 Aleksandar Dobric
Aleksandar Dobric Simone N. De Luca
Simone N. De Luca Huei Jiunn Seow
Huei Jiunn Seow Hao Wang
Hao Wang Kurt Brassington
Kurt Brassington Stanley M. H. Chan
Stanley M. H. Chan Kevin Mou
Kevin Mou Jonathan Erlich
Jonathan Erlich Stella Liong
Stella Liong Stavros Selemidis
Stavros Selemidis Sarah J. Spencer
Sarah J. Spencer Steven Bozinovski
Steven Bozinovski Ross Vlahos*
Ross Vlahos*
- School of Health and Biomedical Sciences, RMIT University, Bundoora, VIC, Australia
Background and Objective: Neurocognitive dysfunction is present in up to ∼61% of people with chronic obstructive pulmonary disease (COPD), with symptoms including learning and memory deficiencies, negatively impacting the quality of life of these individuals. As the mechanisms responsible for neurocognitive deficits in COPD remain unknown, we explored whether chronic cigarette smoke (CS) exposure causes neurocognitive dysfunction in mice and whether this is associated with neuroinflammation and an altered neuropathology.
Methods: Male BALB/c mice were exposed to room air (sham) or CS (9 cigarettes/day, 5 days/week) for 24 weeks. After 23 weeks, mice underwent neurocognitive tests to assess working and spatial memory retention. At 24 weeks, mice were culled and lungs were collected and assessed for hallmark features of COPD. Serum was assessed for systemic inflammation and the hippocampus was collected for neuroinflammatory and structural analysis.
Results: Chronic CS exposure impaired lung function as well as driving pulmonary inflammation, emphysema, and systemic inflammation. CS exposure impaired working memory retention, which was associated with a suppression in hippocampal microglial number, however, these microglia displayed a more activated morphology. CS-exposed mice showed changes in astrocyte density as well as a reduction in synaptophysin and dendritic spines in the hippocampus.
Conclusion: We have developed an experimental model of COPD in mice that recapitulates the hallmark features of the human disease. The altered microglial/astrocytic profiles and alterations in the neuropathology within the hippocampus may explain the neurocognitive dysfunction observed during COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a significant health burden globally and it is currently the third leading cause of death worldwide (Vogelmeier et al., 2020). COPD is characterized by persistent respiratory symptoms and airflow limitations, decreasing the overall quality of life and increases the risk of mortality (Vogelmeier et al., 2020). COPD primarily arises due to the noxious particles and gases from chronic cigarette smoke (CS) which accounts for 95% of cases in industrialized countries (Caramori et al., 2016; Vogelmeier et al., 2020). Over time, CS propagates the oxidative stress burden and pro-inflammatory cell profile within the lungs leading to emphysema (Di Stefano et al., 1998; Hellermann et al., 2002; MacNee, 2005; Sharafkhaneh et al., 2008; Barnes, 2009; Brassington et al., 2021). It is hypothesized that inflammatory mediators and harmful oxidants “spill over” into the systemic circulation, resulting in other chronic comorbid conditions.
COPD-induced neurocognitive dysfunction is associated with reduced quality of life compared with the healthy age-equivalent population, with up to 61% of people experiencing these symptoms (Dodd et al., 2010; Dodd, 2015; Torres-Sánchez et al., 2015). It is reported that people with smoking history and COPD experience neurocognitive deficits, with impairments in working memory (Lv et al., 2020; Nadar et al., 2021), executive functioning (Dodd et al., 2013; Lv et al., 2020), attention (Paul et al., 2006), and delayed recall (Brunette et al., 2021). Moreover, Brunette et al. (2021) elegantly highlighted that people with COPD experiencing neurocognitive dysfunction have difficulties performing normal routine daily activities, further impairing their quality of life. Furthermore, people experiencing episodes of acute exacerbations of COPD have significantly worsened neurocognitive function when compared to people in a stable condition (Dodd et al., 2013; Crisan et al., 2014). Despite the known association between CS and impaired neurocognitive outcomes (Singh et al., 2014; Torres-Sánchez et al., 2015), the underlying mechanisms remain unknown. One possible mechanism could be microglia-dependent neuroinflammation. Under normal physiological conditions, microglia display a highly branched ramified morphology, surveying the microenvironment, modulating neurogenesis and synaptic plasticity, and subsequently influencing cognitive abilities (Nimmerjahn et al., 2005; De Luca et al., 2020a). Upon activation, microglial cells adopt an amoeboid morphology, releasing pro-inflammatory mediators and reactive oxygen species (ROS) (Lull and Block, 2010). However, persistent microglial activation can lead to neuronal and axonal loss (Lehnardt et al., 2003) orchestrating neurocognitive impairments.
We and others have previously shown that sub-chronic CS exposure in mice is sufficient to induce some of the key features of COPD including pulmonary inflammation and oxidative stress as well as comorbidities such as skeletal muscle wasting and vascular endothelial dysfunction (Botelho et al., 2010; Beckett et al., 2013; Chan et al., 2020; Brassington et al., 2021). However, longer durations of CS exposure (3–6 months) in mice are required to cause a decline in lung function, airspace enlargement (emphysema) and airway collagen deposition (Vlahos and Bozinovski, 2014). Our chronic CS exposure model in mice aims to recapitulate key pathological features seen in human disease such as impaired lung function, airway fibrosis, and emphysema. Moreover, there are limited studies investigating neurocognitive dysfunction in COPD and the underlying mechanisms responsible for this comorbidity. Thus, we aimed to investigate whether chronic CS exposure induces neurocognitive dysfunction like that seen in humans with COPD, and whether this is associated with a neuroinflammatory response within the hippocampus, a key region involved in memory formation.
Materials and Methods
Animals
All animal care and experimental procedures followed the ARRIVE Guidelines (Percie du Sert et al., 2020), Australian Code of Practice for the Care of Experimental Animals and RMIT University Animal Ethics Committee requirements (AEC #1521 and #1928). Seven-week-old male BALB/c mice (Animal Resource Centre Pty. Ltd., WA, Australia) were housed in micro-isolator cages at an ambient temperature of 21°C on a 12-h day/night cycle with ad libitum access to water and standard mouse diet.
Cigarette Smoke Exposure
Mice were acclimatized for 1 week prior to experimental commencement. Mice were placed in an 18 L Perspex chamber in a standard fume hood cabinet and exposed to normal fume hood air (sham group; n = 34) or Winfield Red Cigarettes (total particulate matter of ∼419 mg m–3, ∼16 mg of tar, ∼1.2 mg of nicotine, and ∼15 mg of CO; Philip Morris, Melbourne, VIC, Australia; n = 34). CS-exposed mice were exposed to 9 full cigarettes/day, 5 days/week for 24 weeks. Mice were exposed to CS or room air three times a day, with a 2-h break between smoke sessions as described previously (Brassington et al., 2021). We have shown that this CS exposure regime provides similar carboxyhemoglobin levels to human smokers (Vlahos et al., 2006). Following 23 weeks of CS exposure (1 week prior to cull), mice underwent neurocognitive testing followed by lung function and tissue collection.
Neurocognitive Assessment
Novel Object Recognition
To examine whether CS exposure impaired working memory we performed the novel object recognition (NOR) task on mice from cohort 1 (Figure 1). To habituate mice to the arena, mice were allowed to explore the arena (dimensions 60 cm × 60 cm × 60 cm), twice, for 8 min each. The following day mice underwent the testing protocol which involved an 8-min acquisition phase with two identical objects, followed by a 1-h inter-trial-interval, and a retention phase for 8 min with one of the identical objects replaced with a novel object. The arena was thoroughly cleaned with 70% ethanol. Every session was recorded on a video camera and a double-blinded investigator assessed the exploration time of the objects divided by the time spent interacting with the objects. Each interaction involved sniffing and body contact with the objects, however, scoring did not occur when the mice leaned against, stood on, sat on, or faced away from the object. Results from the acquisition trial are expressed as total exploration of both objects and retention trial results are expressed as a Preference Index, calculated as the time spent with the novel object divided by the overall exploration time of the two objects in seconds multiplied by 100 [timenovel / (timenovel + timefamiliar) × 100].
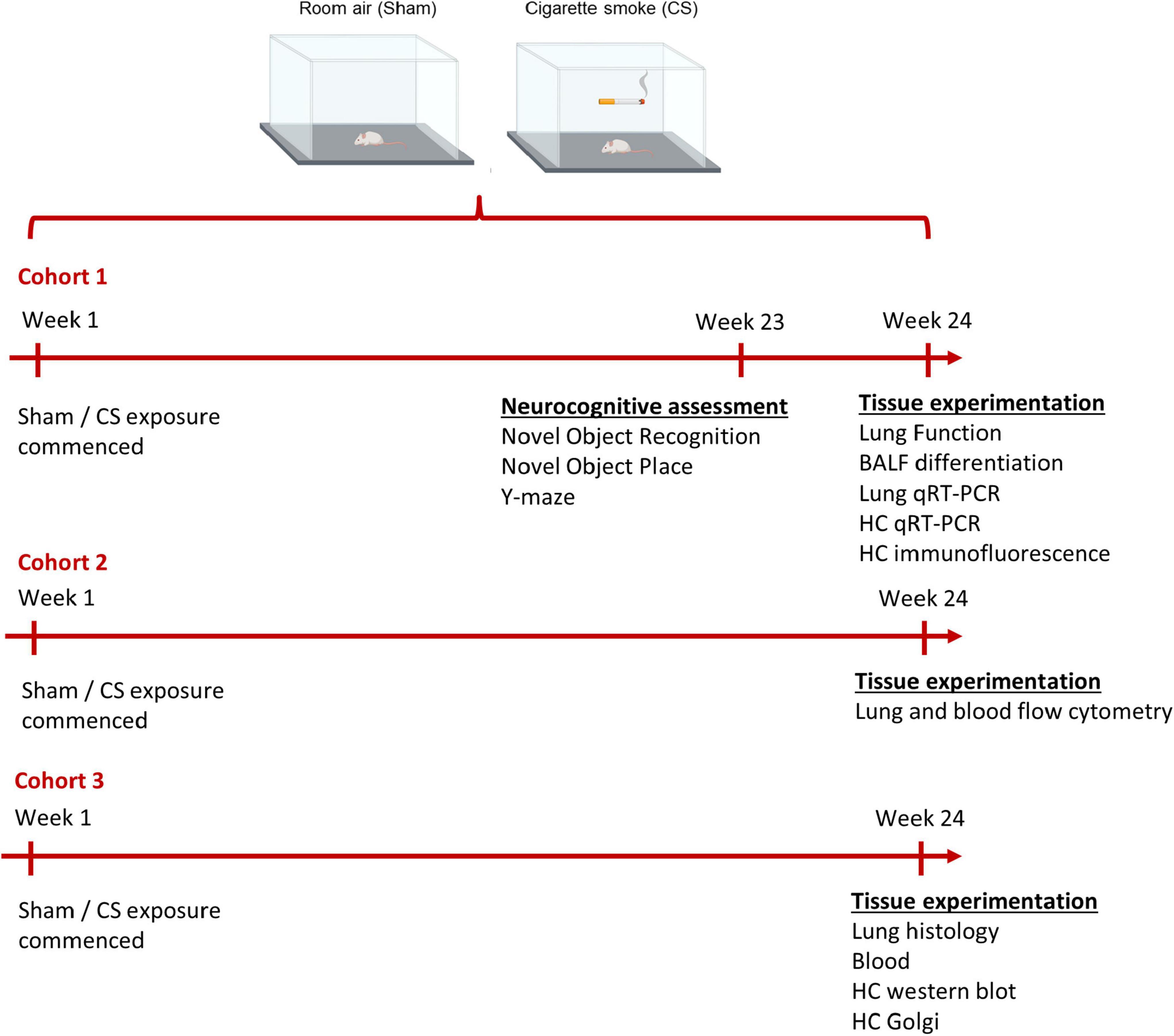
Figure 1. Timeline of experimental design. Mice were exposed to room air (sham) or cigarette smoke (CS; 9 cigarettes/day, 5 days/week) for 24 weeks. Cohort 1: neurocognitive assessments were performed on mice a week prior to protocol conclusion (week 23). Immediately prior to the cull, lung function assessment was performed (week 24). To examine the inflammatory profile within the lungs and hippocampus, we collected bronchoalveolar lavage (BAL) fluid for BAL differentiation and dissected the lungs and left hippocampi for qPCR. The right hippocampi was dissected for immunofluorescence. Cohort 2: lung and cardiac blood from a separate cohort of mice was collected for flow cytometric analysis. Cohort 3: a separate cohort was culled to examine lung pathology, blood brain barrier integrity (left hippocampi), and hippocampal dendritic spines (right hippocampi).
Novel Object Place
To assess spatial memory, we exposed the mice to the novel object placement (NOP) task (cohort 1; Figure 1). Similar to the NOR, the mice underwent a habituation and acquisition phase (as previously described). During the retention phase, one of the identical familiar objects was moved to novel location in the arena. Sessions were recorded on a video camera and assessed by a double-blinded investigator. The exploration time was scored by the amount of time spent interacting with the objects. Results are expressed as total exploration (acquisition phase) and Preference Index (retention phase; as previously described).
Spontaneous Alternation in the Y-Maze
To further assess spatial memory, we tested the mice in the spontaneous alternation in the Y-maze task (cohort 1; Figure 1). Mice were placed at the end of one arm of a symmetric Y maze (arm dimensions: 30 cm length × 11 cm width × 17 cm height) and were allowed to freely explore the arena for 5 min. All sessions were recorded with an overhead webcam and assessed by a double-blinded investigator. The series of arm entries were recorded, and an entry is defined as all four limbs passing the central area connecting the arms. An alternation was defined as the successive entry into three different arms during an overlapping triplet set (e.g., in the sequence 13231232, four alternations were recorded). Data are presented as the percentage of spontaneous alternation [number of alternations / (total number of arm entries − 2)]. After each session, the arena was cleaned with 70% ethanol. The total number of arm entries are presented as an index of ambulatory activity.
Lung Function Testing
To assess pulmonary function, we performed lung function testing using a small-animal ventilator (FlexiVent system, Montreal, QC, Canada) in sham and CS-exposed mice (cohort 1; Figure 1; Wang et al., 2021). Briefly, mice were anaesthetized [125 mg/kg ketamine and 25 mg/kg xylazine intraperitoneal (i.p.)] and a tracheostomy was performed using an 18G cannula, with a suture tightening the wall of the trachea around the cannula. The mouse was subsequently connected to the FlexiVent system to assess different respiratory parameters. The inspiratory capacity (IC) was measured using a deep inflation maneuver. The quasi-static compliance (Cst) was produced from the pressure-volume (PV) loop maneuver, where PV curves and PV loop areas were generated. The forced vital capacity (FVC) was measured via a negative pressure-driven forced expirations (NPFE) maneuver similar to current preclinical literature (Giordano et al., 2019; Starkey et al., 2019), whereas in humans, FVC is driven by voluntary exhalation and air trapping occurs (Devos et al., 2017).
Lung Collection and Histology
A separate cohort of mice were euthanized via i.p. injection by an overdose of sodium pentobarbital (Lethabarb; 240 mg/kg i.p.; Virbac, Sydney, NSW, Australia) to assess lung pathology (cohort 3; Figure 1). Lungs were fixed with neutral buffered formalin (NBF) solution via pressure-inflation techniques (Duan et al., 2012). Briefly, a gravitational pressure from 25 cm above bench level was used as NBF solution filled the lungs for 5 min. Following this period, lungs were excised and stored in NBF solution for 24 h, and then transferred to 70% ethanol. The left lobe of the lungs was sectioned into 4 μm sections and stained with hematoxylin and eosin (H&E) and Masson’s trichrome staining for collagen deposition (characterized by blue stains within the lung parenchyma) at the University of Melbourne Biomedical Sciences Histology Facility (Wang et al., 2021). Sections were imaged on an Olympus slide scanner VS120-SS (Olympus, Japan) at 20× magnification and 5 randomly selected fields within the distal region of the lung were analyzed. The mean linear intercept (Lm) was quantified using a square grid (100 nm × 100 nm) which was created for analysis (VS120-SS). Using ImageJ, the alveolar surface intersecting with each of the horizontal grid lines were marked and distances between these alveolar surfaces was calculated whilst ensuring minimal amounts of blood vessels and airways were present. The Lm was calculated by adding the total distance and the amount of alveolar structures. Collagen deposits were measured within a drawn region of interest (ROI) where positively blue stained areas were present. The amount of collagen present (depicted as % area fraction) was assessed using cellSens Dimension™ software (Olympus).
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) was performed via a surgical tracheotomy and lavaged in situ with 0.4 mL of chilled PBS, followed by further 0.3 mL aliquots of PBS until 1 mL of BAL fluid (BALF) was collected (cohort 1; Figure 1). Cytocentrifuge spots were created by centrifuging the BALF at 400 × g for 10 min. The cytospots were stained with Shandon Kwik-Diff Kit® (ThermoFisher Scientific, Waltham, MA, United States) and Merck’s Hemacolor (eosin and thiazine solutions; Merck, Kenilworth, NJ, United States) as previously described (Brassington et al., 2021). Differential counts were performed, counting a total of 500 cells identifying macrophages, neutrophils, and lymphocytes using standard morphological criteria.
Flow-Cytometry Analysis
A separate cohort of mice (cohort 2; Figure 1) were euthanized by an overdose of sodium pentobarbital (Lethabarb; 240 mg/kg i.p.; Virbac, Sydney, NSW, Australia). The left lobe of the lung was dissected and minced by using scissors and blood was collected via a cardiac puncture. Lungs were digested in an enzymatic digestion buffer (composition Liberase and Hanks balanced salt solution, Sigma-Aldrich, St-Louis, MO, United States) for 45 min with intermittent shakes to make a single-cell suspension. Single cell suspensions were filtered through a 40 μm strainer, and the reaction stopped by adding fluorescence-activated cell-sorting (FACS) buffer. Lung and blood suspensions were centrifuged at 400 × g, and red blood cells were lysed with ACK lysis buffer. Cell viability was determined by incubating with LIVE/DEAD Fixable Violet Dead Cell Stain Kit (ThermoFisher Scientific) for 15 min at 4°C. Cells were then washed with FACS buffer and centrifuged at 400 × g and incubated with cluster differentiation (CD)16/32 (2.4G2) to block Fc-mediated adherence of the antibodies. The white blood cells were stained with respective fluorescent labeled antibodies for flow cytometric analysis for 30 min at 4°C and washed twice with FACS buffer. The antibody panels used for staining, and in their different multicolor combinations were as follows: 1:500 Alexa Flour 700 anti-CD45 (30-F11); 1:500 APC anti-CD3 (145-2C11); 1:200 APC anti-CD4 (GK1.5); 1:1,000 PE-Cy7 anti-CD8 (53-6.7); 1:500 FITC anti-Ly6C (HK1.4); 1:500 APC-Cy7 anti-Ly6G (1A8); 1:500 BV421 anti-CD11b (M1/70); 1:200 FITC anti-NK1.1 (PK136); 1:200 PE-Cy7 anti-CD19 (B4); or live/dead Aqua (L34965). Cells were resuspended in FACS buffer and analyzed on the FACSAria (BD Biosciences, Franklin Lakes, NJ, United States) with FACSDiva software (BD Biosciences). Data analysis was performed using FlowJo software (Tree Star, Inc.). The cells are expressed as percentage of the CD45+ cells (live cells).
Blood Collection and Counts
To identify possible systemic inflammatory responses as a result of chronic CS exposure, blood was collected via the inferior vena cava (cohort 3; Figure 1) and analyzed using the Cell Dyn Emerald Hematology Analyzer (Abbott Core Laboratory, Abbott Park, IL, United States). The blood was warmed to room temperature (RT) and mixed before 9.8 μL of blood was analyzed. Granulocytes, lymphocytes, and total leukocytes were counted via the Emerald Analyzer.
Quantitative Real-Time PCR
Following BALF collection, mice were perfused with PBS and whole lungs and the left hippocampi were dissected and stored at −80°C until required (cohort 1; Figure 1). Lung tissue was crushed and approximately 10 mg was homogenized. Hippocampal samples were thawed and homogenized in 1 mL of TRIzol solution (ThermoFisher Scientific) using the TissueLyser LT® (Qiagen, Valencia, CA, United States). Total RNA was extracted using the RNeasy® Mini Kit (Qiagen). Isolated mRNA was then reverse transcribed to cDNA using a High-Capacity RNA-to-cDNA kit (ThermoFisher Scientific). Real-time PCR reactions were performed using Life Technologies pre-developed TaqMan assay reagents (Table 1) in triplicate using either the endogenous control Rps18 (lung) or Pgk1 (hippocampus) as the internal housekeeping control. We analyzed mRNA expression using the equation 2–ΔΔC(t), where threshold cycle (Ct) fluorescence is first detected significantly above background (Chan et al., 2020). Data are presented as fold expression relative to sham mice.
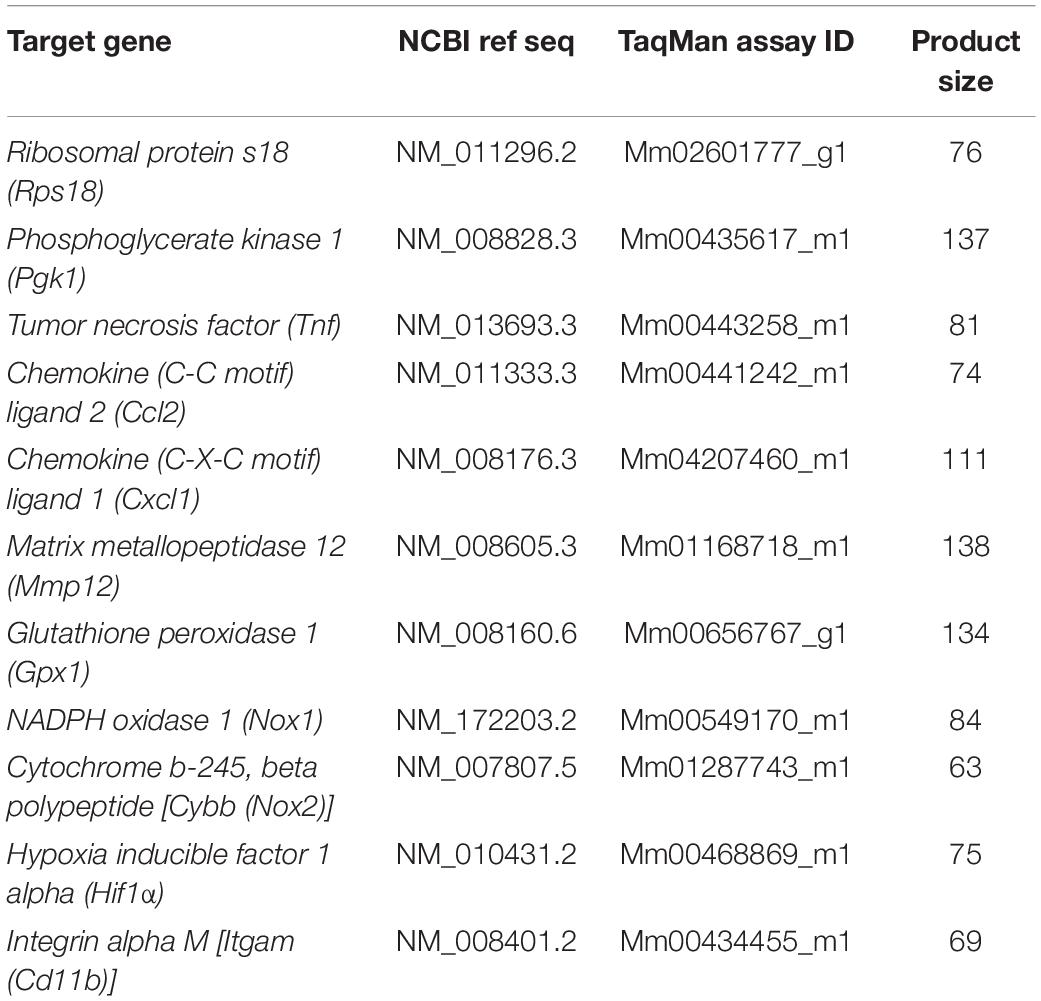
Table 1. TaqMan probes used for qPCR.
Blood–Brain Barrier Integrity
The right hippocampi from cohort 3 (Figure 1) was dissected and homogenized in RIPA lysis buffer. To determine the total amount of protein, a bicinchoninic acid (BCA) assay was performed. Samples were diluted in DEPC water to produce solutions containing 20 μg of protein, mixed with Laemmli sample buffer and heated to 95°C for 10 min. Proteins were separated by their molecular weights using gel electrophoresis (SDS-PAGE; 8–14% bis:acrylamide gel) and the membranes were transferred to methanol-activated polyvinylidene fluoride transfer membranes (Bio-Rad Laboratories Inc., Hercules, CA, United States) via the blotting sandwich technique. Membranes were dried and washed in blocking buffer (5% skim-milk based blocking buffer diluted in TBS-T), followed by incubation with primary antibodies overnight at 4°C [Albumin: 1:1,000, anti-chicken, Abcam (#ab106582). ZO-1: 1:1,000, anti-mouse, ThermoFisher Scientific (#33-9100). Occludin: 1:1,000, anti-rabbit, Cell Signaling Technologies (CST), Danvers, MA, United States (#91131s), β-actin: 1:1,000, anti-rabbit, CST, (#8457)]. Following the overnight incubation, the membranes were washed in TBS-T and incubated in HRP-linked secondary antibodies [Albumin: 1:3,000, anti-chicken IgG HRP-linked, Merck (#A9046). ZO-1: 1:3,000, anti-mouse IgG HRP-linked, CST (#7076s). Occludin and β-actin: 1:3,000, anti-rabbit IgG HRP-linked, CST (#7074s)] at RT for 1 h and prepared for chemiluminescence detection using the chemiluminescence Western Lighting Ultra Solution reagents (Perkin Elmer, Waltham, MA, United States). Detection of IgG did not utilize any primary antibody, however, followed the same procedure when blocking and administering the secondary antibodies. Band intensities were analyzed using the ImageLab software (Bio-Rad Laboratories Inc.) and normalized to β-actin housekeeping antibody.
Immunohistochemistry
The right hemisphere of the brain was immersion-fixed in 4% paraformaldehyde solution for 24 h before cryoprotecting in 20% sucrose solution until further required (cohort 1; Figure 1). Brains were cut into 30 μm coronal sections using a cryostat (Leica Biosystems, Mt Waverly, VIC, Australia) and processed for immunofluorescent staining. Sections were stained for ionized calcium-binding adapter molecule-1 (Iba-1), glial fibrillary acidic protein (GFAP), neuronal nuclei (NeuN), doublecortin (DCX), and synaptophysin. Free-floating sections were washed with PBS-T and blocked with 3% BSA and 0.3% Triton X-100 (Iba-1 and GFAP) or 4% NHS, 3% BSA, and 0.3% Triton X-100 (NeuN, DCX and synaptophysin) for 2 h before submerging the sections in the primary antibody overnight at 4°C [Iba-1; 1:1,000, anti-rabbit, Wako Chemicals, Osaka, Japan (#1022-5). GFAP; 1:500, anti-rabbit, Dako, Bath, United Kingdom (#Z0334). NeuN; 1:1,000, anti-rabbit, Abcam, Cambridge, United Kingdom (#ab104225). DCX; 1:1,000, anti-guinea pig, Abcam (#ab2253). Synaptophysin; 1:2,000, anti-mouse, Sigma-Aldrich (#S5768)]. Following the primary incubation, the sections were transferred into the secondary antibody [Iba-1 and GFAP: 2 h, 1:500, Alexa Fluor 488 goat anti-rabbit, Life Technologies (#A-11008). NeuN: 2 h, 1:400, Alexa Fluor 594 goat anti-rabbit, Life Technologies (#A-11012). DCX: 2 h, 1:400, Alexa Fluor 488 goat anti-guinea pig Life Technologies (#A-11073). Synaptophysin: 2 h, 1:400, Alexa Fluor 488 rabbit anti-mouse, Life Technologies (#A-11001)]. Sections were counterstained with Fluoromount-G™, with DAPI (ThermoFisher Scientific) and imaged at 20× magnification on the Nikon Eclipse E600 microscope (Nikon, Minato City, Tokyo, Japan) and synaptophysin was imaged on the C1 confocal microscope (Nikon).
Microglia (Iba-1), astrocytes (GFAP) and mature neurons (NeuN) were quantified in five regions of the hippocampus including the CA1, CA3, and the hilus, subgranular (SG) and molecular regions of the dentate gyrus (DG) within a ROI, whilst immature neurons (DCX) and synaptophysin were quantified in the subgranular and hilus regions of the DG, respectively. These regions were identified through the Franklin and Paxinos Mouse Brain Atlas and were analyzed by an average of 3–4 sections per brain (between 1.22 and 2.70 mm caudal to the bregma). Microglial numbers were counted on ImageJ software [National Institute of Health (NIH), Bethesda, MD, United States] and the morphology of these cells were analyzed on Imaris software via sholl analysis (Bitplane, Oxford Instruments, Abingdon, United Kingdom). Microglial morphology was assessed in three cells per section and calculated using Imaris software as described previously (Spencer et al., 2019). Astrocyte density, NeuN and synaptophysin were analyzed using imaging analysis software cellSens Dimension™ (Olympus, Tokyo, Japan) by thresholding the positive stains against the background in the thalamus region. Cells stained with DCX were manually counted in the SG region of the DG at 40× magnification.
Golgi Staining and Neuronal Analysis
Dendritic spines on pyramidal neurons were differentiated using the FD Rapid GolgiStain™ Kit (FD Neurotechnologies, Columbia, MD, United States) according to manufacturer’s instructions (cohort 3; Figure 1). We dissected 10 mm-thick brain slices that included the left hippocampi and the brain tissue was rapidly frozen in double distilled milliQ H2O and sectioned onto gelatin-coated slides at 100 μm using a cryostat. Sections were allowed to completely dry at RT in the dark and stained with FD solution and coverslipped with entelen mounting medium.
To analyze apical dendritic spines, images of the pyramidal neurons within the hippocampal CA1 were taken on an upright Olympus BX61 microscope (Olympus) at 100× magnification with a step-width of 0.15 μm using the z-stack option on the imaging software (cellSens Dimension™ software, Olympus). For each mouse, three neurons were assessed across three different sections per animal, up to a total of five animals per group and images were analyzed on the Imaris software (Bitplane). To minimize variability in the analysis of neurons, we only include segments with unbroken secondary apical dendritic branches, the length of the branch was within 20–30 μm, and that the dendrite was not obstructed by other dendrites or background staining. Dendritic spines were assigned to six categories including Filopodia (length >2 μm), long thin (length <2 μm), thin (length <1 μm), stubby (length:width ratio <1), mushroom (length >0.6 μm; width >0.6 μm) and branched (consisting of 2 or more heads, regardless of length and width) (Risher et al., 2014; De Luca et al., 2020b). Data are expressed as the mean number of spines per 10 μm for each mouse.
Data Analysis
All data are presented as the mean + standard error of the mean (SEM) unless otherwise stated; n represents the number of mice per treatment group. Statistical analyses performed between the sham and CS-exposed groups was via Student’s unpaired t-tests. For PV loop assessment, the data was analyzed with a repeated measures analysis of variance (ANOVA). The sholl analysis for microglial morphological assessment was grouped in bins of 5 μm and analyzed with a repeated measures ANOVA. All statistical analyses were performed using GraphPad Prism™ software (Version 9). Statistical significance was assumed when p < 0.05.
Results
Chronic Cigarette Smoke Exposure Induces a Chronic Obstructive Pulmonary Disease Phenotype in Mice
To examine whether chronic CS inhalation in mice recapitulates the human COPD phenotype, we assessed both lung function and lung pathology. CS-exposed mice displayed a significant increase in Cst compared to sham mice, demonstrating reduced elastic recoil due to the destruction of extracellular matrix and alveolar walls (t(14) = 4.937, p = 0.0002; Figure 2A). The lungs of these mice were hyperinflated with increased IC (t(14) = 4.678, p = 0.0004; Figure 2B). In NPFE maneuver, CS-exposed mice presented with a clear difference of 0.27 mL in FVC compared to shams (t(14) = 8.183, p < 0.0001; Figure 2C). Weakened elastic properties and alveolar collapse in CS exposed mice were also supported by the upward shift in the PV loop curve (interaction of exposure by pressure: F(6,84) = 64.50, p < 0.0001; Figure 2D) and the increased PV loop area (t(14) = 7.188, p < 0.0001; Figure 2E). Airway fibrosis was also observed following CS exposure, with elevated levels of collagen deposited around the airways (t(16) = 7.118, p < 0.0001; Figures 2F,G). CS-exposed mice displayed larger alveolar spacing within the lung parenchyma compared to shams (t(16) = 4.574, p = 0.0003; Figures 2H,I).
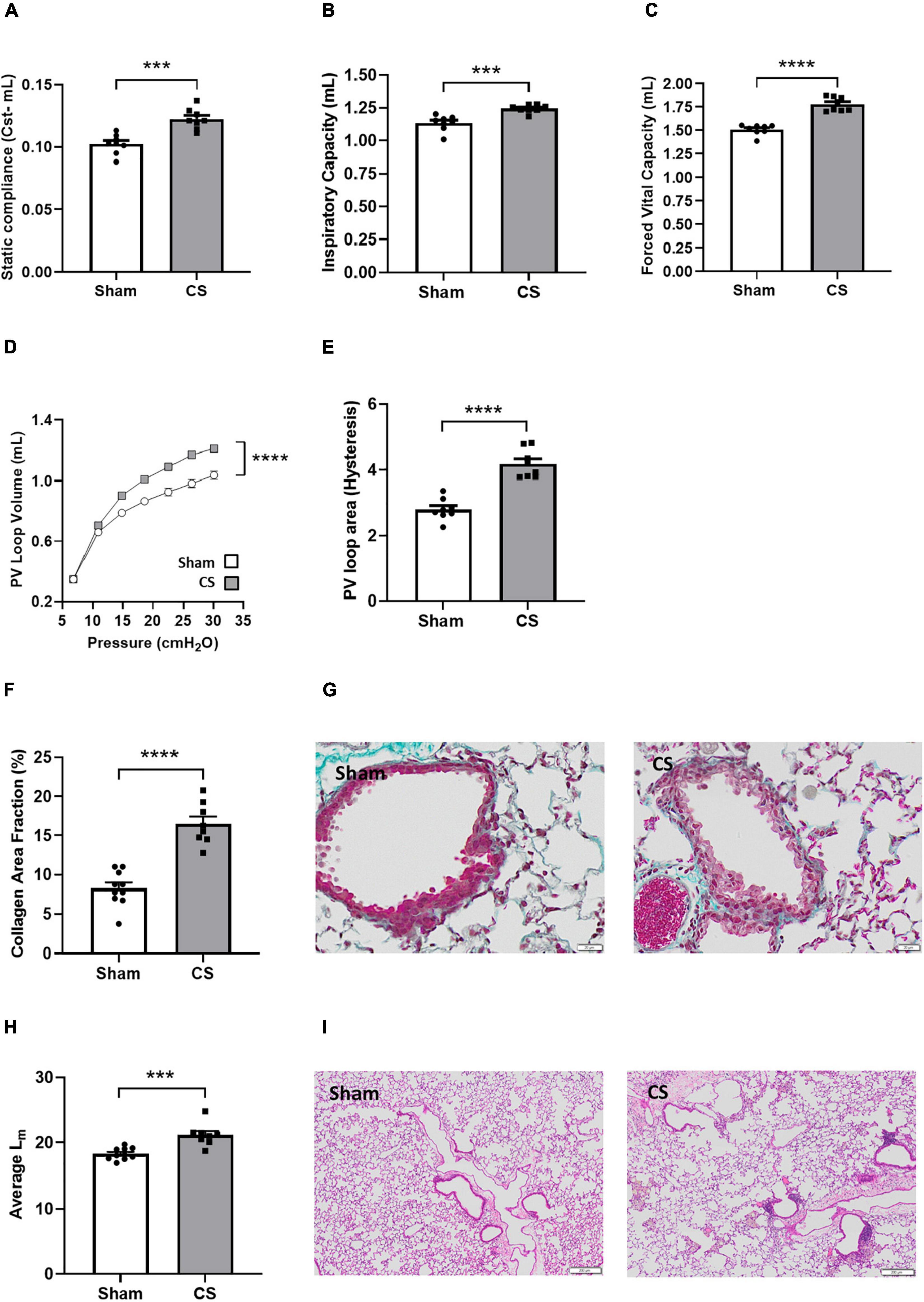
Figure 2. Chronic CS exposure worsened lung function and pathology. Mice were exposed to sham or CS (9 cigarettes/day, 5 days/week for 24 weeks). Lung function parameters assessed included (A) static compliance (Cst) (n = 8), (B) inspiratory capacity, (C) forced vital capacity, (D) pressure-volume (PV) loop, and (E) PV loop area. (F) Percentage area fraction of collagen (n = 8–10) and (G) representative images of collagen-stained (blue) sections. (H) Histological analysis of the mean linear intercept (Lm) (n = 8–10) and (I) representative images of pressure-inflated lung sections. Data was analyzed by Student’s t-test and expressed as mean + SEM with significance being represented as; ***p < 0.001, ****p < 0.0001. PV loop data was analyzed via repeated measures and expressed as mean ± SEM with significance being represented as; ****p < 0.0001.
Cigarette smoke exposure caused an increase in BALF total cells, which was attributed to an increase in macrophages, neutrophils and lymphocytes compared to sham mice (Total number of cells: t(21) = 7.700, p < 0.0001, macrophages: t(21) = 6.044, p < 0.0001, neutrophils: t(20) = 7.726, p < 0.0001, and lymphocytes: t(21) = 3.195, p = 0.0044; Figure 3A). We also found a significant increase in cytokines, proteases and chemokines [tumor necrosis factor (Tnf): t(14) = 7.531, p < 0.0001; chemokine (C-C motif) ligand 2 (Ccl2): t(13) = 5.778, p < 0.0001; chemokine (C-X-C motif) ligand 1 (Cxcl1): t(14) = 11.67, p < 0.0001; matrix metallopeptidase 12 (Mmp12): t(13) = 12.32, p < 0.0001; Figure 3B] and oxidative stress markers in the lungs [NADPH oxidase 1 (Nox1): t(14) = 6.044, p < 0.0001. Cytochrome b-245 beta (Cybb): t(13) = 4.122, p = 0.0012; Figure 3C]. There was a significant increase in the percentage of neutrophils [(CD11b+ Ly6C+) t(14) = 9.308, p < 0.0001; Figures 3D,I] and a decrease in the percentage of patrolling monocytes [(CD11b+ Ly6Clow) t(14) = 4.391, p = 0.0006; Figures 3D,I] and cytotoxic T cells when compared to sham mice (CD3+ CD8+: t(14) = 5.778, p < 0.0001; Figure 3E). CS exposure caused increased leukocyte levels within the blood, particular increases in granulocyte and lymphocyte numbers (leukocytes: t(13) = 2.858; p = 0.135; granulocytes: t(13) = 2.616; p = 0.0213; lymphocytes: t(13) = 3.050; p = 0.0093; Figure 3F). Moreover, we saw a significant increase in patrolling monocytes, and neutrophils in the blood compared to sham mice via flow cytometric analysis [patrolling monocytes (CD11b+ Ly6Clow): t(14) = 3.703, p = 0.0024. neutrophils (CD11b+ Ly6C+): t(14) = 3.583, p = 0.0030; Figures 3G,H].
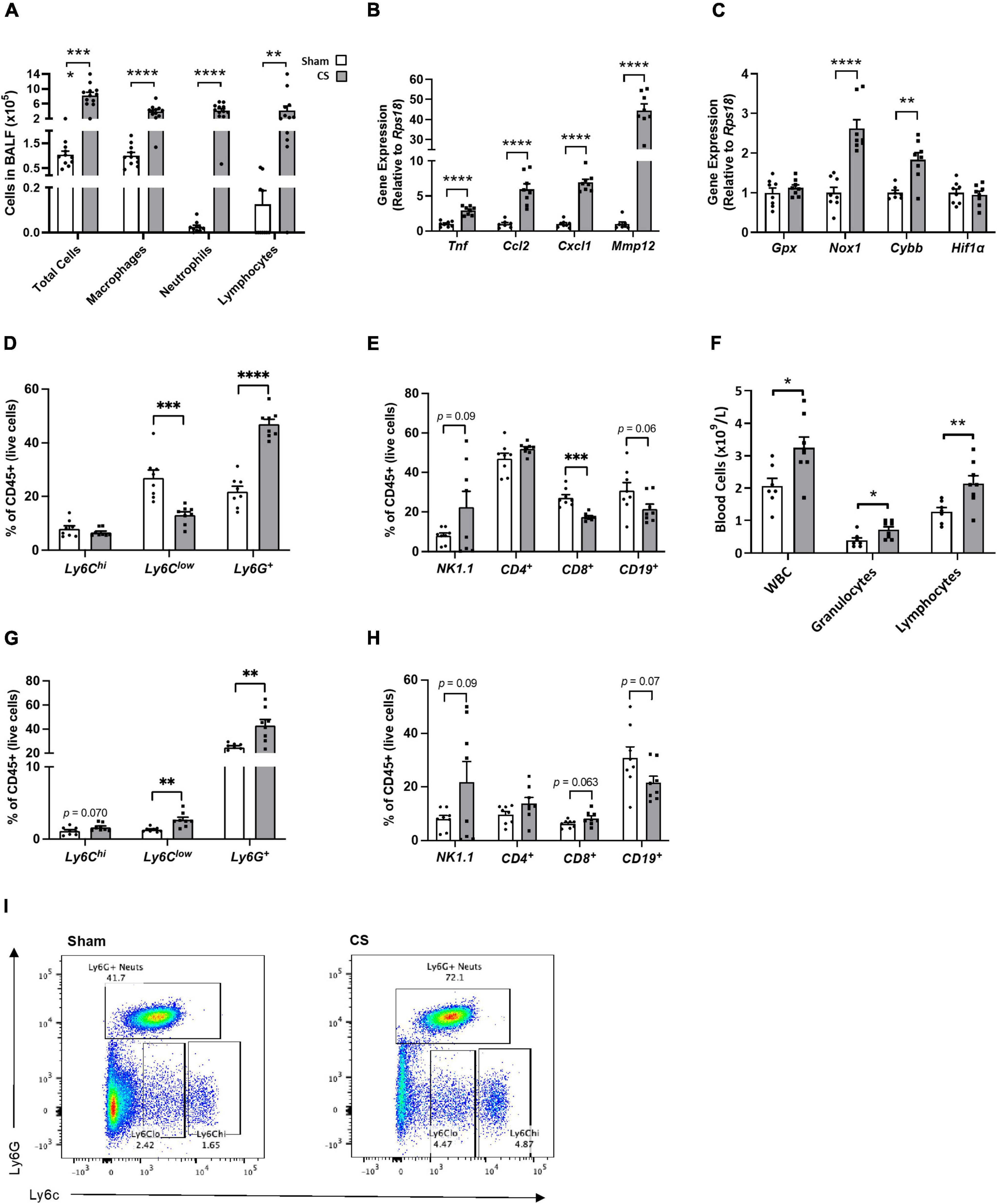
Figure 3. Chronic CS exposure causes lung and systemic inflammation. Mice were exposed to sham or CS (9 cigarettes/day, 5 days/week for 24 weeks). (A) Differential counts in the bronchoalveolar lavage fluid (BALF). (B) Inflammatory and protease mRNA expression in the lung. (C) Oxidative stress mRNA expression in the lung. (D) Percentage of innate immune cells in the lungs. (E) Percentage of adaptive immune cells in lungs. (F) Differential blood cell counts to assess leukocyte, granulocyte, and lymphocyte levels. (G) Percentage of innate immune cells in the blood. (H) Percentage of adaptive immune cells in blood. (I) Flow cytometric lung representative images. Data were analyzed by Student’s t-test and expressed as mean + SEM with significance being represented as; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. n = 7–8.
Cigarette Smoke Exposure Leads to Working Memory Impairment
To understand the influence of CS exposure on neurocognitive function, mice underwent working (NOR) and spatial (NOP/spontaneous alternation Y maze test) memory testing. We observed no differences in object exploration in the NOR trial phase (t(18) = 2.307, p = 0.0332; Figure 4A), however, CS-exposed mice were unable to differentiate between the novel and familiar object during the retention phase (Figures 4B,C), indicative of working memory impairments. However, spatial memory in the NOP remained intact following chronic CS exposure (Figures 4D,E) and we saw a downwards trend in the percentage of spontaneous alternation in the Y maze task (p = 0.07; Figures 4F,G) compared to sham mice.
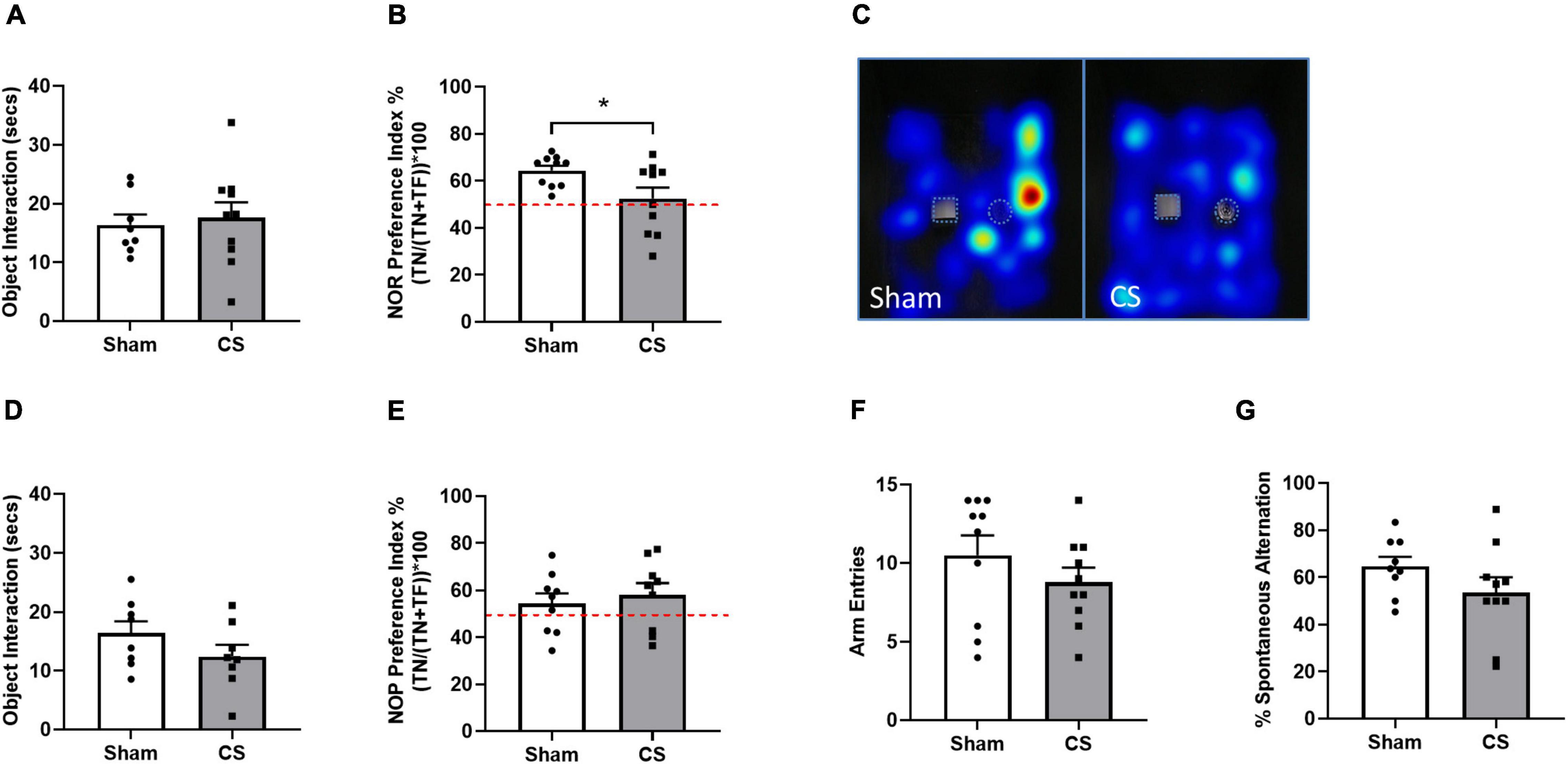
Figure 4. Chronic CS exposure decreased working memory in mice. Behavioral testing was performed on mice 1 week prior to protocol conclusion. (A) Total exploration time (seconds) in novel object recognition (NOR) acquisition phase. (B) NOR Preference Index. (C) Representative heatmaps illustrating familiar object (square) and novel object (circle) exploration in the NOR. (D) Total exploration time (seconds) in novel object placement (NOP) acquisition phase. (E) NOP Preference Index. (F) Arm entries in the Y Maze. (G) Percentage spontaneous alternation in Y Maze. Red dotted line in (B,E) represents a Preference Index of 50%. Data was analyzed by Student’s t-test and expressed as mean + SEM with significance being represented as *p < 0.05. n = 10 for the NOR and NOP and n = 14 for Y maze.
Cigarette Smoke Exposure Disrupts the Integrity of the Blood–Brain Barrier
We performed immunoblotting to determine whether CS exposure disrupted key blood–brain barrier (BBB) proteins. The tight junctional protein ZO-1 was significantly decreased in CS-exposed mice compared to shams (ZO-1: t(11) = 3.2, p = 0.0085; Figures 5A,B). However, occludin, albumin and IgG expression were unaltered in the hippocampus following CS exposure.
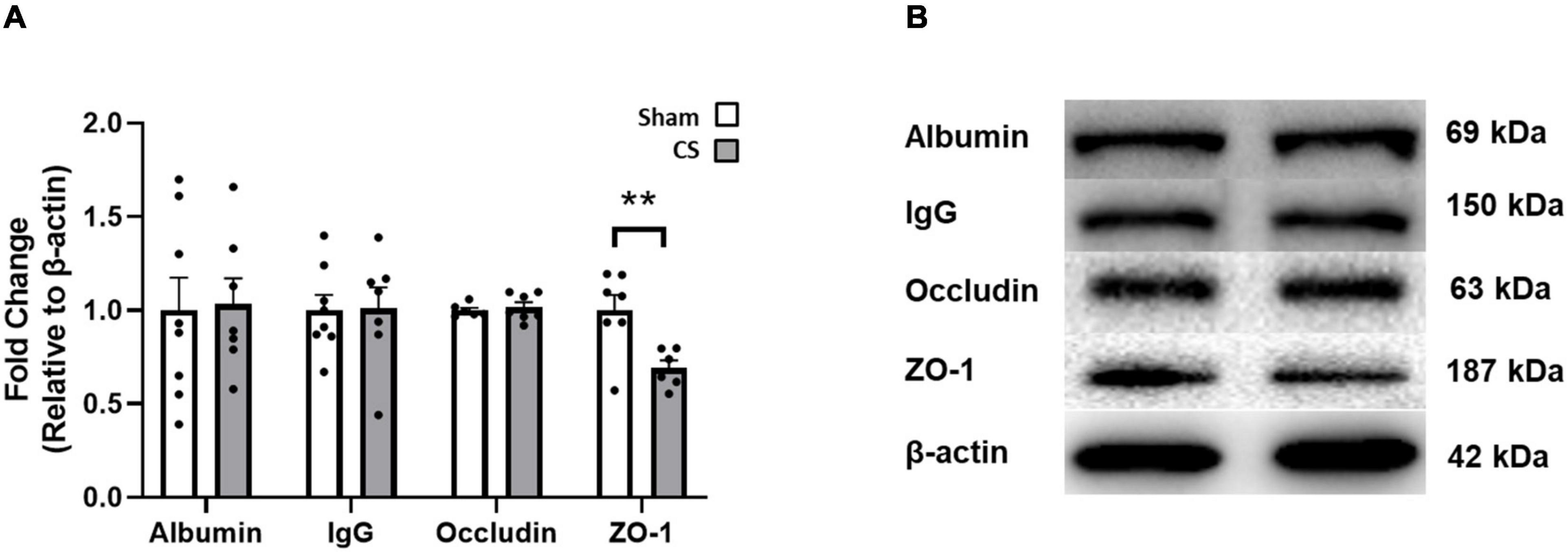
Figure 5. Chronic CS exposure decreased ZO-1 protein in the hippocampus. (A) Blood–brain barrier (BBB) protein levels in the hippocampus. (B) Representative Western blot images of BBB proteins. Data was analyzed by a Student’s t-test and expressed as mean + SEM with significance being represented as **p < 0.01. n = 7–8.
Cigarette Smoke Exposure Induces Hippocampal Microglial Activation
To elucidate whether CS-induced working memory impairments was associated with neuroinflammation, we assessed inflammatory profiles within the hippocampus (Figure 6A). Itgam gene expression was decreased within the hippocampus in CS-exposed mice compared to shams, suggesting a reduction in CD11b-positive myeloid cells in this region (t(14) = 3.698, p = 0.0024; Figure 6B). CS-exposed mice had fewer microglial numbers within the CA3 and the hilus (CA3: t(14) = 2.193, p = 0.0457, hilus: t(14) = 2.672, p = 0.0182; Figure 6C), with no observed differences in the CA1, SG and molecular regions. Chronic CS exposure significantly reduced the overall length of microglial processes in the molecular region of the hippocampus (t(13) = 2.285, p = 0.0397; Figure 6D). Sholl analysis revealed a significant decrease in the average number of branching in the CA3 (F(3,33) = 3.081, p = 0.045; Figures 6F,J) and molecular (F(6,57) = 3.255, p = 0.009; Figures 6I,J) regions but no differences in the CA1 (Figures 6E,J) and SG (Figures 6H,J) regions compared to sham mice. In contrast, microglia in the hilus region were more branched compared to sham mice (F(3,31) = 3.433, p = 0.028; Figures 6G,J).
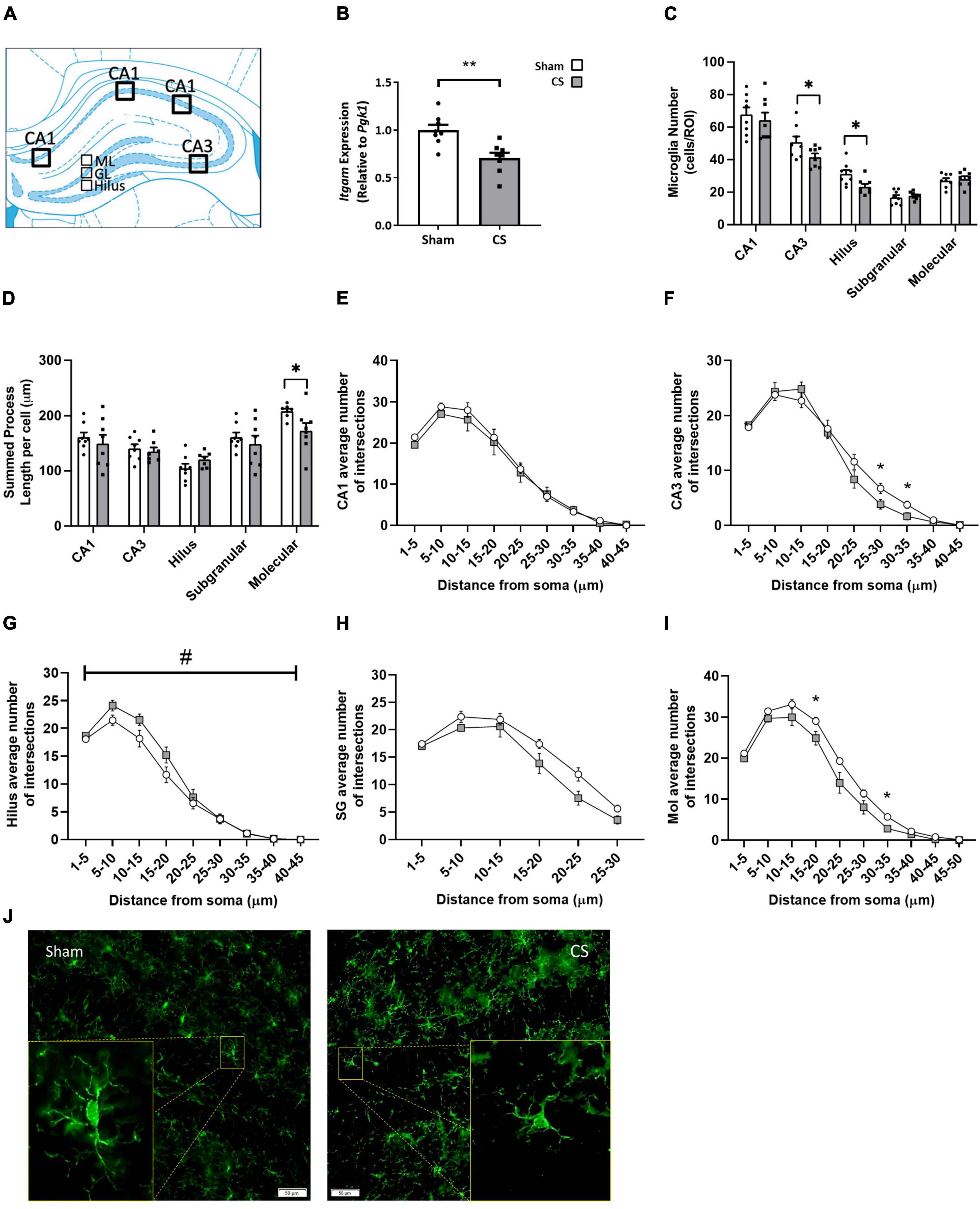
Figure 6. Chronic CS exposure decreased microglial numbers and increased microglial activation in the hippocampus. (A) Schematic diagram of the mouse hippocampus where regions of interest were analyzed. (B) Inflammation in the mouse hippocampi was assessed by qPCR. (C) Microglial (ionized calcium-binding adapter molecule 1 [Iba-1]-positive) numbers in the CA1, CA3, and the dentate gyrus [hilus, subgranular (SG), and molecular regions]. (D) Overall length of microglial processes in all regions of the hippocampus. (E–I) Sholl analysis of microglia in all regions of the hippocampus. (J) Representative images of Iba-1 analysis in the mouse hippocampus. qPCR and microglial number data were analyzed by Student’s t-test and expressed as mean + SEM with significance being represented as; *p < 0.05, **p < 0.01. Sholl analysis data was analyzed via repeated measures and expressed as mean ± SEM with significance being represented as *p < 0.05. The symbol “#” represents interaction between both independent variables when p < 0.05. n = 7–8.
Cigarette Smoke Exposure Altered Hippocampal Astrocyte Profiles and Reduced the Number of Dendritic Spines
To elucidate whether CS-induced working memory impairments are associated with alterations in the neuronal profile, we assessed neurogenesis and synaptogenesis. CS exposure reduced astrocyte density in the CA1, hilus, and SG regions of the hippocampus (CA1: t(13) = 3.349, p = 0.0052; hilus t(13) = 3.928, p = 0.0017; SG: t(14) = 2.203, p = 0.0448; Figures 7A,B) but an increase in density was observed in the molecular region (molecular: t(13) = 6.974; p < 0.0001; Figure 7A) compared to sham mice. We identified no differences in NeuN, a mature neuronal marker (Figure 7C), or the immature neuronal marker, DCX (Figure 7D). Synaptophysin density was decreased in the hilus region following CS exposure (t(14) = 2.911, p = 0.0114; Figures 7E,F). There was a significant reduction in the number of mature stubby spines in the secondary dendrites of CA1 pyramidal neurons (stubby: t(7) = 2.969, p = 0.0208; Figures 7G,H) but no differences were observed in other spine types.
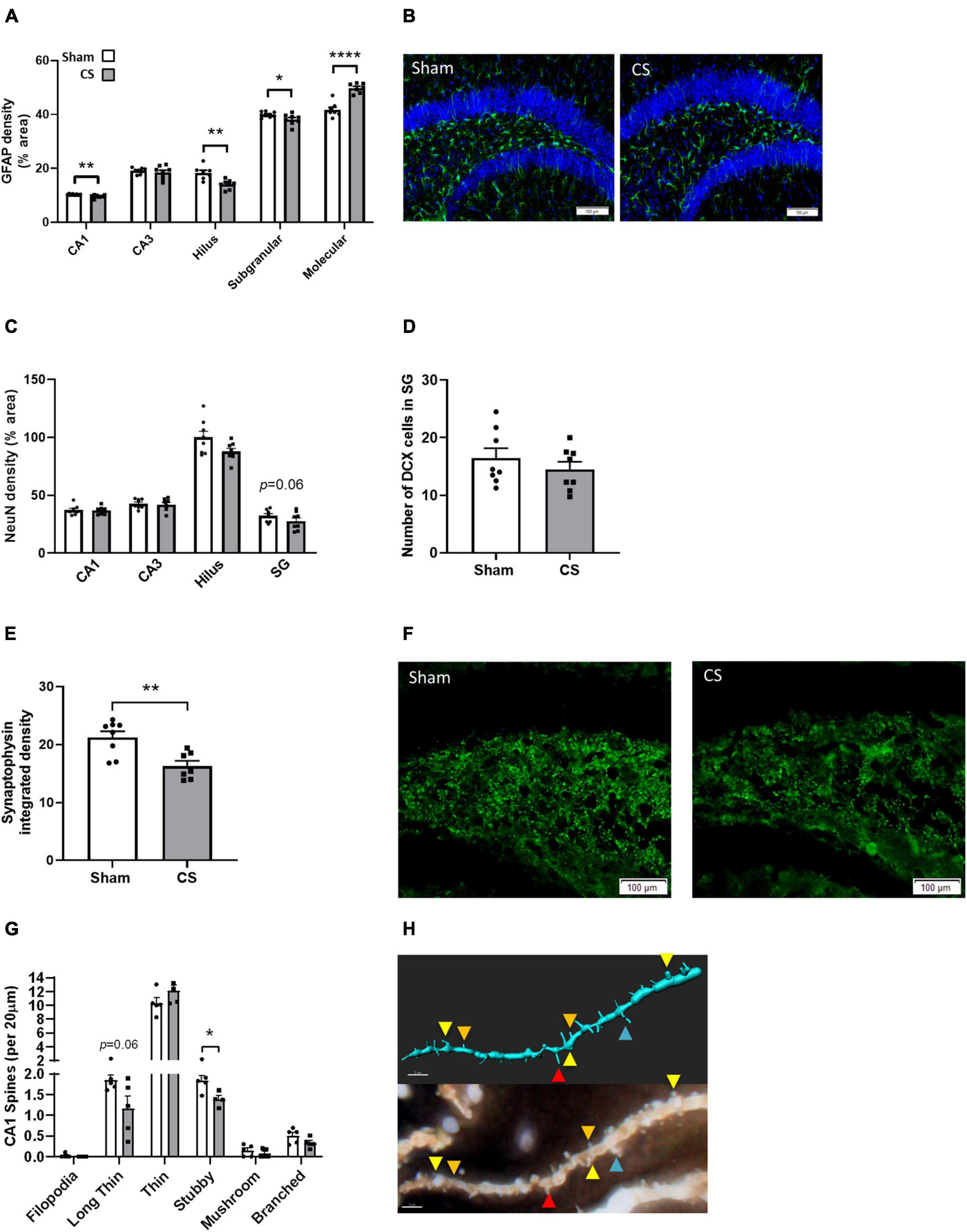
Figure 7. Chronic CS exposure decreased astrocyte numbers and altered synaptogenesis in the hippocampus. (A) Mouse hippocampi were stained with glial fibrillary acidic protein (GFAP) for the assessment of astrocytes in all hippocampal regions. (C) Mature neurons (NeuN-positive cells). (D) Immature neurons DCX-positive cells. (E) Synaptophysin staining in the hilus region of the dentate gyrus. (G) Golgi staining of secondary pyramidal neurons spines in the CA1 (n = 4–5). Representative images of (B) GFAP, (F) synaptophysin, and (H) golgi staining (red, long thin spine; orange, thin spine; yellow, stubby spine; blue, branched spine). Data was analyzed by Student’s t-test and expressed as mean + SEM with significance being represented as; *p < 0.05, **p < 0.01, ****p < 0.001. n = 7–8.
Discussion
Our chronic CS exposure mouse model recapitulated both pulmonary and systemic features of human COPD. This was accompanied by neurocognitive impairments marked by alterations in working memory. Neurocognitive dysfunction was associated with the reduction of BBB tight junctional protein ZO-1, hippocampal neuroinflammation with microglial morphology indicative of a more-activated profile, despite there being fewer microglia in the region. Moreover, CS exposure suppressed hippocampal astrocyte density, reduced synaptophysin and stubby dendritic spine expression.
In conjunction with previously demonstrated lung inflammation, the current study demonstrated an emphysema phenotype alongside impairments in lung function. Altered lung compliance as seen in our CS-exposed mice was associated with more prominent airway spacing between alveolar structures due to alveolar destruction within the lung parenchyma, as well as increased expression of Tnfα and Mmp12 (Chan et al., 2020). This replicates previous literature, where whole-body and nose-only CS exposure present with increase alveolar spacing in the lung tissue (Rinaldi et al., 2012; Shu et al., 2017). Additionally, we established a marked increase in collagen deposition within the lung parenchyma in CS-exposed mice like previous studies (Triantaphyllopoulos et al., 2011), which may inhibit proper repair of lung tissue, thus inducing irreversible lung damage, similar to that seen in people with COPD.
Recent literature has shown that people with COPD are more likely to develop neurocognitive impairments, either globally or in a single domain, with impairments in memory retention, information processing and executive function, than non-COPD smokers and healthy individuals (Dodd, 2015; Morris et al., 2019; Brunette et al., 2021). We have shown that 6 months of CS exposure in BALB/c mice is sufficient to recapitulate the lung profile of human COPD along with working, but not spatial, memory deficits. Similarly, Yang et al. (2019) showed that CS exposure for as little as 10 weeks is sufficient to induce spatial learning and memory impairments. We and others (Yang et al., 2019; Prasedya et al., 2020) have demonstrated that CS exposure induces working memory impairments, however, the mechanisms underlying neurocognitive comorbidities are still largely unknown.
Many studies have suggested an association between cognitive dysfunction and hypoxia-induced neurological damage (Gupta et al., 2013), airway obstruction (Cleutjens et al., 2014, 2017) as well as an elevation in inflammatory mediator expression (Duong et al., 1998). We demonstrate that the neurocognitive impairments following CS exposure may be associated with a loss of BBB integrity and increased neuroinflammatory response within the hippocampus compared to sham mice. Previous literature has shown that BBB endothelial cells and tight junction proteins are affected by pro-inflammatory cytokines following CS exposure (Prasad et al., 2015; Geng et al., 2018). Our study showed that CS-exposed mice had lower levels of the tight-junctional protein ZO-1 in the hippocampus which could contribute to brain damage and neurocognitive impairments. Upon activation, microglia produce pro-inflammatory cytokines and ROS, directly impacting endothelial cell tight junctions, reducing BBB integrity (Geng et al., 2018). Despite reduced astrocyte density within most regions of the hippocampus in our current cohort, CS-exposed mice showed an increase in astrocyte density in the molecular region, which may impact neurogenesis. Krzisch et al. (2015) have elegantly shown that astrocytes of the DG are expected to have greater structural plasticity to allow incorporation of new neurons. Thus, our CS-induced altered astrocyte density may not reduce the number of mature or immature neurons but may inhibit or halt the enseathing of afferent and efferent synapses of adult-born neurons (Krzisch et al., 2015). To access information and memories, a signal is relayed through the molecular region prior to traveling through the other hippocampal regions (Amaral et al., 2007; Roy et al., 2017). The increased astrocyte density in the molecular region of the DG could be assisting in the repair of the DG granule cells, or astrocytes could in fact be activated alongside microglia, potentially causing damage to these cells and hence, inhibiting normal neuronal function (Liddelow et al., 2017). Further investigation into the astrocyte profile is crucial in understanding the function of these cells.
Cigarette smoke-exposed mice displayed reduced expression of Itgam and microglial numbers within the hippocampus, as well as displaying a more activated morphology. A possible explanation could be that the noxious particles in CS are driving both microglial activation and/or a reduction in Itgam expression or cell death. In primary microglial cells, CS condensate induces apoptotic pathways alongside an activated and pro-inflammatory profile (Gao et al., 2014). Interestingly, a clinical study exploring the direct effect of smoking in non-COPD smokers showed decreases in the radiotracer (11C)DAA1106 [which binds to the microglial biomarker translocator proteins (TSPO)], compared to non-smokers (Brody et al., 2017). However, Hillmer et al. (2020) found no evidence of microglial activation in non-COPD smokers. These findings could be due to different radiotracer metabolism or bioavailability [(11C)DAA1106 and 11CPBR28, respectively], or the use of different timeframes (Hillmer et al., 2020). It is noteworthy that these studies only included non-COPD smoking participants, thus, caution must be taken when comparing to the current study. Previous pre-clinical literature assessing the effect of CS on the brain commonly only assessed pro-inflammatory profiles rather than investigating microglial profiles and were performed in whole brain tissue, rather than individual brain regions. It has been shown that as little as 3–6 weeks of CS exposure is sufficient to induce elevated expression of key pro-inflammatory cytokines in the brain (Khanna et al., 2013; Sivandzade et al., 2020), however, these studies do not assess the impact of CS exposure on neurocognition. Memory retention is relayed from the DG to the CA sections, proceeding from CA3 to CA1, then transferred to extra-hippocampal regions of the brain (Amaral et al., 2007). Thus, it could be speculated that the working memory impairments following chronic CS exposure, could be occurring due to altered microglial profiles within the hilus of the DG and the CA3 and specifically reducing microglial activation within these regions could improve CS-induced working memory impairments. Taken together, our current findings reinforce the concept that CS is likely to be responsible for the activation and modulation of microglial cells and this is associated with working memory impairments.
Microglia and astrocytes are integral in maintaining homeostatic neurogenesis, synaptogenesis and axonal development (Sofroniew and Vinters, 2010; Kohman and Rhodes, 2013). Our smoke exposure model induced an amoeboid microglial morphology, which may destabilize the oxidative equilibrium disrupting the neuronal profile within the hippocampus inducing neurocognition impairments. Although we found no differences in hippocampal neuronal populations following CS exposure, we showed a decrease in the pre-synaptic protein, synaptophysin, as well as a reduction in mature stubby spines in pyramidal neurons in the CA1 region, suggesting that CS exposed mice have weakened neuronal connections within the hippocampus. Ramified microglia modulate synaptogenesis to establish neuronal maturity, however, when chronically activated, these microglia may negatively impact the remodeling process. For example, elimination of microglia impaired efficient synaptic formation (Rogers et al., 2011), whilst another study showed ameliorating microglial activation improved working memory retention (Cope et al., 2018). Ho et al. (2012) demonstrated that CS exposure reduced synaptophysin expression and hence, a loss in synaptic integrity within the hilus region of the hippocampus. Overall, the apparent CS-induced increase in neuroinflammation, as well as reduced synaptophysin and mature dendritic spines are all consistent with the disruption of synaptic integrity, potentially orchestrating CS-induced neurocognitive impairments.
Neurocognitive comorbidities of COPD contribute to an increased mortality rate and an increased socioeconomic burden globally, however, there are no effective strategies treating these pathologies. In this study, we show a series of novel findings that CS-induced neurocognitive decline may be, in part, due to alterations in the hippocampal neuroinflammatory profile within the hippocampus. It will be imperative to evaluate whether pharmacologically targeting microglia-related molecular pathways could improve CS-induced cognitive outcomes. For instance, pharmacological inhibition of microglial activation using the tetracycline antibiotic, minocycline, has been shown to improve neurocognitive decline (Jiang et al., 2015; Cope et al., 2018). Moreover, stable COPD participants given a tetracycline analog showed improvements in lung function compared to vehicle-treated COPD participants (Dalvi et al., 2011). Therapeutics targeting oxidative stress may highlight potential targets as mice deficient in the antioxidant, Gpx-1, have elevated lung inflammation following CS exposure compared to wild-type CS-exposed mice (Duong et al., 2010; Oostwoud et al., 2016). Clinically, people with COPD have lower Nrf2 levels, a protein crucial to the upregulation of antioxidants and protection against oxidative stress (Fischer et al., 2015; Yamada et al., 2016). Thus, mitigating the pulmonary inflammation and the potential “spill-over” into the systemic and central circulation could halt neuroinflammation and the associated neurocognitive decline in CS-exposed mice. In-depth investigations of CS concomitant with pharmacological interventions targeting both pulmonary and central inflammation may represent an untapped therapeutic avenue for the development of COPD and the associated neurological comorbidities.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the RMIT University Animal Ethics Committee.
Author Contributions
RV and SD: concept and design. AD, SD, HS, HW, KB, SC, KM, JE, and SL: acquisition of data. AD, SD, RV, HS, HW, KB, SC, KM, JE, SL, SS, SJS, and SB: data analysis and interpretation. SD, HS, KB, SC, and KM: technical assistance. All authors contributed to drafting, editing, and critical revision of the manuscript for intellectual content. RV provided all resources for the work and was the senior investigator, ensuring accuracy and integrity.
Funding
This study was supported by the National Health and Medical Research Council of award to RV (Project Grant ID1139843) and Australian Lung Foundation/Boehringer Ingelheim Fellowship awarded to SD.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AEC, Animal Ethics Committee; ANOVA, analysis of variance; BBB, blood–brain barrier; BAL, bronchoalveolar lavage; BALF, bronchoalveolar lavage fluid; BCA, bicinchoninic acid; Ccl2, chemokine (C-C motif) ligand 2; COPD, chronic obstructive pulmonary disease; CS, cigarette smoke; CD, cluster differentiation; Cxcl1, chemokine (C-X-C motif) ligand 1; Cybb, Cytochrome b-245 beta; DG, dentate gyrus; DCX, doublecortin; FACS, fluorescence-activated cell-sorting; FVC, forced vital capacity; GFAP, glial fibrillary acidic protein; Gpx, glutathione peroxidase 1; HC, hippocampus; H&E, hematoxylin and eosin; IC, inspiratory capacity; Itgam, Integrin alpha M; Iba-1, ionized calcium-binding adapter molecule-1; Lm, Mean linear intercept; Mmp12, matrix metallopeptidase 12; Nox1, NADPH oxidase 1; NPEE, negative pressure-driven forced expirations; NeuN, neuronal nuclei; NBF, neutral buffered formalin; NOP, novel object place; NOR, novel object recognition; Pgk1, phosphoglycerate kinase 1; ROS, reactive oxygen species; Rps18, ribosomal protein s18; RT, room temperature; SG, subgranular; TSPO, translocator proteins; Tnf, tumor necrosis factor.
References
Amaral, D. G., Scharfman, H. E., and Lavenex, P. (2007). The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Prog. Brain Res. 163, 3–22. doi: 10.1016/S0079-6123(07)63001-5
Barnes, P. J. (2009). The Cytokine Network in Chronic Obstructive Pulmonary Disease. Am. J. Res. Cell Mol. Biol. 41, 631–638.
Beckett, E. L., Stevens, R. L., Jarnicki, A. G., Kim, R. Y., Hanish, I., Hansbro, N. G., et al. (2013). A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 131, 752–762. doi: 10.1016/j.jaci.2012.11.053
Botelho, F. M., Gaschler, G. J., Kianpour, S., Zavitz, C. C. J., Trimble, N. J., Nikota, J. K., et al. (2010). Innate Immune Processes Are Sufficient for Driving Cigarette Smoke–Induced Inflammation in Mice. Am. J. Res. Cell Mol. Biol. 42, 394–403. doi: 10.1165/rcmb.2008-0301OC
Brassington, K., Chan, S. M. H., Seow, H. J., Dobric, A., Bozinovski, S., Selemidis, S., et al. (2021). Ebselen reduces cigarette smoke-induced endothelial dysfunction in mice. Br. J. Pharmacol. 178, 1805–1818. doi: 10.1111/bph.15400
Brody, A. L., Hubert, R., Enoki, R., Garcia, L. Y., Mamoun, M. S., Okita, K., et al. (2017). Effect of Cigarette Smoking on a Marker for Neuroinflammation: A [(11)C]DAA1106 Positron Emission Tomography Study. Neuropsychopharmacology 42, 1630–1639. doi: 10.1038/npp.2017.48
Brunette, A. M., Warner, K., Holm, K. E., Meschede, K., Wamboldt, F. S., Kozora, E., et al. (2021). Daily Activities: The Impact of COPD and Cognitive Dysfunction. Arch. Clin. Neuropsychol. 36, acaa090–767. doi: 10.1093/arclin/acaa090
Caramori, G., Casolari, P., Barczyk, A., Durham, A. L., Di Stefano, A., and Adcock, I. (2016). COPD immunopathology. Sem. Immunopathol. 38, 497–515. doi: 10.1007/s00281-016-0561-5
Chan, S. M. H., Cerni, C., Passey, S., Seow, H. J., Bernardo, I., van der Poel, C., et al. (2020). Cigarette Smoking Exacerbates Skeletal Muscle Injury Without Compromising its Regenerative Capacity. Am. J. Res. Cell Mol. Biol. 62, 217–230. doi: 10.1165/rcmb.2019-0106OC
Cleutjens, F., Spruit, M. A., Ponds, R., Dijkstra, J. B., Franssen, F. M. E., Wouters, E. F. M., et al. (2014). Cognitive functioning in obstructive lung disease: results from the United Kingdom biobank. J. Am. Med. Direct. Assoc. 15, 214–219.
Cleutjens, F., Spruit, M. A., Ponds, R., Vanfleteren, L., Franssen, F. M. E., Dijkstra, J. B., et al. (2017). The Impact of Cognitive Impairment on Efficacy of Pulmonary Rehabilitation in Patients With COPD. J. Am. Med. Direct. Assoc. 18, 420–426. doi: 10.1016/j.jamda.2016.11.016
Cope, E. C., LaMarca, E. A., Monari, P. K., Olson, L. B., Martinez, S., Zych, A. D., et al. (2018). Microglia play an active role in obesity-associated cognitive decline. J. Neurosci. 38, 8889–8904. doi: 10.1523/JNEUROSCI.0789-18.2018
Crisan, A. F., Oancea, C., Timar, B., Fira-Mladinescu, O., Crisan, A., and Tudorache, V. (2014). Cognitive impairment in chronic obstructive pulmonary disease. PLoS One 9:e102468. doi: 10.1371/journal.pone.0102468
Dalvi, P. S., Singh, A., Trivedi, H. R., Ghanchi, F. D., Parmar, D. M., and Mistry, S. D. (2011). Effect of doxycycline in patients of moderate to severe chronic obstructive pulmonary disease with stable symptoms. Ann. Thor. Med. 6, 221–226. doi: 10.4103/1817-1737.84777
De Luca, S. N., Miller, A. A., Sominsky, L., and Spencer, S. J. (2020a). Microglial regulation of satiety and cognition. J. Neuroendocrinol. 32:e12838. doi: 10.1111/jne.12838
De Luca, S. N., Soch, A., Sominsky, L., Nguyen, T.-X., Bosakhar, A., and Spencer, S. J. (2020b). Glial remodeling enhances short-term memory performance in Wistar rats. J. Neuroinflam. 17:52. doi: 10.1186/s12974-020-1729-4
Devos, F. C., Maaske, A., Robichaud, A., Pollaris, L., Seys, S., Lopez, C. A., et al. (2017). Forced expiration measurements in mouse models of obstructive and restrictive lung diseases. Res. Res. 18:123. doi: 10.1186/s12931-017-0610-1
Di Stefano, A., Capelli, A., Lusuardi, M., Balbo, P., Vecchio, C., Maestrelli, P., et al. (1998). Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Res. Crit. Care Med. 158, 1277–1285. doi: 10.1164/ajrccm.158.4.9802078
Dodd, J. W. (2015). Lung disease as a determinant of cognitive decline and dementia. Alzheimer’s Res. Ther. 7:32.
Dodd, J. W., Charlton, R. A., van den Broek, M. D., and Jones, P. W. (2013). Cognitive dysfunction in patients hospitalized with acute exacerbation of COPD. Chest 144, 119–127. doi: 10.1378/chest.12-2099
Dodd, J. W., Getov, S. V., and Jones, P. W. (2010). Cognitive function in COPD. Eur. Res. J. 35, 913–922. doi: 10.1183/09031936.00125109
Duan, M., Li, W. C., Vlahos, R., Maxwell, M. J., Anderson, G. P., and Hibbs, M. L. (2012). Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J. Immunol. 189, 946–955. doi: 10.4049/jimmunol.1200660
Duong, C., Seow, H. J., Bozinovski, S., Crack, P. J., Anderson, G. P., and Vlahos, R. (2010). Glutathione peroxidase-1 protects against cigarette smoke-induced lung inflammation in mice. Am. J. Physiol.Lung Cell. Mol. Physiol. 299, L425–L433. doi: 10.1152/ajplung.00038.2010
Duong, T., Acton, P. J., and Johnson, R. A. (1998). The in vitro neuronal toxicity of pentraxins associated with Alzheimer’s disease brain lesions. Brain Res. 813, 303–312. doi: 10.1016/s0006-8993(98)00966-4
Fischer, B. M., Voynow, J. A., and Ghio, A. J. (2015). COPD: balancing oxidants and antioxidants. Int. J. Chron. Obstruct. Pulm. Dis. 10, 261–276. doi: 10.2147/COPD.S42414
Gao, Z., Nissen, J. C., Ji, K., and Tsirka, S. E. (2014). The experimental autoimmune encephalomyelitis disease course is modulated by nicotine and other cigarette smoke components. PLoS One 9:e107979. doi: 10.1371/journal.pone.0107979
Geng, J., Wang, L., Zhang, L., Qin, C., Song, Y., Ma, Y., et al. (2018). Blood-Brain Barrier Disruption Induced Cognitive Impairment Is Associated With Increase of Inflammatory Cytokine. Front. Aging Neurosci. 10:129. doi: 10.3389/fnagi.2018.00129
Giordano, L., Farnham, A., Dhandapani, P. K., Salminen, L., Bhaskaran, J., Voswinckel, R., et al. (2019). Alternative Oxidase Attenuates Cigarette Smoke-induced Lung Dysfunction and Tissue Damage. Am. J. Res. Cell Mol. Biol. 60, 515–522. doi: 10.1165/rcmb.2018-0261OC
Gupta, P. P., Sood, S., Atreja, A., and Agarwal, D. (2013). A comparison of cognitive functions in non-hypoxemic chronic obstructive pulmonary disease (COPD) patients and age-matched healthy volunteers using mini-mental state examination questionnaire and event-related potential. P300 analysis. Lung India 30, 5–11. doi: 10.4103/0970-2113.106119
Hellermann, G. R., Nagy, S. B., Kong, X., Lockey, R. F., and Mohapatra, S. S. (2002). Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Res. Res. 3:15. doi: 10.1186/rr172
Hillmer, A. T., Matuskey, D., Huang, Y., Nabulsi, N., Ropchan, J., Carson, R. E., et al. (2020). Tobacco Smoking in People Is Not Associated with Altered 18-kDa Translocator Protein Levels: A PET Study. J. Nucl. Med. 61, 1200–1204. doi: 10.2967/jnumed.119.237735
Ho, Y.-S., Yang, X., Yeung, S.-C., Chiu, K., Lau, C.-F., Tsang, A. W.-T., et al. (2012). Cigarette Smoking Accelerated Brain Aging and Induced Pre-Alzheimer-Like Neuropathology in Rats. PLoS One 7:e36752. doi: 10.1371/journal.pone.0036752
Jiang, Y., Liu, Y., Zhu, C., Ma, X., Ma, L., Zhou, L., et al. (2015). Minocycline enhances hippocampal memory, neuroplasticity and synapse-associated proteins in aged C57 BL/6 mice. Neurobiol. Learn. Mem. 121, 20–29. doi: 10.1016/j.nlm.2015.03.003
Khanna, A., Guo, M., Mehra, M., and Royal, W. III. (2013). Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J. Neuroimmunol. 254, 69–75. doi: 10.1016/j.jneuroim.2012.09.006
Kohman, R. A., and Rhodes, J. S. (2013). Neurogenesis, inflammation and behavior. Brain Behav. Immun. 27, 22–32. doi: 10.1016/j.bbi.2012.09.003
Krzisch, M., Temprana, S. G., Mongiat, L. A., Armida, J., Schmutz, V., Virtanen, M. A., et al. (2015). Pre-existing astrocytes form functional perisynaptic processes on neurons generated in the adult hippocampus. Brain Struct. Func. 220, 2027–2042. doi: 10.1007/s00429-014-0768-y
Lehnardt, S., Massillon, L., Follett, P., Jensen, F. E., Ratan, R., Rosenberg, P. A., et al. (2003). Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. 100, 8514–8519. doi: 10.1073/pnas.1432609100
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lull, M. E., and Block, M. L. (2010). Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. doi: 10.1016/j.nurt.2010.05.014
Lv, Z., Hu, P., Jiang, Y., Yang, W., Wang, R., Wang, K., et al. (2020). Changes in Spatial Working Memory in Stable Chronic Obstructive Pulmonary Disease: A Retrospective Study. Biomed Res. Int. 2020:7363712. doi: 10.1155/2020/7363712
MacNee, W. (2005). Pathogenesis of Chronic Obstructive Pulmonary Disease. Proc. Am. Thor. Soc. 2, 258–266.
Morris, C., Mitchell, J. W., Moorey, H., Younan, H.-C., Tadros, G., and Turner, A. M. (2019). Memory, attention and fluency deficits in COPD may be a specific form of cognitive impairment. ERJ Open Res. 5, 00229–2018. doi: 10.1183/23120541.00229-2018
Nadar, M. S., Hasan, A. M., and Alsaleh, M. (2021). The negative impact of chronic tobacco smoking on adult neuropsychological function: a cross-sectional study. BMC Public Heal. 21:1278. doi: 10.1186/s12889-021-11287-6
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Oostwoud, L. C., Gunasinghe, P., Seow, H. J., Ye, J. M., Selemidis, S., Bozinovski, S., et al. (2016). Apocynin and ebselen reduce influenza A virus-induced lung inflammation in cigarette smoke-exposed mice. Sci. Rep. 6:20983. doi: 10.1038/srep20983
Paul, R. H., Brickman, A. M., Cohen, R. A., Williams, L. M., Niaura, R., Pogun, S., et al. (2006). Cognitive status of young and older cigarette smokers: data from the international brain database. J. Clin. Neurosci. 13, 457–465. doi: 10.1016/j.jocn.2005.04.012
Percie du Sert, N., Hurst, V., Ahluwalia, A., Alam, S., Avey, M. T., Baker, M., et al. (2020). The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. Exp. Physiol. 105, 1459–1466.
Prasad, S., Sajja, R. K., Park, J. H., Naik, P., Kaisar, M. A., and Cucullo, L. (2015). Impact of cigarette smoke extract and hyperglycemic conditions on blood–brain barrier endothelial cells. Fluids Barriers CNS. 12:18. doi: 10.1186/s12987-015-0014-x
Prasedya, E. S., Ambana, Y., Martyasari, N. W. R., Aprizal, Y., and Nurrijawati Sunarpi. (2020). Short-term E-cigarette toxicity effects on brain cognitive memory functions and inflammatory responses in mice. Toxicol. Res. 36, 267–273. doi: 10.1007/s43188-019-00031-3
Rinaldi, M., Maes, K., De Vleeschauwer, S., Thomas, D., Verbeken, E. K., Decramer, M., et al. (2012). Long-term nose-only cigarette smoke exposure induces emphysema and mild skeletal muscle dysfunction in mice. Dis. Models Mechan. 5, 333–341. doi: 10.1242/dmm.008508
Risher, W. C., Ustunkaya, T., Singh Alvarado, J., and Eroglu, C. (2014). Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS One 9:e107591. doi: 10.1371/journal.pone.0107591
Rogers, J. T., Morganti, J. M., Bachstetter, A. D., Hudson, C. E., Peters, M. M., Grimmig, B. A., et al. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011
Roy, D. S., Kitamura, T., Okuyama, T., Ogawa, S. K., Sun, C., Obata, Y., et al. (2017). Distinct Neural Circuits for the Formation and Retrieval of Episodic Memories. Cell 170, 1000.e–1012.e. doi: 10.1016/j.cell.2017.07.013
Sharafkhaneh, A., Hanania, N. A., and Kim, V. (2008). Pathogenesis of emphysema: from the bench to the bedside. Proc. Am. Thor. Soc. 5, 475–477. doi: 10.1513/pats.200708-126ET
Shu, J., Li, D., Ouyang, H., Huang, J., Long, Z., Liang, Z., et al. (2017). Comparison and evaluation of two different methods to establish the cigarette smoke exposure mouse model of COPD. Sci. Rep. 7:15454. doi: 10.1038/s41598-017-15685-y
Singh, B., Mielke, M. M., Parsaik, A. K., Cha, R. H., Roberts, R. O., Scanlon, P. D., et al. (2014). A prospective study of chronic obstructive pulmonary disease and the risk for mild cognitive impairment. JAMA Neurol. 71, 581–588. doi: 10.1001/jamaneurol.2014.94
Sivandzade, F., Alqahtani, F., Sifat, A., and Cucullo, L. (2020). The cerebrovascular and neurological impact of chronic smoking on post-traumatic brain injury outcome and recovery: an in vivo study. J. Neuroinflam. 17:133. doi: 10.1186/s12974-020-01818-0
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Spencer, S. J., Basri, B., Sominsky, L., Soch, A., Ayala, M. T., Reineck, P., et al. (2019). High-fat diet worsens the impact of aging on microglial function and morphology in a region-specific manner. Neurobiol. Aging 74, 121–134. doi: 10.1016/j.neurobiolaging.2018.10.018
Starkey, M. R., Plank, M. W., Casolari, P., Papi, A., Pavlidis, S., Guo, Y., et al. (2019). IL-22 and its receptors are increased in human and experimental COPD and contribute to pathogenesis. Eur. Res. J. 54:1800174. doi: 10.1183/13993003.00174-2018
Torres-Sánchez, I., Rodríguez-Alzueta, E., Cabrera-Martos, I., López-Torres, I., Moreno-Ramírez, M. P., and Valenza, M. C. (2015). Cognitive impairment in COPD: a systematic review. J. Brasileiro de Pneumol. 41, 182–190. doi: 10.1590/S1806-37132015000004424
Triantaphyllopoulos, K., Hussain, F., Pinart, M., Zhang, M., Li, F., Adcock, I., et al. (2011). A model of chronic inflammation and pulmonary emphysema after multiple ozone exposures in mice. Am. J. Physiol.Lung Cell. Mol. Physiol. 300, L691–L700. doi: 10.1152/ajplung.00252.2010
Vlahos, R., and Bozinovski, S. (2014). Recent advances in pre-clinical mouse models of COPD. Clin. Sci. 126, 253–265. doi: 10.1042/CS20130182
Vlahos, R., Bozinovski, S., Jones, J. E., Powell, J., Gras, J., Lilja, A., et al. (2006). Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L931–L945. doi: 10.1152/ajplung.00201.2005
Vogelmeier, C. F., Criner, G. J., Martinez, F. J., Anzueto, A., Barnes, P. J., Bourbeau, J., et al. (2020). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2020 Report. GOLD Executive Summary. Am. J. Res. Crit. Care Med. 1, 1–141. doi: 10.1164/rccm.200703-456SO
Wang, H., Aloe, C., McQualter, J., Papanicolaou, A., Vlahos, R., Wilson, N., et al. (2021). G-CSFR antagonism reduces mucosal injury and airways fibrosis in a virus-dependent model of severe asthma. Br. J. Pharmacol. 178, 1869–1885. doi: 10.1111/bph.15415
Yamada, K., Asai, K., Nagayasu, F., Sato, K., Ijiri, N., Yoshii, N., et al. (2016). Impaired nuclear factor erythroid 2-related factor 2 expression increases apoptosis of airway epithelial cells in patients with chronic obstructive pulmonary disease due to cigarette smoking. BMC Pulm. Med. 16:27.
Keywords: cigarette smoking, emphysema, neuroinflammation, microglia, cognition, synaptogenesis
Citation: Dobric A, De Luca SN, Seow HJ, Wang H, Brassington K, Chan SMH, Mou K, Erlich J, Liong S, Selemidis S, Spencer SJ, Bozinovski S and Vlahos R (2022) Cigarette Smoke Exposure Induces Neurocognitive Impairments and Neuropathological Changes in the Hippocampus. Front. Mol. Neurosci. 15:893083. doi: 10.3389/fnmol.2022.893083
Received: 10 March 2022; Accepted: 06 April 2022;
Published: 17 May 2022.
Edited by:
Brian Gregory George Oliver, University of Technology Sydney, AustraliaReviewed by:
Hui Chen, University of Technology Sydney, AustraliaChenju Yi, Sun Yat-sen University, China
Yik Lung Chan, University of Technology Sydney, Australia
Copyright © 2022 Dobric, De Luca, Seow, Wang, Brassington, Chan, Mou, Erlich, Liong, Selemidis, Spencer, Bozinovski and Vlahos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ross Vlahos, cm9zcy52bGFob3NAcm1pdC5lZHUuYXU=