 Dongfang Zou1,2,3†
Dongfang Zou1,2,3† Bing Qin4†
Bing Qin4† Jie Wang2,3
Jie Wang2,3 Yiwu Shi2,3
Yiwu Shi2,3 Peng Zhou2,3
Peng Zhou2,3 Yonghong Yi2,3
Yonghong Yi2,3 Jianxiang Liao1*
Jianxiang Liao1* Xinguo Lu1*
Xinguo Lu1*
- 1Epilepsy Center and Department of Neurology, Shenzhen Children’s Hospital, Shenzhen, China
- 2Department of Neurology, Institute of Neuroscience, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 3Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, Guangzhou, China
- 4Epilepsy Center and Department of Neurosurgery, The First Affiliated Hospital of Jinan University, Guangzhou, China
Objective: AFF2 mutations were associated with X-linked intellectual developmental disorder-109 and in males with autism spectrum disorder (ASD). The relationship between AFF2 and epilepsy has not been defined.
Method: Trios-based whole-exome sequencing was performed in a cohort of 372 unrelated cases (families) with partial (focal) epilepsy without acquired causes.
Results: Five hemizygous missense AFF2 mutations were identified in five males with partial epilepsy and antecedent febrile seizures without intellectual disability or other developmental abnormalities. The mutations did not present in the controls of general populations with an aggregate frequency significantly higher than that in the control populations. Previously, intellectual disability-associated AFF2 mutations were genomic rearrangements and CCG repeat expansion mutations mostly, whereas the mutations associated with partial epilepsy were all missense. Missense AFF2 mutations associated with epilepsy fell into the regions from N-terminal to the nuclear localization signal 1 (NLS1), while ASD-associated missense mutations fell in the regions from NLS1 to C-terminal.
Conclusion: AFF2 is potentially a candidate causative gene of X-link partial epilepsy with antecedent febrile seizures. The genotype–phenotype correlation and molecular sub-regional effect of AFF2 help in explaining the mechanisms underlying phenotypic variations.
Introduction
AFF2 (OMIM* 300806) (also known as FMR2 gene), which encodes AF4/FMR2 family member 2, is a transcriptional factor and RNA-binding protein that plays an important role in transcriptional regulation, RNA splicing, mRNA processing, and nuclear speckle organization (Bensaid et al., 2009). AFF2 is highly conserved and abundantly expressed in human brain (Gecz et al., 1997), being essential for brain development. Homozygous AFF2 knock-out mice showed abnormal central nervous system synaptic transmission, abnormal excitatory postsynaptic potential, and premature death.1 Previous studies have identified AFF2 mutations in the etiology of X-linked intellectual developmental disorder-109 (MRX109), a form of mildly to moderately impaired intellectual development associated with learning difficulties, communication deficits, attention problems, hyperactivity, and autistic behavior (Knight et al., 1993, 1996; Mulley et al., 1995; Gecz, 2000), in which epileptic seizures were occasionally observed (Lo Nigro et al., 2000; Lesca et al., 2003). Several point mutations were also identified in patients with autism spectrum disorder (ASD) (Mondal et al., 2012; Jiang et al., 2013; Yuen et al., 2017). The relationship between AFF2 and epilepsy has not been defined. In the present study, trios-based whole-exome sequencing (WES) was performed in a cohort of cases (families) of partial (focal) epilepsy without acquired causes. Five missense mutations of AFF2 were identified in five unrelated individuals of partial epilepsy without intellectual disability or other developmental abnormalities.
Materials and Methods
Participants
A total of 372 cases (families) with partial epilepsy without acquired causes were recruited, including 323 cases from the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University in China between January 2013 and July 2020, and 49 cases from Shenzhen Children’s Hospital and the First Affiliated Hospital of Jinan University in China between January 2018 and July 2020. The probands included 230 males and 142 females and were subjected to trios-based WES for potential genetic etiology of epilepsy. The complete pedigree and clinical data of the probands were collected, including detailed clinical phenotypes, age of seizure onset, seizure type, seizure course and frequency, family history, treatment, prognosis, general and neurological examination, and brain magnetic resonance imaging (MRI). 24-h video electroencephalography (EEG) was performed on all patients, at the age ranging from 4 to 23 years (mean age, 10.2 years), and EEG was analyzed by at least two qualified electroencephalographers. The developmental and intellectual states of all patients (at the age range, 3–24 years; mean age, 10.4 years) were evaluated, including language, fine and gross motor, and adaptive social skills and performance at school or work. Gesell development scale, Wechsler intelligence scale for children-V, and Wechsler adult intelligence scale were utilized in the neuropsychological evaluation of the participants according to their ages. Seizure type and epilepsy syndrome were classified according to the criteria of the Commission on Classification and Terminology of International League of Against Epilepsy (ILAE) (No Author, 1985, 1989; Berg et al., 2010; Scheffer et al., 2017). The inclusion criteria were: (1) diagnosis of partial epilepsy based on ILAE criteria, characterized by focal seizures or focally originated focal to bilateral tonic-clonic seizures; (2) EEG examination presented focal discharges, including unilateral, bilateral, and multiple focal discharges with normal background. The exclusion criteria were: (1) diagnosis of generalized epilepsy based on ILAE criteria; (2) individuals with acquired causes, including brain tumors, head trauma, immune encephalitis, central nervous system infections, and cerebrovascular diseases.
This study was based on the guidelines of the International Committee of Medical Journal Editors concerning patient consent for research or participation. Written informed consent was obtained from all individuals or their legal guardians. The present study was approved by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University.
Whole Exome Sequencing
Blood samples were obtained from all individuals and their parents. Genomic DNA was extracted from the peripheral blood using Qiagen Flexi Gene DNA kit (Qiagen, Hilden, Germany), according to the protocol of the manufacturer. Trio-based WES was conducted on the MGI 2000 platform by BGI-Shenzhen (Shenzhen, China) performing pair-end reads, 100 bp sequencing with 100–150 times average depth and more than 98% coverage of the target region. Deep sequencing data were aligned to the reference GRCh37 build (hg19) and variants were called according to the standard procedures as previously reported (Wang et al., 2018; Cai et al., 2019; Shi et al., 2019). We adopted a case-by-case analytical approach to identify candidate causative mutations in each trio. Firstly, we prioritized the rare variants with a minor allele frequency < 0.005 in the 1,000 Genomes Projects, Exome Aggregation Consortium, and Genome Aggregation Database (gnomAD). Secondly, we retained potentially pathogenic mutations, including frameshift, nonsense, canonical splice site, initiation codon, and missense mutations predicted as being damaging by 21 algorithms in silico prediction.2 Finally and importantly, potential disease-causing variants were screened under five models: (1) epilepsy-associated gene model; (2) de novo autosomal dominant model; (3) autosomal recessive inheritance model, including compound heterozygous and homozygous variants; (4) X-linked inheritance model; (5) co-segregation model. To identify novel epilepsy-associated genes, we put the known epilepsy-associated genes (Wang et al., 2017) aside. The genes with repetitively identified de novo variants, bi-allelic variants, hemizygous variants, and variants with segregations, were selected for further studies to define the gene-disease association. AFF2 appeared as a candidate gene with recurrent hemizygous variants in this cohort of partial epilepsy and was subjected to further analysis in this study. The other potential candidate genes were not included in the present study. The candidate pathogenic variants were validated by Sanger sequencing. All variants in AFF2 were annotated to the reference transcript NM_002025.4. Conservation of the mutated positions was evaluated by generating multiple sequence alignments of different species.
Molecular and Genotype-Phenotype Correlation Analysis
To predict the effect of missense mutations on molecular structure, a protein model was established by Phyre2 (V 2.0).3 PyMOL 1.7 was used to visualize and analyze the three-dimensional protein structure and alteration of hydrogen bonds. Amino acids were annotated to the reference protein NP_002016. To evaluate the genotype-phenotype correlation, we reviewed all the relevant literature about AFF2 mutations reported previously, including point mutations, genomic rearrangements, and CCG repeat expansion variants, as well as all the phenotypes of the mutations. All the mutations were retrieved from PubMed4 and Human Gene Mutation Database5 up to December 2020.
Statistical Analysis
R statistical software (v3.4.1) and SPSS version 22.0 (SPSS, Inc., Chicago, IL, United States) were used for statistical analyses. Fisher’s exact test was applied to access the frequencies of AFF2 mutations in the case cohort and the control populations. A p-value < 0.05 was considered statistically significant.
Results
Identification of AFF2 Mutations
Five hemizygous missense AFF2 variants were identified in five unrelated individuals with partial epilepsy, including c.230A > T/p.N77I, c.391C > T/p.H131Y, c.1540C > T/p.R514C, c.2009G > A/p.R670H, and c.2074C > G/p.P692A. All variants originated from their asymptomatic mothers (Figures 1A,B and Table 1). No AFF2 variants were found in their fathers. The amino acid sequence alignment indicated that N77I, R514C, and R670H were located at residuals of highly conserved across various species (“highly conserved” means the fraction of the AFF2 residues conserved for the used species is 100%); while H131Y and P692A were located at residuals of less conserved according to the sequence alignment (“less conserved” means the fraction is less than 100%) (Figure 1C). None of the five cases was identified to have other pathogenic or likely pathogenic variants in the known genes associated with seizure disorders (Wang et al., 2017), neither other pathogenic nor likely pathogenic variants were found in the de novo autosomal dominant, autosomal recessive inheritance, and co-segregation model.
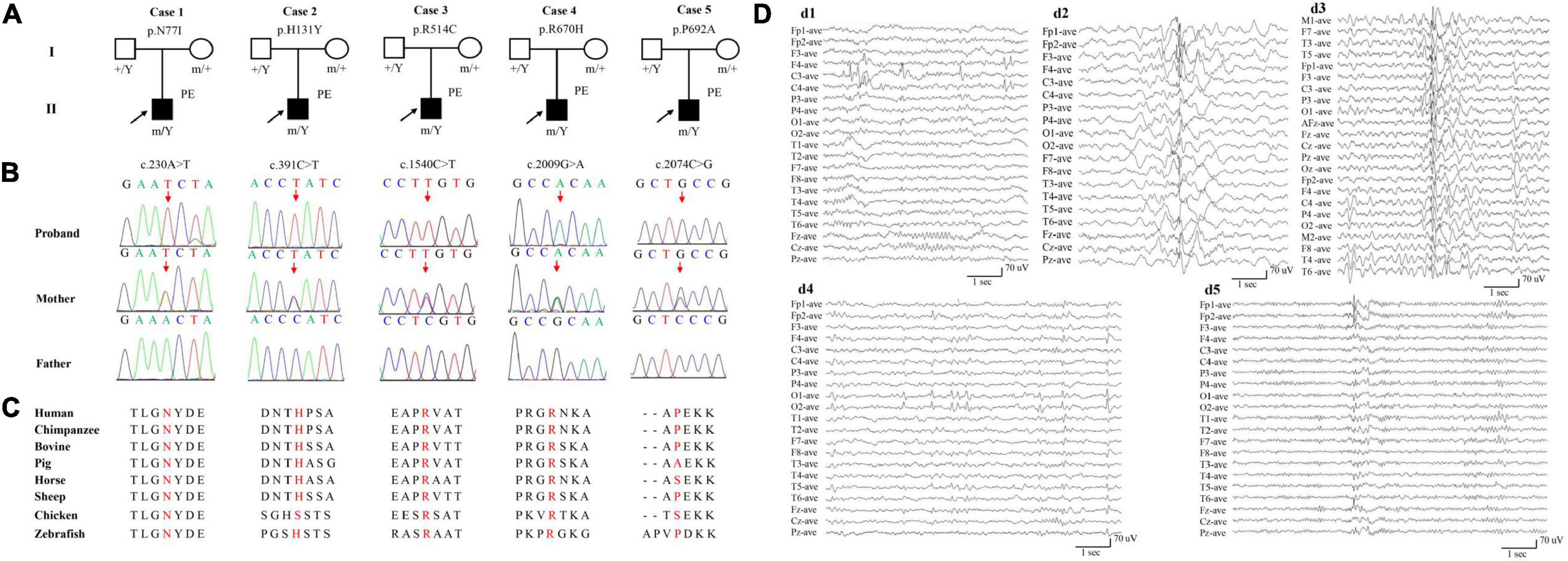
Figure 1. Genetic and electroencephalograms (EEG) of the cases with AFF2 mutations. (A) Pedigrees of the five cases with AFF2 mutations and their corresponding phenotypes. PE, partial epilepsy. (B) DNA sequence chromatogram of the AFF2 mutations. Arrows indicate the positions of the mutations. (C) The amino acid sequence alignment of the five missense mutations shows that residues N77I, R514C, and R670H were highly conserved across various species. Residues H131Y and P692A were less conserved. (D) Changes of interictal EEG in the cases with AFF2 mutations. (d1) Interictal EEG of case 1 showed bilateral frontal-central spike and slow waves (obtained at the age of 8 years). (d2) Interictal EEG of case 2 showed bilateral frontal and anterior-temporal spike and slow waves (at the age of 9 years). (d3) Interictal EEG of case 3 showed spike and slow waves predominant at left hemisphere (at the age of 5 years). (d4) Interictal EEG of case 4 showed sharp waves predominant at bilateral occipital regions (at the age of 3 years). (d5) Interictal EEG of case 5 showed spike and slow waves predominant at bilateral frontal regions (at the age of 17 years).
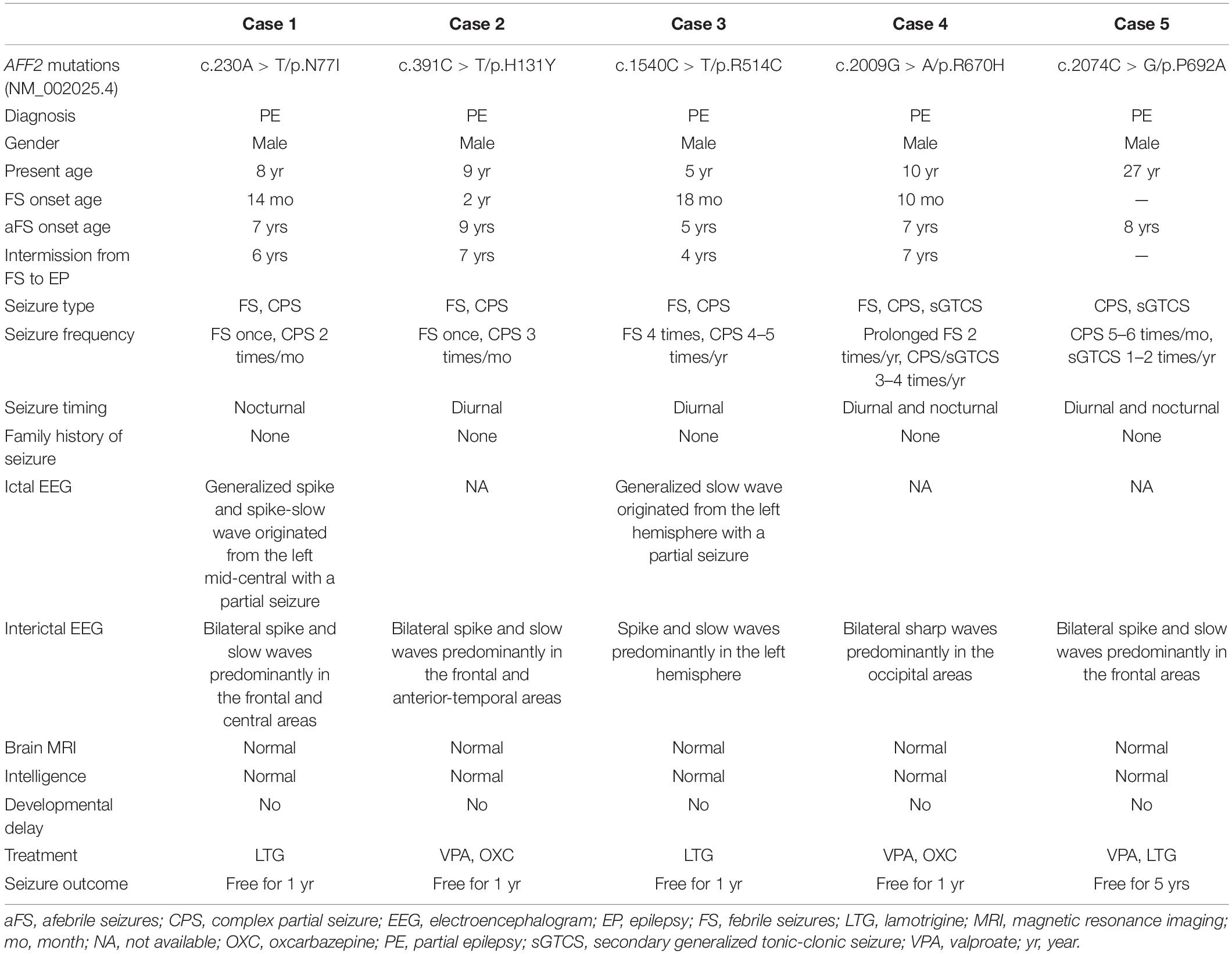
Table 1. Clinical features of the cases with AFF2 mutations.
We compared the aggregate frequency of the five hemizygous AFF2 mutations in this cohort with that in the populations in gnomAD, or controls of gnomAD, according to the aggregate variant analysis method suggested by ClinGen (Strande et al., 2017). Five mutant alleles in a total of 230 alleles (230 males in the 372 cases) were detected in this cohort. Mutation R514C had an extremely low frequency (9.95e-06) but was not present in the controls of gnomAD-East Asian and gnomAD-all population, part of the allele frequency was from the 1000G data. The other four mutations in AFF2 had no allele frequency in gnomAD databases (Table 2). The differences in aggregate frequencies of the mutant alleles between this cohort in male cases and the male controls in gnomAD (general population, East-Asian population) were statistically significant (5/230 vs. 0/46329 in the male controls of gnomAD-all population, p = 2.82 × 10–12; 5/230 vs. 0/3058 in the male controls of gnomAD-East Asian population, p = 1.61 × 10–6, respectively) (Table 2).

Table 2. Analysis of the aggregate frequency of AFF2 mutations identified in this study.
Clinical Features of the Cases With AFF2 Mutations
AFF2 mutations were identified in five unrelated male epilepsy patients with focal seizures and/or focal discharges, ranging in age from 5 to 27 years. The clinical features of the five cases were summarized in Table 1. The age of febrile seizures onset ranged from 10 months to 2 years, and the afebrile seizures started from 5 to 9 years. All the cases presented with infrequent focal impaired awareness seizures, and the cases with mutation R670H (Case 4) and P692A (Case 5) also had focal to bilateral tonic-clonic seizures. Except for the case with mutation P692A (Case 5), the other four cases exhibited antecedent febrile seizures or prolonged febrile seizures, and they developed afebrile seizures later with an intermission of 4–7 years. The interictal EEG recording showed epileptiform discharges, including bilateral, unilateral, and multiple discharges, predominantly at frontal, central, and temporal lobe, predominantly during sleeping. Trends of generalization were also observed in the case with mutation R514C (Case 3) (Figure 1D). The ictal EEG of the case with mutation N77I (Case 1) showed an onset originated from the left mid-central area, and the case with mutation R514C (Case 3) demonstrated a seizure originated from the left hemisphere. Brain MRIs of all the cases were normal. According to the results of the relevant developmental scales, all individuals presented normal development and intellectual ability. The epilepsy of the five cases was controlled with one or two antiepileptic drugs. The cases with mutation N77I (Case 1) and R514C (Case 3) achieved seizure free with monotherapy of lamotrigine (6–7 mg/kg/d). The cases with H131Y (Case 2) and R670H (Case 4) had seizure reductions with valproate (20–25 mg/kg/d) at first and became seizure free after the add-on of oxcarbazepine (20–25 mg/kg/d). The case with P692A (Case 5) had seizure reduction of more than 60% with initial valproate (20 mg/kg/d) and got seizures controlled on the combined use of lamotrigine (5 mg/kg/d).
Molecular Effect of AFF2 Mutations
The length of amino acid of FMR2 is 1311, including two bipartite NLSs (nuclear localization signals)–NLS1 (RKEPRPNIPLAPEKKK) at amino acid position 681–696 and NLS2 (KPAPKGKRKHKPIEVAEKIPEKK) at position 846–868. Additionally, a large serine-rich domain locates at protein position 481–502, which overlaps with the regions involved in transcription activation, and a large alanine-threonine-rich domain is at position 979–1032, being unique to the FMR2 (Gecz et al., 1997). N-terminal domain of the protein includes amino acids 1–541, which is known to have transactivation activity (Hillman and Gecz, 2001), whereas C-terminal domain comprises amino acids 633–1272, being responsible for localization of the protein in nuclear speckles (Bensaid et al., 2009). In the present study, mutations N77I, H131Y, and R514C were located in the N-terminal domain. P692A was located in the NLS1 domain, while R670H was located in the N-terminal frank of NLS1 (Figure 2A).
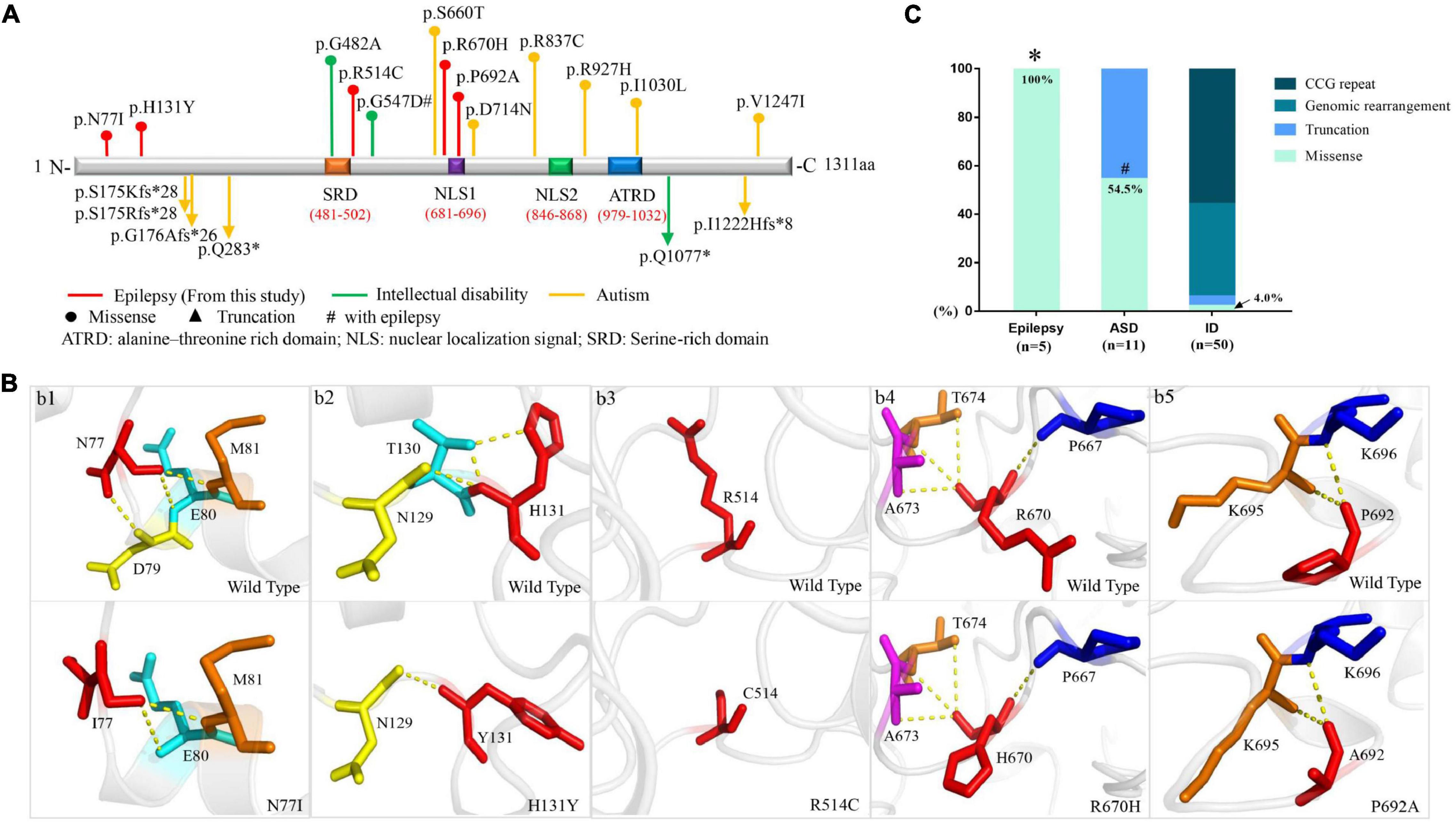
Figure 2. Schematic presentation of FMR2 structure and genotype-phenotype correlation of AFF2. (A) Schematic diagram of missense and destructive AFF2 mutations and their locations on FMR2 protein. Missense mutations were shown at the top of the structural diagram. Destructive mutations were shown at the bottom. (B) Schematic illustration of the changes in hydrogen bonds. The residues where the mutations occurred are shown as red rods. The hydrogen bonds are shown as yellow spheres. (C) Genotypes of AFF2 in epilepsy, autism spectrum disorder (ASD), and intellectual disability (ID). *The proportion of missense mutations in epilepsy is significantly higher than that in ID (p < 0.001) through Fisher’s exact test. #The proportion of missense mutations in ASD is significantly higher than that in ID (p < 0.001) through Fisher’s exact test.
The molecular effect of the five missense mutations was analyzed by protein modeling using PHYRE26 and PyMOL with the templates available (Figure 2B). The protein modeling showed that mutations N77I and H131Y resulted in alteration of hydrogen bonds and potentially affected the protein steric configuration. Originally, residue N77 formed three hydrogen bonds with residue D79, E80, and M81, respectively. When asparagine was replaced by isoleucine at residue N77, the hydrogen bond with D79 was destroyed (Figure 2Bb1). Residue H131 formed two hydrogen bonds with residue T130 and one hydrogen bond with residue N129. When histidine was replaced by tyrosine at residue H131, the hydrogen bonds with residue T130 were destroyed (Figure 2Bb2). There was no hydrogen bond formed with other surrounding residues at residue C514 (Figure 2Bb3). Mutation R670H and P692A showed no hydrogen bond altered at the residues (Figures 2Bb4,b5).
Genotype-Phenotype Correlation and Molecular Sub-Regional Implication
We reviewed the previous studies through PubMed and Human Gene Mutation Database and analyzed the relationship between genotype and phenotype of AFF2 mutations. Previously, 33 AFF2 mutations that were associated with diseases of the central nervous system have been reported (Supplementary Table 1), including 8 missense mutations, 6 destructive (null) mutations (2 nonsense, 1 small deletion, and 3 small insertions), and 19 genomic rearrangements (15 gross deletions, 3 gross insertions, and 1 complex rearrangement). Additionally, 28 cases/families with CCG repeat expansion (≥ 200 repeats or > 5.2 Kb) in 5′-UTR have been published. The missense mutations were identified in 6 cases with ASD (6/8) and 2 cases with intellectual disability (2/8, one of whom accompanied with epilepsy). The destructive mutations were associated with ASD (5/6) mostly, and a case with intellectual disability (1/6). The genomic rearrangements were all associated with intellectual disability, among which 15 cases accompanied with developmental delay (15/19), 9 cases with epilepsy (9/19), and 1 case with ASD (1/19). Similarly, the CCG repeat variants were all associated with intellectual disability (Figure 2C and Supplementary Table 1).
In this study, the 5 cases who presented partial epilepsy all harbored missense mutations (5/5, 100%). In contrast, among the patients with ASD, six cases harbored missense mutations (6/11, 54.5%) and five cases harbored truncated mutations (5/11, 45.5%). In the patients with intellectual disability, missense mutations accounted for 4.0% (2/50), truncating mutation accounted for 2.0% (1/50), genomic rearrangements accounted for 38.0% (19/50), and CCG repeat variants accounted for 56.0% (28/50) of the cases. The proportion of missense mutations in epilepsy without intellectual disability and ASD was 100%, which was significantly higher than that in intellectual disability (p < 0.001) (Figure 2C).
Our previous studies showed that the molecular sub-regional location of the missense mutations was associated with the phenotypic variation and considered to be a critical factor to determine the pathogenicity of variants (Liu et al., 2020; Tang et al., 2020). We thus analyzed the molecular sub-regional implications of AFF2 variants on phenotype variation. The missense mutations with epilepsy in the present study fell into the regions from the N-terminal to the NLS1, whereas the missense mutations with ASD fell in the regions from the NLS1 to the C-terminal, with several mutations overlapped around the NLS1 (Figure 2A).
Discussion
AFF2 is a large gene containing 22 exons and spanning about 500 kb that is located on chromosome Xq28. FMR2, the AFF2 encoded protein, has five annotated isoforms. The longest one is composed of 1,311 amino acids and contains two nuclear localization signals (Gecz et al., 1997). FMR2 acts as a transcriptional factor and RNA-binding protein, playing an essential role for transcriptional regulation and RNA splicing in nuclear speckle (Bensaid et al., 2009). In the present study, five novel missense AFF2 mutations were identified in five male individuals with partial epilepsy and antecedent febrile seizures. The mutations were inherited from their asymptomatic mothers, consistent with an X-linked inheritance pattern. This study suggests that AFF2 is potentially a candidate causative gene of X-link partial epilepsy with antecedent febrile seizures. The molecular sub-regional effect of AFF2 helps in explaining the mechanisms underlying phenotypic variations.
Homozygous knock-out of Aff2 in the mice model resulted in premature death with incomplete penetrance. The survival mice showed impaired learning and memory abilities and increased long-term potentiation—similar features as human intellectual disability (Gu et al., 2002), supporting the view that FMR2 is responsible for intellectual disability. Previous studies showed that intellectual disability associated AFF2 mutations were genomic rearrangements and CCG repeat expansion mostly (Figure 2C and Supplementary Table 1), suggesting a pathogenic role of loss-of-function of AFF2 in intellectual disability. Epileptic seizures were also observed in patients with genomic rearrangements (Wolff et al., 1997; Moore et al., 1999), suggesting that loss-of-function of AFF2 would be potentially associated with epilepsy. In the present study, the patients presented good responses to antiepileptic drugs and become seizure free. These findings suggested that missense AFF2 mutations were potentially associated with epilepsy with favorable outcomes without intellectual disability.
Apart from epilepsy, missense AFF2 mutations have also been identified in patients with ASD (Mondal et al., 2012; Jiang et al., 2013). Further analysis revealed that missense AFF2 mutations with epilepsy in this study mainly fell into the regions from the N-terminal to the NLS1, whereas the missense mutations with ASD mainly fell in the regions from the NLS1 to the C-terminal (Figure 2A). This evidence suggested a molecular sub-regional effect of AFF2 mutations, as that in several genes reported previously (Liu et al., 2020; Tang et al., 2020). FMR2 is a multifunctional protein connecting transcriptional regulation and RNA splicing and plays an important role in regulating gene expression in the cell nucleus (Gecz, 2000). The protein shuttles between speckles and splicing sites, where it can pick up cargo RNAs and transfer them to the nucleolus for subsequent modifications (Bensaid et al., 2009). A previous study showed that FMR2 acted as a potent transcription activator and could regulate transcription via its N-terminal region (Hillman and Gecz, 2001). On the other hand, FMR2 protein is also an RNA binding protein, co-localizing with the splicing factor SC35 in nuclear speckles and is involved in splicing of FMR1 pre-mRNA through its specific interaction with G-quadruplex RNA-forming structure via its C-terminal domain (Lamond and Spector, 2003). The G-quadruplex structure was known to work as an exonic splicing enhancer which could control splicing efficiency (Bensaid et al., 2009). Therefore, the N-terminal region of FMR2 was closely associated with transcription and C-terminal region was able to modulate splicing, providing a molecular basis of the sub-regional effect of AFF2 mutations and potentially explaining the underlying mechanisms of phenotypic variations.
In this study, the cases all presented with partial epilepsy. AFF2 is ubiquitously expressed in multiple human brain tissues (Gecz et al., 1997) and is more abundant in the frontal cortex, anterior cingulate cortex, hippocampus, and the amygdala, which may provide an anatomical basis for the phenotype of partial epilepsy. Moreover, four cases presented with antecedent febrile seizures at the average age of 16.5 months (from 10 months to 2 years old). The intermission from febrile seizures to epilepsy was 4–7 years. It is notable that AFF2 is significantly expressed in the fetal and adult brain (Gecz et al., 1997). The data from Unigene database in NCBI showed that the expression of AFF2 was high in blastocyte and fetus, with an intermission in early life, and then increased again in adults (Supplementary Figure 1). The intermission of gene expression was consistent with the intermission between febrile seizures and epilepsy presented in the patients. The relationship between gene expression and occurrence of phenotype potentially implies clinical significance in evaluation of the course and outcome of the illness.
There were several limitations in this study. First, the functional consequences of these missense variants warrant further validation by experimental studies. Second, previous studies revealed that some of the AFF2 mutations associated with intellectual disability were CCG repeat expansion, which were not included in the present study. Third, a previous study reported that a missense mutation of AFF2 was identified in a 6-year-old boy with focal epilepsy and moderate intellectual disability, but without febrile seizures (Zhang et al., 2015). The study indicated that missense AFF2 mutations could also lead to other phenotypes such as intellectual disability, possibly associated with the molecular sub-regional location. Further studies are required to verify the association. Last, the sample size was limited with geographical limitations. Larger cohorts are required for validation by multicenter research.
Conclusion
This study identified five missense AFF2 mutations in five unrelated males with partial epilepsy and antecedent febrile seizures without intellectual disability or other developmental abnormalities. The frequency of the identified mutant alleles in this cohort was significantly higher than that in the control populations in gnomAD. Further analysis revealed that the AFF2 mutations associated with partial epilepsy in this study were all missense, in contrast, intellectual disability-associated mutations were genomic rearrangements and CCG repeat expansion mostly. These findings suggested AFF2 was potentially a candidate causative gene of X-link partial epilepsy with antecedent febrile seizures. The genotype-phenotype correlation and molecular sub-regional effect of AFF2 help in explaining the mechanisms underlying phenotypic variations.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) are the following: Genbank (https://www.ncbi.nlm.nih.gov/nuccore/) with accession numbers: GenBank MZ958719–MZ958733.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
XL designed the study, administered the project, and revised the manuscript. JL designed the study and contributed to data interpretation. DZ completed collection of the clinical data, analysis of the data, and draft of the manuscript. XL and DZ completed the recruitment of the patients and contributed to data interpretation. BQ, JL, and YY contributed to data analysis and interpretation. JW, PZ, and YS performed data analysis and provided technical assistance. All authors have read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 81871015 and 81870903), the Natural Science Foundation of Guangdong Province (2020A1515010108), the Science and Technology Project of Guangzhou (Grant Nos. 201904020028 and 201904010275), the Science and Technology Project of Guangdong Province (Grant Nos. 2017B09090436 and 2017B030314159), the Multi-Center Clinical Research Fund Project of the Second Affiliated Hospital of Guangzhou Medical University (Grant Nos. DZX-002 and 2020-LCYJ-DZX-03), the Sanming Project of Medicine in Shenzhen (Grant No. SZSM201812005), the Shenzhen Fund for Guangdong Provincial High-level Clinical Key Specialties (Grant No. SZGSP012), and the Shenzhen Key Medical Discipline Construction Fund (Grant No. SZXK033).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are deeply grateful to the patients and clinicians who participated in this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.795840/full#supplementary-material
Footnotes
- ^ http://www.informatics.jax.org/allele/MGI:2182641
- ^ http://varcards.biols.ac.cn/
- ^ http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index
- ^ https://pubmed.ncbi.nlm.nih.gov/
- ^ http://www.hgmd.cf.ac.uk/ac/index.php
- ^ http://www.sbg.bio.ic.ac.uk/p̃hyre2/html/page.cgi?id=index
References
Bensaid, M., Melko, M., Bechara, E. G., Davidovic, L., Berretta, A., Catania, M. V., et al. (2009). FRAXE-associated mental retardation protein (FMR2) is an RNA-binding protein with high affinity for G-quartet RNA forming structure. Nucleic Acids Res. 37, 1269–1279. doi: 10.1093/nar/gkn1058
Berg, A. T., Berkovic, S. F., Brodie, M. J., Buchhalter, J., Cross, J. H., van Emde Boas, W., et al. (2010). Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 51, 676–685. doi: 10.1111/j.1528-1167.2010.02522.x
Cai, K., Wang, J., Eissman, J., Wang, J., Nwosu, G., Shen, W., et al. (2019). A missense mutation in SLC6A1 associated with Lennox-Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp. Neurol. 320:112973. doi: 10.1016/j.expneurol.2019.112973
Cai, N., Bigdeli, T. B., Kretzschmar, W., Li, Y., Liang, J., Song, L., et al. (2015). Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591.
Gecz, J. (2000). The FMR2 gene, FRAXE and non-specific X-linked mental retardation: clinical and molecular aspects. Ann. Hum. Genet. 64, 95–106. doi: 10.1017/S0003480000007983
Gecz, J., Bielby, S., Sutherland, G. R., and Mulley, J. C. (1997). Gene structure and subcellular localization of FMR2, a member of a new family of putative transcription activators. Genomics 44, 201–213. doi: 10.1006/geno.1997.4867
Gu, Y., McIlwain, K. L., Weeber, E. J., Yamagata, T., Xu, B., Antalffy, B. A., et al. (2002). Impaired conditioned fear and enhanced long-term potentiation in Fmr2 knock-out mice. J. Neurosci. 22, 2753–2763. doi: 10.1523/JNEUROSCI.22-07-02753.2002
Hillman, M. A., and Gecz, J. (2001). Fragile XE-associated familial mental retardation protein 2 (FMR2) acts as a potent transcription activator. J. Hum. Genet. 46, 251–259. doi: 10.1007/s100380170074
Jiang, Y. H., Yuen, R. K., Jin, X., Wang, M., Chen, N., Wu, X., et al. (2013). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263. doi: 10.1016/j.ajhg.2013.06.012
Knight, S. J., Flannery, A. V., Hirst, M. C., Campbell, L., Christodoulou, Z., Phelps, S. R., et al. (1993). Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 74, 127–134. doi: 10.1016/0092-8674(93)90300-f
Knight, S. J., Ritchie, R. J., Chakrabarti, L., Cross, G., Taylor, G. R., Mueller, R. F., et al. (1996). A study of FRAXE in mentally retarded individuals referred for fragile X syndrome (FRAXA) testing in the United Kingdom. Am. J. Hum. Genet. 58, 906–913.
Lamond, A. I., and Spector, D. L. (2003). Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell. Biol. 4, 605–612. doi: 10.1038/nrm1172
Lesca, G., Biancalana, V., Brunel, M. J., Quack, B., Calender, A., and Lespinasse, J. (2003). Clinical, cytogenetic, and molecular description of a FRAXE French family. Psychiatr Genet. 13, 43–46. doi: 10.1097/00041444-200303000-00007
Liu, L., Chen, Z. R., Xu, H. Q., Liu, D. T., Mao, Y., Liu, H. K., et al. (2020). DEPDC5 variants associated malformations of cortical development and focal epilepsy with febrile seizure plus/febrile seizures: the role of molecular sub-regional effect. Front. Neurosci. 14:821. doi: 10.3389/fnins.2020.00821
Lo Nigro, C., Faravelli, F., Cavani, S., Perroni, L., Novello, P., Vitali, M., et al. (2000). FRAXE mutation in a mentally retarded subject and in his phenotypically normal twin brother. Eur. J. Hum. Genet. 8, 157–162. doi: 10.1038/sj.ejhg.5200425
Mondal, K., Ramachandran, D., Patel, V. C., Hagen, K. R., Bose, P., Cutler, D. J., et al. (2012). Excess variants in AFF2 detected by massively parallel sequencing of males with autism spectrum disorder. Hum. Mol. Genet. 21, 4356–4364. doi: 10.1093/hmg/dds267
Moore, S. J., Strain, L., Cole, G. F., Miedzybrodzka, Z., Kelly, K. F., and Dean, J. C. (1999). Fragile X syndrome with FMR1 and FMR2 deletion. J. Med. Genet. 36, 565–566.
Mulley, J. C., Yu, S., Loesch, D. Z., Hay, D. A., Donnelly, A., Gedeon, A. K., et al. (1995). FRAXE and mental retardation. J. Med. Genet. 32, 162–169. doi: 10.1136/jmg.32.3.162
No Author (1985). Proposal for classification of epilepsies and epileptic syndromes. Commission on classification and terminology of the international league against Epilepsy. Epilepsia 26, 268–278. doi: 10.1111/j.1528-1157.1985.tb05417.x
No Author (1989). Proposal for revised classification of epilepsies and epileptic syndromes. Commission on classification and terminology of the international league against Epilepsy. Epilepsia 30, 389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x
Scheffer, I. E., Berkovic, S., Capovilla, G., Connolly, M. B., French, J., Guilhoto, L., et al. (2017). ILAE classification of the epilepsies: position paper of the ILAE Commission for classification and terminology. Epilepsia 58, 512–521. doi: 10.1111/epi.13709
Shi, Y. W., Zhang, Q., Cai, K., Poliquin, S., Shen, W., Winters, N., et al. (2019). Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 142, 3028–3044. doi: 10.1093/brain/awz250
Strande, N. T., Riggs, E. R., Buchanan, A. H., Ceyhan-Birsoy, O., DiStefano, M., Dwight, S. S., et al. (2017). Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet. 100, 895–906. doi: 10.1016/j.ajhg.2017.04.015
Tang, B., Li, B., Gao, L. D., He, N., Liu, X. R., Long, Y. S., et al. (2020). Optimization of in silico tools for predicting genetic variants: individualizing for genes with molecular sub-regional stratification. Brief Bioinform. 21, 1776–1786. doi: 10.1093/bib/bbz115
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Wang, J. Y., Zhou, P., Wang, J., Tang, B., Su, T., Liu, X. R., et al. (2018). ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: toward understanding the mechanism underlying phenotypic variation. Neurogenetics 19, 9–16. doi: 10.1007/s10048-017-0528-2
Wolff, D. J., Gustashaw, K. M., Zurcher, V., Ko, L., White, W., Weiss, L., et al. (1997). Deletions in Xq26.3-q27.3 including FMR1 result in a severe phenotype in a male and variable phenotypes in females depending upon the X inactivation pattern. Hum. Genet. 100, 256–261. doi: 10.1007/s004390050501
Yuen, R. K. C., Merico, D., Bookman, M., Howe, J. L., Thiruvahindrapuram, B., Patel, R. V., et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611. doi: 10.1038/nn.4524
Keywords: epilepsy, AFF2 gene, whole-exome sequencing, intellectual disability, autism spectrum disorder
Citation: Zou D, Qin B, Wang J, Shi Y, Zhou P, Yi Y, Liao J and Lu X (2022) AFF2 Is Associated With X-Linked Partial (Focal) Epilepsy With Antecedent Febrile Seizures. Front. Mol. Neurosci. 15:795840. doi: 10.3389/fnmol.2022.795840
Received: 15 October 2021; Accepted: 08 February 2022;
Published: 30 March 2022.
Edited by:
Qian Chen, Massachusetts Institute of Technology, United StatesReviewed by:
Qiongxiang Zhai, Guangdong Academy of Medical Sciences, ChinaDong Zhou, Sichuan University, China
Copyright © 2022 Zou, Qin, Wang, Shi, Zhou, Yi, Liao and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianxiang Liao, bGlhb2ppYW54aWFuZ0B2aXAuc2luYS5jb20=; Xinguo Lu, c3psdXhnMjBAMTYzLmNvbQ==
†These authors have contributed equally to this work