 Zitong Zhao1,2,3†Jian He4†
Zitong Zhao1,2,3†Jian He4† Yibing Chen1,2,3Yuchang Wang1,2,3Chuansen Wang1,2,3
Yibing Chen1,2,3Yuchang Wang1,2,3Chuansen Wang1,2,3 Changwu Tan1,2,3
Changwu Tan1,2,3 Junbo Liao1,2,3
Junbo Liao1,2,3 Gelei Xiao1,2,3*
Gelei Xiao1,2,3*- 1Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, China
- 2Diagnosis and Treatment Center for Hydrocephalus, Xiangya Hospital, Central South University, Changsha, China
- 3National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 4Department of Pediatrics, Xiangya Hospital, Central South University, Changsha, China
Idiopathic normal pressure hydrocephalus (iNPH) is a neurological disorder without a recognized cause. Aquaporins (AQPs) are transmembrane channels that carry water through cell membranes and are critical for cerebrospinal fluid circulation and cerebral water balance. The function of AQPs in developing and maintaining hydrocephalus should be studied in greater detail as a possible diagnostic and therapeutic tool. Recent research indicates that patients with iNPH exhibited high levels of aquaporin 1 and low levels of aquaporin 4 expression, suggesting that these AQPs are essential in iNPH pathogenesis. To determine the source of iNPH and diagnose and treat it, it is necessary to examine and appreciate their function in the genesis and maintenance of hydrocephalus. The expression, function, and regulation of AQPs in iNPH are reviewed in this article, in order to provide fresh targets and suggestions for future research.
Introduction
Normal pressure hydrocephalus (NPH) is a clinical condition characterized by gait abnormalities, cognitive impairment, and urinary incontinence. Clinically, it is often divided into two categories: secondary NPH (sNPH), often secondary to diseases with clear causes, such as craniocerebral trauma, subarachnoid hemorrhage, intracranial infection, encephalitis; and idiopathic NPH (iNPH), which has no definite etiology, mainly occurs in adults, and has a high incidence among the elderly. Misdiagnosis with Alzheimer's, Parkinson's syndrome, or a combination of these is common. The pathophysiology of iNPH is currently unclear. The basic theory of intracranial intravenous system compliance reduction is demonstrated by decreased cerebrospinal fluid (CSF) pulsation and impaired cobweb particle function, influencing CSF flow and absorption. The pathogenesis of iNPH is linked to irregular CSF circulatory dynamics based on circulation theory, inflammatory responses, osmosis abnormalities, and lymphatic drainage disorders based on penetration theory. Aquaporins (AQPs), which are responsible for CSF penetration and lymphatic outflow, are also involved in creating iNPH (Leinonen et al., 2021; Wang Y. et al., 2021).
AQPs are transmembrane channels that specifically transport water through their formation of an integral membrane protein. Totally, 13 AQP members (AQP0 to AQP12) have been found in mammals and amphibians. These proteins are tiny and intact membrane molecules with a molecular weight of about 30 kDa/monomer. There are currently 13 water-channel proteins that can be cloned, and they are found in every organ and gland in the body. The central nervous system contains five AQPs: AQP1, AQP4, AQP7, AQP9, and AQP11, with AQP1 and AQP4 having the greater concentration and AQP4 having the greatest concentration. The role of AQPs in CSF circulation, formation, and absorption of CSF, along with related mechanisms in cerebral edema and hydrocephalus, is being investigated.
Water is transported across primary ventricular fluids via AQPs: between CSF (containing ventricles and subarachnoid space), brain material (intracellular and extracellular), and intra-brain. CSF is a fluid found in the subarachnoid space and ventricles with a total volume of around 150 ml, of which 125 ml is in the substrate (Hodler et al., 2020). Net filtration and adsorption of water and solute in the interstitial space maintains CSF content (Papadopoulos and Verkman, 2013; Iliff et al., 2015; Nakada and Kwee, 2019). CSF enters the brain in the Virchow–Robin space of the para-arterial space and then is absorbed into the venous blood flow in the para-venous space (Iliff et al., 2013; Rasmussen et al., 2018). Whereas, water channels regulate water exchange, AQPs passively allow two-way water transport based on static water pressure and osmotic pressure (Papadopoulos and Verkman, 2013; Rasmussen et al., 2018).
AQP1 and AQP4 are the most abundant proteins in the mammalian central nervous system.
AQP1 is a 28 kD channel-forming integral protein widely distributed in the cell's plasma membrane in the form of tetramers, with each monomer (one AQP molecule) capable of generating a separate functional water channel. Because of its structural characteristics, AQP1 is highly permeable to water molecules, allowing 3,109 water molecules to flow through every second through a single AQP1 molecule. On the CSF side of the choroid plexus (CP), AQP1 is mainly expressed in epithelial cells and, together with carbonic anhydrase and the sodium pump on the basal side, plays a role in the production of CSF.
The most abundant aquaporin in the brain, spinal cord, and the optic nerves is AQP4, which regulates the brain's distribution and balance of water. It is found primarily in the astrocyte end-feet in the blood–brain barrier CSF–astrocyte boundary membranes, and ventricular membranes. As the most abundant water channel, AQP4 governs the water steady state. The AQP4 monomer is 30 kDa in molecular weight and consists of six cross-film spirals and two incomplete cross-film spirals. Each monomer contains a central water hole. AQP4 is expressed as a tetramer in the cell membrane. AQP4 is classified into two subtypes, M1 and M23, and its translation begins earlier than that of methionine M1 (323 amino acids) or methionine M23 (301 amino acids). A quadrangle composed of M23 forms a higher order structure known as an orthogonal array of particles, a crystal-like supermolecular component of a mass membrane (Wang et al., 2022). Although the biological significance of orthogonal arrays of particles has not been determined, it has been hypothesized to increase water permeability, alter cell–cell adhesion, and induce AQP4 polarization. Its highly efficient transmembrane and transcellular water transport channels contribute to the formation and removal of cerebral edema, as well as the generation and absorption of CSF. AQP1 and AQP4 are water channels located in the areas above and are associated with eliminating cerebral edema and hydrocephalus (Castañeyra-Ruiz et al., 2013).
In the current study, patients with iNPH had higher levels of AQP1 expression and lower levels of AQP4 expression, while AQP4 expression has grown with little polarization in chronic and age-related experimental models (de Laurentis et al., 2020).
AQPs may prove to be a useful diagnostic tool. The sole treatment option for hydrocephalus is treatment with a CSF shunt, but there is a risk of fatal complications (Raneri et al., 2017; Hodler et al., 2020). Thus, they are critical for diagnosing hydrocephalus. Diagnosis of iNPH remains challenging. However, detecting AQPs in CSF as biomarkers for identifying iNPH and separating these individuals from those with Alzheimer's disease is impossible, according to recent studies, at least given the detection levels afforded by commercial ELISA kits. AQP1 and AQP4 shedding into the CSF does not appear to occur in any of these illnesses, at least not at measurable quantities by ELISA (Hiraldo-González et al., 2021). A potentially useful method is to provide quantitative CSF samples during aspiration tests (de Laurentis et al., 2020). Although there are no extremely sensitive and specific CSF biomarkers that may predict the efficacy of a CSF shunt intervention, several studies have shown the potential of A42, p-tau, total tau, NfL, and LRG (Nakajima et al., 2021; Thavarajasingam et al., 2022). Additionally, AQPs may be used as therapeutic targets (Rizwan Siddiqui et al., 2018; Ding et al., 2019). Their exact activity enables alternating inhibition and activation of medicines, administered intrauterine or orally. Early intervention in the prodromal phase of iNPH was shown to aid strong cognitive and mobility function as well as social involvement capacity in the long-term experimental investigations. Its ability to maintain long-term cognitive function implies that it can help prevent dementia. Establishing an early diagnosis method for iNPH is desirable in order to provide early intervention for iNPH (Kajimoto et al., 2022).
It is critical to investigate and understand the role of aquaporin in the formation and maintenance of hydrocephalus to ascertain the causes of iNPH, leading to diagnosis and treatment. This article summarizes the current state of research on iNPH and findings on AQPs to identify future targets and research directions.
The pathogenesis of iNPH
The exact cause of iNPH is still unknown, despite the fact that various mechanisms have been theorized to contribute to its development. Previously, it was believed that ventriculomegaly and other neurological abnormalities in iNPH were first caused by a change in the dynamics of the CSF. A proxy for CSF pulsatility may be the aqueduct stroke volume, which is measured using phase-contrast magnetic resonance imaging (PC-MRI) and is defined as the average of the flow volume via the aqueduct during diastole and systole. Aqueduct stroke volume levels in patients with iNPH are higher than in healthy controls (Qvarlander et al., 2017; Yin et al., 2017). Reduced arterial pulsatility and decreased intracranial compliance may be related to increased CSF pulsatility. Under normal circumstances, pressure gradients cause the CSF in the subarachnoid space to be reabsorbed through the arachnoid granulations and brought up into the superior sagittal sinus (Bothwell et al., 2019). In individuals with iNPH, the normal CSF drainage is disturbed. The resistance to CSF outflow (Rout), which has been widely utilized to diagnose iNPH and identify candidates for shunt operations, is pathologically high in these individuals (Malm et al., 2011).
The bulk flow of CSF into the brain is facilitated by the recently discovered glial–lymphatic (glymphatic) pathway along the para-arterial space, the interstitial space, and ultimately into the paravenous space. Intact AQP4 channels, proper arterial pulsation, and proper sleep are all necessary for the glymphatic system to operate normally (Rasmussen et al., 2018). Individuals with iNPH have a compromised glymphatic system. Reduced clearance of neurotoxic chemicals like beta amyloid and hyperphosphorylated tau is a direct result of the glymphatic system disruption (HP tau) (Eide and Ringstad, 2019). Accumulation of these neurotoxic chemicals may cause astrogliosis and neuroinflammation in addition to impairing the physiological functioning of neurons (Angelucci et al., 2019).
In addition, aberrant CSF dynamics cause ventricular hypertrophy, which increases mechanical stress on the parenchyma and blood vessels, leading to hypoperfusion and subsequent hypoxia. Hypoperfusion may also cause a number of pathological abnormalities to brain tissue, such as gliosis, neuroinflammation, and impairments to the blood–brain barrier. They are each acknowledged for their contributions to INPH. In the sections that follow, we'll analyze it in view of CSF overproduction and discharge disorder and investigate how AQP1 and AQP4 may be involved.
AQP1 and hydrocephalus
The polarized expression of AQP1 in humans has also been demonstrated to play a role in CSF secretion. Existing experimental research has demonstrated a strong correlation between the development of hydrocephalus and the overexpression of AQP1. There is still a need for additional research despite evidence in iNPH patients, but numerous investigations have consistently shown that AQP1 controls water molecule distribution. The apical membrane of choroid plexus epithelial cells displays a significant amount of AQP1 expression. CSF production can be decreased by down-regulating AQP1 in the apical membrane. Therefore, specific AQP1 inhibition or down-regulation and strengthening of this beneficial negative feedback can further limit CSF secretion, reduce intracranial pressure, and postpone the pathological progression of hydrocephalus, which may also be a promising therapeutic target.
Regulation of AQP1 on CSF circulation
AQP1 is predominantly present on the apical membrane of plexus epithelial cells (on the CSF side), with a tiny amount on the outside (near the side of the blood vessel). CSF is produced and secreted by CP epithelial cells. Some cells of the chamber membrane can also exude CSF in trace amounts, although the majority is produced by the CP epithelial cells. Regarding its transcellular transport of water molecules during CSF formation, the apical AQP1 has a relatively wide distribution. The synergy between Na, K-ATPase and AQP1 in the CP is presently the critical molecular pathway for water transfer in the ventricle and CSF creation model. Because AQP1 in the apical membrane allows water to flow in the direction of the permeation gradient, it is vital to maintain the permeability of the apical membrane. Reduced AQP1 expression on the CP's surface may account for how water molecules pass through the membrane of transmembrane CP cells and into the ventricle where they create CSF and cause hydrocephalus. According to the study, AQP1's polarized expression pattern is also involved in human CSF generation. AQP1 has been found in astrocytes surrounding cerebral arteries in various neurological disorders, including multiple sclerosis, traumatic brain injury, cerebral infarction, epilepsy, viral infections, and Alzheimer's disease. Healthy CSF contains AQP1, whereas patients with neuro-inflammatory diseases such as bacterial meningitis have elevated AQP1 levels (Blocher et al., 2011). Additionally, AQP1 is located in the blood–brain barrier, which may explain why AQPs are implicated in cerebral edema clearance.
Verkman (2008a) found that AQP1 knockout animals had a 20% decrease in CSF fluid production and a 56% decrease in intracranial pressure compared to wild-type mice. The study discovered that when the AQP1 gene was deleted, aquaporin expression in choroid plexus epithelial cells and CSF secretion decreased in mice, AQP expression in the nephropathic tube decreased, because AQP1 relies on decreased cross-cell reabsorption of primary urine, which results in decreased blood volume, sinus pressure, and CSF reflux. Smith et al. (2007) described an instance of communicating hydrocephalus. CP immunohistochemistry showed that the expression of AQP1 on the apical membrane of the choroid plexus epithelial cells of the ill choroid plexus was lower than that of normal children of the same age.
In conclusion, when hydrocephalus occurs, AQP1 on the to apical membrane of the choroid plexus epithelial cell enters the cytoplasm via endocytosis, decreasing CSF production and secretion and compensating for the decreased CSF secretion to maintain relative intracranial pressure stability, which may be a protective mechanism of the body.
However, the efficacy of this stabilizing mechanism is not without defects, as demonstrated by Jeon et al. (2017), who found (Rasmussen et al., 2018) protein in the CP of acute hydrocephalus rats using a kaolin-induced hydrocephalus paradigm. They reported abrupt reduction of AQP 1 protein expression in the CP of rats with acute hydrocephalus, increasing at later stages, following the same trend as AQP1 mRNA expression. The quick decline in AQP1 expression during the early stages of hydrocephalus is consistent with a decrease in CSF compensatory production, corroborating our theory. On the other hand, later expression levels in the experimental group were considerably higher than in the control group. The researchers believed that this was related to additional mechanisms of CSF secretion, such as neuroendocrine function (AQP1 expression may be enhanced in response to stress-induced neuroendocrine regulators) (Paul et al., 2011). Additionally, this late upregulation of expression must be confirmed. Increased expression of CP AQP1 may increase the number of water channels activated by an atrial natriuretic factor or other cGMP-producing factors in late hydrocephalus, hence decreasing CSF generation. Alternatively, cGMP-producing substances may impede the choroidal Na, K-ATPase action, resulting in decreased fluid secretion (Eide and Ringstad, 2019).
The failure of the stabilizing mechanism, which causes an increase in cerebral fluid and further aggravates stagnant fluid, may be the cause of the later increase in expression. When Wang et al. (2011) examined the brain tissue of kaolin-injected mice to create an animal model of hydrocephalus, they revealed that a substantial number of AQP1 immune granules were present in the apical membrane of CP epithelial cells in the control group. This immunological granule is normally only found in the mitochondria in the cytoplasm, whereas in the experimental group, AQP1 was abundant in both the cytoplasm and the cell membrane. Apart from mitochondria, the cytoplasm contained AQP1 immune gold granules such as vesicles and lysosomes, and almost half of AQP1 entered the cytoplasm via the cytomembrane. Simultaneously, when AQP1 gene knockout animals were compared to wild-type mice, it was revealed that the AQP1 gene knockout mice's baseline ventricular area was much lower than that of wild-type mice. When lateral ventricle injection of kaolin was used to induce hydrocephalus formation, AQP1 mice in the gene deletion group exhibited a 30% reduction in ventricle dilation, a faster weight recovery following surgery, and significantly fewer side effects such as aberrant behavior.
AQP1 in iNPH
There is a dearth of studies on AQP1 markers in patients with iNPH, and Castañeyra-Ruiz et al. (2016) found no significant increase in AQP1 levels in CSF compared to a control group. However, there were fewer cases, and in four of the six cases studied, these levels were significantly higher than those of ten control subjects, and all samples in the first quartile were controls, indicating a significant relationship between controls and low AQP1 values.
The apical membrane of choroid plexus epithelial cells expresses a high level of AQP1, and down-regulation of AQP1 in the apical membrane can result in decreased CSF production. Additionally, AQP1 can decrease blood volume and intracranial pressure by inhibiting primary urine reabsorption in the proximal convoluted tubule. These factors justify further investigation of AQP1 as a potential novel target for the treatment of hydrocephalus. Thus, selective inhibition or down-regulation of AQP1 expression, combined with enhancement of this protective negative feedback loop, can further reduce CSF secretion and intracranial pressure and delay the pathological process of hydrocephalus (Figure 1).
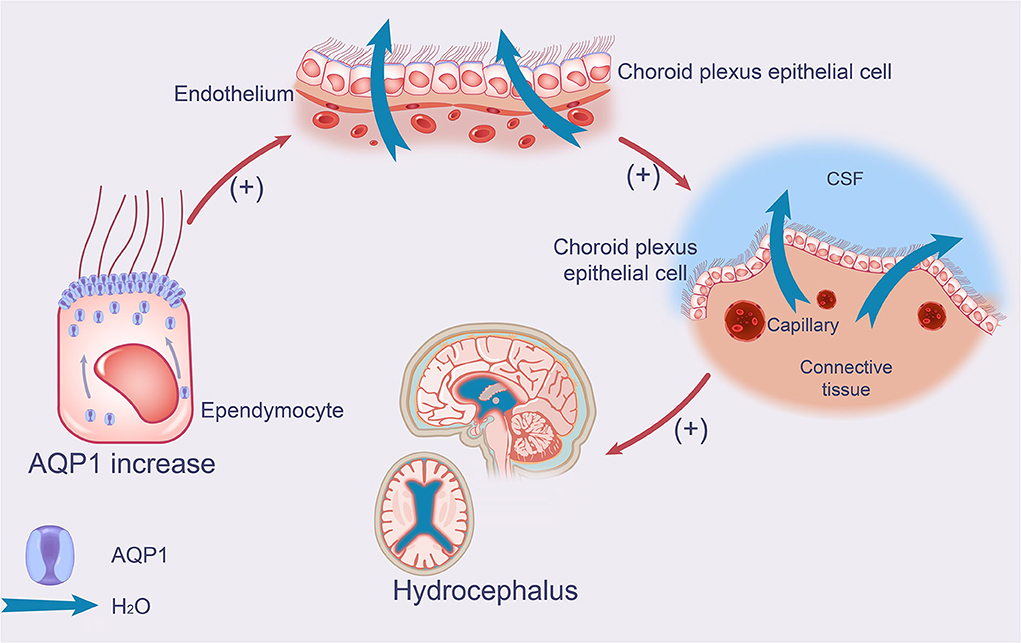
Figure 1. Effect of AQP1 on production of CSF in iNPH. The schematic drawing demonstrates increases in AQP1 and its function in iNPH, which leads to increased CSF production. AQP1 increases the production and secretion of CSF in the apical membrane of choroid plexus epithelial cells by accumulating in the ependymal membrane of ependymal cells. AQP1, aquaporin 1; iNPH, idiopathic normal pressure hydrocephalus; CSF, cerebrospinal fluid.
The expression and regulation mechanism of AQP1
The nervous system contains a high concentration of AQP1, and various variables regulate its expression, including hormones, osmotic pressure, hypoxia, ischemia, and medications. Similar to the regulation of other proteins, cell signal transduction plays a critical role in the regulation of AQP1 expression. Numerous influencing factors can modulate AQP1 expression by activating or inhibiting the appropriate transduction pathway (Wang C. et al., 2021).
Activity regulation of AQP1
Phosphorylation is the most common approach for altering the function of proteins at the protein level. This also applies to AQP1, as protein phosphorylation affects AQP transport, control, and redistribution. As a result, phosphorylation is a critical strategy for modulating AQP1 function. The structural properties of AQP1 demonstrate that the nap structure, ar/R region, and sections of amino acid sequence or domain segments in the center hole all contribute to the protein's transport function; changes to these structures can directly or indirectly modify its permeability. AQP1 comprises four potential phosphorylation sites in humans and mice: S236-PKA phosphate sites, T159/T239-PKC phosphorylation sites, and S262-CKII phosphorylation sites (Zelenina, 2010). There is an additional PKC phosphorylation site in the human AQP1 amino acid sequence that CKI can phosphorylate. Additionally, Cys189 in AQP1 is a Hg2+ binding site, whereas Tyr186 is a TAE binding site, with the former limiting AQP1's water and ion permeability and the latter inhibiting its water permeability.
Several studies have demonstrated that PKA can phosphorylate AQP1 in vitro and that activators of PKA, such as cAMP and forskolin, can increase AQP1's water permeability (Smith et al., 2007). Vasopressin can enhance the water permeability of AQP1-transfected oocytes, and co-incubation and injection of 8-Bromo-cAMP into cells can also increase water permeability, which is hypothesized to occur via the cAMP pathway (Zelenina, 2010). Sakurada et al. (2004) demonstrated that 5% glucose enhanced AQP1 membrane transfer via the PKA pathway, which PKA-specific inhibitors could inhibit. However, several investigations have indicated that PKA does not promote AQP1 permeability in vitro (Wang et al., 2011). Sakurada et al. (2004) and Oshio et al. (2005) found that forskolin may increase CSF in wild-type mice and AQP1−/− mice. It has not been established conclusively that PKA-regulated AQP1 phosphorylation occurs in the brain.
For PKC pathways, Zhang et al. (2007) showed that the water and ion permeability of AQP1 is regulated by PKC's short-term activity, involving two phosphorylation sites, T159 and T239. The most recent study discovered that, in addition to phosphorylation regulation, AQP1 could also be regulated by ubiquitin.
Chemical blockers of AQP1 that are permeable to water are constantly being developed, Hg2+ and TAE are constrained by their toxic effects, and recent studies have demonstrated that the Na+-K+-Cl-co-transport blocker and medullary loop diuretic Bumetanide has a slight inhibitory effect on the water permeability of AQP4. Furthermore, the body can tolerate a high amount (100 mg/kg). This finding paved the way for the development of further molecular compounds based on the structure of Bumetanide. According to Kim et al. (2010), the aryl-sulfonamide AqB013 significantly reduced the water permeability of AQP1 and AQP4, although its therapeutic potential remains unknown.
Gene regulation of AQP1
Changes in the external environment can affect the expression of the AQP1 gene (e.g., high seepage, low oxygen, etc.). AQP1 mRNA expression increased 2 fold in a high seepage environment (220 mM NaCl) but decreased in a low seepage environment (90 mM NaCl) (Bouley et al., 2009). Wang Feng et al. used primary culture of adult rabbit lumbar myelin nuclear cells, immunohistochemistry, and real-time quantitative PCR detection, and found significantly increased AQP1 expression in cells, which may be attributed to the increased expression of AQP1 to enhance oxygen delivery. 2-methoxyestradiol can inhibit hypoxia inducible factor-1α (HIF-1α) and vascular endothelial growth factor, hence influencing the expression of AQP-1.
This signal transduction pathway is critical for the regulation of AQP1 expression. In one study, Schwann cells inflated and AQP1 expression increased in response to hypoxia and HIF-1α may play a role in this process (Zhang et al., 2013), implying that the cGMP pathway regulates AQP1 expression. Ding et al. (2013) mimicked cardiopulmonary reflux using the guanosine cyclase inhibitor ODQ as a control. Compared to the control group, AQP1 mRNA expression fell by 39.7%, indicating that the cGMP pathway regulates AQP1 expression. McCoy and Sontheimer (2010) studied the expression of AQP1 following injury to active astrocytes using epidermal sting tests and discovered that the MEK1/2 pathway inhibitor U0126 might inhibit AQP1 expression. It was further demonstrated that cytokinin activates the protein kinase signaling pathway, inducing AQP expression. Experiments have also verified regulatory effects of JNK (Tie et al., 2012) and related pathways.
In both flanks of the AQP1 gene, a substantial number of cis-regulatory elements have been identified. Although they do not encode any proteins themselves, they are critical for gene regulation. In conjunction with trans-acting factors, these cis-regulatory elements govern the transcription and translation of AQP1 at the gene level. In the promoter region of AQP1, Moon et al. (1997) identified two glucocorticoid response elements. Steroid hormones can activate this response element and increase AQP1 expression in murine leukemia cells.
Additionally, Stoenoiu et al. (2003) discovered that a high dose of corticosteroid (1.5 mg/kg) could stimulate AQP1 mRNA and protein expression in the peritoneum of rats. Simultaneously, the glucocorticoid receptor inhibitor Ru-486 worked on the glucocorticoid receptor and inhibited corticosteroid production. de Arteaga et al. (2011) discovered that high-dose corticosteroid therapy could increase the water level in patients undergoing peritoneal dialysis. These corresponding changes indicate that the glucocorticoid receptor and glucocorticoid response elements in the AQP1 promoter region are intimately related to AQP1 expression.
Translocation of AQP1
Owler et al. (2010) induced hydrocephalus in adult mice by injecting kaolin into the medullary cistern. In the experimental group, AQP1 was detected in the cytoplasm of CP epithelial cells, indicating a shift in the translocation of AQP1. Caveolin is a subunit of the membrane lipid cutting architecture that selectively transfers membrane proteins. It has been suggested that it is closely related to AQP1 translocation (Tie et al., 2012). Kobayashi et al. (2006) extended Western blotting findings to establish co-expression of AQP1 and caveolin in lipid cutting, and AQP1 localization indicated that AQP1 translocation might be associated with caveolin. Tietz et al. (2006) cultured bile duct epithelial cells in vitro, demonstrating that actin and microtubules may be involved in AQP1 translocation.
AQP4 in hydrocephalus
It has been gradually understood that AQP4, a crucial component of CSF transport and interstitial fluid clearance, plays a role in iNPH in the impairment of CSF circulation, astrocyte hyperplasia, and impaired lymphatic circulation in the perivascular space and brain parenchyma. This leads to CSF accumulation, an inability to efficiently remove metabolic waste, and neurodegenerative changes. A promising prospective therapeutic target, AQP4 plays a variety of roles in iNPH. Contrary to other types of hydrocephalus, the expression of AQP4 shows a decrease in iNPH, which may allow treatment routes that affect AQP4 activation to have a more noticeable effect in iNPH than in other types of hydrocephalus (Table 1).
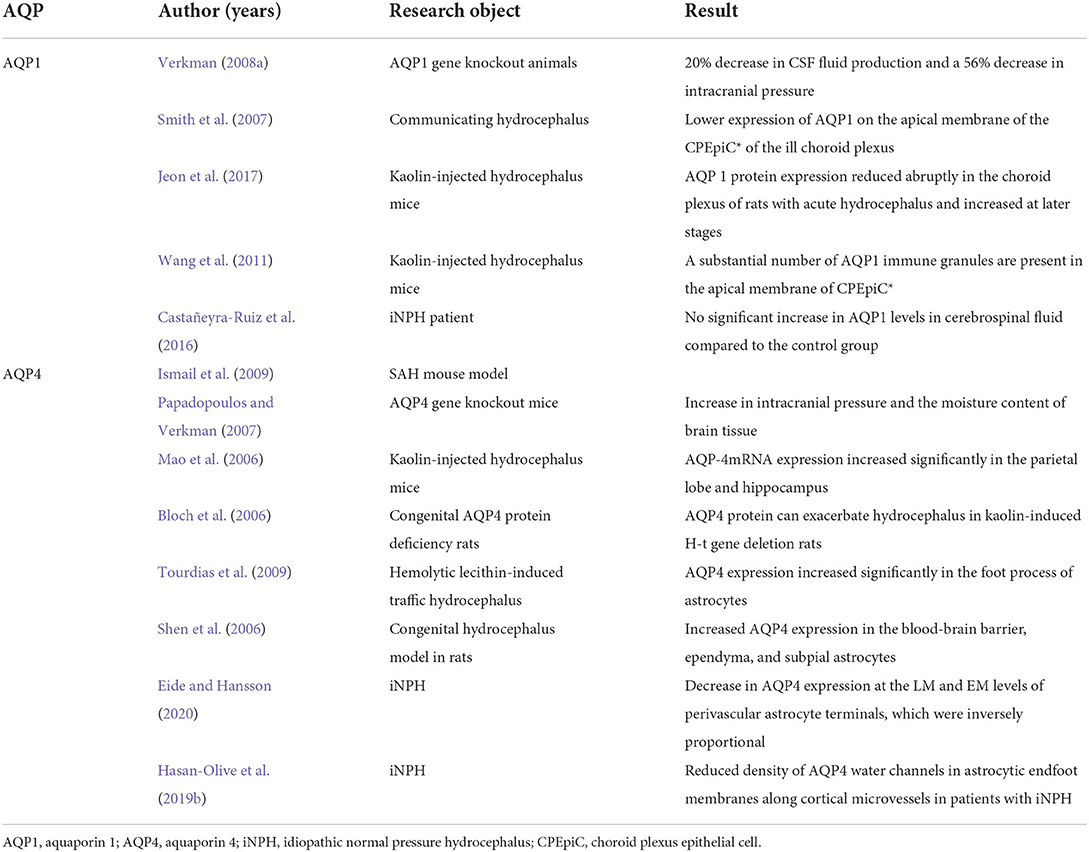
Table 1. Main studies investigating AQP1 and AQP4 in humans.
Regulation of AQP4 on CSF circulation
As another vital member of the water channel family, AQP4 is the most abundant aquaporin subtype in the central nervous system. It is primarily found in the protrusion membrane, glial boundary membrane, and ependyma of the foot processes of astrocytes surrounding capillaries, where it regulates the flow of water into and out of the brain via the CSF and blood. It exerts a considerable regulatory influence on CSF production and absorption (Hara-Chikuma and Verkman, 2008a,b; Verkman, 2008b). In cytotoxic cerebral edema caused by injuries such as water poisoning, cerebral infarction, or meningitis, AQP4-deficient mice had a better prognosis than wild-type mice (Verkman, 2008b). AQP4 deficiency bypasses the whole blood-brain barrier, hence lowering cytotoxic brain edema. In vascular cerebral edema mouse models such as cortical freezing, brain tumors, and intracranial rehydration fluid, AQP4-deficient animals demonstrated significantly more brain swelling than wild-type mice, most likely because AQP4 in wild mice expedited the outflow of water from edema tissue. Additionally, in hydrocephalus, AQP4 regulates CSF absorption (Kusayama et al., 2011). Matthew et al. established the first mouse model of subarachnoid hemorrhage by injecting autologous blood into the basal cistern, simulating how high-pressure blood enters the CSF in human subarachnoid hemorrhage. The results indicated that AQP negative mice had more evident brain swelling and a poorer clinical prognosis than controls, primarily because AQP4 encouraged the evacuation of excess water in brain tissue via the compromised blood–brain barrier (Ismail et al., 2009).
Additional evidence linking AQP4 protein to CSF production and absorption has been revealed. Papadopoulos and Verkman (2007) observed an increase in intracranial pressure and the moisture content of brain tissue in gene knockout mice. When AQP4 knockout mice were compared to wild-type mice, they had significantly reduced water penetration and a larger ventricular volume, implying that AQP4 is required for cross-substantive absorption of CSF. AQP4 demonstrates its impact on the occurrence and progression of hydrocephalus in a number of animal models of the condition. Mao et al. (2006) discovered that AQP4 mRNA expression increased significantly in the parietal lobe and hippocampus when kaolin-induced hydrocephalus was studied in mice. This could be because peripheral AQP4 expression is more easily activated in the brain's parietal lobe, and the parietal lobe and hippocampal regions have a lower water content than other regions. Bloch et al. (2006) also discovered that AQP4 can exacerbate hydrocephalus in kaolin-induced H-t gene deletion rats (congenital AQP4 protein deficiency rats) by decreasing the rate of extracellular fluid clearance across the blood-brain barrier, indicating that AQP4 plays a beneficial role in increasing the blood-brain barrier clearance rate and preventing the accumulation of excess water in the ventricles. Tourdias et al. (2009) calculated the apparent diffusion coefficient in rats with hemolytic lecithin-induced traffic hydrocephalus. After 2 weeks, it was discovered that AQP4 expression increased significantly in the foot processes of astrocytes and that there was a significant correlation between AQP4 levels, apparent diffusion coefficient, and ventricular volume. Shen et al. (2006) used the congenital hydrocephalus model in rats and discovered that 8-week-old rats had increased AQP4 expression in the blood–brain barrier, ependyma, and subpial astrocytes. Additionally, it was hypothesized that increased AQP4 expression might be a compensatory response to CSF absorption in static hydrocephalus. In normal circumstances, CSF circulation is hampered after hydrocephalus. AQP4 expression rises as a result of compensatory trans-parenchymal absorption of CSF, and accumulating CSF is absorbed into the circulation via an AQP4-dependent transcellular pathway.
AQP4 in iNPH
In iNPH, the situation is substantially different, possibly due to a failure of the compensating process or a weakness in specific CSF linkages. Although it is commonly believed that AQP4 levels are elevated in patients with hydrocephalus, it was found that they decreased in iNPH (Eide and Hansson, 2018, 2020; Arighi et al., 2019; Hasan-Olive et al., 2019a,b). Eide and Hansson (2020) found a significant increase in the percentage of fibrin (fibrinogen) areas in iNPH cerebral cortex exosmosis. At the same time, AQP4 expression was decreased in the vicinity of blood vessels. Immunohistochemistry revealed a decrease in AQP4 expression at the light and electron microscopy levels of perivascular astrocyte terminals, which were inversely proportional.
In the research of Hasan-Olive et al. (2019b), AQP4 immunogold cytochemistry study of cortical brain samples from 30 patients with iNPH and 12 control participants revealed lower AQP4 density in the perivascular astroglial end-feet of patients. In brains from individuals with aneurysms, epilepsy, and cancer, astroglial AQP4 density facing the neuropil remained unaltered, as did perivascular AQP4 density. This link could point to a role for inflammation-induced AQP4 depolarization in iNPH pathogenesis, in which reduced perivascular AQP4 expression limits glymphatic fluid flux, increasing beta amyloid buildup and ventriculomegaly. In iNPH, the expression of AQP4 is decreased in the vicinity of blood vessels, resulting in issues with lymphatic circulation, waste disposal, and neurodegeneration (Hasan-Olive et al., 2019b).
There is no apparent explanation for why AQP4 expression differs so much between common hydrocephalus and iNPH. Recent research suggests that this phenomenon may be linked to inflammation. It was related to lymphatic malfunction in the early stages of inflammation by Reeves et al. (2020). In fact, investigations in both humans (Sosvorova et al., 2015) and animals (Sosvorova et al., 2015; Chaudhry et al., 2017) have identified a connection between perivascular space inflammation and the development of iNPH. In iNPH, the neuroinflammatory response is highly noticeable (Lolansen et al., 2021), and the level of inflammatory cytokines indicated by TNF in the CSF fluid is dramatically altered (Wang et al., 2020). However, we can demonstrate that AQP4 plays a role in CSF transport and interstitial fluid clearance, and that its reduction in iNPH has an effect on CSF circulation and lymphatic metabolic abnormalities.
Effect of AQP4 on the hyperplasia of astrocytes
The AQP4 protein is favorably linked with astrocyte development. Tomas-Camardiel et al. (2005) described an intracerebroventricular injection of lipopolysaccharide as a model of hemorrhagic brain edema. It was revealed that activated astrocytes expressed a high quantity of AQP4. Saadoun et al. (2009) discovered that pricking the cortex decreased astrocyte growth in AQP4 rats with congenital gene deficits, indicating that AQP4 promoted astrocyte proliferation. Zhou et al. (2008) also discovered that the absence of AQP4 caused a significant decrease in the expression of GFAP in the surrounding brain microvessels when examining rats with congenital AQP4 deficits, and GFAP was positively linked with the degree of astrocyte proliferation.
GFAP interacts closely with membrane anchoring proteins, allowing GFAP fibers to pull the membrane back when they retract in response to changes in signaling molecules. That is, as GFAP fibers retract, squeezing water out of the cytoplasm of the processes, the interactions between the anchor proteins and GFAP fibers cause volemic reduction (Wang and Hatton, 2009). The expression of volume-regulating proteins and their interactions with membrane anchoring proteins determine the volemic influence on GFAP's guiding role. AQP4 expression is synchronized with the levels and spatial distribution of GFAP, as expected for a volume-regulating protein (Wang and Hatton, 2009; Li et al., 2020). Furthermore, Wang X. et al. (2021) found TGN-020's ability to prevent GFAP retraction surrounding vasopressin neurons during hyposmotic challenge (20 min) and hyperosmotic stress suggesting that AQP4 plays a role in GFAP retraction. AQP4 is functionally connected with several ion channels and transporters, and its opening for water flux is accompanied by changes in ion concentration in astrocytes, which might affect GFAP metabolism and assembly, as previously discussed. By coupling with transient receptor potential vanilloid 4, Benfenati et al. (2011) and Stokum et al. (2018) discovered that AQP4 can increase intracellular Ca2+; however this coupling is not required (Mola et al., 2016). Increased intracellular Ca2+ levels are required for GFAP depolymerization via protein kinase A and other signaling pathways (Petković et al., 2017), suggesting that AQP4 might reduce GFAP filament extension.
However, astrocyte growth also contributes to alterations in brain tissue function after hydrocephalus. When hydrocephalus occurs, the ventricle system extends the ventricular wall due to the accumulation of CSF. Astrocyte proliferation also occurs surrounding the ventricle, creating compensatory enlargement of the ventricle, causing CSF extravasation into the brain tissue around the ventricle, causing brain white matter edema and white matter tissue deterioration. When the CSF content is further increased, the elasticity of the brain tissue reduces due to compression, the cerebral cortex gets thinner, leading to brain atrophy, and these pathological changes result in neurological dysfunction, indicating that AQP4 stimulates GFAP and astrocyte proliferation, which may be the source of the changes in neurological function following hydrocephalus.
In patients with iNPH, Eide and Hansson (2018, 2020) detected nerve cell degeneration, astrocyte proliferation, and decreased expression of AQP4 and Dp71. In comparison to reference patients, cortical biopsies of individuals with iNPH revealed increased astrocyte proliferation and lower expression of AQP4 and Dp71 in astrocyte perivascular terminals in the foot and certain surrounding neural canals. The degree of astrocyte proliferation was substantially linked with a decrease in AQP4 and Dp71 levels in the astrocytes' perivascular terminal feet. Additionally, all patients with iNPH have bulging nerve cell bodies and neurodegeneration. While astrocytosis is associated with activated macrophages and microglia, the inflammatory marker CD68 did not differ between iNPH and REF. Dp71 is the major dystrophin subtype in the brain, connecting the cytoskeleton, cell membrane, and extracellular matrix dystrophin-related protein complex (DAPC) to anchor AQP4 and ion channels, with a preference for the lipid raft surrounding the astrocytes' terminal foot. It is required for adequate fluid transport between the cerebrovascular system, the interstitial fluid space, and astrocytes. AQP4 appears to be polarized toward the terminal foot of perivascular astrocytes in response to the components of the DAPC (Nicchia et al., 2008; Enger et al., 2012; Camassa et al., 2015). As Eide and Hansson (2018) previously established, dystrophin deficiency causes instability in the interface between the cytoskeleton and the mass membrane, impairing signal transduction. Experiments causing the loss or displacement of AQP4 or DAPC can result in aberrant brain water flux and disruption of intermediate metabolism, which can eventually result in the slow development of neurodegeneration (Nagelhus and Ottersen, 2013; Pavlin et al., 2017). Patients with iNPH who reacted to CSF fluid shunts had aberrant intracranial throbbing pressure, indicating impaired compliance. The primary histopathological findings were the proliferation of astrocytes and the subsequent drop of AQP4 and Dp71 at the astrocytes' perivascular terminal foot. According to Eide, alterations in the AQP4 and Dp71 complexes are related to sub-ischemia, often in tissue (Eide and Hansson, 2018).
Astrocytosis may contribute to the decreased intracranial compliance (pressure-volume reserve capacity) observed in patients with iNPH, as indicated by higher pulse wave intracranial pressure (Eide and Sorteberg, 2010, 2016; Eidsvaag et al., 2017). Mechanical resistance and hardness are known to be significantly increased by iNPH (Lu et al., 2011). Glia, including astrocytes, account for more than half of the total number of cells and brain volume in adults (Zhan et al., 2020). Fibrin (fibrinogen) has been shown to excite astrocytes and small astrocytes, implying that they may contribute to the progression of neurodegenerative changes and cognitive deficiencies (Figure 2) (Cortes-Canteli et al., 2010; Schachtrup et al., 2010; Merlini et al., 2019).
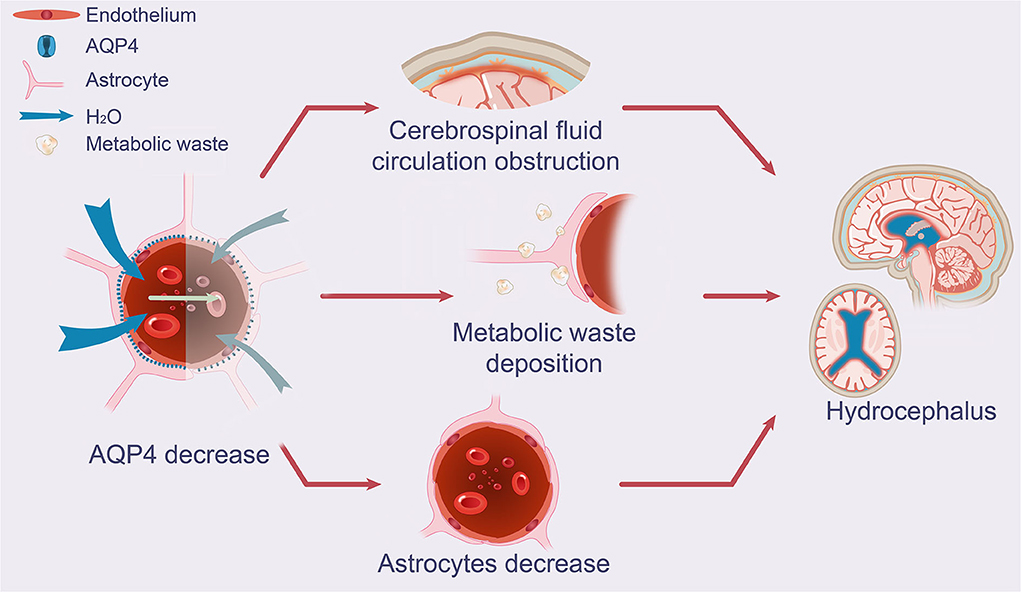
Figure 2. Effect of AQP4 on potential glymphatic dysfunction and growth of astrocytes in iNPH. The schematic drawing demonstrates decreases in AQP4 and its function in iNPH, which leads to diminished CSF periarterial inflow, CSF-ISF exchange, and perivenous outflow of CSF mediated by AQP4 depolarization, as seen in patients with iNPH. Increased concentrations of neuronal metabolic waste products in the brain interstitial space result from the decreased exchange of CSF and ISF. A decrease in AQP4 in the end-feet of astrocytes near arteries was substantially associated with astrocyte growth and CSF circulation obstruction. AQP4, aquaporin 4; iNPH, idiopathic normal pressure hydrocephalus; CSF, cerebrospinal fluid; ISF, interstitial fluid.
Effect of AQP4 on lymphatic system drainage disorder
In contrast to other types of hydrocephalus, the decrease in AQP4 in iNPH and Alzheimer's disease is related to the progressive loss of perivascular units responsible for water exchange (Reeves et al., 2020). This finding may indicate a different sort of hydrocephalus, which would account for the normal intracranial pressure and low AQP4 levels. Indeed, there may be no loss of brain parenchyma in traffic hypertension hydrocephalus but rather an accumulation of CSF due to constriction of the perivascular space (Blasco et al., 2014; Schmidt et al., 2016). As a result, as intracranial pressure increases, AQP4 levels may rise, first in the blood, then in the CSF (Castañeyra-Ruiz et al., 2013; Blasco et al., 2014). On the other hand, iNPH does not result in an increase in intracranial pressure despite the increasing buildup of amyloid beta, loss of brain parenchyma, and progressive lymphatic system dysfunction. Brain atrophy could be the cause. Hence, patients with iNPH may have low AQP4 concentrations in their CSF due to a lack of AQP4 expression and the loss of functional vascular units (Eide and Hansson, 2018, 2020; Arighi et al., 2019; Hasan-Olive et al., 2019a,b).
In rodents, AQP4 deficiency results in water build-up in the brain parenchyma (Haj-Yasein et al., 2011; Vindedal et al., 2016), decreased perivascular cerebrospinal and interstitial fluid circulation, and poor amyloid beta clearance (Iliff et al., 2012). These findings are equally perplexing as those regarding the blood–brain barrier, as AQP4 defective mice demonstrate normal blood–brain barrier integrity (Saadoun et al., 2009; Eilert-Olsen et al., 2012). As a result, a decrease in AQP4 surrounding blood vessels may directly affect the flow of fluid around blood vessels and the brain's steady state.
According to MRI studies, individuals with iNPH showed decreased lymphatic function, higher accumulation, and a reduced clearance rate for the intrauterine injectable contrast agent gadobutrol (Ringstad et al., 2017; Eide and Ringstad, 2019). MRI revealed reduced vascular side gaps and lymphatic circulation in patients with iNPH (Eide and Ringstad, 2019). Gadobutrol circulates through the brain's large soft meningeal arteries. In individuals with iNPH, cerebral blood flow perfusion and brain metabolic levels were decreased (Eide and Stanisic, 2010; Calcagni et al., 2013; Ziegelitz et al., 2016).
Hasan-Olive et al. (2019b) confirmed for the first time that AQP4 expression in patients with iNPH dropped dramatically in astrocytic end-foot membranes facing capillary endothelial cells than in end-foot membranes facing neuropil. Reduced AQP4 around blood vessels may impair fluid and lymphatic circulation in the brains of patients with iNPH. Given the strategic location of the AQP4 water channel between the blood and the interstitial fluid, it is fair to assume that a CSF shunt will improve liquid flow through the AQP4 water channel. This increases the availability of nutrients and oxygen, as well as waste elimination, in the brains of patients with iNPH (Tan et al., 2021).
The expression and regulation mechanism of AQP4
Numerous studies have been conducted to elucidate the numerous endogenous mechanisms of AQP4 control, as well as the potential for pharmacological modulation. Four distinct regulation mechanisms have been extensively studied: microRNA (miRNA)-mediated regulation of gene expression, phosphorylation-mediated regulation of AQP4 channel gating/transport, heavy metal-ion-mediated regulation of water permeability, and small molecular inhibitor-mediated regulation of water permeability.
MiRNA targeting of AQP4 has become an area of research and development in AQP4 regulation (Gomes et al., 2018). The miRNA is an endogenous single-stranded RNA sequence obtained from most of its inclusions that function primarily in the degradation or inhibition of mRNA translation (Eide and Sorteberg, 2010, 2016). Recently, numerous studies have established a relationship between specific miRNAs and therapeutic AQP4s (miR-224/miR-19a, miR-29b, miRNA-145, miRNA-320a, miRNA-130a, and miRNA-130b).
Phosphorylation is an effective post-translational modification method. It can modulate protein function or expression by influencing protein structure, protein-protein interaction, and protein transport labels (Nesverova and Tornroth-Horsefield, 2019). Phosphorylation of AQP4 is required for its transport and subcellular localization and may be involved in channel gating (Kadohira et al., 2008; Nesverova and Tornroth-Horsefield, 2019). While the majority of recent research has been on S111, S180, and S276, there are still more putative phosphorylation sites, including S285, S315, S316, S321, and S322, whose function is unknown (Lundby et al., 2012; Nesverova and Tornroth-Horsefield, 2019). However, tests in oocytes utilizing mutant rAQP4 protein showed that phosphorylation of the COOH terminal serine residues S315, S316, S321, and S322 did not affect transport or channel gating (Assentoft et al., 2014). The S315 phosphorylation location discovered in rats is not conserved in human AQP4 (Lundby et al., 2012) as serine is converted to glutamine. The effect of phosphorylation on AQP4 remains an intriguing area of research, with numerous processes and routes yet unknown.
The Kidins220–SNX27–retromer–AQP4 pathway was discovered as a regulatory mechanism for brain AQP4 expression and its participation in brain ventricular enlargement and hydrocephalus by Puerto et al. We detected an unexpected relationship between SNX27-retromer downregulation and AQP4 lysosomal degradation caused by the loss of Kidins220, as well as a dramatic drop in Kidins220 and AQP4 expression at the ependymal barrier in patients with iNPH (Del Puerto et al., 2021).
Mercury is at least one of the metals that regulates the majority of AQPs (Ximenes-da-Silva, 2016; Abir-Awan et al., 2019). It inhibits practically all AQPs except AQP6 and acts predominantly on AQPs (Yasui et al., 1999). Experiments with protein liposomes demonstrated that the Hg2+ binding site on AQP4 is located within the cell, which means that Hg2+ cannot reach its direct binding site on AQP4 in oocytes or most cell models and, thus, cannot regulate its activity (Yukutake et al., 2008; Yukutake and Yasui, 2010). On the other hand, the mercury ion can covalently attach to the C178 residue of the D ring of rAQP4's intracellular domain, exposing the Hg2+ binding site to the external solution and perhaps causing AQP4's conformational change (Yukutake et al., 2008; Ximenes-da-Silva, 2016). In vivo, however, the effect was not inhibition but a significant increase in AQP4 protein production, most seen in reactive astrocytes.
Additionally, zinc and copper were found to be inhibitors of rAQP4 in protein liposomes (Yukutake et al., 2009). Not all metal ions that affect AQP4 activity operate directly; some act indirectly by initiating phosphorylation pathways, leading to AQP4 expression or water permeability changes. These characteristics are shared by other materials such as lead (Gunnarson et al., 2005), manganese (Rao et al., 2010), and copper (Qing et al., 2009).
A small-molecule inhibitor is a compound that inhibits AQP4 without the need for metal ions. It is critical to identify small molecule AQP4 antagonists as AQP4 activity or expression is associated with various illnesses, and small molecule inhibitors can inhibit AQP4 without generating metal ion toxicity. Experimental verification tests have been performed on acetazolamide, validic acid, topiramate, ethoxzolamide, zonisamide, phenytoin, lamotrigine, and sumatriptan, among other drugs.
The methodological diversity is one of the most perplexing features of AQP4 regulation. Although in vitro techniques vary considerably, various miRNAs, heavy metal ions, and two small molecule inhibitors, acetazolamide and TGN-020, all demonstrate promise in regulating AQP4.
Conclusions
While the pathogenesis of iNPH remains a complicated subject, AQPs appear to be a feasible avenue for treatment. As AQP4 and AQP1, which have been studied for over a decade, are highly expressed in brain tissue and participate in the pathophysiological processes underlying related brain diseases, their critical role in maintaining the balance of brain tissue water in the nervous system and CSF circulation is undisputed. Numerous experimental studies have corroborated the pattern of increased AQP1 and decreased AQP4 protein expression in iNPH (Table 2).

Table 2. AQP1 and AQP4 in iNPH.
AQP1, through its polar distribution, mainly plays a role in promoting CSF production, maintaining the balance of hydrocephalus, and participating in pathological processes. When CSF levels are elevated in iNPH, defective internal protective mechanisms permit AQP1 to continue to play a role in boosting generation, suggesting that we can govern linked brain illnesses by regulating AQP1. In the three levels of AQP1 regulation, the most effective control is the translation level of transcription adjustment and AQP1 itself. Transposing during the pathologic process complicates its management. Thus, in the future, we will require a more accurate understanding and in-depth investigations of the regulation mechanisms of AQP1 expression to develop a more effective method for AQP1 regulation. At the same time, the mechanisms causing changes in its polar distribution requires further investigation; the causes of abnormalities in AQP1-related protection mechanisms—the mechanism of iNPH occurrence—from a more comprehensive perspective are necessary for a deeper level understanding essential for the diagnosis and treatment.
AQP4 contributes to the development of many neurological illnesses through various biological effects, including an increase in the pace of blood-brain barrier clearance and a decrease in excess water storage in ventricles. AQP4 expression is increased in other types of hydrocephaly to drain extra CSF. However, its expression is frequently reduced in patients with iNPH, resulting in poor lymphatic circulation, waste clearance, and consequent neurodegeneration. As a result, AQP4 may be a critical therapeutic target in the treatment of neurological disorders, and with the advancement of research, the study of AQP4 gene polymorphisms and a range of neurological diseases is also advancing.
The role of AQPs in iNPH has gradually come to the attention of more researchers, with the majority of research focusing on AQP4 and AQP1, and the mechanisms of damage in iNPH have been gradually revealed, although there is still a need for further exploration. Applications in the diagnosis and treatment of iNPH have also received an increasing amount of interest. However, as evidenced by recent studies, it appears to be impossible to detect AQP1 and AQP4 in CSF to serve as diagnostic markers. As far as enzyme-linked immunodetection assays are concerned, it appears that AQP1 and AQP4 will not enter CSF. Additional opportunities, such as targeted imaging enhancement, are also worth investigating. Following acetazolamide, the AQP4 activator EPO has emerged as a potential therapy option and has so far shown promising results in animal models. Despite the fact that AQP1 inhibitors have been cited in a few publications, there has been no advancement in their use at this time. In subsequent stages of this research, there are numerous points to consider as well. The distribution of AQP1 and AQP4 in the brain is not confined to a single location. While AQP4 was discovered in the CP, AQP1 was also discovered in capillary astrocytes. The issue remains of how to direct medications to the precise locations where they will be most beneficial. The usefulness of using AQPs in targeted treatments for neurological illnesses has been made more evident.
Author contributions
ZZ and JH collected the related manuscript. ZT, JH, YC, YW, CW, CT, and JL drafted and revised the manuscript. GX participated in the design of the review, helped to draft, and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (No. 82171347), Hunan Provincial Natural Science Foundation of China (No. 2019JJ50949), the Scientific Research Project of Hunan Provincial Health Commission of China (No. 202204040024), and the Students Innovations in Central South University of China (No. S2022105330687).
Acknowledgments
I would like to express my gratitude to all those who helped us during the writing of this manuscript and thanks to all the peer reviewers for their opinions and suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abir-Awan, M., Kitchen, P., Salman, M. M., Conner, M. T., Conner, A. C., and Bill, R. M. (2019). Inhibitors of mammalian aquaporin water channels. Int. J. Mol. Sci. 20. doi: 10.3390/ijms20071589
Angelucci, F., Cechová, K., Pruša, R., and Hort, J. (2019). Amyloid beta soluble forms and plasminogen activation system in alzheimer's disease: consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci. Ther. 25, 303–313. doi: 10.1111/cns.13082
Arighi, A., Di Cristofori, A., Fenoglio, C., Borsa, S., D'Anca, M., Fumagalli, G. G., et al. (2019). Cerebrospinal fluid level of aquaporin4: a new window on glymphatic system involvement in neurodegenerative disease? J. Alzheimers. Dis. 69, 663–669. doi: 10.3233/JAD-190119
Assentoft, M., Larsen, B. R., Olesen, E. T. B., Fenton, R. A., and MacAulay, N. (2014). Aqp4 plasma membrane trafficking or channel gating is not significantly modulated by phosphorylation at cooh-terminal serine residues. Am. J. Physiol,. Cell Physiol. 307, C957–C965. doi: 10.1152/ajpcell.00182.2014
Benfenati, V., Caprini, M., Dovizio, M., Mylonakou, M. N., Ferroni, S., Ottersen, O. P., et al. (2011). An aquaporin-4/transient receptor potential vanilloid 4 (aqp4/trpv4) complex is essential for cell-volume control in astrocytes. P. Natl. Acad. Sci. U. S. A. 108, 2563–2568. doi: 10.1073/pnas.1012867108
Blasco, E., Martorell, J., De la Fuente, C., and Pumarola, M. (2014). Immunohistochemical study of aquaporins in an african grey parrot (psittacus erithacus) with hydrocephalus. J. Avian Med. Surg. 28, 309–315. doi: 10.1647/2013-059
Bloch, O., Auguste, K. I., Manley, G. T., and Verkman, A. S. (2006). Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J. Cereb. Blood Flow Metab. 26, 1527–1537. doi: 10.1038/sj.jcbfm.9600306
Blocher, J., Eckert, I., Elster, J., Wiefek, J., Eiffert, H., and Schmidt, H. (2011). Aquaporins aqp1 and aqp4 in the cerebrospinal fluid of bacterial meningitis patients. Neurosci. Lett. 504, 23–27. doi: 10.1016/j.neulet.2011.08.049
Bothwell, S. W., Janigro, D., and Patabendige, A. (2019). Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS 16, 9. doi: 10.1186/s12987-019-0129-6
Bouley, R., Palomino, Z., Tang, S. S., Nunes, P., Kobori, H., Lu, H. A., et al. (2009). Angiotensin ii and hypertonicity modulate proximal tubular aquaporin 1 expression. Am. J. Physiol. Renal Physiol. 297, F1575–F1586. doi: 10.1152/ajprenal.90762.2008
Calcagni, M. L., Taralli, S., Mangiola, A., Indovina, L., Lavalle, M., De Bonis, P., et al. (2013). Regional cerebral metabolic rate of glucose evaluation and clinical assessment in patients with idiopathic normal-pressure hydrocephalus before and after ventricular shunt placement: a prospective analysis. Clin. Nucl. Med. 38, 426–431. doi: 10.1097/RLU.0b013e31828e949b
Camassa, L., Lunde, L. K., Hoddevik, E. H., Stensland, M., Boldt, H. B., De Souza, G. A., et al. (2015). Mechanisms underlying aqp4 accumulation in astrocyte endfeet. Glia. 63, 2073–2091. doi: 10.1002/glia.22878
Castañeyra-Ruiz, L., González-Marrero, I., Carmona-Calero, E. M., Abreu-Gonzalez, P., Lecuona, M., Brage, L., et al. (2016). Cerebrospinal fluid levels of tumor necrosis factor alpha and aquaporin 1 in patients with mild cognitive impairment and idiopathic normal pressure hydrocephalus. Clin. Neurol. Neurosurg. 146, 76–81. doi: 10.1016/j.clineuro.2016.04.025
Castañeyra-Ruiz, L., González-Marrero, I., González-Toledo, J. M., Castañeyra-Ruiz, A., de Paz-Carmona, H., Castañeyra-Perdomo, A., et al. (2013). Aquaporin-4 expression in the cerebrospinal fluid in congenital human hydrocephalus. Fluids Barriers CNS 10, 18. doi: 10.1186/2045-8118-10-18
Chaudhry, S. R., Stoffel-Wagner, B., Kinfe, T. M., Guresir, E., Vatter, H., Dietrich, D., et al. (2017). Elevated systemic il-6 levels in patients with aneurysmal subarachnoid hemorrhage is an unspecific marker for post-sah complications. Int. J. Mol. Sci. 18. doi: 10.3390/ijms18122580
Cortes-Canteli, M., Paul, J., Norris, E. H., Bronstein, R., Ahn, H. J., Zamolodchikov, D., et al. (2010). Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to alzheimer's disease. Neuron 66, 695–709. doi: 10.1016/j.neuron.2010.05.014
de Arteaga, J., Ledesma, F., Garay, G., Chiurchiu, C., de la Fuente, J., Douthat, W., et al. (2011). High-dose steroid treatment increases free water transport in peritoneal dialysis patients. Nephrol. Dial. Transplant. 26, 4142–4145. doi: 10.1093/ndt/gfr533
de Laurentis, C., Cristaldi, P., Arighi, A., Cavandoli, C., Trezza, A., Sganzerla, E. P., et al. (2020). Role of aquaporins in hydrocephalus: what do we know and where do we stand? A systematic review. J. Neurol. 268, 4078–4094. doi: 10.1007/s00415-020-10122-z
Del Puerto, A., Pose-Utrilla, J., Simón-García, A., López-Menéndez, C., Jiménez, A. J., Porlan, E., et al. (2021). Kidins220 deficiency causes ventriculomegaly via snx27-retromer-dependent aqp4 degradation. Mol. Psychiatr. 26, 6411–6426. doi: 10.1038/s41380-021-01127-9
Ding, F. B., Yan, Y. M., Bao, C. R., Huang, J. B., Mei, J., Liu, H., et al. (2013). The role of aquaporin 1 activated by cgmp in myocardial edema caused by cardiopulmonary bypass in sheep. Cell. Physiol. Biochem. 32, 1320–1330. doi: 10.1159/000354530
Ding, Y., Zhang, T., Wu, G., McBride, D. W., Xu, N., Klebe, D. W., et al. (2019). Astrogliosis inhibition attenuates hydrocephalus by increasing cerebrospinal fluid reabsorption through the glymphatic system after germinal matrix hemorrhage. Exp. Neurol. 320, 113003. doi: 10.1016/j.expneurol.2019.113003
Eide, P. K., and Hansson, H. (2018). Astrogliosis and impaired aquaporin-4 and dystrophin systems in idiopathic normal pressure hydrocephalus. Neuropath. Appl. Neuro. 44, 474–490. doi: 10.1111/nan.12420
Eide, P. K., and Hansson, H. A. (2020). Blood-brain barrier leakage of blood proteins in idiopathic normal pressure hydrocephalus. Brain Res. 1727, 146547. doi: 10.1016/j.brainres.2019.146547
Eide, P. K., and Ringstad, G. (2019). Delayed clearance of cerebrospinal fluid tracer from entorhinal cortex in idiopathic normal pressure hydrocephalus: a glymphatic magnetic resonance imaging study. J. Cereb. Blood Flow Metab. 39, 1355–1368. doi: 10.1177/0271678X18760974
Eide, P. K., and Sorteberg, W. (2010). Diagnostic intracranial pressure monitoring and surgical management in idiopathic normal pressure hydrocephalus: a 6-year review of 214 patients. Neurosurgery. 66, 80–91. doi: 10.1227/01.NEU.0000363408.69856.B8
Eide, P. K., and Sorteberg, W. (2016). Outcome of surgery for idiopathic normal pressure hydrocephalus: role of preoperative static and pulsatile intracranial pressure. World Neurosurg. 86, 186–193. doi: 10.1016/j.wneu.2015.09.067
Eide, P. K., and Stanisic, M. (2010). Cerebral microdialysis and intracranial pressure monitoring in patients with idiopathic normal-pressure hydrocephalus: association with clinical response to extended lumbar drainage and shunt surgery. J. Neurosurg. 112, 414–424. doi: 10.3171/2009.5.JNS09122
Eidsvaag, V. A., Hansson, H., Heuser, K., Nagelhus, E. A., and Eide, P. K. (2017). Brain capillary ultrastructure in idiopathic normal pressure hydrocephalus: relationship with static and pulsatile intracranial pressure. J. Neuropath. Exp. Neur. 76, 1034–1045. doi: 10.1093/jnen/nlx091
Eilert-Olsen, M., Haj-Yasein, N. N., Vindedal, G. F., Enger, R., Gundersen, G. A., Hoddevik, E. H., et al. (2012). Deletion of aquaporin-4 changes the perivascular glial protein scaffold without disrupting the brain endothelial barrier. Glia 60, 432–440. doi: 10.1002/glia.22277
Enger, R., Gundersen, G. A., Haj-Yasein, N. N., Eilert-Olsen, M., Thoren, A. E., Vindedal, G. F., et al. (2012). Molecular scaffolds underpinning macroglial polarization: an analysis of retinal muller cells and brain astrocytes in mouse. Glia 60, 2018–2026. doi: 10.1002/glia.22416
Gomes, A., Da, S. I., Rodrigues, C., Castro, R. E., and Soveral, G. (2018). The emerging role of micrornas in aquaporin regulation. Front Chem. 6, 238. doi: 10.3389/fchem.2018.00238
Gunnarson, E., Axehult, G., Baturina, G., Zelenin, S., Zelenina, M., and Aperia, A. (2005). Lead induces increased water permeability in astrocytes expressing aquaporin 4. Neuroscience 136, 105–114. doi: 10.1016/j.neuroscience.2005.07.027
Haj-Yasein, N. N., Vindedal, G. F., Eilert-Olsen, M., Gundersen, G. A., Skare, O., Laake, P., et al. (2011). Glial-conditional deletion of aquaporin-4 (aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. U. S. A. 108, 17815–17820. doi: 10.1073/pnas.1110655108
Hara-Chikuma, M., and Verkman, A. S. (2008a). Prevention of skin tumorigenesis and impairment of epidermal cell proliferation by targeted aquaporin-3 gene disruption. Mol. Cell. Biol. 28, 326–332. doi: 10.1128/MCB.01482-07
Hara-Chikuma, M., and Verkman, A. S. (2008b). Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J. Mol. Med. 86, 221–231. doi: 10.1007/s00109-007-0272-4
Hasan-Olive, M. M., Enger, R., Hansson, H. A., Nagelhus, E. A., and Eide, P. K. (2019a). Pathological mitochondria in neurons and perivascular astrocytic endfeet of idiopathic normal pressure hydrocephalus patients. Fluids Barriers CNS 16, 39. doi: 10.1186/s12987-019-0160-7
Hasan-Olive, M. M., Enger, R., Hansson, H. A., Nagelhus, E. A., and Eide, P. K. (2019b). Loss of perivascular aquaporin-4 in idiopathic normal pressure hydrocephalus. Glia 67, 91–100. doi: 10.1002/glia.23528
Hiraldo-González, L., Trillo-Contreras, J. L., García-Miranda, P., Pineda-Sánchez, R., Ramírez-Lorca, R., Rodrigo-Herrero, S., et al. (2021). Evaluation of aquaporins in the cerebrospinal fluid in patients with idiopathic normal pressure hydrocephalus. PLoS ONE 16, e258165. doi: 10.1371/journal.pone.0258165
Hodler, J., Kubik-Huch, R. A., and von Schulthess, G. K. (2020). Diseases of the Brain, Head and Neck, Spine 2020-2023: Diagnostic Imaging. Cham: Springer International Publishing AG. doi: 10.1007/978-3-030-38490-6
Iliff, J. J., Goldman, S. A., and Nedergaard, M. (2015). Implications of the discovery of brain lymphatic pathways. Lancet Neurol. 14, 977–979. doi: 10.1016/S1474-4422(15)00221-5
Iliff, J. J., Lee, H., Yu, M., Feng, T., Logan, J., Nedergaard, M., et al. (2013). Brain-wide pathway for waste clearance captured by contrast-enhanced mri. J. Clin. Invest. 123, 1299–1309. doi: 10.1172/JCI67677
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates csf flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 4, 111r−147r. doi: 10.1126/scitranslmed.3003748
Ismail, M., Bokaee, S., Morgan, R., Davies, J., Harrington, K. J., and Pandha, H. (2009). Inhibition of the aquaporin 3 water channel increases the sensitivity of prostate cancer cells to cryotherapy. Br. J. Cancer 100, 1889–1895. doi: 10.1038/sj.bjc.6605093
Jeon, T., Park, K., Park, S., Hwang, J., and Hwang, S. K. (2017). Expression of aquaporin 1 and 4 in the choroid plexus and brain parenchyma of kaolin-induced hydrocephalic rats. Korean J. Neurotrauma 13, 68–75. doi: 10.13004/kjnt.2017.13.2.68
Kadohira, I., Abe, Y., Nuriya, M., Sano, K., Tsuji, S., Arimitsu, T., et al. (2008). Phosphorylation in the c-terminal domain of aquaporin-4 is required for golgi transition in primary cultured astrocytes. Biochem. Biophys. Res. Commun. 377, 463–468. doi: 10.1016/j.bbrc.2008.09.155
Kajimoto, Y., Kameda, M., Kambara, A., Kuroda, K., Tsuji, S., Nikaido, Y., et al. (2022). Impact of early intervention for idiopathic normal pressure hydrocephalus on long-term prognosis in prodromal phase. Front. Neurol. 13, 866352. doi: 10.3389/fneur.2022.866352
Kim, J. H., Lee, Y. W., Park, K. A., Lee, W. T., and Lee, J. E. (2010). Agmatine attenuates brain edema through reducing the expression of aquaporin-1 after cerebral ischemia. J. Cereb. Blood Flow Metab. 30, 943–949. doi: 10.1038/jcbfm.2009.260
Kobayashi, H., Yokoo, H., Yanagita, T., Satoh, S., Kis, B., Deli, M., et al. (2006). Induction of aquaporin 1 by dexamethasone in lipid rafts in immortalized brain microvascular endothelial cells. Brain Res. 1123, 12–19. doi: 10.1016/j.brainres.2006.09.066
Kusayama, M., Wada, K., Nagata, M., Ishimoto, S., Takahashi, H., Yoneda, M., et al. (2011). Critical role of aquaporin 3 on growth of human esophageal and oral squamous cell carcinoma. Cancer Sci. 102, 1128–1136. doi: 10.1111/j.1349-7006.2011.01927.x
Leinonen, V., Kuulasmaa, T., and Hiltunen, M. (2021). Inph-the mystery resolving. EMBO Mol. Med. 13, e13720. doi: 10.15252/emmm.202013720
Li, D., Liu, X., Liu, T., Liu, H., Tong, L., Jia, S., et al. (2020). Neurochemical regulation of the expression and function of glial fibrillary acidic protein in astrocytes. Glia 68, 878–897. doi: 10.1002/glia.23734
Lolansen, S. D., Rostgaard, N., Oernbo, E. K., Juhler, M., Simonsen, A. H., and MacAulay, N. (2021). Inflammatory markers in cerebrospinal fluid from patients with hydrocephalus: a systematic literature review. Dis. Markers 2021, 8834822. doi: 10.1155/2021/8834822
Lu, Y., Iandiev, I., Hollborn, M., Körber, N., Ulbricht, E., Hirrlinger, P. G., et al. (2011). Reactive glial cells: increased stiffness correlates with increased intermediate filament expression. FASEB J. 25, 624–631. doi: 10.1096/fj.10-163790
Lundby, A., Secher, A., Lage, K., Nordsborg, N. B., Dmytriyev, A., Lundby, C., et al. (2012). Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 3, 876. doi: 10.1038/ncomms1871
Malm, J., Jacobsson, J., Birgander, R., and Eklund, A. (2011). Reference values for csf outflow resistance and intracranial pressure in healthy elderly. Neurology 76, 903–909. doi: 10.1212/WNL.0b013e31820f2dd0
Mao, X., Enno, T. L., and Del, B. M. (2006). Aquaporin 4 changes in rat brain with severe hydrocephalus. Eur. J. Neurosci. 23, 2929–2936. doi: 10.1111/j.1460-9568.2006.04829.x
McCoy, E., and Sontheimer, H. (2010). Mapk induces aqp1 expression in astrocytes following injury. Glia 58, 209–217. doi: 10.1002/glia.20916
Merlini, M., Rafalski, V. A., Rios Coronado, P. E., Gill, T. M., Ellisman, M., Muthukumar, G., et al. (2019). Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an alzheimer's disease model. Neuron 101, 1099–1108. doi: 10.1016/j.neuron.2019.01.014
Mola, M. G., Sparaneo, A., Gargano, C. D., Spray, D. C., Svelto, M., Frigeri, A., et al. (2016). The speed of swelling kinetics modulates cell volume regulation and calcium signaling in astrocytes: a different point of view on the role of aquaporins. Glia 64, 139–154. doi: 10.1002/glia.22921
Moon, C., King, L. S., and Agre, P. (1997). Aqp1 expression in erythroleukemia cells: genetic regulation of glucocorticoid and chemical induction. Am. J. Physiol. 273, C1562–C1570. doi: 10.1152/ajpcell.1997.273.5.C1562
Nagelhus, E. A., and Ottersen, O. P. (2013). Physiological roles of aquaporin-4 in brain. Physiol. Rev. 93, 1543–1562. doi: 10.1152/physrev.00011.2013
Nakada, T., and Kwee, I. L. (2019). Fluid dynamics inside the brain barrier: current concept of interstitial flow, glymphatic flow, and cerebrospinal fluid circulation in the brain. Neuroscientist 25, 155–166. doi: 10.1177/1073858418775027
Nakajima, M., Kawamura, K., Akiba, C., Sakamoto, K., Xu, H., Kamohara, C., et al. (2021). Differentiating comorbidities and predicting prognosis in idiopathic normal pressure hydrocephalus using cerebrospinal fluid biomarkers: a review. Croat. Med. J. 62, 387–398. doi: 10.3325/cmj.2021.62.387
Nesverova, V., and Tornroth-Horsefield, S. (2019). Phosphorylation-dependent regulation of mammalian aquaporins. Cells 8, 82. doi: 10.3390/cells8020082
Nicchia, G. P., Rossi, A., Nudel, U., Svelto, M., and Frigeri, A. (2008). Dystrophin-dependent and -independent aqp4 pools are expressed in the mouse brain. Glia 56, 869–876. doi: 10.1002/glia.20661
Oshio, K., Watanabe, H., Song, Y., Verkman, A. S., and Manley, G. T. (2005). Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel aquaporin-1. FASEB J. 19, 76–78. doi: 10.1096/fj.04-1711fje
Owler, B. K., Pitham, T., and Wang, D. (2010). Aquaporins: relevance to cerebrospinal fluid physiology and therapeutic potential in hydrocephalus. Cerebrospinal Fluid Res. 7, 15. doi: 10.1186/1743-8454-7-15
Papadopoulos, M. C., and Verkman, A. S. (2007). Aquaporin-4 and brain edema. Pediatr. Nephrol. 22, 778–784. doi: 10.1007/s00467-006-0411-0
Papadopoulos, M. C., and Verkman, A. S. (2013). Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 14, 265–277. doi: 10.1038/nrn3468
Paul, L., Madan, M., Rammling, M., Chigurupati, S., Chan, S. L., and Pattisapu, J. V. (2011). Expression of aquaporin 1 and 4 in a congenital hydrocephalus rat model. Neurosurgery 68, 462–473. doi: 10.1227/NEU.0b013e3182011860
Pavlin, T., Nagelhus, E. A., Brekken, C., Eyjolfsson, E. M., Thoren, A., Haraldseth, O., et al. (2017). Loss or mislocalization of aquaporin-4 affects diffusion properties and intermediary metabolism in gray matter of mice. Neurochem. Res. 42, 77–91. doi: 10.1007/s11064-016-2139-y
Petković, F., Campbell, I. L., Gonzalez, B., and Castellano, B. (2017). Reduced cuprizone-induced cerebellar demyelination in mice with astrocyte-targeted production of il-6 is associated with chronically activated, but less responsive microglia. J. Neuroimmunol. 310, 97–102. doi: 10.1016/j.jneuroim.2017.07.003
Qing, W. G., Dong, Y. Q., Ping, T. Q., Lai, L. G., Fang, L. D., Min, H. W., et al. (2009). Brain edema after intracerebral hemorrhage in rats: the role of iron overload and aquaporin 4. J. Neurosurg. 110, 462–468. doi: 10.3171/2008.4.JNS17512
Qvarlander, S., Ambarki, K., Wåhlin, A., Jacobsson, J., Birgander, R., Malm, J., et al. (2017). Cerebrospinal fluid and blood flow patterns in idiopathic normal pressure hydrocephalus. Acta Neurol. Scand. 135, 576–584. doi: 10.1111/ane.12636
Raneri, F., Zella, M. A. S., Di Cristofori, A., Zarino, B., Pluderi, M., and Spagnoli, D. (2017). Supplementary tests in idiopathic normal pressure hydrocephalus: a single-center experience with a combined lumbar infusion test and tap test. World Neurosurg. 100, 567–574. doi: 10.1016/j.wneu.2017.01.003
Rao, K. V. R., Jayakumar, A. R., Reddy, P. V. B., Tong, X., Curtis, K. M., and Norenberg, M. D. (2010). Aquaporin-4 in manganese-treated cultured astrocytes. Glia 58, 1490–1499. doi: 10.1002/glia.21023
Rasmussen, M. K., Mestre, H., and Nedergaard, M. (2018). The glymphatic pathway in neurological disorders. Lancet Neurol. 17, 1016–1024. doi: 10.1016/S1474-4422(18)30318-1
Reeves, B. C., Karimy, J. K., Kundishora, A. J., Mestre, H., Cerci, H. M., Matouk, C., et al. (2020). Glymphatic system impairment in alzheimer's disease and idiopathic normal pressure hydrocephalus. Trends Mol. Med. 26, 285–295. doi: 10.1016/j.molmed.2019.11.008
Ringstad, G., Vatnehol, S., and Eide, P. K. (2017). Glymphatic mri in idiopathic normal pressure hydrocephalus. Brain 140, 2691–2705. doi: 10.1093/brain/awx191
Rizwan Siddiqui, M., Attar, F., Mohanty, V., Kim, K. S., Shekhar Mayanil, C., and Tomita, T. (2018). Erythropoietin-mediated activation of aquaporin-4 channel for the treatment of experimental hydrocephalus. Childs Nervous Syst. ChNS 34, 2195–2202. doi: 10.1007/s00381-018-3865-z
Saadoun, S., Tait, M. J., Reza, A., Davies, D. C., Bell, B. A., Verkman, A. S., et al. (2009). Aqp4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience 161, 764–772. doi: 10.1016/j.neuroscience.2009.03.069
Sakurada, T., Kuboshima, S., Ogimoto, G., Fujino, T., Sato, T., Maeba, T., et al. (2004). Aquaporin-1 is recruited to the plasma membrane by hyperosmotic stimuli via a protein kinase a-dependent pathway in rat peritoneal mesothelial cells. Adv. Perit. Dial. 20, 37–42.
Schachtrup, C., Ryu, J. K., Helmrick, M. J., Vagena, E., Galanakis, D. K., Degen, J. L., et al. (2010). Fibrinogen triggers astrocyte scar formation by promoting the availability of active tgf-beta after vascular damage. J. Neurosci. 30, 5843–5854. doi: 10.1523/JNEUROSCI.0137-10.2010
Schmidt, M. J., Rummel, C., Hauer, J., Kolecka, M., Ondreka, N., McClure, V., et al. (2016). Increased csf aquaporin-4, and interleukin-6 levels in dogs with idiopathic communicating internal hydrocephalus and a decrease after ventriculo-peritoneal shunting. Fluids Barriers CNS 13, 12. doi: 10.1186/s12987-016-0034-1
Shen, X. Q., Miyajima, M., Ogino, I., and Arai, H. (2006). Expression of the water-channel protein aquaporin 4 in the h-tx rat: possible compensatory role in spontaneously arrested hydrocephalus. J. Neurosurg 105, 459–464. doi: 10.3171/ped.2006.105.6.459
Smith, Z. A., Moftakhar, P., Malkasian, D., Xiong, Z., Vinters, H. V., and Lazareff, J. A. (2007). Choroid plexus hyperplasia: surgical treatment and immunohistochemical results. Case Rep. 107, 255–262. doi: 10.3171/PED-07/09/255
Sosvorova, L., Mohapl, M., Vcelak, J., Hill, M., Vitku, J., and Hampl, R. (2015). The impact of selected cytokines in the follow-up of normal pressure hydrocephalus. Physiol. Res. 64, S283–S290. doi: 10.33549/physiolres.933069
Stoenoiu, M. S., Ni, J., Verkaeren, C., Debaix, H., Jonas, J. C., Lameire, N., et al. (2003). Corticosteroids induce expression of aquaporin-1 and increase transcellular water transport in rat peritoneum. J. Am. Soc. Nephrol. 14, 555–565. doi: 10.1097/01.ASN.0000053420.37216.9E
Stokum, J. A., Kwon, M. S., Woo, S. K., Tsymbalyuk, O., Vennekens, R., Gerzanich, V., et al. (2018). Sur1-trpm4 and aqp4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia 66, 108–125. doi: 10.1002/glia.23231
Tan, C., Wang, X., Wang, Y., Wang, C., Tang, Z., Zhang, Z., et al. (2021). The pathogenesis based on the glymphatic system, diagnosis, and treatment of idiopathic normal pressure hydrocephalus. Clin. Interv. Aging 16, 139–153. doi: 10.2147/CIA.S290709
Thavarajasingam, S. G., El-Khatib, M., Vemulapalli, K. V., Iradukunda, H. A. S., Laleye, J., Russo, S., et al. (2022). Cerebrospinal fluid and venous biomarkers of shunt-responsive idiopathic normal pressure hydrocephalus: a systematic review and meta-analysis. Acta Neurochir. 164, 1719–1746. doi: 10.1007/s00701-022-05154-5
Tie, L., Lu, N., Pan, X. Y., Pan, Y., An, Y., Gao, J. W., et al. (2012). Hypoxia-induced up-regulation of aquaporin-1 protein in prostate cancer cells in a p38-dependent manner. Cell. Physiol. Biochem. 29, 269–280. doi: 10.1159/000337608
Tietz, P. S., McNiven, M. A., Splinter, P. L., Huang, B. Q., and Larusso, N. F. (2006). Cytoskeletal and motor proteins facilitate trafficking of aqp1-containing vesicles in cholangiocytes. Biol. Cell. 98, 43–52. doi: 10.1042/BC20040089
Tomas-Camardiel, M., Venero, J. L., Herrera, A. J., De Pablos, R. M., Pintor-Toro, J. A., Machado, A., et al. (2005). Blood-brain barrier disruption highly induces aquaporin-4 mrna and protein in perivascular and parenchymal astrocytes: protective effect by estradiol treatment in ovariectomized animals. J. Neurosci. Res. 80, 235–246. doi: 10.1002/jnr.20443
Tourdias, T., Dragonu, I., Fushimi, Y., Deloire, M. S., Boiziau, C., Brochet, B., et al. (2009). Aquaporin 4 correlates with apparent diffusion coefficient and hydrocephalus severity in the rat brain: a combined mri-histological study. Neuroimage 47, 659–666. doi: 10.1016/j.neuroimage.2009.04.070
Verkman, A. S. (2008a). Mammalian aquaporins: diverse physiological roles and potential clinical significance. Expert Rev. Mol. Med. 10, e13. doi: 10.1017/S1462399408000690
Verkman, A. S. (2008b). A cautionary note on cosmetics containing ingredients that increase aquaporin-3 expression. Exp. Dermatol. 17, 871–872. doi: 10.1111/j.1600-0625.2008.00698.x
Vindedal, G. F., Thoren, A. E., Jensen, V., Klungland, A., Zhang, Y., Holtzman, M. J., et al. (2016). Removal of aquaporin-4 from glial and ependymal membranes causes brain water accumulation. Mol. Cell. Neurosci. 77, 47–52. doi: 10.1016/j.mcn.2016.10.004
Wang, C., Wang, X., Tan, C., Wang, Y., Tang, Z., Zhang, Z., et al. (2021). Novel therapeutics for hydrocephalus: insights from animal models. CNS Neurosci. Ther. 27, 1012–1022. doi: 10.1111/cns.13695
Wang, D., Nykanen, M., Yang, N., Winlaw, D., North, K., Verkman, A. S., et al. (2011). Altered cellular localization of aquaporin-1 in experimental hydrocephalus in mice and reduced ventriculomegaly in aquaporin-1 deficiency. Mol. Cell. Neurosci. 46, 318–324. doi: 10.1016/j.mcn.2010.10.003
Wang, X., Li, T., Liu, Y., Jia, S., Liu, X., Jiang, Y., et al. (2021). Aquaporin 4 differentially modulates osmotic effects on vasopressin neurons in rat supraoptic nucleus. Acta physiologica 232, e13672. doi: 10.1111/apha.13672
Wang, Y., Chen, R., Wa, Y., Ding, S., Yang, Y., Liao, J., et al. (2022). Tumor immune microenvironment and immunotherapy in brain metastasis from non-small cell lung cancer. Front. Immunol. 13, 829451. doi: 10.3389/fimmu.2022.829451
Wang, Y., and Hatton, G. I. (2009). Astrocytic plasticity and patterned oxytocin neuronal activity: dynamic interactions. J. Neurosci. 29, 1743–1754. doi: 10.1523/JNEUROSCI.4669-08.2009
Wang, Y., Wang, X., Tan, C., Wang, C., Tang, Z., Zhang, Z., et al. (2021). Hydrocephalus after aneurysmal subarachnoid hemorrhage: epidemiology, pathogenesis, diagnosis, and management. Signa Vitae 17, 4–17. doi: 10.22514/sv.2021.021
Wang, Z., Zhang, Y., Hu, F., Ding, J., and Wang, X. (2020). Pathogenesis and pathophysiology of idiopathic normal pressure hydrocephalus. CNS Neurosci. Ther. 26, 1230–1240. doi: 10.1111/cns.13526
Ximenes-da-Silva, A. (2016). Metal ion toxins and brain aquaporin-4 expression: an overview. Front. Neurosci. 10, 233. doi: 10.3389/fnins.2016.00233
Yasui, M., Hazama, A., Kwon, T. H., Nielsen, S., Guggino, W. B., and Agre, P. (1999). Rapid gating and anion permeability of an intracellular aquaporin. Nature. 402, 184–187. doi: 10.1038/46045
Yin, L. K., Zheng, J. J., Zhao, L., Hao, X. Z., Zhang, X. X., Tian, J. Q., et al. (2017). Reversed aqueductal cerebrospinal fluid net flow in idiopathic normal pressure hydrocephalus. Acta Neurol. Scand. 136, 434–439. doi: 10.1111/ane.12750
Yukutake, Y., Hirano, Y., Suematsu, M., and Yasui, M. (2009). Rapid and reversible inhibition of aquaporin-4 by zinc. Biochemistry 48, 12059–12061. doi: 10.1021/bi901762y
Yukutake, Y., Tsuji, S., Hirano, Y., Adachi, T., Takahashi, T., Fujihara, K., et al. (2008). Mercury chloride decreases the water permeability of aquaporin-4-reconstituted proteoliposomes. Biol. Cell 100, 355–363. doi: 10.1042/BC20070132
Yukutake, Y., and Yasui, M. (2010). Regulation of water permeability through aquaporin-4. Neuroscience 168, 885–891. doi: 10.1016/j.neuroscience.2009.10.029
Zelenina, M. (2010). Regulation of brain aquaporins. Neurochem. Int. 57, 468–488. doi: 10.1016/j.neuint.2010.03.022
Zhan, C., Xiao, G., Zhang, X., Chen, X., Zhang, Z., and Liu, J. (2020). Decreased mir-30a promotes tgf-beta1-mediated arachnoid fibrosis in post-hemorrhagic hydrocephalus. Transl. Neurosci. 11, 60–74. doi: 10.1515/tnsci-2020-0010
Zhang, J., Xiong, Y., Lu, L. X., Wang, H., Zhang, Y. F., Fang, F., et al. (2013). Aqp1 expression alterations affect morphology and water transport in schwann cells and hypoxia-induced up-regulation of aqp1 occurs in a hif-1alpha-dependent manner. Neuroscience 252, 68–79. doi: 10.1016/j.neuroscience.2013.08.006
Zhang, W., Zitron, E., Homme, M., Kihm, L., Morath, C., Scherer, D., et al. (2007). Aquaporin-1 channel function is positively regulated by protein kinase c. J. Biol. Chem. 282, 20933–20940. doi: 10.1074/jbc.M703858200
Zhou, J., Kong, H., Hua, X., Xiao, M., Ding, J., and Hu, G. (2008). Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 19, 1–5. doi: 10.1097/WNR.0b013e3282f2b4eb
Ziegelitz, D., Arvidsson, J., Hellstrom, P., Tullberg, M., Wikkelso, C., and Starck, G. (2016). Pre-and postoperative cerebral blood flow changes in patients with idiopathic normal pressure hydrocephalus measured by computed tomography (ct)-perfusion. J. Cereb. Blood Flow Metab. 36, 1755–1766. doi: 10.1177/0271678X15608521
Keywords: iNPH, AQP1, AQP4, hydrocephalus, pathogenesis
Citation: Zhao Z, He J, Chen Y, Wang Y, Wang C, Tan C, Liao J and Xiao G (2022) The pathogenesis of idiopathic normal pressure hydrocephalus based on the understanding of AQP1 and AQP4. Front. Mol. Neurosci. 15:952036. doi: 10.3389/fnmol.2022.952036
Received: 24 May 2022; Accepted: 12 August 2022;
Published: 20 September 2022.
Edited by:
Adjanie Patabendige, Edge Hill University, United KingdomReviewed by:
Yu-Feng Wang, Harbin Medical University, ChinaAndrea Quattrone, University Magna Graecia, Italy
Nobutaka Hattori, Juntendo University, Japan
Ville Leinonen, Kuopio University Hospital, Finland
Copyright © 2022 Zhao, He, Chen, Wang, Wang, Tan, Liao and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gelei Xiao, eGlhb2dlbGVpQGNzdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship