 Hong-Jun Yan1*†
Hong-Jun Yan1*† Qiang Guo
Qiang Guo Sheng Luo
Sheng Luo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 05 January 2023
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.1081097
This article is part of the Research Topic Biomarkers, Mechanisms, and Treatment Strategies for Epilepsy View all 8 articles
The KCNK4 gene, predominantly distributed in neurons, plays an essential role in controlling the resting membrane potential and regulating cellular excitability. Previously, only two variants were identified to be associated with human disease, facial dysmorphism, hypertrichosis, epilepsy, intellectual/developmental delay, and gingival overgrowth (FHEIG) syndrome. In this study, we performed trio-based whole exon sequencing (WES) in a cohort of patients with epilepsy. Two de novo likely pathogenic variants were identified in two unrelated cases with heterogeneous phenotypes, including one with Rolandic epilepsy and one with the FHEIG syndrome. The two variants were predicted to be damaged by the majority of in silico algorithms. These variants showed no allele frequencies in controls and presented statistically higher frequencies in the case cohort than that in controls. The FHEIG syndrome-related variants were all located in the region with vital functions in stabilizing the conductive conformation, while the Rolandic epilepsy-related variant was distributed in the area with less impact on the conductive conformation. This study expanded the genetic and phenotypic spectrum of KCNK4. Phenotypic variations of KCNK4 are potentially associated with the molecular sub-regional effects. Carbamazepine/oxcarbazepine and valproate may be effective antiepileptic drugs for patients with KCNK4 variants.
The KCNK4 gene (OMIM* 605720), located at chromosomal locus 11q13.1, is highly expressed in the brain, predominantly in the cerebral cortex and hippocampus. It encodes the potassium two pore domain channels subfamily K member 4 (K2P4.1), a lipid-and mechano-sensitive K+ ion channel with 393 amino acids. The K2P4.1 channel, mainly distributed in the membrane of neurons, plays an essential role in controlling the resting membrane potential and regulating cellular excitability (Brohawn et al., 2012). Previously, only two KCNK4 variants were identified to be associated with human disease (Bauer et al., 2018; Mariani et al., 2021). The association between KCNK4 and human disease remains to be further researched with more cases.
Ion channelopathies shared overlapping phenotypes in genes encoding similar ion subunits(Wei et al., 2017). The KCNK4 gene was previously reported to be associated with facial dysmorphism, hypertrichosis, epilepsy, intellectual/developmental delay, and gingival overgrowth syndrome (FHEIG, OMIM# 618381) (Bauer et al., 2018). It is a syndromic neurodevelopmental disorder that shared notable phenotypic overlaps with the syndrome caused by variants of potassium channel-encoding genes KCNN3 and KCNH1 (Gripp et al., 2021). With increasing variants identified, the phenotypic spectrums of KCNN3 and KCNH1 are expanding, from syndromic neurodevelopmental disorder to pure epilepsy (Leu et al., 2020; Gripp et al., 2021). There remains room to explore the phenotypic spectrum of KCNK4.
In this study, we performed trio-based WES in a cohort of patients with epilepsy without acquired causes. Two novel likely pathogenic KCNK4 variants were identified in two unrelated cases, including one with Rolandic epilepsy and one with the FHEIG syndrome. The molecular structure alteration was analyzed by 3D protein modeling. We further reviewed all KCNK4 variants and analyzed the molecular sub-regional effects. The present study expanded the phenotypic and genetic spectrum of KCNK4.
A total of 156 children with epilepsy without any acquired causes were recruited from Guangdong 999 Brain Hospital from June 2020 to March 2021. Clinical information of the affected individuals was collected, including age at onset, type, and frequency of seizures, family history, systemic and neurological findings, and effective antiepileptic drugs. The structural abnormalities were detected by brain magnetic resonance imaging (MRI) scans. Long-term video electroencephalography (EEG) monitoring records were performed with electrodes being arranged according to the international standard of the 10–20 reduced montage system. The EEG results were reviewed by at least two qualified electroencephalographers.
This study adhered to the principles of the International Committee of Medical Journal Editors concerning patient consent for research or participation and received approval from the ethics committee of the Guangdong 999 Brain Hospital. Written informed consent was provided by the patient’s legal guardians.
Blood samples of the probands and their parents were collected for identifying the source of variants. Genomic DNAs were extracted from blood samples using the Qiagen Flexi Gene DNA kit (Qiagen, Hilden, Germany). WES was performed using a NextSeq500 sequencing instrument (Illumina, San Diego, California, United States) following previously described standard procedures (Wang et al., 2018; Luo et al., 2022). The sequencing data were generated by massively parallel sequencing with an average depth of >125x and >98% coverage of the capture region on the chip for obtaining high-quality reads that were mapped to the Genome Reference Consortium Human genome build 37 by Burrows-Wheeler alignment. Single-nucleotide point variants and indels were called with the Genome Analysis Toolkit.
Protein modeling was performed by using the Iterative Threading ASSEmbly Refinement software (I-TASSER1; Yang and Zhang, 2015). The confidence of each model was quantitatively measured by a C-score in the range of [−5, 2]. PyMOL Molecular Graphics System (Version 2.3.2; Schrödinger, LLC; New York, United States) was used for three-dimensional protein structure visualization and analysis. The consequences of all the missense variants were predicted by 21 in silico tools, including CADD, GERP, SIFT, Fathmm-MKL, fitCons, LRT, M_CAP, polyphen2_HDIV, PolyPhen2_HVAR, MutationTaster, MetaSVM, MetaLR, GenoCanyon, VEST3, Eigen, ClinPred, REVEL, phastCons, phyloP, SiPhy, and PROVEAN. The scores of these tools were obtained from the VarCards database (Li et al., 2018). The Varsite web server was used to analyze the changes in amino acid and hydrophobicity2 (Laskowski et al., 2020). The pathogenicity of variants was evaluated by the American College of Medical Genetics and Genomics guideline (ACMG) criterion (Richards et al., 2015).
R (version 4.0.3) was used for data processing. The two-sided Fisher exact test was used in the aggregate frequency analysis, which was recommended by ClinGen to compare the frequencies of the KCNK4 variants between the case-cohort and controls (Consortium, 2015). p < 0.05 was considered statistically significant.
Two heterozygous missense variants (c.416G > A/ p.Gly139Glu and c.698C > T/p.Pro233Leu) in the KCNK4 gene were identified in two unrelated patients, including one with FHEIG syndrome and one with Rolandic epilepsy (Figures 1A,B; Table 1). The two variants were de novo, consistent with an autosomal dominant inheritance pattern.
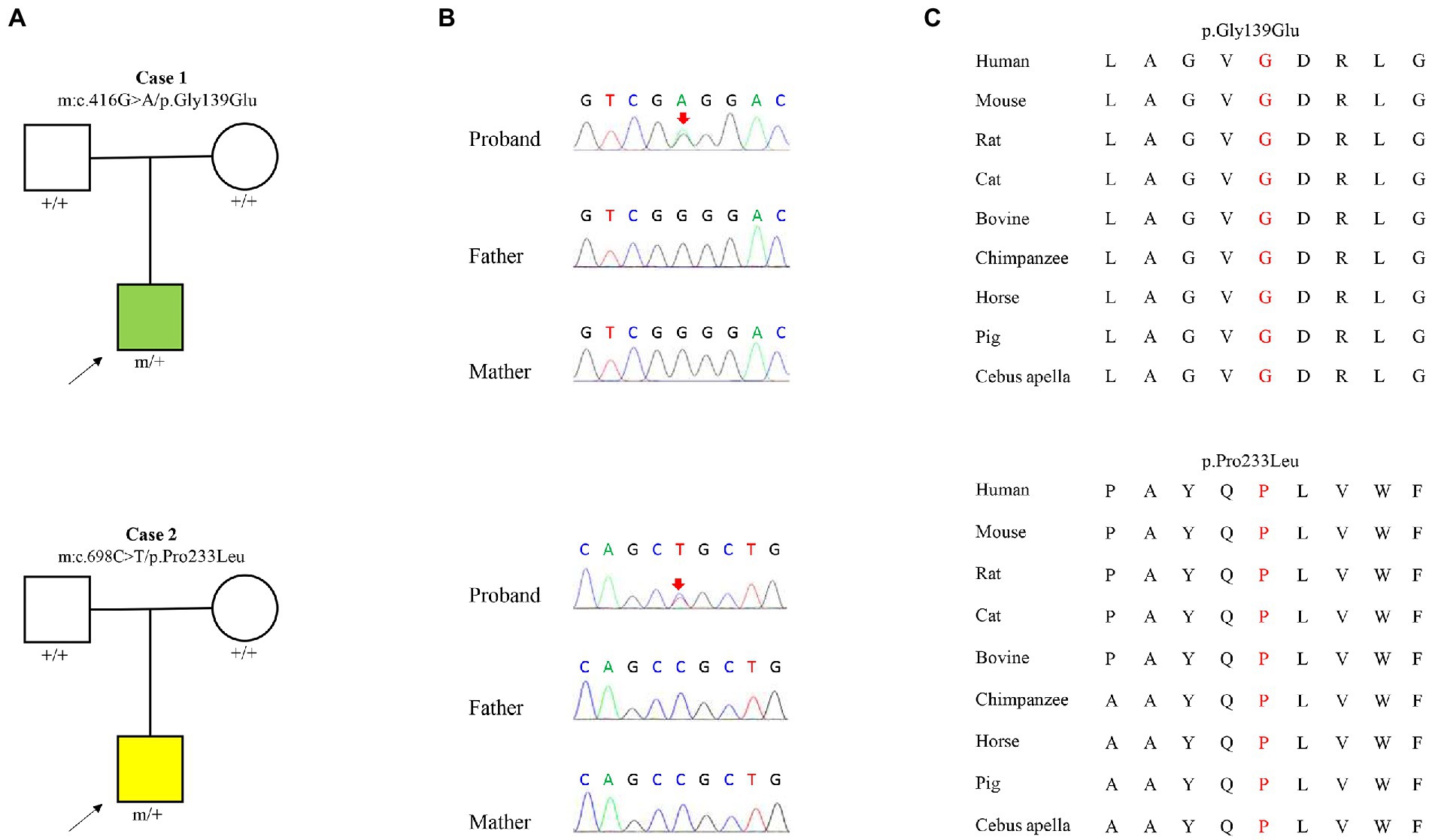
Figure 1. Genetic data of cases with KCNK4 variants. (A) Pedigrees of the two cases with KCNK4 variants. (B) DNA sequence chromatogram of the KCNK4 variants. Arrows indicate the positions of the mutations. (C) The amino acid sequence alignment of the six missense mutations shows that the two variants are highly conserved across species.
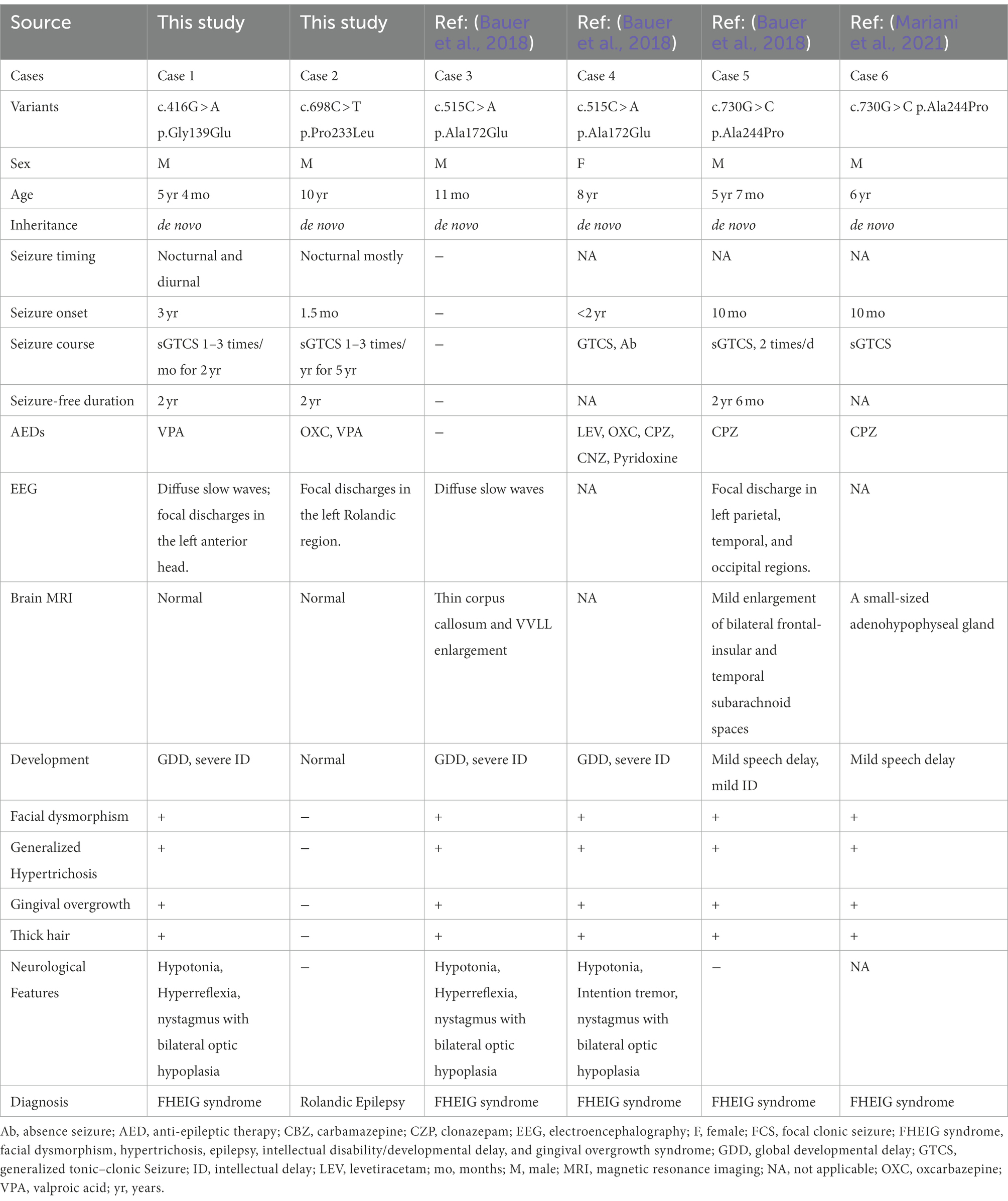
Table 1. Clinical features of individuals with KCNK4 variants.
The two variant was not presented in any controls, including the population in the gnomAD database, the ExAC database, the 1000 genome database, and the Epi25 WES Brown database (Table 2). The aggregate frequency of KCNK4 variants identified in this cohort was statistically higher than that in controls, including the gnomAD-all populations (2/312 vs. 0/282912, p = 1.21 × 10−6), the gnomAD-East Asian (2/312 vs. 0/19954, p = 2.36 × 10−4), the ExAC Population (2/312 vs. 0/121416, p = 6.55 × 10−6), the ESP 6500 Population (2/312 vs. 0/13006, p = 5.47 × 10−4), the 1000 Genomes Population (2/312 vs. 0/5008, p = 3.43 × 10−3), and the Epi25 WES Brower Population (2/312 vs. 0/35068, p = 7.752 × 10−5).
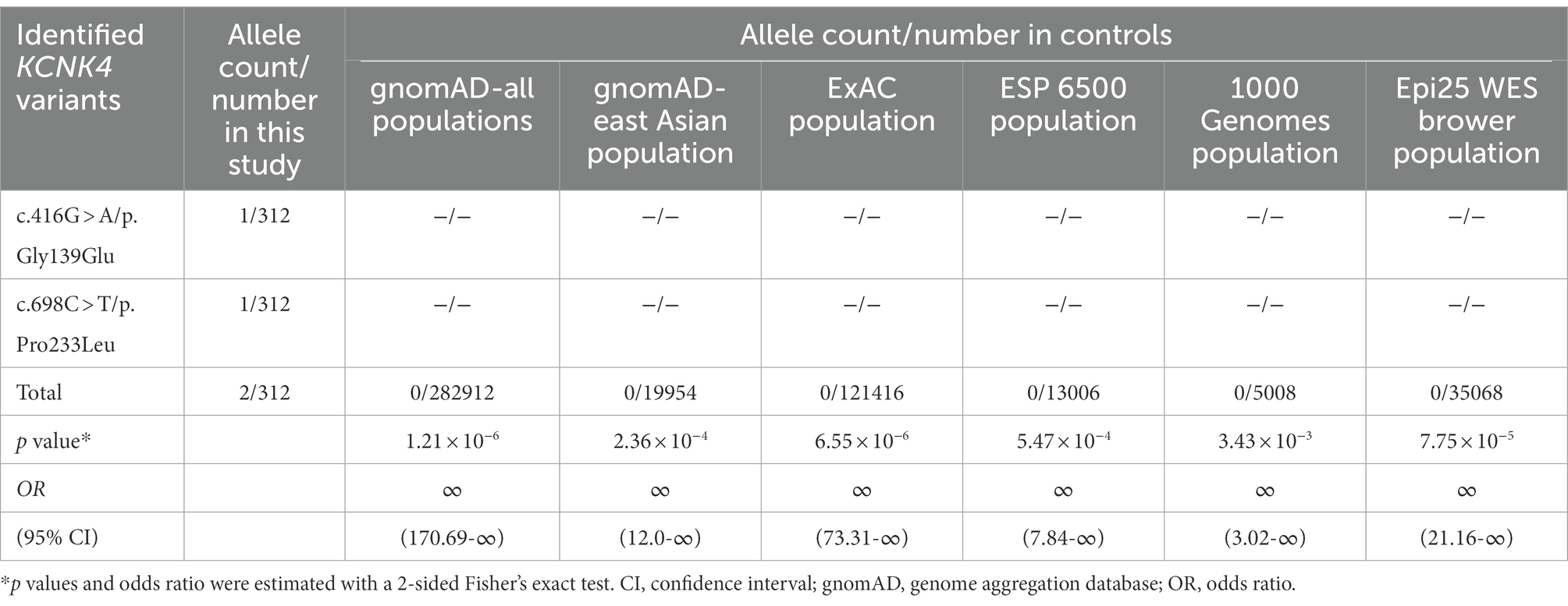
Table 2. Aggregate analysis of the frequency of KCNK4 variants identified in this study.
The KCNK4 variants identified in this study were predicted to be “Damaged” by numerous in silico tools, (16/17 algorithms for p.Gly139Glu and 12/17 algorithms for p.Pro233Leu) (Table 3). The two variants were also predicted to be “Conserved” by four in silico algorithms, including GERP, phastCons, phyloP, and SiPhy. Amino acid sequence alignment suggested that the variants were located in residue with high conservation across diverse species (Figure 1C). According to the ACMG guideline, the two variants were rated as “likely pathogenic” due to de novo origin (PS2), absence in controls (PM2), and being predicted to be damaged by multiple in silico tools (PP3; Table 3).
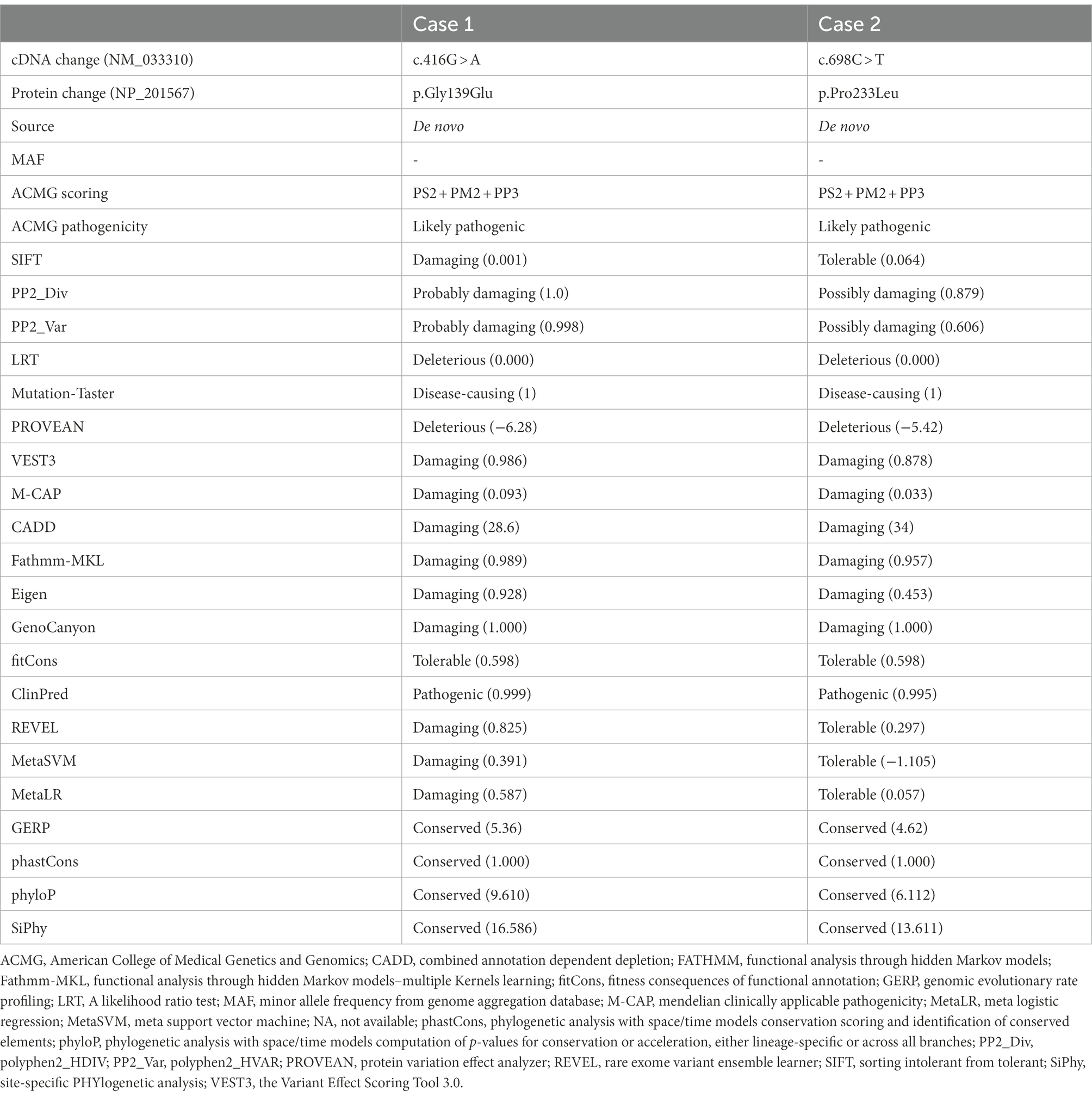
Table 3. Genetic characteristics and ACMG scoring of the KCNK4 variants identified in this study.
No pathogenic or likely pathogenic variants in other epilepsy-related genes were identified in the two patients (Zhou et al., 2018).
The detailed clinical information of the patients were summarized in Table 1. The missense variant p.Gly139Glu was identified in a boy with FHEIG syndrome. The first seizure occurred when he was 3 years old, mainly presenting as focal to bilateral tonic–clonic seizure (FBTCS). Thereafter, seizures were attacked 1–3 times per month. Since treatment with valproate (24.5 mg/kg/day) and oxcarbazepine (10 mg/kg/day), he became seizure-free. He also exhibits global developmental delay, microcephalus, hypertrichosis, and gingival overgrowth, which are highly consistent with the FHEIG syndrome. Electroencephalogram (EEG) showed diffuse 4–5 Hz slow waves and spikes-slow waves in the right anterior head.
The boy with the p.Pro233Leu variant presented a mild phenotype of Rolandic epilepsy. He suffered from FBTCS 1–3 times per year since 1.5-year-old. Seizure-free was achieved after being treated with valproate (21 mg/kg/day) and oxcarbazepine (9.1 mg/kg/day). He presented a purely epileptic phenotype without any other phenotype. EEG showed epileptic discharge in the Rolandic area with right-side dominance (Figure 2).
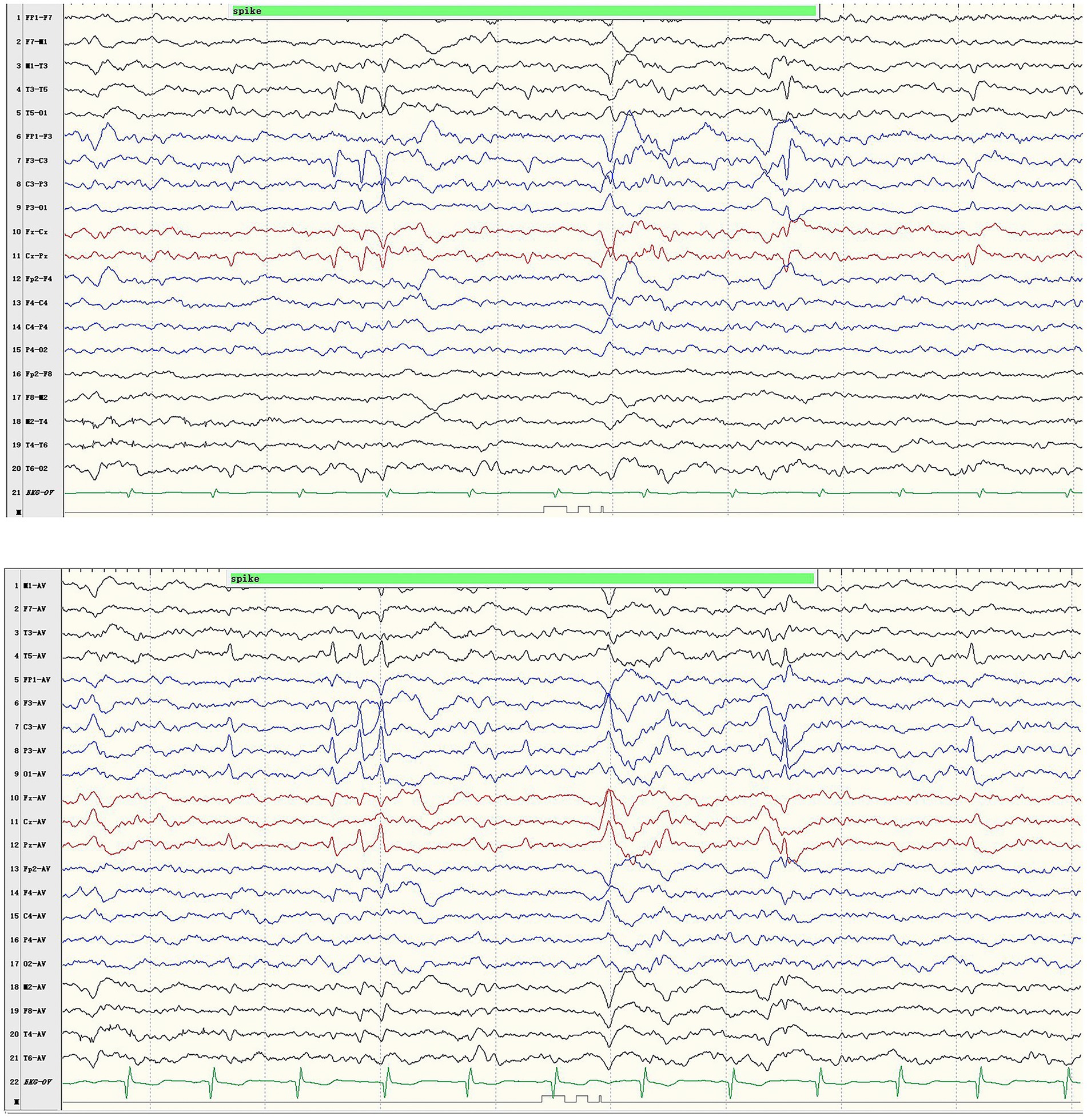
Figure 2. Representative EEG results of case 1. Ictal EEG recorded focal discharges in the centrotemporal area, prominently on the left side.
The two patients were born to unrelated parents after an uneventful pregnancy. The brain MRI detected no abnormalities.
The KCNK4 gene encodes a highly conservated lipid-and mechano-sensitive K+ ion channel with 393 amino acids. It contains four transmembrane segments (TM 1–4) and two pore domains (P 1 and P 2; Figure 3A). The variant p.Gly139Glu was located in the TM2 segment, while the variant p.Pro233Leu was located in the linker between the P2 domain and TM4 segment. As shown in protein modeling, the two variants were not predicted to alter any hydrogen bonds forming with the surrounding residues (Figures 3B,C). The substitution of Glutamic acid in residues Gly139 alters the polarity of amino acids (Figure 3B), while the substitution of Leucine in residue Pro233 seems not to affect the polarity/charge (Figure 3C). The two variants are all expected to cause hydrophobicity changes that may lead to functional abnormities, based on the Fauchère and Pliska hydrophobicity scale calculating fifteen physicochemical parameters of the amino acid side chain (Fauchere et al., 1988; Figure 3D).
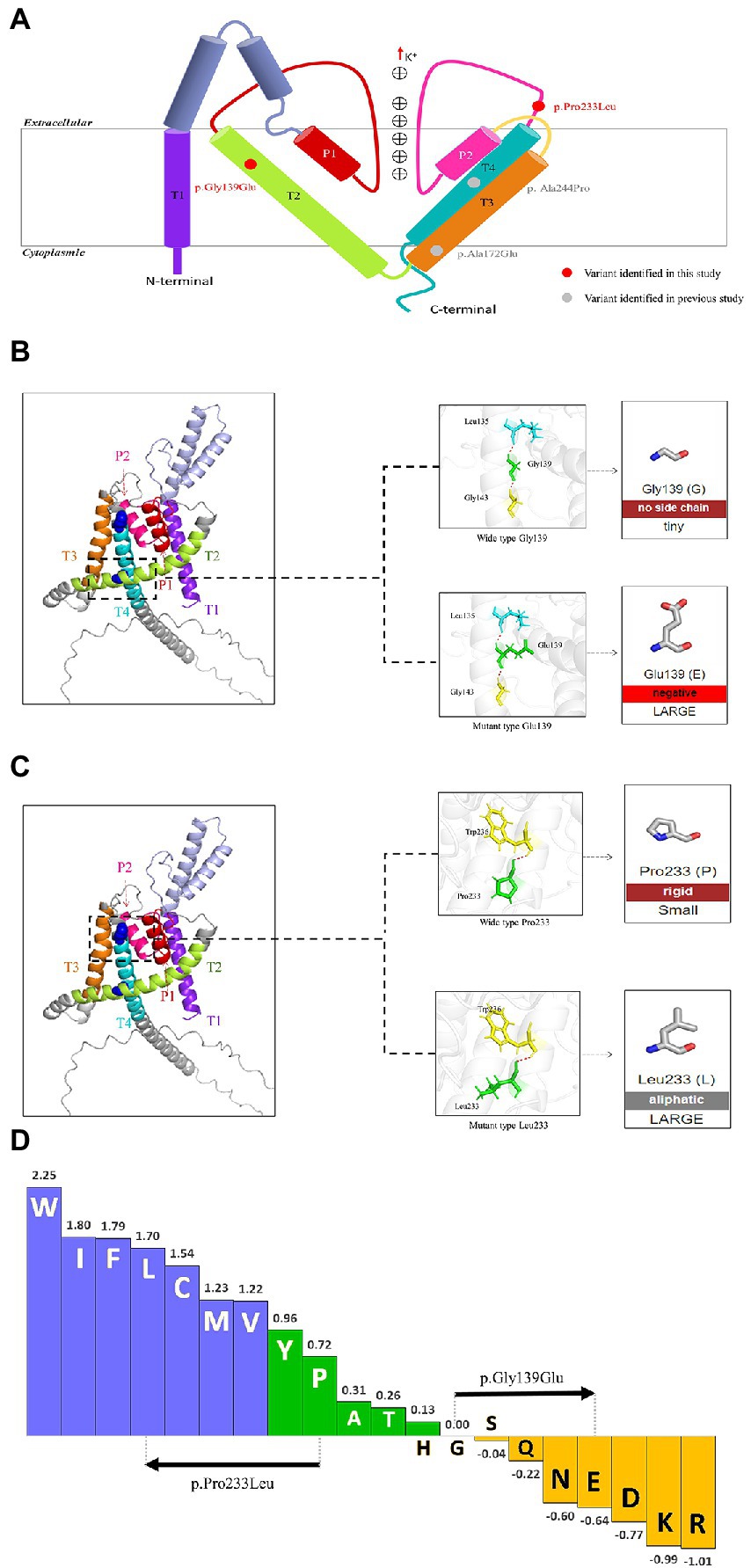
Figure 3. Schematic illustration of KCNK4 variants (A) Schematic of the location of all KCNK4 variants. Variants identified in this study were shown in red color. Variants reported previously were shown in gray color. (B) Protein modeling of the p.Gly139Glu variants. The dashed line box in the left panel indicated the wild-type structure of K2P4.1 channels of p.Gly139Glu. The substitution of Glutamic acid in residue Gly139 was not predicted to alter hydrogen bonds but the polarity of amino acids. (C) Protein modeling of the p.Pro233Leu variants. The dashed line box in the left panel indicated the wild-type structure of K2P4.1 channels of the p.Pro233Leu. The substitution of Leucine in residue Pro233 seems not to affect the hydrogen bonds or polarity/charge. (D) Fauchère and Pliska hydrophobicity scale exhibited the hydrophobicity of 20 amino acids. Abscissa: from left to right, hydrophobicity gradually decreased. Blue amino acids were hydrophobic, green amino acids were neutral, and yellow amino acids were hydrophilic. Amino acids with high positive values are more hydrophobic, whereas amino acids with low negative values are more hydrophilic.
The KCNK4 gene encodes the K2P4.1 channel, which is a highly conserved voltage-insensitive potassium channel. The K2P4.1 channel, predominantly distributed in neurons, plays a vital role in controlling the resting membrane potential and subsequently regulating neuronal excitability (Brohawn et al., 2012). To date, only two KCNK4 mutations were found to be associated with human disease, the FHEIG syndrome. In this study, we identified two de novo likely pathogenic variants of KCNK4 in two unrelated cases. The two patients exhibited distinct clinical phenotypes, including one with Rolandic epilepsy and one with FHEIG syndrome. The variants were novel and not reported previously, with no frequencies in any controls. The aggregative frequencies were significantly higher in the epilepsy cohort than that in the controls. This study suggested that the KCNK4 gene was potentially associated with epilepsy without other neurological phenotypes. The phenotypic spectrum of KCNK4 potentially ranged from mild epilepsy such as Rolandic epilepsy to severe syndromic neurodevelopment disorder.
In mice, knockout Kcnk4 exhibited no abnormalities in the nervous system and no significant susceptibility to pharmacologically induced seizures3 (Heurteaux et al., 2004). In the normal control of the gnomAD database, 12 frameshift and 13 stop-gained variants were detected. The probability of being loss-of-function intolerant (pLI) for the KCNK4 gene is 0.01, indicating that loss-of-function variants of KCNK4 were not susceptible to natural selection (Karczewski et al., 2020). On the other hand, the pathogenic KCNK4 mutations reported previously were all missense, which were functionally validated to be gain-of-function (GOF; Bauer et al., 2018). Thus, it is possible that GOF is the pathogenic mechanism and heterozygous missense variants were the main pathogenic genotype for KCNK4. The variants identified in this study were all missense, located in highly conserved residues across species, predicted to be damaging by numerous algorithms, and categorized as likely pathogenic by the ACMG criteria. The variants presented limited impact on protein structure (Figure 3), potentially reflecting the less possibility of loss-of-function. Therefore, the two variants are potentially considered to be disease-causing. However, the functional alteration of the two variants remains to be functionally validated.
Previously, two KCNK4 mutations were reported to be associated with the FHEIG syndrome (Bauer et al., 2018; Mariani et al., 2021). Clinically, the FHEIG syndrome included not only neurological symptoms (epilepsy, development delays, and intellectual disability) but also non-neurological symptoms (facial dysmorphism, hypertrichosis, and gingival overgrowth) (Bauer et al., 2018). The KCNK4 gene is expressed almost exclusively in the brain4. Generally, tissue-specific expression is the basis of gene function and subsequently the clinical phenotype. Such pleiotropy of KCNK4 is unexpected and low persuasive, and the pure epileptical phenotypes of KCNK4 are receivable (such as the Rolandic epilepsy in Case 1). Nevertheless, the recurrence of the FHEIG syndrome in Case 2 provided further clinical evidence in supporting the association between KCNK4 and the FHEIG syndrome. The mechanisms of multisystem symptoms of KCNK4 are of great challenge and warrant further studies.
Potassium channels play vital roles in neurodevelopmental diseases, particularly in epilepsy. Previously, a series of potassium-encoding genes were initially reported to be the severest phenotypes, such as developmental and epileptic encephalopathy (KCNA2, KCNT1, KCNT2, and KCNB1; Barcia et al., 2012; Torkamani et al., 2014; Syrbe et al., 2015; Gururaj et al., 2017) and syndromic neurodevelopment disorder (KCNN3 and KCNH1) (Kortum et al., 2015; Bauer et al., 2019). With increasing identification of variants, these genes were recently found to be associated with mild phenotypes, suggesting the possible phenotypic spectrum for potassium-encoding genes. In this study, we reported the first variant that is associated with Rolandic epilepsy, expanding the phenotypic spectrum of KCNK4.
Previous studies suggested that the phenotypic variation was potentially associated with the location of variants, which was known as the molecular sub-regional effect (Tang et al., 2020; Liao et al., 2022). Functional K2P4.1 channels contain four transmembrane segments (TM 1–4) and two pore domains (P 1–2), among which TM4 and TM2-TM3 segment in stabilizing the conductive conformation. Rotation of TM4 about a central hinge seals the intramembrane opening in the conductive state, preventing the lipid block of the cavity and permitting ion entry (Miller and Long, 2012). Additional rotation of TM4 by the TM2-TM3 segment further stabilizes the conductive conformation (Miller and Long, 2012). It is noted that the variants of patients with FHEIG syndrome (case 1 and cases previously reported) are all located in the TM2-TM3 segment or TM4, whereas the variant of the patient with Rolandic epilepsy is distributed in the linker between P2 and TM4 (case 2; Figure 3A). It is speculated that the variants in the area with the function of conductive conformation led to the severe phenotype of FHEIG syndrome, while variants in other areas are possibly associated with mild phenotypes such as Rolandic epilepsy. This potential molecular sub-regional effect may be one of the possible explanations for the phenotypic variation, which needed more cases to confirm.
To date, a total of four KCNK4 variants (including two from this study) were identified in six patients to be associated with human disease (Table 1). The patients with an identical variant also presented with subtle heterogeneous phenotypes, which might be partially the result of environmental factors and/or modified genes that warrants further investigation. From the perspective of prognosis, almost all patients responded well to carbamazepine/oxcarbazepine and valproate, indicating the potential clinical implication of treatment (Table 1).
In summary, this study expanded the genetic and phenotypic spectrum of KCNK4-related disease, providing further clinical evidence in supporting the pathogenic role of KCNK4. The phenotypic variation was potentially associated with the molecular sub-regional effect. Carbamazepine/oxcarbazepine and valproate were effective antiepileptic drugs for patients with KCNK4 variants.
The datasets generated for this study can be found in the Genebank repository with the following accession numbers: BankIt2652432 Seq1 OP997444, BankIt2652432 Seq2 OP997445.
The studies involving human participants were reviewed and approved by the Ethics Committee of the Guangdong 999 Brain Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
SL and H-JY designed the study and administered the project and revised and write the manuscript with the contribution of all authors. H-JY, QG, and J-HZ completed the collection of the clinical data. Y-yH and LJ analyzed the data.
This research was funded by Guangzhou Science and Technology Planning Project (202102080024).
We deeply appreciated the participation of patients and clinicians in this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. ^https://zhanglab.ccmb.med.umich.edu/I-TASSER/
2. ^https://www.ebi.ac.uk/thornton-srv/databases/VarSite
3. ^http://www.informatics.jax.org/marker/MGI:1298234
4. ^https://www.proteinatlas.org/ENSG00000182450-KCNK4/tissue
Barcia, G., Fleming, M. R., Deligniere, A., Gazula, V. R., Brown, M. R., Langouet, M., et al. (2012). De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44, 1255–1259. doi: 10.1038/ng.2441
Bauer, C. K., Calligari, P., Radio, F. C., Caputo, V., Dentici, M. L., Falah, N., et al. (2018). Mutations in KCNK4 that affect gating cause a recognizable neurodevelopmental syndrome. Am. J. Hum. Genet. 103, 621–630. doi: 10.1016/j.ajhg.2018.09.001
Bauer, C. K., Schneeberger, P. E., Kortum, F., Altmuller, J., Santos-Simarro, F., Baker, L., et al. (2019). Gain-of-function mutations in KCNN3 encoding the small-conductance ca(2+)-activated K(+) channel SK3 cause Zimmermann-Laband syndrome. Am. J. Hum. Genet. 104, 1139–1157. doi: 10.1016/j.ajhg.2019.04.012
Brohawn, S. G., del Marmol, J., and MacKinnon, R. (2012). Crystal structure of the human K2P TRAAK, a lipid-and mechano-sensitive K+ ion channel. Science 335, 436–441. doi: 10.1126/science.1213808
Consortium, C. (2015). Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591. doi: 10.1038/nature14659
Fauchere, J. L., Charton, M., Kier, L. B., Verloop, A., and Pliska, V. (1988). Amino acid side chain parameters for correlation studies in biology and pharmacology. Int. J. Pept. Protein Res. 32, 269–278. doi: 10.1111/j.1399-3011.1988.tb01261.x
Gripp, K. W., Smithson, S. F., Scurr, I. J., Baptista, J., Majumdar, A., Pierre, G., et al. (2021). Syndromic disorders caused by gain-of-function variants in KCNH1, KCNK4, and KCNN3-a subgroup of K(+) channelopathies. Eur. J. Hum. Genet. 29, 1384–1395. doi: 10.1038/s41431-021-00818-9
Gururaj, S., Palmer, E. E., Sheehan, G. D., Kandula, T., Macintosh, R., Ying, K., et al. (2017). A De novo mutation in the sodium-activated Potassium Channel KCNT2 alters ion selectivity and causes epileptic encephalopathy. Cell Rep. 21, 926–933. doi: 10.1016/j.celrep.2017.09.088
Heurteaux, C., Guy, N., Laigle, C., Blondeau, N., Duprat, F., Mazzuca, M., et al. (2004). TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 23, 2684–2695. doi: 10.1038/sj.emboj.7600234
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kortum, F., Caputo, V., Bauer, C. K., Stella, L., Ciolfi, A., Alawi, M., et al. (2015). Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 47, 661–667. doi: 10.1038/ng.3282
Laskowski, R. A., Stephenson, J. D., Sillitoe, I., Orengo, C. A., and Thornton, J. M. (2020). VarSite: disease variants and protein structure. Protein Sci. 29, 111–119. doi: 10.1002/pro.3746
Leu, C., Bautista, J. F., Sudarsanam, M., Niestroj, L. M., Stefanski, A., Ferguson, L., et al. (2020). Neurological disorder-associated genetic variants in individuals with psychogenic nonepileptic seizures. Sci. Rep. 10:15205. doi: 10.1038/s41598-020-72101-8
Li, J., Shi, L., Zhang, K., Zhang, Y., Hu, S., Zhao, T., et al. (2018). VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46, D1039–D1048. doi: 10.1093/nar/gkx1039
Liao, W. P., Chen, Q., Jiang, Y. W., Luo, S., and Liu, X. R. (2022). Editorial: sub-molecular mechanism of genetic epilepsy. Front. Mol. Neurosci. 15:958747. doi: 10.3389/fnmol.2022.958747
Luo, S., Liu, Z. G., Wang, J., Luo, J. X., Ye, X. G., Li, X., et al. (2022). Recessive LAMA5 variants associated with partial epilepsy and spasms in infancy. Front. Mol. Neurosci. 15:825390. doi: 10.3389/fnmol.2022.825390
Miller, A. N., and Long, S. B. (2012). Crystal structure of the human two-pore domain potassium channel K2P1. Science 335(6067), 432–436. doi: 10.1126/science.1213274
Mariani, P., Zhurakivska, K., Santoro, R., Laino, G., Russo, D., and Laino, L. (2021). Hereditary gingival fibromatosis associated with the missense mutation of the KCNK4 gene. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 131, e175–e182. doi: 10.1016/j.oooo.2020.08.006
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Syrbe, S., Hedrich, U. B. S., Riesch, E., Djemie, T., Muller, S., Moller, R. S., et al. (2015). De novo loss-or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 47, 393–399. doi: 10.1038/ng.3239
Tang, B., Li, B., Gao, L. D., He, N., Liu, X. R., Long, Y. S., et al. (2020). Optimization of in silico tools for predicting genetic variants: individualizing for genes with molecular sub-regional stratification. Brief. Bioinform. 21, 1776–1786. doi: 10.1093/bib/bbz115
Torkamani, A., Bersell, K., Jorge, B. S., Bjork, R. L., Friedman, J. R., Bloss, C. S., et al. (2014). De novo KCNB1 mutations in epileptic encephalopathy. Ann. Neurol. 76, 529–540. doi: 10.1002/ana.24263
Wang, J. Y., Zhou, P., Wang, J., Tang, B., Su, T., Liu, X. R., et al. (2018). ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: toward understanding the mechanism underlying phenotypic variation. Neurogenetics 19, 9–16. doi: 10.1007/s10048-017-0528-2
Wei, F., Yan, L. M., Su, T., He, N., Lin, Z. J., Wang, J., et al. (2017). Ion Channel genes and epilepsy: functional alteration, pathogenic potential, and mechanism of epilepsy. Neurosci. Bull. 33, 455–477. doi: 10.1007/s12264-017-0134-1
Yang, J., and Zhang, Y. (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. doi: 10.1093/nar/gkv342
Keywords: KCNK4, FHEIG syndrome, rolandic epilepsy, molecular sub-regional effects, phenotypic variations
Citation: Yan H-J, He Y-y, Jin L, Guo Q, Zhou J-H and Luo S (2023) Expanding the phenotypic spectrum of KCNK4: From syndromic neurodevelopmental disorder to rolandic epilepsy. Front. Mol. Neurosci. 15:1081097. doi: 10.3389/fnmol.2022.1081097
Edited by:
Shaunik Sharma, The University of Iowa, United StatesReviewed by:
Saima Siddiqi, Institute of Biomedical and Genetic Engineering (IBGE), PakistanCopyright © 2023 Yan, He, Jin, Guo, Zhou and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheng Luo, ✉ c2hlbmdsdW9AMTg5LmNu; Hong-Jun Yan, ✉ eWhqODQ0MTI4NUBxcS5jb20=
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.