
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci., 24 February 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.691534
This article is part of the Research TopicAlternative Splicing in Brain FunctionView all 15 articles
 Ying-Qian Lu1,2†Jian-Min Chen1†Han Lin1Shu-Yan Feng3Chun-Hui Che1,2Chang-Yun Liu1,2
Ying-Qian Lu1,2†Jian-Min Chen1†Han Lin1Shu-Yan Feng3Chun-Hui Che1,2Chang-Yun Liu1,2 Hua-Pin Huang1,2*
Hua-Pin Huang1,2* Zhang-Yu Zou1,2*
Zhang-Yu Zou1,2*
TANK-binding kinase 1 (TBK1) has been identified as a causative gene of amyotrophic lateral sclerosis (ALS) in the Caucasian population in 2015. Here, we sequenced for TBK1 variants in a cohort of 15 familial ALS (fALS) and 275 sporadic ALS (sALS) of Chinese origin by targeted next-generation sequencing. We identified one likely benign missense variant (p. Ser398Pro), two missense variants of uncertain significance (p. Ile37Leu and p. Tyr677Asn), and two novel heterozygous variants in introns of TBK1, c.1522-3T > G and c.2066 + 4A > G. We performed splicing assays through minigene plasmids and RNA pull-down assay to determine that the two substitutions of nucleotides disrupted the binding of the important splicing regulator hnRNPA1 and promoted aberrant pre-mRNA splicing modes. The c.1522-3T > G variant promoted nearly 50.0% of abnormal transcripts (3 different types of insertions and deletions (indels) in junction of intron 13-exon 14) and the c.2066 + 4A > G variant inhibited about 75.0% inclusion of exon 19, both causing premature stop codon and producing TBK1 protein without CCD2. Immunofluorescence analysis showed that the expression of TBK1 with intronic variants was lower since less TBK1 distribution was observed in HEK293T cells. Both patients carrying TBK1 c.1522-3T > G and c.2066 + 4A > G variants developed a rapidly progressive ALS, with a survival of 31 and 10 months, respectively. The frequency of loss of function (LoF) variants in TBK1 was 0.73% in sALS in our cohort. We emphasize that intronic sequencing and pre-mRNA splicing analysis cannot be ignored to demonstrate the complex mutational spectrum and pathogenesis of ALS.
Amyotrophic lateral sclerosis (ALS) is a devastating neurological disease characterized by degeneration of upper and lower motor neurons (Chou and Norris, 1993). ALS patients are characterized by progressive muscle weakness and wasting, eventually leading to respiratory failure. Typically, patients die within 3∼5 years after symptom onset due to respiratory failure (Brown and Al-Chalabi, 2017). The incidence rate of ALS is about 2/100,000 (Rosenbohm et al., 2018).
Approximately 10% of ALS are familial ALS (fALS) and the remaining 90% are sporadic ALS (sALS) (Al-Chalabi et al., 2016). To date, mutations in more than 30 genes have been reported to contribute to the pathogenesis of ALS. The first ALS-associated gene was superoxide dismutase-1 (SOD1) and was described in 1993 (Rosen et al., 1993). ALS-associated mutations in SOD1 are identified in 12–20% of fALS patients and 1–2% of sALS patients. SOD1 is the most common gene associated with ALS in Asian populations (Zou et al., 2017). In 2008, the second contributor to ALS, TAR DNA binding protein (TARDBP), was discovered as an important causative gene of ALS (Gitcho et al., 2008). TARDBP mutations account for ∼3.3% of fALS patients and ∼0.5% of sALS (Zou et al., 2017). Fused in sarcoma (FUS) gene is the most common gene detected in juvenile and pediatric ALS, and the p.P525L mutation particularly tends to be associated with an aggressive and early onset form (Kwiatkowski et al., 2009; Lattante et al., 2013; Gonzalez et al., 2021). Approximately, FUS mutations could be found in 5% of fALS patients and 1% of sALS patients (Andersen and Al-Chalabi, 2011). A hexanucleotide (GGGGCC-) repeat expansion in the chromosome 9 open reading frame 72 (C9orf72) gene is the most frequent genetic cause of ALS in Caucasian populations. A C9orf72 repeat expansion was the genetic cause in up to 35% of fALS patients and about 5% of sALS patients (Renton et al., 2011; Zou et al., 2017). Recently, many new genes have been found as causative or highly associated with ALS, like TBK1, MATR3, CHCHD10, TUBA4A, NEK1, and C21orf2 (Chia et al., 2018).
TBK1 (TANK-binding kinase 1) gene has been identified as a causative gene of ALS in the Caucasian population in 2015 (Cirulli et al., 2015). TBK1 is a multifunctional serine/threonine protein kinase that plays key roles in various cellular processes through phosphorylating a wide range of substrates, including neuroinflammation, innate immunity, ubiquitin-proteasome systems, cell proliferation, and autophagy pathways involving other genes also associated with ALS, such as SQSTM1/p62, OPTN, and VCP (Oakes et al., 2017; Zhao et al., 2018). TBK1 includes four domains: a serine/threonine kinase domain (KD), a ubiquitin-like domain (ULD), and two coiled-coil domains (CCD1 and CCD2) (Le Ber et al., 2015). More than 90 mutations in TBK1, such as non-sense mutations, frameshift, out-frame, and splice-site, have been reported to result in loss of function (LoF) of the TBK1 and lead to ALS or frontotemporal dementia (FTD) (van der Zee et al., 2017).
In this study, we aimed to screen variants of TBK1 in a cohort of Chinese ALS patients through targeted next-generation sequencing. Moreover, we further performed in vitro functional experiments to address the pathogenic potential of the variants.
Our study included 275 sporadic ALS (sALS) patients and 15 familial ALS (fALS) probands. The characteristics of the ALS patients were summarized in Table 1. The patients were enrolled from Fujian Medical Union Hospital and Henan Provincial People’s Hospital between January 2017 and December 2018. The diagnosis of ALS was made according to the El Escorial revised criteria (Brooks et al., 2000). Patients with clinically definite, probable, or probable laboratory-supported ALS were recruited. fALS patients were diagnosed if one or more first- or second-degree relatives developed ALS (Ludolph et al., 2015). Written informed consent was obtained from every subject to use their DNA for genetic analysis. This study was approved by the ethics committee of Fujian Medical University Union Hospital and Henan Provincial People’s Hospital. The control group included 1,000 ethnicity matched controls who had performed whole exome sequencing in AmCare Genomics Lab, Guangzhou, China.

Table 1. Characteristics of the ALS patients in this study.
Genomic DNA of each subject was extracted from peripheral blood leukocytes using a TIANamp Genomic DNA Kit (Tiangen) according to the manufacturer’s instructions. Variants in TBK1 were screened by targeted next-generation sequencing on Illumina Hiseq sequencer (Illumina Inc., San Diego, CA). The targeted regions included all exons with intronic 50bp flanking sites of the TBK1, then followed by Sanger sequencing for mutation site verification on an ABI 3730 Genetic Analyzer (Foster City, CA, United States). As a result of sequencing, the mean on-target coverage was 560X with an average percentage of targets covered greater or equal to 100X of 99.8%. The identified variants were Sanger sequenced for confirmation. Patients carrying TBK1 variants were also screened for mutations in other known ALS related genes SOD1, TARDBP, FUS, VAPB, SPG11, VCP, PFN1, ANG, ALS2, DAO, UBQLN2, SIGMAR1, SETX, FIG4, DCTN1, OPTN, SQSTM1, CHCHD10, MATR3, hnRNPA1, hnRNPA2B1, ANXA11, KIF5A, TIA1, CCNF, NEK1, CHCHD10, TUBA4A, as well as the presence of the GGGGCC expansions in the C9orf72 gene.
Gene variants were evaluated by their absence or frequency in the public single-nucleotide polymorphism database (dbSNP), 1,000 genome, and ExAc (Exome Aggregation Consortium). The SIFT,1 PolyPhen-22 and Mutation Taster3 were used to assess the functional effects of the missense variants. The pathogenicity analysis was conducted according to the ACMG standard. The two intronic variants, c.1522-3T > G and c.2066 + 4A > G, were predicted to have changes of serine/arginine-rich splicing factors (SRSFs) using the program “ESEfinder” (version 3.0)4 (Cartegni et al., 2003).
The minigene plasmid was designed to determine the pathogenesis of c.1522-3T > G (named V-minigene 1), containing the last 146 bp of intron 12, full-length sequences from exon 13 to exon 15 (including the intron 13 and intron 14), and the first 229 bp of intron 15 of the TBK1 gene. The corresponding wild-type minigene plasmid (the position c.1522-3 was T) was named W-minigene 1. The minigene plasmid designed to determine the pathogenesis of c.2066 + 4A > G (named V-minigene 2) contained the last 267 bp of intron 16, full-length sequences from exon 17 to exon 20 (including the intron 17, intron 18, and intron 19), and the first 350 bp of intron 20 of the TBK1 gene; the corresponding wild type minigene plasmid (the position c.2066 + 4 was A) was named W-minigene 2. The genomic sequences were PCR amplified using a KOD-plus-Neo Kit (TOYOBO) from human DNA with primers (P1-F/R and P2-F/R for V/W-minigene 1, P3-F/R and P4-F/R for V/W-minigene 2) listed in Supplementary Table 1. Then the PCR fragments were cloned into a pCMV-cDNA-EF1a-EGFP-BGH-expressing plasmid using a ClonExpress MultiS One Step Cloning Kit (Vazyme Biotech). The DNA sequence of FLAG was cloned and added in the front of the TBK1 gene in the minigene plasmids for immunofluorescence analysis. The identity of all the minigene plasmids was verified by Sanger sequencing. T-A cloning plasmids were constructed using a pMD™ 19-T Vector Kit (Takara).
The HEK293T and HeLa cell lines were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) (Gibco) containing 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin (PS) (Thermo Fisher Scientific). Cells were dissociated using 0.05% Trypsin/EDTA (Gibco) and were cultured in a 37°C incubator within 5% CO2 atmosphere. All of the plasmids were transfected using Lipofectamine 3000 Reagent (Thermo Fisher Scientific) according to the standard instructions.
Total RNA of HEK293T and HeLa cells transfected with minigene plasmids was extracted 2 days later using TRIzol reagent (Invitrogen) following the manufacturer’s recommendations. For cDNA synthesis, we used 1 μg of each RNA sample per 20 μl reverse transcription reaction using a HiScript II Q RT SuperMix Kit (Vazyme Biotech) according to the manufacturer’s instructions. RT-PCR was performed using a KOD-plus-Neo Kit (TOYOBO) with primers (RT1-F/R for V/W-minigene 1 and RT2-F/R for V/W-minigene 2) listed in Supplementary Table 1 (the upstream primers RT1/2-F were the same). To distinguish the pre-mRNA splicing isoforms originating from HEK293T (or HeLa) cells and minigene plasmids, the upstream primers for RT-PCR were designed specifically to bind to the back-bone of minigene plasmids and would not bind to the sequences of HEK293T (or HeLa) cells (Figure 1A). Then Sanger sequencing was performed to determine the detailed pre-mRNA splicing modes using the upstream primers. The splicing assays were repeated three times independently.
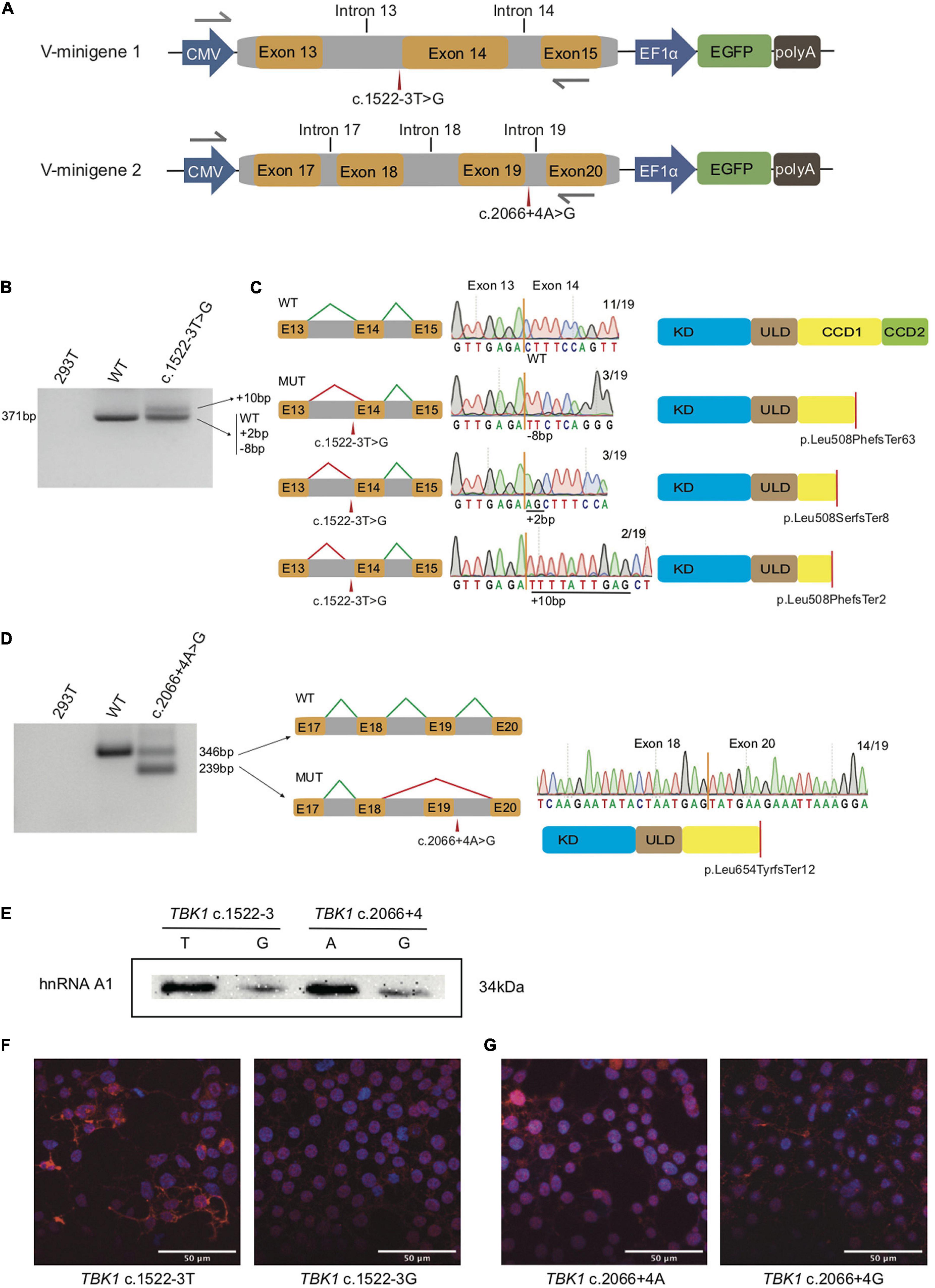
Figure 1. Splicing modes verification and functional characterization of the two TBK1 intronic variants. (A) Construction of V-minigene 1 (bearing TBK1 c.1522-3T > G variant) and V-minigene 2 (bearing TBK1 c.2066 + 4A > G variant) plasmids. The minigene plasmids contain CMV promotor, consecutive genomic DNA, EF1α promotor and EGFP sequences. Yellow boxes indicate exons, gray boxes indicate introns, gray arrows indicate primers. (B) Agarose gel electrophoresis of RT-PCR products, lane 1: cDNA from HEK293T cells (blank control), lane 2: cDNA from W-minigene 1 plasmid (wild type control, bearing TBK1 c.1522-3T), lane 3: cDNA from V-minigene 1 plasmid (bearing TBK1 c.1522-3T > G variant). (C) Left: schematics of normal splicing mode (WT) and aberrant splicing modes (MUT) of V-minigene 1 plasmid; yellow boxes indicate exons, gray boxes indicate introns, green lines show normal splicing modes, red lines show aberrant splicing modes, E13: exon 13, E14: exon 14, E15: exon 15. Middle: T-A clone results of RT-PCR products from V-minigene 1 plasmid. Right: Schematics of TBK1 protein corresponding to the former splicing modes; red lines indicate positions of premature stop codon. (D) Left: agarose gel electrophoresis of RT-PCR products, lane 1: cDNA from HEK293T cells (blank control), lane 2: cDNA from W-minigene 2 plasmid (wild type control, bearing TBK1 c.2066 + 4A), lane 3: cDNA from V-minigene 2 plasmid (TBK1 c.2066 + 4A > G variant). Middle: schematic of normal splicing mode (WT) and aberrant splicing mode (MUT) of V-minigene 2 plasmid; green lines show normal splicing modes, red lines show aberrant splicing modes, yellow boxes indicate exons, gray boxes indicate introns, E17: exon 17, E18: exon 18, E19: exon 19, E20: exon 20. Right: T-A clone results of RT-PCR products from V-minigene 2 plasmid and schematic of TBK1 protein corresponding to the MUT splicing mode. (E) RNA pull-down assay and western blot analysis. Synthetic RNAs were labeled with biotin and incubated with nuclear extract from HeLa cells, then the RNA-binding protein complexes were interacted with anti-hnRNPA1 antibody specifically and separated by SDS-PAGE. (F,G) Immunofluorescence analysis of subcellular distribution of TBK1 in HEK293T cells overexpressed by TBK1 cDNA carrying c.1522-3T or c.1522-3G (F) and TBK1 cDNA carrying c.2066 + 4A or c.2066 + 4G (G) was visualized by anti-FLAG antibody followed by anti-mouse Alexa Fluor 594 (red)-conjugated secondary antibody and DAPI (blue) staining. Scale bar: 50 μm.
RNA pull-down assay was performed with a Pierce™ Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. RNA TBK1 r.1522-3U (5′-UUUGGUUUUAUUUAGCUUUCCAGUU-3′), TBK1 r.1522-3G (5′- UUUGGUUUUAUUGAGCUUUCCAGUU-3′), TBK1 r. 2066 + 4A (5′- GACUCUUGGGUAAGAAACUCAUC AU-3′), and TBK1 r. 2066 + 4G (5′- GACUCUUGGGUAG GAAACUCAUCAU-3′) were synthesized by Sangon Biotech and were biotin labeled at 3′ End using a Pierce RNA 3′ End Desthiobiotinylation Kit (Thermo Fisher Scientific). The biotin-labeled RNAs were added to streptavidin magnetic beads and incubated for 1 h at 4°C with agitation. After washing, HeLa nuclear extract was added to the RNA-beads mixture and incubated for 2 h at 4°C with agitation. RNA-binding protein complexes were analyzed by SDS-PAGE and immunoblotting with anti-hnRNPA1 antibody (Abcam). The experiments were repeated three times independently.
HEK293T cells cultured on glass coverslips were fixed with 4% PFA diluted in PBS for 30 min at room temperature and then incubated with primary anti-FLAG antibody (1:1,000, Sigma) followed by Alexa Fluor 594-conjugated anti-mouse secondary antibody (1:500, Sigma). The primary and secondary antibodies were both diluted in PBS containing 1% BSA and 0.3% Tx-100. At last, samples were briefly stained with DAPI (1:2,500, Sigma). Images were captured with Leica TCS SP8 confocal microscopy.
Three heterozygous missense variants, c.109A > C (p.Ile37Leu), c.1192T > C (p.Ser398Pro), and c.2029T > A (p.Tyr677Asn) in the exons of TBK1 gene (Figure 2A and Table 2), were each identified in one sALS patient. The p.Ser398Pro and p.Tyr677Asn variants had been reported in dbSNP (rs781434264 and rs1163013930), while the p.Ile37Leu variant was novel. Two novel heterozygous variants, c.1522-3T > G and c.2066 + 4A > G, located in intron 13 and intron 19 of TBK1, respectively, were each identified in one sALS case (Figure 2A and Table 2). No TBK1 variants were found in the fALS patients. No variants of other ALS-related genes were identified in the five sALS cases. No parental DNA samples of these patients were available for further analysis. All five TBK1 variants were absent from the population database 1000genome and ExAC, ExAC, as well as 1,000 ethnicity matched controls. The novel variant p. Ile37Leu was predicted to be “damaging” and “benign” with SIFT and Polyphen-2, respectively; and the variant was predicted to be “disease causing” with Mutation Taster. The p. Ser398Pro and p.Tyr677Asn variants were both predicted to be “tolerable” with SIFT and “benign” with Polyphen-2. With Mutation Taster, the p. Ser398Pro and p. Tyr677Asn variants were predicted to be “polymorphism” and “disease causing,” respectively. The p. Ile37Leu and p. Tyr677Asn variants were interpreted as variants of uncertain significance (VUS) while the p.Ser398Pro variant was likely benign according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines (Table 2).
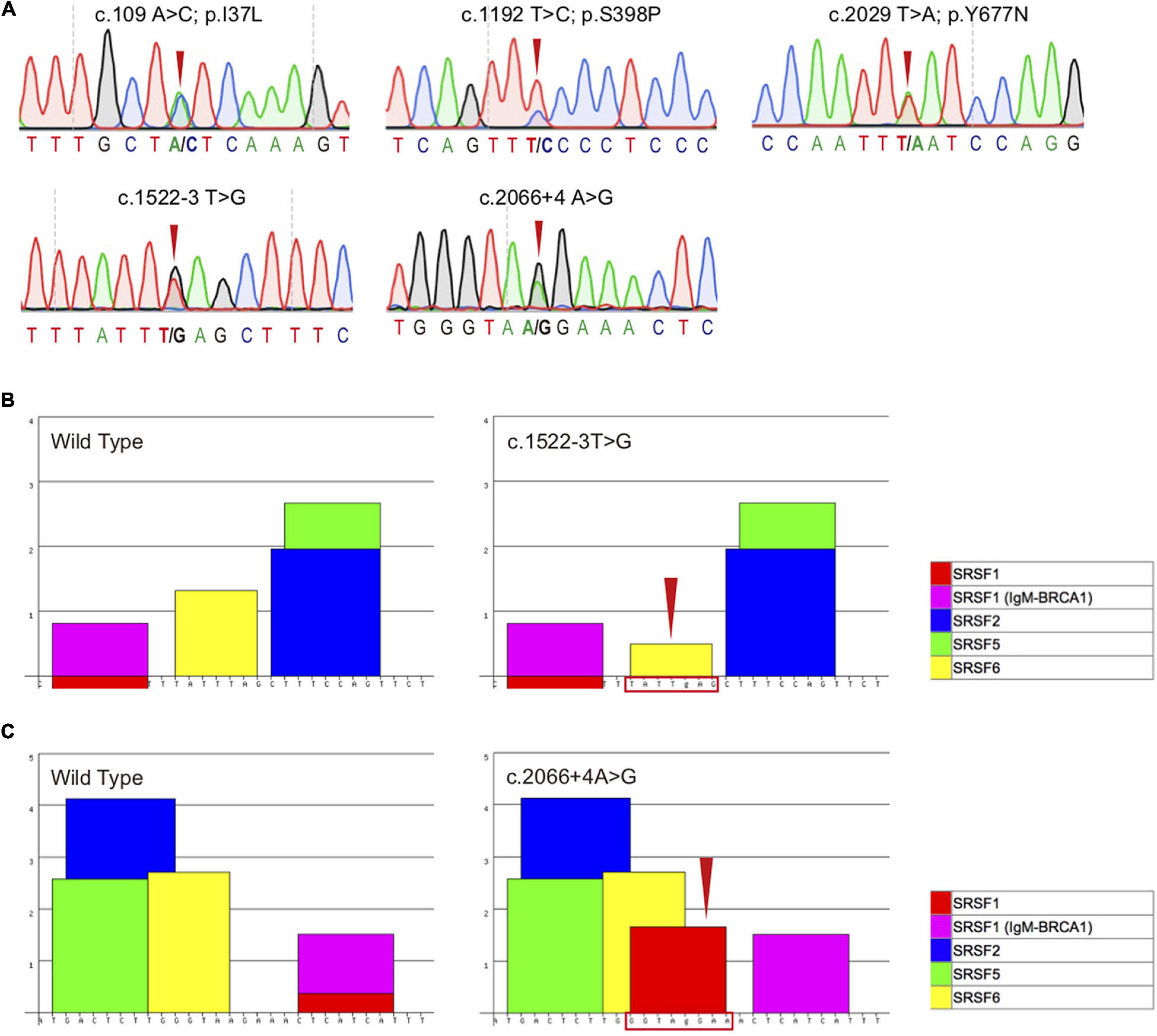
Figure 2. Sequencing chromatogram and bioinformatics analysis of the TBK1 variants. (A) Sequencing chromatograms of the five TBK1 variants (c.109A > C, c.1192T > C, c.2029T > A, c.1522-3T > G and c.2066 + 4A > G) detected in this study. (B) Bioinformatics analysis of the wild type sequences and mutated sequences with the TBK1 c.1522-3T > G variant using ESEfinder 3.0, the program recognized that the TATTTAG (the binding site of SRSF6) (left) transformed to TATTgAG (right, indicated by red arrow and frame). (C) Bioinformatics analysis of the wild type sequences and mutated sequences with the TBK1 c.2066 + 4A > G variant using ESEfinder 3.0, the program recognized that a new motif GGTAgGAA (the binding site of SRSF1) was generated (right, indicated by red arrow and frame).
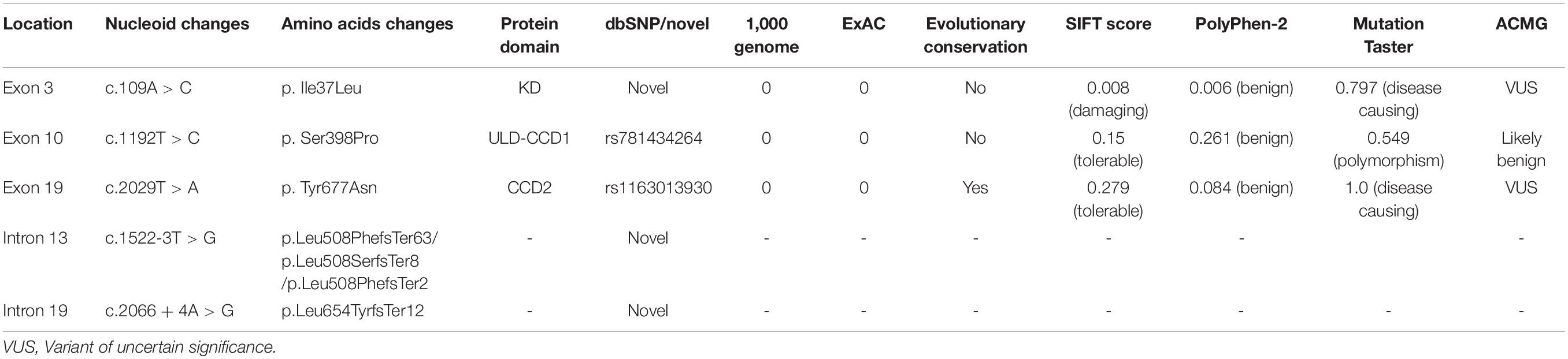
Table 2. Genetic profile of the TBK1 variants identified in this study.
As adjacent to the splicing sites, both TBK1 c.1522-3T > G and c.2066 + 4A > G variants were likely to be pathogenic based on the potential interference in the splicing regulator. To determine the pathogenic significance, we conducted bioinformatics analysis of the variants using the ESEfinder 3.0. The ESEfinder 3.0 predicted that the c.1522-3T > G variant transformed the serine/arginine-rich splicing factor 6 (SRSF6) (TATTTAG termed to TATTgAG) and the c.2066 + 4A > G variant generated a new SRSF1 (GGTAgGAA) (Figures 2B,C). It thus appeared that the preexisting splicing modes might be interfered.
In the light of the prediction, we performed in vitro splicing assays using minigene plasmids harboring wild type sequence (named W-minigene 1 and W-minigene 2) or the intronic variants (named V-minigene 1 and V-minigene 2) of TBK1 to mimic the endogenous splicing processes (Figure 1A). These minigene plasmids were transfected into HEK293T cells, then total RNA was extracted and reverse transcription followed by PCR (RT-PCR) was performed to assess the splicing modes following a previous study (Lin et al., 2020). We employed TA clone and Sanger sequencing to confirm the detailed splicing modes.
There was more than one kind of RT-PCR product from the cDNA of V-minigene 1 plasmid (Figure 1B). In addition to the wild type splicing mode (termed WT), Sanger sequencing revealed that the minigene plasmid harboring c.1522-3T > G variant (V-minigene 1) promoted another 3 types of aberrant splicing modes (termed MUT) (Figure 1C). As the results of TA clone, 3 (15.79%) showed deletion of 8 nucleotides, 3 (15.79%) showed insertion of 2 nucleotides, and 2 (10.53%) showed insertion of 10 nucleotides in all the 19 clones (Figure 1C). The aberrant splicing modes all caused premature stop codon (p. Leu508PhefsTer63, p. Leu508SerfsTer8, and p. Leu508PhefsTer2) of TBK1 protein (Figure 1C). The minigene plasmid bearing c.2066 + 4A > G variant (V-minigene 2) promoted 2 types of splicing modes, one was the wild type splicing mode (termed WT) and the other one was skipping of exon 19 (termed MUT). The splicing mode also caused premature stop codon (p. Leu654TyrfsTer12) of TBK1 protein (Figure 1D). TA clone and Sanger sequencing results revealed that 14/19 (73.68%) of clones showed exclusion of exon 19 (Figure 1D). To ascertain whether splicing modes of TBK1 could be interfered with by different conditions of cells, we repeated the in vitro splicing assays three times at different times independently in HEK293T cells. Splicing assays were also repeated three times in HeLa cells to avoid the interference of different cell types in splicing modes. The TA clone and Sanger sequencing results showed that splicing modes of TBK1 had no changes in different cell conditions or types (Supplementary Figure 1).
We performed an RNA pull-down assay to confirm changes of the SRSF1 and SRSF6 binding at the sequences around the two TBK1 intronic variants. Biotin-labeled RNAs were incubated with HeLa nuclear extract to form RNA-binding protein complexes. Then, the presence of SRSF1 or SRSF6 in the pull-down materials was assessed by western blot analysis. As a result, the SRSF1 and SRSF6 binding at the sequences around the intronic variants had no significant differences with the binding at the reference sequences (data did not show). We considered that this may be the transformation or generating of SRSF1/SRSF6 was predicted in the website, which is just indicative of the possibility of aberrant splicing. We then identified the hnRNPA1 binding at the region around the two TBK1 variants. The hnRNPA1 was one of the key splicing regulators bound at the splicing sites usually. Compared to the wild type sequences (c.1522-3U and c.2066 + 4A), RNAs bearing the variants (c.1522-3G and c.2066 + 4G) showed obviously lower binding of hnRNPA1 (Figure 1E). These findings indicated that hnRNPA1 binding at the sequences around the intronic variants was disrupted with the generation of new negative splicing-regulatory elements possibly, which promotes aberrant splicing. We also performed immunofluorescence analysis showing the subcellular distribution of TBK1 (overexpressed by TBK1 cDNA with c.1522-3T/G and TBK1 cDNA with c.2066 + 4A/G) visualized by anti-FLAG antibody in HEK293T cells. HEK293T cells overexpressed with TBK1 cDNA bearing variants showed less TBK1 distribution both in the nucleus and cytoplasm than the cells overexpressed with wild type TBK1 cDNA (Figures 1F,G). The results indicated that the truncated TBK1 protein produced by TBK1 intronic variants had a lower level of expression than the wild type TBK1 protein.
Patient 1 (bearing TBK1 c.1522-3T > G variant) was a 71-year-old woman referred due to dysarthria for 6 months and weakness of left hand for 2 months. Neurological examination revealed severe dysarthria, obvious atrophy of the tongue with fibrillation. Muscle strength was grade 4 in muscles of the left hand and grade 5 in other limbs. Fasciculations were observed in the left upper limb. Deep tendon reflexes were increased in the left upper limb. Palm-chin reflex was elicited bilaterally. She scored 43/48 on ALS Functional Rating Scale-Revised (ALSFRS-R) and 28/30 on Montreal Cognitive Assessment testing. Electromyography demonstrated acute denervation in all limbs and thoracic muscles, as well as chronic reinnervation in all limbs and sternocleidomastoid muscles. Laboratory testing for diabetes, vasculitis, and anti-double-stranded DNA, anti-Ro/SSA, anti-La/SSB, anti-nuclear antibodies, and anti-neuronal antibodies were negative. Brain MRI showed an increased symmetrical fluid attenuation inversion recovery (FLAIR) signal intensity in the posterior limb of the internal capsule. One year after onset, she developed dysphagia and weakness of the right hand. She began to have difficulties in walking and had a weight loss of 10 kilograms due to severe dysphagia 2 years after onset. She refused to perform percutaneous endoscopic gastrostomy and died 31 months after onset.
Patient 2 (bearing TBK1 c.2066 + 4A > G variant) was a 60-year-old man who presented with weakness of the left upper and lower limbs at the age of 59 years. The symptom progressed quickly to involve the right arm 5 months later. The patient had some difficulties in climbing upstairs and began to develop dysarthria and slight choking when eating. On examination 6 months after disease onset, obvious atrophy of muscles of upper and lower limbs and the tongue with fasciculations were noticed. Deep tendon reflexes were increased in all limbs but Babinski’s sign was not elicited. He was cognitively normal. The initial assessment of the ALSFRS-R score was 34/48 with an estimated progression rate of 2.3 points/month since symptom onset. Electromyography demonstrated acute and chronic neurogenic changes in the upper limb, lower limbs thoracic, and sternocleidomastoid muscles. Laboratory testing for diabetes, vasculitis, anti-nuclear antibodies, and anti-neuronal antibodies were negative. Brain MRI was normal. The patient refused to use percutaneous endoscopic gastrostomy or nasogastric tube feeding and died due to asphyxia caused by choking on food10 months after onset.
In this study, we identified five TBK1 variants by targeted next-generation sequencing in a Chinese cohort of 275 sALS patients and 15 fALS probands, including three missense variants (p.Ile37Leu, p.Ser398Pro, p.Tyr677Asn) and two novel intronic mutations (c.1522-3T > G, c.2066 + 4A > G). Online in silico programs were used to predict the potential impacts of the three missense variants on the structure and function of TBK1 protein with SIFT, Polyphen-2, and Mutation Taster. The p.Ile37Leu and p.Tyr677Asn were VUS while c.1192T > C (p. Ser398Pro) was a likely benign missense variant according to ACMG guidelines. However, the results of in silico programs should be interpreted with cautions and further experiments will need to determine the pathogenicity of these missense TBK1 variants.
The two novel intronic mutations (c.1522-3T > G, c.2066 + 4A > G) did not affect the function of TBK1 protein via amino-acid substitution. Instead, they interfered with normal pre-mRNA splicing due to disruption of the hnRNPA1 binding and forming aberrant splicing modes. Our results demonstrated that the c.1522-3T > G mutation promoted nearly 50% of abnormal transcripts with 3 different types of insertions and deletions (indels) in the junction of intron 13-exon 14 and the c.2066 + 4A > G mutation inhibited ∼75.0% inclusion of exon 19. The aberrant splicing modes caused premature stop codon and shifted the codon triplets of the genetic codes of mRNA, resulting in truncated protein without CCD2. Protein-truncated mutations in TBK1 caused the reduction of TBK1 transcripts and/or protein levels, which was an indication of pathogenicity (Pottier et al., 2015). We showed that the expression level of TBK1 with intronic mutations was lower than the wild type TBK1 through immunofluorescence analysis. CCD2 of TBK1 is involved in binding to and phosphorylating OPTN, which is an important regulator of autophagy (Swift et al., 2021). Previous research showed that altering the TBK1 downstream regulatory pathways might be the pathogenic mechanism of most TBK1 mutations (Freischmidt et al., 2015; Chia et al., 2018). Some TBK1 mutations may contribute to disease pathogenesis by disrupting mitophagic flux or inducing mitochondrial stress, preventing mitophagy components from performing cellular roles. Moreover, deficient mitophagy might stimulate innate immune pathways and promote the accumulation of toxic aggregates (Harding et al., 2021). Another research suggested that TBK1 haploinsufficiency might contribute to ALS/FTD through vesicular trafficking, a new molecular pathway different from previous thought (Lu et al., 2021). The pathogenic mechanisms above could explain the pathogenicity of these two intronic mutations in the two ALS patients.
In this study, the frequency of variants and LoF variants in TBK1 in sALS was 1.82% (5/275) and 0.73% (2/275), respectively. In previous studies in the Chinese population, the frequency of LoF TBK1 mutations in sporadic ALS ± FTD was between 0 and 0.62% (none in 271 cases, one out of 162 patients, one out of 207 patients, and one out of 608 cases, respectively) (Liu et al., 2021). Combined with the results of our cohort, the frequency of LoF mutations in sporadic ALS ± FTD was 0.38% (5/1,323) in the Chinese population. Only one TBK1 LoF mutation was identified in familial ALS ± FTD in the Chinese population (0.77%, 1/130) (Liu et al., 2021). The frequency of TBK1 LoF mutations in sporadic ALS ± FTD in the Chinese population is similar to the frequency of the Japanese population (0.42%, 3/713), lower than the frequency of the Korean population (0.78%, 1/129) and the European populations (1.7%, 11/665) (van der Zee et al., 2017). The frequency of TBK1 LoF mutations in fALS in the Chinese population is lower than the frequency of the European population (1.0%, 7/699) (de Majo et al., 2018).
The reported clinical features of ALS associated with TBK1 mutations are variable in the age of onset, progression of the disease, survival duration, and presence of cognitive impairment and extrapyramidal symptoms (Van Mossevelde et al., 2016). The ALS patients carrying TBK1 intronic mutations in the study showed typical clinical features of ALS without cognitive impairment. Their symptoms progressed relatively quickly and the survival duration was only 31 and 10 months, respectively. We supposed that both patients with intronic mutations manifesting severe clinical symptoms may be attributed to the indels in junction of intron-exon or the skipping of exons result from aberrant splicing modes, which produced TBK1 protein containing truncated CCD1 without CCD2. Besides, the patient with TBK1 c.2066 + 4A > G mutation showed more severe clinical features than the one with c.1522-3T > G mutation may be due to the higher proportion of the abnormal TBK1 transcripts.
In summary, we identified one likely benign missense variant, two missense VUS, and two novel intronic mutations of TBK1 in a Chinese ALS cohort. Combined with other studies in the Chinese population, the frequencies of LoF mutations in sporadic ALS ± FTD and familial ALS ± FTD were 0.38% and 0.77%, respectively. The pathogenic mechanisms of the two novel mutations were considered as disruption of the interaction between TBK1/OPTN due to truncated TBK1 protein result from aberrant splicing and TBK1 haploinsufficiency. Several other molecular pathways might also involve in the pathogenicity of the TBK1 mutations. We confirmed that substitution of a single nucleotide in the splicing regulators can result in partial or full-length exon or intronic sequence inclusion or exclusion, generating aberrant splicing and producing truncated protein. The findings also emphasize the importance of intronic sequencing in detecting variants and the need to combine with an analysis of pre-mRNA splicing modes in determining the pathogenic mechanisms of ALS. ASOs or gene-editing technology such as CRISPR/Cas9 may be potential treatment strategies worthy for further explorations by masking or disrupting the negative splicing-regulatory elements to correct the aberrant splicing (Jin et al., 2020; Li et al., 2020).
The data presented in the study can be found in the link below: https://www.ncbi.nlm.nih.gov/sra/PRJNA750742.
The studies involving human participants were reviewed and approved by the Fujian Medical University Union Hospital and Henan Provincial People’s Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Fujian Medical University Union Hospital and Henan Provincial People’s Hospital. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Z-YZ and H-PH designed and conceived the study. Z-YZ and Y-QL performed the analysis of mutations in all the patients. Y-QL and J-MC performed the functional experiments and analysis of data. Y-QL wrote the manuscript. Z-YZ critically revised the manuscript. All remaining authors participated in analysis of data, discussion of the final manuscript.
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 81671271 and 81974199), the Joint Funds for the Innovation of Science and Technology, Fujian province (Grant No. 2017Y9002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patients and their families for cooperation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.691534/full#supplementary-material
Supplementary Figure 1 | Splicing modes of TBK1 with intronic variants in HEK293T and HeLa cells. (A) T-A clone results of normal splicing mode (WT) and three aberrant splicing modes (deletion of 8bp, insertion of 2bp, and insertion of 10bp) of TBK1 with c.1522-3T > G in HEK293T and HeLa cells, respectively. Fractions below roman numerals I, II, and III showed the proportion of the four splicing modes (WT and aberrant) respectively, from three independent repetitions. (B) Schematic of normal splicing mode (WT) and aberrant splicing mode (MUT) of TBK1 with c.2066 + 4A > G in HEK293T and HeLa cells, respectively. Green lines show normal splicing modes, red lines show aberrant splicing modes, yellow boxes indicate exons, gray boxes indicate introns. E17: exon 17, E18: exon 18, E19: exon 19, E20: exon 20. Fractions below roman numerals I, II, and III showed the proportion of the two splicing modes (WT and MUT) respectively, from three independent repetitions.
Al-Chalabi, A., Hardiman, O., Kiernan, M. C., Chio, A., Rix-Brooks, B., and van den Berg, L. H. (2016). Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 15, 1182–1194. doi: 10.1016/S1474-4422(16)30199-5
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi: 10.1038/nrneurol.2011.150
Brooks, B. R., Miller, R. G., Swash, M., Munsat, T. L., and World Federation of Neurology Research Group on Motor Neuron Diseases (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. Mot. Neuron. Disord. 1, 293–299. doi: 10.1080/146608200300079536
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172.
Cartegni, L., Wang, J., Zhu, Z., Zhang, M. Q., and Krainer, A. R. (2003). ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 31, 3568–3571. doi: 10.1093/nar/gkg616
Chia, R., Chio, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. doi: 10.1016/S1474-4422(17)30401-5
Chou, S. M., and Norris, F. H. (1993). Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve 16, 864–869. doi: 10.1002/mus.880160810
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
de Majo, M., Topp, S. D., Smith, B. N., Nishimura, A. L., Chen, H. J., Gkazi, A. S., et al. (2018). ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 71, 266.e261–266.e210. doi: 10.1016/j.neurobiolaging.2018.06.015
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Muller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi: 10.1002/ana.21344
Gonzalez, A., Mannen, T., Cagatay, T., Fujiwara, A., Matsumura, H., Niesman, A. B., et al. (2021). Mechanism of karyopherin-beta2 binding and nuclear import of ALS variants FUS(P525L) and FUS(R495X). Sci. Rep. 11:3754. doi: 10.1038/s41598-021-83196-y
Harding, O., Evans, C. S., Ye, J., Cheung, J., Maniatis, T., and Holzbaur, E. L. F. (2021). ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. U.S.A. 118:e2025053118. doi: 10.1073/pnas.2025053118
Jin, M., Li, J. J., Xu, G. R., Wang, N., and Wang, Z. Q. (2020). Cryptic exon activation causes dystrophinopathy in two Chinese families. Eur. J. Hum. Genet. 28, 947–955. doi: 10.1038/s41431-020-0578-z
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lattante, S., Rouleau, G. A., and Kabashi, E. (2013). TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat. 34, 812–826. doi: 10.1002/humu.22319
Le Ber, I., De Septenville, A., Millecamps, S., Camuzat, A., Caroppo, P., Couratier, P., et al. (2015). TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol. Aging 36, 3116.e5–3116.e8. doi: 10.1016/j.neurobiolaging.2015.08.009
Li, J. J., Lin, X., Tang, C., Lu, Y. Q., Hu, X., Zuo, E., et al. (2020). Disruption of splicing-regulatory elements using CRISPR/Cas9 to rescue spinal muscular atrophy in human iPSCs and mice. Natl. Sci. Rev. 7, 92–101. doi: 10.1093/nsr/nwz131
Lin, X., Chen, H., Lu, Y. Q., Hong, S., Hu, X., Gao, Y., et al. (2020). Base editing-mediated splicing correction therapy for spinal muscular atrophy. Cell Res. 30, 548–550. doi: 10.1038/s41422-020-0304-y
Liu, X., He, J., Chen, L., Zhang, N., Tang, L., Liu, X., et al. (2021). TBK1 variants in Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging 97, 149.e9–149.e15.
Lu, Y., Almeida, S., and Gao, F. B. (2021). TBK1 haploinsufficiency in ALS and FTD compromises membrane trafficking. Acta Neuropathol. 142, 217–221. doi: 10.1007/s00401-021-02331-1
Ludolph, A., Drory, V., Hardiman, O., Nakano, I., Ravits, J., Robberecht, W., et al. (2015). A revision of the El Escorial criteria - 2015. Amyotroph. Lateral. Scler Frontotemporal. Degener. 16, 291–292. doi: 10.3109/21678421.2015.1049183
Oakes, J. A., Davies, M. C., and Collins, M. O. (2017). TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10:5. doi: 10.1186/s13041-017-0287-x
Pottier, C., Bieniek, K. F., Finch, N., van de Vorst, M., Baker, M., Perkersen, R., et al. (2015). Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92. doi: 10.1007/s00401-015-1436-x
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62.
Rosenbohm, A., Liu, M., Nagel, G., Peter, R. S., Cui, B., Li, X., et al. (2018). Phenotypic differences of amyotrophic lateral sclerosis (ALS) in China and Germany. J. Neurol. 265, 774–782. doi: 10.1007/s00415-018-8735-9
Swift, I. J., Bocchetta, M., Benotmane, H., Woollacott, I. O., Shafei, R., and Rohrer, J. D. (2021). Variable clinical phenotype in TBK1 mutations: case report of a novel mutation causing primary progressive aphasia and review of the literature. Neurobiol. Aging 99, 100.e9–100.e15. doi: 10.1016/j.neurobiolaging.2020.08.014
van der Zee, J., Gijselinck, I., Van Mossevelde, S., Perrone, F., Dillen, L., Heeman, B., et al. (2017). TBK1 mutation spectrum in an extended european patient cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Hum. Mutat. 38, 297–309. doi: 10.1002/humu.23161
Van Mossevelde, S., van der Zee, J., Gijselinck, I., Engelborghs, S., Sieben, A., Van Langenhove, T., et al. (2016). Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort. Brain 139, 452–467. doi: 10.1093/brain/awv358
Zhao, P., Wong, K. I, Sun, X., Reilly, S. M., Uhm, M., Liao, Z., et al. (2018). TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell 172, 731.e12–743.e12. doi: 10.1016/j.cell.2018.01.007
Keywords: amyotrophic lateral sclerosis (ALS), TANK-binding kinase 1 (TBK1), targeted next-generation sequencing, intronic variants, aberrant splicing
Citation: Lu Y-Q, Chen J-M, Lin H, Feng S-Y, Che C-H, Liu C-Y, Huang H-P and Zou Z-Y (2022) Novel Intronic Mutations of TBK1 Promote Aberrant Splicing Modes in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 15:691534. doi: 10.3389/fnmol.2022.691534
Received: 06 April 2021; Accepted: 20 January 2022;
Published: 24 February 2022.
Edited by:
Michele Papa, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Piergiorgio La Rosa, Sapienza University of Rome, ItalyCopyright © 2022 Lu, Chen, Lin, Feng, Che, Liu, Huang and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhang-Yu Zou, Zm11enp5QGZqbXUuZWR1LmNu; Hua-Pin Huang, aGgtcEAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.