
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 26 April 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.822129
This article is part of the Research Topic Molecular Advances and Applications of Machine Learning in Understanding Autism and Comorbid Psychiatric Disorders View all 16 articles
 Xiaofan Yang1,2
Xiaofan Yang1,2 Hongqiang Yin3,4
Hongqiang Yin3,4 Xiaojing Wang2Yueqing Sun2Xianli Bian5Gaorui Zhang6Anning Li6Aihua Cao1
Xiaojing Wang2Yueqing Sun2Xianli Bian5Gaorui Zhang6Anning Li6Aihua Cao1 Baomin Li1
Baomin Li1 Darius Ebrahimi-Fakhari7
Darius Ebrahimi-Fakhari7 Zhuo Yang3
Zhuo Yang3 Miriam H. Meisler8,9
Miriam H. Meisler8,9 Qiji Liu2,10*
Qiji Liu2,10*Mutations in the SCN8A gene encoding the voltage-gated sodium channel α-subunit Nav1. 6 have been reported in individuals with epilepsy, intellectual disability and features of autism spectrum disorder. SCN8A is widely expressed in the central nervous system, including the cerebellum. Cerebellar dysfunction has been implicated in autism spectrum disorder. We investigated conditional Scn8a knockout mice under C57BL/6J strain background that specifically lack Scn8a expression in cerebellar Purkinje cells (Scn8aflox/flox, L7Cre+ mice). Cerebellar morphology was analyzed by immunohistochemistry and MR imaging. Mice were subjected to a battery of behavioral tests including the accelerating rotarod, open field, elevated plus maze, light-dark transition box, three chambers, male-female interaction, social olfaction, and water T-maze tests. Patch clamp recordings were used to evaluate evoked action potentials in Purkinje cells. Behavioral phenotyping demonstrated that Scn8aflox/flox, L7Cre+ mice have impaired social interaction, motor learning and reversal learning as well as increased repetitive behavior and anxiety-like behaviors. By 5 months of age, Scn8aflox/flox, L7Cre+ mice began to exhibit cerebellar Purkinje cell loss and reduced molecular thickness. At 9 months of age, Scn8aflox/flox, L7Cre+ mice exhibited decreased cerebellar size and a reduced number of cerebellar Purkinje cells more profoundly, with evidence of additional neurodegeneration in the molecular layer and deep cerebellar nuclei. Purkinje cells in Scn8aflox/flox, L7Cre+ mice exhibited reduced repetitive firing. Taken together, our experiments indicated that loss of Scn8a expression in cerebellar Purkinje cells leads to cerebellar degeneration and several ASD-related behaviors. Our study demonstrated the specific contribution of loss of Scn8a in cerebellar Purkinje cells to behavioral deficits characteristic of ASD. However, it should be noted that our observed effects reported here are specific to the C57BL/6 genome type.
Autism spectrum disorder (ASD) is a behaviorally defined pervasive neurodevelopmental disorder, characterized by persistent impairment of social communication, restricted interests and repetitive behaviors (Varghese et al., 2017). In addition to these core symptoms, there may be psychiatric or neurological comorbidities, of which attention-deficit/hyperactivity disorder (ADHD), anxiety, depression and epilepsy are most common (Lord et al., 2020). It is estimated that the prevalence of ASD is 1–2% in the general population (Wisniowiecka-Kowalnik and Nowakowska, 2019). Over the past decade, genomic technologies have enabled rapid progress in the identification of genes linked to ASD (Abrahams and Geschwind, 2008; Arnett et al., 2019). Although much effort has centered on the genetic delineation of syndromic forms of ASD, the underlying molecular mechanisms remain incompletely understood (Hampson and Blatt, 2015).
Several lines of evidence implicate cerebellar dysfunction in the development of ASD. Post-mortem studies have demonstrated a reduced number and density of cerebellar Purkinje cells (PC) in patients with ASD, and isolated cerebellar injury has been associated with a higher incidence of ASD (Wang et al., 2014; Hampson and Blatt, 2015). The cerebellum has been consistently implicated in several monogenetic syndromes associated with ASD (Fatemi and Folsom, 2015; Sundberg and Sahin, 2015; Varghese et al., 2017). Recent studies suggest that PC dysfunction caused by mutations in Tsc1, Tsc2, Shank2, and PTEN during a critical developmental period may contribute to behavioral deficits relevant to ASDs (Tsai et al., 2012, 2018; Reith et al., 2013; Cupolillo et al., 2016; Peter et al., 2016).
The Scn8a gene encodes the voltage-gated sodium channel α subunit Nav1.6. Nav1.6 is enriched at the neuronal axon initial segment and nodes of Ranvier, where it promotes neuronal excitability by participating in the initiation and propagation of action potentials (O'Brien and Meisler, 2013; Meisler et al., 2016). Nav1.6 is expressed throughout the central nervous system, including high expression in cerebellum, particularly in PCs (Schaller and Caldwell, 2003; Chen et al., 2009). In humans, pathogenic SCN8A variants are associated with a wide spectrum of phenotypes, from benign familial infantile seizures to developmental and epileptic encephalopathies (Gardella and Moller, 2019; Gertler and Carvill, 2019; Meisler, 2019). While prior work focused on the role of SCN8A in epileptic encephalopathies, there is evidence in humans and mice that variants in SCN8A are also associated with intellectual disability and neuropsychiatric abnormalities (Trudeau et al., 2006; McKinney et al., 2008; Blanchard et al., 2015; Butler et al., 2017; Wagnon et al., 2017; Liu et al., 2019; Meisler et al., 2021). Mice with heterozygous loss of Scn8a expression display enhanced emotionality in contextual fear conditioning, open field test and the light dark box test (McKinney et al., 2008). Conditional PC-specific Scn8a knockout mice were previously found to exhibit ataxia, impaired coordination, and deficits in delay eyeblink conditioning and Morris water maze tests (Levin et al., 2006; Woodruff-Pak et al., 2006) which are frequently observed in animal models with autistic features (Tsai et al., 2012; Reith et al., 2013; Piochon et al., 2014; Kloth et al., 2015; Cupolillo et al., 2016). Similar to findings in PC-specific Tsc1 and PTEN knockout mice, PC-specific Scn8a knockout mice had a lower frequency of spontaneous firing of PC indicative of lower excitability (Raman et al., 1997; Khaliq et al., 2003; Levin et al., 2006).
Motivated by these studies implicating the PC of Scn8a mutant mice in ASD related traits, we used the Cre-loxP recombination system to generate conditional knockout mice in which Scn8a inactivation is restricted to PC. We found that PC-specific Scn8a knockout mice of C57BL/6J strain background exhibited late-onset cerebellar degeneration and deficits in motor coordination and social interaction, increased repetitive behavior, anxiety and abnormal activity of PC, demonstrating the specific contribution of PC to these Scn8a-dependent phenotypes.
All animal experimental procedures were performed in accordance with the animal protocol approved by the Institutional Animal Care and Use Committee at the School of Medicine, Shandong University. Scn8aflox/flox mice (Levin and Meisler, 2004) have been maintained on a C57BL/6J strain background since 2004 in the Meisler lab at the University of Michigan, and were imported to Shandong University in 2017. L7/PCP2-Cre transgenic mice (stock number: J006207) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China), where they had also been maintained on the C57BL/6J strain background. Mice were housed in a 12-h light/dark cycle (lights on 7:00 AM) with controlled temperature and humidity and ventilated with a dedicated system. All mice had ad libitum access to sterile food and water. We crossed Scn8aflox/flox, L7Cre+ female mice and Scn8aflox/flox,L7Cre− male mice to generate Scn8aflox/flox,L7Cre− (control) and Scn8aflox/flox L7Cre+ (mutant) mice on the C57BL/6J strain background using the previously described breeding scheme (Levin et al., 2006).
Mice were intraperitoneally anesthetized with 4% chloral hydrate and transcardially perfused with PBS and then 4% paraformaldehyde (PFA). Brains were extracted, postfixed overnight in 4% PFA, dehydrated, embedded in paraffin, and sectioned at 3 μm. The sections were submerged into EDTA antigenic retrieval buffer (pH 8.0) and microwaved for antigenic retrieval. Sections were blocked with 10% goat serum and 0.5% Triton X-100 in 1× PBS for 20 min. Slides were incubated in primary antibody solution overnight at 4°C. Sections were washed in 3× PBS and incubated with secondary antibody for 1 h at room temperature, then washed in 3× PBS and incubated with DAPI. Finally, anti-fluorescence quencher was used to seal the sections (Wuhan goodbio technology CO., LTD, Wuhan, China). Fluorescence images were acquired using a Nikon Eclipse Ti-SR Inverted Microscope. Images were then processed and analyzed using CaseViewer software (3D Histech Ltd, Budapest, Hungary).
The following primary antibodies were used: Calbindin (1:100; ab82812, Abcam), Nav1.6 (1:100; ab65166, Abcam), Caspase3 (1:250; GB11532, Wuhan goodbio technology CO., LTD, Wuhan, China). Secondary antibodies were: goat anti-mouse 488 (1:400; GB25301, Wuhan goodbio, Wuhan, China), goat anti-rabbit 488 (1:400; GB25303, Wuhan goodbio, Wuhan, China), goat anti-mouse Cy3-IgG (1:300; GB21301, Wuhan goodbio, Wuhan, China), goat anti-rabbit Cy3-IgG (1:300; GB21303, Wuhan goodbio, Wuhan, China).
MRIs were collected on a 3-tesla MRI scanner (GE Healthcare, USA) using a standard birdcage head coil. Before the functional scans, high-resolution anatomical scans were acquired for each subject [repetition time (TR) = 2,100 ms, echo time (TE) = 111, field of view (FOV) = 10 × 10 cm, 190 slices, slice thickness = 1 mm] for later coregistration with functional maps. PD/T2 (TR = 2,500 ms, TE = 11.1/90, flip angle = 90°, FOV = 5 cm, slice thickness = 1 mm).
Six to eight week old mice were anesthetized with urethane (1.2 g/kg, i.p.) and decapitated. Parasagittal cerebellar slices were prepared with vibratome (VT 1000S, Leica, Germany) with a thickness of 300 μm. The slices were incubated in ACSF (saturated with 95% O2 to 5% CO2) for at least 1 h before recording.
After incubation, the slices were promptly transferred to the recording chamber placed on the staged of a modified upright Olympus microscope and continuously perfused with ACSF (95% O2 to 5% CO2). The patch electrodes (3–7 MΩ) were pulled on a multistage micropipette puller (P-97, Sutter Instrument, USA), and the pipette solution contained (in mM): KCl 140, MgCl2 2, EGTA 10, HEPES 10, Mg-ATP 2, buffered to pH = 7.4 with KOH. After the whole-cell clamp configuration was formatted, the cells were stabilized for 5 min before recording. Then the PCs were depolarized by current steps to evoke action potential at a holding potential of −70 mV.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was carried out on cerebellar slices according to the manufacturer's instructions (Roche). TUNEL-positive and DAPI-positive nuclei were examined using a fluorescence light inverted microscope (Nikon Eclipse TI-SR). The ratio of TUNEL-positive to total DAPI positive cells was calculated in six visual fields at ×100 magnification.
All behavioral tests were performed during the light cycle between 07:00 and 19:00. Male and female 5–9-week old mice were used. Similar numbers of male and female mice for each genotype were included. It is possible that genotype effects may have been underestimated or overlooked if they were sex dependent or if the baseline differences between female and male mice increased the variance in the data. Therefore, analysis of variance (ANOVA) models was used to test for the sex dependence of the genotype effects. A three-way repeated measures ANOVA with between-subject factors for genotype and sex and a repeated measure for training day was applied to accelerating rotarod and water-T maze data; a three-way ANOVA with factors for genotype, sex and pairing group was used for three-chambered test, and a two-way ANOVA with factors for genotype and sex was utilized for open-field test, light-dark transition box, elevated plus maze, grooming and male-female interaction. These analyses did not show any measures in which there was a significant effect of sex or a sex-genotype interaction. All behavioral assays were performed with the examiner blind to mouse genotypes. All the videos were analyzed by Smart software (Pan Lab, Harvard Apparatus).
The gait of Scn8aflox/flox L7Cre+ mutant mice was compared with Scn8aflox/flox L7Cre− control mice by footprint analysis as previously described (Carter et al., 1999). Briefly, to obtain footprints the fore and hind paws of the mice were coated with red and black non-toxic, water-soluble paint, respectively. Footprint patterns were analyzed using a runway (50 cm × 9 cm wide) with white paper on the bottom. The average length and width of the steps were measured.
Motor coordination and balance were tested with the accelerating rotarod (Panlab, Harvard apparatus) as described previously (Buitrago et al., 2004). Animals were tested over 5 consecutive days, each day consisting of 3–5 trials. The mice were placed on a 3 cm diameter rod which began rotating at 4 rpm and accelerated to 40 rpm over a period of 2 min. Latency to fall was recorded. Animals were tested at 5–6 weeks of age.
Exploratory locomotor activity was measured in an open field as previously described (Burne et al., 2005). Each mouse was placed in an opaque open field (30 cm × 30 cm × 30 cm), under dim light. Mice were placed in the chamber for a 15 min period. Distance traveled in 1 min time bins was recorded using a centrally placed video camera and automated video tracking software (Smart software, Pan Lab/Harvard Apparatus). To assess anxiety-related behaviors, the number of entries in the center zone and percent of time in the center of the chamber was also recorded (Bailey and Crawley, 2009). Measurements were taken from animals aged 6 weeks.
The elevated plus maze is a plus-shaped apparatus consisted of two open arms 8 × 25 cm and two closed arms (8 × 25 × 25 cm) with 8 × 8 cm central area, elevated 50 cm from the floor. Mice were placed in the central area facing one of the open arms, and allowed to freely explore the maze for 5 min. The number of entries and time spent in open or enclosed arms was measured as a parameter of anxiety-like behavior using an overhead camera and tracking system (SMART® Panlab, Harvard Apparatus).
To further measure anxiety-like responses, the light-dark box test was performed as described previously (Tang et al., 2017). The light/dark box was constructed of plexiglass (45 × 27 × 27 cm) consisting of two chambers, a black chamber (18 × 27 cm) and a light chamber (27 × 27 cm). Mice were placed into the dark box and allowed to freely move between the light box and dark box for 5 min. The amount of time spent in the dark side and the total number of transitions between the light and dark sides were recorded.
The automated three-chambered social approach task is commonly employed as a standard test for assaying sociability in mice (Yang et al., 2011). The apparatus consists of a rectangular, three-chambered box made from clear polycarbonate. Retractable doorways within the two dividing walls allowed access to the side chambers. The number of entries and time spent in the chambers were automatically recorded using an overhead camera and tracking system (Smart software, Pan Lab/Harvard Apparatus). The subject mouse was allowed to habituate in the apparatus for 20 min before the sociability test, first for 10 min in the central chamber, followed by 10 min of free exploration in the entire empty arena with both doors open. In the social interaction testing period, a novel object (an inverted wire mesh cup) was placed in one of the side chambers and a novel mouse (with different genetic background matched to the subject mouse by sex and age) was placed inside an identical inverted wire cup in the other side chamber. In the social novelty testing period, another novel mouse was placed inside the empty wire cup. The apparatus was cleaned with 70% ethanol and water between subjects. Time spent interacting with the novel animal and with the object was recorded by an examiner with a stopwatch (Crawley, 2007). Animals were tested between 7 and 9 weeks of age.
We evaluated the ability of the mouse to detect novel odors and social odors as previously described (Yang et al., 2011; Tsai et al., 2012). Animals were placed in an empty, clean observation cage containing a thin layer of clean bedding and a hole on the flat filter top lid for inserting a cotton-tipped swab. Mice were habituated for 30 min with a clean cotton swab and then presented sequentially with non-social odors and social odors. Odors were presented in three consecutive trials per odorant stimulus (2 min per trial) in the following order: water, almond extract (1:100), banana extract (1:100), social odor 1, social odor 2. Social odors were created by swabbing the cotton tip in a zigzag fashion in previously soiled bedding from cages containing unfamiliar gender and age-matched animals the experimental animal had not interacted with. Time spent sniffing the swab with each presentation for each 2 min trial was measured by an investigator with a stopwatch. Measurements were taken from animals aged 7–8 weeks.
The procedure was adapted from a previously described protocol (Cupolillo et al., 2016). The test for male-female interaction was performed in a clean testing cage (Plexiglass box, 25 × 40 × 18.5 cm). Each male mouse was habituated to the testing cage for 15 min, after which an unfamiliar female of the same genotype was placed into the testing cage with a single layer of corncob bedding. An experimenter blind to the mouse genotype measured the cumulative time (by means of a stopwatch) that the male mouse spent in close contact with the female. Social interaction behavior included close following at the same speed behind the female, touching, nose-to-nose sniffing, anogenital sniffing and/or mouthing and licking the fur of the female. The cumulative time was measured (using a stopwatch) by the investigator and calculated as total time spent in contact. Animals were tested between 7 and 8 weeks of age.
Mice were scored for spontaneous repetitive self-grooming behavior as previously described (Silverman et al., 2010a). Each mouse was placed individually into a standard mouse shoebox observation cage with no bedding and a flat filter top lid. After habituation for 10 min, animals were observed for another 10 min. Two mice were scored simultaneously by a trained observer, who was blind to mouse genotype. Cumulative grooming time in the observation period was recorded using stopwatches. Measurements were taken from animals aged 5–6 weeks.
To measure reversal learning, the water T-maze was performed as described (Bednar et al., 2002; Tsai et al., 2018). A transparent platform submerged about 1 cm below the surface of the water at one of the short arms of the T-maze and served as an escape for the animals. After 1 day consisting of a habituation swim trial (60 s) with no platform present, mice were given 15 trials a day for 3 consecutive days to learn the location of the platform. After 15 trials on day 4, the platform was changed to the other short arm of the maze. Mice were then tested for 15 additional trials (reversal day 1). Then for 2 subsequent days (reversal days 2 and 3), mice were given 15 trials per day. The number of correct trials and the number of trials required to achieve five consecutive correct trials were recorded. Measurements were taken from animals aged 8–9 weeks.
Data are expressed as mean ± s.e.m., and statistical analysis was carried out using GraphPad Prism 8 software (GraphPad Software Inc., La Jolla, CA, USA). Statistical analyses included Student's T-test (paired or unpaired), one-way ANOVA followed by Tukey's post-hoc analysis, two-way repeated measures ANOVA followed by Bonferroni's post-hoc analysis. P < 0.05 was considered statistically significant.
We generated Purkinje cell specific Scn8a knockout mice of genotype Scn8aflox/flox,Cre+ (PC Scn8a mutant mice) according to the breeding scheme (Levin et al., 2006). As shown in Supplementary Figure S1, PC Scn8a mutant mice had reduced expression of Nav 1.6 in Purkinje cells compared with control mice, whereas expression remained high in granule cells and neurons (stellate cells and basket cells) of the molecular layer in PC mutant mice.
Consistent with earlier reports (Sprunger et al., 1999; Levin et al., 2006), cerebellar malformations were not detected in both 2 and 4-month-old PC Scn8a mutant mice. Calbindin staining indicated that the Purkinje cells in the mutant mice retained the regular orientation of dendrites present in control animals (Supplementary Figure S2). However, calbindin staining was noted in the granule cell layer of PC Scn8a mutant mice (Supplementary Figure S2, arrows). This pattern of labeling has previously been described in Scn8amed−jo (A1071T) mice (Dick et al., 1985) and Scn8aflox/flox,L7Cre+ mice (Levin et al., 2006) and may reflect axonal swelling. We revealed that PC density, size of soma, and thickness of the molecular layer were normal at both 2 and 4 months of age (Figure 1). However, by 5 months of age, there was a reduction in the density of Purkinje cells and the thickness of molecular cell (P < 0.01), but the size of Purkinje cells retained normal. At 9 months of age there was a significant reduction in the thicknesses of the molecular layer and both the size and the density of Purkinje cells was significantly reduced (P < 0.001. n > 3 per group, two-way ANOVA, Bonferroni's post-hoc analysis). Overall brain volume was decreased at 9 months of age, as evident in the whole-mount brain (Figure 2A). MRI measurements revealed reduced area of the cerebellum of the mutant mice in both axial and coronal views (Figures 2B–D).
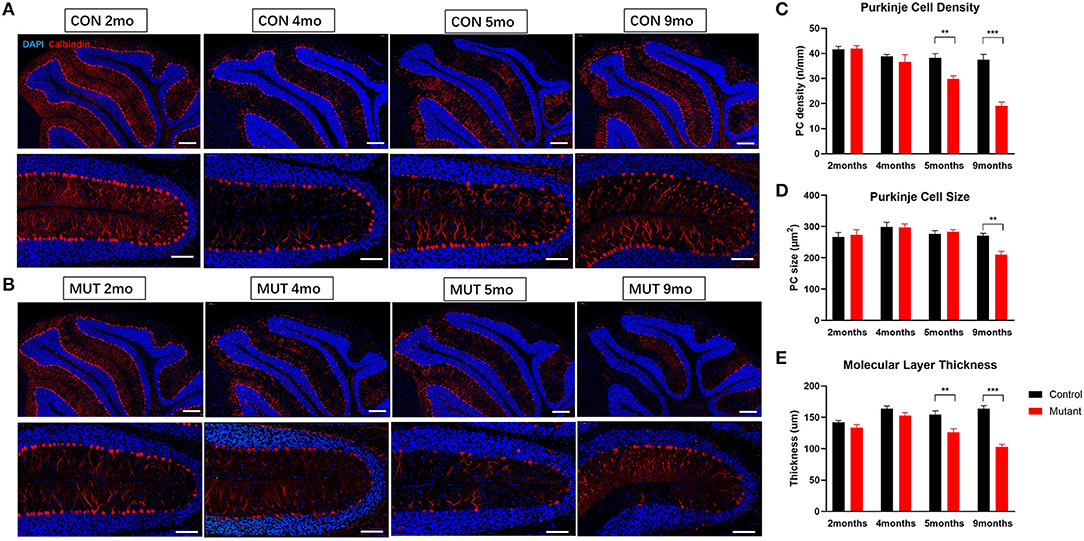
Figure 1. PC Scn8a mutant mice exhibit histological changes in cerebellum. (A,B) Abnormal PC morphology and gradual reduced PC number and molecular layer thickness in Scn8a mutant cerebellum (Scale bar upper row 200 μm. bottom row 100 μm). (C,D) The quantification of PC density and the quantification of PC soma area was performed in the IV–V. PC loss was not apparent until 5 months of age, and showed ongoing loss till 9 months (C). At 9 months, mutant Purkinje cell size decreased significantly (D). (E) The thickness of the molecular layer (ML) was normal in mutant cerebellum at 4 months of age, whereas it decreased afterward at 5 months (n ≥ 3 per group. Data shown are means ± SEM. **P < 0.01, ***P < 0.001, two-way ANOVA, Bonferroni's post-hoc analysis).
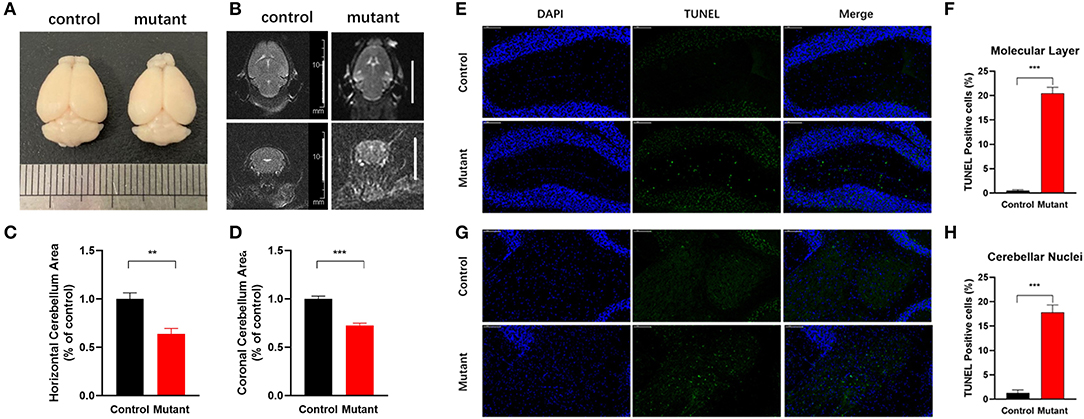
Figure 2. Reduced cerebellar volume and increased apoptosis in Scn8a PC mutant cerebellum. (A) Representative whole-mount images of control and PC Scn8a mutant brains at 9 months of age. The mutant cerebellum appears smaller than control. (B) Magnetic resonance imaging T2 axial and coronal images of the brain showing cerebellar volume loss in the mutant mice at 9–10 months of age (scale bar 10 mm). (C,D) Measurement of the cerebellar area of Scn8a mutant and control mice using T2-weighted 3.0 T MR imaging. Quantitative analysis revealed a significant reduction in cerebellar area in mutant animals in both axial (C) and coronal area (D) (Mean ± SEM; n = 5 per group; **P < 0.01, ***P < 0.001, Student's unpaired t-test). (E,G) Sections of cerebellum of control and PC Scn8a mutant mice at 9–10 months of age labeled with TUNEL showing apoptosis in the molecular cell layer and cerebellar nuclei (Scale bar: 100 μm). (F,H) Percentage of TUNEL-positive cells per unit area of cerebellar molecular layer and deep cerebellar nuclei in the PC Scn8a mice compared with controls (n = 4 for each group, ***P < 0.001, Student's unpaired t-test).
To determine whether neurodegeneration could account for the observed cerebellar atrophy, we adopted TUNEL staining to quantify apoptosis. The number of TUNEL-positive cells was significantly increased in PC Scn8a mutant mice in both the molecular layer (Figures 2E,F) and cerebellar deep nuclei (Figures 2G,H) (P < 0.001; n = 4 for mutants and control, unpaired student's t-test).
It was previously demonstrated that partial or complete loss of Nav1.6 from cerebellar Purkinje cells reduces excitability and repetitive firing (Raman et al., 1997; Khaliq et al., 2003; Levin et al., 2006). To examine excitability of PCs in our mice, evoked APs were recorded by whole-cell patch clamp. Repetitive firing spikes were evoked by current injection of 200 pA with 500 ms duration. The frequency of repetitive firing was inhibited in Scn8a mutant PCs compared with that in control (Supplementary Figures S3A,B). The mean frequency of repetitive firing at each current injection from 50 to 400 pA was significantly reduced in Scn8a mutant PCs (Supplementary Figure S3C). These results confirm the previous evidence that loss of Scn8a inhibits the excitability of PCs (Levin et al., 2006).
By 6–8 weeks of age, PC Scn8a mutant mice developed a mildly ataxic, waddling gait that appeared to be caused by poor hindlimb coordination. However, at 6 weeks of age, mutant and control mice had comparable fore-base width and no difference in stride length and hind-base width (Figures 3A,B) (P > 0.05, Student's unpaired t-test, control 6wk n = 10, mutant 6wk n = 10). However, at 12 weeks of age, PC Scn8a mutant mice displayed a narrower stride length (P = 0.022) and a wider hind-base width (P = 0.007) (control n = 6, mutant n = 9), demonstrating a deficit in motor coordination.
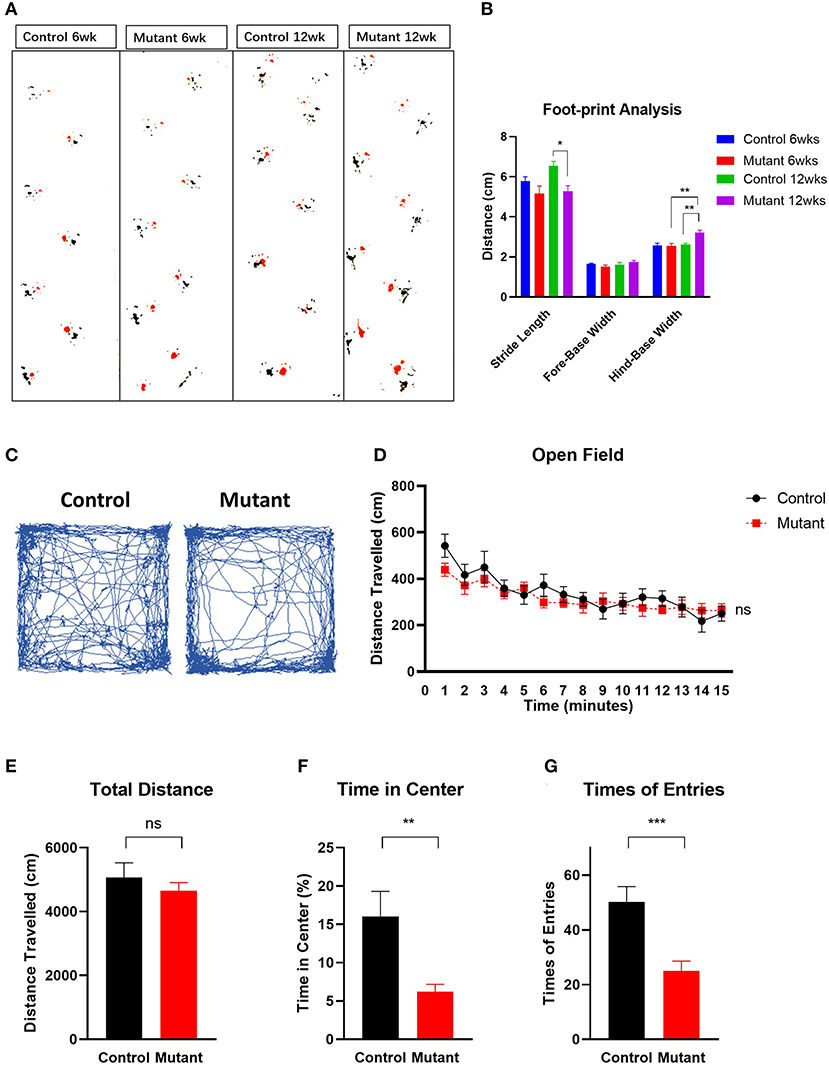
Figure 3. Motor coordination impairment and locomotor activity in PC Scn8a mutant mice. (A) Representative walking footprint patterns. (B) Gait footprint analysis at 6 and 12 weeks was evaluated for stride length, fore-base width and hind-base width. Mutant mice displayed ataxic gait with reduced stride length and increased stride width at 12 weeks of age (n > 6 per group. *P < 0.05, **P < 0.01; ***P < 0.001, two-way ANOVA, Bonferroni's post-hoc analysis). (C) Representative traces of mouse activity in an open field during a 15-min recording time. (D) Locomotor activity in the open field was comparable between control (n = 9) and mutant mice (n = 11) in a 15 min time bin (P = 0.4223). Time in minutes is shown on the X-axis. (E) The total distance moved in the open field test was not significantly different between controls and mutants (P > 0.05). (F,G) Mutant mice spent less time (F) in the center of the open field (P = 0.0063), and entered the center of the open field less often (G) than the controls (P < 0.001). Data shown are means ± SEM. ns, not significant. *P < 0.05, **P < 0.01, ***P < 0.001, two-way ANOVA, Bonferroni's post-hoc analysis for (D). Student unpaired t-test for (E–G).
Consistent with previous work (Levin et al., 2006), mutant mice demonstrated impaired performance on the rotarod test (Supplementary Figure S4). At 5 weeks of age, the latency to fall was significantly different between wildtype and mutant mice [two-way repeated measures ANOVA F(1,18) = 27.99, Bonferroni post-hoc, P < 0.0001, n = 9 for control, n = 11 for mutant]. While control mice remained on the rod longer during each of 5 daily sessions of training, mutant mice had shorter latency to fall on day 1 (P < 0.001) and did not improve with time. These findings further demonstrate impaired motor coordination and poor motor learning in mice with ongoing PC loss.
Patients harboring loss of function variants of Scn8a exhibit neuropsychological abnormalities including emotional instability, anxiety and attention deficit hyperactivity disorders (Trudeau et al., 2006; Wagnon et al., 2017). We therefore carried out behavioral testing of 6-week-old PC Scn8a mutant mice to assess their anxiety level.
The open field exploration test is a behavioral assay widely used to evaluate locomotor responses to novel environments in rodents. Representative tracks are shown in Figure 3C. During a 15-min test, the distance traveled by mutant mice in the open field during a 1 min time bin was similar to controls [Figure 3D, two-way ANOVA, Bonferroni's post-hoc analysis, F(1,17) = 0.6760, P = 0.4223]. The total distance traveled was also similar in controls and mutants [Figure 3E, Student unpaired t-test, t(17) = 0.822, P > 0.05]. However, PC Scn8a mutants spent significantly less time in the center of the open field [Figure 3F, Student unpaired t-test, t(17) = 3.254, P = 0.0047] and had fewer entries into the center [Figure 3G, Student unpaired t-test, t(17) = 3.982, P < 0.001] (control n = 8, mutant n = 11).
As an independent test of anxiety, we used the light/dark box transition task (Bourin and Hascoet, 2003). The number of entries into the light compartment [t(25) = 5.297, P < 0.0001] and the time spent in the bright area [t(25) = 4.129, P < 0.001] were significantly decreased in PC Scn8a mutant mice, indicating increased innate anxiety-like behavior (control n = 15, mutant n = 13 Student's unpaired t-test) (Figures 4A–C).
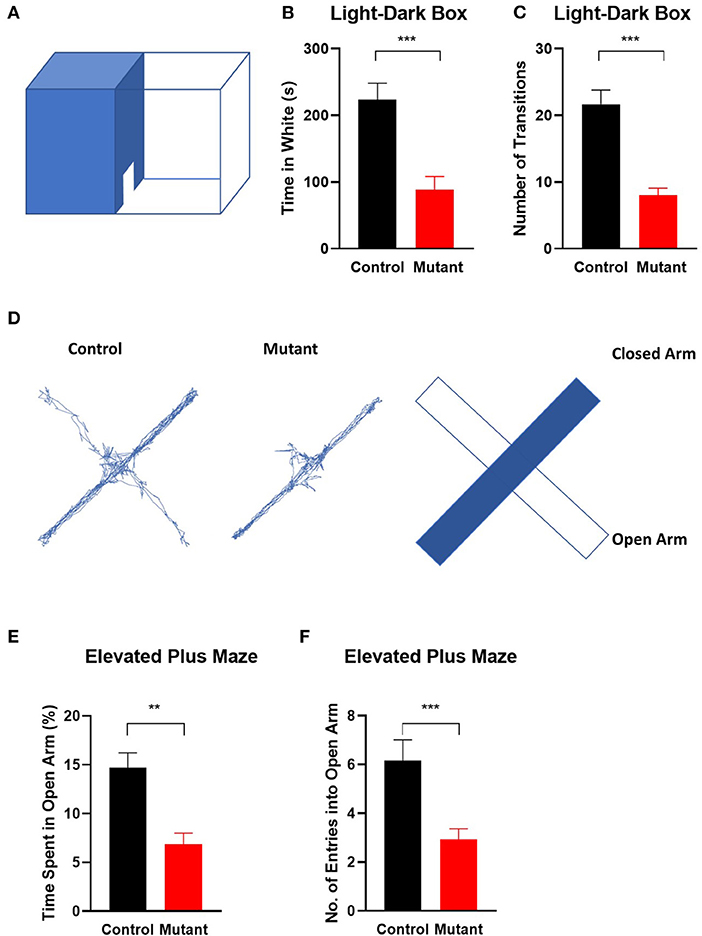
Figure 4. Elevation of anxiety in Scn8a PC mutant mice. (A) Schematic of light-dark transition box. In the light–dark preference test, PC Scn8a mutant mice spent significantly less time in the light room (B) and entered it less often than controls (C). (n = 15 for control and n = 13 for mutant). (D) Representative elevated plus maze tracking plots and diagram illustrates locations of open and closed arms of the maze. Mutant mice demonstrated decreased time spent in the open arms (E), and less frequently enter into the open arms (F) of the elevated plus maze (Control: n = 13 Mutant: n = 16). **P < 0.01, ***P < 0.001. All P-values by the Student's unpaired t-test.
To further assess anxiety, we analyzed behavior on an elevated plus maze (Hogg, 1996) (Figures 4D–F). As in the open field, mutant mice demonstrated decreased total time spent in the open arms [t(27) = 4.191, P < 0.001], and a reduced number of entries into the open arms of the elevated plus maze [t(27) = 3.532, P = 0.0015] (control n = 13 and mutant n = 16, Student's unpaired t-test).
Taken together, these assays indicate that mutant mice display increased anxiety compared with control littermates.
A three-chambered apparatus was used to measure social approach and social novelty (Figure 5). Compared with control mice, PC Scn8a mutant mice spent less time in the side chamber with the novel mouse and more time with the novel object (Figure 5B, P > 0.05). Control mice spent significantly more time with the novel mouse than with the novel object [P < 0.001, chamber × genotype Interaction F(1,31) = 3.984, P = 0.0548, chamber effect F(1,31) = 19.10, P = 0.0001, genotype effect F(1,31) = 7.637, P = 0.0095, n = 16 for control, n = 17 for mutant, two-way repeated measures ANOVA, Bonferroni's post-hoc analysis].
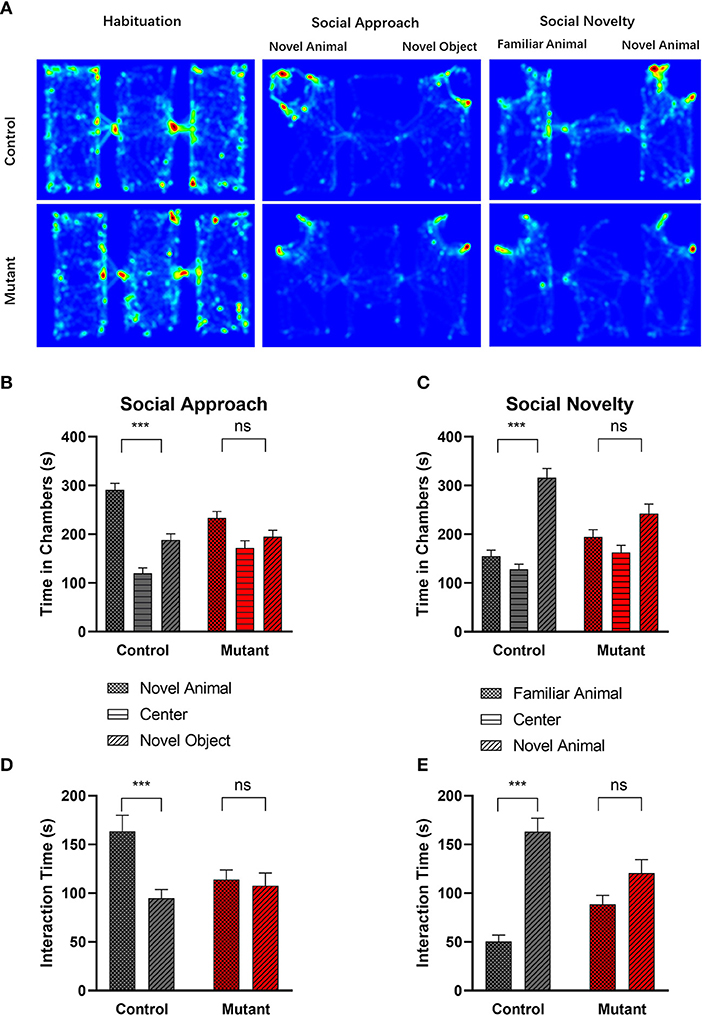
Figure 5. Abnormal social behavior in PC Scn8a mutant mice. (A) Representative heat maps showing time spent by Control mice (upper panel) and PC Scn8a mutant mice (lower panel) at each location of the three-chambered apparatus during the test. (B,D) In the social approach test, WT mice spent more time in the chamber with the stranger animal than in the chamber with the inanimate object (B), and spent more time interacting with novel animal (D) in comparison with a novel object, whereas PC Scn8a mutant mice show no preference. (C,E) PC Scn8a mutant mice show no preference for the chamber with the novel animal in comparison with that with a familiar animal in an assay of social novelty (C), and spent similar time interacting with both the novel and familiar animals (E). This is in distinct contrast to control mice. Control: n = 16; Mutants: n = 17, ***P < 0.001, two-way ANOVA, Bonferroni's post-hoc analysis.
We examined the amount of time each experimental mouse spent interacting with the novel mouse or the object through sniffing [Figure 5D, Genotype × chamber interaction, F(1,31) = 7.420, P = 0.010; Chamber effect F(1,31) = 11.17, P = 0.0022; Genotype effect F(1,31) = 1.035, P = 0.3168, two-way repeated measures ANOVA, Bonferroni's post-hoc analysis]. While control mice showed more interest in interacting with the novel mouse (P < 0.001), mutant mice spent a comparable amount of time sniffing the novel mouse and the novel object (P > 0.05).
When the inanimate object was replaced with another novel mouse, control mice spent more time in the chamber containing a novel mouse than in the chamber with the familiar mouse (P < 0.001), demonstrating normal sociability [genotype × chamber interaction F(1,31) = 6.596, P = 0.0153, Chamber effect F(1,31) = 22.62, P < 0.0001, Genotype effect F(1,31) = 3.479, P = 0.0717, two-way repeated measures ANOVA, Bonferroni's post-hoc analysis] (Figure 5C). In contrast, the PC Scn8a mutants failed to show any preference between the two social chambers (P > 0.05). Control mice also spent more time sniffing the novel animal over the familiar animal (Figure 5E, P < 0.001), while mutant mice failed to show such preference (P > 0.05) [genotype × chamber interaction F(1,31) = 9.777, P = 0.0038, chamber effect F(1,31) = 33.51, P < 0.0001, genotype effect F(1,31) = 0.02699, P = 0.8706, two-way repeated measures ANOVA, Bonferroni's post-hoc analysis].
It is thought that impaired discrimination of social olfactory cues may contribute to social deficits in mice (Silverman et al., 2010b). We used a social olfaction assay to assess interaction with social olfactory cues (Figure 6A). When mice were first presented with three non-social odor cues (water, almond extract and banana extract), there were no significant differences between mutant and wildtype mice (P > 0.05, n = 11 Control and 11 mutants), demonstrating normal olfactory function. When social odors (social A and social B) were applied, PC Scn8a mutant mice spent less time sniffing the cotton tips than control mice, suggesting a lack of interest in social odor stimuli [n = 11 per group, two-way repeated measures ANOVA F(1,20) = 15.25 Bonferroni post-hoc, P < 0.0001 for Social A1, P = 0.001 for Social A2, P < 0.0001 for Social B1, P = 0.036 for Social B2].
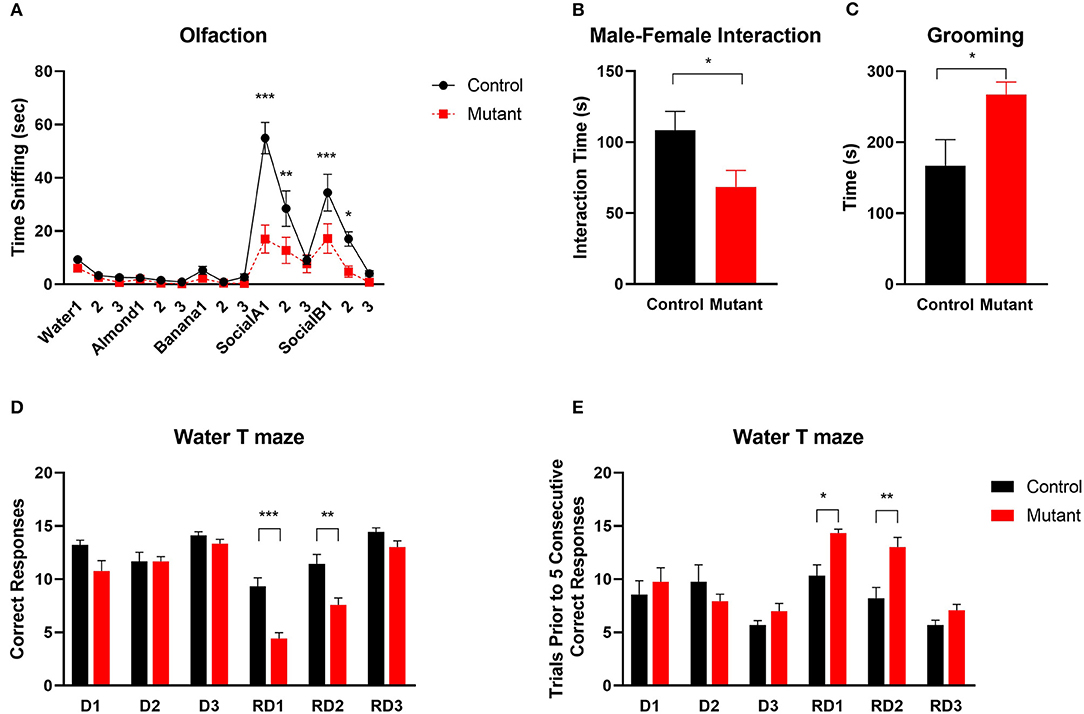
Figure 6. Abnormal social and repetitive behaviors and impaired reversal learning in PC Scn8a mutant mice. (A) Mutant mice spent comparable time sniffing non-social stimuli whereas less time sniffing social odor cues (n = 11 per group). (B) Reduced sociability of KO mice is indicated by reduced time interacting with a female conspecific (Control: n = 8; Mutant: n = 10). (C) PC Scn8a mutant mice spent more time self-grooming (Control: n = 10; Mutant: n = 11). (D,E) On Day 1 to Day 3, mutants display normal acquisition learning of the escape platform location in the water T maze as indicated by total correct trials (D) and trials needed for 5 consecutive correct responses (E). However, on reversal day 1 and day 2, PC Scn8a mutant mice have significantly fewer correct trials (D), and take more trials to achieve five consecutive correct responses (Control: n = 9, Mutant: n = 12). RD, Reversal Day. Data shown are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Two-way ANOVA, Bonferroni post-hoc analysis in (A), Student's unpaired t-test in (B–E).
Social behavior was further tested by assessing social interaction with a female conspecific mouse. Compared with control mice, there was a significant reduction in the time spent sniffing, allogrooming, mounting, or following the female by male PC Scn8a mutant mice [Student's t-test t(18) = 2.23, P = 0.038; Control n = 8; Mutant n = 12] (Figure 6B).
One of the diagnostic behavioral symptoms of autism is repetitive behavior. We analyzed grooming as an index for stereotyped, repetitive behaviors. PC Scn8a mutant mice engaged in much longer bouts of self-grooming than control mice (Figure 6C) [n = 14 per group, Student's t-test t(26) = 2.48, P = 0.019].
ASD patients often show inflexible and rigid behavior and thinking (Bralten et al., 2018). To evaluate behavioral flexibility, we used the reversal water-T maze assay (Figures 6D,E). During the first 3 days of training, while the platform was in the right arm of the maze, both groups of mice performed equally well and learned the location of submerged platform. On the first reversal day with the platform moved to the left arm of the maze, PC Scn8a mutant mice incorrectly visited the right arm more frequently than control mice. Mutant mice also needed more trials to achieve 5 consecutive correct responses. Comparable results were observed on Reversal day 2. [Reversal Day 1, number of correct trials: Student's t-test t(19) = 5.097, P < 0.0001; number of trials prior to five consecutive correct trials: Student's t-test t(19) = 3.221, P = 0.0045; Reversal Day 2: number of correct trials: Student's t-test t(19) = 3.48, P = 0.0025; number of trials prior to five consecutive correct trials: Student's t-test t(19) = 3.467, P = 0.0026]. By the third reversal day, the mutant mice behaved similarly to the control mice in number of correct trials and number of trials prior to 5 consecutive correct responses.
The first link between human disease and SCN8A mutation was obtained in 2006 in a study of a small pedigree in which heterozygous carriers of a loss-of-function SCN8A mutation exhibited a range of phenotypes including ataxia, cognitive deficits, and emotional instability (Trudeau et al., 2006). Since the development of next generation sequencing, many pathogenic variants of SCN8A have been identified in patients with a spectrum of neurodevelopmental disorders, and some genotype-phenotype correlations have emerged. SCN8A missense variants with gain-of-function channel properties are associated with developmental epileptic encephalopathy with early onset of severe seizures, hypotonia, and paroxysmal dyskinesia. In contrast, loss of function mutations of SCN8A can cause autism or intellectual disability without seizures (Larsen et al., 2015; Gertler and Carvill, 2019; Liu et al., 2019; Meisler et al., 2021). In a recent study by Wong et al. (2021), they generated a novel Scn8a mouse model carrying the human R1620L mutation (with both gain- and loss-of-function effects) which exhibited a range of behavioral abnormalities, including hyperactivity, impaired learning and memory and social deficits. These findings suggest that SCN8A dysfunction may contribute to other neurological and neuropsychiatric disorders. Neuropsychiatric comorbidities were not previously studied in Purkinje cell specific Scn8a knockout model, and the role of reduced Nav1.6 in cerebellar function was incompletely characterized.
In this study, we provide evidence that SCN8A in cerebellar PCs has a key role in mechanisms involved in ASDs. We show that mice of C57BL/6J strain background with selective disruption of Scn8a in PCs display behavioral traits related to neuropsychiatric abnormalities such as ASDs and anxiety, associated with graded loss of PCs and progressive cerebellar atrophy.
One of the main symptoms of cerebellar dysfunction in humans is ataxia (Schniepp et al., 2017). Our results revealed that PC Scn8a mutant mice exhibit deficits in motor coordination and motor learning in the rotarod test. Gait analysis demonstrated wide base and ataxic gait. Similar features are also seen in patients with loss of function mutation of SCN8A (Trudeau et al., 2006), and in patients with ASD and other murine models of ASD (Fatemi et al., 2012; Tsai et al., 2012; Reith et al., 2013; Cupolillo et al., 2016).
PC Scn8a mutant mice were previously shown to be impaired in delay eyeblink conditioning (Woodruff-Pak et al., 2006), an additional connection to cerebellar dysfunction and autism. Eyeblink conditioning is affected in the general ASD patient population, and is viewed as a biomarker for ASD (Oristaglio et al., 2013; Welsh and Oristaglio, 2016). Eyeblink-conditioning defects appear more often in mouse autism models than in non-autism-like phenotypes (Kloth et al., 2015). Given the similarity of the PC Scn8a mutant mice to other autism models with regards to motor deficit, delay eye-blink conditioning impairment and electrophysiological changes in Purkinje cells, it is not surprising that PC Scn8a mutant mice also exhibit ASD-relevant social deficits.
Purkinje cell loss is the most consistent presentation in post mortem studies of ASD patients, with 35–95% fewer cerebellar Purkinje cells in ASD brains than controls (Whitney et al., 2009; Wegiel et al., 2014; Mosconi et al., 2015). Purkinje cells are recognized as key cells mediating autism-like phenotypes in mice (Fatemi et al., 2012; Tsai et al., 2012; Reith et al., 2013; Cupolillo et al., 2016). PC Scn8a mutant mice also displayed abnormalities in the composition of the cerebellum. The thickness of molecular layer as well as the density of Purkinje cells were comparable at early age. However, Purkinje cell beginning to lose between 4 and 5 months of age, with an ongoing and significantly decreased in both Purkinje cell density and thickness of molecular layer in PC Scn8a mutant mice by 9 months of age (Figure 1). Progressive PC loss is accompanied by cerebellar atrophy indicated by both gross anatomy and MR imaging of cerebellum.
Previous studies demonstrated a relationship between cerebellar function and behavior including sociability (novel mouse vs. novel object), social preference (familiar mouse vs. novel mouse), social odor preference (conspecific urine vs. water), and male-female interaction (Tsai et al., 2012; Cupolillo et al., 2016). Our demonstration of social deficits in three-chambered tests were highly consistent with earlier studies in other cerebellar genetic mouse models of autism (Tsai et al., 2012; Reith et al., 2013). PC Scn8a mutant mice also displayed reduced responses to female social cues in a male-female reciprocal social interaction context. Mutant mice spent significantly less time sniffing the social odors than control mice in the context of olfaction test.
Autism and anxiety disorders are frequently comorbid with each other (Adams et al., 2020; Baribeau et al., 2020, 2021), and autistic and anxious traits are highly correlated (Ha et al., 2016; Tatsukawa et al., 2019). Likewise, co-existing anxiety-like behaviors were observed in the Scn8a PC mutant mice. Anxiety-like behaviors in the mutant mice included reduced exploration behavior in the center in the open field test and reduced non-social anxiety in the elevated plus maze and light-dark transition test. Similar anxiety-like behaviors also presented in autistic mice with Purkinje cell-specific deficiency of Shank2 (Ha et al., 2016) and Pianp mice (Winkler et al., 2020). McKinney et al. (2008) investigated mice heterozygous for a null mutation of Scn8a (Scn8a+/−) and demonstrated avoidance of well-lit, open environments as well as pronounced stress-induced coping behavior. Impaired Purkinje cell firing was also demonstrated in heterozygous Scn8a+/− null mice (Raman et al., 1997; Khaliq et al., 2003). Our data on the PC-specific KO of Scn8a suggest that the enhanced anxiety-like behaviors in Scn8a heterozygous null mice may be mediated by altered output signaling from the cerebellum.
It has been observed that cognitive inflexibility and cerebellar pathology co-occur in psychiatric disorders (e.g., autism, schizophrenia, addiction). Recent studies using lurcher mutant mouse, which lose 100% of their Purkinje cells postnatally or lurcher mouse chimeras, which lose varying numbers of Purkinje cells, suggested impairment in behavioral flexibility, reversal learning and increased repetitive behaviors, as well as higher level cognitive processes (Dickson et al., 2010, 2017; Martin et al., 2010). Inflexibility to change in routine can be viewed as a form of perseverative behavior, and assessments of reversal learning are used as a behavioral endpoint in studies of rodent models. In water T-maze tests, the initial acquisition of the behavior was normal but PC Scn8a mutant mice exhibited impaired reversal learning. Autistic patients often exhibit repetitive behaviors that refuse to change (Hollander et al., 2003). This feature can be recapitulated by rodent grooming behaviors (Crawley, 2004). PC Scn8a mutant mice displayed increased repetitive behavior, including self-grooming. All these results, together with previous findings in lurcher mutant mouse and lucher chimeras, indicated that cerebellar pathology may play a causal role in the generation of repetitive behaviors and cognitive inflexibility.
Previous research found the levels of Bcl-2 and P53 protein, two important markers of apoptosis, were increased in cerebellum of autistic subjects compared with controls (Araghi-Niknam and Fatemi, 2003). Dong et al. (2018) demonstrated elevation of ER stress signals, oxidative stress, and apoptosis in the molecular layer of the autistic cerebellum. Deep cerebellar nuclear cells to which Purkinje cells project are abnormal in ASD, showing enlargement during childhood followed by reduction in size and number during adolescence and adulthood (Bauman, 1991). Using the TUNEL assay, we found increased cell apoptosis in molecular layer of PC Scn8a mutant mice. Interestingly, we also noted obvious cell apoptosis in neurons of the deep cerebellar nuclei.
In Purkinje cells lacking Nav1.6 channels, both spontaneous firing activity and high frequency discharge are impaired (Raman et al., 1997; Khaliq et al., 2003; Levin et al., 2006). We also confirmed the reduced excitability in Scn8a mutant PCs. Since PC provide the only output from the cerebellar cortex, the reduced firing of Purkinje cells observed in many ASD-like mouse models (Tsai et al., 2012; Cupolillo et al., 2016; Yang et al., 2020) may disinhibit the downstream deep cerebellar nuclei, which gate the outgoing information to the thalamus, basal ganglia, and neocortex (Sundberg and Sahin, 2015), potentially influencing integrative networks (Bostan and Strick, 2018). Neurons of the deep cerebellar nuclei receive major basal and driven inhibition from PC, but they are also spontaneously active, producing action potentials even without excitation (Zheng and Raman, 2011). Therefore, we suggest that loss of Scn8a in Purkinje cells might disrupt Purkinje-mediated inhibition and increase firing in the deep cerebellar nuclei. The resulting elevated spontaneous activity of cerebellar nuclei could result in oxidative stress and induced apoptosis. Further research is needed to elucidate the mechanism connecting Purkinje cell loss to cerebellar apoptosis.
However, it should be noted that our study was performed on a single inbred strain, C57BL/6J. Therefore, our understanding of these effects is limited to this single genome type. However, recent studies showed that results from studies that utilize diverse genetic backgrounds are a better model of complex disease across individuals and are more likely to generalize across patient populations (Neuner et al., 2019). Therefore, further research is needed to validate the effects of loss of Scn8a expression in Purkinje cells on cognitive and autistic behavior changes using different inbred strains.
We have demonstrated that mice with loss of Scn8a in Purkinje cells provide a new model of features of ASD. Mice of C57BL/6J strain with homozygous loss-of-function of Nav1.6 channels in Purkinje cells exhibit motor deficits and autistic traits such as deficits in social interaction, stereotyped behaviors and anxiety-like behaviors, demonstrating an ASD-like phenotype with strong resemblance to other Purkinje cell-dependent models of ASD (Fatemi et al., 2012; Tsai et al., 2012; Reith et al., 2013; Cupolillo et al., 2016). Together with previously reports of reduced spontaneous and repetitive firing (Raman et al., 1997; Levin et al., 2006), learning deficits in the rotarod test (Levin et al., 2006) and delay eyeblink conditioning impairment (Woodruff-Pak et al., 2006), these studies support PC Scn8a mutant mice as a model that reproduces many symptoms of patients with SCN8A loss of function mutations.
Our study has demonstrated the specific contribution of loss of Scn8a in cerebellar Purkinje cells to behavioral deficits characteristic of ASD. These results provide novel insights into mechanisms contributing to the pathogenesis of ASDs. Future research to unravel the pathogenesis underlying molecular and cellular alterations will improve our understanding of the still enigmatic fields of ASD and anxiety, and open new avenues for molecular diagnosis and therapy.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by Ethics Committee of the School of Basic Medical Sciences of Shandong University.
XY and QL designed the experiments and synthesized and analyzed data. HY performed the electrophysiology study and analyzed data. XY, BL, and AC performed behavioral studies. AL and GZ performed and analyzed MRI data. XW, YS, and XB contributed to animal husbandry. XY and XW performed immunohistochemistry experiments and analyzed data. XY and MM organized the manuscript. DE-F, ZY, MM, and QL reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by National Natural Science Foundation of China (Grant Nos. 81601195, 81671114, 81741055, and 81873878), Natural Science Foundation of Shandong Province (Grant Nos. ZH2016HB38 and ZR2021MH229), and Open Research Fund of National Health Commission Key Laboratory of Birth Defects Prevention (ZD202101).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to Dr. Jingwei Liu from Qilu Hospital of Shandong University for helpful discussions and comments on the manuscript. We thank Dr. Young Park of the University of Michigan for breeding, maintenance and shipping of the Scn8a floxed mice. We thank the Translational Medicine Core Facility of Shandong University for consultation and instrument availability that supported this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.822129/full#supplementary-material
Supplementary Figure S1. Sagittal cryosection of cerebellar Nav1.6 staining (green) and calbindin staining (red) showing the deficiency of Nav1.6 expression in PC of the Scn8a mutant mice. Nav1.6 staining in control Purkinje cells are indicated by yellow arrows. Scale bar 50 μm.
Supplementary Figure S2. Scattered calbindin staining in the granule cell layer along the proximal segment of PC axons of Purkinje Scn8a mutant mice (arrows). Scale bar 100 μm.
Supplementary Figure S3. Reduced excitability of Scn8a mutant PCs. (A,B) The evoked action potential in control and Scn8a mutant PCs in response to depolarization of 200 pA. (C) Comparison of the mean frequency of repetitive firing at command potentials (n = 10 for both control and Scn8a mutant groups. All data were presented as mean ± SEM, ***P < 0.001).
Supplementary Figure S4. Motor coordination and motor learning assessed on an accelerating rotarod. PC Scn8a mutants displayed decreased time on an accelerating rotarod and no improvement with training (Control: n = 9, Mutant: n = 11. P < 0.001, two-way ANOVA, Bonferroni's post-hoc analysis).
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Adams, D., Clark, M., and Simpson, K. (2020). The relationship between child anxiety and the quality of life of children, and parents of children, on the autism spectrum. J. Autism Dev. Disord. 50, 1756–1769. doi: 10.1007/s10803-019-03932-2
Araghi-Niknam, M., and Fatemi, S. H. (2003). Levels of Bcl-2 and P53 are altered in superior frontal and cerebellar cortices of autistic subjects. Cell. Mol. Neurobiol. 23, 945–952. doi: 10.1023/B:CEMN.0000005322.27203.73
Arnett, A. B., Trinh, S., and Bernier, R. A. (2019). The state of research on the genetics of autism spectrum disorder: methodological, clinical and conceptual progress. Curr. Opin. Psychol. 27, 1–5. doi: 10.1016/j.copsyc.2018.07.004
Bailey, K. R., and Crawley, J. N. (2009). “Chapter 5: Anxiety-related behaviors in mice,” in Methods of Behavior Analysis in Neuroscience, ed J. J. Buccafusco (Boca Raton, FL: Press/Taylor & Francis).
Baribeau, D. A., Vigod, S., Pullenayegum, E., Kerns, C. M., Mirenda, P., Smith, I. M., et al. (2020). Repetitive behavior severity as an early indicator of risk for elevated anxiety symptoms in autism spectrum disorder. J. Am. Acad. Child. Adolesc. Psychiatry 59, 890–899 e893. doi: 10.1016/j.jaac.2019.08.478
Baribeau, D. A., Vigod, S., Pullenayegum, E., Kerns, C. M., Mirenda, P., Smith, I. M., et al. (2021). Co-occurring trajectories of anxiety and insistence on sameness behaviour in autism spectrum disorder. Br. J. Psychiatry 218, 20–27. doi: 10.1192/bjp.2020.127
Bauman, M. L. (1991). Microscopic neuroanatomic abnormalities in autism. Pediatrics 87, 791–796. doi: 10.1542/peds.87.5.791
Bednar, I., Paterson, D., Marutle, A., Pham, T. M., Svedberg, M., Hellstrom-Lindahl, E., et al. (2002). Selective nicotinic receptor consequences in APP(SWE) transgenic mice. Mol. Cell. Neurosci. 20, 354–365. doi: 10.1006/mcne.2002.1112
Blanchard, M. G., Willemsen, M. H., Walker, J. B., Dib-Hajj, S. D., Waxman, S. G., Jongmans, M. C., et al. (2015). De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J. Med. Genet. 52, 330–337. doi: 10.1136/jmedgenet-2014-102813
Bostan, A. C., and Strick, P. L. (2018). The basal ganglia and the cerebellum: nodes in an integrated network. Nat. Rev. Neurosci. 19, 338–350. doi: 10.1038/s41583-018-0002-7
Bourin, M., and Hascoet, M. (2003). The mouse light/dark box test. Eur. J. Pharmacol. 463, 55–65. doi: 10.1016/S0014-2999(03)01274-3
Bralten, J., Van Hulzen, K. J., Martens, M. B., Galesloot, T. E., Arias Vasquez, A., Kiemeney, L. A., et al. (2018). Autism spectrum disorders and autistic traits share genetics and biology. Mol. Psychiatry 23, 1205–1212. doi: 10.1038/mp.2017.98
Buitrago, M. M., Schulz, J. B., Dichgans, J., and Luft, A. R. (2004). Short and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol. Learn. Mem. 81, 211–216. doi: 10.1016/j.nlm.2004.01.001
Burne, T. H., Mcgrath, J. J., Eyles, D. W., and Mackay-Sim, A. (2005). Behavioural characterization of vitamin D receptor knockout mice. Behav. Brain Res. 157, 299–308. doi: 10.1016/j.bbr.2004.07.008
Butler, K. M., Da Silva, C., Shafir, Y., Weisfeld-Adams, J. D., Alexander, J. J., Hegde, M., et al. (2017). De novo and inherited SCN8A epilepsy mutations detected by gene panel analysis. Epilepsy Res. 129, 17–25. doi: 10.1016/j.eplepsyres.2016.11.002
Carter, R. J., Lione, L. A., Humby, T., Mangiarini, L., Mahal, A., Bates, G. P., et al. (1999). Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J. Neurosci. 19, 3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999
Chen, K., Godfrey, D. A., Ilyas, O., Xu, J., and Preston, T. W. (2009). Cerebellum-related characteristics of Scn8a-mutant mice. Cerebellum 8, 192–201. doi: 10.1007/s12311-009-0110-z
Crawley, J. N. (2004). Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment. Retard. Dev. Disabil. Res. Rev. 10, 248–258. doi: 10.1002/mrdd.20039
Crawley, J. N. (2007). Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 17, 448–459. doi: 10.1111/j.1750-3639.2007.00096.x
Cupolillo, D., Hoxha, E., Faralli, A., De Luca, A., Rossi, F., Tempia, F., et al. (2016). Autistic-like traits and cerebellar dysfunction in purkinje cell pten knock-out mice. Neuropsychopharmacology 41, 1457–1466. doi: 10.1038/npp.2015.339
Dick, D. J., Boakes, R. J., and Harris, J. B. (1985). A cerebellar abnormality in the mouse with motor end-plate disease. Neuropathol. Appl. Neurobiol. 11, 141–147. doi: 10.1111/j.1365-2990.1985.tb00011.x
Dickson, P. E., Cairns, J., Goldowitz, D., and Mittleman, G. (2017). Cerebellar contribution to higher and lower order rule learning and cognitive flexibility in mice. Neuroscience 345, 99–109. doi: 10.1016/j.neuroscience.2016.03.040
Dickson, P. E., Rogers, T. D., Del Mar, N., Martin, L. A., Heck, D., Blaha, C. D., et al. (2010). Behavioral flexibility in a mouse model of developmental cerebellar Purkinje cell loss. Neurobiol. Learn. Mem. 94, 220–228. doi: 10.1016/j.nlm.2010.05.010
Dong, D., Zielke, H. R., Yeh, D., and Yang, P. (2018). Cellular stress and apoptosis contribute to the pathogenesis of autism spectrum disorder. Autism Res. 11, 1076–1090. doi: 10.1002/aur.1966
Fatemi, S. H., Aldinger, K. A., Ashwood, P., Bauman, M. L., Blaha, C. D., Blatt, G. J., et al. (2012). Consensus paper: pathological role of the cerebellum in autism. Cerebellum 11, 777–807. doi: 10.1007/s12311-012-0355-9
Fatemi, S. H., and Folsom, T. D. (2015). GABA receptor subunit distribution and FMRP-mGluR5 signaling abnormalities in the cerebellum of subjects with schizophrenia, mood disorders, and autism. Schizophr. Res. 167, 42–56. doi: 10.1016/j.schres.2014.10.010
Gardella, E., and Moller, R. S. (2019). Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia 60(Suppl. 3), S77–S85. doi: 10.1111/epi.16319
Gertler, T. S., and Carvill, G. L. (2019). SCN8A: when neurons are so excited, they just can't hide it. Epilepsy Curr. 19, 269–271. doi: 10.1177/1535759719858338
Ha, S., Lee, D., Cho, Y. S., Chung, C., Yoo, Y. E., Kim, J., et al. (2016). Cerebellar Shank2 regulates excitatory synapse density, motor coordination, and specific repetitive and anxiety-like behaviors. J. Neurosci. 36, 12129–12143. doi: 10.1523/JNEUROSCI.1849-16.2016
Hampson, D. R., and Blatt, G. J. (2015). Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 9, 420. doi: 10.3389/fnins.2015.00420
Hogg, S. (1996). A review of the validity and variability of the elevated plus-maze as an animal model of anxiety. Pharmacol. Biochem. Behav. 54, 21–30. doi: 10.1016/0091-3057(95)02126-4
Hollander, E., King, A., Delaney, K., Smith, C. J., and Silverman, J. M. (2003). Obsessive-compulsive behaviors in parents of multiplex autism families. Psychiatry Res. 117, 11–16. doi: 10.1016/S0165-1781(02)00304-9
Khaliq, Z. M., Gouwens, N. W., and Raman, I. M. (2003). The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J. Neurosci. 23, 4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003
Kloth, A. D., Badura, A., Li, A., Cherskov, A., Connolly, S. G., Giovannucci, A., et al. (2015). Cerebellar associative sensory learning defects in five mouse autism models. Elife 4, e06085. doi: 10.7554/eLife.06085.017
Larsen, J., Carvill, G. L., Gardella, E., Kluger, G., Schmiedel, G., Barisic, N., et al. (2015). The phenotypic spectrum of SCN8A encephalopathy. Neurology 84, 480–489. doi: 10.1212/WNL.0000000000001211
Levin, S. I., Khaliq, Z. M., Aman, T. K., Grieco, T. M., Kearney, J. A., Raman, I. M., et al. (2006). Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar purkinje neurons and granule cells. J. Neurophysiol. 96, 785–793. doi: 10.1152/jn.01193.2005
Levin, S. I., and Meisler, M. H. (2004). Floxed allele for conditional inactivation of the voltage-gated sodium channel Scn8a (NaV1.6). Genesis 39, 234–239. doi: 10.1002/gene.20050
Liu, Y., Schubert, J., Sonnenberg, L., Helbig, K. L., Hoei-Hansen, C. E., Koko, M., et al. (2019). Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 142, 376–390. doi: 10.1093/brain/awy326
Lord, C., Brugha, T. S., Charman, T., Cusack, J., Dumas, G., Frazier, T., et al. (2020). Autism spectrum disorder. Nat. Rev. Dis. Primers 6, 5. doi: 10.1038/s41572-019-0138-4
Martin, L. A., Goldowitz, D., and Mittleman, G. (2010). Repetitive behavior and increased activity in mice with Purkinje cell loss: a model for understanding the role of cerebellar pathology in autism. Eur. J. Neurosci. 31, 544–555. doi: 10.1111/j.1460-9568.2009.07073.x
McKinney, B. C., Chow, C. Y., Meisler, M. H., and Murphy, G. G. (2008). Exaggerated emotional behavior in mice heterozygous null for the sodium channel Scn8a (Nav1.6). Genes Brain Behav. 7, 629–638. doi: 10.1111/j.1601-183X.2008.00399.x
Meisler, M. H. (2019). SCN8A encephalopathy: mechanisms and models. Epilepsia 60(Suppl. 3), S86–S91. doi: 10.1111/epi.14703
Meisler, M. H., Helman, G., Hammer, M. F., Fureman, B. E., Gaillard, W. D., Goldin, A. L., et al. (2016). SCN8A encephalopathy: research progress and prospects. Epilepsia 57, 1027–1035. doi: 10.1111/epi.13422
Meisler, M. H., Hill, S. F., and Yu, W. (2021). Sodium channelopathies in neurodevelopmental disorders. Nat. Rev. Neurosci. 22, 152–166. doi: 10.1038/s41583-020-00418-4
Mosconi, M. W., Wang, Z., Schmitt, L. M., Tsai, P., and Sweeney, J. A. (2015). The role of cerebellar circuitry alterations in the pathophysiology of autism spectrum disorders. Front. Neurosci. 9, 296. doi: 10.3389/fnins.2015.00296
Neuner, S. M., Heuer, S. E., Huentelman, M. J., O'connell, K. M. S., and Kaczorowski, C. C. (2019). Harnessing genetic complexity to enhance translatability of Alzheimer's disease mouse models: a path toward precision medicine. Neuron 101, 399–411 e395. doi: 10.1016/j.neuron.2018.11.040
O'Brien, J. E., and Meisler, M. H. (2013). Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front. Genet. 4, 213. doi: 10.3389/fgene.2013.00213
Oristaglio, J., Hyman West, S., Ghaffari, M., Lech, M. S., Verma, B. R., Harvey, J. A., et al. (2013). Children with autism spectrum disorders show abnormal conditioned response timing on delay, but not trace, eyeblink conditioning. Neuroscience 248, 708–718. doi: 10.1016/j.neuroscience.2013.06.007
Peter, S., Ten Brinke, M. M., Stedehouder, J., Reinelt, C. M., Wu, B., Zhou, H., et al. (2016). Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 7, 12627. doi: 10.1038/ncomms12627
Piochon, C., Kloth, A. D., Grasselli, G., Titley, H. K., Nakayama, H., Hashimoto, K., et al. (2014). Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat. Commun. 5, 5586. doi: 10.1038/ncomms6586
Raman, I. M., Sprunger, L. K., Meisler, M. H., and Bean, B. P. (1997). Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19, 881–891. doi: 10.1016/S0896-6273(00)80969-1
Reith, R. M., Mckenna, J., Wu, H., Hashmi, S. S., Cho, S. H., Dash, P. K., et al. (2013). Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 51, 93–103. doi: 10.1016/j.nbd.2012.10.014
Schaller, K. L., and Caldwell, J. H. (2003). Expression and distribution of voltage-gated sodium channels in the cerebellum. Cerebellum 2, 2–9. doi: 10.1080/14734220309424
Schniepp, R., Mohwald, K., and Wuehr, M. (2017). Gait ataxia in humans: vestibular and cerebellar control of dynamic stability. J. Neurol. 264, 87–92. doi: 10.1007/s00415-017-8482-3
Silverman, J. L., Tolu, S. S., Barkan, C. L., and Crawley, J. N. (2010a). Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology 35, 976–989. doi: 10.1038/npp.2009.201
Silverman, J. L., Yang, M., Lord, C., and Crawley, J. N. (2010b). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502. doi: 10.1038/nrn2851
Sprunger, L. K., Escayg, A., Tallaksen-Greene, S., Albin, R. L., and Meisler, M. H. (1999). Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Hum. Mol. Genet. 8, 471–479. doi: 10.1093/hmg/8.3.471
Sundberg, M., and Sahin, M. (2015). Cerebellar development and autism spectrum disorder in tuberous sclerosis complex. J. Child Neurol. 30, 1954–1962. doi: 10.1177/0883073815600870
Tang, G. B., Zeng, Y. Q., Liu, P. P., Mi, T. W., Zhang, S. F., Dai, S. K., et al. (2017). The histone H3K27 demethylase UTX regulates synaptic plasticity and cognitive behaviors in mice. Front. Mol. Neurosci. 10, 267. doi: 10.3389/fnmol.2017.00267
Tatsukawa, T., Raveau, M., Ogiwara, I., Hattori, S., Miyamoto, H., Mazaki, E., et al. (2019). Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol. Autism 10, 15. doi: 10.1186/s13229-019-0265-5
Trudeau, M. M., Dalton, J. C., Day, J. W., Ranum, L. P., and Meisler, M. H. (2006). Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J. Med. Genet. 43, 527–530. doi: 10.1136/jmg.2005.035667
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tsai, P. T., Rudolph, S., Guo, C., Ellegood, J., Gibson, J. M., Schaeffer, S. M., et al. (2018). Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep. 25 357–367 e354. doi: 10.1016/j.celrep.2018.09.039
Varghese, M., Keshav, N., Jacot-Descombes, S., Warda, T., Wicinski, B., Dickstein, D. L., et al. (2017). Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. 134, 537–566. doi: 10.1007/s00401-017-1736-4
Wagnon, J. L., Barker, B. S., Ottolini, M., Park, Y., Volkheimer, A., Valdez, P., et al. (2017). Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet. 3, e170. doi: 10.1212/NXG.0000000000000170
Wang, S. S., Kloth, A. D., and Badura, A. (2014). The cerebellum, sensitive periods, and autism. Neuron 83, 518–532. doi: 10.1016/j.neuron.2014.07.016
Wegiel, J., Flory, M., Kuchna, I., Nowicki, K., Ma, S. Y., Imaki, H., et al. (2014). Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol. Commun. 2, 141. doi: 10.1186/s40478-014-0141-7
Welsh, J. P., and Oristaglio, J. T. (2016). Autism and classical eyeblink conditioning: performance changes of the conditioned response related to autism spectrum disorder diagnosis. Front. Psychiatry 7, 137. doi: 10.3389/fpsyt.2016.00137
Whitney, E. R., Kemper, T. L., Rosene, D. L., Bauman, M. L., and Blatt, G. J. (2009). Density of cerebellar basket and stellate cells in autism: evidence for a late developmental loss of Purkinje cells. J. Neurosci. Res. 87, 2245–2254. doi: 10.1002/jnr.22056
Winkler, M., Biswas, S., Berger, S. M., Kuchler, M., Preisendorfer, L., Choo, M., et al. (2020). Pianp deficiency links GABAB receptor signaling and hippocampal and cerebellar neuronal cell composition to autism-like behavior. Mol. Psychiatry 25, 2979–2993. doi: 10.1038/s41380-019-0519-9
Wisniowiecka-Kowalnik, B., and Nowakowska, B. A. (2019). Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J. Appl. Genet. 60, 37–47. doi: 10.1007/s13353-018-00480-w
Wong, J. C., Grieco, S. F., Dutt, K., Chen, L., Thelin, J. T., Inglis, G., et al. (2021). Autistic-like behavior, spontaneous seizures, and increased neuronal excitability in a Scn8a mouse model. Neuropsychopharmacology 46, 2011–2020. doi: 10.1038/s41386-021-00985-9
Woodruff-Pak, D. S., Green, J. T., Levin, S. I., and Meisler, M. H. (2006). Inactivation of sodium channel Scn8A (Na-sub(v)1.6) in Purkinje neurons impairs learning in Morris water maze and delay but not trace eyeblink classical conditioning. Behav. Neurosci. 120, 229–240. doi: 10.1037/0735-7044.120.2.229
Yang, M., Silverman, J. L., and Crawley, J. N. (2011). Automated three-chambered social approach task for mice. Curr. Protoc. Neurosci. Chapter 8, Unit 8.26. doi: 10.1002/0471142301.ns0826s56
Yang, Y. M., Arsenault, J., Bah, A., Krzeminski, M., Fekete, A., Chao, O. Y., et al. (2020). Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol. Psychiatry 25, 2017–2035. doi: 10.1038/s41380-018-0240-0
Keywords: Purkinje cell, SCN8A, mouse, autism, cerebellum, anxiety
Citation: Yang X, Yin H, Wang X, Sun Y, Bian X, Zhang G, Li A, Cao A, Li B, Ebrahimi-Fakhari D, Yang Z, Meisler MH and Liu Q (2022) Social Deficits and Cerebellar Degeneration in Purkinje Cell Scn8a Knockout Mice. Front. Mol. Neurosci. 15:822129. doi: 10.3389/fnmol.2022.822129
Received: 25 November 2021; Accepted: 18 February 2022;
Published: 26 April 2022.
Edited by:
Rainer Schwarting, University of Marburg, GermanyReviewed by:
Yuri Bozzi, University of Trento, ItalyCopyright © 2022 Yang, Yin, Wang, Sun, Bian, Zhang, Li, Cao, Li, Ebrahimi-Fakhari, Yang, Meisler and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiji Liu, bGl1cWlqaUBzZHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.