 Clara Rossetti
Clara Rossetti Antoine Cherix
Antoine Cherix Laetitia F. Guiraud1,2‡
Laetitia F. Guiraud1,2‡ Jean-René Cardinaux
Jean-René Cardinaux
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 15 February 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.810641
This article is part of the Research Topic Bidirectional Communication between Synapses and Nucleus in Brain Physiology and Disease View all 7 articles
Depression and obesity are major public health concerns, and there is mounting evidence that they share etiopathophysiological mechanisms. The neurobiological pathways involved in both mood and energy balance regulation are complex, multifactorial and still incompletely understood. As a coactivator of the pleiotropic transcription factor cAMP response element-binding protein (CREB), CREB-regulated transcription coactivator 1 (CRTC1) has recently emerged as a novel regulator of neuronal plasticity and brain functions, while CRTC1 dysfunction has been associated with neurodegenerative and psychiatric diseases. This review focuses on recent evidence emphasizing the critical role of CRTC1 in the neurobiology of depression and comorbid obesity. We discuss the role of CRTC1 downregulation in mediating chronic stress-induced depressive-like behaviors, and antidepressant response in the light of the previously characterized Crtc1 knockout mouse model of depression. The putative role of CRTC1 in the alteration of brain energy homeostasis observed in depression is also discussed. Finally, we highlight rodent and human studies supporting the critical involvement of CRTC1 in depression-associated obesity.
According to the World Health Organization, more than 300 million people suffer from depression worldwide (World Health Organization, 2017). This mood disorder is now the leading cause of disability and a major contributor to the overall global burden of disease. A high suicide risk is associated with severe depression and more than 800,000 people commit suicide each year. Current antidepressant therapies are mostly targeting monoamines neurotransmission with a limited efficacy. Several weeks of treatment are needed before mood improvement occurs, and approximately 30% of patients do not respond to at least two consecutive antidepressant treatments (Caraci et al., 2018). Treatment-resistant depression may benefit from the recent development of rapid-acting antidepressants targeting glutamate neurotransmission, such as the N-methyl-D-aspartate (NMDA) receptor blocker ketamine (Duman et al., 2016, 2019b; Zanos and Gould, 2018; Krystal et al., 2019; Shinohara et al., 2021). However, the dissociative and potentially addictive side effects of ketamine have been stimulating the development of new antidepressant drugs based on ketamine, but without its drawbacks (Ragguett et al., 2019). Unfortunately, many clinical trials failed to show a significant advantage for antidepressant medication over inert placebo (Kraus et al., 2019; Wilkinson and Sanacora, 2019). These failures might also reflect insufficient clinical subtyping of major depressive disorder (MDD), which would certainly benefit from a better understanding of the neurobiology of depression that remains elusive despite decades of intensive research (Akil et al., 2018).
Overweight and obesity are major public health problems that continue to increase both in developing and developed countries. Epidemiological evidence strongly supports the existence of a bidirectional relationship between depression and abdominal (visceral) obesity, which is the main risk factor for metabolic syndrome (Milaneschi et al., 2019). Clinically, obesity and metabolic syndrome are associated with the atypical depression subtype of MDD characterized by neurovegetative symptoms consisting of lethargy, fatigue, excessive sleepiness, increased food intake, weight gain and depressive symptoms that are lowest in the morning and worsen as the day progresses (Gold, 2015). Among the shared biological pathways that may mechanistically explain the atypical depression–obesity link, increased chronic inflammation is emerging as the central pathophysiological process involved (Milaneschi et al., 2019).
The monoamine hypothesis of depression is based on a deficit of monoamines (serotonin and noradrenaline) that would be corrected by antidepressants. However, current antidepressants require weeks of treatment to produce a clinical response, which suggests that their effects do not only rely on enhanced monoamines transmission, but rather to long-term changes in monoamines signaling and neuronal circuitry. Therefore, research efforts have been focusing on the long-term molecular changes that underlie depression and antidepressant treatments, and led to the neurotrophic and network hypotheses of depression, which proposed that impaired mechanisms of neuroplasticity are a core pathophysiological feature of MDD. Neuroplasticity is a fundamental mechanism of neuronal adaptation involving several forms of plasticity, such as adult hippocampal neurogenesis, neuronal survival and maturation, synaptogenesis, and structural plasticity (e.g., number of spines and complexity of dendritic arborization). Decreased neuroplasticity in the hippocampus and prefrontal cortex, and the resulting problems in information processing within relevant neural networks might thus underlie mood disorders (Castren, 2005; Pittenger and Duman, 2008; Marsden, 2013; Boku et al., 2018). Indeed, chronic stress strongly contributes to the development of MDD by affecting neuroplasticity and connectivity at several levels (Pittenger and Duman, 2008; Albert, 2019; Duman et al., 2019a). The transcription factor cAMP response element-binding protein (CREB) and one of its target genes, brain-derived neurotrophic factor (BDNF) are critically involved in the concept of altered neuroplasticity in MDD (Carlezon et al., 2005; Blendy, 2006; Castren and Hen, 2013; Levy et al., 2018; Umemori et al., 2018; Xiao et al., 2018; Castren and Monteggia, 2021). Moreover, there is evidence in rodents and humans that the CREB-BDNF pathway is also implicated in obesity (Rios, 2013; Lin et al., 2014; Marosi and Mattson, 2014; Xu and Xie, 2016; Amare et al., 2017). BDNF and several other neuroplasticity genes are regulated by CREB and a coactivator called CREB-Regulated Transcription Coactivator 1 (CRTC1) (Zhou et al., 2006; Kovacs et al., 2007). Recent studies suggest that CRTC1 dysregulation may be involved in the etiopathogenesis of many brain disorders, including MDD (Saura and Cardinaux, 2017). This review will focus on the increasing evidence of the key role of the transcription coactivator CRTC1 in the central control of mood and eating behavior, and its possible involvement in depression and comorbid obesity.
The CRTC family includes three members in mammals, with CRTC2 and CRTC3 expressed in most tissues, and CRTC1 mainly found in the brain (Kovacs et al., 2007; Li et al., 2009; Watts et al., 2011). Several comprehensive reviews already provided a detailed description of the signaling pathways regulating CRTC1 and its roles in brain physiology and pathology (Altarejos and Montminy, 2011; Escoubas et al., 2017; Saura and Cardinaux, 2017; Uchida and Shumyatsky, 2018; Parra-Damas and Saura, 2019). In this section, we summarize the mechanisms of CRTC1 regulation and its main functions in the brain, with a particular emphasis on CRTC1’s role in neuroplasticity.
As mentioned in the introduction, CRTC1 is a coactivator of CREB, a transcription factor playing pleiotropic roles in the nervous system (Carlezon et al., 2005; Barco and Marie, 2011). The canonical mode of CREB activation involves its phosphorylation at Ser133 by multiple signaling pathways, and the ensuing recruitment of CBP/p300 coactivators that activate transcription by acetylating nucleosomal histones and by interacting with factors of the general transcription machinery (Mayr and Montminy, 2001; Lonze and Ginty, 2002; Carlezon et al., 2005; Josselyn and Nguyen, 2005; Benito and Barco, 2010). However, this model was challenged by several studies suggesting that CREB can be activated without Ser133 phosphorylation (Brindle et al., 1995; Impey et al., 1996; Briand et al., 2015). The discovery of the CRTC family of coactivators brought elements of explanation about how CREB-mediated transcription could be enhanced independently of Ser133 phosphorylation, and suggested the existence of a non-canonical, alternative way of CREB target genes activation (Conkright et al., 2003; Iourgenko et al., 2003; Altarejos and Montminy, 2011; Parra-Damas et al., 2017b; Feldmann et al., 2019). De facto, the N-terminal domain of CRTCs interacts with the dimerization and DNA-binding bZIP domain of CREB in a phosphorylation-independent manner. Once recruited to gene promoters, CRTCs strongly activate CREB-mediated transcription through their C-terminal transactivation domain. CREB is nonetheless not constitutively activated by CRTCs, as their phosphorylation state regulates their cytoplasmic versus nuclear localization (Bittinger et al., 2004; Screaton et al., 2004; Sonntag et al., 2017). In resting conditions, they are phosphorylated and sequestered in the cytoplasm by scaffolding 14-3-3 proteins and their nuclear translocation requires the concomitant activation of calcium and cAMP signaling pathways (Figure 1). Upon elevated intracellular calcium levels, CRTCs are dephosphorylated by the Ca2+-dependent protein phosphatase 2B (PP2B)/calcineurin leading to their dissociation from 14–3–3 proteins and subsequent nuclear translocation. Coincidently, increased cAMP levels stimulate calcium-mediated dephosphorylation by inhibiting salt-inducible kinases (SIKs) that phosphorylate CRTCs (Altarejos and Montminy, 2011).
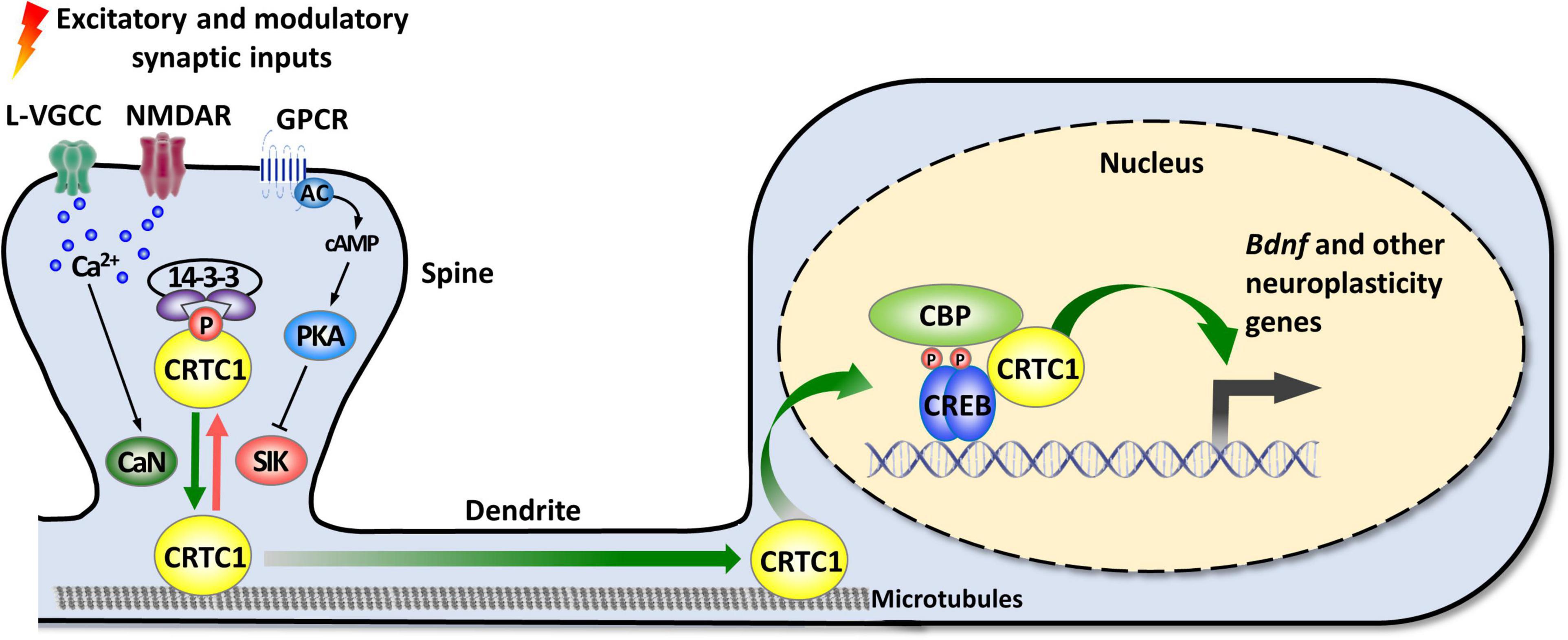
Figure 1. Activity-dependent synapse-to-nucleus translocation of CRTC1 mediates the activation of neuroplasticity gene transcription. CRTC1 is sequestered in dendritic spines under basal conditions via a phosphorylation-dependent association with 14-3-3 proteins. Simultaneous activation of calcium and cAMP pathways (by L-VGCC or NMDAR, and GPCR-activated AC) triggers release from 14-3-3 proteins by, respectively, activating the phosphatase calcineurin (CaN) and inhibiting kinases of the AMPK family (SIK). Dephosphorylated CRTC1 migrates into the nucleus and is recruited to the promoter via an interaction with the bZIP domain of CREB, thus promoting expression of Bdnf and other neuroplasticity genes. AC, adenylate cyclase; AMPK, AMP-activated protein kinase; CaN, calcineurin; CBP, CREB-binding protein; CREB, cAMP response-element binding protein; CRTC1, CREB-regulated transcription coactivator 1; GPCR, G protein-coupled receptor; L-VGCC, L-type voltage-gated calcium channels; NMDAR, N-methyl-D-aspartate receptor; PKA, protein kinase A; SIK, salt-inducible kinase.
In neurons, synaptic activity-induced CRTC1 translocation from synapses to the nucleus is critical for the transcription-dependent phase of neuronal plasticity (Zhou et al., 2006; Kovacs et al., 2007; Li et al., 2009; Finsterwald et al., 2010; Ch’ng et al., 2012, 2015; Nonaka et al., 2014; Parra-Damas et al., 2014, 2017a). Activity-dependent transport of CRTC1 from dendritic spines to the nucleus requires local elevation of calcium triggered by activation of glutamate receptors and L-type voltage-gated calcium channels, leading to calcineurin activation and dephosphorylation of three conserved serine residues (S64, S151, and S245) in the amino-terminal third of CRTC1, which contains a nuclear localization signal (Ch’ng et al., 2015). Dephosphorylated CRTC1 is released from 14-3-3 proteins and actively translocated to the nucleus via a dynein motor protein-mediated retrograde transport along microtubules. Nuclear CRTC1 upregulates the expression of genes that have been involved in synaptic plasticity and memory formation. These neuroplasticity genes include, among others, the neurotrophic factor Bdnf, the immediate-early genes c-fos, Zif268/Egr1, and Arc, the orphan nuclear receptors Nr4a1 and Nr4a2, the brain-specific growth factor Fgf1, and autophagy genes involved in synaptic turnover and late-phase long-term synaptic depression (Zhou et al., 2006; Espana et al., 2010; Breuillaud et al., 2012; Ch’ng et al., 2012, 2015; Nonaka et al., 2014; Parra-Damas et al., 2014, 2017a; Fukuchi et al., 2015; Uchida et al., 2017; Pan et al., 2021). In agreement with CRTC1’s role in the induction of these neuroplasticity genes, accumulating evidence supports its involvement in neuronal plasticity processes, such as activity- and BDNF-induced dendritic growth of developing cortical neurons (Li et al., 2009; Finsterwald et al., 2010), maintenance of hippocampal late-phase LTP (Zhou et al., 2006; Kovacs et al., 2007; Uchida et al., 2017), and long-term memory formation (Sekeres et al., 2012; Nonaka et al., 2014; Parra-Damas et al., 2014, 2017a; Uchida et al., 2017; Zhang et al., 2019; Shu et al., 2020; Yan et al., 2021). Of particular importance, increasing evidence suggests that CRTC1 dysregulation may be implicated in the etiopathogenesis of neurodegenerative diseases, such as Huntington’s, Parkinson’s, and Alzheimer’s disease, as well as psychiatric disorders (Boulting et al., 2021), including mood disorders [reviewed in Saura and Cardinaux (2017)].
In line with CRTC1’s role in neuroplasticity processes, as well as the involvement of CREB and BDNF in rodent models of depression, increasing evidence suggests that CRTC1 is implicated in the pathogenesis of MDD (summarized in Table 1). Originally, we developed a CRTC1 knockout mouse model (Breuillaud et al., 2009), and performed its in-depth phenotypic characterization (Breuillaud et al., 2012). This led us to provide the first preclinical evidence of the involvement of CRTC1 in mood regulation and antidepressant response. Indeed, Crtc1–/– mice exhibit depressive-like neurobehavioral endophenotypes, including increased behavioral despair in the forced swim test (FST) and in the repeated open-space forced swim test (OSFST), social withdrawal, decreased sexual motivation, psychomotor retardation, and increased emotional response to stressful events (Breuillaud et al., 2012). High-performance liquid chromatography (HPLC) monitoring of the levels of monoamines and metabolites in the prefrontal cortex, hippocampus, hypothalamus, and nucleus accumbens revealed significantly lower levels of the dopamine metabolites 3,4 dihydroxy-phenylacetic acid (DOPAC) and homovanillic acid (HVA), as well as of the serotonin metabolite 5-hydroxyindole acetic acid (5-HIAA) in the prefrontal cortex of Crtc1–/– male mice as compared to wild-type littermates. Decreased levels of these metabolites are thought to reflect a reduced dopamine and serotonin release in the prefrontal cortex that was previously associated with aggressive behaviors, impulsivity, and depressive-like behaviors. Moreover, Crtc1–/– male mice have a decreased expression of several neuroplasticity genes in the prefrontal cortex and hippocampus, including Bdnf and its receptor TrkB, as well as the nuclear receptors Nr4a1-3, thus suggesting impaired neuroplasticity processes in these brain structures. Interestingly, we showed that Crtc1–/– mice are resistant to the chronic antidepressant effects of the selective serotonin reuptake inhibitor fluoxetine, as well as the tricyclic antidepressant desipramine, in a behavioral despair paradigm (OSFST), which suggests that CRTC1 is required for conventional antidepressant therapy (Breuillaud et al., 2012; Meylan et al., 2016b). Supporting the blunted behavioral response to chronic desipramine, this antidepressant does not increase Bdnf expression in the prefrontal cortex of Crtc1–/– mice in contrast to its upregulation in wild-type mice. Moreover, we showed that chronic systemic administration of the histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA, also known as vorinostat) partially rescues the depressive-like behavior of Crtc1–/– mice and restores Bdnf expression in the prefrontal cortex (Meylan et al., 2016b). The rationale behind this epigenetic intervention was that CRTC1 helps phosphorylated CREB to recruit the histone acetyltransferase CBP and that the absence of CRTC1 decreases CBP recruitment and histone acetylation of neuroplasticity gene promoters involved in mood regulation. Reduction of CBP recruitment and histone acetylation in Crtc1–/– mice can thus be partially overcome by HDAC inhibition, which restores the expression of a subset of genes by acting downstream of CRTC1. These findings are in line with the current understanding of the involvement of epigenetic processes in the pathogenesis of MDD and antidepressant action, as well as with the recent evidence of the therapeutic potential of HDAC inhibitors (Vialou et al., 2013; Misztak et al., 2018; Uchida et al., 2018).
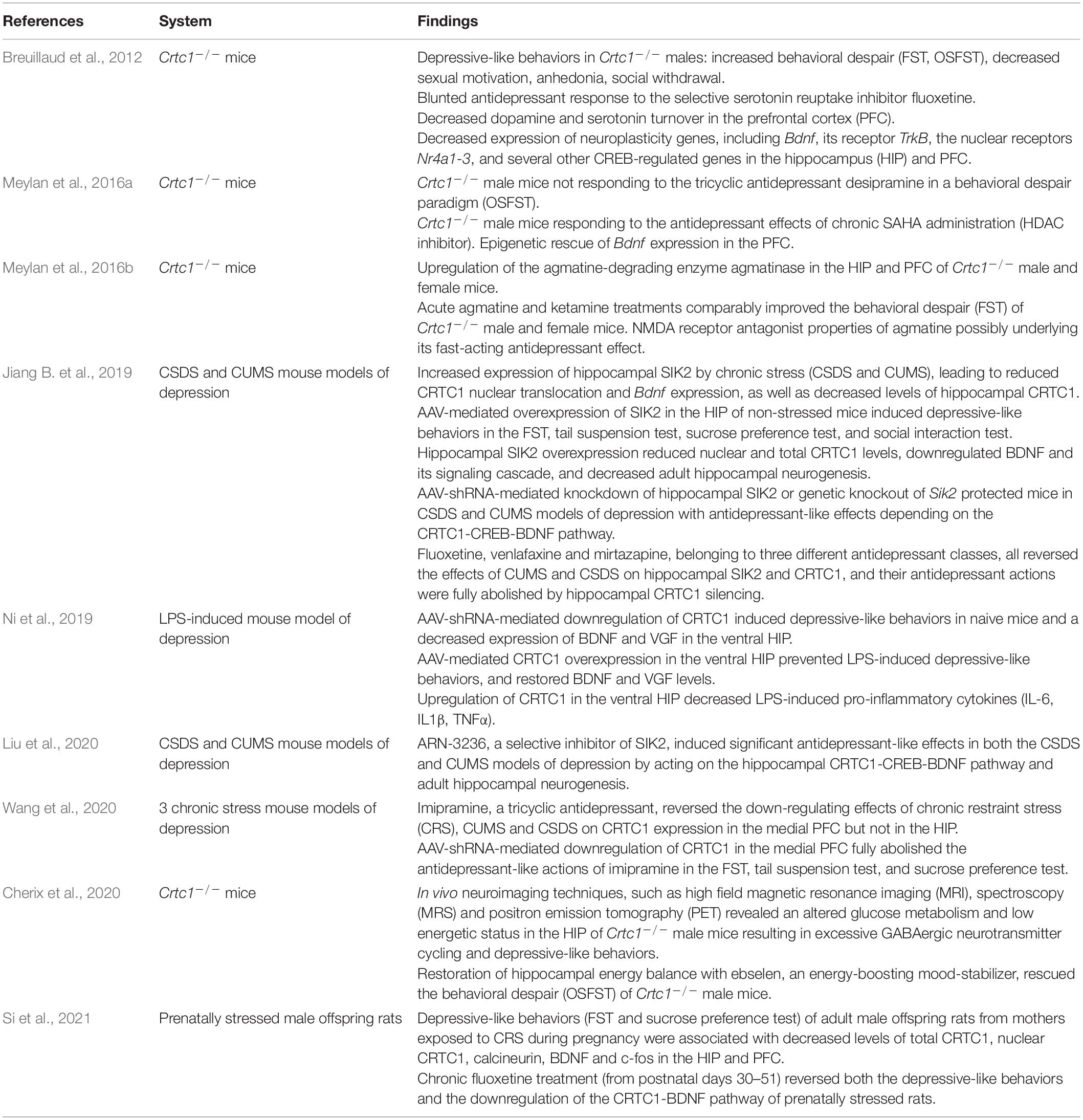
Table 1. Summary of studies implicating CRTC1 in the pathophysiology of major depressive disorder (MDD).
Using a cDNA microarray approach, we identified differentially expressed genes in the cortex of Crtc1–/– mice, among which we found an upregulation of agmatinase, the enzyme degrading the arginine-decarboxylation product agmatine (Meylan et al., 2016a). Increasing evidence suggests that agmatine is an important neuromodulator, possibly even serving as a neurotransmitter, as it is stored in synaptic vesicles and released upon depolarization, selectively recaptured and finally degraded by agmatinase to form putrescine, a key precursor for polyamine synthesis (Li et al., 1994; Reis and Regunathan, 2000; Uzbay, 2012; Piletz et al., 2013). Agmatine is primarily found in neurons of brain regions that subserve cognition, processing of emotions, visceral and neuroendocrine control, and pain perception, as the distribution of agmatine-containing neurons is concentrated in the cerebral cortex, hippocampus, amygdala, septum, bed nucleus of the stria terminalis, periventricular thalamic and hypothalamic nuclei, as well as several areas of the midbrain and brainstem (Reis and Regunathan, 2000). Agmatine has been colocalized with other neurotransmitters, notably glutamate in synaptic terminals in hippocampal CA1 region (Seo et al., 2011). It also binds with high affinity several postsynaptic membrane receptors, such as α2-adrenergic receptors, imidazoline I1 and I2 receptors, nicotinic receptors and serotoninergic 5HT-2A and 5HT-3 receptors, and blocks glutamate NMDA receptor channels in a similar way as the rapid-acting antidepressant ketamine (Yang and Reis, 1999; Reis and Regunathan, 2000; Halaris and Piletz, 2007; Uzbay, 2012; Neis et al., 2016a,b, 2018; Barua et al., 2019; Camargo and Rodrigues, 2019; Valverde et al., 2021). Moreover, agmatine is a key modulator of nitric oxide and polyamine overproduction during the “Polyamine Stress Response” that is important for proper adaptive responses of quiescent cells to stressful conditions (Piletz et al., 2013). Accumulating evidence implicates the agmatinergic system in the etiopathogenesis of mood disorders (Laube and Bernstein, 2017; Weiss et al., 2018; Watts et al., 2019) and other central nervous system disorders (Neis et al., 2017). Post-mortem studies showed a strong upregulation of agmatinase levels in hippocampal GABAergic interneurons of brain tissues from depressed subjects (Bernstein et al., 2012), and decreased agmatine levels in the cortex of individuals who died by suicide (Chen et al., 2018). Acute and chronic stress reduce agmatine levels in rodent forebrain and exogenous agmatine administration has rapid antidepressant effect in animal models of depression (Zomkowski et al., 2002; Freitas et al., 2016). Consistent with their common property to block glutamate NMDA receptors, we found that acute agmatine and ketamine treatments comparably improve the depressive-like phenotype of male and female Crtc1–/– mice in the forced swim test (Meylan et al., 2016a). Current understanding of the mechanisms underlying ketamine’s rapid antidepressant action involves direct and indirect downstream consequences of NMDA glutamate receptor antagonism (Duman et al., 2019b; Krystal et al., 2019). The indirect hypothesis of the antidepressant actions of ketamine implies that ketamine initially blocks NMDA receptors on GABA inhibitory interneurons leading to “indirect” disinhibition of glutamate transmission and enhanced stimulation of AMPA glutamate receptors on excitatory neurons. AMPA receptor activation triggers a signaling cascade that raises BDNF levels. TrkB receptors activated by the local release of BDNF stimulate the activation of the molecular target of rapamycin complex 1 (mTORC1), which, in turn, catalyzes local protein synthesis necessary for increasing dendritic spine formation and restoring synaptic connectivity. The second hypothesis of ketamine’s action is based on the direct inhibition of NMDA receptors located on excitatory neurons, and the involvement of the eukaryotic elongation factor 2 (eEF2) kinase pathway. Ketamine blockade of NMDA receptors at rest leads to suppression of eEF2 kinase activity and subsequent decreased eEF2 phosphorylation that activates molecular target of rapamycin (mTOR)-regulated local protein synthesis. The resulting increased synthesis of BDNF and other postsynaptic proteins stimulates the shuttling of AMPA glutamate receptors to the synapse, which enhances synaptic efficacy (Autry et al., 2011). In our study, we found that agmatine rapidly increases BDNF levels only in the prefrontal cortex of wild-type females, and decreases eEF2 phosphorylation in the prefrontal cortex of male and female wild-type mice, indicating that agmatine might function as a fast-acting antidepressant with NMDA receptor antagonist properties. In contrast to wild-type mice, the acute antidepressant effects of agmatine do not seem to rely on eEF2 dephosphorylation-dependent increase of BDNF synthesis in Crtc1–/– mice. These findings support a causal role of the deregulated agmatinergic system in the Crtc1–/– mouse model of depression, as it has been suggested for human MDD. On the other hand, the mechanistic relationship between agmatinase upregulation and Crtc1 deficiency is not clear yet and deserves more investigation.
Recently, several studies highlighted the importance of CRTC1 in stress- and inflammation-induced depressive-like behaviors, thus extending our pioneering findings on the role of CRTC1 in MDD (Davis, 2019; Jiang B. et al., 2019; Ni et al., 2019; Liu et al., 2020; Wang et al., 2020; Si et al., 2021). In a quite extensive study, the SIK2-CRTC1-CREB-BDNF pathway was proved highly instrumental in mediating chronic stress-induced depressive-like behaviors and antidepressant response in C57BL/6J mice (Jiang B. et al., 2019). Both chronic unpredictable mild stress (CUMS) and chronic social defeat stress (CSDS) markedly increase hippocampal Sik2 expression, which reduces CRTC1 nuclear translocation and Bdnf expression, as well as total protein levels of hippocampal CRTC1. Adeno-associated virus (AAV)-mediated overexpression of SIK2 in the hippocampus of non-stressed mice induces depressive-like behaviors in the FST, tail suspension test, sucrose preference test, and social interaction test, thus mimicking the effects of chronic stress. Hippocampal SIK2 overexpression lowers nuclear and total CRTC1 levels, downregulates BDNF and its signaling cascade, and decreases adult hippocampal neurogenesis. Conversely, AAV-short hairpin RNA (shRNA)-mediated knockdown of hippocampal SIK2 or genetic knockout of Sik2 protect mice against both chronic stress models of depression with antidepressant-like effects that require the downstream CRTC1-CREB-BDNF pathway. Finally, fluoxetine, venlafaxine and mirtazapine, belonging to three different antidepressant classes, all reverse the effects of CUMS and CSDS on hippocampal SIK2 and CRTC1, and their antidepressant actions are fully abolished by hippocampal CRTC1 silencing. Two follow-up studies from the same laboratory then showed that the antidepressant effects of imipramine depend on CRTC1 in the medial prefrontal cortex (Wang et al., 2020), and that ARN-3236, a selective inhibitor of SIK2, induced significant antidepressant-like effects in both the CSDS and CUMS models of depression by acting on the hippocampal CRTC1-CREB-BDNF pathway (Liu et al., 2020). Moreover, it was recently shown that prenatally stressed offspring male rats display depressive-like behaviors associated with decreased levels of CRTC1 and BDNF in the hippocampus and prefrontal cortex, which are restored by a chronic fluoxetine treatment (Si et al., 2021). Taken together, these findings strengthen the pivotal role of hippocampal and prefrontal CRTC1 in the pathogenesis of MDD and in antidepressant response.
Another study convincingly highlighted CRTC1’s key role in lipopolysaccharide (LPS)-induced depressive-like behaviors (Ni et al., 2019). Systemic administration of LPS causes chronic neuroinflammation and depressive-like behaviors in mice (Dantzer et al., 2008). This animal model is relevant for the human neuroinflammation hypothesis of MDD that is associated with the atypical depression subtype (Woelfer et al., 2019). Starting from the observation that a concentration of LPS inducing depressive-like behaviors in ICR mice also decreases hippocampal CRTC1 protein levels, Ni et al. (2019) used AAV-mediated silencing or upregulation of CRTC1 in the ventral hippocampus to determine its role in LPS-induced depressive-like behaviors. AAV-shRNA-mediated downregulation of CRTC1 induces depressive-like behaviors in naive mice and a decreased expression of BDNF and VGF in the ventral hippocampus. Conversely, ventral hippocampal AAV-mediated CRTC1 overexpression prevents depressive-like behaviors induced by a single i.p. injection of LPS, and restores BDNF and VGF levels. Interestingly, upregulation of CRTC1 in the ventral hippocampus interferes with neuroinflammatory processes, as shown by the dampened accumulation of LPS-induced pro-inflammatory cytokines, such as interleukin-6, interleukin 1-β and tumor necrosis factor α. These compelling findings further support the essential role of CRTC1 in MDD pathogenesis, and suggest that CRTC1 coactivates genes controlling neuroinflammation through still unknown mechanisms (Davis, 2019; Ni et al., 2019).
Lately, ketamine has attracted much attention for its rapid antidepressant effects in patients with treatment-resistant depression (Kraus et al., 2019). Ketamine and a few other compounds acting on glutamate neurotransmission are the only antidepressant drugs in development that are able to relieve MDD symptoms within hours after a single dose, the effects of which lasting for up to a week. Unfortunately, ketamine is also a well-known psychedelic drug with an addictive potential, and almost nothing is known about the risks of its long-term use. Current research is thus focusing on the development of ketamine-like molecules having the same fast-acting antidepressant benefits without the undesirable side effects (Zanos et al., 2018; Ragguett et al., 2019; Wilkinson and Sanacora, 2019). This preclinical development of novel pharmacotherapies requires a better understanding of the molecular and cellular mechanisms underlying the antidepressant actions of ketamine. A recent study showed that ketamine’s acute antidepressant effects precede its action on spine formation in the prefrontal cortex, indicating that spinogenesis is not required for the rapid (less than 12 h) behavioral response (Moda-Sava et al., 2019). In contrast, prefrontal cortical restoration of lost spines by a single ketamine injection is necessary for its sustained (2–7 days) antidepressant effects. BDNF and its receptor TrkB, as well as VGF, have been implicated in ketamine’s antidepressant effects (Autry et al., 2011; Bjorkholm and Monteggia, 2016; Ma et al., 2017; Song et al., 2017; Zanos and Gould, 2018; Jiang C. et al., 2019). However, their respective function in the acute and/or sustained effects of ketamine is not completely understood. The involvement of CRTC1 in the regulation of BDNF and VGF expression (Zhou et al., 2006; Parra-Damas et al., 2014; Fukuchi et al., 2015; Ni et al., 2019), as well as the decreased brain BDNF levels in Crtc1–/– mice (Breuillaud et al., 2012; Meylan et al., 2016a), suggest that ketamine’s antidepressant effects might rely on CRTC1, as well. We previously showed that the immobility of Crtc1–/– males and females is significantly decreased 30 min after a single injection of ketamine (3 mg/kg) in the FST paradigm [Supplementary Figure 2 in Meylan et al. (2016a)], thus indicating that CRTC1 is not required for the acute antidepressant effects of ketamine. Future investigations are nevertheless needed to determine whether CRTC1 is involved in the neuroplasticity-related sustained long-term antidepressant effects of ketamine. In summary, our findings, as well as several subsequent studies, highlight the pivotal role of the CRTC1-CREB-BDNF pathway in preclinical animal models of MDD, and provide evidence of its involvement in antidepressant response.
Several lines of evidence suggest that altered energy metabolism underlies mood disorders, both at the brain and peripheral level (Ostergaard et al., 2018). The rationale being that normal neuronal function is challenged in MDD due to an imbalance in energy homeostasis, through either an impaired glycolytic (Videbech, 2000) or mitochondrial ATP production (Klinedinst and Regenold, 2015). This imbalance is exacerbated by excessive energy demand, typically occurring during stress exposure (Picard et al., 2018). Several magnetic resonance spectroscopy (MRS) and positron-emission tomography (PET) studies have identified key brain regions with abnormal metabolic activity. However, it appears that systemic metabolic mechanisms play a role as well, associating several endocrine dysregulations and inflammatory processes with impaired neuroenergetics. An example of this association is the relation of metabolic syndrome to atypical depression, as highlighted by their high comorbidity (Marazziti et al., 2014). Metabolic syndrome is a cluster of health conditions including abdominal obesity, high blood pressure and insulin resistance, which reflect impaired energy utilization and storage. In fact, MDD has been associated with impaired glucose homeostasis, and in particular insulin resistance [for a systematic review and meta-analysis see Kan et al. (2013)], which can occur in the brain itself (Watson et al., 2018; Lyra et al., 2019). Brain insulin resistance has thus become a therapeutic target for treating MDD and its associated symptoms (Hamer et al., 2019). For instance, insulin sensitizers have proven antidepressant efficacy in animal models (Zhao et al., 2016) and patients (Sepanjnia et al., 2012; Kemp et al., 2014) leading to improved glucose metabolism (Lin et al., 2015). Among the numerous possible causes, stress appears to be a key modulator, if not initiator, of such metabolic disturbances. Notably, insulin resistance is a recognized consequence of allostatic overload triggered by stress (Rasgon and McEwen, 2016) and has been associated with depressive phenotype (Li et al., 2013; van der Kooij et al., 2018; Yang et al., 2018).
Control of energy homeostasis is complex and involves several energy-sensing molecules. Intracellular detectors like AMPK (AMP-activated kinase), SIRT1 (Sirtuin 1) or HIF (Hypoxia-inducible factor), provide a rapid response to fluctuations in ATP, NADH and oxygen concentrations, and have been implicated in MDD (Ostergaard et al., 2018). Intercellular signaling, in a paracrine, endocrine or neurotransmission fashion (e.g., through monoamines, glucocorticoids or insulin), is another key mechanism for regulating metabolic activity based on energy demand. As such, the cAMP-CREB pathway is central to metabolic homeostasis by stimulating tissue-specific gene expression, through its extracellular metabotropic signaling sensitivity. CRTCs, as co-activators of CREB, play a central modulatory role in this cascade (Altarejos and Montminy, 2011). Hepatic CRTC2 promotes gluconeogenic response through opposite action of insulin or glucagon on SIK2. During short-term fasting, glucagon-induced rise in hepatic cAMP triggers the dephosphorylation of CRTC2, whereas upon feeding, the insulin-dependent activation of Akt leads to CRTC2 phosphorylation and cytoplasmic retention (Altarejos and Montminy, 2011). CRTC2 also holds a regulatory role on insulin secretion in the β-cells of the pancreatic islets (Screaton et al., 2004). In contrast, CRTC3 is mainly found in adipose tissue, where it enhances insulin resistance and obesity (Song et al., 2010). Finally, in skeletal muscles, the CRTC-CREB pathway seems to play a role in mitochondrial biogenesis through the expression of peroxisome proliferator activated receptor-γ (PPARγ) co-activator 1α (PGC1α) (Wu et al., 2006). CRTC1 is primarily expressed in the central nervous system and particularly abundant in limbic areas of mammalian brain. Neuronal CRTC-1 is controlling mitochondrial metabolism in Caenorhabditis elegans (Burkewitz et al., 2015). Likewise, the SIK-CRTC1 pathway was identified as an important cascade for controlling neuronal energy homeostasis in Drosophila (Choi et al., 2011; Shen et al., 2016). Studies in rodents highlight the importance of the neuronal CRTC1-CREB pathway in translating metabotropic neurotransmission signaling into synaptic plasticity (Saura and Cardinaux, 2017). While it seems clear that synapse formation and plasticity require stimulation of energy metabolism, the mechanistic pathway involving CRTC1 remains to be discovered in the mammalian brain. The role of CRTC1 in controlling energy homeostasis appears to be complex and multifactorial, involving both peripheral and central control. In fact, despite being primarily expressed in the brain, CRTC1 plays a role in peripheral tissues as well (Kim et al., 2015; Kim, 2016; Schumacher et al., 2016; Gao et al., 2018; Morhenn et al., 2019). Deletion of Crtc1 gene in mice induces insulin resistance and obesity, together with a depressive-like phenotype (Altarejos et al., 2008; Breuillaud et al., 2009, 2012; Rossetti et al., 2017). Using in vivo MRS at high field, we have observed that this phenotype translates into measurable neurochemical alterations in the hippocampus of Crtc1–/– mice (Cherix et al., 2020). Noteworthy, we identified lower levels of phosphocreatine relative to creatine, an indication that ATP production or consumption is impaired. These results suggest that CRTC1 is required for normal energy homeostasis in the brain and could mechanistically underlie similar observations arising from MRS studies in human depression (Moore et al., 1997; Harper et al., 2017). Finally, the strong resemblance between the Crtc1–/– mouse phenotype and metabolic syndrome might provide new avenues for exploring how energy deregulations relate to human MDD (Rasgon and McEwen, 2016; Watson et al., 2018).
Epidemiological evidence clearly suggests a comorbid association between depression and obesity (Amare et al., 2017; Milaneschi et al., 2019). Indeed, recent cross-sectional and longitudinal meta-analyses have demonstrated a positive and bidirectional relationship between these two pathological conditions. Moreover, these meta-analyses indicate that the MDD-obesity comorbidity occurs in both adulthood and adolescence, that it is not related to sociodemographic and lifestyle factors, and cannot be completely explained by the body weight increase induced by antidepressant medications. Compared to the general depression-obesity association, even stronger comorbidity was observed in atypical depressed patients, a subgroup of MDD patients showing sustained food intake during depressive episodes, increased visceral fat deposition and adverse metabolic profile (Xu et al., 2011). In keeping with this observation, a recent investigation suggests that the association between atypical depressive symptoms and obesity-related traits may arise from shared pathophysiological mechanisms (Milaneschi et al., 2017).
The wide distribution of CRTC1 in the rodent brain has been related to several functions spanning from synaptic plasticity, learning and memory, emotional processing and central energy-balance regulation (Saura and Cardinaux, 2017). In accordance with this broad function, the genetic suppression of CRTC1 in the mouse was found to induce depressive-like symptoms and obesity (Altarejos et al., 2008; Breuillaud et al., 2009, 2012; Rossetti et al., 2017). Although the human CRTC1 polymorphisms studied so far were not clearly associated with depression, they were associated with obesity markers [body mass index (BMI), fat mass] in psychiatric cohorts and in individuals with MDD (Choong et al., 2013; Quteineh et al., 2016). With regard to obesity, further human genetic investigations showed that the CRTC1 locus links fat mass to cardiometabolic diseases (Lu et al., 2016) and that genetic and epigenetic control of CRTC1 transcription affects fat distribution and eating behavior (Booij, 2019; Delacretaz et al., 2019; Rohde et al., 2019; Wang et al., 2021). Altogether, these observations strengthen the hypothesis that CRTC1 may represent a pivotal transcription coactivator regulating both MDD and obesity etiological pathways (see Tables 1, 2).
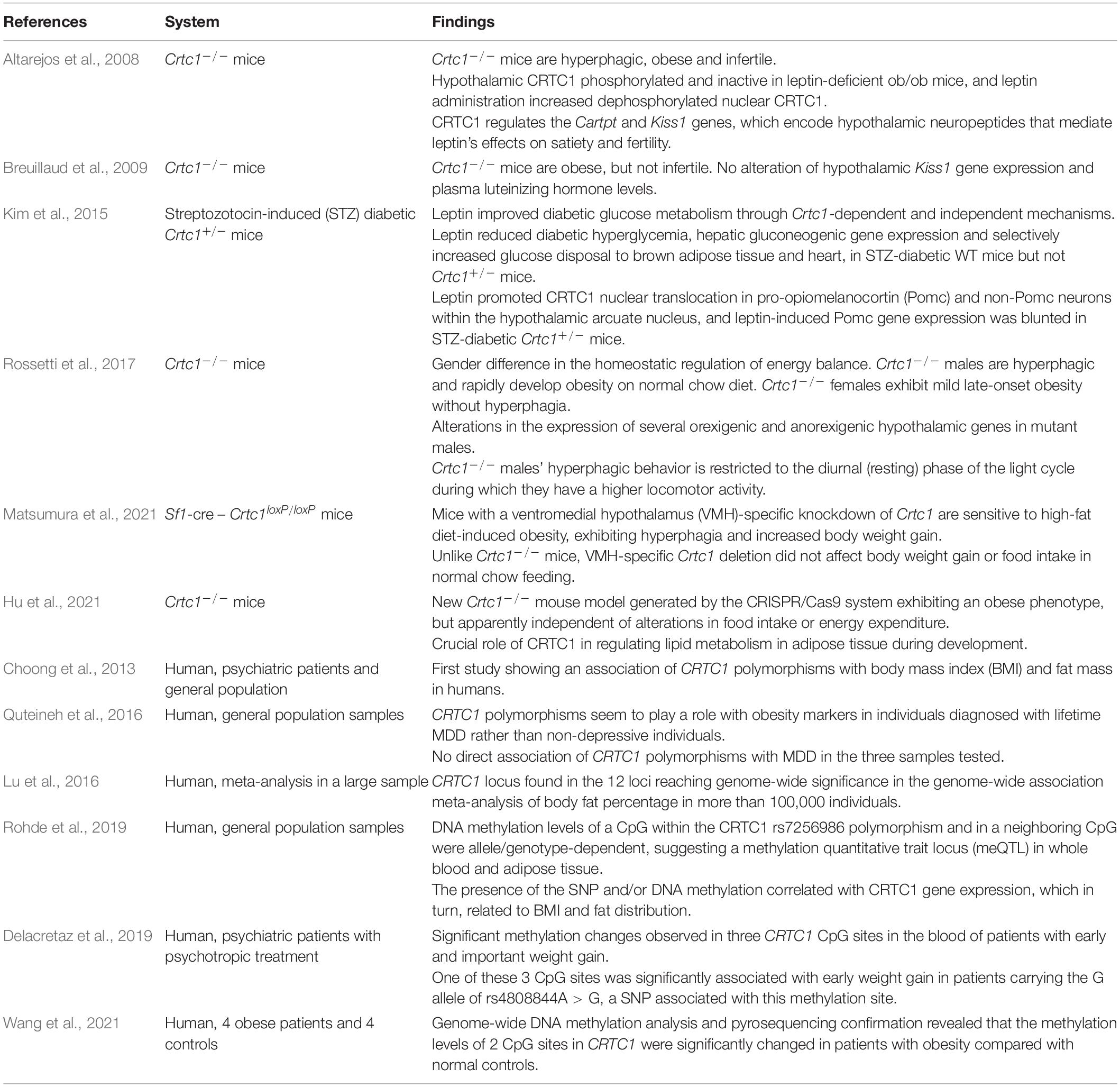
Table 2. Summary of studies implicating CRTC1 in obesity.
Several human studies evaluated the association of CRTC1 single nucleotide polymorphisms (SNPs) with obesity markers in the general population. To our knowledge, this association was first studied by Choong et al. (2013), who showed no significant association of the CRTC1 SNP rs6510997 (a proxy of the rs3746266 G allele) with BMI, weight or waist circumference in the general population. Similarly, CRTC1 locus did not reach genome-wide significance for BMI in the study of Locke et al. (2015), despite the very large sample size. Conversely, CRTC1 SNP rs757318 was found, for the first time, to be associated with Body Fat percentage (BF%) and BMI in a genome-wide association study (GWAS) aiming at establishing a link between adiposity and cardiometabolic disease risk (Lu et al., 2016). This study reported that CRTC1 SNP rs757318 had a more pronounced effect on BF% than on BMI, suggesting that BMI is an heterogeneous and less precise marker for adiposity, as it depends on both lean and fat mass. Interestingly, CRTC1 locus showed a significant sex-specific interaction with a two to threefold larger effect in women than in men. However, the BF%-increasing CRTC1 rs757318 allele was not associated with any of the cardiometabolic traits analyzed in this study. An interaction between CRTC1 SNP and sex was also observed in a human study that examined whether the CRTC1 polymorphism was associated with obesity markers in subjects with lifetime depression. Here, the CRTC1 SNP rs6510997 was found to be negatively associated with BMI in women with a lifetime diagnosis of depression (Quteineh et al., 2016). Taken together these observations suggest that: (1) the association of CRTC1 locus with obesity markers is stronger when reliable markers of adiposity (such as BF%) are used, (2) it may exist a sex-specific effect, even though, the use of different CRTC1 SNPs and different sample populations (general population versus psychiatric cohorts) complicate the interpretation of these GWAS studies. Moreover, due to the lack of knowledge about the biological effect of the tested SNPs on CRTC1’s function in humans (enhancement or decrease of CRTC1 activity in the brain), it is difficult to know whether the sex difference in energy balance regulation observed in Crtc1–/– mice is a specific feature of rodents.
Besides these human studies, the greatest contribution to the understanding of the regulatory role of CRTC1 in energy balance comes from animal experiments. A first study described the effects of the suppression of CRTC1 in the central control of food intake and energy expenditure (Altarejos et al., 2008). Crtc1–/– mice exhibited hyperphagia and body weight gain from early adult age with consequent development of obesity. The obese state of these mutant mice was also accompanied by reduced locomotor activity, lower energy expenditure and impaired sensitivity to leptin and insulin. However, it should be emphasized that there are gender differences regarding the vulnerability to develop obesity in this mouse model. Indeed, we showed that Crtc1–/– females are not hyperphagic and are less prone to gain weight as compared to mutant males, whose hyperphagia appears to be related to altered circadian locomotor activity (Rossetti et al., 2017). Moreover, CRTC1 seems necessary to protect against hepatic steatosis, which is strongly associated with obesity and metabolic syndrome, and to modulate leptin’s glucoregulatory actions in insulin-dependent diabetes (Kim et al., 2015; Kim, 2016). Whether CRTC1 is solely involved in the central control of energy balance or whether it also plays a role in peripheral organs is still unclear. A new Crtc1–/– mouse model generated by the CRISPR/Cas9 system also exhibits an obese phenotype, but it appears to be independent of alterations in food intake or energy expenditure (Hu et al., 2021). In this complete knockout mouse model, CRTC1 seems to be rather implicated in the regulation of lipid metabolism in adipose tissue during development. The creation of Crtc1 conditional knockout (cKO) mouse models should greatly help to decipher the specific role of CRTC1 in various brain regions and cell types. To date, there has been only one article reporting the use of a Crtc1 cKO mouse model (Matsumura et al., 2021). This study showed that mice with a ventromedial hypothalamus-specific knockdown of Crtc1 are sensitive to high-fat diet-induced obesity, exhibiting hyperphagia and increased body weight gain, but have a normal feeding behavior with a control chow diet.
Energy balance regulation depends on the interaction of many brain pathways that finely adapt food intake and energy expenditure to maintain energy homeostasis. After feeding, the release of leptin from adipocytes promotes satiety by acting on arcuate neurons in the hypothalamus (Myers et al., 2009; Pan and Myers, 2018). The activity of leptin depends on the stimulation of hypothalamic neurons releasing anorexigenic peptides and on the concomitant inhibition of neurons that secrete orexigenic peptides (Schwartz et al., 2000; Williams and Elmquist, 2012; Yeo and Heisler, 2012). The current working hypothesis suggests that CRTC1 contributes to leptin anorexigenic activity by modulating the expression of genes that participate in energy homeostasis. Accordingly, experimental evidence shows that leptin is able to activate the CRTC1/CREB pathway through different manners. This adipokine promotes CREB phosphorylation through the activation of the JAK2/STAT3 intracellular cascade (Catalano et al., 2009). In addition, leptin enhances CRTC1 nuclear translocation (Altarejos et al., 2008), possibly through the inhibition of AMP-activated kinase (Minokoshi et al., 2004) and increased activity of anorexigenic POMC neurons (Cowley et al., 2001). On the other hand, disrupted leptin signaling in leptin–deficient ob/ob mice is associated with increased amounts of phosphorylated, inactive CRTC1 in the cytoplasm of hypothalamic neurons (Altarejos et al., 2008).
Among the CRTC1/CREB-regulated genes, Cart (cocaine-amphetamine related transcript) is of particular importance for the suppression of food consumption, as it mediates leptin activity in the hypothalamus. In line with these molecular interactions, obese leptin-deficient ob/ob mice have a lower expression of Cart in the hypothalamus, and peripheral leptin injection normalizes hypothalamic CART peptide levels in these obese mice (Duan et al., 2007). Moreover, the capacity of CRTC1 to mediate the stimulating effect of leptin on Cart expression is supported by the increased plasmatic levels of leptin, reduced leptin receptor (LepRb) mRNA expression and lower Cart transcription in the arcuate nucleus of obese Crtc1–/– mice. Conversely, younger hyperphagic but not obese Crtc1–/– mice that are still leptin-sensitive, show comparable number of phospho-STAT3 positive cells in the arcuate nucleus after leptin infusion and similar leptin receptor (LepRb) and Cart mRNA levels as compared to lean wild-type mice (Altarejos et al., 2008; Rossetti et al., 2017). These data validate the functional interplay between leptin and CRTC1/CREB pathway, but point out the fact that the impaired Cart expression is probably just a consequence of leptin resistance and obesity, and that the lack of CRTC1 has likely broader effects on energy balance regulation by affecting other relevant genes.
In this regard, BDNF is an interesting candidate. Although first implicated in the etiology of MDD, this member of the neurotrophin family of growth factors was later recognized as a key component of mechanisms regulating energy intake and expenditure (Rios, 2013; Marosi and Mattson, 2014; Xu and Xie, 2016). According to a physiological role of BDNF in energy control, the expression of Bdnf and its receptor TrkB is sensitive to the nutritional state. Indeed, food deprivation reduces BDNF mRNA expression in the rat ventromedial hypothalamus, whereas glucose injection rapidly induces Bdnf and TrkB transcription in the same region (Xu et al., 2003; Unger et al., 2007). Complementary studies showed that the central injections of BDNF in rodents (i.c.v. or directly in the ventromedial or paraventricular hypothalamus) lead to reduced food intake, increased energy expenditure and body weight loss (Toriya et al., 2010). On the contrary, different genetic manipulations in mice, resulting in lower expression of either Bdnf or its TrkB receptor, cause hyperphagia and induce signs of metabolic syndrome including leptin and insulin resistance, dyslipidemia and hyperglycemia (Kernie et al., 2000; Rios et al., 2001). Finally, BDNF is also playing a role in the hedonic regulation of food intake involving the mesolimbic dopamine system. Consumption of palatable, high-fat food (HFF) influences Bdnf and TrkB expression in the dopaminergic neurons of the ventral tegmental area (VTA), whereas BDNF depletion in the VTA affects the reward function of the mesolimbic dopamine system and leads to excessive intake of palatable HFF, but not of standard chow, and to increased body weight under HFF conditions (Cordeira et al., 2010). This study suggests that an alteration of BDNF signaling may interfere with the activity of the mesolimbic dopamine pathway, leading to reward deficiency and compensatory overeating of palatable food.
In parallel to this preclinical research, human genetic studies also support the involvement of BDNF in energy balance regulation. The functional loss of one copy of the BDNF gene reduces serum BDNF concentration, increase ad libitum food intake and provokes severe early-onset obesity (Gray et al., 2006). Moreover, a missense mutation in the NTRK2 gene (human TrkB receptor gene) that prevents the regular activity of the receptor was identified in patients exhibiting overweight and severe obesity (Yeo et al., 2004). Finally, a functional polymorphism of the BDNF gene (BDNF Val66Met), which impedes the correct secretion and signaling of BDNF, was correlated with obesity predisposition in children and adolescents (Beckers et al., 2008; Skledar et al., 2012).
Consistent with the importance of the CRTC1/CREB pathway for Bdnf transcription, Crtc1–/– mice have lower mRNA levels of both Bdnf and TrkB in different brain regions as compared to wild-type mice. Precisely, the analysis of the multiple Bdnf splice variants in the hypothalamus of these mice revealed a significant reduction in the Bdnf mRNA for the exon I and IV (Breuillaud et al., 2012). A recent study using mice with selective knockdown of BDNF production from either promoter I, II, IV, or VI showed that disruption of BDNF from promoter I or II, but not IV or VI, induces hyperphagic obesity (McAllan et al., 2018). The energy balance dysregulation observed in Crtc1–/– mice might thus be related to a decrease of Bdnf exon I mRNA in the hypothalamus. Collectively, these findings suggest that BDNF could represent one of the most important CRTC1/CREB downstream genes that contribute to the energy homeostasis. However, a direct causality between Crtc1 deficiency and reduced hypothalamic Bdnf transcripts in the development of obesity is still elusive, and therefore, further molecular and behavioral researches are required.
Although less studied than Bdnf, another potential interesting CRTC1/CREB target gene is the neuron-derived orphan nuclear receptor 1 (Nor-1, also known as Nr4a3). This gene controls food intake in the arcuate nucleus by integrating leptin and glucocorticoid signaling (Kim et al., 2013). Interestingly, its expression in the arcuate nucleus is downregulated both in young and old Crtc1–/– mice (Rossetti et al., 2017).
Over the last decade, a compelling number of studies showed that chronodisruption (or circadian rhythm alteration) in humans severely increases the risk for developing both psychiatric and metabolic diseases (Barandas et al., 2015; Albrecht, 2017; Logan and McClung, 2019; Xie et al., 2019). Accordingly, the deletion of genes belonging to the mouse circadian clock machinery, notably Bmal1 and Clock, leads to hyperphagia, metabolic alterations and obesity (Rudic et al., 2004; Turek et al., 2005; Albrecht, 2012). Many environmental and genetic factors contribute to the maintenance of the circadian rhythm and allow the optimization of multiple biological functions along the daily night and day cycle (Dietrich and Horvath, 2013; Challet, 2019). It is long time known that photic inputs activate the CREB pathway in neurons of the suprachiasmatic nucleus (SCN) of the hypothalamus. This effect is likely due to variations in Ca2+ and cAMP levels in the SCN neurons induced by increased glutamate release from the fibers of the retino-hypothalamic tract that connects neural cells in the retina to the SCN cells (Ginty et al., 1993; Obrietan et al., 1999). This hypothesis is in accordance with the fact that both cAMP and Ca2+ levels oscillate within the SCN (Prosser and Gillette, 1991; Ikeda et al., 2003). In line with the role of CREB in the light-evoked circadian clock entrainment in SCN neurons, CREB-phosphorylation oscillates along the day in the mouse SCN and is higher during the first part of the light cycle (Travnickova-Bendova et al., 2002). More recently, it was shown that the dephosphorylation of CRTC1 and its subsequent nuclear accumulation peaks during the light part of the daily cycle (Table 3). This effect was specific for CRTC1, because CRTC2 did not show any variation (Sakamoto et al., 2013). Besides, the interaction of the CRTC1/CREB pathway with the circadian system is bidirectional, because complementary evidence exists about the control of some core clock genes by this transcriptional pathway. The suppression of CREB functionality in a tetracycline-inducible CREB repressor mouse strain leads to a significant reduction of both the expression of the circadian clock proteins PERIOD1 and PERIOD2 and the clock output hormones AVP and VIP in the SCN (Lee et al., 2010). Moreover, CRTC1 is involved in the negative feedback of the clock-resetting process that follows a light phase shift (jet lag) (Jagannath et al., 2013). In the SCN, light-activated CRTC1 increases SIK1 that phosphorylates CRTC1 and inactivates it. With this feedback mechanism, CRTC1 limits the transcription of Period1 that promotes clock resetting. Thereby, CRTC1 may slow down clock resetting and prevent an abrupt desynchrony between the master clock located in the SCN and the other peripheral clocks. This buffering system could protect the SCN clock (master clock) from inappropriate phase shifting caused not only by exposure to sudden changes in light intensity during the night, but also by abnormal Zeitgeber stimuli (such as food intake, physical activity, and temperature changes) that are all known to affect its resetting. Because the SCN master clock controls the other clocks located in other brain regions and in peripheral organs, an abrupt shift of the SCN clock would severely compromise normal physiological functions of the organism [see discussion in Jagannath et al. (2013)]. More recently, Jagannath et al. (2021) also highlighted the role of the CREB/CRTC1 pathway in adenosine-mediated integration of both light and sleep signaling to regulate circadian timing in mice. Another study in Drosophila showed that CRTC mutation affects the circadian oscillation of clock genes expression producing a phase delay especially in PERIOD and TIMLESS proteins. This work highlights that the interaction between CRTC homologs and the circadian clock machinery has likely an ancestral origin and selectively evolved among different species (Kim et al., 2016).

Table 3. Summary of studies implicating CRTC1 in circadian rhythms regulation.
Although the effective participation of CRTC1 in the entrainment of circadian clock in vivo and the consequent effect on the circadian energy homeostasis are not completely understood, the behavioral observation of Crtc1–/– mice suggested that the obesity of mutant males is due to overeating during the resting phase of the light cycle and to circadian alteration of spontaneous locomotor activity (Rossetti et al., 2017). Remarkably, the depressive-like phenotype of Crtc1–/– mice and their concomitant altered circadian rhythms and feeding behavior strongly suggest that CRTC1 is an important player in the central regulation of mood, circadian rhythms, and energy balance – all of which are dysregulated in MDD (see Figure 2).
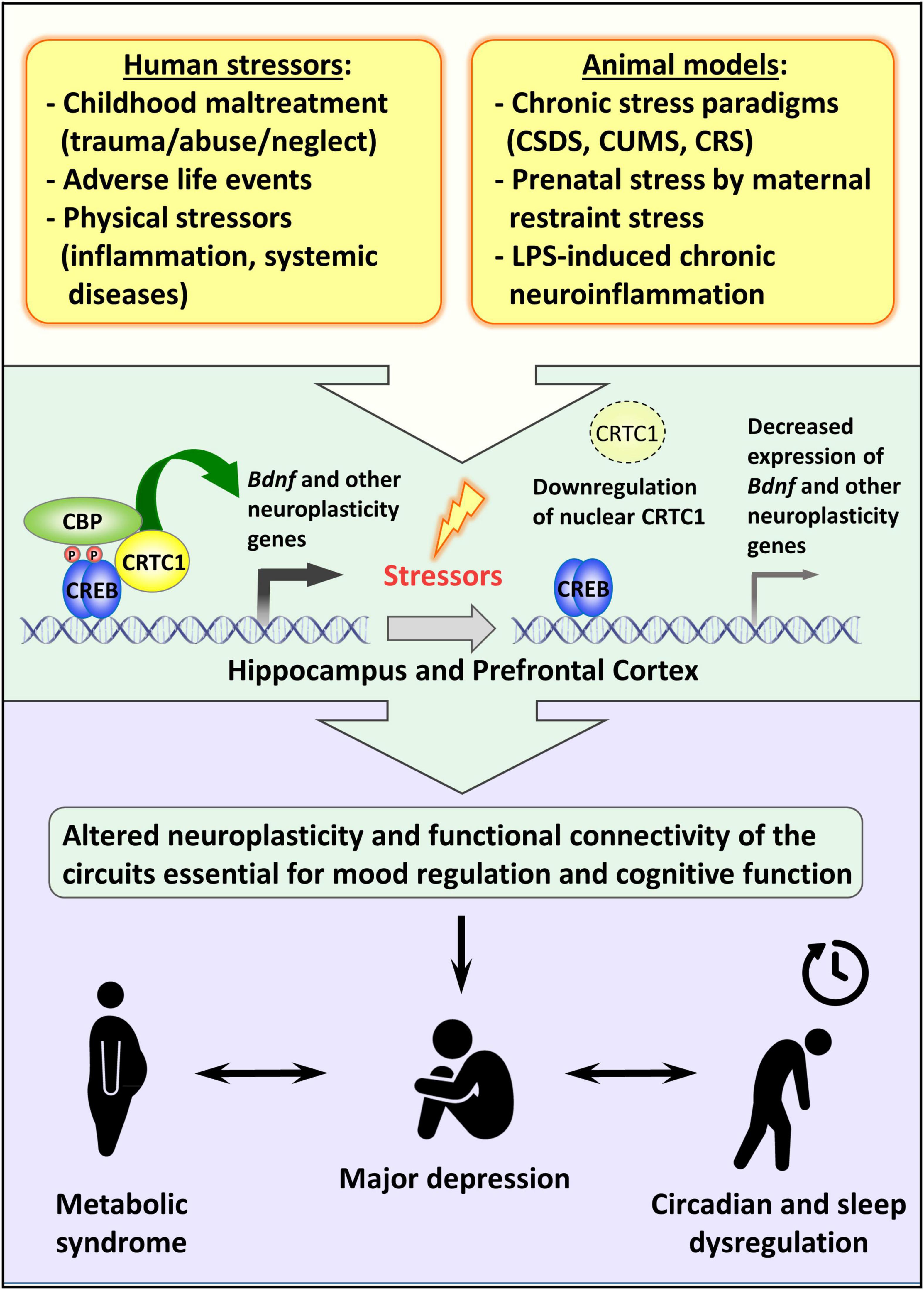
Figure 2. Possible role of CRTC1 in the pathogenesis of depression and associated disorders. Preclinical animal models of depression suggest that life stressors decrease CRTC1 levels in the prefrontal cortex and hippocampus, which triggers an altered neuroplasticity and functional connectivity related to the pathogenesis of depression and associated metabolic syndrome and chronodisruption.
This review highlighted the increasing evidence of the pivotal role of the transcription coactivator CRTC1 in MDD and comorbid obesity. Mice lacking CRTC1 exhibit depressive-like endophenotypes (Breuillaud et al., 2012; Meylan et al., 2016a,b) and an altered regulation of feeding behavior resulting in hyperphagic obesity only in males (Rossetti et al., 2017). Interestingly, this gender-specific alteration of energy balance is associated with a dysregulated circadian locomotor activity. More research is needed to understand why Crtc1–/– female mice are less affected and whether the altered circadian activity of the males correlates, for instance, with a reduced average duration of sleep and modified sleep patterns. Possibly related to these metabolic and circadian alterations, MRS studies with Crtc1–/– male mice suggest that CRTC1 plays a critical role in regulating brain neuroenergetics. A possible limitation to the face validity of the Crtc1 knockout mouse model of depression should, however, be considered, as the constitutive lack of CRTC1 may have neurodevelopmental effects that may influence the adulthood phenotypes. Future studies with Crtc1 conditional knockout mouse models that may be combined with viral approaches should address this issue and assess a putative role of CRTC1 during neurodevelopment. Human CRTC1 polymorphisms were associated with obesity markers (BMI, fat mass and distribution, eating behavior) in psychiatric cohorts and in the general population (Choong et al., 2013; Lu et al., 2016; Quteineh et al., 2016; Rohde et al., 2019). However, these polymorphisms were not directly associated with depression and further investigations are required to find a possible link between CRTC1 and MDD in humans.
Recent reports highlighted the instrumental role of ventral hippocampal CRTC1 in mediating chronic stress- and LPS-induced depressive-like behaviors, and antidepressant response in mice (Jiang B. et al., 2019; Ni et al., 2019). Jiang B. et al. (2019) compellingly shed light on the specific increased hippocampal expression of SIK2 triggered by chronic stress in two different paradigms (CUMS and CSDS). The higher levels of SIK2 increase CRTC1 phosphorylation and its retention in the cytoplasm, thus leading to a less active CRTC1-CREB-BDNF pathway and depressive-like symptoms. In the LPS-induced sickness behavior model of depression, Ni et al. (2019), uncovered the important regulatory role of hippocampal CRTC1 in controlling neuroinflammation and the CRTC1-CREB-BDNF-VGF pathway. Future research should characterize the molecular mechanisms of SIK2 induction in various chronic stress paradigms. The effects of CRTC1, BDNF, and VGF downregulation on hippocampal neuroplasticity should also be better defined and correlated with depressive-like behaviors. We and others showed that CRTC1 is required for conventional antidepressants response, but it is still unclear whether CRTC1 is involved in the sustained long-term effects of the novel rapid-acting antidepressants targeting glutamate neurotransmission. Finally, it remains to be determined whether the same CRTC1-regulated genes are jointly involved in MDD-related neuroplasticity processes, brain energy metabolism, circadian rhythms, and central control of energy balance, or whether different subsets of CRTC1 target genes are independently implicated. The transcription factor CREB is considered as a primary hub for activity-driven neuronal gene expression (Benito et al., 2011). Likewise, as a coactivator of CREB and possibly other transcription factors, CRTC1 could be a primary hub for a network of genes involved in the central regulation of mood, circadian rhythms, and energy balance. Future studies aiming at deciphering the underlying mechanisms through which downregulated CRTC1 functions commonly impinge on mood, circadian rhythms and energy balance regulation should lead to better insight into the etiopathogenesis of MDD and comorbid obesity, and may provide new therapeutic targets.
CR, AC, LG, and J-RC drafted and edited the manuscript. All authors read and approved the manuscript.
This work was supported by a grant from the Swiss National Science Foundation (31003A-170126).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank present and past lab members for their scientific contribution to the content of this review. We are also grateful to P. J. Magistretti, O. Halfon, and K. J. von Plessen for their support. We apologize to authors whose original work could not be cited because of space limitations.
Akil, H., Gordon, J., Hen, R., Javitch, J., Mayberg, H., Mcewen, B., et al. (2018). Treatment resistant depression: a multi-scale, systems biology approach. Neurosci. Biobehav. Rev. 84, 272–288. doi: 10.1016/j.neubiorev.2017.08.019
Albert, P. R. (2019). Adult neuroplasticity: a new “cure” for major depression? J. Psychiatry Neurosci. 44, 147–150. doi: 10.1503/jpn.190072
Albrecht, U. (2012). Timing to perfection: the biology of central and peripheral circadian clocks. Neuron 74, 246–260. doi: 10.1016/j.neuron.2012.04.006
Albrecht, U. (2017). Molecular mechanisms in mood regulation involving the circadian clock. Front. Neurol. 8:30. doi: 10.3389/fneur.2017.00030
Altarejos, J. Y., Goebel, N., Conkright, M. D., Inoue, H., Xie, J., Arias, C. M., et al. (2008). The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat. Med. 14, 1112–1117. doi: 10.1038/nm.1866
Altarejos, J. Y., and Montminy, M. (2011). CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151. doi: 10.1038/nrm3072
Amare, A. T., Schubert, K. O., Klingler-Hoffmann, M., Cohen-Woods, S., and Baune, B. T. (2017). The genetic overlap between mood disorders and cardiometabolic diseases: a systematic review of genome wide and candidate gene studies. Transl. Psychiatry 7:e1007. doi: 10.1038/tp.2016.261
Autry, A. E., Adachi, M., Nosyreva, E., Na, E. S., Los, M. F., Cheng, P. F., et al. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. doi: 10.1038/nature10130
Barandas, R., Landgraf, D., Mccarthy, M. J., and Welsh, D. K. (2015). Circadian clocks as modulators of metabolic comorbidity in psychiatric disorders. Curr. Psychiatry Rep. 17:98. doi: 10.1007/s11920-015-0637-2
Barco, A., and Marie, H. (2011). Genetic approaches to investigate the role of CREB in neuronal plasticity and memory. Mol. Neurobiol. 44, 330–349. doi: 10.1007/s12035-011-8209-x
Barua, S., Kim, J. Y., Kim, J. Y., Kim, J. H., and Lee, J. E. (2019). Therapeutic effect of agmatine on neurological disease: focus on ion channels and receptors. Neurochem. Res. 44, 735–750. doi: 10.1007/s11064-018-02712-1
Beckers, S., Peeters, A., Zegers, D., Mertens, I., Van Gaal, L., and Van Hul, W. (2008). Association of the BDNF Val66Met variation with obesity in women. Mol. Genet. Metab. 95, 110–112. doi: 10.1016/j.ymgme.2008.06.008
Benito, E., and Barco, A. (2010). CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 33, 230–240. doi: 10.1016/j.tins.2010.02.001
Benito, E., Valor, L. M., Jimenez-Minchan, M., Huber, W., and Barco, A. (2011). cAMP response element-binding protein is a primary hub of activity-driven neuronal gene expression. J. Neurosci. 31, 18237–18250. doi: 10.1523/JNEUROSCI.4554-11.2011
Bernstein, H. G., Stich, C., Jager, K., Dobrowolny, H., Wick, M., Steiner, J., et al. (2012). Agmatinase, an inactivator of the putative endogenous antidepressant agmatine, is strongly upregulated in hippocampal interneurons of subjects with mood disorders. Neuropharmacology 62, 237–246. doi: 10.1016/j.neuropharm.2011.07.012
Bittinger, M. A., Mcwhinnie, E., Meltzer, J., Iourgenko, V., Latario, B., Liu, X., et al. (2004). Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr. Biol. 14, 2156–2161. doi: 10.1016/j.cub.2004.11.002
Bjorkholm, C., and Monteggia, L. M. (2016). BDNF - a key transducer of antidepressant effects. Neuropharmacology 102, 72–79. doi: 10.1016/j.neuropharm.2015.10.034
Blendy, J. A. (2006). The role of CREB in depression and antidepressant treatment. Biol. Psychiatry 59, 1144–1150. doi: 10.1016/j.biopsych.2005.11.003
Boku, S., Nakagawa, S., Toda, H., and Hishimoto, A. (2018). Neural basis of major depressive disorder: beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 72, 3–12. doi: 10.1111/pcn.12604
Booij, L. (2019). Genetic and epigenetic regulation of CRTC1 in human eating behaviour and fat distribution: Methodological and clinical insights and considerations. EBioMedicine 45, 15–16. doi: 10.1016/j.ebiom.2019.06.019
Boulting, G. L., Durresi, E., Ataman, B., Sherman, M. A., Mei, K., Harmin, D. A., et al. (2021). Activity-dependent regulome of human GABAergic neurons reveals new patterns of gene regulation and neurological disease heritability. Nat. Neurosci. 24, 437–448. doi: 10.1038/s41593-020-00786-1
Breuillaud, L., Halfon, O., Magistretti, P. J., Pralong, F. P., and Cardinaux, J. R. (2009). Mouse fertility is not dependent on the CREB coactivator Crtc1. Nat. Med. 15, 989–990; authorrely991. doi: 10.1038/nm0909-989
Breuillaud, L., Rossetti, C., Meylan, E. M., Merinat, C., Halfon, O., Magistretti, P. J., et al. (2012). Deletion of CREB-regulated transcription coactivator 1 induces pathological aggression, depression-related behaviors, and neuroplasticity genes dysregulation in mice. Biol. Psychiatry 72, 528–536. doi: 10.1016/j.biopsych.2012.04.011
Briand, L. A., Lee, B. G., Lelay, J., Kaestner, K. H., and Blendy, J. A. (2015). Serine 133 phosphorylation is not required for hippocampal CREB-mediated transcription and behavior. Learn. Memory 22, 109–115. doi: 10.1101/lm.037044.114
Brindle, P., Nakajima, T., and Montminy, M. (1995). Multiple protein kinase A-regulated events are required for transcriptional induction by cAMP. Proc. Natl. Acad. Sci. U.S.A. 92, 10521–10525. doi: 10.1073/pnas.92.23.10521
Burkewitz, K., Morantte, I., Weir, H. J., Yeo, R., Zhang, Y., Huynh, F. K., et al. (2015). Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell 160, 842–855. doi: 10.1016/j.cell.2015.02.004
Camargo, A., and Rodrigues, A. L. S. (2019). Novel targets for fast antidepressant responses: possible role of endogenous neuromodulators. Chronic Stress (Thousand Oaks) 3:2470547019858083. doi: 10.1177/2470547019858083
Caraci, F., Calabrese, F., Molteni, R., Bartova, L., Dold, M., Leggio, G. M., et al. (2018). International union of basic and clinical pharmacology CIV: the neurobiology of treatment-resistant depression: from antidepressant classifications to novel pharmacological targets. Pharmacol. Rev. 70, 475–504. doi: 10.1124/pr.117.014977
Carlezon, W. A. Jr., Duman, R. S., and Nestler, E. J. (2005). The many faces of CREB. Trends Neurosci. 28, 436–445. doi: 10.1016/j.tins.2005.06.005
Castren, E., and Hen, R. (2013). Neuronal plasticity and antidepressant actions. Trends Neurosci. 36, 259–267. doi: 10.1016/j.tins.2012.12.010
Castren, E., and Monteggia, L. M. (2021). Brain-derived neurotrophic factor signaling in depression and antidepressant action. Biol. Psychiatry 90, 128–136. doi: 10.1016/j.biopsych.2021.05.008
Catalano, S., Giordano, C., Rizza, P., Gu, G., Barone, I., Bonofiglio, D., et al. (2009). Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J. Cell Physiol. 218, 490–500. doi: 10.1002/jcp.21622
Ch’ng, T. H., Desalvo, M., Lin, P., Vashisht, A., Wohlschlegel, J. A., and Martin, K. C. (2015). Cell biological mechanisms of activity-dependent synapse to nucleus translocation of CRTC1 in neurons. Front. Mol. Neurosci. 8:48. doi: 10.3389/fnmol.2015.00048
Ch’ng, T. H., Uzgil, B., Lin, P., Avliyakulov, N. K., O’dell, T. J., and Martin, K. C. (2012). Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150, 207–221. doi: 10.1016/j.cell.2012.05.027
Challet, E. (2019). The circadian regulation of food intake. Nat. Rev. Endocrinol. 15, 393–405. doi: 10.1038/s41574-019-0210-x
Chen, G. G., Almeida, D., Fiori, L., and Turecki, G. (2018). Evidence of reduced agmatine concentrations in the cerebral cortex of suicides. Int. J. Neuropsychopharmacol. 21, 895–900. doi: 10.1093/ijnp/pyy058
Cherix, A., Poitry-Yamate, C., Lanz, B., Zanoletti, O., Grosse, J., Sandi, C., et al. (2020). Deletion of Crtc1 leads to hippocampal neuroenergetic impairments associated with depressive-like behavior. bioRxiv [Preprint]. doi: 10.1101/2020.11.05.370221
Choi, S., Kim, W., and Chung, J. (2011). Drosophila salt-inducible kinase (SIK) regulates starvation resistance through cAMP-response element-binding protein (CREB)-regulated transcription coactivator (CRTC). J. Biol. Chem. 286, 2658–2664. doi: 10.1074/jbc.C110.119222
Choong, E., Quteineh, L., Cardinaux, J. R., Gholam-Rezaee, M., Vandenberghe, F., Dobrinas, M., et al. (2013). Influence of CRTC1 polymorphisms on body mass index and fat mass in psychiatric patients and the general adult population. JAMA Psychiatry 70, 1011–1019. doi: 10.1001/jamapsychiatry.2013.187
Conkright, M. D., Canettieri, G., Screaton, R., Guzman, E., Miraglia, L., Hogenesch, J. B., et al. (2003). TORCs: transducers of regulated CREB activity. Mol. Cell 12, 413–423. doi: 10.1016/j.molcel.2003.08.013
Cordeira, J. W., Frank, L., Sena-Esteves, M., Pothos, E. N., and Rios, M. (2010). Brain-derived neurotrophic factor regulates hedonic feeding by acting on the mesolimbic dopamine system. J. Neurosci. 30, 2533–2541. doi: 10.1523/JNEUROSCI.5768-09.2010
Cowley, M. A., Smart, J. L., Rubinstein, M., Cerdan, M. G., Diano, S., Horvath, T. L., et al. (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. doi: 10.1038/35078085
Dantzer, R., O’connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. doi: 10.1038/nrn2297
Davis, R. L. (2019). Understanding depression: the hippocampus might hold the answer in a CREB-regulated transcription coactivator: an editorial for ‘AAV-mediated over-expression of CRTC1 in the hippocampal dentate gyrus ameliorates lipopolysaccharide-induced depression-like behavior in mice’ on page 111. J. Neurochem. 149, 9–11. doi: 10.1111/jnc.14659
Delacretaz, A., Glatard, A., Dubath, C., Gholam-Rezaee, M., Sanchez-Mut, J. V., Graff, J., et al. (2019). Psychotropic drug-induced genetic-epigenetic modulation of CRTC1 gene is associated with early weight gain in a prospective study of psychiatric patients. Clin. Epigenetics 11:198. doi: 10.1186/s13148-019-0792-0
Dietrich, M. O., and Horvath, T. L. (2013). Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends Neurosci. 36, 65–73. doi: 10.1016/j.tins.2012.12.005
Duan, J., Choi, Y. H., Hartzell, D., Della-Fera, M. A., Hamrick, M., and Baile, C. A. (2007). Effects of subcutaneous leptin injections on hypothalamic gene profiles in lean and ob/ob mice. Obesity (Silver Spring) 15, 2624–2633. doi: 10.1038/oby.2007.314
Duman, R. S., Aghajanian, G. K., Sanacora, G., and Krystal, J. H. (2016). Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med. 22, 238–249. doi: 10.1038/nm.4050
Duman, R. S., Sanacora, G., and Krystal, J. H. (2019a). Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 102, 75–90. doi: 10.1016/j.neuron.2019.03.013
Duman, R. S., Shinohara, R., Fogaca, M. V., and Hare, B. (2019b). Neurobiology of rapid-acting antidepressants: convergent effects on GluA1-synaptic function. Mol. Psychiatry 24, 1816–1832. doi: 10.1038/s41380-019-0400-x
Escoubas, C. C., Silva-Garcia, C. G., and Mair, W. B. (2017). Deregulation of CRTCs in aging and age-related disease risk. Trends Genet. 33, 303–321. doi: 10.1016/j.tig.2017.03.002
Espana, J., Valero, J., Minano-Molina, A. J., Masgrau, R., Martin, E., Guardia-Laguarta, C., et al. (2010). beta-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J. Neurosci. 30, 9402–9410. doi: 10.1523/JNEUROSCI.2154-10.2010
Feldmann, K. G., Chowdhury, A., Becker, J. L., Mcalpin, N., Ahmed, T., Haider, S., et al. (2019). Non-canonical activation of CREB mediates neuroprotection in a Caenorhabditis elegans model of excitotoxic necrosis. J. Neurochem. 148, 531–549. doi: 10.1111/jnc.14629
Finsterwald, C., Fiumelli, H., Cardinaux, J. R., and Martin, J. L. (2010). Regulation of dendritic development by BDNF requires activation of CRTC1 by glutamate. J. Biol. Chem. 285, 28587–28595. doi: 10.1074/jbc.M110.125740
Freitas, A. E., Neis, V. B., and Rodrigues, A. L. S. (2016). Agmatine, a potential novel therapeutic strategy for depression. Eur. Neuropsychopharmacol. 26, 1885–1899. doi: 10.1016/j.euroneuro.2016.10.013
Fukuchi, M., Tabuchi, A., Kuwana, Y., Watanabe, S., Inoue, M., Takasaki, I., et al. (2015). Neuromodulatory effect of Galphas- or Galphaq-coupled G-protein-coupled receptor on NMDA receptor selectively activates the NMDA receptor/Ca2+/calcineurin/cAMP response element-binding protein-regulated transcriptional coactivator 1 pathway to effectively induce brain-derived neurotrophic factor expression in neurons. J. Neurosci. 35, 5606–5624. doi: 10.1523/JNEUROSCI.3650-14.2015
Gao, W. W., Tang, H. V., Cheng, Y., Chan, C. P., Chan, C. P., and Jin, D. Y. (2018). Suppression of gluconeogenic gene transcription by SIK1-induced ubiquitination and degradation of CRTC1. Biochim. Biophys. Acta 1861, 211–223. doi: 10.1016/j.bbagrm.2018.01.021
Ginty, D. D., Kornhauser, J. M., Thompson, M. A., Bading, H., Mayo, K. E., Takahashi, J. S., et al. (1993). Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science 260, 238–241. doi: 10.1126/science.8097062
Gold, P. W. (2015). The organization of the stress system and its dysregulation in depressive illness. Mol. Psychiatry 20, 32–47. doi: 10.1038/mp.2014.163
Gray, J., Yeo, G. S., Cox, J. J., Morton, J., Adlam, A. L., Keogh, J. M., et al. (2006). Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55, 3366–3371. doi: 10.2337/db06-0550
Halaris, A., and Piletz, J. (2007). Agmatine : metabolic pathway and spectrum of activity in brain. CNS Drugs 21, 885–900. doi: 10.2165/00023210-200721110-00002
Hamer, J. A., Testani, D., Mansur, R. B., Lee, Y., Subramaniapillai, M., and Mcintyre, R. S. (2019). Brain insulin resistance: a treatment target for cognitive impairment and anhedonia in depression. Exp. Neurol. 315, 1–8. doi: 10.1016/j.expneurol.2019.01.016
Harper, D. G., Jensen, J. E., Ravichandran, C., Perlis, R. H., Fava, M., Renshaw, P. F., et al. (2017). Tissue type-specific bioenergetic abnormalities in adults with major depression. Neuropsychopharmacology 42, 876–885. doi: 10.1038/npp.2016.180
Hu, Y., Lv, J., Fang, Y., Luo, Q., He, Y., Li, L., et al. (2021). Crtc1 deficiency causes obesity potentially via regulating PPARgamma pathway in white adipose. Front. Cell Dev. Biol. 9:602529. doi: 10.3389/fcell.2021.602529
Ikeda, M., Sugiyama, T., Wallace, C. S., Gompf, H. S., Yoshioka, T., Miyawaki, A., et al. (2003). Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron 38, 253–263. doi: 10.1016/s0896-6273(03)00164-8
Impey, S., Mark, M., Villacres, E. C., Poser, S., Chavkin, C., and Storm, D. R. (1996). Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 16, 973–982. doi: 10.1016/s0896-6273(00)80120-8
Iourgenko, V., Zhang, W., Mickanin, C., Daly, I., Jiang, C., Hexham, J. M., et al. (2003). Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 100, 12147–12152. doi: 10.1073/pnas.1932773100
Jagannath, A., Butler, R., Godinho, S. I., Couch, Y., Brown, L. A., Vasudevan, S. R., et al. (2013). The CRTC1-SIK1 pathway regulates entrainment of the circadian clock. Cell 154, 1100–1111. doi: 10.1016/j.cell.2013.08.004
Jagannath, A., Varga, N., Dallmann, R., Rando, G., Gosselin, P., Ebrahimjee, F., et al. (2021). Adenosine integrates light and sleep signalling for the regulation of circadian timing in mice. Nat. Commun. 12:2113. doi: 10.1038/s41467-021-22179-z
Jiang, B., Wang, H., Wang, J. L., Wang, Y. J., Zhu, Q., Wang, C. N., et al. (2019). Hippocampal salt-inducible kinase 2 plays a role in depression via the CREB-regulated transcription coactivator 1-cAMP response element binding-brain-derived neurotrophic factor pathway. Biol. Psychiatry 85, 650–666. doi: 10.1016/j.biopsych.2018.10.004
Jiang, C., Lin, W. J., and Salton, S. R. (2019). Role of a VGF/BDNF/TrkB autoregulatory feedback loop in rapid-acting antidepressant efficacy. J. Mol. Neurosci. 68, 504–509. doi: 10.1007/s12031-018-1124-0
Josselyn, S. A., and Nguyen, P. V. (2005). CREB, synapses and memory disorders: past progress and future challenges. Curr. Drug Targets CNS Neurol. Disord. 4, 481–497. doi: 10.2174/156800705774322058
Kan, C., Silva, N., Golden, S. H., Rajala, U., Timonen, M., Stahl, D., et al. (2013). A systematic review and meta-analysis of the association between depression and insulin resistance. Diabetes Care 36, 480–489. doi: 10.2337/dc12-1442
Kemp, D. E., Schinagle, M., Gao, K., Conroy, C., Ganocy, S. J., Ismail-Beigi, F., et al. (2014). PPAR-gamma agonism as a modulator of mood: proof-of-concept for pioglitazone in bipolar depression. CNS Drugs 28, 571–581. doi: 10.1007/s40263-014-0158-2
Kernie, S. G., Liebl, D. J., and Parada, L. F. (2000). BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 19, 1290–1300. doi: 10.1093/emboj/19.6.1290
Kim, G. H., Szabo, A., King, E. M., Ayala, J., Ayala, J. E., and Altarejos, J. Y. (2015). Leptin recruits Creb-regulated transcriptional coactivator 1 to improve hyperglycemia in insulin-deficient diabetes. Mol. Metab. 4, 227–236. doi: 10.1016/j.molmet.2014.12.006
Kim, H. (2016). The transcription cofactor CRTC1 protects from aberrant hepatic lipid accumulation. Sci. Rep. 6:37280. doi: 10.1038/srep37280
Kim, M., Lee, H., Hur, J.-H., Choe, J., and Lim, C. (2016). CRTC potentiates light-independent timeless transcription to sustain circadian rhythms in Drosophila. Sci. Rep. 6:32113. doi: 10.1038/srep32113
Kim, S. G., Lee, B., Kim, D. H., Kim, J., Lee, S., Lee, S. K., et al. (2013). Control of energy balance by hypothalamic gene circuitry involving two nuclear receptors, neuron-derived orphan receptor 1 and glucocorticoid receptor. Mol. Cell Biol. 33, 3826–3834. doi: 10.1128/MCB.00385-13
Klinedinst, N. J., and Regenold, W. T. (2015). A mitochondrial bioenergetic basis of depression. J. Bioenerg. Biomembr. 47, 155–171. doi: 10.1007/s10863-014-9584-6
Kovacs, K. A., Steullet, P., Steinmann, M., Do, K. Q., Magistretti, P. J., Halfon, O., et al. (2007). TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 104, 4700–4705. doi: 10.1073/pnas.0607524104
Kraus, C., Kadriu, B., Lanzenberger, R., Zarate, C. A. Jr., and Kasper, S. (2019). Prognosis and improved outcomes in major depression: a review. Transl. Psychiatry 9:127. doi: 10.1038/s41398-019-0460-3
Krystal, J. H., Abdallah, C. G., Sanacora, G., Charney, D. S., and Duman, R. S. (2019). Ketamine: a paradigm shift for depression research and treatment. Neuron 101, 774–778. doi: 10.1016/j.neuron.2019.02.005
Laube, G., and Bernstein, H. G. (2017). Agmatine: multifunctional arginine metabolite and magic bullet in clinical neuroscience? Biochem. J. 474, 2619–2640. doi: 10.1042/BCJ20170007
Lee, B., Li, A., Hansen, K. F., Cao, R., Yoon, J. H., and Obrietan, K. (2010). CREB influences timing and entrainment of the SCN circadian clock. J. Biol. Rhythms 25, 410–420. doi: 10.1177/0748730410381229
Levy, M. J. F., Boulle, F., Steinbusch, H. W., Van Den Hove, D. L. A., Kenis, G., and Lanfumey, L. (2018). Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology (Berl) 235, 2195–2220. doi: 10.1007/s00213-018-4950-4
Li, G., Regunathan, S., Barrow, C. J., Eshraghi, J., Cooper, R., and Reis, D. J. (1994). Agmatine: an endogenous clonidine-displacing substance in the brain. Science 263, 966–969. doi: 10.1126/science.7906055
Li, L., Li, X., Zhou, W., and Messina, J. L. (2013). Acute psychological stress results in the rapid development of insulin resistance. J. Endocrinol. 217, 175–184. doi: 10.1530/JOE-12-0559
Li, S., Zhang, C., Takemori, H., Zhou, Y., and Xiong, Z. Q. (2009). TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J. Neurosci. 29, 2334–2343. doi: 10.1523/JNEUROSCI.2296-08.2009
Lin, K. W., Wroolie, T. E., Robakis, T., and Rasgon, N. L. (2015). Adjuvant pioglitazone for unremitted depression: clinical correlates of treatment response. Psychiatry Res. 230, 846–852. doi: 10.1016/j.psychres.2015.10.013
Lin, L., Hales, C. M., Garber, K., and Jin, P. (2014). Fat mass and obesity-associated (FTO) protein interacts with CaMKII and modulates the activity of CREB signaling pathway. Hum. Mol. Genet. 23, 3299–3306. doi: 10.1093/hmg/ddu043
Liu, Y., Tang, W., Ji, C., Gu, J., Chen, Y., Huang, J., et al. (2020). The Selective SIK2 inhibitor ARN-3236 produces strong antidepressant-like efficacy in mice via the hippocampal CRTC1-CREB-BDNF pathway. Front. Pharmacol. 11:624429. doi: 10.3389/fphar.2020.624429
Locke, A. E., Kahali, B., Berndt, S. I., Justice, A. E., Pers, T. H., Day, F. R., et al. (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206. doi: 10.1038/nature14177
Logan, R. W., and McClung, C. A. (2019). Rhythms of life: circadian disruption and brain disorders across the lifespan. Nat. Rev. Neurosci. 20, 49–65. doi: 10.1038/s41583-018-0088-y
Lonze, B. E., and Ginty, D. D. (2002). Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623. doi: 10.1016/s0896-6273(02)00828-0
Lu, Y., Day, F. R., Gustafsson, S., Buchkovich, M. L., Na, J., Bataille, V., et al. (2016). New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat. Commun. 7:10495. doi: 10.1038/ncomms10495
Lyra, E. S. N. M., Lam, M. P., Soares, C. N., Munoz, D. P., Milev, R., and De Felice, F. G. (2019). Insulin resistance as a shared pathogenic mechanism between depression and type 2 diabetes. Front. Psychiatry 10:57. doi: 10.3389/fpsyt.2019.00057
Ma, Z., Zang, T., Birnbaum, S. G., Wang, Z., Johnson, J. E., Zhang, C. L., et al. (2017). TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat. Commun. 8:1668. doi: 10.1038/s41467-017-01709-8
Marazziti, D., Rutigliano, G., Baroni, S., Landi, P., and Dell’osso, L. (2014). Metabolic syndrome and major depression. CNS Spectr. 19, 293–304. doi: 10.1017/S1092852913000667
Marosi, K., and Mattson, M. P. (2014). BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol. Metab. 25, 89–98. doi: 10.1016/j.tem.2013.10.006
Marsden, W. N. (2013). Synaptic plasticity in depression: molecular, cellular and functional correlates. Prog. Neuropsychopharmacol. Biol. Psychiatry 43, 168–184. doi: 10.1016/j.pnpbp.2012.12.012
Matsumura, S., Ishikawa, F., Sasaki, T., Terkelsen, M. K., Ravnskjaer, K., Jinno, T., et al. (2021). Loss of CREB coactivator CRTC1 in SF1 cells leads to hyperphagia and obesity by high-fat diet but not normal chow diet. Endocrinology 162:bqab076. doi: 10.1210/endocr/bqab076
Mayr, B., and Montminy, M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609. doi: 10.1038/35085068
McAllan, L., Maynard, K. R., Kardian, A. S., Stayton, A. S., Fox, S. L., Stephenson, E. J., et al. (2018). Disruption of brain-derived neurotrophic factor production from individual promoters generates distinct body composition phenotypes in mice. Am. J. Physiol. Endocrinol. Metab. 315, E1168–E1184. doi: 10.1152/ajpendo.00205.2018
Meylan, E. M., Breuillaud, L., Seredenina, T., Magistretti, P. J., Halfon, O., Luthi-Carter, R., et al. (2016a). Involvement of the agmatinergic system in the depressive-like phenotype of the Crtc1 knockout mouse model of depression. Transl. Psychiatry 6:e852. doi: 10.1038/tp.2016.116
Meylan, E. M., Halfon, O., Magistretti, P. J., and Cardinaux, J. R. (2016b). The HDAC inhibitor SAHA improves depressive-like behavior of CRTC1-deficient mice: possible relevance for treatment-resistant depression. Neuropharmacology 107, 111–121. doi: 10.1016/j.neuropharm.2016.03.012
Milaneschi, Y., Lamers, F., Peyrot, W. J., Baune, B. T., Breen, G., Dehghan, A., et al. (2017). Genetic association of major depression with atypical features and obesity-related immunometabolic dysregulations. JAMA Psychiatry 74, 1214–1225. doi: 10.1001/jamapsychiatry.2017.3016
Milaneschi, Y., Simmons, W. K., Van Rossum, E. F. C., and Penninx, B. W. (2019). Depression and obesity: evidence of shared biological mechanisms. Mol. Psychiatry 24, 18–33. doi: 10.1038/s41380-018-0017-5
Minokoshi, Y., Alquier, T., Furukawa, N., Kim, Y. B., Lee, A., Xue, B., et al. (2004). AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574. doi: 10.1038/nature02440
Misztak, P., Panczyszyn-Trzewik, P., and Sowa-Kucma, M. (2018). Histone deacetylases (HDACs) as therapeutic target for depressive disorders. Pharmacol. Rep. 70, 398–408. doi: 10.1016/j.pharep.2017.08.001
Moda-Sava, R. N., Murdock, M. H., Parekh, P. K., Fetcho, R. N., Huang, B. S., Huynh, T. N., et al. (2019). Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364:eaat8078. doi: 10.1126/science.aat8078
Moore, C. M., Christensen, J. D., Lafer, B., Fava, M., and Renshaw, P. F. (1997). Lower levels of nucleoside triphosphate in the basal ganglia of depressed subjects: a phosphorous-31 magnetic resonance spectroscopy study. Am. J. Psychiatry 154, 116–118. doi: 10.1176/ajp.154.1.116
Morhenn, K., Quentin, T., Wichmann, H., Steinmetz, M., Prondzynski, M., Sohren, K. D., et al. (2019). Mechanistic role of the CREB-regulated transcription coactivator 1 in cardiac hypertrophy. J. Mol. Cell Cardiol. 127, 31–43. doi: 10.1016/j.yjmcc.2018.12.001
Myers, M. G. Jr., Munzberg, H., Leinninger, G. M., and Leshan, R. L. (2009). The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 9, 117–123. doi: 10.1016/j.cmet.2008.12.001
Neis, V. B., Bettio, L. B., Moretti, M., Rosa, P. B., Olescowicz, G., Fraga, D. B., et al. (2018). Single administration of agmatine reverses the depressive-like behavior induced by corticosterone in mice: comparison with ketamine and fluoxetine. Pharmacol. Biochem. Behav. 173, 44–50. doi: 10.1016/j.pbb.2018.08.005
Neis, V. B., Bettio, L. E. B., Moretti, M., Rosa, P. B., Ribeiro, C. M., Freitas, A. E., et al. (2016a). Acute agmatine administration, similar to ketamine, reverses depressive-like behavior induced by chronic unpredictable stress in mice. Pharmacol. Biochem. Behav. 150, 108–114. doi: 10.1016/j.pbb.2016.10.004
Neis, V. B., Moretti, M., Bettio, L. E., Ribeiro, C. M., Rosa, P. B., Goncalves, F. M., et al. (2016b). Agmatine produces antidepressant-like effects by activating AMPA receptors and mTOR signaling. Eur. Neuropsychopharmacol. 26, 959–971. doi: 10.1016/j.euroneuro.2016.03.009
Neis, V. B., Rosa, P. B., Olescowicz, G., and Rodrigues, A. L. S. (2017). Therapeutic potential of agmatine for CNS disorders. Neurochem. Int. 108, 318–331. doi: 10.1016/j.neuint.2017.05.006
Ni, S., Huang, H., He, D., Chen, H., Wang, C., Zhao, X., et al. (2019). Adeno-associated virus-mediated over-expression of CREB-regulated transcription coactivator 1 in the hippocampal dentate gyrus ameliorates lipopolysaccharide-induced depression-like behaviour in mice. J. Neurochem. 149, 111–125. doi: 10.1111/jnc.14670
Nonaka, M., Kim, R., Fukushima, H., Sasaki, K., Suzuki, K., Okamura, M., et al. (2014). Region-specific activation of CRTC1-CREB signaling mediates long-term fear memory. Neuron 84, 92–106. doi: 10.1016/j.neuron.2014.08.049
Obrietan, K., Impey, S., Smith, D., Athos, J., and Storm, D. R. (1999). Circadian regulation of cAMP response element-mediated gene expression in the suprachiasmatic nuclei. J. Biol. Chem. 274, 17748–17756. doi: 10.1074/jbc.274.25.17748
Ostergaard, L., Jorgensen, M. B., and Knudsen, G. M. (2018). Low on energy? An energy supply-demand perspective on stress and depression. Neurosci. Biobehav. Rev. 94, 248–270. doi: 10.1016/j.neubiorev.2018.08.007
Pan, W. W., and Myers, M. G. Jr. (2018). Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 19, 95–105. doi: 10.1038/nrn.2017.168
Pan, Y., He, X., Li, C., Li, Y., Li, W., Zhang, H., et al. (2021). Neuronal activity recruits the CRTC1/CREB axis to drive transcription-dependent autophagy for maintaining late-phase LTD. Cell Rep. 36:109398. doi: 10.1016/j.celrep.2021.109398
Parra-Damas, A., Chen, M., Enriquez-Barreto, L., Ortega, L., Acosta, S., Perna, J. C., et al. (2017a). CRTC1 function during memory encoding is disrupted in neurodegeneration. Biol. Psychiatry 81, 111–123. doi: 10.1016/j.biopsych.2016.06.025
Parra-Damas, A., Rubio-Ferrarons, L., Shen, J., and Saura, C. A. (2017b). CRTC1 mediates preferential transcription at neuronal activity-regulated CRE/TATA promoters. Sci. Rep. 7:18004. doi: 10.1038/s41598-017-18215-y
Parra-Damas, A., and Saura, C. A. (2019). Synapse-to-nucleus signaling in neurodegenerative and neuropsychiatric disorders. Biol. Psychiatry 86, 87–96. doi: 10.1016/j.biopsych.2019.01.006
Parra-Damas, A., Valero, J., Chen, M., Espana, J., Martin, E., Ferrer, I., et al. (2014). Crtc1 activates a transcriptional program deregulated at early Alzheimer’s disease-related stages. J. Neurosci. 34, 5776–5787. doi: 10.1523/JNEUROSCI.5288-13.2014
Picard, M., Mcewen, B. S., Epel, E. S., and Sandi, C. (2018). An energetic view of stress: focus on mitochondria. Front. Neuroendocrinol. 49:72–85. doi: 10.1016/j.yfrne.2018.01.001
Piletz, J. E., Aricioglu, F., Cheng, J. T., Fairbanks, C. A., Gilad, V. H., Haenisch, B., et al. (2013). Agmatine: clinical applications after 100 years in translation. Drug Discov. Today 18, 880–893. doi: 10.1016/j.drudis.2013.05.017
Pittenger, C., and Duman, R. S. (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33, 88–109. doi: 10.1038/sj.npp.1301574
Prosser, R. A., and Gillette, M. U. (1991). Cyclic changes in cAMP concentration and phosphodiesterase activity in a mammalian circadian clock studied in vitro. Brain Res. 568, 185–192. doi: 10.1016/0006-8993(91)91396-i
Quteineh, L., Preisig, M., Rivera, M., Milaneschi, Y., Castelao, E., Gholam-Rezaee, M., et al. (2016). Association of CRTC1 polymorphisms with obesity markers in subjects from the general population with lifetime depression. J. Affect. Disord. 198, 43–49. doi: 10.1016/j.jad.2016.03.031
Ragguett, R. M., Tamura, J. K., and Mcintyre, R. S. (2019). Keeping up with the clinical advances: depression. CNS Spectr. 24, 25–37. doi: 10.1017/S1092852919001159
Rasgon, N. L., and McEwen, B. S. (2016). Insulin resistance-a missing link no more. Mol. Psychiatry 21, 1648–1652. doi: 10.1038/mp.2016.162
Reis, D. J., and Regunathan, S. (2000). Is agmatine a novel neurotransmitter in brain? Trends Pharmacol. Sci. 21, 187–193. doi: 10.1016/s0165-6147(00)01460-7
Rios, M. (2013). BDNF and the central control of feeding: accidental bystander or essential player? Trends Neurosci. 36, 83–90. doi: 10.1016/j.tins.2012.12.009
Rios, M., Fan, G., Fekete, C., Kelly, J., Bates, B., Kuehn, R., et al. (2001). Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol. Endocrinol. 15, 1748–1757. doi: 10.1210/mend.15.10.0706
Rohde, K., Keller, M., La Cour Poulsen, L., Ronningen, T., Stumvoll, M., Tonjes, A., et al. (2019). (Epi)genetic regulation of CRTC1 in human eating behaviour and fat distribution. EBioMedicine 44, 476–488. doi: 10.1016/j.ebiom.2019.05.050
Rossetti, C., Sciarra, D., Petit, J. M., Eap, C. B., Halfon, O., Magistretti, P. J., et al. (2017). Gender-specific alteration of energy balance and circadian locomotor activity in the Crtc1 knockout mouse model of depression. Transl. Psychiatry 7:1269. doi: 10.1038/s41398-017-0023-4
Rudic, R. D., Mcnamara, P., Curtis, A. M., Boston, R. C., Panda, S., Hogenesch, J. B., et al. (2004). BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2:e377. doi: 10.1371/journal.pbio.0020377
Sakamoto, K., Norona, F. E., Alzate-Correa, D., Scarberry, D., Hoyt, K. R., and Obrietan, K. (2013). Clock and light regulation of the CREB coactivator CRTC1 in the suprachiasmatic circadian clock. J. Neurosci. 33, 9021–9027. doi: 10.1523/JNEUROSCI.4202-12.2013
Saura, C. A., and Cardinaux, J. R. (2017). Emerging roles of CREB-regulated transcription coactivators in brain physiology and pathology. Trends Neurosci. 40, 720–733. doi: 10.1016/j.tins.2017.10.002
Schumacher, Y., Aparicio, T., Ourabah, S., Baraille, F., Martin, A., Wind, P., et al. (2016). Dysregulated CRTC1 activity is a novel component of PGE2 signaling that contributes to colon cancer growth. Oncogene 35, 2602–2614. doi: 10.1038/onc.2015.283
Schwartz, M. W., Woods, S. C., Porte, D. Jr., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671. doi: 10.1038/35007534
Screaton, R. A., Conkright, M. D., Katoh, Y., Best, J. L., Canettieri, G., Jeffries, S., et al. (2004). The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119, 61–74. doi: 10.1016/j.cell.2004.09.015
Sekeres, M. J., Mercaldo, V., Richards, B., Sargin, D., Mahadevan, V., Woodin, M. A., et al. (2012). Increasing CRTC1 function in the dentate gyrus during memory formation or reactivation increases memory strength without compromising memory quality. J. Neurosci. 32, 17857–17868. doi: 10.1523/JNEUROSCI.1419-12.2012
Seo, S., Liu, P., and Leitch, B. (2011). Spatial learning-induced accumulation of agmatine and glutamate at hippocampal CA1 synaptic terminals. Neuroscience 192, 28–36. doi: 10.1016/j.neuroscience.2011.07.007
Sepanjnia, K., Modabbernia, A., Ashrafi, M., Modabbernia, M. J., and Akhondzadeh, S. (2012). Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: randomized double-blind placebo-controlled trial. Neuropsychopharmacology 37, 2093–2100. doi: 10.1038/npp.2012.58
Shen, R., Wang, B., Giribaldi, M. G., Ayres, J., Thomas, J. B., and Montminy, M. (2016). Neuronal energy-sensing pathway promotes energy balance by modulating disease tolerance. Proc. Natl. Acad. Sci. U.S.A. 113, E3307–E3314. doi: 10.1073/pnas.1606106113
Shinohara, R., Aghajanian, G. K., and Abdallah, C. G. (2021). Neurobiology of the rapid-acting antidepressant effects of ketamine: impact and opportunities. Biol. Psychiatry 90, 85–95. doi: 10.1016/j.biopsych.2020.12.006
Shu, H., Wang, M., Song, M., Sun, Y., Shen, X., Zhang, J., et al. (2020). Acute nicotine treatment alleviates LPS-induced impairment of fear memory reconsolidation through AMPK activation and CRTC1 upregulation in hippocampus. Int. J. Neuropsychopharmacol. 23, 687–699. doi: 10.1093/ijnp/pyaa043
Si, Y., Xue, X., Liu, S., Feng, C., Zhang, H., Zhang, S., et al. (2021). CRTC1 signaling involvement in depression-like behavior of prenatally stressed offspring rat. Behav. Brain Res. 399:113000. doi: 10.1016/j.bbr.2020.113000
Skledar, M., Nikolac, M., Dodig-Curkovic, K., Curkovic, M., Borovecki, F., and Pivac, N. (2012). Association between brain-derived neurotrophic factor Val66Met and obesity in children and adolescents. Prog. Neuropsychopharmacol. Biol. Psychiatry 36, 136–140. doi: 10.1016/j.pnpbp.2011.08.003
Song, M., Martinowich, K., and Lee, F. S. (2017). BDNF at the synapse: why location matters. Mol. Psychiatry 22, 1370–1375. doi: 10.1038/mp.2017.144
Song, Y., Altarejos, J., Goodarzi, M. O., Inoue, H., Guo, X., Berdeaux, R., et al. (2010). CRTC3 links catecholamine signalling to energy balance. Nature 468, 933–939. doi: 10.1038/nature09564
Sonntag, T., Moresco, J. J., Vaughan, J. M., Matsumura, S., Yates, J. R. III, and Montminy, M. (2017). Analysis of a cAMP regulated coactivator family reveals an alternative phosphorylation motif for AMPK family members. PLoS One 12:e0173013. doi: 10.1371/journal.pone.0173013
Toriya, M., Maekawa, F., Maejima, Y., Onaka, T., Fujiwara, K., Nakagawa, T., et al. (2010). Long-term infusion of brain-derived neurotrophic factor reduces food intake and body weight via a corticotrophin-releasing hormone pathway in the paraventricular nucleus of the hypothalamus. J. Neuroendocrinol. 22, 987–995. doi: 10.1111/j.1365-2826.2010.02039.x
Travnickova-Bendova, Z., Cermakian, N., Reppert, S. M., and Sassone-Corsi, P. (2002). Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. U.S.A. 99, 7728–7733. doi: 10.1073/pnas.102075599
Turek, F. W., Joshu, C., Kohsaka, A., Lin, E., Ivanova, G., and Mcdearmon, E. (2005). Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308, 1043–1045. doi: 10.1126/science.1108750
Uchida, S., and Shumyatsky, G. P. (2018). Synaptically localized transcriptional regulators in memory formation. Neuroscience 370, 4–13. doi: 10.1016/j.neuroscience.2017.07.023
Uchida, S., Teubner, B. J., Hevi, C., Hara, K., Kobayashi, A., Dave, R. M., et al. (2017). CRTC1 nuclear translocation following learning modulates memory strength via exchange of chromatin remodeling complexes on the Fgf1 gene. Cell Rep. 18, 352–366. doi: 10.1016/j.celrep.2016.12.052
Uchida, S., Yamagata, H., Seki, T., and Watanabe, Y. (2018). Epigenetic mechanisms of major depression: targeting neuronal plasticity. Psychiatry Clin. Neurosci. 72, 212–227. doi: 10.1111/pcn.12621
Umemori, J., Winkel, F., Didio, G., Llach Pou, M., and Castren, E. (2018). iPlasticity: induced juvenile-like plasticity in the adult brain as a mechanism of antidepressants. Psychiatry Clin. Neurosci. 72, 633–653. doi: 10.1111/pcn.12683
Unger, T. J., Calderon, G. A., Bradley, L. C., Sena-Esteves, M., and Rios, M. (2007). Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J. Neurosci. 27, 14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007
Uzbay, T. I. (2012). The pharmacological importance of agmatine in the brain. Neurosci. Biobehav. Rev. 36, 502–519. doi: 10.1016/j.neubiorev.2011.08.006
Valverde, A. P., Camargo, A., and Rodrigues, A. L. S. (2021). Agmatine as a novel candidate for rapid-onset antidepressant response. World J. Psychiatry 11, 981–996. doi: 10.5498/wjp.v11.i11.981
van der Kooij, M. A., Jene, T., Treccani, G., Miederer, I., Hasch, A., Voelxen, N., et al. (2018). Chronic social stress-induced hyperglycemia in mice couples individual stress susceptibility to impaired spatial memory. Proc. Natl. Acad. Sci. U.S.A. 115, E10187–E10196. doi: 10.1073/pnas.1804412115
Vialou, V., Feng, J., Robison, A. J., and Nestler, E. J. (2013). Epigenetic mechanisms of depression and antidepressant action. Annu. Rev. Pharmacol. Toxicol. 53, 59–87. doi: 10.1146/annurev-pharmtox-010611-134540
Videbech, P. (2000). PET measurements of brain glucose metabolism and blood flow in major depressive disorder: a critical review. Acta Psychiatr. Scand. 101, 11–20. doi: 10.1034/j.1600-0447.2000.101001011.x
Wang, C., Wang, M., and Ma, J. (2021). Analysis of genome-wide DNA methylation patterns in obesity. Endocr. J. 68, 1439–1453. doi: 10.1507/endocrj.EJ20-0734
Wang, Y. J., Liu, L., Wang, Y., Wang, J. L., Gao, T. T., Wang, H., et al. (2020). Imipramine exerts antidepressant-like effects in chronic stress models of depression by promoting CRTC1 expression in the mPFC. Brain Res. Bull. 164, 257–268. doi: 10.1016/j.brainresbull.2020.08.028
Watson, K., Nasca, C., Aasly, L., Mcewen, B., and Rasgon, N. (2018). Insulin resistance, an unmasked culprit in depressive disorders: promises for interventions. Neuropharmacology 136, 327–334. doi: 10.1016/j.neuropharm.2017.11.038
Watts, A. G., Sanchez-Watts, G., Liu, Y., and Aguilera, G. (2011). The distribution of messenger RNAs encoding the three isoforms of the transducer of regulated cAMP responsive element binding protein activity in the rat forebrain. J. Neuroendocrinol. 23, 754–766. doi: 10.1111/j.1365-2826.2011.02178.x
Watts, D., Pfaffenseller, B., Wollenhaupt-Aguiar, B., Paul Gea, L., Cardoso, T. A., and Kapczinski, F. (2019). Agmatine as a potential therapeutic intervention in bipolar depression: the preclinical landscape. Expert. Opin. Ther. Targets 23, 327–339. doi: 10.1080/14728222.2019.1581764
Weiss, T., Bernard, R., Bernstein, H. G., Veh, R. W., and Laube, G. (2018). Agmatine modulates spontaneous activity in neurons of the rat medial habenular complex-a relevant mechanism in the pathophysiology and treatment of depression? Transl. Psychiatry 8:201. doi: 10.1038/s41398-018-0254-z
Wilkinson, S. T., and Sanacora, G. (2019). A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Discov. Today 24, 606–615. doi: 10.1016/j.drudis.2018.11.007
Williams, K. W., and Elmquist, J. K. (2012). From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat. Neurosci. 15, 1350–1355. doi: 10.1038/nn.3217
Woelfer, M., Kasties, V., Kahlfuss, S., and Walter, M. (2019). The role of depressive subtypes within the neuroinflammation hypothesis of major depressive disorder. Neuroscience 403, 93–110. doi: 10.1016/j.neuroscience.2018.03.034
World Health Organization. (2017). Depression and Other Common Mental Disorders. WHO Reference Number: WHO/MSD/MER/2017.2. Geneva: World Health Organization.
Wu, Z., Huang, X., Feng, Y., Handschin, C., Gullicksen, P. S., Bare, O., et al. (2006). Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. U.S.A. 103, 14379–14384. doi: 10.1073/pnas.0606714103
Xiao, X., Zhang, C., Grigoroiu-Serbanescu, M., Wang, L., Li, L., Zhou, D., et al. (2018). The cAMP responsive element-binding (CREB)-1 gene increases risk of major psychiatric disorders. Mol. Psychiatry 23, 1957–1967. doi: 10.1038/mp.2017.243
Xie, Y., Tang, Q., Chen, G., Xie, M., Yu, S., Zhao, J., et al. (2019). New insights into the circadian rhythm and its related diseases. Front. Physiol. 10:682. doi: 10.3389/fphys.2019.00682
Xu, B., Goulding, E. H., Zang, K., Cepoi, D., Cone, R. D., Jones, K. R., et al. (2003). Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 6, 736–742. doi: 10.1038/nn1073
Xu, B., and Xie, X. (2016). Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 17, 282–292. doi: 10.1038/nrn.2016.24
Xu, Q., Anderson, D., and Lurie-Beck, J. (2011). The relationship between abdominal obesity and depression in the general population: a systematic review and meta-analysis. Obes. Res. Clin. Pract. 5, e267–e360. doi: 10.1016/j.orcp.2011.04.007
Yan, P., Xue, Z., Li, D., Ni, S., Wang, C., Jin, X., et al. (2021). Dysregulated CRTC1-BDNF signaling pathway in the hippocampus contributes to Abeta oligomer-induced long-term synaptic plasticity and memory impairment. Exp. Neurol. 345:113812. doi: 10.1016/j.expneurol.2021.113812
Yang, H., Li, W., Meng, P., Liu, Z., Liu, J., and Wang, Y. (2018). Chronic unpredictable mild stress aggravates mood disorder, cognitive impairment, and brain insulin resistance in diabetic rat. Evid. Based Compl. Alternat. Med. 2018:2901863. doi: 10.1155/2018/2901863
Yang, X. C., and Reis, D. J. (1999). Agmatine selectively blocks the N-methyl-D-aspartate subclass of glutamate receptor channels in rat hippocampal neurons. J. Pharmacol. Exp. Ther. 288, 544–549.
Yeo, G. S., Connie Hung, C. C., Rochford, J., Keogh, J., Gray, J., Sivaramakrishnan, S., et al. (2004). A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 7, 1187–1189. doi: 10.1038/nn1336
Yeo, G. S., and Heisler, L. K. (2012). Unraveling the brain regulation of appetite: lessons from genetics. Nat. Neurosci. 15, 1343–1349. doi: 10.1038/nn.3211
Zanos, P., and Gould, T. D. (2018). Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 23, 801–811. doi: 10.1038/mp.2017.255
Zanos, P., Thompson, S. M., Duman, R. S., Zarate, C. A. Jr., and Gould, T. D. (2018). Convergent mechanisms underlying rapid antidepressant action. CNS Drugs 32, 197–227. doi: 10.1007/s40263-018-0492-x
Zhang, X., Shen, X., Dong, J., Liu, W. C., Song, M., Sun, Y., et al. (2019). Inhibition of reactive astrocytes with fluorocitrate ameliorates learning and memory impairment through upregulating crtc1 and synaptophysin in ischemic stroke rats. Cell Mol. Neurobiol. 39, 1151–1163. doi: 10.1007/s10571-019-00709-0
Zhao, Q., Wu, X., Yan, S., Xie, X., Fan, Y., Zhang, J., et al. (2016). The antidepressant-like effects of pioglitazone in a chronic mild stress mouse model are associated with PPARgamma-mediated alteration of microglial activation phenotypes. J Neuroinflammation 13:259. doi: 10.1186/s12974-016-0728-y
Zhou, Y., Wu, H., Li, S., Chen, Q., Cheng, X. W., Zheng, J., et al. (2006). Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One 1:e16. doi: 10.1371/journal.pone.0000016
Keywords: major depressive disorder, obesity, circadian rhythms, neuroplasticity, CREB, CRTC1, BDNF
Citation: Rossetti C, Cherix A, Guiraud LF and Cardinaux J-R (2022) New Insights Into the Pivotal Role of CREB-Regulated Transcription Coactivator 1 in Depression and Comorbid Obesity. Front. Mol. Neurosci. 15:810641. doi: 10.3389/fnmol.2022.810641
Received: 07 November 2021; Accepted: 07 January 2022;
Published: 15 February 2022.
Edited by:
Carlos A. Saura, Universidad Autónoma de Barcelona, SpainReviewed by:
Peter Hamilton, Virginia Commonwealth University, United StatesCopyright © 2022 Rossetti, Cherix, Guiraud and Cardinaux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-René Cardinaux, SmVhbi1SZW5lLkNhcmRpbmF1eEBjaHV2LmNo
†Present address: Antoine Cherix, Wellcome Centre for Integrative Neuroimaging, WIN-FMRIB, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.