 Alecia Biel1
Alecia Biel1 Anthony S. Castanza2
Anthony S. Castanza2 Ryan Rutherford1
Ryan Rutherford1 Summer R. Fair1
Summer R. Fair1 Lincoln Chifamba1
Lincoln Chifamba1 Jason C. Wester3
Jason C. Wester3 Mark E. Hester1,3,4*
Mark E. Hester1,3,4* Robert F. Hevner2*
Robert F. Hevner2*
- 1The Steve and Cindy Rasmussen Institute for Genomic Medicine, Abigail Wexner Research Institute at Nationwide Children’s Hospital, Columbus, OH, United States
- 2Department of Pathology, University of California, San Diego, San Diego, CA, United States
- 3Department of Neuroscience, The Ohio State University College of Medicine, Columbus, OH, United States
- 4Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH, United States
AUTS2 syndrome is a genetic disorder that causes intellectual disability, microcephaly, and other phenotypes. Syndrome severity is worse when mutations involve 3’ regions (exons 9-19) of the AUTS2 gene. Human AUTS2 protein has two major isoforms, full-length (1259 aa) and C-terminal (711 aa), the latter produced from an alternative transcription start site in exon 9. Structurally, AUTS2 contains the putative “AUTS2 domain” (∼200 aa) conserved among AUTS2 and its ohnologs, fibrosin, and fibrosin-like-1. Also, AUTS2 contains extensive low-complexity sequences and intrinsically disordered regions, features typical of RNA-binding proteins. During development, AUTS2 is expressed by specific progenitor cell and neuron types, including pyramidal neurons and Purkinje cells. AUTS2 localizes mainly in cell nuclei, where it regulates transcription and RNA metabolism. Some studies have detected AUTS2 in neurites, where it may regulate cytoskeletal dynamics. Neurodevelopmental functions of AUTS2 have been studied in diverse model systems. In zebrafish, auts2a morphants displayed microcephaly. In mice, excision of different Auts2 exons (7, 8, or 15) caused distinct phenotypes, variously including neonatal breathing abnormalities, cerebellar hypoplasia, dentate gyrus hypoplasia, EEG abnormalities, and behavioral changes. In mouse embryonic stem cells, AUTS2 could promote or delay neuronal differentiation. Cerebral organoids, derived from an AUTS2 syndrome patient containing a pathogenic missense variant in exon 9, exhibited neocortical growth defects. Emerging technologies for analysis of human cerebral organoids will be increasingly useful for understanding mechanisms underlying AUTS2 syndrome. Questions for future research include whether AUTS2 binds RNA directly, how AUTS2 regulates neurogenesis, and how AUTS2 modulates neural circuit formation.
Introduction to AUTS2 Syndrome
The AUTS2 gene (autism-susceptibility-gene-2) was first identified in humans by genetic analysis of monozygotic twins with autism and chromosomal translocation t(7;20) (Sultana et al., 2002). Human AUTS2 was further revealed as a 1.2-Mb gene on chromosome 7q11.22, which encodes a 1259-aa full-length protein, and a 711-aa C-terminal isoform (Beunders et al., 2013). Sequence analysis of AUTS2 detected motifs such as proline-rich regions and histidine repeats, but no recognizable structural domains. As of 2021, more than 60 patients with pathogenic AUTS2 variants have been reported, and the AUTS2 syndrome has been well characterized as a neurodevelopmental and somatic malformation disorder with diverse phenotypes. The most common phenotypes are intellectual disability (ID) and microcephaly (Beunders et al., 2016).
Although the AUTS2 gene was named for autism susceptibility, many AUTS2 syndrome patients have an outgoing personality in childhood (Beunders et al., 2016). Rather, the most frequent trait is ID (or developmental delay), mild to severe, in virtually all patients. The major traits and their frequency are ID (98%), microcephaly (65%), feeding difficulties (62%), attention deficit hyperactivity disorder (ADHD) (54%), and autistic traits (52%) (Beunders et al., 2013, 2016; Sanchez-Jimeno et al., 2021). Hypotonia (38%) and spasticity (37%), inverse disorders of neuromuscular reflexes, are also relatively frequent. A small minority have epilepsy (7%). As observed by neuroimaging, structural brain anomalies occur in 27% of AUTS2 patients (Sanchez-Jimeno et al., 2021). The reported brain malformations include corpus callosum hypoplasia, cerebellar hypoplasia, small posterior fossa, and Chiari malformation type 1 (Liu et al., 2021; Fair et al., 2022). Somatic developmental problems are also numerous in AUTS2 syndrome. These include growth defects, such as low birth weight (20%) and short stature (43%); musculoskeletal anomalies, such as kyphosis/scoliosis (24%) and tight heel cords (37%); and facial dysmorphisms, such as hypertelorism (44%) and micrognathia/retrognathia (36%). Thus, in the most severe cases, AUTS2 syndrome can affect many organs.
As a gauge of overall severity, an “AUTS2 syndrome severity score” representing the sum of 32 traits was formulated (Beunders et al., 2013). Interestingly, the severity scores were observed more severe in patients with whole gene deletions or C-terminal mutations (exons 9–19), and less severe in patients with N-terminal mutations (exons 1–8) (Beunders et al., 2013; Sanchez-Jimeno et al., 2021). The hypothesis that the C-terminal part of the protein mediates major AUTS2 functions was further supported by Auts2a knockdown experiments in zebrafish, in which expression of the C-terminal isoform rescued the microcephaly phenotype (Beunders et al., 2013). The C-terminal isoform is comprised of a proline-rich region (exons 9–13), the putative “AUTS2 domain” of ∼200 aa (exons 14–19), and disordered regions (exon 19), but does not include the HX repeat encoded by the first part of exon 9 (described in more detail below). Indeed, the alternative tss, corresponding to position 1,597 of full-length AUTS2 cDNA (Beunders et al., 2013), lies within the HX repeat encoded by cDNA positions 1,575–1,626. The translation start site for AUTS2-C corresponds to position 1,666 of full-length cDNA, and thus does not include the HX repeat.
AUTS2 syndrome exhibits phenotypic overlap with several other genetic causes of ID, including Rubinstein-Taybi syndrome (CREBBP or EP300) (Fergelot et al., 2016), NONO syndrome (Sewani et al., 2020), TBR1 syndrome (Nambot et al., 2020), and FBRSL1 syndrome (Ufartes et al., 2020). The similarities among these disorders reflect their related roles in genetic pathways and binding interactions. Specifically, expression of Auts2 is regulated by transcription factor TBR1 (Bedogni et al., 2010); AUTS2 protein interacts with NONO protein (Castanza et al., 2021); AUTS2 binds and regulates EP300 mRNA (Castanza et al., 2021); and FBRSL1, an RNA-binding protein (Baltz et al., 2012), is the closest homolog of AUTS2 in the genome (Sellers et al., 2020).
The AUTS2 Gene Family and Functions
When AUTS2 was first described, its functions could not be predicted on the basis of homology to known proteins. Indeed, only partial sequence homology was found to two genes with unknown functions, designated CG9056 and FLJ11618 (Sultana et al., 2002). Subsequent research identified CG9056 as Drosophila tay bridge (tay), and FLJ11618 as human fibrosin (FBRS). Both genes, it is now known, indeed belong to the same diversified superfamily of genes as AUTS2 (Sellers et al., 2020).
More narrowly, the “AUTS2 gene family” consists of the three most closely related genes, AUTS2, FBRSL1, and FBRS. These genes are ohnologs—defined as paralogs produced by two-rounds of gene duplication from a single ancestral gene during the evolution of jawed vertebrates ∼450 million years ago (Sacerdot et al., 2018; Sellers et al., 2020). Accordingly, AUTS2, FBRSL1, and FBRS genes are found in all vertebrates. By sequence analysis, AUTS2 appears most closely related to the ancestral AUTS2 precursor (aAUTS2p) gene; also, AUTS2 and FBRSL1 are closer to each other than to FBRS (Sellers et al., 2020).
The ancestral aAUTS2p gene arose in early bilaterian animals ∼650 million years ago, and its derivatives are found in the Nephrozoa clade (Sellers et al., 2020). Thus, non-Chordate bilaterians, such as flies and amphioxus, contain only one AUTS2-related gene. In Drosophila, that gene is tay (tay bridge), which regulates neuronal development in the protocerebral bridge and motoneurons (Poeck et al., 2008; Molnar and de Celis, 2013; Molnar et al., 2018). However, tay is relatively divergent from aAUTS2p, and shows only patchy homology to mammalian AUTS2 (Sellers et al., 2020). The most highly conserved sequence in AUTS2-related proteins from diverse vertebrate and invertebrate species aligns with exon 14 of human AUTS2, which encodes part of the predicted “AUTS2 domain.” Also highly conserved is the “HX repeat” or “HQHT repeat” region encoded in the first half of AUTS2 exon 9.
While much progress has been made comprehending evolution of the AUTS2 gene family, no consensus conserved functions of the proteins have been determined. Several AUTS2 superfamily members localize in cell nuclei, including AUTS2 (Bedogni et al., 2010), FBRSL1 (Ufartes et al., 2020), and tay bridge (Molnar and de Celis, 2013). Among these, FBRSL1 was previously identified as an mRNA binding protein (Baltz et al., 2012). Further studies of the AUTS2 gene family and superfamily will be necessary to shed light on conserved and divergent functions of these proteins.
Structure of the Human AUTS2 Gene, Transcripts, and Protein Isoforms
Human AUTS2 spans 1,195,032 base pairs on chromosome 7q11.22 (UCSD Genome Browser, assembly hg381). Exons 1–7 are separated by long introns (16–301 kb), while exons 7–19 have shorter introns (∼1–3 kb) (Figure 1A and Supplementary Table 1). Full-length AUTS2 cDNA (NCBI CCDS5539.1) encodes a protein of 1259 amino acids (NP_056385.1), here designated isoform AUTS2-FL (Figure 1B). Many variant transcripts have been annotated from sequencing projects, but the best documented variant is generated from an in-frame alternative transcription start site (tss) in exon 9, to produce the C-terminal isoform of AUTS2 (Beunders et al., 2013), here designated AUTS2-C (Figures 1A,B). The AUTS2-C transcript also utilizes an alternative splice junction between exons 9–10 to incorporate an additional 7 amino acids, totaling 711 aa. Both AUTS2-FL and AUTS2-C are highly basic (pI = 9.41).
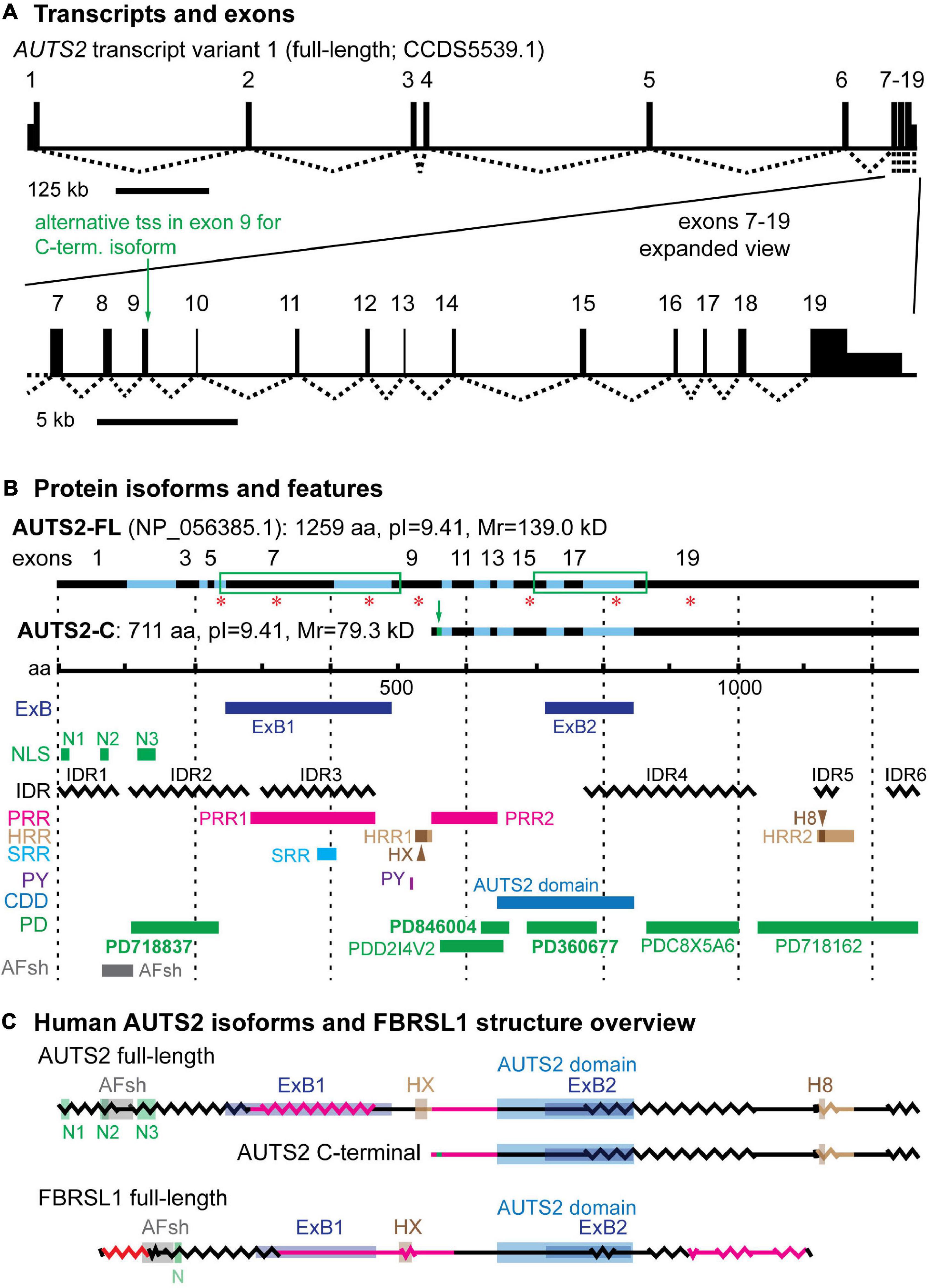
Figure 1. AUTS2 transcripts and protein isoforms. (A) The full-length AUTS2 transcript has 19 exons. An alternative transcription start site (tss) in exon 9 produces mRNA for the C-terminal protein isoform. (B) The full-length and C-terminal isoforms are indicated with source exons in black (odd) and blue (even). Green boxes enclose splice junctions in which amino acids were encoded across the junctions, comprising exon blocs. Red asterisks indicate source exons that encoded a non-integer number of amino acids. Features mapped from protein sequence: ExB, exon bloc; NLS, nuclear localization sequence; IDR, intrinsically disordered region; PRR, Pro-rich repeat; HRR, His-rich repeat; HX, HX repeat; H8, polyhistidine (8×) repeat; SRR, Ser-rich repeat; PY, PY protein binding motif; CDD, conserved domain database; PD, ProDom predicted domains (domain names in bold also identified in FBRSL1); AFsh, AUTS2-FBRSL1 short homology region. (C) Both major AUTS2 isoforms have a high content of IDRs (zigzag lines) and regions enriched in amino acids Pro, His, or Ser (red lines). The C-terminal isoform includes the AUTS2 domain, but lacks N-terminal features such as the HX repeat.
Sequence analysis has revealed several features of AUTS2 protein that suggest possible functions (Figure 1B). Among the most salient features of AUTS2 are its high contents of predicted intrinsically disordered regions (IDRs) and low complexity sequences (LCSs). One recent analysis found that predicted IDRs comprise 64.4% of AUTS2-FL (Sellers et al., 2020). Among LCSs, AUTS2 contains two proline-rich regions (PRRs), two histidine-rich regions (HRRs), and one serine-rich region (SRR) (Sultana et al., 2002; and Expasy ScanProsite) (Figure 1B). HRR1 contains an HX (HQ and HT) repeat (aa 525–542), while HRR2 contains an 8-aa stretch of only histidines. Together, putative IDRs and LCSs cover ∼74.2% of AUTS2-FL. This is significant because IDRs and short repetitive amino acid motifs are typical features of mRNA-binding proteins (Castello et al., 2012; Hentze et al., 2018). Consistent with this analysis, AUTS2 was recently found to bind RNA-protein complexes, and possibly RNA directly (Castanza et al., 2021).
Sequence analysis also indicates that AUTS2 is likely to be localized in cell nuclei. AUTS2-FL has multiple nuclear localization sequences (NLSs) in the 5′ region, and NucPred indicates 100% likelihood of intranuclear localization. AUTS2-C does not have discrete NLSs, but its overall sequence predicts 70% likelihood of intranuclear localization (NucPred). In addition, AUTS2-FL, but not AUTS2-C, contains a PY motif (PPPY), which may bind WW-domains to interact with other proteins. PY motifs interact with Nedd4 family E3 ubiquitin ligases for proteolysis (Hatstat et al., 2021), and with other proteins for signal transduction or transcription (Lin et al., 2019).
The domain structure of AUTS2 is unknown. The protein contains no recognized structural domain sequences, and its structure has not been studied experimentally. Previous studies have used bioinformatics to propose AUTS2-FL structures with three domains (Sellers et al., 2020) or five domains (Castanza et al., 2021), but these remain speculative. Sequence analysis of AUTS2 using NCBI Conserved Domains Database identifies an ∼200-aa “AUTS2 domain” (pfam15336), predicted on the basis of sequence conservation across the AUTS2 gene family (AUTS2, FBRSL1, and FBRS) (Castanza et al., 2021). The AUTS2 domain is present in both AUTS2-FL and AUTS2-C isoforms (Figure 1B). Sequence analysis of AUTS2-FL with ProDom identified six possible domains, three of which are also found in FBRSL1 (Supplementary Figure 1 and Supplementary Table 2). All of the putative domains identified by ProDom are annotated as likely poly(A)-RNA binding proteins. Finally, comparison of AUTS2 and FBRSL1 identifies a highly conserved 46 aa sequence (73.9% identity, no gaps) in the N-terminal part of AUTS2-FL, here designated the AUTS2/FBRSL1 short homology (AFsh) region (Figure 1B, Supplementary Figure 1B, and Supplementary Table 3).
Other interesting features of AUTS2 include two blocs of exons, ExB1 and ExB2 (comprised of exons 6–9 and 15–19, respectively), each connected by splice junctions that encode amino acids across the junctions; as well as several exons that encode non-integer numbers of amino acids (Figure 1B). These features imply reduced likelihood of alternative splicing involving those exons and may indicate important functions for the encoded protein sequences. Indeed, ExB1 encodes PRR1, SRR, and IDR regions, potentially for RNA binding; ExB2 encodes part of the AUTS2 domain. Interestingly, ExB1 and ExB2 are conserved in FBRSL1 as exons 6–9 and 13–17 of that gene (Supplementary Figure 1 and Supplementary Table 3). For ExB1, the encoded protein sequences are relatively divergent between AUTS2 and FBRSL1, mostly due to insertions and deletions involving PRR1. In contrast, ExB2 is better conserved at the protein level, and encodes most of the putative AUTS2 domain in both FBRSL1 and AUTS2.
To further explore potential AUTS2 protein structures, we used RoseTTAfold2 (Baek et al., 2021) to computationally predict structures of AUTS2, FBRSL1, and the AUTS2 domains of each protein (Supplementary Figures 2–5). None of the RoseTTAfold models yielded high-confidence results, most likely because no known structures are available for proteins of similar sequence. However, all of the models showed extensive stretches of disordered, open structure. Indeed, one possibility is that outside the putative AUTS2 domain, AUTS2 and FBRSL1 lack classic well-structured domains, and instead utilize disordered sequences for RNA binding. Putting all features together, both AUTS2 and FBRSL1 are seen as proteins with extensive IDRs and LCSs, punctuated by conserved motifs and the putative AUTS2 domain (Figure 1C and Supplementary Figure 1C).
AUTS2 Expression Patterns and Intracellular Localization
The expression of AUTS2 mRNA and protein isoforms has been studied in multiple tissues and cell types (Table 1). In humans, AUTS2 mRNA is expressed at relatively high levels in fetal and adult brain, skeletal muscle, and kidney; and at lower levels in several other tissues (Sultana et al., 2002). In mice, Auts2 mRNA is expressed in many areas of the developing brain and spinal cord (Figure 2A) (Bedogni et al., 2010). In developing cerebral cortex, Auts2 is expressed mainly in the cortical plate, where postmitotic neurons are located, although lower levels of mRNA are also detected in progenitor compartments (ventricular zone and subventricular zone) (Bedogni et al., 2010). Moreover, AUTS2 protein has been detected in neurogenic cortical progenitor cells (Castanza et al., 2021). From E16.5 to the first postnatal week in mice, Auts2 mRNA is expressed in an intracortical gradient from high rostral to low caudal, suggesting a possible role in cortical patterning (Bedogni et al., 2010). AUTS2 protein is localized mainly in the nuclei of neurons, but not glial cells; in cerebral cortex, AUTS2 is expressed by pyramidal neurons, but not GABAergic interneurons (Bedogni et al., 2010; Gao et al., 2014; Castanza et al., 2021). AUTS2 protein may additionally be present in neuronal cytoplasm, including neurites and growth cones (Hori et al., 2014). In postnatal mouse cerebellum, AUTS2 protein is detected at high levels in Purkinje cells and Golgi neurons (Figures 2B,C) (Yamashiro et al., 2020).
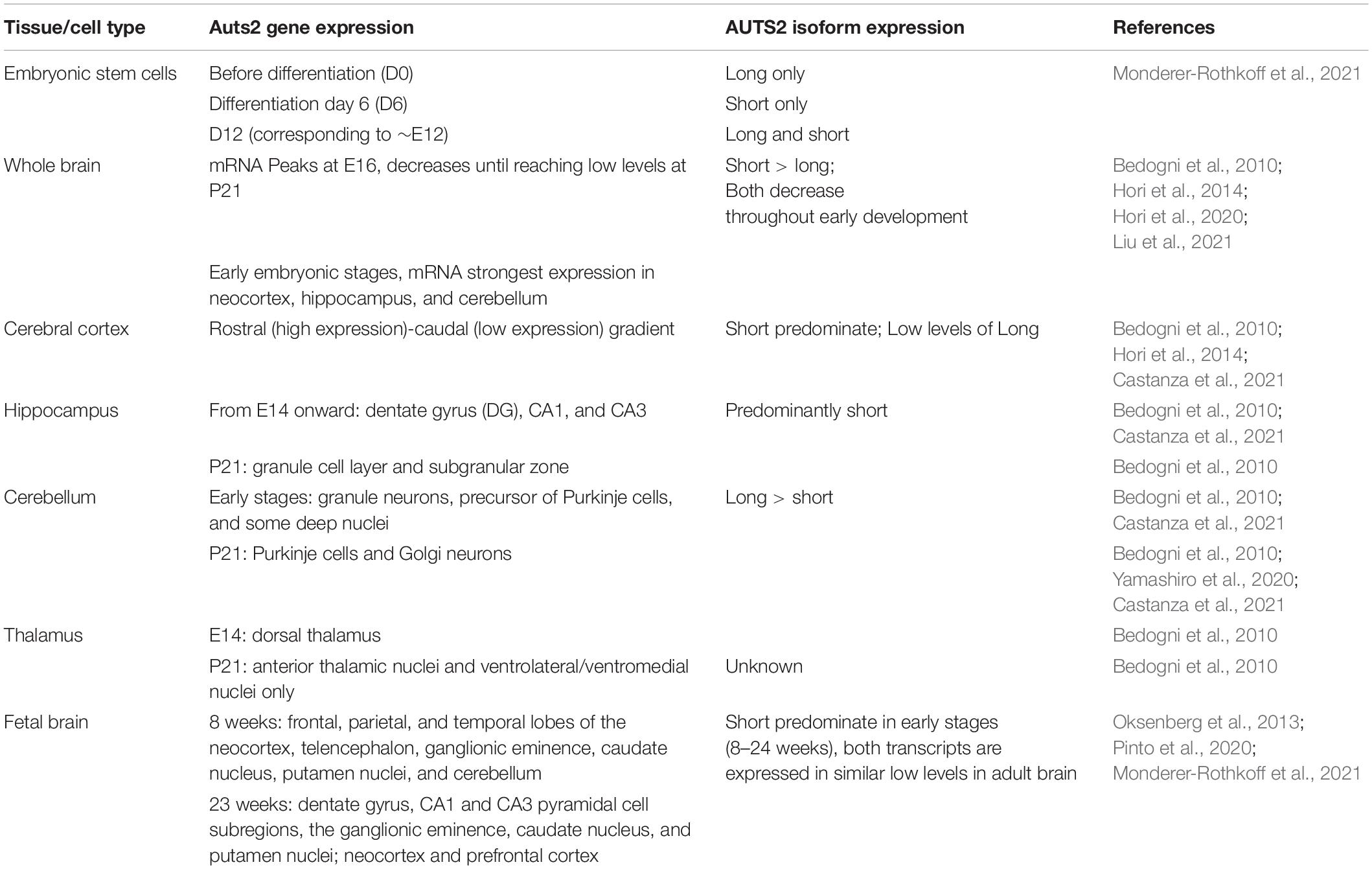
Table 1. AUTS2 expression in developing brain and cultured cells.
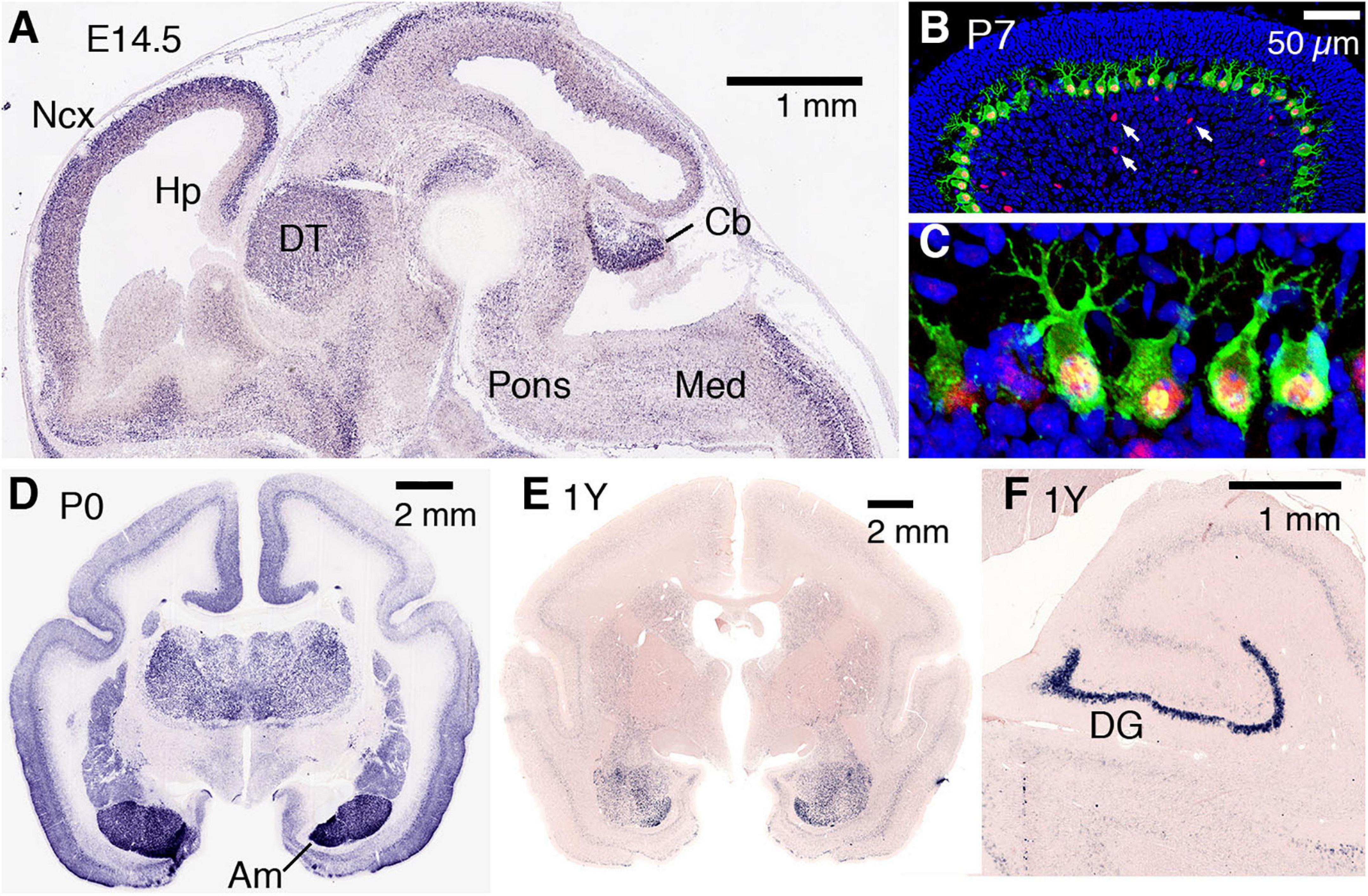
Figure 2. AUTS2 expression in developing mouse and marmoset brain. (A) By in situ hybridization, Auts2 expression is observed in multiple areas of E14.5 mouse brain, including cerebral neocortex (Ncx), hippocampus (Hp), dorsal thalamus (DT), pons, cerebellum (Cb), and medulla (Med). Sagittal section from Genepaint (https://gp3.mpg.de/). (B,C) In P7 mouse cerebellum, AUTS2 protein (red) localizes in the nuclei of calbindin + (green) Purkinje cells, and in scattered Golgi neurons (arrows). Panel (C) magnified 5× from (B). (D) In neonatal marmosets, AUTS2 is expressed in many brain regions, with highest levels in amygdala (Am). (E,F) In 1-year-old (young adult) marmosets, AUTS2 levels remain high in amygdala and dentate gyrus (DG). Data for panels (D–F) from the Marmoset Gene Atlas (https://gene-atlas.brainminds.riken.jp/).
Recently, the open Marmoset Gene Atlas3 has published AUTS2 expression results in the developing and adult non-human primate brain. As shown by in situ hybridization, AUTS2 mRNA is expressed in many regions of the developing marmoset brain, with particularly high levels in the amygdala (Figure 2D). In adult marmosets, AUTS2 mRNA levels remain high in the amygdala, and in granule neurons of the hippocampal dentate gyrus (Figures 2E,F).
Molecular Functions and Interactions of AUTS2 Protein
Transcriptional Activation
Molecular functions of AUTS2 have been elucidated in the context of AUTS2 interacting molecules, complexes, and chromatin (Figure 3). The first proposed function of AUTS2, as a transcriptional activator, was determined on the basis of its interactions with other regulators of transcription, and its distribution in active open chromatin. In human embryonic kidney (HEK) cells induced to express AUTS2, AUTS2 associated with non-canonical forms of polycomb repressive complex 1 (PRC1), which is a complex that contains PCGF3/5, RING1A/B, and RYBP/YAF2, but no CBX proteins (Gao et al., 2014). Canonical PRC1 is an epigenetic repressor that ubiquitinates histones. In contrast, non-canonical PRC1 (ncPRC1) was found to activate gene expression via AUTS2-mediated recruitment of P300, a histone lysine acetyltransferase (Figure 3A) (Gao et al., 2014). Consistent with this function, ChIP-seq indicated that in developing brain, AUTS2 localizes to actively transcribed chromatin, usually within ±5 kb of transcriptional start sites (Gao et al., 2014; Oksenberg et al., 2014; Liu et al., 2021). Subsequently, WDR68 was identified as an additional component of the AUTS2-containing ncPRC1 complex (Wang et al., 2018). On the other hand, two groups have reported that AUTS2-FL interacts with ncPRC1, but AUTS2-C does not (Geng et al., 2021; Monderer-Rothkoff et al., 2021). In a study of mutations in AUTS2 syndrome, the interaction of AUTS2 with P300 in HEK 293 cells was found to be disrupted by mutations involving the HX repeat (Liu et al., 2021). Importantly, the HX repeat encompasses the alternative tss for AUTS2-C, and indeed a small deletion in the HX repeat was found to eliminate expression of AUTS2-C (Martinez-Delgado et al., 2021). Significantly, most of the above-mentioned AUTS2-protein interactions and disruptions were studied in HEK cells after AUTS2 overexpression (Gao et al., 2014; Wang et al., 2018; Geng et al., 2021), or in yeast two-hybrid assays (Monderer-Rothkoff et al., 2021). Further studies are needed to confirm that AUTS2 interacts with proteins such as PCGF3/5, RING1B, and P300 at physiological levels in cortical neurons in vivo.
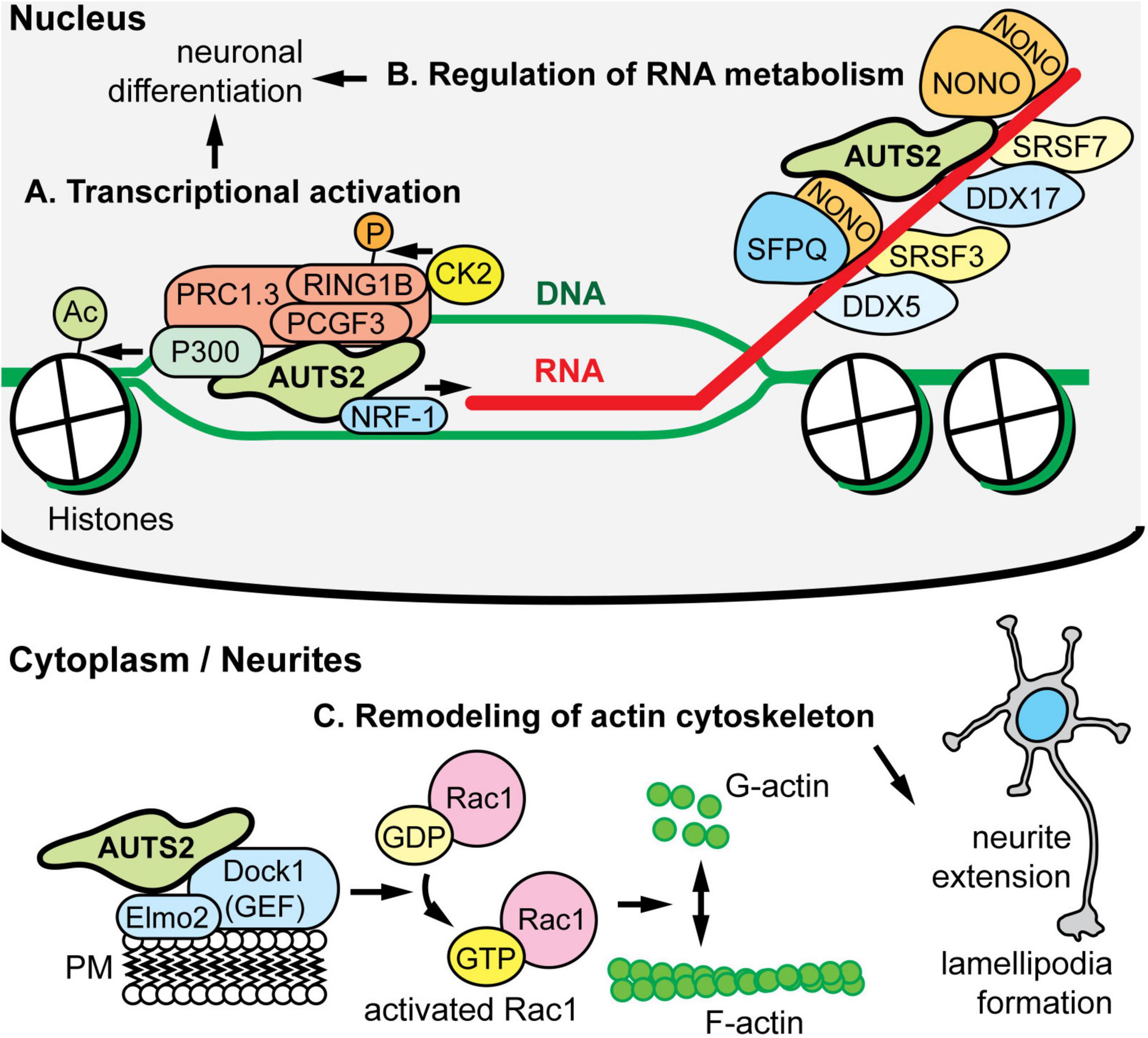
Figure 3. AUTS2 molecular functions and interactions. (A) In HEK293 cells, AUTS2 associates with variant forms of polycomb repressive complex 1 (vPRC1), which also contains PCGF3/5, RING1B, and other PRC1 subunits (PMID: 25519132). In turn, AUTS2 recruits P300, a histone acetyltransferase that opens chromatin, and binds NRF1, a transcriptional activator (PMID: 34637754). Also, vPRC1 recruits CK2, a protein kinase that phosphorylates and inactivates RING1B, a ubiquitin ligase and core subunit of PRC1 for chromatin inactivation. The net effect of AUTS2 is to convert PRC1 from a repressor to an activator of transcription. (B) In developing cerebral cortex, AUTS2 associates with multiple RNA-binding proteins (RBPs), including the scaffolds NONO and SFPQ; splicing factors SRSF3 and SRSF7; and RNA helicases DDX5 and DDX7 (PMID: 34013328). Also, AUTS2 co-immunoprecipitated multiple RNA species, suggesting that AUTS2 binds RNA directly or indirectly. (C) In HEK293 cells, AUTS2 associates with a guanine nucleotide exchange factor (GEF) complex containing DOCK1 (also known as DOCK180) and ELMO2 (PMID: 25533347). Through this interaction, AUTS2 is thought to cause activation of Rac1 (a small G protein), remodeling of the cytoskeleton, neurite elongation, and lamellipodia formation. AUTS2 was also found to interact with another GEF, P-REX1; and may regulate multiple small G proteins.
RNA Metabolism
Another proposed intranuclear function of AUTS2 is to regulate RNA metabolism by associating with RNA-binding protein (RBP) complexes, and possibly with RNA directly. Immunoprecipitation (IP) of AUTS2 from developing mouse cortex followed by mass spectrometry (IP-MS) revealed that AUTS2 interacts with multiple RBPs in vivo, including scaffolds NONO and SFPQ, splicing factors such as SRSF3/7, and RNA helicases DDX5/17 (Figure 3A) (Castanza et al., 2021). Also, in yeast two-hybrid assays, AUTS2 was reported to interact with splicing factor SF3B1 (Monderer-Rothkoff et al., 2021). In neonatal mouse neocortex, AUTS2 IP followed by RNA sequencing (RIP-seq) detected abundant transcripts, including many, such as Ep300, that are dysregulated in Auts2 conditional mutant mice (described below) (Castanza et al., 2021). The hypothesis that AUTS2 may bind RNA directly is further supported by its high content of IDRs and LCSs, characteristic of RBPs (Castello et al., 2012; Hentze et al., 2018) (Figure 1). Furthermore, FBRSL1—the closest homolog (ohnolog) of AUTS2—has been identified as a poly(A)-RBP (Baltz et al., 2012). More studies of RNA regulation by AUTS2 are needed to determine if AUTS2 interacts directly with RNA, or only indirectly through protein complexes.
Cytoskeletal Dynamics
Outside the nucleus, AUTS2-FL was found to interact with guanine nucleotide exchange factors (GEFs) such as P-Rex1 and the Dock1/Elmo2 complex (Figure 3B) (Hori et al., 2014). These GEFs activate Rac1, a small G protein “molecular switch” that controls cytoskeletal organization. By interacting with GEFs, AUTS2 was proposed to enhance Rac1 activation and thus control neurite outgrowth, cell migration, and the formation of lamellipodia and filopodia (Hori et al., 2014). For future studies, it would be useful to see if these AUTS2-GEF interactions, found in HEK cells after overexpression, also occur under physiologic conditions in neurons in vivo.
Inhibition of BMP Signaling to Promote Neuronal Differentiation
In HEK cells and in mESCs differentiated to radial glia-like neuronal progenitor cells, both AUTS2-FL and AUTS2-C interacted with WDR68 and SKI to form a novel AUTS2-WDR68-SKI (AWS) complex (Geng et al., 2021). The AWS complex recruited CUL4, a ubiquitin E3 ligase, to mediate proteolytic degradation of phosphorylated SMAD1/5/9, and thereby inhibit BMP pathway signaling to promote neuronal differentiation (Geng et al., 2021). Further studies of this proposed mechanism are needed to evaluate its relevance to cortical neuron differentiation in vivo, since the aforementioned studies were done in cultured HEK cells or mESCs, after overexpression of AUTS2 or proposed interacting molecules.
Neurodevelopmental Functions of AUTS2 in Animal Model Systems
Zebrafish
The first animal studies of AUTS2 neurodevelopmental functions were conducted in zebrafish, using splice-blocking and translation-blocking morpholinos to knock down auts2a (Beunders et al., 2013; Oksenberg et al., 2013). In auts2a morphants, AUTS2 deficiency resulted in microcephaly, decreased neurogenesis, and other growth defects (Table 2). These phenotypes were rescued by expression of either AUTS2-FL or AUTS2-C, indicating that important AUTS2 functions are retained in the C-terminal region (Beunders et al., 2013). Interestingly, one study in zebrafish observed micrognathia/retrognathia in auts2a morphants, replicating a phenotype observed in humans (Beunders et al., 2013). Jaw growth may potentially be a conserved function of AUTS2, related to the evolutionary amplification of AUTS2 ohnologs in gnathostomes (Sellers et al., 2020). However, since zebrafish lack laminated cerebral cortex, and morphants often exhibit off-target effects (Vogan, 2015), the utility of zebrafish for studying human brain development is very limited. Moreover, zebrafish have a second gene, auts2b, also expressed in developing brain (Kondrychyn et al., 2017).
Table 2. Animal models of AUTS2 syndrome.
Mice
Several mouse models of AUTS2 deficiency have been produced by gene targeting of different Auts2 exons (Table 2). The structure of the Auts2 gene in mice (1.1 Mb, chromosome 5) is similar as in humans, and likewise comprises 19 exons (Castanza et al., 2021). The alternative TSS in exon 9 is active in mice and produces a C-terminal AUTS2 isoform similar to that in humans (Hori et al., 2014). In addition, mice have another alternative TSS in exon 7, which uses a translational start site in exon 8 and produces a slightly longer C-terminal isoform (Hori et al., 2014). Analyses of protein and RNA indicate that AUTS2-FL and AUTS2-C isoforms are both expressed in developing mouse brain, with AUTS2-C isoforms predominating in embryonic cerebral cortex, and AUTS2-FL in cerebellum (Gao et al., 2014; Hori et al., 2014; Molyneaux et al., 2015; Castanza et al., 2021).
In mice lacking Auts2 exon 7 (Auts2del7/del7) in the nervous system (Nes-Cre), mutants had normal birth weight but grew more slowly than controls postnatally, and exhibited behavioral abnormalities such as decreased ultrasonic vocalizations (Table 2) (Gao et al., 2014). No structural brain defects were reported, and only one gene (Dynll1) was significantly dysregulated among a panel of 9 candidate Auts2 target genes assayed by RT-PCR. Importantly, excision of exon 7 did not disrupt the exon 9 TSS, and AUTS2-C was presumably still expressed, although this was not tested (Gao et al., 2014). Thus, this model likely caused only partial loss of AUTS2 function, related to the full-length isoform.
In another mouse model, mutants lacking Auts2 exon 8 (Auts2del8/del8) throughout the organism died in the neonatal period (Table 2) (Hori et al., 2014). The neonatal brains showed no abnormalities by macroscopic examination or histology. In this model, the alternative TSS in exon 9 was again undisturbed, and AUTS2-C protein was actually increased in the cerebral cortex of Auts2del8/del8 mice, possibly as a compensatory response to AUTS2-FL depletion (Hori et al., 2014). Conditional excision of exon 8 in cerebral cortex pyramidal neurons during development (Emx1-Cre) or adulthood (CaMKIIa-CreERT2) permitted longer postnatal survival and additional studies (Hori et al., 2020). In these models, increased numbers of dendritic spines were observed on pyramidal neurons in the hippocampus (CA1) and neocortical layers 2–3 of mutant mice, along with increased numbers of miniature excitatory postsynaptic currents (mEPSCs), as well as behavioral abnormalities (Hori et al., 2020). Conditional deletion of Auts2 exon 8 in the developing cerebellum and caudal midbrain (En1-Cre) caused cerebellar hypoplasia, with defects of Purkinje cell maturation and synaptogenesis, plus behavioral abnormalities (Yamashiro et al., 2020). The latter results accord with previous evidence that AUTS2-FL is the main isoform in developing cerebellum (Castanza et al., 2021). Importantly, the cerebellar hypoplasia in mice (Yamashiro et al., 2020) recapitulates the cerebellar hypoplasia seen in some AUTS2 syndrome patients with missense or microdeletion variants in exon 9 (Liu et al., 2021; Fair et al., 2022).
In the course of producing Auts2del8, an unexpected loss-of-function allele (Auts2neo) that reduces both AUTS2-FL and AUTS2-C was also produced (Table 2) (Hori et al., 2014). Like Auts2del8/del8, the Auts2neo/neo genotype caused neonatal lethality. Heterozygous Auts2neo/+ mice, reported to have ∼50% reduction of AUTS2-FL and AUTS2-C isoforms, showed behavioral abnormalities (Hori et al., 2015). Since human AUTS2 mutations are heterozygous (Beunders et al., 2013; Sanchez-Jimeno et al., 2021), Auts2neo/+ mice model the human AUTS2 syndrome in this regard.
Recently, a conditional exon 15 allele (Auts2del15) was produced to interfere with expression of both AUTS2-FL and AUTS2-C isoforms (Table 2) (Castanza et al., 2021). Mice lacking exon 15 (Auts2del15/del15) throughout the CNS (Nes-Cre) died neonatally with severe breathing abnormalities, documented by plethysmography. The erratic breathing implicated abnormal development of brainstem respiratory centers and raised the possibility that AUTS2 might be a vulnerability gene for sudden infant death syndrome. Excision of exon 15 only in the developing cortex (Emx1-Cre) allowed for survival to adulthood, and revealed defects of cortical structure, function, and gene expression (Castanza et al., 2021). Notably, the dentate gyrus (DG) of hippocampus was hypoplastic, with virtual absence of hilar mossy neurons and reduced numbers of granule neurons. Also, EEG recordings showed abnormal spiking activity. While the neocortex appeared structurally intact, genes involved in neocortical patterning (such as Tshz2) and laminar identity (such as Wnt7b and Pcdh20) were significantly dysregulated, as determined by RNA-seq. Significantly, many RNA transcripts that interacted with AUTS2 in normal neocortex (by RIP-seq) were also dysregulated in Auts2del15/del15 mutant neocortex (by RNA-seq). Indeed, mRNA dysregulation correlated more strongly with binding of the RNA to AUTS2 protein (RIP-seq), than with binding of AUTS2 to cognate genes in chromatin (ChIP-seq). These distinctions support the hypothesis that AUTS2 regulates gene expression mainly by modulating RNA metabolism.
Interestingly, the transcriptome analysis of Auts2del15/del15 neocortex (Castanza et al., 2021) revealed dysregulation of mRNAs for proteins that were previously reported to interact with AUTS2 protein. Specifically, transcripts Ep300 and Prex1 not only bound AUTS2, but also were significantly decreased in Auts2del15/del15 neocortex. Ep300 and Prex1 encode P300 and P-Rex1, respectively, both of which reportedly interacted with AUTS2 upon overexpression in HEK cells (Gao et al., 2014; Hori et al., 2014). These findings suggest that overexpression of AUTS2 might induce elevated levels of Ep300 and Prex1 mRNA, and consequently, likewise elevated levels of P300 and P-Rex1 proteins. Overexpression of P300 and P-Rex1 with AUTS2 might lead to non-physiologic interactions in HEK cells, which are extremely different from neurons in cerebral cortex. Similarly, the activation of Rac1 observed after AUTS2 overexpression in N1E-115 mouse neuroblastoma cells (the “AUTS2-Rac1 pathway”) may also be explained, at least in part, by AUTS2 regulation of transcripts Prex1 and Rasgrf2, which were significantly reduced in Auts2del15/del15 neocortex. Both Prex1 and Rasgrf2 encode GEFs that activate Rac1 (Yoshizawa et al., 2005; Li et al., 2006). Thus, AUTS2 overexpression in HEK cells might indirectly induce elevated levels of GEFs via RNA regulation, and thereby activate Rac1.
Finally, a novel mouse Auts2 allele with conditional excision of exon 9 (Auts2del9) was recently described (Geng et al., 2021). Excision of exon 9 was reported to deplete both AUTS2-FL and AUTS2-C, although no details of the generation of these mice, nor documentation of the protein depletion, were provided. Homozygous excision of exon 9 (Auts2del9/del9) in the central nervous system (Nes-Cre) was reported to cause early embryonic lethality. Heterozygous exon 9 excision was found to impair neuronal gene expression, as determined by RT-PCR of cultured neocortical cells (Geng et al., 2021). No brain abnormalities were reported. Further studies are needed to validate this model.
Mouse Embryonic Stem Cell Models
The question of whether AUTS2 regulates neurogenesis has also been studied using mouse embryonic stem cells (mESCs) treated with in vitro differentiation protocols. One group reported that AUTS2 fosters the differentiation of mESCs to motor neurons (MNs) (Liu et al., 2021). Interestingly, a reduction of spinal MNs was reported in auts2a morphant zebrafish (Oksenberg et al., 2013); however, no MN defects have been reported in human AUTS2 patients or mouse models. In another study, AUTS2 was found necessary for the differentiation of mESCs to mixed neuron types (Russo et al., 2018). For cortical-type neurons, AUTS2 facilitated the differentiation of mESCs to cortical neurons in one study (Geng et al., 2021), but delayed mESC differentiation to cortical neurons in another (Monderer-Rothkoff et al., 2021). WDR68, implicated as an essential binding partner for transcriptional activation by AUTS2, also appeared necessary for the differentiation of mESCs to cortical neurons (Wang et al., 2018). While defects of neurogenesis presumably cause microcephaly in human AUTS2 syndrome, mESCs have so far shown variable results and limited utility for understanding AUTS2 syndrome phenotypes.
Cerebral Organoids as a Model to Investigate Molecular Mechanisms Underlying AUTS2 Syndrome
Mouse models of AUTS2 deficiency have furthered our understanding of AUTS2 functions during neurodevelopment. However, the role of AUTS2 during human cortical development, which involves a greater expansion of radial glia cell and neuronal diversity compared to mice, has not been investigated until recently with cerebral organoids (COs) (Fair et al., 2022). COs, derived from human pluripotent stem cells, are three-dimensional in vitro models that can be utilized to investigate the complex and human-specific features of early brain development (Lancaster et al., 2013; Di Lullo and Kriegstein, 2017; Pasca, 2018; Pollen et al., 2019; Qian et al., 2019; Setia and Muotri, 2019). As COs have been extensively reviewed elsewhere, we will focus on how COs can be used to investigate the mechanisms of aberrant human corticogenesis underlying various neurodevelopmental disorders (NDs), emphasizing studies relevant to AUTS2 syndrome.
Recently, the first COs derived from an AUTS2 syndrome patient, with a pathogenic missense variant in exon 9, were generated and studied (Fair et al., 2022). Patient COs displayed reduced growth analogous to microcephaly, deficits in neural progenitor cell (NPC) proliferation, and abnormal neuronal differentiation, which were rescued by CRISPR-Cas9 gene editing of the variant to the wild-type allele (Figure 4). These data highlight essential roles for AUTS2 during early human cortical development as well as identified proliferative deficits and reduced WNT-β-Catenin signaling in NPCs, which may underlie microcephaly in AUTS2 syndrome. Gene expression signatures of defective neuritogenesis were also observed, similar findings which are consistent with previous mouse studies. Future investigation is necessary to understand how AUTS2 functions in various progenitor cells (e.g., apical, intermediate, and basal), how it regulates neuronal differentiation, and how it controls the formation of neuronal circuits. The development and comparison of additional brain region-specific COs coupled with advanced technological tools to investigate various patient AUTS2 variants can start to uncover neurobiological functions that contribute to the clinical heterogeneity observed in AUTS2 syndrome patients (Figure 4).
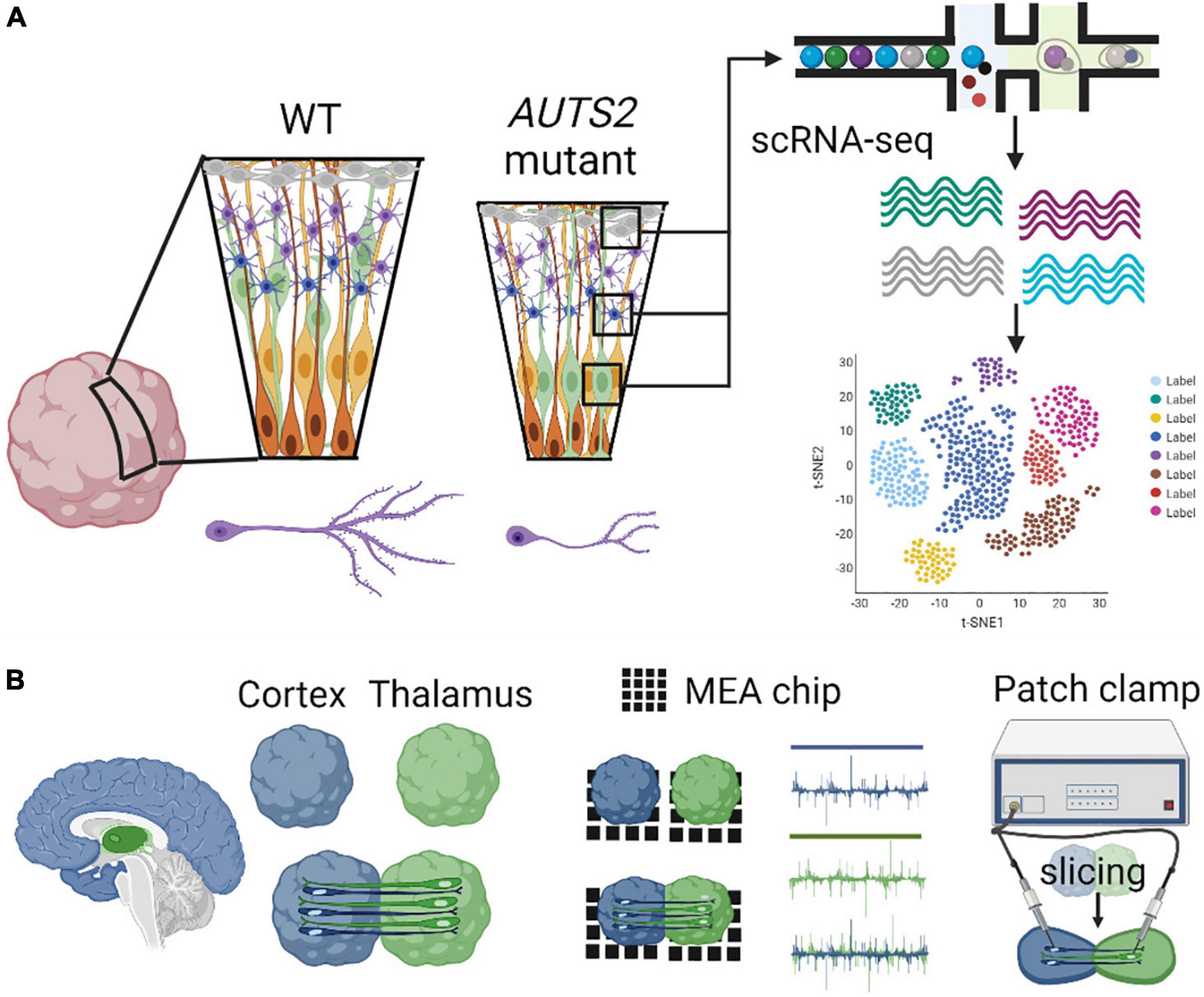
Figure 4. Modeling AUTS2 syndrome using cerebral organoids. (A) AUTS2 mutant cerebral organoids have proliferative deficits in neural progenitor cells and impaired neuritogenesis in cortical neurons (left). Utilization of scRNA-seq to determine cell type specific changes in gene expression within diverse progenitor, neuronal, and glial cell populations (right). (B) Generation of assembloids through the fusion of brain region specific organoids (cortical-thalamic assembloid shown, left) investigating neural circuitry using MEA and patch-clamp electrophysiology (right).
Investigating Mechanisms Underlying Aberrant Corticogenesis in AUTS2 Deficient Cerebral Organoids
Disruptions in the AUTS2 gene are associated with microcephaly in human patients (Beunders et al., 2013; Sanchez-Jimeno et al., 2021). However, current mouse models of Auts2 deficiency have not been able to recapitulate a microcephalic phenotype (Gao et al., 2014; Hori et al., 2014; Castanza et al., 2021). Understanding the etiology of microcephaly in AUTS2 and other neurodevelopmental disorders has been limited by the primary use of mouse models which do not adequately recapitulate the manner and severity in which microcephaly arises (Gabriel et al., 2020; Nieto-Estevez and Hsieh, 2020). A major developmental difference occurs during neurogenesis between humans and mice, particularly in the generation of the subventricular zone (SVZ). Although both humans and rodents undergo extensive SVZ growth, humans have a more complex, organized, and expansive SVZ subdivided into the outer and inner SVZ (Borrell and Gotz, 2014; Florio and Huttner, 2014; Florio et al., 2018). This compartmentalization allows for a more heterogeneous population of neural precursor cells with dynamic proliferative capabilities (such as cell cycle length and mode of division). Subsequently, the cortical plate is massively expanded in humans and becomes highly folded (undergoes gyrification) whereas the mouse brain is smooth (lissencephalic) (Kelava and Lancaster, 2016; Gabriel et al., 2020). Thus, major variations exist between human and rodent brain development resulting in dramatic differences in brain composition, size, and complexity.
As a complementary system to mouse models, COs can be used to dissect the underlying mechanisms of microcephaly while also helping to elucidate the fundamental mechanism of normal human brain development (Gopalakrishnan, 2019; Setia and Muotri, 2019; Gabriel et al., 2020). Microcephaly is thought to arise from a common disease mechanism ultimately owing to dysregulation of the cell cycle that disrupts the timing of this carefully orchestrated neurogenesis (Jean et al., 2020). It is hypothesized that human brain development occurs in distinct stages of cell proliferation and subsequent differentiation, whereas mice may undergo both proliferation and differentiation simultaneously (Geschwind and Rakic, 2013; Florio and Huttner, 2014; Gabriel et al., 2020). Furthermore, mouse models of candidate microcephaly genes typically require homozygous inactivation of the causal gene or significant mutant gene overexpression in order to induce a phenotype. However, primary microcephaly in humans is caused predominately by gene point mutations and truncations (Barrera et al., 2010; Buchman et al., 2010; Lizarraga et al., 2010; Alkuraya et al., 2011; McIntyre et al., 2012; Insolera et al., 2014). For example, mouse models of microcephaly associated CDK5RAP2 disease requires a complete knockout of CDK5RAP2 and result in apoptosis as the primary cause of neuronal progenitor cell depletion and microcephaly (Barrera et al., 2010; Buchman et al., 2010; Lizarraga et al., 2010). Conversely, a patient-derived CO model containing a heterozygous truncating mutation in the CDK5RAP2 gene recapitulated severe microcephaly and resulted in fewer neurons and smaller progenitor zones (Lancaster et al., 2013). Further investigation of radial glial spindle orientations revealed that patient COs displayed a greater percentage of oblique and vertical divisions compared to controls. This observation in patient COs correlated with premature neural differentiation, which was further supported by increased BrdU+/doublecortin positive cells.
Similarly, a recent study modeling a pathogenic AUTS2 missense variant in COs revealed significantly reduced organoid growth (Fair et al., 2022). AUTS2 mutant COs showed dysregulated cell cycle control and reduced symmetrical (horizontal) cellular division, which correlated with premature neuronal differentiation in comparison to control COs. Increased asymmetrical progenitor divisions and premature neuronal differentiation are also common features observed in other CO models containing mutant microcephaly genes (Gabriel et al., 2020). These data support the role of AUTS2 in regulating the highly intricate transcriptional program of neuronal differentiation and perturbations in this process could underlie cognitive deficits in patients.
Although mice do not have an OSVZ, both OSVZ formation and DG morphogenesis involve extensive cell migration and proliferation (Molnár et al., 2019; Nelson et al., 2020). Castanza et al. (2021) used a conditional knock-out model to inactivate Auts2 specifically in the cerebral cortex and found that the dentate gyrus had a reduced size that was associated with decreased neurogenesis, a common feature of AUTS2-associated microcephaly. It is possible that similar molecular mechanisms that perturbs DG neurogenesis in AUTS2 deficient mice, may contribute to neocortical growth defects in human AUTS2 syndrome patients.
Genomics Technologies to Study Molecular Features of AUTS2 Deficiency in Cerebral Organoids
Single cell (sc) RNA-seq, combined with immunohistochemical, spatial transcriptomics, and chromatin immunoprecipitation techniques can provide important morphological context to reconstruct the organization of disease-related expression patterns (Rao et al., 2021). Early rosette structures in COs represent ventricular zone (VZ)-like structures that are comprised of stratified progenitors, which undergo distinct stromal translocations critical to their progression through the cell cycle. Apical neural progenitors divide symmetrically within VZ-like structures in COs, and at the onset of neurogenesis, they shift to asymmetric divisions forming another apical neural progenitor and either a neuron or an intermediate progenitor (IP) cell, which is a type of basal neural progenitor. Although apical neural progenitors can directly give rise to neurons, most neurogenesis in the human neocortex occurs indirectly through subsequent IP division and neural differentiation (Pebworth et al., 2021). In contrast to rodents, humans and larger mammals contain an abundant type of basal progenitor called outer radial glial (ORG) cells, which also give rise to IP cells and are formed in the OSVZ. COs are invaluable models of human brain development since they contain an outer subventricular zone (oSVZ), which are abundant in ORG (Kadoshima et al., 2013; Lancaster et al., 2013) (Figure 4).
Recent work identified altered morphological and functional properties of apical neural progenitors in AUTS2 deficient COs (Fair et al., 2022). Further investigation of AUTS2 function using immunofluorescence techniques and sc-RNA seq will provide deeper understanding for the role of AUTS in IP and ORG populations in human corticogenesis and in AUTS2 syndrome. Correlating spatial progenitor zone context with cell type specific transcriptomic signatures may provide further mechanistic insight into whether downstream targets of AUTS2 are affected in specific progenitor zone populations in COs. Additionally, combining sc and epigenetic sequencing methods may also provide critical insights into altered regions of chromatin accessibility leading to deficits in transcriptional control caused by AUTS2 deficiency.
Although ChIP-seq is a widely used method to evaluate global active and repressed chromatin states, it is limited by low signal-to-noise ratios and is not compatible with low cellular input–a potentially significant challenge for organoid applications (Lewis et al., 2021). However, newer chromatin mapping methods such as CUT&RUN and CUT&Tag can be combined with scRNA-seq to target and profile specific chromatin signatures with single cell resolution (Kaya-Okur et al., 2019; Yu et al., 2022). Single-cell chromatin profiling of AUTS2 syndrome organoid models could extend our understanding for the role of the PRC1-AUTS2 complex in epigenetically heterogeneous cell populations. Such studies could also be valuable to understand the function of AUTS2 variants in regard to cell-type specific regulatory elements, especially those related to cell cycle control, cell fate inheritance, and timing of neuronal differentiation.
Recently, sc-RNA seq and ATAC-seq analyses uncovered a dynamic period of chromatin remodeling during the development of human forebrain organoids (Trevino et al., 2020). Dynamic epigenetic changes were identified within these organoids during human cortical neurogenesis, driven by specific transcription factors (particularly those associated with astrocyte maturation and interneuron specification). Furthermore, direct comparisons with human tissue confirmed that in vivo forebrain regulatory programs largely map with those seen in forebrain organoids cultured for over 20 months. Interestingly, during forebrain organoid development, 81% of Simons Foundation Autism Research Initiative (SFARI) genes were found to be expressed (Trevino et al., 2020). Further analysis identified SFARI genes as significantly enriched for enhancer-gene linkages predominantly in glial progenitor and mature neural cell types. These data provide a foundation for future investigation of potential disease mechanisms in ASD and hold implications for understanding molecular and cellular mechanisms underlying AUTS2 syndrome.
Electrophysiological Tools to Investigate the Role of AUTS2 in Synaptic Transmission
Several studies have shown that loss of Auts2 results in dysregulated regional and laminar neuronal differentiation in mouse models (Hori et al., 2020; Castanza et al., 2021; Monderer-Rothkoff et al., 2021) and more recently in COs (Fair et al., 2022). However, the neurodevelopmental impact of altered differentiation on synaptic physiology and how loss of AUTS2 leads to epilepsy in AUTS2 syndrome remains unclear. One study showed that the elimination of dendritic spines was impaired in Auts2-knocked-down hippocampal neurons (Hori et al., 2020). As a result, an excess of dendritic spines was observed and was suggested to drive the increase of excitatory inputs within Auts2 mutant hippocampal slices as demonstrated through electrophysiological experiments (Hori et al., 2020). Further studies revealed alterations in synaptic gene regulation in both Auts2 mutant mouse hippocampus and frontal cortex (Hori et al., 2020; Castanza et al., 2021), and in a CO model containing an AUTS2 pathogenic variant (Fair et al., 2022). All of these studies suggest AUTS2 functions at excitatory synapses, however, the mechanisms of AUTS2 in establishing synaptic maturation and regulating synaptic homeostasis during neurodevelopment remains largely unknown.
To investigate synaptic functions in vitro, conventional patch-clamp recording techniques remain the gold standard for providing high temporal resolution of electrical activities in neurons. Whole-cell voltage-clamp recordings on acute hippocampal slices from conditional AUTS2 deficient (Auts2del8/del8; Emx1-Cre) mice (P33–P44) revealed an increased frequency of miniature excitatory postsynaptic currents (mEPSCs) with no change in amplitude, suggesting an increase of functional excitatory synapses (Table 2). Further investigations at earlier developmental time points and across different regions in Auts2 deficient brains would provide a more complete understanding of Auts2 function in synaptic transmission.
Although patch-clamp techniques offer multiple advantages to probe synaptic function in neuronal cultures, its limited throughput nature does not allow for probing neuronal connectivity and investigating neural network dynamics. To address this limitation, multi-electrode array (MEA) platforms have been extensively utilized to record extracellular field potentials from a diverse and large number of neurons in vitro to investigate neural network properties. The application of MEAs in studying the effects of AUTS2 deficiency on neuronal network dynamics has not been explored, and thus would provide further mechanistic understanding of AUTS2 function in synaptic transmission (Figure 4).
Several studies have investigated electrical properties and neuronal connectivity within CO models (Quadrato et al., 2017; Trujillo et al., 2019; Fair et al., 2020). MEAs provide the unique advantage of non-invasively measuring electrical activity of neuronal networks within developing COs, although their utilization to study mechanisms underlie ASD is underexplored. Thus, investigation of spontaneous electrical activities and potential alterations in neural network properties in AUTS2 deficient COs would lead to greater understanding of mechanisms underlying epilepsy in AUTS2 syndrome.
Generation of Brain Region-Specific Cerebral Organoids and Assembloids to Understand the Role of AUTS2 During Neurodevelopment
Animal studies have shown that Auts2 is dynamically regulated during brain development and is expressed from a gradient in a high rostral to low caudal pattern within the developing neocortex (Bedogni et al., 2010). As previously described, two main AUTS2 isoforms are differentially regulated during brain development, although the regulatory mechanisms and functions of these isoforms are not well established (Table 1). Owing to this complex and heterogenous expression profile, it is important to understand the precise role of these isoforms within different brain regions throughout development. Numerous protocols exist to generate either brain region specific or whole brain organoids. In this section, we will discuss application of brain region specific organoid and assembloid models that can be leveraged to investigate Auts2 function during neurodevelopment.
Postnatal AUTS2 expression includes high expression levels frontal cortex, cerebellum, and hippocampus (Figure 2). However, it is unclear whether postnatal AUTS2 expression is essential to maintain proper function within these brain regions (Table 1). One hypothesis is that AUTS2 deficiency within the developing hippocampus may underlie ID in AUTS2 syndrome patients. Hippocampal organoids serve as in vitro models for investigating human hippocampus development and hippocampus-related diseases and consist of hippocampal granule- and pyramidal-like neurons that form a functional electrical network (Sakaguchi et al., 2015; Pomeshchik et al., 2020). Recently used to model Alzheimer’s disease (Sakaguchi et al., 2015; Pomeshchik et al., 2020), hippocampal organoids can also be applied to understand the role of AUTS2 during hippocampus development. As previously described, Castanza et al. (2021) showed dentate gyrus hypoplasia in Emx1-Cre Auts2del15/del15 mice. Utilization of hippocampal organoids can yield important mechanistic insights into the role of AUTS2 in progenitor proliferation, migration, and differentiation within human hippocampal development.
Cerebellar organoids can also be leveraged to understand the role of AUTS2 during cerebellar development. Mouse models of AUTS2 deficiency show cerebellar hypoplasia, impaired dendrite maturation in Purkinje cells, increased parallel fiber synapse formation, and decreased number of excitatory presynaptic synapses from climbing fiber innervations (Yamashiro et al., 2020). Previous work utilizing a polarized cerebellar organoid model identified similarities in human cerebellar ontogenesis regarding the layered neural-tube-like structure with dorsoventral and apicobasal polarities (Muguruma et al., 2015; Silva et al., 2020; Nayler et al., 2021). Cerebellar organoids derived from AUTS2 syndrome patients could be generated to further understand multiple aspects of cerebellar development, such as cellular differentiation, gene expression changes within specific cell types (i.e., sc RNA-seq focusing on Purkinje cells, granular cells, etc.), and investigating E/I balance in the cerebellum utilizing MEA or patch clamp electrophysiology (Figure 4).
Though the development of in vitro tools to model and study long-range human brain circuits has remained a challenging endeavor, novel platforms are emerging based on the fusion of regionally-specified brain organoids called assembloids. For instance, the neural circuitry between the human thalamus and cerebral cortex can be mimicked and studied in a cortico-thalamic assembloid (Gobbo et al., 2021) (Figure 4). Assembloids are becoming increasingly important, serving as a functional tool to investigate the underlying causes of synaptic deficits underlying neurodevelopmental disorders. A variety of assembloids are being developed to investigate multi-synaptic circuitry in human models, such as cortico-thalamic, cortico-striatal, cortico-cortico assembloids (Xiang et al., 2019; Andersen et al., 2020; Chen et al., 2020; Marton and Pasca, 2020). Cortico-thalamic and cortico-striatal dysfunction has been associated with neurodevelopmental disorders and associated co-morbidities such as epilepsy (Shepherd, 2013; He et al., 2021). AUTS2 is highly expressed in the dorsal thalamus and in the striatum (Bedogni et al., 2010), to study neural circuitry between the frontal cortex and these brain regions. To investigate the role of AUTS2 within the prefrontal-thalamic circuit, an assembloid model could be recapitulated between forebrain and thalamic organoids, and several methodologies such as patch clamping or MEAs could be used to investigate alterations in synaptic transmission occurring in assembloid circuits (Figure 4).
The utilization of patient AUTS2 variant iPSCs to create assembloids and brain region specific organoids, such as hippocampal and cerebellar organoids, will provide invaluable tools to understand mechanisms underlying AUTS2 syndrome at multiple levels of analysis including molecular, cellular, and functional levels, as well as provide a novel screening platform for developing therapeutics.
Molecular Tools for Analyzing the Role of AUTS2 in Neural Circuit Development and Maintenance
Auts2 conditional knockout mouse models are invaluable for dissecting the circuit mechanisms that may contribute to phenotypes observed in AUTS2 syndrome. As noted above, mouse models for Auts2 deficiency do not demonstrate microcephaly (Castanza et al., 2021). However, loss of Auts2 causes changes in molecular expression that suggest disrupted neuronal differentiation, which should lead to altered circuit architecture (Castanza et al., 2021; Monderer-Rothkoff et al., 2021). Indeed, in some models, the density of dendritic spines on excitatory neocortical and hippocampal pyramidal cells is abnormally high, with a concomitant increase in the frequency of miniature excitatory postsynaptic currents (mEPSCs) (Hori et al., 2020). This suggests there may be an imbalance in the ratio of excitation to inhibition (E/I) within neural circuits of Auts2 mouse models. Congruent with this hypothesis, abnormal voltage spikes similar to epileptic interictal discharges occur spontaneously in the hippocampi of Auts2 cKO mice (Castanza et al., 2021). Disruptions in E/I balance are commonly observed across several animal models of neurodevelopmental disorders (Sohal and Rubenstein, 2019). However, it is important to determine how loss of Auts2 alters the developmental trajectory of specific neuronal subtypes and circuits, to guide precision therapeutic approaches.
In mouse neocortex, AUTS2 is largely restricted to excitatory projection neurons (Castanza et al., 2021). These cells can be parsed into three major subclasses based on their long-range axonal targets: (1) intratelencephalic (IT) that project across the corpus callosum, (2) pyramidal tract (PT) that project to subcortical structures, and (3) corticothalamic (CT) that project to thalamic nuclei (Harris and Shepherd, 2015). In mice, these three subclasses form unique local circuits among each other and with neighboring inhibitory interneurons (Lee S.H. et al., 2014; Harris and Shepherd, 2015; Wester et al., 2019). Thus, it is important to determine if specific subtypes of projection neurons are uniquely susceptible to loss of Auts2.
Based on molecular expression data, current evidence suggests AUTS2 may be involved in development of all 3 cortical projection neuron types. First, AUTS2 is strongly expressed in superficial cortical layers (Bedogni et al., 2010; Castanza et al., 2021), which are populated exclusively by IT-type cells. Second, AUTS2 is also expressed in layer 5, which contains PT-type neurons (Castanza et al., 2021). Third, Auts2 expression is activated by Tbr1 (Bedogni et al., 2010), which is a transcription factor that promotes the differentiation of IT-, CT-, and PT-type neurons (Hevner et al., 2002; Han et al., 2011; McKenna et al., 2011; Srinivasan et al., 2012). Fourth, AUTS2 can also be co-expressed with CTIP2 (Bedogni et al., 2010), which is a transcription factor prominently associated with PT-type cells. However, a subset of IT-type neurons does express CTIP2 (Harb et al., 2016), which may account for this observation.
Current tools in mice can directly test how loss of Auts2 differentially alters neocortical circuits involving IT-, CT-, and PT-type neurons. The Emx1-Cre mouse line expresses Cre recombinase in progenitors of neocortical and hippocampal projection neurons as early as embryonic day (E) 10.5 (Gorski et al., 2002) and is a powerful strategy to eliminate Auts2 expression from the forebrain when crossed to Auts2 floxed mice (Castanza et al., 2021). On this genetic background, IT-, CT-, and PT-type cells can be identified by injecting retrograde tracers or viral vectors into non-overlapping target structures. Using this strategy, it will be possible to determine which subtypes receive abnormally high excitatory synaptic input, and if it originates from neighboring projection neurons in local circuits or from long-range afferents outside of cortex, such as thalamus. Furthermore, Auts2 can be conditionally deleted from specific subclasses. For example, to target CT-type cells, the Ntsr1-Cre line can be used to express Cre in these neurons as early as embryonic day 16.5 (Gong et al., 2007). Superficial IT-type neurons can be targeted by in utero electroporation of a plasmid encoding Cre into Auts2 floxed mice after E14.5. Several tools exist to investigate if loss of Auts2 from projection neurons alters their synaptic connectivity with neighboring inhibitory interneurons. For example, conditional Auts2 knockout mice can be crossed to those expressing fluorescent reporters in specific interneuron subtypes (e.g., the PV-tdTomato mouse line (Kaiser et al., 2016). Alternatively, AAV vectors are available that encode fluorophores and channelrhodopsin (ChR2) under the control of an inhibitory interneuron-specific promoter [e.g., AAV-mDlx-NLS-mRuby (Chan et al., 2017)].
In the hippocampus, AUTS2 is expressed throughout the canonical tri-synaptic circuit that includes the entorhinal cortex, dentate gyrus, CA3, and CA1 (Bedogni et al., 2010; Castanza et al., 2021). Thus, loss of Auts2 may disrupt information processing at multiple stages within this structure. In the dentate gyrus, AUTS2 is expressed in two different populations of excitatory projection neurons: granule cells (GCs) and hilar mossy neurons (HMNs). Importantly, forebrain knockout of Auts2 leads to a significant reduction in the number of HMNs (Castanza et al., 2021). HMNs project within the dentate gyrus both ipsilaterally and contralaterally to regulate the output of GCs, which in turn target region CA3 (Scharfman and Myers, 2016; Danielson et al., 2017). Although HMNs provide excitatory synaptic input to CGs, their primary role may be to disynaptically inhibit GCs via their input to a diverse set of inhibitory interneurons (Jinde et al., 2012; Scharfman and Myers, 2016). Indeed, loss of HMNs is common in temporal lobe epilepsy (Bui et al., 2018) and may be responsible for the interictal-like events observed by Castanza et al. (2021). Several tools are available to study how loss of Auts2 alters circuits in the dentate gyrus, perhaps leading to disinhibition and hyperexcitability. HMNs can be selectively targeted for expression of fluorophores and ChR2 by injection of retrograde viral vectors into the contralateral dentate gyrus (Danielson et al., 2017). Furthermore, as described above, local inhibitory interneurons can also be selectively targeted using transgenic and viral strategies. Combining these tools should allow the investigation of synaptic connections among HMNs, GCs, and interneurons on the background of conditional forebrain knockout of Auts2. An alternative strategy to selectively target HMNs is to use Crlr-Cre or Drd2-Cre mouse lines (Gangarossa et al., 2012; Jinde et al., 2012), however, the temporal expression patterns of Cre are not well characterized and may not allow for conditional knockout of Auts2 during early developmental periods. Finally, loss of Auts2 may lead to hippocampal hyperexcitability via increases in excitatory synapse formation onto CA1 projection neurons (Hori et al., 2020). The laminar structure of the hippocampus should allow future studies to determine if increased excitatory drive to CA1 originates primarily from CA3 or extrahippocampal inputs (entorhinal cortex or thalamus). This can be accomplished by stimulating axonal projections traveling through either the stratum radiatum or the stratum lacunosum moleculare, respectively.
Finally, mouse models offer an opportunity to investigate if communication among different brain structures is affected by loss of Auts2. Indeed, several brain structures that prominently express AUTS2 directly target each other for synaptic connections. These include the prefrontal cortex (PFC), ventrolateral/ventromedial (VL/VM) thalamic nuclei, cerebellum, and hippocampus (Bedogni et al., 2010; Castanza et al., 2021). For example, deep cerebellar nuclei project to directly to VL/VM thalamus (Gornati et al., 2018), which in turn projects to the PFC (Collins et al., 2018). Furthermore, CT- and PT-type projection neurons in PFC provide feedback synaptic input to VM thalamus (Collins et al., 2018). Finally, excitatory hippocampal CA1 projection neurons directly target the PFC (Lee A.T. et al., 2014). An important open question is whether loss of Auts2 alters the afferent input strength of these connections. This can be addressed by injecting AAVs encoding ChR2 into each of these structures to assess synaptic connectivity and physiology via optogenetic assisted circuit mapping (Petreanu et al., 2007; Collins et al., 2018).
Future Directions and Key Questions
As this review has illustrated, multiple functions have been proposed for AUTS2 (Figure 3) on the basis of studies in multiple model systems (Table 2). Other members of the AUTS2 gene family (FBRSL1, FBRS) and superfamily (such as tay) likewise appear to have overlapping, but diversified functions. Importantly, many AUTS2 functions and interactions with other proteins remain to be confirmed, or have been studied in heterologous cell types (such as HEK cells). Considering these caveats, a paramount future goal will be to better define the functions and binding partners of AUTS2 in neurons from cerebral organoids. With several lines of evidence suggesting that AUTS2 is an RNA-binding protein, this possibility can be investigated using methods such as eCLIP. With the advent of methods to produce human COs and assembloids, future studies can go beyond animal models to evaluate AUTS2 molecular functions, binding partners, and neurodevelopmental roles in developing human neural tissues.
Author Contributions
RH and AB produced figures and supplementary figures. All authors wrote and edited the manuscript.
Funding
This work was supported by NINDS grants R01NS085081 and R01NS092339 (RH), by the Nationwide Foundation Pediatric Innovation Fund (MH), and by NIMH grant R01MH124870 and SFARI Pilot Award #724187 (JW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figure 4 contains schematics that were created with BioRender.com.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.858582/full#supplementary-material
Supplementary Figure 1 | FBRSL1 transcript and protein. (A) The full-length FBRSL1 transcript has 17 exons. (B) Protein source exons are indicated in black (odd) and blue (even). Green boxes enclose splice junctions in which amino acids were encoded across the junctions, comprising exon blocs. Red asterisks indicate source exons that encoded a non-integer number of amino acids. Features mapped from protein sequence: ExB, exon bloc; NLS, nuclear localization sequence; IDR, intrinsically disordered region; PRR, Pro-rich repeat; HRR, His-rich repeat; HX, HX repeat; RRR, Arg-rich repeat; CDD, conserved domain database; PD, ProDom predicted domains (domain names in bold also identified in AUTS2); AFsh, AUTS2-FBRSL1 short homology region. (C) FBRSL has a high content of IDRs (zigzag lines) and regions enriched in amino acids Pro, His, or Arg (red lines).
Supplementary Figure 2 | Predictive modeling of AUTS2 (1–1,199) secondary and tertiary structure. The AUTS2 protein sequence (aa 1–1,199) was modeled using RoseTTAfold. (The C-terminal aa 1,200–1,259 were omitted because only sequences up to 1,199 aa can be analyzed). Left panels show predicted structures, and right panels show error estimates for each aa position, for models 1–5. Overall confidence was low (0.19 on scale 0–1), most likely because no structures of similar sequences were available. All of the models show multiple long stretches of disordered secondary structure (most obvious in model 5), with intervening alpha helices. Models not shown to the same scale.
Supplementary Figure 3 | Predictive modeling of FBRSL1 secondary and tertiary structure. The complete FBRSL1 protein sequence (aa 1–1,045) was modeled using RoseTTAfold. Left panels show predicted structures, and right panels show error estimates for each aa position, for models 1–5. Overall confidence was low (0.17 on scale 0–1), most likely because no structures of similar sequences were available. All of the models show multiple long stretches of disordered secondary structure (most obvious in model 4), with intervening alpha helices. Models not shown to the same scale.
Supplementary Figure 4 | Predictive modeling of the AUTS2 domain of AUTS2 protein. The AUTS2 domain of AUTS2 protein (aa 645–844) was modeled using RoseTTAfold. Left panels show predicted structures, and right panels show error estimates for each aa position, for models 1–5. Overall confidence was low (0.45 on scale 0–1), most likely because no structures of similar sequences were available. All of the models show stretches of disordered secondary structure with intervening alpha helices. Models not shown to the same scale.
Supplementary Figure 5 | Predictive modeling of the AUTS2 domain of FBRSL1 protein. The AUTS2 domain of FBRSL1 protein (aa 587–784) was modeled using RoseTTAfold. Left panels show predicted structures, and right panels show error estimates for each aa position, for models 1–5. Overall confidence was low (0.42 on scale 0–1), most likely because no structures of similar sequences were available. All of the models show stretches of disordered secondary structure with intervening alpha helices. Models not shown to the same scale.
Supplementary Table 1 | AUTS2 cDNA and protein features. Human AUTS2 cDNA (CCDS5539.1) and AUTS2 protein (NP_056385.1) features.
Supplementary Table 2 | FBRSL1 cDNA and protein features. Human FBRSL1 cDNA (CCDS45010.1) and FBRSL1 protein (NP_001136113.1) features.
Supplementary Table 3 | Comparison of AUTS2 and FBRSL1 cDNA and protein features. Exon homologies, amino acid sequence homologies, and contents of IDRs and LCSs.
Footnotes
References
Alkuraya, F. S., Cai, X., Emery, C., Mochida, G. H., Al-Dosari, M. S., Felie, J. M., et al. (2011). Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am. J. Hum. Genet. 88, 536–547. doi: 10.1016/j.ajhg.2011.04.003
Andersen, J., Revah, O., Miura, Y., Thom, N., Amin, N. D., Kelley, K. W., et al. (2020). Generation of functional human 3D Cortico-motor assembloids. Cell 183, 1913–1929.e26. doi: 10.1016/j.cell.2020.11.017
Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876. doi: 10.1126/science.abj8754
Baltz, A. G., Munschauer, M., Schwanhausser, B., Vasile, A., Murakawa, Y., Schueler, M., et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 46, 674–690. doi: 10.1016/j.molcel.2012.05.021
Barrera, J. A., Kao, L. R., Hammer, R. E., Seemann, J., Fuchs, J. L., and Megraw, T. L. (2010). CDK5RAP2 regulates centriole engagement and cohesion in mice. Dev. Cell 18, 913–926. doi: 10.1016/j.devcel.2010.05.017
Bedogni, F., Hodge, R. D., Nelson, B. R., Frederick, E. A., Shiba, N., Daza, R. A., et al. (2010). Autism susceptibility candidate 2 (Auts2) encodes a nuclear protein expressed in developing brain regions implicated in autism neuropathology. Gene Expr. Patterns 10, 9–15. doi: 10.1016/j.gep.2009.11.005
Beunders, G., van de Kamp, J., Vasudevan, P., Morton, J., Smets, K., Kleefstra, T., et al. (2016). A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J. Med. Genet. 53, 523–532. doi: 10.1136/jmedgenet-2015-103601
Beunders, G., Voorhoeve, E., Golzio, C., Pardo, L. M., Rosenfeld, J. A., Talkowski, M. E., et al. (2013). Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 92, 210–220. doi: 10.1016/j.ajhg.2012.12.011
Borrell, V., and Gotz, M. (2014). Role of radial glial cells in cerebral cortex folding. Curr. Opin. Neurobiol. 27, 39–46. doi: 10.1016/j.conb.2014.02.007
Buchman, J. J., Tseng, H. C., Zhou, Y., Frank, C. L., Xie, Z., and Tsai, L. H. (2010). Cdk5rap2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron 66, 386–402. doi: 10.1016/j.neuron.2010.03.036
Bui, A. D., Nguyen, T. M., Limouse, C., Kim, H. K., Szabo, G. G., Felong, S., et al. (2018). Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science 359, 787–790. doi: 10.1126/science.aan4074
Castanza, A. S., Ramirez, S., Tripathi, P. P., Daza, R. A. M., Kalume, F. K., Ramirez, J. M., et al. (2021). AUTS2 regulates RNA metabolism and dentate gyrus development in mice. Cereb. Cortex 26, 4808–4824. doi: 10.1093/cercor/bhab124
Castello, A., Fischer, B., Eichelbaum, K., Horos, R., Beckmann, B. M., Strein, C., et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406. doi: 10.1016/j.cell.2012.04.031
Chan, K. Y., Jang, M. J., Yoo, B. B., Greenbaum, A., Ravi, N., Wu, W. L., et al. (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179. doi: 10.1038/nn.4593
Chen, A., Guo, Z., Fang, L., and Bian, S. (2020). Application of fused organoid models to study human brain development and neural disorders. Front. Cell. Neurosci. 14:133. doi: 10.3389/fncel.2020.00133
Collins, D. P., Anastasiades, P. G., Marlin, J. J., and Carter, A. G. (2018). Reciprocal circuits linking the prefrontal cortex with dorsal and ventral thalamic nuclei. Neuron 98, 366–379.e4. doi: 10.1016/j.neuron.2018.03.024
Danielson, N. B., Turi, G. F., Ladow, M., Chavlis, S., Petrantonakis, P. C., Poirazi, P., et al. (2017). In vivo imaging of dentate gyrus mossy cells in behaving mice. Neuron 93, 552–559.e4. doi: 10.1016/j.neuron.2016.12.019
Di Lullo, E., and Kriegstein, A. R. (2017). The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 18, 573–584. doi: 10.1038/nrn.2017.107
Fair, S. R., Julian, D., Hartlaub, A. M., Pusuluri, S. T., Malik, G., Summerfied, T. L., et al. (2020). Electrophysiological maturation of cerebral organoids correlates with dynamic morphological and cellular development. Stem Cell Rep. 15, 855–868. doi: 10.1016/j.stemcr.2020.08.017
Fair, S. R., Schwind, W., Julian, D., Biel, A., Ramadesikan, S., Westfall, J., et al. (2022). Cerebral organoids containing an AUTS2 missense variant model microcephaly. medRxiv [Preprint]. doi: 10.1101/2022.02.23.22271091
Fergelot, P., Van Belzen, M., Van Gils, J., Afenjar, A., Armour, C. M., Arveiler, B., et al. (2016). Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by EP300 mutations. Am. J. Med. Genet. A 170, 3069–3082. doi: 10.1002/ajmg.a.37940
Florio, M., Heide, M., Pinson, A., Brandl, H., Albert, M., Winkler, S., et al. (2018). Evolution and cell-type specificity of human-specific genes preferentially expressed in progenitors of fetal neocortex. eLife 7:e32332. doi: 10.7554/eLife.32332
Florio, M., and Huttner, W. B. (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141, 2182–2194. doi: 10.1242/dev.090571
Gabriel, E., Ramani, A., Altinisik, N., and Gopalakrishnan, J. (2020). Human brain organoids to decode mechanisms of microcephaly. Front. Cell. Neurosci. 14:115. doi: 10.3389/fncel.2020.00115
Gangarossa, G., Longueville, S., De Bundel, D., Perroy, J., Herve, D., Girault, J. A., et al. (2012). Characterization of dopamine D1 and D2 receptor-expressing neurons in the mouse hippocampus. Hippocampus 22, 2199–2207. doi: 10.1002/hipo.22044
Gao, Z., Lee, P., Stafford, J. M., von Schimmelmann, M., Schaefer, A., and Reinberg, D. (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354. doi: 10.1038/nature13921
Geng, Z., Wang, Q., Miao, W., Wolf, T., Chavez, J., Giddings, E., et al. (2021). AUTS2 controls neuronal lineage choice through a novel PRC1-independent complex and BMP inhibition. bioRxiv [Preprint]. doi: 10.1101/2021.06.29.450402
Geschwind, D. H., and Rakic, P. (2013). Cortical evolution: judge the brain by its cover. Neuron 80, 633–647. doi: 10.1016/j.neuron.2013.10.045
Gobbo, D., Scheller, A., and Kirchhoff, F. (2021). From physiology to pathology of cortico-thalamo-cortical oscillations: astroglia as a target for further research. Front. Neurol. 12:661408. doi: 10.3389/fneur.2021.661408
Gong, S., Doughty, M., Harbaugh, C. R., Cummins, A., Hatten, M. E., Heintz, N., et al. (2007). Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J. Neurosci. 27, 9817–9823. doi: 10.1523/JNEUROSCI.2707-07.2007
Gopalakrishnan, J. (2019). The emergence of stem cell-based brain organoids: trends and challenges. Bioessays 41:e1900011. doi: 10.1002/bies.201900011
Gornati, S. V., Schafer, C. B., Eelkman Rooda, O. H. J., Nigg, A. L., De Zeeuw, C. I., and Hoebeek, F. E. (2018). Differentiating cerebellar impact on thalamic nuclei. Cell Rep. 23, 2690–2704. doi: 10.1016/j.celrep.2018.04.098
Gorski, J. A., Talley, T., Qiu, M., Puelles, L., Rubenstein, J. L., and Jones, K. R. (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 22, 6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002
Han, W., Kwan, K. Y., Shim, S., Lam, M. M., Shin, Y., Xu, X., et al. (2011). TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc. Natl. Acad. Sci. U.S.A. 108, 3041–3046. doi: 10.1073/pnas.1016723108
Harb, K., Magrinelli, E., Nicolas, C. S., Lukianets, N., Frangeul, L., Pietri, M., et al. (2016). Area-specific development of distinct projection neuron subclasses is regulated by postnatal epigenetic modifications. eLife 5:e09531. doi: 10.7554/eLife.09531
Harris, K. D., and Shepherd, G. M. (2015). The neocortical circuit: themes and variations. Nat. Neurosci. 18, 170–181. doi: 10.1038/nn.3917
Hatstat, A. K., Ahrendt, H. D., Foster, M. W., Mayne, L., Moseley, M. A., Englander, S. W., et al. (2021). Characterization of small-molecule-induced changes in Parkinson’s-Related Trafficking via the Nedd4 Ubiquitin Signaling Cascade. Cell Chem. Biol. 28, 14–25.e9. doi: 10.1016/j.chembiol.2020.10.008
He, C., Cortes, J. M., Kang, X., Cao, J., Chen, H., Guo, X., et al. (2021). Individual-based morphological brain network organization and its association with autistic symptoms in young children with autism spectrum disorder. Hum. Brain Mapp. 42, 3282–3294. doi: 10.1002/hbm.25434
Hentze, M. W., Castello, A., Schwarzl, T., and Preiss, T. (2018). A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341. doi: 10.1038/nrm.2017.130
Hevner, R. F., Miyashita-Lin, E., and Rubenstein, J. L. (2002). Cortical and thalamic axon pathfinding defects in Tbr1, Gbx2, and Pax6 mutant mice: evidence that cortical and thalamic axons interact and guide each other. J. Comp. Neurol. 447, 8–17. doi: 10.1002/cne.10219
Hori, K., Nagai, T., Shan, W., Sakamoto, A., Abe, M., Yamazaki, M., et al. (2015). Heterozygous Disruption of Autism susceptibility candidate 2 Causes impaired emotional control and cognitive memory. PLoS One 10:e0145979. doi: 10.1371/journal.pone.0145979
Hori, K., Nagai, T., Shan, W., Sakamoto, A., Taya, S., Hashimoto, R., et al. (2014). Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 9, 2166–2179. doi: 10.1016/j.celrep.2014.11.045
Hori, K., Yamashiro, K., Nagai, T., Shan, W., Egusa, S. F., Shimaoka, K., et al. (2020). AUTS2 regulation of synapses for proper synaptic inputs and social communication. iScience 23, 101183. doi: 10.1016/j.isci.2020.101183
Insolera, R., Bazzi, H., Shao, W., Anderson, K. V., and Shi, S. H. (2014). Cortical neurogenesis in the absence of centrioles. Nat. Neurosci. 17, 1528–1535. doi: 10.1038/nn.3831
Jean, F., Stuart, A., and Tarailo-Graovac, M. (2020). Dissecting the genetic and etiological causes of primary microcephaly. Front. Neurol. 11:570830. doi: 10.3389/fneur.2020.570830
Jinde, S., Zsiros, V., Jiang, Z., Nakao, K., Pickel, J., Kohno, K., et al. (2012). Hilar mossy cell degeneration causes transient dentate granule cell hyperexcitability and impaired pattern separation. Neuron 76, 1189–1200. doi: 10.1016/j.neuron.2012.10.036
Kadoshima, T., Sakaguchi, H., Nakano, T., Soen, M., Ando, S., Eiraku, M., et al. (2013). Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. U.S.A. 110, 20284–20289. doi: 10.1073/pnas.1315710110
Kaiser, T., Ting, J. T., Monteiro, P., and Feng, G. (2016). Transgenic labeling of parvalbumin-expressing neurons with tdTomato. Neuroscience 321, 236–245. doi: 10.1016/j.neuroscience.2015.08.036
Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., et al. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10:1930. doi: 10.1038/s41467-019-09982-5
Kelava, I., and Lancaster, M. A. (2016). Stem cell models of human brain development. Cell Stem Cell 18, 736–748. doi: 10.1016/j.stem.2016.05.022
Kondrychyn, I., Robra, L., and Thirumalai, V. (2017). Transcriptional complexity and distinct expression patterns of auts2 Paralogs in Danio rerio. G3 2577–2593. doi: 10.1534/g3.117.042622
Lancaster, M. A., Renner, M., Martin, C. A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. doi: 10.1038/nature12517
Lee, S. H., Kwan, A. C., and Dan, Y. (2014). Interneuron subtypes and orientation tuning. Nature 508, E1–E2. doi: 10.1038/nature13128
Lee, A. T., Vogt, D., Rubenstein, J. L., and Sohal, V. S. (2014). A class of GABAergic neurons in the prefrontal cortex sends long-range projections to the nucleus accumbens and elicits acute avoidance behavior. J. Neurosci. 34, 11519–11525. doi: 10.1523/JNEUROSCI.1157-14.2014
Lewis, E., Kaushik, K., Sandoval, L., Antony, I., Dietmann, S., and Kroll, K. (2021). Epigenetic regulation during human cortical development: seq-ing answers from the brain to the organoid. Neurochem. Int. 147:105039. doi: 10.1016/j.neuint.2021.105039
Li, S., Tian, X., Hartley, D. M., and Feig, L. A. (2006). Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J. Neurosci. 26, 1721–1729. doi: 10.1523/JNEUROSCI.3990-05.2006
Lin, Z., Yang, Z., Xie, R., Ji, Z., Guan, K., and Zhang, M. (2019). Decoding WW domain tandem-mediated target recognitions in tissue growth and cell polarity. eLife 8:e49439. doi: 10.7554/eLife.49439
Liu, S., Aldinger, K. A., Cheng, C. V., Kiyama, T., Dave, M., McNamara, H. K., et al. (2021). NRF1 association with AUTS2-Polycomb mediates specific gene activation in the brain. Mol. Cell 81:4757. doi: 10.1016/j.molcel.2021.10.023
Lizarraga, S. B., Margossian, S. P., Harris, M. H., Campagna, D. R., Han, A. P., Blevins, S., et al. (2010). Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137, 1907–1917. doi: 10.1242/dev.040410
Martinez-Delgado, B., Lopez-Martin, E., Lara-Herguedas, J., Monzon, S., Cuesta, I., Juliá, M., et al. (2021). De novo small deletion affecting transcription start site of short isoform of AUTS2 gene in a patient with syndromic neurodevelopmental defects. Am. J. Med. Genet. A 185, 877–883. doi: 10.1002/ajmg.a.62017
Marton, R. M., and Pasca, S. P. (2020). Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol. 30, 133–143. doi: 10.1016/j.tcb.2019.11.004
McIntyre, R. E., Lakshminarasimhan Chavali, P., Ismail, O., Carragher, D. M., Sanchez-Andrade, G., Forment, J. V., et al. (2012). Disruption of mouse Cenpj, a regulator of centriole biogenesis, phenocopies Seckel syndrome. PLoS Genet. 8:e1003022. doi: 10.1371/journal.pgen.1003022
McKenna, W. L., Betancourt, J., Larkin, K. A., Abrams, B., Guo, C., Rubenstein, J. L., et al. (2011). Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. J. Neurosci. 31, 549–564. doi: 10.1523/JNEUROSCI.4131-10.2011
Molnar, C., and de Celis, J. F. (2013). Tay bridge is a negative regulator of EGFR signalling and interacts with Erk and Mkp3 in the Drosophila melanogaster wing. PLoS Genet. 9:e1003982. doi: 10.1371/journal.pgen.1003982
Molnar, C., Estrada, B., and de Celis, J. F. (2018). Tay bridge and extracellular-regulated kinase activity are required for motoneuron function in the Drosophila neural system. Genes Brain Behav. 17:e12470. doi: 10.1111/gbb.12470
Molnár, Z., Clowry, G. J., Sestan, N., Alzu’bi, A., Bakken, T., Hevner, R. F., et al. (2019). New insights into the development of the human cerebral cortex. J. Anat. 235, 432–451. doi: 10.1111/joa.13055
Molyneaux, B. J., Goff, L. A., Brettler, A. C., Chen, H. H., Hrvatin, S., Rinn, J. L., et al. (2015). DeCoN: genome-wide analysis of in vivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron 85, 275–288. doi: 10.1016/j.neuron.2014.12.024
Monderer-Rothkoff, G., Tal, N., Risman, M., Shani, O., Nissim-Rafinia, M., Malki-Feldman, L., et al. (2021). AUTS2 isoforms control neuronal differentiation. Mol. Psychiatry 26, 666–681. doi: 10.1038/s41380-019-0409-1
Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., and Sasai, Y. (2015). Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 10, 537–550. doi: 10.1016/j.celrep.2014.12.051
Nambot, S., Faivre, L., Mirzaa, G., Thevenon, J., Bruel, A. L., Mosca-Boidron, A. L., et al. (2020). De novo TBR1 variants cause a neurocognitive phenotype with ID and autistic traits: report of 25 new individuals and review of the literature. Eur. J. Hum. Genet. 28, 770–782. doi: 10.1038/s41431-020-0571-6
Nayler, S., Agarwal, D., Curion, F., Bowden, R., and Becker, E. B. E. (2021). High-resolution transcriptional landscape of xeno-free human induced pluripotent stem cell-derived cerebellar organoids. Sci. Rep. 11:12959. doi: 10.1038/s41598-021-91846-4
Nelson, B. R., Hodge, R. D., Daza, R. A., Tripathi, P. P., Arnold, S. J., Millen, K. J., et al. (2020). Intermediate progenitors support migration of neural stem cells into dentate gyrus outer neurogenic niches. eLife 9:e53777. doi: 10.7554/eLife.53777
Nieto-Estevez, V., and Hsieh, J. (2020). Human brain organoid models of developmental epilepsies. Epilepsy Curr. 20, 282–290. doi: 10.1177/1535759720949254
Oksenberg, N., Haliburton, G. D., Eckalbar, W. L., Oren, I., Nishizaki, S., Murphy, K., et al. (2014). Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl. Psychiatry 4:e431. doi: 10.1038/tp.2014.78
Oksenberg, N., Stevison, L., Wall, J. D., and Ahituv, N. (2013). Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 9:e1003221. doi: 10.1371/journal.pgen.1003221
Pasca, S. P. (2018). The rise of three-dimensional human brain cultures. Nature 553, 437–445. doi: 10.1038/nature25032
Pebworth, M. P., Ross, J., Andrews, M., Bhaduri, A., and Kriegstein, A. R. (2021). Human intermediate progenitor diversity during cortical development. Proc. Natl. Acad. Sci. U.S.A. 118, e2019415118. doi: 10.1073/pnas.2019415118
Petreanu, L., Huber, D., Sobczyk, A., and Svoboda, K. (2007). Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat. Neurosci. 10, 663–668. doi: 10.1038/nn1891
Pinto, D., Pagnamenta, A., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2020). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Poeck, B., Triphan, T., Neuser, K., and Strauss, R. (2008). Locomotor control by the central complex in Drosophila-An analysis of the tay bridge mutant. Dev. Neurobiol. 68, 1046–1058. doi: 10.1002/dneu.20643
Pollen, A. A., Bhaduri, A., Andrews, M. G., Nowakowski, T. J., Meyerson, O. S., Mostajo-Radji, M. A., et al. (2019). Establishing cerebral organoids as models of human-specific brain evolution. Cell 176, 743–756.e17. doi: 10.1016/j.cell.2019.01.017
Pomeshchik, Y., Klementieva, O., Gil, J., Martinsson, I., Hansen, M. G., de Vries, T., et al. (2020). Human iPSC-derived hippocampal spheroids: an innovative tool for stratifying Alzheimer disease patient-specific cellular phenotypes and developing therapies. Stem Cell Rep. 15, 256–273. doi: 10.1016/j.stemcr.2020.06.001
Qian, X., Song, H., and Ming, G. L. (2019). Brain organoids: advances, applications and challenges. Development 146:dev166074. doi: 10.1242/dev.166074
Quadrato, G., Nguyen, T., Macosko, E. Z., Sherwood, J. L., Min Yang, S., Berger, D. R., et al. (2017). Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53. doi: 10.1038/nature22047
Rao, A., Barkley, D., França, G. S., and Yanai, I. (2021). Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220. doi: 10.1038/s41586-021-03634-9
Russo, D., Della Ragione, F., Rizzo, R., Sugiyama, E., Scalabri, F., Hori, K., et al. (2018). Glycosphingolipid metabolic reprogramming drives neural differentiation. EMBO J. 37:e97674. doi: 10.15252/embj.201797674
Sacerdot, C., Louis, A., Bon, C., Berthelot, C., and Roest Crollius, H. (2018). Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 19:166. doi: 10.1186/s13059-018-1559-1
Sakaguchi, H., Kadoshima, T., Soen, M., Narii, N., Ishida, Y., Ohgushi, M., et al. (2015). Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 6:8896. doi: 10.1038/ncomms9896
Sanchez-Jimeno, C., Blanco-Kelly, F., Lopez-Grondona, F., Losada-Del Pozo, R., Moreno, B., Rodrigo-Moreno, M., et al. (2021). Attention deficit hyperactivity and autism spectrum disorders as the core symptoms of AUTS2 syndrome: description of five new patients and update of the frequency of manifestations and genotype-phenotype correlation. Genes 12:1360. doi: 10.3390/genes12091360
Scharfman, H. E., and Myers, C. E. (2016). Corruption of the dentate gyrus by “dominant” granule cells: implications for dentate gyrus function in health and disease. Neurobiol. Learn. Mem. 129, 69–82. doi: 10.1016/j.nlm.2015.09.005
Sellers, R. A., Robertson, D. L., and Tassabehji, M. (2020). Ancestry of the AUTS2 family-A novel group of polycomb-complex proteins involved in human neurological disease. PLoS One 15:e0232101. doi: 10.1371/journal.pone.0232101
Setia, H., and Muotri, A. R. (2019). Brain organoids as a model system for human neurodevelopment and disease. Semin. Cell Dev. Biol. 95, 93–97. doi: 10.1016/j.semcdb.2019.03.002
Sewani, M., Nugent, K., Blackburn, P. R., Tarnowski, J. M., Hernandez-Garcia, A., Amiel, J., et al. (2020). Further delineation of the phenotypic spectrum associated with hemizygous loss-of-function variants in NONO. Am. J. Med. Genet. A 182, 652–658. doi: 10.1002/ajmg.a.61466
Shepherd, G. M. (2013). Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 14, 278–291. doi: 10.1038/nrn3469
Silva, T. P., Fernandes, T. G., Nogueira, D. E. S., Rodrigues, C. A. V., Bekman, E. P., Hashimura, Y., et al. (2020). Scalable generation of mature cerebellar organoids from human pluripotent stem cells and characterization by immunostaining. J. Vis. Exp. 160:e61143. doi: 10.3791/61143
Sohal, V. S., and Rubenstein, J. L. R. (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. doi: 10.1038/s41380-019-0426-0
Srinivasan, K., Leone, D. P., Bateson, R. K., Dobreva, G., Kohwi, Y., Kohwi-Shigematsu, T., et al. (2012). A network of genetic repression and derepression specifies projection fates in the developing neocortex. Proc. Natl. Acad. Sci. U.S.A. 109, 19071–19078. doi: 10.1073/pnas.1216793109
Sultana, R., Yu, C. E., Yu, J., Munson, J., Chen, D., Hua, W., et al. (2002). Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 80, 129–134. doi: 10.1006/geno.2002.6810
Trevino, A. E., Sinnott-Armstrong, N., Andersen, J., Yoon, S. J., Huber, N., Pritchard, J. K., et al. (2020). Chromatin accessibility dynamics in a model of human forebrain development. Science 367:eaay1645. doi: 10.1126/science.aay1645
Trujillo, C. A., Gao, R., Negraes, P. D., Gu, J., Buchanan, J., Preissl, S., et al. (2019). Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell 25, 558–569.e7. doi: 10.1016/j.stem.2019.08.002
Ufartes, R., Berger, H., Till, K., Salinas, G., Sturm, M., Altmuller, J., et al. (2020). De novo mutations in FBRSL1 cause a novel recognizable malformation and intellectual disability syndrome. Hum. Genet. 139, 1363–1379. doi: 10.1007/s00439-020-02175-x
Wang, Q., Geng, Z., Gong, Y., Warren, K., Zheng, H., Imamura, Y., et al. (2018). WDR68 is essential for the transcriptional activation of the PRC1-AUTS2 complex and neuronal differentiation of mouse embryonic stem cells. Stem Cell Res. 33, 206–214. doi: 10.1016/j.scr.2018.10.023
Wester, J. C., Mahadevan, V., Rhodes, C. T., Calvigioni, D., Venkatesh, S., Maric, D., et al. (2019). Neocortical projection neurons instruct inhibitory interneuron circuit development in a lineage-dependent manner. Neuron 102, 960–975.e6. doi: 10.1016/j.neuron.2019.03.036
Xiang, Y., Tanaka, Y., Cakir, B., Patterson, B., Kim, K. Y., Sun, P., et al. (2019). hESC-derived thalamic organoids form reciprocal projections when fused with cortical organoids. Cell Stem Cell 24, 487–497.e7. doi: 10.1016/j.stem.2018.12.015
Yamashiro, K., Hori, K., Lai, E. S. K., Aoki, R., Shimaoka, K., Arimura, N., et al. (2020). AUTS2 governs cerebellar development, purkinje cell maturation, motor function and social communication. iScience 23:101820. doi: 10.1016/j.isci.2020.101820
Yoshizawa, M., Kawauchi, T., Sone, M., Nishimura, Y. V., Terao, M., Chihama, K., et al. (2005). Involvement of a Rac activator, P-Rex1, in neurotrophin-derived signaling and neuronal migration. J. Neurosci. 25, 4406–4419. doi: 10.1523/JNEUROSCI.4955-04.2005
Keywords: intellectual disability, microcephaly, RNA-binding protein, AUTS2 syndrome, FBRSL1, dentate gyrus hypoplasia, cerebellar hypoplasia, cerebral organoids
Citation: Biel A, Castanza AS, Rutherford R, Fair SR, Chifamba L, Wester JC, Hester ME and Hevner RF (2022) AUTS2 Syndrome: Molecular Mechanisms and Model Systems. Front. Mol. Neurosci. 15:858582. doi: 10.3389/fnmol.2022.858582
Received: 20 January 2022; Accepted: 01 March 2022;
Published: 31 March 2022.
Edited by:
Sumru Bayin, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Esther B. E. Becker, University of Oxford, United KingdomAshwin S. Shetty, Harvard University, United States
Copyright © 2022 Biel, Castanza, Rutherford, Fair, Chifamba, Wester, Hester and Hevner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark E. Hester, TWFyay5IZXN0ZXJATmF0aW9ud2lkZUNoaWxkcmVucy5vcmc=; Robert F. Hevner, cmhldm5lckBoZWFsdGgudWNzZC5lZHU=, cmhldm5lckB1Y3NkLmVkdQ==