 Yueran Cui
Yueran Cui Haiyang Yu
Haiyang Yu Zhongqi Bu
Zhongqi Bu Lulu Wen
Lulu Wen Lili Yan
Lili Yan Juan Feng
Juan Feng- Department of Neurology, Shengjing Hospital of China Medical University, Shenyang, China
Neuroinflammation is initiated with an aberrant innate immune response in the central nervous system (CNS) and is involved in many neurological diseases. Inflammasomes are intracellular multiprotein complexes that can be used as platforms to induce the maturation and secretion of proinflammatory cytokines and pyroptosis, thus playing a pivotal role in neuroinflammation. Among the inflammasomes, the nucleotide-binding oligomerization domain-, leucine-rich repeat- and pyrin domain-containing 3 (NLRP3) inflammasome is well-characterized and contributes to many neurological diseases, such as multiple sclerosis (MS), Alzheimer's disease (AD), and ischemic stroke. MS is a chronic autoimmune disease of the CNS, and its hallmarks include chronic inflammation, demyelination, and neurodegeneration. Studies have demonstrated a relationship between MS and the NLRP3 inflammasome. To date, the pathogenesis of MS is not fully understood, and clinical studies on novel therapies are still underway. Here, we review the activation mechanism of the NLRP3 inflammasome, its role in MS, and therapies targeting related molecules, which may be beneficial in MS.
Introduction
Neuroinflammation is caused by innate immune response; however, the dysregulation of innate immunity can be deleterious to the nervous system (Leite et al., 2022). It has been found that inflammasomes participate in innate immunity and link innate immunity with inflammation (Awad et al., 2018). Inflammasomes are cytosolic multiprotein platforms that exert their effects through pattern-recognition receptors (PRRs), thus inducing the maturation and secretion of proinflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18) (Kayagaki et al., 2015). PRRs form inflammasomes and act as sensors. To date, there are a few known PRRs, such as nucleotide-binding oligomerization domain-like receptor family members NLRP1, NLRP3, NLRC4, absent-in-melanoma 2 (AIM2), and pyrin, and they sense different kinds of stimuli (Broz and Dixit, 2016). Among inflammasomes, the nucleotide-binding oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing 3 (NLRP3) inflammasome is reported to sense a wide range of stimuli and involve in a variety of diseases.
NLRP3 inflammasome was initially characterized in Muckle-Wells syndrome, which is an autoinflammatory disease (Martinon et al., 2002). As a common inflammasome, the NLRP3 inflammasome senses a wide range of danger- and pathogen-associated molecular patterns (DAMPs and PAMPs, respectively), and participates in a myriad of immune and inflammatory diseases (Peñin-Franch et al., 2022). There are three components of the NLRP3 inflammasome: NLRP3 protein (sensor), apoptosis-associated specklike protein containing a caspase-activation and recruitment domain (ASC) (adaptor), and pro-caspase-1 (effector) (Broz and Dixit, 2016) (Figure 1). NLRP3 protein itself comprises three domains: a C-terminal leucine-rich repeats (LRRs) domain, a central nucleotide binding and oligomerization domain (NACHT), and an N-terminal pyrin domain (PYD) (Kumar et al., 2011). Under normal conditions, LRRs and NACHT domains bind to each other, and NLRP3 protein is unable to bind ASC (Shao et al., 2015). Once stimulated, the two domains separate, and the NLRP3 protein binds to ASC via the PYD. ASC has two domains: a C-terminal caspase activation and recruitment domain (CARD) and an N-terminal PYD (Dick et al., 2016). When interacting with the NLRP3 protein, ASC can bind to pro-caspase-1 through the CARD, promoting the production of cleaved caspase-1, which in turn induces the maturation and release of IL-1β and IL-18 and the occurrence of pyroptosis (Shao et al., 2015; Liu et al., 2016). Pyroptosis, a form of programmed cell death that differs from apoptosis and necrosis, induces membrane rupture, leading to the release of inflammatory factors (Shi et al., 2017).
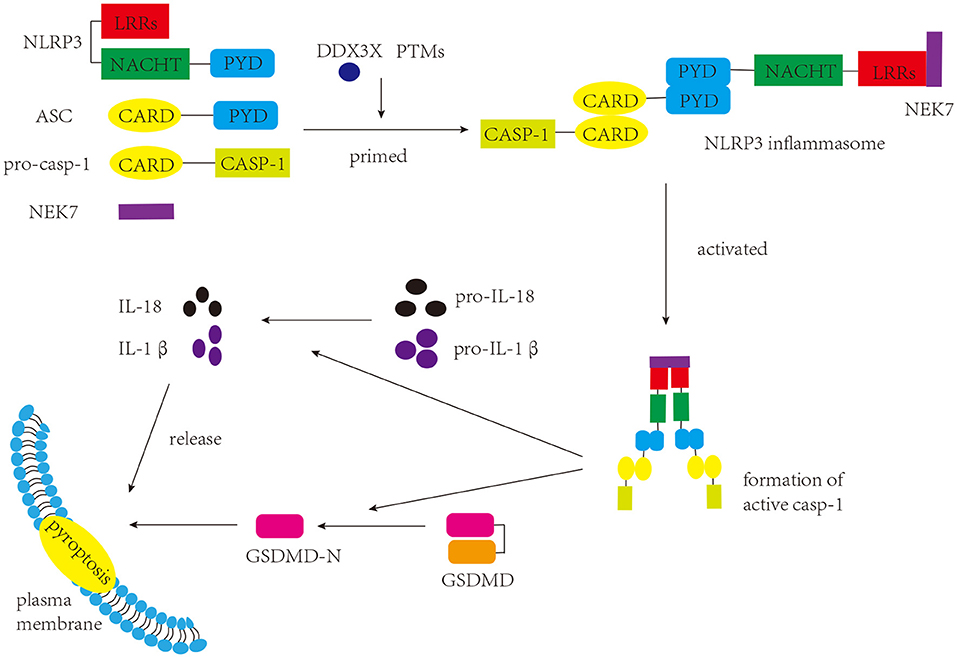
Figure 1. Components and function of NLRP3 inflammasome. The NLRP3 inflammasome consists of three components: NLRP3, ASC, and pro-caspase-1. Once stimulated, the assembly and activation of the NLRP3 inflammasome lead to the production of active caspase-1. Caspase-1 cleaves GSDMD to produce GSDMD-N, which interacts with the plasma membrane and induces pyroptosis. Caspase-1 can also promote the maturation of pro-IL-1β and pro-IL-18 into IL-1β and IL-18, respectively, which are either secreted or released during pyroptosis.
Multiple sclerosis (MS) is a common autoimmune, neurodegenerative disease of the central nervous system (CNS) (Calahorra et al., 2022). More than two million people around the world suffer from it, which brings a huge social and economic burden to patients (Browne et al., 2014; Kobelt et al., 2017). Clinically, MS is classified into four phenotypes: clinically isolated syndrome, primary progressive MS, secondary progressive MS, and progressive relapsing MS. There is no clear distinction among the different phenotypes. MS diagnosis requires a combination of clinical manifestations, imaging studies, and laboratory tests. Among the diagnostic techniques, MRI is very important (Brownlee et al., 2015). The clinical manifestations of MS patients are diverse and mainly include ataxia, cognitive dysfunction, and visual impairment (Pinke et al., 2020), which may be related to the complexity of its etiology. Currently, the exact etiology of MS is unclear and research has proposed several possibilities, which include genetic susceptibility and single nucleotide polymorphisms (SNP) of immune system-related genes, Epstein-Barr virus (EBV) infection, smoking, and reduced levels of Vitamin D (Hafler et al., 2007; Healy et al., 2009; Endriz et al., 2017; de Oliveira et al., 2020). In addition, dysfunction of the intestinal flora, a research hotspot in recent years, has also been reported to be related to MS (Pröbstel and Baranzini, 2018). According to studies, myelin-specific autoreactive CD4+ T cells may be activated in the periphery during early stages of MS. CD4+ T cells, B cells, and macrophages cross the blood-brain barrier and migrate into the CNS after destruction of the blood-brain barrier. Furthermore, antigen-presenting cells (APCs) induce differentiation of CD4+ T cells into the Th1 and Th17 cells, which exacerbate inflammatory response and contribute to the progression of MS (Shao et al., 2021). As the disease progresses, the infiltration of peripheral immune cells decreases, whereas neurodegenerative changes increase in the brain (Serafini et al., 2004; Rottlaender and Kuerten, 2015). Currently, clinical treatment for MS is not ideal. Disease-modifying therapies have been shown to be beneficial to MS patients and usually focus on recovery from relapses and slowing the progression. However, these therapies have serious side effects and do not inhibit the progressive increase in neurological disability. Therefore, more research is needed to develop effective treatment for MS patients.
Mechanisms of NLRP3 Inflammasome Activation
NLRP3 inflammasome is the most widely studied inflammasome. However, the mechanism underlying its activation remains to be elucidated. The canonical activation pathway includes a priming step (signal 1) and an activating step (signal 2) (Figure 2) (Bauernfeind et al., 2009; Kelley et al., 2019). In the priming step, stimuli interact with the Toll-like receptor (TLR)-adaptor molecule myeloid differentiation primary response 88 (MyD88), interleukin-1 receptor (IL-1R) and/or cytokine receptors, such as the tumor necrosis factor receptor (TNFR), thus activating the nuclear factor kappa B (NF-kB) pathway, which increases the expression levels of NLRP3 protein and pro-IL-1β (Latz et al., 2013). Post-translational modifications (PTMs) of NLRP3 protein are crucial to NLRP3 inflammasome activation. Once the NLRP3 protein is stimulated by signal 2, it begins to undergo deubiquitination and phosphorylation, which allows it to oligomerize and form the NLRP3 inflammasome (He et al., 2016a; Jo et al., 2016). In the activating step, many DAMPs and PAMPs can promote the assembly and activation of the NLRP3 inflammasome, which causes the formation of cleaved caspase-1, inducing the release of IL-1β and IL-18 and pyroptosis (Kayagaki et al., 2015; Shi et al., 2015; Liu et al., 2016). Several regulatory mechanisms are involved in the canonical activation pathway. For example, VSIG4 is reported to mediate the inhibition of Nlrp3 and Il-1β at the transcriptional level (Huang et al., 2019). In addition, NEK7 and DDX3X are thought to bind to the NLRP3 protein, which is crucial for NLRP3 inflammasome formation. DDX3X is an ATPase/RNA helicase of the DEAD-box family that directly binds to the NLRP3 protein. Research has reported that the knockdown of Ddx3x in peritoneal macrophages inhibits NLRP3 inflammasome activation and pyroptosis (Samir et al., 2019). Studies found that AKT could block the interaction between DDX3X and NLRP3 by phosphorylating DDX3X. Alternatively, AKT could also directly interact with NLRP3, thus inhibiting NLRP3 oligomerization (Zhao et al., 2020; Guo et al., 2021). NEK7 binds and bridges the NLRP3 protein, triggering NLRP3 inflammasome activation, which is affected by the status of NLRP3 S803, a site that is phosphorylated after priming and later dephosphorylated after activation (Sharif et al., 2019; Niu et al., 2021). A recent study showed that TRIM65, an E3 Ubiquitin Ligase, can interact with the NACHT domain of NLRP3 and promote ubiquitination of lys48 and lys63 of NLRP3, thus inhibiting the NEK7-NLRP3 interaction and suppressing the activation of NLRP3 inflammasome (Tang et al., 2021). In addition, misshapen (Msn)/NIK-related kinase 1 (MINK1) interacts with the NLRP3 LRR domain, inducing the phosphorylation of Ser725 and priming the activation of the NLRP3 inflammasome (Zhu et al., 2021). Moreover, research has demonstrated a negative feedback regulation between caspase-1 and MyD88, which may suggest self-regulation of the body (Avbelj et al., 2021). NLRP3 inflammasome activation can promote the formation of GSDMD-N, inducing pyroptosis. Gao et al. reported that TRIM21, a tripartite motif protein, binds to GSDMD via its PRY-SPRY domain, and positively regulates GSDMD-dependent pyroptosis. It maintains stable expression of GSDMD in resting cells, while inducing GSDMD-N aggregation during pyroptosis (Gao et al., 2022).
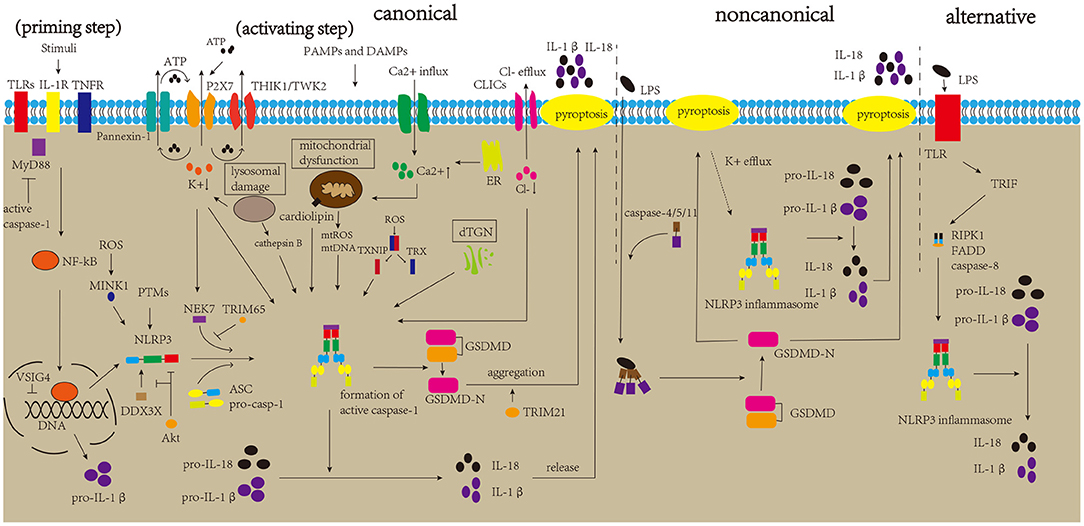
Figure 2. Mechanisms of NLRP3 inflammasome activation. The canonical activation pathway of the NLRP3 inflammasome includes priming and activating step. In the priming step, stimulatory factors can bind to TLRs and other molecules, promoting the production of NLRP3 and pro-IL-1β. After priming, DAMPs and PAMPs can generate major activation signals, such as ion fluctuations, mitochondrial dysfunction, lysosomal damage, and dTGN, which affect the assembly and activation of the NLRP3 inflammasome. The activated NLRP3 inflammasome triggers the maturation of pro-caspase-1 into active caspase-1. Caspase-1 can induce the maturation and release of IL-1β and IL-18 and pyroptosis, thereby aggravating neuroinflammation. The non-canonical pathway is mediated by caspase-4/5/11, which directly sense cytosolic LPS, and K+ efflux due to pyroptosis can trigger NLRP3 inflammasome activation. The alternative pathway is mediated by the TLR4-TRIF-RIPK1-FADD-caspase-8 signaling pathway, triggering NLRP3 inflammasome activation.
Several stimuli can activate the NLRP3 inflammasome, and it is impossible for NLRP3 inflammasome to directly bind to the stimulatory factors. These stimuli may generate major activation signals, triggering activation of the NLRP3 inflammasome. Many related studies have proposed some major activation signals, such as ion fluctuations, dysfunction of mitochondria, lysosomal disruption, and Golgi dispersal (Song et al., 2017; Shao et al., 2021). This is discussed in detail in the following sections.
The ion fluctuations involve K+, Ca2+, and Cl–. There is copious extracellular ATP due to tissue damage and immune cells activation during inflammation, and high concentrations of extracellular ATP activate P2X7R, which induces K+ efflux and then recruits Pannexin-1 to release intracellular ATP (Gombault et al., 2012; Bartlett et al., 2014; Di Virgilio et al., 2017). Moreover, two K+ channels, two-pore domain weak inwardly rectifying K+ channel 2 (TWIK2) and tandem pore domain halothane-inhibited K+ channel 1 (THIK1), have been reported to response to ATP, and regulate NLRP3 inflammasome activation (Di et al., 2018; Drinkall et al., 2022). Subsequently, the triggered K+ efflux can promote NEK7 to interact with NLRP3 via the catalytic domain of NEK7 and the LRR domain of NLRP3 (He et al., 2016b). Research has reported that a decrease in cytosolic K+ level triggers the activation of the NLRP3 inflammasome (Muñoz-Planillo et al., 2013). A type of K+ channel inhibitor has also been found to greatly inhibit NLRP3 inflammasome activation (Lamkanfi and Dixit, 2009). Ca2+ fluctuations have a crucial effect on the activation of NLRP3 inflammasome by triggering mitochondrial dysfunction. In detail, influent Ca2+ and the Ca2+ released from the stimulated endoplasmic reticulum can be transported into the mitochondria through uniporters to depolarize the mitochondrial membrane, activating voltage-gated channels of the mitochondria. Subsequent entry of many ions and metabolites trigger the production and release of reactive oxygen species (ROS) (Horng, 2014; Shenker et al., 2015). A recent study demonstrated that the transient receptor potential vanilloid type 1 (TRPV1) channel could regulate the activation of NLRP3 inflammasome by affecting Ca2+ influx. Moreover, its deletion suppressed NLRP3 inflammasome activation, thereby alleviating experimental autoimmune encephalomyelitis (EAE, a common animal model of MS) (Zhang et al., 2021). BAPTA-AM, a Ca2+ chelator, suppresses activation of the NLRP3 inflammasome (Wu et al., 2019). In addition, NLRP3 agonists have been found to induce Ca2+ signaling, triggering NLRP3 inflammasome activation (Murakami et al., 2012). A previous study suggested that the substitution of extracellular Cl- with gluconate increases the maturation and secretion of IL-1β (Verhoef et al., 2005). Levels of cytosolic Cl- were found to be reduced upon NLRP3 inflammasome activation, and inhibition of the Cl- channel could suppress NLRP3 inflammasome activation (Compan et al., 2012; Daniels et al., 2016). Mitochondrial dysfunction plays an important role in the activation of the NLRP3 inflammasome. Research has reported that stimulation of the NLRP3 inflammasome causes mitochondrial dysfunction, which leads to the production and release of mitochondrial ROS (mtROS) and the release of mitochondrial DNA (mtDNA) into the cytoplasm. Subsequently, mtROS can oxidize mtDNA, and mtDNA can be recognized by the NLRP3 inflammasome, leading to its activation (Shimada et al., 2012; Zhong et al., 2018; De Gaetano et al., 2021). Additionally, cardiolipin, which is located in the mitochondria, triggers NLRP3 inflammasome activation (Iyer et al., 2013). Under normal conditions, the thioredoxin interaction protein (TXNIP) and thioredoxin (TRX) bind to each other. Once the level of ROS increases, TRX oxidizes itself to remove ROS, along with the separation of TRX and TXNIP. The separated TXNIP can bind to NLRP3, triggering NLRP3 inflammasome activation. ROS have also been shown to promote the interaction between MINK1 and NLRP3 (Zhu et al., 2021). In addition, the ensuing inflammatory response recruits immune cells to increase ROS production, which suggests a feedback loop between ROS and the NLRP3 inflammasome (Franchi et al., 2009; Dominic et al., 2022). However, a few studies have reported that ROS are not necessary for this activation (Gabelloni et al., 2013; Gurung et al., 2015); therefore, the exact mechanism of mitochondrial dysfunction in the activation of the NLRP3 inflammasome requires further studies. Lysosomal rupture triggers the release of lysosomal contents, thereby triggering NLRP3 inflammasome activation (Halle et al., 2008; Hornung et al., 2008). Among lysosomal contents, cathepsin B is considered important for NLRP3 inflammasome activation. CA-074-Me suppresses NLRP3 inflammasome activation by inhibiting cathepsin B (Hornung et al., 2008). Besides that, it has been reported that ruptured lysosomes could trigger K+ efflux, thus promoting NLRP3 inflammasome activation (Muñoz-Planillo et al., 2013). Moreover, research has demonstrated that stimuli can induce the disassembly of the trans-Golgi network (TGN), and dispersed TGN (dTGN) can recruit NLRP3 proteins via ionic bonding between phosphatidylinositol-4-phosphate (PtdIns4P, negatively charged, on the dTGN) and a polybasic region (on the NLRP3 protein), then inducing the assembly and activation of the NLRP3 inflammasome (Chen and Chen, 2018; Shao et al., 2021).
Apart from the canonical activation pathway, NLRP3 inflammasome is also activated via non-canonical and alternative pathways (Figure 2). Caspase-11 in mice was found to directly sense cytosolic lipopolysaccharide (LPS) and lead to the formation of pores in the plasma membrane, inducing pyroptosis and K+ efflux, thus triggering non-canonical NLRP3 inflammasome activation without the involvement of caspase-1 (Kayagaki et al., 2011; Hagar et al., 2013; Ramirez et al., 2018). Similarly, studies have demonstrated that caspase-4 and caspase-5 in humans promote non-canonical activation by directly binding to cytosolic LPS (Shi et al., 2014; Casson et al., 2015). Besides that, the involvement of caspase-8 in NLRP3 alternative activation also draws our attention, which is mediated by the TLR4-TRIF-RIPK1-FADD-caspase-8 signaling pathway (Gurung et al., 2014; Gurung and Kanneganti, 2015; Gaidt et al., 2016). Recently, Zhang et al. reported the important role of the caspase-8-dependent inflammasome in CNS inflammation (Zhang et al., 2018).
There is still a lot of unknown information about the mechanisms of NLRP3 inflammasome activation. For example, the temporal and spatial distribution of NLRP3 inflammasome after activation, the contradictory results of major activation signals in NLRP3 inflammasome activation, induction of activation after the generation of major activation signals, and the mechanistic details of non-canonical and alternative activation pathways.
NLRP3 Inflammasome in the Pathogenesis/Pathophysiology of MS
Gene Polymorphisms of NLRP3 Signaling Pathway Molecules in MS
The NLRP3 inflammasome has been found to exert important effects in a myriad of diseases (Bonomini et al., 2019; Cheng et al., 2020; Yu et al., 2020). Occurrence of MS is closely related to the NLRP3 inflammasome (Gris et al., 2010; Olcum et al., 2020). Recent research has revealed that alterations in genes of NLRP3-related molecules are associated with susceptibility to MS. A study analyzed the association of SNPs of NLRP3 (rs-10754558, rs-35829419, rs-3806265, rs-4612666) with susceptibility to MS and revealed the pivotal role of NLRP3 polymorphisms in MS (Imani et al., 2018). Moreover, functional genetic variants in NLRP3 (Q705K) are associated with the severity of MS (Soares et al., 2019). In addition to NLRP3 genes, alterations in the gene expression of PYCARD and CASP1 were reported in MS patients who initially presented with the clinically isolated syndrome, which showed an association between them and MS (Hagman et al., 2015). Mutations in the downstream cytokines have also been studied. Research has found that there is no relationship between the polymorphisms of IL-1β−511 (rs16944) and IL-1β +3,953 (rs1143634) and the risk of MS; however, further studies revealed that early-onset MS had a higher association with heterozygosity of rs16944 than with homozygosity of rs16944 in IL-1β (Huang et al., 2013; Isik et al., 2013). Soares et al. analyzed SNPs distribution in different clinical phenotypes of MS and found that variants of−511C>T could result in more frequent in MS (Soares et al., 2019). There are conflicting results regarding the role of IL-18 polymorphisms in MS. Research has revealed that polymorphisms in IL18−607 C/A are not significant, while polymorphisms at position−137 are related to MS risk. However, another study found that polymorphisms of the IL-18−137C/G gene were insignificant, while IL-18−607C/A gene polymorphisms were a potential genetic risk factor for susceptibility to MS (Karakas Celik et al., 2014; Orhan et al., 2016). A recent study analyzed gene polymorphisms of IL-18 and reported that the genotype of rs1946518 was markedly different between MS patients and the control group (Jahanbani-Ardakani et al., 2019).
NLRP3 Inflammasome-Related Molecules in MS
NLRP3 inflammasome-related molecules are involved in the pathogenesis of MS. Studies have reported increased expression levels of NLRP3 and IL-1β genes in MS plaques and elevated levels of ASC, caspase-1, and IL-18 in the sera of MS patients (Keane et al., 2018; Voet et al., 2018). In EAE, the expression levels of NLRP3 mRNA and protein were increased (Gris et al., 2010; Ke et al., 2017), and compared to the wild-type mice, Nlrp3−/− mice showed reduced numbers of Th1 and Th17 cells in the spinal cord and peripheral lymphoid tissues and markedly mild EAE (Gris et al., 2010; Inoue et al., 2012a). Recently, it was reported that thymic stromal lymphopoietin (TSLP) could directly induce NLRP3 expression through phosphorylation of Janus kinase (JAK) 2, and Tslpr−/− mice showed decreased NLRP3 expression and EAE scores (Yu et al., 2021). Zhang et al. (2018) reported that a deficiency of microglial ASC could attenuate the expansion of T cells and the infiltration of neutrophil, which suggests the important role of ASC in EAE. In addition, the expression of caspase-1 was found to increase at the mRNA and protein levels (Ke et al., 2017; Bai et al., 2018), and inhibition of caspase-1 could suppress inflammasomes activation, thus attenuating the severity of EAE (Ahmed et al., 2002; McKenzie et al., 2018). Furthermore, a previous study analyzed the severity of EAE in Asc−/− and caspase-1−/− models and demonstrated that Asc−/− mice showed a decreased number of MOG-specific T cells in the lymph nodes, delayed disease progression, and reduced clinical scores (Shaw et al., 2010). In addition, Asc−/− mice were more protected from EAE progression than caspase-1−/− mice.
IL-1β and IL-18 in MS
Many studies have focused on the involvement of IL-1β and IL-18 in MS (Figure 3). As mentioned above, in the early stage of MS, the loss of integrity of CNS barriers can cause the infiltration of peripheral immune cells, and IL-1β promotes the destruction of CNS barriers (Kermode et al., 1990; Paul and Bolton, 1995; Paré et al., 2018). Briefly, immune mediators cause dysregulation of junctional components (tight junctions and adherens junctions) of CNS barriers, and the secretion of proinflammatory cytokines further triggers the infiltration of peripheral immune cells (Alvarez et al., 2011). Both CNS astrocytes and endothelial cells are involved in this process, and activated astrocytes can release proinflammatory cytokines that damage the tight junctions of endothelial cells. In addition, chemokines released by activated astrocytes can recruit leukocytes into the CNS, which is involved in the development of MS (Dong and Benveniste, 2001; Ching et al., 2007; Argaw et al., 2012). These proinflammatory cytokines and chemokines can also activate microglia (Correale and Farez, 2015). Microglia, which can be activated by IL-1β, are considered to be a kind of APCs that activate infiltrated CD4+ T cells, amplifying neuroinflammation. Besides that, microglia secrete chemokines that recruit immune cells. In turn, infiltrated CD4+ T cells secrete proinflammatory cytokines to activate microglia (Ferrari et al., 2004; Mallucci et al., 2015). According to existing research, microglia, astrocyte, and CD4+ T cells are the major cell types involved in NLRP3 inflammasome activation in MS. Studies have shown that the proportion of microglia expressing NLRP3 and IL-1β is positively associated with the degree of demyelination in MS patients (Malhotra et al., 2020). Furthermore, after activation, the NLRP3 inflammasome in activated microglia converts astrocytes to the neurotoxic A1 phenotype in MS, aggravating cognitive deficits (Hou B. et al., 2020). Tibolone and sinomenine were reported to alleviate astrocytic and microglial reactions and mobilization, respectively, decreasing NLRP3 inflammasome activation and EAE severity, which further supports the pathogenic role of the NLRP3 inflammasome in MS (Kiasalari et al., 2021; Mancino et al., 2022). Excitatory neurotransmitters can induce excitatory postsynaptic currents (EPSCs), which promote neurotoxicity and are involved in MS. IL-1β can increase the level of EPSCs in corticostriatal slice cultures and cerebellum and contribute to the severity of EAE (Gentile et al., 2013; Mandolesi et al., 2013). In addition, it was found that IL-1β could promote the expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) and the differentiation of Th17 cells, thus being involved in the pathogenesis of MS (Chung et al., 2009; Russi et al., 2018). However, a previous study found elevated levels of the IL-1β protein only in the spinal cord but not in the brain (McKenzie et al., 2018). In addition to IL-1β, the expression level of the IL-18 mRNA was increased in the CNS during EAE, and increased IL-18 levels could aggravate demyelination and neurodegeneration in EAE (Jander and Stoll, 1998; Shi et al., 2000; Jha et al., 2010). IL-18 triggers γδ and CD4+ T cells to release innate IL-17, thereby amplyfing autoimmunity (Lalor et al., 2011). Moreover, research has demonstrated that Il-18−/− mice are resistant to EAE, and the functions of natural killer (NK) cells and the production of autoreactive Th1 cells are impaired (Shi et al., 2000). Although studies on MS and EAE have shown the important role for IL-18 in the disease, the results are still conflicting. In one study, the symptoms of Il-18−/− mice were significantly alleviated, while in another study, the alleviation was not obvious, which may be related to the method of disease induction and generation of animal models (Gutcher et al., 2006; Gris et al., 2010). These studies indicate that IL-1β and IL-18 participate in the course of MS; however, further studies are needed to understand the exact mechanisms and target them for the treatment of MS.
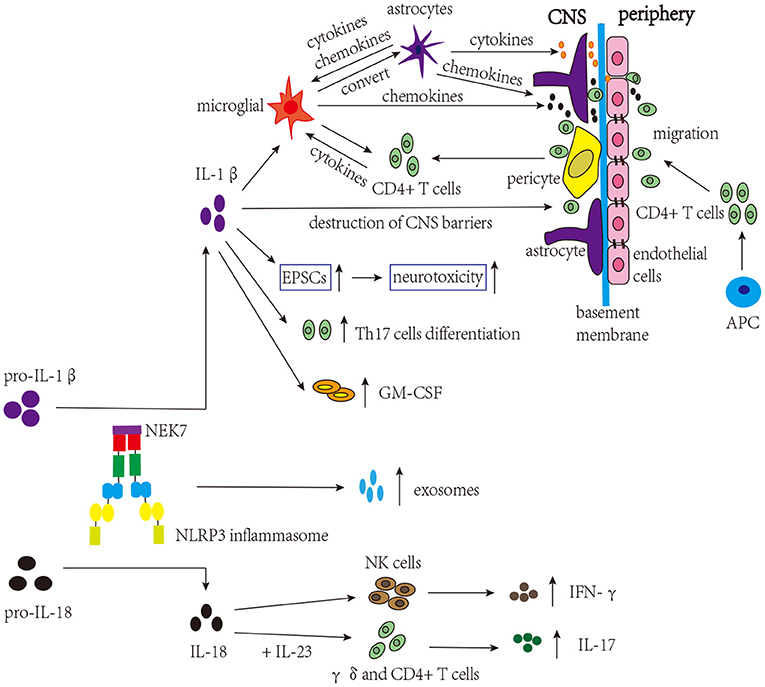
Figure 3. NLRP3 inflammasome-related molecules are involved in the pathogenesis of MS. The NLRP3 inflammasome induces the migration of CD4+ T cells into the CNS and release of exosomes. IL-1β activates microglia, which present self-antigens to activate infiltrated CD4+ T cells, thereby amplifying neuroinflammation. Microglia can convert astrocytes to cytotoxic A1 phenotype and release chemokines to recruit CD4+ T cells. Activated astrocytes release proinflammatory cytokines and chemokines, affecting microglia, tight junctions of endothelial cells, and the infiltration of CD4+ T cells. IL-1β promotes the destruction of CNS barriers, facilitating the infiltration of CD4+ T cells. IL-1β can also increase the levels of EPSCs, contributing to the severity of MS. Moreover, IL-1β can increase the expression of GM-CSF and trigger the differentiation of Th17 cells, which are involved in the pathogenesis of MS. In addition, combined with IL-23, IL-18 can trigger γδ and CD4 + T cells to release innate IL-17, amplifying autoimmunity. IL-18 can also induce the production of IFN-γ by NK cells.
NLRP3 Inflammasome and T Cells in MS
Research has demonstrated that the change in the population of T cells is perhaps not important in MS, whereas their migration into the CNS, induced by NLRP3 inflammasome, mediates the development of MS (Inoue et al., 2012a,b). As an important component of innate immunity, the NLRP3 inflammasome connects innate and adaptive immunity by promoting the production of IL-1β and IL-18, increasing the infiltration of peripheral immune cells into the CNS, and affecting the function of T and B cells. A previous study has proposed that Th cells need to be primed by APCs containing NLRP3 inflammasome before their migration into the CNS, and the study found several related results: (1). The number of immune cells migrating into the CNS was reduced in Asc−/− and Nlrp3−/− mice. (2). The expression levels of migration-related genes were increased by the NLRP3 inflammasome in Th cells and APCs. More specifically, increased levels of OPN, CCR2, and CXCR6 were found in Th cells, and the expression of α4β1 integrin, CCL2, CCL7, CCL8, and CXCL16 was increased in APCs. Interestingly, these components can bind with matched ligands or receptors (OPN-α4β1 integrin; CCR2-CCL2, CCL7, CCL8, and CXCL16; CXCR6-CXCL16) (Inoue et al., 2012a). Therefore, targeting immune cell migration is probably an effective treatment for MS. After migration, primed T cells play an important role in the course of the disease, and the NLRP3 inflammasome, in both T cells and microglia, has been reported to induce T cells to release proinflammatory cytokines involved in the pathogenesis of MS (Inoue et al., 2012a; Olcum et al., 2020).
NLRP3 Inflammasome and B Cells in MS
Although most studies suggest that MS is a kind of immune disease predominantly involving T cells, growing research has demonstrated that B cells are also crucial in the development of the disease (Li et al., 2018). The mechanism of action of B cells in MS includes four aspects: antigen presentation, cytokine production, antibody production, and ectopic lymphogenesis (Lazibat et al., 2018). B cells can present antigens to T cells, which is important for T cells activation, and the interaction between B and T cells is direct and two-way (Dalakas, 2008; Lazibat et al., 2018). Moreover, cytokines produced by B cells also exert effects on MS; for instance, B cells increase the expression of IL-6, activating Th17 responses in MS (Li et al., 2018). There are no antibodies in the CNS of healthy individuals; therefore, the presence of antibodies in the cerebrospinal fluid (CSF) of MS patients suggests that B cells are involved in the development of MS. Immunoglobulin G (IgG) and immunoglobulin M (IgM) oligoclonal bands could be used as biomarkers of MS and to predict the course of MS (Villar et al., 2005). Ectopic lymphogenesis refers to the formation of ectopic lymphoid structures, which are similar to follicles in MS patients and contain B cells; they are associated with EBV infection and may be involved in the development of MS (Serafini et al., 2004). Research has demonstrated a relationship between NLRP3 inflammasome and B cells. A recent study has reported that B cell-activating factor (BAFF) activates the NLRP3 inflammasome in B cells by triggering the binding of cIAP-TRAF2 to components of the NLRP3 inflammasome and by promoting K+ efflux and production of Src activity-dependent ROS (Lim et al., 2020). β-glucan and CpG, two major microbial antigens, have been reported to trigger NLRP3 inflammasome activation in circulating B cells. It was found that Dectin-1 activation via SYK and mammalian target of rapamycin (mTOR) participates in this process (Ali et al., 2017). Although there are many studies on the relationship between NLRP3 inflammasome and B cells, their relationship in MS is not clear, and further studies are needed to clarify the underlying mechanism.
NLRP3 Inflammasome and Exosomes in MS
In recent years, increasing attention has been paid to the role of exosomes, in tumors and autoimmune diseases, as important messengers in cell-to-cell communication (Prada et al., 2013; Paolicelli et al., 2019). It was revealed that exosomes could be selectively loaded with immunomodulatory proteins, which make them vital media for transmitting biological signals (Cypryk et al., 2018). Studies have found that damage to the CNS can induce inflammasomes activation, and exosome secretion is related to inflammasome activity. Therefore, exosomes induced by inflammasomes can promote the inflammatory response in recipient cells by transporting inflammasome-related components and other substances (de Rivero Vaccari et al., 2016; Hezel et al., 2017; Zhang et al., 2017). Zhang et al. (2017) found that inflammasome-derived exosomes could activate the NF-kB signaling pathway in recipient cells, thereby promoting an inflammatory response. Furthermore, recent research has suggested that NLRP3 inflammasome activation in microglia could promote the secretion of exosomes and facilitate the transmission of α-synuclein in Parkinson's Disease (PD) (Si et al., 2021). However, a recent study found that exosomes loaded protective molecules can protect against cardiovascular damage (Pearce et al., 2021). The above studies indicate that exosomes may be protective or damaging, which is closely related to the environment and condition of the cells from which they originate. Besides that, the effects of exosomes on inflammasomes are also being studied. Research has demonstrated that exosomes/microvesicles from periodontal ligament stem cells of MS patients can inhibit NLRP3 inflammasome activation and attenuate the severity of EAE (Soundara Rajan et al., 2017). In detail, the effects of exosomes on NLRP3 inflammasome activation depend on the origin of exosomes, including inhibition (from plasma, mesenchymal stem cells, and macrophages) and promotion (from cancer cells) (Li et al., 2021). For example, Yan et al. (2020) reported that exosomes derived from human umbilical cord mesenchymal stem cells could inhibit NLRP3 inflammasome activation by releasing circular RNA homeodomain-interacting protein kinase 3 (circHIPK3). In addition, cancer-derived exosomes have been found to promote NLRP3 inflammasome activation in macropahges (Liang et al., 2020). However, the relationship between the NLRP3 inflammasome and exosomes in MS is not well-understood, and more studies are needed. Alteration of the components of these exosomes or inhibition of their transfer may be promising therapies for the treatment of MS.
NLRP3 Inflammasome and Intestinal Flora in MS
The intestinal flora has been a research hotspot in recent years. It develops from birth, is affected by a variety of internal and external factors, and reaches a relatively stable state of composition (Mueller et al., 2015). Recent studies have found that dyregulation of the intestinal flora affects the development of many diseases, including CNS disorders (Ochoa-Repáraz et al., 2009; Sommer and Bäckhed, 2013). This suggests the presence of communication between the intestine and CNS, which we refer to as gut-brain axis (GBA). GBA is a complicated bidirectional communication among the microbiota, gut and CNS, involving the microbiota and its metabolites, immune system, endocrine system, and nervous system (Rutsch et al., 2020). Gut microbiota can act on intestinal epithelial cells to regulate their growth and differentiation, expression of tight junction proteins, and permeability of mucous membranes, thereby maintaining the integrity of the intestinal epithelial barrier. Furthermore, the release of effector molecules following activation of the intestinal inflammasome by the intestinal flora can affect the CNS via the vagus nerve (Sharma et al., 2010; Rao et al., 2017). Many studies have suggested a relationship between intestinal flora and MS. The composition of intestinal flora in MS patients is different from that in healthy individuals. Studies have demonstrated that the abundance of Bacteroides decreased and that of Pseudomonas increased in the intestinal flora of MS patients, while the protein levels of NLRP3, ASC and cleaved caspase-1 increased in the CNS, and the mRNA levels of ASC and caspase-1 increased in the circulation (Pellegrini et al., 2020). It has been reported that overactivation of the NLRP3 inflammasome can promote the shift of intestinal flora to a proinflammatory phenotype, which in turn promotes neuroinflammation and neurodegeneration (Pellegrini et al., 2020). Mackay et al. reported that consumption of a high-fiber diet, which affects the host microbiota, increases the levels of IL-18 in the serum through GPR43 and GPR109A receptors, which are expressed on colonic epithelial cells, and trigger K+ efflux and Ca2+ mobilization in an NLRP3-dependent manner (Macia et al., 2015). Mechanistically, certain metabolites produced by the microbiota in the body can enter the bloodstream and directly affect peripheral immune cells. Alternatively, these metabolites can enter the brain through the bloodstream and affect immune cells in the CNS (Wikoff et al., 2009; Rothhammer et al., 2016), which is regulated by the NLRP3 inflammasome as an important component that bridges innate and adaptive immunity. However, the exact mechanism of interplay between the microbiota and NLRP3 inflammasome is currently unknown, as is the mechanism of the subsequent effects of this interaction on the CNS. A study proposed that the effect of the interaction between the microbiota and NLRP3 inflammasome on the CNS may be through regulation of the plasma level of the circHIPK2, which in turn affects astrocyte function (Zhang et al., 2019). Furthermore, the existence of GBA is further supported by a study showing improvement in severe constipation and other MS symptoms after fecal microbiota transplantation in three MS patients (Rutsch et al., 2020). Besides that, the use of probiotics or antibiotic cocktails could lead to attenuation of EAE (Colpitts et al., 2017; Camara-Lemarroy et al., 2018). Pertussis toxin is required for the induction of EAE and is a major virulence factor of Bordetella pertussis. A study demonstrated that pertussis toxin could trigger the activation of TLR4 and induce the formation of a pyrin-dependent inflammasome, which only leads to mild EAE in pyrin-, ASC-, or caspase-1-deficient models (Dumas et al., 2014). The above studies have shown a connection among intestinal flora, inflammasomes, and MS; however, the underlying mechanism is still unclear, and further research is needed to clarify the molecules, pathways, and organs involved. This will be an attractive research area for the development of MS treatment in the future.
NLRP3 Inflammasome as a Biomarker in MS
Studies have shown that NLRP3 inflammasome-related components can be used as biomarkers for MS. For example, NLRP3 protein was found to be overexpressed in the monocytes of MS (Malhotra et al., 2020) and neuromyelitis optica spectrum disorders patients (Peng et al., 2019), compared to those of healthy individuals. In addition, elevated levels of ASC and caspase-1 were found in the sera of MS patients, and ASC was considered to be more sensitive and specific for the diagnosis of MS severity owing to its 90% specificity (Keane et al., 2018). A recent study showed that serum caspase-1 levels were elevated during relapse in MS patients, suggesting that caspase-1 may act as a biomarker of MS disease activity (Beheshti et al., 2020).
As the downstream effectors of the NLRP3 inflammasome, IL-1β and IL-18 can be used as potential biomarkers for MS. The results of studies on the expression level of IL-1β in the sera and CSF of MS patients are contradictory, and the level of IL-1β was increased or not detected in different studies (Maimone et al., 1991; Peter et al., 1991; Dujmovic et al., 2009). However, the expression of IL-1β in the CSF is associated with cortical pathology in the early stage of MS and causes neurodegeneration, following neuroinflammation (Seppi et al., 2014). One study reported increased serum IL-18 levels in MS patients, and several other studies also reported increase in IL-18 levels in the sera and CSF of MS patients (Losy and Niezgoda, 2001; Chen et al., 2012; Jahanbani-Ardakani et al., 2019).
In recent years, the neurofilament protein has been considered a promising biomarker for MS. The neurofilament protein is a cytoskeletal protein expressed in neurons, and it can be released into the blood and CSF after the damage of neurons (Khalil et al., 2018; van Lieverloo et al., 2019). Serum neurofilament protein is a useful biomarker for progressive MS and pediatric MS, and its level is associated with the risk of disability in MS (Kapoor et al., 2020; Manouchehrinia et al., 2020; Reinert et al., 2020). It consists of three isotypes: a neurofilament light (NfL) chain, neurofilament intermediate (NfM) chain, and neurofilament heavy (NfH) chain. Elevated NfL levels in the CSF occur in all stages of MS, and their levels are correlated with the MS severity score (Salzer et al., 2010; Teunissen and Khalil, 2012). In addition, interleukin-1 receptor antagonist (IL-1RA) was considered a novel biomarker in MS patients and was associated with NfL in a separate cohort (Blandford et al., 2021). Neurofilament degradation is one of the main pathological damage caused by chronic exposure to n-hexane in humans (Huang, 2008; Wang et al., 2008). A recent study found that glibenclamide ameliorated 2,5-hexanedione (the toxic metabolite of n-hexane)-induced axon degeneration by inhibiting NLRP3 inflammasome activation, thus suppressing NfL reduction in rats (Hou L. et al., 2020). This suggests a relationship between the NLRP3 inflammasome and NfL; however, their exact relationship in MS is not fully understood, and further studies are needed.
Targeting NLRP3 Inflammasome-Related Pathways for the Treatment of MS
In this section, we briefly summarize therapies targeting NLRP3 inflammasome-related pathways for the treatment of MS (Table 1).
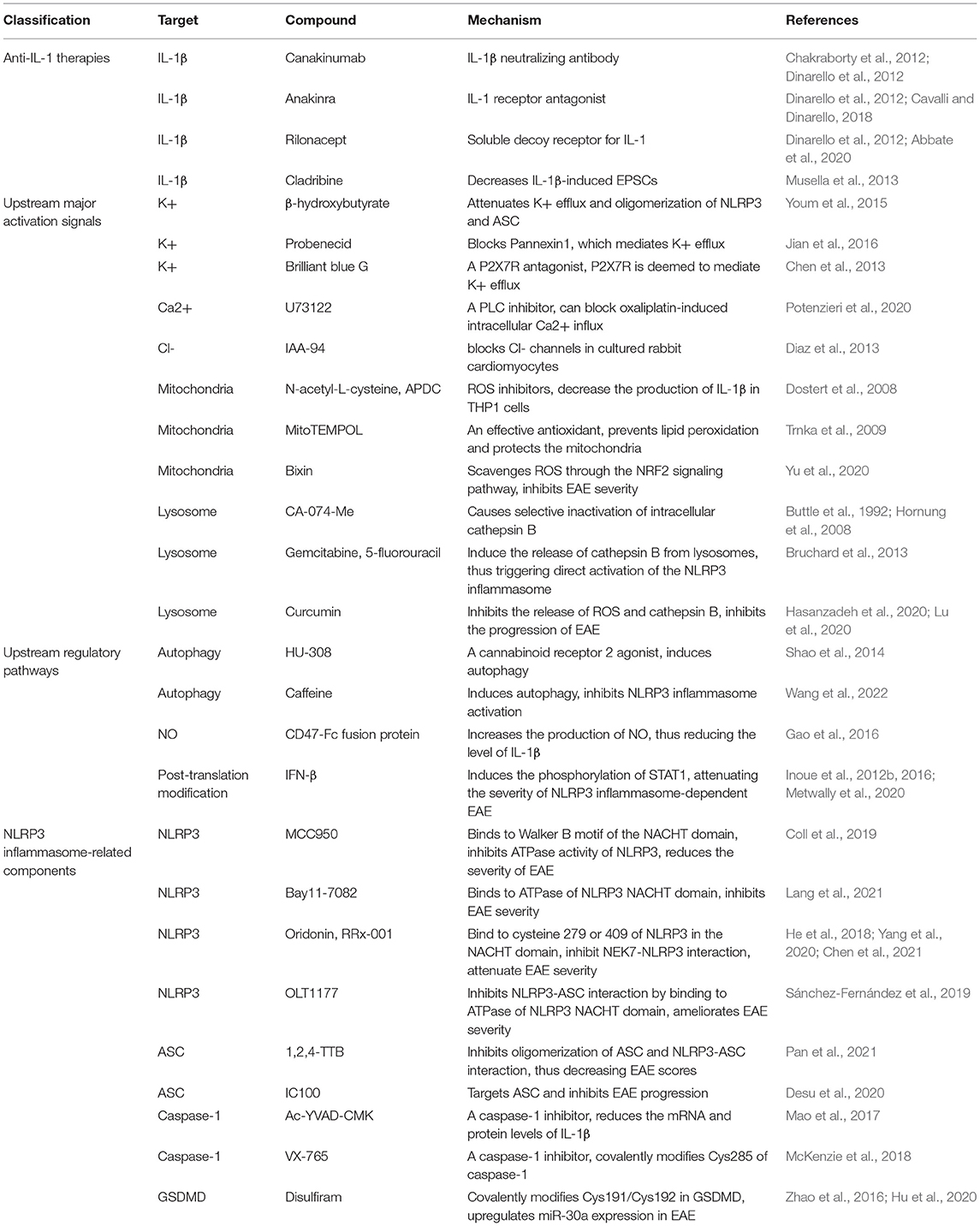
Table 1. Compounds targeting NLRP3 inflammasome-related pathways.
Anti-IL-1 Therapies
IL-1β contributes to the progression of MS. Related therapies that have been studied to date include IL-1β-specific neutralizing antibody (canakinumab), IL-1 receptor antagonist (anakinra), and soluble decoy receptor for IL-1α and IL-1β (rilonacept), and the first two have been used in clinical practice (Chakraborty et al., 2012; Dinarello et al., 2012; Cavalli and Dinarello, 2018; Abbate et al., 2020). In addition, studies have demonstrated that cladribine can attenuate IL-1β-induced EPSCs and is beneficial for the treatment of MS (Musella et al., 2013). However, the use of anti-IL-1 therapies is costly, and their penetration across brain tissues is poor. Moreover, IL-1β is not the only downstream cytokine of the NLRP3 inflammasome; therefore, targeting upstream molecules of the NLRP3 inflammasome is perhaps more effective than anti-IL-1 therapies.
Targeting Upstream Major Activation Signals
It has been reported that β-hydroxybutyrate can attenuate K+ efflux and suppress oligomerization of NLRP3 and ASC, thus inhibiting NLRP3 inflammasome activation (Youm et al., 2015). Pannexin1 and P2X7R mediate K+ efflux. Probenecid, which blocks Pannexin1, attenuates NLRP3 inflammasome activation (Jian et al., 2016). Brilliant blue G is a P2X7R antagonist, and it suppresses neuroinflammation in subarachnoid hemorrhage animal models, which may be associated with the inhibition of K+ efflux (Chen et al., 2013). Therapies targeting Ca2+ and Cl- have also been studied, including U73122 and IAA-94 (Diaz et al., 2013; Potenzieri et al., 2020). However, therapies targeting ion fluctuations can produce unavoidable side effects caused by the dysregulation of ion-related reactions. Therefore, although therapies targeting ion fluctuations are effective in the experiment, their usage in clinical practice may be limited. ROS scavengers, including N-acetyl-L-cysteine (2R,4R)-4-aminopyrro lidine-2,4-dicarboxylate (APDC), and Mito-TEMPO, were found to inhibit ROS production and attenuate NLRP3 inflammasome activation (Dostert et al., 2008; Trnka et al., 2009). In addition, Yu et al. found that Bixin could scavenge ROS through the NRF2 signaling pathway and attenuate the severity of EAE (Yu et al., 2020). Studies have suggested that mtROS can damage NADPH oxidase, causing inflammasome activation (Dostert et al., 2008; Bordt and Polster, 2014). However, similar to ion fluctuations, mtROS are also involved in many biological reactions; thus, the inhibition of mtROS cause side effects, and therapies targeting mtROS require further study. It has been reported that cathepsin B released from lysosomes plays a role in NLRP3 inflammasome activation, and CA-074-Me attenuates NLRP3 inflammasome activation by inhibiting cathepsin B (Buttle et al., 1992; Hornung et al., 2008). In addition, gemcitabine and 5-fluorouracil have been shown to contribute to the activation of NLRP3 inflammasome, which is related to cathepsin B (Bruchard et al., 2013). Recently, research has demonstrated that curcumin can inhibit the release of ROS and cathepsin B, thereby inhibiting the progression of EAE (Hasanzadeh et al., 2020; Lu et al., 2020).
Targeting Upstream Regulatory Pathways
In addition to major activation signals, other regulatory mechanisms are involved in NLRP3 inflammasome activation. HU-308, a cannabinoid receptor 2 agonist, was reported to induce autophagy, thus attenuating NLRP3 inflammasome activation and severity of EAE (Shao et al., 2014). Moreover, a recent study found that caffeine can reduce NLRP3 inflammasome activation by inducing autophagy, thus attenuating the severity of EAE (Wang et al., 2022). It was found that the CD47-Fc fusion protein could reduce the production of NLRP3 inflammasome-induced IL-1β by promoting the production of NO, aiding to the treatment of EAE (Gao et al., 2016). In addition, as a first-line drug in the treatment of MS, IFN-β can induce the phosphorylation of STAT1, which mainly exerts therapeutic effects on NLRP3 inflammasome-dependent EAE (Inoue et al., 2012b, 2016; Metwally et al., 2020). However, the reactions involved in these regulatory mechanisms are more complicated, and side effects will inevitably occur, which means that therapies targeting molecules upstream of the NLRP3 inflammasome may not be as potent as directly targeting NLRP3 inflammasome-related components.
Targeting NLRP3 Inflammasome-Related Components
MCC950, a classic NLRP3 inhibitor, can bind to the Walker B motif of the NACHT domain of NLRP3 and inhibit ATPase activity, thereby reducing the severity of EAE (Coll et al., 2019). Furthermore, Bay11-7082 was found to interact with the ATPase of the NLRP3 NACHT domain and reduce EAE severity (Lang et al., 2021). The NEK7-NLRP3 interaction is important for activation of the NLRP3 inflammasome. Research has demonstrated that oridonin and RRx-001 can inhibit this interaction by binding to cysteine 279 of the NACHT domain of NLRP3 or covalently binding to cysteine 409 of NLRP3, thereby attenuating EAE severity (He et al., 2018; Yang et al., 2020; Chen et al., 2021). Besides that, it was reported that OLT1177 could interact with the ATPase of the NLRP3 NACHT domain, inhibiting NLRP3-ASC interaction and ameliorating EAE severity (Sánchez-Fernández et al., 2019). 1,2,4-TTB was found to inhibit the oligomerization of ASC and NLRP3-ASC interactions and reduce EAE scores (Pan et al., 2021). Recently, IC100 has been reported to target ASC and inhibit EAE progression (Desu et al., 2020). Caspase-1 is an effector of NLRP3 inflammasome activation, and several caspase-1 inhibitors have been reported. For example, Ac-YVAD-CMK, a caspase-1 inhibitors, decreases the mRNA and protein levels of IL-1β and improves the symptoms of PD models (Mao et al., 2017). Moreover, VX-765 can covalently modify Cys285 of caspase-1 for the treatment of inflammatory diseases (McKenzie et al., 2018). Research has reported that the level of GSDMD, which is crucial in pyroptosis, can be inhibited by disulfiram by upregulating miR-30a expression (Zhao et al., 2016). A recent study further showed that disulfiram covalently modified Cys191/Cys192 in GSDMD, thus blocking pore formation (Hu et al., 2020).
Taken together, there are many studies on therapies targeting NLRP3 inflammasome-related pathways. However, only a few drugs can be used clinically, and there are no drugs that can effectively cure MS. Therefore, further research is necessary, and studies on the NLRP3 inflammasome-related pathway can shed light on the treatment of MS patients.
The Involvement of the NLRP3 Inflammasome in Neurodegenerative Diseases
In addition to MS, the NLRP3 inflammasome has important roles in neurodegenerative diseases such as Alzheimer's disease (AD), PD, and amyotrophic lateral sclerosis (ALS).
NLRP3 Inflammasome in AD
AD is a neurodegenerative disease with a very high prevalence, characterized by progressive loss of neurons and accumulation of extracellular amyloid beta (Aβ) (Piancone et al., 2021). The clinical manifestations of AD include dementia and cognitive dysfunction, which imposes a large burden on patients and their families (Swanton et al., 2018). Currently, the pathogenesis of AD is thought to be associated with the presence of amyloid plaques and neurofibrillary tangles, while neuroinflammation has also been found to be involved (DeTure and Dickson, 2019). Growing evidence suggests that the NLRP3 inflammasome is involved in the pathogenesis of AD. In 2008, a study reported that fibrillar Aβ could promote NLRP3 inflammasome activation, which was confirmed in subsequent studies (Halle et al., 2008). For example, amylin receptors participate in Aβ-induced NLRP3 inflammasome activation. Furthermore, Aβ can increase the level of ROS, which trigger NLRP3 inflammasome activation (Fu et al., 2017; Aminzadeh et al., 2018). Clinically, elevated expression levels of active caspase-1 and IL-1β can be found in AD patients (Blum-Degen et al., 1995; Heneka et al., 2013), which is related to the activation of the NLRP3 inflammasome. Further mechanistic studies were performed in the AD models. A study found that the levels of neuroinflammation and Aβ accumulation were reduced with concomitant improvement of neuronal function in NLRP3-deficient AD models (Heneka, 2017). Besides that, deletion of NLRP3 or caspase-1 in AD models was shown to decrease the levels of IL-1β and caspase-1 while promoting microglial differentiation to the M2 phenotype (Dempsey et al., 2017). The above studies have demonstrated the role of NLRP3 inflammasome activation in AD, but the underlying mechanisms still need to be explored.
NLRP3 Inflammasome in PD
PD is a common neurodegenerative disease characterized by a progressive loss of dopaminergic (DA) neurons in the substantia nigra and the formation of intracellular Lewy bodies by α-synuclein (Petrucci et al., 2014). The clinical manifestations of PD include typical motor symptoms and other non-motor symptoms; however, the underlying etiology is currently not clear. A previous study suggested the important role of the NLRP3 inflammasome in the pathogenesis of PD in vitro (Codolo et al., 2013), which is supported by a growing number of studies. In animal models, the levels of NLRP3 inflammasome-related molecules are increased, while deletion of NLRP3 could reduce the loss of DA neurons and improve motor dysfunction (Qiao et al., 2018; Lee et al., 2019). The involvement of the NLRP3 inflammasome in the pathogenesis of PD has also been reported in PD patients. A study reported increased expression levels of NLRP3 inflammasome-related molecules in PD patients; however, in another study, it was found that although there was a difference in the expression level of NLRP3 in the sera of PD patients compared to those of controls, the levels of IL-1β and IL-18 were not significantly different (Zhang et al., 2016; Fan et al., 2020). Considering the important role of α-synuclein in the pathogenesis of PD, several studies have explored the relationship between α-synuclein and the NLRP3 inflammasome. Research has shown that α-synuclein can promote NLRP3 inflammasome activation in PD models, and further studies have revealed that α-synuclein increases the levels of cathepsin B and ROS, thus triggering NLRP3 inflammasome activation (Codolo et al., 2013; Chatterjee et al., 2020). Further studies are needed to explore the underlying mechanisms of the NLRP3 inflammasome in the pathogenesis of PD, which may shed light on the treatment of PD patients.
NLRP3 Inflammasome in ALS
ALS, a prevalent neurodegenerative disease, is characterized by the loss of motor neurons (Feng et al., 2021). ALS patients clinically present with a continuous aggravation of motor function loss until death. Mutations in Cu/Zn superoxide dismutase 1 (SOD1) have been found to be associated with ALS (Rosen et al., 1993). The etiology of ALS remains unclear, but the role of neuroinflammation and the NLRP3 inflammasome in ALS has received attention in recent years. Studies have found increased levels of NLRP3 and caspase-1 in astrocytes and the brains of ALS patients (Haidet-Phillips et al., 2011; Kadhim et al., 2016). Studies have used G93A-SOD1 mutation models to simulate the pathogenesis of ALS. Research has revealed increased levels of NLRP3 inflammasome-related molecules and IL-1β in models, and genes knockout of caspase-1 and IL-1β can inhibit neuroinflammation (Johann et al., 2015; Lall and Baloh, 2017). Further studies are needed to explain these results and clarify the pathogenesis of ALS.
Conclusions
MS is an immune-mediated, demyelinating, neurodegenerative disease that occurs in the CNS. It predominantly affects young women and causes a huge social and economic burden. Nowadays, the underlying mechanism of MS is not fully understood, and there are still some limitations in the related diagnostics methods and therapies. In recent years, the role of inflammasomes in MS has received widespread attention. For the most widely studied NLRP3 inflammasome, we briefly introduced its composition, mechanisms of activation, and related research in MS, which may help us better understand its involvement in MS.
However, there are several unclear aspects of the NLRP3 inflammasome. For example, NLRP3 inflammasome activation is widely regulated, and it is necessary to clarify each step of the cascade of activation. Although there is a close relationship between the NLRP3 inflammasome and EAE, NLRP3 inflammasome-independent EAE also exists (Arnason, 1999). For example, 300 μg heat-killed mycobacteria was found to successfully induce EAE in Asc−/− or Nlrp3−/− animal models, while normal 200 μg Mtb mycobacteria could not (Inoue et al., 2012b). In addition, a previous study reported that aggressive immunization induces EAE in caspase-1−/− mice (Furlan et al., 1999). Therefore, the intensity of immunization has a great influence on the induction of EAE, which also suggests the important role of environmental factors in the heterogeneity of MS. A study proposed a possible mechanism for the development of NLRP3 inflammasome-independent EAE, which involves membrane-bound lymphotoxin-β receptor (LTβR) and CXC chemokine receptor 2 (CXCR2), but the exact mechanism is yet to be studied in depth (Inoue et al., 2016). Besides that, studies have shown that IFN-β, a long-used clinical first-line drug, is not therapeutically effective for some types of MS and EAE. Further mechanistic studies have identified IFN-β to have a therapeutic effect on NLRP3-dependent EAE models, which also reflects the heterogeneity of MS (Inoue et al., 2012b). Moreover, EAE mainly simulates MS through immune induction; however, there is no such artificial induction method of MS in patients which can fully simulate the heterogeneity and complexity of MS. Therefore, new and better animal models need to be developed. At present, relevant research on the role of the NLRP3 inflammasome in MS is mainly at the level of cells and animal models, and has not yet involved humans. Therefore, further in-depth studies are urgently required.
Author Contributions
JF was responsible to conceive, design, and revise this review. YC wrote the original manuscript. HY and ZB completed the figures and tables. LW and LY searched and collected the related references. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Outstanding Scientific Fund of Shengjing Hospital (to JF), Key Research and Development Program of Liaoning Province (No. 2020JH2/10300047 to JF), and National Natural Science Foundation of China (No. 81771271 to JF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer TH declared a shared parent affiliation with the authors to the handling editor at the time of review.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully thank members of Feng's lab for their intellectual input.
References
Abbate, A., Toldo, S., Marchetti, C., Kron, J., Van Tassell, B. W., and Dinarello, C. A. (2020). Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126, 1260–1280. doi: 10.1161/CIRCRESAHA.120.315937
Ahmed, Z., Doward, A. I., Pryce, G., Taylor, D. L., Pocock, J. M., Leonard, J. P., et al. (2002). A role for caspase-1 and−3 in the pathology of experimental allergic encephalomyelitis: inflammation versus degeneration. Am. J. Pathol. 161, 1577–1586. doi: 10.1016/S0002-9440(10)64436-7
Ali, M. F., Dasari, H., Van Keulen, V. P., and Carmona, E. M. (2017). Canonical stimulation of the NLRP3 inflammasome by fungal antigens links innate and adaptive B-lymphocyte responses by modulating IL-1beta and IgM production. Front. Immunol. 8, 1504. doi: 10.3389/fimmu.2017.01504
Alvarez, J. I., Cayrol, R., and Prat, A. (2011). Disruption of central nervous system barriers in multiple sclerosis. Biochim. Biophys. Acta 1812, 252–264. doi: 10.1016/j.bbadis.2010.06.017
Aminzadeh, M., Roghani, M., Sarfallah, A., and Riazi, G. H. (2018). TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer's disease. Int. Immunopharmacol. 54, 78–85. doi: 10.1016/j.intimp.2017.10.024
Argaw, A. T., Asp, L., Zhang, J., Navrazhina, K., Pham, T., Mariani, J. N., et al. (2012). Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Invest. 122, 2454–2468. doi: 10.1172/JCI60842
Arnason, B. G. (1999). Immunologic therapy of multiple sclerosis. Annu. Rev. Med. 50, 291–302. doi: 10.1146/annurev.med.50.1.291
Avbelj, M., Hafner-Bratkovič, I., Lainšček, D., Manček-Keber, M., Peternelj, T. T., Panter, G., et al. (2021). Cleavage-mediated regulation of Myd88 signaling by inflammasome-activated caspase-1. Front. Immunol. 12, 790258. doi: 10.3389/fimmu.2021.790258
Awad, F., Assrawi, E., Louvrier, C., Jumeau, C., Georgin-Lavialle, S., Grateau, G., et al. (2018). Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol. Ther. 187, 133–149. doi: 10.1016/j.pharmthera.2018.02.011
Bai, X.-Y., Wang, X.-F., Zhang, L.-S., Du, P.-C., Cao, Z., and Hou, Y. (2018). Tetramethylpyrazine ameliorates experimental autoimmune encephalomyelitis by modulating the inflammatory response. Biochem. Biophys. Res. Commun. 503, 1968–1972. doi: 10.1016/j.bbrc.2018.07.143
Bartlett, R., Stokes, L., and Sluyter, R. (2014). The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol. Rev. 66, 638–675. doi: 10.1124/pr.113.008003
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. doi: 10.4049/jimmunol.0901363
Beheshti, M., Salehi, Z., Abolfazli, R., Shirzad, H., and Izad, M. (2020). Increased level of caspase-1 in the serum of relapsing-remitting multiple sclerosis (RRMS) patients. Iran. J. Allergy Asthma Immunol. 19, 534–538. doi: 10.18502/ijaai.v19i5.4470
Blandford, S. N., Galloway, D. A., Williams, J. B., Arsenault, S., Brown, J., MacLean, G., et al. (2021). Interleukin-1 receptor antagonist: an exploratory plasma biomarker that correlates with disability and provides pathophysiological insights in relapsing-remitting multiple sclerosis. Mult. Scler. Relat. Disord. 52, 103006. doi: 10.1016/j.msard.2021.103006
Blum-Degen, D., Müller, T., Kuhn, W., Gerlach, M., Przuntek, H., and Riederer, P. (1995). Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo Parkinson's disease patients. Neurosci. Lett. 202, 17–20. doi: 10.1016/0304-3940(95)12192-7
Bonomini, F., Dos Santos, M., Veronese, F., and Rezzani, R. (2019). NLRP3 inflammasome modulation by melatonin supplementation in chronic pristane-induced lupus nephritis. Int. J. Mol. Sci. 20, 3466. doi: 10.3390/ijms20143466
Bordt, E. A., and Polster, B. M. (2014). NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic. Biol. Med. 76, 34–46. doi: 10.1016/j.freeradbiomed.2014.07.033
Browne, P., Chandraratna, D., Angood, C., Tremlett, H., Baker, C., Taylor, B. V., et al. (2014). Atlas of multiple sclerosis 2013: a growing global problem with widespread inequity. Neurology 83, 1022–1024. doi: 10.1212/WNL.0000000000000768
Brownlee, W. J., Swanton, J. K., Altmann, D. R., Ciccarelli, O., and Miller, D. H. (2015). Earlier and more frequent diagnosis of multiple sclerosis using the McDonald criteria. J. Neurol. Neurosurg. Psychiatry 86, 584–585. doi: 10.1136/jnnp-2014-308675
Broz, P., and Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Bruchard, M., Mignot, G., Derangère, V., Chalmin, F., Chevriaux, A., Végran, F., et al. (2013). Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64. doi: 10.1038/nm.2999
Buttle, D. J., Murata, M., Knight, C. G., and Barrett, A. J. (1992). CA074 methyl ester: a proinhibitor for intracellular cathepsin B. Arch. Biochem. Biophys. 299, 377–380. doi: 10.1016/0003-9861(92)90290-d
Calahorra, L., Camacho-Toledano, C., Serrano-Regal, M. P., Ortega, M. C., and Clemente, D. (2022). Regulatory cells in multiple sclerosis: from blood to brain. Biomedicines 10, 335. doi: 10.3390/biomedicines10020335
Camara-Lemarroy, C., Metz, L., and Yong, V. (2018). Focus on the gut-brain axis: Multiple sclerosis, the intestinal barrier and the microbiome. World J. Gastroenterol. 24, 4217–4223. doi: 10.3748/wjg.v24.i37.4217
Casson, C. N., Yu, J., Reyes, V. M., Taschuk, F. O., Yadav, A., Copenhaver, A. M., et al. (2015). Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 112, 6688–6693. doi: 10.1073/pnas.1421699112
Cavalli, G., and Dinarello, C. A. (2018). Anakinra therapy for non-cancer inflammatory diseases. Front. Pharmacol. 9, 1157. doi: 10.3389/fphar.2018.01157
Chakraborty, A., Tannenbaum, S., Rordorf, C., Lowe, P. J., Floch, D., Gram, H., et al. (2012). Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1β monoclonal antibody. Clin. Pharmacokinet. 51, e1–18. doi: 10.2165/11599820-000000000-00000
Chatterjee, K., Roy, A., Banerjee, R., Choudhury, S., Mondal, B., Halder, S., et al. (2020). Inflammasome and α-synuclein in Parkinson's disease: A cross-sectional study. J. Neuroimmunol. 338, 577089. doi: 10.1016/j.jneuroim.2019.577089
Chen, J., and Chen, Z. J. (2018). PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76. doi: 10.1038/s41586-018-0761-3
Chen, S., Ma, Q., Krafft, P. R., Hu, Q., Rolland, W., Sherchan, P., et al. (2013). P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol. Dis. 58, 296–307. doi: 10.1016/j.nbd.2013.06.011
Chen, Y., He, H., Lin, B., Chen, Y., Deng, X., Jiang, W., et al. (2021). RRx-001 ameliorates inflammatory diseases by acting as a potent covalent NLRP3 inhibitor. Cell. Mol. Immunol. 18, 1425–1436. doi: 10.1038/s41423-021-00683-y
Chen, Y.-C., Chen, S.-D., Miao, L., Liu, Z.-G., Li, W., Zhao, Z.-X., et al. (2012). Serum levels of interleukin (IL)-18, IL-23 and IL-17 in Chinese patients with multiple sclerosis. J. Neuroimmunol. 243, 56–60. doi: 10.1016/j.jneuroim.2011.12.008
Cheng, Y., Pan, X., Wang, J., Li, X., Yang, S., Yin, R., et al. (2020). Fucoidan inhibits NLRP3 inflammasome activation by enhancing p62/SQSTM1-dependent selective autophagy to alleviate atherosclerosis. Oxid. Med. Cell. Longev. 2020, 3186306. doi: 10.1155/2020/3186306
Ching, S., Zhang, H., Belevych, N., He, L., Lai, W., Pu, X.-a., et al. (2007). Endothelial-specific knockdown of interleukin-1 (IL-1) type 1 receptor differentially alters CNS responses to IL-1 depending on its route of administration. J. Neurosci. 27, 10476–10486. doi: 10.1523/JNEUROSCI.3357-07.2007
Chung, Y., Chang, S. H., Martinez, G. J., Yang, X. O., Nurieva, R., Kang, H. S., et al. (2009). Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30, 576–587. doi: 10.1016/j.immuni.2009.02.007
Codolo, G., Plotegher, N., Pozzobon, T., Brucale, M., Tessari, I., Bubacco, L., et al. (2013). Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8, e55375. doi: 10.1371/journal.pone.0055375
Coll, R. C., Hill, J. R., Day, C. J., Zamoshnikova, A., Boucher, D., Massey, N. L., et al. (2019). MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 15, 556–559. doi: 10.1038/s41589-019-0277-7
Colpitts, S., Kasper, E., Keever, A., Liljenberg, C., Kirby, T., Magori, K., et al. (2017). A bidirectional association between the gut microbiota and CNS disease in a biphasic murine model of multiple sclerosis. Gut Microbes 8, 561–573. doi: 10.1080/19490976.2017.1353843
Compan, V., Baroja-Mazo, A., López-Castejón, G., Gomez, A. I., Martínez, C. M., Angosto, D., et al. (2012). Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37, 487–500. doi: 10.1016/j.immuni.2012.06.013
Correale, J., and Farez, M. (2015). The role of astrocytes in multiple sclerosis progression. Front. Neurol. 6, 180. doi: 10.3389/fneur.2015.00180
Cypryk, W., Nyman, T. A., and Matikainen, S. (2018). From inflammasome to exosome-does extracellular vesicle secretion constitute an inflammasome-dependent immune response? Front. Immunol. 9, 2188. doi: 10.3389/fimmu.2018.02188
Dalakas, M. C. (2008). B cells as therapeutic targets in autoimmune neurological disorders. Nat. Clin. Pract. Neurol. 4, 557–567. doi: 10.1038/ncpneuro0901
Daniels, M. J. D., Rivers-Auty, J., Schilling, T., Spencer, N. G., Watremez, W., Fasolino, V., et al. (2016). Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat. Commun. 7, 12504. doi: 10.1038/ncomms12504
De Gaetano, A., Solodka, K., Zanini, G., Selleri, V., Mattioli, A. V., Nasi, M., et al. (2021). Molecular mechanisms of mtDNA-mediated inflammation. Cells 10, 2898. doi: 10.3390/cells10112898
de Oliveira, L. R. C., Mimura, L. A. N., Fraga-Silva, T. F. C., Ishikawa, L. L. W., Fernandes, A. A. H., Zorzella-Pezavento, S. F. G., et al. (2020). Calcitriol prevents neuroinflammation and reduces blood-brain barrier disruption and local macrophage/microglia activation. Front. Pharmacol. 11, 161. doi: 10.3389/fphar.2020.00161
de Rivero Vaccari, J. P., Brand, F., Adamczak, S., Lee, S. W., Perez-Barcena, J., Wang, M. Y., et al. (2016). Exosome-mediated inflammasome signaling after central nervous system injury. J. Neurochem. 136(Suppl. 1), 39–48. doi: 10.1111/jnc.13036
Dempsey, C., Rubio Araiz, A., Bryson, K. J., Finucane, O., Larkin, C., Mills, E. L., et al. (2017). Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 61, 306–316. doi: 10.1016/j.bbi.2016.12.014
Desu, H. L., Plastini, M., Illiano, P., Bramlett, H. M., Dietrich, W. D., de Rivero Vaccari, J. P., et al. (2020). IC100: a novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J. Neuroinflamm. 17, 143. doi: 10.1186/s12974-020-01826-0
DeTure, M. A., and Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer's disease. Mol. Neurodegener. 14, 32. doi: 10.1186/s13024-019-0333-5
Di Virgilio, F., Dal Ben, D., Sarti, A. C., Giuliani, A. L., and Falzoni, S. (2017). The P2X7 receptor in infection and inflammation. Immunity 47, 15–31. doi: 10.1016/j.immuni.2017.06.020
Di, A., Xiong, S., Ye, Z., Malireddi, R. K. S., Kometani, S., Zhong, M., et al. (2018). The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49, 56–65.e54. doi: 10.1016/j.immuni.2018.04.032
Diaz, R. J., Fernandes, K., Lytvyn, Y., Hawrylyshyn, K., Harvey, K., Hossain, T., et al. (2013). Enhanced cell-volume regulation in cyclosporin A cardioprotection. Cardiovasc. Res. 98, 411–419. doi: 10.1093/cvr/cvt056
Dick, M. S., Sborgi, L., Rühl, S., Hiller, S., and Broz, P. (2016). ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun. 7, 11929. doi: 10.1038/ncomms11929
Dinarello, C. A., Simon, A., and van der Meer, J. W. M. (2012). Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652. doi: 10.1038/nrd3800
Dominic, A., Le, N. T., and Takahashi, M. (2022). Loop between NLRP3 inflammasome and reactive oxygen species. Antioxid. Redox Signal. 36, 784–796. doi: 10.1089/ars.2020.8257
Dong, Y., and Benveniste, E. (2001). Immune function of astrocytes. Glia 36, 180–190. doi: 10.1002/glia.1107
Dostert, C., Pétrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677. doi: 10.1126/science.1156995
Drinkall, S., Lawrence, C. B., Ossola, B., Russell, S., Bender, C., Brice, N. B., et al. (2022). The two pore potassium channel THIK-1 regulates NLRP3 inflammasome activation. Glia. 70, 1301–1316. doi: 10.1002/glia.24174
Dujmovic, I., Mangano, K., Pekmezovic, T., Quattrocchi, C., Mesaros, S., Stojsavljevic, N., et al. (2009). The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: the elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J. Neuroimmunol. 207, 101–106. doi: 10.1016/j.jneuroim.2008.11.004
Dumas, A., Amiable, N., de Rivero Vaccari, J., Chae, J., Keane, R., Lacroix, S., et al. (2014). The inflammasome pyrin contributes to pertussis toxin-induced IL-1β synthesis, neutrophil intravascular crawling and autoimmune encephalomyelitis. PLoS Pathog. 10, e1004150. doi: 10.1371/journal.ppat.1004150
Endriz, J., Ho, P. P., and Steinman, L. (2017). Time correlation between mononucleosis and initial symptoms of MS. Neurol. Neuroimmunol. Neuroinflamm. 4, e308. doi: 10.1212/nxi.0000000000000308
Fan, Z., Pan, Y. T., Zhang, Z. Y., Yang, H., Yu, S. Y., Zheng, Y., et al. (2020). Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson's disease. J. Neuroinflammation 17, 11. doi: 10.1186/s12974-019-1670-6
Feng, Y. S., Tan, Z. X., Wu, L. Y., Dong, F., and Zhang, F. (2021). The involvement of NLRP3 inflammasome in the treatment of neurodegenerative diseases. Biomed. Pharmacother. 138, 111428. doi: 10.1016/j.biopha.2021.111428
Ferrari, C. C., Depino, A. M., Prada, F., Muraro, N., Campbell, S., Podhajcer, O., et al. (2004). Reversible demyelination, blood-brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain. Am. J. Pathol. 165, 1827–1837. doi: 10.1016/S0002-9440(10)63438-4
Franchi, L., Eigenbrod, T., Muñoz-Planillo, R., and Nuñez, G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247. doi: 10.1038/ni.1703
Fu, W., Vukojevic, V., Patel, A., Soudy, R., MacTavish, D., Westaway, D., et al. (2017). Role of microglial amylin receptors in mediating beta amyloid (Aβ)-induced inflammation. J. Neuroinflamm. 14, 199. doi: 10.1186/s12974-017-0972-9
Furlan, R., Martino, G., Galbiati, F., Poliani, P. L., Smiroldo, S., Bergami, A., et al. (1999). Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J. Immunol. 163, 2403–2409.
Gabelloni, M. L., Sabbione, F., Jancic, C., Fuxman Bass, J., Keitelman, I., Iula, L., et al. (2013). NADPH oxidase derived reactive oxygen species are involved in human neutrophil IL-1β secretion but not in inflammasome activation. Eur. J. Immunol. 43, 3324–3335. doi: 10.1002/eji.201243089
Gaidt, M., Ebert, T., Chauhan, D., Schmidt, T., Schmid-Burgk, J., Rapino, F., et al. (2016). Human monocytes engage an alternative inflammasome pathway. Immunity 44, 833–846. doi: 10.1016/j.immuni.2016.01.012
Gao, Q., Zhang, Y., Han, C., Hu, X., Zhang, H., Xu, X., et al. (2016). Blockade of CD47 ameliorates autoimmune inflammation in CNS by suppressing IL-1-triggered infiltration of pathogenic Th17 cells. J. Autoimmun. 69, 74–85. doi: 10.1016/j.jaut.2016.03.002
Gao, W., Li, Y., Liu, X., Wang, S., Mei, P., Chen, Z., et al. (2022). TRIM21 regulates pyroptotic cell death by promoting Gasdermin D oligomerization. Cell Death Differ. 29, 439–450. doi: 10.1038/s41418-021-00867-z
Gentile, A., Rossi, S., Studer, V., Motta, C., De Chiara, V., Musella, A., et al. (2013). Glatiramer acetate protects against inflammatory synaptopathy in experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 8, 651–663. doi: 10.1007/s11481-013-9436-x
Gombault, A., Baron, L., and Couillin, I. (2012). ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 3, 414. doi: 10.3389/fimmu.2012.00414
Gris, D., Ye, Z., Iocca, H. A., Wen, H., Craven, R. R., Gris, P., et al. (2010). NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 185, 974–981. doi: 10.4049/jimmunol.0904145
Guo, X., Chen, S., Yu, W., Chi, Z., Wang, Z., Xu, T., et al. (2021). AKT controls NLRP3 inflammasome activation by inducing DDX3X phosphorylation. FEBS Lett. 595, 2447–2462. doi: 10.1002/1873-3468.14175
Gurung, P., Anand, P. K., Malireddi, R. K. S., Vande Walle, L., Van Opdenbosch, N., Dillon, C. P., et al. (2014). FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192, 1835–1846. doi: 10.4049/jimmunol.1302839
Gurung, P., and Kanneganti, T.-D. (2015). Novel roles for caspase-8 in IL-1β and inflammasome regulation. Am. J. Pathol. 185, 17–25. doi: 10.1016/j.ajpath.2014.08.025
Gurung, P., Lukens, J. R., and Kanneganti, T.-D. (2015). Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 21, 193–201. doi: 10.1016/j.molmed.2014.11.008
Gutcher, I., Urich, E., Wolter, K., Prinz, M., and Becher, B. (2006). Interleukin 18-independent engagement of interleukin 18 receptor-alpha is required for autoimmune inflammation. Nat. Immunol. 7, 946–953. doi: 10.1038/ni1377
Hafler, D. A., Compston, A., Sawcer, S., Lander, E. S., Daly, M. J., De Jager, P. L., et al. (2007). Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 357, 851–862. doi: 10.1056/NEJMoa073493
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K., and Miao, E. A. (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. doi: 10.1126/science.1240988
Hagman, S., Kolasa, M., Basnyat, P., Helminen, M., Kahonen, M., Dastidar, P., et al. (2015). Analysis of apoptosis-related genes in patients with clinically isolated syndrome and their association with conversion to multiple sclerosis. J. Neuroimmunol. 280, 43–48. doi: 10.1016/j.jneuroim.2015.02.006
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828. doi: 10.1038/nbt.1957
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hasanzadeh, S., Read, M. I., Bland, A. R., Majeed, M., Jamialahmadi, T., and Sahebkar, A. (2020). Curcumin: an inflammasome silencer. Pharmacol. Res. 159, 104921. doi: 10.1016/j.phrs.2020.104921
He, H., Jiang, H., Chen, Y., Ye, J., Wang, A., Wang, C., et al. (2018). Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 9, 2550. doi: 10.1038/s41467-018-04947-6
He, Y., Hara, H., and Núñez, G. (2016a). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021. doi: 10.1016/j.tibs.2016.09.002
He, Y., Zeng, M. Y., Yang, D., Motro, B., and Núñez, G. (2016b). NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357. doi: 10.1038/nature16959
Healy, B. C., Ali, E. N., Guttmann, C. R., Chitnis, T., Glanz, B. I., Buckle, G., et al. (2009). Smoking and disease progression in multiple sclerosis. Arch. Neurol. 66, 858–864. doi: 10.1001/archneurol.2009.122
Heneka, M. T. (2017). Inflammasome activation and innate immunity in Alzheimer's disease. Brain Pathol. 27, 220–222. doi: 10.1111/bpa.12483
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hezel, M. E. v., Nieuwland, R., Bruggen, R. v., and Juffermans, N. P. (2017). The ability of extracellular vesicles to induce a pro-inflammatory host response. Int. J. Mol. Sci. 18, 1285. doi: 10.3390/ijms18061285
Horng, T. (2014). Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 35, 253–261. doi: 10.1016/j.it.2014.02.007
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856. doi: 10.1038/ni.1631
Hou, B., Zhang, Y., Liang, P., He, Y., Peng, B., Liu, W., et al. (2020). Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 11, 377. doi: 10.1038/s41419-020-2565-2
Hou, L., Yang, J., Li, S., Huang, R., Zhang, D., Zhao, J., et al. (2020). Glibenclamide attenuates 2,5-hexanedione-induced neurotoxicity in the spinal cord of rats through mitigation of NLRP3 inflammasome activation, neuroinflammation and oxidative stress. Toxicol. Lett. 331, 152–158. doi: 10.1016/j.toxlet.2020.06.002
Hu, J. J., Liu, X., Xia, S., Zhang, Z., Zhang, Y., Zhao, J., et al. (2020). FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 21, 736–745. doi: 10.1038/s41590-020-0669-6
Huang, C. (2008). Polyneuropathy induced by n-hexane intoxication in Taiwan. Acta Neurol. Taiwan 17, 3–10.
Huang, J., Xie, Z.-K., Lu, R.-B., and Xie, Z.-F. (2013). Association of interleukin-1 gene polymorphisms with multiple sclerosis: a meta-analysis. Inflamm. Res. 62, 97–106. doi: 10.1007/s00011-012-0556-1
Huang, X., Feng, Z., Jiang, Y., Li, J., Xiang, Q., Guo, S., et al. (2019). VSIG4 mediates transcriptional inhibition of Nlrp3 and Il-1β in macrophages. Sci. Adv. 5, eaau7426. doi: 10.1126/sciadv.aau7426
Imani, D., Azimi, A., Salehi, Z., Rezaei, N., Emamnejad, R., Sadr, M., et al. (2018). Association of nod-like receptor protein-3 single nucleotide gene polymorphisms and expression with the susceptibility to relapsing-remitting multiple sclerosis. Int. J. Immunogenet. 45, 329–336. doi: 10.1111/iji.12401
Inoue, M., Chen, P.-H., Siecinski, S., Li, Q.-J., Liu, C., Steinman, L., et al. (2016). An interferon-β-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat. Neurosci. 19, 1599–1609. doi: 10.1038/nn.4421
Inoue, M., Williams, K. L., Gunn, M. D., and Shinohara, M. L. (2012a). NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 109, 10480–10485. doi: 10.1073/pnas.1201836109
Inoue, M., Williams, K. L., Oliver, T., Vandenabeele, P., Rajan, J. V., Miao, E. A., et al. (2012b). Interferon-β therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci. Signal. 5, ra38. doi: 10.1126/scisignal.2002767
Isik, N., Arman, A., Canturk, I. A., Gurkan, A. C., Candan, F., Aktan, S., et al. (2013). Multiple sclerosis: association with the interleukin-1 gene family polymorphisms in the Turkish population. Int. J. Neurosci. 123, 711–718. doi: 10.3109/00207454.2013.795563
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Jahanbani-Ardakani, H., Alsahebfosoul, F., Etemadifar, M., and Abtahi, S.-H. (2019). Interleukin 18 polymorphisms and its serum level in patients with multiple sclerosis. Ann. Indian Acad. Neurol. 22, 474–476. doi: 10.4103/aian.AIAN_515_18
Jander, S., and Stoll, G. (1998). Differential induction of interleukin-12, interleukin-18, and interleukin-1beta converting enzyme mRNA in experimental autoimmune encephalomyelitis of the Lewis rat. J. Neuroimmunol. 91, 93–99. doi: 10.1016/s0165-5728(98)00162-3
Jha, S., Srivastava, S. Y., Brickey, W. J., Iocca, H., Toews, A., Morrison, J. P., et al. (2010). The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J. Neurosci. 30, 15811–15820. doi: 10.1523/JNEUROSCI.4088-10.2010
Jian, Z., Ding, S., Deng, H., Wang, J., Yi, W., Wang, L., et al. (2016). Probenecid protects against oxygen-glucose deprivation injury in primary astrocytes by regulating inflammasome activity. Brain Res. 1643, 123–129. doi: 10.1016/j.brainres.2016.05.002
Jo, E. K., Kim, J. K., Shin, D. M., and Sasakawa, C. (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 13, 148–159. doi: 10.1038/cmi.2015.95
Johann, S., Heitzer, M., Kanagaratnam, M., Goswami, A., Rizo, T., Weis, J., et al. (2015). NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63, 2260–2273. doi: 10.1002/glia.22891
Kadhim, H., Deltenre, P., Martin, J. J., and Sébire, G. (2016). In-situ expression of Interleukin-18 and associated mediators in the human brain of sALS patients: hypothesis for a role for immune-inflammatory mechanisms. Med. Hypotheses 86, 14–17. doi: 10.1016/j.mehy.2015.11.022
Kapoor, R., Smith, K., Allegretta, M., Arnold, D., Carroll, W., Comabella, M., et al. (2020). Serum neurofilament light as a biomarker in progressive multiple sclerosis. Neurology 95, 436–444. doi: 10.1212/wnl.0000000000010346
Karakas Celik, S., Öz, Z. S., Dursun, A., Unal, A., Emre, U., Cicek, S., et al. (2014). Interleukin 18 gene polymorphism is a risk factor for multiple sclerosis. Mol. Biol. Rep. 41, 1653–1658. doi: 10.1007/s11033-013-3013-5
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Ke, P., Shao, B.-Z., Xu, Z.-Q., Chen, X.-W., Wei, W., and Liu, C. (2017). Activating α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of β-arrestin-1. CNS Neurosci. Ther. 23, 875–884. doi: 10.1111/cns.12758
Keane, R., Dietrich, W., and de Rivero Vaccari, J. (2018). Inflammasome proteins as biomarkers of multiple sclerosis. Front. Neurol. 9, 135. doi: 10.3389/fneur.2018.00135
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20, 3328. doi: 10.3390/ijms20133328
Kermode, A. G., Thompson, A. J., Tofts, P., MacManus, D. G., Kendall, B. E., Kingsley, D. P., et al. (1990). Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Pathogenetic and clinical implications. Brain 113(Pt 5), 1477–1489. doi: 10.1093/brain/113.5.1477
Khalil, M., Teunissen, C., Otto, M., Piehl, F., Sormani, M., Gattringer, T., et al. (2018). Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 14, 577–589. doi: 10.1038/s41582-018-0058-z
Kiasalari, Z., Afshin-Majd, S., Baluchnejadmojarad, T., Azadi-Ahmadabadi, E., Fakour, M., Ghasemi-Tarie, R., et al. (2021). Sinomenine alleviates murine experimental autoimmune encephalomyelitis model of multiple sclerosis through inhibiting NLRP3 inflammasome. J. Mol. Neurosci. 71, 215–224. doi: 10.1007/s12031-020-01637-1
Kobelt, G., Thompson, A., Berg, J., Gannedahl, M., and Eriksson, J. (2017). New insights into the burden and costs of multiple sclerosis in Europe. Multiple Scler. 23, 1123–1136. doi: 10.1177/1352458517694432
Kumar, H., Kawai, T., and Akira, S. (2011). Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30, 16–34. doi: 10.3109/08830185.2010.529976
Lall, D., and Baloh, R. H. (2017). Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest. 127, 3250–3258. doi: 10.1172/jci90607
Lalor, S. J., Dungan, L. S., Sutton, C. E., Basdeo, S. A., Fletcher, J. M., and Mills, K. H. G. (2011). Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J. Immunol. 186, 5738–5748. doi: 10.4049/jimmunol.1003597
Lamkanfi, M., and Dixit, V. M. (2009). Inflammasomes: guardians of cytosolic sanctity. Immunol. Rev. 227, 95–105. doi: 10.1111/j.1600-065X.2008.00730.x
Lang, Y., Chu, F., Liu, L., Zheng, C., Li, C., Shen, D., et al. (2021). Potential role of BAY11-7082, a NF-κB blocker inhibiting experimental autoimmune encephalomyelitis in C57BL/6J mice via declining NLRP3 inflammasomes. Clin. Exp. Immunol. doi: 10.1093/cei/uxab022
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Lazibat, I., Rubinic Majdak, M., and Zupanic, S. (2018). Multiple sclerosis: new aspects of immunopathogenesis. Acta Clin. Croat. 57, 352–361. doi: 10.20471/acc.2018.57.02.17
Lee, E., Hwang, I., Park, S., Hong, S., Hwang, B., Cho, Y., et al. (2019). MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 26, 213–228. doi: 10.1038/s41418-018-0124-5
Leite, J. A., Cavalcante-Silva, L. H. A., Ribeiro, M. R., de Morais Lima, G., Scavone, C., and Rodrigues-Mascarenhas, S. (2022). Neuroinflammation and neutrophils: modulation by ouabain. Front. Pharmacol. 13, 824907. doi: 10.3389/fphar.2022.824907
Li, R., Patterson, K. R., and Bar-Or, A. (2018). Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 19, 696–707. doi: 10.1038/s41590-018-0135-x
Li, Z., Chen, X., Tao, J., Shi, A., Zhang, J., and Yu, P. (2021). Exosomes regulate NLRP3 inflammasome in diseases. Front Cell Dev Biol 9, 802509. doi: 10.3389/fcell.2021.802509
Liang, M., Chen, X., Wang, L., Qin, L., Wang, H., Sun, Z., et al. (2020). Cancer-derived exosomal TRIM59 regulates macrophage NLRP3 inflammasome activation to promote lung cancer progression. J. Exp. Clin. Cancer Res. 39, 176. doi: 10.1186/s13046-020-01688-7
Lim, K. H., Chen, L. C., Hsu, K., Chang, C. C., Chang, C. Y., Kao, C. W., et al. (2020). BAFF-driven NLRP3 inflammasome activation in B cells. Cell Death Dis. 11, 820. doi: 10.1038/s41419-020-03035-2
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. doi: 10.1038/nature18629
Losy, J., and Niezgoda, A. (2001). IL-18 in patients with multiple sclerosis. Acta Neurol. Scand. 104, 171–173. doi: 10.1034/j.1600-0404.2001.00356.x
Lu, L., Qi, S., Chen, Y., Luo, H., Huang, S., Yu, X., et al. (2020). Targeted immunomodulation of inflammatory monocytes across the blood-brain barrier by curcumin-loaded nanoparticles delays the progression of experimental autoimmune encephalomyelitis. Biomaterials 245, 119987. doi: 10.1016/j.biomaterials.2020.119987
Macia, L., Tan, J., Vieira, A. T., Leach, K., Stanley, D., Luong, S., et al. (2015). Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 6, 6734. doi: 10.1038/ncomms7734
Maimone, D., Gregory, S., Arnason, B. G., and Reder, A. T. (1991). Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J. Neuroimmunol. 32, 67–74. doi: 10.1016/0165-5728(91)90073-g
Malhotra, S., Costa, C., Eixarch, H., Keller, C. W., Amman, L., Martínez-Banaclocha, H., et al. (2020). NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 143, 1414–1430. doi: 10.1093/brain/awaa084
Mallucci, G., Peruzzotti-Jametti, L., Bernstock, J., and Pluchino, S. (2015). The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog. Neurobio 127, 1–22. doi: 10.1016/j.pneurobio.2015.02.003
Mancino, D. N. J., Lima, A., Roig, P., García Segura, L. M., De Nicola, A. F., and Garay, L. I. (2022). Tibolone restrains neuroinflammation in mouse experimental autoimmune encephalomyelitis. J. Neuroendocrinol. 34, e13078. doi: 10.1111/jne.13078
Mandolesi, G., Musella, A., Gentile, A., Grasselli, G., Haji, N., Sepman, H., et al. (2013). Interleukin-1β alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosc. 33, 12105–12121. doi: 10.1523/JNEUROSCI.5369-12.2013
Manouchehrinia, A., Stridh, P., Khademi, M., Leppert, D., Barro, C., Michalak, Z., et al. (2020). Plasma neurofilament light levels are associated with risk of disability in multiple sclerosis. Neurology 94, e2457–e2467. doi: 10.1212/wnl.0000000000009571
Mao, Z., Liu, C., Ji, S., Yang, Q., Ye, H., Han, H., et al. (2017). The NLRP3 inflammasome is involved in the pathogenesis of Parkinson's disease in rats. Neurochem. Res. 42, 1104–1115. doi: 10.1007/s11064-017-2185-0
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi: 10.1016/s1097-2765(02)00599-3
McKenzie, B. A., Mamik, M. K., Saito, L. B., Boghozian, R., Monaco, M. C., Major, E. O., et al. (2018). Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 115, E6065–E6074. doi: 10.1073/pnas.1722041115
Metwally, H., Tanaka, T., Li, S., Parajuli, G., Kang, S., Hanieh, H., et al. (2020). Noncanonical STAT1 phosphorylation expands its transcriptional activity into promoting LPS-induced IL-6 and IL-12p40 production. Sci. Signal. 13, eaay0574. doi: 10.1126/scisignal.aay0574
Mueller, N., Bakacs, E., Combellick, J., Grigoryan, Z., and Dominguez-Bello, M. (2015). The infant microbiome development: mom matters. Trends Mol. Med. 21, 109–117. doi: 10.1016/j.molmed.2014.12.002
Muñoz-Planillo, R., Kuffa, P., Martínez-Colón, G., Smith, B. L., Rajendiran, T. M., and Núñez, G. (2013). K? efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. doi: 10.1016/j.immuni.2013.05.016
Murakami, T., Ockinger, J., Yu, J., Byles, V., McColl, A., Hofer, A. M., et al. (2012). Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 109, 11282–11287. doi: 10.1073/pnas.1117765109
Musella, A., Mandolesi, G., Gentile, A., Rossi, S., Studer, V., Motta, C., et al. (2013). Cladribine interferes with IL-1β synaptic effects in experimental multiple sclerosis. J. Neuroimmunol. 264. doi: 10.1016/j.jneuroim.2013.08.009
Niu, T., De Rosny, C., Chautard, S., Rey, A., Patoli, D., Groslambert, M., et al. (2021). NLRP3 phosphorylation in its LRR domain critically regulates inflammasome assembly. Nat. Commun. 12, 5862. doi: 10.1038/s41467-021-26142-w
Ochoa-Repáraz, J., Mielcarz, D., Ditrio, L., Burroughs, A., Foureau, D., Haque-Begum, S., et al. (2009). Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol. 183, 6041–6050. doi: 10.4049/jimmunol.0900747
Olcum, M., Tastan, B., Kiser, C., Genc, S., and Genc, K. (2020). Microglial NLRP3 inflammasome activation in multiple sclerosis. Adv. Protein Chem. Struct. Biol. 119, 247–308. doi: 10.1016/bs.apcsb.2019.08.007
Orhan, G., Eruyar, E., Mungan, S. Ö., Ak, F., and Karahalil, B. (2016). The association of IL-18 gene promoter polymorphisms and the levels of serum IL-18 on the risk of multiple sclerosis. Clin. Neurol. Neurosurg. 146, 96–101. doi: 10.1016/j.clineuro.2016.04.027
Pan, R. Y., Kong, X. X., Cheng, Y., Du, L., Wang, Z. C., Yuan, C., et al. (2021). 1,2,4-Trimethoxybenzene selectively inhibits NLRP3 inflammasome activation and attenuates experimental autoimmune encephalomyelitis. Acta Pharmacol. Sin. 42, 1769–1779. doi: 10.1038/s41401-021-00613-8
Paolicelli, R. C., Bergamini, G., and Rajendran, L. (2019). Cell-to-cell communication by extracellular vesicles: focus on microglia. Neuroscience 405, 148–157. doi: 10.1016/j.neuroscience.2018.04.003
Paré, A., Mailhot, B., Lévesque, S. A., Juzwik, C., Ignatius Arokia Doss, P. M., Lécuyer, M.-A., et al. (2018). IL-1β enables CNS access to CCR2 monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 115, E1194–E1203. doi: 10.1073/pnas.1714948115
Paul, C., and Bolton, C. (1995). Inhibition of blood-brain barrier disruption in experimental allergic encephalomyelitis by short-term therapy with dexamethasone or cyclosporin A. Int. J. Immunopharmacol. 17, 497–503. doi: 10.1016/0192-0561(95)00034-y
Pearce, L., Davidson, S. M., and Yellon, D. M. (2021). Does remote ischaemic conditioning reduce inflammation? A focus on innate immunity and cytokine response. Basic Res. Cardiol. 116, 12. doi: 10.1007/s00395-021-00852-0
Pellegrini, C., Antonioli, L., Calderone, V., Colucci, R., Fornai, M., and Blandizzi, C. (2020). Microbiota-gut-brain axis in health and disease: is NLRP3 inflammasome at the crossroads of microbiota-gut-brain communications? Prog. Neurobiol. 191, 101806. doi: 10.1016/j.pneurobio.2020.101806
Peng, Y., Chen, J., Dai, Y., Jiang, Y., Qiu, W., Gu, Y., et al. (2019). NLRP3 level in cerebrospinal fluid of patients with neuromyelitis optica spectrum disorders: Increased levels and association with disease severity. Mult. Scler. Relat. Disord. 39, 101888. doi: 10.1016/j.msard.2019.101888
Peñin-Franch, A., García-Vidal, J. A., Martínez, C. M., Escolar-Reina, P., Martínez-Ojeda, R. M., Gómez, A. I., et al. (2022). Galvanic current activates the NLRP3 inflammasome to promote type I collagen production in tendon. Elife 11, e73675. doi: 10.7554/eLife.73675
Peter, J. B., Boctor, F. N., and Tourtellotte, W. W. (1991). Serum and CSF levels of IL-2, sIL-2R, TNF-alpha, and IL-1 beta in chronic progressive multiple sclerosis: expected lack of clinical utility. Neurology 41, 121–123. doi: 10.1212/wnl.41.1.121
Petrucci, S., Consoli, F., and Valente, E. M. (2014). Parkinson Disease Genetics: A “Continuum” from Mendelian to Multifactorial Inheritance. Curr. Mol. Med. 14, 1079–1088. doi: 10.2174/1566524014666141010155509
Piancone, F., La Rosa, F., Marventano, I., Saresella, M., and Clerici, M. (2021). The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 26(4). doi: 10.3390/molecules26040953
Pinke, K. H., Zorzella-Pezavento, S. F. G., de Campos Fraga-Silva, T. F., Mimura, L. A. N., de Oliveira, L. R. C., Ishikawa, L. L. W., et al. (2020). Calming down mast cells with ketotifen: a potential strategy for multiple sclerosis therapy? Neurotherapeutics 17, 218–234. doi: 10.1007/s13311-019-00775-8
Potenzieri, A., Riva, B., and Genazzani, A. A. (2020). Unexpected Ca-mobilization of oxaliplatin via H1 histamine receptors. Cell Calcium 86, 102128. doi: 10.1016/j.ceca.2019.102128
Prada, I., Furlan, R., Matteoli, M., and Verderio, C. (2013). Classical and unconventional pathways of vesicular release in microglia. Glia 61, 1003–1017. doi: 10.1002/glia.22497
Pröbstel, A.-K., and Baranzini, S. E. (2018). The role of the gut microbiome in multiple sclerosis risk and progression: towards characterization of the “MS microbiome”. Neurotherapeutics 15, 126–134. doi: 10.1007/s13311-017-0587-y
Qiao, C., Zhang, Q., Jiang, Q., Zhang, T., Chen, M., Fan, Y., et al. (2018). Inhibition of the hepatic Nlrp3 protects dopaminergic neurons via attenuating systemic inflammation in a MPTP/p mouse model of Parkinson's disease. J. Neuroinflammation 15, 193. doi: 10.1186/s12974-018-1236-z
Ramirez, M., Poreba, M., Snipas, S., Groborz, K., Drag, M., and Salvesen, G. (2018). Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J. Biol. Chem. 293, 7058–7067. doi: 10.1074/jbc.RA117.001329
Rao, S., Schieber, A. M. P., O'Connor, C. P., Leblanc, M., Michel, D., and Ayres, J. S. (2017). Pathogen-mediated inhibition of anorexia promotes host survival and transmission. Cell 168, 503–516.e512. doi: 10.1016/j.cell.2017.01.006
Reinert, M., Benkert, P., Wuerfel, J., Michalak, Z., Ruberte, E., Barro, C., et al. (2020). Serum neurofilament light chain is a useful biomarker in pediatric multiple sclerosis. Neurol Neuroimmunol. Neuroinflamm. 7, e749. doi: 10.1212/nxi.0000000000000749
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rothhammer, V., Mascanfroni, I. D., Bunse, L., Takenaka, M. C., Kenison, J. E., Mayo, L., et al. (2016). Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597. doi: 10.1038/nm.4106
Rottlaender, A., and Kuerten, S. (2015). Stepchild or prodigy? Neuroprotection in multiple sclerosis (MS) research. Int. J. Mol. Sci. 16, 14850–14865. doi: 10.3390/ijms160714850
Russi, A. E., Walker-Caulfield, M. E., and Brown, M. A. (2018). Mast cell inflammasome activity in the meninges regulates EAE disease severity. Clin. Immunol. 189, 14–22. doi: 10.1016/j.clim.2016.04.009
Rutsch, A., Kantsjo, J. B., and Ronchi, F. (2020). The gut-brain axis: how microbiota and host inflammasome influence brain physiology and pathology. Front. Immunol. 11, 604179. doi: 10.3389/fimmu.2020.604179
Salzer, J., Svenningsson, A., and Sundström, P. (2010). Neurofilament light as a prognostic marker in multiple sclerosis. Mult. Scler. 16, 287–292. doi: 10.1177/1352458509359725
Samir, P., Kesavardhana, S., Patmore, D. M., Gingras, S., Malireddi, R. K. S., Karki, R., et al. (2019). DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594. doi: 10.1038/s41586-019-1551-2
Sánchez-Fernández, A., Skouras, D. B., Dinarello, C. A., and López-Vales, R. (2019). OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front. Immunol. 10, 2578. doi: 10.3389/fimmu.2019.02578
Seppi, D., Puthenparampil, M., Federle, L., Ruggero, S., Toffanin, E., Rinaldi, F., et al. (2014). Cerebrospinal fluid IL-1β correlates with cortical pathology load in multiple sclerosis at clinical onset. J. Neuroimmunol. 270, 56–60. doi: 10.1016/j.jneuroim.2014.02.014
Serafini, B., Rosicarelli, B., Magliozzi, R., Stigliano, E., and Aloisi, F. (2004). Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 14, 164–174. doi: 10.1111/j.1750-3639.2004.tb00049.x
Shao, B.-Z., Wei, W., Ke, P., Xu, Z.-Q., Zhou, J.-X., and Liu, C. (2014). Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci. Ther. 20, 1021–1028. doi: 10.1111/cns.12349
Shao, B.-Z., Xu, Z.-Q., Han, B.-Z., Su, D.-F., and Liu, C. (2015). NLRP3 inflammasome and its inhibitors: a review. Front. Pharmacol. 6, 262. doi: 10.3389/fphar.2015.00262
Shao, S., Chen, C., Shi, G., Zhou, Y., Wei, Y., Fan, N., et al. (2021). Therapeutic potential of the target on NLRP3 inflammasome in multiple sclerosis. Pharmacol. Ther. 227, 107880. doi: 10.1016/j.pharmthera.2021.107880
Sharif, H., Wang, L., Wang, W. L., Magupalli, V. G., Andreeva, L., Qiao, Q., et al. (2019). Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343. doi: 10.1038/s41586-019-1295-z
Sharma, R., Young, C., and Neu, J. (2010). Molecular modulation of intestinal epithelial barrier: contribution of microbiota. J. Biomed. Biotechnol. 2010, 305879. doi: 10.1155/2010/305879
Shaw, P. J., Lukens, J. R., Burns, S., Chi, H., McGargill, M. A., and Kanneganti, T. D. (2010). Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J. Immunol. 184, 4610–4614. doi: 10.4049/jimmunol.1000217
Shenker, B. J., Ojcius, D. M., Walker, L. P., Zekavat, A., Scuron, M. D., and Boesze-Battaglia, K. (2015). Aggregatibacter actinomycetemcomitans cytolethal distending toxin activates the NLRP3 inflammasome in human macrophages, leading to the release of proinflammatory cytokines. Infect. Immun. 83, 1487–1496. doi: 10.1128/iai.03132-14
Shi, F. D., Takeda, K., Akira, S., Sarvetnick, N., and Ljunggren, H. G. (2000). IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-gamma by NK cells. J. Immunol. 165, 3099–3104. doi: 10.4049/jimmunol.165.6.3099
Shi, J., Gao, W., and Shao, F. (2017). Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254. doi: 10.1016/j.tibs.2016.10.004
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J., Li, P., et al. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. doi: 10.1038/nature13683
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Si, X., Fang, Y., Li, L., Gu, L., Yin, X., Yan, Y., et al. (2021). From inflammasome to Parkinson's disease: does the NLRP3 inflammasome facilitate exosome secretion and exosomal alpha-synuclein transmission in Parkinson's disease? Exp. Neurol. 336, 113525. doi: 10.1016/j.expneurol.2020.113525
Soares, J. L., Oliveira, E. M., and Pontillo, A. (2019). Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult. Scler. Relat. Disord. 29, 26–34. doi: 10.1016/j.msard.2019.01.023
Sommer, F., and Bäckhed, F. (2013). The gut microbiota–masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238. doi: 10.1038/nrmicro2974
Song, L., Pei, L., Yao, S., Wu, Y., and Shang, Y. (2017). NLRP3 inflammasome in neurological diseases, from functions to therapies. Front. Cell. Neurosci. 11, 63. doi: 10.3389/fncel.2017.00063
Soundara Rajan, T., Giacoppo, S., Diomede, F., Bramanti, P., Trubiani, O., and Mazzon, E. (2017). Human periodontal ligament stem cells secretome from multiple sclerosis patients suppresses NALP3 inflammasome activation in experimental autoimmune encephalomyelitis. Int. J. Immunopathol. Pharmacol. 30, 238–252. doi: 10.1177/0394632017722332
Swanton, T., Cook, J., Beswick, J. A., Freeman, S., Lawrence, C. B., and Brough, D. (2018). Is targeting the inflammasome a way forward for neuroscience drug discovery? SLAS Discov. 23, 991–1017. doi: 10.1177/2472555218786210
Tang, T., Li, P., Zhou, X., Wang, R., Fan, X., Yang, M., et al. (2021). The E3 ubiquitin ligase TRIM65 negatively regulates inflammasome activation through promoting ubiquitination of NLRP3. Front. Immunol. 12, 741839. doi: 10.3389/fimmu.2021.741839
Teunissen, C., and Khalil, M. (2012). Neurofilaments as biomarkers in multiple sclerosis. Mult. Scler. 18, 552–556. doi: 10.1177/1352458512443092
Trnka, J., Blaikie, F. H., Logan, A., Smith, R. A. J., and Murphy, M. P. (2009). Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic. Res. 43, 4–12. doi: 10.1080/10715760802582183
van Lieverloo, G., Wieske, L., Verhamme, C., Vrancken, A., van Doorn, P., Michalak, Z., et al. (2019). Serum neurofilament light chain in chronic inflammatory demyelinating polyneuropathy. J. Peripher. Nerv. Syst. 24, 187–194. doi: 10.1111/jns.12319
Verhoef, P. A., Kertesy, S. B., Lundberg, K., Kahlenberg, J. M., and Dubyak, G. R. (2005). Inhibitory effects of chloride on the activation of caspase-1, IL-1beta secretion, and cytolysis by the P2X7 receptor. J. Immunol. 175, 7623–7634. doi: 10.4049/jimmunol.175.11.7623
Villar, L. M., Sádaba, M. C., Roldán, E., Masjuan, J., González-Porqué, P., Villarrubia, N., et al. (2005). Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. J. Clin. Investig. 115, 187–194. doi: 10.1172/jci200522833
Voet, S., Mc Guire, C., Hagemeyer, N., Martens, A., Schroeder, A., Wieghofer, P., et al. (2018). A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 9, 2036. doi: 10.1038/s41467-018-04376-5
Wang, H. Q., Song, K. Y., Feng, J. Z., Huang, S. Y., Guo, X. M., Zhang, L., et al. (2022). Caffeine inhibits activation of the NLRP3 inflammasome via autophagy to attenuate microglia-mediated neuroinflammation in experimental autoimmune encephalomyelitis. J. Mol. Neurosci. 72, 97–112. doi: 10.1007/s12031-021-01894-8
Wang, Q., Hou, L., Zhang, C., Song, F., and Xie, K. (2008). Changes of cytoskeletal proteins in nerve tissues and serum of rats treated with 2,5-hexanedione. Toxicology 244, 166–178. doi: 10.1016/j.tox.2007.11.009
Wikoff, W. R., Anfora, A. T., Liu, J., Schultz, P. G., Lesley, S. A., Peters, E. C., et al. (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U.S.A. 106, 3698–3703. doi: 10.1073/pnas.0812874106
Wu, X., Ren, G., Zhou, R., Ge, J., and Chen, F. H. (2019). The role of Ca(2+) in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis. Lab. Invest. 99, 499–513. doi: 10.1038/s41374-018-0135-3
Yan, B., Zhang, Y., Liang, C., Liu, B., Ding, F., Wang, Y., et al. (2020). Stem cell-derived exosomes prevent pyroptosis and repair ischemic muscle injury through a novel exosome/circHIPK3/ FOXO3a pathway. Theranostics 10, 6728–6742. doi: 10.7150/thno.42259
Yang, X., Cao, X., Wei, X., and Zheng, P. (2020). Oridonin ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-κB signaling in T lymphocytes. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 36, 492–498.
Youm, Y.-H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Yu, X., Lv, J., Wu, J., Chen, Y., Chen, F., and Wang, L. (2021). The autoimmune encephalitis-related cytokine TSLP in the brain primes neuroinflammation by activating the JAK2-NLRP3 axis. Clin. Exp. Immunol. 207, 113–122. doi: 10.1093/cei/uxab023
Yu, Y., Wu, D., Li, J., Deng, S., Liu, T., Zhang, T., et al. (2020). Bixin attenuates experimental autoimmune encephalomyelitis by suppressing TXNIP/NLRP3 inflammasome activity and activating NRF2 signaling. Front. Immunol. 11, 593368. doi: 10.3389/fimmu.2020.593368
Zhang, C. J., Jiang, M., Zhou, H., Liu, W., Wang, C., Kang, Z., et al. (2018). TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and promotes neuroinflammation. J. Clin. Invest. 128, 5399–5412. doi: 10.1172/jci121901
Zhang, P., Shao, X. Y., Qi, G. J., Chen, Q., Bu, L. L., Chen, L. J., et al. (2016). Cdk5-dependent activation of neuronal inflammasomes in Parkinson's disease. Mov. Disord. 31, 366–376. doi: 10.1002/mds.26488
Zhang, Y., Hou, B., Liang, P., Lu, X., Wu, Y., Zhang, X., et al. (2021). TRPV1 channel mediates NLRP3 inflammasome-dependent neuroinflammation in microglia. Cell Death Dis. 12, 1159. doi: 10.1038/s41419-021-04450-9
Zhang, Y., Huang, R., Cheng, M., Wang, L., Chao, J., Li, J., et al. (2019). Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome 7, 116. doi: 10.1186/s40168-019-0733-3
Zhang, Y., Liu, F., Yuan, Y., Jin, C., Chang, C., Zhu, Y., et al. (2017). Inflammasome-Derived Exosomes Activate NF-κB Signaling in Macrophages. J. Proteome Res. 16, 170–178. doi: 10.1021/acs.jproteome.6b00599
Zhao, M., Sun, D., Guan, Y., Wang, Z., Sang, D., Liu, M., et al. (2016). Disulfiram and diphenhydramine hydrochloride upregulate miR-30a to suppress IL-17-associated autoimmune inflammation. J. Neurosci. 36, 9253–9266. doi: 10.1523/jneurosci.4587-15.2016
Zhao, W., Shi, C. S., Harrison, K., Hwang, I. Y., Nabar, N. R., Wang, M., et al. (2020). AKT regulates NLRP3 inflammasome activation by phosphorylating NLRP3 serine 5. J. Immunol. 205, 2255–2264. doi: 10.4049/jimmunol.2000649
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X.-J., et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. doi: 10.1038/s41586-018-0372-z
Keywords: inflammation, NLRP3 inflammasome, multiple sclerosis, biomarker, treatment
Citation: Cui Y, Yu H, Bu Z, Wen L, Yan L and Feng J (2022) Focus on the Role of the NLRP3 Inflammasome in Multiple Sclerosis: Pathogenesis, Diagnosis, and Therapeutics. Front. Mol. Neurosci. 15:894298. doi: 10.3389/fnmol.2022.894298
Received: 11 March 2022; Accepted: 05 May 2022;
Published: 25 May 2022.
Edited by:
Kiran Bhaskar, University of New Mexico, United StatesReviewed by:
Jong-Seok Moon, Soonchunhyang University, South KoreaTianhao Hu, The First Affiliated Hospital of China Medical University, China
Lyu Yi Qing, King Saud University, Saudi Arabia
Copyright © 2022 Cui, Yu, Bu, Wen, Yan and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Feng, anVhbmZlbmdAY211LmVkdS5jbg==