 Andrew P. K. Wodrich
Andrew P. K. Wodrich Andrew W. Scott1†
Andrew W. Scott1† Arvind Kumar Shukla
Arvind Kumar Shukla Brent T. Harris
Brent T. Harris Edward Giniger
Edward Giniger- 1National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, United States
- 2Interdisciplinary Program in Neuroscience, Georgetown University, Washington, DC, United States
- 3College of Medicine, University of Kentucky, Lexington, KY, United States
- 4Department of Pathology, Georgetown University, Washington, DC, United States
- 5Department of Neurology, Georgetown University, Washington, DC, United States
Aging and age-related neurodegeneration are both associated with the accumulation of unfolded and abnormally folded proteins, highlighting the importance of protein homeostasis (termed proteostasis) in maintaining organismal health. To this end, two cellular compartments with essential protein folding functions, the endoplasmic reticulum (ER) and the mitochondria, are equipped with unique protein stress responses, known as the ER unfolded protein response (UPRER) and the mitochondrial UPR (UPRmt), respectively. These organellar UPRs play roles in shaping the cellular responses to proteostatic stress that occurs in aging and age-related neurodegeneration. The loss of adaptive UPRER and UPRmt signaling potency with age contributes to a feed-forward cycle of increasing protein stress and cellular dysfunction. Likewise, UPRER and UPRmt signaling is often altered in age-related neurodegenerative diseases; however, whether these changes counteract or contribute to the disease pathology appears to be context dependent. Intriguingly, altering organellar UPR signaling in animal models can reduce the pathological consequences of aging and neurodegeneration which has prompted clinical investigations of UPR signaling modulators as therapeutics. Here, we review the physiology of both the UPRER and the UPRmt, discuss how UPRER and UPRmt signaling changes in the context of aging and neurodegeneration, and highlight therapeutic strategies targeting the UPRER and UPRmt that may improve human health.
Introduction
Maintaining protein homeostasis (hereafter: proteostasis) within cells is essential for cellular health, especially in post-mitotic cells such as neurons that cannot dilute the ill-effects of misfolded, unfolded, or aggregated proteins through cell division. Several cellular compartments are more intimately in contact with proteins through their role as sites of protein synthesis and folding and are, therefore, more prone to encountering abnormal protein species over the lifetime of a cell. Two such compartments, the endoplasmic reticulum (ER) and the mitochondria, must maintain tight regulation of proteostasis on their specialized cellular functions and thus are equipped with dedicated protein stress responses. These organelles harbor molecular machinery to rapidly detect and mitigate proteotoxic stress through conserved signaling pathways that elicit defined transcriptional responses to promote proteostasis and sustain organellar and cellular health. Collectively, these elaborate networks of proteotoxic stress detection, signaling, and alleviation are termed the ER unfolded protein response (UPRER) and the mitochondrial UPR (UPRmt), respectively. An accumulation of UPR research combining in vitro, in vivo, and human post-mortem data has revealed that these processes are intimately entwined with both aging and neurodegeneration. Here, we will review the core machinery responsible for UPRER and UPRmt function; how these components are affected with age and in age-related neurodegenerative disease (NDD); and how their genetic or pharmacological manipulation can, in turn, influence these biological processes to shape organismal health.
Physiology of the Unfolded Protein Response
Work primarily done over the past three decades has elucidated how the organellar UPRs, the UPRER and the UPRmt, are activated; how the activation of these UPRs induces targeted cellular responses; and how the UPR-inducing stresses are resolved. Here, we focus on the core biological pathways that define the UPRER and UPRmt, emphasizing the machinery relevant to aging and neurodegeneration. Thus, we will unfortunately omit the work of many, which can be reviewed elsewhere for the UPRER (Hetz et al., 2020; Metcalf et al., 2020) and the UPRmt (Kenny and Germain, 2017; Münch, 2018; Shpilka and Haynes, 2018). Please note that when referencing gene or protein names in a specific organism, we will use the nomenclature relevant to the organism; when referencing a gene or protein generally, we will use the human nomenclature. For a complete list of aging- and NDD-associated genes and their orthologs, please see Table 1.
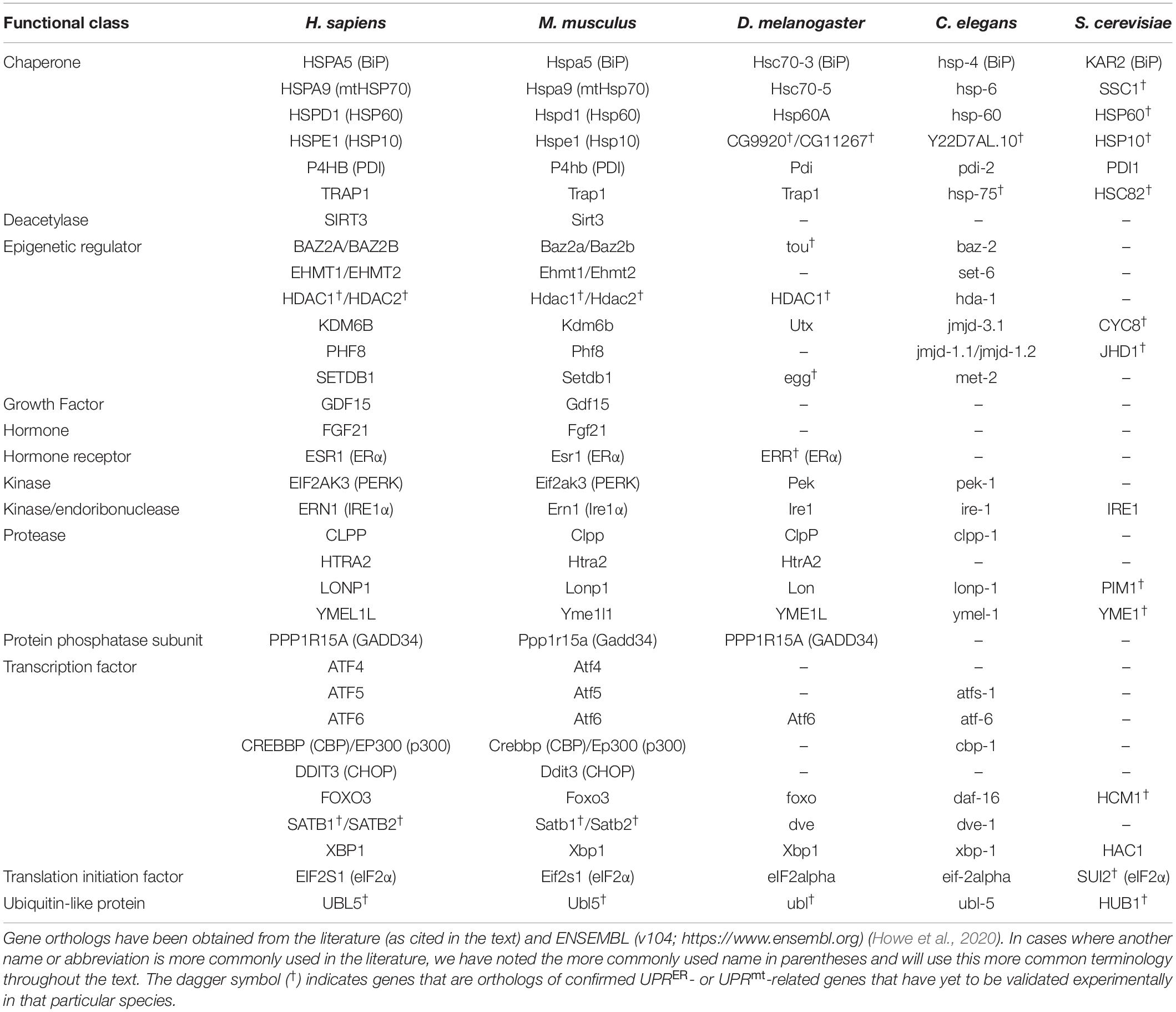
Table 1. UPRER- and UPRmt-related genes with relevance in aging or neurodegeneration and their orthologs in humans (Homo sapiens), house mice (Mus musculus), fruit flies (Drosophila melanogaster), nematodes (Caenorhabditis elegans), and yeast (Saccharomyces cerevisiae).
Physiology of the Endoplasmic Reticulum Unfolded Protein Response
Mechanism of the Endoplasmic Reticulum Unfolded Protein Response
Endoplasmic Reticulum Unfolded Protein Response Activation
The first mechanistic step of the UPRER is the sensing of ER stress. The protein folding environment within the ER is sensitive to a range of internal and external stressors that can trigger ER stress. Hypoxia, redox or calcium imbalance, nutrient deprivation and viral challenge, as well as errors of translation and expression of genetic mutants, can all disrupt ER homeostasis and lead to the accumulation of misfolded proteins within the ER lumen (Wang and Kaufman, 2016). The convergence of diverse stimuli on proteotoxic stress establishes it as the primary trigger of the UPRER, though recent work has also demonstrated that impaired lipid metabolism can activate the UPRER in a manner distinct from protein misfolding (Hou et al., 2014).
Endoplasmic Reticulum Unfolded Protein Response Effectors and Effects
Upon sensing ER stress, the second step of the UPRER process is to resolve the homeostatic disruption. To facilitate this, the UPRER is coordinated through three distinct transmembrane proteins that initiate parallel transcriptional and translational responses: inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6) (see Figure 1). Each protein contains a luminal stress-sensing domain within the ER, along with a cytosolic fragment that facilitates transduction of the stress signal to the nucleus. The sensors are typically kept dormant under homeostatic conditions through the binding of ER chaperone binding immunoglobulin protein (BiP) to their luminal domains. However, upon ER stress, BiP preferentially associates with misfolded or unfolded proteins rather than IREα, PERK, or ATF6 leading to activation of UPRER signaling pathways (Bertolotti et al., 2000; Shen J. et al., 2005).
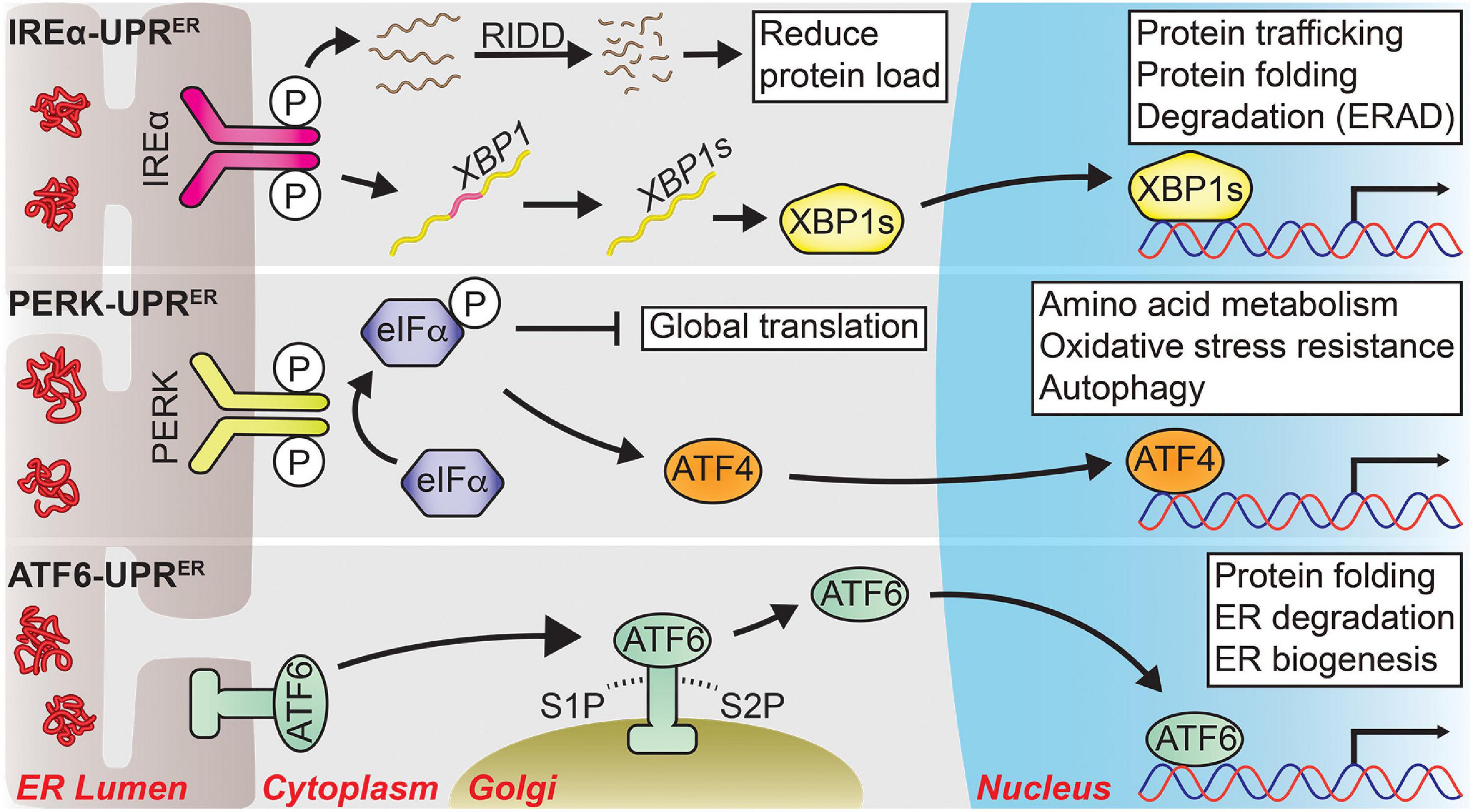
Figure 1. The three major signal transduction pathways of the UPRER. Following ER stress, three distinct branches are activated that shape the UPRER. IRE1α-UPRER: Once activated via its dimerization and autophosphorylation, IRE1α cleaves a select group of mRNAs and miRNAs to drive their degradation through a process known as regulated IRE1-dependent decay (RIDD), reducing the total protein folding load on the ER. IRE1α also facilitates the unconventional splicing of XBP1 mRNA into its spliced form, a potent transcription factor known as XBP1s, which drives the expression of genes tied to protein quality control to restore ER homeostasis. PERK-UPRER: PERK also dimerizes and autophosphorylates upon ER stress, which then phosphorylates eIF2α to attenuate global translation. The mRNA of transcription factor ATF4 is preferentially translated following eIF2α phosphorylation, allowing it to upregulate genes involved in amino acid metabolism, oxidative stress resistance, autophagy, and apoptosis. ATF6-UPRER: ER stress unmasks several Golgi-localization signals within ATF6 that allow it to translocate to the Golgi body. There, it is sequentially cleaved by site-1 protease (S1P) and site-2 protease (S2P) from its full-length form (ATF6p90) into its transcriptionally active form (ATF6p50), which initiates the transcription of UPR target genes pertaining to protein quality control and ER biogenesis to promote ER secretory capacity. Solid arrows represent direct actions.
IRE1α, a protein kinase/endoribonuclease that was first identified in yeast as required for protection against ER stress, sits atop the most evolutionarily conserved arm of the UPRER (hereafter: IRE1a-UPRER) (Cox et al., 1993; Mori et al., 1993). Following its activation via dimerization and subsequent autophosphorylation, IRE1α processes the mRNA encoding X-box binding protein 1 (XBP1), removing a small intron that facilitates the translation of XBP1 into its active form as a potent transcription factor (XBP1s) (Sidrauski and Walter, 1997; Shen et al., 2001; Yoshida et al., 2001; Calfon et al., 2002). XBP1s then translocates to the nucleus where it drives the induction of genes involved in protein trafficking, folding, and degradation (Lee et al., 2003a; Shen X. et al., 2005; Acosta-Alvear et al., 2007). XBP1s-induced protein degradation occurs largely through ER-associated degradation (ERAD), which targets ER-localized proteins for cytosolic degradation via the proteasome (Hwang and Qi, 2018). Independent of XBP1s activation, IRE1α also cleaves a subset of mRNAs and microRNAs (miRNAs) that leads to their rapid degradation through a process known as regulated IRE1-dependent decay (RIDD) (Hollien et al., 2009; Hollien and Weissman, 2006; Upton et al., 2012). ER-localized transcripts are preferentially targeted for degradation as a means to reduce the total translation and protein-folding burden placed on the ER (Maurel et al., 2014).
The second arm of the UPRER is governed by the protein kinase PERK (hereafter: PERK-UPRER). Like IRE1α, PERK undergoes dimerization and autophosphorylation following ER stress. Following its activation, PERK acts through its kinase domain to attenuate translation via the phosphorylation of eukaryotic initiation factor 2α (eIF2α) to prevent the influx of newly synthesized proteins into the ER (Harding et al., 1999). However, this translational shutdown also permits a select group of mRNA transcripts to undergo translation; one of which is activating transcription factor 4 (ATF4) (Lu et al., 2004; Vattem and Wek, 2004). Following activation, ATF4 induces the expression of genes tied to amino acid metabolism and oxidative stress resistance (Harding et al., 2003), but during persistent ER stress can also promote apoptotic signaling through an interaction with downstream transcription factor C/EBP homologous protein (CHOP) (Ma et al., 2002; Han et al., 2013). The ATF4/CHOP interaction also forms part of a negative feedback loop through upregulation of the protein phosphatase 1 (PP1) regulatory subunit GADD34, which forms a complex with PP1 to dephosphorylate eIF2α and restore translational capacity following the resolution of ER stress (Novoa et al., 2001).
The third arm of the UPRER is regulated through ATF6 (hereafter: ATF6-UPRER). Upon ER stress, BiP dissociates from ATF6, unmasking two Golgi-localization signals that cause ATF6 to translocate to the Golgi body (Shen et al., 2002). Here, the full-length form of ATF6, ATP6p90, is sequentially cleaved by site-1 protease (S1P) and site-2 protease (S2P), releasing the cytosolic ATF6p50 fragment that translocates to the nucleus to function as a transcription factor (Haze et al., 1999; Ye et al., 2000). ATF6p50 then induces the expression of genes pertaining to protein quality control and ER biogenesis to improve ER secretory capacity (Wu et al., 2007; Yamamoto et al., 2007; Bommiasamy et al., 2009). Of note, the transcriptional program induced by ATF6p50 displays a degree of overlap with that of XBP1s. Indeed, ATF6p50 and XBP1s exhibit crosstalk via the formation of heterodimers, likely serving to fine tune the transcriptional output following ER stress (Yamamoto et al., 2007; Shoulders et al., 2013).
Through the combined IRE1α/PERK/ATF6 signaling axes, the adaptive UPRER phase represents a rapid response to promote protein quality control and allow the resumption of important cellular functions performed by the ER.
Non-cell Autonomous Endoplasmic Reticulum Unfolded Protein Response Signaling
A host of recent work in Caenorhabditis elegans has highlighted an additional novel aspect of UPRER biology – the ability to communicate ER stress across multiple tissue types to coordinate a whole-body organismal response. Expression of xbp-1s specifically in worm neurons leads to UPRER activation in the intestine and confers greater organismal resistance against ER stress (Taylor and Dillin, 2013). This method of intercellular communication relies on the release of small clear vesicles (SCVs) carrying neuronally derived stress signals to distal tissue (Taylor and Dillin, 2013). Intriguingly, expression of xbp-1s in glial cells elicits a similar non-cell autonomous mode of intestinal UPRER activation, though via a different mechanism involving neuropeptide signaling (Frakes et al., 2020).
Further investigation of the non-cell autonomous mechanism in nematodes has revealed that neuronally expressed xbp-1s regulates intestinal stress resistance via several distinct methods, including the upregulation of lysosomal activity to improve proteostasis (Imanikia et al., 2019a) and alterations of lipid metabolism (Imanikia et al., 2019b; Daniele et al., 2020). Additionally, it was demonstrated that distinct neuronal populations apply these mechanisms differentially; dopaminergic xbp-1s expression drives lipid remodeling, while serotonergic xbp-1s expression induces chaperones to promote protein quality control (Higuchi-Sanabria et al., 2020). Finally, upon ER or starvation stress, xbp-1 is spliced in two interneurons required for tyramine synthesis, which drives intestinal UPRER activation and modulates changes in reproductive and feeding behavior (Özbey et al., 2020).
Taken together, these studies serve to highlight the mechanistic complexity of the UPRER; stress signals can produce distinct responses depending on where in the CNS it is received, while a pan-neuronal response likely integrates multiple stress signals to drive a more comprehensive modulation of ER function to maintain organismal homeostasis. Importantly, distal UPRER activation appears to be conserved through evolution; activation of Xbp1s in mouse proopiomelanocortin (Pomc) neurons regulates metabolic alterations through UPRER activation in the liver (Williams et al., 2014; Brandt et al., 2018). Thus, the non-cell autonomous aspect of UPRER signaling appears to play a key role in orchestrating the whole-body adaptive response to ER stress.
Acute Versus Chronic Endoplasmic Reticulum Unfolded Protein Response Activation
Should the adaptive phase of the UPRER prove insufficient in restoring ER proteostasis, chronic UPRER activation can drive cells toward an apoptotic fate. Much like the adaptive response, UPRER-mediated apoptotic signaling consists of the integration of several interweaved pathways.
The canonical intrinsic apoptotic pathway, regulated by the B cell lymphoma 2 (Bcl2) family of proteins and characterized by the permeabilization of the mitochondrial outer membrane (MOM) and release of pro-apoptotic factors including cytochrome c (Pihán et al., 2017), can be initiated through separate means by both PERK and IRE1α-mediated signaling. PERK/ATF4-mediated activation of CHOP can inhibit the negative regulator of cell death Bcl2 (McCullough et al., 2001) and activate pro-apoptotic factor BIM (Puthalakath et al., 2007), with both outcomes converging on pro-apoptotic signaling through core Bcl2 proteins BAX and BAK (Pihán et al., 2017). Additionally, together with ATF4, CHOP can promote cell death by enhancing protein synthesis and reactive oxygen species (ROS) production under chronic ER stress (McCullough et al., 2001; Marciniak et al., 2004; Han et al., 2013).
Sustained IRE1α RNAse activity leads to the decay of a select group of precursor miRNAs that typically repress the translation of initiator protease Caspase-2 (Upton et al., 2012). This allows Caspase-2 to cleave BID and induce BAX/BAK-mediated apoptosis (Wei et al., 2001; Bonzon et al., 2006). However, a separate study did not observe Caspase-2 activation following ER stress, nor its requirement for ER stress-induced cell death (Sandow et al., 2014), suggesting that this mechanism requires further investigation. Under chronic ER stress, RIDD also induces thioredoxin-interacting protein (TXNIP) via the degradation of its negative regulator microRNA-17, leading to activation of the NLRP3 inflammasome and its subsequent downstream hallmarks, sterile inflammation and pyroptotic cell death (Lerner et al., 2012). Additionally, activated IRE1α can form a complex with adaptor protein TRAF2 and ASK1 to initiate a JNK-mediated signaling cascade that ultimately drives cell death (Urano et al., 2000; Nishitoh et al., 2002).
Both pro-survival and pro-death signals are integrated through the UPRER to decide cell fate, but how this balance is tipped to favor a specific fate remains an important question in the field (Hetz et al., 2020). Indeed, UPR-induced apoptosis may prove beneficial under certain physiological contexts as the removal of damaged or dysfunctional cells can prevent the triggering of a detrimental inflammatory response (Tabas and Ron, 2011), and sustained ER stress can contribute to a range of pathological outcomes if left unresolved (Wang and Kaufman, 2016).
Physiology of the Mitochondrial Unfolded Protein Response
Mechanism of the Mitochondrial Unfolded Protein Response
Mitochondrial Unfolded Protein Response Activation
Mitochondrial proteostasis failure is the most thoroughly studied trigger of the UPRmt, and it can arise from (1) accumulated misfolded proteins, (2) impaired mitochondrial translation, (3) an imbalance in the ratio of mitochondria-derived to nuclear-derived proteins in the mitochondria (hereafter: mito-nuclear imbalance), or (4) impaired mitochondrial protein import. Early work in the field using monkey kidney cells showed that misfolded mitochondrial proteins cause an increase in protease and chaperone transcript and protein levels (Zhao, 2002). Subsequent work in invertebrates and mammals has shown that loss of mitochondrial proteases such as LONP1 or mitochondrial chaperones mtHSP70, HSP60, or TRAP1 causes accumulation of misfolded proteins and the activation of the UPRmt (Yoneda et al., 2004; Münch and Harper, 2016). Impaired mitochondrial translation can also activate the UPRmt, likely due to reduced protein levels and the accumulation of truncated proteins (Dogan et al., 2014; Chung et al., 2017). Another proteostasis-related trigger of the UPRmt is mito-nuclear imbalance, which impairs the ability for multi-protein complexes to form creating unfolded protein stress (Houtkooper et al., 2013; Mouchiroud et al., 2013; Yuan et al., 2020). Lastly, impaired mitochondrial protein import functions as perhaps the major sensor activating the UPRmt because mitochondrial protein import requires ATP, mtHSP70, and an intact mitochondrial membrane potential and thus is rapidly perturbed in response to most mitochondrial stresses, however minor (Shpilka and Haynes, 2018). This has been demonstrated in C. elegans or human cell lines; knockdown of TIM23, which encodes a major translocase located on the mitochondrial inner membrane (MIM), reduces protein import and activates the UPRmt (Rainbolt et al., 2013).
Electron transport chain (ETC) dysfunction can also initiate the UPRmt. Mild inhibition of genes encoding mitochondrial ETC components in C. elegans activates the UPRmt transcriptional regulators atfs-1, dve-1, and ubl-5 (Haynes et al., 2007; Nargund et al., 2012). Furthermore, decreasing the expression of mitochondrial ETC complex subunits or reducing ETC complex function has been shown to induce the expression of UPRmt-related chaperones in C. elegans, Drosophila, and mammals (Yoneda et al., 2004; Hamilton et al., 2005; Benedetti et al., 2006; Owusu-Ansah et al., 2013; Pulliam et al., 2014). Additionally, a mito-nuclear imbalance of ETC proteins can result in these unfolded proteins accumulating within the mitochondrial matrix and activating the UPRmt (Martinus et al., 1996; Yoneda et al., 2004; Nargund et al., 2012; Houtkooper et al., 2013; Liu et al., 2014).
Lastly, changes in mtDNA can also induce the UPRmt. Loss of mtDNA increases expression of nuclear-encoded mitochondrial chaperones but not cytosolic chaperones suggesting an activation of the UPRmt (Martinus et al., 1996). Additionally, harboring even a small amount of damaged mtDNA is sufficient to induce the UPRmt (Gitschlag et al., 2016; Lin et al., 2016). Furthermore, mutations in mtDNA helicases increase the expression of UPRmt-related proteins suggesting that changes to the structure of mtDNA, in addition to the sequence of mtDNA, can activate the UPRmt (Yoneda et al., 2004; Khan et al., 2014; Forsström et al., 2019).
Mitochondrial Unfolded Protein Response Effectors and Effects
There are three parallel – and potentially overlapping – mechanisms linking mitochondrial stresses to nuclear transcriptional responses relevant to aging and neurodegeneration (see Figure 2). The classical UPRmt mechanism, primarily researched in worms and subsequently shown to be highly conserved in mammals, is primarily driven by the nuclear localization of the transcription factor atfs-1 in worms or the transcription factors CHOP, ATF4, and ATF5 in mammals (hereafter: ATF5-UPRmt) (Shpilka and Haynes, 2018). Two other arms of the UPRmt have been shown to be at least partially independent of the classical UPRmt axis. One signals through the mitochondrial NAD+-dependent deacetylase SIRT3 and the other is dependent upon the activation of the hormone receptor Estrogen Receptor alpha (ERα) (Papa and Germain, 2011, 2014). Activation of either of these alternative UPRmt arms induces differing cellular effects from those of the classical arm of the UPRmt.
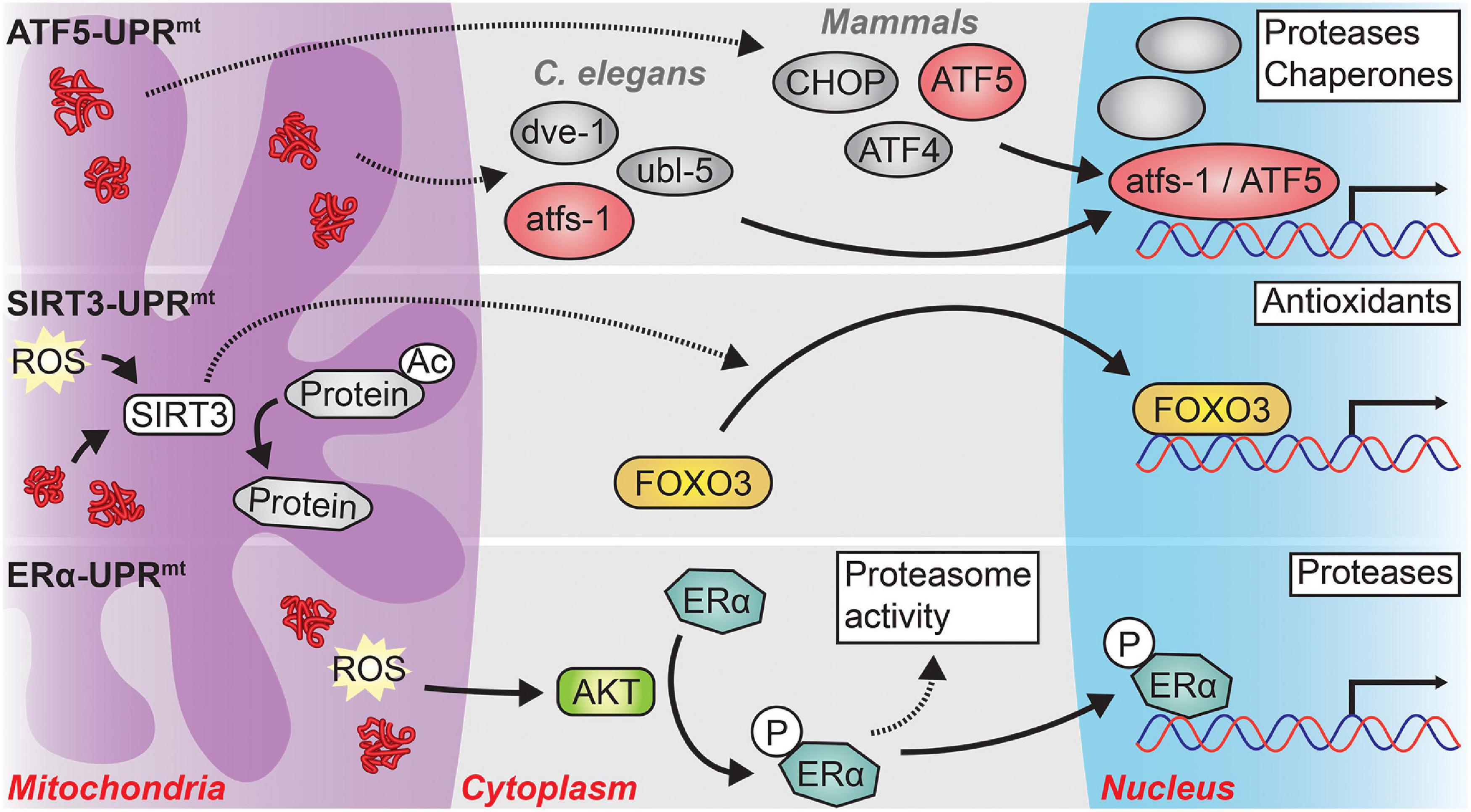
Figure 2. The three major signaling pathways of the UPRmt. In response to mitochondrial stress, three distinct branches of the UPRmt may be activated, depending on the type and location of the mitochondrial stress. ATF5-UPRmt: In C. elegans, protein stress in the mitochondrial matrix causes the cytosolic accumulation of atfs-1, or its mammalian ortholog ATF5. In concert with the transcription factor dve-1 and the ubiquitin-like protein ubl-5, atfs-1 translocates to the nucleus where it induces the transcription of proteases and chaperones to relieve mitochondrial protein stress. A similar process occurs in mammals, albeit with the requirement of two transcription factors, CHOP and ATF4, in addition to ATF5. The precise interactions among atfs-1, dve-1, and ubl-5 as well as among ATF5, CHOP, and ATF4 remain unclear. SIRT3-UPRmt: Mitochondrial matrix reactive oxygen species (ROS) or protein stress activates SIRT3 which then directly deacetylates numerous mitochondrial proteins and indirectly causes the nuclear localization of the transcription factor FOXO3. FOXO3 then induces an antioxidant transcriptional program to combat high levels of oxidative stress in the mitochondria. ERα-UPRmt: Misfolded proteins and ROS located within the mitochondrial intermembrane space (IMS) activate the kinase AKT. AKT phosphorylates Estrogen Receptor alpha (ERα) which then increases the activity of the proteasome and functions as a transcription factor in the nucleus to induce the expression of IMS-specific proteases. Solid arrows represent direct actions while dashed arrows represent indirect actions or actions with unclear mechanisms.
The most thoroughly studied arm of the UPRmt is the ATF5-UPRmt. In both worms and mammals, the ATF5-UPRmt modulates both transcription and chromatin organization to counter mitochondrial stress (Haynes et al., 2010; Nargund et al., 2012; Fiorese et al., 2016). In C. elegans, atfs-1, which contains a nuclear localization sequence and a mitochondrial targeting sequence, is normally imported into the mitochondria and rapidly degraded by the mitochondrial matrix protease lonp-1 (Haynes et al., 2010; Nargund et al., 2012, 2015). However, under conditions of mitochondrial stress, import of atfs-1 into the mitochondria is precluded, and atfs-1 is trafficked to the nucleus where it coordinates with other factors, including ubl-5 and dve-1, to elicit the UPRmt transcriptional program (Benedetti et al., 2006; Haynes et al., 2007, 2010; Nargund et al., 2012, 2015). In mammals, ATF5 plays a similar role to that of atfs-1; however, the transcription factors CHOP and ATF4 are also required for the induction of the UPRmt, although it remains unclear precisely how they interact to do so (Aldridge et al., 2007; Horibe and Hoogenraad, 2007; Fiorese et al., 2016; Quirós et al., 2017).
In addition to transcriptional changes, the ATF5-UPRmt modifies the epigenetic landscape to promote an open chromatin state at UPRmt genes and repress transcription of genes antagonistic to the UPRmt. In C. elegans, the ATF5-UPRmt promotes global heterochromatin formation through the methylation of H3K9 by met-2 and the nuclear co-factor lin-65 (Andersen and Horvitz, 2007; Tian et al., 2016). Despite this global chromatin silencing, other chromatin regions become more open, in part due to stabilization by the transcription factor dve-1 (Tian et al., 2016). Simultaneously, the demethylases jmjd-1.2 and jmjd-3.1, or the mammalian equivalents PHF8 and KDM6B, create transcriptionally active chromatin marks in a process that is dependent upon CREB binding protein 1 (cbp-1), the ortholog of the mammalian CBP/p300 (Merkwirth et al., 2016; Li et al., 2021).
For invertebrates as well as mammals, the translational and epigenetic changes induced by the mitochondrial stress signaled through the ATF5-UPRmt result in (1) an upregulation of the expression of mitochondrial chaperones, proteases, protein importers, and ETC components; (2) a reduction in global protein translation in the cytosol; (3) a decrease in protein translation within mitochondria; and (4) a reprogramming of mitochondrial metabolism (Zhao, 2002; Yoneda et al., 2004; Aldridge et al., 2007; Baker et al., 2012; Nargund et al., 2012, 2015; Houtkooper et al., 2013; Gitschlag et al., 2016; Münch and Harper, 2016; Borch Jensen et al., 2018; Molenaars et al., 2020; Yuan et al., 2020). Cumulatively, these effects reduce proteotoxic stress in the mitochondrial matrix by increasing protein folding and degradation capacity, reducing protein burden within the mitochondria, and shifting metabolic demand away from the mitochondria, presumably to allow for the restoration of proper ETC function.
As opposed to the ATF5-UPRmt, which responds primarily to proteotoxic stress within the mitochondrial matrix, the ERα arm of the UPRmt (hereafter: ERα-UPRmt) responds to proteotoxic stress within the mitochondrial inner membrane space (IMS) (Radke et al., 2008; Papa and Germain, 2011). Proteotoxic stress and ROS within the IMS leads to the activation ERα in an AKT-dependent fashion (Papa and Germain, 2011). ERα increases the activity of the cytosolic ubiquitin-proteasome system and translocates to the nucleus where it functions as a transcription factor to induce the expression of IMS-specific proteases such as HTRA2; both processes reduce protein stress in the mitochondrial IMS (Radke et al., 2008; Papa and Germain, 2011). Interestingly, it appears that if the ERα-UPRmt is not working, the ATF5-UPRmt will eventually be activated (Papa and Germain, 2011). This suggests that there may be direct spillover of protein stress from the IMS to the matrix to trigger the ATF5-UPRmt.
The last arm of the UPRmt signals through SIRT3 and the transcription factor FOXO3 (hereafter: SIRT3-UPRmt) (Papa and Germain, 2014; Marcus and Andrabi, 2018). SIRT3-UPRmt signaling occurs both by SIRT3-dependent deacetylation of mitochondrial targets and SIRT3/FOXO3-dependent transcriptional changes (Schwer et al., 2002; Lombard et al., 2007; Sundaresan et al., 2009). The SIRT3/FOXO3-dependent signaling induces an antioxidant transcriptional program to combat high levels of ROS within the mitochondria (Papa and Germain, 2014; Marcus and Andrabi, 2018). As an example of the dual nature of this arm, expression of misfolding-prone proteins or pharmacological inhibition of ETC complexes causes a ROS-dependent activation of SIRT3 which then deacetylates a number of downstream targets leading to the nuclear-localization of FOXO3 (Papa and Germain, 2014).
Despite significant overlap between the ATF5-UPRmt and SIRT3-UPRmt, there is significant evidence that the SIRT3-UPRmt is a bona fide unique and parallel arm of the UPRmt that is separable from the ATF5-UPRmt (Münch, 2018). CHOP, a transcription factor required for the mammalian ATF5-UPRmt, is dispensable for the expression of antioxidants in response to mitochondrial stress (Papa and Germain, 2014). Additionally, ETC complex I and III inhibitors – known to increase levels of mitochondrial ROS (mtROS) – increase levels of SIRT3 and FOXO3 suggesting that ROS are sufficient to induce the SIRT3-dependent UPRmt (Papa and Germain, 2014).
The overlap between the ATF5-UPRmt and SIRT3-UPRmt likely arises due to the multiple actions of ROS within the mitochondria. It is likely that ROS per se activates the SIRT3-UPRmt while ROS-dependent protein damage and aggregation stimulates the ATF5-UPRmt. In this way, the SIRT3-UPRmt is the primary response to imbalances in redox homeostasis within the mitochondria while the ATF5-UPRmt is activated in circumstances where elevated ROS levels are too high or persist too long causing oxidative damage. Furthermore, the SIRT3-UPRmt appears to maintain ROS within a homeostatic window and allow for critical ROS-dependent physiological signaling to occur without excess oxidative damage occurring due to promiscuous ROS interactions with mitochondrial macromolecules (Kenny and Germain, 2017). Indeed, as evidence of such, a transient increase in mtROS results in the persistent, steady-state decrease in ROS levels 48-h later (Schmeisser et al., 2013). This suggests that mtROS spikes induce the expression of antioxidant machinery that installs a new redox homeostasis setpoint and reduces total steady-state levels of mtROS.
Non-cell Autonomous Mitochondrial Unfolded Protein Response Signaling
Similar to the non-cell autonomous effect observed with the UPRER, the induction of the UPRmt also exerts non-cell autonomous effects on distant tissues. In C. elegans, signaling pathways dependent upon the Wnt ligand egl-20, the neuropeptide flp-2, the neurotransmitter serotonin, and the G-protein coupled receptor fshr-1 have been linked to non-cell autonomous signaling in response to neuronal mitochondrial dysfunction (Berendzen et al., 2016; Shao et al., 2016; Zhang et al., 2018; Kim and Sieburth, 2020). It also appears to be the case in mammals that secreted factors, termed mitokines, relay mitochondrial stress to distant tissues (Durieux et al., 2011). Numerous mitochondrial stresses from impaired protein translation to mtDNA deletions can be signaled to distant tissues through the secretion of FGF21 and GDF15 (Suomalainen et al., 2011; Chung et al., 2017; Forsström et al., 2019; Kang et al., 2021). In many cases, however, it remains unclear exactly how these individual mitokines induce the wide-ranging transcriptional changes characteristic of the UPRmt. It appears that much of the non-cell autonomous signaling of mitochondrial dysfunction originates from neurons, perhaps because neuronal mitochondria, as hubs of metabolism in a metabolically sensitive cell population, function as sentinels of metabolic stress to prime the whole organism for a high stress environment (Durieux et al., 2011; Tian et al., 2016). However, it should be noted that non-neuronal tissues such as gonads or muscles also have a role in non-cell autonomous UPRmt signaling which underscores the usefulness of a rapidly inducible transcriptional program that can modulate the capacity of cellular mitochondria to engage in energy production, biosynthesis, calcium homeostasis, and immune signaling (Owusu-Ansah et al., 2013; Lan et al., 2019).
Acute Versus Chronic Mitochondrial Unfolded Protein Response Activation
Like with the UPRER, acute activation of the UPRmt results in distinct outcomes from that of chronic UPRmt activation. The UPRmt can induce acute, pro-survival transcriptional and epigenetic changes that alleviate the inciting mitochondrial stress (Münch, 2018). However, in cases where the instigating stress cannot be resolved, a prolonged response occurs that can cause detrimental outcomes such as reduced fecundity and the propagation of deleterious mtDNA (Rauthan et al., 2013; Gitschlag et al., 2016; Lin et al., 2016). Experimental approaches seem to offer conflicting evidence for whether chronic UPRmt is beneficial or detrimental for organismal fitness. Whereas some reports suggest mild, constitutive RNAi knockdown of components of the ETC extends lifespan in fruit flies, others suggest that partial inactivation of ETC genes does not extend lifespan (Rera et al., 2010; Owusu-Ansah et al., 2013). While knockdown of ETC complex subunits is a known trigger of the UPRmt, it remains possible that other mechanisms account for the differences in organism longevity in these studies. However, what is apparent is that long-term hyperactivation of the UPRmt is detrimental for both cellular and organismal health (Rauthan et al., 2013; Ishizawa et al., 2019). This is perhaps best illustrated by the lack of genetic mutations that exist that chronically activate the UPRmt; these mutations presumably have evolutionary trade-offs that, in the long-term, favor neither the cell nor the organism (Lamech and Haynes, 2015). That being said, it does appear that some low basal level of UPRmt activation is required for proper cellular health, perhaps as a buffer against subtle forms of mitochondrial stress (Shpilka and Haynes, 2018).
The Unfolded Protein Response in Aging
It has become increasingly apparent from research conducted over the past few years that aging, the functional decline of biological systems that occurs over time eventually leading to both cellular and organismal dysfunction and death, is driven significantly by the loss of proteostasis and mitochondrial dysfunction (López-Otín et al., 2013; Hipp et al., 2014; Piper and Partridge, 2018; Singh et al., 2019). However, it remains unclear what role the UPRER and UPRmt, as responses to age-dependent proteostasis collapse and mitochondrial dysfunction, play in compensating for these aging-related changes to preserve cellular function and organismal fitness. Here, we discuss how the functionality of the UPRER and UPRmt are affected with age and how manipulations targeting the UPRER and UPRmt can influence organismal aging in model systems.
The Endoplasmic Reticulum Unfolded Protein Response in Aging
Changes to the Endoplasmic Reticulum Unfolded Protein Response in Aging
Studies in several model systems have provided evidence of changes to the UPRER with aging. Work in C. elegans has demonstrated that the ability to respond to ER stress through the UPRER is not only weakened with age, but that this decline also appears to be an early event during the aging process (Ben-Zvi et al., 2009; Taylor and Dillin, 2013). ER-localized protein chaperones, including BiP and protein disulfide isomerase (PDI), undergo oxidative damage with age in mouse liver, contributing to the age-associated increase in misfolded protein products within the ER (Rabek et al., 2003; Nuss et al., 2008). Within the aged mouse brain, the ability to induce adaptive UPRER signaling following sleep deprivation is diminished, while evidence of dysregulated UPRER homeostasis with age is also observed in two neuronal subpopulations, orexinergic and noradrenergic neurons, that govern wakefulness (Naidoo et al., 2008, 2011). In aged rat brains, induction of the IRE1α-UPRER and the ATF6-UPRER is suppressed following proteasome inhibition, coinciding with elevated levels of pro-apoptotic factors CHOP, BAX, and BAK (Paz Gavilán et al., 2009). A similar pattern is also observed in other tissues from aged rats; lung, liver, kidney, and spleen all experience loss of ATF4 and BiP, whilst CHOP and phosphorylated JNK are elevated (Hussain and Ramaiah, 2007). Finally, a recent in vitro study demonstrated that senescent human lung fibroblasts remain capable of sensing ER stress effectively but lose the ability to coordinate the subsequent transcriptional response through XBP1s and ATF6 (Sabath et al., 2020). Collectively, these studies show that the UPRER loses the ability to mount the adaptive response with age. In contrast, pro-apoptotic signaling potency is retained, partly due to a lowering of the homeostatic threshold that determines the switch from acute to chronic UPRER signaling in aging cells.
Manipulations to the Endoplasmic Reticulum Unfolded Protein Response Affect Aging
The established decline of the UPRER with age suggests that manipulations aimed at maintaining adaptive UPRER functionality throughout the organism’s lifespan may prove beneficial to healthy aging. Indeed, a study on long-lived naked mole rats uncovered protein quality control maintenance as a key determinant of healthy aging (Pérez et al., 2009b). In this regard, model organisms have provided excellent experimental systems to dissect the role of UPRER signaling in aging.
Inositol-Requiring Enzyme 1α-Endoplasmic Reticulum Unfolded Protein Response in Aging
The most conserved pathway within the UPRER, IRE1α/XBP1 signaling has been shown to regulate aging from yeast to mammals. IRE1α and yeast XBP1 homolog HAC1 are both required for yeast replicative lifespan extension following several UPRER manipulations, either through the specific deletion of UPRER target genes which leads to constitutive UPRER signaling (Labunskyy et al., 2014), or via the enhancement of basal UPRER activity (Cui et al., 2015). In nematodes, constitutive neuronal, glial, or intestinal expression of xbp-1s restores UPRER functionality with age and is sufficient to extend lifespan (Taylor and Dillin, 2013; Frakes et al., 2020). Of note, ubiquitous expression of xbp-1s does not extend lifespan, while its expression specifically in muscle cells decreases lifespan (Taylor and Dillin, 2013), suggesting that constitutive XBP1s activity can prove detrimental in certain tissues.
The IRE1α-UPRER also influences organismal lifespan in conjunction with other well-established aging pathways. Both IRE1α and XBP1 are required for enhanced longevity through the reduction of the insulin/IGF-1 like signaling (IIS) pathway, whereby xbp-1 coordinates with daf-16, the C. elegans ortholog of FOXO3, to upregulate genes involved in longevity and ER stress resistance (Henis-Korenblit et al., 2010). In yeast, HAC1 activity is necessary for lifespan extension via dietary restriction (DR) (Choi et al., 2013). The IRE1α-UPRER also contributes to dietary-induced lifespan modulation in nematodes, where it is required for the longevity effects observed under both DR conditions and in hypoxia-inducible factor-1 (hif-1) mutants under high nutrient conditions (Chen et al., 2009). Additionally, IRE1α is essential for DR-induced lifespan extension in conjunction with transcription factor pha-4, whereby DR in early life promotes ERAD function to improve proteostasis with age (Matai et al., 2019). Finally, low-nutrient conditions activate xbp-1 in tyramine-producing neurons to alter reproductive and feeding behavior in parallel with intestinal UPRER activation to extend lifespan (Özbey et al., 2020). Of note, Xbp1 can also promote distal UPRER-mediated metabolic adaptation following food sensing in fruit flies (Luis et al., 2016) and mice (Williams et al., 2014; Brandt et al., 2018), highlighting the evolutionary conservation of this nutrient sensing role for the IRE1α-UPRER. Dietary interventions have been well-established as modulators of organismal health with age (Fontana and Partridge, 2015), and evidence garnered in model organisms has placed the UPRER, in particular the IRE1α-UPRER, as a key signaling pathway within this form of lifespan regulation.
Protein Kinase RNA-Like Endoplasmic Reticulum Kinase-Endoplasmic Reticulum Unfolded Protein Response in Aging
Dysregulation of the PERK-UPRER is a common feature with age (Hussain and Ramaiah, 2007; Paz Gavilán et al., 2009; Sabath et al., 2020), and manipulations targeting this pathway have focused mainly on suppressing aberrant PERK signaling. Early work observed that genetic deletion of the nematode PERK ortholog pek-1 does not affect lifespan (Henis-Korenblit et al., 2010). In contrast, a more recent study demonstrated that a single amino acid substitution in the pek-1 kinase domain prolongs lifespan (Derisbourg et al., 2021), which may reflect the difference in pek-1 mutations between studies. In addition, abolishing the activation of eIF2α via a phospho-deficient mutation also confers lifespan extension (Derisbourg et al., 2021), suggesting that downstream targeting of the PERK pathway can also promote longevity.
Work in Drosophila has demonstrated that ER stress triggers intestinal expression of the Drosophila PERK ortholog Pek via both cell autonomous and non-cell autonomous mechanisms, which then regulates intestinal stem cell (ISC) regeneration to maintain proliferative homeostasis (Wang et al., 2015). However, sustained Pek activity in ISCs, as seen in aged flies, shortens lifespan through gut dysplasia, serving to highlight the differential outcomes on lifespan attained between acute and chronic PERK signaling (Wang et al., 2015). In mice, reducing PERK activity can also prove beneficial; knockdown of Perk in the CA1 hippocampus region in aging mice reverses age-related declines in memory and neuronal excitability (Sharma et al., 2018). However, the outcome of neuronal PERK inhibition in mammals appears cell-specific; deletion of Perk specifically in mouse dopaminergic neurons leads to a range of both cognitive and motor phenotypes via the dysregulation of de novo translation and dopamine release (Longo et al., 2021). Taken together, these murine studies demonstrate the complexity involved in inhibiting PERK activity; reducing PERK function has the potential to affect both the beneficial acute and deleterious chronic modes of signaling. Indeed, the long-lived C. elegans phospho-deficient eIF2α mutant was not impaired in either global translation capacity or ER stress resistance, suggesting that the fine-tuning of PERK signaling, rather than ablation, has the potential to slow aging (Derisbourg et al., 2021).
Activating Transcription Factor 6-Endoplasmic Reticulum Unfolded Protein Response in Aging
The ATF6-UPRER is the least studied branch of the UPRER in the context of aging. Like the IRE1α-UPRER, evidence suggests that the ATF6-UPRER loses adaptive signaling potency with age (Paz Gavilán et al., 2009; Sabath et al., 2020), and deletion of Atf6 renders mice sensitive to sources of chronic ER stress (Wu et al., 2007; Yamamoto et al., 2007). However, recent work in nematodes suggests that the relationship between the ATF6-UPRER and age is more complex. Burkewitz et al. (2020) demonstrated that atf-6 in C. elegans is dispensable for resistance to proteotoxic stress, and that its loss confers longevity via the downregulation of its conserved transcriptional target calreticulin, a regulator of ER calcium homeostasis. This result is in apparent contradiction with the perceived role of ATF6 as a largely protective UPRER effector. Together with the observation that calreticulin is elevated with age in an atf6-dependent manner, this study suggests that ATF6 signaling can prove detrimental with age under basal conditions, independent of its canonical UPRER role in maintaining proteostasis (Burkewitz et al., 2020).
Non-cell Autonomous Endoplasmic Reticulum Unfolded Protein Response Signaling and Aging
Given the recently uncovered importance of non-cell autonomous UPRER signaling in regulating whole-body organismal homeostasis, it is no surprise that this mechanism also plays a role in the aging process. In C. elegans, neuronal xbp-1s expression leads to intestinal UPRER activation, conferring extended lifespan and improved ER stress resistance with age (Taylor and Dillin, 2013). However, if distal communication to the intestine is disrupted via the intestinal knockdown of xbp-1s or inhibition of neuronal secretory regulator unc-13, the beneficial effects on longevity and stress resistance are lost (Taylor and Dillin, 2013). Similarly, the enhancements to lifespan and proteostasis attained via glia-intestine non-cell autonomous UPRER signaling are both abolished in worms defective in neuropeptide release (Frakes et al., 2020). The specific messenger molecules required for non-cell autonomous UPRER activation in nematodes, however, remain unknown in both the neuronal and glial paradigms. Identifying these mediators, and whether they themselves are conserved, are vital steps in uncovering how UPRER regulates aging in a whole-body manner. Additionally, recent studies in mice have provided initial evidence of mammalian UPRER signaling across distal tissues; Xbp1s expression in Pomc neurons drives UPRER activation in the liver to promote metabolic homeostasis, protecting mice from diet-induced obesity (Williams et al., 2014; Brandt et al., 2018).
In line with these observations, the intestinal pathways initiated downstream of neuronal UPRER signaling are also required to promote healthy aging. Impairing these UPRER-mediated changes, namely increased lysosomal activity and lipid remodeling, block the lifespan extension conferred by neuronal xbp-1s expression (Imanikia et al., 2019a,b; Daniele et al., 2020). Interestingly, neuronal or intestinal expression of xbp-1s also enhances lysosome activity and proteostasis in distal muscle cells, while expression of xbp-1s specifically in muscle cells leads to a downregulation of lysosomal genes and reduced lifespan (Imanikia et al., 2019a). Thus, via modulation of two critical ER functions, protein and lipid homeostasis, distal UPRER signaling can, in turn, benefit organismal health. Additionally, these findings also position the intestine as a critical regulator of organism-wide aging. This is in agreement with a host of recent studies demonstrating the role of the gut, in particular gut microbiota, as a key regulator of organismal aging (Smith et al., 2017; Donaldson et al., 2020; Boehme et al., 2021; Shukla et al., 2021). Whether gut microbial composition contributes to the non-cell autonomous UPRER regulation of aging is at this stage largely unknown and provides an important target for investigation moving forward.
The Mitochondrial Unfolded Protein Response in Aging
Changes to the Mitochondrial Unfolded Protein Response in Aging
Through observational investigations, activation of the UPRmt has been found to correlate positively with longevity across the animal kingdom. Experiments conducted in C. elegans have shown that the expression level of mitochondrial chaperones and proteases progressively increases across the lifespan and further studies have shown that worms with higher levels of basal UPRmt activation have longer lifespans (Sheng et al., 2021; Zhang et al., 2021). In mammals, analyses of cell cultures derived from long-lived mouse strains show upregulation of the UPRmt genes Hsp60 and Lonp1 (Ozkurede and Miller, 2019). Furthermore, Houtkooper and colleagues, by analyzing the natural variation of gene transcript levels in multiple strains of an inbred population of mice, show that conditions known to activate the UPRmt are correlated with longevity (Houtkooper et al., 2013). In total, these data suggest that the activation of the UPRmt with age is beneficial and extends lifespan.
However, data also exist suggesting that activation of the UPRmt is not correlated with lifespan extension. Higher plasma concentrations of the UPRmt-inducing mitokines FGF21 and GDF15 are associated with shorter lifespans, poorer handgrip strength, and increased insulin insensitivity in elderly adults (Conte et al., 2019). These plasma mitokines may represent a response to increased mitochondrial stress; however, the UPRmt-promoting effects of these mitokines may not be enacted due to decreased age-related inducibility of the UPRmt leading to a failure to alleviate the inciting mitochondrial stress and restore cellular homeostasis (Sheng et al., 2021).
Manipulations to the Mitochondrial Unfolded Protein Response Affect Aging
Direct manipulations of mitochondrial stress and components of the UPRmt have shed important insights into the role that UPRmt activation plays in aging. Overall, it appears that signaling through the ATF5-UPRmt and SIRT3-UPRmt, either independently, together, or in concert with other cellular pathways, slows aging and extends organismal lifespan.
Activating Transcription Factor 5-Mitochondrial Unfolded Protein Response in Aging
There is a large body of literature supporting the assertion that the transcriptional changes induced by the ATF5-UPRmt work to promote longevity and healthy aging across species. Multiple studies have shown that the lifespan extension observed in numerous mitochondrial ETC mutants is dependent upon signaling through atfs-1 or its downstream factors ubl-5 and dve-1 (Durieux et al., 2011; Wu et al., 2018; Gao et al., 2019). Impairment of mitochondrial protein translation extends C. elegans lifespan via a mechanism that is dependent upon the creation of a mito-nuclear imbalance within the mitochondria and signaling through the ATF5-UPRmt; this mechanism also appears to be conserved in mice (Houtkooper et al., 2013). Furthermore, as evidence of ATF5-UPRmt-dependent healthy aging, Owusu-Ansah and colleagues showed that flies with impaired ETC complex I function retain more motor function as they age compared to controls; this delay in age-related functional decline is partially lost upon knockdown of ATF5-UPRmt-dependent protease and chaperones (Owusu-Ansah et al., 2013).
However, data do exist that challenge the notion that ATF5-UPRmt-dependent transcriptional changes alter longevity. A critical study from the Kaeberlein lab provides multiple pieces of data that suggest that the ATF5-UPRmt is not involved in lifespan extension. They show that the activation of the UPRmt in the absence of mitochondrial stress does not extend lifespan in C. elegans and that atfs-1 is neither necessary nor sufficient for lifespan extension (Bennett et al., 2014). Collectively, these are surprising results, but the possibility remains that the pro-longevity effects of UPRmt activation are due to non-atfs-1-dependent mechanisms; that is, UPRmt-induced lifespan extension is separable from atfs-1-induced transcriptional changes. Indeed, studies that show atfs-1 playing a role in UPRmt-dependent lifespan extension only show a partial reduction in longevity upon deletion or knockdown of atfs-1; this suggests the involvement of other UPRmt or non-UPRmt pathways in promulgating mitochondrial stress-induced lifespan extension (Owusu-Ansah et al., 2013; Wu et al., 2018). Additionally, a recent study showed that knockdown of the ATP synthase component atp-3 in early adulthood causes atfs-1-dependent activation of the UPRmt and reduces lifespan through a mechanism dependent upon the formation of a mitochondrial permeability transition pore (mPTP), a non-specific pore in the MIM that allows for increased movement of contents between the mitochondrial matrix and IMS (Angeli et al., 2021). Manipulations that prevent the formation of the mPTP restore lifespan and partially ameliorate the UPRmt activation (Angeli et al., 2021). It may be that the persistent opening of the mPTP in the context of atp-3 manipulation is a strong pathological event that counteracts any beneficial effects of UPRmt activation (Pérez and Quintanilla, 2017). Lastly, some data suggest that genetic manipulations that induce mitochondrial stress do not extend lifespan (Durieux et al., 2011; Bennett et al., 2014). Thus, the lack of evidence of the universality of ATF5-UPRmt-induced lifespan extension points to the complexity of the relationship between ATF5-UPRmt-related cellular manipulations and organismal lifespan.
Epigenetic modifications induced by the ATF5-UPRmt seem to promote longevity. Numerous epigenetic modulators have been shown to play roles in maintaining the activation of the UPRmt and facilitating lifespan extension from worms to humans. These include the histone demethylases jmjd-1.2/PHF8, jmjd-3.1/KDM6B, the epigenetic reader baz-2/BAZ2B, the histone methyltransferase set-6/EHMT1, the acetyltransferase cbp-1/CBP/p300, and the histone deacetylase hda-1 (Merkwirth et al., 2016; Shao et al., 2020; Yuan et al., 2020; Li et al., 2021). Additionally, overexpression of histone H4 is sufficient to extend lifespan in C. elegans (Sural et al., 2020). To our knowledge, there exists no data showing that epigenetic changes associated with activation of the UPRmt are detrimental for longevity. In conclusion, the long-lasting induction of the UPRmt after the resolution of mitochondrial stress points to the existence of epigenetic changes that maintain the UPRmt-induced transcriptional state to induce lifespan extension (Durieux et al., 2011; Zhang et al., 2021).
ERα-Mitochondrial Unfolded Protein Response in Aging
Of the three identified arms of the UPRmt, the ERα-UPRmt arm is least implicated in longevity modulation. Evidence exists that administration of 17α-estradiol, which acts through ERα, can extend lifespan and that these effects are partly mediated through metabolic changes; however, it is yet to be shown whether these effects are mediated by the same ERα signaling pathways that are also activated by mitochondrial stress (Strong et al., 2016; Stout et al., 2017; Garratt et al., 2018; Mann et al., 2020). Additionally, increased expression of ERα appears to mediate some of the effects of lifespan extending manipulations such as caloric restriction (Yaghmaie et al., 2005; Morselli et al., 2014). It should be noted that multiple studies of the role of UPRmt activation on longevity have demonstrated marked reproductive changes with UPRmt activation (Dillin et al., 2002; Rea et al., 2007; Durieux et al., 2011; Baqri et al., 2014; Zhang et al., 2021). While not explicitly linked to ERα, there is certainly a relationship, however unclear, among UPRmt activation, lifespan extension, and reproductive capacity.
SIRT3-Mitochondrial Unfolded Protein Response in Aging
The SIRT3-UPRmt appears to play a role in lifespan extension, but whether these effects are due to direct SIRT3-dependent deacetylation of mitochondrial proteins or SIRT3/FOXO3-dependent transcriptional changes is not yet clear. SIRT3 was first suggested as a longevity gene after the identification of multiple single nucleotide polymorphisms within SIRT3 that are associated with lifespan extension (Hurst et al., 2002; Rose et al., 2003; Bellizzi et al., 2005). Later mechanistic studies have converged on Sirt3-dependent deacetylation of mitochondrial proteins that are involved in metabolism, proteostasis, and antioxidant defense as critical for lifespan extension in mammals (Hebert et al., 2013). Additionally, Sirt3 post-transcriptionally modifies the key UPRmt effectors Hsp10 and Lonp1; however, it is yet unclear whether this post-transcriptional modification plays a role in longevity modulation (Gibellini et al., 2014; Lu et al., 2015). While some data suggest that the deacetylation of mitochondrial proteins by SIRT3 may play a role in lifespan extension, it is entirely unclear whether the FOXO3-dependent transcriptional axis of the SIRT3-UPRmt is involved in controlling lifespan. Although nuclear accumulation of daf-16, the C. elegans ortholog of FOXO3, is known to contribute to lifespan extension in worms, it remains unclear whether this occurs through the SIRT3-UPRmt or another parallel pro-longevity pathway (Lin et al., 2001). Additionally, data showing that daf-16 is dispensable for mtROS-induced lifespan extension argue that SIRT3/FOXO3-dependent transcriptional changes do not contribute to lifespan extension (Schmeisser et al., 2013). Additional research is warranted in this area to determine whether the transcriptional axis of the SIRT3-UPRmt is involved in lifespan extension.
Signaling Through Multiple Arms of the Mitochondrial Unfolded Protein Response in Aging
Alterations in ETC complexes and ROS levels appear to activate the UPRmt and affect longevity. However, these changes do not fit cleanly into a paradigm where they only activate a single arm of the UPRmt; that is, there are mitochondrial stresses that appear to simultaneously signal through multiple arms of the UPRmt to influence longevity. There is extensive evidence that manipulations of the ETC modulate longevity. Specifically, decreased expression of ETC complex subunits has been shown to extend lifespan in model organisms ranging from C. elegans to mice (Dillin et al., 2002; Dell’Agnello et al., 2007; Copeland et al., 2009). Importantly, it appears that the magnitude of the knockdown of ETC complex components is critical for the lifespan extending effect: severe disruptions of the ETC shorten lifespan while mild ETC disruptions extend lifespan (Rea et al., 2007). This suggests that the mitochondrial stress associated with changes in the ETC causes a mitohormetic extension of lifespan (Tapia, 2006). The link between changes in the ETC and longevity appears to be tied to ROS production and UPRmt activation (Lee et al., 2003b,2010b; Yang and Hekimi, 2010a; Rauthan et al., 2015). For example, mild knockdown of an ETC complex I component increases lifespan in Drosophila through a redox-dependent induction of the UPRmt, although the precise branch of the UPRmt that this effect was mediated through is not well defined (Owusu-Ansah et al., 2013). Mechanistically, data are lacking to determine specifically how ETC disruption affects lifespan, but it is likely that manipulations of the ETC alter lifespan by affecting both the SIRT3-UPRmt and ATF5-UPRmt. Because SIRT3 is a redox-sensitive protein, disruption of ETC complexes I and III, which are known to be the primary sources of ROS in the mitochondria, may activate the SIRT3-UPRmt to modulate aging (Lennicke and Cochemé, 2021). Simultaneously, ETC manipulations could create a mito-nuclear imbalance or oxidative protein damage which activates the ATF5-UPRmt to alter longevity.
It should be noted, however, that there are manipulations of the ETC that are known to activate the UPRmt without extending lifespan. For example, manipulating the expression of ETC complex II subunits does not result in long-lived worms despite the activation of the UPRmt (Ishii et al., 1998; Durieux et al., 2011; Kuang and Ebert, 2012). This may be because complex II is the only ETC complex that is solely encoded by nuclear DNA; this may suggest that the mito-nuclear imbalance, and the subsequent activation of the ATF5-UPRmt, is a critical factor in the lifespan extension (Kuang and Ebert, 2012; Houtkooper et al., 2013). It may also represent the fact that the inclusion of complex II as part of the ETC is a historical artifact; indeed, succinate dehydrogenase, as a key player in the TCA cycle, has more in common with other TCA enzymes than it does with the other ETC complexes (Munkácsy and Rea, 2014). Additionally, ETC manipulations that extend lifespan in one setting may not extend lifespan in another; for example, RNAi knockdown of a complex IV component in intestine and neurons extends lifespan in C. elegans, but muscle-specific knockdown of the same complex IV component does not (Durieux et al., 2011).
As discussed previously, ROS appear to modulate aging through both the ATF5-UPRmt and SIRT3-UPRmt. For example, mice with high concentrations of ROS at young ages tend to live longer than mice with lower levels of ROS; additionally, these long-lived mice show elevated levels of proteins related to the ATF5-UPRmt that persist throughout life, even after ROS levels normalize in adulthood (Latorre-Pellicer et al., 2016). Regarding the SIRT3-UPRmt, it has been shown that transient increases in ROS increase the expression of antioxidant enzymes and extend lifespan in C. elegans; although this study did not explicitly investigate whether the ROS-induced antioxidant gene expression was dependent upon the SIRT3-UPRmt, the similarity of the end effects leads to the speculation that transient ROS spikes may extend lifespan through the SIRT3-UPRmt (Zarse et al., 2012). Additionally, the lifespan increase of ETC mutants due to the activation of the ATF5-UPRmt is separable from the lifespan increase due to elevated levels of mtROS (Yang and Hekimi, 2010b). This suggests that increased ROS, in part, extends lifespan through pathways parallel to the ATF5-UPRmt. Again, it may be that this parallel pathway is the SIRT3-UPRmt, although this has not yet been examined. Lastly, elevated levels of mtROS increase lifespan while elevated levels of cytosolic ROS decrease lifespan providing further evidence of the potentially beneficial effects of mtROS, perhaps due to the ability of mtROS to activate the UPRmt (Schaar et al., 2015).
There are, however, numerous reports that suggest ROS can activate the UPRmt without leading to lifespan extension. In five different C. elegans lines with RNAi-mediated knockdown of mitochondrial proteins, the levels of oxidative stress within the animals does not correlate with lifespan (Rea et al., 2007). Additionally, treatment of long-lived C. elegans mitochondrial mutants with ROS-scavenging antioxidants fails to reduce lifespan despite the strong activation of the UPRmt (Durieux et al., 2011; Houtkooper et al., 2013). This suggests that another UPRmt trigger such as mito-nuclear imbalance rather than ROS explains the lifespan extension in these animals. In mice, overexpression of antioxidant genes largely has no effect on lifespan suggesting that oxidative stress and damage does not play a role in mammalian aging; however, the overexpression of antioxidants may impair the ability of a transient ROS spike to induce the UPRmt to extend lifespan (Pérez et al., 2009a). Collectively, these studies suggest that the relationship among ROS, UPRmt activation, and lifespan extension remains murky. It is likely that some combination of the timing, magnitude, and type of ROS spike is important for lifespan extension, but further research is required to determine the precise conditions that facilitate ROS- and UPRmt-dependent lifespan extension and why.
The Mitochondrial Unfolded Protein Response Interacts With Other Cellular Pathways in Aging
There also exist mitochondrial stress-dependent pathways that modulate aging by mechanisms not typically associated with UPRmt activation. For example, mitochondrial stress has been shown to extend lifespan, in part, through anti-viral defense mechanisms, non-canonical apoptosis signaling, cytosolic stress responses pathways, and reduced cytosolic translation (Baker et al., 2012; Delaney et al., 2013; Yee et al., 2014; Labbadia et al., 2017; Matilainen et al., 2017; Mao et al., 2020; Molenaars et al., 2020). Collectively, these data suggest that parallel cellular mechanisms play roles in the lifespan extension associated with mild mitochondrial stress and the activation of the UPRmt. Furthermore, these data may explain some discrepancies regarding the role of the UPRmt in longevity modulation.
Non-cell Autonomous Mitochondrial Unfolded Protein Response Signaling and Aging
Mitochondrial stress in one tissue can activate the UPRmt in tissues not experiencing mitochondrial stress, and this non-cell autonomous stress signaling has recently been shown to be involved, at least in part, in lifespan extension. To achieve this effect, it appears that two tissues are critical: neurons as the cells of stress sensing and the intestine as the tissue in which the UPRmt is activated. Durieux et al. (2011) first demonstrated that mitochondrial stress in C. elegans neurons non-cell autonomously activates the UPRmt in the intestine and that this signaling plays a role in lifespan extension. Other work has confirmed that this mitochondrial stress pathway from neurons to intestine promotes longevity (Tian et al., 2016; Zhu et al., 2020). Interestingly, this non-cell autonomous signaling mirrors the non-cell autonomous link between neurons and the intestine that is implicated in longevity in response to ER stress, as discussed previously (Taylor and Dillin, 2013; Frakes et al., 2020). It appears, moreover, that there are other inter-tissue communication networks that signal mitochondrial stress and influence longevity. For example, translational repression of cytochrome c in the gonad causes the non-cell autonomous activation of the UPRmt in the intestine to extend lifespan (Lan et al., 2019). However, the interconnected signaling networks that exist between reproductive and neuronal cells raises the potential that the gonad is responding to signals originating further upstream in the nervous system (Miller et al., 2020). Collectively, these data allow speculation that mitochondria in one cell type, perhaps neurons, coordinate the rate of aging for an organism through non-cell autonomous mechanisms and that the intestine may be the major tissue responsible for executing these effects. If this proves to be at least partly true, it must also be considered whether the gut plays a role in longevity not because of some intrinsic property of intestinal cells but because of the influence of the gut microbiome (Shukla et al., 2021).
Temporal Effects of Mitochondrial Unfolded Protein Response Activation on Aging
Across multiple studies investigating the role of UPRmt activation on longevity, a noteworthy trend has emerged: activation of the UPRmt during development appears to confer longevity benefits while initiation of the UPRmt during adulthood does not. Induction of the UPRmt during larval development is sufficient to extend lifespan and maintain the induction of the UPRmt into adulthood (Dillin et al., 2002; Rea et al., 2007; Lee et al., 2010b; Durieux et al., 2011; Borch Jensen et al., 2018; Bazopoulou et al., 2019). It appears that mitochondrial stress during development causes epigenetic changes that induce persistent UPRmt-associated gene changes into adulthood (Merkwirth et al., 2016; Tian et al., 2016; Zhu et al., 2020). However, induction of the UPRmt only during adulthood does not affect lifespan and may have detrimental effects, perhaps because of age-related epigenetic alterations that prevent access to UPRmt-induced genes (Dillin et al., 2002; Rea et al., 2007; Durieux et al., 2011; Labbadia and Morimoto, 2015; Conte et al., 2019).
Conclusion
Collectively, data from human post-mortem tissue and animal models suggest that both the UPRER and the UPRmt change significantly at older ages. With increasing age, both the UPRER and the UPRmt lose adaptive signaling potency potentially leading to a detrimental state where the UPR is unable to resolve the stresses brought about by age-related cellular dysfunction (Sheng et al., 2021). It appears that modulation of the UPRER and the UPRmt can have differing effects on age-related functional and physiological changes. For the UPRER, manipulations can prove either beneficial or deleterious depending upon the molecular and tissue targets being investigated, and thus more research is needed to define these specific contexts. For the UPRmt, it appears that activation through the ATF5-UPRmt and SIRT3-UPRmt promotes healthy aging and lifespan extension, particularly if induced in juveniles. However, whether activation of these arms of the UPRmt is sufficient to extend lifespan remains unclear due, in part, to the technical challenges of isolating UPRmt signaling from the inciting stressors and other activated parallel stress response pathways.
The Unfolded Protein Response in Neurodegenerative Disease
The direct physiological and functional consequences of aging are not the only drivers of age-dependent mortality; aging is also the greatest risk factor for the majority of neurodegenerative diseases (NDDs), including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) (Hou et al., 2019). Two common features shared amongst the age-related NDDs are the accumulation of proteinaceous aggregates and mitochondrial dysfunction (Hou et al., 2019). Consequently, recent studies have begun to examine how stress response pathways like the UPRER and UPRmt mitigate or aggravate cellular dysfunction in age-related NDDs. Here, we discuss the relationship between the organellar UPRs and several prominent age-related NDDs, including evidence from human post-mortem tissue of UPR activation and experimental data from model organisms demonstrating how the activation of the UPRs may alter the course of neurodegenerative pathology.
The Endoplasmic Reticulum Unfolded Protein Response in Neurodegenerative Disease
The Endoplasmic Reticulum Unfolded Protein Response in Alzheimer’s Disease
Alzheimer’s disease is a NDD that primarily affects cognitive function, and represents the most common cause of dementia worldwide (Schneider et al., 2009). Histologically, AD is largely defined by the accumulation of extracellular amyloid-β (Aβ)-containing plaques and intracellular abnormal tau neurofibrillary tangles (NFTs), both of which are recognized as key, though not the only, biomarkers and possible drivers of AD pathology (Knopman et al., 2021).
A host of post-mortem studies on human brain tissue have provided evidence of UPRER activity in AD. In the affected brain regions of AD patients, the molecular chaperone BiP is elevated in neurons at an early stage of AD pathology, suggesting that the UPRER is activated early as a protective response in AD (Hoozemans et al., 2005). Phosphorylated IRE1α (p-IRE1α) is increased in the hippocampus of AD patients (Hoozemans et al., 2009) and has been shown to correlate with AD progression (Duran-Aniotz et al., 2017). The phosphorylated forms of PERK and eIF2α (p-PERK and p-eIF2α, respectively) are also associated with regions with AD pathology (Hoozemans et al., 2009), with p-PERK in particular concentrating in neurons with high levels of phosphorylated tau (p-tau) aggregation and markers of early NFT-induced degeneration (Unterberger et al., 2006; Hoozemans et al., 2009). Thus, a close association between IRE1α and PERK, two master regulators of the UPRER, and the presence of p-tau appears a common feature in post-mortem AD studies. Indeed, separate studies examining brain tissue from other sporadic tauopathies, including Pick disease and progressive supranuclear palsy, have also demonstrated the correlation of both p-IRE1α and p-PERK with p-tau (Nijholt et al., 2012; Stutzbach et al., 2013). Downstream of PERK, elevated levels of ATF4, GADD34, and CHOP have also been observed in AD tissue (Yoon et al., 2012; Baleriola et al., 2014; Honjo et al., 2015). Collectively, these studies suggest that UPRER activation represents an early event in AD pathogenesis, raising the question of whether the shift of UPRER signaling from adaptive to chronic with disease progression may contribute to disease progression.
Studies utilizing animal models of AD have further emphasized both positive and negative roles of the IRE1α-UPRER and PERK-UPRER in AD pathogenesis. In Drosophila, Xbp1 is upregulated following transgenic expression of either Aβ or mutant human tau and confers neuroprotection in both models (Loewen and Feany, 2010; Casas-Tinto et al., 2011). In line with this neuroprotective role, hippocampal expression of Xbp1s in a triple transgenic mouse model of AD (3xTg-AD) rescues structural abnormalities and memory deficits (Cissé et al., 2017). Confusingly, however, the IRE1α-UPRER can also drive AD pathology; conditional knockout of IRE1α in a widely used mouse model containing five human familial AD mutations (5xFAD) restores cognitive function and reduces Aβ deposition (Duran-Aniotz et al., 2017). Likewise, xbp-1 deficiency in nematodes reduces Aβ aggregation and toxicity (Safra et al., 2013). Thus, activity of the IRE1α-UPRER can prove either beneficial or detrimental in AD models. This could reflect both the differences in disease model and at what stage the manipulation occurs in disease progression, which in turn may dictate whether IRE1α/XBP1 activation ameliorates or hastens neurodegenerative decline.
Elevated activity of the PERK-UPRER is a common feature in AD patient tissue; therefore, manipulations aimed at inhibiting PERK signaling have largely been the focus in the context of AD. Suppressing eIF2α phosphorylation via the genetic deletion of Perk rescues deficits in spatial memory and synaptic plasticity in a double transgenic APP/PS1 mouse model of AD (Ma et al., 2013; Yang et al., 2016). Similarly, Perk haploinsufficiency protects against memory impairment and cholinergic neuronal loss in 5xFAD mice, concomitant with a reduction in Aβ plaque burden (Devi and Ohno, 2014). Further downstream of PERK, ATF4 may also contribute to AD pathology; knockdown of Atf4 in axons exposed to oligomeric Aβ prevents the spreading of a degenerative signal across the mouse brain and subsequent neuronal cell loss, in part through the suppression of CHOP-mediated apoptotic signaling (Baleriola et al., 2014). Taken together, the results point to PERK signaling in AD as largely detrimental, suggesting that suppressing PERK activity may prove beneficial as an intervention in AD disease.
Little is known regarding whether the ATF6-UPRER contributes to AD pathogenesis, though recent work has provided insight into this question. Atf6 expression is reduced in the APP/PS1 mouse model, and overexpression of Atf6 rescues spatial memory impairment and decreases Aβ plaque load in these mice (Du et al., 2020). Thus, the upregulation of ATF6 appears to function as a protective response in AD, though more work in independent models will be required to confirm this role.
The Endoplasmic Reticulum Unfolded Protein Response in Parkinson’s Disease
Parkinson’s disease is a progressive NDD most obviously associated with motor symptoms including bradykinesia, tremor, and rigidity, and is the second most prevalent age-related NDD (Poewe et al., 2017). Neuropathological hallmarks of the disease include the selective loss of dopaminergic neurons within the substantia nigra and the intracellular accumulation of proteinaceous aggregates containing α-synuclein (α-syn), referred to as Lewy bodies (Bloem et al., 2021).
In post-mortem PD tissue, increased p-PERK and p-eIF2α immunoreactivity is observed in dopaminergic neurons of the substantia nigra, and p-PERK presence correlates with neurons positive for α-syn accumulation (Hoozemans et al., 2007). In addition, α-syn aggregates accumulate within ER microsome fractions of human PD brain tissue (Colla et al., 2012). Therefore, UPRER activation in PD appears largely in response to the deposition of α-syn aggregates, which is in agreement with in vitro and in vivo experiments demonstrating that α-syn accumulates within the ER and can activate the UPRER via a physical interaction with BiP (Bellucci et al., 2011).
Neurotoxin-induced experimental models of PD have primarily been used to dissect the role of UPRER signaling in PD pathophysiology. Viral overexpression of Xbp1s protects against dopaminergic neuron loss in a mouse model of PD induced by treatment with MPP+, an inhibitor of ETC complex I (Sado et al., 2009). A similar neuroprotective role for Xbp1s was shown in mice following treatment with 6-hydroxydopamine (6-OHDA), a generator of ROS that is acutely toxic to dopaminergic neurons (Valdés et al., 2014). Intriguingly, while reducing Xbp1 in the adult mouse brain triggers chronic ER stress and is detrimental for neuronal survival, developmental deletion of Xbp1 is in fact neuroprotective (Valdés et al., 2014). This developmental ablation of Xbp1 triggers mild ER stress and subsequently drives an adaptive UPRER program, including enhancing autophagy, that protects dopaminergic neurons from cell death (Valdés et al., 2014). A similar form of UPRER/autophagy-mediated protection was independently observed in both fly and mouse PD models following low dosage treatment with tunicamycin (Fouillet et al., 2012), suggesting that early ER stress can prime the UPRER for later PD-associated challenges, in part through the upregulation of autophagy. In contrast to the largely protective role of XBP1 in PD, Ire1 expression in fly dopaminergic neurons drives JNK- and autophagy-dependent neuronal death, while its reduction ameliorates α-syn induced neurodegeneration (Yan et al., 2019). This deleterious mode of IRE1α signaling appears independent of XBP1, which therefore might represent the switch to chronic IRE1α activity in PD. Taken together, these data highlight that XBP1 expression downstream of IRE1α is neuroprotective and may represent a possible therapeutic target but that other XBP1-independent IRE1α signaling pathways have the potential to act in a deleterious fashion in PD.
The role of the other two UPRER arms in PD pathogenesis has been less-well studied genetically. Work in murine models of PD has demonstrated that deletion of the PERK downstream apoptotic regulator Chop protects against 6-OHDA-induced dopaminergic neuron death (Silva et al., 2005), suggesting that the pro-apoptotic arm of PERK signaling is induced and is detrimental for dopaminergic neuron survival. Conversely, the role of ATF6 appears neuroprotective in the context of PD. Following MPP+ treatment, Atf6 promotes astrocytic activation and the upregulation of chaperones and ERAD genes to prevent dopaminergic neurodegeneration (Egawa et al., 2011; Hashida et al., 2012). Taken together with the previously discussed protective function of XBP1 in PD, signaling through both the PERK-UPRER and the ATF6-UPRER appear to augment neuroprotective programs in PD models. This conclusion is supported by a recent study demonstrating that viral-mediated delivery of an XBP1/ATF6 fusion protein promotes degradation of α-syn aggregates in vivo and protects dopaminergic neurons following 6-OHDA treatment (Vidal et al., 2021). Therefore, the promotion of UPRER genes downstream of XBP1 and ATF6, including chaperones and degradative pathways, displays neuroprotective potential in PD pathogenesis.
The Endoplasmic Reticulum Unfolded Protein Response in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
Amyotrophic lateral sclerosis is a NDD characterized cellularly by motor neuron death and clinically by muscle weakness and atrophy leading to paralysis and death typically within 2–5 years of disease onset (Harris, 2014). Additionally, approximately 40% of ALS patients present with cognitive and behavioral impairments with half of these patients being diagnosed with behavioral variant FTD (Masrori and Van Damme, 2020). Indeed, ALS and FTD display significant clinical and genetic overlap and are now considered to be on a spectrum; lesions in several genes, including TAR-DNA binding protein 43 (TDP-43) and chromosome 9 open reading frame 72 (c9ORF72), can cause ALS, FTD, or both (Abramzon et al., 2020). The majority of ALS/FTD subtypes can be characterized by the presence of cytoplasmic proteinaceous aggregates positive for TDP-43 (Sreedharan et al., 2008). However, in other subtypes different protein species can be present such as misfolded superoxide dismutase (SOD1) or dipeptide repeat proteins (DPRs) produced from a mutant form of c9ORF72, the highest frequency familial ALS/FTD causing lesion (Hardiman et al., 2017).
The activation of UPRER chaperones appears to be a common feature in ALS patient samples. Both BiP and PDI have been observed in ALS spinal cord tissue (Ilieva et al., 2007; Sasaki, 2010; Montibeller et al., 2020), as have UPRER transcription factors XBP1 (Hetz et al., 2009) and CHOP (Ito et al., 2009). Furthermore, BiP and PDI are represented among a cluster of chaperones upregulated in ALS peripheral blood mononuclear cells (Nardo et al., 2011). Finally, a recent transcriptome analysis of c9ORF72 ALS patient tissue identified widespread alterations to genes involved in the UPRER in both cerebellum and cortex samples (Prudencio et al., 2015). UPRER activation is also associated with FTD; p-IRE1α, p-PERK, and p-eIF2α are elevated in c9ORF72 FTD patient samples, observed in close association with DPR inclusions (Gami-Patel et al., 2021). Therefore, the presence of markers of UPRER activation appear across the ALS/FTD continuum, suggesting that UPRER activity may represent a common pathological feature amongst the range of lesions known to cause these diseases.
Experimental models of ALS/FTD have been used to investigate the contribution of upregulated UPRER components to disease pathology. Knockdown of IRE1α or Xbp1 reduces mutant SOD1 aggregation in motor neurons both in vitro and in mutant SOD1 mice, subsequently extending lifespan and reducing apoptotic cell death (Hetz et al., 2009). Intriguingly, Xbp1 deficiency impairs ERAD function but upregulates autophagy, suggesting that compensatory degradative pathways are activated in this paradigm (Hetz et al., 2009). Initial studies using mutant SOD1 mice demonstrated that the PERK-UPRER acts protectively in ALS pathogenesis (Wang et al., 2011, 2014a), though a more recent study using several strains of mutant SOD1 mice demonstrated that manipulating the PERK pathway had no effect on disease onset or progression, which could reflect relative differences in mutant SOD1 expression levels between mice strains (Dzhashiashvili et al., 2019). However, downstream of PERK, deleting Atf4 in mutant SOD1 mice reduces developmental viability but ameliorates disease phenotypes, possibly via the reduction of apoptotic effector CHOP (Matus et al., 2013). Thus, the true contribution of the PERK-UPRER to mutant SOD1 forms of ALS/FTD remains up for debate, whilst its participation in other forms of ALS/FTD pathology is largely unexplored.
The Mitochondrial Unfolded Protein Response in Neurodegenerative Disease
The Mitochondrial Unfolded Protein Response in Alzheimer’s Disease
Data from both animal models and humans show that the UPRmt is intimately involved in AD pathobiology. Genes involved in the UPRmt, such as YMEL1L, which encodes a mitochondrial AAA + protease and HSP60, which encodes a chaperone, are upregulated in post-mortem brain tissue obtained from patients with AD (Beck et al., 2016; Sorrentino et al., 2017). Upregulation of UPRmt-related genes is also recapitulated in the nervous systems of animal models of AD (Sorrentino et al., 2017; Shen et al., 2020). Based on data showing that Aβ accumulation co-occurs with mitochondrial import impairments, Sorrentino et al. (2017) argue that Aβ may impair mitochondrial import efficiency to trigger the UPRmt. Additionally, the finding that resveratrol, an activator of sirtuins, reduces Aβ toxicity in a ubl-5-dependent fashion suggests that Aβ may activate both the ATF5-UPRmt and SIRT3-UPRmt (Regitz et al., 2016). Regardless of the mechanism by which the UPRmt is activated in AD, it is clear from multiple independent studies that the activation is largely beneficial. Brief exposure of human neuroblastoma-derived SH-SY5Y cells to Aβ fragments causes the accumulation of the mitochondrial proteases LONP1, HTRA2, and CLPP and as well as the mitochondrial chaperone HSP60, indicating that the UPRmt has been activated (Shen et al., 2020). This induction of the UPRmt prevents Aβ-induced cellular toxicity which suggests that acute UPRmt activation may represent a beneficial compensatory cellular response (Shen et al., 2020). Additionally, the overexpression of atfs-1 in a C. elegans model of AD further induces the UPRmt and improves healthspan suggesting that the induction of the UPRmt offsets some of the pathological effects of Aβ aggregation (Sorrentino et al., 2017). Lastly, inhibition of the UPRmt worsens cellular features commonly associated with AD such as an increased Aβ42:Aβ40 ratio and increased phosphorylated tau in an AD-like human cerebral organoid model (Pérez et al., 2020).
The Mitochondrial Unfolded Protein Response in Parkinson’s Disease
Mitochondrial dysfunction is a key player in the pathophysiology of PD, and many genetic and toxic causes of PD are also known to activate the UPRmt (Pellegrino and Haynes, 2015; Bloem et al., 2021). To our knowledge, no one has yet completed a comprehensive study of post-mortem brain tissue from PD patients examining multiple markers of UPRmt activation. However, transcript and protein levels of UPRmt-related proteases and chaperones are increased in human cell lines exposed to the PD-causing toxin MPP+ suggesting that the UPRmt may indeed be activated in PD (Cai et al., 2020). Bolstering evidence that the UPRmt plays a role in PD in humans is the finding that some PD patients have mutations in HTRA2, a key player in the ERα-UPRmt (Strauss et al., 2005; Unal Gulsuner et al., 2014). In animal and cell models of PD, the activation of the UPRmt has been shown to exert both protective and damaging effects. In MPP+-treated SH-SY5Y cells, further activation of the UPRmt reduces cell death (Hu et al., 2021). Additionally, in genetic worm models of PD, inactivation of the UPRmt decreases lifespan and worsens dopaminergic neuron loss (Cooper et al., 2017). Furthermore, overexpression of the UPRmt component CLPP, which encodes a mitochondrial matrix protease, in induced pluripotent stem cell-derived neurons from PD patients reduces pathogenic α-syn phosphorylation, reduces mitochondrial oxidative stress, and restores neuron morphology (Hu et al., 2019). In contrast, Martinez et al. (2017) found that chronic over-activation of the UPRmt is detrimental for lifespan and healthspan in C. elegans overexpressing a mutant form of α-syn. It is likely that this chronic UPRmt hyperactivation exerts detrimental cellular effects in concert with aggregates of α-syn. Mechanistically, it appears that aggregation-prone α-syn accumulates within mitochondria to activate the UPRmt directly (Hu et al., 2019). Overall, it appears that the UPRmt is beneficial under many circumstances, save those where the UPRmt is chronically over-activated.
The Mitochondrial Unfolded Protein Response in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
Mounting evidence suggests that the UPRmt is activated in the context of ALS/FTD-related mutant TDP-43, SOD1, CHCHD10, and FUS; however, it remains unclear what role this UPRmt activation plays in the ALS/FTD pathobiology (Riar et al., 2017; Deng et al., 2018; Anderson et al., 2019; Pharaoh et al., 2019; Wang et al., 2019; Straub et al., 2021). Mutant TDP-43 induces the UPRmt, likely through inhibition of ETC complex I leading to reduced mitochondrial ATP synthesis, in both human cell lines and transgenic Drosophila models of ALS/FTD (Wang et al., 2019). In these models, downregulation of the mitochondrial protease LONP1 increased TDP-43 protein levels and exacerbated TDP-43-induced mitochondrial dysfunction and neurodegeneration suggesting that the UPRmt acts to counter the detrimental effects of TDP-43 (Wang et al., 2019). In transgenic mice expressing mutant human SOD1, the ATF5-UPRmt appears to be activated in the early stages of ALS disease in mouse spinal cord (Pharaoh et al., 2019). Additionally, mutant SOD1, which is known to accumulate in the mitochondrial IMS to cause mitochondrial dysfunction, appears to upregulate ERα-UPRmt effectors such as the IMS protease HTRA2 (Riar et al., 2017). Mutations in CHCHD10 cause mitochondrial dysfunction, impaired mitochondrial proteostasis, and the activation of the UPRmt (Bannwarth et al., 2014; Anderson et al., 2019; Straub et al., 2021). The loss of CHCHD10 appears to activate both the ATF5-UPRmt and SIRT3-UPRmt as evidenced by enhanced activation of the transcription factors ATF4, ATF5, and FOXO3 (Straub et al., 2021). Lastly, in cell lines and flies, the overexpression of the FUS leads to disruption of the ATP synthase complex and activation of the UPRmt, both of which contribute to the FUS-induced neurodegeneration (Deng et al., 2018). These disparate findings potentially suggest that different ALS/FTD-related mutations have differing effects on the UPRmt, although it cannot be ruled out that differences in disease models account for the contradictory effects of UPRmt activation on cellular survival.
Conclusion
The inability of cells and tissues to maintain proteostasis, including mitochondrial proteostasis, appears to be central to the pathophysiology of many age-related NDDs (Hipp et al., 2014; Hou et al., 2019). Thus, it is no surprise that evidence of UPRER and UPRmt activation is a common feature in patient post-mortem nervous system tissue. Whether this activation represents protective or detrimental signaling is complex. Activation of the UPRER by virally delivered XBP1 or XBP1/ATF6 fusion protein appears to be protective across multiple NDD contexts (Sado et al., 2009; Zuleta et al., 2012; Cissé et al., 2017; Vidal et al., 2021). However, it is also clear that dysregulated UPRER signaling can drive pathogenesis under certain disease states, and so future work aimed at better defining both protective and detrimental UPRER signatures in disease models will be critical moving forward to allow targeting of therapies to the appropriate disease populations. Disease stage and context also appear to define the contribution of the UPRmt to disease onset and progression. The currently available data paint a picture where the UPRmt is acutely activated early in the course of age-related NDD to preserve cellular health. At this stage, it is likely that moderate levels of proteostasis impairment trigger the UPRmt to preserve mitochondrial and cellular integrity and health. This view is bolstered by data suggesting that inducing the accumulation of proteins within the mitochondria does not lead to cell death but does activate the UPRmt (Poveda-Huertes et al., 2020). However, data also suggest that continued activation or hyper-activation of the UPRmt, likely in response to chronic or severe proteotoxic stress that occurs with the progression of age-related NDD, can elicit detrimental effects (Kenny and Germain, 2017; Martinez et al., 2017; Hou et al., 2019). Thus, the relationship between the organellar UPRs and NDD is complex, and more work is needed to understand these relationships more completely.
Therapeutics and the Unfolded Protein Response
Modification of UPRER and UPRmt signaling has been shown to affect aging and age-related NDDs. Pharmacological agents targeting the UPRER and UPRmt may, therefore, hold promise as therapeutics (see Table 2). Here, we review several potential therapeutic strategies seeking to alter UPRER and UPRmt signaling to elicit beneficial effects; we also discuss the next steps needed to translate these pre-clinical findings into clinical therapeutics. For a more comprehensive review of therapeutic strategies targeting UPR function, including chemical chaperones, we refer readers to a recent review (Marciniak et al., 2021).
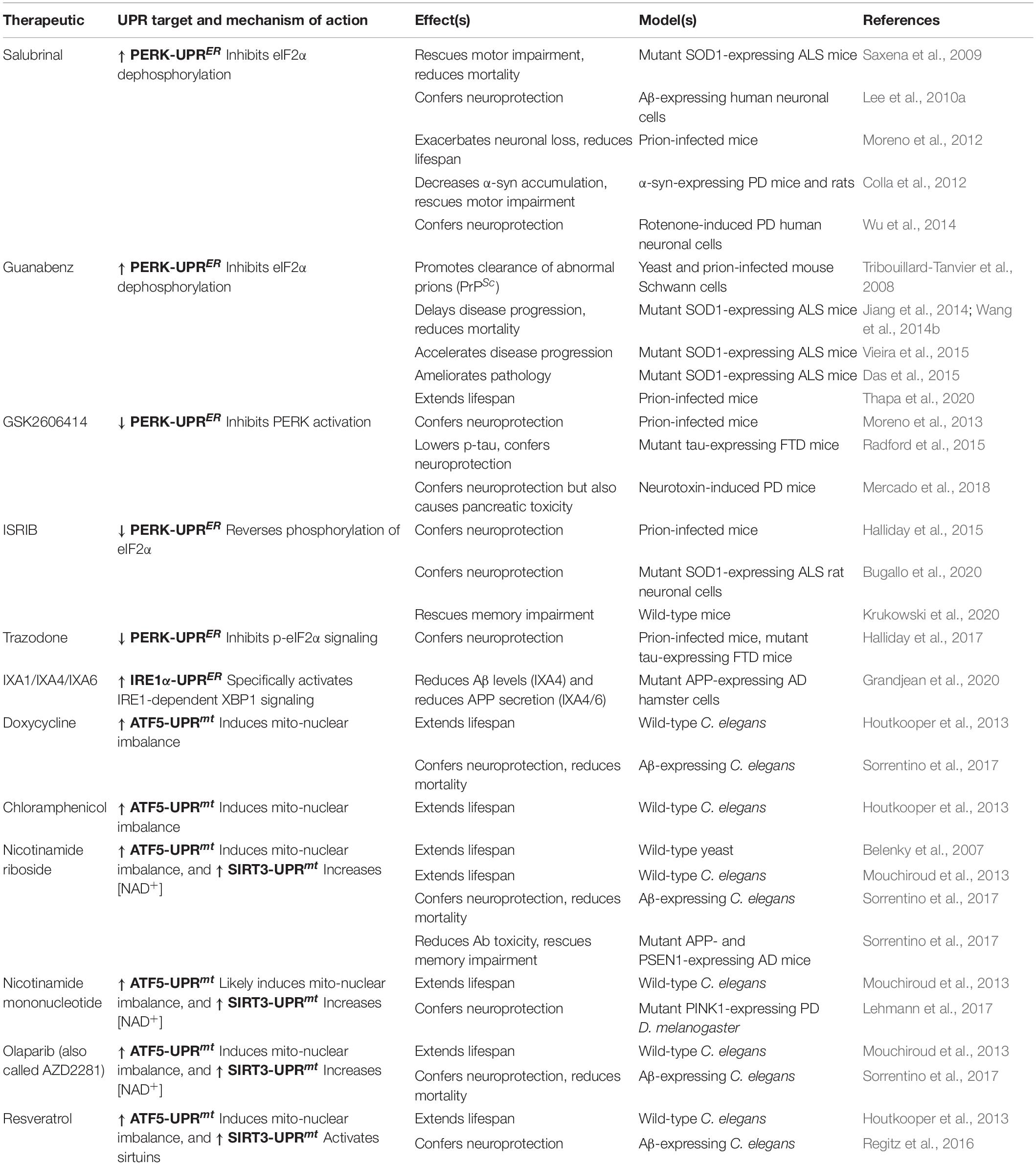
Table 2. Therapeutics targeting either the UPRER or the UPRmt, their mechanisms of action, and their effects in various model systems.
Therapeutics Targeting the Endoplasmic Reticulum Unfolded Protein Response
Therapeutics Targeting the Inositol-Requiring Enzyme 1α-Endoplasmic Reticulum Unfolded Protein Response
Pharmacological targeting of the IRE1α-UPRER has received little attention in the context of aging and NDD. However, a recent study screening for compounds that activate XBP1 splicing through IRE1α identified three promising options. IXA1, IXA4, and IXA6 selectively activate XBP1 signaling without affecting other functions of IRE1α, and IXA4 and IXA6 reduce levels of Aβ by improving proteostasis (Grandjean et al., 2020). Thus, selective pharmacological targeting of XBP1 may offer a therapeutic strategy to activate protective UPRER signaling and further testing of these strategies in NDD models is warranted.
Therapeutics Targeting the Protein Kinase RNA-Like Endoplasmic Reticulum Kinase-Endoplasmic Reticulum Unfolded Protein Response
In contrast to the IRE1α-UPRER, pharmacological modulation of the PERK-UPRER signaling pathway has been investigated extensively. Enhancing PERK-eIF2α signaling through the inhibition of eIF2α dephosphorylation has provided mixed results, with evidence that it can either protect (Saxena et al., 2009; Vaccaro et al., 2013; Jiang et al., 2014; Wang et al., 2014b; Das et al., 2015) or exacerbate (Moreno et al., 2012; Vieira et al., 2015) NDDs pathology in different animal models. Pharmacological inhibition of PERK kinase activity has also demonstrated neuroprotection in mice (Moreno et al., 2013; Radford et al., 2015; Mercado et al., 2018), but also can cause pancreatic toxicity as a secondary effect (Mercado et al., 2018), highlighting the need to further characterize the functional consequences of PERK kinase inhibition to organismal health. Indeed, partial inhibition of signaling downstream of PERK has proven effective at ameliorating NDD pathology without causing secondary toxicity (Halliday et al., 2015, 2017; Bugallo et al., 2020), and has also been demonstrated to ameliorate natural age-related cognitive decline in old mice (Krukowski et al., 2020), suggesting that fine-tuning PERK signaling as opposed to total inhibition may prove a better therapeutic strategy to target aging and age-related NDD.
Therapeutics Targeting the Activating Transcription Factor 6-Endoplasmic Reticulum Unfolded Protein Response
Similar to the IRE1α-UPRER, pharmacological strategies targeting ATF6 in aging and neurodegeneration have not yet been thoroughly explored. A recent screen to identify small regulators of ER proteostasis identified a number of regulators of ATF6-dependent transcriptional pathways (Plate et al., 2016). One compound in particular, named AA147, demonstrated effectiveness in reducing the secretion and aggregation of several proteotoxic species, including amyloidogenic immunoglobulin light chain (ALLC) (Plate et al., 2016). Moving forward, it will be of interest to determine whether these compounds are also capable of reducing protein inclusions characteristic of many NDDs.
Therapeutics Targeting the Mitochondrial Unfolded Protein Response
Therapeutics Targeting the Activating Transcription Factor 6-Mitochondrial Unfolded Protein Response
Two therapeutic strategies appear capable of activating the ATF5-UPRmt to avert the deleterious effects of aging and neurodegeneration. First, antibiotics that inhibit mitochondrial protein synthesis, namely doxycycline and chloramphenicol, have been shown to upregulate the expression of ATF5-dependent UPRmt genes and extend lifespan in C. elegans (Houtkooper et al., 2013). Furthermore, repression of protein synthesis can also protect against Aβ and α-syn aggregation and neurotoxicity by inducing the expression of UPRmt-linked genes (Sorrentino et al., 2017; Dastidar et al., 2020). Second, drugs that increase the concentration of NAD+, or mimic this effect, appear to activate the ATF5-UPRmt and SIRT3-UPRmt to extend lifespan and protect against NDD-related cellular and organismal decline in various model systems (Belenky et al., 2007; Houtkooper et al., 2013; Mouchiroud et al., 2013; Regitz et al., 2016; Lehmann et al., 2017; Sorrentino et al., 2017).
Therapeutics Targeting the Estrogen Receptor Alpha-Mitochondrial Unfolded Protein Response
Currently, there exist no drugs targeting the ERα-UPRmt that have been specifically investigated in the context of aging or neurodegeneration. As current data suggest that the ERα-UPRmt does not play a role in modulating aging nor neurodegeneration, more research is needed in this area to assess whether pharmacological targeting of this pathway is a worthwhile endeavor.
Therapeutics Targeting the SIRT3-Mitochondrial Unfolded Protein Response
As discussed previously, many NAD+ modulators not only activate the ATF5-UPRmt but also the SIRT3-UPRmt. As drugs that increase the cellular NAD+ concentration appear to extend lifespan and protect against neurodegeneration in model systems, future research investigating whether activation of the SIRT3-UPRmt independently of the ATF5-UPRmt achieves these same effects could aid in the design of more targeted therapeutics with the potential to increase resistance to the ill-effects of aging and NDD.
Conclusion
Taken together, current data suggest that targeting either the UPRER or UPRmt may hold promise as a therapeutic strategy to combat the ill-effects of aging and NDDs. However, many issues remain to be solved before therapeutic strategies targeting the UPRER and UPRmt can be translated to humans. First, it remains a key question how best to harness beneficial UPR signaling without inducing the pathological effects of chronic hyperactivity, though recent studies of the UPRER support the idea that fine-tuning UPR activity may provide a better template than directly targeting the master regulators. Second, it remains to be seen whether different neuronal subtypes respond similarly to modulators of the UPRs; activation of either of the organellar UPRs may be beneficial in some neurons but harmful in others. Third, as both the UPRER and UPRmt are composed of multiple, parallel signaling pathways, more research is needed to assess how individual pathways may compensate for drug-induced changes in another. Fourth, activation of the UPRER or UPRmt appears to be beneficial in some contexts and detrimental in others. Therefore, UPR-modifying therapies will rely upon the identification of biomarkers that can differentiate beneficial from detrimental circumstances. Lastly, numerous reports have suggested that there are non-cell autonomous effects of activating the UPR in one tissue; understanding how the activation of the UPR in multiple tissues simultaneously affects organismal health will be important when considering if UPR modulators represent an avenue to stave off the detrimental effects of aging and NDD.
Conclusion
The decline of proteostasis and mitochondrial dysfunction are both closely tied to the deterioration of organismal health, and it is clear from the data collected thus far that the UPRER and UPRmt both have complex relationships with aging and age-related NDDs. These relationships share common features as both UPRs experience a functional decline with age, while evidence of their activation is frequently observed in post-mortem NDD patient tissue. Additionally, genetic studies in model organisms demonstrate that manipulations of either UPR can influence aging and neurodegeneration, though these changes remain complex and context dependent. Activating specific components of either mechanism can prove beneficial in ameliorating disease phenotypes and extending lifespan under some circumstances, though sustained activation results in dysregulated signaling and can instead exacerbate pathology. Thus, how to best harness beneficial UPR functionality, while avoiding the consequences of sustained activity, remains a critical question when considering the therapeutic potential of UPR modulation for aging and age-related NDDs.
Author Contributions
APKW and AWS conducted the literature search and wrote the first draft of the manuscript. APKW designed the figures. All authors contributed to conceptualizing, reviewing, and editing the manuscript, approved the submission of the manuscript.
Funding
This work was supported by the Basic Neuroscience Program of the Intramural Research Program of the National Institute of Neurological Disorders and Stroke (NINDS)/National Institutes of Health (NIH) (Z01 NS003106).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
α-syn, alpha synuclein; A β, amyloid beta; AD, Alzheimer’s disease; ALS/FTD, amyotrophic lateral sclerosis/frontotemporal dementia; ATF4, activating transcription factor 4; ATF5, activating transcription factor 5; ATF6, activating transcription factor 6; Bcl2, B cell lymphoma 2; BiP, binding immunoglobulin protein; cbp-1, CREB binding protein 1; CHOP, C/EBP homologous protein; DPR, dipeptide repeat; DR, dietary restriction; eIF2 α, elongation initiation factor alpha; ER, endoplasmic reticulum; ER α, estrogen receptor alpha; ERAD, ER-associated degradation; ETC, electron transport chain; IMS, inner membrane space; IRE1 α, inositol-requiring enzyme 1 alpha; ISC, intestinal stem cell; MIM, mitochondrial inner membrane; miRNA, microRNA; MOM, mitochondrial outer membrane; mPTP, mitochondrial permeability transition pore; mtROS, mitochondrial reactive oxygen species; NDD, neurodegenerative disease; PD, Parkinson’s disease; PDI, protein disulfide isomerase; PERK, protein kinase RNA-like endoplasmic reticulum kinase; Pomc, proopiomelanocortin; PP1, protein phosphatase 1; RIDD, regulated IRE1-dependent decay; ROS, reactive oxygen species; S1P, site-1 protease; S2P, site-2 protease; SCV, small clear vesicle; SOD1, superoxide dismutase; UPR, unfolded protein response; UPRER, endoplasmic reticulum unfolded protein response, UPRmt, mitochondrial unfolded protein response; XBP1, X-box binding protein 1; XBP1s, X-box binding protein 1, spliced.
References
Abramzon, Y. A., Fratta, P., Traynor, B. J., and Chia, R. (2020). The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 14:42. doi: 10.3389/fnins.2020.00042
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., et al. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66. doi: 10.1016/j.molcel.2007.06.011
Aldridge, J. E., Horibe, T., and Hoogenraad, N. J. (2007). Discovery of genes activated by the mitochondrial Unfolded Protein Response (mtUPR) and cognate promoter elements. PLoS One 2:e0000874. doi: 10.1371/journal.pone.0000874
Andersen, E. C., and Horvitz, H. R. (2007). Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development 134, 2991–2999. doi: 10.1242/dev.009373
Anderson, C. J., Bredvik, K., Burstein, S. R., Davis, C., Meadows, S. M., Dash, J., et al. (2019). ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 138, 103–121. doi: 10.1007/s00401-019-01989-y
Angeli, S., Foulger, A., Chamoli, M., Peiris, T. H., Gerencser, A., Shahmirzadi, A. A., et al. (2021). The mitochondrial permeability transition pore activates the mitochondrial unfolded protein response and promotes aging. eLife 10, 1–23. doi: 10.7554/eLife.63453
Baker, B. M., Nargund, A. M., Sun, T., and Haynes, C. M. (2012). Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 8:e1002760. doi: 10.1371/journal.pgen.1002760
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Bannwarth, S., Ait-El-Mkadem, S., Chaussenot, A., Genin, E. C., Lacas-Gervais, S., Fragaki, K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345. doi: 10.1093/brain/awu138
Baqri, R. M., Pietron, A. V., Gokhale, R. H., Turner, B. A., Kaguni, L. S., Shingleton, A. W., et al. (2014). Mitochondrial chaperone TRAP1 activates the mitochondrial UPR and extends healthspan in Drosophila. Mech. Ageing Dev. 141-142, 35–45. doi: 10.1016/j.mad.2014.09.002
Bazopoulou, D., Knoefler, D., Zheng, Y., Ulrich, K., Oleson, B. J., Xie, L., et al. (2019). Developmental ROS individualizes organismal stress resistance and lifespan. Nature 576, 301–305. doi: 10.1038/s41586-019-1814-y
Beck, J. S., Mufson, E. J., and Counts, S. E. (2016). Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 13, 610–614. doi: 10.2174/1567205013666151221145445
Belenky, P., Racette, F. G., Bogan, K. L., McClure, J. M., Smith, J. S., and Brenner, C. (2007). Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 Pathways to NAD+. Cell 129, 473–484. doi: 10.1016/j.cell.2007.03.024
Bellizzi, D., Rose, G., Cavalcante, P., Covello, G., Dato, S., De Rango, F., et al. (2005). A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 85, 258–263. doi: 10.1016/j.ygeno.2004.11.003
Bellucci, A., Navarria, L., Zaltieri, M., Falarti, E., Bodei, S., Sigala, S., et al. (2011). Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 116, 588–605. doi: 10.1111/j.1471-4159.2010.07143.x
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P., and Ron, D. (2006). Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229–239. doi: 10.1534/genetics.106.061580
Bennett, C. F., Vander Wende, H., Simko, M., Klum, S., Barfield, S., Choi, H., et al. (2014). Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 5, 1–10. doi: 10.1038/ncomms4483
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. U.S.A. 106, 14914–14919. doi: 10.1073/pnas.0902882106
Berendzen, K. M., Durieux, J., Shao, L. W., Tian, Y., Kim, H. E., Wolff, S., et al. (2016). Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166, 1553–1563. doi: 10.1016/j.cell.2016.08.042
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Bloem, B. R., Okun, M. S., and Klein, C. (2021). Parkinson’s disease. Lancet 397, 2284–2303. doi: 10.1016/S0140-6736(21)00218-X
Boehme, M., Guzzetta, K. E., Bastiaanssen, T. F. S., van de Wouw, M., Moloney, G. M., Gual-Grau, A., et al. (2021). Microbiota from young mice counteracts selective age-associated behavioral deficits. Nat. Aging 1, 666–676. doi: 10.1038/s43587-021-00093-9
Bommiasamy, H., Back, S. H., Fagone, P., Lee, K., Meshinchi, S., Vink, E., et al. (2009). ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 122, 1626–1636. doi: 10.1242/jcs.045625
Bonzon, C., Bouchier-Hayes, L., Pagliari, L. J., Green, D. R., and Newmeyer, D. D. (2006). Caspase-2–induced apoptosis requires bid cleavage: a physiological role for bid in heat shock–induced death. Mol. Biol. Cell 17, 2150–2157. doi: 10.1091/mbc.e05-12-1107
Borch Jensen, M., Qi, Y., Riley, R., Rabkina, L., and Jasper, H. (2018). PGAM5 promotes lasting FoxO activation after developmental mitochondrial stress and extends lifespan in Drosophila. eLife 7, 1–22. doi: 10.7554/eLife.37316
Brandt, C., Nolte, H., Henschke, S., Engström Ruud, L., Awazawa, M., Morgan, D. A., et al. (2018). Food perception primes hepatic ER homeostasis via melanocortin-dependent control of mTOR activation. Cell 175, 1321–1335. doi: 10.1016/j.cell.2018.10.015
Bugallo, R., Marlin, E., Baltanás, A., Toledo, E., Ferrero, R., Vinueza-Gavilanes, R., et al. (2020). Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 11:397. doi: 10.1038/s41419-020-2601-2
Burkewitz, K., Feng, G., Dutta, S., Kelley, C. A., Steinbaugh, M., Cram, E. J., et al. (2020). Atf-6 regulates lifespan through ER-mitochondrial calcium homeostasis. Cell Rep. 32:108125. doi: 10.1016/j.celrep.2020.108125
Cai, Y., Shen, H., Weng, H., Wang, Y., Cai, G., Chen, X., et al. (2020). Overexpression of PGC-1α influences the mitochondrial unfolded protein response (mtUPR) induced by MPP+ in human SH-SY5Y neuroblastoma cells. Sci. Rep. 10, 1–12. doi: 10.1038/s41598-020-67229-6
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Casas-Tinto, S., Zhang, Y., Sanchez-Garcia, J., Gomez-Velazquez, M., Rincon-Limas, D. E., and Fernandez-Funez, P. (2011). The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum. Mol. Genet. 20, 2144–2160. doi: 10.1093/hmg/ddr100
Chen, D., Thomas, E. L., and Kapahi, P. (2009). HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 5:e1000486. doi: 10.1371/journal.pgen.1000486
Choi, K.-M., Kwon, Y.-Y., and Lee, C.-K. (2013). Characterization of global gene expression during assurance of lifespan extension by caloric restriction in budding yeast. Exp. Gerontol. 48, 1455–1468. doi: 10.1016/j.exger.2013.10.001
Chung, H. K., Ryu, D., Kim, K. S., Chang, J. Y., Kim, Y. K., Yi, H. S., et al. (2017). Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 216, 149–165. doi: 10.1083/jcb.201607110
Cissé, M., Duplan, E., Lorivel, T., Dunys, J., Bauer, C., Meckler, X., et al. (2017). The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 22, 1562–1575. doi: 10.1038/mp.2016.152
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012). Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/jneurosci.5367-11.2012
Conte, M., Ostan, R., Fabbri, C., Santoro, A., Guidarelli, G., Vitale, G., et al. (2019). Human aging and longevity are characterized by high levels of mitokines. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 74, 600–607. doi: 10.1093/gerona/gly153
Cooper, J. F., Machiela, E., Dues, D. J., Spielbauer, K. K., Senchuk, M. M., and Van Raamsdonk, J. M. (2017). Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 7:16441. doi: 10.1038/s41598-017-16637-2
Copeland, J. M., Cho, J., Lo, T., Hur, J. H., Bahadorani, S., Arabyan, T., et al. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory Chain. Curr. Biol. 19, 1591–1598. doi: 10.1016/j.cub.2009.08.016
Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. doi: 10.1016/0092-8674(93)90648-a
Cui, H.-J., Liu, X.-G., McCormick, M., Wasko, B. M., Zhao, W., He, X., et al. (2015). PMT1 deficiency enhances basal UPR activity and extends replicative lifespan of Saccharomyces cerevisiae. Age 37:46. doi: 10.1007/s11357-015-9788-7
Daniele, J. R., Higuchi-Sanabria, R., Durieux, J., Monshietehadi, S., Ramachandran, V., Tronnes, S. U., et al. (2020). UPRER promotes lipophagy independent of chaperones to extend life span. Sci. Adv. 6:eaaz1441. doi: 10.1126/sciadv.aaz1441
Das, I., Krzyzosiak, A., Schneider, K., Wrabetz, L., D’Antonio, M., Barry, N., et al. (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242. doi: 10.1126/science.aaa4484
Dastidar, S. G., Pham, M. T., Mitchell, M. B., Yeom, S. G., Jordan, S., Chang, A., et al. (2020). 4E-BP1 protects neurons from misfolded protein stress and Parkinson’s disease toxicity by inducing the mitochondrial unfolded protein response. J. Neurosci. 40, 8734–8745. doi: 10.1523/JNEUROSCI.0940-20.2020
Delaney, J. R., Ahmed, U., Chou, A., Sim, S., Carr, D., Murakami, C. J., et al. (2013). Stress profiling of longevity mutants identifies Afg3 as a mitochondrial determinant of cytoplasmic mRNA translation and aging. Aging Cell 12, 156–166. doi: 10.1111/acel.12032
Dell’Agnello, C., Leo, S., Agostino, A., Szabadkai, G., Tiveron, C. C., Zulian, A. A., et al. (2007). Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444. doi: 10.1093/hmg/ddl477
Deng, J., Wang, P., Chen, X., Cheng, H., Liu, J., Fushimi, K., et al. (2018). FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. U.S.A. 115, E9678–E9686. doi: 10.1073/pnas.1806655115
Derisbourg, M. J., Wester, L. E., Baddi, R., and Denzel, M. S. (2021). Mutagenesis screen uncovers lifespan extension through integrated stress response inhibition without reduced mRNA translation. Nat. Commun. 12:1678. doi: 10.1038/s41467-021-21743-x
Devi, L., and Ohno, M. (2014). PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 35, 2272–2281. doi: 10.1016/j.neurobiolaging.2014.04.031
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. doi: 10.1126/science.1077780
Dogan, S. A., Pujol, C., Maiti, P., Kukat, A., Wang, S., Hermans, S., et al. (2014). Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 19, 458–469. doi: 10.1016/j.cmet.2014.02.004
Donaldson, D. S., Pollock, J., Vohra, P., Stevens, M. P., and Mabbott, N. A. (2020). Microbial stimulation reverses the age-related decline in M Cells in aged mice. iScience 23:101147. doi: 10.1016/j.isci.2020.101147
Du, Y., Liu, X., Zhu, X., Liu, Y., Wang, X., and Wu, X. (2020). Activating transcription factor 6 reduces Aβ1–42 and restores memory in Alzheimer’s disease model mice. Int. J. Neurosci. 130, 1015–1023. doi: 10.1080/00207454.2020.1715977
Duran-Aniotz, C., Cornejo, V. H., Espinoza, S., Ardiles, ÁO., Medinas, D. B., Salazar, C., et al. (2017). IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 134, 489–506. doi: 10.1007/s00401-017-1694-x
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. doi: 10.1016/j.cell.2010.12.016
Dzhashiashvili, Y., Monckton, C. P., Shah, H. S., Kunjamma, R. B., and Popko, B. (2019). The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis. 127, 527–544. doi: 10.1016/j.nbd.2019.03.024
Egawa, N., Yamamoto, K., Inoue, H., Hikawa, R., Nishi, K., Mori, K., et al. (2011). The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J. Biol. Chem. 286, 7947–7957. doi: 10.1074/jbc.M110.156430
Fiorese, C. J., Schulz, A. M., Lin, Y. F., Rosin, N., Pellegrino, M. W., and Haynes, C. M. (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043. doi: 10.1016/j.cub.2016.06.002
Fontana, L., and Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. doi: 10.1016/j.cell.2015.02.020
Forsström, S., Jackson, C. B., Carroll, C. J., Kuronen, M., Pirinen, E., Pradhan, S., et al. (2019). Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 30, 1040–1054. doi: 10.1016/j.cmet.2019.08.019
Fouillet, A., Levet, C., Virgone, A., Robin, M., Dourlen, P., Rieusset, J., et al. (2012). ER stress inhibits neuronal death by promoting autophagy. Autophagy 8, 915–926. doi: 10.4161/auto.19716
Frakes, A. E., Metcalf, M. G., Tronnes, S. U., Bar-Ziv, R., Durieux, J., Gildea, H. K., et al. (2020). Four glial cells regulate ER stress resistance and longevity via neuropeptide signaling in C. elegans. Science 367, 436–440. doi: 10.1126/science.aaz6896
Gami-Patel, P., van Dijken, I., Meeter, L. H., Melhem, S., Morrema, T. H. J., Scheper, W., et al. (2021). Unfolded protein response activation in C9orf72 frontotemporal dementia is associated with dipeptide pathology and granulovacuolar degeneration in granule cells. Brain Pathol. 31, 163–173. doi: 10.1111/bpa.12894
Gao, K., Li, Y., Hu, S., and Liu, Y. (2019). SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. eLife 8, 1–24. doi: 10.7554/eLife.41792
Garratt, M., Lagerborg, K. A., Tsai, Y. M., Galecki, A., Jain, M., and Miller, R. A. (2018). Male lifespan extension with 17-α estradiol is linked to a sex-specific metabolomic response modulated by gonadal hormones in mice. Aging Cell 17, 1–14. doi: 10.1111/acel.12786
Gibellini, L., Pinti, M., Beretti, F., Pierri, C. L., Onofrio, A., Riccio, M., et al. (2014). Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 18, 76–81. doi: 10.1016/j.mito.2014.08.001
Gitschlag, B. L., Kirby, C. S., Samuels, D. C., Gangula, R. D., Mallal, S. A., and Patel, M. R. (2016). Homeostatic responses regulate selfish mitochondrial genome dynamics in C. elegans. Cell Metab. 24, 91–103. doi: 10.1016/j.cmet.2016.06.008
Grandjean, J. M. D., Madhavan, A., Cech, L., Seguinot, B. O., Paxman, R. J., Smith, E., et al. (2020). Pharmacologic IRE1/XBP1s activation confers targeted ER proteostasis reprogramming. Nat. Chem. Biol. 16, 1052–1061. doi: 10.1038/s41589-020-0584-z
Halliday, M., Radford, H., Sekine, Y., Moreno, J., Verity, N., le Quesne, J., et al. (2015). Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6:e1672. doi: 10.1038/cddis.2015.49
Halliday, M., Radford, H., Zents, K. A. M., Molloy, C., Moreno, J. A., Verity, N. C., et al. (2017). Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783. doi: 10.1093/brain/awx074
Hamilton, B., Dong, Y., Shindo, M., Liu, W., Odell, I., Ruvkun, G., et al. (2005). A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 19, 1544–1555. doi: 10.1101/gad.1308205
Han, J., Back, S. H., Hur, J., Lin, Y.-H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490. doi: 10.1038/ncb2738
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 3:17071. doi: 10.1038/nrdp.2017.71
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. doi: 10.1038/16729
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. doi: 10.1016/s1097-2765(03)00105-9
Harris, B. T. (2014). “Amyotrophic lateral sclerosis,” in Pathobiology of Human Disease, eds L. M. McManus and R. N. Mitchell (San Diego, CA: Academic Press), 2036–2044.
Hashida, K., Kitao, Y., Sudo, H., Awa, Y., Maeda, S., Mori, K., et al. (2012). ATF6alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. PLoS One 7:e47950. doi: 10.1371/journal.pone.0047950
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480. doi: 10.1016/j.devcel.2007.07.016
Haynes, C. M., Yang, Y., Blais, S. P., Neubert, T. A., and Ron, D. (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529–540. doi: 10.1016/j.molcel.2010.01.015
Haze, K., Hiderou, Y., Hideki, Y., Takashi, Y., and Kazutoshi, M. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
Hebert, A. S., Dittenhafer-Reed, K. E., Yu, W., Bailey, D. J., Selen, E. S., Boersma, M. D., et al. (2013). Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell. 49, 186–199. doi: 10.1016/j.molcel.2012.10.024
Henis-Korenblit, S., Zhang, P., Hansen, M., McCormick, M., Lee, S.-J., Cary, M., et al. (2010). Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc. Natl. Acad. Sci. U.S.A. 107, 9730–9735. doi: 10.1073/pnas.1002575107
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
Hetz, C., Zhang, K., and Kaufman, R. J. (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438. doi: 10.1038/s41580-020-0250-z
Higuchi-Sanabria, R., Durieux, J., Kelet, N., Homentcovschi, S., de los Rios Rogers, M., Monshietehadi, S., et al. (2020). Divergent nodes of non-autonomous UPRER signaling through serotonergic and dopaminergic neurons. Cell Rep. 33:108489. doi: 10.1016/j.celrep.2020.108489
Hipp, M. S., Park, S. H., and Hartl, U. U. (2014). Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514. doi: 10.1016/j.tcb.2014.05.003
Hollien, J., Lin, J. H., Li, H., Stevens, N., Walter, P., and Weissman, J. S. (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331. doi: 10.1083/jcb.200903014
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. doi: 10.1126/science.1129631
Honjo, Y., Ayaki, T., Tomiyama, T., Horibe, T., Ito, H., Mori, H., et al. (2015). Increased GADD34 in oligodendrocytes in Alzheimer’s disease. Neurosci. Lett. 602, 50–55. doi: 10.1016/j.neulet.2015.06.052
Hoozemans, J. J. M., van Haastert, E. S., Eikelenboom, P., de Vos, R. A. I., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 354, 707–711. doi: 10.1016/j.bbrc.2007.01.043
Hoozemans, J. J. M., van Haastert, E. S., Nijholt, D. A. T., Rozemuller, A. J. M., Eikelenboom, P., and Scheper, W. (2009). The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 174, 1241–1251. doi: 10.2353/ajpath.2009.080814
Hoozemans, J. J. M., Veerhuis, R., Van Haastert, E. S., Rozemuller, J. M., Baas, F., Eikelenboom, P., et al. (2005). The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 110, 165–172. doi: 10.1007/s00401-005-1038-0
Horibe, T., and Hoogenraad, N. J. (2007). The Chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One 2:e0000835. doi: 10.1371/journal.pone.0000835
Hou, N. S., Gutschmidt, A., Choi, D. Y., Pather, K., Shi, X., Watts, J. L., et al. (2014). Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, E2271–E2280. doi: 10.1073/pnas.1318262111
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. doi: 10.1038/nature12188
Howe, K. L., Achuthan, P., Allen, J., Allen, J., Alvarez-Jarreta, J., Amode, M. R., et al. (2020). Ensembl 2021. Nucleic Acids Res. 49, D884–D891. doi: 10.1093/nar/gkaa942
Hu, D., Liu, Z., and Qi, X. (2021). UPRmt activation protects against MPP+-induced toxicity in a cell culture model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 569, 17–22. doi: 10.1016/j.bbrc.2021.06.079
Hu, D., Sun, X., Liao, X., Zhang, X., Zarabi, S., Schimmer, A., et al. (2019). Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol. 137, 939–960. doi: 10.1007/s00401-019-01993-2
Hurst, L. D., Williams, E. J. B., and Pál, C. (2002). Natural selection promotes the conservation of linkage of co-expressed genes. Trends Genet. 18, 604–606. doi: 10.1016/S0168-9525(02)02813-5
Hussain, S. G., and Ramaiah, K. V. A. (2007). Reduced eIF2α phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 355, 365–370. doi: 10.1016/j.bbrc.2007.01.156
Hwang, J., and Qi, L. (2018). Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43, 593–605. doi: 10.1016/j.tibs.2018.06.005
Ilieva, E. V., Ayala, V., Jové, M., Dalfó, E., Cacabelos, D., Povedano, M., et al. (2007). Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 130, 3111–3123. doi: 10.1093/brain/awm190
Imanikia, S., Özbey, N. P., Krueger, C., Casanueva, M. O., and Taylor, R. C. (2019a). Neuronal XBP-1 activates intestinal lysosomes to improve proteostasis in C. elegans 7*. Curr. Biol. 29:14. doi: 10.1016/j.cub.2019.06.031
Imanikia, S., Sheng, M., Castro, C., Griffin, J. L., and Taylor, R. C. (2019b). XBP-1 remodels lipid metabolism to extend longevity. Cell Rep. 28, 581–589. doi: 10.1016/j.celrep.2019.06.057
Ishii, N., Tsuda, M., Yasuda, K., Yanase, S., and Suzuki, K. (1998). A mutation in succinate dehydrogenase cytochrome. Nat. Lett. 394, 694–697. doi: 10.1038/29331
Ishizawa, J., Zarabi, S. F., Davis, R. E., Halgas, O., Nii, T., Jitkova, Y., et al. (2019). Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell 35, 721.e9–737.e9. doi: 10.1016/j.ccell.2019.03.014
Ito, Y., Yamada, M., Tanaka, H., Aida, K., Tsuruma, K., Shimazawa, M., et al. (2009). Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 36, 470–476. doi: 10.1016/j.nbd.2009.08.013
Jiang, H. Q., Ren, M., Jiang, H. Z., Wang, J., Zhang, J., Yin, X., et al. (2014). Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 277, 132–138. doi: 10.1016/j.neuroscience.2014.03.047
Kang, S. G., Choi, M. J., Jung, S. B., Chung, H. K., Chang, J. Y., Kim, J. T., et al. (2021). Differential roles of GDF15 and FGF21 in systemic metabolic adaptation to the mitochondrial integrated stress response. iScience 24:102181. doi: 10.1016/j.isci.2021.102181
Kenny, T. C., and Germain, D. (2017). From discovery of the CHOP axis and targeting ClpP to the identification of additional axes of the UPRmt driven by the estrogen receptor and SIRT3. J. Bioenerg. Biomembr. 49, 297–305. doi: 10.1007/s10863-017-9722-z
Khan, N. A., Auranen, M., Paetau, I., Pirinen, E., Euro, L., Forsström, S., et al. (2014). Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 6, 721–731. doi: 10.1002/emmm.201403943
Kim, S., and Sieburth, D. (2020). FSHR-1/GPCR regulates the mitochondrial unfolded protein response in Caenorhabditis elegans. Genetics 214, 409–418. doi: 10.1534/genetics.119.302947
Knopman, D. S., Amieva, H., Petersen, R. C., Chételat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Prim. 7:33. doi: 10.1038/s41572-021-00269-y
Krukowski, K., Nolan, A., Frias, E. S., Boone, M., Ureta, G., Grue, K., et al. (2020). Small molecule cognitive enhancer reverses age-related memory decline in mice. eLife 9:e62048. doi: 10.7554/eLife.62048
Kuang, J., and Ebert, P. R. (2012). The failure to extend lifespan via disruption of complex II is linked to preservation of dynamic control of energy metabolism. Mitochondrion 12, 280–287. doi: 10.1016/j.mito.2011.10.003
Labbadia, J., Brielmann, R. M., Neto, M. F., Lin, Y. F., Haynes, C. M., and Morimoto, R. I. (2017). Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep. 21, 1481–1494. doi: 10.1016/j.celrep.2017.10.038
Labbadia, J., and Morimoto, R. I. (2015). Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell. 59, 639–650. doi: 10.1016/j.molcel.2015.06.027
Labunskyy, V. M., Gerashchenko, M. V., Delaney, J. R., Kaya, A., Kennedy, B. K., Kaeberlein, M., et al. (2014). Lifespan extension conferred by endoplasmic reticulum secretory pathway deficiency requires induction of the unfolded protein response. PLoS Genet. 10:e1004019. doi: 10.1371/journal.pgen.1004019
Lamech, L. T., and Haynes, C. M. (2015). The unpredictability of prolonged activation of stress response pathways. J. Cell Biol. 209, 781–787. doi: 10.1083/jcb.201503107
Lan, J., Rollins, J. A., Zang, X., Wu, D., Zou, L., Wang, Z., et al. (2019). Translational regulation of non-autonomous mitochondrial stress response promotes longevity. Cell Rep. 28, 1050.e6–1062.e6. doi: 10.1016/j.celrep.2019.06.078
Latorre-Pellicer, A., Moreno-Loshuertos, R., Lechuga-Vieco, A. V., Sánchez-Cabo, F., Torroja, C., Acín-Pérez, R., et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. doi: 10.1038/nature18618
Lee, A.-H., Iwakoshi, N. N., and Glimcher, L. H. (2003a). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003
Lee, S. S., Lee, R. Y. N., Fraser, A. G., Kamath, R. S., Ahringer, J., and Ruvkun, G. (2003b). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48. doi: 10.1038/ng1056
Lee, S. J., Hwang, A. B., and Kenyon, C. (2010b). Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136. doi: 10.1016/j.cub.2010.10.057
Lee, D. Y., Lee, K.-S., Lee, H. J., Kim, D. H., Noh, Y. H., Yu, K., et al. (2010a). Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS One 5:e10489. doi: 10.1371/journal.pone.0010489
Lehmann, S., Loh, S. H. Y., and Martins, L. M. (2017). Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 6, 141–147. doi: 10.1242/bio.022186
Lennicke, C., and Cochemé, H. M. (2021). Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 81, 3691–3707. doi: 10.1016/j.molcel.2021.08.018
Lerner, A. G., Upton, J.-P., Praveen, P. V. K., Ghosh, R., Nakagawa, Y., Igbaria, A., et al. (2012). IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264. doi: 10.1016/j.cmet.2012.07.007
Li, T. Y., Sleiman, M. B., Li, H., Gao, A. W., Mottis, A., Bachmann, A. M., et al. (2021). The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat. Aging 1, 165–178. doi: 10.1038/s43587-020-00025-z
Lin, K., Hsin, H., Libina, N., and Kenyon, C. (2001). Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28, 139–145. doi: 10.1038/88850
Lin, Y. F., Schulz, A. M., Pellegrino, M. W., Lu, Y., Shaham, S., and Haynes, C. M. (2016). Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419. doi: 10.1038/nature17989
Liu, Y., Samuel, B. S., Breen, P. C., and Ruvkun, G. (2014). Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508, 406–410. doi: 10.1038/nature13204
Loewen, C. A., and Feany, M. B. (2010). The unfolded protein response protects from tau neurotoxicity in vivo. PLoS One 5:e13084. doi: 10.1371/journal.pone.0013084
Lombard, D. B., Alt, F. W., Cheng, H.-L., Bunkenborg, J., Streeper, R. S., Mostoslavsky, R., et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814. doi: 10.1128/MCB.01636-07
Longo, F., Mancini, M., Ibraheem, P. L., Aryal, S., Mesini, C., Patel, J. C., et al. (2021). Cell-type-specific disruption of PERK-eIF2α signaling in dopaminergic neurons alters motor and cognitive function. Mol. Psychiatry 26, 6427–6450. doi: 10.1038/s41380-021-01099-w
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lu, P. D., Harding, H. P., and Ron, D. (2004). Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33. doi: 10.1083/jcb.200408003
Lu, Z., Chen, Y., Aponte, A. M., Battaglia, V., Gucek, M., and Sack, M. N. (2015). Prolonged fasting identifies heat shock protein 10 as a sirtuin 3 substrate: elucidating a new mechanism linking mitochondrial protein acetylation to fatty acid oxidation enzyme folding and function. J. Biol. Chem. 290, 2466–2476. doi: 10.1074/jbc.M114.606228
Luis, N. M., Wang, L., Ortega, M., Deng, H., Katewa, S. D., Li, P. W., et al. (2016). Intestinal IRE1 is required for increased triglyceride metabolism and longer lifespan under dietary restriction. Cell Rep. 17, 1207–1216. doi: 10.1016/j.celrep.2016.10.003
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer’s disease–related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Ma, Y., Brewer, J. W., Alan Diehl, J., and Hendershot, L. M. (2002). Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318, 1351–1365. doi: 10.1016/s0022-2836(02)00234-6
Mann, S. N., Hadad, N., Holte, M. N., Rothman, A. R., Sathiaseelan, R., Mondal, S. A., et al. (2020). Health benefits attributed to 17α- estradiol, a lifespan-extending compound, are mediated through estrogen receptor α. eLife 9, 1–30. doi: 10.7554/eLife.59616
Mao, K., Breen, P., and Ruvkun, G. (2020). Mitochondrial dysfunction induces RNA interference in C. elegans through a pathway homologous to the mammalian RIG-I antiviral response. PLoS Biol. 18:e3000996. doi: 10.1371/journal.pbio.3000996
Marciniak, S. J., Chambers, J. E., and Ron, D. (2021). Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. [Epub ahead of print]. doi: 10.1038/s41573-021-00320-3
Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. doi: 10.1101/gad.1250704
Marcus, J. M., and Andrabi, S. A. (2018). Sirt3 regulation under cellular stress: making sense of the ups and downs. Front. Neurosci. 12:799. doi: 10.3389/fnins.2018.00799
Martinez, B. A., Petersen, D. A., Gaeta, A. L., Stanley, S. P., Caldwell, G. A., and Caldwell, K. A. (2017). Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of parkinson’s disease. J. Neurosci. 37, 11085–11100. doi: 10.1523/JNEUROSCI.1294-17.2017
Martinus, R. D., Garth, G. P., Webster, T. L., Cartwright, P., Naylor, D. J., Høj, P. B., et al. (1996). Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 240, 98–103. doi: 10.1111/j.1432-1033.1996.0098h.x
Masrori, P., and Van Damme, P. (2020). Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. 27, 1918–1929. doi: 10.1111/ene.14393
Matai, L., Sarkar, G. C., Chamoli, M., Malik, Y., Kumar, S. S., Rautela, U., et al. (2019). Dietary restriction improves proteostasis and increases life span through endoplasmic reticulum hormesis. Proc. Natl. Acad. Sci. U.S.A. 116, 17383–17392. doi: 10.1073/pnas.1900055116
Matilainen, O., Sleiman, M. S. B., Quiros, P. M., Garcia, S. M. D. A., and Auwerx, J. (2017). The chromatin remodeling factor ISW-1 integrates organismal responses against nuclear and mitochondrial stress. Nat. Commun. 8:1818. doi: 10.1038/s41467-017-01903-8
Matus, S., Lopez, E., Valenzuela, V., Nassif, M., and Hetz, C. (2013). Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS One 8:e66672. doi: 10.1371/journal.pone.0066672
Maurel, M., Chevet, E., Tavernier, J., and Gerlo, S. (2014). Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39, 245–254. doi: 10.1016/j.tibs.2014.02.008
McCullough, K. D., Martindale, J. L., Klotz, L.-O., Aw, T.-Y., and Holbrook, N. J. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001
Mercado, G., Castillo, V., Soto, P., López, N., Axten, J. M., Sardi, S. P., et al. (2018). Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 112, 136–148. doi: 10.1016/j.nbd.2018.01.004
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223. doi: 10.1016/j.cell.2016.04.012
Metcalf, M. G., Higuchi-Sanabria, R., Garcia, G., Tsui, C. K., and Dillin, A. (2020). Beyond the cell factory: homeostatic regulation of and by the UPRER. Sci. Adv. 6:eabb9614. doi: 10.1126/sciadv.abb9614
Miller, H. A., Dean, E. S., Pletcher, S. D., and Leiser, S. F. (2020). Cell non-autonomous regulation of health and longevity. eLife 9, 1–28. doi: 10.7554/ELIFE.62659
Molenaars, M., Janssens, G. E., Williams, E. G., Jongejan, A., Lan, J., Rabot, S., et al. (2020). A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metab. 31, 549–563. doi: 10.1016/j.cmet.2020.01.011
Montibeller, L., Tan, L. Y., Kim, J. K., Paul, P., and de Belleroche, J. (2020). Tissue-selective regulation of protein homeostasis and unfolded protein response signalling in sporadic ALS. J. Cell Mol. Med. 24, 6055–6069. doi: 10.1111/jcmm.15170
Moreno, J. A., Halliday, M., Molloy, C., Radford, H., Verity, N., Axten, J. M., et al. (2013). Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5:206ra138. doi: 10.1126/scitranslmed.3006767
Moreno, J. A., Radford, H., Peretti, D., Steinert, J. R., Verity, N., Martin, M. G., et al. (2012). Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511. doi: 10.1038/nature11058
Mori, K., Ma, W., Gething, M.-J., and Sambrook, J. (1993). A transmembrane protein with a cdc2+CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756. doi: 10.1016/0092-8674(93)90521-q
Morselli, E., Fuente-Martin, E., Finan, B., Kim, M., Frank, A., Garcia-Caceres, C., et al. (2014). Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep. 9, 633–645. doi: 10.1016/j.celrep.2014.09.025
Mouchiroud, L., Houtkooper, R. H., Moullan, N., Katsyuba, E., Ryu, D., Cantó, C., et al. (2013). The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. doi: 10.1016/j.cell.2013.06.016
Münch, C. (2018). The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 16:81. doi: 10.1186/s12915-018-0548-x
Münch, C., and Harper, J. W. (2016). Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534, 710–713. doi: 10.1038/nature18302
Munkácsy, E., and Rea, S. L. (2014). The paradox of mitochondrial dysfunction and extended longevity. Exp. Gerontol. 56, 221–233. doi: 10.1016/j.exger.2014.03.016
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi: 10.1523/jneurosci.5685-07.2008
Naidoo, N., Zhu, J., Zhu, Y., Fenik, P., Lian, J., Galente, R., et al. (2011). Endoplasmic reticulum stress in wake-active neurons progresses with aging. Aging Cell 10, 640–649. doi: 10.1111/j.1474-9726.2011.00699.x
Nardo, G., Pozzi, S., Pignataro, M., Lauranzano, E., Spano, G., Garbelli, S., et al. (2011). Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One 6:e25545. doi: 10.1371/journal.pone.0025545
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P., and Haynes, C. M. (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPRmt. Mol. Cell. 58, 123–133. doi: 10.1016/j.molcel.2015.02.008
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. doi: 10.1126/science.1223560
Nijholt, D. A., van Haastert, E. S., Rozemuller, A. J., Scheper, W., and Hoozemans, J. J. (2012). The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 226, 693–702. doi: 10.1002/path.3969
Nishitoh, H., Matsuzawa, A., Tobiume, K., Saegusa, K., Takeda, K., Inoue, K., et al. (2002). ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355. doi: 10.1101/gad.992302
Novoa, I., Zeng, H., Harding, H. P., and Ron, D. (2001). Feedback Inhibition of the Unfolded Protein Response by GADD34-Mediated Dephosphorylation of eIF2α. J. Cell Biol. 153, 1011–1022. doi: 10.1083/jcb.153.5.1011
Nuss, J. E., Choksi, K. B., DeFord, J. H., and Papaconstantinou, J. (2008). Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 365, 355–361. doi: 10.1016/j.bbrc.2007.10.194
Owusu-Ansah, E., Song, W., and Perrimon, N. (2013). Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712. doi: 10.1016/j.cell.2013.09.021
Özbey, N. P., Imanikia, S., Krueger, C., Hardege, I., Morud, J., Sheng, M., et al. (2020). Tyramine acts downstream of neuronal XBP-1s to coordinate inter-tissue UPRER activation and behavior in C. elegans. Dev. Cell 55, 754.e9–770.e9. doi: 10.1016/j.devcel.2020.10.024
Ozkurede, U., and Miller, R. A. (2019). Improved mitochondrial stress response in long-lived Snell dwarf mice. Aging Cell 18, 1–11. doi: 10.1111/acel.13030
Papa, L., and Germain, D. (2011). Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 124, 1396–1402. doi: 10.1242/jcs.078220
Papa, L., and Germain, D. (2014). SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 34, 699–710. doi: 10.1128/mcb.01337-13
Paz Gavilán, M., Pintado, C., Gavilán, E., Jiménez, S., Ríos, R. M., Vitorica, J., et al. (2009). Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 8, 654–665. doi: 10.1111/j.1474-9726.2009.00519.x
Pellegrino, M. W., and Haynes, C. M. (2015). Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 13:22. doi: 10.1186/s12915-015-0129-1
Pérez, M. J., Ivanyuk, D., Panagiotakopoulou, V., Di Napoli, G., Kalb, S., Brunetti, D., et al. (2020). Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 26, 5733–5750. doi: 10.1038/s41380-020-0807-4
Pérez, M. J., and Quintanilla, R. A. (2017). Development or disease: duality of the mitochondrial permeability transition pore. Dev. Biol. 426, 1–7. doi: 10.1016/j.ydbio.2017.04.018
Pérez, V. I., Buffenstein, R., Masamsetti, V., Leonard, S., Salmon, A. B., Mele, J., et al. (2009b). Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc. Natl. Acad. Sci. U.S.A. 106, 3059–3064. doi: 10.1073/pnas.0809620106
Pérez, V. I., Bokov, A., Remmen, H. V., Mele, J., Ran, Q., Ikeno, Y., et al. (2009a). Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 1790, 1005–1014. doi: 10.1016/j.bbagen.2009.06.003
Pharaoh, G., Sataranatarajan, K., Street, K., Hill, S., Gregston, J., Ahn, B., et al. (2019). Metabolic and stress response changes precede disease onset in the spinal cord of mutant SOD1 ALS mice. Front. Neurosci. 13:487. doi: 10.3389/fnins.2019.00487
Pihán, P., Carreras-Sureda, A., and Hetz, C. (2017). BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Diff. 24, 1478–1487. doi: 10.1038/cdd.2017.82
Piper, M. D. W., and Partridge, L. (2018). Drosophila as a model for ageing. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2707–2717. doi: 10.1016/j.bbadis.2017.09.016
Plate, L., Cooley, C. B., Chen, J. J., Paxman, R. J., Gallagher, C. M., Madoux, F., et al. (2016). Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 5:e15550. doi: 10.7554/eLife.15550
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3:17013. doi: 10.1038/nrdp.2017.13
Poveda-Huertes, D., Matic, S., Marada, A., Habernig, L., Licheva, M., Myketin, L., et al. (2020). An early mtUPR: redistribution of the nuclear transcription factor Rox1 to mitochondria protects against intramitochondrial proteotoxic aggregates. Mol. Cell. 77, 180–188. doi: 10.1016/j.molcel.2019.09.026
Prudencio, M., Belzil, V. V., Batra, R., Ross, C. A., Gendron, T. F., Pregent, L. J., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182. doi: 10.1038/nn.4065
Pulliam, D. A., Deepa, S. S., Liu, Y., Hill, S., Lin, A. L., Bhattacharya, A., et al. (2014). Complex IV-deficient Surf1-/- mice initiate mitochondrial stress responses. Biochem. J. 462, 359–371. doi: 10.1042/BJ20140291
Puthalakath, H., O’Reilly, L. A., Gunn, P., Lee, L., Kelly, P. N., Huntington, N. D., et al. (2007). ER stress triggers apoptosis by activating BH3-only protein bim. Cell 129, 1337–1349. doi: 10.1016/j.cell.2007.04.027
Quirós, P. M., Prado, M. A., Zamboni, N., D’Amico, D., Williams, R. W., Finley, D., et al. (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045. doi: 10.1083/jcb.201702058
Rabek, J. P., Boylston Iii, W. H., and Papaconstantinou, J. (2003). Carbonylation of ER chaperone proteins in aged mouse liver. Biochem. Biophys. Res. Commun. 305, 566–572. doi: 10.1016/s0006-291x(03)00826-x
Radford, H., Moreno, J. A., Verity, N., Halliday, M., and Mallucci, G. R. (2015). PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–642. doi: 10.1007/s00401-015-1487-z
Radke, S., Chander, H., Schäfer, P., Meiss, G., Krüger, R., Schulz, J. B., et al. (2008). Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 283, 12681–12685. doi: 10.1074/jbc.C800036200
Rainbolt, T. K., Atanassova, N., Genereux, J. C., and Wiseman, R. L. (2013). Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 18, 908–919. doi: 10.1016/j.cmet.2013.11.006
Rauthan, M., Ranji, P., Abukar, R., and Pilon, M. (2015). A mutation in Caenorhabditis elegans NDUF-7 activates the mitochondrial stress response and prolongs lifespan via ROS and CED-4. G3 Genes Genomes Genet. 5, 1639–1648. doi: 10.1534/g3.115.018598
Rauthan, M., Ranji, P., Pradenas, N. A., Pitot, C., and Pilon, M. (2013). The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc. Natl. Acad. Sci. U.S.A. 110, 5981–5986. doi: 10.1073/pnas.1218778110
Rea, S. L., Ventura, N., and Johnson, T. E. (2007). Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 5:e0050259. doi: 10.1371/journal.pbio.0050259
Regitz, C., Fitzenberger, E., Mahn, F. L., Dußling, L. M., and Wenzel, U. (2016). Resveratrol reduces amyloid-beta (Aβ1–42)-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur. J. Nutr. 55, 741–747. doi: 10.1007/s00394-015-0894-1
Rera, M., Monnier, V., and Tricoire, H. (2010). Mitochondrial electron transport chain dysfunction during development does not extend lifespan in Drosophila melanogaster. Mech. Ageing Dev. 131, 156–164. doi: 10.1016/j.mad.2010.01.004
Riar, A. K., Burstein, S. R., Palomo, G. M., Arreguin, A., Manfredi, G., and Germain, D. (2017). Sex specific activation of the ERα axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum. Mol. Genet. 26, 1318–1327. doi: 10.1093/hmg/ddx049
Rose, G., Dato, S., Altomare, K., Bellizzi, D., Garasto, S., Greco, V., et al. (2003). Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 38, 1065–1070. doi: 10.1016/S0531-5565(03)00209-2
Sabath, N., Levy-Adam, F., Younis, A., Rozales, K., Meller, A., Hadar, S., et al. (2020). Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. U.S.A. 117, 31902–31913. doi: 10.1073/pnas.2018138117
Sado, M., Yamasaki, Y., Iwanaga, T., Onaka, Y., Ibuki, T., Nishihara, S., et al. (2009). Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res. 1257, 16–24. doi: 10.1016/j.brainres.2008.11.104
Safra, M., Ben-Hamo, S., Kenyon, C., and Henis-Korenblit, S. (2013). The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans. J. Cell Sci. 126, 4136–4146. doi: 10.1242/jcs.123000
Sandow, J. J., Dorstyn, L., O’Reilly, L. A., Tailler, M., Kumar, S., Strasser, A., et al. (2014). ER stress does not cause upregulation and activation of caspase-2 to initiate apoptosis. Cell Death Diff. 21, 475–480. doi: 10.1038/cdd.2013.168
Sasaki, S. (2010). Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 346–355. doi: 10.1097/NEN.0b013e3181d44992
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schaar, C. E., Dues, D. J., Spielbauer, K. K., Machiela, E., Cooper, J. F., Senchuk, M., et al. (2015). Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 11:e1004972. doi: 10.1371/journal.pgen.1004972
Schmeisser, S., Priebe, S., Groth, M., Monajembashi, S., Hemmerich, P., Guthke, R., et al. (2013). Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol. Metab. 2, 92–102. doi: 10.1016/j.molmet.2013.02.002
Schneider, J. A., Arvanitakis, Z., Leurgans, S. E., and Bennett, D. A. (2009). The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 66, 200–208. doi: 10.1002/ana.21706
Schwer, B., North, B. J., Frye, R. A., Melanie, O., and Verdin, E. (2002). The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide–dependent deacetylase. J. Cell Biol. 158, 647–657. doi: 10.1083/jcb.200205057
Shao, L. W., Niu, R., and Liu, Y. (2016). Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. 26, 1182–1196. doi: 10.1038/cr.2016.118
Shao, L. W., Peng, Q., Dong, M., Gao, K., Li, Y., Li, Y., et al. (2020). Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 11, 1–12. doi: 10.1038/s41467-020-18501-w
Sharma, V., Ounallah-Saad, H., Chakraborty, D., Hleihil, M., Sood, R., Barrera, I., et al. (2018). Local inhibition of PERK enhances memory and reverses age-related deterioration of cognitive and neuronal properties. J. Neurosci. 38, 648–658. doi: 10.1523/jneurosci.0628-17.2017
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 3, 99–111. doi: 10.1016/s1534-5807(02)00203-4
Shen, J., Snapp, E. L., Lippincott-Schwartz, J., and Prywes, R. (2005). Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 25, 921–932. doi: 10.1128/MCB.25.3.921-932.2005
Shen, X., Ellis, R. E., Sakaki, K., and Kaufman, R. J. (2005). Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 1:e0010037. doi: 10.1371/journal.pgen.0010037
Shen, X., Ellis, R. E., Lee, K., Liu, C.-Y., Yang, K., Solomon, A., et al. (2001). Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893–903. doi: 10.1016/s0092-8674(01)00612-2
Shen, Y., Ding, M., Xie, Z., Liu, X., Yang, H., Jin, S., et al. (2020). Activation of mitochondrial unfolded protein response in SHSY5Y expressing APP Cells and APP/PS1 mice. Front. Cell. Neurosci. 13:568. doi: 10.3389/fncel.2019.00568
Sheng, Y., Yang, G., Markovich, Z., Han, S. M., and Xiao, R. (2021). Distinct temporal actions of different types of unfolded protein responses during aging. J. Cell. Physiol. 236, 5069–5079. doi: 10.1002/jcp.30215
Shoulders, M. D., Ryno, L. M., Genereux, J. C., Moresco, J. J., Tu, P. G., Wu, C., et al. (2013). Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 3, 1279–1292. doi: 10.1016/j.celrep.2013.03.024
Shpilka, T., and Haynes, C. M. (2018). The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 19, 109–120. doi: 10.1038/nrm.2017.110
Shukla, A. K., Johnson, K., and Giniger, E. (2021). Common features of aging fail to occur in Drosophila raised without a bacterial microbiome. iScience 24, 102703–102703. doi: 10.1016/j.isci.2021.102703
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039. doi: 10.1016/s0092-8674(00)80369-4
Silva, R. M., Ries, V., Oo, T. F., Yarygina, O., Jackson-Lewis, V., Ryu, E. J., et al. (2005). CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 95, 974–986. doi: 10.1111/j.1471-4159.2005.03428.x
Singh, P. P., Demmitt, B. A., Nath, R. D., and Brunet, A. (2019). The genetics of aging: a vertebrate perspective. Cell 177, 200–220. doi: 10.1016/j.cell.2019.02.038
Smith, P., Willemsen, D., Popkes, M., Metge, F., Gandiwa, E., Reichard, M., et al. (2017). Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. eLife 6:e27014. doi: 10.7554/eLife.27014
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D’Amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193. doi: 10.1038/nature25143
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stout, M. B., Steyn, F. J., Jurczak, M. J., Camporez, J. P. G., Zhu, Y., Hawse, J. R., et al. (2017). 17A -estradiol alleviates age-related metabolic and inflammatory dysfunction in male mice without inducing feminization. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 72, 3–15. doi: 10.1093/gerona/glv309
Straub, I. R., Weraarpachai, W., and Shoubridge, E. A. (2021). Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 30, 687–705. doi: 10.1093/hmg/ddab078
Strauss, K. M., Martins, L. M., Plun-Favreau, H., Marx, F. P., Kautzmann, S., Berg, D., et al. (2005). Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 14, 2099–2111. doi: 10.1093/hmg/ddi215
Strong, R., Miller, R. A., Antebi, A., Astle, C. M., Bogue, M., Denzel, M. S., et al. (2016). Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 15, 872–884. doi: 10.1111/acel.12496
Stutzbach, L. D., Xie, S. X., Naj, A. C., Albin, R., Gilman, S., Lee, V. M. Y., et al. (2013). The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 1:31. doi: 10.1186/2051-5960-1-31
Sundaresan, N. R., Gupta, M., Kim, G., Rajamohan, S. B., Isbatan, A., and Gupta, M. P. (2009). Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 119, 2758–2771. doi: 10.1172/JCI39162
Suomalainen, A., Elo, J. M., Pietiläinen, K. H., Hakonen, A. H., Sevastianova, K., Korpela, M., et al. (2011). FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 10, 806–818. doi: 10.1016/S1474-4422(11)70155-7
Sural, S., Liang, C. Y., Wang, F. Y., Ching, T. T., and Hsu, A. L. (2020). HSB-1/HSF-1 pathway modulates histone H4 in mitochondria to control mtDNA transcription and longevity. Sci. Adv. 6, 1–18. doi: 10.1126/sciadv.aaz4452
Tabas, I., and Ron, D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. doi: 10.1038/ncb0311-184
Tapia, P. C. (2006). Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med. Hypotheses 66, 832–843. doi: 10.1016/j.mehy.2005.09.009
Taylor, R. C., and Dillin, A. (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. doi: 10.1016/j.cell.2013.05.042
Thapa, S., Abdelaziz, D. H., Abdulrahman, B. A., and Schatzl, H. M. (2020). Sephin1 reduces prion infection in prion-infected cells and animal model. Mol. Neurobiol. 57, 2206–2219. doi: 10.1007/s12035-020-01880-y
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165, 1197–1208. doi: 10.1016/j.cell.2016.04.011
Tribouillard-Tanvier, D., Béringue, V., Desban, N., Gug, F., Bach, S., Voisset, C., et al. (2008). Antihypertensive drug guanabenz is active in vivo against both yeast and mammalian prions. PLoS One 3:e1981. doi: 10.1371/journal.pone.0001981
Unal Gulsuner, H., Gulsuner, S., Mercan, F. N., Onat, O. E., Walsh, T., Shahin, H., et al. (2014). Mitochondrial serine protease HTRA2 p.G399S in a kindred with essential tremor and Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 111, 18285–18290. doi: 10.1073/pnas.1419581111
Unterberger, U., Höftberger, R., Gelpi, E., Flicker, H., Budka, H., and Voigtländer, T. (2006). Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J. Neuropathol. Exp. Neurol. 65, 348–357. doi: 10.1097/01.jnen.0000218445.30535.6f
Upton, J.-P., Wang, L., Han, D., Wang, E. S., Huskey, N. E., Lim, L., et al. (2012). IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science 338, 818–822. doi: 10.1126/science.1226191
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. doi: 10.1126/science.287.5453.664
Vaccaro, A., Patten, S. A., Aggad, D., Julien, C., Maios, C., Kabashi, E., et al. (2013). Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 55, 64–75. doi: 10.1016/j.nbd.2013.03.015
Valdés, P., Mercado, G., Vidal, R. L., Molina, C., Parsons, G., Court, F. A., et al. (2014). Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. U.S.A. 111, 6804–6809. doi: 10.1073/pnas.1321845111
Vattem, K. M., and Wek, R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 11269–11274. doi: 10.1073/pnas.0400541101
Vidal, R. L., Sepulveda, D., Troncoso-Escudero, P., Garcia-Huerta, P., Gonzalez, C., Plate, L., et al. (2021). Enforced dimerization between XBP1s and ATF6f enhances the protective effects of the UPR in models of neurodegeneration. Mol. Ther. 29, 1862–1882. doi: 10.1016/j.ymthe.2021.01.033
Vieira, F. G., Ping, Q., Moreno, A. J., Kidd, J. D., Thompson, K., Jiang, B., et al. (2015). Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS One 10:e0135570. doi: 10.1371/journal.pone.0135570
Wang, L., Popko, B., and Roos, R. P. (2011). The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 20, 1008–1015. doi: 10.1093/hmg/ddq546
Wang, L., Popko, B., and Roos, R. P. (2014a). An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 23, 2629–2638. doi: 10.1093/hmg/ddt658
Wang, L., Popko, B., Tixier, E., and Roos, R. P. (2014b). Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 71, 317–324. doi: 10.1016/j.nbd.2014.08.010
Wang, L., Ryoo, H. D., Qi, Y., and Jasper, H. (2015). PERK limits Drosophila lifespan by promoting intestinal stem cell proliferation in response to ER stress. PLoS Genet. 11:e1005220. doi: 10.1371/journal.pgen.1005220
Wang, M., and Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. doi: 10.1038/nature17041
Wang, P., Deng, J., Dong, J., Liu, J., Bigio, E. H., Mesulam, M., et al. (2019). TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 15:e1007947. doi: 10.1371/journal.pgen.1007947
Wei, M. C., Zong, W.-X., Cheng, E. H.-Y., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., et al. (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. doi: 10.1126/science.1059108
Williams, K. W., Liu, T., Kong, X., Fukuda, M., Deng, Y., Berglund, E. D., et al. (2014). Xbp1s in pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 20, 471–482. doi: 10.1016/j.cmet.2014.06.002
Wu, J., Rutkowski, D. T., Dubois, M., Swathirajan, J., Saunders, T., Wang, J., et al. (2007). ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364. doi: 10.1016/j.devcel.2007.07.005
Wu, L., Luo, N., Zhao, H.-R., Gao, Q., Lu, J., Pan, Y., et al. (2014). Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 1549, 52–62. doi: 10.1016/j.brainres.2014.01.003
Wu, Z., Senchuk, M. M., Dues, D. J., Johnson, B. K., Cooper, J. F., Lew, L., et al. (2018). Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 16:147. doi: 10.1186/s12915-018-0615-3
Yaghmaie, F., Saeed, O., Garan, S. A., Freitag, W., Timiras, P. S., and Sternberg, H. (2005). Caloric restriction reduces cell loss and maintains estrogen receptor-alpha immunoreactivity in the pre-optic hypothalamus of female B6D2F1 mice. Neuro Endocrinol. Lett. 26, 197–203.
Yamamoto, K., Sato, T., Matsui, T., Sato, M., Okada, T., Yoshida, H., et al. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 13, 365–376. doi: 10.1016/j.devcel.2007.07.018
Yan, C., Liu, J., Gao, J., Sun, Y., Zhang, L., Song, H., et al. (2019). IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis. 10:800. doi: 10.1038/s41419-019-2039-6
Yang, W., and Hekimi, S. (2010a). A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 8:e1000556. doi: 10.1371/journal.pbio.1000556
Yang, W., and Hekimi, S. (2010b). Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9, 433–447. doi: 10.1111/j.1474-9726.2010.00571.x
Yang, W., Zhou, X., Zimmermann, H. R., Cavener, D. R., Klann, E., and Ma, T. (2016). Repression of the eIF2α kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 41, 19–24. doi: 10.1016/j.neurobiolaging.2016.02.005
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Davé, U. P., Prywes, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 6, 1355–1364. doi: 10.1016/S1097-2765(00)00133-7
Yee, C., Yang, W., and Hekimi, S. (2014). The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 157, 897–909. doi: 10.1016/j.cell.2014.02.055
Yoneda, T., Benedetti, C., Urano, F., Clark, S. G., Harding, H. P., and Ron, D. (2004). Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 117, 4055–4066. doi: 10.1242/jcs.01275
Yoon, S. O., Park, D. J., Ryu, J. C., Ozer, H. G., Tep, C., Shin, Y. J., et al. (2012). JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 75, 824–837. doi: 10.1016/j.neuron.2012.06.024
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Yuan, J., Chang, S. Y., Yin, S. G., Liu, Z. Y., Cheng, X., Liu, X. J., et al. (2020). Two conserved epigenetic regulators prevent healthy ageing. Nature 579, 118–122. doi: 10.1038/s41586-020-2037-y
Zarse, K., Schmeisser, S., Groth, M., Priebe, S., Beuster, G., Kuhlow, D., et al. (2012). Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 15, 451–465. doi: 10.1016/j.cmet.2012.02.013
Zhang, Q., Wang, Z., Zhang, W., Wen, Q., Li, X., Zhou, J., et al. (2021). The memory of neuronal mitochondrial stress is inherited transgenerationally via elevated mitochondrial DNA levels. Nat. Cell Biol. 23, 870–880. doi: 10.1038/s41556-021-00724-8
Zhang, Q., Wu, X., Chen, P., Liu, L., Xin, N., Tian, Y., et al. (2018). The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 174, 870–883. doi: 10.1016/j.cell.2018.06.029
Zhao, Q. (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419. doi: 10.1093/emboj/cdf445
Zhu, D., Wu, X., Zhou, J., Li, X., Huang, X., Li, J., et al. (2020). NuRD mediates mitochondrial stress–induced longevity via chromatin remodeling in response to acetyl-CoA level. Sci. Adv. 6, 1–13. doi: 10.1126/sciadv.abb2529
Zuleta, A., Vidal, R. L., Armentano, D., Parsons, G., and Hetz, C. (2012). AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington’s disease. Biochem. Biophys. Res. Commun. 420, 558–563. doi: 10.1016/j.bbrc.2012.03.033
Keywords: unfolded protein response (UPR), mitochondrial unfolded protein response (UPRmt), endoplasmic reticulum unfolded protein response, aging, neurodegeneration
Citation: Wodrich APK, Scott AW, Shukla AK, Harris BT and Giniger E (2022) The Unfolded Protein Responses in Health, Aging, and Neurodegeneration: Recent Advances and Future Considerations. Front. Mol. Neurosci. 15:831116. doi: 10.3389/fnmol.2022.831116
Received: 08 December 2021; Accepted: 26 January 2022;
Published: 25 February 2022.
Edited by:
Neha Gogia, Yale University, United StatesReviewed by:
Narendra Nath Jha, Columbia University Irving Medical Center, United StatesVidyadhara D. J., Yale University, United States
Nirinjini Naidoo, University of Pennsylvania, United States
Copyright © 2022 Wodrich, Scott, Shukla, Harris and Giniger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward Giniger, Z2luaWdlcmVAbmluZHMubmloLmdvdg==
†These authors have contributed equally to this work and share first authorship