 Parul Goel
Parul Goel Sasanka Chakrabarti2
Sasanka Chakrabarti2- 1Department of Biochemistry, Shri Atal Bihari Vajpayee Government Medical College Chhainsa, Faridabad, India
- 2Department of Biochemistry, Maharishi Markandeshwar Institute of Medical Sciences and Research, Maharishi Markandeshwar (Deemed to be University), Ambala, India
- 3Department of Community Medicine and School of Public Health, Post Graduate Institute of Medical Education and Research, Chandigarh, India
- 4Department of Surgery, Maharishi Markandeshwar Institute of Medical Sciences and Research, Maharishi Markandeshwar (Deemed to be University), Ambala, India
Regulated cell death (RCD) is an ordered and tightly orchestrated set of changes/signaling events in both gene expression and protein activity and is responsible for normal development as well as maintenance of tissue homeostasis. Aberrant activation of this pathway results in cell death by various mechanisms including apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy-dependent cell death. Such pathological changes in neurons alone or in combination have been observed in the pathogenesis of various neurodegenerative diseases including Alzheimer’s disease (AD). Pathological hallmarks of AD focus primarily on the accumulation of two main protein markers: amyloid β peptides and abnormally phosphorylated tau proteins. These protein aggregates result in the formation of A-β plaques and neuro-fibrillary tangles (NFTs) and induce neuroinflammation and neurodegeneration over years to decades leading to a multitude of cognitive and behavioral deficits. Autopsy findings of AD reveal massive neuronal death manifested in the form of cortical volume shrinkage, reduction in sizes of gyri to up to 50% and an increase in the sizes of sulci. Multiple forms of cell death have been recorded in neurons from different studies conducted so far. However, understanding the mechanism/s of neuronal cell death in AD patients remains a mystery as the trigger that results in aberrant activation of RCD is unknown and because of the limited availability of dying neurons. This review attempts to elucidate the process of Regulated cell death, how it gets unregulated in response to different intra and extracellular stressors, various forms of unregulated cell death, their interplay and their role in pathogenesis of Alzheimer’s Disease in both human and experimental models of AD. Further we plan to explore the correlation of both amyloid-beta and Tau with neuronal loss as seen in AD.
Introduction
Alzheimer’s disease is a chronic neurodegenerative disease of the elderly people recognized clinically as gradual impairment in memory and cognition along with various disorders in the emotional domain (Sperling et al., 2011; Scheltens et al., 2016; Qiu et al., 2022). It is considered the sixth leading cause of death in the United States where the death from AD increased by 71% from 2000 to 2013 (Alzheimer’s Association, 2016). Estimated in 2015, the burden of dementia worldwide constitutes over 46 million affected people and a societal loss of United States dollar 818 billion, and AD accounts for the vast majority of such patients with dementia (Prince et al., 2015). The disease usually presents in a sexagenarian and follows a progressive and irreversible course. As the memory and cognitive skills continue to get worsened, the patients eventually depend on caregivers extensively and finally succumb to the illness within 4–8 years after diagnosis. Changes in the brain related to AD, however, begin in the preclinical AD stage several decades before the manifestation of clinical dementia (Dubois et al., 2016). The vast majority of AD subjects suffer from the sporadic form of the disease, while in less than 5% of cases the disease appears in the familial form with defined mutations in APP, PS1, and PS2 genes (Reitz and Mayeux, 2014). Neuropathologically, the AD brain is characterized by extra-cellular depositions of oligomerized and fibrilar amyloid beta peptide 42 (Aβ42) surrounded by dystrophic neurites called neuritic plaques, as well as intra-neuronal formation of neurofibrillary tangles composed of phosphorylated tau protein (Serrano-Pozo et al., 2011; Chakrabarti et al., 2015). Associated with these changes diffuse neuronal loss, synaptic degeneration and reactive gliosis could be seen in many brain areas concerned with memory and cognition (Serrano-Pozo et al., 2011; Chakrabarti et al., 2015).
Aging is considered as the primary risk factor for many neurodegenerative diseases including AD, and after 65 years the age-specific prevalence of AD almost doubles every 5 years (Harman, 2006; Qiu et al., 2022). The factors responsible for making certain individuals progress from non-pathological brain aging to complex cascade of AD pathogenesis are largely unknown, but the commonality of underlying mechanisms in these two processes are interesting (Podtelezhnikov et al., 2011; Poddar et al., 2019). Apart from aging, multiple genetic and modifiable risk factors also contribute to the pathogenesis of sporadic AD (Reitz and Mayeux, 2014; Qiu et al., 2022). For more than two decades, the dominant hypothesis attempted to explain the pathogenesis of AD by the multiple toxic effects of oligomerized Aβ42 on neurons, glia and different subcellular components in the brain (amyloid cascade hypothesis), but the shortcomings of this hypothesis are becoming increasingly apparent (Korczyn, 2008; Herrup, 2015). It appears that at the cellular level in the AD brain, mitochondrial dysfunction, oxidative stress, abnormal accumulation of proteins (Aβ42, tau, etc.) with associated toxic effects and inflammatory response by microglia interact in a concerted manner mediating the pathogenesis of AD (Chakrabarti et al., 2015; Ganguly et al., 2017; Guo et al., 2020). This review will, however, not cover the different aspects of AD pathogenesis, but focus on a limited aspect of the latter scrutinizing the available evidence on the nature and mechanisms of neuronal death in AD, highlighting the knowledge gaps and providing new insights into this complex process.
Neuronal death in Alzheimer’s disease brain
The high degree of neuronal death apparent in various areas of the brain in AD contrasts with relatively little neuronal loss in non-pathological brain aging (West of Scotland Coronary Prevention Group, 1996). The neuronal loss in AD begins in the preclinical stage and then progresses during the prodromal phase or MCI and AD dementia; an early loss could be in the entorhinal cortex, basal nucleus of Meynert, locus ceruleus, but later the neuronal loss is diffusely present in multiple brain areas like the hippocampus, amygdala, temporal lobe and frontal and parietal cortices (Gómez-Isla et al., 1996, 1997; Serrano-Pozo et al., 2011; Arendt et al., 2015; DeTure and Dickson, 2019). It is not certain if the neuronal loss begins in one brain area and then progresses to other areas or starts simultaneously in different regions. The neuronal loss correlates well with the regional distribution of NFTs, but within a defined region the neuronal loss is more conspicuous than the presence of NFTs (Serrano-Pozo et al., 2011). Interestingly, synaptic degeneration might precede neuronal loss, and both these processes contribute to memory impairment in AD patients (Arendt et al., 2015). Although multiple studies have shown that the cognitive deficits correlate with the extent of NFT formation, it is even better correlated with the degree of neuronal loss and synaptic degeneration (Serrano-Pozo et al., 2011; Arendt et al., 2015). In multiple studies the neuronal loss has been estimated in transgenic AD models (carrying mutated familial AD genes or Tau gene) in brain areas affected in clinical AD especially the hippocampus; in single APP transgenic or double or triple transgenic (APP/PS1 or APP/PS2 or APP/PS1/TAU, etc.) experimental animals a slight to substantial neuronal loss was observed (Schmitz et al., 2004; Wirths and Bayer, 2010; Wirths and Zampar, 2020). However, the neuronal death is much less pronounced than amyloid-beta pathology or tauopathy or reactive gliosis in the brains of AD transgenic animals (Wirths and Zampar, 2020). Before analyzing further this aspect of neuronal death in AD, it would be worthwhile to provide a bird’s eye view of different pathways of cell death and salient features of neuronal differentiation and death in the brain.
Cell death types: A snapshot
Cell death can be activated in response to various forms of internal or cellular stress, and primarily it is classified into two major groups- accidental cell death and regulated cell death (RCD). The physiological form of regulated cell death is programmed cell death (Cui et al., 2021). RCD has been sub-classified over the years from the early characterization of apoptosis in more than ten sub-types of ingenious names (Tang et al., 2019). However, we will limit ourselves to a discussion on some of these pathways that appear to be more relevant in the context of AD.
Apoptosis
Apoptosis is a type of regulated cell death (RCD) characterized by morphological features that include cell shrinkage, chromatin condensation, nuclear fragmentation, plasma membrane blebbing and the formation of membrane-bound apoptotic bodies (Elmore, 2007; Toné et al., 2007; Del Re et al., 2019). The pathway of apoptosis can be triggered by the binding of extracellular death ligands (TNF-α, FASL, TRAIL, etc.) to death receptors (extrinsic pathway); on the other hand, mitochondrial membrane depolarization as a result of cellular injury (DNA damage, metabolic stress, ROS) might initiate the intrinsic pathway of apoptotic death (Elmore, 2007; Galluzzi et al., 2012; Tang et al., 2019). In both the pathways a group of proteolytic enzymes known as caspases, derived from the inactive forms called pro-caspases, are involved, and some of these are initiator caspases (caspase 8, caspase 9, caspase 10) and others effector caspases (caspase 3, caspase 6, caspase 7) (Elmore, 2007; Del Re et al., 2019). The effector caspases degrade various protein substrates to cause the cytomorphological and functional alterations of apoptosis; the chromatin fragmentation is because of activation of CAD following the degradation of ICAD which keeps the CAD as an inactive complex (Elmore, 2007; Cui et al., 2021). However, a caspase-independent intrinsic apoptotic pathway is also known (Galluzzi et al., 2012). The mitochondria play a central role in the intrinsic pathway of apoptosis. The mitochondrial outer membrane permeabilization (MOMP) mediated by pro-apoptotic proteins BAX and BAD of the Bcl-2 family is a key event of intrinsic apoptosis, but mitochondrial inner membrane permeability alterations and loss of transmembrane potential are also involved in this process (Elmore, 2007; Galluzzi and Kroemer, 2008; Wang and Youle, 2009; Averill-Bates et al., 2018; Tang et al., 2019; Cui et al., 2021). These membrane changes lead to the release of cytochrome c, AIF, endoG, Smac/DIABLO, etc., from the mitochondria causing activation of effector caspases and apoptotic death (Averill-Bates et al., 2018; Del Re et al., 2019; Tang et al., 2019).
In the extrinsic pathway, the activation of initiator caspase 8 occurs which might further activate executioner caspase like caspase 3 leading to apoptotic death; alternatively, a cross-talk between extrinsic and intrinsic pathways takes place in which caspase 8 acts on BID forming the truncated BID (tBID) which might lead to mitochondrial MOMP with subsequent initiation of intrinsic apoptotic cascade (Elmore, 2007; Tang et al., 2019).
Necrosis and necroptosis
Traditionally, necrosis is considered an unregulated cell death process characterized by cell swelling, loss of membrane integrity, dissipation of ionic gradients, and release of intracellular contents into the extracellular environment; the necrosis usually triggers an inflammatory response presumably through the release of DAMPs or ‘damage-associated molecular patterns’ from the dead cells (Vanlangenakker et al., 2012; Feoktistova and Leverkus, 2015; Sachet et al., 2017). Necrosis can occur due to overwhelming external physicochemical stress such as freezing, burning, mechanical trauma, and this is often categorized as accidental or primary necrosis; a process of secondary necrosis has also been suggested as the outcome of apoptosis under certain conditions where the clearance of apoptotic cells by phagocytes is absent or impaired (Silva, 2010; Vanlangenakker et al., 2012; Feoktistova and Leverkus, 2015; Sachet et al., 2017). The necrosis generally lacks a definite cellular signaling pathway, but some forms of necrosis follow well-defined mechanisms such as the one driven by mitochondrial permeability transition pore (mPTP) activation (Kinnally et al., 2011; Feoktistova and Leverkus, 2015). On the other hand, necroptosis refers to a regulated form of cell death with morphological features of necrosis but with a defined signaling cascade involving RIP kinases (RIPK1 and RIPK3), necrosome formation and activation of MLKL (Berghe et al., 2010; Galluzzi et al., 2018). Necroptosis can be induced by the ligand-binding to several death receptors (TNFR1, TRAIL-R1 and -R2, FAS), interferon receptors and other receptors like stocktickerRIG1, STING sensing pathogen-associated molecular patterns; in addition activated TLRs or interferon α-receptor and certain viruses can trigger necroptosis without involving RIPK1 (Tummers and Green, 2017; Choi et al., 2019; Tang et al., 2019). In the typical necroptotic pathway such as the one induced by TNF-α, the ligand binding initiates a multimeric complex formation with RIPK1 (complex I) followed by the formation of another complex called complex II within which autophosphorylation of RIPK1 occurs at serine 161; the activated RIPK1 then phosphorylates RIPK3 and forms a complex with it called necrosome (Vanlangenakker et al., 2012; Su et al., 2016; Choi et al., 2019). A subsequent phosphorylation of MLKL by RIPK3 causes the oligomerization of MLKL that culminates in lytic cell death (Vanlangenakker et al., 2012; Su Z. et al., 2016; Choi et al., 2019). It is important to realize that the same stimuli that activate the extrinsic pathway of apoptosis might also trigger necroptosis, and this presumably occurs when caspase 8 is deficient or its activation is impaired (Choi et al., 2019; Tang et al., 2019). Likewise, the deficiency of FADD or cIAPs might also direct the cell to follow necroptosis instead of apoptosis (Vanlangenakker et al., 2012; Choi et al., 2019). Necroptosis can be inhibited by necrostatin which is a specific inhibitor of RIPK1.
Ferroptosis
The ferroptotic mode of cell death is iron-dependent and induced by diverse triggers; it involves an accumulation of ROS and lipid peroxidation products and a depletion of reduced glutathione inside the cells followed by cell death, and the phenomenon is prevented by iron-chelators, vitamin E, butylated hydroxytoluene, and several other inhibitors of lipid peroxidation like ferrostatin-1 and liproxstatin-1 (Stockwell et al., 2017; Li J. et al., 2020). Morphologically, ferroptosis is associated with shrunken mitochondria with loss of cristae and rupture of the outer mitochondrial membrane, chromatin condensation, cytoplasmic swelling and cell membrane rupture (Li J. et al., 2020). The term ferroptosis was initially identified as a mode of non-apoptotic death triggered by erastin in fibrosarcoma cells; erastin inhibits the transport of cystine through cystine/glutamate antiport system leading to a deficient intracellular antioxidant status as a result of decreased content of reduced glutathione (Dixon et al., 2012). Many other molecules have been shown to induce ferroptosis which includes glutathione peroxidase 4 (Gpx4) inhibitor like RSL3, BSO or buthionine sulfoximine causing cellular glutathione depletion, compounds increasing intracellular iron content and inhibitors of mitochondrial electron transport chain (Cao and Dixon, 2016; Xie et al., 2016; Xu et al., 2019). Genetic studies have identified further that apart from Gpx and cystine/glutamate antiport, cellular iron transporters, iron-responsive element binding proteins, ferritin degradation, acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidyl acyltransferase 3, NRF-2 pathway, etc., are important modulators of ferroptosis which have been implicated in various pathological conditions including cancer and neurodegenerative diseases (Cao and Dixon, 2016; Xu et al., 2019; Li J. et al., 2020). Despite initial ambiguities, disruption of the plasma membrane and ROS production from NADPH oxidase, mitochondrial respiratory complexes and iron-catalyzed Fenton’s reaction are all considered important features of ferroptosis (Dixon et al., 2012; Xie et al., 2016; Xu et al., 2019).
Autophagy
Autophagy or macroautophagy is primarily a mechanism by which damaged and dysfunctional organelles or subcellular components, protein aggregates are degraded by the cell by forming a double-membrane structure (autophagosome) which fuses with the lysosomes for the final degradation of its content by the lysosomal hydrolytic enzymes (Green and Llambi, 2015; Araújo Pereira et al., 2016; Cui et al., 2021). There is some debate about whether autophagy is a pro-survival event or a cell death pathway; probably the autophagy in certain contexts functions as a cell death pathway with typical morphological features of cytoplasmic vacuolization and lysosomal degradation (Green and Llambi, 2015). Though initially characterized in yeast, many autophagy related (Atg) proteins have been identified in mammalian system which through complex interactions help in the formation and maturation of autophagosome and its subsequent fusion with lysosomes (Green and Llambi, 2015; Araújo Pereira et al., 2016; Galluzzi et al., 2017). Thus Beclin1 (Atg6), Vsp34, Vsp15 form an initiation complex which with the help of several Atg proteins finally causes conjugation of LC3 with phosphatidyl ethanolamine forming LC3II which assists in the fusion of autophagosomes with lysosomes resulting in the formation of autolysosomes; the latter process also requires other proteins especially the transmembrane LAMP2B present in the lysosomal membrane (Badadani, 2012; Green and Llambi, 2015; Galluzzi et al., 2017). Various cellular stressors such as nutrient deprivation, hypoxia, ER-stress and absence of growth factors can regulate autophagy by modulating the activities of PKA, AMPK or mTORC1 (Parzych and Klionsky, 2014; Galluzzi et al., 2017). A special type of autophagy called microautophagy simply involves the direct engulfment of autophagic substrates by invagination of the lysosomal membrane and the subsequent degradation of the captured cargo by the hydrolytic enzymes (Parzych and Klionsky, 2014; Galluzzi et al., 2017). Chaperone-mediated autophagy or CMA is distinct from the macroautophagy and involves the removal of specific proteins containing a KFERQ-like pentapeptide motif which can bind the molecular chaperone such as the heat shock cognate 70 kDa protein or hsc70; several co-chaperones like hsp90, hsp40, Bag-1 co-operate with this process (Bejarano and Cuervo, 2010; Galluzzi et al., 2017). The hsc70 facilitates the translocation of the CMA substrate proteins through lysosomal membranes with the help of the LAMP2A receptor (Bejarano and Cuervo, 2010; Galluzzi et al., 2017).
Pyroptosis
This is a form of lytic cell death, morphologically distinct from apoptosis and necrosis, which is associated with inflammation; the formation of inflammasomes, the activation of inflammatory caspases such as caspase-1/4/5/11, cleavage of the Gasdermin family of proteins especially Gasdermin-D, maturation and release of various inflammatory cytokines IL-1β and IL-18 at different phases of pyroptosis (Tan et al., 2014; Fang et al., 2020; Cui et al., 2021; Pian et al., 2021). Pyroptosis can take place either independent of inflammasome formation or by an inflammasome dependent mechanisms. When the process is independent of inflammasome formation, there is an early activation of caspase-3 which cleaves Gasdermin-E, and the N-terminal portion of Gasdermin-E oligomerizes to form a pore through the cell membrane initiating the pyroptotic death of the cells and further, the N-terminal portion of Gasdermin-E can activate the inflammasome-mediated pyroptosis (Tan et al., 2021). In other contexts, the granzyme proteases might be activated to cleave Gasdermin-B and its N-terminal portion may again form membrane pores (Tan et al., 2021). On the other hand, pyroptosis occurs through the mediation of inflammasomes either following a canonical pathway or a non-canonical pathway. In the canonical pathway, several members of NOD-like receptors (NLRP1, NLRP3, NLRC), pro-caspase-1 and other proteins such as apoptosis-associated spec-like protein containing a CARD (ASC) form a multimeric complex of inflammasome where pro-caspase-1 is cleaved to active caspase-1; the active caspase-1 helps in the maturation of IL-1β and IL-18 by proteolytic cleavage and also releases the N-terminal portion of Gasdermin-D which oligomerizes to form membrane-spanning pores (Hsu et al., 2021; Pian et al., 2021). In the non-canonical pathway inflammatory caspases (caspase-4/5/11) are activated by intracellular lipopolysaccharide or LPS, and the active caspases then cleave Gasdermin-D triggering the formation of membrane-pores (Pian et al., 2021; Tan et al., 2021). The inflammasome mediated pyroptosis is initiated by the cytosolic receptors that recognize pathogen-associated molecular patterns or danger-associated molecular patterns in the form of bacterial or viral or fungal peptides, lipopolysaccharides, β-glucan, nucleic acids or toxins or sometimes endogenous molecules (Hsu et al., 2021; Pian et al., 2021; Tan et al., 2021). A body of experimental evidence has shown that pyroptosis is important in the context of innate immunity, inflammatory response and tumor suppression and cancer therapy; additionally, it might be a key mechanism of many diseases like cerebral ischemia, myocardial infarction, neurodegenerative diseases and others where inflammation is an important component of pathology (Tan et al., 2014; Fang et al., 2020; Cui et al., 2021).
Neuronal death during development and disease
Regulated cell death signaling cascades in the brain are active during the embryonic developmental phase as well as in the early post-natal period; these pathways operate in both mitotic precursors of neurons and post-mitotic differentiated neurons in a spatially and temporally restricted manner in removing the surplus neurons, providing shape to the CNS and establishing the normal neural architecture and circuitry (Kim and Sun, 2011). The major RCD mechanism responsible for normal neural development in both embryonic and post-natal phase is apoptosis (Yamaguchi and Miura, 2015). However, in the adult brain; the cell death pathways become highly restricted, because the mature neurons utilize multiple complex mechanisms to inhibit the activation of cell death pathways (Kole et al., 2013; Hollville et al., 2019). However, the triggers and pathological changes that cause reversal of events that normally promote the survival of mature neurons are yet to be fully established. Kole et al. (2013) further stated that return to immature neuronal phenotype might be a fundamental mechanism in various neurodegenerative disorders known for progressive and widespread neuronal death. In a recent study by Mertens et al. (2021), an age-equivalent neuronal model was generated by direct conversion of fibroblasts of AD patients into induced neurons and transcriptome from such induced neurons (AD iNs) displayed immature and upregulated expression of progenitor- like signaling pathways and downregulation of properties of mature neurons. Further, these iNs reflected a hypo-mature state as they exhibited various markers of stress, de-differentiation and cell cycle re-entry, and based on these observations the authors concluded that these changes are responsible for the many pathological changes seen in neurons in AD brains. Using neurons from EOFAD patients; Caldwell et al. (2020) generated human induced pluripotent stem cell (hiPSC) and demonstrated their dedifferentiation to precursor-like state with markers of ectoderm and non-ectoderm lineages.
García-Osta et al. (2022) in their recent review highlighted that in response to various external stimuli (Aβ42, p-tau, insulin resistance) the neurons try to re-enter mitosis but there is abortive re-entry into the cell cycle that ultimately results in neuronal degeneration. Re-expression of cyclins in post mitotic neurons represents reactivation of the cell cycle and re-entry of terminally differentiated neurons; which is implicated in the pathogenesis of AD (Sharma et al., 2017). Evidence from post mortem AD brains and (iPSC)-derived neurons indicate that in the early stages of AD; neurons show various signs of de-differentiation and return to progenitor-like fates, re-enter cell cycle, become dysfunctional and ultimately lose their resilience (Koffie et al., 2011; Arendt, 2012; Kole et al., 2013). Further recent evidence from -omics data on AD patients reveals abnormal activation of various cell cycle components to be responsible for neuronal death in AD (Hooper et al., 2020). Increased levels of Cyclin E in the hippocampal region of AD brains than in age-matched controls suggest that in AD, the cell cycle is driven through the G1/S phase and this was found to match with plaque burden in the neocortex region (Bonda et al., 2010). Furthermore, in various studies on animal models, improvement in symptoms of AD was noted after administration of CDKIs (Senderowicz, 1999; Leggio et al., 2016; Wilkaniec et al., 2018; Zhang et al., 2019; Allnutt et al., 2020; Malhotra et al., 2021). To obviate the disease symptoms in such disorders; various biomolecules – Flavopiridol, Roscovitine, and Olomoucine as CDKs inhibitor, flavonoids – Resveratrol, Apigenin and Epigallocatechin-gallate for inducing cell cycle arrest at different checkpoints is used as drug for the treatment of AD and other neurodegenerative diseases (Sharma et al., 2017; Malhotra et al., 2021). Modi et al. (2015) in their study on transgenic mouse model of AD used miR-34a to regulate neuronal cell cycle by suppressing cyclin D1 thus highlighting the importance of cell cycle suppression for survival of terminally differentiated neurons.
Traxler et al. (2021) in their recent review highlighted that under pathogenic metabolic states there is a loss of resilience and re-activation of cell death mechanisms in neurons unlike the somatic cells which shift to malignant transformation. Neurons rely heavily on mitochondrial oxidative phosphorylation for energy as they require a large amount of energy to maintain ion gradients and generate action potentials with negligible proportion coming from glycolysis (Mergenthaler et al., 2013; Zhang et al., 2014; Zheng et al., 2016). However, based on several lines of evidence Traxler et al. (2021) in their review concluded that the metabolic switch from oxidative phosphorylation to glycolysis as a primary source of energy in neurons renders them susceptible to cell death as they reverse to precursor-like state. A simple activation of glycolysis in mature neurons has been suggested to act as a trigger for neuronal cell death (Herrero-Mendez et al., 2009). This metabolic switch leads to neuronal de-differentiation whereby the mature neurons return to immaturity and reach such a point at which they become apoptosis competent again (Kole et al., 2013; Su et al., 2016). Post-mortem brain tissue from AD patients further showed signs of metabolic state switch from oxidative phosphorylation to glycolysis in neurons and thus support the fact of neuronal de-dedifferentiation (Marinaro et al., 2020).
Though multiple evidence from different studies shows increased expression of immaturity markers in mature neurons derived from brains of patients with AD and other neurodegenerative diseases (Nagy et al., 1997; Jin et al., 2004; Koffie et al., 2011; Kole et al., 2013); this is not the case as contrary findings have been observed in different sets of experiments (Hagihara et al., 2019). Tobin et al. (2019) observed no difference in immaturity markers in AD vs. control brains while on the other hand decreased expression of immaturity markers in AD brains was noted by another group (Moreno-Jiménez et al., 2019). Both these observations raise strong doubts on the hypothesis of increase/upregulation in dematuration/cell cycle re-entry in AD patients. Based on these completely different set of observations, Hagihara et al. (2019) in a review concluded that re-expression of immaturity/cell cycle markers on mature neurons must be interpreted with caution.
Types of neuronal death in Alzheimer’s disease
Post-mortem studies
Apoptosis
The previous report by Lassmann et al. (1995) illustrates 30 times higher DNA fragmentation in brain tissue from AD cases than from age-matched controls. Smale et al. (1995) observed increased apoptosis in the hippocampal area in brains of AD patients as compared to age-matched non-AD brains. Further apoptosis was observed in both neurons and astrocytes. Levels of various apoptotic, pro-apoptotic and anti-apoptotic proteins were examined in cytosolic and membranous fractions of temporal cortex from AD patients and control brains and increased levels of Bcl-2 alpha, Bcl-xL, Bcl-x beta, Bak and Bad in membranous fraction, Bcl-x beta in cytosolic fraction were observed (Kitamura et al., 1998). In another study, Troncoso et al. (1996) observed TUNEL positivity in neurons, glia and microglial cells in AD brains than age-matched controls and highlighted the laminar pattern of this process in the neocortical region. On the other hand; Selznick et al. (1999) in their study noted no association of senile plaques and NFTs in cortical and hippocampal brain sections from AD patients with caspase-3 activation and concluded non-significant role of apoptosis in neuronal death seen in AD. In comparison of neurons from AD and age-matched controls, Stadelmann et al. (1999) observed caspase-3 immunoreactivity in AD neurons and in GVD granules and concluded that in AD, neuronal apoptosis progresses at a slow rate. Contrary to these findings; Su et al. (2001) studied elevated caspase 3 immuno neurons and astrocytes from AD brains and also noted its co-localization with senile plaques and NFT. Matsui et al. (2006) observed elevated mRNA levels of caspase 7 and 8 in the temporal neocortex of AD brains in comparison to control while no significant difference was detected in mRNA levels of caspase 3 and 9. The authors also noted the correlation of Aβ42 accumulation with transcriptional activation of apoptotic cascade components (Matsui et al., 2006). Based on the observation of immunoreactivity of Caspase 6 in neuritic plaques and NFTs, Albrecht et al. (2009) highlighted the role of caspase-6 in pathophysiology of AD. Burguillos et al. (2011) observed activation of caspase-8 and caspase-3/7 in microglia from the frontal cortex of AD brains and concluded that caspase signaling controls both microglia activation and neurotoxicity. Neuroinflammation mediated by microglia plays a significant role in neurotoxicity. Immunohistochemical analysis of hippocampal tissue sections from AD cases and controls by Rohn et al. (2001) revealed immunolabeling with CASP-8p18 in neurons of AD patients suggesting the role of caspase-8 in activation of caspase 3 inside the neurons of AD patients. Anderson et al. (1996) performed double labeling experiments on AD tissue to explore the association of c-Jun with DNA strand breaks and noted strong co-localization between these two markers, however gel electrophoresis of DNA isolated from AD and control brain tissue did not reveal any evidence of apoptotic or necrotic cell death mechanism in AD. Sajan et al. (2007) compared the expression of 14 apoptotic genes between normal and AD human hippocampi and noted upregulation in gene expression for c-Fos and BAK in AD patients suggesting the role of these genes in apoptotic cascades of AD. Further Lee et al. (2012) highlighted caspase-independent mode of neuronal death in AD (AIF-induced apoptosis) by studying the expression of AIF protein in different regions of the brain with increased disease progression and noted increased AIF immunoreactivity within the nucleus of hippocampal pyramidal CA1 neurons in early-stage and CA2 in the late stage of AD compared to age-matched, healthy control brains. Colurso et al. (2003) observed a significant degree of DNA fragmentation, dense plaque accumulation and moderate correlation between the two in brain tissues from AD cases compared to controls thus highlighting beta-amyloid as one of the triggers to provoke cell injury in AD. In contrast, no apoptotic morphology was noted in any areas of brain in AD patients, while many ISEL positive nuclei and glia-like cells and necrotic morphology were noted (Lucassen et al., 1997). This set of contrasting observations raises doubts about apoptosis as the main mechanism responsible for neuronal cell death in AD. Table 1 depicts cell death pathways responsible for neuronal death in postmortem AD brains with evidence from various studies.
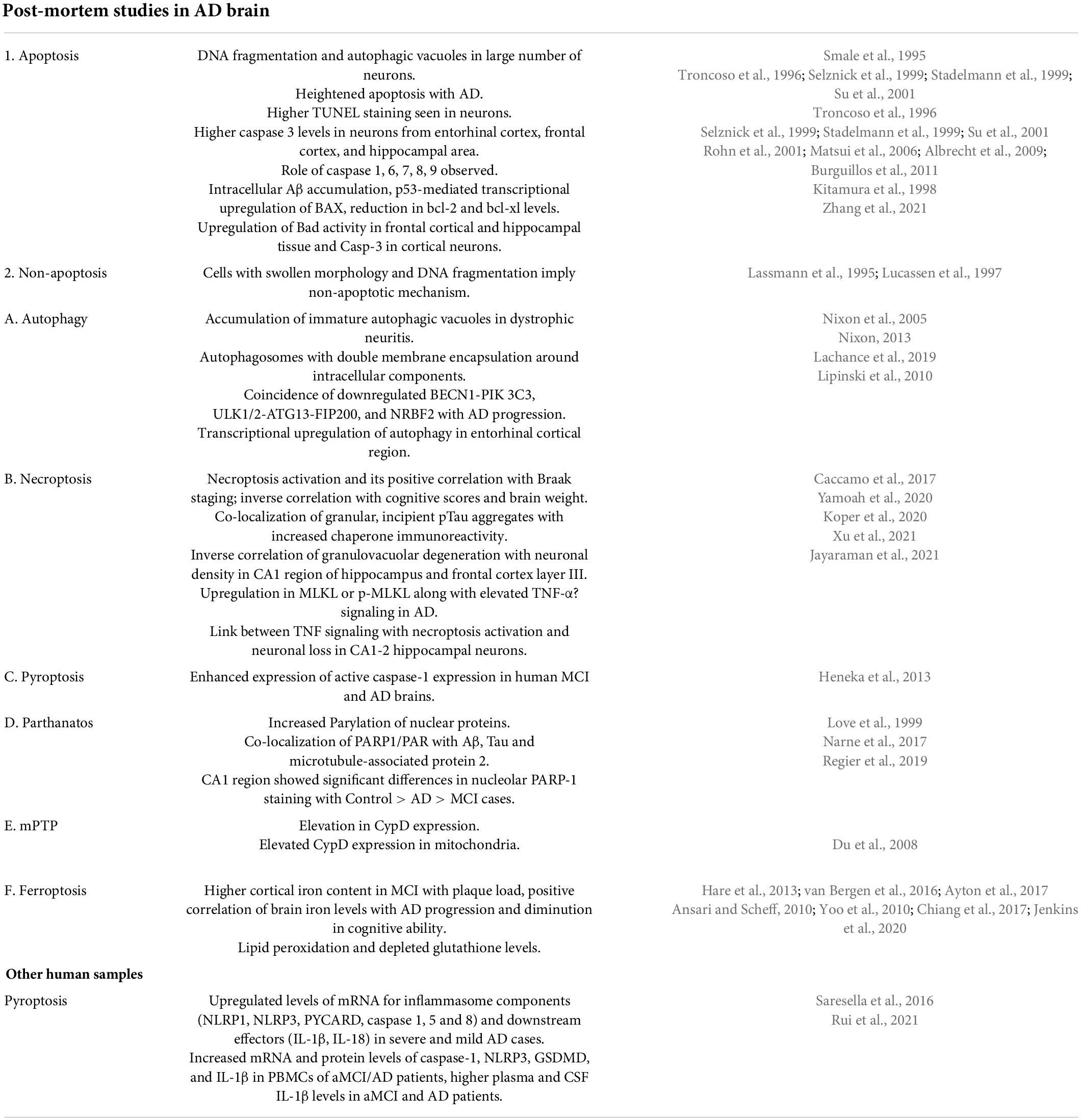
Table 1. Cell death pathways responsible for neuronal death in postmortem AD brains with evidence from various studies.
Necroptosis
Tanaka et al. (2020) in their recent study on MCI vs. AD patients observed an increased necrosis rate in brain tissues from MCI cases as compared to symptomatic AD cases, thus implying that YAP (Yes-associated protein) deprivation-induced neuronal necrosis in early AD stages much before aggregation of extracellular Amyloid beta. In their study on post-mortem human AD brains, Caccamo et al. (2017) reported necroptosis activation and its positive correlation with Braak staging while an inverse correlation was noted with cognitive scores and brain weight. Similar results of necroptosis detection in granulovacuolar degeneration, its association with neuronal loss and correlation with tauopathy and abnormal aggregation of ER chaperones and RNA binding proteins were seen in AD by Yamoah et al. (2020). Koper et al. (2020) in their recent study conducted on brains of preclinical AD patients observed inverse correlation of GVDn+ (granulovacuolar degeneration) with neuronal density in CA1 region of the hippocampus which is early affected and also with late affected frontal cortex layer III and concluded that necrosome represents necroptosis-related neuron death in AD. Consistent with these observations, Xu et al. (2021) also demonstrated upregulated levels of MLKL or p-MLKL in brains of AD patients and concluded that in AD; elevated TNF-α signaling induces necroptosis activation in neurons. Similar findings of link between TNF signaling with activation of necroptosis and loss of neurons in CA1-2 hippocampal neurons have been reported by Anusha Jayaraman et al. (2021) in their recent study on post-mortem brain tissue from AD patients. Based on their observations, the authors hypothesized that in AD; neuronal cell death by TNF-α mediated necroptosis is an early event in disease progression and this signaling pathway is amenable to various therapeutic interventions. In a very recent study, Salvadores et al. (2022) demonstrated association of necroptosis effector pMLKL with Aβ pathology and correlation of Aβo with various markers of necroptosis activation.
Autophagy
Nixon et al. (2005) observed accumulation of immature autophagic vacuoles (AV) in dystrophic neurites from AD brain samples by using an electron microscope. A striking increase in classic autophagosomes with double membrane encapsulation around heterogeneous intracellular components was reported in the brains of patients with early stages of AD by the same group several years later (Nixon, 2013). Véronik Lachance, however, reported downregulation of various autophagy kinase complex components, e.g., BECN1-PIK 3C3, ULK1/2-ATG13-FIP200, and NRBF2 and this coincided with clinical dementia progression (CDR) in AD patients (Lachance et al., 2019). Genome-wide analysis of AD patients indicated transcriptional upregulation of autophagy in entorhinal cortex region of brains of AD patients (Lipinski et al., 2010).
Parthanatosis
There is increasing evidence of the involvement of PARP-1 in causing a neuronal loss in various neurological diseases, including AD (Fatokun et al., 2014; Narne et al., 2017). Nuclear proteins Parylation was demonstrated in AD human brains (Love et al., 1999). Co-localization of PARP1/PAR with both Aβ, Tau and also microtubule-associated protein 2, which is a dendritic marker was observed in the brain of AD patients (Narne et al., 2017). Regier et al. (2019) examined the expression of PARP-1 in the hippocampi of MCI individuals and compared the same with both control and AD cases and noted decreased PARP-1 immunohistochemical staining in nucleolar region of hippocampal pyramidal cells (CA1 region) in AD and MCI patients than controls and hypothesized that disruption of both form and function of nucleolus in progression of cognitive impairment in MCI and AD cases. Further, the authors suggested decreased immunohistochemical staining of nucleolar PARP-1 in hippocampal pyramidal cells could be considered an early marker of impairment in cognition in AD patients (Regier et al., 2019).
Mitochondrial permeability transition pore
Cells can die as a result of mPT and this is another distinctive form of cell death. In an Aβ-rich environment seen in AD, the role of Aβ in formation of mPTP is speculated. CypD is a crucial regulator of mPTP and increased CypD expression favors mPTP activation (Jia and Du, 2021). Hippocampal and Cortical regions of brains of AD patients demonstrated elevated expression of CypD in mitochondria than in those from non-AD brains (Du et al., 2008). A positive association between increased expression of CypD and Aβ was noted by Du and Yan (2010).
Ferroptosis
With aging, the cerebral iron levels increase (Bartzokis et al., 2007; Acosta-Cabronero et al., 2016) and this is related to impairment in cognition (Daugherty et al., 2015; Ghadery et al., 2015). Supporting this hypothesis; Tao et al. (2014) in a meta-analysis reported significantly elevated Iron levels in multiple cortical regions of brain tissue from 300 AD cases across 19 studies. Further higher content of cortical iron was observed in MCI patients with plaque load and these brain iron levels positively correlated with progression of AD and diminution of cognition (Hare et al., 2013; van Bergen et al., 2016; Ayton et al., 2017). Iron not only increases toxic Aβ plaque formation (Becerril-Ortega et al., 2014; Ayton et al., 2017; Everett et al., 2018), hyperphosphorylation of tau (Sayre et al., 2000; Hare et al., 2013) but also induces oxidative damage in neurons directly (Thirupathi and Chang, 2019). Lipid peroxidation and depletion in levels of glutathione were noted in post-mortem studies on AD brains (Ansari and Scheff, 2010; Yoo et al., 2010; Chiang et al., 2017; Jenkins et al., 2020). Ashraf et al. (2020), however, demonstrated Iron dyshomeostasis, elevated ferritin levels and the absence of elevated elemental iron in AD patients.
Experimental evidence
Apoptosis
Results from a recent study by Zhang et al. (2021) show upregulation in the pro-apoptotic activity of Bad in frontal cortical and hippocampal tissues and caspase-3 in cortical neurons in AD brains from mice AD models. The authors also observed restoration of behavioral alteration and cognitive decline in 5X-FAD mice after loss of Bad. This loss of Bad inhibited both neuronal apoptosis and neuronal degeneration and based on their observations, the authors highlighted the role of Bad as a genetic link between apoptosis in neurons and pathology of AD (Zhang et al., 2021). Selznick et al. (1999) in their study on APPsw transgenic mouse noted no association between amyloid-beta deposition with caspase-3 activation contradictory to findings in vitro. Qian et al. (2015) studied the effect of caspase-8 knockdown on apoptosis induced by β amyloid in PC12 cells and noted significant inhibition in proliferation of PC12 cells, while caspase 8 siRNA transfection led to a reduction in apoptosis after treatment with β-amyloid. Table 2 depicts cell death pathways responsible for neuronal death in experimental studies on AD models postmortem AD brains with evidence from various studies.

Table 2. Cell death pathways responsible for neuronal death in experimental studies on AD models with evidence from various studies.
Necroptosis
Caccamo et al. (2017) in their study on a mouse model of AD, observed a reduction in cell loss when necroptosis activation was lowered and hypothesized that new therapeutic strategies blocking necroptosis activation hold strong potential in prevention and treatment of AD. Another group demonstrated that necrostatin-1 treatment in APP/PS1 double transgenic mice led to reduced levels of Aβ-oligomer, hyperphosphorylated tau and amyloid plaque, alleviated brain cell death and also ameliorated cognitive impairments (Yang et al., 2017). Upregulated levels of necroptosis markers (MLKL and p-MLKL) were noted in AD mouse models of APP/PS1 and 5xFAD mice by Xu et al. (2021) and they hypothesized that elevated TNF-α signaling in AD induce activation of neuronal necroptosis.
Autophagy
Several studies conducted on various AD models reveal that compromised mitophagy contributes to AD pathology by increasing oxidative stress, synaptic dysfunction, loss of neurons and decline in cognition. Accumulation of autophagosomes in neuronal dendrites was noted by Yu et al. (2005) in PS-1/APP double transgenic mice, however, this abnormal gathering of autophagosomes was found to occur before the formation of amyloid plaque. Similarly, another study reported accumulation of immature AVs in hippocampal neurons of AD mice much before the actual neuronal and synaptic loss (Sanchez-Varo et al., 2012). Correia et al. (2015) stated that deficiency(ies) in autophagosome pathway precedes the formation of Aβ plaques or neurofibrillary tangles in AD. Lachance et al. (2019) demonstrated that deletion of NRBF2 (a component of the BECN1-PIK3C3 complex) reduced autophagy, led to impairment of memory in mice, altered long-term potentiation (LTP) and promoted accumulation of Aβ. Furthermore, overexpression of NRBF2 in the hippocampus rescued impaired autophagy and memory deficits and also reduced β-amyloid levels and improved memory in an AD mouse model (Lachance et al., 2019).
Pyroptosis
Mounting evidence suggests activation of NLRP3 inflammasome in AD and its role in causing pathological changes in AD mice through the pyroptosis pathway (Heneka et al., 2013; Abbott, 2018). While upregulation of NLRP3, a key player of pyroptosis was observed in neurons of AD mouse models, its knockdown led to reduced neuronal death and also reversed cognitive impairment (Tan et al., 2014). Aβ and hyperphosphorylated tau (the pathological hallmark of AD) have been shown to activate the inflammasomes (NLRP1, AIM2 and NLRP3) and lead to GSDMD-mediated neuronal pyroptosis both in vitro and in vivo (Heneka et al., 2013; Tan et al., 2014; Han et al., 2020). Li Y. et al. (2020) evaluated the reciprocity between hyperphosphorylated tau and pyroptosis by using in vitro (FSK or STZ-treated PC12 cells) and in vivo (ICV-FSK or STZ rat) models and based on their observations, the authors concluded that inflammasome-mediated pyroptosis is one of the underlying mechanism responsible for neurotoxicity induced by hyperphosphorylated tau in both the AD models used. Rui et al. (2021) in a recent study observed increased levels of both mRNA and proteins of caspase-1, NLRP3, GSDMD, and IL-1β in PBMCs of aMCI/AD patients as compared to controls. Plasma and CSF IL-1β levels were found to be significantly higher in aMCI and AD patients and it correlated negatively with Aβ1–42 levels in CSF and with MMSE and MoCA scores. Based on these findings the authors inferred that there is peripheral activation of NLRP3/caspase-1/GSDMD signaling pathway in aMCI and AD patients and concluded that targeting inflammasome induced pyroptosis might serve as a novel approach to mitigate neuroinflammation seen in these patients (Rui et al., 2021). Shen et al. (2021) noted higher levels of GSDMD, t-tau and tau 181p in CSF samples of AD patients as compared to controls and highlighted good diagnostic accuracy of GSDMD in AD. Further, Saresella et al. (2016) in their study on PBMC from MCI, moderate and severe AD and healthy controls observed upregulated mRNA levels for various components of inflammasome and IL-1β, IL-18 in patients with severe and mild AD. To further support this set of observations; Heneka et al. (2013) in their study on APP/PS1 mice demonstrated that NLRP3–/– and Casp1–/– mice were protected from spatial memory loss and other AD sequelae.
Parthanatosis
Li and Jiang (2018) in their study on hippocampal mice neurons; demonstrated that amyloid β led to hippocampal neurotoxicity by inducing oxidative stress-mediated PARP1 activation, transient receptor potential melastatin-related 2 (TRPM2) activation and further causing Ca2+ influx and mitochondrial dysfunction. Martire et al. (2013) in their series of experiments on neuroblastoma cells noted that treatment with Aβ25–35 led to enhanced PARP activity which was seen to be prevented by pre-treatment of cells with MC2050 (PARP-1 inhibitor). Similar results were observed in CHO cells and brain specimens from TgCRND8 transgenic mice, thus confirming the activation of PARP-1 by Aβ25–35 induced DNA damage. Further Aβ25–35 activated NF-kB, upregulated p53 protein levels along with a decrease in Bcl-2 protein (Martire et al., 2013). Turunc Bayrakdar et al. (2014) in their study on four groups of Sprague–Dawley rats control, Aβ(1–42), Aβ(1–42) + NA (nicotinamide 100 and 500 mg/kg), noted overexpression of p53, PARP-1, NF-κB, Bax levels and decreased Bcl-2 levels in Aβ(1–42)-treated group while upregulation in Bcl-2 and downregulated PARP-1, NF-κB, p53, and Bax levels was noted with NA treatment. Crossing of PARP-1 (−/−) mouse with AD transgenic mouse could prevent the development of synaptic damage, cognitive dysfunction and also activation of microglia. Furthermore, it was observed that when PARP-1 (−/−) mice were injected with Aβ or when PARP-1 was inhibited with PJ34; lower microglial activation was induced as compared to controls. Based on these observations, the authors concluded that PARP-1 inhibition or KO improved synaptic damage, cognitive dysfunction and microglial activation (Kauppinen et al., 2011).
Mitochondrial permeability transition pore
The formation of mPTP is a mitochondrial response to both intramitochondrial as well as intracellular stresses (Jia and Du, 2021). In a study by Hansson Petersen et al. (2008) on isolated rat mitochondria; co-localization of extracellularly applied Aβ was observed with mitochondrial markers in IMM, thus suggesting that Aβ was taken by the cells. An increase in Cyp D levels was noted in an AD mouse model with overexpression of mutant human form of APP; highlighting the fact that Aβ acts as a mediator connecting AD to mPTP (Du et al., 2008). Cyp D deficiency in transgenic mice overexpressing APP and Aβ improved both synaptic and mitochondrial function and learning/memory (Du et al., 2011). The interplay between intramitochondrial Aβ and CypD results in the formation of mPTP; which reduces mitochondrial potential, decreases mitochondrial respiratory function, and increases formation of free radicals that causes oxidative stress and releases pro-apoptotic proteins (Du et al., 2008; 2011; Yao et al., 2011; Rao et al., 2014). Research using mAPP model of mice has established that mPTP formation is accompanied by increased CypD expression in cortical mitochondria with translocation to IMM, Ca2+ dyshomeostasis and mitochondrial swelling (Du and ShiDu Yan, 2010; Hung et al., 2010; Rao et al., 2014). Further to support this observation, it was noted that CypD ablation with cyclosporine A (CsA), mitigated these detrimental effects (Du et al., 2008; Yao et al., 2011). Similarly, significant improvement in learning/memory and mitochondrial function was observed by Du et al. (2008, 2011) in their study on CypD deficient mice thus highlighting the fact that Cyp-D ablation in mice protects them from behavioral and mitochondrial dysfunction throughout their lives. Mitochondria derived from CypD-deficient mAPP mouse model (mAPP/Ppif−/−) exhibited a reduction in mitochondrial swelling as well as permeability transition after induction by Ca2+ compared with mAPP mice. In a recent study on a transgenic mouse model of cerebral β-amyloidosis, it was demonstrated that accumulation of Aβ oligomers provokes Ca2+ overload in mitochondria via the MCU Complex (mitochondrial Ca2+ uniporter), thus contributing to neuronal death (Calvo-Rodriguez et al., 2020). In another study; Jadiya et al. (2019) revealed that loss of efflux from MCU leads to mCa2+ overload which accelerates the development of AD by promoting generation of superoxide, metabolic dysfunction and neuronal cell death.
Ferroptosis
In a series of experiments by Hambright et al. (2017) on GPX4BIKO mice, defects in both spatial learning and memory function were observed and were related to lipid peroxidation and increased neuroinflammation while liproxstatin-1 treatment improved neurodegeneration in these mice. Bao et al. (2021) reported downregulation of Ferropotin1 (Fpn-mammalian non-heme iron exporter) in brains of APPswe/PS1dE9 mice and genetic deletion of Fpn led to AD-like atrophy in the hippocampus region along with various memory deficits.
Considering the complex cellular and architectural diversity of brain, it is very reasonable to foresee that not one but multiple forms of cell death occur simultaneously and are responsible for AD pathogenesis (Moujalled et al., 2021). Complex interplay among these RCD cascades and it is this crosstalk between these processes that stand out as the main cause of neuronal death in AD and other neurodegenerative diseases (Saleem, 2021). Adrienne M. Gorman in a review stated that more than one type of cell death mechanism can be induced by multiple intracellular and extracellular cell death stimuli depending on several conditions such as duration and severity of stress, the health status (redox levels, integrity of mitochondria) within the cell milieu, etc. (Gorman, 2008). To maintain cellular homeostasis in response to several stressors, the neurons respond by enhancing cross-talk between various cell death pathways. Initially as a pro-survival mechanism to stressors; the cells perform autophagy and avoid cell death but when the level of stressors increases beyond a limit, autophagy is blocked while apoptotic cascades are initiated (Wu et al., 2014). Cell death becomes the only solution when the stress signals cross a certain threshold level where recovery becomes impossible. Autophagy has been reported to act as a switch that regulates cell death by shifting between necrosis and apoptosis (Goodall et al., 2016). In recent studies many key players like Beclin1, Atg1, p53, RIPK1, LC3, p62, TRIB3 play a pivotal role in switching between these death paradigms by toggling from one to the other cell death mechanism, cross communicating between various cell death pathways thus creating a complex scenario of cell death in various neurodegenerative diseases including AD (Saleem, 2021). Beclin-1 which is autophagy associated protein has been shown to interact with Bcl-2, undergo modifications and induce apoptosis and inhibit autophagy (Wang et al., 2018). Studies demonstrate induction of autophagy by pro-apoptotic protein, p53 upon proteasomal inhibition (Du et al., 2009; Dong et al., 2012). The role of p62 which is an autophagy flux marker has been suggested in apoptosis. Another protein Trib-3 (pseudo-kinase) has been shown to induce both autophagy and apoptosis. Similarly, TNF-α might cause simultaneous activation of both RIPK1-dependent apoptosis and necroptosis with interactions between each pathway (Geng et al., 2017). Considering the complex cellular and architectural diversity of the brain, it is very reasonable to foresee that not one but multiple forms of cell death occur simultaneously and are responsible for AD pathogenesis (Moujalled et al., 2021). Figure 1 depicts the schematic representation of cell death mechanisms responsible for neuronal death in AD.
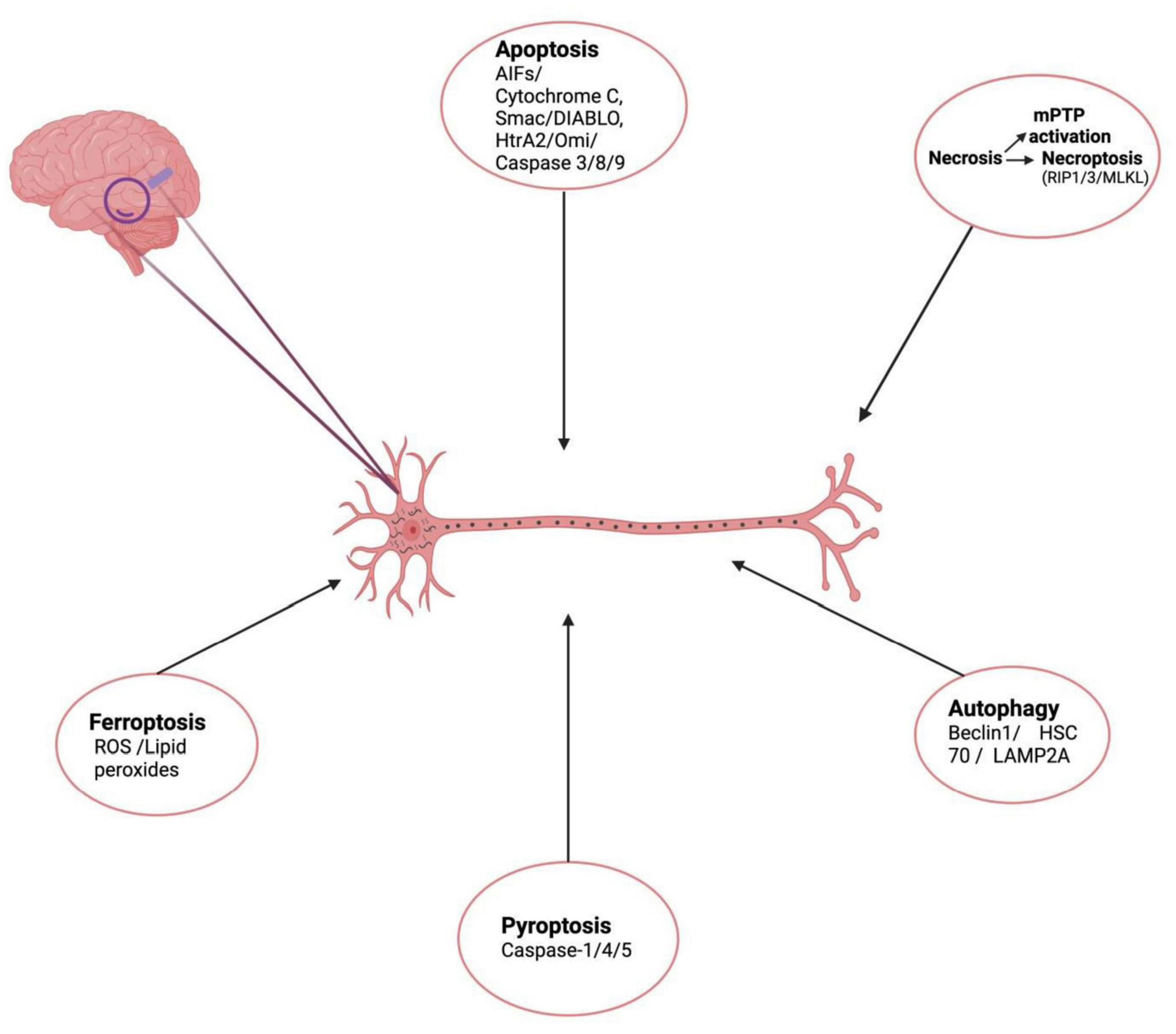
Figure 1. Schematic representation of cell death mechanisms responsible for neuronal death in AD. (Created with BioRender.com.) Apoptosis – initiated inner mitochondrial membrane changes leads to release of cytochrome c, AIF, Smac/DIABLO, HtrA2/Omi, activates effector caspases and causes apoptotic cell death. Necroptosis – is a form of RCD involving RIPK1, RIPK3, necrosome formation and MLKL activation. Another form of necrosis mPTP leading to rupture of outer membrane and non-specific release of intermembrane space proteins into cytosol, causing cell death. Autophagy – another cell death mechanism mediated by Beclin 1, HSC70, LAMP2A leads to formation of LC3II autolysosomes causing cell death. Pyroptosis – lytic form of cell death associated with inflammasomes formation, activation caspase-1/4/5 Gasdermin-D cleavage and release of inflammatory cytokines is also responsible for neuronal cell death in AD. Ferroptosis – Iron-dependent mode of cell death, induced by diverse triggers involves ROS, lipid peroxide accumulation depleted glutathione levels.
Results from a study on age-associated changes in OXYS rats at different ages reveal that the nature of alterations in various RCD pathways as outlined above depends on the stage of AD. Based on their observations, the authors concluded that during late stage of AD-like pathology; apoptosis and necroptosis gets activated because of a decline in autophagy-mediated proteostasis (Telegina et al., 2019). Since the development of AD starts significantly before the patient/s present clinically; Morsch et al. (1999) proposed that though RCD activation and neuron loss continue to take place in AD, still neurons with AD pathology continue to survive for decades. Subsequently, it is therefore projected that besides RCD activation, there is an activation of compensatory pathways within the neurons to counteract the various neurotoxic effects of the disease (Bloss et al., 2008; Okonkwo et al., 2012; Talboom et al., 2019). Multiple such cell survival pathways which promotes cell repair, inhibit cell death and respond to cytotoxic stress might be involved in compensation. These pathways include the PI3K/AKT that promotes cell survival and inhibits apoptosis (Heras-Sandoval et al., 2014; Morita et al., 2015), the MAPK pathway (Kim and Choi, 2015), and NF- κB pathway that promote cell survival by various direct and indirect mechanisms (Nikoletopoulou et al., 2013; Kim and Choi, 2015; Pires et al., 2018). These signaling pathways might on one hand promote cell survival and mitigate cell stress in AD but on the other hand; their hyperactivation might lead to neuropathology as seen in AD (Heras-Sandoval et al., 2014; Kim and Choi, 2015; Shi et al., 2016).
Triggers for neuronal death in Alzheimer’s disease
Protein aggregates
Cell death emerges as the final solution for a neuron when multiple stresses pile up to such an extent that is much beyond the healing capacity of the cell and this point acts as a trigger for activation of RCD signaling cascades ultimately resulting in the development of a multitude of neurodegenerative diseases including AD. Most of neurodegenerative diseases are linked with formation/accumulation of pathological protein(s) to the extent of formation of high-order aggregates (Soto, 2003; Blokhuis et al., 2013; Michel et al., 2016). These higher-order aggregates act as stressors and induce a plethora of cytotoxic mechanisms including upregulation in ROS levels, synaptic dysfunction, excitotoxicity, impaired degradation of protein systems, ER stress, inflammation, cell cycle re-entry, DNA damage and mitochondrial dysfunction ultimately culminating in neuronal cell death (Seward et al., 2013; Currais et al., 2016; Chi et al., 2018).
Amyloid beta and its role in neuronal cell death
Hardy and Selkoe in the year 2002 proposed the “amyloid cascade hypothesis” as the main theoretical construct for AD in which Aβ peptide was postulated as the direct causative agent responsible for progressive neurodegeneration. (De Strooper and Karran, 2016) Recently the “Aβ oligomer hypothesis” has supplanted this “amyloid cascade hypothesis” and provides a wealth of insights on the fact that it is AβOs and not amyloid plaques that play a central role in AD pathogenesis (Selkoe and Hardy, 2016). The collective evidence from various behavioral, neuropathological, and cell biological studies available in literature reveal that elevated levels of AβOs in the brain have pathogenic consequences (Cline et al., 2018). The oligomeric and fibrillar species of Aβ formed after its aggregation is toxic. After the initial transduction; downstream the AβOs have multiple effects on different subcellular organelles and have multiple effects including mitochondrial effects, ER stress, and autophagy/lysosomal dysfunction (Cline et al., 2018). Impairment in Aβ processing pathways leads to its deposition which adversely affects a large number of pathways such as neurotransmitter release, intracellular signaling cascades, autophagy regulation, lipid metabolism, and synaptic function culminating in neuronal death (Serpell et al., 2000). Soluble Aβ oligomers and insoluble aggregates of Aβ bind and activate microglia and astrocytes and stimulate a low level of chronic neuroinflammation. Furthermore, APP accumulation leads to an increase in intracellular calcium, which causes excitotoxicity, as well as an increase in pro-apoptotic molecules and heat shock proteins (Mattson et al., 1992). Multiple mechanisms induced by intraneuronal Aβ oligomers have been postulated for the etiopathogenesis of AD including ER stress-induced apoptosis (Nishitsuji et al., 2009), endosomal/lysosomal leakage, mitochondrial dysfunction (Umeda et al., 2011), oxidative stress (Tabner et al., 2005; De Felice et al., 2007), deterioration of synapses (Lacor et al., 2007; Shankar et al., 2007). Further Zhao et al. (2008, 2009) demonstrated that Aβ oligomers induce insulin resistance in AD brains while brain insulin and IGF-1 receptors dysfunction further lead to Aβ aggregation and synaptic loss. Ferretti et al. (2012) suggested neuroinflammation as one of the earliest pathological responses to intracellular Aβ-oligomers accumulation. Tomiyama et al. (2010) in their experiment on APP transgenic mice with E693Δ mutation observed that intraneuronal Aβ oligomers accumulation not only led to impairment of hippocampal synaptic plasticity and memory, downregulation of presynaptic protein synaptophysin but also abnormal phosphorylation of tau, activation of microglia and astrocyte with neuronal loss, thus highlighting that Aβ oligomers activate multiple causal mechanisms responsible for the pathogenesis of AD.
Tau
The second core pathology in AD is the formation of neurofibrillary tangles (NFT) that are formed from tau hyperphosphorylation. Tau protein gets phosphorylated at multiple sites in AD and tau gets removed from the microtubule resulting in collapse of microtubule structure and disruption of various cellular processes. Further p-tau aggregates and forms paired helical fragments and eventually, NFTs are formed. This pathogenic form of Tau at the molecular level acts as a stressor for neurons in multiple ways (Chang et al., 2018). But how tau can directly induce neuronal death is yet unanswered. Hyperphosphorylated tau accumulation alters microtubule stability, resulting in two outcomes: (1) tau interacts with synaptogyrin-3 (a pre-synaptic protein) leading to defects in synaptic release, synaptic loss and neuronal dysfunction (McInnes et al., 2018) and (2) activation of retrograde neurodegeneration and neuronal death. Tau aggregation has also been found to be linked to stocktickerDNA fragmentation, implying a relationship to regulated neuronal cell death. Further at the post-synaptic level; Tau stabilizes NMDA receptors and has been shown to elicit excitotoxicity (Ittner et al., 2010, 2016). Another study discovered that phosphorylation of numerous particular Tau residues causes cyclin D1 and BrdU incorporation to increase (Zhao et al., 2003). Prolonged NMDA receptor activation incites Ca2+, activates calpain and causes mitochondrial dysfunction leading to cell death (Bano and Ankarcrona, 2018). Furthermore, evidence from several studies indicate that tau pathology affected several systems within the cell including signaling system (Cabrales Fontela et al., 2017; Zhou et al., 2017; Bardai et al., 2018), transport system, cytoskeletal system (Fulga et al., 2007) and also disturbed mitochondrial integrity (DuBoff et al., 2012) resulting in extensive damage within the neurons.
Furthermore in a recent in vivo study; Pereira et al. (2021) demonstrated toxic effects of amyloid-β and tau on synaptic function and axonal integrity respectively in the etiopathogenesis of AD. The authors showed that early stages of AD are characterized by synaptic damage induced by amyloid deposits, impaired memory and changes in functional connectivity while tau-associated damage to axons, cognitive decline and anatomical connectivity reduction is seen in late stages of AD.
Inflammation and microglial response
Inflammation in Alzheimer’s disease plays a role in both the onset as well as progression of the disease. The presence of amyloid β has been proposed to be the primary driver for microglia activation as intracellular Aβ accumulation triggers a neuron-derived inflammatory reaction, characterized by mobilization of activated microglia toward damaged site (Welikovitch et al., 2020). A buildup of glial cells that normally keep brain debris-free results in chronic inflammation. In AD, microglia fail to remove debris and various protein aggregates including amyloid β, become chronically activated and release various pro-inflammatory and toxic products including cytokines, ROS, NO, etc. The expression of various inflammatory cytokines is higher in the vicinity of NFT and Aβ peptide deposits (Akiyama et al., 2000). Multiple studies reveal inflammatory response and activation of microglia in both post mortem tissues obtained from AD patients (Fan et al., 2015; Stefaniak and O’Brien, 2016; Passamonti et al., 2019), and in various preclinical AD models (Heppner et al., 2015; Hoeijmakers et al., 2016; Villegas-Llerena et al., 2016). Hong et al. (2016) in their study on AD mouse models demonstrated that the challenge of synaptotoxic Aβ oligomers results in inappropriate activation of microglia in adult CNS which phagocytose the synapses and causes synaptic loss and concluded that immune-related pathways and microglia act as early mediators for loss of synapse and its dysfunction much before plaque formation. Further several Genome-wide association studies state role of microglia-related molecular pathways in the etiopathogenesis of AD. TREM2 mutations on microglia are linked with an increased AD risk (Guerreiro et al., 2013; Jonsson et al., 2013). Passamonti et al. (2019) in a recent study provided clear evidence of the association of impairment in large-scale network connectivity with in vivo neuro-inflammation in AD and the degree of this association correlated with a cognitive deficit in the subjects enrolled in the study. Cognitive scores of patients correlated to brain connectivity pattern which was related to microglia activation (Passamonti et al., 2019). Rangaraju et al. (2018) compared the relevance of homeostatic and Disease-associated microglia (DAM) and noted positive association of pro-inflammatory DAM proteins with neuropathology while there was high representation of human AD risk genes within homeostatic microglia.
Open questions and conclusion
This review has clearly brought out the knowledge-gap in our understanding of the mechanisms of neuronal death in clinical cases of AD. While clearly defined RCD pathways have been demonstrated in a large number of studies in experimental AD models, it is questionable if these can be extrapolated directly to clinical AD. This is particularly because many of these studies employed transgenic AD animals which could not be considered as representatives of vast majority of AD patients who suffer from the sporadic form of the disease. On the other hand, post-mortem studies present the end-points of the pathology and fail to establish the ante-mortem scenario of neuronal death in the AD brain; this is further complicated by the post-mortem artifacts and various limitations of biochemical and immunohistochemical methods applicable to clinical samples to identify clearly the different RCD pathways. It will not be possible to resolve these issues easily, and there are other unanswered questions. For example, in experimental studies in vitro the same trigger depending on the context often leads to different RCD pathways, and there also exists extensive cross-talk among these pathways. Likewise, the inflammation, mitochondrial dysfunction and oxidative stress, the key elements of AD pathology, are linked to multiple RCD pathways. Thus, it cannot be ruled out that different neuronal populations in the AD brain may follow different pathways of death. The development of suitable imaging techniques to identify neuronal death in the ante-mortem brain can provide some answers to these questions. Finally, it is important to point out that the attempts to develop neuroprotective therapy for AD have not really targeted RCD pathways; it has mostly targeted the triggers like amyloid beta, oxidative stress, inflammation, etc., by various means without much success in clinical trials. It remains an open question if specific inhibitors of apoptosis, necroptosis or ferroptosis would provide clinical benefit in AD.
Author contributions
PG and SC provided the idea and mainly wrote the manuscript. PG and KG contributed to the search and assessment of the available literature. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank Ms Yashi Jain for her support in creating the figure using Biorender software.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbott, A. (2018). Is’ friendly fire’ in the brain provoking Alzheimer’s disease? Nature 556, 426–428. doi: 10.1038/d41586-018-04930-7
Acosta-Cabronero, J., Betts, M. J., Cardenas-Blanco, A., Yang, S., and Nestor, P. J. (2016). In vivo MRI mapping of brain iron deposition across the adult lifespan. J. Neurosci. 36, 364–374. doi: 10.1523/JNEUROSCI.1907-15.2016
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/s0197-4580(00)00124-x
Albrecht, S., Bogdanovic, N., Ghetti, B., Winblad, B., and LeBlanc, A. C. (2009). Caspase-6 activation in familial Alzheimer disease brains carrying amyloid precursor protein or presenilin I or presenilin II mutations. J. Neuropathol. Exp. Neurol. 68, 1282–1293. doi: 10.1097/NEN.0b013e3181c1da10
Allnutt, A. B., Waters, A. K., Kesari, S., and Yenugonda, V. M. (2020). Physiological and pathological roles of Cdk5: potential directions for therapeutic targeting in neurodegenerative disease. ACS Chem. Neurosci. 11, 1218–1230. doi: 10.1021/acschemneuro.0c00096
Alzheimer’s Association (2016). 2016 Alzheimer’s Disease Facts and Figures. Alzheimers Dement 12, 459–509. doi: 10.1016/j.jalz.2016.03.001
Anderson, A. J., Su, J. H., and Cotman, C. W. (1996). DNA damage and apoptosis in Alzheimer’s disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J. Neurosci. 16, 1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996
Ansari, M. A., and Scheff, S. W. (2010). Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 69, 155–167. doi: 10.1097/NEN.0b013e3181cb5af4
Araújo Pereira, P., Alvim-Soares, A., Aparecida Camargos, Bicalho, M., Nunes de Moraes, E., Malloy-Diniz, L., et al. (2016). Lack of association between genetic polymorphism of circadian genes (PER2, PER3, CLOCK and OX2R) with late onset depression and alzheimer’s disease in a sample of a Brazilian population (circadian genes, late-onset depression and Alzheimer’s disease). Curr. Alzheimer Res. 13, 1397–1406. doi: 10.2174/15672050136661606030
Arendt, T. (2012). Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mole. Neurobiol. 46, 125–135. doi: 10.1007/s12035-012-8262-0
Arendt, T., Brückner, M. K., Morawski, M., Jäger, C., and Gertz, H. J. (2015). Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropatholog. Comm. 3, 1–11. doi: 10.1186/s40478-015-0187-1
Ashraf, A., Jeandriens, J., Parkes, H. G., and So, P. W. (2020). Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol. 32:101494. doi: 10.1016/j.redox.2020.101494
Averill-Bates, D., Grondin, M. A., and Ouellet, F. (2018). Activation of apoptosis signaling pathways by reactive oxygen species. Cryobiology 80:170. doi: 10.1016/j.bbamcr.2016.09.012
Ayton, S., Fazlollahi, A., Bourgeat, P., Raniga, P., Ng, A., Lim, Y. Y., et al. (2017). Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain 140, 2112–2119. doi: 10.1093/brain/awx137
Badadani, M. (2012). Autophagy mechanism, regulation, functions, and disorders. Internat. Schol. Res. Not. 2012:97064. doi: 10.5402/2012/927064
Bano, D., and Ankarcrona, M. (2018). Beyond the critical point: an overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci. Lett. 663, 79–85. doi: 10.1016/j.neulet.2017.08.048
Bao, W. D., Pang, P., Zhou, X. T., Hu, F., Xiong, W., Chen, K., et al. (2021). Loss of ferroportin induces memory impairment by promoting ferroptosis in alzheimer’s disease. Cell Death Diff. 28, 1548–1562. doi: 10.1038/s41418-020-00685-9
Bardai, F. H., Ordonez, D. G., Bailey, R. M., Hamm, M., Lewis, J., and Feany, M. B. (2018). Lrrk promotes tau neurotoxicity through dysregulation of actin and mitochondrial dynamics. PLoS Biol. 16:e2006265. doi: 10.1371/journal.pbio.2006265
Bartzokis, G., Tishler, T. A., Lu, P. H., Villablanca, P., Altshuler, L. L., Carter, M., et al. (2007). Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol. Aging 28, 414–423. doi: 10.1016/j.neurobiolaging.2006.02.005
Becerril-Ortega, J., Bordji, K., Freret, T., Rush, T., and Buisson, A. (2014). Iron overload accelerates neuronal amyloid-beta production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiol. Aging 35, 2288–2301. doi: 10.1016/j.neurobiolaging.2014.04.019
Bejarano, E., and Cuervo, A. M. (2010). Chaperone-mediated autophagy. Proc. Am. Thor. Soc. 7, 29–39. doi: 10.1513/pats.200909-102JS
Berghe, T. V., Vanlangenakker, N., Parthoens, E., Deckers, W., Devos, M., Festjens, N., et al. (2010). Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Diff. 17, 922–930. doi: 10.1038/cdd.2009.184
Blokhuis, A. M., Groen, E. J., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6
Bloss, C. S., Delis, D. C., Salmon, D. P., and Bondi, M. W. (2008). Decreased cognition in children with risk factors for Alzheimer’s disease. Biolog. Psychiatry 64, 904–906. doi: 10.1016/j.biopsych.2008.07.004
Bonda, D. J., Lee, H. P., Kudo, W., Zhu, X., Smith, M. A., and Lee, H. G. (2010). Pathological implications of cell cycle re-entry in Alzheimer disease. Exp. Rev. Mole. Med. 12:e19. doi: 10.1017/S146239941000150X
Burguillos, M. A., Deierborg, T., Kavanagh, E., Persson, A., Hajji, N., Garcia-Quintanilla, A., et al. (2011). Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324. doi: 10.1038/nature09788
Cabrales Fontela, Y., Kadavath, H., Biernat, J., Riedel, D., Mandelkow, E., and Zweckstetter, M. (2017). Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nat. Comm. 8:1981. doi: 10.1038/s41467-017-02230-8
Caccamo, A., Branca, C., Piras, I. S., Ferreira, E., Huentelman, M. J., Liang, W. S., et al. (2017). Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20, 1236–1246. doi: 10.1038/nn.4608
Caldwell, A. B., Liu, Q., Schroth, G. P., Galasko, D. R., Yuan, S. H., Wagner, S. L., et al. (2020). Dedifferentiation and neuronal repression define familial Alzheimer’s disease. Sci. Adv. 6:eaba5933. doi: 10.1126/sciadv.aba5933
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Comm. 11, 1–7. doi: 10.1038/s41467-020-16074-2
Cao, J. Y., and Dixon, S. J. (2016). Mechanisms of ferroptosis. Cell. Mole. Life Sci. 73, 2195–2209. doi: 10.1007/s00018-016-2194-1
Chakrabarti, S., Khemka, V. K., Banerjee, A., Chatterjee, G., Ganguly, A., and Biswas, A. (2015). Metabolic risk factors of sporadic Alzheimer’s disease: implications in the pathology, pathogenesis and treatment. Aging Dis. 6:282. doi: 10.14336/AD.2014.002
Chang, H. Y., Sang, T. K., and Chiang, A. S. (2018). Untangling the Tauopathy for Alzheimer’s disease and parkinsonism. J. Biomed. Sci. 25, 1–11. doi: 10.1186/s12929-018-0457-x
Chi, H., Chang, H. Y., and Sang, T. K. (2018). Neuronal cell death mechanisms in major neurodegenerative diseases. Internat. J. Mole. Sci. 19:3082. doi: 10.3390/ijms19103082
Chiang, G. C., Mao, X., Kang, G., Chang, E., Pandya, S., Vallabhajosula, S., et al. (2017). Relationships among cortical glutathione levels, brain amyloidosis, and memory in healthy older adults investigated in vivo with (1)H-MRS and Pittsburgh compound-B PET. AJNR. Am. J. Neuroradiol. 38, 1130–1137. doi: 10.3174/ajnr.A5143
Choi, M. E., Price, D. R., Ryter, S. W., and Choi, A. (2019). Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight 4:e128834. doi: 10.1172/jci.insight.128834
Cline, E. N., Bicca, M. A., Viola, K. L., and Klein, W. L. (2018). The amyloid-β oligomer hypothesis: beginning of the third decade. J. Alzheimer’s Dis. 64, S567–S610. doi: 10.3233/JAD-179941
Correia, S. C., Resende, R., Moreira, P. I., and Pereira, C. M. (2015). Alzheimer’s disease-related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol. 34, 261–273. doi: 10.1089/dna.2014.2757
Cui, J., Zhao, S., Li, Y., Zhang, D., Wang, B., Xie, J., et al. (2021). Regulated cell death: discovery, features and implications for neurodegenerative diseases. Cell Comm. Signal. 19:120. doi: 10.1186/s12964-021-00799-8
Currais, A., Fischer, W., Maher, P., and Schubert, D. (2016). Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 31, 5–10. doi: 10.1096/fj.201601184
Daugherty, A. M., Haacke, E. M., and Raz, N. (2015). Striatal iron content predicts its shrinkage and changes in verbal working memory after two years in healthy adults. J. Neurosci. 35, 6731–6743. doi: 10.1523/JNEUROSCI.4717-14.2015
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.M607483200
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Del Re, D. P., Amgalan, D., Linkermann, A., Liu, Q., and Kitsis, R. N. (2019). Fundamental mechanisms of regulated cell death and implications for heart disease. Physiolog. Rev. 99, 1765–1817. doi: 10.1152/physrev.00022.2018
DeTure, M. A., and Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer’s disease. Mole. Neurodegen. 14, 1–18. doi: 10.1186/s13024-019-0333-5
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dong, X. X., Wang, Y. R., Qin, S., Liang, Z. Q., Liu, B. H., Qin, Z. H., et al. (2012). p53 mediates autophagy activation and mitochondria dysfunction in kainic acid-induced excitotoxicity in primary striatal neurons. Neuroscience 207, 52–64. doi: 10.1016/j.neuroscience.2012.01.018
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Du, H., Guo, L., Zhang, W., Rydzewska, M., and Yan, S. (2011). Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 32, 398–406. doi: 10.1016/j.neurobiolaging.2009.03.003
Du, H., and ShiDu Yan, S. (2010). Unlocking the door to neuronal woes in alzheimer’s disease: aβ and mitochondrial permeability transition pore. Pharmaceuticals 3, 1936–1948. doi: 10.3390/ph3061936
Du, H., and Yan, S. S. (2010). Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochimica et biophysica acta 1802, 198–204. doi: 10.1016/j.bbadis.2009.07.005
Du, Y., Yang, D., Li, L., Luo, G., Li, T., Fan, X., et al. (2009). An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy 5, 663–675. doi: 10.4161/auto.5.5.8377
DuBoff, B., Götz, J., and Feany, M. B. (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632. doi: 10.1016/j.neuron.2012.06.026
Dubois, B., Hampel, H., Feldman, H. H., Scheltens, P., Aisen, P., Andrieu, S., et al. (2016). Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement 12, 292–323. doi: 10.1016/j.jalz.2016.02.002
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Everett, J., Collingwood, J. F., Tjendana-Tjhin, V., Brooks, J., Lermyte, F., Plascencia-Villa, G., et al. (2018). Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale 10, 11782–11796. doi: 10.1039/C7NR06794A
Fan, Z., Okello, A. A., Brooks, D. J., and Edison, P. (2015). Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 138(Pt 12), 3685–3698. doi: 10.1093/brain/awv288
Fang, Y., Tian, S., Pan, Y., Li, W., Wang, Q., Tang, Y., et al. (2020). Pyroptosis: a new frontier in cancer. Biomed. Pharm. 121:109595. doi: 10.1016/j.biopha.2019.109595
Fatokun, A. A., Dawson, V. L., and Dawson, T. M. (2014). Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016. doi: 10.1111/bph.12416
Feoktistova, M., and Leverkus, M. (2015). Programmed necrosis and necroptosis signalling. FEBS J. 282, 19–31. doi: 10.1111/febs.13120
Ferretti, M. T., Bruno, M. A., Ducatenzeiler, A., Klein, W. L., and Cuello, A. C. (2012). Intracellular Aβ-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol. Aging 33, 1329–1342. doi: 10.1016/j.neurobiolaging.2011.01.007
Fulga, T. A., Elson-Schwab, I., Khurana, V., Steinhilb, M. L., Spires, T. L., Hyman, B. T., et al. (2007). Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 9, 139–148. doi: 10.1038/ncb1528
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi: 10.15252/embj.201796697
Galluzzi, L., and Kroemer, G. (2008). Necroptosis: a specialized pathway of programmed necrosis. Cell 135, 1161–1163. doi: 10.1016/j.cell.2008.12.004
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Diff. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., et al. (2012). Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Diff. 19, 107–120. doi: 10.1038/cdd.2011.96
Ganguly, G., Chakrabarti, S., Chatterjee, U., and Saso, L. (2017). Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Design Dev. Therapy 11:797. doi: 10.2147/DDDT.S130514
García-Osta, A., Dong, J., Moreno-Aliaga, M. J., and Ramirez, M. J. (2022). p27, The Cell Cycle and Alzheimer’s Disease. Internat. J. Mole. Sci. 23:1211. doi: 10.3390/ijms23031211
Geng, J., Ito, Y., Shi, L., Amin, P., Chu, J., Ouchida, A. T., et al. (2017). Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Comm. 8:359. doi: 10.1038/s41467-017-00406-w
Ghadery, C., Pirpamer, L., Hofer, E., Langkammer, C., Petrovic, K., Loitfelder, M., et al. (2015). R2* mapping for brain iron: associations with cognition in normal aging. Neurobiol. Aging 36, 925–932. doi: 10.1016/j.neurobiolaging.2014.09.013
Gómez-Isla, T., Price, J. L., McKeel, D. W. Jr., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996
Gómez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Goodall, M. L., Fitzwalter, B. E., Zahedi, S., Wu, M., Rodriguez, D., Mulcahy-Levy, J. M., et al. (2016). The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev. Cell 37, 337–349. devcel.2016.04.018 doi: 10.1016/j
Gorman, A. M. (2008). Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J. Cell. Mole. Med. 12, 2263–2280. doi: 10.1111/j.1582-4934.2008.00402.x
Green, D. R., and Llambi, F. (2015). Cell death signaling. Cold Spr. Harb. Persp. Biol. 7:a006080. doi: 10.1101/cshperspect.a006080
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. New Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guo, T., Zhang, D., Zeng, Y., Huang, T. Y., Xu, H., and Zhao, Y. (2020). Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mole. Neurodegen. 15, 1–37. doi: 10.1186/s13024-020-00391-7
Hagihara, H., Murano, T., Ohira, K., Miwa, M., Nakamura, K., and Miyakawa, T. (2019). Expression of progenitor cell/immature neuron markers does not present definitive evidence for adult neurogenesis. Mole. Brain 12, 1–6. doi: 10.1186/s13041-019-0522-8
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi: 10.1016/j.redox.2017.01.021
Han, C., Yang, Y., Guan, Q., Zhang, X., Shen, H., Sheng, Y., et al. (2020). New mechanism of nerve injury in Alzheimer’s disease: β-amyloid-induced neuronal pyroptosis. J. Cell. Mole. Med. 24, 8078–8090. doi: 10.1111/jcmm.15439
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U S A 105, 13145–13150. doi: 10.1073/pnas.0806192105
Hare, D., Ayton, S., Bush, A., and Lei, P. (2013). A delicate balance: iron metabolism and diseases of the brain. Front. Aging Neurosci. 5:34. doi: 10.3389/fnagi.2013.00034
Harman, D. (2006). Alzheimer’s disease pathogenesis: role of aging. Ann. N Y Acad. Sci. 1067, 454–460. doi: 10.1196/annals.1354.065
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Heras-Sandoval, D., Pérez-Rojas, J. M., Hernández-Damián, J., and Pedraza-Chaverri, J. (2014). The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 26, 2694–2701. doi: 10.1016/j.cellsig.2014.08.019
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S., and Bolaños, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Herrup, K. (2015). The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799. doi: 10.1038/nn.4017
Hoeijmakers, L., Heinen, Y., van Dam, A. M., Lucassen, P. J., and Korosi, A. (2016). Microglial priming and alzheimer’s disease: a possible role for (early) immune challenges and epigenetics? Front. Hum. Neurosci. 10:398. doi: 10.3389/fnhum.2016.00398
Hollville, E., Romero, S. E., and Deshmukh, M. (2019). Apoptotic cell death regulation in neurons. FEBS J. 286, 3276–3298. doi: 10.1111/febs.14970
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hooper, C., Coley, N., De Souto Barreto, P., Payoux, P., Salabert, A. S., Andrieu, S., et al. (2020). Cortical β-amyloid in older adults is associated with multidomain interventions with and without omega 3 polyunsaturated fatty acid supplementation. J. Prev. Alzheimer’s Dis. 7, 128–134. doi: 10.14283/jpad.2020.4
Hsu, S. K., Li, C. Y., Lin, I. L., Syue, W. J., Chen, Y. F., Cheng, K. C., et al. (2021). Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics 11, 8813–8835. doi: 10.7150/thno.62521
Hung, C. H., Ho, Y. S., and Chang, R. C. (2010). Modulation of mitochondrial calcium as a pharmacological target for Alzheimer’s disease. Ageing Res. Rev. 9, 447–456. doi: 10.1016/j.arr.2010.05.003
Ittner, A., Chua, S. W., Bertz, J., Volkerling, A., van der Hoven, J., Gladbach, A., et al. (2016). Site-Specific phosphorylation of tau Inhibits amyloid-beta toxicity in Alzheimer’s mice. Science 354, 904–908. doi: 10.1007/s00401-020-02191-1
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jadiya, P., Kolmetzky, D. W., Tomar, D., Di Meco, A., Lombardi, A. A., Lambert, J. P., et al. (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Comm. 10, 1–14. doi: 10.1038/s41467-019-11813-6
Jayaraman, A., Htike, T. T., James, R., Picon, C., and Reynolds, R. (2021). TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathol. Comm. 9:159. doi: 10.1186/s40478-021-01264-w
Jenkins, N. L., James, S. A., Salim, A., Sumardy, F., Speed, T. P., Conrad, M., et al. (2020). Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife 9:e56580. doi: 10.7554/eLife.56580
Jia, K., and Du, H. (2021). Mitochondrial permeability transition: a pore intertwines brain aging and alzheimer’s disease. Cells 10:649. doi: 10.3390/cells10030649
Jin, K., Peel, A. L., Mao, X. O., Xie, L., Cottrell, B. A., Henshall, D. C., et al. (2004). Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 101, 343–347. doi: 10.1073/pnas.2634794100
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. New Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kauppinen, T. M., Suh, S. W., Higashi, Y., Berman, A. E., Escartin, C., Won, S. J., et al. (2011). Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid β. J. Neuroinflamm. 8:152. doi: 10.1186/1742-2094-8-152
Kim, E. K., and Choi, E. J. (2015). Compromised MAPK signaling in human diseases: an update. Archiv. Toxicol. 89, 867–882. doi: 10.1007/s00204-015-1472-2
Kim, W. R., and Sun, W. (2011). Programmed cell death during postnatal development of the rodent nervous system. Devel. Growth Diff. 53, 225–235. doi: 10.1111/j.1440-169X.2010.01226.x
Kinnally, K. W., Peixoto, P. M., Ryu, S. Y., and Dejean, L. M. (2011). Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochimica et Biophysica Acta (BBA) 1813, 616–622. doi: 10.1016/j.bbamcr.2010.09.013
Kitamura, Y., Shimohama, S., Kamoshima, W., Ota, T., Matsuoka, Y., Nomura, Y., et al. (1998). Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer’s disease. Brain Res. 780, 260–269.
Koffie, R. M., Hyman, B. T., and Spires-Jones, T. L. (2011). Alzheimer’s disease: synapses gone cold. Mole. Neurodegen. 6:63. doi: 10.1186/1750-1326-6-63
Kole, A. J., Annis, R. P., and Deshmukh, M. (2013). Mature neurons: equipped for survival. Cell Death Dis. 4, e689–e689. doi: 10.1038/cddis.2013.220
Koper, M. J., Van Schoor, E., Ospitalieri, S., Vandenberghe, R., Vandenbulcke, M., von Arnim, C., et al. (2020). Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 139, 463–484. doi: 10.1007/s00401-019-02103-y
Korczyn, A. D. (2008). The amyloid cascade hypothesis. Alzheimers Dement 4, 176–178. doi: 10.1016/j.jalz.2007.11.008
Lachance, V., Wang, Q., Sweet, E., Choi, I., Cai, C. Z., Zhuang, X. X., et al. (2019). Autophagy protein NRBF2 has reduced expression in Alzheimer’s brains and modulates memory and amyloid-beta homeostasis in mice. Mole. Neurodegen. 14:43. doi: 10.1186/s13024-019-0342-4
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., et al. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807. doi: 10.1523/JNEUROSCI.3501-06.2007
Lassmann, H., Bancher, C., Breitschopf, H., Wegiel, J., Bobinski, M., Jellinger, K., et al. (1995). Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol. 89, 35–41. doi: 10.1007/BF00294257
Lee, J. H., Cheon, Y. H., Woo, R. S., Song, D. Y., Moon, C., and Baik, T. K. (2012). Evidence of early involvement of apoptosis inducing factor-induced neuronal death in Alzheimer brain. Anat. Cell Biol. 45, 26–37. doi: 10.5115/acb.2012.45.1.26
Leggio, G. M., Catania, M. V., Puzzo, D., Spatuzza, M., Pellitteri, R., Gulisano, W., et al. (2016). The antineoplastic drug flavopiridol reverses memory impairment induced by Amyloid-ß1-42 oligomers in mice. Pharmacol. Res. 106, 10–20. doi: 10.1016/j.phrs.2016.02.007
Li, J., Cao, F., Yin, H. L., Huang, Z. J., Lin, Z. T., Mao, N., et al. (2020). Ferroptosis: past, present and future. Cell Death Dis. 11, 1–13. doi: 10.1038/s41419-020-2298-2
Li, X., and Jiang, L. H. (2018). Multiple molecular mechanisms form a positive feedback loop driving amyloid β42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis. 9:195. doi: 10.1038/s41419-018-0270-1
Li, Y., Xu, P., Shan, J., Sun, W., Ji, X., Chi, T., et al. (2020). Interaction between hyperphosphorylated tau and pyroptosis in forskolin and streptozotocin induced AD models. Biomed. Pharmacother. 121:109618. doi: 10.1016/j.biopha.2019.109618
Lipinski, M. M., Zheng, B., Lu, T., Yan, Z., Py, B. F., Ng, A., et al. (2010). Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 107, 14164–14169. doi: 10.1073/pnas.1009485107
Love, L., Barber, R., and Wilcock, G. (1999). Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 122, 247–253. doi: 10.1093/brain/122.2.247
Lucassen, P. J., Chung, W. C. J., Kamphorst, W., and Swaab, D. F. N. (1997). DNA damage distribution in the human brain as shown by in situ end labeling; area-spe differences in aging and Alzheimer disease in the absence of apoptotic morphology. J. Neuropathol. Exp. Neurol. 56, 887–900. doi: 10.1097/00005072-199708000-00007
Malhotra, N., Gupta, R., and Kumar, P. (2021). Pharmacological relevance of CDK inhibitors in Alzheimer’s disease. Neurochem. Int. 148:105115. doi: 10.1016/j.neuint.2021.105115
Marinaro, F., Haneklaus, M., Zhang, Z., Strano, A., Evans, L., Handfield, L.-F., et al. (2020). Molecular and cellular pathology of monogenic Alzheimer’s disease at single cell resolution. bioRxiv 2020:202317. doi: 10.1101/2020.07.14.202317
Martire, S., Fuso, A., Rotili, D., Tempera, I., Giordano, C., De Zottis, I., et al. (2013). PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS One 8:e72169. doi: 10.1371/journal.pone.0072169
Matsui, T., Ramasamy, K., Ingelsson, M., Fukumoto, H., Conrad, C., Frosch, M. P., et al. (2006). Coordinated expression of caspase 8, 3 and 7 mRNA in temporal cortex of Alzheimer disease: relationship to formic acid extractable abeta42 levels. J. Neuropathol. Exp. Neurol. 65, 508–515. doi: 10.1097/01.jnen.0000229238.05748.12
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., and Rydel, R. E. (1992). beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12, 376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992
McInnes, J., Wierda, K., Snellinx, A., Bounti, L., Wang, Y. C., Stancu, I. C., et al. (2018). Synaptogyrin-3 mediates presynaptic dysfunction induced by tau. Neuron 97, 823–835. doi: 10.1016/j.neuron.2018.01.022
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Mertens, J., Herdy, J. R., Traxler, L., Schafer, S. T., Schlachetzki, J., Böhnke, L., et al. (2021). Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer’s patients. Cell Stem Cell 28, 1533.e–1548.e. doi: 10.1016/j.stem.2021.04.004
Michel, P. P., Hirsch, E. C., and Hunot, S. (2016). Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 90, 675–691. doi: 10.1016/j.neuron.2016.03.038
Modi, P. K., Jaiswal, S., and Sharma, P. (2015). Regulation of neuronal cell cycle and apoptosis by microRNA 34a. Mole. Cell. Biol. 36, 84–94. doi: 10.1128/MCB.00589-15
Moreno-Jiménez, E. P., Flor-García, M., Terreros-Roncal, J., Rábano, A., Cafini, F., Pallas-Bazarra, N., et al. (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 25, 554–560. doi: 10.1038/s41591-019-0375-9
Morita, M., Gravel, S. P., Hulea, L., Larsson, O., Pollak, M., St-Pierre, J., et al. (2015). mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14, 473–480. doi: 10.4161/15384101.2014.991572
Morsch, R., Simon, W., and Coleman, P. D. (1999). Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 188–197. doi: 10.1097/00005072-199902000-00008
Moujalled, D., Strasser, A., and Liddell, J. R. (2021). Molecular mechanisms of cell death in neurological diseases. Cell Death Diff. 28, 2029–2044. doi: 10.1038/s41418-021-00814-y
Nagy, Z., Esiri, M. M., and Smith, A. D. (1997). Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions. Acta Neuropathol. 93, 294–300. doi: 10.1007/s004010050617
Narne, P., Pandey, V., Simhadri, P. K., and Phanithi, P. B. (2017). Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons. Semin. Cell Dev. Biol. 63, 154–166. doi: 10.1016/j.semcdb.2016.11.007
Nikoletopoulou, V., Markaki, M., Palikaras, K., and Tavernarakis, N. (2013). Crosstalk between apoptosis, necrosis and autophagy. Biochimica et biophysica acta 1833, 3448–3459. doi: 10.1016/j.bbamcr.2013.06.001
Nishitsuji, K., Tomiyama, T., Ishibashi, K., Ito, K., Teraoka, R., Lambert, M. P., et al. (2009). The E693Delta mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am. J. Pathol. 174, 957–969. doi: 10.2353/ajpath.2009.080480
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Okonkwo, O. C., Xu, G., Dowling, N. M., Bendlin, B. B., Larue, A., Hermann, B. P., et al. (2012). Family history of Alzheimer disease predicts hippocampal atrophy in healthy middle-aged adults. Neurology 78, 1769–1776. doi: 10.1212/WNL.0b013e3182583047
Parzych, K. R., and Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Red. Signal. 20, 460–473. doi: 10.1089/ars.2013.5371
Passamonti, L., Tsvetanov, K. A., Jones, P. S., Bevan-Jones, W. R., Arnold, R., Borchert, R., et al. (2019). Neuroinflammation and functional connectivity in alzheimer’s disease: interactive influences on cognitive performance. J. Neurosci. 39, 7218–7226. doi: 10.1523/JNEUROSCI.2574-18.2019
Pereira, J. B., Janelidze, S., Ossenkoppele, R., Kvartsberg, H., Brinkmalm, A., Mattsson-Carlgren, N., et al. (2021). Untangling the association of amyloid-β and tau with synaptic and axonal loss in Alzheimer’s disease. Brain 144, 310–324. doi: 10.1093/brain/awaa395
Pian, Y., Zhang, X., Liu, N., Tang, L., Cong, P., and Chen, X. (2021). Pyroptosis: mechanisms and diseases. Sign. Transduct. Targ. Ther. 6:1. doi: 10.1038/s41392-021-00507-5
Pires, B., Silva, R., Ferreira, G. M., and Abdelhay, E. (2018). NF-kappaB: two sides of the same coin. Genes 9:24. doi: 10.3390/genes9010024
Poddar, J., Pradhan, M., Ganguly, G., and Chakrabarti, S. (2019). Biochemical deficits and cognitive decline in brain aging: intervention by dietary supplements. J. Chem. Neuroanat. 95, 70–80. doi: 10.1016/j.jchemneu.2018.04.002
Podtelezhnikov, A. A., Tanis, K. Q., Nebozhyn, M., Ray, W. J., Stone, D. J., and Loboda, A. P. (2011). Molecular insights into the pathogenesis of Alzheimer’s disease and its relationship to normal aging. PLoS One 6:e29610. doi: 10.1371/journal.pone.0029610
Prince, M., Wimo, A., Guerchet, M., Ali, G. C., Wu, Y. T., and Prina, M. (2015). World Alzheimer report: The global impact of dementia. An analysis of prevalence, incidence, cost and trends. London: Alzheimer’s Disease International (ADI).
Qian, M. C., Liu, J., Yao, J. S., Wang, W. M., Yang, J. H., Wei, L. L., et al. (2015). Caspase-8 Mediates amyloid-β-induced apoptosis in differentiated PC12 Cells. J. Mole. Neurosci. 56, 491–499. doi: 10.1007/s12031-015-0498-5
Qiu, C., Kivipelto, M., and Von Strauss, E. (2022). Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dial. Clin. Neurosci. 2022:2. doi: 10.31887/DCNS.2009.11.2/cqiu
Rangaraju, S., Dammer, E. B., Raza, S. A., Rathakrishnan, P., Xiao, H., Gao, T., et al. (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mole. Neurodegen. 13:24. doi: 10.1186/s13024-018-0254-8
Rao, V. K., Carlson, E. A., and Yan, S. S. (2014). Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochimica et biophysica acta 1842, 1267–1272. doi: 10.1016/j.bbadis.2013.09.003
Regier, M., Liang, J., Choi, A., Verma, K., Libien, J., and Hernández, A. (2019). Evidence for decreased nucleolar PARP-1 as an early marker of cognitive impairment. Neural. Plast. 2019:4383258. doi: 10.1155/2019/4383258
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharm. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
Rohn, T. T., Head, E., Nesse, W. H., Cotman, C. W., and Cribbs, D. H. (2001). Activation of Caspase-8 in the Alzheimer’s Disease brain. Neurobiol. Dis. 8, 1006–1016. doi: 10.1006/nbdi.2001.0449
Rui, W., Xiao, H., Fan, Y., Ma, Z., Xiao, M., Li, S., et al. (2021). Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer’s disease. J. Neuroinflam. 18:280. doi: 10.1186/s12974-021-02329-2
Sachet, M., Liang, Y. Y., and Oehler, R. (2017). The immune response to secondary necrotic cells. Apoptosis 22, 1189–1204. doi: 10.1007/s10495-017-1413-z
Sajan, F. D., Martiniuk, F., Marcus, D. L., Frey, W. H., Hite, R., Bordayo, E. Z., et al. (2007). Apoptotic gene expression in Alzheimer’s disease hippocampal tissue. Am. J. Alzheimer’s Dis. 22, 319–328. doi: 10.1177/1533317507302447
Saleem, S. (2021). Apoptosis, autophagy, necrosis and their multi galore crosstalk in neurodegeneration. Neuroscience 469:16. doi: 10.1016/j.neuroscience.2021.06.023
Salvadores, N., Moreno-Gonzalez, I., Gamez, N., Quiroz, G., Vegas- Gomez, L., Escandon, M., et al. (2022). Aβ oligomers trigger necroptosis-mediated neurodegeneration via microglia activation in Alzheimer’s disease. Acta Neuropathol. Commun. 10:31. doi: 10.1186/s40478-022-01332-9
Sanchez-Varo, R., Trujillo-Estrada, L., Sanchez-Mejias, E., Torres, M., Baglietto-Vargas, D., Moreno-Gonzalez, I., et al. (2012). Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 123, 53–70. doi: 10.1007/s00401-011-0896-x
Saresella, M., La Rosa, F., Piancone, F., Zoppis, M., Marventano, I., Calabrese, E., et al. (2016). The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mole. Neurodegen. 11:23. doi: 10.1186/s13024-016-0088-1
Sayre, L. M., Perry, G., Harris, P. L., Liu, Y., Schubert, K. A., and Smith, M. A. (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J. Neurochem. 74, 270–279. doi: 10.1046/j.1471-4159.2000.0740270.x
Scheltens, P., Blennow, K., Breteler, M. M. B., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s Disease. Lancet 388, 505–517. doi: 10.1016/s0140-6736(15)01124-1
Schmitz, C., Rutten, B. P., Pielen, A., Schäfer, S., Wirths, O., Tremp, G., et al. (2004). Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 164, 1495–1502. doi: 10.1016/S0002-9440(10)63235-X
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Selznick, L. A., Holtzman, D. M., Han, B. H., Gökden, M., Srinivasan, A. N., and Johnson, E. M. Jr., et al. (1999). In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J. Neuropathol. Exp. Neurol. 58, 1020–1026. doi: 10.1097/00005072-199909000-00012
Senderowicz, A. (1999). Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Investig. New Drugs 17, 313–320. doi: 10.1023/A:1006353008903
Serpell, L. C., Berriman, J., Jakes, R., Goedert, M., and Crowther, R. A. (2000). Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. U S A 97, 4897–4902. doi: 10.1073/pnas.97.9.4897
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spr. Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Seward, M. E., Swanson, E., Norambuena, A., Reimann, A., Cochran, J. N., Li, R., et al. (2013). Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 126(Pt 5), 1278–1286. doi: 10.1242/jcs.1125880
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Sharma, R., Kumar, D., Jha, N. K., Jha, S. K., Ambasta, R. K., and Kumar, P. (2017). Re-expression of cell cycle markers in aged neurons and muscles: whether cells should divide or die? Biochimica et biophysica acta 1863, 324–336. doi: 10.1016/j.bbadis.2016.09.010
Shi, Z. M., Han, Y. W., Han, X. H., Zhang, K., Chang, Y. N., Hu, Z. M., et al. (2016). Upstream regulators and downstream effectors of NF-κB in Alzheimer’s disease. J. Neurol. Sci. 366, 127–134. doi: 10.1016/j.jns.2016.05.022
Silva, M. T. (2010). Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 584, 4491–4499. doi: 10.1016/j.febslet.2010.10.046
Smale, G., Nichols, N. R., Brady, D. R., Finch, C. E., and Horton, W. E. Jr. (1995). Evidence for apoptotic cell death in Alzheimer’s disease. Exp. Neurol. 133, 225–230. doi: 10.1006/exnr.1995.1025
Soto, C. (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. doi: 10.1038/nrn1007
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia 7, 280–292. doi: 10.1016/j.jalz.2011.03.005
Stadelmann, C., Deckwerth, T. L., Srinivasan, A., Bancher, C., Brück, W., Jellinger, K., et al. (1999). Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am. J. Pathol. 155, 1459–1466. doi: 10.1016/S0002-9440(10)65460-0
Stefaniak, J., and O’Brien, J. (2016). Imaging of neuroinflammation in dementia: a review. J. Neurol. Neurosurg. Psychiat. 87, 21–28. doi: 10.1136/jnnp-2015-311336
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Su, J. H., Zhao, M., Anderson, A. J., Srinivasan, A., and Cotman, C. W. (2001). Activated-caspase-3 expression in Alzheimer’s and aged control brain: Correlation with Alzheimer pathology. Brain Res. 898, 350–357. doi: 10.1016/S0006-8993(01)02018-2
Su, X., Wellen, K. E., and Rabinowitz, J. D. (2016). Metabolic control of methylation and acetylation Acetylation and deacetylation. Curr. Opin. Chem. Biol. 30, 52–60. doi: 10.1016/j.cbpa.2015.10.030
Su, Z., Yang, Z., Xie, L., DeWitt, J. P., and Chen, Y. (2016). Cancer therapy in the necroptosis era. Cell Death Diff. 23, 748–756. doi: 10.1186/s12943-019-1029-8
Tabner, B. J., El-Agnaf, O. M., Turnbull, S., German, M. J., Paleologou, K. E., Hayashi, Y., et al. (2005). Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J. Biol. Chem. 280, 35789–35792. doi: 10.1074/jbc.C500238200
Talboom, J. S., Håberg, A., De Both, M. D., Naymik, M. A., Schrauwen, I., Lewis, C. R., et al. (2019). Family history of Alzheimer’s disease alters cognition and is modified by medical and genetic factors. eLife 8:e46179. doi: 10.7554/eLife.46179
Tan, M. S., Tan, L., Jiang, T., Xhu, X. C., Wang, H. F., and Jia, C. D. (2014). Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 5:e1382. doi: 10.1038/nrmicro2070
Tan, Y., Chen, Q., Li, X., Zeng, Z., Xiong, W., Li, G., et al. (2021). Pyroptosis: a new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 40:153. doi: 10.1186/s13046-021-01959-x
Tanaka, H., Homma, H., Fujita, K., Kondo, K., Yamada, S., Jin, X., et al. (2020). YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat. Comm. 11:507. doi: 10.1038/s41467-020-14353-6
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P., and Kroemer, G. (2019). The molecular machinery of regulated cell death. Cell Res. 29, 347–364. doi: 10.1038/s41422-019-0164-5
Tao, Y., Wang, Y., Rogers, J. T., and Wang, F. (2014). Perturbed iron distribution in Alzheimer’s disease serum, cerebrospinal fluid, and selected brain regions: a systematic review and meta-analysis. J. Alzheimer’s Dis. 42, 679–690. doi: 10.3233/JAD-140396
Telegina, D. V., Suvorov, G. K., Kozhevnikova, O. S., and Kolosova, N. G. (2019). Mechanisms of neuronal death in the cerebral Cortex during aging and development of Alzheimer’s disease-like pathology in rats. Internat. J. Mole. Sci. 20:5632. doi: 10.3390/ijms20225632
Thirupathi, A., and Chang, Y. Z. (2019). Brain iron metabolism and CNS diseases. Adv. Exp. Med. Biol. 1173, 1–19. doi: 10.1007/978-981-13-9589-5_1
Tobin, M. K., Musaraca, K., Disouky, A., Shetti, A., Bheri, A., Honer, W. G., et al. (2019). Human hippocampal neurogenesis persists in aged adults and alzheimer’s disease patients. Cell Stem Cell 24, 974.e–982.e. doi: 10.1016/j.stem.2019.05.003
Tomiyama, T., Matsuyama, S., Iso, H., Umeda, T., Takuma, H., Ohnishi, K., et al. (2010). A mouse model of amyloid β oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 30, 4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010
Toné, S., Sugimoto, K., Tanda, K., Suda, T., Uehira, K., Kanouchi, H., et al. (2007). Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp Cell Res. 313, 3635–3644. doi: 10.1016/j.yexcr.2007.06.018
Traxler, L., Lagerwall, J., Eichhorner, S., Stefanoni, D., D’Alessandro, A., and Mertens, J. (2021). Metabolism navigates neural cell fate in development, aging and neurodegeneration. Dis. Mod. Mech. 14:dmm048993. doi: 10.1242/dmm.048993
Troncoso, J. C., Sukhov, R. R., Kawas, C. H., and Koliatsos, V. E. (1996). In situ labeling of dying cortical neurons in normal aging and in Alzheimer’s disease: correlations with senile plaques and disease progression. J. Neuropathol. Exp. Neurol. 55, 1134–1142. doi: 10.1097/00005072-199611000-00004
Tummers, B., and Green, D. R. (2017). Caspase-8: regulating life and death. Immunol. Rev. 277, 76–89. doi: 10.1111/imr.12541
Turunc Bayrakdar, E., Uyanikgil, Y., Kanit, L., Koylu, E., and Yalcin, A. (2014). Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ(1-42)-induced rat model of Alzheimer’s disease. Free Rad. Res. 48, 146–158. doi: 10.3109/10715762.2013.857018
Umeda, T., Tomiyama, T., Sakama, N., Tanaka, S., Lambert, M. P., Klein, W. L., et al. (2011). Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 89, 1031–1042. doi: 10.1002/jnr.22640
van Bergen, J. M., Li, X., Hua, J., Schreiner, S. J., Steininger, S. C., Quevenco, F. C., et al. (2016). Colocalization of cerebral iron with amyloid beta in mild cognitive impairment. Sci. Rep. 6:35514. doi: 10.1038/srep35514
Vanlangenakker, N., Vanden Berghe, T., and Vandenabeele, P. (2012). Many stimuli pull the necrotic trigger, an overview. Cell Death Diff. 19, 75–86. doi: 10.1038/cdd.2011.164
Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., Hardy, J., and Pocock, J. M. (2016). Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 36, 74–81. doi: 10.1016/j.conb.2015.10.004
Wang, C., and Youle, R. J. (2009). The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118. doi: 10.1146/annurev-genet-102108-134850
Wang, X. T., Wu, X. D., Lu, Y. X., Sun, Y. H., Zhu, H. H., Liang, J. B., et al. (2018). Egr-1 is involved in coronary microembolization-induced myocardial injury via Bim/Beclin-1 pathway-mediated autophagy inhibition and apoptosis activation. Aging 10, 3136–3147. doi: 10.18632/aging.101616
Welikovitch, L. A., Do Carmo, S., Maglóczky, Z., Malcolm, J. C., Lõke, J., Klein, W. L., et al. (2020). Early intraneuronal amyloid triggers neuron-derived inflammatory signaling in APP transgenic rats and human brain. Proc. Natl. Acad. Sci. U S A 117, 6844–6854. doi: 10.1073/pnas.1914593117
West of Scotland Coronary Prevention Group (1996). West of Scotland Coronary Prevention Study: identification of high-risk groups and comparison with other cardiovascular intervention trials. Lancet 348, 1339–1342.
Wilkaniec, A., Gąssowska-Dobrowolska, M., Strawski, M., Adamczyk, A., and Czapski, G. A. (2018). Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid beta toxicity. J. Neuroinflam. 15, 1–18. doi: 10.1186/s12974-017-1027-y
Wirths, O., and Bayer, T. A. (2010). Neuron loss in transgenic mouse models of Alzheimer’s disease. Internat. J. Alzheimer’s Dis. 2010:723782. doi: 10.4061/2010/723782
Wirths, O., and Zampar, S. (2020). Neuron loss in Alzheimer’s disease: translation in transgenic mouse models. Internat. J. Mole. Sci. 21:8144.
Wu, H., Che, X., Zheng, Q., Wu, A., Pan, K., Shao, A., et al. (2014). Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Internat. J. Biol. Sci. 10, 1072–1083. doi: 10.7150/ijbs.9719
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Diff. 23, 369–379. doi: 10.1038/cdd.2015.158
Xu, C., Wu, J., Wu, Y., Ren, Z., Yao, Y., Chen, G., et al. (2021). TNF-α-dependent neuronal necroptosis regulated in Alzheimer’s disease by coordination of RIPK1-p62 complex with autophagic UVRAG. Theranostics 11, 9452–9469. doi: 10.7150/thno.62376
Xu, T., Ding, W., Ji, X., Ao, X., Liu, Y., Yu, W., et al. (2019). Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mole. Med. 23, 4900–4912. doi: 10.1111/jcmm.14511
Yamaguchi, Y., and Miura, M. (2015). Programmed cell death in neurodevelopment. Devel. Cell 32, 478–490. s doi: 10.1016/j.devcel.2015.s01.019
Yamoah, A., Tripathi, P., Sechi, A., Koehler, C., Guo, H., Chandrasekar, A., et al. (2020). Aggregates of RNA binding proteins and ER chaperones linked to exosomes in granulovacuolar degeneration of the Alzheimer’s disease brain. J. Alzheimer’s Dis. 75, 139–156. doi: 10.3233/JAD-190722
Yang, S. H., Lee, D. K., Shin, J., Lee, S., Baek, S., Kim, J., et al. (2017). Nec-1 alleviates cognitive impairment with reduction of Aβ and tau abnormalities in APP/PS 1 mice. EMBO Mole. Med. 9, 61–77. doi: 10.15252/emmm.201606566
Yao, J., Du, H., Yan, S., Fang, F., Wang, C., Lue, L. F., et al. (2011). Inhibition of amyloid-β (Aβ) peptide-binding alcohol dehydrogenase-Aβ interaction reduces Aβ accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 31, 2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011
Yoo, M. H., Gu, X., Xu, X. M., Kim, J. Y., Carlson, B. A., Patterson, A. D., et al. (2010). Delineating the role of glutathione peroxidase 4 in protecting cells against lipid hydroperoxide damage and in Alzheimer’s disease. Antioxid. Red. Signal. 12, 819–827. doi: 10.1089/ars.2009.2891
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J. H., et al. (2005). Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 171, 87–98. doi: 10.1083/jcb.200505082
Zhang, D., Tang, Z., Huang, H., Zhou, G., Cui, C., Weng, Y., et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. doi: 10.1038/s41586-019-1678-1
Zhang, L., Qian, Y., Li, J., Zhou, X., Xu, H., Yan, J., et al. (2021). BAD-mediated neuronal apoptosis and neuroinflammation contribute to Alzheimer’s disease pathology. Iscience 24:102942. doi: 10.1016/j.isci.2021.102942
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’Keeffe, S., et al. (2014). An RNA sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhao, W. Q., De Felice, F. G., Fernandez, S., Chen, H., Lambert, M. P., Quon, M. J., et al. (2008). Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 22, 246–260. doi: 10.1096/fj.06-7703com
Zhao, W. Q., Lacor, P. N., Chen, H., Lambert, M. P., Quon, M. J., Krafft, G. A., et al. (2009). Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta}. J. Biol. Chem. 284, 18742–18753. doi: 10.1074/jbc.M109.011015
Zhao, Z., Ho, L., Suh, J., Qin, W., Pyo, H., Pompl, P., et al. (2003). A role of P301L tau mutant in anti-apoptotic gene expression, cell cycle and apoptosis. Mol. Cell Neurosci. 24, 367–379. doi: 10.1016/S1044-7431(03)00175-1
Zheng, X., Boyer, L., Jin, M., Mertens, J., Kim, Y., Ma, L., et al. (2016). Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 5:e13374. doi: 10.7554/eLife.13374.034
Keywords: Alzheimer’s disease, apoptosis, necroptosis, autophagy, ferroptosis
Citation: Goel P, Chakrabarti S, Goel K, Bhutani K, Chopra T and Bali S (2022) Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 15:937133. doi: 10.3389/fnmol.2022.937133
Received: 05 May 2022; Accepted: 18 July 2022;
Published: 25 August 2022.
Edited by:
Ramesh Kandimalla, Indian Institute of Chemical Technology (CSIR), IndiaReviewed by:
Suvarna Prasad, All India Institute of Medical Sciences, Deoghar (AIIMS Deoghar), IndiaKahkasha, All India Institute of Medical Sciences, Deoghar (AIIMS Deoghar), India
Debapriya Bandyopadhyay, All India Institute of Medical Sciences Bhubaneswar, India
Copyright © 2022 Goel, Chakrabarti, Goel, Bhutani, Chopra and Bali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Parul Goel, cGFydWwwMDZAZ21haWwuY29t