 Kate E. Hills
Kate E. Hills Kostas Kostarelos
Kostas Kostarelos Robert C. Wykes
Robert C. Wykes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 27 June 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.903115
This article is part of the Research TopicInsights in Brain Disease Mechanisms: 2021View all 15 articles
Glioblastoma (GBM) is the most common and advanced form of primary malignant tumor occurring in the adult central nervous system, and it is frequently associated with epilepsy, a debilitating comorbidity. Seizures are observed both pre- and post-surgical resection, indicating that several pathophysiological mechanisms are shared but also prompting questions about how the process of epileptogenesis evolves throughout GBM progression. Molecular mutations commonly seen in primary GBM, i.e., in PTEN and p53, and their associated downstream effects are known to influence seizure likelihood. Similarly, various intratumoral mechanisms, such as GBM-induced blood-brain barrier breakdown and glioma-immune cell interactions within the tumor microenvironment are also cited as contributing to network hyperexcitability. Substantial alterations to peri-tumoral glutamate and chloride transporter expressions, as well as widespread dysregulation of GABAergic signaling are known to confer increased epileptogenicity and excitotoxicity. The abnormal characteristics of GBM alter neuronal network function to result in metabolically vulnerable and hyperexcitable peri-tumoral tissue, properties the tumor then exploits to favor its own growth even post-resection. It is evident that there is a complex, dynamic interplay between GBM and epilepsy that promotes the progression of both pathologies. This interaction is only more complicated by the concomitant presence of spreading depolarization (SD). The spontaneous, high-frequency nature of GBM-associated epileptiform activity and SD-associated direct current (DC) shifts require technologies capable of recording brain signals over a wide bandwidth, presenting major challenges for comprehensive electrophysiological investigations. This review will initially provide a detailed examination of the underlying mechanisms that promote network hyperexcitability in GBM. We will then discuss how an investigation of these pathologies from a network level, and utilization of novel electrophysiological tools, will yield a more-effective, clinically-relevant understanding of GBM-related epileptogenesis. Further to this, we will evaluate the clinical relevance of current preclinical research and consider how future therapeutic advancements may impact the bidirectional relationship between GBM, SDs, and seizures.
Glioblastoma (GBM) is an astrocytic-origin neoplasm categorized as a WHO grade IV glioma (Ohgaki and Kleihues, 2013). It is generally regarded as the most aggressive form of primary brain cancer in adults, evidenced by its notoriously high mortality rate. Median survival after primary diagnosis is ~14 months when following standard treatment regimens (Goodwin and Laterra, 2010; Lim et al., 2011; Szopa et al., 2017; Khandwala et al., 2021). Long-term survival is so exceedingly rare that long-term survivor status is achieved only 2.5 years post-diagnosis (Thakkar et al., 2014). The presence and progression of GBM is often associated with seizures, a debilitating comorbidity otherwise known as tumor-associated epilepsy (TAE). Epilepsy associated with GBM is regarded as a “secondary focal epilepsy” as it progressively develops secondary to the nature and presence of the primary lesion (de Curtis et al., 2015). The high chance of seizure recurrence in GBM, primarily due to the presence of the neoplasm, means that patients are often diagnosed with and treated for epilepsy following initial seizure occurrence.
Seizures manifest as a presenting symptom in two-thirds of GBM patients, with the average incidence of epilepsy throughout the disease course varying between 30% and 62% (Bruna et al., 2013; Kerkhof and Vecht, 2013; Kerkhof et al., 2013; Dührsen et al., 2019). Seizures in GBM are varied in semiology. The most common types seen in the clinic are focal to bilateral tonic clonic, and either simple or complex focal seizures (Kerkhof et al., 2013; Liang et al., 2016; Toledo et al., 2017). The initial focal onset of the seizures indicates that the transition to seizure is influenced by the location of the tumor, and in general GBM patients with tumors located in cortical regions are at the highest risk of experiencing seizures (Chaichana et al., 2009; Englot et al., 2012; Ertürk Çetin et al., 2017). Lesions situated in the temporal lobe, frontal lobe, and parietal lobe are particularly epileptogenic (Liang et al., 2016). Patients with temporal lobe involvement are thought to be most likely to develop pre-operative seizures due to the high epileptogenicity of mesial temporal lobe structures, as observed in epilepsy syndromes such as temporal lobe epilepsy (TLE). GBMs in cerebellar or brainstem regions are thought to carry a reduced risk of seizures, but these tumors are rare regardless (van Breemen et al., 2007; Lee J. W. et al., 2010; Zhang et al., 2020).
In this review, we will first consider the mechanisms that underlie epileptogenesis in GBM and the impact of their resulting bidirectional relationship on disease progression. We focus on both “intratumoral” and “peri-tumoral” mechanisms that involve epigenetic changes, neuroinflammation, BBB dysfunction, and extensive alterations to glutamatergic and GABAergic neurotransmission. We then recognize in our review that our extensive knowledge gap has implications for clinical translation, and address the challenges faced in current preclinical research. We also then discuss the potential influence of anti-tumorigenic and anti-seizure treatments on GBM-associated epileptogenesis. Finally, we evaluate how approaching these pathologies from a network level is a necessary direction for translational future research.
There are four main stages associated with clinical GBM progression: pre-resection, post-resection, recurrence, and end-of-life. The occurrence of seizures relative to these defined periods, i.e., their timing, has potential implications for disease progression and prognosis. The occurrence of seizures as a presenting symptom has historically had a more favorable relationship to prognosis, vs. their appearance post-primary diagnosis. This former connection is primarily based on the hypothesis that seizures provoke earlier tumor discovery and initiation of anti-tumorigenic treatment, translating to increased survival. Yet, recent clinical evidence has indicated a significant association between new-onset seizures post-GBM diagnosis and mortality in IDH-wildtype GBM patients (Climans et al., 2020; Wasade et al., 2020; Mastall et al., 2021). This disparity suggests that the mechanisms underlying seizure occurrence at these respective time points may slightly differ in their progression. Along the same vein, the recurrence or new-onset occurrence of seizures post-resection is associated with more negative outcomes. In particular, seizure recurrence following first-line anti-tumor therapy and successful seizure control is closely associated with disease progression (Chaichana et al., 2009; Vecht et al., 2014).
It is commonplace that patients with pre-operative seizures will experience seizure re-emergence post-resection. If GBM is considered the singular “driving force” of seizure generation then it is reasonable to suggest that removal of the bulk tumor should result in complete seizure cessation. However, this is more often not the case. Those with lesions previously situated in the frontal lobe carry the highest risk of post-operative seizures; a switch with temporal lobe lesions, which are associated with the highest pre-operative seizure likelihood (Liang et al., 2016). Interestingly, patients with no previous history of TAE have also been known to develop new-onset seizures post-tumor resection. Liang et al. (2016) found that 45% of their GBM cohort without any prior seizure history developed new-onset epilepsy 12 months post-resection. It is well established that epileptogenesis is an ongoing process that continues after seizure onset, and in the case of GBM, this process takes place in the peri-tumoral brain tissue as opposed to the tumor itself (Figure 1; Köhling et al., 2006). Seizure generation may continue or newly arise post-tumor removal, as the epileptogenic loci is often still present in the remaining “normal” parenchyma surrounding the resection cavity, otherwise known as the peri-cavity area. Furthermore, the infiltrative nature of GBM confers that small populations of tumor cells will be present in the peri-tumoral border post-resection, and these may continue to promote the cycle of epileptogenesis. The influence of tumor resection on, and the potential post-resection mechanisms underlying seizure generation will be discussed further in-depth later in our surgical resection and recurrence section.

Figure 1. Seizure occurrence and pathoanatomical changes relative to disease stages in glioblastoma. The different stages of glioblastoma progression are associated with their own relationship to seizure occurrence, of which focal and focal-to-bilateral are the most common semiology. When the bulk tumor is present (glioblastoma), the area where tumor meets “normal brain” is known as the peri-tumoral border (in red). The network organization of this area changes progressively with disease evolution. Upon resection, the peri-tumoral border is now termed the peri-cavity area. Now independent of the glioblastoma, this area may become intrinsically epileptic. When the tumor recurs, areas of the peri-cavity area now merge with the new peri-tumoral border (orange/red). At the endstage of the disease, the peri-cavity area and peri-tumoral border are simultaneously present (orange/red). Therefore, there are a multitude of mechanisms generating seizure activity both in the new peri-tumoral border and the epileptic peri-cavity area.
The disease’s end-of-life phase often includes the development of new-onset seizures in patients with no history of TAE, or recurrence of seizures in patients with previously effective anti-epileptic regimens (Oberndorfer et al., 2008; Pace et al., 2013; Koekkoek et al., 2014). The non-linear appearance of seizures with GBM progression suggests that the mechanisms by which seizures manifest prior to, or following diagnosis (pre-surgical resection) may be different to those resulting in post-resection recurrent or new-onset seizures. And these may be different still from those occurring in the final phase of the disease. The presence of seizures post-resection infers that whilst GBM appears to initially influence the development of epilepsy, the mechanisms by which seizures are continuously generated may eventually become independent of the tumor.
The importance of seizure occurrence relative to disease stage has only recently been delineated, and this reflects the historical inconsistencies within the literature when reporting seizure occurrence in GBM. The extent of the relationship between seizures and prognosis has been clouded by studies grouping GBM-associated seizures in investigations of low-grade gliomas. This is exacerbated by the lack of distinction between IDH-mutant vs. IDH-wildtype tumors in studies. It is evident that the mechanisms underlying network hyperexcitability, and their effect on disease progression, are extremely varied between tumors despite the umbrella classification of “glioma”. The inclusion of GBM-associated seizures in studies of lower-grade gliomas leads to misinterpretation of the role seizures play in GBM progression. Further to this, the IDH subtypes of GBM show distinct relationships to seizure occurrence and prognosis. Higher rates of seizures are often associated with the IDH-mutant GBMs that are known to occur in younger patients, evolve from lower-grade tumors, and have a more favorable prognosis (Alzial et al., 2022). Accordingly, the mechanisms contributing to seizure generation in this patient population (IDH-mutant) are thought to be markedly different to those occurring in IDH-wildtype patients (Liubinas et al., 2014; Berendsen et al., 2016; Toledo et al., 2017; Wasade et al., 2020). Therefore, it is crucial that future studies differentiate not only between low and high-grade gliomas but also subtypes.
The rapid growth of GBM and the presence of areas of hemorrhage or necrosis confers that there is a high probability of acute tissue damage, the consequences of which could promote epileptogenesis. Yet, clinical studies have not observed a strong correlation between tumor volume, seizures, and mass effect. Generally, smaller tumor volume is associated with seizure presence, and so other tumor-driven mechanisms may provoke epileptogenesis (Chaichana et al., 2009; Henker et al., 2019). GBM’s disorganized, uncontrolled growth inevitably leads to mutations, and consequently, there is substantial genetic heterogeneity inter- and even intratumorally (Szopa et al., 2017). Collective mutations in tumor suppressor genes PTEN (Wang et al., 1997; Han et al., 2016), TP53 (Dittmer et al., 1993; Nagpal et al., 2006; Zhang et al., 2018), and NF1 (Pearson and Regad, 2017; Soomro et al., 2017; Szopa et al., 2017) are common in primary GBM. Disruptions to their associated downstream signaling pathways are heavily implicated in tumorigenesis. Interestingly, PTEN, NF1, and TP53 are all also intrinsically linked to epilepsy. Recent experimental studies have selected them for deletion/mutation to produce genetic rodent models of GBM that reliably produce epileptogenic tumors (Hatcher et al., 2020; Hu et al., 2022). Their commonality in both GBM and epilepsy indicates that whilst the tumor itself may not be electrographically active, its intrinsic molecular properties produce an environment that is both vulnerable to and promotes mechanisms of hyperexcitability.
Physiologically, PTEN negatively regulates PI3K/AKT/mTOR pathway. However, mutated or deleted forms of PTEN are present in GBM resulting in the disinhibition of PI3K/AKT and hyperactivation of mTOR signaling (Endersby and Baker, 2008; Venkatesan et al., 2016). PTEN mutations have been associated with poorer prognosis in GBM and are also known to play a role in epilepsy syndromes. Targeted conditional deletion of PTEN in mice results in spontaneous seizures with post-mortem brains displaying the hallmark pathologies of temporal lobe epilepsy (Nishio et al., 2000; Backman et al., 2001; Luikart et al., 2011; Pun et al., 2012; Williams et al., 2015). NF1, and its protein neurofibromin 1, are known to negatively regulate Ras signaling through GTPase activity (Pearson and Regad, 2017; Soomro et al., 2017). GBM-induced loss-of-function mutations in NF1 disinhibits the Ras/MAPK pathway, leading to hyperactivation of mTOR and unchecked cell proliferation, favoring tumor progression. Targeted knockout of the NF1 gene is associated with decreased latency to epilepsy and greater seizure severity in mice (Sabetghadam et al., 2020). Its mutated form is associated with increased seizure presence in patients with neurofibromatosis-1 (Sorrentino et al., 2021). Mutations in TP53, also known as p53, disrupt its ability to induce the mechanisms of cell cycle arrest, senescence, and apoptosis that prevent proliferation and migration of damaged or neoplastic cells (Kastenhuber and Lowe, 2017). Gain-of-function (GOF) mut-p53 is a protein highly expressed in GBM that enhances pro-invasive signaling by upregulating the activity of receptor tyrosine kinases e.g., MET and EGFR (Zhang et al., 2018). Increased expression of p53, particularly within the hippocampus, is found in both experimental models and resected tissue samples from patients with drug-resistant TLE (Morrison et al., 1996; Engel et al., 2007). In seizures, overexpression of p53 is associated with greater activation of apoptotic processes and neuronal cell death, further contributing to network imbalances and potentiating mechanisms of excitability (Morrison et al., 1996). Importantly, elevated p53 expression has been previously correlated with epileptogenic GBMs (Toledo et al., 2017). It is worth noting that TP53 mutations alone are not sufficient to induce the formation of GBM, and require other genes such as PTEN to also be mutated to drive GBM progression (Zheng et al., 2008).
Whilst there are overt epileptogenic molecular mutations potentiated by GBM growth as discussed above, the epileptogenic potential of a tumor may also be influenced by the particular subpopulation of glioma cells that constitute the tumor. An interesting investigation into this relationship is seen in Lin et al. (2017). They utilized their CRISPR-Cas9 in-utero electroporation (IUE) GBM model to identify glioma-analogs of astrocyte subpopulations specifically involved in the emergence of GBM-associated seizures. One such identified was astrocyte population C which increased linearly with tumor progression. This trend was tightly correlated with increased invasion and importantly, seizure onset. Interestingly, astrocyte population C was found to be highly enriched for the expression of synaptic genes, and also for a number of established epilepsy-associated genes. This indicates an additional genetic glial basis for epileptogenesis in GBM. These findings also confer that some of the most intrinsic properties of the tumor, the genes enriching the glioma cells, directly influence the development of epilepsy.
GBM is one of the most vascularized tumors in humans and its associated neoangiogenesis promotes blood-brain barrier (BBB) disruption. This breakdown allows infiltration of serum proteins into the brain parenchyma, encourages vasogenic oedema, and increases intracranial pressure (Noell et al., 2012). Pathologic BBB disruption is readily apparent in GBM patients using gadolinium contrast MRI, whereby the leaky BBB enables gadolinium to diffuse into the tissue and presents as ring-enhancing lesions (Wolburg et al., 2012; Dubois et al., 2014). Morphologically, blood vessels involved in the GBM blood-tumor barrier are typically non-uniform with altered pericyte coverage, fenestrations, and reduced tight junctions. For in-depth reviews focused on tumor progression and its impact on the BBB, please see Dubois et al. (2014) and Arvanitis et al. (2020). It is notable that BBB dysfunction is observed in both human and animal studies following acute seizures, and its leakage is particularly associated with TLE (van Vliet et al., 2007; Marchi et al., 2012). As a corollary, there may be additional effects on the BBB in GBM due to the effect of epileptogenesis on pericytes, please see Löscher and Friedman (2020) and Yamanaka et al. (2021) for further discussion of this work. Interestingly, epileptogenesis secondary to BBB injury largely involves the activation of inflammatory responses and reactive astrogliosis, attributes observed in the GBM microenvironment.
The exposure of the brain to blood serum components such as fibrinogen, albumin, glutamate, and K+ following GBM-induced BBB disruption contributes to changes in neuronal excitability. The transformation of astrocytes under these conditions further provokes epileptogenic changes. The action of albumin at transforming growth factor beta (TGF-β) receptors induces changes in the expression of critical inwardly rectifying potassium channels (Kir; Ivens et al., 2007; Marchi et al., 2012) and water channels i.e., aquaporin 4 (AQP4; Ikeshima-Kataoka, 2016). In human and animal models of epilepsy syndromes, it has been shown that serum-exposure can lead to the direct downregulation or mislocalization of Kir4.1 astrocytic K+ channels consequently resulting in disrupted K+ homeostasis (Campbell et al., 2012; Buckingham and Robel, 2013; Pallud et al., 2013). Kir4.1 is responsible for buffering extracellular K+ and maintaining a negative resting membrane potential (RMP; Olsen and Sontheimer, 2004; Hibino et al., 2010). Therefore, GBM-induced disruption would likely result in RMP dysregulation of astrocyte processes, accumulation of extracellular K+ and consequently a lowered neuronal firing threshold (Table 1; Olsen and Sontheimer, 2004). GBM-induced AQP4 dysregulation and redistribution is known to be associated with increased peri-tumoral vasogenic oedema and aberrant BBB function (Warth et al., 2007).
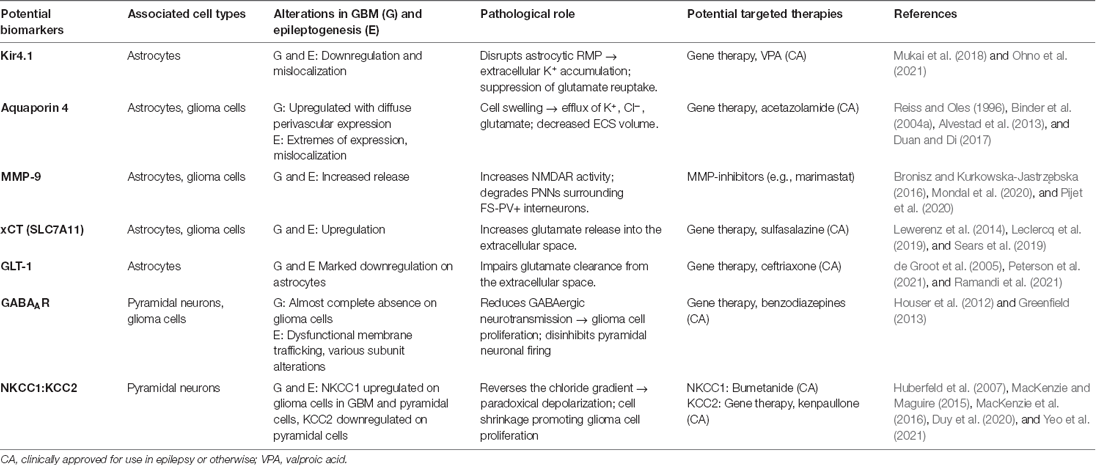
Table 1. Potential biomarkers in GBM-associated epilepsy, their relationship to each pathology, role, and potential targeted therapies.
Notably, Kir4.1-mediated K+ buffering also plays a crucial role in glutamate homeostasis as the channel tends to colocalize with GLT-1 and AQP4 at tripartite synapses (Nagelhus et al., 2004; Olsen and Sontheimer, 2008). Glutamate reuptake is suppressed in the case of Kir4.1 inhibition, due to disruption of ionic gradients and astrocytic RMP, with epileptogenic consequences. These occur secondary to greater extracellular glutamate accumulation, as will be discussed later in glutamate release, reuptake, and post-synaptic action (Kucheryavykh et al., 2007; Armbruster et al., 2021). Both downregulation and mislocalization of Kir4.1 has been demonstrated in patient-derived GBM samples and cell lines (Zurolo et al., 2012; Madadi et al., 2021). As well as significant electrophysiological changes including a depolarized astrocytic RMP and large outward K+ currents (Olsen and Sontheimer, 2004).
Accumulation of extracellular K+ is known to be a key initiating event in the generation of spreading depolarization (SD). SD is characterized by a near-complete regional depolarization of neurons and glial cells that is accompanied by the collapse of transmembrane ion gradients and followed by a temporary suppression of neuronal activity (Pietrobon and Moskowitz, 2014; Ayata and Lauritzen, 2015). SDs are primarily self-terminating in healthy brain tissue as the resources required to recover from major disruptions to transmembrane ion gradients are readily available. However, in a metabolically compromised brain, such as one with GBM, there is decreased availability of these resources. Consequently, repolarization and recovery is significantly delayed to a point that may exacerbate existing tissue damage (Ayata and Lauritzen, 2015). The complex relationship between seizures and SDs is well documented, seizures create an excitotoxic environment conducive to SD generation and vice versa (Berger et al., 2008; Kramer et al., 2017). The BBB disruption and subsequent steady increase in K+ accumulation that occurs within the GBM peri-tumoral border contributes to a progressive reduction in the threshold for seizures and SDs (Figure 2).

Figure 2. The vicious cycle of hyperexcitability in glioblastoma (GBM) progression. How epileptogenesis is initiated, is influenced by, and contributes to GBM growth. (1) GBM growth and its interaction with neuronal/glial cells initiates the cycle to epileptogenesis. (2) Upregulation of xCT on glioma cells and downregulation of KCC2 on surrounding pyramidal neurons creates an initial imbalance between excitatory and inhibitory neurotransmission. (3) Pro-inflammatory cytokines and MMPs present in the GBM TME act to degrade the PNNs surrounding GABAergic interneurons. (3) Once epileptogenesis has been initiated, further proinflammatory cytokine, K+, and glutamate release exacerbates BBB disruption and encourages network reorganization creating a microenvironment conducive to SDs. (4) The function of Kir4.1, AQP4 and GLT-1 transporters is disrupted by recurrent seizures and SDs, and via actions of GBM. (5) This dysfunction continues in a cyclical fashion whereby seizures promote more seizures and SDs, and the associated pathological downstream signaling actively potentiates glioma cell proliferation, and vice versa. BBB, blood brain-barrier; TME, tumor microenvironment; MMPs, matrix metalloproteinases; PNNs, perineuronal nets; SDs, spreading depolarizations; APQ4, aquaporin 4.
Upregulation and redistribution of AQP4 is widely reported in GBM (Figure 2). Interestingly significantly increased diffuse and perivascular expression of AQP4 on glioma cells is seen in resected tumor tissue from GBM patients with seizures vs. those without (Isoardo et al., 2012). Dysregulation of astrocytic AQP4 expression is known to contribute to epileptogenesis in traditional epilepsy syndromes (Binder and Steinhäuser, 2006; Binder et al., 2012). AQP4-knockout mice demonstrate increased latency to seizure generation when challenged with pentylenetetrazole (PTZ). This is largely attributed to expansion of the extracellular space (ECS), but seizures showed a more severe phenotype once eventually generated (Binder et al., 2004a, 2006). Conversely, significant AQP4 upregulation is observed in TLE and in the chronically epileptic brain following intra-hippocampal kainic acid (IHKA) epilepsy induction (Lee et al., 2004; Hubbard et al., 2016). This suggests that extremes of AQP4 expression contribute to epileptogenesis (Binder et al., 2004a, b). GBM-induced upregulation of AQP4 expression may lead to cell swelling, compensatory efflux of K+, Cl−, and glutamate, and decreased ECS volume (Table 1). The net effect of which would favor neuronal depolarization and potentiation of excitatory activity throughout the surrounding neuronal network (Binder et al., 2012). Therefore, GBM-induced alterations to both AQP4 and Kir4.1 aggressively promote microenvironment changes conducive to both seizures and SDs.
The extreme infiltrative and epileptogenic nature of GBM can be, at least in part, attributed to its master manipulation of the immune system. GBMs are primarily immunosuppressive tumors, and this is primarily due to the high presence of tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and regulatory T cells (Tregs). These mediators are crucial in maintaining GBM’s extreme immunosuppressive phenotype and are also directly linked to cancer cell proliferation and the recruitment of surrounding non-neoplastic cells (Fanelli et al., 2021). However, whilst the net effect of the tumor microenvironment (TME) is immunosuppressive, this does not preclude the existence of underlying pro-inflammatory actions. The peri-tumoral environment is inundated with a variety of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α), chemokines, and extracellular matrix remodeling enzymes (MMP-2, MMP-9; Figure 2; DeCordova et al., 2020). Reactive astrocytes, which also produce said mediators (Guan et al., 2018), are also increasingly present. In particular, IL-1β and IL-6 are well recognized to contribute to oncogenesis and their expression is abundantly found in GBM cell lines and tissues (Lu et al., 2007; Yeung et al., 2012; Liu et al., 2021), IL-6, in particular, is associated with poorer prognosis (Hori et al., 2019). IL-1β signaling stimulates activation of NF-kB, ERK, and p38 MAPK pathway signaling, as well as promoting the release of IL-6 and IL-8 from GBM cells (Yeung et al., 2013). In turn, the release of IL-8 is postulated to induce TNF-α secretion from TAMs (Wei et al., 2021).
Interestingly, the pro-inflammatory molecules seen within the TME are historically associated with seizures (Rana and Musto, 2018). It is well recognized that neuroinflammation and associated BBB breakdown are necessary for epileptogenesis (Marchi et al., 2007). Experimental models of TLE, and resected hippocampal tissue from TLE patients both display upregulated expressions of pro-inflammatory molecules, reactive astrocytosis, and activated microglia (Ravizza et al., 2008). Transgenic mice overexpressing IL-6 and TNF-α demonstrate increased seizure susceptibility (Campbell et al., 1993; Probert et al., 1997; Samland et al., 2003), and treatment with IL-1β prior to kainic acid administration potentiates seizure duration (Vezzani et al., 1999). IL-1β exerts its pro-convulsive actions through IL-1R (IL-1 receptor)-mediated phosphorylation of the NMDAR GluN2B subunit (Viviani et al., 2003). TNF-α upregulates Ca2+-permeable AMPAR (CP-AMPAR) membrane expression and trafficking, enabling greater Ca2+ influx with cytotoxic effects. It also acts to increase the internalization of GABAA receptors (GABAARs), subsequently reducing the influence of GABAergic inhibitory neurotransmission (Stellwagen et al., 2005). IL-1β and TNF-α are also known to increase glutaminase activity (Bezzi et al., 2001; Ye et al., 2013), which results in enhanced glutamate release whilst also decreasing its re-uptake (Hu et al., 2000; Yeung et al., 2013; Rana and Musto, 2018). All alterations to network function that can be interpreted as epileptogenic.
Concomitant to pro-inflammatory cytokine secretion, GBM cells release matrix metalloproteinase 9 (MMP-9). Chronic MMP-9 production is associated with BBB disruption, independent of the insult, and mediates its pathological effects by decreasing the efficiency of BBB tight junctions (Asahi et al., 2001). MMP-9 also impacts both excitatory and inhibitory neurotransmission. It acts to increase the activity of NMDARs through integrin-β1 signaling (Michaluk et al., 2009), and induces the degradation of perineuronal nets (PNNs) that surround fast-spiking parvalbumin positive (FS-PV+) inhibitory interneurons (Figure 2; Tewari et al., 2018). These alterations increase the excitability of the peri-tumoral network, and when occurring in conjunction with the epileptogenic mechanisms mentioned in the following section, result in seizure generation. These are just a few mechanisms by which GBM promotes network hyperexcitability to the point of recurrent seizure generation. Following which the seizures themselves potentiate tumor progression and increased excitability in a malignant feed-forward nature.
In the GBM peri-tumoral environment, there is a breakdown in glutamate homeostasis and subsequent pathological network signaling creates an environment favorable to seizures and SDs. In GBM, glutamate is primarily released into the synaptic cleft by the cystine/glutamate antiporter system (system xc-), also known by its active subunit xCT (Buckingham et al., 2011). System xc- primarily mediates the exchange of intracellular L-glutamate for extracellular L-cystine, a precursor for the antioxidant glutathione (GSH). Its expression is known to be significantly upregulated in the tumor tissue of epileptic GBM patients (Lin et al., 2020). Upregulated expression of system xc- in GBM cells leads to greater concentrations of glutamate in the extracellular space (Table 1). This promotes hyperexcitability through activation of post-synaptic glutamatergic receptors on peri-tumoral neurons, subsequently initiating processes of depolarization and excitotoxicity (Figure 2; Buckingham et al., 2011; Neal et al., 2016). As a corollary, increased xCT expression has been associated with shorter progression-free survival and poorer overall survival in GBM patients, as well as a more infiltrative phenotype on MRI (Takeuchi et al., 2013). Interestingly, the expression of xCT in glioma patients has been found to significantly correlate with epileptic seizures at GBM diagnosis, even denoted as an “independent biomarker” (Sørensen et al., 2018). Notably, epileptiform activity can be reduced by treatment of both epileptogenic GBM-bearing acute brain slices and mice with sulfasalazine (SAS), an FDA-approved system xc- antagonist (Table 1; Buckingham et al., 2011; Campbell et al., 2012; Robert et al., 2015; Hatcher et al., 2020). There are no current clinical trials looking at SAS as a treatment for GBM-related epilepsy. Yet, a phase 2 randomized clinical study evaluating SAS as a primary anti-tumor treatment in recurrent GBM was conducted 2005–2007. This trial was terminated early due to unreasonable occurrence of adverse events and lack of tumor response. However, this study was conducted in a severely ill, neurologically impaired patient population. They also neglected to assess the anti-epileptic potential of SAS as an adjuvant treatment to standard anti-tumor regimens and therefore this avenue would still be of great interest (Robe et al., 2009).
The disruption in peri-tumoral glutamate homeostasis is thought to be further exacerbated by GBM-induced downregulation of astrocytic glutamate transporter 1 (GLT-1; Table 1). This impairs clearance of glutamate from the synaptic cleft and indirectly acts to potentiate its actions at post-synaptic NMDARs/AMPARs (Robert and Sontheimer, 2014). GLT-1 (also known as EAAT2) is responsible for 80%–90% of all extracellular glutamate reuptake activity (Lin et al., 2012; Takahashi et al., 2015), but both GBM tumor tissue and cell lines show an almost complete absence of the transporter (Ye et al., 1999; de Groot et al., 2005). Initial experimental evidence suggests that GLT-1 expression is significantly downregulated in both tumoral and peri-tumoral tissue from GBM patients with seizures vs. those without (Yuen et al., 2012). Interestingly, parallels are seen in TLE patients with hippocampal sclerosis and drug-refractory seizure phenotypes where GLT-1 expression is markedly reduced (Proper et al., 2002; Sarac et al., 2009). Additionally, GLT-1 knockout mice are known to develop lethal spontaneous seizures (Tanaka et al., 1997), and GLT-1 conditional knock-out mice demonstrate increased susceptibility to SD (Aizawa et al., 2020). A reduction in GLT-1 expression favors SD propagation by allowing a more rapid increase in extracellular glutamate concentration during the course of the phenomenon (Figure 2). Direct links are yet to be fully elucidated in the literature, but current findings support the theory that the pathological mechanisms underlying seizures and SDs may be in part initiated by GBM-induced GLT-1 downregulation.
The pathological downstream signaling cascades that occur secondary to extracellular glutamate accumulation in GBM are primarily mediated by post-synaptic NMDARs and AMPARs. Glutamate-mediated prolonged NMDAR and CP-AMPAR activation triggers a wide variety of intracellular events, that in turn initiate processes of Ca2+-mediated excitotoxicity and apoptosis (Zhu et al., 2000; Buckingham and Robel, 2013; Henley and Wilkinson, 2016). In both peri-tumoral human and murine GBM tissue, there is increased phosphorylation of the NMDAR GluN2B subunit, a widely used measure of NMDAR activation (Gao et al., 2014). Notably, a relative increase in the NMDAR GluN2B subunit confers slower receptor decay times enabling a greater volume of Ca2+ influx through the receptor, thereby increasing the risk of Ca2+-mediated excitotoxicity and neuronal death. NMDARs are also implicated in the propagation of SDs, therefore any alterations to NMDAR activation may potentiate SD propagation and alter their waveform (Masvidal-Codina et al., 2021).
There are marked decreases in the editing of the AMPAR GluA2 subunit and increases in the expression of GluA1 subunits in human GBM samples and cell lines (Maas et al., 2001; de Groot et al., 2008; Venkataramani et al., 2019). This indicates that there is Ca2+ influx through, and increased activity at, post-synaptic ionotropic glutamate receptors that trends towards increased neuronal excitability. Epileptiform activity originating from GBM-bearing tissue can be completely abolished by the administration of NMDAR antagonist APV (Buckingham et al., 2011; Campbell et al., 2012), and AMPAR antagonists perampanel and cyanquixaline (CNQX; Venkataramani et al., 2019; Venkatesh et al., 2019; Lange et al., 2020; Mayer et al., 2020). When occurring in GBM, the surrounding cell death occurring secondary to overactivation of NMDARs/AMPARs could encourage greater tumor growth by “making space” and further exacerbate the network imbalance fuelling excitability.
The generation of seizures in peri-tumoral neuronal networks inevitably disrupts ionic homeostasis. This primarily manifests as increased extracellular K+ concentration and intracellular Na+, Cl−, and Ca2+ concentrations, as well as a decrease in intracellular pH (Raimondo et al., 2015). Moreover, recurrent seizures provoke excess glutamate release from pre-synaptic neurons. Parallels can be drawn between the signaling pathways initiated by seizures/SDs and those potentiated by GBM. The dysfunctional actions of glutamate transporters and receptors also create an environment conducive to glioma cell proliferation, migration, and invasion. Blockade of xCT with SAS consistently reduces GBM glioma cell migration in multiple cell lines, an anti-tumor effect corroborated in vivo (Lyons et al., 2007). GLT-1 downregulation also contributes to GBM oncogenesis, in physiological conditions it plays a role in growth suppression therefore the absence of this transporter on glioma cells allows their growth to proceed unchecked. Adeno-associated virus (AAV)-mediated reintroduction of GLT-1 into GBM cells mitigated in vitro growth. Furthermore, flank implantation of U87 cells transfected ex vivo with AAV-GLT-1 into nude mice was associated with significantly decreased tumor growth (de Groot et al., 2005). As such, evidence suggests that glutamate released by GBM glioma cells may be able to promote pro-migration/invasion and proliferative effects through seizure-induced alterations to the functionality of GLT-1, NMDARs, and AMPARs.
GBM induces major perturbations to inhibitory GABAergic neurotransmission. These disruptions originate from a loss of GABAergic synaptic density on pyramidal neurons, reductions in GABAAR expression, and changes to the Cl− gradient. GBM-induced degradation of the PNNs surrounding FS-PV+ inhibitory interneurons significantly decreases their frequency of action potential firing (Figure 2; Tewari et al., 2018). Significant selective loss of FS-PV+ interneurons is also observed within the peri-tumoral cortex, secondary to excitoxicity associated with increased extracellular glutamate accumulation (Tewari et al., 2018). Cumulatively, this manifests as reduced GABAergic neurotransmission and disinhibition of pyramidal neurons within the peri-tumoral network. GBM also induces a reduction in the density of GABA binding sites, such that functional GABAARs are postulated to be almost completely absent on glioma cells (Labrakakis et al., 1998; Smits et al., 2012; Jung et al., 2019). Experimental studies have demonstrated that the majority of GBM cells, from both human GBM samples and cell lines, do not exhibit any electrophysiological response when challenged with the application of GABA. This is suggestive of a lack of functional GABA receptors (Labrakakis et al., 1998). Whilst glioma cells appear to lack functional GABAARs, they are still postulated to be present on surrounding peri-tumoral neurons allowing them to play a role in epileptogenic neurotransmission (Campbell et al., 2015; Huberfeld and Vecht, 2016).
GBM-induced alterations to the expression ratio of Cl− transporters KCC2 and NKCC1 influence GABAergic neurotransmission by extruding and accumulating intracellular Cl− ([Cl−]i), respectively. They are primarily responsible for the pathological reversal of the Cl− gradient that occurs in GBM (Liu et al., 2020; Zhang et al., 2021). Higher KCC2 expression ensures a low [Cl−]i concentration such that GABA binding elicits Cl− influx and cell hyperpolarization, potentiating inhibitory GABAergic signaling. Whereas increased NKCC1 expression in mature neurons results in a greater concentration of [Cl−]i and reversal of the chloride gradient, promoting Cl− efflux upon GABA binding and cell depolarization (Plotkin et al., 1997; Hübner et al., 2001). Various epilepsy syndromes commonly demonstrate alterations in both Cl− cotransporters (Liu et al., 2020). Significant downregulation of KCC2 is known to occur within the GBM peri-tumoral cortex (Figure 2), correlating with greater [Cl−]i concentration and a more positive GABA equilibrium potential (Conti et al., 2011; Pallud et al., 2014; MacKenzie et al., 2016). This has also been shown to significantly correlate with decreased survival in GBM patients (Campbell et al., 2015). Peri-tumoral tissues from patients with epileptogenic GBMs show significantly increased expression of NKCC1 vs. non-epileptogenic patients (Conti et al., 2011; Garzon-Muvdi et al., 2012; Pallud et al., 2014; Schiapparelli et al., 2017; Luo et al., 2020). Epileptic discharges from resected human tissue were suppressed by the application of FDA-approved NKCC1 antagonist bumetanide (Table 1; Pallud et al., 2014). Further to this, latency to seizure onset was increased in GBM-bearing mice treated with continuous infusion of bumetanide (MacKenzie et al., 2016). Evidently, the Cl− dysregulation and consequent defective GABAergic signaling in GBM facilitates the generation of epileptiform activity. GBM-induced changes in Cl− transporter expression may be potentially triggered by overstimulation of NMDARs and consequent dephosphorylation of KCC2 residue serine 940 (Ser940); a residue responsible for transporter function and stability in the membrane (Lee H. H. C. et al., 2010; Lee et al., 2011; MacKenzie et al., 2016). Blockade of excess glutamate release from glioma cells with SAS prevents KCC2 downregulation in GBM cells (MacKenzie et al., 2016), implicating increased glutamate signaling in Cl− gradient dysregulation and aberrant GABAergic neurotransmission. Additionally, BDNF release from glioma cells or locally activated microglia is known to elicit the downregulation of KCC2 and upregulation of NKCC1 on peri-tumoral neurons (Pallud et al., 2013).
Defective GABAergic neurotransmission also confers a growth advantage in GBM. Increased GABAergic signaling via GABAARs has been shown to negatively regulate glioma cell proliferation (Blanchart et al., 2017). Therefore, the observed reduction in functional GABAARs expression on glioma cells would result in decreased GABAergic neurotransmission and favor tumor growth (Table 1; Labrakakis et al., 1998). NKCC1 is also known to regulate glioma cell volume changes by increasing [Cl−]i, which results in Cl− efflux upon GABA binding and cell shrinkage to promote glioma cell proliferation, migration, and invasion of surrounding parenchyma (Table 1; Schiapparelli et al., 2017). Both knockdown of NKCC1 and inhibition with bumetanide significantly reduces GBM cell migration in vitro and in vivo, and overexpression of the transporter markedly increases GBM cell invasion (Haas and Sontheimer, 2010; Garzon-Muvdi et al., 2012; Schiapparelli et al., 2017).
The majority of current GBM literature focuses on either the complex GBM microenvironment or the development of novel therapeutics. Ergo, studies on the epileptological aspect of GBM are extremely limited, and even less have investigated the impact of anti-tumorigenic treatment strategies on the relationships between epilepsy, SDs, and tumor growth. Experimental studies of GBM-associated epilepsy primarily employ GBM cell lines (human, mouse, or rat) or human primary cultures to generate intracranial GBM tumors. Wherein a tumor cell suspension is intracranially injected into brain areas associated with seizure vulnerability and/or propagation (Table 2). Genetic manipulation of epilepsy-associated known oncogenes via IUE CRISPR/Cas9-mediated deletion or utilization of inducible-Cre-LoxP transgenic models has recently been employed to aid the investigation into genetic mechanisms of GBM-associated epileptogenesis (Table 2; Hatcher et al., 2020; Jin et al., 2021). The majority of existing studies utilize transplantation methods (Köhling et al., 2006; Bouckaert et al., 2020), with many seminal articles employing xenografting (Table 2; Buckingham et al., 2011; Campbell et al., 2012, 2015). The use of mice lacking an intact immune system, e.g., SCID mice, in investigations of GBM-associated epileptogenicity is controversial as the neuroinflammatory environment created and maintained by the tumor is a key contributor to the development of network hyperexcitability. As the primary aim of using animal models is to accurately recapitulate the disease as seen in humans, it stands to reason that GBM should be modeled in an immunocompetent animal to incorporate all aspects of the immune system that may play any role in seizure generation or GBM progression.
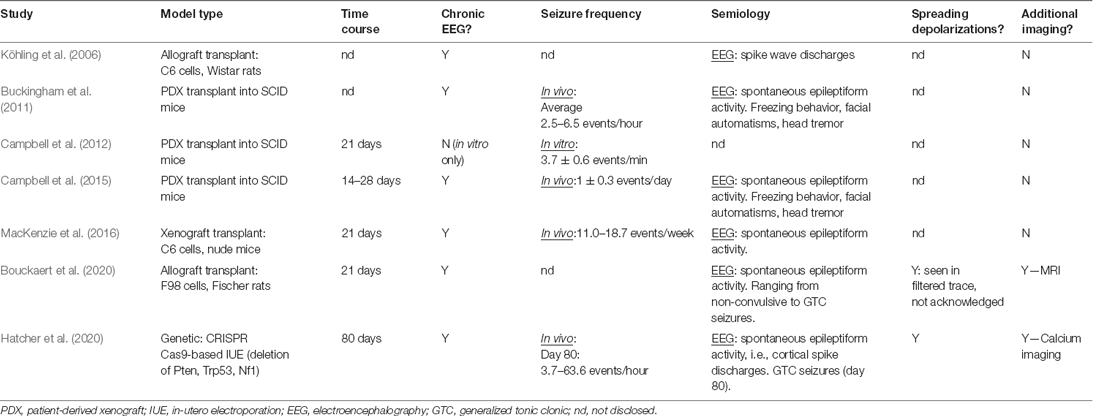
Table 2. Seminal experimental studies, with a primary objective of GBM-associated epilepsy investigation.
The paucity of the literature including seizures as a measured variable in oncological studies is a substantial obstacle to collating information about mechanisms underlying GBM-related epileptogenesis. Where the experimental focus is oncogenesis or novel treatment development, the presence of seizures is often overlooked or only mentioned as a general indicator of a humane endpoint. Experimental animals typically express complex or simple focal seizures characterized by subtle behavioral changes such as freezing periods, facial automatisms, head tremors, or exaggerated startle responses to audiogenic stimuli (Köhling et al., 2006; Buckingham et al., 2011; Campbell et al., 2015). These events are completely spontaneous and may never occur in the presence of the researcher. Therefore, seizures could be easily missed if their investigation is not an objective of the study. However, arguably, neglecting to include seizures as a monitored variable in therapeutic studies has implications for clinical translation. The network changes occurring secondary to seizures and SDs in GBM undoubtedly impact tumor growth, disease progression, and potential risk of recurrence. Testing novel treatments in a model of GBM that does not account for seizure presence may confound results by omitting to include a potentially important influencer of therapeutic efficacy that affects a substantial portion of the patient population. Examples of how clinically-used anti-tumor treatments affect seizures in GBM have been examined in clinical trials (Chalifoux and Elisevich, 1997; Climans et al., 2020), but the literature primarily focuses on low-grade gliomas.
The Stupp regimen is the standard of care for GBM, consisting of resection followed 6 weeks later by post-surgical ionizing radiotherapy (RT) with concomitant TMZ chemotherapy and maintained by 20 weeks of adjuvant TMZ (Stupp et al., 2005, 2009; Fernandes et al., 2017). RT induces an acute inflammatory reaction to mediate its anti-tumor effects but, as it targets the vulnerable peri-cavity environment, this may paradoxically contribute to network hyperexcitability (Baskar et al., 2014). RT has been historically associated with improved seizure control in low-grade gliomas (Chalifoux and Elisevich, 1997; Rudà et al., 2013). However, extensive investigations have not yet been reported for GBM and therefore, seizure outcomes may differ. A prospective interventional trial evaluating the effect of RT on seizure activity in high grade (IV) gliomas is currently ongoing (Rades et al., 2021). Results from this study will aid understanding and inform future decisions pertaining to the application of individual anti-tumorigenic treatments in GBM patients with seizures. TMZ is an oral alkylating chemotherapeutic that combats tumor growth and progression through DNA methylation and cell cycle arrest. The efficacy of TMZ in GBM patients is correlated with increased methylation of the MGMT promoter (Perry et al., 2017). Interestingly this has also been associated with increased post-operative seizure control in one clinical study (Feyissa et al., 2019). This appears to indicate that increased susceptibility to TMZ treatment is associated with greater seizure control and better survival outcomes. A reduction in seizure burden in TMZ-treated MGMT-methylated epileptogenic GBM patients confers a proportional decrease in the activity of associated epileptogenic downstream signaling cascades. These pathways are known to encourage tumor growth, and so their interruption would restrict the ability of enduring tumor cells to progress into fully-fledged recurrent GBMs. However, other recent evidence suggests that the addition of TMZ to RT has a minimal effect on seizure presence in elderly GBM patients vs. RT alone (Climans et al., 2020). Climans et al. (2020) also demonstrated that decreased overall survival was significantly associated with seizures. This paradoxically supports the hypothesis that continuous seizure presence and associated excitotoxicity may increase the likelihood of tumor recurrence, and subsequently decrease the overall survival of epileptogenic GBM patients relative to patients without seizures. However, these conclusions were derived from previously acquired clinical trial data and MGMT methylation nor IDH-status were not taken into account in the reanalysis. Moreover, seizure outcomes were not reliably recorded in the original study potentially confounding results (Perry et al., 2017; Climans et al., 2020).
The prevalence of seizures in GBM necessitates the use of anti-seizure drugs (ASDs) as the constant presence of the tumor increases the likelihood of seizure recurrence. GBM-associated seizures present unique challenges as they are often poorly controlled, requiring poly-therapy. However, there are several potential contraindications related to the use of ASDs in GBM, including increased risk of enhanced toxicity and undesirable interactions with anti-cancer treatments. Generally, non-enzyme inducing agents are first-line ASD options, i.e., levetiracetam (LEV), as they do not interfere with anti-tumor treatment strategies (Rosati et al., 2010; Kerkhof et al., 2013). Experimental evidence indicates that certain ASDs may have anti-tumorigenic effects, supporting a connection between seizures and mortality in GBM. Historically, valproic acid (VPA) treatment has been associated with a potential survival benefit in GBM patients (Kerkhof et al., 2013; Krauze et al., 2015; Lu et al., 2018). VPA is employed as an ASD as it disrupts GABA degradation and blocks voltage-gated Na+ and Ca2+ channels. Its anti-tumor effects are mediated through its actions as a histone deacetylase (HDAC) inhibitor and potentiation of the cytotoxicity induced by temozolomide (TMZ; Van Nifterik et al., 2012). Application of VPA to a variety of, but not all, GBM cell lines slowed cell cycle progression and inhibits proliferation (Knüpfer et al., 1998; Chavez-Blanco et al., 2006). However, the beneficial effect of VPA on GBM progression in the clinic is extremely variable. A recent study inferred that wild-type p53 status is necessary for VPA to enhance the anti-neoplastic effect of TMZ, demonstrating a potential reason for inconsistencies seen within the literature (Tsai et al., 2021).
In recent years, standards of care have shifted to favor LEV for anti-seizure treatment in GBM, and this has its own association with anti-cancer effects (Roh et al., 2020). It is currently favored as a therapeutic dose that can be achieved quickly in a clinic and there is a low likelihood of myelosuppression, hepatotoxicity, or hyponatremia (Hovinga, 2001). LEV binds to synaptic vesicle protein 2A, interfering with neurotransmitter release and reducing epileptogenic excitatory neurotransmission (Lynch et al., 2004). Evidence also indicates that LEV exerts a significant inhibitory effect on O6-methylguanine-DNA methyltransferase (MGMT) in GBM cell lines, conferring reduced resistance to TMZ (Bobustuc et al., 2010). LEV also increases MGMT transcription in normal astrocytes exhibiting a neuroprotective effect (Bobustuc et al., 2010). LEV treatment concurrent with standard chemo-radiation protocol has been associated with increased overall survival in IDH-wildtype GBM patients (Roh et al., 2020; Pallud et al., 2022).
A third-generation ASD, perampanel (PER), recently gained attention as it was shown to significantly reduce the seizure frequency of previously drug-refractory GBM-related seizures (Vecht et al., 2017; Dunn-Pirio et al., 2018; Izumoto et al., 2018; Coppola et al., 2020; Maschio et al., 2020). Its anti-seizure effects are mediated by its actions as a non-competitive AMPARs antagonist (Hanada et al., 2011; Frampton, 2015; Augustin et al., 2018; Brito da Silva et al., 2021). PER exhibits significant anti-proliferative effects on multiple GBM cell lines at various concentrations, and appears to reduce the overall concentration of extracellular glutamate (Lange et al., 2019; Salmaggi et al., 2021). Initial evidence suggests a correlation between PER treatment and tumor volume reduction on MRI-FLAIR (Izumoto et al., 2018). However, more clinical investigations are required before conclusions can be drawn as to the full extent of PER’s anti-tumor effects and any synergistic interactions it has with chemo-radiation regimens. Notably, PER is associated with various serious adverse effects, primarily psychiatric changes that are often the primary cause of discontinuation due to their prevalence (Rohracher et al., 2018; Villanueva et al., 2022). Administration of PER concomitant with anti-tumorigenic treatment regimens in a patient population already vulnerable to psychiatric changes, secondary to the GBM, must be carefully considered as adverse effects may be more frequent or severe.
It is of note that ASDs are not disease-modifying drugs, they do not prevent epileptogenesis but do reduce seizure burden. To our knowledge there are no known treatments that act to prevent epileptogenesis in the context of GBM, however, please see Terrone et al. (2020) for a comprehensive discussion of this work in other types of epilepsy.
Despite a wide research field dedicated to anti-GBM therapeutics, no significant positive advances have been made in prognosis for over a decade. As such, novel immunotherapeutic approaches such as immune checkpoint inhibitors (ICI), chimeric antigen receptor T cell (CAR-T) therapy, myeloid-targeted therapies, and tumor vaccines, have been the focus of GBM literature in recent years. GBM mediates an immunosuppressive TME by upregulating the expression of immune checkpoint molecules to reduce T cell activation and proliferation. This confers an almost intrinsic resistance to traditional anti-cancer treatments, making immune checkpoint molecules attractive therapeutic targets. In particular, increased expression of CTLA-4 is seen on Tregs and cytotoxic T lymphocytes, and PD-L1 on tumor cells and TAMs (Yu and Quail, 2021). Correspondingly, initial in vivo evidence demonstrated that the anti-PD-1 ICI nivolumab was remarkably effective at eradicating GL261 tumors. However, whilst essentially dominating the preclinical ICI research field, nivolumab’s effect failed to translate clinically (Yu and Quail, 2021). Interestingly seizures were the second most common neurological adverse event observed in clinical trials (Lim et al., 2017). ICIs provoke an adaptive immune response in the peri-cavity area to increase the activity and infiltration of cytotoxic T cells and pro-inflammatory cytokine secretion. As such, adverse effects have been attributed to “overshooting” T cell activation against antigens expressed on surrounding non-neoplastic cells (Roth et al., 2021). This mistargeting could act to promote hyperexcitability in peri-cavity neuronal networks post-resection, the time period at which systemic treatments are typically employed. Interestingly, an adaptive immune response characterized by the infiltration of cytotoxic CD8+ T cells and associated pro-inflammatory cytokine secretion is known to give rise to neuronal cell death and seizure induction in Rasmussen Encephalitis (Varadkar et al., 2014). Furthermore, the epileptogenic potential of ICIs is evidenced in reports of paraneoplastic encephalitis, and associated seizures, directly linked to the administration of anti-PD-1 (Shi et al., 2020) and anti-CTLA-4 (ipilimumab; Roth et al., 2021). It stands to reason that if a potentially epileptogenic ICI is employed in a seizure-vulnerable patient population, e.g., GBM, this may indirectly encourage tumor progression secondary to increased epileptiform activity and paradoxically result in decreased therapeutic efficacy. Unfortunately, there are no reports of the impact of ICIs on seizure presence and how this affects outcomes in GBM specifically. Therefore, the use of particular ICIs in the treatment of epileptogenic GBMs should be further investigated preclinically to validate their safe use in the clinic.
CAR-T therapy is another T-cell mediated immunotherapy, wherein a patient’s own T cells are harvested and engineered ex vivo to express chimeric antigen receptors (CARs) that recognize an identified tumor antigen, e.g., EGFRvIII, before being re-transplanted (Sterner and Sterner, 2021). CAR-T therapy application in GBM presents significant challenges namely due to intratumoral heterogeneity in antigen expression and the high likelihood of target antigens also being expressed in normal tissue (Maggs et al., 2021). Pathological overactive T-cell responses in CAR-T therapy commonly manifest as seizure-associated toxicities such as cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS; Shimabukuro-Vornhagen et al., 2018). Headaches, hallucinations, hemiparesis, and seizures are some of the most prominent behavioral manifestations observed when CRS and ICANS occur secondary to brain-targeted CAR-T therapy (Shimabukuro-Vornhagen et al., 2018). The release of IL-1, TNF-α, IFN-β, and IL-6 are postulated to play key roles in both CRS and ICANS pathophysiology as they trigger an almost uncontrolled activation of microglia (Shimabukuro-Vornhagen et al., 2018; Siegler and Kenderian, 2020). In ICANS, activated CAR-T cells also provoke the release of pro-inflammatory cytokines IL-1 and IL-6, which subsequently activate endothelial cells of the BBB to drive its breakdown. The potentiation of significant neuroinflammation in peri-cavity tissue, when coupled with BBB breakdown, mirrors an environment seen in GBM-associated seizure generation. Furthermore, CAR-T cells can remain in circulation for a prolonged time post-infusion, and so continued downstream pro-inflammatory signaling may progressively encourage epileptogenesis in the vulnerable peri-cavity area. Whilst CRS/ICANS and the acute neurological toxicity associated with them are predominantly reversible, the potentiation of epileptogenic processes has permanent consequences and may result in paradoxical resistance to therapy secondary to seizure-potentiated tumor growth.
The therapeutic potential of macrophage reprogramming/repolarization has attracted a great deal of interest in recent years, but it also represents an area of uncertainty in its unknown relationship to epileptogenesis. The majority of TAMs are monocyte-derived macrophages (MDMs) that infiltrate the brain through the BBB and they account for up to 50% of the tumor mass (Parmigiani et al., 2021). M1-polarized macrophages display primarily pro-inflammatory, anti-tumor attributes making them an attractive therapeutic target. Whereas M2-polarized macrophages exhibit predominantly anti-inflammatory features with pro-tumor effects and are the subtype primarily associated with the GBM TME (Ma et al., 2021). Repolarization of TAMs within the TME focuses on therapeutic pro-inflammatory actions either in the tumor proper, in the absence of resection, or in peri-cavity tissue, negating the need for systemic application. However, this may have implications for epileptogenic GBMs as this area is highly vulnerable to the inflammation-induced alterations in NMDARs, AMPARs, and ion homeostasis that promote hyperexcitability. The anti-tumor consequences of macrophage repolarization are mediated through increased production of inducible nitric oxide synthase (iNOS), TNF-α, IL-1β, IL-6, and chemokines associated with cytotoxic T cell recruitment (Duan and Luo, 2021). As previously stated, these are all mediators known to contribute to epileptogenesis. Whilst there are no reported investigations into macrophage reprogramming in epileptogenic GBMs, studies have acknowledged seizures as a common side effect in cytosine-guanine oligonucleotide (CpG-ODN) mediated MDM repolarization (Carpentier et al., 2006, 2010). Intracranial infusion of litenimod (CpG-28) in a cohort of 34 recurrent GBM patients was associated with seizures of either generalized or focal semiology in 14 patients (Carpentier et al., 2010). The downstream actions of M1-polarized macrophages and aforementioned evidence suggests that TAM reprogramming may possess at least an acute epileptogenic capability, and indicates a clear need for further preclinical investigation.
Whilst it is clearly important that seizure presence and prevalence be a monitored variable in preclinical studies of novel anti-tumorigenic treatment regimens, it ultimately returns to a balance of risk-benefit when translating these therapeutics into the clinic. If these therapies effectively eradicate the GBM, despite increasing the risk of epilepsy, then this would be tolerable. However, if these therapies only extend survival by a few months and this time is associated with debilitating seizures, would this still be an acceptable outcome?
Another equally important challenge in preclinical research is the focus on pre-resective models of GBM, all current epileptological GBM research has been performed in pre-resection models. These represent a valuable method to investigate underlying mechanisms but may not be as clinically relevant. The majority of GBM patients are eligible for resective surgery to remove the bulk tumor, and as a corollary, the most common clinical scenario involves a post-resection brain. The post-resection environment demonstrates a substantially altered neural network, even vs. pre-resection, and as such the mechanisms of hyperexcitability and seizure generation may also be altered (Figure 3). Resection models of GBM have been developed (Kauer et al., 2011; Momiyama et al., 2013; Bianco et al., 2017; Otvos et al., 2021; Tang et al., 2021), but their use has not been reported in the investigation of GBM-related epileptogenesis. Therefore, there is currently no evidence indicative of how mechanisms of excitability and seizure generation are altered in a post-resection brain. Both pre-resective and recurrent GBMs display considerable intratumoral heterogeneity, but recurrent GBMs may also have a slightly different molecular phenotype than the tumor it originated from (Campos et al., 2016). The recurrent tumor acquires new genetic aberrations as it grows (Kim et al., 2015), potentially resulting in different reactions to anti-tumor or anti-epileptic therapeutic regimens used to treat the pre-resective tumor. This has downstream effects on the ability to make research-informed decisions regarding anti-seizure treatment regimens post-resection in the clinic, and demonstrates an incomplete understanding of the impact seizures have on disease progression.

Figure 3. Changes in seizure characteristics during disease progression and treatment. There are a multitude of mechanisms that alter seizure characteristics and phenotype in GBM. The molecular mutations and consequently activated signaling pathways change throughout its progression and as it enters new areas of the brain. Upon resection, these mechanisms are altered again and often accompanied by a period of seizure cessation. Seizures may then be produced in the peri-cavity region due to pre-existing network disruption or tumor regrowth, or in an entirely different region due to secondary epileptogenesis.
The recurrence of seizures following GBM resection could be attributed to a phenomenon known as secondary epileptogenesis (Figure 3), wherein an extended epileptogenic network enables epileptogenesis to continue following the removal of a primary focus (Morrell et al., 1956; Morrell, 1985; Scholly et al., 2017). Morrell (1985) defined secondary epileptogenesis as a three-stage process in which an actively discharging epileptogenic region gradually induces paroxysmal activity in the cellular components of distally connected normal neuronal network(s). In this context, GBM can be considered the primary epileptogenic foci and the generation of seizures is predominantly dependent on its presence (stage 1). Following GBM resection, or removal of the primary focus, there is a period of temporary seizure cessation whilst the network re-adjusts to further reorganization, aligning with stage 2. Finally, consistent with stage 3, underlying epileptiform activity in a secondary previously-connected focus persists independently to manifest as post-resection seizures. This is a hypothesis not previously considered in experimental or clinical literature, and evidently the controversial existence of secondary epileptogenesis requires greater attention in the context of GBM.
New-onset seizures or their recurrence post-tumor resection is common, and have been independently associated with tumor recurrence, but evidence also suggests a correlation to the extent of resection (EOR). Generally, surgical treatment of GBM can range from a minimally invasive surgical biopsy to a craniotomy involving varying degrees of “total” tumor resection (McGirt et al., 2009; Brown et al., 2016; Tan et al., 2020). Increased EOR is independently associated with increased survival rates (McGirt et al., 2009), and this also correlates with seizure freedom. Patients who have undergone gross total tumor resection (GTR; >80%) are much more likely to be seizure-free post-operatively than sub-total resection (SubTR; <70%; Liang et al., 2016). Still significantly higher rates of seizure freedom are seen with supra-total resection (SupraTR; >100%) in the form of anterior temporal lobectomy (ATL; Borger et al., 2021). Comprehensive investigations into the effect of seizures on risk of tumor recurrence and how this is impacted by EOR have not yet been reported. However, interesting initial evidence from a medium-sized cohort study appears to indicate that patients with post-operative seizures may exhibit less time to tumor recurrence vs. those without, independent of EOR (Liang et al., 2016). It is well recognized that the peri-tumoral area is a primary source of epileptiform activity and hyperexcitability, as previously discussed. It has been postulated that, as this area often goes un-resected, post-resection seizures may continue to originate from the pre-existing epileptogenic locus in the original peri-tumoral border. The process of epileptogenesis and associated tumorigenic network alterations may potentially continue in this peri-cavity area even independent of tumor presence. Increased use of intraoperative electrocorticography (ECoG) in GBM patients with pre-operative seizure history has the potential to increase maximal resection margins to include greater portions of peri-tumoral epileptogenic loci. This would increase the likelihood of post-operative seizure freedom and potentially influence tumor recurrence risk.
Additionally, the cycle of epileptogenesis may be potentiated post-resection by the small populations of neoplastic cells that invariably persist in the “normal” parenchyma surrounding the resection cavity. These remaining glioma cells promote network reorganization of the peri-cavity area through further BBB breakdown (Figure 4A), and continued dysfunctional glutamatergic (Figure 4B), and GABAergic signaling (Figure 4C). Additionally, their release of MMP-9 works to degrade FS-PV+PNNs (Figure 4C). Therefore, the hyperexcitable peri-cavity area in epileptogenic GBM patients is highly likely to actively be undergoing epileptogenesis secondary to the actions of these enduring glioma cells. These seizures- and glioma cell-driven aberrances will result in further progressive reorganization of peri-cavity neuronal networks, working in pathological synergy to promote recurrence. The multiple month latency to seizure generation and tumor recurrence following initial GBM resection appears to fit with this timeline (Liang et al., 2016). It is pertinent to mention that the cumulative risk of de novo post-operative epilepsy is generally increased following all craniotomy procedures, but this is markedly higher in patients undergoing craniotomies for GBM resection (13.9% vs. 23.6% respectively; Giraldi et al., 2021).

Figure 4. Epileptogenic molecular mechanisms post-GBM resection. (A) The neuroinflammatory environment and BBB disruption originally potentiated by the tumor can also be transiently exacerbated by the resective surgery itself. Serum albumin infiltration evokes Kir4.1 downregulation on reactive astrocytes, enabling K+ accumulation. AQP4 expression is increased on glioma cells, decreasing the ECS. (B) Increased xCT expression on glioma cells and decreased GLT-1 expression on astrocytes leads to extracellular glutamate accumulation in the peri-cavity region. It’s post-synaptic action at NMDARs and CP-AMPARs increases neuronal excitability. (C) The remaining glioma cells continue to release MMP-9 which induces PNN degradation. GABAAR expression is almost absent on glioma cells to aid their proliferation. NKCC1 is upregulated and KCC2 downregulated on surrounding pyramidal neurons, reversing the Cl− gradient. The subsequent decreased inhibitory influence disinhibits pyramidal neuronal firing enabling seizure generation. ECS, extracellular space; BBB, blood-brain barrier; NMDAR, NMDA receptor; CP-AMPARs, Ca2+-permeable AMPARs; MMPs, matrix metalloproteinases; PNNs, perineuronal nets.
By nature, the seizures that occur in models of GBM-related epilepsy are spontaneous and so represent unique challenges. A comprehensive investigation of epileptogenesis in GBM models requires continuous electrophysiological monitoring of epileptiform activity. This is typically performed through chronic implantation of electroencephalographic devices, as it is impossible to predict seizure occurrence without pro-convulsant intervention by pharmaceutical or stimulatory means. Furthermore, the lack of overt behavioral manifestations of seizure presence in experimental animals renders video monitoring insufficient in the absence of accompanying EEG recordings. Equipment to record epileptiform activity long-term is widely accessible and readily employed in experimental epilepsy studies, commonly as tethered recordings or subcutaneous battery implantation to power the recording headstage (Wykes et al., 2012). However, these technologies are largely unsuitable for use in a progressive GBM model as they primarily employ a single or few electrodes, rather than configurations that allow high-density mapping. If a rigid electrode is invasively implanted into the brain, this could result in mechanical and physical damage secondary to the tumor growth, and a progressive attenuation of the signal as the recording site is enveloped by the tumor. Therefore, to effectively monitor epileptiform activity in GBM, any electrophysiological methods must include some form of multi-electrode array to account for the progressive expansion of the peri-tumoral border. Furthermore, whilst some electrophysiological techniques, i.e., epicortical arrays, can be used in conjunction with imaging techniques, conventional electrodes are poorly suited due to the presence of overwhelming artifact or magnetic interference with electronics. This represents an obstacle in current preclinical research, wherein the restricted ability to employ live imaging methods in conjunction with ECoG monitoring hinders the investigation of seizures and SDs in GBM.
The ideal preclinical scenario would incorporate MRI-compatible multi-electrode, flexible, penetrating arrays that can be placed at the peri-tumoral border, and are capable of electrographically mapping the changes to network excitability associated with progressive tumor growth over multiple areas of the brain. The closest preclinical example of this is seen in Hatcher et al. (2020) wherein in vivo Ca2+ fluorescence imaging was used in conjunction with EEG monitoring in a CRISPR-Cas9 IUE model of GBM-associated epilepsy, with great success. However, Ca2+ imaging is a complex technique not readily adopted nor clinically translatable. This hurdle presents challenges in both transplantation and genetic models of GBM-related epilepsy as tumor volume and growth rates can vary between animals. As a corollary, previous conclusions reached about the causal relationship between volume or growth patterns and the development of epileptiform activity are more illustrative than certain statements.
The presence of SDs in GBM-associated epilepsy further complicates electrophysiological investigations. The massive, abrupt change in activity seen in SD results in cortical electrical potential changes over a wide spectral range. This primarily manifests as slow potentials approaching the DC (or 0 Hz) level. Negative potential shift is an important identifier of SD but is not measured using standard AC-coupled (>0.5 Hz) highpass filtered electrophysiological methods. This can be partially overcome by the use of amplifiers capable of recording at lower high-pass filter limits (>0.02 Hz), or by using a DC-coupled system. However, substantial attenuation of the amplitude and distortion of the SD waveform is evident when using passive metal-based electrodes, as well as issues with drift and amplifier saturation. The presence of SD has not yet been investigated intra- or post-operatively in GBM patients, epileptogenic or otherwise, and only very few experimental studies acknowledge its presence (Bouckaert et al., 2020; Hatcher et al., 2020). Current detection of SDs in humans is mediated using platinum electrodes. They allow for the unfiltered recording of DC shifts and have a relatively high signal-to-noise ratio (SNR), but the unfortunate presence of artifacts and transients makes them unsuitable for accurate monitoring and mapping of SD in GBM. Experimental studies employ solution-filled glass micropipettes and silver or silver chloride (Ag/AgCl) wires to detect SDs (Hatcher et al., 2020), but these offer little to no spatial resolution. Additionally, the inherent toxicity of Ag/AgCl makes it inappropriate for use in clinical studies.
There is a clear need for novel means of electrophysiological recording that can sustain continuous and simultaneous recording of both high frequency synchronous epileptiform activity and SD DC-shifts across the brain. Importantly, these technologies need to be compatible with a variety of imaging modalities in order to draw direct conclusions relating to the interaction between tumor presence and seizure generation. The application of novel technologies currently in development for preclinical investigations of epileptogenesis in GBM are crucial to improving our understanding. One such example of which is graphene-based solution-gated field-effect transistors (gSGFETs). gSGFETs have the potential to fill this technology gap as they offer the means to record SDs, either independently or concurrently with high-frequency activity, both epi- and intra-cortically with high spatiotemporal fidelity (Hébert et al., 2018; Masvidal-Codina et al., 2019, 2021; Bonaccini Calia et al., 2021). The potential for SDs to exacerbate tissue damage and promote mechanisms of hyperexcitability in GBM could have consequences for disease progression, prognosis, and resistance to therapies.
It is clear that GBMs create a pro-epileptogenic environment via a multitude of mechanisms, and thus the associated seizures present unique clinical challenges. However, it is also known that some GBM patients do not present with, and never experience tumor-associated seizures or epilepsy. Therefore, we may gain more mechanistic insight into what the important essential factors are for the development of epilepsy by understanding why some patients do not develop epilepsy despite an aggressive tumor environment.
The answer may lie somewhere in the complex interaction between genetic influences and acquired mechanisms of epilepsy. There is a precedent of diseases that are associated with epilepsy where seizures manifest only following treatment. In neurocysticercosis, intracranial cysts evade detection by the immune system through various mechanisms, and it is only once they are treated that seizures are generated (Carpio et al., 1998; Prodjinotho et al., 2020; Espino et al., 2022). This master evasion of the immune system is an aspect mirrored in GBM. In the GBM TME of some patients, the tumor may promote such extensive anti-inflammatory signaling to suppress the immune system, that it indirectly transiently discourages epileptogenesis. This may also offer a further potential explanation for new-onset seizures post-resection. Wherein the removal of the tumor may provoke such an aggressive inflammatory immune response that it potentiates seizure generation.
Recent experimental evidence suggests that tumor-localized expansion of certain pathological glioma subpopulations enriched for epilepsy-associated genes may confer hyperexcitability secondary to neosynaptogenesis (Lin et al., 2017). Therefore, if the proportional expression of certain glioma subpopulations in a tumor differ to not favor this particular population, then the changes incurred by the aforementioned peri-tumoral mechanisms may not be cumulatively sufficient to evoke hyperexcitability and ultimately initiate epileptogenesis. This is a link between genetic and acquired mechanisms of GBM-associated epilepsy not previously delineated (Lin et al., 2017). The intratumoral heterogeneity of GBM, and its unique microenvironment, support this theory. This is indicative that many different pro-epileptogenic mechanisms must work in pathological synergy in order for seizures to occur. Further investigations are needed to unravel the interactions between genetic predisposition and acquired mechanisms of epilepsy in GBM as this is clearly an area that could provide great insight into the determinants of seizure presence in a population of GBM patients. Investigations in this vein would also support the transition to more personalized therapeutics, as is an important current area of research in GBM.
Ultimately, a better understanding of the relationship between GBM, epilepsy, and SDs will stem from further experimental investigations. Whilst clinical studies have provided initial evidence connecting tumor location to seizure presence, the reluctance of oncology studies to stray from the standard striatal injection site precludes further preclinical investigation. Incorporating a variety of injection sites, i.e., mesial temporal or frontal lobe structures, into implantation models would help to further our understanding of how GBM growth in different brain regions predisposes a patient to comorbidities. Different areas of the brain are considerably different in how their networks function. Therefore, certain networks may communicate in ways that have a more favorable influence on the relationship between tumor growth and secondary epileptogenesis. Greater insight into how tumor location specifically affects the process of epileptogenesis will undoubtedly have clinical significance. Patients identified as predisposed to developing seizures may receive slightly altered anti-tumorigenic regimens that also simultaneously address the underlying network dysfunction occurring during tumor-associated epileptogenesis, instead of just using an adjuvant ASD.
It is important to emphasize that novel technologies need to be developed to comprehensively investigate the impact tumor location has on epileptogenesis. The ability to map seizure and SD propagation across multiple brain areas with high fidelity is an aspect of electrophysiology that has eluded the research community up until very recently. Initial experimental evidence shows that cortical involvement is progressive in preclinical GBM-epilepsy models (Hatcher et al., 2020). Therefore, the availability of both epi-dural and intra-cortical arrays is crucial to spatially map the relationship of epileptiform activity with the expansion of the peri-tumoral border. Evidently, the spontaneous nature of epileptiform activity and SDs in this disease model necessitates 24/7 electrophysiological recording. However, chronic monitoring is a substantial experimental and technological investment and therefore is not readily performed. The inaccessibility, and unavailability, of this technology is a major obstacle to investigating seizures as a measured variable in oncology studies. Many novel anti-tumorigenic treatments appear to have pro-epileptogenic potential, but it is only with increased accessibility of electrophysiological technologies that seizures will be included and monitored in these studies. The field of GBM research overwhelmingly focuses on the development of novel treatment options. Therefore, it is crucially important that the effect seizures and SDs may have on therapeutic efficacy is evaluated to best inform their clinical success.
KH, KK, and RW developed the concept and designed the initial manuscript. KH wrote the manuscript and produced the figures. RW and KK assisted with manuscript revisions. All authors contributed to the article and approved the submitted version.
This review is supported by the Medical Research Council (MRC), grant reference MR/W019752/1. KH is sponsored by the Graphene NOWNANO PhD programme at the University of Manchester. RW is supported by a Senior Research Fellowship awarded by the Worshipful Company of Pewterers.
KK would like to declare he is co-founder and holds equity in INBRAIN Neuroelectronics (Barcelona, Spain).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the institutions and funding agencies for their support. We also thank Dr. Thomas Kisby and Ms. Maria Stylianou for their assistance. All figures were created with Biorender.com.
Aizawa, H., Sun, W., Sugiyama, K., Itou, Y., Aida, T., Cui, W., et al. (2020). Glial glutamate transporter GLT-1 determines susceptibility to spreading depression in the mouse cerebral cortex. Glia 68, 2631–2642. doi: 10.1002/glia.23874
Alvestad, S., Hammer, J., Hoddevik, E. H., Skare, Ø., Sonnewald, U., Amiry-Moghaddam, M., et al. (2013). Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. 105, 30–41. doi: 10.1016/j.eplepsyres.2013.01.006
Alzial, G., Renoult, O., Paris, F., Gratas, C., Clavreul, A., and Pecqueur, C. (2022). Wild-type isocitrate dehydrogenase under the spotlight in glioblastoma. Oncogene 41, 613–621. doi: 10.1038/s41388-021-02056-1
Armbruster, M., Naskar, S., Garcia, J., Sommer, M., Kim, E., Adam, Y., et al. (2021). Neuronal activity drives pathway-specific depolarization of astrocyte distal processes. bioRxiv [Preprint]. 2021.07.03.450922. doi: 10.1101/2021.07.03.450922
Arvanitis, C. D., Ferraro, G. B., and Jain, R. K. (2020). The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 20, 26–41. doi: 10.1038/s41568-019-0205-x
Asahi, M., Wang, X., Mori, T., Sumii, T., Jung, J. C., Moskowitz, M. A., et al. (2001). Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 21, 7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001
Augustin, K., Williams, S., Cunningham, M., Devlin, A. M., Friedrich, M., Jayasekera, A., et al. (2018). Perampanel and decanoic acid show synergistic action against AMPA receptors and seizures. Epilepsia 59, e172–e178. doi: 10.1111/epi.14578
Ayata, C., and Lauritzen, M. (2015). Spreading Depression, Spreading Depolarizations and the Cerebral Vasculature. Physiol. Rev. 95, 953–993. doi: 10.1152/physrev.00027.2014
Backman, S. A., Stambolic, V., Suzuki, A., Haight, J., Elia, A., Pretorius, J., et al. (2001). Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling lhermitte-duclos disease. Nat. Genet. 29, 396–403. doi: 10.1038/ng782
Baskar, R., Dai, J., Wenlong, N., Yeo, R., and Yeoh, K.-W. (2014). Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 1:24. doi: 10.3389/fmolb.2014.00024
Berendsen, S., Varkila, M., Kroonen, J., Seute, T., Snijders, T. J., Kauw, F., et al. (2016). Prognostic relevance of epilepsy at presentation in glioblastoma patients. Neuro Oncol. 18, 700–706. doi: 10.1093/neuonc/nov238
Berger, M., Speckmann, E.-J., Pape, H. C., and Gorji, A. (2008). Spreading depression enhances human neocortical excitability in vitro. Cephalalgia 28, 558–562. doi: 10.1111/j.1468-2982.2008.01556.x
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq, E., et al. (2001). CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710. doi: 10.1038/89490
Bianco, J., Bastiancich, C., Joudiou, N., Gallez, B., des Rieux, A., and Danhier, F. (2017). Novel model of orthotopic U-87 MG glioblastoma resection in athymic nude mice. J. Neurosci. Methods 284, 96–102. doi: 10.1016/j.jneumeth.2017.04.019
Binder, D. K., Nagelhus, E. A., and Ottersen, O. P. (2012). Aquaporin-4 and epilepsy. Glia 60, 1203–1214. doi: 10.1002/glia.22317
Binder, D. K., Oshio, K., Ma, T., Verkman, A. S., and Manley, G. T. (2004a). Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 15, 259–262. doi: 10.1097/00001756-200402090-00009
Binder, D. K., Papadopoulos, M. C., Haggie, P. M., and Verkman, A. S. (2004b). in vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J. Neurosci. 24, 8049–8056. doi: 10.1523/JNEUROSCI.2294-04.2004
Binder, D. K., and Steinhäuser, C. (2006). Functional changes in astroglial cells in epilepsy. Glia 54, 358–368. doi: 10.1002/glia.20394
Binder, D. K., Yao, X., Zador, Z., Sick, T. J., Verkman, A. S., and Manley, G. T. (2006). Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 53, 631–636. doi: 10.1002/glia.20318
Blanchart, A., Fernando, R., Häring, M., Assaife-Lopes, N., Romanov, R. A., Andäng, M., et al. (2017). Endogenous GABAA receptor activity suppresses glioma growth. Oncogene 36, 777–786. doi: 10.1038/onc.2016.245
Bobustuc, G. C., Baker, C. H., Limaye, A., Jenkins, W. D., Pearl, G., Avgeropoulos, N. G., et al. (2010). Levetiracetam enhances p53-mediated MGMT inhibition and sensitizes glioblastoma cells to temozolomide. Neuro Oncol. 12, 917–927. doi: 10.1093/neuonc/noq044
Bonaccini Calia, A., Masvidal-Codina, E., Smith, T. M., Schäfer, N., Rathore, D., Rodríguez-Lucas, E., et al. (2021). Full-bandwidth electrophysiology of seizures and epileptiform activity enabled by flexible graphene microtransistor depth neural probes. Nat. Nanotechnol. 17, 301–309. doi: 10.1038/s41565-021-01041-9
Borger, V., Hamed, M., Ilic, I., Potthoff, A.-L., Racz, A., Schäfer, N., et al. (2021). Seizure outcome in temporal glioblastoma surgery: lobectomy as a supratotal resection regime outclasses conventional gross-total resection. J. Neurooncol. 152, 339–346. doi: 10.1007/s11060-021-03705-x
Bouckaert, C., Germonpré, C., Verhoeven, J., Chong, S.-A., Jacquin, L., Mairet-Coello, G., et al. (2020). Development of a rat model for glioma-related epilepsy. Int. J. Mol. Sci. 21:6999. doi: 10.3390/ijms21196999
Brito da Silva, A., Pennifold, J., Henley, B., Chatterjee, K., Bateman, D., Whittaker, R. W., et al. (2021). The AMPA receptor antagonist perampanel suppresses epileptic activity in human focal cortical dysplasia. Epilepsia Open [Preprint]. doi: 10.1002/epi4.12549
Bronisz, E., and Kurkowska-Jastrzębska, I. (2016). Matrix metalloproteinase 9 in epilepsy: the role of neuroinflammation in seizure development. Mediators Inflamm. 2016:7369020. doi: 10.1155/2016/7369020
Brown, T. J., Brennan, M. C., Li, M., Church, E. W., Brandmeir, N. J., Rakszawski, K. L., et al. (2016). Association of the extent of resection with survival in glioblastoma: a systematic review and meta-analysis. JAMA Oncol. 2, 1460–1469. doi: 10.1001/jamaoncol.2016.1373
Bruna, J., Miró, J., and Velasco, R. (2013). Epilepsy in glioblastoma patients: basic mechanisms and current problems in treatment. Expert Rev. Clin. Pharmacol. 6, 333–344. doi: 10.1586/ecp.13.12
Buckingham, S. C., Campbell, S. L., Haas, B. R., Montana, V., Robel, S., Ogunrinu, T., et al. (2011). Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 17, 1269–1274. doi: 10.1038/nm.2453
Buckingham, S. C., and Robel, S. (2013). Glutamate and tumor-associated epilepsy: glial cell dysfunction in the peritumoral environment. Neurochem. Int. 63, 696–701. doi: 10.1016/j.neuint.2013.01.027
Campbell, I. L., Abraham, C. R., Masliah, E., Kemper, P., Inglis, J. D., Oldstone, M. B., et al. (1993). Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. U S A 90, 10061–10065. doi: 10.1073/pnas.90.21.10061
Campbell, S. L., Buckingham, S. C., and Sontheimer, H. (2012). Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 53, 1360–1370. doi: 10.1111/j.1528-1167.2012.03557.x
Campbell, S. L., Robel, S., Cuddapah, V. A., Robert, S., Buckingham, S. C., Kahle, K. T., et al. (2015). GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 63, 23–36. doi: 10.1002/glia.22730
Campos, B., Olsen, L. R., Urup, T., and Poulsen, H. S. (2016). A comprehensive profile of recurrent glioblastoma. Oncogene 35, 5819–5825. doi: 10.1038/onc.2016.85
Carpentier, A., Laigle-Donadey, F., Zohar, S., Capelle, L., Behin, A., Tibi, A., et al. (2006). Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 8, 60–66. doi: 10.1215/S1522851705000475
Carpentier, A., Metellus, P., Ursu, R., Zohar, S., Lafitte, F., Barrié, M., et al. (2010). Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 12, 401–408. doi: 10.1093/neuonc/nop047
Carpio, A., Escobar, A., and Hauser, W. A. (1998). Cysticercosis and epilepsy: a critical review. Epilepsia 39, 1025–1040. doi: 10.1111/j.1528-1157.1998.tb01287.x
Chaichana, K. L., Parker, S. L., Olivi, A., and Quiñones-Hinojosa, A. (2009). Long-term seizure outcomes in adult patients undergoing primary resection of malignant brain astrocytomas. Clinical article. J. Neurosurg. 111, 282–292. doi: 10.3171/2009.2.JNS081132
Chalifoux, R., and Elisevich, K. (1997). Effect of ionizing radiation on partial seizures attributable to malignant cerebral tumors. Stereotact. Funct. Neurosurg. 67, 169–182. doi: 10.1159/000099446
Chavez-Blanco, A., Perez-Plasencia, C., Perez-Cardenas, E., Carrasco-Legleu, C., Rangel-Lopez, E., Segura-Pacheco, B., et al. (2006). Antineoplastic effects of the DNA methylation inhibitor hydralazine and the histone deacetylase inhibitor valproic acid in cancer cell lines. Cancer Cell Int. 6:2. doi: 10.1186/1475-2867-6-2
Climans, S. A., Brandes, A. A., Cairncross, J. G., Ding, K., Fay, M., Laperriere, N., et al. (2020). Temozolomide and seizure outcomes in a randomized clinical trial of elderly glioblastoma patients. J. Neurooncol. 149, 65–71. doi: 10.1007/s11060-020-03573-x
Conti, L., Palma, E., Roseti, C., Lauro, C., Cipriani, R., de Groot, M., et al. (2011). Anomalous levels of Cl− transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia 52, 1635–1644. doi: 10.1111/j.1528-1167.2011.03111.x
Coppola, A., Zarabla, A., Maialetti, A., Villani, V., Koudriavtseva, T., Russo, E., et al. (2020). Perampanel confirms to be effective and well-tolerated as an add-on treatment in patients with brain tumor-related epilepsy (PERADET Study). Front. Neurol. 11:592. doi: 10.3389/fneur.2020.00592
de Curtis, M., Librizzi, L., and Avanzini, G. (2015). “Chapter 6 - Mechanisms of Focal Epileptogenesis,” in Epilepsy and Brain Tumors, eds H. B. Newton, and M. Maschio (Amsterdam: Academic Press), 101–109
de Groot, J. F., Liu, T. J., Fuller, G., and Yung, W. K. A. (2005). The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res. 65, 1934–1940. doi: 10.1158/0008-5472.CAN-04-3626
de Groot, J. F., Piao, Y., Lu, L., Fuller, G. N., and Yung, W. K. A. (2008). Knockdown of GluR1 expression by RNA interference inhibits glioma proliferation. J. Neurooncol. 88, 121–133. doi: 10.1007/s11060-008-9552-2
DeCordova, S., Shastri, A., Tsolaki, A. G., Yasmin, H., Klein, L., Singh, S. K., et al. (2020). Molecular heterogeneity and immunosuppressive microenvironment in glioblastoma. Front. Immunol 11:1402. doi: 10.3389/fimmu.2020.01402
Dittmer, D., Pati, S., Zambetti, G., Chu, S., Teresky, A. K., Moore, M., et al. (1993). Gain of function mutations in p53. Nat. Genet. 4, 42–46. doi: 10.1038/ng0593-42
Duan, L., and Di, Q. (2017). Acetazolamide suppresses multi-drug resistance-related protein 1 and p-glycoprotein expression by inhibiting aquaporins expression in a mesial temporal epilepsy rat model. Med. Sci. Monit. 23, 5818–5825. doi: 10.12659/msm.903855
Duan, Z., and Luo, Y. (2021). Targeting macrophages in cancer immunotherapy. Signal Transduct. Target Ther. 6:127. doi: 10.1038/s41392-021-00506-6
Dubois, L. G., Campanati, L., Righy, C., D’Andrea-Meira, I., de Sampaio e Spohr, C. L., Porto-Carreiro, I., et al. (2014). Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci. 8:418. doi: 10.3389/fncel.2014.00418
Dührsen, L., Sauvigny, T., Ricklefs, F. L., Mende, K.-C., Schaper, M., Matschke, J., et al. (2019). Seizures as presenting symptom in patients with glioblastoma. Epilepsia 60, 149–154. doi: 10.1111/epi.14615
Dunn-Pirio, A. M., Woodring, S., Lipp, E., Herndon, J. E., Healy, P., Weant, M., et al. (2018). Adjunctive perampanel for glioma-associated epilepsy. Epilepsy Behav. Case Rep. 10, 114–117. doi: 10.1016/j.ebcr.2018.09.003
Duy, P. Q., He, M., He, Z., and Kahle, K. T. (2020). Preclinical insights into therapeutic targeting of KCC2 for disorders of neuronal hyperexcitability. Expert Opin. Ther. Targets 24, 629–637. doi: 10.1080/14728222.2020.1762174
Endersby, R., and Baker, S. J. (2008). PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene 27, 5416–5430. doi: 10.1038/onc.2008.239
Engel, T., Murphy, B. M., Schindler, C. K., and Henshall, D. C. (2007). Elevated p53 and lower MDM2 expression in hippocampus from patients with intractable temporal lobe epilepsy. Epilepsy Res. 77, 151–156. doi: 10.1016/j.eplepsyres.2007.09.001
Englot, D. J., Berger, M. S., Chang, E. F., and Garcia, P. A. (2012). Characteristics and treatment of seizures in patients with high-grade glioma: a review. Neurosurg. Clin. N. Am. 23, 227–235. doi: 10.1016/j.nec.2012.01.009
Ertürk Çetin, Ö., Işler, C., Uzan, M., and Özkara, Ç. (2017). Epilepsy-related brain tumors. Seizure 44, 93–97. doi: 10.1016/j.seizure.2016.12.012
Espino, P. H., Couper, R. G., and Burneo, J. G. (2022). An update on Neurocysticercosis-related epilepsy. Clin. Neurol. Neurosurg. 213:107139. doi: 10.1016/j.clineuro.2022.107139
Fanelli, G. N., Grassini, D., Ortenzi, V., Pasqualetti, F., Montemurro, N., Perrini, P., et al. (2021). Decipher the glioblastoma microenvironment: the first milestone for new groundbreaking therapeutic strategies. Genes (Basel). 12:445. doi: 10.3390/genes12030445
Fernandes, C., Costa, A., Osorio, L., Costa Lago, R., Linhares, P., Carvalho, B., et al. (2017). “Chapter 11 Current Standards of Care in Glioblastoma Therapy,” in Glioblastoma [Internet], ed S. De Vleeschouwer (Brisbane, QLD: Codon Publications).
Feyissa, A. M., Worrell, G. A., Tatum, W. O., Chaichana, K. L., Jentoft, M. E., Guerrero Cazares, H., et al. (2019). Potential influence of IDH1 mutation and MGMT gene promoter methylation on glioma-related preoperative seizures and postoperative seizure control. Seizure 69, 283–289. doi: 10.1016/j.seizure.2019.05.018
Frampton, J. E. (2015). Perampanel: a review in drug-resistant epilepsy. Drugs 75, 1657–1668. doi: 10.1007/s40265-015-0465-z
Gao, X., Wang, H., Cai, S., Saadatzadeh, M. R., Hanenberg, H., Pollok, K. E., et al. (2014). Phosphorylation of NMDA 2B at S1303 in human glioma peritumoral tissue: implications for glioma epileptogenesis. Neurosurg. Focus 37:E17. doi: 10.3171/2014.9.FOCUS14485
Garzon-Muvdi, T., Schiapparelli, P., ap Rhys, C., Guerrero-Cazares, H., Smith, C., Kim, D.-H., et al. (2012). Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 10:e1001320. doi: 10.1371/journal.pbio.1001320
Giraldi, L., Vinsløv Hansen, J., Wohlfahrt, J., Fugleholm, K., Melbye, M., Munch, T. N., et al. (2021). Postoperative de novo epilepsy after craniotomy: a nationwide register-based cohort study. J. Neurol. Neurosurg. Psychiatry 93, 436–444. doi: 10.1136/jnnp-2021-326968
Goodwin, C. R., and Laterra, J. (2010). Neuro-oncology: unmasking the multiforme in glioblastoma. Nat. Rev. Neurol. 6, 304–305. doi: 10.1038/nrneurol.2010.67
Greenfield, L. J. Jr. (2013). Molecular mechanisms of antiseizure drug activity at GABAA receptors. Seizure 22, 589–600. doi: 10.1016/j.seizure.2013.04.015
Guan, X., Hasan, M. N., Maniar, S., Jia, W., and Sun, D. (2018). Reactive astrocytes in glioblastoma multiforme. Mol. Neurobiol. 55, 6927–6938. doi: 10.1007/s12035-018-0880-8
Haas, B. R., and Sontheimer, H. (2010). Inhibition of the sodium-potassium-chloride cotransporter isoform-1 reduces glioma invasion. Cancer Res. 70, 5597–5606. doi: 10.1158/0008-5472.CAN-09-4666
Han, F., Hu, R., Yang, H., Liu, J., Sui, J., Xiang, X., et al. (2016). PTEN gene mutations correlate to poor prognosis in glioma patients: a meta-analysis. Onco Targets Ther. 9, 3485–3492. doi: 10.2147/OTT.S99942
Hanada, T., Hashizume, Y., Tokuhara, N., Takenaka, O., Kohmura, N., Ogasawara, A., et al. (2011). Perampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia 52, 1331–1340. doi: 10.1111/j.1528-1167.2011.03109.x
Hatcher, A., Yu, K., Meyer, J., Aiba, I., Deneen, B., and Noebels, J. L. (2020). Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model. J. Clin. Invest. 130, 2286–2300. doi: 10.1172/JCI133316
Hébert, C., Masvidal-Codina, E., Suarez-Perez, A., Calia, A. B., Piret, G., Garcia-Cortadella, R., et al. (2018). Flexible graphene solution-gated field-effect transistors: efficient transducers for micro-electrocorticography. Adv. Funct. Mater. 28:1703976. doi: 10.1002/adfm.201703976
Henker, C., Kriesen, T., Scherer, M., Glass, Ä., von Deimling, A., Bendszus, M., et al. (2019). Association between tumor compartment volumes, the incidence of pretreatment seizures and statin-mediated protective effects in glioblastoma. Neurosurgery 85, E722–E729. doi: 10.1093/neuros/nyz079
Henley, J. M., and Wilkinson, K. A. (2016). Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 17, 337–350. doi: 10.1038/nrn.2016.37
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: their structure, function and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Hori, T., Sasayama, T., Tanaka, K., Koma, Y.-i., Nishihara, M., Tanaka, H., et al. (2019). Tumor-associated macrophage related interleukin-6 in cerebrospinal fluid as a prognostic marker for glioblastoma. J. Clin. Neurosci. 68, 281–289. doi: 10.1016/j.jocn.2019.07.020
Houser, C., Zhang, N., and Peng, N. (2012). “Alterations in the distribution of gabaa receptors in epilepsy,” in Jasper’s Basic Mechanisms of the Epilepsies, 4th Edn., eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: Oxford University Press).
Hovinga, C. A. (2001). Levetiracetam: a novel antiepileptic drug. Pharmacotherapy 21, 1375–1388. doi: 10.1592/phco.21.17.1375.34432
Hu, S., Kao, H.-Y., Yang, T., and Wang, Y. (2022). Early and Bi-hemispheric seizure onset in a rat glioblastoma multiforme model. Neurosci. Lett. 766:136351. doi: 10.1016/j.neulet.2021.136351
Hu, S., Sheng, W. S., Ehrlich, L. C., Peterson, P. K., and Chao, C. C. (2000). Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7, 153–159. doi: 10.1159/000026433
Hubbard, J. A., Szu, J. I., Yonan, J. M., and Binder, D. K. (2016). Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4). expression in a model of epilepsy. Exp. Neurol. 283, 85–96. doi: 10.1016/j.expneurol.2016.05.003
Huberfeld, G., and Vecht, C. J. (2016). Seizures and gliomas–towards a single therapeutic approach. Nat. Rev. Neurol. 12, 204–216. doi: 10.1038/nrneurol.2016.26
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., et al. (2007). Perturbed chloride homeostasis and gabaergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007
Hübner, C. A., Lorke, D. E., and Hermans-Borgmeyer, I. (2001). Expression of the Na-K-2Cl-cotransporter NKCC1 during mouse development. Mech. Dev. 102, 267–269. doi: 10.1016/s0925-4773(01)00309-4
Ikeshima-Kataoka, H. (2016). Neuroimmunological Implications of AQP4 in Astrocytes. Int. J. Mol. Sci. 17:1306. doi: 10.3390/ijms17081306
Isoardo, G., Morra, I., Chiarle, G., Audrito, V., Deaglio, S., Melcarne, A., et al. (2012). Different aquaporin-4 expression in glioblastoma multiforme patients with and without seizures. Mol. Med. 18, 1147–1151. doi: 10.2119/molmed.2012.00015
Ivens, S., Kaufer, D., Flores, L. P., Bechmann, I., Zumsteg, D., Tomkins, O., et al. (2007). TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547. doi: 10.1093/brain/awl317
Izumoto, S., Miyauchi, M., Tasaki, T., Okuda, T., Nakagawa, N., Nakano, N., et al. (2018). Seizures and tumor progression in glioma patients with uncontrollable epilepsy treated with perampanel. Anticancer Res. 38, 4361–4366. doi: 10.21873/anticanres.12737
Jin, F., Jin-Lee, H., and Johnson, A. (2021). “Mouse models of experimental glioblastoma,” in Gliomas, ed W. Debinski (Brisbane, QLD: Exon Publications).
Jung, E., Alfonso, J., Osswald, M., Monyer, H., Wick, W., and Winkler, F. (2019). Emerging intersections between neuroscience and glioma biology. Nat. Neurosci. 22, 1951–1960. doi: 10.1038/s41593-019-0540-y
Kastenhuber, E. R., and Lowe, S. W. (2017). Putting p53 in context. Cell 170, 1062–1078. doi: 10.1016/j.cell.2017.08.028
Kauer, T. M., Figueiredo, J.-L., Hingtgen, S., and Shah, K. (2011). Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 15, 197–204. doi: 10.1038/nn.3019
Kerkhof, M., Dielemans, J. C. M., van Breemen, M. S., Zwinkels, H., Walchenbach, R., Taphoorn, M. J., et al. (2013). Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro Oncol. 15, 961–967. doi: 10.1093/neuonc/not057
Kerkhof, M., and Vecht, C. J. (2013). Seizure characteristics and prognostic factors of gliomas. Epilepsia 54, 12–17. doi: 10.1111/epi.12437
Khandwala, K., Mubarak, F., and Minhas, K. (2021). The many faces of glioblastoma: pictorial review of atypical imaging features. Neuroradiol. J. 34, 33–41. doi: 10.1177/1971400920965970
Kim, H., Zheng, S., Amini, S. S., Virk, S. M., Mikkelsen, T., Brat, D. J., et al. (2015). Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 25, 316–327. doi: 10.1101/gr.180612.114
Knüpfer, M. M., Hernáiz-Driever, P., Poppenborg, H., Wolff, J. E., and Cinatl, J. (1998). Valproic acid inhibits proliferation and changes expression of CD44 and CD56 of malignant glioma cells in vitro. Anticancer Res. 18, 3585–3589.
Koekkoek, J. A. F., Dirven, L., Reijneveld, J. C., Postma, T. J., Grant, R., Pace, A., et al. (2014). Epilepsy in the end of life phase of brain tumor patients: a systematic review. Neurooncol. Pract. 1, 134–140. doi: 10.1093/nop/npu018
Köhling, R., Senner, V., Paulus, W., and Speckmann, E.-J. (2006). Epileptiform activity preferentially arises outside tumor invasion zone in glioma xenotransplants. Neurobiol. Dis. 22, 64–75. doi: 10.1016/j.nbd.2005.10.001
Kramer, D. R., Fujii, T., Ohiorhenuan, I., and Liu, C. Y. (2017). Interplay between cortical spreading depolarization and seizures. Stereotact. Funct. Neurosurg. 95, 1–5. doi: 10.1159/000452841
Krauze, A. V., Myrehaug, S. D., Chang, M. G., Holdford, D. J., Smith, S., Shih, J., et al. (2015). A phase 2 study of concurrent radiation therapy, temozolomide and the histone deacetylase inhibitor valproic acid for patients with glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 92, 986–992. doi: 10.1016/j.ijrobp.2015.04.038
Kucheryavykh, Y. V., Kucheryavykh, L. Y., Nichols, C. G., Maldonado, H. M., Baksi, K., Reichenbach, A., et al. (2007). Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55, 274–281. doi: 10.1002/glia.20455
Labrakakis, C., Patt, S., Hartmann, J., and Kettenmann, H. (1998). Functional GABAA receptors on human glioma cells. Eur. J. Neurosci. 10, 231–238. doi: 10.1046/j.1460-9568.1998.00036.x
Lange, F., Hartung, J., Liebelt, C., Boisserée, J., Resch, T., Porath, K., et al. (2020). Perampanel add-on to standard radiochemotherapy in vivo promotes neuroprotection in a rodent f98 glioma model. Front. Neurosci. 14:598266. doi: 10.3389/fnins.2020.598266
Lange, F., Weßlau, K., Porath, K., Hörnschemeyer, J., Bergner, C., Krause, B. J., et al. (2019). AMPA receptor antagonist perampanel affects glioblastoma cell growth and glutamate release in vitro. PLoS One 14:e0211644. doi: 10.1371/journal.pone.0211644
Leclercq, K., Liefferinge, J. V., Albertini, G., Neveux, M., Dardenne, S., Mairet-Coello, G., et al. (2019). Anticonvulsant and antiepileptogenic effects of system xc- inactivation in chronic epilepsy models. Epilepsia 60, 1412–1423. doi: 10.1111/epi.16055
Lee, H. H. C., Deeb, T. Z., Walker, J. A., Davies, P. A., and Moss, S. J. (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 14, 736–743. doi: 10.1038/nn.2806
Lee, T. S., Eid, T., Mane, S., Kim, J. H., Spencer, D. D., Ottersen, O. P., et al. (2004). Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol. 108, 493–502. doi: 10.1007/s00401-004-0910-7
Lee, H. H. C., Jurd, R., and Moss, S. J. (2010). Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 45, 173–179. doi: 10.1016/j.mcn.2010.06.008
Lee, J. W., Wen, P. Y., Hurwitz, S., Black, P., Kesari, S., Drappatz, J., et al. (2010). Morphological characteristics of brain tumors causing seizures. Arch. Neurol. 67, 336–342. doi: 10.1001/archneurol.2010.2
Lewerenz, J., Baxter, P., Kassubek, R., Albrecht, P., Van Liefferinge, J., Westhoff, M.-A., et al. (2014). Phosphoinositide 3-kinases upregulate system x−c via eukaryotic initiation factor 2α and activating transcription factor 4 - a pathway active in glioblastomas and epilepsy. Antioxid. Redox. Signal. 20, 2907–2922. doi: 10.1089/ars.2013.5455
Liang, S., Zhang, J., Zhang, S., and Fu, X. (2016). Epilepsy in adults with supratentorial glioblastoma: incidence and influence factors and prophylaxis in 184 patients. PLoS One 11:e0158206. doi: 10.1371/journal.pone.0158206
Lim, S. K., Llaguno, S. R. A., McKay, R. M., and Parada, L. F. (2011). Glioblastoma multiforme: a perspective on recent findings in human cancer and mouse models. BMB Rep. 44, 158–164. doi: 10.5483/BMBRep.2011.44.3.158
Lim, M., Omuro, A., Vlahovic, G., Reardon, D. A., Sahebjam, S., Cloughesy, T., et al. (2017). Nivolumab (nivo) in combination with radiotherapy (RT) ± temozolomide (TMZ). Updated safety results from CheckMate 143 in pts with methylated or unmethylated newly diagnosed glioblastoma (GBM). Ann. Oncol. 28: v109. doi: 10.1093/annonc/mdx366
Lin, C.-L. G., Kong, Q., Cuny, G. D., and Glicksman, M. A. (2012). Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med. Chem. 4, 1689–1700. doi: 10.4155/fmc.12.122
Lin, W., Wang, C., Liu, G., Bi, C., Wang, X., Zhou, Q., et al. (2020). SLC7A11/xCT in cancer: biological functions and therapeutic implications. Am. J. Cancer Res. 10, 3106–3126.
Lin, C.-C. J., Yu, K., Hatcher, A., Huang, T.-W., Lee, H. K., Carlson, J., et al. (2017). Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405. doi: 10.1038/nn.4493
Liu, H., Sun, Y., Zhang, Q., Jin, W., Gordon, R. E., Zhang, Y., et al. (2021). Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep. 36:109718. doi: 10.1016/j.celrep.2021.109718
Liu, R., Wang, J., Liang, S., Zhang, G., and Yang, X. (2020). Role of NKCC1 and KCC2 in epilepsy: from expression to function. Front. Neurol. 10:1407. doi: 10.3389/fneur.2019.01407
Liubinas, S. V., D’Abaco, G. M., Moffat, B. M., Gonzales, M., Feleppa, F., Nowell, C. J., et al. (2014). IDH1 mutation is associated with seizures and protoplasmic subtype in patients with low-grade gliomas. Epilepsia 55, 1438–1443. doi: 10.1111/epi.12662
Löscher, W., and Friedman, A. (2020). Structural, molecular and functional alterations of the blood-brain barrier during epileptogenesis and epilepsy: a cause, consequence, or both? Int. J. Mol. Sci. 21:591. doi: 10.3390/ijms21020591
Lu, V. M., Texakalidis, P., McDonald, K. L., Mekary, R. A., and Smith, T. R. (2018). The survival effect of valproic acid in glioblastoma and its current trend: a systematic review and meta-analysis. Clin. Neurol. Neurosurg. 174, 149–155. doi: 10.1016/j.clineuro.2018.09.019
Lu, T., Tian, L., Han, Y., Vogelbaum, M., and Stark, G. R. (2007). Dose-dependent cross-talk between the transforming growth factor-β and interleukin-1 signaling pathways. Proc. Natl. Acad. Sci. U S A 104, 4365–4370. doi: 10.1073/pnas.0700118104
Luikart, B. W., Schnell, E., Washburn, E. K., Bensen, A. L., Tovar, K. R., and Westbrook, G. L. (2011). Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J. Neurosci. 31, 4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011
Luo, L., Guan, X., Begum, G., Ding, D., Gayden, J., Hasan, M. N., et al. (2020). Blockade of cell volume regulatory protein NKCC1 increases TMZ-induced glioma apoptosis and reduces astrogliosis. Mol. Cancer Ther. 19, 1550–1561. doi: 10.1158/1535-7163.MCT-19-0910
Lynch, B. A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S. M., Matagne, A., et al. (2004). The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. U S A 101, 9861–9866. doi: 10.1073/pnas.0308208101
Lyons, S. A., Chung, W. J., Weaver, A. K., Ogunrinu, T., and Sontheimer, H. (2007). Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 67, 9463–9471. doi: 10.1158/0008-5472.CAN-07-2034
Ma, J., Chen, C. C., and Li, M. (2021). Macrophages/microglia in the glioblastoma tumor microenvironment. Int. J. Mol. Sci. 22:5775. doi: 10.3390/ijms22115775
Maas, S., Patt, S., Schrey, M., and Rich, A. (2001). Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. U S A 98, 14687–14692. doi: 10.1073/pnas.251531398
MacKenzie, G., and Maguire, J. (2015). Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res. 109, 13–27. doi: 10.1016/j.eplepsyres.2014.10.003
MacKenzie, G., O’Toole, K. K., Moss, S. J., and Maguire, J. (2016). Compromised GABAergic inhibition contributes to tumor-associated epilepsy. Epilepsy Res. 126, 185–196. doi: 10.1016/j.eplepsyres.2016.07.010
Madadi, A., Wolfart, J., Lange, F., Brehme, H., Linnebacher, M., Bräuer, A. U., et al. (2021). Correlation between Kir4.1 expression and barium-sensitive currents in rat and human glioma cell lines. Neurosci. Lett. 741:135481. doi: 10.1016/j.neulet.2020.135481
Maggs, L., Cattaneo, G., Dal, A. E., Moghaddam, A. S., and Ferrone, S. (2021). CAR T cell-based immunotherapy for the treatment of glioblastoma. Front. Neurosci. 15:662064. doi: 10.3389/fnins.2021.662064
Marchi, N., Angelov, L., Masaryk, T., Fazio, V., Granata, T., Hernandez, N., et al. (2007). Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 48, 732–742. doi: 10.1111/j.1528-1167.2007.00988.x
Marchi, N., Granata, T., Ghosh, C., and Janigro, D. (2012). Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia 53, 1877–1886. doi: 10.1111/j.1528-1167.2012.03637.x
Maschio, M., Zarabla, A., Maialetti, A., Giannarelli, D., Koudriavtseva, T., Villani, V., et al. (2020). Perampanel in brain tumor-related epilepsy: observational pilot study. Brain Behav. 10:e01612. doi: 10.1002/brb3.1612
Mastall, M., Wolpert, F., Gramatzki, D., Imbach, L., Becker, D., Schmick, A., et al. (2021). Survival of brain tumour patients with epilepsy. Brain 144, 3322–3327. doi: 10.1093/brain/awab188
Masvidal-Codina, E., Illa, X., Dasilva, M., Calia, A. B., Dragojević, T., Vidal-Rosas, E. E., et al. (2019). High-resolution mapping of infraslow cortical brain activity enabled by graphene microtransistors. Nat. Mater. 18, 280–288. doi: 10.1038/s41563-018-0249-4
Masvidal-Codina, E., Smith, T. M., Rathore, D., Gao, Y., Illa, X., Prats-Alfonso, E., et al. (2021). Characterization of optogenetically-induced cortical spreading depression in awake mice using graphene micro-transistor arrays. J. Neural Eng. 18:55002. doi: 10.1088/1741-2552/abecf3
Mayer, J., Kirschstein, T., Resch, T., Porath, K., Krause, B. J., Köhling, R., et al. (2020). Perampanel attenuates epileptiform phenotype in C6 glioma. Neurosci. Lett. 715:134629. doi: 10.1016/j.neulet.2019.134629
McGirt, M. J., Chaichana, K. L., Gathinji, M., Attenello, F. J., Than, K., Olivi, A., et al. (2009). Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J. Neurosurg. 110, 156–162. doi: 10.3171/2008.4.17536
Michaluk, P., Mikasova, L., Groc, L., Frischknecht, R., Choquet, D., and Kaczmarek, L. (2009). Matrix metalloproteinase-9 controls NMDA receptor surface diffusion through integrin beta1 signaling. J. Neurosci. 29, 6007–6012. doi: 10.1523/JNEUROSCI.5346-08.2009
Momiyama, M., Hiroshima, Y., Suetsugu, A., Tome, Y., Mii, S., Yano, S., et al. (2013). Enhanced resection of orthotopic red-fluorescent-protein-expressing human glioma by fluorescence-guided surgery in nude mice. Anticancer Res. 33, 107–111.
Mondal, S., Adhikari, N., Banerjee, S., Amin, S. A., and Jha, T. (2020). Matrix metalloproteinase-9 (MMP-9). and its inhibitors in cancer: a minireview. Eur. J. Med. Chem. 194:112260. doi: 10.1016/j.ejmech.2020.112260
Morrell, F. (1985). Secondary epileptogenesis in man. Arch. Neurol. 42, 318–335. doi: 10.1001/archneur.1985.04060040028009
Morrell, F., Roberts, L., and Jasper, H. H. (1956). Effect of focal epileptogenic lesions and their ablation upon conditioned electrical responses of the brain in the monkey. Electroencephalogr. Clin. Neurophysiol. 8, 217–236. doi: 10.1016/0013-4694(56)90115-8
Morrison, R. S., Wenzel, H. J., Kinoshita, Y., Robbins, C. A., Donehower, L. A., and Schwartzkroin, P. A. (1996). Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J. Neurosci. 16, 1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996
Mukai, T., Kinboshi, M., Nagao, Y., Shimizu, S., Ono, A., Sakagami, Y., et al. (2018). Antiepileptic drugs elevate astrocytic kir4.1 expression in the rat limbic region. Front. Pharmacol. 9:845. doi: 10.3389/fphar.2018.00845
Nagelhus, E. A., Mathiisen, T. M., and Ottersen, O. P. (2004). Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 129, 905–913. doi: 10.1016/j.neuroscience.2004.08.053
Nagpal, J., Jamoona, A., Gulati, N. D., Mohan, A., Braun, A., Murali, R., et al. (2006). Revisiting the role of p53 in primary and secondary glioblastomas. Anticancer Res. 26, 4633–4639.
Neal, A., Yuen, T., Bjorksten, A. R., Kwan, P., O’Brien, T. J., and Morokoff, A. (2016). Peritumoural glutamate correlates with post-operative seizures in supratentorial gliomas. J. Neurooncol. 129, 259–267. doi: 10.1007/s11060-016-2169-y
Nishio, S., Morioka, T., Hisada, K., and Fukui, M. (2000). Temporal lobe epilepsy: a clinicopathological study with special reference to temporal neocortical changes. Neurosurg. Rev. 23, 84–89. doi: 10.1007/pl00021698
Noell, S., Ritz, R., Wolburg-Buchholz, K., Wolburg, H., and Fallier-Becker, P. (2012). An Allograft glioma model reveals the dependence of aquaporin-4 expression on the brain microenvironment. PLoS One 7:e36555. doi: 10.1371/journal.pone.0036555
Oberndorfer, S., Lindeck-Pozza, E., Lahrmann, H., Struhal, W., Hitzenberger, P., and Grisold, W. (2008). The end-of-life hospital setting in patients with glioblastoma. J. Palliat. Med. 11, 26–30. doi: 10.1089/jpm.2007.0137
Ohgaki, H., and Kleihues, P. (2013). The definition of primary and secondary glioblastoma. Clin Cancer Res. 19, 764–772. doi: 10.1158/1078-0432.CCR-12-3002
Ohno, Y., Kunisawa, N., and Shimizu, S. (2021). Emerging roles of astrocyte kir4.1 channels in the pathogenesis and treatment of brain diseases. Int. J. Mol. Sci. 22:10236. doi: 10.3390/ijms221910236
Olsen, M. L., and Sontheimer, H. (2004). Mislocalization of Kir channels in malignant glia. Glia 46, 63–73. doi: 10.1002/glia.10346
Olsen, M. L., and Sontheimer, H. (2008). Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J. Neurochem. 107, 589–601. doi: 10.1111/j.1471-4159.2008.05615.x
Otvos, B., Alban, T. J., Grabowski, M. M., Bayik, D., Mulkearns-Hubert, E. E., Radivoyevitch, T., et al. (2021). Preclinical modeling of surgery and steroid therapy for glioblastoma reveals changes in immunophenotype that are associated with tumor growth and outcome. Clin. Cancer Res. 27, 2038–2049. doi: 10.1158/1078-0432.CCR-20-3262
Pace, A., Villani, V., Di Lorenzo, C., Guariglia, L., Maschio, M., Pompili, A., et al. (2013). Epilepsy in the end-of-life phase in patients with high-grade gliomas. J. Neurooncol. 111, 83–86. doi: 10.1007/s11060-012-0993-2
Pallud, J., Capelle, L., and Huberfeld, G. (2013). Tumoral epileptogenicity: how does it happen? Epilepsia 54, 30–34. doi: 10.1111/epi.12440
Pallud, J., Huberfeld, G., Dezamis, E., Peeters, S., Moiraghi, A., Gavaret, M., et al. (2022). Effect of levetiracetam use duration on overall survival of isocitrate dehydrogenase wildtype glioblastoma in adults: an observational study. Neurology 98, e125–e140. doi: 10.1212/WNL.0000000000013005
Pallud, J., Le Van Quyen, M., Bielle, F., Pellegrino, C., Varlet, P., Cresto, N., et al. (2014). Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 6:244ra89. doi: 10.1126/scitranslmed.3008065
Parmigiani, E., Scalera, M., Mori, E., Tantillo, E., and Vannini, E. (2021). Old stars and new players in the brain tumor microenvironment. Front. Cell. Neurosci. 15:709917. doi: 10.3389/fncel.2021.709917
Pearson, J. R. D., and Regad, T. (2017). Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2:17040. doi: 10.1038/sigtrans.2017.40
Perry, J. R., Laperriere, N., O’Callaghan, C. J., Brandes, A. A., Menten, J., Phillips, C., et al. (2017). Short-Course radiation plus temozolomide in elderly patients with glioblastoma. N. Engl. J. Med. 376, 1027–1037. doi: 10.1056/NEJMoa1611977
Peterson, A. R., Garcia, T. A., Cullion, K., Tiwari-Woodruff, S. K., Pedapati, E. V., and Binder, D. K. (2021). Targeted overexpression of glutamate transporter-1 reduces seizures and attenuates pathological changes in a mouse model of epilepsy. Neurobiol. Dis. 157:105443. doi: 10.1016/j.nbd.2021.105443
Pietrobon, D., and Moskowitz, M. A. (2014). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 15, 379–393. doi: 10.1038/nrn3770
Pijet, B., Konopka, A., Rejmak, E., Stefaniuk, M., Khomiak, D., Bulska, E., et al. (2020). The matrix metalloproteinase inhibitor marimastat inhibits seizures in a model of kainic acid-induced status epilepticus. Sci. Rep. 10:21314. doi: 10.1038/s41598-020-78341-y
Plotkin, M. D., Snyder, E. Y., Hebert, S. C., and Delpire, E. (1997). Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J. Neurobiol. 33, 781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5
Probert, L., Akassoglou, K., Kassiotis, G., Pasparakis, M., Alexopoulou, L., and Kollias, G. (1997). TNF-α transgenic and knockout models of CNS inflammation and degeneration. J. Neuroimmunol. 72, 137–141. doi: 10.1016/s0165-5728(96)00184-1
Prodjinotho, U. F., Lema, J., Lacorcia, M., Schmidt, V., Vejzagic, N., Sikasunge, C., et al. (2020). Host immune responses during Taenia solium neurocysticercosis infection and treatment. PLoS Negl. Trop. Dis. 14:e0008005. doi: 10.1371/journal.pntd.0008005
Proper, E. A., Hoogland, G., Kappen, S. M., Jansen, G. H., Rensen, M. G. A., Schrama, L. H., et al. (2002). Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125, 32–43. doi: 10.1093/brain/awf001
Pun, R. Y. K., Rolle, I. J., Lasarge, C. L., Hosford, B. E., Rosen, J. M., Uhl, J. D., et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034. doi: 10.1016/j.neuron.2012.08.002
Rades, D., Witteler, J., Olbrich, D., Trillenberg, P., Schild, S. E., Tvilsted, S., et al. (2021). A prospective interventional study evaluating seizure activity during a radiotherapy course for high-grade gliomas (SURF-ROGG). BMC Cancer 21:386. doi: 10.1186/s12885-021-08121-y
Raimondo, J. V., Burman, R. J., Katz, A. A., and Akerman, C. J. (2015). Ion dynamics during seizures. Front. Cell. Neurosci. 9:419. doi: 10.3389/fncel.2015.00419
Ramandi, D., Salmani, M. E., Moghimi, A., Lashkarbolouki, T., and Fereidoni, M. (2021). Pharmacological upregulation of GLT-1 alleviates the cognitive impairments in the animal model of temporal lobe epilepsy. PLoS One 16:e0246068. doi: 10.1371/journal.pone.0246068
Rana, A., and Musto, A. E. (2018). The role of inflammation in the development of epilepsy. J. Neuroinflammation 15:144. doi: 10.1186/s12974-018-1192-7
Ravizza, T., Gagliardi, B., Noé, F., Boer, K., Aronica, E., and Vezzani, A. (2008). Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 29, 142–160. doi: 10.1016/j.nbd.2007.08.012
Reiss, W. G., and Oles, K. S. (1996). Acetazolamide in the treatment of seizures. Ann. Pharmacother. 30, 514–519. doi: 10.1177/106002809603000515
Robe, P. A., Martin, D. H., Nguyen-Khac, M. T., Artesi, M., Deprez, M., Albert, A., et al. (2009). Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of Sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 9:372. doi: 10.1186/1471-2407-9-372
Robert, S. M., Buckingham, S. C., Campbell, S. L., Robel, S., Holt, K. T., Ogunrinu-Babarinde, T., et al. (2015). SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 7:289ra86. doi: 10.1126/scitranslmed.aaa8103
Robert, S. M., and Sontheimer, H. (2014). Glutamate transporters in the biology of malignant gliomas. Cell. Mol. Life Sci. 71, 1839–1854. doi: 10.1007/s00018-013-1521-z
Roh, T. H., Moon, J. H., Park, H. H., Kim, E. H., Hong, C.-K., Kim, S. H., et al. (2020). Association between survival and levetiracetam use in glioblastoma patients treated with temozolomide chemoradiotherapy. Sci. Rep. 10:10783. doi: 10.1038/s41598-020-67697-w
Rohracher, A., Zimmermann, G., Villanueva, V., Garamendi, I., Sander, J. W., Wehner, T., et al. (2018). Perampanel in routine clinical use across Europe: pooled, multicenter, observational data. Epilepsia 59, 1727–1739. doi: 10.1111/epi.14520
Rosati, A., Buttolo, L., Stefini, R., Todeschini, A., Cenzato, M., and Padovani, A. (2010). Efficacy and safety of levetiracetam in patients with glioma: a clinical prospective study. Arch. Neurol. 67, 343–346. doi: 10.1001/archneurol.2009.335
Roth, P., Winklhofer, S., Müller, A. M. S., Dummer, R., Mair, M. J., Gramatzki, D., et al. (2021). Neurological complications of cancer immunotherapy. Cancer Treat. Rev. 97:102189. doi: 10.1016/j.ctrv.2021.102189
Rudà, R., Magliola, U., Bertero, L., Trevisan, E., Bosa, C., Mantovani, C., et al. (2013). Seizure control following radiotherapy in patients with diffuse gliomas: a retrospective study. Neuro Oncol. 15, 1739–1749. doi: 10.1093/neuonc/not109
Sabetghadam, A., Wu, C., Liu, J., Zhang, L., and Reid, A. Y. (2020). Increased epileptogenicity in a mouse model of neurofibromatosis type 1. Exp. Neurol. 331:113373. doi: 10.1016/j.expneurol.2020.113373
Salmaggi, A., Corno, C., Maschio, M., Donzelli, S., D’Urso, A., Perego, P., et al. (2021). Synergistic effect of perampanel and temozolomide in human glioma cell lines. J. Pers. Med. 11:390. doi: 10.3390/jpm11050390
Samland, H., Huitron-Resendiz, S., Masliah, E., Criado, J., Henriksen, S. J., and Campbell, I. L. (2003). Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J. Neurosci. Res. 73, 176–187. doi: 10.1002/jnr.10635
Sarac, S., Afzal, S., Broholm, H., Madsen, F. F., Ploug, T., and Laursen, H. (2009). Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 117, 291–301. doi: 10.1111/j.1600-0463.2009.02443.x
Schiapparelli, P., Guerrero-Cazares, H., Magaña-Maldonado, R., Hamilla, S. M., Ganaha, S., Goulin Lippi Fernandes, E., et al. (2017). NKCC1 regulates migration ability of glioblastoma cells by modulation of actin dynamics and interacting with cofilin. EBioMedicine 21, 94–103. doi: 10.1016/j.ebiom.2017.06.020
Scholly, J., Staack, A. M., Kahane, P., Scavarda, D., Régis, J., Hirsch, E., et al. (2017). Hypothalamic hamartoma: epileptogenesis beyond the lesion? Epilepsia 58, 32–40. doi: 10.1111/epi.13755
Sears, S. M. S., Hewett, J. A., and Hewett, S. J. (2019). Decreased epileptogenesis in mice lacking the System xc^- transporter occurs in association with a reduction in AMPA receptor subunit GluA1. Epilepsia Open 4, 133–143. doi: 10.1002/epi4.12307
Shi, S., Jaoube, J. A., Kanwar, R., Jin, M. C., Amorin, A., Varanasi, V., et al. (2020). Neurological adverse effects due to programmed death 1 (PD-1) inhibitors. J. Neurooncol. 148, 291–297. doi: 10.1007/s11060-020-03514-8
Shimabukuro-Vornhagen, A., Gödel, P., Subklewe, M., Stemmler, H. J., Schlößer, H. A., Schlaak, M., et al. (2018). Cytokine release syndrome. J. Immunother. Cancer 6:56. doi: 10.1186/s40425-018-0343-9
Siegler, E. L., and Kenderian, S. S. (2020). Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T Cell therapy: insights into mechanisms and novel therapies. Front. Immunol. 11:1973. doi: 10.3389/fimmu.2020.01973
Smits, A., Jin, Z., Elsir, T., Pedder, H., Nistér, M., Alafuzoff, I., et al. (2012). GABA-A channel subunit expression in human glioma correlates with tumor histology and clinical outcome. PLoS One 7:e37041. doi: 10.1371/journal.pone.0037041
Soomro, S. H., Ting, L. R., Qing, Y. Y., and Ren, M. (2017). Molecular biology of glioblastoma: classification and mutational locations. J. Pak. Med. Assoc. 67, 1410–1414.
Sørensen, M. F., Heimisdóttir, S. B., Sørensen, M. D., Mellegaard, C. S., Wohlleben, H., Kristensen, B. W., et al. (2018). High expression of cystine-glutamate antiporter xCT (SLC7A11) is an independent biomarker for epileptic seizures at diagnosis in glioma. J. Neurooncol. 138, 49–53. doi: 10.1007/s11060-018-2785-9
Sorrentino, U., Bellonzi, S., Mozzato, C., Brasson, V., Toldo, I., Parrozzani, R., et al. (2021). Epilepsy in NF1: Epidemiologic, genetic and clinical features. a monocentric retrospective study in a cohort of 784 patients. Cancers (Basel) 13:6336. doi: 10.3390/cancers13246336
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Sterner, R. C., and Sterner, R. M. (2021). CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11:69. doi: 10.1038/s41408-021-00459-7
Stupp, R., Hegi, M. E., Mason, W. P., van den Bent, M. J., Taphoorn, M. J. B., Janzer, R. C., et al. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466. doi: 10.1016/S1470-2045(09)70025-7
Stupp, R., Mason, W. P., van den Bent, M. J., Weller, M., Fisher, B., Taphoorn, M. J. B., et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. doi: 10.1056/NEJMoa043330
Szopa, W., Burley, T. A., Kramer-Marek, G., and Kaspera, W. (2017). Diagnostic and therapeutic biomarkers in glioblastoma: current status and future perspectives. Biomed. Res. Int. 2017:8013575. doi: 10.1155/2017/8013575
Takahashi, K., Foster, J. B., and Lin, C.-L. G. (2015). Glutamate transporter EAAT2: regulation, function and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 72, 3489–3506. doi: 10.1007/s00018-015-1937-8
Takeuchi, S., Wada, K., Toyooka, T., Shinomiya, N., Shimazaki, H., Nakanishi, K., et al. (2013). Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery 72, 33–41. doi: 10.1227/NEU.0b013e318276b2de
Tan, A. C., Ashley, D. M., López, G. Y., Malinzak, M., Friedman, H. S., and Khasraw, M. (2020). Management of glioblastoma: state of the art and future directions. CA Cancer J. Clin. 70, 299–312. doi: 10.3322/caac.21613
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. doi: 10.1126/science.276.5319.1699
Tang, B., Foss, K., Lichtor, T., Phillips, H., and Roy, E. (2021). Resection of orthotopic murine brain glioma. Neuroimmunol. Neuroinflammation 8, 64–69. doi: 10.20517/2347-8659.2020.28
Terrone, G., Balosso, S., Pauletti, A., Ravizza, T., and Vezzani, A. (2020). Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology 167:107742. doi: 10.1016/j.neuropharm.2019.107742
Tewari, B. P., Chaunsali, L., Campbell, S. L., Patel, D. C., Goode, A. E., and Sontheimer, H. (2018). Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat. Commun. 9:4724. doi: 10.1038/s41467-018-07113-0
Thakkar, J. P., Dolecek, T. A., Horbinski, C., Ostrom, Q. T., Lightner, D. D., Barnholtz-Sloan, J. S., et al. (2014). Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 23, 1985–1996. doi: 10.1158/1055-9965.EPI-14-0275
Toledo, M., Sarria-Estrada, S., Quintana, M., Maldonado, X., Martinez-Ricarte, F., Rodon, J., et al. (2017). Epileptic features and survival in glioblastomas presenting with seizures. Epilepsy Res. 130, 1–6. doi: 10.1016/j.eplepsyres.2016.12.013
Tsai, H.-C., Wei, K.-C., Chen, P.-Y., Huang, C.-Y., Chen, K.-T., Lin, Y.-J., et al. (2021). Valproic acid enhanced temozolomide-induced anticancer activity in human glioma through the p53-PUMA apoptosis pathway. Front. Oncol. 11:722754. doi: 10.3389/fonc.2021.722754
van Breemen, M. S. M., Wilms, E. B., and Vecht, C. J. (2007). Epilepsy in patients with brain tumours: epidemiology, mechanisms and management. Lancet Neurol. 6, 421–430. doi: 10.1016/S1474-4422(07)70103-5
Van Nifterik, K. A., Van den Berg, J., Slotman, B. J., Lafleur, M. V. M., Sminia, P., and Stalpers, L. J. A. (2012). Valproic acid sensitizes human glioma cells for temozolomide and γ-radiation. J. Neurooncol. 107, 61–67. doi: 10.1007/s11060-011-0725-z
van Vliet, E. A., da Costa Araújo, S., Redeker, S., van Schaik, R., Aronica, E., and Gorter, J. A. (2007). Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130, 521–534. doi: 10.1093/brain/awl318
Varadkar, S., Bien, C. G., Kruse, C. A., Jensen, F. E., Bauer, J., Pardo, C. A., et al. (2014). Rasmussen’s encephalitis: clinical features, pathobiology and treatment advances. Lancet Neurol. 13, 195–205. doi: 10.1016/S1474-4422(13)70260-6
Vecht, C., Duran-Peña, A., Houillier, C., Durand, T., Capelle, L., and Huberfeld, G. (2017). Seizure response to perampanel in drug-resistant epilepsy with gliomas: early observations. J. Neurooncol. 133, 603–607. doi: 10.1007/s11060-017-2473-1
Vecht, C. J., Kerkhof, M., and Duran-Pena, A. (2014). Seizure prognosis in brain tumors: new insights and evidence-based management. Oncologist 19, 751–759. doi: 10.1634/theoncologist.2014-0060
Venkataramani, V., Tanev, D. I., Strahle, C., Studier-Fischer, A., Fankhauser, L., Kessler, T., et al. (2019). Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538. doi: 10.1038/s41586-019-1564-x
Venkatesan, S., Lamfers, M. L. M., Dirven, C. M. F., and Leenstra, S. (2016). Genetic biomarkers of drug response for small-molecule therapeutics targeting the RTK/Ras/PI3K, p53 or Rb pathway in glioblastoma. CNS Oncol. 5, 77–90. doi: 10.2217/cns-2015-0005
Venkatesh, H. S., Morishita, W., Geraghty, A. C., Silverbush, D., Gillespie, S. M., Arzt, M., et al. (2019). Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545. doi: 10.1038/s41586-019-1563-y
Vezzani, A., Conti, M., De Luigi, A., Ravizza, T., Moneta, D., Marchesi, F., et al. (1999). Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J. Neurosci. 19, 5054–5065. doi: 10.1523/JNEUROSCI.19-12-05054.1999
Villanueva, V., D’Souza, W., Goji, H., Kim, D. W., Liguori, C., McMurray, R., et al. (2022). PERMIT study: a global pooled analysis study of the effectiveness and tolerability of perampanel in routine clinical practice. J. Neurol. 269, 1957–1977. doi: 10.1007/s00415-021-10751-y
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., et al. (2003). Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
Wang, S. I., Puc, J., Li, J., Bruce, J. N., Cairns, P., Sidransky, D., et al. (1997). Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 57, 4183–4186.
Warth, A., Simon, P., Capper, D., Goeppert, B., Tabatabai, G., Herzog, H., et al. (2007). Expression pattern of the water channel aquaporin-4 in human gliomas is associated with blood-brain barrier disturbance but not with patient survival. J. Neurosci. Res. 85, 1336–1346. doi: 10.1002/jnr.21224
Wasade, V. S., Viarasilpa, T., Balki, I., Osman, G., Gaddam, A., Dharaiya, D., et al. (2020). Effect of seizure timing on long-term survival in patients with brain tumor. Epilepsy Behav. 111:107307. doi: 10.1016/j.yebeh.2020.107566
Wei, Q., Singh, O., Ekinci, C., Gill, J., Li, M., Mamatjan, Y., et al. (2021). TNFα secreted by glioma associated macrophages promotes endothelial activation and resistance against anti-angiogenic therapy. Acta Neuropathol. Commun. 9:67. doi: 10.1186/s40478-021-01163-0
Williams, M. R., DeSpenza Jr, T., Li, M., Gulledge, A. T., and Luikart, B. W. (2015). Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J. Neurosci. 35, 943–959. doi: 10.1523/JNEUROSCI.3144-14.2015
Wolburg, H., Noell, S., Fallier-Becker, P., Mack, A. F., and Wolburg-Buchholz, K. (2012). The disturbed blood-brain barrier in human glioblastoma. Mol. Aspects Med. 33, 579–589. doi: 10.1016/j.mam.2012.02.003
Wykes, R. C., Heeroma, J. H., Mantoan, L., Zheng, K., MacDonald, D. C., Deisseroth, K., et al. (2012). Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci. Transl. Med. 4:161ra152. doi: 10.1126/scitranslmed.3004190
Yamanaka, G., Takata, F., Kataoka, Y., Kanou, K., Morichi, S., Dohgu, S., et al. (2021). The neuroinflammatory role of pericytes in epilepsy. Biomedicines 9:759. doi: 10.3390/biomedicines9070759
Ye, L., Huang, Y., Zhao, L., Li, Y., Sun, L., Zhou, Y., et al. (2013). IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J. Neurochem. 125, 897–908. doi: 10.1111/jnc.12263
Ye, Z.-C., Rothstein, J. D., and Sontheimer, H. (1999). Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 19, 10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999
Yeo, M., Chen, Y., Jiang, C., Chen, G., Wang, K., Chandra, S., et al. (2021). Repurposing cancer drugs identifies kenpaullone which ameliorates pathologic pain in preclinical models via normalization of inhibitory neurotransmission. Nat. Commun. 12:6208. doi: 10.1038/s41467-021-26270-3
Yeung, Y. T., Bryce, N. S., Adams, S., Braidy, N., Konayagi, M., McDonald, K. L., et al. (2012). p38 MAPK inhibitors attenuate pro-inflammatory cytokine production and the invasiveness of human U251 glioblastoma cells. J. Neurooncol. 109, 35–44. doi: 10.1007/s11060-012-0875-7
Yeung, Y. T., McDonald, K. L., Grewal, T., and Munoz, L. (2013). Interleukins in glioblastoma pathophysiology: implications for therapy. Br. J. Pharmacol. 168, 591–606. doi: 10.1111/bph.12008
Yu, M. W., and Quail, D. F. (2021). Immunotherapy for glioblastoma: current progress and challenges. Front. Immunol. 12:676301. doi: 10.3389/fimmu.2021.676301
Yuen, T. I., Morokoff, A. P., Bjorksten, A., D’Abaco, G., Paradiso, L., Finch, S., et al. (2012). Glutamate is associated with a higher risk of seizures in patients with gliomas. Neurology 79, 883–889. doi: 10.1212/WNL.0b013e318266fa89
Zhang, Y., Dube, C., Gibert Jr, M., Cruickshanks, N., Wang, B., Coughlan, M., et al. (2018). The p53 pathway in glioblastoma. Cancers (Basel) 10:297. doi: 10.3390/cancers10090297
Zhang, X., Zheng, L., Duan, J., Li, Z., and Tang, Y. (2020). Clinical characteristics of brain tumor-related epilepsy and factors influencing the identification of epilepsy-associated tumors. Acta Epileptol. 2:25. doi: 10.1186/s42494-020-00034-w
Zhang, S., Zhou, J., Zhang, Y., Liu, T., Friedel, P., Zhuo, W., et al. (2021). The structural basis of function and regulation of neuronal cotransporters NKCC1 and KCC2. Commun. Biol. 4:226. doi: 10.1038/s42003-021-01750-w
Zheng, H., Ying, H., Yan, H., Kimmelman, A. C., Hiller, D. J., Chen, A.-J., et al. (2008). p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455, 1129–1133. doi: 10.1038/nature07443
Zhu, J. J., Esteban, J. A., Hayashi, Y., and Malinow, R. (2000). Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat. Neurosci. 3, 1098–1106. doi: 10.1038/80614
Keywords: glioma, peritumoral border, epilepsy, seizures, spreading depolarizations
Citation: Hills KE, Kostarelos K and Wykes RC (2022) Converging Mechanisms of Epileptogenesis and Their Insight in Glioblastoma. Front. Mol. Neurosci. 15:903115. doi: 10.3389/fnmol.2022.903115
Received: 23 March 2022; Accepted: 25 May 2022;
Published: 27 June 2022.
Edited by:
Detlev Boison, Rutgers, The State University of New Jersey, United StatesReviewed by:
Chuanzuo Yang, Zhengzhou University, ChinaCopyright © 2022 Hills, Kostarelos and Wykes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert C. Wykes, cm9iLnd5a2VzQG1hbmNoZXN0ZXIuYWMudWs=
†ORCID: Kate E. Hills, orcid.org/0000-0003-2685-3100; Kostas Kostarelos, orcid.org/0000-0002-2224-6672; Robert C. Wykes, orcid.org/0000-0002-6141-6822
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.