
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci., 26 April 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.809810
This article is part of the Research TopicIon Channels and Transporters in Epilepsy: From Genes and Mechanisms to Disease-Targeted TherapiesView all 15 articles
 Tiantian Xiao1†
Tiantian Xiao1† Xiang Chen1†Yan Xu2Huiyao Chen3Xinran Dong3
Xiang Chen1†Yan Xu2Huiyao Chen3Xinran Dong3 Lin Yang4
Lin Yang4 Bingbing Wu3Liping Chen5
Bingbing Wu3Liping Chen5 Long Li6Deyi Zhuang7Dongmei Chen8
Long Li6Deyi Zhuang7Dongmei Chen8 Yuanfeng Zhou2*
Yuanfeng Zhou2* Huijun Wang3*
Huijun Wang3* Wenhao Zhou1,3
Wenhao Zhou1,3Background: KCNQ2-related disorder is typically characterized as neonatal onset seizure and epileptic encephalopathy. The relationship between its phenotype and genotype is still elusive. This study aims to provide clinical features, management, and prognosis of patients with novel candidate variants of the KCNQ2 gene.
Methods: We enrolled patients with novel variants in the KCNQ2 gene from the China Neonatal Genomes Project between January 2018 and January 2021. All patients underwent next-generation sequencing tests and genetic data were analyzed by an in-house pipeline. The pathogenicity of variants was classified according to the guideline of the American College of Medical Genetics. Each case was evaluated by two geneticists back to back. Patients' information was acquired from clinical records.
Results: A total of 30 unrelated patients with novel variants in the KCNQ2 gene were identified, including 19 patients with single-nucleotide variants (SNVs) and 11 patients with copy number variants (CNVs). For the 19 SNVs, 12 missense variants and 7 truncating variants were identified. Of them, 36.8% (7/19) of the KCNQ2 variants were located in C-terminal regions, 15.7% (3/19) in segment S2, and 15.7% (3/19) in segment S4. Among them, 18 of 19 patients experienced seizures in the early neonatal period. However, one patient presented neurodevelopmental delay (NDD) as initial phenotype when he was 2 months old, and he had severe NDD when he was 3 years old. This patient did not present seizure but had abnormal electrographic background activity and brain imaging. Moreover, for the 11 patients with CNVs, 20q13.3 deletions involving EEF1A2, KCNQ2, and CHRNA4 genes were detected. All of them presented neonatal-onset seizures, responded to antiepileptic drugs, and had normal neurological development.
Conclusion: In this study, patients with novel KCNQ2 variants have variable phenotypes, whereas patients with 20q13.3 deletion involving EEF1A2, KCNQ2, and CHRNA4 genes tend to have normal neurological development.
KCNQ2 encodes the Kv7.2 subunit of potassium channels. It is located in the neuronal axon initial segment, which plays a critical role in spike initiation (Pan et al., 2006). In the Kcnq2-conditional knock-out mice model, the pyramidal neurons located in layer 2/3 (L2/3) were hyperactivated (Niday et al., 2017). Therefore, the KCNQ2 gene is essential for the regulation of neuronal excitability. In human beings, pathogenic variants in the KCNQ2 gene could cause benign neonatal seizures and epileptic encephalopathy. Seizure onset usually occurs in the neonatal period. The clinical features of KCNQ2-related disorders have a large spectrum of phenotypes, ranging from KCNQ2-related benign familial neonatal epilepsy (KCNQ2-BFNE) to KCNQ2-related neonatal epileptic encephalopathy (KCNQ2-NEE) (Numis et al., 2014). Other rare phenotypes, including myokymia, benign familial infantile seizures (BFIS), and infantile spasms, have also been reported in KCNQ2-related disorders. The studies reveal that the electroencephalogram (EEG) is characterized as burst-suppression and multifocal epileptic activity (Kato et al., 2013; Lee et al., 2021). Most patients do not present structural abnormality in brain imaging. However, some studies reveal that patients can have thin corpus callosum and abnormal signals in globus pallidus in magnetic resonance imaging (MRI) (Weckhuysen et al., 2012). Regarding management, the response to antiepileptic drugs (AED) also varies (Kuersten et al., 2020). Moreover, the prognostic spectrum is broad in KCNQ2-related disorders. Phenotype severity could range from seizure freedom spontaneously to mental developmental delay (Dalen Meurs-van der Schoor et al., 2014). With heterogeneous clinical features, treatment responses, and prognosis, researchers tried to investigate the relationship between the genotype and the phenotype. However, the clear correlation is unknown. In this study, we aim to explore novel candidate variants of KCNQ2 and provide the related clinical features, AED, and prognosis as well. This information can provide evidence on clinical management in patients with suspected KCNQ2-related disorders.
In this retrospective study, from January 2018 to January 2021, we enrolled patients with novel pathogenic or likely pathogenic variants of KCNQ2 or copy number variants (CNVs) covering the KCNQ2 gene from the China Neonatal Genomes Project (CNGP). All variants were classified according to the guideline of the American College of Medical Genetics (Richards et al., 2015) (Supplementary Table S1). These variants were checked in the Epilepsy Gene project (updated in July 2014; http://www.wzgenomics.cn/EpilepsyGene/), the RIKEE project (updated in December 2015; https://www.rikee.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), the Human Gene Mutation Database (HGMD, updated in November 2021, http://www.hgmd.cf.ac.uk).
Clinical data were extracted from medical records, including clinical features, MRI, or EEG findings, and follow-up information in the clinic. The last follow-up was performed by phone call if possible. The study was conducted following the Declaration of Helsinki (as revised in 2013). The Children's Hospital of Fudan University ethics committee approved this study since the study began (No. 2020-227). Pretest counseling was performed by physicians and geneticists. Informed consent was obtained from the patients' parents.
Sequences were generated using the Agilent ClearSeq Inherited Disease Kit, Illumina Cluster, and SBS Kit and performed on an Illumina HiSeq 2000/2500 platform. The detected variants were confirmed using polymerase chain reaction (PCR) and PCR-amplified DNA products, which were subjected to direct automated sequencing (3500XL Genetic Analyzer, Applied Biosystems). De novo variants were confirmed by parental evaluation via Sanger sequencing. We performed HMZDelFinder (Gambin et al., 2017) and CANOES (Backenroth et al., 2014) for the CNV detection. Each case was evaluated by two geneticists back to back. The annotation and filtrations of both SNVs and CNVs have been described in a published work (Dong et al., 2020).
From January 2018 to June 2021, we identified 30 patients with pathogenic variations in the KCNQ2 gene by the in-house pipeline, including 19 single-nucleotide variants (SNVs) and 11 CNVs. Among the 19 SNVs, one was classified as pathogenic variant, and 18 were likely pathogenic variants (Supplementary Table S1). These variants had not been reported with the detailed clinical phenotypes in the public database. Among them, 12 missense variants, four frameshift variants, two stop-gained variants, and one splicing variant were identified (Figure 1). Nine variants were confirmed as de novo variants by Sanger sequencing their parents (Table 1). We identified patient 8 with the variant of c.1623_1631+5del of the KCNQ2 gene. His father carried a 24% mosaic in his blood, without a seizure history or any neurological phenotype.
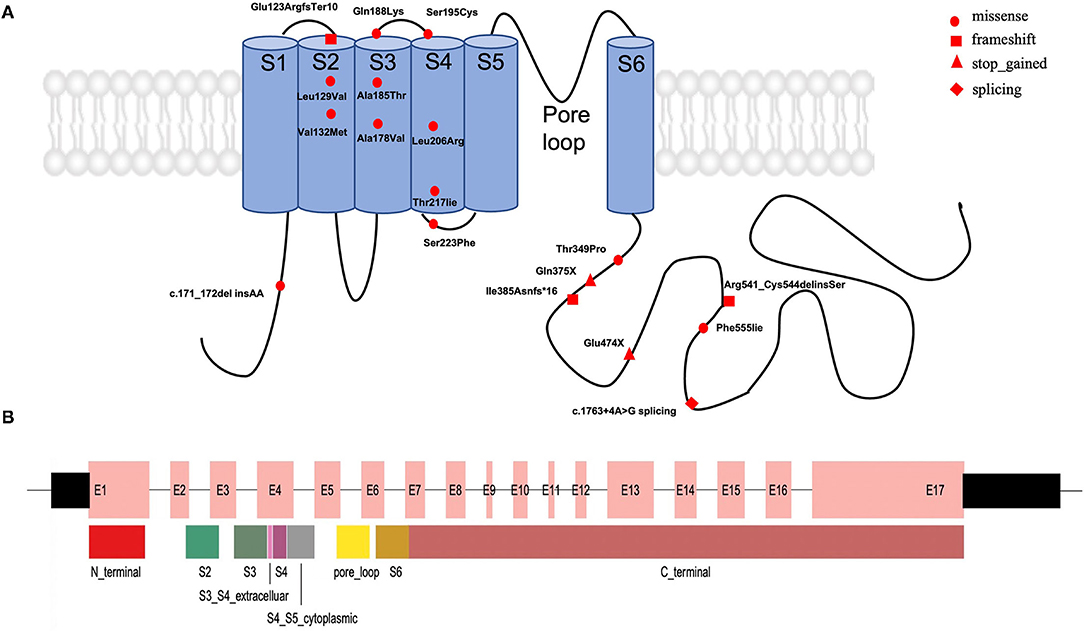
Figure 1. The distribution of the 19 novel variations in the KCNQ2 gene. (A) Approximate locations of the 19 novel KCNQ2 variants. KCNQ2 protein has six transmembrane domains (blue). The fourth segment acts as the voltage sensor, and the loop between the fifth and sixth domains forms the ion pore in the Kv7.2 potassium channel. (B) The distribution of exons and protein domain in the KCNQ2 gene. The wide box represents the coding region in the 17 exons. S1: segment S1, S2: segment S2, S3: segment S3, S4: segment S4, S5: segment S5, S6: segment S6, S3_S4_extracelluar: the extracellular region between segment S3 and segment S4; S4_S5_cytoplasmic: the cytoplasmic region between segment S4 and segment S5; pore_loop: the loop between the fifth and sixth domains forms the ion pore in a K7.2 potassium channel.
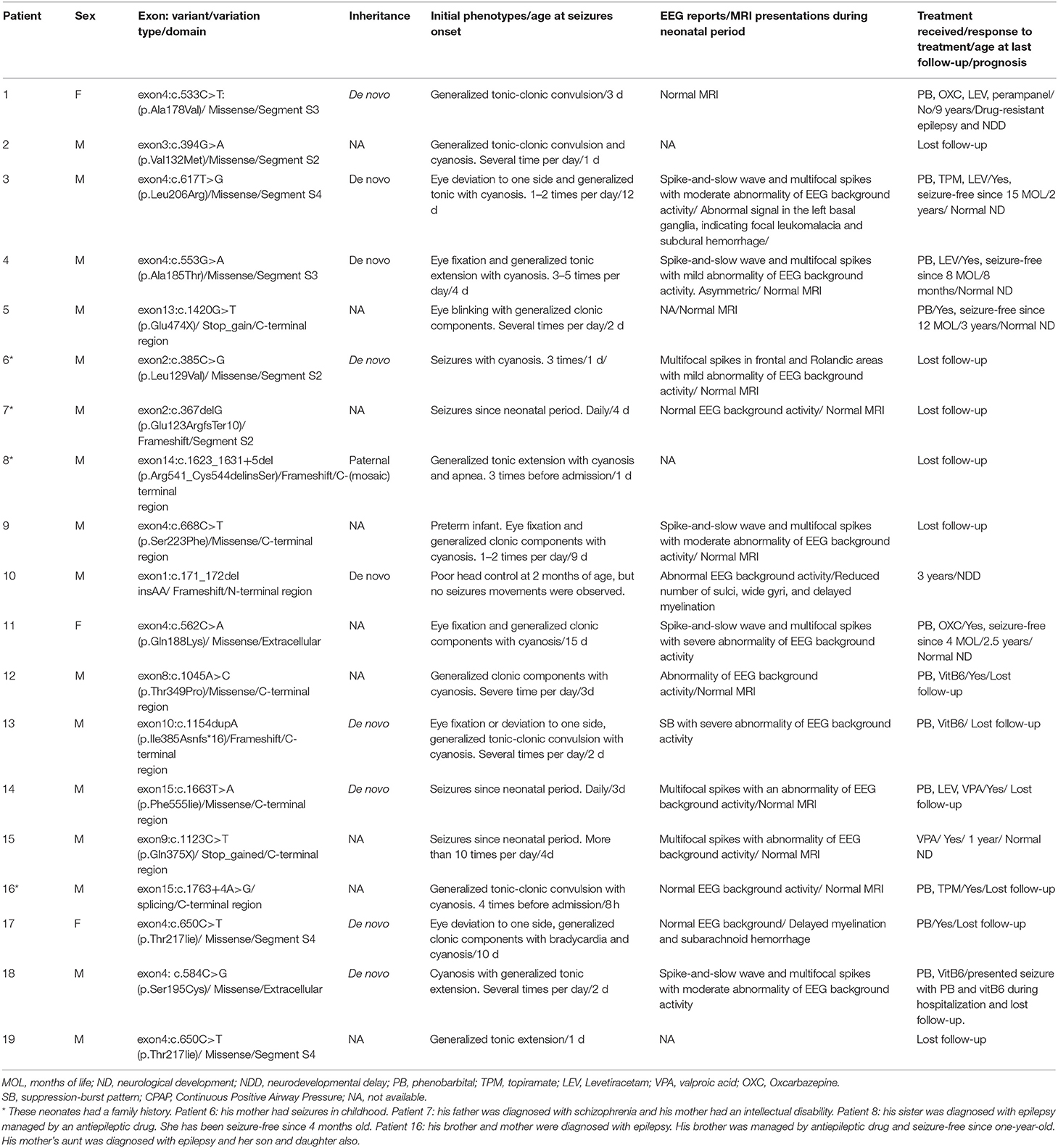
Table 1. Novel variants in KCNQ2 gene identified in 19 neonates with KCNQ2-related disorders.
For the 19 variants, 36.8% (7/19) of the KCNQ2 variants were located in C-terminal regions, 15.7% (3/19) in segment S2, and 15.7% (3/19) in segment S4. The variant of c.171_172delinsAA located in the N-terminal region, two variants (exon4:c.562C>A and exon4:c.584C>G) in the extracellular region, three variants (exon3:c.394G>A, exon2:c.385C>G, and exon2:c.367delG) in segment S2, two variants (exon4:c. 533C>T and exon4:c.553G>A) in segment S3, three variants (exon4:c.617T>G, exon4:c.650C>T; and exon4:c.650C>T) in segment S4, one variant (exon4:c.668C>T) in cytoplasmic domain between segment S4 and segment S5, and seven variants (exon13:c.1420G>T, exon14:c.1623_1631+5del, exon8:c.1045A>C, exon10c.1154dupA, exon15:c.1663T>A, exon9:c.1123C>T, and exon15:c.1763+4A>G) in C-terminal region (Figure 1).
We also detected 11 patients with 20q13.3 deletion. The size of deletion ranges from 59 kb to 1.8 Mb. In this region, three genes including KCNQ2, EEF1A2, and CHRNA4 were related to dominant epileptic encephalopathy, and KCNQ2 is the key gene. Seven patients had a continuous deletion of EEF1A2, KCNQ2, and CHRNA4 genes; three had a deletion of CHRNA4 and KCNQ2 genes; one had a deletion of KCNQ2 and EEF1A2 genes.
Seizures are the dominant and initial features (29/30, 96.7%) in this cohort (Tables 1, 2). The onset time of seizures ranged from 8 h of life to 15 days of life. The most common EEG finding is spike-and-slow wave and multifocal spikes with mild-to-severe abnormality of EEG background activity. Eight patients had positive MRI findings, showing abnormal signal in the left basal ganglia (patient 3), hypoplasia of the brain (patient 10), delayed myelination (patient 17), left ventricle enlargement (patient 20 and patient 21), an abnormal signal in the right frontal lobe (patient 24), and dilation of bilateral ventricles (patient 27 and patient 28). All patients with 20q13.3 deletion presented tonic seizures or tonic-clonic seizures during the neonatal period. Moreover, there were no significant different motor manifestations or imaging findings between the groups with SNVs and CNVs in the neonatal period.
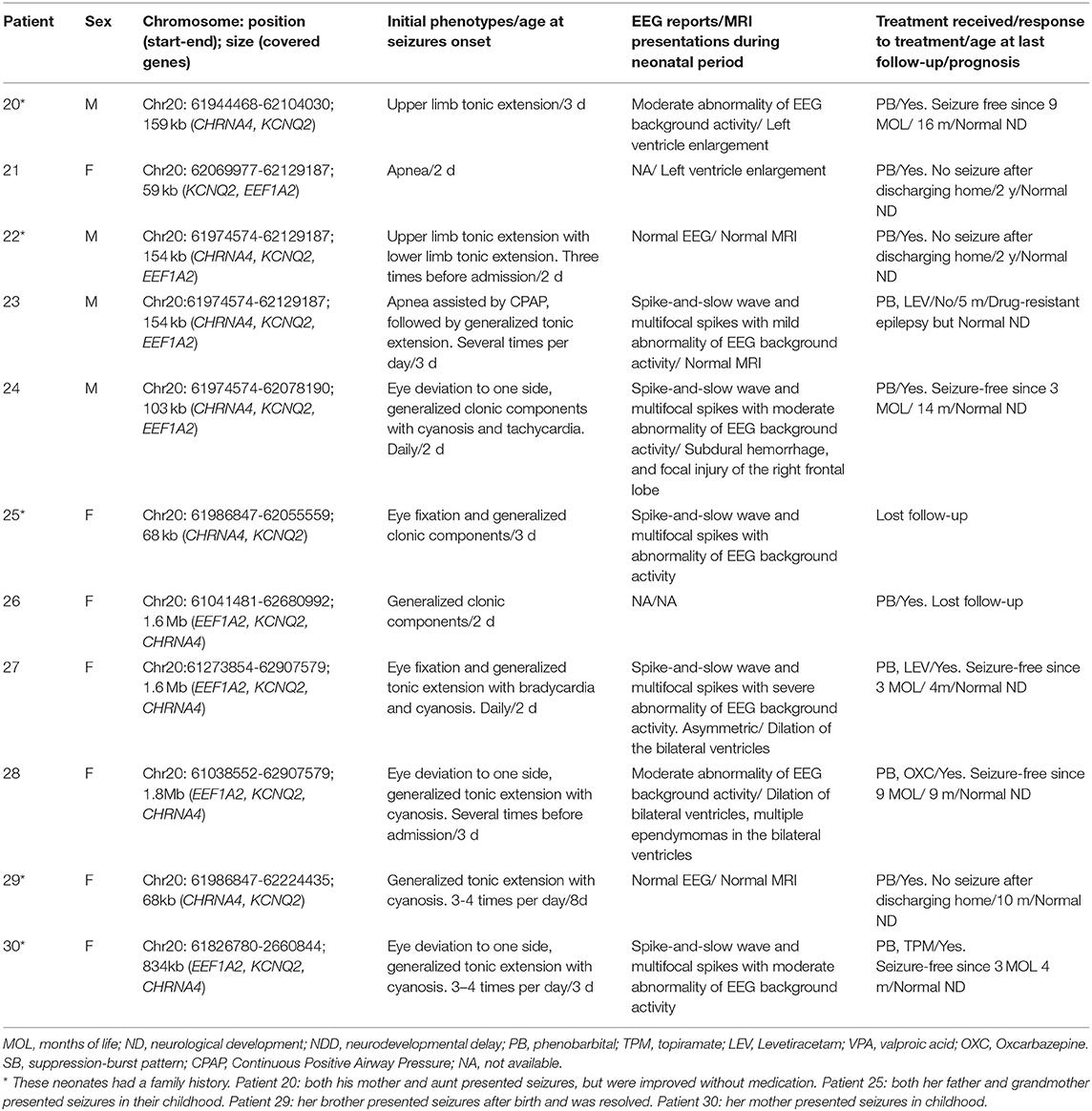
Table 2. Novel deletion in KCNQ2 gene identified in 11 neonates with KCNQ2-related disorders.
Among them, we found a male term patient (patient 10) presented with motor developmental delay as the initial phenotype when he was 2 months old. He was born uneventfully. He was diagnosed with pneumonia after birth and admitted to the neonatal department. He presented poor head control at 2 months of age, and he was referred to a local children's hospital. He could sit unsupported until 10 months old and was diagnosed with motor developmental delay. Cranial MRI showed a reduced number of sulci, wide gyri, and delayed myelination. The EEG finding is abnormal. As the EEG was performed after the onset of motor developmental delay disorder, whether the EEG was positive at the early stage was not available. The seizure and tremor phenotype of this patient is negative (information from his mother).
The overall prognosis was favorable for the patients with follow-up in the clinic. Among the 19 patients with SNVs, nine patients were responsive to AED and seizure-free by 2 years old, one patient (patient 1, segment S3, p.Ala178Val) had drug-resistant epilepsy, one patient (patient 10) did not present seizure, and eight patients lost follow-up. Among the 9 patients who were seizure-free, the regions where the variants were located included two segment S4 regions (patient 3, p.Leu206Arg; patient 17, p.Thr217Iie), 1 segment S3 (patient 4, p.Ala185Thr), one extracellular region (patient 11, p.Gln188Lys), and 5 C-terminal regions (patient 5, p.Glu474X; patient 12, p.Thr349Pro; patient 14, p.Phe555Iie; patient 15, p.Gln375X; and patient 16, exon15:c.1763+4A>G). Regarding the prognosis, two patients (patient 1 and patient 10) presented neurodevelopmental delay (NDD), nine patients had normal neurological development, and eight patients lost follow-up.
Among the 11 patients with 20q13.3 deletion, nine patients were responsive to AED, and eight of them were seizure-free by 2 years old. The remaining two patients, one (patient 23) had drug-resistant epilepsy, and one (patient 25) lost follow-up. Regarding the prognosis, one (patient 25) had NDD. Different from the variable outcomes of patients with SNVs, all patients with 20q13.3 deletions with available information had normal neurological development.
We report 30 unrelated patients with novel variants in the KCNQ2 gene, including 19 SNVs and 11 CNVs. For SNVs, missense was the most common mutation type (63.2%, 12/19), and 36.8% (7/19) of the KCNQ2 variants were located in C-terminal regions in our cohort. Mosaic parents in the KCNQ2 gene were reported in the literature, the mosaic state of asymptomatic parents is from 5% to 28% (Milh et al., 2015). One father with 30% mosaicism had a neurological phenotype (Weckhuysen et al., 2012). This information may indicate that mosaicism could not be ignored in epileptic encephalopathy. Furthermore, parental carrier testing should be considered regarding suffering. The next baby may still have a chance to inherit the pathogenic variant and will be affected.
In our study, all but one patient (patient 10 with the variant of c.171_172delinsAA) presented seizures in the neonatal period. Patient 10 presented motor developmental delay as an initial clinical feature. Ten EEG findings showed multifocal epileptiform with an abnormality of background activity. However, the clear phenotype–genotype correlation is unknown (Malerba et al., 2020). Previous studies indicated that KCNQ2 missense variants were associated with severe epilepsy phenotype and poor neurological outcomes because of dominant-negative effects (Orhan et al., 2014), whereas truncating variants were likely to be KCNQ2-BFNE (Soldovieri et al., 2007). Research suggested that the phenotype of patients was not only related to the mutation type but also associated with the affected regions of KCNQ2 (Goto et al., 2019). For example, missense variants in segment S6 and its nearby regions are likely to result in poor neurological outcomes (Goto et al., 2019). However, in our study, patient 1 with missense variant located in segment S3 had NDD, but patient 4 also with missense variant located in segment S3 had normal neurological development (aged 8 months old). Other patients with missense variants located in segment S4 also had normal neurological development. Therefore, the characteristics of pathogenic variants are still difficult to be linked to their clinical characteristics.
Consistent with previous studies, the patients who were responsive to AED could have variants located in segment S2 (Soldovieri et al., 2019), pore-loop domain (Weckhuysen et al., 2012; Pisano et al., 2015; Gomis-Perez et al., 2019), segment S4 (Weckhuysen et al., 2012; Pisano et al., 2015), segment S6 (Abidi et al., 2015; Pisano et al., 2015), C-terminal region (Weckhuysen et al., 2012; Pisano et al., 2015; Lee et al., 2017; Gomis-Perez et al., 2019), and extracellular region (Weckhuysen et al., 2012). Moreover, one patient (patient 4) with a variant located in segment S3 was responsive to AED and was seizure-free since he was 8 months old.
Neurodevelopmental delay often onset after seizure in KCNQ2-related disorders. In this study, we reported one patient had NDD as the initial phenotype. Then, the EEG was abnormal. No tremor or seizure was observed in this patient. This patient carried a de novo frameshift variant (c.171_172delinsAA). This variant was ranked as a likely pathogenic variant (Supplementary Table S1). Apart from NDD, the KCNQ2 gene is also related to autism (Millichap et al., 2016; Long et al., 2019). This study is reported in patients and was proved by the animal model (Kim et al., 2020). Therefore, the KCNQ2 gene may also be the candidate gene in patients with social behavior abnormalities in clinical genetic counseling.
The 20q13.3 microdeletion syndrome is characterized as seizure, brain abnormalities, NDD, and psychological problems (Pascual et al., 2013). There are variable phenotypes of 20q13.33 deletion (Kurahashi et al., 2009; Traylor et al., 2010; Mefford et al., 2012). The severe neurological phenotypes include learning disability, hyperlaxity, and strabismus (Béna et al., 2007). In this study, we reported a mild phenotype in 11 patients with 20q13.3 deletion involving EEF1A2, KCNQ2 and CHRNA4 genes. These clinical features are similar to BFNE caused by KCNQ2 variations and are different from those of autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE) caused by CHRNA4 variations (Steinlein et al., 1995) and developmental and epileptic encephalopathy 33 (DEE33) caused by EEF1A2 variations (Carvill et al., 2020). Moreover, the dosage sensitivity curations of the above three genes in the ClinGen (https://search.clinicalgenome.org/kb/gene-dosage?page=1&size=25&search=) suggested that KCNQ2 gene had sufficient evidence for haploinsufficiency and was ranked as the top 1 causative gene based on gnomAD pLI score and gnomAD predicted loss-of-function, whereas the other two genes were not yet evaluated. Therefore, the KCNQ2 gene is considered the causative gene of the patients with 20q13.3 deletions in our study.
Consistent with a previous study (Okumura et al., 2015), 20q13.3 deletions are restricted to just KCNQ2 and CHRNA4 genes are likely to result in KCNQ2-BFNE, and one case with 20q13.3 deletion involving EEF1A2, KCNQ2, and CHRNA4 had normal psychomotor development (Okumura et al., 2015). However, the studies indicated that patients with NDD had a larger deletion of the KCNQ2 gene (Kurahashi et al., 2009; Traylor et al., 2010; Mefford et al., 2012; Pascual et al., 2013; Okumura et al., 2015). Patient 27 and patient 28 had a large deletion (>1 Mb). They were seizure-free and had normal neurological development. However, both of them were <1 year old at the last visit. Therefore, long-term follow-up will be necessary to determine precise phenotypes. These patients had a milder phenotype than some patients with one single-nucleotide KCNQ2 pathogenic variant. The underlying reason is elusive and needs to be investigated.
Our study has limitations. The follow-up information was absent in some patients because patients did not present for follow-up in the clinic consistently. Therefore, we cannot diagnose the KCNQ2-BFNE or KCNQ2-NEE in some patients according to the current information. The EEG and MRI findings were not available because some patients were enrolled from other hospitals, and they could not perform EEG or MRI. Third, our study ended in January 2021. Some patients were <1 year old. Therefore, it will be essential to follow these families up to assess neurological development.
In conclusion, we reported 30 unrelated patients with novel variations in the KCNQ2 gene, including SNVs and CNVs. The clinical features and prognosis are heterogeneous in patients with SNVs. However, patients with 20q13.3 deletions restricted to KCNQ2, CHRNA4, and EEF1A2 genes have similar to the phenotypes of BFNE. These findings could assist clinicians in diagnosing and predicting the prognosis of KCNQ2-related disorders.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Children's Hospital of Fudan University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
TX, XC, YZ, HW, and WZ: conception and design. HW and WZ: administrative support. TX, XC, YX, LY, BW, LC, LL, DZ, and DC: provision of study materials or patients. TX, XC, HC, XD, and HW: collection and assembly of data. XC, XD, and LY: data analysis and interpretation. All authors: manuscript writing and final approval of manuscript.
The work was funded by the Clinical Research Plan of SHDC (No. SHDC2020CR4085).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors sincerely thank all the family members for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.809810/full#supplementary-material
Abidi, A., Devaux, J. J., Molinari, F., Alcaraz, G., Michon, F. X., Sutera-Sardo, J., et al. (2015). A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol Dis. 80, 80–92. doi: 10.1016/j.nbd.2015.04.017
Backenroth, D., Homsy, J., Murillo, L. R., Glessner, J., Lin, E., Brueckner, M., et al. (2014). CANOES: detecting rare copy number variants from whole exome sequencing data. Nucleic Acids Res. 42, e97. doi: 10.1093/nar/gku345
Béna, F., Bottani, A., Marcelli, F., Sizonenko, L. D., Conrad, B., and Dahoun, S. (2007). A de novo 1.1-1.6 Mb subtelomeric deletion of chromosome 20q13.33 in a patient with learning difficulties but without obvious dysmorphic features. Am. J. Med. Genet. A. 143a, 1894–1899. doi: 10.1002/ajmg.a.31789
Carvill, G. L., Helbig, K. L., Myers, C. T., Scala, M., Huether, R., Lewis, S., et al. (2020). Damaging de novo missense variants in EEF1A2 lead to a developmental and degenerative epileptic-dyskinetic encephalopathy. Hum Mutat 41, 1263–1279. doi: 10.1002/humu.24015
Dalen Meurs-van der Schoor, C., van Weissenbruch, M., van Kempen, M., Bugiani, M., Aronica, E., Ronner, H., et al. (2014). Severe Neonatal Epileptic Encephalopathy and KCNQ2 Mutation: Neuropathological Substrate? Front Pediatr 2, 136. doi: 10.3389/fped.2014.00136
Dong, X., Liu, B., Yang, L., Wang, H., Wu, B., Liu, R., et al. (2020). Clinical exome sequencing as the first-tier test for diagnosing developmental disorders covering both CNV and SNV: a Chinese cohort. J. Med. Genet. 57, 558–566. doi: 10.1136/jmedgenet-2019-106377
Gambin, T., Akdemir, Z. C., Yuan, B., Gu, S., Chiang, T., Carvalho, C. M. B., et al. (2017). Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 45, 1633–1648. doi: 10.1093/nar/gkw1237
Gomis-Perez, C., Urrutia, J., Marce-Grau, A., Malo, C., Lopez-Laso, E., Felipe-Rucian, A., et al. (2019). Homomeric Kv7.2 current suppression is a common feature in KCNQ2 epileptic encephalopathy. Epilepsia. 60, 139–148. doi: 10.1111/epi.14609
Goto, A., Ishii, A., Shibata, M., Ihara, Y., Cooper, E. C., and Hirose, S. (2019). Characteristics of KCNQ2 variants causing either benign neonatal epilepsy or developmental and epileptic encephalopathy. Epilepsia. 60, 1870–1880. doi: 10.1111/epi.16314
Kato, M., Yamagata, T., Kubota, M., Arai, H., Yamashita, S., Nakagawa, T., et al. (2013). Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. 54, 1282–1287. doi: 10.1111/epi.12200
Kim, E. C., Patel, J., Zhang, J., Soh, H., Rhodes, J. S., Tzingounis, A. V., et al. (2020). Heterozygous loss of epilepsy gene KCNQ2 alters social, repetitive and exploratory behaviors. Genes. Brain Behav. 19, e12599. doi: 10.1111/gbb.12599
Kuersten, M., Tacke, M., Gerstl, L., Hoelz, H., Stülpnagel, C. V., and Borggraefe, I. (2020). Antiepileptic therapy approaches in KCNQ2 related epilepsy: A systematic review. Eur. J. Med Genet. 63, 103628. doi: 10.1016/j.ejmg.2019.02.001
Kurahashi, H., Wang, J. W., Ishii, A., Kojima, T., Wakai, S., Kizawa, T., et al. (2009). Deletions involving both KCNQ2 and CHRNA4 present with benign familial neonatal seizures. Neurology. 73, 1214–1217. doi: 10.1212/WNL.0b013e3181bc0158
Lee, I. C., Chang, M. Y., Liang, J. S., and Chang, T. M. (2021). Ictal and interictal electroencephalographic findings can contribute to early diagnosis and prompt treatment in KCNQ2-associated epileptic encephalopathy. J. Formos Med. Assoc. 120(1 Pt 3), 744–754. doi: 10.1016/j.jfma.2020.08.014
Lee, I. C., Yang, J. J., Liang, J. S., Chang, T. M., and Li, S. Y. (2017). KCNQ2-Associated Neonatal Epilepsy: Phenotype Might Correlate With Genotype. J. Child Neurol. 32, 704–711. doi: 10.1177/0883073817701873
Long, S., Zhou, H., Li, S., Wang, T., Ma, Y., Li, C., et al. (2019). The Clinical and Genetic Features of Co-occurring Epilepsy and Autism Spectrum Disorder in Chinese Children. Front Neurol 10, 505. doi: 10.3389/fneur.2019.00505
Malerba, F., Alberini, G., Balagura, G., Marchese, F., Amadori, E., Riva, A., et al. (2020). Genotype-phenotype correlations in patients with de novo KCNQ2 pathogenic variants. Neurol Genet. 6, e528. doi: 10.1212/NXG.0000000000000528
Mefford, H. C., Cook, J., and Gospe, S. M. Jr. (2012). Epilepsy due to 20q13.33 subtelomere deletion masquerading as pyridoxine-dependent epilepsy. Am. J. Med Genet. A. 158A, 3190–3195. doi: 10.1002/ajmg.a.35633
Milh, M., Lacoste, C., Cacciagli, P., Abidi, A., Sutera-Sardo, J., Tzelepis, I., et al. (2015). Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am. J. Med. Genet. A 167A, 2314–2318. doi: 10.1002/ajmg.a.37152
Millichap, J. J., Park, K. L., Tsuchida, T., Ben-Zeev, B., Carmant, L., Flamini, R., et al. (2016). KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2, e96. doi: 10.1212/NXG.0000000000000096
Niday, Z., Hawkins, V. E., Soh, H., Mulkey, D. K., and Tzingounis, A. V. (2017). Epilepsy-Associated KCNQ2 Channels Regulate Multiple Intrinsic Properties of Layer 2/3 Pyramidal Neurons. J. Neurosci. 37, 576–586. doi: 10.1523/JNEUROSCI.1425-16.2016
Numis, A. L., Angriman, M., Sullivan, J. E., Lewis, A. J., Striano, P., Nabbout, R., et al. (2014). KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology. 82, 368–370. doi: 10.1212/WNL.0000000000000060
Okumura, A., Atsushi, I., Shimojima, K., Kurahashi, H., Yoshitomi, S., lmai, K., et al. (2015). Phenotypes of children with 20q13.3 microdeletion affecting KCNQ2 and CHRNA4. Epileptic Disord. 17, 165–171. doi: 10.1684/epd.2015.0746
Orhan, G., Bock, M., Schepers, D., Ilina, E. I., Reichel, S. N., Loffler, H., et al. (2014). Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 75, 382–394. doi: 10.1002/ana.24080
Pan, Z., Kao, T., Horvath, Z., Lemos, J., Sul, J. Y., Cranstoun, S. D., et al. (2006). A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosc.i 26, 2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006
Pascual, F. T., Wierenga, K. J., and Ng, Y. T. (2013). Contiguous deletion of KCNQ2 and CHRNA4 may cause a different disorder from benign familial neonatal seizures. Epilepsy Behav. Case Rep. 1, 35–38. doi: 10.1016/j.ebcr.2013.01.004
Pisano, T., Numis, A. L., Heavin, S. B., Weckhuysen, S., Angriman, M., Suls, A., et al. (2015). Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. 56, 685–691. doi: 10.1111/epi.12984
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Soldovieri, M. V., Ambrosino, P., Mosca, I., Miceli, F., Franco, C., Canzoniero, L. M. T., et al. (2019). Epileptic encephalopathy in a patient with a novel variant In The Kv7.2 S2 transmembrane segment: clinical, genetic, and functional features. Int. J. Mol. Sci. 20. doi: 10.3390/ijms20143382
Soldovieri, M. V., Miceli, F., Bellini, G., Coppola, G., Pascotto, A., and Taglialatela, M. (2007). Correlating the clinical and genetic features of benign familial neonatal seizures (BFNS) with the functional consequences of underlying mutations. Channels (Austin). 1, 228–233. doi: 10.4161/chan.4823
Steinlein, O. K., Mulley, J. C., Propping, P., Wallace, R. H., Phillips, H. A., Sutherland, G. R., et al. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 11, 201–203. doi: 10.1038/ng1095-201
Traylor, R. N., Bruno, D. L., Burgess, T., Wildin, R., Spencer, A., Ganesamoorthy, D., et al. (2010). A genotype-first approach for the molecular and clinical characterization of uncommon de novo microdeletion of 20q13.33. PLoS ONE. 5, e12462. doi: 10.1371/journal.pone.0012462
Keywords: KCNQ2, Kv7.2, newborn, epilepsy, epileptic encephalopathy
Citation: Xiao T, Chen X, Xu Y, Chen H, Dong X, Yang L, Wu B, Chen L, Li L, Zhuang D, Chen D, Zhou Y, Wang H and Zhou W (2022) Clinical Study of 30 Novel KCNQ2 Variants/Deletions in KCNQ2-Related Disorders. Front. Mol. Neurosci. 15:809810. doi: 10.3389/fnmol.2022.809810
Received: 05 November 2021; Accepted: 02 March 2022;
Published: 26 April 2022.
Edited by:
Jing Peng, Central South University, ChinaReviewed by:
Weiping Liao, Second Affiliated Hospital of Guangzhou Medical University, ChinaCopyright © 2022 Xiao, Chen, Xu, Chen, Dong, Yang, Wu, Chen, Li, Zhuang, Chen, Zhou, Wang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanfeng Zhou, eXVhbmZlbmd6aG91OTlAMTYzLmNvbQ==; Huijun Wang, aHVpanVud2FuZ0BmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.