example, TFAM levels in brain lysates of HD patients present significant grade-dependent reductions, with a 15% drop in Grade 2 HD, 32% in Grade 3 HD, and 41% in Grade 4 HD. The decreased TFAM levels are accompanied by mitochondrial deletion as well as aberrant mitochondrial morphogenesis and dynamics (Kim et al., 2010). Besides the lower TFAM levels seen in patients with neurological diseases, TFAM gene mutations or polymorphisms, primarily from two TFAM mutations, rs1937, and rs2306604, are also supposed to increase the risk of PD (Gaweda-Walerych et al., 2010; Gatt et al., 2013), AD (Günther et al., 2004; Zhang et al., 2011; Lillenes et al., 2017), and HD progression (Lillenes et al., 2017).
In mice, heterozygous TFAM mutations (TFAM+/−) result in a reduction in mtDNA copy number, but homozygous TFAM mutations (TFAM−/−) are embryonically lethal (Larsson et al., 1998). Moreover, the generated CaMKII neuron-specific Tfam knockout mice through the Cre-loxP system show a remarkably reduction of mtDNA copy number and mtRNA levels in the neocortex at 2 and 4 months of age, respectively. The neuropathological consequences of the diminished mitochondrial genome in Tfam knockout mice are progressive nerve cell loss, apoptosis, gliosis in the neocortex and hippocampus, and a low induction of antioxidant defenses (Sörensen et al., 2001). Additional studies show that TFAM knockout in neurons results in reduced mtDNA expression and respiratory chain deficiency in midbrain DA neurons, which, in turn, leads to a Parkinson’s-like neurodegenerative phenotype with adult-onset of slowly progressive impairment of motor function accompanied by the formation of intraneuronal inclusions and dopamine nerve cell death (Ekstrand et al., 2007). These investigations reveal the crucial roles of TFAM in the maintenance of the mitochondrial genome, and that TFAM deletion in neurons would render neurons more sensitive to any impairments, leading to neurodegenerative phenotypes.
Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α)
PGC-1α is a transcriptional coactivator and a master inducible upstream regulator of mitochondrial biogenesis functioning in both the nucleus and mitochondria (Wu et al., 1999; Aquilano et al., 2010; Safdar et al., 2011). In the nucleus, PGC-1α exerts its pleiotropic effects in mediating mitochondrial biogenesis by binding and working together with other transcription factors, such as peroxisome proliferator-activated receptors (PPARs), nuclear respiratory factors (NRF1 and NRF2), and cAMP response element-binding protein (CREB; Schreiber et al., 2004; Arany et al., 2005; Wu et al., 2006; Nirwane and Majumdar, 2018; Liu et al., 2021). For example, PGC-1α activates the transcriptional factor NRF1, which subsequently binds to the specific promoter site of TFAM and regulates its expression in the nucleus. Elevated TFAM expression eventually promotes the transcription of the mtDNA (Wu et al., 1999). Interestingly, PGC-1α was also found to reside in the mitochondria and form a multiprotein complex with sirtuin 1 and TFAM, suggesting that they may play a coordinating role in the regulation of mitochondrial biogenesis (Aquilano et al., 2010).
PGC-1α expression is closely correlated with the survival of a variety of neurons, including cholinergic, glutamatergic, dopaminergic, and GABAergic synapses in various regions of the CNS (Zhao et al., 2011; Arnold et al., 2014; Bartley et al., 2015; Jiang et al., 2016; Panes et al., 2022). And growing evidence reveals that impaired PGC-1α expression and/or function is a frequent underlying cause of mitochondrial malfunction in neurological illnesses like PD, HD, and ALS (Johri et al., 2013; Bayer et al., 2017; Yang et al., 2020; Piccinin et al., 2021). For example, PGC-1 levels are found to be lower in HD postmortem brains (Reddy et al., 2009). Investigation of a large cohort of PD patients and age-matched controls by multiplexed probe sequencing reveals that two PGC-1α variants (rs6821591 CC and rs2970848 GG) are associated with the risk of PD onset (Clark et al., 2011). Therefore, it is essential to maintain PGC-1α level in a proper range and its normal function.
Mitochondrial RNA polymerase (POLRMT)
After TFAM binds to a position upstream of the transcription start site to initiate mtDNA transcription, TFAM recruits the POLRMT to the promoters, followed by the recruitment of another transcription factor TFB2M to fully assemble the transcription initiation complex (Shi et al., 2012; Barshad et al., 2018). POLRMT is an RNA polymerase executing transcription of the mtDNA. Additionally, during mtDNA replication, POLRMT is also required to initiate mtDNA replication by synthesizing the RNA primers (Fusté et al., 2010; Posse et al., 2019). POLRMT comprises four primary domains: the N-terminal extension (also known as the “tether helix”), a pentatricopeptide repeat (PPR) domain, the NTD, and the CTD (Ringel et al., 2011). The N-terminal extension domain is involved in the interactions between POLRMT and TFAM, which is crucial for anchoring the active site of POLRMT near the transcription start site, whereas the NTD is involved in the contact of POLRMT with TFB2M. which leads to the melting of the DNA duplex and the formation of the open initiation complex, where de novo RNA synthesis can begin (Hillen et al., 2017; Oláhová et al., 2021).
Eight patients with diverse mutations in the POLRMT gene at different points of sequence, including p.P566S, p.S1193F, p.D870N, p.P742_P747del, p.H250D, p.S611F, p.F641L, p.C925X, p.P810S, p.F149X, p.R1013C, p.G881_K883del, cause a broad range of neurological manifestations, such as developmental delays including fine motor, gross motor, language, social/behavioral, and thinking/intellectual. These POLRMT mutations induce a defect in mitochondrial mRNA synthesis but without mtDNA deletions or copy number abnormalities (Oláhová et al., 2021).
Dysregulation of Mitochondrial Import Machinery
In mammalian mitochondria, 99% of proteins are encoded by nuclear genes and synthesized on cytosolic ribosomes as precursor proteins, most of which are subsequently recognized and imported into mitochondria through a common entry gate, the translocase of TOM complex, and then sorted to the destined subcompartments of mitochondria via five major protein import pathways (Figure 6; Schmidt et al., 2010; Wiedemann and Pfanner, 2017): (1) The classical import pathway is termed the “presequence pathway”, which is characterized by cleavable N-terminal mitochondrial targeting sequences (MTSs). The vast majority of matrix proteins and many inner membrane-localized proteins are dependent on the presequence pathway for mitochondrial entry (Abe et al., 2000; Vögtle et al., 2009). After passing through the TOM complex, those presequence-carrying precursors are subsequently imported by presequence translocase of the inner membrane TIM23 (Mokranjac and Neupert, 2015). The presequence translocase-associated motor (PAM) drives protein translocation into the matrix, where the MTSs are proteolytically removed by an MPP (Hawlitschek et al., 1988). In contrast to the presequence pathway, the precursor proteins of the other four import pathways do not carry cleavable MTS, but instead possess different kinds of internal MTSs (Schmidt et al., 2010; Wiedemann and Pfanner, 2017); (2) SAM pathway: the precursors of β-barrel proteins are translocated through TOM and bind the small TIM chaperones TIM9/10 in the intermembrane space, where the sorting and assembly machinery (SAM) inserts them into the outer membrane (Paschen et al., 2003; Wiedemann et al., 2003); (3) The mitochondrial IMS import and assembly (MIA) pathway: the cysteine-rich precursors are kept in a reduced state in the cytosol, imported by the TOM complex, and oxidized by the MIA system (Chacinska et al., 2004); (4) Carrier pathway: precursors of the multispanning hydrophobic carrier proteins of the inner membrane are imported via TOM, small TIM chaperones TIM9/10, and the carrier translocase TIM22 (Sirrenberg et al., 1996, 1998); and (5) The mitochondrial import (MIM) pathway: a number of proteins with α-helical transmembrane segments are inserted into the outer membrane by the MIM complex (Becker et al., 2008; Popov-Čeleketić et al., 2008).
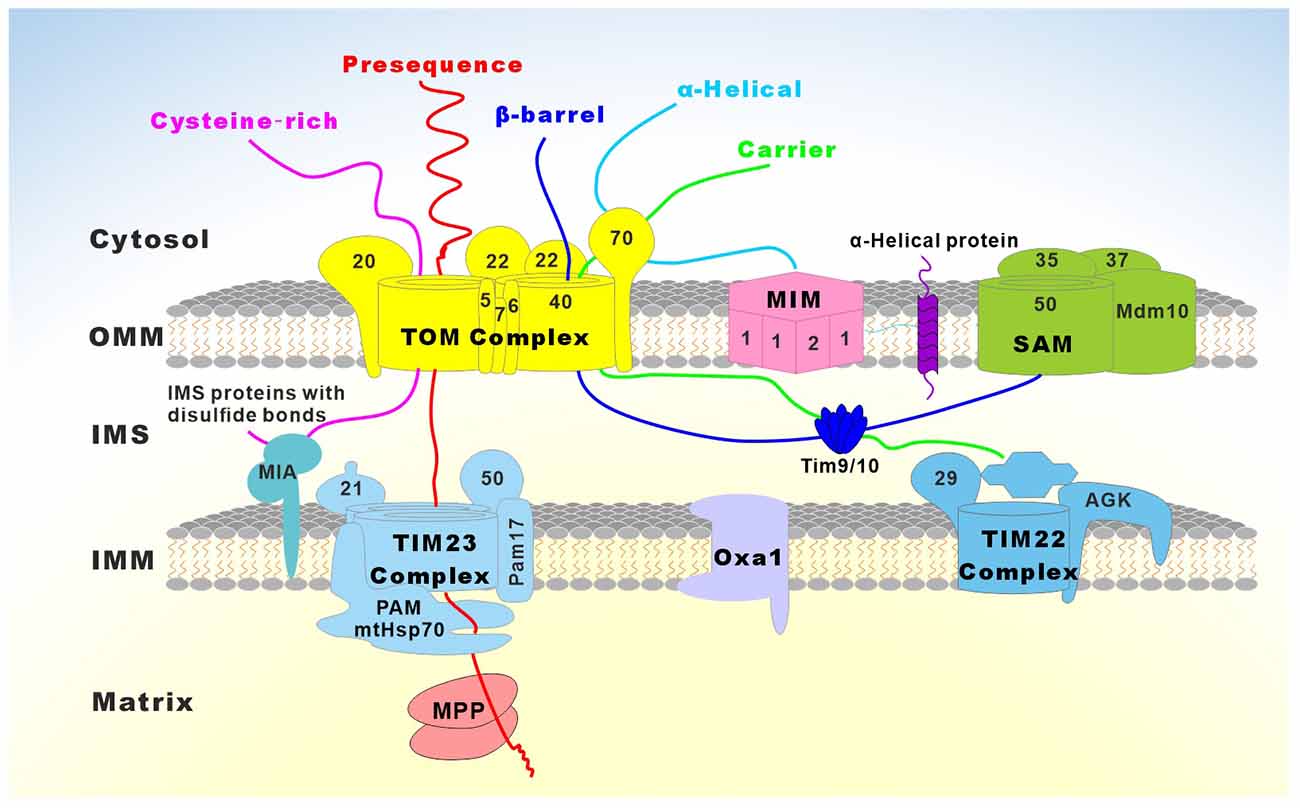
Figure 6. Overview of the major mitochondrial protein import pathways. Mitochondrial precursor proteins are synthesized in the cytosol, transported to the mitochondria, and directed to the correct mitochondrial compartments. Five major protein import pathways have been identified so far: (1) Presequence-carrying preproteins are imported through the translocase of the outer mitochondrial membrane (TOM) and the translocase of the inner mitochondrial membrane (TIM23) complexes. Proteins containing a hydrophobic sorting signal are released into the inner mitochondrial membrane (IMM), while hydrophilic proteins are imported into the matrix with the help of the presequence translocase-associated motor (PAM) complex. Membrane potential across the IMM is essential for the entry of presequences into the matrix. The presequences are cleaved by the mitochondrial processing peptidase (MPP) and additional proteolytic processing occurs by intermediate cleaving peptidases. Presequence-carrying precursors that are integrated into the IMM are either directly released from the TIM23 complex or transported into the matrix, followed by further insertion into the IMM with the help of Oxa1; (2) Cysteine-rich proteins of the intermembrane space (IMS) are imported by TOM and the mitochondrial IMS import and assembly (MIA) system, which inserts disulfide bonds in the imported proteins; (3) The precursors of β-barrel proteins are translocated through the TOM complex to the TIM9/10 in the IMS and are inserted into the outer mitochondrial membrane (OMM) by the sorting and assembly machinery (SAM); (4) The precursors of metabolite carriers of the IMM are imported via the TOM complex, small TIM9/10 chaperones, and the carrier translocase TIM22 complex; and (5) Some proteins with α-helical transmembrane segments are inserted into the OMM by the mitochondrial import (MIM) complex.
Mitochondrial importing machinery is essential for protein translocation and is deeply integrated into a functional network of mitochondrial bioenergetics, dynamics, and morphology, as well as protein quality control and interaction with other organelles. Thus, mitochondrial import defects can potentially induce all types of mitochondrial dysfunction. As a result, an increasing number of cases of defects in proteins involved in mitochondrial import have been associated with a spectrum of disorders, including different kinds of neuropathies (Nicolas et al., 2019; Palmer et al., 2021; Figure 6 and Table 4).
Table 4. Neurological diseases caused by protein variants involved in mitochondrial import machinery.
Tom70
Tom70 is an adaptor protein of the TOM complex that loosely attaches to the TOM complex and plays a major role in the import of non-cleavable hydrophobic precursor proteins such as the carrier precursors (Brix et al., 1999; Young et al., 2003). Tom70 binds the precursors-chaperone complex, facilitating the transfer of precursors to the channel-forming protein Tom40, where the precursors are translocated across the outer membrane (Hill et al., 1998; Wiedemann and Pfanner, 2017). Two children with distinct point mutations in Tom70, p.T607I, and p.I554F, present common features including significant white matter abnormalities, hypotonia, hyperreflexia, dystonia, and cognitive deficits. Causative investigation of Tom70-induced neurological abnormalities in Drosophila reveals that homozygous Tom70 null flies are pupal lethal. Tom70 knockdown impairs synaptic transmission and results in degenerative eye phenotype, indicating that Tom70 is required in neuronal maintenance (Dutta et al., 2020). Another study on a patient with Tom70 heterozygous mutations [(p.T265M) and (p.A582V)] found OXPHOS complex impairments, particularly complex IV (Wei et al., 2020b).
Tim50
Tim50 is a component of the Tim23 translocase complex in the IMM. Tim50 exposes its domains to the IMS and functions as a presequence receptor that binds preproteins emerging on the intermembrane space side of Tom40-Tom22 (Lytovchenko et al., 2013; Rahman et al., 2014). By cooperating with the regulatory subunit Tim21, Tim50 interacts with the N-terminal intermembrane space domain of Tim23, the channel-forming protein of the TIM23 complex, and regulates the open or closed state of the Tim23 channel. In the absence of preproteins, Tim50 helps keep the Tim23 channel in a closed state. Upon binding of a presequence, the Tim23 channel is activated and opened (Truscott et al., 2001; Meinecke et al., 2006). Using whole-exome sequencing, researchers discovered that two homozygous missense mutations in the TIMM50 gene, p.R217W, and p.T252M, cause severe intellectual disability and seizure disorder in four patients (Shahrour et al., 2017). Moreover, a compound heterozygous mutation in TIMM50 (p.S112X and p.G190A) in an infant patient causes rapidly progressive and severe encephalopathy. These mutations induce the reduction of the Tim50 level, accompanied by lower steady-state levels of several components of OXPHOS and enhanced ROS production (Reyes et al., 2018).
Tim22
Tim22 is a channel-forming protein of the TIM22 complex, mediating the insertion of precursor proteins into the inner membrane in a Δψ-driven manner (Wiedemann and Pfanner, 2017). Tim22 with the p.V33L mutation disrupts the formation of the translocase complex and compromises levels of metabolite carrier proteins within the inner membrane, which may cause an imbalance of mitochondrial metabolites and lead to a secondary dysfunction of OXPHOS. A case of compound heterozygous variants in Tim22 (a point mutation variant p.V33L and a premature truncated variant p.Y25X) in a young patient presents delayed myelination, neuromuscular illness with hypotonia, gastroesophageal reflux disease, consistently elevated lactate in the serum, and cerebrospinal fluid (CSF; Pacheu-Grau et al., 2018).
Tim8a
Tim8 is a member of the small Tim chaperones that dynamically interacts with the TIM22 complex to transfer the precursor proteins for sorting and insertion into the inner membrane by preventing protein aggregation within the aqueous IMS (Bauer et al., 1999). In humans, there are two Tim8 isoforms, Tim8a and Tim8b, which have been discovered to play a role in the assembly of complex IV (Gentle et al., 2007; Kang et al., 2019). Compared to Tim8b, Tim8a is more strongly expressed in the human brain (Kang et al., 2019). In accordance with its distribution, diverse mutations in the TIMM8A gene (p.M1L, p.E24X p.Q28X, p.Q38X, p.M39fs, p.C43fs, p.K50fs, p.C66W, p.R80X) cause Mohr-Tranebjaerg syndrome (also called deafness-dystonia-optic neuronopathy [DDON]), an X-linked recessive neurodegenerative disorder characterized by progressive sensorineural hearing loss, dystonia, cortical blindness, and dysphagia (Tranebjærg et al., 1995, 2000; Jin et al., 1996; Ujike et al., 2001; Neighbors et al., 2020).
Oxa1
Oxa1 is a member of the YidC/Alb3/Oxa1 insertase family and is implicated in protein insertion machinery by exporting proteins from the matrix into the inner membrane (Hennon et al., 2015). A large number of inner membrane-localized proteins, such as the TIM22 complex, CI, CIV, and F1Fo-ATP Synthase, are dependent on the Oxa1 involved import machinery (Bonnefoy et al., 1994; Altamura et al., 1996; Stiburek et al., 2007). Due to the crucial role of Oxa1 in the assembly of OXPHOS complexes, biallelic variants in OXA1L (p.S170Qfs*18 and p.C207F) in a patient caused a profoundly decreased level of OXPHOS complexes I, IV, and V, which leads to severe illness with hypotonia, severe encephalopathy, and developmental delay in early childhood (Thompson et al., 2018).
MPP
MPP is a heterodimeric peptidase that is comprised of MPPα and MPPβ residing in the matrix (Hawlitschek et al., 1988; Taylor et al., 2001; Gakh et al., 2002). MPP proteolytically cleaves off the N-terminal presequences of most precursor proteins that are fully translocated to the matrix as well as precursors in transit to the inner membrane or the intermembrane space (Vögtle et al., 2009; Wiedemann and Pfanner, 2017). The larger MPP subunit MPPα is likely to be engaged in substrate recognition, whereas the smaller MPP subunit MPPβ is the catalytic subunit that removes the MTS of freshly imported precursor proteins (Braun et al., 1992; Greene et al., 2012). In humans, mutations in the MPPα encoding gene PMPCA, including the homozygous missense mutation (p.A377T) and the compound heterozygous mutations (p.S96L and p.G515R), have been uncovered in patients with non-progressive and slowly progressive cerebellar ataxias. Moreover, all patients showed impaired processing of frataxin, a well-known substrate of MPP, highlighting the tight association of MPP with ataxia-related illness (Choquet et al., 2016). Besides MPPα, biallelic mutations in the encoding gene of MPPβ, PMPCB (p.R175C/p.A201P, p.V177G/p.R175H, p.I422T, p.E396D), caused a complex and severe neurological phenotype of neurodegeneration and cerebellar atrophy in early childhood (Vögtle et al., 2018). Both PMPCA and PMPCB gene mutations result in a strong increase in the intermediate form of frataxin, a conserved mitochondrial protein that is implicated in the Fe-S cluster assembly in mitochondria. The reduction of mature frataxin protein impairs the biogenesis of Fe-S clusters, such as Fe-S cluster-containing OXPHOS complexes (Stemmler et al., 2010; Choquet et al., 2016; Vögtle et al., 2018).
Dysregulation of Mitochondrial Ion Channels
Mitochondrial ion channels are a group of integral membrane proteins mediating ionic fluxes across the mitochondrial membranes, which are driven by electrochemical gradients (Δμion; O’Rourke, 2007), such as the voltage-dependent anion channel of the outer membrane (VDAC) in the OMM and the mitochondrial calcium uniporter (MCU) in the IMM. Despite the opening of mitochondrial ion channels may be transient and tightly regulated, these ion channels are critical for diverse mitochondrial functions and cell survival (O’Rourke, 2007; Szabo and Zoratti, 2014; Urbani et al., 2021). Growing evidence reveals that malfunction of mitochondrial ion channels are linked to aging and diseases like PD and AD (Liao et al., 2017; Surmeier et al., 2017; Shoshan-Barmatz et al., 2018; Ashrafuzzaman, 2022).
In normal conditions, the IMM is quite impermeable to ions in order to maintain the efficiency of the OXPHOS (Javadov and Karmazyn, 2007). Accumulation of ROS, imbalance in Ca2+ concentration, depolarization of mitochondrial membrane potential, or mutation in the mtDNA may cause a sudden increase in the IMM permeability to ions and other solutes up to 1,500 Da non-selectively. This permeability transition (PT) is caused by the opening of the permeability transition pores (PTP; Halestrap et al., 1998; Rasola and Bernardi, 2011). PTPs are large-conductance (~1 nS) pores formed by multi-proteins, such as the mitochondrial adenine nucleotide translocase (ANT) in the IMM and the VDAC in the OMM. Non-specific entry of water, ions, and solutes into mitochondria due to the opening of PTP leads to mitochondrial swelling. Although slight increases in matrix volume within the physiological range can stimulate mitochondrial function and metabolism, excessive mitochondrial swelling caused by persistent PTP opening initiates an irreversible cascade of events, including the depolarized inner membrane potential, the uncoupled OXPHOS, excess ROS production, crista unfolding, the release of stored Ca2+ and other apoptogenic proteins (such as Cyt c) into the cytosol. These events ultimately result in neuronal death (Javadov et al., 2018). In response to oxidative stress, the PTP opening also plays a causal role in the disassembling of the ETC supercomplexes (Jang et al., 2017). In PINK1-deficient midbrain neurons, the severely inhibited Ca2+ extrusion mediates Ca2+ overload inside the mitochondria and excessive ROS generation, which causes PTP opening and eventually midbrain neuronal death (Gandhi et al., 2009; Schapira, 2013). This neuronal loss is thought to play a central role in the neurodegeneration of PD (Ludtmann and Abramov, 2018; Figure 7).
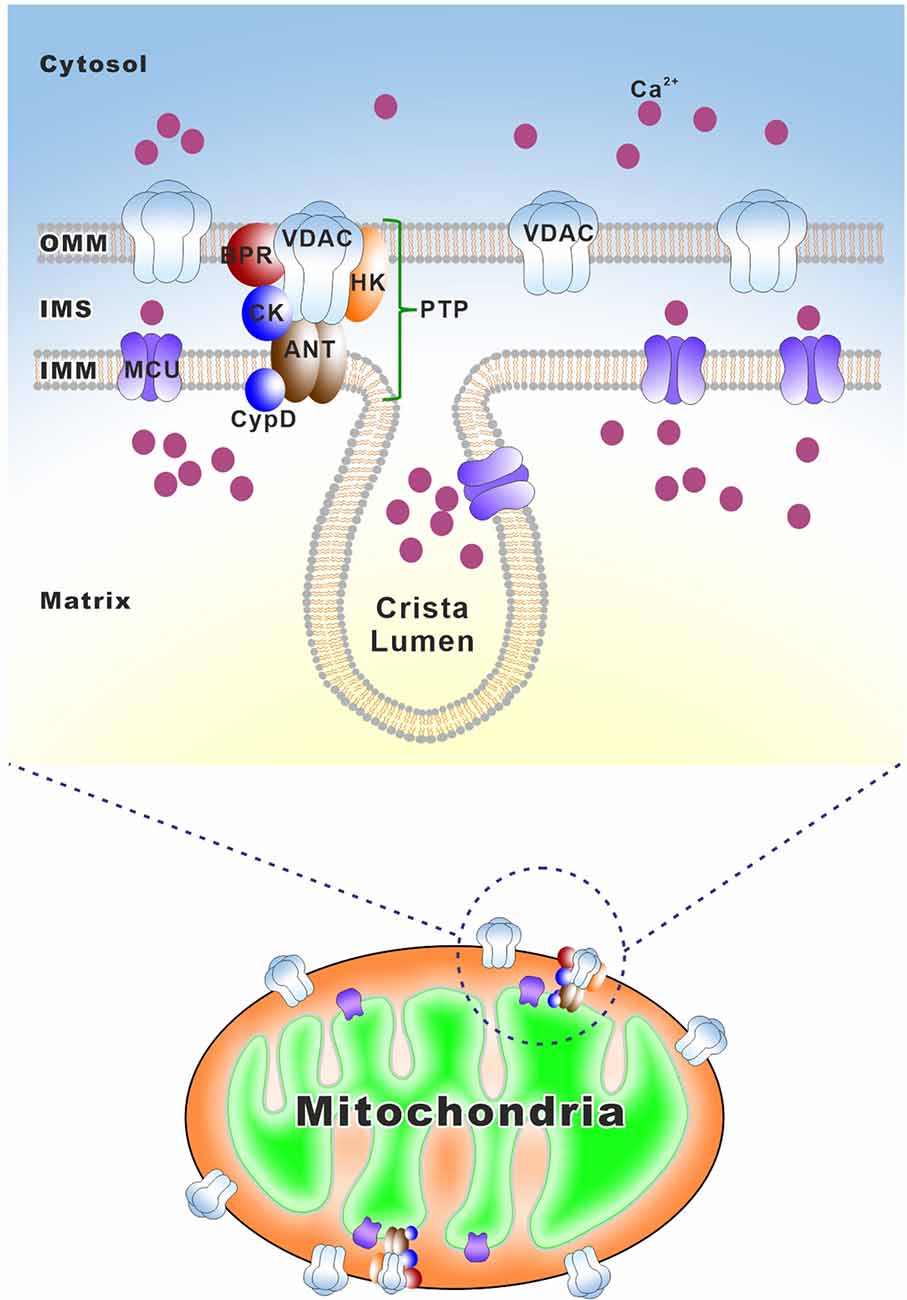
Figure 7. Schematic overview of mitochondrial ion channels that are implicated in neurodegeneration. Mitochondrial ion channels are a group of integral membrane proteins mediating ionic fluxes across the mitochondrial membranes. VDAC is a voltage-dependent anion channel residing in the OMM and MCU is a mitochondrial calcium uniporter in the IMM. PTP is a mitochondrial permeability transition pore that allows mitochondria to undergo a sudden increase of permeability, including VDAC in the OMM and the adenine nucleotide translocase (ANT) in the IMM. Cyclophilin D (CypD) in the matrix is a sensitizer of PTP. Peripheral benzodiazepine receptor (BPR), hexokinase (HK), and creatine kinase (CK) are the other components of PTP.
VDAC
VDAC is a highly conserved pore-forming protein located in the OMM. It is a large-conductance anion channel, providing the major pathway for transmembrane fluxes of ions and metabolites across the OMM (Shoshan-Barmatz et al., 2010; Rosencrans et al., 2021). VDAC is the most abundant OMM protein accounting for ~50% of the overall protein content and thus is responsible for 90% of the OMM permeability (Gonçalves et al., 2007; Morgenstern et al., 2017). In addition, VDAC associates with over 100 other proteins, such as proteins involved in Ca2+ homeostasis and apoptosis (Shoshan-Barmatz et al., 2017). Three VDAC isoforms (VDAC1, VDAC2, and VDAC3) are found in mammalian cells, and VDAC1 is the most widely expressed, followed by VDAC2 and VDAC3 (Cesar and Wilson, 2004; Messina et al., 2012). VDAC1 is linked to the toxicity of pathogenic proteins in neurodegenerative disorders, including phosphorylated tau, Aβ, α-synuclein, and γ-secretase (Magri and Messina, 2017). Furthermore, significant changes in the VDAC1 level were found in neurodegenerative illnesses such as AD, PD, ALS, and HD (Risiglione et al., 2021). For example, an aberrant VDAC1 level was found in patients with AD or Down’s syndrome (César Rosa and de Cerqueira César, 2016).
MCU
MCU is a calcium-selective ion channel in the IMM and is responsible for low-affinity Ca2+ uptake into the mitochondrial matrix (Baughman et al., 2011). The MCU complex is composed of several components, including pore-forming MCU protein, MICU1 (calcium uptake protein 1, mitochondria), MICU2/3 (calcium uptake protein 2/3, mitochondria), EMRE (essential MCU regulator, mitochondria), MCUR1 (mitochondrial calcium uniporter regulator 1), and MCUb (calcium uniporter regulatory subunit MCUb, mitochondria). MCU functions as a critical pore-forming and calcium-conducting subunit, and the rest components work as regulatory subunits (Reed and Bygrave, 1974; Liao et al., 2017). MICU1 and MICU2/3 sense matrix calcium via their conserved calcium-binding EF-hand domains (Perocchi et al., 2010).
The mitochondrial calcium level regulates the mitochondrial activity and cellular ATP supply. Impaired Ca2+ homeostasis leads to mitochondrial malfunction, which promotes neurodegeneration (Kawahara and Kuroda, 2000; Wang et al., 2013; Ryan et al., 2020). For example, Aβ peptide treatment elevated the mitochondrial calcium level in cortical neurons, resulting in neurodegeneration. Inhibition of MCU activity with a specific MCU inhibitor can stop this process, indicating that the toxicity of Aβ may be partly due to its capacity to interfere with the homeostasis of mitochondrial calcium (Hedskog et al., 2013). In a multiplex consanguineous family, a homozygous truncating mutation in MICU2 (c.42G4A, p.W14*) was found to fully segregate with a neurodevelopmental disorder characterized by severe cognitive impairment, spasticity, and white matter involvement. Patient-derived MICU2-deficient cells present impaired mitochondrial Ca2+ homeostasis with abnormal regulation of inner mitochondrial membrane potential as well as an increase in the vulnerability of mitochondria to oxidative stress (Shamseldin et al., 2017).
Conclusions
A growing number of neurological illnesses are found to be linked with mitochondrial protein dysfunction. In recent years, many novel mitochondrial proteins and mutations have been identified and characterized, providing a better understanding of both mitochondrial function and dysfunction. In this review, we summarized the cellular roles of mitochondrial proteins involved in mitochondrial bioenergetics, dynamics, mitophagy, genome maintenance, importing machinery, and ion channels, as well as how aberrant mitochondrial proteins contribute to the pathogenesis of neurological disorders.
In addition to the mitochondrial protein deficits, mounting evidence reveals that numerous abnormal non-mitochondrial proteins also target mitochondria, causing mitochondrial neurotoxicity and, consequently, the onset of neurological diseases. Therefore, modulating mitochondrial function, especially regulating the mitochondrial proteome, could be a promising therapeutic avenue in treating various neurological disorders. However, many challenges remain. Although we are increasingly able to establish the genetic diagnosis in patients with mitochondrial diseases, there are still patients for whom the diagnosis is difficult. Furthermore, complications of mitochondrial disease and clinical phenotype variations in patients with similar levels of heteroplasmy are common. Thus, identifying the cause of this variation is critical for designing potentially preventative treatment strategies.
Author Contributions
LW and ZY prepared the original draft, the figure and tables. XH, SP, and CY participated in the preparation of the manuscript. HZ supervised and revised the manuscript. QW, ZZ, and XC reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Academy of Finland (HZ; decision No. 323670), Jane and Aatos Erkko Foundation (HZ), and Guangxi distinguished expert funding (HZ). LW is supported by the National Natural Science Foundation of China (No. 32000719), the fellowship of China Postdoctoral Science Foundation (No. 2021M702362), and the “Post-Doctor Research project, West China Hospital, Sichuan University” (No. 2020HXBH010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ababneh, N. A., Barham, R., Al-kurdi, B., Ali, D., Al, S., Ismail, M. A., et al. (2022). Generation of a human induced pluripotent stem cell (iPSC) line (JUCTCi019-A) from a patient with Charcot-Marie-Tooth disease type 2A2 (CMT2A2) due to a heterozygous missense substitution c.2119C>T (p.Arg707Trp) in MFN2 gene. Stem Cell Res. 62:102786. doi: 10.1016/j.scr.2022.102786
Abdoh, R. M. (2019). Homozygote c. 1789c>t (p.arg597trp) polg gene mutation in consanguineous moroccan patient with predominant motor phenotype of sando syndrome. J. Neurol. Sci. 405:65. doi: 10.1016/j.jns.2019.10.1684
Abdulhag, U. N., Soiferman, D., Schueler-Furman, O., Miller, C., Shaag, A., Elpeleg, O., et al. (2015). Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 23, 159–164. doi: 10.1038/ejhg.2014.85
Abe, Y., Shodai, T., Muto, T., Mihara, K., Torii, H., Nishikawa, S., et al. (2000). Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100, 551–560. doi: 10.1016/s0092-8674(00)80691-1
Abramov, A. Y., and Angelova, P. R. (2019). Cellular mechanisms of complex I-associated pathology. Biochem. Soc. Trans. 47, 1963–1969. doi: 10.1042/BST20191042
Abrams, A. J., Fontanesi, F., Tan, N. B. L., Buglo, E., Campeanu, I. J., Rebelo, A. P., et al. (2018). Insights into the genotype-phenotype correlation and molecular function of SLC25A46. Hum. Mutat. 39, 1995–2007. doi: 10.1002/humu.23639
Abrams, A. J., Hufnagel, R. B., Rebelo, A., Zanna, C., Patel, N., Gonzalez, M. A., et al. (2015). Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 47, 926–932. doi: 10.1038/ng.3354
Adjobo-Hermans, M. J. W., de Haas, R., Willems, P. H. G. M., Wojtala, A., van Emst-de Vries, S. E., Wagenaars, J. A., et al. (2020). NDUFS4 deletion triggers loss of NDUFA12 in Ndufs4−/− mice and Leigh syndrome patients: a stabilizing role for NDUFAF2. Biochim. Biophys. Acta - Bioenerg. 1861:148213. doi: 10.1016/j.bbabio.2020.148213
Agarwal, A., Goyal, K., Mathur, P., and Mathur, A. (2020). Encephalopathy due to mutation in mitochondrial fission factor gene. Indian J. Child Health 07, 184–187. doi: 10.32677/IJCH.2020.v07.i04.012
Allkanjari, K., and Baldock, R. A. (2021). Beyond base excision repair: an evolving picture of mitochondrial DNA repair. Biosci. Rep. 41:BSR20211320. doi: 10.1042/BSR20211320
Alston, C. L., Davison, J. E., Meloni, F., van der Westhuizen, F. H., He, L., Hornig-Do, H.-T., et al. (2012). Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J. Med. Genet. 49, 569–577. doi: 10.1136/jmedgenet-2012-101146
Altamura, N., Capitanio, N., Bonnefoy, N., Papa, S., and Dujardin, G. (1996). The Saccharomyces cerevisiae OXA1 gene is required for the correct assembly of cytochrome c oxidase and oligomycin-sensitive ATP synthase. FEBS Lett. 382, 111–115. doi: 10.1016/0014-5793(96)00165-2
Alvarez, V., Corao, A. I., Sánchez-Ferrero, E., De Mena, L., Alonso-Montes, C., Huerta, C., et al. (2008). Mitochondrial transcription factor A (TFAM) gene variation in Parkinson’s disease. Neurosci. Lett. 432, 79–82. doi: 10.1016/j.neulet.2007.12.010
Amati-Bonneau, P., Milea, D., Bonneau, D., Chevrollier, A., Ferré, M., Guillet, V., et al. (2009). OPA1-associated disorders: phenotypes and pathophysiology. Int. J. Biochem. Cell Biol. 41, 1855–1865. doi: 10.1016/j.biocel.2009.04.012
Amiri, M., and Hollenbeck, P. J. (2008). Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev. Neurobiol. 68, 1348–1361. doi: 10.1002/dneu.20668
Andrews, B., Carroll, J., Ding, S., Fearnley, I. M., and Walker, J. E. (2013). Assembly factors for the membrane arm of human complex I. Proc. Natl. Acad. Sci. U S A 110, 18934–18939. doi: 10.1073/pnas.1319247110
Angelova, P. R., and Abramov, A. Y. (2018). Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 592, 692–702. doi: 10.1002/1873-3468.12964
Antonicka, H. (2003). Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 12, 2693–2702. doi: 10.1093/hmg/ddg284
Aquilano, K., Vigilanza, P., Baldelli, S., Pagliei, B., Rotilio, G., Ciriolo, M. R., et al. (2010). Peroxisome Proliferator-activated Receptor γ Co-activator 1α (PGC-1α) and Sirtuin 1 (SIRT1) reside in mitochondria. J. Biol. Chem. 285, 21590–21599. doi: 10.1074/jbc.M109.070169
Arano, T., and Imai, Y. (2015). “Mitophagy regulated by the PINK1-parkin pathway,” in Cell Death - Autophagy, Apoptosis and Necrosis, ed. T. M. Ntuli (London: IntechOpen), 113–131. doi: 10.5772/61284
Arany, Z., He, H., Lin, J., Hoyer, K., Handschin, C., Toka, O., et al. (2005). Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab. 1, 259–271. doi: 10.1016/j.cmet.2005.03.002
Ardissone, A., Invernizzi, F., Nasca, A., Moroni, I., Farina, L., and Ghezzi, D. (2015). Mitochondrial leukoencephalopathy and complex II deficiency associated with a recessive SDHB mutation with reduced penetrance. Mol. Genet. Metab. Rep. 5, 51–54. doi: 10.1016/j.ymgmr.2015.10.006
Arnold, A.-S., Gill, J., Christe, M., Ruiz, R., McGuirk, S., St-Pierre, J., et al. (2014). Morphological and functional remodelling of the neuromuscular junction by skeletal muscle PGC-1α. Nat. Commun. 5:3569. doi: 10.1038/ncomms4569
Ashrafuzzaman, M. (2022). Mitochondrial ion channels in aging and related diseases. Curr. Aging Sci. 15, 97–109. doi: 10.2174/1874609815666220119094324
Assouline, Z., Jambou, M., Rio, M., Bole-Feysot, C., de Lonlay, P., Barnerias, C., et al. (2012). A constant and similar assembly defect of mitochondrial respiratory chain complex I allows rapid identification of NDUFS4 mutations in patients with Leigh syndrome. Biochim. Biophys. Acta 1822, 1062–1069. doi: 10.1016/j.bbadis.2012.01.013
Atamna, H., and Boyle, K. (2006). Amyloid-β peptide binds with heme to form a peroxidase: Relationship to the cytopathologies of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 103, 3381–3386. doi: 10.1073/pnas.0600134103
Atay, Z., Bereket, A., Turan, S., Haliloglu, B., Memisoglu, A., Khayat, M., et al. (2013). A novel homozygous TMEM70 mutation results in congenital cataract and neonatal mitochondrial encephalo-cardiomyopathy. Gene 515, 197–199. doi: 10.1016/j.gene.2012.11.044
Atwal, P. S. (2013). Mutations in the complex III assembly factor tetratricopeptide 19 gene TTC19 are a rare cause of leigh syndrome. JIMD Rep. 14, 43–45. doi: 10.1007/8904_2013_282
Bakare, A. B., Lesnefsky, E. J., and Iyer, S. (2021). Leigh syndrome: a tale of two genomes. Front. Physiol. 12:12:693734. doi: 10.3389/fphys.2021.693734
Baker, M. J., Lampe, P. A., Stojanovski, D., Korwitz, A., Anand, R., Tatsuta, T., et al. (2014). Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 33, 578–593. doi: 10.1002/embj.201386474
Baker, R. A., Priestley, J. R. C., Wilstermann, A. M., Reese, K. J., and Mark, P. R. (2019). Clinical spectrum of BCS1L mitopathies and their underlying structural relationships. Am. J. Med. Genet. Part A 179, 373–380. doi: 10.1002/ajmg.a.61019
Balsa, E., Marco, R., Perales-Clemente, E., Szklarczyk, R., Calvo, E., Landázuri, M. O., et al. (2012). NDUFA4 Is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 16, 378–386. doi: 10.1016/j.cmet.2012.07.015
Ban, R., Liu, Z., Shimura, M., Tong, X., Wang, J., Yang, L., et al. (2021). Biallelic COA7-variants leading to developmental regression with progressive spasticity and brain atrophy in a chinese patient. Front. Genet. 12:685035. doi: 10.3389/fgene.2021.685035
Barel, O., Shorer, Z., Flusser, H., Ofir, R., Narkis, G., Finer, G., et al. (2008). Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am. J. Hum. Genet. 82, 1211–1216. doi: 10.1016/j.ajhg.2008.03.020
Barghuti, F., Elian, K., Gomori, J. M., Shaag, A., Edvardson, S., Saada, A., et al. (2008). The unique neuroradiology of complex I deficiency due to NDUFA12L defect. Mol. Genet. Metab. 94, 78–82. doi: 10.1016/j.ymgme.2007.11.013
Barshad, G., Marom, S., Cohen, T., and Mishmar, D. (2018). Mitochondrial DNA transcription and its regulation: an evolutionary perspective. Trends Genet. 34, 682–692. doi: 10.1016/j.tig.2018.05.009
Bartley, A. F., Lucas, E. K., Brady, L. J., Li, Q., Hablitz, J. J., Cowell, R. M., et al. (2015). Interneuron transcriptional dysregulation causes frequency-dependent alterations in the balance of inhibition and excitation in hippocampus. J. Neurosci. 35, 15276–15290. doi: 10.1523/JNEUROSCI.1834-15.2015
Bauer, M. F., Gempel, K., Reichert, A. S., Rappold, G. A., Lichtner, P., Gerbitz, K.-D., et al. (1999). Genetic and structural characterization of the human mitochondrial inner membrane translocase. J. Mol. Biol. 289, 69–82. doi: 10.1006/jmbi.1999.2751
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Bayer, H., Lang, K., Buck, E., Higelin, J., Barteczko, L., Pasquarelli, N., et al. (2017). ALS-causing mutations differentially affect PGC-1α expression and function in the brain vs. peripheral tissues. Neurobiol. Dis. 97, 36–45. doi: 10.1016/j.nbd.2016.11.001
Becker, T., Pfannschmidt, S., Guiard, B., Stojanovski, D., Milenkovic, D., Kutik, S., et al. (2008). Biogenesis of the mitochondrial TOM complex. J. Biol. Chem. 283, 120–127. doi: 10.1074/jbc.M706997200
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Bezawork-Geleta, A., Rohlena, J., Dong, L., Pacak, K., and Neuzil, J. (2017). Mitochondrial complex II: at the crossroads. Trends Biochem. Sci. 42, 312–325. doi: 10.1016/j.tibs.2017.01.003
Birch-Machin, M. A., Taylor, R. W., Cochran, B., Ackrell, B. A. C., and Turnbull, D. M. (2000). Late-onset optic atrophy, ataxia and myopathy associated with a mutation of a complex II gene. Ann. Neurol. 48, 330–335. doi: 10.1002/1531-8249(200009)48:3<330::AID-ANA7>3.0.CO;2-A
Bogenhagen, D. F., and Clayton, D. A. (2003). The mitochondrial DNA replication bubble has not burst. Trends Biochem. Sci. 28, 357–360. doi: 10.1016/S0968-0004(03)00132-4
Böhm, M., Pronicka, E., Karczmarewicz, E., Pronicki, M., Piekutowska-Abramczuk, D., Sykut-Cegielska, J., et al. (2006). Retrospective, multicentric study of 180 children with cytochrome c oxidase deficiency. Pediatr. Res. 59, 21–26. doi: 10.1203/01.pdr.0000190572.68191.13
Bonneau, D., Lenaers, G., Procaccio, V., Amati-Bonneau, P., and Reynier, P. (2015). Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Acta Ophthalmol. 93, e301–e301. doi: 10.1111/j.1755-3768.2015.0128
Bonnefoy, N., Chalvet, F., Hamel, P., Slonimski, P. P., and Dujardin, G. (1994). OXA1, a saccharomyces cerevisiae nuclear gene whose sequence is conserved form prokaryotes to eukaryotes controls cytochrome oxidase biogenesis. J. Mol. Biol. 239, 201–212. doi: 10.1006/jmbi.1994.1363
Borna, N. N., Kishita, Y., Sakai, N., Hamada, Y., Kamagata, K., Kohda, M., et al. (2020). Leigh syndrome due to NDUFV1 mutations initially presenting as LBSL. Genes (Basel) 11:1325. doi: 10.3390/genes11111325
Bourgeron, T., Rustin, P., Chretien, D., Birch-Machin, M., Bourgeois, M., Viegas-Péquignot, E., et al. (1995). Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 11, 144–149. doi: 10.1038/ng1095-144
Bowmaker, M., Yang, M. Y., Yasukawa, T., Reyes, A., Jacobs, H. T., Huberman, J. A., et al. (2003). Mammalian mitochondrial DNA replicates bidirectionally from an initiation zone. J. Biol. Chem. 278, 50961–50969. doi: 10.1074/jbc.M308028200
Braun, H. P., Emmermann, M., Kruft, V., and Schmitz, U. K. (1992). The general mitochondrial processing peptidase from potato is an integral part of cytochrome c reductase of the respiratory chain. EMBO J. 11, 3219–3227. doi: 10.1002/j.1460-2075.1992.tb05399.x
Bredesen, D. E., Rao, R. V., and Mehlen, P. (2006). Cell death in the nervous system. Nature 443, 796–802. doi: 10.1038/nature05293
Breuer, M. E., Koopman, W. J., Koene, S., Nooteboom, M., Rodenburg, R. J., Willems, P. H., et al. (2013). The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol. Dis. 51, 27–34. doi: 10.1016/j.nbd.2012.03.007
Brix, J., Rüdiger, S., Bukau, B., Schneider-Mergener, J., and Pfanner, N. (1999). Distribution of binding sequences for the mitochondrial import receptors Tom20, Tom22 and Tom70 in a presequence-carrying preprotein and a non-cleavable preprotein. J. Biol. Chem. 274, 16522–16530. doi: 10.1074/jbc.274.23.16522
Brown, M. D., Voljavec, A. S., Lott, M. T., Torroni, A., Yang, C. C., and Wallace, D. C. (1992). Mitochondrial DNA complex I and III mutations associated with Leber’s hereditary optic neuropathy. Genetics 130, 163–173. doi: 10.1093/genetics/130.1.163
Brzezinski, P., Moe, A., and Ädelroth, P. (2021). Structure and mechanism of respiratory III-IV supercomplexes in bioenergetic membranes. Chem. Rev. 121, 9644–9673. doi: 10.1021/acs.chemrev.1c00140
Bugiani, M. (2005). Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J. Med. Genet. 42, e28–e28. doi: 10.1136/jmg.2004.029926
Burska, D., Stiburek, L., Krizova, J., Vanisova, M., Martinek, V., Sladkova, J., et al. (2021). Homozygous missense mutation in UQCRC2 associated with severe encephalomyopathy, mitochondrial complex III assembly defect and activation of mitochondrial protein quality control. Biochim. Biophys. Acta Mol. Basis Dis. 1867:166147. doi: 10.1016/j.bbadis.2021.166147
Cai, Q., and Jeong, Y. Y. (2020). Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 9:150. doi: 10.3390/cells9010150
Campbell, C. T., Kolesar, J. E., and Kaufman, B. A. (2012). Mitochondrial transcription factor a regulates mitochondrial transcription initiation, DNA packaging and genome copy number. Biochim. Biophys. Acta Gene Regul. Mech. 1819, 921–929. doi: 10.1016/j.bbagrm.2012.03.002
Capiau, S., Smet, J., De Paepe, B., Yildiz, Y., Arslan, M., Stevens, O., et al. (2022). Clinical heterogeneity in MT-ATP6 pathogenic variants: same genotype—different onset. Cells 11:489. doi: 10.3390/cells11030489
Cardaci, S., Zheng, L., MacKay, G., van den Broek, N. J. F., MacKenzie, E. D., Nixon, C., et al. (2015). Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 17, 1317–1326. doi: 10.1038/ncb3233
Carelli, V., Musumeci, O., Caporali, L., Zanna, C., La Morgia, C., Del Dotto, V., et al. (2015a). Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 78, 21–38. doi: 10.1002/ana.24410
Carelli, V., Sabatelli, M., Carrozzo, R., Rizza, T., Schimpf, S., Wissinger, B., et al. (2015b). “Behr syndrome” with OPA1 compound heterozygote mutations. Brain 138, e321–e321. doi: 10.1093/brain/awu234
Carroll, J., Fearnley, I. M., Skehel, J. M., Shannon, R. J., Hirst, J., and Walker, J. E. (2006). Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 281, 32724–32727. doi: 10.1074/jbc.M607135200
Caruso Bavisotto, C., Scalia, F., Marino Gammazza, A., Carlisi, D., Bucchieri, F., Conway de Macario, E., et al. (2019). Extracellular vesicle-mediated cell-cell communication in the nervous system: focus on neurological diseases. Int. J. Mol. Sci. 20:434. doi: 10.3390/ijms20020434
Castro, J. P., Wardelmann, K., Grune, T., and Kleinridders, A. (2018). Mitochondrial chaperones in the brain: safeguarding brain health and metabolism? Front. Endocrinol. (Lausanne) 9:196. doi: 10.3389/fendo.2018.00196
Catarino, C. B., Ahting, U., Gusic, M., Iuso, A., Repp, B., Peters, K., et al. (2017). Characterization of a Leber’s hereditary optic neuropathy (LHON) family harboring two primary LHON mutations m.11778G > A and m.14484T > C of the mitochondrial DNA. Mitochondrion 36, 15–20. doi: 10.1016/j.mito.2016.10.002
Cecchini, G. (2003). Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 72, 77–109. doi: 10.1146/annurev.biochem.72.121801.161700
César Rosa, J., and de Cerqueira César, M. (2016). Role of hexokinase and VDAC in neurological disorders. Curr. Mol. Pharmacol. 9, 320–331. doi: 10.2174/1874467209666160112123036
Cesar, M. D. C., and Wilson, J. E. (2004). All three isoforms of the voltage-dependent anion channel (VDAC1, VDAC2 and VDAC3) are present in mitochondria from bovine, rabbit and rat brain. Arch. Biochem. Biophys. 422, 191–196. doi: 10.1016/j.abb.2003.12.030
Chacinska, A., Pfannschmidt, S., Wiedemann, N., Kozjak, V., Sanjuán Szklarz, L. K., Schulze-Specking, A., et al. (2004). Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 23, 3735–3746. doi: 10.1038/sj.emboj.7600389
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L. J., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chang, C.-R., and Blackstone, C. (2010). Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 1201, 34–39. doi: 10.1111/j.1749-6632.2010.05629.x
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, H., McCaffery, J. M., and Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562. doi: 10.1016/j.cell.2007.06.026
Cho, D.-H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science (80) 324, 102–105. doi: 10.1126/science.1171091
Chocron, E. S., Munkácsy, E., and Pickering, A. M. (2019). Cause or casualty: the role of mitochondrial DNA in aging and age-associated disease. Biochim. Biophys. Acta - Mol. Basis Dis. 1865, 285–297. doi: 10.1016/j.bbadis.2018.09.035
Choquet, K., Zurita-Rendón, O., La Piana, R., Yang, S., Dicaire, M.-J., Boycott, K. M., et al. (2016). Autosomal recessive cerebellar ataxia caused by a homozygous mutation in PMPCA. Brain 139, e19–e19. doi: 10.1093/brain/awv363
Chu, C. T. (2019). Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol. Dis. 122, 23–34. doi: 10.1016/j.nbd.2018.07.015
Čížková, A., Stránecký, V., Mayr, J. A., Tesařová, M., Havlíčková, V., Paul, J., et al. (2008). TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 40, 1288–1290. doi: 10.1038/ng.246
Claeys, K. G., Abicht, A., Häusler, M., Kleinle, S., Wiesmann, M., Schulz, J. B., et al. (2016). Novel genetic and neuropathological insights in neurogenic muscle weakness, ataxia and retinitis pigmentosa (NARP). Muscle Nerve 54, 328–333. doi: 10.1002/mus.25125
Clark, J., Reddy, S., Zheng, K., Betensky, R. A., and Simon, D. K. (2011). Association of PGC-1alphapolymorphisms with age of onset and risk of Parkinson’s disease. BMC Med. Genet. 12:69. doi: 10.1186/1471-2350-12-69
Clayton, D. A. (1982). Replication of animal mitochondrial DNA. Cell 28, 693–705. doi: 10.1016/0092-8674(82)90049-6
Clayton, D. A. (2000). Transcription and replication of mitochondrial DNA. Hum. Reprod. 15, 11–17. doi: 10.1093/humrep/15.suppl_2.11
Coenen, M. J. H., Smeitink, J. A. M., Pots, J. M., van Kaauwen, E., Trijbels, F. J. M., Hol, F. A., et al. (2006). Sequence analysis of the structural nuclear encoded subunits and assembly genes of cytochrome c oxidase in a cohort of 10 isolated complex IV—deficient patients revealed five mutations. J. Child Neurol. 21, 508–511. doi: 10.1177/08830738060210062501
Cogliati, S., Cabrera-Alarcón, J. L., and Enriquez, J. A. (2021). Regulation and functional role of the electron transport chain supercomplexes. Biochem. Soc. Trans. 49, 2655–2668. doi: 10.1042/BST20210460
Coskun, P., Wyrembak, J., Schriner, S. E., Chen, H.-W., Marciniack, C., LaFerla, F., et al. (2012). A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta Gen. Subj. 1820, 553–564. doi: 10.1016/j.bbagen.2011.08.008
Craven, L., Alston, C. L., Taylor, R. W., and Turnbull, D. M. (2017). Recent advances in mitochondrial disease. Annu. Rev. Genomics Hum. Genet. 18, 257–275. doi: 10.1146/annurev-genom-091416-035426
Čunátová, K., Pajuelo Reguera, D., Houštěk, J., Mráček, T., and Pecina, P. (2020). Role of cytochrome c oxidase nuclear-encoded subunits in health and disease. Physiol. Res. 69, 947–965. doi: 10.33549/physiolres.934446
Dallabona, C., Abbink, T. E. M., Carrozzo, R., Torraco, A., Legati, A., van Berkel, C. G. M., et al. (2016). LYRM7 mutations cause a multifocal cavitating leukoencephalopathy with distinct MRI appearance. Brain 139, 782–794. doi: 10.1093/brain/awv392
Danis, D., Brennerova, K., Skopkova, M., Kurdiova, T., Ukropec, J., Stanik, J., et al. (2018). Mutations in SURF1 are important genetic causes of Leigh syndrome in Slovak patients. Endocr. Regul. 52, 110–118. doi: 10.2478/enr-2018-0013
Dautant, A., Meier, T., Hahn, A., Tribouillard-Tanvier, D., di Rago, J.-P., and Kucharczyk, R. (2018). ATP synthase diseases of mitochondrial genetic origin. Front. Physiol. 9:329. doi: 10.3389/fphys.2018.00329
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid. Redox Signal. 10, 621–634. doi: 10.1089/ars.2007.1934
de Lonlay, P., Valnot, I., Barrientos, A., Gorbatyuk, M., Tzagoloff, A., Taanman, J.-W., et al. (2001). A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet. 29, 57–60. doi: 10.1038/ng706
De Meirleir, L. (2004). Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J. Med. Genet. 41, 120–124. doi: 10.1136/jmg.2003.012047
De Michele, G., Sorrentino, P., Nesti, C., Rubegni, A., Ruggiero, L., Peluso, S., et al. (2018). Reversible valproate-induced subacute encephalopathy associated with a MT-ATP8 variant in the mitochondrial genome. Front. Neurol. 9:728. doi: 10.3389/fneur.2018.00728
Del Dotto, V., Fogazza, M., Carelli, V., Rugolo, M., and Zanna, C. (2018). Eight human OPA1 isoforms, long and short: What are they for? Biochim. Biophys. Acta Bioenerg. 1859, 263–269. doi: 10.1016/j.bbabio.2018.01.005
Delettre, C., Lenaers, G., Griffoin, J.-M., Gigarel, N., Lorenzo, C., Belenguer, P., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210. doi: 10.1038/79936
Detmer, S. A., and Chan, D. C. (2007). Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 176, 405–414. doi: 10.1083/jcb.200611080
Díaz-García, C. M., Mongeon, R., Lahmann, C., Koveal, D., Zucker, H., and Yellen, G. (2017). Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 26, 361–374.e4. doi: 10.1016/j.cmet.2017.06.021
Dikov, D., and Reichert, A. S. (2011). How to split up: lessons from mitochondria. EMBO J. 30, 2751–2753. doi: 10.1038/emboj.2011.219
Ding, W.-X., and Yin, X.-M. (2012). Mitophagy: mechanisms, pathophysiological roles and analysis. Biol. Chem. 393, 547–564. doi: 10.1515/hsz-2012-0119
Dohrn, M. F., Heller, C., Zengeler, D., Obermaier, C. D., Biskup, S., Weis, J., et al. (2022). Heterozygous POLG variant Ser1181Asn co-segregating in a family with autosomal dominant axonal neuropathy, proximal muscle fatigability, ptosis and ragged red fibers. Neurol. Res. Pract. 4:5. doi: 10.1186/s42466-022-00169-w
Dong, H.-L., Ma, Y., Yu, H., Wei, Q., Li, J.-Q., Liu, G.-L., et al. (2021). Bi-allelic loss of function variants in COX20 gene cause autosomal recessive sensory neuronopathy. Brain 144, 2457–2470. doi: 10.1093/brain/awab135
Donmez, I., and Patel, S. S. (2006). Mechanisms of a ring shaped helicase. Nucleic Acids Res. 34, 4216–4224. doi: 10.1093/nar/gkl508
Dosekova, P., Dubiel, A., Karlowicz, A., Zietkiewicz, S., Rydzanicz, M., Habalova, V., et al. (2020). Whole exome sequencing identifies a homozygous POLG2 missense variant in an adult patient presenting with optic atrophy, movement disorders, premature ovarian failure and mitochondrial DNA depletion. Eur. J. Med. Genet. 63:103821. doi: 10.1016/j.ejmg.2019.103821
Doxaki, C., and Palikaras, K. (2021). Neuronal mitophagy: friend or foe? Front. Cell Dev. Biol. 8:1864. doi: 10.3389/fcell.2020.611938
Dröse, S., and Brandt, U. (2012). Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 1007, 145–169. doi: 10.1007/978-1-4614-3573-0_6
Duno, M., Wibrand, F., Baggesen, K., Rosenberg, T., Kjaer, N., and Frederiksen, A. L. (2013). A novel mitochondrial mutation m.8989G>C associated with neuropathy, ataxia, retinitis pigmentosa — The NARP syndrome. Gene 515, 372–375. doi: 10.1016/j.gene.2012.12.066
Dutta, D., Briere, L. C., Kanca, O., Marcogliese, P. C., Walker, M. A., High, F. A., et al. (2020). De novo mutations in TOMM70, a receptor of the mitochondrial import translocase, cause neurological impairment. Hum. Mol. Genet. 29, 1568–1579. doi: 10.1093/hmg/ddaa081
Ebanks, B., Ingram, T. L., and Chakrabarti, L. (2020). ATP synthase and Alzheimer’s disease: putting a spin on the mitochondrial hypothesis. Aging (Albany. NY) 12, 16647–16662. doi: 10.18632/aging.103867
Echaniz-Laguna, A., Ghezzi, D., Chassagne, M., Mayencon, M., Padet, S., Melchionda, L., et al. (2013). SURF1 deficiency causes demyelinating Charcot-Marie-Tooth disease. Neurology 81, 1523–1530. doi: 10.1212/WNL.0b013e3182a4a518
Ekstrand, M. I. (2004). Mitochondrial transcription factor a regulates mtDNA copy number in mammals. Hum. Mol. Genet. 13, 935–944. doi: 10.1093/hmg/ddh109
Ekstrand, M. I., Terzioglu, M., Galter, D., Zhu, S., Hofstetter, C., Lindqvist, E., et al. (2007). Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. U S A 104, 1325–1330. doi: 10.1073/pnas.0605208103
El-Hattab, A. W., Suleiman, J., Almannai, M., and Scaglia, F. (2018). Mitochondrial dynamics: biological roles, molecular machinery and related diseases. Mol. Genet. Metab. 125, 315–321. doi: 10.1016/j.ymgme.2018.10.003
Fahrner, J. A., Liu, R., Perry, M. S., Klein, J., and Chan, D. C. (2016). A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. Part A 170, 2002–2011. doi: 10.1002/ajmg.a.37721
Fairman-Williams, M. E., Guenther, U.-P., and Jankowsky, E. (2010). SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 20, 313–324. doi: 10.1016/j.sbi.2010.03.011
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Farías, G. G., Guardia, C. M., De Pace, R., Britt, D. J., and Bonifacino, J. S. (2017). BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci. U S A 114, E2955–E2964. doi: 10.1073/pnas.1616363114
Farooqui, A. A. (2019). Neurochemical aspects of neurological disorders. Curcumin Neurol. Psychiatr. Disord. 2019, 1–22. doi: 10.1016/B978-0-12-815461-8.00001-3
Feichtinger, R. G., Brunner-Krainz, M., Alhaddad, B., Wortmann, S. B., Kovacs-Nagy, R., Stojakovic, T., et al. (2017). Combined respiratory chain deficiency and UQCC2 mutations in neonatal encephalomyopathy: defective supercomplex assembly in complex III deficiencies. Oxid. Med. Cell. Longev. 2017, 1–11. doi: 10.1155/2017/7202589
Ferguson, S. M., and De Camilli, P. (2012). Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13, 75–88. doi: 10.1038/nrm3266
Fernández-Vizarra, E., and Zeviani, M. (2015). Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front. Genet. 6:134. doi: 10.3389/fgene.2015.00134
Ferré, M., Amati-Bonneau, P., Tourmen, Y., Malthièry, Y., and Reynier, P. (2005). eOPA1: an online database for OPA1 mutations. Hum. Mutat. 25, 423–428. doi: 10.1002/humu.20161
Fiedorczuk, K., and Sazanov, L. A. (2018). Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. 28, 835–867. doi: 10.1016/j.tcb.2018.06.006
Filiou, M. D., and Sandi, C. (2019). Anxiety and brain mitochondria: a bidirectional crosstalk. Trends Neurosci. 42, 573–588. doi: 10.1016/j.tins.2019.07.002
Finsterer, J., Fiorini, A. C., Scorza, C. A., and Scorza, F. A. (2018). CMT2 due to homozygous MFN2 variants is a multiorgan mitochondrial disorder. Eur. J. Paediatr. Neurol. 22, 889–891. doi: 10.1016/j.ejpn.2018.04.012
Finsterer, J., and Winklehner, M. (2021). Adult-onset, isolated respiratory chain complex-IV deficiency with mild manifestations. Polish J. Pathol. 72, 185–189. doi: 10.5114/pjp.2021.109523
Formosa, L. E., Dibley, M. G., Stroud, D. A., and Ryan, M. T. (2018). Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 76, 154–162. doi: 10.1016/j.semcdb.2017.08.011
Fox, T. D. (2012). Mitochondrial protein synthesis, import and assembly. Genetics 192, 1203–1234. doi: 10.1534/genetics.112.141267
Fragaki, K., Chaussenot, A., Serre, V., Acquaviva, C., Bannwarth, S., Rouzier, C., et al. (2019). A novel variant m. 8561C>T in the overlapping region of MT-ATP6 and MT-ATP8 in a child with early-onset severe neurological signs. Mol. Genet. Metab. Rep. 21:100543. doi: 10.1016/j.ymgmr.2019.100543
Franco-Iborra, S., Plaza-Zabala, A., Montpeyo, M., Sebastian, D., Vila, M., and Martinez-Vicente, M. (2021). Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 17, 672–689. doi: 10.1080/15548627.2020.1728096
Franco-Iborra, S., Vila, M., and Perier, C. (2018). Mitochondrial quality control in neurodegenerative diseases: focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. 12:342. doi: 10.3389/fnins.2018.00342
Fratter, C., Gorman, G. S., Stewart, J. D., Buddles, M., Smith, C., Evans, J., et al. (2010). The clinical, histochemical and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology 74, 1619–1626. doi: 10.1212/WNL.0b013e3181df099f
Fu, H., Hardy, J., and Duff, K. E. (2018). Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 21, 1350–1358. doi: 10.1038/s41593-018-0221-2
Fullerton, M., McFarland, R., Taylor, R. W., and Alston, C. L. (2020). The genetic basis of isolated mitochondrial complex II deficiency. Mol. Genet. Metab. 131, 53–65. doi: 10.1016/j.ymgme.2020.09.009
Fusté, J. M., Wanrooij, S., Jemt, E., Granycome, C. E., Cluett, T. J., Shi, Y., et al. (2010). Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol. Cell 37, 67–78. doi: 10.1016/j.molcel.2009.12.021
Gakh, O., Cavadini, P., and Isaya, G. (2002). Mitochondrial processing peptidases. Biochim. Biophys. Acta Mol. Cell Res. 1592, 63–77. doi: 10.1016/s0167-4889(02)00265-3
Galber, C., Carissimi, S., Baracca, A., and Giorgio, V. (2021). The ATP synthase deficiency in human diseases. Life 11:325. doi: 10.3390/life11040325
Gandhi, S. (2006). PINK1 protein in normal human brain and Parkinson’s disease. Brain 129, 1720–1731. doi: 10.1093/brain/awl114
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Gandre-Babbe, S., and van der Bliek, A. M. (2008). The novel tail-anchored membrane protein mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 19, 2402–2412. doi: 10.1091/mbc.e07-12-1287
Gatt, A. P., Jones, E. L., Francis, P. T., Ballard, C., and Bateman, J. M. (2013). Association of a polymorphism in mitochondrial transcription factor a (TFAM) with Parkinson’s disease dementia but not dementia with Lewy bodies. Neurosci. Lett. 557, 177–180. doi: 10.1016/j.neulet.2013.10.045
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U S A 105, 11364–11369. doi: 10.1073/pnas.0802076105
Gaweda-Walerych, K., Safranow, K., Maruszak, A., Bialecka, M., Klodowska-Duda, G., Czyzewski, K., et al. (2010). Mitochondrial transcription factor a variants and the risk of Parkinson’s disease. Neurosci. Lett. 469, 24–29. doi: 10.1016/j.neulet.2009.11.037
Genov, G. R., Douthwaite, J. L., Lahdenperä, A. S. K., Gibson, D. C., and Phipps, R. J. (2020). Enantioselective remote C-H activation directed by a chiral cation. Science (80-) 367, 1246–1251. doi: 10.1126/science.aba1120
Gentle, I. E., Perry, A. J., Alcock, F. H., Likic, V. A., Dolezal, P., Ng, E. T., et al. (2007). Conserved motifs reveal details of ancestry and structure in the small TIM chaperones of the mitochondrial intermembrane space. Mol. Biol. Evol. 24, 1149–1160. doi: 10.1093/molbev/msm031
Gerber, S., Charif, M., Chevrollier, A., Chaumette, T., Angebault, C., Kane, M. S., et al. (2017). Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 140, 2586–2596. doi: 10.1093/brain/awx219
Ghezzi, D., Arzuffi, P., Zordan, M., Da Re, C., Lamperti, C., Benna, C., et al. (2011). Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat. Genet. 43, 259–263. doi: 10.1038/ng.761
Ghezzi, D., Goffrini, P., Uziel, G., Horvath, R., Klopstock, T., Lochmüller, H., et al. (2009). SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 41, 654–656. doi: 10.1038/ng.378
Ghezzi, D., Saada, A., D’Adamo, P., Fernandez-Vizarra, E., Gasparini, P., Tiranti, V., et al. (2008). FASTKD2 Nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 83, 415–423. doi: 10.1016/j.ajhg.2008.08.009
Giacomello, M., Pyakurel, A., Glytsou, C., and Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224. doi: 10.1038/s41580-020-0210-7
Giannoccaro, M. P., La Morgia, C., Rizzo, G., and Carelli, V. (2017). M itochondrial DNA and primary mitochondrial dysfunction in P arkinson’s disease. Mov. Disord. 32, 346–363. doi: 10.1002/mds.26966
Gonçalves, R. P., Buzhynskyy, N., Prima, V., Sturgis, J. N., and Scheuring, S. (2007). Supramolecular assembly of VDAC in native mitochondrial outer membranes. J. Mol. Biol. 369, 413–418. doi: 10.1016/j.jmb.2007.03.063
González-Rodríguez, P., Zampese, E., Stout, K. A., Guzman, J. N., Ilijic, E., Yang, B., et al. (2021). Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599, 650–656. doi: 10.1038/s41586-021-04059-0
Greene, A. W., Grenier, K., Aguileta, M. A., Muise, S., Farazifard, R., Haque, M. E., et al. (2012). Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385. doi: 10.1038/embor.2012.14
Griffin, E. E., and Chan, D. C. (2006). Domain interactions within fzo1 oligomers are essential for mitochondrial fusion. J. Biol. Chem. 281, 16599–16606. doi: 10.1074/jbc.M601847200
Grillo, A. S., Bitto, A., and Kaeberlein, M. (2021). The NDUFS4 knockout mouse: a dual threat model of childhood mitochondrial disease and normative aging. Methods Mol. Biol. 2277, 143–155. doi: 10.1007/978-1-0716-1270-5_10
Gruschke, S., Römpler, K., Hildenbeutel, M., Kehrein, K., Kühl, I., Bonnefoy, N., et al. (2012). The Cbp3-Cbp6 complex coordinates cytochrome b synthesis with bc1 complex assembly in yeast mitochondria. J. Cell Biol. 199, 137–150. doi: 10.1083/jcb.201206040
Günther, C., Hadeln, K. v., Müller-Thomsen, T., Alberici, A., Binetti, G., Hock, C., et al. (2004). Possible association of mitochondrial transcription factor a (TFAM) genotype with sporadic Alzheimer disease. Neurosci. Lett. 369, 219–223. doi: 10.1016/j.neulet.2004.07.070
Guo, R., Gu, J., Zong, S., Wu, M., and Yang, M. (2018). Structure and mechanism of mitochondrial electron transport chain. Biomed. J. 41, 9–20. doi: 10.1016/j.bj.2017.12.001
Guo, Y., Zhang, Y., Li, F., Liu, P., Liu, Y., Yang, C., et al. (2018). The biochemical characterization of a missense mutation m.8914C>T in ATP6 gene associated with mitochondrial encephalomyopathy. Int. J. Dev. Neurosci. 71, 172–174. doi: 10.1016/j.ijdevneu.2018.09.007
Gustafsson, C. M., Falkenberg, M., and Larsson, N.-G. (2016). Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 85, 133–160. doi: 10.1146/annurev-biochem-060815-014402
Habibzadeh, P., Inaloo, S., Silawi, M., Dastsooz, H., Farazi Fard, M. A., Sadeghipour, F., et al. (2019). A novel TTC19 mutation in a patient with neurological, psychological and gastrointestinal impairment. Front. Neurol. 10:944. doi: 10.3389/fneur.2019.00944
Hadrava Vanova, K., Kraus, M., Neuzil, J., and Rohlena, J. (2020). Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 25, 26–32. doi: 10.1080/13510002.2020.1752002
Hakonen, A. H., Goffart, S., Marjavaara, S., Paetau, A., Cooper, H., Mattila, K., et al. (2008). Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum. Mol. Genet. 17, 3822–3835. doi: 10.1093/hmg/ddn280
Hakonen, A. H., Heiskanen, S., Juvonen, V., Lappalainen, I., Luoma, P. T., Rantamäki, M., et al. (2005). Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient european origin. Am. J. Hum. Genet. 77, 430–441. doi: 10.1086/444548
Halestrap, A. P., Kerr, P. M., Javadov, S., and Woodfield, K.-Y. (1998). Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim. Biophys. Acta Bioenerg. 1366, 79–94. doi: 10.1016/s0005-2728(98)00122-4
Hallmann, K., Kudin, A. P., Zsurka, G., Kornblum, C., Reimann, J., Stüve, B., et al. (2016). Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain 139, 338–345. doi: 10.1093/brain/awv357
Han, S., Jeong, Y. Y., Sheshadri, P., Su, X., and Cai, Q. (2020). Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 21:e49801. doi: 10.15252/embr.201949801
Han, X.-J., Hu, Y.-Y., Yang, Z.-J., Jiang, L.-P., Shi, S.-L., Li, Y.-R., et al. (2017). Amyloid β-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 16, 4521–4528. doi: 10.3892/mmr.2017.7203
Harding, O., Evans, C. S., Ye, J., Cheung, J., Maniatis, T., and Holzbaur, E. L. F. (2021). ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. U S A 118:e2025053118. doi: 10.1073/pnas.2025053118
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Hatano, T., Kubo, S., Sato, S., and Hattori, N. (2009). Pathogenesis of familial Parkinson’s disease: new insights based on monogenic forms of Parkinson’s disease. J. Neurochem. 111, 1075–1093. doi: 10.1111/j.1471-4159.2009.06403.x
Hatano, Y., Li, Y., Sato, K., Asakawa, S., Yamamura, Y., Tomiyama, H., et al. (2004). NovelPINK1 mutations in early-onset parkinsonism. Ann. Neurol. 56, 424–427. doi: 10.1002/ana.20251
Hattori, N., Kitada, T., Matsumine, H., Asakawa, S., Yamamura, Y., Yoshino, H., et al. (1998a). Molecular genetic analysis of a novelParkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in theParkin gene in affected individuals. Ann. Neurol. 44, 935–941. doi: 10.1002/ana.410440612
Hattori, N., Matsumine, H., Asakawa, S., Kitada, T., Yoshino, H., Elibol, B., et al. (1998b). Point mutations (Thr240Arg and Gln311Stop; correction of Thr240Arg and Ala311Stop) in the Parkin gene. Biochem. Biophys. Res. Commun. 249, 754–758. doi: 10.1006/bbrc.1998.9134
Haun, F., Nakamura, T., Shiu, A. D., Cho, D.-H., Tsunemi, T., Holland, E. A., et al. (2013). S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid. Redox Signal. 19, 1173–1184. doi: 10.1089/ars.2012.4928
Hawlitschek, G., Schneider, H., Schmidt, B., Tropschug, M., Hartl, F.-U., and Neupert, W. (1988). Mitochondrial protein import: identification of processing peptidase and of PEP, a processing enhancing protein. Cell 53, 795–806. doi: 10.1016/0092-8674(88)90096-7
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U S A 110, 7916–7921. doi: 10.1073/pnas.1300677110
Heidari, E., Rasoulinezhad, M., Pak, N., Reza Ashrafi, M., Heidari, M., Banwell, B., et al. (2021). Defective complex III mitochondrial respiratory chain due to a novel variant in CYC1 gene masquerades acute demyelinating syndrome or Leber hereditary optic neuropathy. Mitochondrion 60, 12–20. doi: 10.1016/j.mito.2021.07.001
Helman, G., Caldovic, L., Whitehead, M. T., Simons, C., Brockmann, K., Edvardson, S., et al. (2016). Magnetic resonance imaging spectrum of succinate dehydrogenase-related infantile leukoencephalopathy. Ann. Neurol. 79, 379–386. doi: 10.1002/ana.24572
Hennon, S. W., Soman, R., Zhu, L., and Dalbey, R. E. (2015). YidC/Alb3/Oxa1 family of insertases. J. Biol. Chem. 290, 14866–14874. doi: 10.1074/jbc.R115.638171
Higuchi, Y., Okunushi, R., Hara, T., Hashiguchi, A., Yuan, J., Yoshimura, A., et al. (2018). Mutations in COA7 cause spinocerebellar ataxia with axonal neuropathy. Brain 141, 1622–1636. doi: 10.1093/brain/awy104
Hikmat, O., Isohanni, P., Keshavan, N., Ferla, M. P., Fassone, E., Abbott, M., et al. (2021). Expanding the phenotypic spectrum of BCS1L-related mitochondrial disease. Ann. Clin. Transl. Neurol. 8, 2155–2165. doi: 10.1002/acn3.51470
Hikmat, O., Tzoulis, C., Chong, W. K., Chentouf, L., Klingenberg, C., Fratter, C., et al. (2017). The clinical spectrum and natural history of early-onset diseases due to DNA polymerase gamma mutations. Genet. Med. 19, 1217–1225. doi: 10.1038/gim.2017.35
Hill, K., Model, K., Ryan, M. T., Dietmeier, K., Martin, F., Wagner, R., et al. (1998). Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins. Nature 395, 516–521. doi: 10.1038/26780
Hillen, H. S., Morozov, Y. I., Sarfallah, A., Temiakov, D., and Cramer, P. (2017). Structural basis of mitochondrial transcription initiation. Cell 171, 1072–1081.e10. doi: 10.1016/j.cell.2017.10.036
Hoefs, S. J. G., Dieteren, C. E. J., Rodenburg, R. J., Naess, K., Bruhn, H., Wibom, R., et al. (2009). Baculovirus complementation restores a novel NDUFAF2 mutation causing complex I deficiency. Hum. Mutat. 30, E728–E736. doi: 10.1002/humu.21037
Hogarth, K. A., Costford, S. R., Yoon, G., Sondheimer, N., and Maynes, J. T. (2018). DNM1L variant alters baseline mitochondrial function and response to stress in a patient with severe neurological dysfunction. Biochem. Genet. 56, 56–77. doi: 10.1007/s10528-017-9829-2
Holper, L., Ben-Shachar, D., and Mann, J. J. (2019). Psychotropic and neurological medication effects on mitochondrial complex I and IV in rodent models. Eur. Neuropsychopharmacol. 29, 986–1002. doi: 10.1016/j.euroneuro.2019.06.010
Houlden, H., and Singleton, A. B. (2012). The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 124, 325–338. doi: 10.1007/s00401-012-1013-5
Howarth, C., Gleeson, P., and Attwell, D. (2012). Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab. 32, 1222–1232. doi: 10.1038/jcbfm.2012.35
Hristova, V. A., Beasley, S. A., Rylett, R. J., and Shaw, G. S. (2009). Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J. Biol. Chem. 284, 14978–14986. doi: 10.1074/jbc.M808700200
Hsieh, P., Wang, C., Tsai, C., Yeh, Y., Lee, Y. S., and Wu, Y. (2019). POLG R964C and GBA L444P mutations in familial Parkinson’s disease: case report and literature review. Brain Behav. 9:e01281. doi: 10.1002/brb3.1281
Hudson, G., Keers, S., Man, P. Y. W., Griffiths, P., Huoponen, K., Savontaus, M.-L., et al. (2005). Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am. J. Hum. Genet. 77, 1086–1091. doi: 10.1086/498176
Iakovenko, E. V., Fedotova, E. Y., and Illarioshkin, S. N. (2020). Progressive external ophthalmoplegia. Russ. Neurol. J. 24, 4–13. doi: 10.1007/s11910-016-0652-7
Iapadre, G., Morana, G., Vari, M. S., Pinto, F., Lanteri, P., Tessa, A., et al. (2018). A novel homozygous MFN2 mutation associated with severe and atypical CMT2 phenotype. Eur. J. Paediatr. Neurol. 22, 563–567. doi: 10.1016/j.ejpn.2017.12.020
Ichikawa, K., Tsuyusaki, Y., Shimbo, H., and Goto, T. (2019). Late-onset Leigh syndrome with m.9176T>C mutation in the mitochondrial ATPase 6 gene. Pediatr. Int. 61, 1055–1056. doi: 10.1111/ped.13991
Imai, A., Fujita, S., Kishita, Y., Kohda, M., Tokuzawa, Y., Hirata, T., et al. (2016). Rapidly progressive infantile cardiomyopathy with mitochondrial respiratory chain complex V deficiency due to loss of ATPase 6 and 8 protein. Int. J. Cardiol. 207, 203–205. doi: 10.1016/j.ijcard.2016.01.026
Inak, G., Rybak-Wolf, A., Lisowski, P., Pentimalli, T. M., Jüttner, R., Glažar, P., et al. (2021). Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat. Commun. 12:1929. doi: 10.1038/s41467-021-22117-z
Invernizzi, F., Tigano, M., Dallabona, C., Donnini, C., Ferrero, I., Cremonte, M., et al. (2013). A homozygous mutation in LYRM7/MZM1L associated with early onset encephalopathy, lactic acidosis and severe reduction of mitochondrial complex III activity. Hum. Mutat. 34, 1619–1622. doi: 10.1002/humu.22441
Ishihara, N., Eura, Y., and Mihara, K. (2004). Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 117, 6535–6546. doi: 10.1242/jcs.01565
Ishikawa, K., Yamamoto, S., Hattori, S., Nishimura, N., Mito, T., Matsumoto, H., et al. (2019). Acquired expression of mutant mitofusin 2 causes progressive neurodegeneration and abnormal behavior. J. Neurosci. 39, 1588–1604. doi: 10.1523/JNEUROSCI.2139-18.2018
Iverson, T. M., Maklashina, E., and Cecchini, G. (2012). Structural basis for malfunction in Complex II. J. Biol. Chem. 287, 35430–35438. doi: 10.1074/jbc.R112.408419
Iwata, S., Lee, J. W., Okada, K., Lee, J. K., Iwata, M., Rasmussen, B., et al. (1998). Complete structure of the 11-subunit bovine mitochondrial cytochrome bc 1 complex. Science (80-) 281, 64–71. doi: 10.1126/science.281.5373.64
Jackson, C. B., Nuoffer, J.-M., Hahn, D., Prokisch, H., Haberberger, B., Gautschi, M., et al. (2014). Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J. Med. Genet. 51, 170–175. doi: 10.1136/jmedgenet-2013-101932
Jain-Ghai, S., Cameron, J. M., Al Maawali, A., Blaser, S., MacKay, N., Robinson, B., et al. (2013). Complex II deficiency-A case report and review of the literature. Am. J. Med. Genet. Part A 161, 285–294. doi: 10.1002/ajmg.a.35714
Janer, A., Prudent, J., Paupe, V., Fahiminiya, S., Majewski, J., Sgarioto, N., et al. (2016). SLC 25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 8, 1019–1038. doi: 10.15252/emmm.201506159
Jang, S., Lewis, T. S., Powers, C., Khuchua, Z., Baines, C. P., Wipf, P., et al. (2017). Elucidating mitochondrial electron transport chain supercomplexes in the heart during ischemia-reperfusion. Antioxid. Redox Signal. 27, 57–69. doi: 10.1089/ars.2016.6635
Javadov, S., Chapa-Dubocq, X., and Makarov, V. (2018). Different approaches to modeling analysis of mitochondrial swelling. Mitochondrion 38, 58–70. doi: 10.1016/j.mito.2017.08.004
Javadov, S., and Karmazyn, M. (2007). Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell. Physiol. Biochem. 20, 1–22. doi: 10.1159/000103747
Jiang, H., Kang, S.-U., Zhang, S., Karuppagounder, S., Xu, J., Lee, Y.-K., et al. (2016). Adult conditional knockout of PGC-1α leads to loss of dopamine neurons. eNeuro 3:ENEURO.018316.2016. doi: 10.1523/ENEURO.0183-16.2016
Jimah, J. R., and Hinshaw, J. E. (2019). Structural insights into the mechanism of dynamin superfamily proteins. Trends Cell Biol. 29, 257–273. doi: 10.1016/j.tcb.2018.11.003
Jin, H., May, M., Tranebjærg, L., Kendall, E., Fontán, G., Jackson, J., et al. (1996). A novel X-linked gene, DDP, shows mutations in families with deafness (DFN-1), dystonia, mental deficiency and blindness. Nat. Genet. 14, 177–180. doi: 10.1038/ng1096-177
Jobling, R. K., Assoum, M., Gakh, O., Blaser, S., Raiman, J. A., Mignot, C., et al. (2015). PMPCA mutations cause abnormal mitochondrial protein processing in patients with non-progressive cerebellar ataxia. Brain 138, 1505–1517. doi: 10.1093/brain/awv057
Johnson, A. A., Tsai, Y., Graves, S. W., and Johnson, K. A. (2000). Human mitochondrial DNA polymerase holoenzyme: reconstitution and characterization. Biochemistry 39, 1702–1708. doi: 10.1021/bi992104w
Johri, A., Chandra, A., and Flint Beal, M. (2013). PGC-1α, mitochondrial dysfunction and Huntington’s disease. Free Radic. Biol. Med. 62, 37–46. doi: 10.1016/j.freeradbiomed.2013.04.016
Jonckheere, A. I., Renkema, G. H., Bras, M., van den Heuvel, L. P., Hoischen, A., Gilissen, C., et al. (2013). A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 136, 1544–1554. doi: 10.1093/brain/awt086
Jonckheere, A. I., Smeitink, J. A. M., and Rodenburg, R. J. T. (2012). Mitochondrial ATP synthase: architecture, function and pathology. J. Inherit. Metab. Dis. 35, 211–225. doi: 10.1007/s10545-011-9382-9
Joshi, M., Anselm, I., Shi, J., Bale, T. A., Towne, M., Schmitz-Abe, K., et al. (2016). Mutations in the substrate binding glycine-rich loop of the mitochondrial processing peptidase-α protein (PMPCA) cause a severe mitochondrial disease. Mol. Case Stud. 2:a000786. doi: 10.1101/mcs.a000786
Kahlhöfer, F., Kmita, K., Wittig, I., Zwicker, K., and Zickermann, V. (2017). Accessory subunit NUYM (NDUFS4) is required for stability of the electron input module and activity of mitochondrial complex I. Biochim. Biophys. Acta Bioenerg. 1858, 175–181. doi: 10.1016/j.bbabio.2016.11.010
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kang, Y., Anderson, A. J., Jackson, T. D., Palmer, C. S., De Souza, D. P., Fujihara, K. M., et al. (2019). Function of hTim8a in complex IV assembly in neuronal cells provides insight into pathomechanism underlying Mohr-Tranebjærg syndrome. eLife 8:e48848. doi: 10.7554/eLife.48828
Kang, I., Chu, C. T., and Kaufman, B. A. (2018). The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. 592, 793–811. doi: 10.1002/1873-3468.12989
Kanungo, S., Morton, J., Neelakantan, M., Ching, K., Saeedian, J., and Goldstein, A. (2018). Mitochondrial disorders. Ann. Transl. Med. 6, 475–475. doi: 10.21037/atm.2018.12.13
Kato, H., Lu, Q., Rapaport, D., and Kozjak-Pavlovic, V. (2013). Tom70 is essential for PINK1 import into mitochondria. PLoS One 8:e58435. doi: 10.1371/journal.pone.0058435
Kaufman, B. A., Durisic, N., Mativetsky, J. M., Costantino, S., Hancock, M. A., Grutter, P., et al. (2007). The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell 18, 3225–3236. doi: 10.1091/mbc.e07-05-0404
Kaur, P., Sharma, S., Kadavigere, R., Girisha, K. M., and Shukla, A. (2020). Novel variant p.(Ala102Thr) in SDHB causes mitochondrial complex II deficiency: case report and review of the literature. Ann. Hum. Genet. 84, 345–351. doi: 10.1111/ahg.12377
Kawahara, M., and Kuroda, Y. (2000). Molecular mechanism of neurodegeneration induced by Alzheimer’s β-amyloid protein: Channel formation and disruption of calcium homeostasis. Brain Res. Bull. 53, 389–397. doi: 10.1016/s0361-9230(00)00370-1
Kawajiri, S., Saiki, S., Sato, S., Sato, F., Hatano, T., Eguchi, H., et al. (2010). PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 584, 1073–1079. doi: 10.1016/j.febslet.2010.02.016
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/BJ20140334
Keeney, P. M., and Bennett, J. P. (2010). ALS spinal neurons show varied and reduced mtDNA gene copy numbers and increased mtDNA gene deletions. Mol. Neurodegener. 5:21. doi: 10.1186/1750-1326-5-21
Kenvin, S., Torregrosa-Muñumer, R., Reidelbach, M., Pennonen, J., Turkia, J. J., Rannila, E., et al. (2022). Threshold of heteroplasmic truncating MT-ATP6 mutation in reprogramming, Notch hyperactivation and motor neuron metabolism. Hum. Mol. Genet. 31, 958–974. doi: 10.1093/hmg/ddab299
Kim, J., Moody, J. P., Edgerly, C. K., Bordiuk, O. L., Cormier, K., Smith, K., et al. (2010). Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 19, 3919–3935. doi: 10.1093/hmg/ddq306
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Koch, J., Feichtinger, R. G., Freisinger, P., Pies, M., Schrödl, F., Iuso, A., et al. (2016). Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 53, 270–278. doi: 10.1136/jmedgenet-2015-103500
Korhonen, J. A., Gaspari, M., and Falkenberg, M. (2003). TWINKLE has 5’-3’ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 278, 48627–48632. doi: 10.1074/jbc.M306981200
Korhonen, J. A., Pande, V., Holmlund, T., Farge, G., Pham, X. H., Nilsson, L., et al. (2008). Structure-function defects of the TWINKLE linker region in progressive external ophthalmoplegia. J. Mol. Biol. 377, 691–705. doi: 10.1016/j.jmb.2008.01.035
Kori, A., Hori, I., Tanaka, T., Aoyama, K., Ito, K., Hattori, A., et al. (2019). Transition from Leigh syndrome to MELAS syndrome in a patient with heteroplasmic MT-ND3 m.10158T>C. Brain Dev. 41, 803–807. doi: 10.1016/j.braindev.2019.05.006
Korwitz, A., Merkwirth, C., Richter-Dennerlein, R., Tröder, S. E., Sprenger, H.-G., Quirós, P. M., et al. (2016). Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J. Cell Biol. 212, 157–166. doi: 10.1083/jcb.201507022
Kose, M., Canda, E., Kagnici, M., Aykut, A., Adebali, O., Durmaz, A., et al. (2020). SURF1 related Leigh syndrome: clinical and molecular findings of 16 patients from Turkey. Mol. Genet. Metab. Rep. 25:100657. doi: 10.1016/j.ymgmr.2020.100657
Kotecha, S., and Kairamkonda, V. (2019). Mitochondrial respiratory chain complex IV deficiency presenting as neonatal respiratory distress syndrome. BMJ Case Rep. 12:e229668. doi: 10.1136/bcr-2019-229668
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kucharczyk, R., Salin, B., and di Rago, J.-P. (2009). Introducing the human Leigh syndrome mutation T9176G into Saccharomyces cerevisiae mitochondrial DNA leads to severe defects in the incorporation of Atp6p into the ATP synthase and in the mitochondrial morphology. Hum. Mol. Genet. 18, 2889–2898. doi: 10.1093/hmg/ddp226
Kukat, C., Davies, K. M., Wurm, C. A., Spåhr, H., Bonekamp, N. A., Kühl, I., et al. (2015). Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc. Natl. Acad. Sci. U S A 112, 11288–11293. doi: 10.1073/pnas.1512131112
Kunii, M., Doi, H., Higashiyama, Y., Kugimoto, C., Ueda, N., Hirata, J., et al. (2015). A Japanese case of cerebellar ataxia, spastic paraparesis and deep sensory impairment associated with a novel homozygous TTC19 mutation. J. Hum. Genet. 60, 187–191. doi: 10.1038/jhg.2015.7
Kushnareva, Y. E., Gerencser, A. A., Bossy, B., Ju, W.-K., White, A. D., Waggoner, J., et al. (2013). Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. 20, 353–365. doi: 10.1038/cdd.2012.128
Kytövuori, L., Lipponen, J., Rusanen, H., Komulainen, T., Martikainen, M. H., Majamaa, K., et al. (2016). A novel mutation m.8561C>G in MT-ATP6/8 causing a mitochondrial syndrome with ataxia, peripheral neuropathy, diabetes mellitus and hypergonadotropic hypogonadism. J. Neurol. 263, 2188–2195. doi: 10.3390/s22114165
Lake, N. J., Compton, A. G., Rahman, S., and Thorburn, D. R. (2016). Leigh syndrome: one disorder, more than 75 monogenic causes. Ann. Neurol. 79, 190–203. doi: 10.1002/ana.24551
Larsson, N.-G. (2010). Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 79, 683–706. doi: 10.1146/annurev-biochem-060408-093701
Larsson, N.-G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., et al. (1998). Mitochondrial transcription factor A is necessary for mtDNA maintance and embryogenesis in mice. Nat. Genet. 18, 231–236. doi: 10.1038/ng0398-231
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Lazarou, M., Thorburn, D. R., Ryan, M. T., and McKenzie, M. (2009). Assembly of mitochondrial complex I and defects in disease. Biochim. Biophys. Acta Mol. Cell Res. 1793, 78–88. doi: 10.1016/j.bbamcr.2008.04.015
Lee, I.-C., and Chiang, K.-L. (2021). Clinical diagnosis and treatment of leigh syndrome based on SURF1: genotype and phenotype. Antioxidants 10:1950. doi: 10.3390/antiox10121950
Lee, J. S., Yoo, T., Lee, M., Lee, Y., Jeon, E., Kim, S. Y., et al. (2020). Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clin. Genet. 97, 586–594. doi: 10.1111/cge.13713
Lefort, N., Yi, Z., Bowen, B., Glancy, B., De Filippis, E. A., Mapes, R., et al. (2009). Proteome profile of functional mitochondria from human skeletal muscle using one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. J. Proteomics 72, 1046–1060. doi: 10.1016/j.jprot.2009.06.011
Legros, F., Lombès, A., Frachon, P., and Rojo, M. (2002). Mitochondrial fusion in human cells is efficient, requires the inner membrane potential and is mediated by mitofusins. Mol. Biol. Cell 13, 4343–4354. doi: 10.1091/mbc.e02-06-0330
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Lenaz, G. (2012). Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 924, 93–136. doi: 10.1007/978-94-007-2869-1_5
Leroy, E., Anastasopoulos, D., Konitsiotis, S., Lavedan, C., and Polymeropoulos, M. H. (1998). Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson’s disease. Hum. Genet. 103, 424–427. doi: 10.1007/s004390050845
Lesch, K.-P., Selch, S., Renner, T. J., Jacob, C., Nguyen, T. T., Hahn, T., et al. (2011). Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol. Psychiatry 16, 491–503. doi: 10.1038/mp.2010.29
Lewis, T. L., Kwon, S.-K., Lee, A., Shaw, R., and Polleux, F. (2018). MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 9:5008. doi: 10.1038/s41467-018-07416-2
Li, J.-L., Lin, T.-Y., Chen, P.-L., Guo, T.-N., Huang, S.-Y., Chen, C.-H., et al. (2021a). Mitochondrial function and Parkinson’s disease: from the perspective of the electron transport chain. Front. Mol. Neurosci. 14:797833. doi: 10.3389/fnmol.2021.797833
Li, Q., Madden, J. A., Lin, J., Shi, J., Rosen, S. M., Schmitz-Abe, K., et al. (2021b). Reanalysis of exome data identifies novel SLC25A46 variants associated with leigh syndrome. J. Pers. Med. 11:1277. doi: 10.3390/jpm11121277
Li, Z., Peng, Y., Hufnagel, R. B., Hu, Y.-C., Zhao, C., Queme, L. F., et al. (2017). Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice. Hum. Mol. Genet. 26, 3776–3791. doi: 10.1093/hmg/ddx262
Li, Z., Slone, J., Wu, L., and Huang, T. (2019). “Neurodegenerative diseases associated with mutations in SLC25A46,” in Recent Advances in Neurodegeneration, ed A. Borreca (London: IntechOpen), 1–21. doi: 10.5772/intechopen.79992
Liao, Y., Dong, Y., and Cheng, J. (2017). The function of the mitochondrial calcium uniporter in neurodegenerative disorders. Int. J. Mol. Sci. 18:248. doi: 10.3390/ijms18020248
Lillenes, M. S., Støen, M., Günther, C.-C., Selnes, P., Stenset, V. T. V., Espeseth, T., et al. (2017). Mitochondrial transcription factor a (TFAM) rs1937 and AP endonuclease 1 (APE1) rs1130409 alleles are associated with reduced cognitive performance. Neurosci. Lett. 645, 46–52. doi: 10.1016/j.neulet.2017.02.062
Lim, S. C., Smith, K. R., Stroud, D. A., Compton, A. G., Tucker, E. J., Dasvarma, A., et al. (2014). A founder mutation in PET100 causes isolated complex IV deficiency in lebanese individuals with leigh syndrome. Am. J. Hum. Genet. 94, 209–222. doi: 10.1016/j.ajhg.2013.12.015
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Lin, S., Fasham, J., Al-Hijawi, F., Qutob, N., Gunning, A., Leslie, J. S., et al. (2021). Consolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency. Eur. J. Hum. Genet. 29, 1570–1576. doi: 10.1038/s41431-021-00887-w
Lin, J., Wu, P.-H., Tarr, P. T., Lindenberg, K. S., St-Pierre, J., Zhang, C., et al. (2004). Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135. doi: 10.1016/j.cell.2004.09.013
Liu, Z., Ding, Y., Du, A., Zhang, B., Zhao, G., and Ding, M. P. (2008). A novel Twinkle (PEO1) gene mutation in a Chinese family with adPEO. Mol. Vis. 14, 1995–2001. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2579934/.
Liu, L., Li, Y., Wang, J., Zhang, D., Wu, H., Li, W., et al. (2021). Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 22:e50629. doi: 10.15252/embr.202050629
Lohman, T. M., Tomko, E. J., and Wu, C. G. (2008). Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat. Rev. Mol. Cell Biol. 9, 391–401. doi: 10.1038/nrm2394
Longo, F., Benedetti, S., Zambon, A. A., Sora, M. G. N., Di Resta, C., De Ritis, D., et al. (2020). Impaired turnover of hyperfused mitochondria in severe axonal neuropathy due to a novel DRP1 mutation. Hum. Mol. Genet. 29, 177–188. doi: 10.1093/hmg/ddz211
Lonnqvist, T., Paetau, A., Valanne, L., and Pihko, H. (2009). Recessive twinkle mutations cause severe epileptic encephalopathy. Brain 132, 1553–1562. doi: 10.1093/brain/awp045
López-Doménech, G., Howden, J. H., Covill-Cooke, C., Morfill, C., Patel, J. V., Bürli, R., et al. (2021). Loss of neuronal Miro1 disrupts mitophagy and induces hyperactivation of the integrated stress response. EMBO J. 40:e100715. doi: 10.15252/embj.2018100715
Losón, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49 and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lou, G., Palikaras, K., Lautrup, S., Scheibye-Knudsen, M., Tavernarakis, N., and Fang, E. F. (2020). Mitophagy and neuroprotection. Trends Mol. Med. 26, 8–20. doi: 10.1016/j.molmed.2019.07.002
Lu, B., and Guo, S. (2020). Mechanisms linking mitochondrial dysfunction and proteostasis failure. Trends Cell Biol. 30, 317–328. doi: 10.1016/j.tcb.2020.01.008
Lücking, C., Abbas, N., Dürr, A., Bonifati, V., Bonnet, A.-M., de Broucker, T., et al. (1998). Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile Parkinsonism. The European consortium on genetic susceptibility in Parkinson’s disease and the French Parkinson’s disease genetics study group. Lancet 352, 1355–1356. doi: 10.1097/HCR.0000000000000719
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). Association between early-onset Parkinson’s disease and mutations in the Parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Ludtmann, M. H. R., and Abramov, A. Y. (2018). Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 663, 86–90. doi: 10.1016/j.neulet.2017.08.044
Luoma, P., Melberg, A., Rinne, J. O., Kaukonen, J. A., Nupponen, N. N., Chalmers, R. M., et al. (2004). Parkinsonism, premature menopause and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. Lancet 364, 875–882. doi: 10.1016/S0140-6736(04)16983-3
Lussey-Lepoutre, C., Hollinshead, K. E. R., Ludwig, C., Menara, M., Morin, A., Castro-Vega, L.-J., et al. (2015). Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat. Commun. 6:8784. doi: 10.1038/ncomms9784
Lyalina, S., Percha, B., LePendu, P., Iyer, S. V., Altman, R. B., and Shah, N. H. (2013). Identifying phenotypic signatures of neuropsychiatric disorders from electronic medical records. J. Am. Med. Inform. Assoc. 20, e297–e305. doi: 10.1136/amiajnl-2013-001933
Lytovchenko, O., Melin, J., Schulz, C., Kilisch, M., Hutu, D. P., and Rehling, P. (2013). Signal recognition initiates reorganization of the presequence translocase during protein import. EMBO J. 32, 886–898. doi: 10.1038/emboj.2013.23
MacVicar, T., and Langer, T. (2016). OPA1 processing in cell death and disease - the long and short of it. J. Cell Sci. 129, 2297–2306. doi: 10.1242/jcs.159186
Maday, S., Twelvetrees, A. E., Moughamian, A. J., and Holzbaur, E. L. F. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. doi: 10.1016/j.neuron.2014.10.019
Madruga, E., Maestro, I., and Martínez, A. (2021). Mitophagy modulation, a new player in the race against ALS. Int. J. Mol. Sci. 22:740. doi: 10.3390/ijms22020740
Magistretti, P. J., and Allaman, I. (2015). A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. doi: 10.1016/j.neuron.2015.03.035
Magistretti, P. J., and Allaman, I. (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 19, 235–249. doi: 10.1038/nrn.2018.19
Magri, A., and Messina, A. (2017). Interactions of VDAC with proteins involved in neurodegenerative aggregation: an opportunity for advancement on therapeutic molecules. Curr. Med. Chem. 24, 4470–4487. doi: 10.2174/0929867324666170601073920
Malek, M. H., Hüttemann, M., and Lee, I. (2018). Mitochondrial structure, function and dynamics: the common thread across organs, disease and aging. Oxid. Med. Cell. Longev. 2018:1863414. doi: 10.1155/2018/1863414
Malpartida, A. B., Williamson, M., Narendra, D. P., Wade-Martins, R., and Ryan, B. J. (2021). Mitochondrial dysfunction and mitophagy in Parkinson’s disease: from mechanism to therapy. Trends Biochem. Sci. 46, 329–343. doi: 10.1016/j.tibs.2020.11.007
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. doi: 10.1126/science.1075762
Marcelino, L. A., and Thilly, W. G. (1999). Mitochondrial mutagenesis in human cells and tissues. Mutat. Res. Repair 434, 177–203. doi: 10.1016/s0921-8777(99)00028-2
Marin, S. E., Mesterman, R., Robinson, B., Rodenburg, R. J., Smeitink, J., and Tarnopolsky, M. A. (2013). Leigh syndrome associated with mitochondrial complex I deficiency due to novel mutations In NDUFV1 and NDUFS2. Gene 516, 162–167. doi: 10.1016/j.gene.2012.12.024
Martin, I., Dawson, V. L., and Dawson, T. M. (2011). Recent advances in the genetics of Parkinson’s disease. Annu. Rev. Genomics Hum. Genet. 12, 301–325. doi: 10.1146/annurev-genom-082410-101440
Martín, M. A., Blázquez, A., Gutierrez-Solana, L. G., Fernández-Moreira, D., Briones, P., Andreu, A. L., et al. (2005). Leigh syndrome associated with mitochondrial complex I deficiency due to a novel mutation in the NDUFS1 gene. Arch. Neurol. 62, 659–661. doi: 10.1001/archneur.62.4.659
Martinez Lyons, A., Ardissone, A., Reyes, A., Robinson, A. J., Moroni, I., Ghezzi, D., et al. (2016). COA7 (C1orf163/RESA1) mutations associated with mitochondrial leukoencephalopathy and cytochrome c oxidase deficiency. J. Med. Genet. 53, 846–849. doi: 10.1136/jmedgenet-2016-104194
Martinez-Vicente, M. (2017). Neuronal mitophagy in neurodegenerative diseases. Front. Mol. Neurosci. 10:64. doi: 10.3389/fnmol.2017.00064
Massa, V., Fernandez-Vizarra, E., Alshahwan, S., Bakhsh, E., Goffrini, P., Ferrero, I., et al. (2008). Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 82, 1281–1289. doi: 10.1016/j.ajhg.2008.05.002
Mata, I. F. (2004). Parkin genetics: one model for Parkinson’s disease. Hum. Mol. Genet. 13, 127R–133. doi: 10.1093/hmg/ddh089
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Matsumine, H., Saito, M., Shimoda-Matsubayashi, S., Tanaka, H., Ishikawa, A., Nakagawa-Hattori, Y., et al. (1997). Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2–27. Am. J. Hum. Genet. 60, 588–596.
Mayr, J. A., Havlickova, V., Zimmermann, F., Magler, I., Kaplanova, V., Jesina, P., et al. (2010). Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 ε subunit. Hum. Mol. Genet. 19, 3430–3439. doi: 10.1093/hmg/ddq254
Mei, S., Huang, X., Cheng, L., Peng, S., Zhu, T., Chen, L., et al. (2019). A missense mutation in OPA1 causes dominant optic atrophy in a Chinese family. J. Ophthalmol. 2019:1424928. doi: 10.1155/2019/1424928
Meinecke, M., Wagner, R., Kovermann, P., Guiard, B., Mick, D. U., Hutu, D. P., et al. (2006). Tim50 maintains the permeability barrier of the mitochondrial inner membrane. Science 312, 1523–1526. doi: 10.1126/science.1127628
Melchionda, L., Damseh, N. S., Abu Libdeh, B. Y., Nasca, A., Elpeleg, O., Zanolini, A., et al. (2014). A novel mutation in TTC19 associated with isolated complex III deficiency, cerebellar hypoplasia and bilateral basal ganglia lesions. Front. Genet. 5:397. doi: 10.3389/fgene.2014.00397
Merkwirth, C., Martinelli, P., Korwitz, A., Morbin, M., Brönneke, H. S., Jordan, S. D., et al. (2012). Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 8:e1003021. doi: 10.1371/journal.pgen.1003021
Messina, A., Reina, S., Guarino, F., and De Pinto, V. (2012). VDAC isoforms in mammals. Biochim. Biophys. Acta 1818, 1466–1476. doi: 10.1016/j.bbamem.2011.10.005
Meunier, B., Fisher, N., Ransac, S., Mazat, J.-P., and Brasseur, G. (2013). Respiratory complex III dysfunction in humans and the use of yeast as a model organism to study mitochondrial myopathy and associated diseases. Biochim. Biophys. Acta 1827, 1346–1361. doi: 10.1016/j.bbabio.2012.11.015
Miryounesi, M., Fardaei, M., Tabei, S. M., and Ghafouri-Fard, S. (2016). Leigh syndrome associated with a novel mutation in the COX15 gene. J. Pediatr. Endocrinol. Metab. 29, 741–744. doi: 10.1515/jpem-2015-0396
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666. doi: 10.1016/j.neuron.2017.09.055
MITCHELL, P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. doi: 10.1038/191144a0
Miyadera, H., Shiomi, K., Ui, H., Yamaguchi, Y., Masuma, R., Tomoda, H., et al. (2003). Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc. Natl. Acad. Sci. U S A 100, 473–477. doi: 10.1073/pnas.0237315100
Mnatsakanyan, N., and Jonas, E. A. (2020). The new role of F1Fo ATP synthase in mitochondria-mediated neurodegeneration and neuroprotection. Exp. Neurol. 332:113400. doi: 10.1016/j.expneurol.2020.113400
Mokranjac, D., and Neupert, W. (2015). Cell biology: architecture of a protein entry gate. Nature 528, 201–202. doi: 10.1038/nature16318
Morgenstern, M., Stiller, S. B., Lübbert, P., Peikert, C. D., Dannenmaier, S., Drepper, F., et al. (2017). Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 19, 2836–2852. doi: 10.1016/j.celrep.2017.06.014
Morland, C., Lauritzen, K. H., Puchades, M., Holm-Hansen, S., Andersson, K., Gjedde, A., et al. (2015). The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: expression and action in brain. J. Neurosci. Res. 93, 1045–1055. doi: 10.1002/jnr.23593
Mouton-Liger, F., Jacoupy, M., Corvol, J.-C., and Corti, O. (2017). PINK1/Parkin-dependent mitochondrial surveillance: from pleiotropy to Parkinson’s disease. Front. Mol. Neurosci. 10:120. doi: 10.3389/fnmol.2017.00120
Muñoz, J. P., Ivanova, S., Sánchez-Wandelmer, J., Martínez-Cristóbal, P., Noguera, E., Sancho, A., et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361. doi: 10.1038/emboj.2013.168
Na, J., and Lee, Y. (2022). Genotype-phenotype analysis of MT-ATP6 -associated Leigh syndrome. Acta Neurol. Scand. 145, 414–422. doi: 10.1111/ane.13566
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D.-F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Narendra, D., Tanaka, A., Suen, D.-F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Nasca, A., Legati, A., Baruffini, E., Nolli, C., Moroni, I., Ardissone, A., et al. (2016). Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum. Mutat. 37, 898–903. doi: 10.1002/humu.23033
Natarajan, S., Ramaswamy, G., Kannan, L., Gunasekeran, V., and Kathirvelu, G. (2021). Acute devastating multifocal cavitating leukoencephalopathy in a six-year-old girl due to missense mutation in LYRM7 gene. Pediatr. Neurol. 117, 44–46. doi: 10.1016/j.pediatrneurol.2020.12.010
Ndi, M., Marin-Buera, L., Salvatori, R., Singh, A. P., and Ott, M. (2018). Biogenesis of the bc1 complex of the mitochondrial respiratory chain. J. Mol. Biol. 430, 3892–3905. doi: 10.1016/j.jmb.2018.04.036
Neeve, V. C. M., Samuels, D. C., Bindoff, L. A., van den Bosch, B., Van Goethem, G., Smeets, H., et al. (2012). What is influencing the phenotype of the common homozygous polymerase-γ mutation p.Ala467Thr? Brain 135, 3614–3626. doi: 10.1093/brain/aws298
Neighbors, A., Moss, T., Holloway, L., Yu, S., Annese, F., Skinner, S., et al. (2020). Functional analysis of a novel mutation in the TIMM8A gene that causes deafness-dystonia-optic neuronopathy syndrome. Mol. Genet. Genomic Med. 8:e1121. doi: 10.1002/mgg3.1121
Nicolas, E., Tricarico, R., Savage, M., Golemis, E. A., and Hall, M. J. (2019). Disease-associated genetic variation in human mitochondrial protein import. Am. J. Hum. Genet. 104, 784–801. doi: 10.1016/j.ajhg.2019.03.019
Nirwane, A., and Majumdar, A. (2018). Understanding mitochondrial biogenesis through energy sensing pathways and its translation in cardio-metabolic health. Arch. Physiol. Biochem. 124, 194–206. doi: 10.1080/13813455.2017.1391847
Nogueira, C., Barros, J., Sá, M. J., Azevedo, L., Taipa, R., Torraco, A., et al. (2013). Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency. Neurogenetics 14, 153–160. doi: 10.1007/s10048-013-0361-1
Nolfi-Donegan, D., Braganza, A., and Shiva, S. (2020). Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production and methods of measurement. Redox Biol. 37:101674. doi: 10.1016/j.redox.2020.101674
Nolte, D., Kang, J.-S., Hofmann, A., Schwaab, E., Krämer, H. H., and Müller, U. (2021). Mutations in MT-ATP6 are a frequent cause of adult-onset spinocerebellar ataxia. J. Neurol. 268, 4866–4873. doi: 10.1007/s00415-021-10607-5
Nortley, R., and Attwell, D. (2017). Control of brain energy supply by astrocytes. Curr. Opin. Neurobiol. 47, 80–85. doi: 10.1016/j.conb.2017.09.012
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
O’Rourke, B. (2007). Mitochondrial ion channels. Annu. Rev. Physiol. 69, 19–49. doi: 10.1146/annurev.physiol.69.031905.163804
Oktay, Y., Güngör, S., Zeltner, L., Wiethoff, S., Schöls, L., Sonmezler, E., et al. (2020). Confirmation of TACO1 as a leigh syndrome disease gene in two additional families. J. Neuromuscul. Dis. 7, 301–308. doi: 10.3233/JND-200510
Oláhová, M., Peter, B., Szilagyi, Z., Diaz-Maldonado, H., Singh, M., Sommerville, E. W., et al. (2021). POLRMT mutations impair mitochondrial transcription causing neurological disease. Nat. Commun. 12:1135. doi: 10.1038/s41467-021-21279-0
Olichon, A., ElAchouri, G., Baricault, L., Delettre, C., Belenguer, P., and Lenaers, G. (2007). OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. 14, 682–692. doi: 10.1038/sj.cdd.4402048
Oliveira, M. T., and Kaguni, L. S. (2011). Reduced stimulation of recombinant DNA polymerase γ and mitochondrial DNA (mtDNA) helicase by variants of mitochondrial single-stranded DNA-binding protein (mtSSB) correlates with defects in mtDNA replication in animal cells. J. Biol. Chem. 286, 40649–40658. doi: 10.1074/jbc.M111.289983
Onishi, M., Yamano, K., Sato, M., Matsuda, N., and Okamoto, K. (2021). Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40:e104705. doi: 10.15252/embj.2020104705
Oquendo, C. E. (2004). Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J. Med. Genet. 41, 540–544. doi: 10.1136/jmg.2003.017426
Ortigoza-Escobar, J. D., Oyarzabal, A., Montero, R., Artuch, R., Jou, C., Jiménez, C., et al. (2016). Ndufs4 related Leigh syndrome: a case report and review of the literature. Mitochondrion 28, 73–78. doi: 10.1016/j.mito.2016.04.001
Osellame, L. D., Blacker, T. S., and Duchen, M. R. (2012). Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 26, 711–723. doi: 10.1016/j.beem.2012.05.003
Ostergaard, E., Weraarpachai, W., Ravn, K., Born, A. P., Jønson, L., Duno, M., et al. (2015). Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity and short statur. J. Med. Genet. 52, 203–207. doi: 10.1136/jmedgenet-2014-102914
Otero, M. G., Tiongson, E., Diaz, F., Haude, K., Panzer, K., Collier, A., et al. (2018). Novel pathogenic COX20 variants causing dysarthria, ataxia and sensory neuropathy. Ann. Clin. Transl. Neurol. 6, 154–160. doi: 10.1002/acn3.661
Otera, H., Wang, C., Cleland, M. M., Setoguchi, K., Yokota, S., Youle, R. J., et al. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158. doi: 10.1083/jcb.201007152
Pacheu-Grau, D., Callegari, S., Emperador, S., Thompson, K., Aich, A., Topol, S. E., et al. (2018). Mutations of the mitochondrial carrier translocase channel subunit TIM22 cause early-onset mitochondrial myopathy. Hum. Mol. Genet. 27, 4135–4144. doi: 10.1093/hmg/ddy305
Pagliuso, A., Cossart, P., and Stavru, F. (2018). The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 75, 355–374. doi: 10.1007/s00018-017-2603-0
Pagnamenta, A. T., Hargreaves, I. P., Duncan, A. J., Taanman, J.-W., Heales, S. J., Land, J. M., et al. (2006). Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol. Genet. Metab. 89, 214–221. doi: 10.1016/j.ymgme.2006.05.003
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Palmer, C. S., Anderson, A. J., and Stojanovski, D. (2021). Mitochondrial protein import dysfunction: mitochondrial disease, neurodegenerative disease and cancer. FEBS Lett. 595, 1107–1131. doi: 10.1002/1873-3468.14022
Panda, I., Ahmad, I., Sagar, S., Zahra, S., Shamim, U., Sharma, S., et al. (2020). Encephalopathy due to defective mitochondrial and peroxisomal fission 2 caused by a novel MFF gene mutation in a young child. Clin. Genet. 97, 933–937. doi: 10.1111/cge.13740
Panes, J., Wendt, A., Ramirez-Molina, O., Castro, P., and Fuentealba, J. (2022). Deciphering the role of PGC-1α in neurological disorders: from mitochondrial dysfunction to synaptic failure. Neural Regen. Res. 17, 237–245. doi: 10.4103/1673-5374.317957
Papa, S., and De Rasmo, D. (2013). Complex I deficiencies in neurological disorders. Trends Mol. Med. 19, 61–69. doi: 10.1016/j.molmed.2012.11.005
Papa, S., Martino, P. L., Capitanio, G., Gaballo, A., De Rasmo, D., Signorile, A., et al. (2012). The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 942, 3–37. doi: 10.1007/978-94-007-2869-1_1
Parada-Garza, J. D., López-Valencia, G., Miranda-García, L. A., Pérez-García, G., and Ruiz-Sandoval, J. L. (2020). MRI findings in SANDO variety of the ataxia-neuropathy spectrum with a novel mutation in POLG (c.3287G>T): a case report. Neuromuscul. Disord. 30, 590–592. doi: 10.1016/j.nmd.2020.04.008
Parey, K., Wirth, C., Vonck, J., and Zickermann, V. (2020). Respiratory complex I — structure, mechanism and evolution. Curr. Opin. Struct. Biol. 63, 1–9. doi: 10.1016/j.sbi.2020.01.004
Parfait, B., Chretien, D., Rötig, A., Marsac, C., Munnich, A., and Rustin, P. (2000). Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum. Genet. 106, 236–243. doi: 10.1007/s004390051033
Park, C. B., and Larsson, N.-G. (2011). Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 193, 809–818. doi: 10.1083/jcb.201010024
Paschen, S. A., Waizenegger, T., Stan, T., Preuss, M., Cyrklaff, M., Hell, K., et al. (2003). Evolutionary conservation of biogenesis of β-barrel membrane proteins. Nature 426, 862–866. doi: 10.1038/nature02208
Patel, S. S., and Picha, K. M. (2000). Structure and function of hexameric helicases. Annu. Rev. Biochem. 69, 651–697. doi: 10.1146/annurev.biochem.69.1.651
Patro, S., Ratna, S., Yamamoto, H. A., Ebenezer, A. T., Ferguson, D. S., Kaur, A., et al. (2021). ATP synthase and mitochondrial bioenergetics dysfunction in Alzheimer’s disease. Int. J. Mol. Sci. 22:11185. doi: 10.3390/ijms222011185
Pellino, G., Faggioli, R., Galuppi, A., Leon, A., Fusco, C., Tugnoli, V., et al. (2021). Mitofusin 2: the missing link between mtDNA maintenance defects and neurotransmitter disorders. Mitochondrion 61, 159–164. doi: 10.1016/j.mito.2021.09.011
Pepperberg, D. R. (2019). Amyloid-β-dependent inactivation of the mitochondrial electron transport chain at low transmembrane potential: an ameliorating process in hypoxia-associated neurodegenerative disease? J. Alzheimer’s Dis. 72, 663–675. doi: 10.3233/JAD-190476
Perocchi, F., Gohil, V. M., Girgis, H. S., Bao, X. R., McCombs, J. E., Palmer, A. E., et al. (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296. doi: 10.1038/nature09358
Peter, B., and Falkenberg, M. (2020). TWINKLE and other human mitochondrial DNA helicases: structure, function and disease. Genes (Basel) 11:408. doi: 10.3390/genes11040408
Peter, B., Farge, G., Pardo-Hernandez, C., Tångefjord, S., and Falkenberg, M. (2019). Structural basis for adPEO-causing mutations in the mitochondrial TWINKLE helicase. Hum. Mol. Genet. 28, 1090–1099. doi: 10.1093/hmg/ddy415
Pfanner, N., Warscheid, B., and Wiedemann, N. (2019). Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20, 267–284. doi: 10.1038/s41580-018-0092-0
Pfeffer, G., Blakely, E. L., Alston, C. L., Hassani, A., Boggild, M., Horvath, R., et al. (2012). Adult-onset spinocerebellar ataxia syndromes due to MTATP6 mutations. J. Neurol. Neurosurg. Psychiatry 83, 883–886. doi: 10.1136/jnnp-2012-302568
Pham, A. H., Meng, S., Chu, Q. N., and Chan, D. C. (2012). Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 21, 4817–4826. doi: 10.1093/hmg/dds311
Piccinin, E., Sardanelli, A. M., Seibel, P., Moschetta, A., Cocco, T., and Villani, G. (2021). PGC-1s in the spotlight with Parkinson’s disease. Int. J. Mol. Sci. 22:3487. doi: 10.3390/ijms22073487
Pickles, S., Vigié, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Pillai, N. R., AlDhaheri, N. S., Ghosh, R., Lim, J., Streff, H., Nayak, A., et al. (2019). Biallelic variants in COX4I1 associated with a novel phenotype resembling Leigh syndrome with developmental regression, intellectual disability and seizures. Am. J. Med. Genet. A 179, 2138–2143. doi: 10.1002/ajmg.a.61288
Pinke, G., Zhou, L., and Sazanov, L. A. (2020). Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat. Struct. Mol. Biol. 27, 1077–1085. doi: 10.1038/s41594-020-0503-8
Pitceathly, R. D. S., Rahman, S., Wedatilake, Y., Polke, J. M., Cirak, S., Foley, A. R., et al. (2013). NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 3, 1795–1805. doi: 10.1016/j.celrep.2013.05.005
Poole, A. C., Thomas, R. E., Yu, S., Vincow, E. S., and Pallanck, L. (2010). The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5:e10054. doi: 10.1371/journal.pone.0010054
Popov-Čeleketić, J., Waizenegger, T., and Rapaport, D. (2008). Mim1 functions in an oligomeric form to facilitate the integration of Tom20 into the mitochondrial outer membrane. J. Mol. Biol. 376, 671–680. doi: 10.1016/j.jmb.2007.12.006
Posse, V., Al-Behadili, A., Uhler, J. P., Clausen, A. R., Reyes, A., Zeviani, M., et al. (2019). RNase H1 directs origin-specific initiation of DNA replication in human mitochondria. PLoS Genet. 15:e1007781. doi: 10.1371/journal.pgen.1007781
Pozo Devoto, V. M., and Falzone, T. L. (2017). Mitochondrial dynamics in Parkinson’s disease: a role for α-synuclein? Dis. Model. Mech. 10, 1075–1087. doi: 10.1242/dmm.026294
Pradeepkiran, J. A., and Reddy, P. H. (2020). Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 64:101191. doi: 10.1016/j.arr.2020.101191
Pyle, A. M. (2008). Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 37, 317–336. doi: 10.1146/annurev.biophys.37.032807.125908
Qa, Y., Yan, L., Yu, C., Guo, X., Zhou, X., Hu, X., et al. (2016). Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 215, 621–629. doi: 10.1083/jcb.201609019
Quintana, A., Kruse, S. E., Kapur, R. P., Sanz, E., and Palmiter, R. D. (2010). Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. U S A 107, 10996–11001. doi: 10.1073/pnas.1006214107
Quintana, A., Zanella, S., Koch, H., Kruse, S. E., Lee, D., Ramirez, J. M., et al. (2012). Fatal breathing dysfunction in a mouse model of Leigh syndrome. J. Clin. Invest. 122, 2359–2368. doi: 10.1172/JCI62923
Rahman, S., and Copeland, W. C. (2019). POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 15, 40–52. doi: 10.1038/s41582-018-0101-0
Rahman, B., Kawano, S., Yunoki-Esaki, K., Anzai, T., and Endo, T. (2014). NMR analyses on the interactions of the yeast Tim50 C-terminal region with the presequence and Tim50 core domain. FEBS Lett. 588, 678–684. doi: 10.1016/j.febslet.2013.12.037
Rajakulendran, S., Pitceathly, R. D. S., Taanman, J.-W., Costello, H., Sweeney, M. G., Woodward, C. E., et al. (2016). A clinical, neuropathological and genetic study of homozygous A467T POLG-related mitochondrial disease. PLoS One 11:e0145500. doi: 10.1371/journal.pone.0145500
Ramachandran, A., Basu, U., Sultana, S., Nandakumar, D., and Patel, S. S. (2017). Human mitochondrial transcription factors TFAM and TFB2M work synergistically in promoter melting during transcription initiation. Nucleic Acids Res. 45, 861–874. doi: 10.1093/nar/gkw1157
Rangaraju, V., Lewis, T. L., Hirabayashi, Y., Bergami, M., Motori, E., Cartoni, R., et al. (2019). Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J. Neurosci. 39, 8200–8208. doi: 10.1523/JNEUROSCI.1157-19.2019
Rasola, A., and Bernardi, P. (2011). Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50, 222–233. doi: 10.1016/j.ceca.2011.04.007
Rebelo, A. P., Saade, D., Pereira, C. V., Farooq, A., Huff, T. C., Abreu, L., et al. (2018). SCO2 mutations cause early-onset axonal Charcot-Marie-Tooth disease associated with cellular copper deficiency. Brain 141, 662–672. doi: 10.1093/brain/awx369
Reddy, P. H., and Oliver, D. M. (2019). Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8:488. doi: 10.3390/cells8050488
Reddy, P. H., Mao, P., and Manczak, M. (2009). Mitochondrial structural and functional dynamics in Huntington’s disease. Brain Res. Rev. 61, 33–48. doi: 10.1016/j.brainresrev.2009.04.001
Reed, K. C., and Bygrave, F. L. (1974). The inhibition of mitochondrial calcium transport by lanthanides and Ruthenium Red. Biochem. J. 140, 143–155. doi: 10.1042/bj1400143
Renaldo, F., Amati-Bonneau, P., Slama, A., Romana, C., Forin, V., Doummar, D., et al. (2012). MFN2, a new gene responsible for mitochondrial DNA depletion. Brain 135, e223–e223. doi: 10.1093/brain/aws111
Renkema, G. H., Visser, G., Baertling, F., Wintjes, L. T., Wolters, V. M., van Montfrans, J., et al. (2017). Mutated PET117 causes complex IV deficiency and is associated with neurodevelopmental regression and medulla oblongata lesions. Hum. Genet. 136, 759–769. doi: 10.1007/s00439-017-1794-7
Reyes, A., Melchionda, L., Burlina, A., Robinson, A. J., Ghezzi, D., and Zeviani, M. (2018). Mutations in TIMM50 compromise cell survival in OxPhos-dependent metabolic conditions. EMBO Mol. Med. 10:e8698. doi: 10.15252/emmm.201708698
Ringel, R., Sologub, M., Morozov, Y. I., Litonin, D., Cramer, P., and Temiakov, D. (2011). Structure of human mitochondrial RNA polymerase. Nature 478, 269–273. doi: 10.1038/nature10435
Riordan-Eva, P., and Harding, A. E. (1995a). Leber’s hereditary optic neuropathy: the clinical relevance of different mitochondrial DNA mutations. J. Med. Genet. 32, 81–87. doi: 10.1136/jmg.32.2.81
Riordan-Eva, P., and Harding, A. E. (1995b). Leber’s hereditary optic neuropathy: the clinical relevance of different mitochondrial DNA mutations. J. Med. Genet. 32, 81–87. doi: 10.1136/jmg.32.2.81
Risiglione, P., Zinghirino, F., Di Rosa, M. C., Magrì, A., and Messina, A. (2021). Alpha-synuclein and mitochondrial dysfunction in Parkinson’s disease: the emerging role of VDAC. Biomolecules 11:718. doi: 10.3390/biom11050718
Rodenburg, R. J. (2016). Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 1857, 938–945. doi: 10.1016/j.bbabio.2016.02.012
Rogaeva, E., Johnson, J., Lang, A. E., Gulick, C., Gwinn-Hardy, K., Kawarai, T., et al. (2004). Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch. Neurol. 61, 1898–1904. doi: 10.1001/archneur.61.12.1898
Rohé, C. F., Montagna, P., Breedveld, G., Cortelli, P., Oostra, B. A., and Bonifati, V. (2004). Homozygous PINK1 C-terminus mutation causing early-onset parkinsonism. Ann. Neurol. 56, 427–431. doi: 10.1002/ana.20247
Ropp, P. A., and Copeland, W. C. (1996). Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase γ. Genomics 36, 449–458. doi: 10.1006/geno.1996.0490
Rosencrans, W. M., Rajendran, M., Bezrukov, S. M., and Rostovtseva, T. K. (2021). VDAC regulation of mitochondrial calcium flux: from channel biophysics to disease. Cell Calcium 94:102356. doi: 10.1016/j.ceca.2021.102356
Ruffoli, R., Bartalucci, A., Frati, A., and Fornai, F. (2015). Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 9:341. doi: 10.3389/fncel.2015.00341
Ryan, K. C., Ashkavand, Z., and Norman, K. R. (2020). The role of mitochondrial calcium homeostasis in Alzheimer’s and related diseases. Int. J. Mol. Sci. 21:9153. doi: 10.3390/ijms21239153
Safdar, A., Little, J. P., Stokl, A. J., Hettinga, B. P., Akhtar, M., and Tarnopolsky, M. A. (2011). Exercise increases mitochondrial PGC-1α content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J. Biol. Chem. 286, 10605–10617. doi: 10.1074/jbc.EC118.002682
Salama, M., and Mohamed, W. M. Y. (2015). NDUFA12L mitochondrial complex-I assembly factor: implications for taupathies. Appl. Transl. Genomics 5, 37–39. doi: 10.1016/j.atg.2015.05.003
Sánchez-Caballero, L., Guerrero-Castillo, S., and Nijtmans, L. (2016). Unraveling the complexity of mitochondrial complex I assembly: a dynamic process. Biochim. Biophys. Acta 1857, 980–990. doi: 10.1136/archdischild-2021-323390
Santarelli, R., Rossi, R., Scimemi, P., Cama, E., Valentino, M. L., La Morgia, C., et al. (2015). OPA1-related auditory neuropathy: site of lesion and outcome of cochlear implantation. Brain 138, 563–576. doi: 10.1093/brain/awu378
Santel, A., and Fuller, M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114, 867–874. doi: 10.1242/jcs.114.5.867
Sazanov, L. A. (2015). A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 16, 375–388. doi: 10.1038/nrm3997
Scacco, S., Petruzzella, V., Budde, S., Vergari, R., Tamborra, R., Panelli, D., et al. (2003). Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J. Biol. Chem. 278, 44161–44167. doi: 10.1074/jbc.M307615200
Scarffe, L. A., Stevens, D. A., Dawson, V. L., and Dawson, T. M. (2014). Parkin and PINK1: much more than mitophagy. Trends Neurosci. 37, 315–324. doi: 10.1016/j.tins.2014.03.004
Schapira, A. H. V. (2013). Calcium dysregulation in Parkinson’s disease. Brain 136, 2015–2016. doi: 10.1093/brain/awt180
Schlehe, J. S., Journel, M. S. M., Taylor, K. P., Amodeo, K. D., and LaVoie, M. J. (2013). The mitochondrial disease associated protein Ndufaf2 is dispensable for Complex-1 assembly but critical for the regulation of oxidative stress. Neurobiol. Dis. 58, 57–67. doi: 10.1016/j.nbd.2013.05.007
Schmidt, O., Pfanner, N., and Meisinger, C. (2010). Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 11, 655–667. doi: 10.1038/nrm2959
Schreiber, S. N., Emter, R., Hock, M. B., Knutti, D., Cardenas, J., Podvinec, M., et al. (2004). The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. U S A 101, 6472–6477. doi: 10.1073/pnas.0308686101
Schubert, A. F., Gladkova, C., Pardon, E., Wagstaff, J. L., Freund, S. M. V., Steyaert, J., et al. (2017). Structure of PINK1 in complex with its substrate ubiquitin. Nature 552, 51–56. doi: 10.1038/nature24645
Ścieżyńska, A., Ruszkowska, E., Szulborski, K., Rydz, K., Wierzbowska, J., Kosińska, J., et al. (2017). Processing of OPA1 with a novel N-terminal mutation in patients with autosomal dominant optic atrophy: escape from nonsense-mediated decay. PLoS One 12:e0183866. doi: 10.1371/journal.pone.0183866
Seager, R., Lee, L., Henley, J. M., and Wilkinson, K. A. (2020). Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 4:NS20200008. doi: 10.1042/NS20200008
Sferruzza, G., Del Bondio, A., Citterio, A., Vezzulli, P., Guerrieri, S., Radaelli, M., et al. (2021). U-fiber leukoencephalopathy due to a novel mutation in the TACO1 gene. Neurol. Genet. 7:e573. doi: 10.1212/NXG.0000000000000573
Shahrour, M. A., Staretz-Chacham, O., Dayan, D., Stephen, J., Weech, A., Damseh, N., et al. (2017). Mitochondrial epileptic encephalopathy, 3-methylglutaconic aciduria and variable complex V deficiency associated with TIMM50 mutations. Clin. Genet. 91, 690–696. doi: 10.1111/cge.12855
Shamseldin, H. E., Alshammari, M., Al-Sheddi, T., Salih, M. A., Alkhalidi, H., Kentab, A., et al. (2012). Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 49, 234–241. doi: 10.1136/jmedgenet-2012-100836
Shamseldin, H. E., Alasmari, A., Salih, M. A., Samman, M. M., Mian, S. A., Alshidi, T., et al. (2017). A null mutation in MICU2 causes abnormal mitochondrial calcium homeostasis and a severe neurodevelopmental disorder. Brain 140, 2806–2813. doi: 10.1093/brain/awx237
Sheng, Z.-H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Sheng, B., Wang, X., Su, B., Lee, H., Casadesus, G., Perry, G., et al. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 120, 419–429. doi: 10.1111/j.1471-4159.2011.07581.x
Shi, Y., Dierckx, A., Wanrooij, P. H., Wanrooij, S., Larsson, N.-G., Wilhelmsson, L. M., et al. (2012). Mammalian transcription factor A is a core component of the mitochondrial transcription machinery. Proc. Natl. Acad. Sci. U S A 109, 16510–16515. doi: 10.1073/pnas.1119738109
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2:1002. doi: 10.1038/srep01002
Shim, J. S., Song, M.-Y., Yim, S.-V., Lee, S.-E., and Park, K.-S. (2017). Global analysis of ginsenoside Rg1 protective effects in β-amyloid-treated neuronal cells. J. Ginseng Res. 41, 566–571. doi: 10.1016/j.jgr.2016.12.003
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. doi: 10.1038/77060
Shoshan-Barmatz, V., De Pinto, V., Zweckstetter, M., Raviv, Z., Keinan, N., and Arbel, N. (2010). VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285. doi: 10.1016/j.mam.2010.03.002
Shoshan-Barmatz, V., N. Maldonado, E., and Krelin, Y. (2017). VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 1, 11–36. doi: 10.15698/cst2017.10.104
Shoshan-Barmatz, V., Nahon-Crystal, E., Shteinfer-Kuzmine, A., and Gupta, R. (2018). VDAC1, mitochondrial dysfunction and Alzheimer’s disease. Pharmacol. Res. 131, 87–101. doi: 10.1016/j.phrs.2018.03.010
Signes, A., and Fernandez-Vizarra, E. (2018). Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 62, 255–270. doi: 10.1042/EBC20170098
Sim, C. H., Lio, D. S. S., Mok, S. S., Masters, C. L., Hill, A. F., Culvenor, J. G., et al. (2006). C-terminal truncation and Parkinson’s disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum. Mol. Genet. 15, 3251–3262. doi: 10.1093/hmg/ddl398
Singleton, M. R., Dillingham, M. S., and Wigley, D. B. (2007). Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50. doi: 10.1146/annurev.biochem.76.052305.115300
Sinha, S., and Manoj, N. (2019). Molecular evolution of proteins mediating mitochondrial fission-fusion dynamics. FEBS Lett. 593, 703–718. doi: 10.1002/1873-3468.13356
Sirrenberg, C., Bauer, M. F., Guiard, B., Neupert, W., and Brunner, M. (1996). Import of carrier proteins into the mitochondrial inner membrane mediated by Tim22. Nature 384, 582–585. doi: 10.1038/384582a0
Sirrenberg, C., Endres, M., Fölsch, H., Stuart, R. A., Neupert, W., and Brunner, M. (1998). Carrier protein import into mitochondria mediated by the intermembrane proteins Tim10/Mrs11 and Tim12/Mrs5. Nature 391, 912–915. doi: 10.1038/36136
Smirnova, E., Griparic, L., Shurland, D.-L., and van der Bliek, A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. doi: 10.1091/mbc.12.8.2245
Sokoloff, L. (1973). Metabolism of ketone bodies by the brain. Annu. Rev. Med. 24, 271–280. doi: 10.1146/annurev.me.24.020173.001415
Song, P., Guan, Y., Chen, X., Wu, C., Qiao, A., Jiang, H., et al. (2021). Frameshift mutation of Timm8a1 gene in mouse leads to an abnormal mitochondrial structure in the brain, correlating with hearing and memory impairment. J. Med. Genet. 58, 619–627. doi: 10.1136/jmedgenet-2020-106925
Šonský, I., Vodićka, P., Vodičková Kepková, K., and Hansíková, H. (2021). Mitophagy in Huntington’s disease. Neurochem. Int. 149:105147. doi: 10.1016/j.neuint.2021.105147
Sörensen, L., Ekstrand, M., Silva, J., Lindqvist, E., Xu, B., Rustin, P., et al. (2001). Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J. Neurosci. 21, 8082–8090. doi: 10.1523/JNEUROSCI.21-20-08082.2001
Sotelo-Hitschfeld, T., Niemeyer, M. I., Machler, P., Ruminot, I., Lerchundi, R., Wyss, M. T., et al. (2015). Channel-Mediated Lactate Release by K+-Stimulated Astrocytes. J. Neurosci. 35, 4168–4178. doi: 10.1523/JNEUROSCI.5036-14.2015
Spelbrink, J. N., Li, F.-Y., Tiranti, V., Nikali, K., Yuan, Q.-P., Tariq, M., et al. (2001). Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 28, 223–231. doi: 10.1038/90058
Spikes, T. E., Montgomery, M. G., and Walker, J. E. (2020). Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl. Acad. Sci. U S A 117, 23519–23526. doi: 10.1073/pnas.2013998117
Stemmler, T. L., Lesuisse, E., Pain, D., and Dancis, A. (2010). Frataxin and mitochondrial FeS cluster biogenesis. J. Biol. Chem. 285, 26737–26743. doi: 10.1074/jbc.R110.118679
Stephan, K., and Ott, M. (2020). Timing of dimerization of the bc complex during mitochondrial respiratory chain assembly. Biochim. Biophys. Acta Bioenerg. 1861:148177. doi: 10.1016/j.bbabio.2020.148177
Stiburek, L., Fornuskova, D., Wenchich, L., Pejznochova, M., Hansikova, H., and Zeman, J. (2007). Knockdown of human oxa1l impairs the biogenesis of F1Fo-ATP synthase and NADH:ubiquinone oxidoreductase. J. Mol. Biol. 374, 506–516. doi: 10.1016/j.jmb.2007.09.044
Su, B., Wang, X., Zheng, L., Perry, G., Smith, M. A., and Zhu, X. (2010). Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 1802, 135–142. doi: 10.1016/j.bbadis.2009.09.013
Sun, F., Huo, X., Zhai, Y., Wang, A., Xu, J., Su, D., et al. (2005). Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121, 1043–1057. doi: 10.1016/j.cell.2005.05.025
Sun, Y., Xue, W., Song, Z., Huang, K., and Zheng, L. (2016). Restoration of Opa1-long isoform inhibits retinal injury-induced neurodegeneration. J. Mol. Med. 94, 335–346. doi: 10.1007/s00109-015-1359-y
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028
Surmeier, D. J., Schumacker, P. T., Guzman, J. D., Ilijic, E., Yang, B., and Zampese, E. (2017). Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 483, 1013–1019. doi: 10.1016/j.bbrc.2016.08.168
Szabo, I., and Zoratti, M. (2014). Mitochondrial channels: ion fluxes and more. Physiol. Rev. 94, 519–608. doi: 10.1152/physrev.00021.2013
Taanman, J.-W. (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123. doi: 10.1016/s0005-2728(98)00161-3
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi: 10.1074/jbc.M607279200
Tamiya, G., Makino, S., Hayashi, M., Abe, A., Numakura, C., Ueki, M., et al. (2014). A mutation of COX6A1 causes a recessive axonal or mixed form of charcot-marie-tooth disease. Am. J. Hum. Genet. 95, 294–300. doi: 10.1002/humu.24393
Tanaka, K. (2020). The PINK1-Parkin axis: an overview. Neurosci. Res. 159, 9–15. doi: 10.1016/j.neures.2020.01.006
Tay, S. K. H., Sacconi, S., Ohran Akman, H., Morales, J. F., Morales, A., De Vivo, D. C., et al. (2005). Unusual clinical presentations in four cases of Leigh disease, cytochrome C oxidase deficiency and SURF1 gene mutations. J. Child Neurol. 20, 670–674. doi: 10.1177/08830738050200080701
Taylor, S. W., Fahy, E., Zhang, B., Glenn, G. M., Warnock, D. E., Wiley, S., et al. (2003). Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21, 281–286. doi: 10.1038/nbt793
Taylor, A. B., Smith, B. S., Kitada, S., Kojima, K., Miyaura, H., Otwinowski, Z., et al. (2001). Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9, 615–625. doi: 10.1016/s0969-2126(01)00621-9
Terzenidou, M. E., Segklia, A., Kano, T., Papastefanaki, F., Karakostas, A., Charalambous, M., et al. (2017). Novel insights into SLC25A46-related pathologies in a genetic mouse model. PLoS Genet. 13:e1006656. doi: 10.1371/journal.pgen.1006656
Thomas, B., von Coelln, R., Mandir, A. S., Trinkaus, D. B., Farah, M. H., Leong Lim, K., et al. (2007). MPTP and DSP-4 susceptibility of substantia nigra and locus coeruleus catecholaminergic neurons in mice is independent of parkin activity. Neurobiol. Dis. 26, 312–322. doi: 10.1016/j.nbd.2006.12.021
Thompson, K., Mai, N., Oláhová, M., Scialó, F., Formosa, L. E., Stroud, D. A., et al. (2018). OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol. Med. 10:e9060. doi: 10.15252/emmm.201809060
Tilokani, L., Nagashima, S., Paupe, V., and Prudent, J. (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360. doi: 10.1042/EBC20170104
Tranebjærg, L., Hamel, B. C. J., Gabreels, F. J. M., Renier, W. O., and Ghelue, M. V. (2000). A de novo missense mutation in a critical domain of the X-linked DDP gene causes the typical deafness-dystonia-optic atrophy syndrome. Eur. J. Hum. Genet. 8, 464–467. doi: 10.1038/sj.ejhg.5200483
Tranebjærg, L., Jensen, P. K. A., Van Ghelue, M., Vnencak-Jones, C. L., Sund, S., Elgjo, K., et al. (2001). Neuronal cell death in the visual cortex is a prominent feature of the X-linked recessive mitochondrial deafness-dystonia syndrome caused by mutations in the TIMM8a gene. Ophthalmic Genet. 22, 207–223. doi: 10.1076/opge.22.4.207.2220
Tranebjærg, L., Schwartz, C., Eriksen, H., Andreasson, S., Ponjavic, V., Dahl, A., et al. (1995). A new X linked recessive deafness syndrome with blindness, dystonia, fractures and mental deficiency is linked to Xq22. J. Med. Genet. 32, 257–263. doi: 10.1136/jmg.32.4.257
Trempe, J.-F., Sauvé, V., Grenier, K., Seirafi, M., Tang, M. Y., Ménade, M., et al. (2013). Structure of Parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455. doi: 10.1126/science.1237908
Trevisan, T., Pendin, D., Montagna, A., Bova, S., Ghelli, A. M., and Daga, A. (2018). Manipulation of mitochondria dynamics reveals separate roles for form and function in mitochondria distribution. Cell Rep. 23, 1742–1753. doi: 10.1016/j.celrep.2018.04.017
Truscott, K. N., Kovermann, P., Geissler, A., Merlin, A., Meijer, M., Driessen, A. J. M., et al. (2001). A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Struct. Biol. 8, 1074–1082. doi: 10.1038/nsb726
Tucker, E. J., Wanschers, B. F. J., Szklarczyk, R., Mountford, H. S., Wijeyeratne, X. W., van den Brand, M. A. M., et al. (2013). Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 9:e1004034. doi: 10.1371/journal.pgen.1004034
Tuppen, H. A. L., Blakely, E. L., Turnbull, D. M., and Taylor, R. W. (2010). Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 1797, 113–128. doi: 10.1016/j.bbabio.2009.09.005
Tysnes, O.-B., and Storstein, A. (2017). Epidemiology of Parkinson’s disease. J. Neural Transm. (Vienna) 124, 901–905. doi: 10.1007/s00702-017-1686-y
Tzoulis, C., Tran, G. T., Schwarzlmüller, T., Specht, K., Haugarvoll, K., Balafkan, N., et al. (2013). Severe nigrostriatal degeneration without clinical parkinsonism in patients with polymerase gamma mutations. Brain 136, 2393–2404. doi: 10.1093/brain/awt103
Ujike, H., Tanabe, Y., Takehisa, Y., Hayabara, T., and Kuroda, S. (2001). A family with X-linked dystonia-deafness syndrome with a novel mutation of the DDP gene. Arch. Neurol. 58:1004. doi: 10.1001/archneur.58.6.1004
Ullah, F., Rauf, W., Khan, K., Khan, S., Bell, K. M., de Oliveira, V. C., et al. (2021). A recessive variant in TFAM causes mtDNA depletion associated with primary ovarian insufficiency, seizures, intellectual disability and hearing loss. Hum. Genet. 140, 1733–1751. doi: 10.1007/s00439-021-02380-2
Urbani, A., Prosdocimi, E., Carrer, A., Checchetto, V., and Szabò, I. (2021). Mitochondrial ion channels of the inner membrane and their regulation in cell death signaling. Front. Cell Dev. Biol. 8:620081. doi: 10.3389/fcell.2020.620081
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Valente, E. M., Bentivoglio, A. R., Dixon, P. H., Ferraris, A., Ialongo, T., Frontali, M., et al. (2001). Localization of a novel locus for autosomal recessive early-onset Parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900. doi: 10.1086/319522
Valera-Alberni, M., Joffraud, M., Miro-Blanch, J., Capellades, J., Junza, A., Dayon, L., et al. (2021). Crosstalk between Drp1 phosphorylation sites during mitochondrial remodeling and their impact on metabolic adaptation. Cell Rep. 36:109565. doi: 10.1016/j.celrep.2021.109565
Valnot, I. (2000). A mutation in the human heme A: farnesyltransferase gene (COX10 ) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 9, 1245–1249. doi: 10.1093/hmg/9.8.1245
Van Coster, R., Seneca, S., Smet, J., Van Hecke, R., Gerlo, E., Devreese, B., et al. (2003). Homozygous Gly555Glu mutation in the nuclear-encoded 70 kDa flavoprotein gene causes instability of the respiratory chain complex II. Am. J. Med. Genet. 120A, 13–18. doi: 10.1002/ajmg.a.10202
Van Goethem, G., Mercelis, R., Lofgren, A., Seneca, S., Ceuterick, C., Martin, J. J., et al. (2003). Patient homozygous for a recessive POLG mutation presents with features of MERRF. Neurology 61, 1811–1813. doi: 10.1212/01.wnl.0000098997.23471.65
Van Hove, J. L. K., Cunningham, V., Rice, C., Ringel, S. P., Zhang, Q., Chou, P.-C., et al. (2009). Finding twinkle in the eyes of a 71-year-old lady: a case report and review of the genotypic and phenotypic spectrum of TWINKLE-related dominant disease. Am. J. Med. Genet. Part A 149A, 861–867. doi: 10.1002/ajmg.a.32731
Vercellino, I., and Sazanov, L. A. (2022). The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 23, 141–161. doi: 10.1038/s41580-021-00415-0
Vodopivec, I., Cho, T. A., Rizzo, J. F., Frosch, M. P., and Sims, K. B. (2016). Mitochondrial encephalopathy and optic neuropathy due to m.10158 MT-ND3 Complex I mutation presenting in an adult patient. Neurologist 21, 61–65. doi: 10.1097/NRL.0000000000000084
Vögtle, F.-N., Brändl, B., Larson, A., Pendziwiat, M., Friederich, M. W., White, S. M., et al. (2018). Mutations in PMPCB encoding the catalytic subunit of the mitochondrial presequence protease cause neurodegeneration in early childhood. Am. J. Hum. Genet. 102, 557–573. doi: 10.1097/HCR.0000000000000719
Vögtle, F. N., Wortelkamp, S., Zahedi, R. P., Becker, D., Leidhold, C., Gevaert, K., et al. (2009). Global analysis of the mitochondrial N-Proteome identifies a processing peptidase critical for protein stability. Cell 139, 428–439. doi: 10.1097/HCR.0000000000000719
Wakabayashi, J., Zhang, Z., Wakabayashi, N., Tamura, Y., Fukaya, M., Kensler, T. W., et al. (2009). The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816. doi: 10.1083/jcb.200903065
Walker, J. E. (1992). The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q. Rev. Biophys. 25, 253–324. doi: 10.1017/s003358350000425x
Wan, J., Steffen, J., Yourshaw, M., Mamsa, H., Andersen, E., Rudnik-Schöneborn, S., et al. (2016). Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain 139, 2877–2890. doi: 10.1093/brain/aww212
Wang, J.-Q., Chen, Q., Wang, X., Wang, Q.-C., Wang, Y., Cheng, H.-P., et al. (2013). Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in huntington disease. J. Biol. Chem. 288, 3070–3084. doi: 10.1074/jbc.M112.407726
Wang, Y., Liu, N., and Lu, B. (2019). Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 25, 859–875. doi: 10.1111/cns.13140
Wang, H., Song, P., Du, L., Tian, W., Yue, W., Liu, M., et al. (2011). Parkin Ubiquitinates Drp1 for proteasome-dependent degradation. J. Biol. Chem. 286, 11649–11658. doi: 10.1074/jbc.M110.144238
Wang, W., Sun, Y., Lin, Y., Xu, X., Zhao, D., Ji, K., et al. (2021). A novel nonsense variant in MT-CO3 causes MELAS syndrome. Neuromuscul. Disord. 31, 558–565. doi: 10.1016/j.nmd.2021.02.020
Wang, K., Takahashi, Y., Gao, Z.-L., Wang, G.-X., Chen, X.-W., Goto, J., et al. (2009). Mitochondrial ND3 as the novel causative gene for Leber hereditary optic neuropathy and dystonia. Neurogenetics 10, 337–345. doi: 10.1007/s10048-009-0194-0
Wang, H., Wang, L., Yang, J., Yin, L., Lan, L., Li, J., et al. (2019). Phenotype prediction of Mohr-Tranebjaerg syndrome (MTS) by genetic analysis and initial auditory neuropathy. BMC Med. Genet. 20:11. doi: 10.1186/s12881-018-0741-3
Wanschers, B. F. J., Szklarczyk, R., van den Brand, M. A. M., Jonckheere, A., Suijskens, J., Smeets, R., et al. (2014). A mutation in the human CBP4 ortholog UQCC3 impairs complex III assembly, activity and cytochrome b stability. Hum. Mol. Genet. 23, 6356–6365. doi: 10.1093/hmg/ddu357
Ware, S. M., El-Hassan, N., Kahler, S. G., Zhang, Q. Y.-W., and Miller, E.. (2009). Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes. J. Med. Genet. 46, 308–314. doi: 10.1136/jmg.2008.063149
Wasiak, S., Zunino, R., and McBride, H. M. (2007). Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 177, 439–450. doi: 10.1083/jcb.200610042
Waterham, H. R., Koster, J., van Roermund, C. W. T., Mooyer, P. A. W., Wanders, R. J. A., and Leonard, J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 356, 1736–1741. doi: 10.1056/NEJMoa064436
Wauer, T., Swatek, K. N., Wagstaff, J. L., Gladkova, C., Pruneda, J. N., Michel, M. A., et al. (2015). Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 34, 307–325. doi: 10.15252/embj.201489847
Wei, X., Du, M., Li, D., Wen, S., Xie, J., Li, Y., et al. (2020a). Mutations in FASTKD2 are associated with mitochondrial disease with multi-OXPHOS deficiency. Hum. Mutat. 41, 961–972. doi: 10.1002/humu.23985
Wei, X., Du, M., Xie, J., Luo, T., Zhou, Y., Zhang, K., et al. (2020b). Mutations in TOMM70 lead to multi-OXPHOS deficiencies and cause severe anemia, lactic acidosis and developmental delay. J. Hum. Genet. 65, 231–240. doi: 10.1038/s10038-019-0714-1
Weisschuh, N., Schimpf-Linzenbold, S., Mazzola, P., Kieninger, S., Xiao, T., Kellner, U., et al. (2021). Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: identification and classification of 48 novel variants. PLoS One 16:e0253987. doi: 10.1371/journal.pone.0253987
Weraarpachai, W., Antonicka, H., Sasarman, F., Seeger, J., Schrank, B., Kolesar, J. E., et al. (2009). Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 41, 833–837. doi: 10.1038/ng.390
Whitley, B. N., Lam, C., Cui, H., Haude, K., Bai, R., Escobar, L., et al. (2018). Aberrant Drp1-mediated mitochondrial division presents in humans with variable outcomes. Hum. Mol. Genet. 27, 3710–3719. doi: 10.1093/hmg/ddy287
Wiedemann, N., Kozjak, V., Chacinska, A., Schönfisch, B., Rospert, S., Ryan, M. T., et al. (2003). Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature 424, 565–571. doi: 10.1038/nature01753
Wiedemann, N., and Pfanner, N. (2017). Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 86, 685–714. doi: 10.1146/annurev-biochem-060815-014352
Williams, P. A., Piechota, M., von Ruhland, C., Taylor, E., Morgan, J. E., and Votruba, M. (2012). Opa1 is essential for retinal ganglion cell synaptic architecture and connectivity. Brain 135, 493–505. doi: 10.1093/brain/awr330
Wirth, C., Brandt, U., Hunte, C., and Zickermann, V. (2016). Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 1857, 902–914. doi: 10.1016/j.bbabio.2016.02.013
Wu, Z., Huang, X., Feng, Y., Handschin, C., Feng, Y., Gullicksen, P. S., et al. (2006). Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1α transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. U S A 103, 14379–14384. doi: 10.1073/pnas.0606714103
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. doi: 10.1016/S0092-8674(00)80611-X
Xia, D., Esser, L., Tang, W.-K., Zhou, F., Zhou, Y., Yu, L., et al. (2013). Structural analysis of cytochrome bc1 complexes: implications to the mechanism of function. Biochim. Biophys. Acta 1827, 1278–1294. doi: 10.1016/j.bbabio.2012.11.008
Xia, D., Yu, C.-A., Kim, H., Xia, J.-Z., Kachurin, A. M., Zhang, L., et al. (1997). Crystal structure of the cytochrome bc 1 complex from bovine heart mitochondria. Science 277, 60–66. doi: 10.1126/science.277.5322.60
Xiyang, Y.-B., Liu, R., Wang, X.-Y., Li, S., Zhao, Y., Lu, B.-T., et al. (2020). COX5A plays a vital role in memory impairment associated with brain aging via the BDNF/ERK1/2 signaling pathway. Front. Aging Neurosci. 12:215. doi: 10.3389/fnagi.2020.00215
Xu, D., Yang, P., Yang, Z.-J., Li, Q.-G., Ouyang, Y.-T., Yu, T., et al. (2021). Blockage of Drp1 phosphorylation at Ser579 protects neurons against Aβ1-42-induced degeneration. Mol. Med. Rep. 24:657. doi: 10.3892/mmr.2021.12296
Yamada, T., Adachi, Y., Fukaya, M., Iijima, M., and Sesaki, H. (2016). Dynamin-related protein 1 deficiency leads to receptor-interacting protein kinase 3-mediated necroptotic neurodegeneration. Am. J. Pathol. 186, 2798–2802. doi: 10.1016/j.ajpath.2016.06.025
Yamano, K., and Youle, R. J. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi: 10.4161/auto.24633
Yang, Z., Wang, L., Yang, C., Pu, S., Guo, Z., Wu, Q., et al. (2022). Mitochondrial membrane remodeling. Front. Bioeng. Biotechnol. 9:786806. doi: 10.3389/fbioe.2021.786806
Yang, X., Xu, S., Qian, Y., He, X., Chen, S., and Xiao, Q. (2020). Hypermethylation of the gene coding for PGC-1α in peripheral blood leukocytes of patients with Parkinson’s disease. Front. Neurosci. 14:97. doi: 10.3389/fnins.2020.00097
Yang, J., Zhu, Y., Tong, Y., Chen, L., Liu, L., Zhang, Z., et al. (2009). Confirmation of the mitochondrial ND1 gene mutation G3635A as a primary LHON mutation. Biochem. Biophys. Res. Commun. 386, 50–54. doi: 10.1016/j.bbrc.2009.05.127
Yankovskaya, V., Horsefield, R., TÖrnroth, S., Luna-Chavez, C., Miyoshi, H., Léger, C., et al. (2003). Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299, 700–704. doi: 10.1126/science.1079605
Yin, Z., Burger, N., Kula-Alwar, D., Aksentijević, D., Bridges, H. R., Prag, H. A., et al. (2021). Structural basis for a complex I mutation that blocks pathological ROS production. Nat. Commun. 12:707. doi: 10.1038/s41467-021-20942-w
Yoon, G., Malam, Z., Paton, T., Marshall, C. R., Hyatt, E., Ivakine, Z., et al. (2016). Lethal disorder of mitochondrial fission caused by mutations in DNM1L. J. Pediatr. 171, 313–316.e1-2. doi: 10.1016/j.jpeds.2015.12.060
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Young, J. C., Hoogenraad, N. J., and Hartl, F. U. (2003). Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112, 41–50. doi: 10.1016/s0092-8674(02)01250-3
Young, M. J., Humble, M. M., DeBalsi, K. L., Sun, K. Y., and Copeland, W. C. (2015). POLG2 disease variants: analyses reveal a dominant negative heterodimer, altered mitochondrial localization and impaired respiratory capacity. Hum. Mol. Genet. 24, 5184–5197. doi: 10.1093/hmg/ddv240
Zaha, K., Matsumoto, H., Itoh, M., Saitsu, H., Kato, K., Kato, M., et al. (2016). DNM1L -related encephalopathy in infancy with Leigh syndrome-like phenotype and suppression-burst. Clin. Genet. 90, 472–474. doi: 10.1111/cge.12805
Zanna, C., Ghelli, A., Porcelli, A. M., Karbowski, M., Youle, R. J., Schimpf, S., et al. (2008). OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 131, 352–367. doi: 10.1093/brain/awm335
Zeng, T., Liao, L., Guo, Y., Liu, X., Xiong, X., Zhang, Y., et al. (2020). Concurrent OPA1 mutation and chromosome 3q deletion leading to Behr syndrome: a case report. BMC Pediatr. 20:420. doi: 10.1186/s12887-020-02309-0
Zeviani, M., Servidei, S., Gellera, C., Bertini, E., DiMauro, S., and DiDonato, S. (1989). An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 339, 309–311. doi: 10.1038/339309a0
Zhang, K., Li, H., and Song, Z. (2014). Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 15, 576–585. doi: 10.1002/embr.201338240
Zhang, J., Liu, M., Zhang, Z., Zhou, L., Kong, W., Jiang, Y., et al. (2019). Genotypic spectrum and natural history of cavitating leukoencephalopathies in childhood. Pediatr. Neurol. 94, 38–47. doi: 10.1016/j.pediatrneurol.2019.01.002
Zhang, L., Shi, W., Song, L., Zhang, X., Cheng, L., Wang, Y., et al. (2015). A recurrent deletion mutation in OPA1 causes autosomal dominant optic atrophy in a Chinese family. Sci. Rep. 4:6936. doi: 10.1038/srep06936
Zhang, Z., Huang, L., Shulmeister, V. M., Chi, Y.-I., Kim, K. K., Hung, L.-W., et al. (1998). Electron transfer by domain movement in cytochrome bc1. Nature 392, 677–684. doi: 10.1038/33612
Zhang, Z., Liu, L., Jiang, X., Zhai, S., and Xing, D. (2016). The essential role of Drp1 and its regulation by S-Nitrosylation of Parkin in dopaminergic neurodegeneration: implications for Parkinson’s disease. Antioxid. Redox Signal. 25, 609–622. doi: 10.1089/ars.2016.6634
Zhang, Q., Yu, J.-T., Wang, P., Chen, W., Wu, Z.-C., Jiang, H., et al. (2011). Mitochondrial transcription factor A (TFAM) polymorphisms and risk of late-onset Alzheimer’s disease in Han Chinese. Brain Res. 1368, 355–360. doi: 10.1016/j.brainres.2010.10.074
Zhao, W., Varghese, M., Yemul, S., Pan, Y., Cheng, A., Marano, P., et al. (2011). Peroxisome proliferator activator receptor gamma coactivator-1α (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 6:51. doi: 10.1186/1750-1326-6-51
Zheng, Y., Zhang, X., and Chen, Z. (2019). Mitochondrial transport serves as a mitochondrial quality control strategy in axons: implications for central nervous system disorders. CNS Neurosci. Ther. 25, 876–886. doi: 10.1111/cns.13122
Zhou, L., and Sazanov, L. A. (2019). Structure and conformational plasticity of the intact Thermus thermophilus V/A-type ATPase. Science 365:eaaw9144. doi: 10.1126/science.aaw9144
Zhu, J., Vinothkumar, K. R., and Hirst, J. (2016). Structure of mammalian respiratory complex I. Nature 536, 354–358. doi: 10.1038/nature19095
Zimmer, E. R., Parent, M. J., Souza, D. G., Leuzy, A., Lecrux, C., Kim, H.-I., et al. (2017). [18F]FDG PET signal is driven by astroglial glutamate transport. Nat. Neurosci. 20, 393–395. doi: 10.1038/nn.4492
Zinovkina, L. A. (2018). Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry (Mosc) 83, 233–249. doi: 10.1134/S0006297918030045
Zong, S., Wu, M., Gu, J., Liu, T., Guo, R., and Yang, M. (2018). Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 28, 1026–1034. doi: 10.1038/s41422-018-0071-1
Zorzano, A., and Claret, M. (2015). Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction. Front. Aging Neurosci. 7:101. doi: 10.3389/fnagi.2015.00101
Keywords: mitochondrial proteins, neurological diseases, pathogenesis, mitochondrial bioenergetics, mitochondrial dynamics, mitophagy, mtDNA maintenance, mitochondrial import machinery
Citation: Wang L, Yang Z, He X, Pu S, Yang C, Wu Q, Zhou Z, Cen X and Zhao H (2022) Mitochondrial protein dysfunction in pathogenesis of neurological diseases. Front. Mol. Neurosci. 15:974480. doi: 10.3389/fnmol.2022.974480
Received: 21 June 2022; Accepted: 08 August 2022;
Published: 07 September 2022.
Edited by:
Lorena Perrone, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Sara Baldelli, Università telematica San Raffaele, ItalyVeronica Bazzani, University of Udine, Italy
Copyright © 2022 Wang, Yang, He, Pu, Yang, Wu, Zhou, Cen and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongxia Zhao, aG9uZ3hpYS56aGFvQGhlbHNpbmtpLmZp
† These authors have contributed equally to this work