 Jay S. Schneider*
Jay S. Schneider* Garima Singh
Garima Singh- Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA, United States
The precise mechanisms initiating and perpetuating the cellular degeneration in Parkinson’s disease (PD) remain unclear. There is decreased expression of the main brain gangliosides, and GM1 ganglioside in particular, in the PD brain along with decreased expression of the genes coding for the glycosyltranferase and the sialyltransferase responsible for the synthesis of these brain gangliosides. However, potentially important pathogenic mechanisms contributing to the neurodegeneration in PD may also include altered levels of expression of genes involved in glycosylation, sialylation and sphingolipid synthesis and metabolism. Although various studies have described pathological lipid and glycolipid changes in PD brain, there have been limited studies of expression of glycobiology-related genes in PD brain. The current study was performed as an initial attempt to gain new information regarding potential changes in glycoprotein and glycolipid-related genes in PD by investigating the gene expression status for select glycosyltransferases, sialyltransferases, sialidases, sphingosine kinases, and lysosomal enzymes in the substantia nigra and putamen from patients with PD and neurologically normal controls. Results showed altered expression of glycosyltransferase genes (B3GALT2 and B4GALT1) potentially involved in microglial activation and neuroinflammation, sphingosine-1-phosphate (S1P) modulators (SPHK1, SPHK2, and SGPL1) involved in sphingolipid synthesis and metabolism, polysialyltransferase genes (ST8SIA2 and ST8SIA4) that encode enzymes responsible for polysialic acid (polySia) biosynthesis, and the sialidase NEU4, expression of which has been linked to the clearance of storage materials from lysosomes. The data presented here underscore the complexity of the glycolipid/sphingolipid dysregulation in the PD brain and continued and expanded study of these processes may not only provide a greater understanding of the complex roles of aberrant glycosylation sialylation, and sphingolipid synthesis/metabolism in the pathophysiology of PD but may identify potential druggable targets for PD therapeutics.
Introduction
Parkinson’s disease (PD) is a complex progressive neurodegenerative disorder primarily characterized by the loss of nigrostriatal dopamine-producing neurons in the substantia nigra. Although the majority of cases of PD are idiopathic in origin, several mechanisms have been proposed to explain the onset and progression of the neurodegeneration in PD including mitochondrial dysfunction, increased oxidative stress and oxidative damage, α-synuclein aggregation and associated toxicity, and lysosomal and autophagic dysfunction, among other potential contributing factors (Doria et al., 2016). In addition, various abnormalities in the content and composition of various lipids, including gangliosides, have been reported in PD brain [ex. (den Jager, 1969; Riekkinen et al., 1975; Seyfried et al., 2018)]. Recently, dysregulation of ceramide synthesis and metabolism have been suggested to play a role in PD-associated neurodegeneration (Plotegher et al., 2019) and lipdomic studies have reported PD-specific lipid alterations detected in brain and in plasma that have been suggested to promote PD-associated neurodegeneration at least in part through promoting α-synuclein aggregation, neuroinflammation, and dysfunction of autophagic processing [see Alecu and Bennett (2019) for review].
In addition to changes in lipid content and metabolism in the PD brain, some studies have also reported altered glycosylation and sialylation in PD brain (Videira and Castro-Caldas, 2018; Wilkinson et al., 2021). Altered levels of sialylation and fucosylation have also been reported in serum samples from PD patients and interestingly, mainly in male patients (Varadi et al., 2019). Additionally, many proteins are synthesized in the endoplasmic reticulum (ER) where many of them undergo glycoslylation and functionalization. ER stress has been suggested to be one of the pathological mechanisms contributing to PD (Mercado et al., 2013; Tsujii et al., 2015), and aberrant glycosylation has been suggested to contribute to an overload of the ER in PD brain with underglycosylated proteins (Videira and Castro-Caldas, 2018). Oxidative stress and inflammation have also been suggested to potentially trigger abnormal glycosylation in PD (Videira and Castro-Caldas, 2018).
Although various studies have described pathological lipid and glycolipid changes in PD brain, there have been limited studies of expression of glycobiology-related genes in PD brain (see (Alecu and Bennett, 2019) for review) and how dysregulation of the expression of these genes may contribute to PD-like neurodegeneration. In contrast, genes related to glycobiology have been examined in Huntington’s disease (HD) transgenic mice as well as in the caudate nucleus from human HD subjects (Desplats et al., 2007) where a number of glycosyltransferases and sialyltransferases were found to be significantly changed compared to normal controls. In particular, ganglioside metabolism genes ST3GAL5, ST8SIA3, B4GALNT1, and ST3GAL2 had significantly decreased expression in HD caudate compared to control caudate (Desplats et al., 2007). We previously described significantly decreased expression of gene B3GALT4 and ST3GAL2 in residual dopaminergic neurons in the PD substantia nigra (Schneider, 2018), consistent with an earlier finding of decreased expression of the main brain gangliosides (GM1, GD1a, GD1b, and GT1b) in PD substantia nigra (Seyfried et al., 2018). The current study was performed to gain additional information regarding potential changes in glycoprotein and glycolipid metabolism in PD by investigating the gene expression status for select glycosyltransferases (B3GALT4, B4GALT1, B4GALNT1, and B4GALT5), sialyltransferases (ST6GALNAC4, ST8SIA2, ST8SIA4, and NCAM1 (substrate for ST8SIA2, ST8SIA4), sialidases (NEU1, NEU3, and NEU4), sphingosine kinases (S1P modulators) (SPHK1, SPHK2, and SGPL1) (S1P lyase), and lysosomal enzymes (GBA (β-glucocerebroside), GLB1 (β-galactosidase)) in the substantia nigra and putamen in patients with PD and neurologically normal controls. These genes were chosen to expand upon limited previous preliminary data that suggested potential abnormal expression levels of some glycosyltransferases, sialyltransferases, sialidases, and S1P modulators in PD brain (Schneider and Singh, 2019).
Materials and methods
Human brain tissue collection
Coded/anonymous substantia nigra-containing tissue blocks were obtained through the NIH NeuroBioBank and sourced from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, the Harvard Brain Tissue Resource Center, which is supported in part by HHSN-271-2013-00030C, and from the Human Brain and Spinal Fluid Resource Center, VA, West Los Angeles Healthcare Center, 11301 Wilshire Blvd., Los Angeles, CA, which is sponsored by NINDS/NIMH, the National Multiple Sclerosis Society, and the Department of Veterans Affairs. Coded/anonymous putamen samples were obtained solely from the Human Brain and Spinal Fluid Resource Center, VA, West Los Angeles Healthcare Center. The clinical diagnosis of Parkinson’s disease was confirmed at autopsy by presence of gross depigmentation of the SN and microscopic confirmation of SN cell loss and presence of Lewy bodies in the SN and normal findings in other brain regions sampled. Frozen tissue blocks containing the SN were stored at −80°C and warmed to −20°C for dissection of samples. Dissected substantia nigra samples (containing the pars compacta region (SNc)) and dissected putamen samples (taken from dorsal putamen) were placed in sterile Eppendorf tubes and were rapidly refrozen in powdered dry ice. Standard BL2 procedures for handling human tissues were observed. Coded/anonymous non-neurological disease control tissues were obtained from the same sources mentioned above. Subject characteristics are described in Table 1 regarding substantia nigra samples and Table 2 regarding putamen samples.
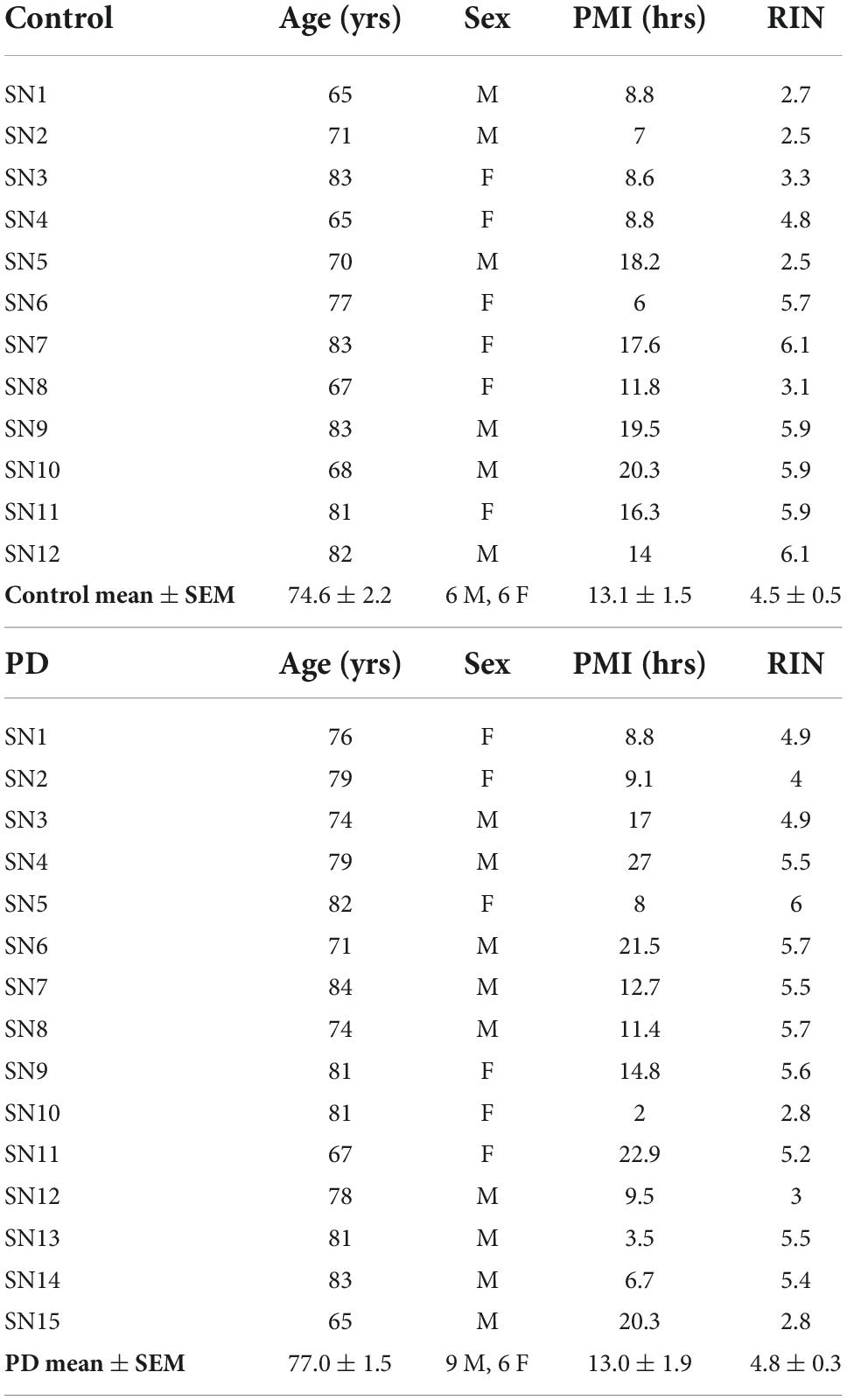
Table 1. Subject and tissue characteristics: Substantia nigra.
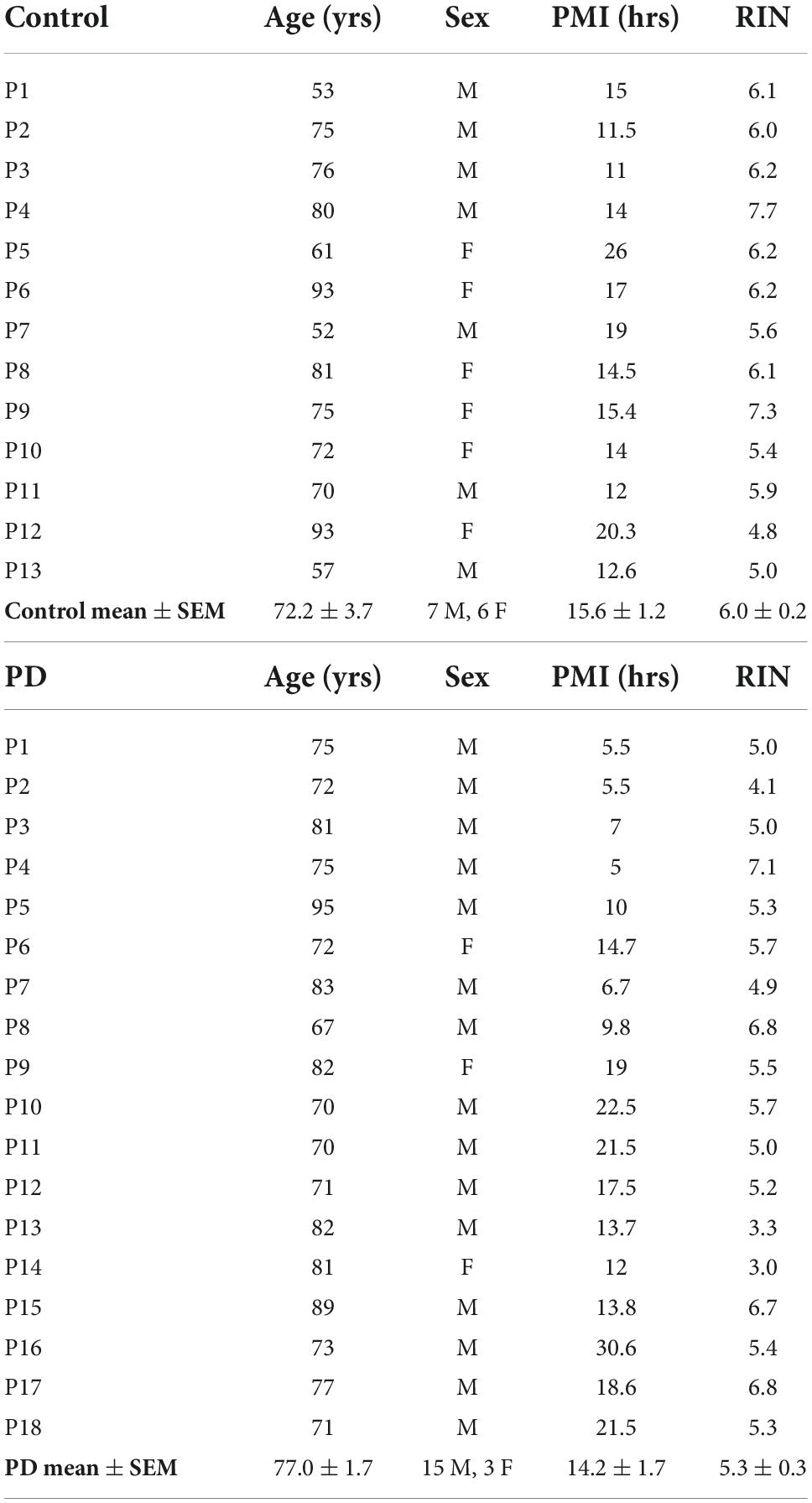
Table 2. Subject and tissue characteristics: Putamen.
Ribonucleic acid isolation and quantitative real-time polymerase chain reaction
Ribonucleic acid (RNA) was extracted from the frozen SN and putamen samples using Zymo Direct-zol RNA miniprep Plus. During RNA isolation the DNase digestion step was performed with RNase-free DNase I (included in the kit) to eliminate genomic DNA contamination. To determine RNA quality, all the RNA samples were analyzed on an Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano kit per the manufacturer’s instructions. The RNA integrity (RIN) numbers for the samples are reported in Tables 1, 2. cDNA was prepared using NEB Protoscript II First strand cDNA synthesis, and Real-Time PCR was performed using a Roche LightCycler 480 with Roche LightCycler 480 SYBR Green I Mastermix. Real-Time PCR was carried out using commercially sourced and validated primers from GeneGlobe Qiagen against human genes B3GALT2, B4GALT1, B4GALT5, B4GALNT1, GLB1, GBA, NCAM1, NEU1, NEU3, NEU4, SGPL1, SPHK1, SPHK2, ST6GALNAC4, ST8SIA2, and ST8SIA4 (GeneGlobe IDs are provided in Supplementary Table 1). The ΔΔCt method was used to calculate mRNA expression change relative to GAPDH (housekeeping gene) expression.
Statistical analyses
Raw data were subjected to outlier analysis using Grubbs test to identify and remove values that were significant outliers from the other values in each dataset. Statistical analyses were then performed using unpaired t-test using GraphPad Prism software (v9) with significance for gene expression change set at P < 0.05 (GraphPad Software, San Diego, California USA). Data were converted to fold change relative to control for graphical presentation.
Results
Subjects and controls were well matched for age, post-mortem interval (PMI), and RNA integrity number (RIN) for both substantia nigra (Table 1) and putamen (Table 2) samples. There were no statistically significant differences between subjects and controls on any of these measures for either tissue type. Male/female ratios for substantia nigra samples were 50:50 for controls and 60:40 for subjects with PD. For putamen samples, male/female ratios were 54:46 for controls and 83:17 for subjects with PD. There were no significant sex-related differences in any of the gene expression data in either brain structure (data not shown).
Significant differences in gene expression were found in the substantia nigra from patients with PD compared to controls. Fold changes relative to control are shown in Figure 1 for the genes assayed. Gene expression for glycosyltransferases B3GALT2 and B4GALT1, the S1P modulator SGPL1, and the lysosomal enzyme GBA were significantly upregulated in PD substantia nigra. Alternatively, gene expression for polysialyltransferases ST8SIA2 and ST8SIA4, sialidase NEU4, and sphingosine kinases SPHK1 and SPHK2 were significantly downregulated in PD substantia nigra.
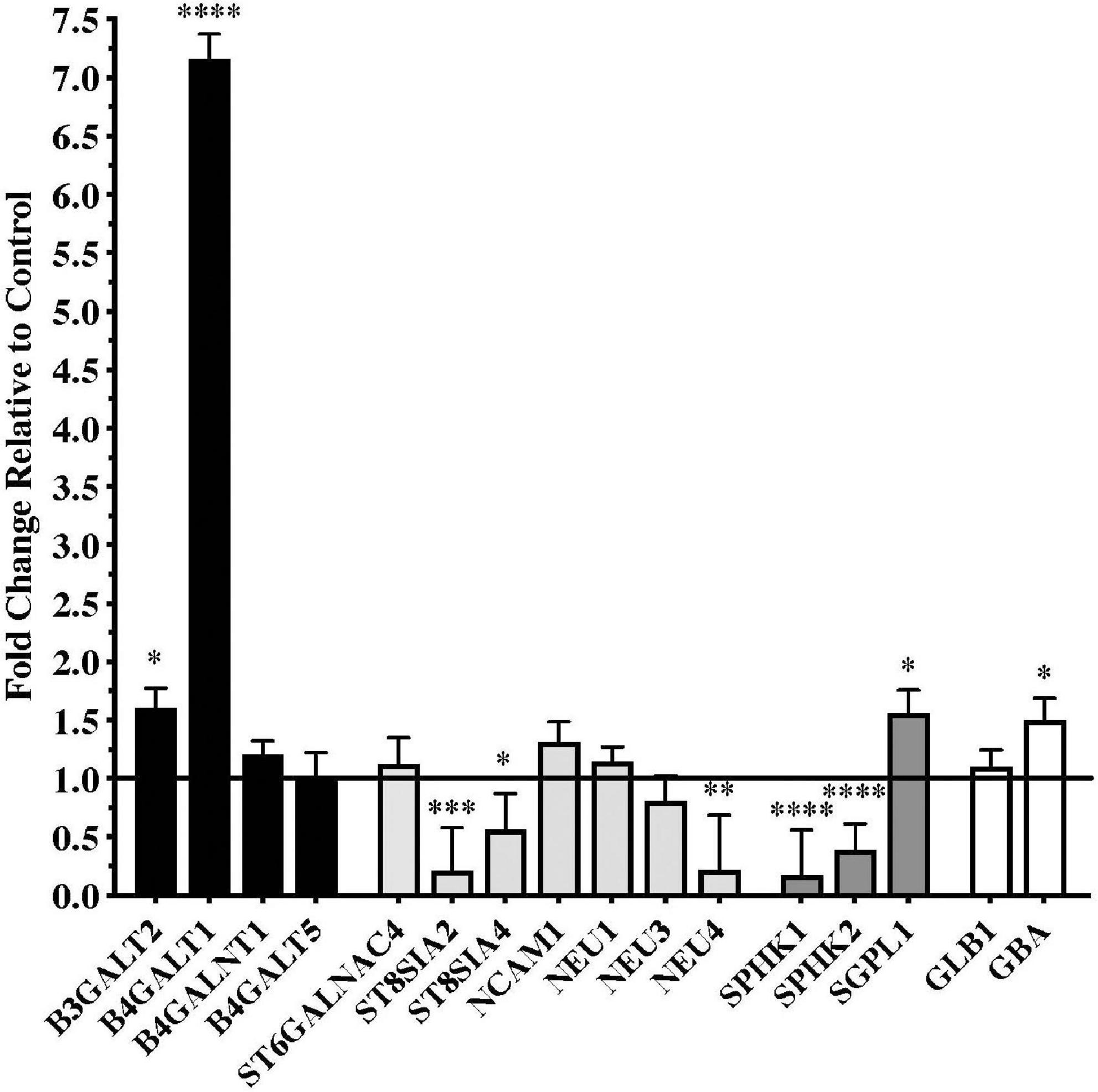
Figure 1. Gene expression changes in substantia nigra from patients with Parkinson’s disease (N = 15) relative to normal, age-matched controls (N = 12). Data are presented as fold change relative to control. Gene expression for glycosyltransferases B3GALT2 and B4GALT1 was significantly higher in patients with PD than in normal controls, while polysialyltransferase genes ST8SIA2 and ST8SIA4 were significantly down-regulated in the PD substantia nigra along with the gene for sialidase NEU4. Genes involved with sphingodine-1-phosphate (S1P) metabolism were significantly dysregulated in PD substantia nigra with expression of sphingosine kinases necessary for S1P synthesis, SPHK1 and SPHK2, significantly down-regulated and gene expression for S1P lyase, SGPL1, involved in the degradation of S1P, was significantly up-regulated. Gene expression for glucosidase beta acid 1 (GBA) was also significantly up-regulated in the PD substantia nigra. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 vs. control.
Gene expression data from the putamen were overall more variable than data from the substantia nigra. Although a number of genes trended toward being downregulated in the PD samples, none of these reached statistical significance compared to the controls (Figure 2). Only one gene, the lysosomal enzyme GLB1, was significantly upregulated in the PD putamen (Figure 2).
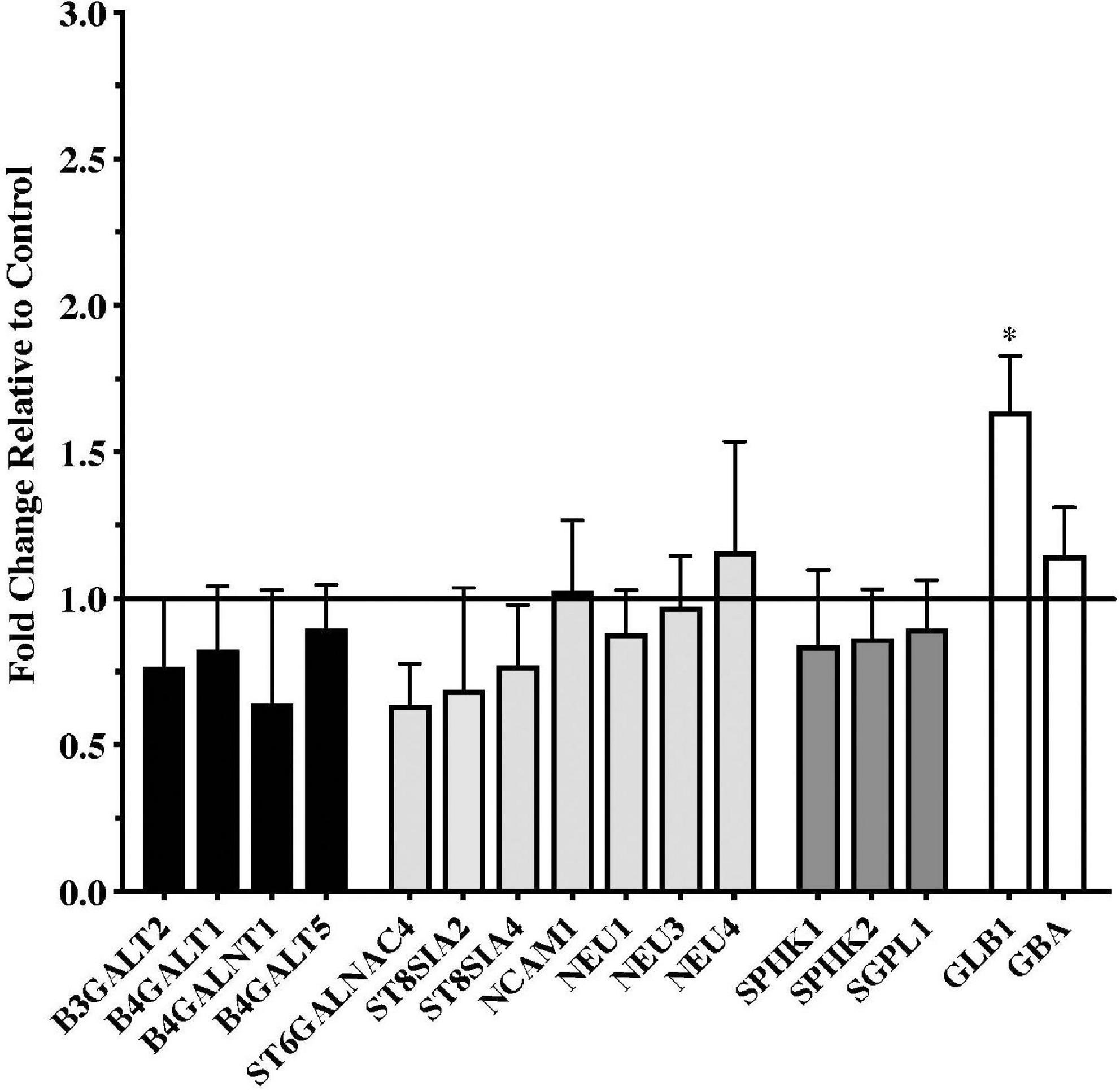
Figure 2. Gene expression changes in putamen from patients with Parkinson’s disease (N = 18) relative to normal, age-matched controls (N = 13). Data are presented as fold change relative to control. In contrast to the numerous changes in gene expression observed in the PD substantia nigra, expression of only one of the genes examined, the lysosomal hydrolytic enzyme β-Galactosidase (GLB1), was significantly altered in the PD putamen. *P < 0.05 vs. control.
Discussion
Over the last several years, there has been an increasing appreciation for the role that gangliosides and sphingolipids in general may play in the pathogenesis and progression of neurodegenerative diseases (Maglione et al., 2010; Di Pardo and Maglione, 2018; Lansbury, 2022) and PD in particular (Chiricozzi et al., 2020; Ledeen et al., 2022; Schneider, 2022). However, less attention has been paid to the roles that possible alterations in expression of genes involved in glycosylation, sialylation, and S1P regulation may play in the development and progression of PD. The current results show significant changes in gene expression of several key molecules involved in glycosylation, sialylation, and other process relevant to sphingolipid structure and function in the PD substantia nigra.
Glycosyltransferases
Glycosyltransferases are enzymes that catalyze the addition of polysaccharides to proteins, lipids, or nucleic acids to form glycoconjugates during glycosylation, a critical posttranslational process (Lv et al., 2017). Glycosyltransferases play important roles in the nervous system where they not only promote the development of neurons and glial cells and mediate the development of the myelin sheath (Angata et al., 2006; Lv et al., 2017) but are also critically involved in processes relevant to neurodegeneration including inflammation and microglial function, mitochondrial function, and autophagic processing (Chen et al., 2009; Videira and Castro-Caldas, 2018; Wang et al., 2020). There are various structural classes of glycosyltransferases that relate to their diverse biological functions. We previously showed that gene expression for the glycosyltransferase B3GALT4, an important enzyme in the synthesis of brain gangliosides GM1 and GD1b, was significantly reduced in residual dopaminergic neurons in the PD substantia nigra (Schneider, 2018). Currently, we show that B3GALT2 and B4GALT1 gene expression is significantly increased in the PD substantia nigra, compared to age-matched controls. This may be potentially significant for the expression and potentiation of PD-related pathology as protein glycosylation regulated by B4Galt1 has been suggested to be related to microglial activation and neuroinflammatory responses (Yang et al., 2020), with increased expression of B4Galt1 related to increased microglial inflammatory responses (Yang et al., 2020). Increased levels of B3GALT2 are also associated with neuroinflammation and knockdown of B3GALT2 reduced levels of inflammatory cytokines TNFα and IL-6 (Lv et al., 2017). This is significant as microglia activation and neuroinflammatory processes have been suggested to play important roles in the pathophysiology of PD (McGeer et al., 1988; Mogi et al., 1994; Ouchi et al., 2005; Gerhard et al., 2006; Zhang et al., 2017). Further, high levels of B4GALT1 have been suggested to suppress autophagic processes (Wang et al., 2020). Impaired autophagic processing is believed to play a significant role in the accumulation of toxic α-synuclein aggregates in dopaminergic neurons in the substantia nigra and the ensuing neurodegeneration (Karabiyik et al., 2017; Miki et al., 2018; Hou et al., 2020). In contrast to the significant changes observed in the substantia nigra, there were no significant changes in glycosyltransferase gene expression in the PD putamen.
Sialyltransferases and sialidases
Sialic acids are acidic sugars mostly found as terminal residues in glycan structures of glycoconjugates including glycoproteins and glycolipids (Rawal and Zhao, 2021). The highest levels of sialic acids are expressed in the brain where they regulate a diverse range of processes including neuronal sprouting, plasticity, myelination and myelin stability (Rawal and Zhao, 2021). Sialylation is the process mediated by sialytransferase enzymes, though which sialic acid is added to a glycoconjugate. Removal of sialic acid from sialoglycan is mediated by lysosomal, cytoplasmic, or plasma membrane bound sialidase enzymes (Schnaar et al., 2014; Rawal and Zhao, 2021). Gangliosides, sialylated glycosphingolipids that contain over 75% of the brain’s sialic acid, are the most abundant sialoglycans in the nervous system (Schnaar et al., 2014). We previously showed that gene expression for the sialyltransferase ST3GAL2, the enzyme responsible for the synthesis of brain gangliosides GD1a from GM1 and GT1b from GD1b, was significantly reduced in residual dopaminergic neurons in the PD substantia nigra (Schneider, 2018).
The current study also shows that there are significantly decreased levels of gene expression for two polysialyltransferases, ST8SIA2 and ST8SIA4, genes that encode enzymes responsible for polysialic acid (polySia) biosynthesis. PolySia plays important roles in brain development with neural cell adhesion molecule (NCAM) as the major polySia acceptor protein (Schnaar et al., 2014). While ST8SIA2 and ST8SIA4 are important for the addition of polySia to NCAM, gene expression for NCAM1 was not altered in the PD substantia nigra. In developing and mature brains, polySia plays roles in modulating the function of neurotrophic factors including BDNF and FGF2, NMDA and AMPA receptors, and potentially also influences dopamine and norepinephrine neurotransmission through regulating the interactions of these transmitters with their receptors (Sato et al., 2016). In addition to these functions, polySia plays a role in inhibiting innate immunity reactions, inflammation, and microglia activation (Liao et al., 2021). Thus, abnormal polysialyation could play an important role in various physiological mechanisms of relevance to the development or progression of PD. The expression of polySia is highly correlated with gene expression of ST8SIA2 and ST8SIA4 (Sato and Hane, 2018). ST8SIA2 has been implicated in myelin formation and ST8SIA2 deficiency leads to myelin deficits, thinning axons, and age-related white matter degeneration (Szewczyk et al., 2017). Interestingly, St8sia2–/– mice have reduced polysialylation and display schizophrenic-like behaviors including cognitive and behavioral deficits and it was proposed that genetic variation in ST8SIA2 in humans may have the potential to confer a neurodevelopmental predisposition to schizophrenia (Krocher et al., 2015). It is not possible to know if the ST8SIA2 gene down-regulation observed in the PD substantia nigra is a consequence of the disease or whether defects in the polytransferase gene expressions detected in our study predispose to the development of PD. While both sialyltransferases are present in the adult brain albeit at relatively low levels, ST8SIA4 appears to be the predominant polysiayltransferase in the adult brain where it has been suggested to be important for neuronal plasticity (Curto et al., 2019), with ST8SIA4 deficiency related to memory deficits in mice (Nacher et al., 2010). While the functional significance of the decreased expression of ST8SIA2 and ST8SIA4 genes in the PD substantia nigra are not entirely clear at this time, their dysregulation may signal a more widespread impairment in sialo-conjugate metabolism that is worthy of further study.
In addition to the decreases in mRNA expression of the polysialyltransferases discussed above, sialylation could also be influenced by the decrease expression of the sialidase gene NEU4. There are four main mammalian sialidases, NEU1, NEU2, NEU3, and NEU4 (Glanz et al., 2019). NEU1 is a lysosomal sialidase that participates in lysosomal exocytosis, NEU2 is primarily a cytoplasmic sialidase and plays a role in neuronal differentiation, NEU3 is a plasma membrane sialidase involved in ganglioside metabolism and regulation of transmembrane signaling, and NEU4 is located to lysosomes, mitochondria, and endoplasmic reticulum, has broad substrate specificity against sialylated glycoconjugates, and its expression has been linked to the clearance of storage materials from lysosomes, among other functions (Miyagi and Yamaguchi, 2012). In the present study, NEU4 gene expression was significantly decreased in PD substantia nigra. In Neu4–/– mice, NEU4 has been shown to be a ganglioside metabolizing enzyme, increasing relative amounts of GD1a ganglioside while substantially decreasing GM1 levels (Seyrantepe et al., 2008). Additionally, NEU4 has been suggested to regulate neuronal function through the degradation of polySia and may also play a role on immune function in microglia (Seyrantepe et al., 2008; Takahashi et al., 2012). Together, the data presented in the current paper suggest a dysregulation of sialylation in the PD substantia nigra that could have multiple negative influences on dopaminergic neuronal function and survival. In contrast to the significant changes observed in the substantia nigra, there were no significant changes in sialylation-related gene expression in the PD putamen.
Sphingosine-1-phosphate metabolism
Sphingosine-1-phosphate (S1P) is one the most potent signaling lipids, regulating several molecular events underlying cellular homeostasis and viability (Di Pardo and Maglione, 2018). S1P is normally synthesized by sphingosine kinase-1 and sphingosine kinase-2 (SPHK1 and SPHK2) and degraded by S1P phosphate phosphatase (SGPP) or S1P lyase (SGPL1). A balance between S1P synthesis and degradation is required for cellular homeostasis and normal cell functions (Di Pardo and Maglione, 2018). Decreased SPHK1/2 levels and increased SGPL1 levels are expected to decrease S1P levels, potentially impairing autophagic mechanisms, down-regulating pro-survival pathways, and promoting neurodegeneration (Di Pardo et al., 2019). Up-regulation of SGPL1 and reduced expression of SPHK1, with a subsequent decrease in S1P, has been associated with neurodegeneration in Alzheimer’s disease (He et al., 2010; Ceccom et al., 2014; Couttas et al., 2014) and has also been described in animal models of Huntington’s disease (HD) as well as in post-mortem brain tissues from patients with HD (Di Pardo et al., 2017). Interestingly, in HD transgenic mice, abnormally increased SGPL1 expression was observed at a very early stage of disease while SPHK1 and SPHK2 levels were not affected, suggesting that the process of dysregulation of S1P metabolism may begin very early in the disease process with alterations in expression SPHK1 and SPHK2 appearing as the disease progresses (Di Pardo et al., 2017). Importantly, pharmacological interventions aimed at modulating S1P metabolism were neuroprotective, suggesting modulation of S1P-metabolizing enzymes as potential druggable therapeutic targets for neuroprotection (Di Pardo et al., 2017). Although previous studies have demonstrated alterations in S1P metabolism using cellular and animal models of PD (Pyszko and Strosznajder, 2014; Sivasubramanian et al., 2015; Badawy et al., 2018; Motyl and Strosznajder, 2018; Zhang et al., 2018; Pepin et al., 2020) we believe the current report is the first to demonstrate this in post-mortem tissue from patients with PD. In contrast to the significant changes observed in the substantia nigra, there were no significant changes in expression of genes related to S1P metabolism in the PD putamen.
Lysosomal enzymes
Of the lysosomal-related genes examined, only GBA was affected in the PD substantia nigra and only GLB1 was affected in the PD putamen. GBA encodes for the lysosomal hydrolase β-glucocerebrosidase (GCase), that catalyzes the conversion of glucosylceramide into glucose and ceramide. Our finding of increased expression of GBA mRNA in the PD substantia nigra was surprising as others have reported decreased GBA gene expression in the substantia nigra in patients with sporadic PD (Chiasserini et al., 2015) and reduced GBA gene expression in brain regions with and without pathological synuclein aggregates and in early and late-stage sporadic PD (Murphy et al., 2014). The activity of GBA can be a ceramide source (Giussani et al., 2014) and ceramides play important roles in modulating membrane protein dynamics and signaling as well as modulating processes related to autophagy and mitochondrial-mediated apoptosis (Ferrazza et al., 2016). However, it is a decrease in GBA activity that is typically associated with increased ceramide levels and inhibition of autophagy and accumulation of synuclein. It is uncertain what the significance of an increase in GBA expression might be and how this may affect ceramide metabolism and accumulation as the relationship between GBA and ceramide levels is complex (Kurzawa-Akanbi et al., 2021).
β-Galactosidase (GLB1), a lysosomal hydrolytic enzyme, catalyzes the degradation of galactosylceramide to galactose and ceramide within the lysosome and GLB1 mutation causes a deficiency in β-galactosidase-1 resulting in abnormal lysosomal accumulation of GM1 (GM1 gangliosidosis). GLB1 has not been studied extensively in PD and the significance of the increase in GLB1 gene expression in the PD putamen is uncertain at this point.
Study limitations
There are some potential limitations of the current study. This study utilized whole tissue extracts of substantia nigra and putamen and thus interpretation of potential gene expression changes such as the ones observed in substantia nigra homogenates from PD brain could be complicated due to loss of dopaminergic neurons and signals from other cell types (i.e., microglia). While it is not possible to know in which cell types in the substantia nigra the observed gene expression changes originated, we observed both increases and decreases in expression of specific genes and thus our data are likely not attributed solely to neuronal loss in the PD substantia nigra. There was only one gene in the PD putamen that showed a significant change in expression. This may reflect the relative contributions of the genes assessed to the pathological process that occur in the substantia nigra and not in the putamen, although the putamen gene expression data were more variable than the data derived from the substantia nigra, potentially obscuring some significant gene expression changes in the putamen. The reasons for the higher level of variability in levels of gene expression in the putamen are not entirely clear but could relate at least in part to the anatomy of the putamen and the samples made available to us for this study. The human putamen is a very large structure and although we made an effort to take all samples from the dorsal putamen, the samples came from different rostro-caudal levels of the putamen and it is possible that there are sub-regionally specific patterns of expression of the genes examined in this study in different regions of the putamen. Regional heterogeneity in expression of various neuropeptides and in dopamine innervation and gradients of dopamine transporter loss in the PD putamen are well known and this regional heterogeneity may also apply to the expression of genes currently examined. Also, a relatively small number of patient samples were examined in the current study and only a relatively small number of genes were examined. Based on the consistency of the gene expression changes observed in the substantia nigra, it is unlikely that the data are related to a potentially different gene mutation status of different patients. However, additional studies using a larger number of cases with verified gene mutation status and examining a more extensive array of genes are indicated.
Conclusion
In summary, the current study shows significant changes in gene expression for several key molecules involved in glycosylation, sialylation, and S1P metabolism in the PD substantia nigra. Abnormal regulation of these processes has also been described in other neurodegenerative diseases including Alzheimer’s disease and Huntington’s disease, suggesting that dysregulation of processes involving glycosylation, sialylation, and sphingolipid metabolism such as those described here may transcend different brain disorders and neurodegenerative diseases.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
JS: conceptualization and writing—original draft. GS: collection of data. JS and GS: writing—review and editing and formal analyses. Both authors contributed to the article and approved the submitted version.
Funding
This study was supported by a grant from Qilu Pharmaceutical Co., Ltd. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
The authors thank Vikrant Singh for initial work on the isolation of RNA from the clinical samples used in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.1078854/full#supplementary-material
References
Alecu, I., and Bennett, S. A. L. (2019). Dysregulated lipid metabolism and its role in alpha-synucleinopathy in Parkinson’s disease. Front. Neurosci. 13:328. doi: 10.3389/fnins.2019.00328
Angata, K., Lee, W., Mitoma, J., Marth, J. D., and Fukuda, M. (2006). Cellular and molecular analysis of neural development of glycosyltransferase gene knockout mice. Methods Enzymol. 417, 25–37. doi: 10.1016/S0076-6879(06)17003-2
Badawy, S. M. M., Okada, T., Kajimoto, T., Hirase, M., Matovelo, S. A., Nakamura, S., et al. (2018). Extracellular alpha-synuclein drives sphingosine 1-phosphate receptor subtype 1 out of lipid rafts, leading to impaired inhibitory G-protein signaling. J. Biol. Chem. 293, 8208–8216. doi: 10.1074/jbc.RA118.001986
Ceccom, J., Loukh, N., Lauwers-Cances, V., Touriol, C., Nicaise, Y., Gentil, C., et al. (2014). Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol. Commun. 2:12. doi: 10.1186/2051-5960-2-12
Chen, J., Wang, H., Yang, H., Huang, X., Zhu, J., Hu, L., et al. (2009). Beta-1,4-galactosyltransferase-I participates in lipopolysaccharide induced reactive microgliosis. Neurotoxicology 30, 1107–1113. doi: 10.1016/j.neuro.2009.06.003
Chiasserini, D., Paciotti, S., Eusebi, P., Persichetti, E., Tasegian, A., Kurzawa-Akanbi, M., et al. (2015). Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 10:15. doi: 10.1186/s13024-015-0010-2
Chiricozzi, E., Lunghi, G., Di Biase, E., Fazzari, M., Sonnino, S., and Mauri, L. (2020). GM1 ganglioside is a key factor in maintaining the mammalian neuronal functions avoiding neurodegeneration. Int. J. Mol. Sci. 21:868. doi: 10.3390/ijms21030868
Couttas, T. A., Kain, N., Daniels, B., Lim, X. Y., Shepherd, C., Kril, J., et al. (2014). Loss of the neuroprotective factor sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2:9. doi: 10.1186/2051-5960-2-9
Curto, Y., Alcaide, J., Rockle, I., Hildebrandt, H., and Nacher, J. (2019). Effects of the genetic depletion of polysialyltransferases on the structure and connectivity of interneurons in the adult prefrontal cortex. Front. Neuroanat. 13:6. doi: 10.3389/fnana.2019.00006
den Jager, W. A. (1969). Sphingomyelin in Lewy inclusion bodies in Parkinson’s disease. Arch. Neurol. 21, 615–619. doi: 10.1001/archneur.1969.00480180071006
Desplats, P. A., Denny, C. A., Kass, K. E., Gilmartin, T., Head, S. R., Sutcliffe, J. G., et al. (2007). Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol. Dis. 27, 265–277. doi: 10.1016/j.nbd.2007.05.003
Di Pardo, A., Amico, E., Basit, A., Armirotti, A., Joshi, P., Neely, M. D., et al. (2017). Defective sphingosine-1-phosphate metabolism is a druggable target in Huntington’s disease. Sci. Rep. 7:5280. doi: 10.1038/s41598-017-05709-y
Di Pardo, A., and Maglione, V. (2018). Sphingolipid metabolism: A New therapeutic opportunity for brain degenerative disorders. Front. Neurosci. 12:249. doi: 10.3389/fnins.2018.00249
Di Pardo, A., Pepe, G., Castaldo, S., Marracino, F., Capocci, L., Amico, E., et al. (2019). Stimulation of sphingosine kinase 1 (SPHK1) is beneficial in a Huntington’s disease pre-clinical model. Front. Mol. Neurosci. 12:100. doi: 10.3389/fnmol.2019.00100
Doria, M., Maugest, L., Moreau, T., Lizard, G., and Vejux, A. (2016). Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 101, 393–400. doi: 10.1016/j.freeradbiomed.2016.10.008
Ferrazza, R., Cogo, S., Melrose, H., Bubacco, L., Greggio, E., Guella, G., et al. (2016). LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 478, 1141–1146. doi: 10.1016/j.bbrc.2016.08.082
Gerhard, A., Pavese, N., Hotton, G., Turkheimer, F., Es, M., Hammers, A., et al. (2006). In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 21, 404–412. doi: 10.1016/j.nbd.2005.08.002
Giussani, P., Tringali, C., Riboni, L., Viani, P., and Venerando, B. (2014). Sphingolipids: Key regulators of apoptosis and pivotal players in cancer drug resistance. Int. J. Mol. Sci. 15, 4356–4392. doi: 10.3390/ijms15034356
Glanz, V. Y., Myasoedova, V. A., Grechko, A. V., and Orekhov, A. N. (2019). Sialidase activity in human pathologies. Eur. J. Pharmacol. 842, 345–350. doi: 10.1016/j.ejphar.2018.11.014
He, X., Huang, Y., Li, B., Gong, C. X., and Schuchman, E. H. (2010). Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 31, 398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
Hou, X., Watzlawik, J. O., Fiesel, F. C., and Springer, W. (2020). Autophagy in Parkinson’s disease. J. Mol. Biol. 432, 2651–2672. doi: 10.1016/j.jmb.2020.01.037
Karabiyik, C., Lee, M. J., and Rubinsztein, D. C. (2017). Autophagy impairment in Parkinson’s disease. Essays Biochem. 61, 711–720. doi: 10.1042/EBC20170023
Krocher, T., Malinovskaja, K., Jurgenson, M., Aonurm-Helm, A., Zharkovskaya, T., Kalda, A., et al. (2015). Schizophrenia-like phenotype of polysialyltransferase ST8SIA2-deficient mice. Brain Struct. Funct. 220, 71–83. doi: 10.1007/s00429-013-0638-z
Kurzawa-Akanbi, M., Tammireddy, S., Fabrik, I., Gliaudelyte, L., Doherty, M. K., Heap, R., et al. (2021). Altered ceramide metabolism is a feature in the extracellular vesicle-mediated spread of alpha-synuclein in Lewy body disorders. Acta Neuropathol. 142, 961–984. doi: 10.1007/s00401-021-02367-3
Lansbury, P. (2022). The sphingolipids clearly play a role in Parkinson’s disease, but nature has made it complicated. Mov. Disord. 37, 1985–1989. doi: 10.1002/mds.29204
Ledeen, R., Chowdhury, S., Lu, Z. H., Chakraborty, M., and Wu, G. (2022). Systemic deficiency of GM1 ganglioside in Parkinson’s disease tissues and its relation to the disease etiology. Glycoconj. J. 39, 75–82. doi: 10.1007/s10719-021-10025-9
Liao, H., Winkler, J., Wissfeld, J., Shahraz, A., Klaus, C., and Neumann, H. (2021). Low molecular weight polysialic acid prevents lipopolysaccharide-induced inflammatory dopaminergic neurodegeneration in humanized SIGLEC11 transgenic mice. Glia 69, 2845–2862. doi: 10.1002/glia.24073
Lv, Y., Ren, L., Fu, Y., Huang, K., and Bi, J. (2017). Role of beta-1,3-galactosyltransferase 2 in trigeminal neuronal sensitization induced by peripheral inflammation. Neuroscience 349, 17–26. doi: 10.1016/j.neuroscience.2017.02.043
Maglione, V., Marchi, P., Di Pardo, A., Lingrell, S., Horkey, M., Tidmarsh, E., et al. (2010). Impaired ganglioside metabolism in Huntington’s disease and neuroprotective role of GM1. J. Neurosci. 30, 4072–4080. doi: 10.1523/JNEUROSCI.6348-09.2010
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/wnl.38.8.1285
Mercado, G., Valdes, P., and Hetz, C. (2013). An ERcentric view of Parkinson’s disease. Trends Mol. Med. 19, 165–175. doi: 10.1016/j.molmed.2012.12.005
Miki, Y., Shimoyama, S., Kon, T., Ueno, T., Hayakari, R., Tanji, K., et al. (2018). Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 63, 33–43. doi: 10.1016/j.neurobiolaging.2017.11.006
Miyagi, T., and Yamaguchi, K. (2012). Mammalian sialidases: Physiological and pathological roles in cellular functions. Glycobiology 22, 880–896. doi: 10.1093/glycob/cws057
Mogi, M., Harada, M., Riederer, P., Narabayashi, H., Fujita, K., and Nagatsu, T. (1994). Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 165, 208–210. doi: 10.1016/0304-3940(94)90746-3
Motyl, J., and Strosznajder, J. B. (2018). Sphingosine kinase 1/sphingosine-1-phosphate receptors dependent signalling in neurodegenerative diseases. The promising target for neuroprotection in Parkinson’s disease. Pharmacol. Rep. 70, 1010–1014. doi: 10.1016/j.pharep.2018.05.002
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137(Pt 3), 834–848. doi: 10.1093/brain/awt367
Nacher, J., Guirado, R., Varea, E., Alonso-Llosa, G., Rockle, I., and Hildebrandt, H. (2010). Divergent impact of the polysialyltransferases ST8SiaII and ST8SiaIV on polysialic acid expression in immature neurons and interneurons of the adult cerebral cortex. Neuroscience 167, 825–837. doi: 10.1016/j.neuroscience.2010.02.067
Ouchi, Y., Yoshikawa, E., Sekine, Y., Futatsubashi, M., Kanno, T., Ogusu, T., et al. (2005). Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 57, 168–175. doi: 10.1002/ana.20338
Pepin, E., Jalinier, T., Lemieux, G. L., Massicotte, G., and Cyr, M. (2020). Sphingosine-1-phosphate receptors modulators decrease signs of neuroinflammation and prevent Parkinson’s disease symptoms in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model. Front. Pharmacol. 11:77. doi: 10.3389/fphar.2020.00077
Plotegher, N., Bubacco, L., Greggio, E., and Civiero, L. (2019). Ceramides in Parkinson’s disease: From recent evidence to new hypotheses. Front. Neurosci. 13:330. doi: 10.3389/fnins.2019.00330
Pyszko, J. A., and Strosznajder, J. B. (2014). The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia Neuropathol. 52, 260–269. doi: 10.5114/fn.2014.45567
Rawal, P., and Zhao, L. (2021). Sialometabolism in brain health and Alzheimer’s disease. Front. Neurosci. 15:648617. doi: 10.3389/fnins.2021.648617
Riekkinen, P., Rinne, U. K., Pelliniemi, T. T., and Sonninen, V. (1975). Interaction between dopamine and phospholipids. Studies of the substantia nigra in Parkinson disease patients. Arch. Neurol. 32, 25–27. doi: 10.1001/archneur.1975.00490430047006
Sato, C., and Hane, M. (2018). Mental disorders and an acidic glycan-from the perspective of polysialic acid (PSA/polySia) and the synthesizing enzyme, ST8SIA2. Glycoconj. J. 35, 353–373. doi: 10.1007/s10719-018-9832-9
Sato, C., Hane, M., and Kitajima, K. (2016). Relationship between ST8SIA2, polysialic acid and its binding molecules, and psychiatric disorders. Biochim. Biophys. Acta 1860, 1739–1752. doi: 10.1016/j.bbagen.2016.04.015
Schnaar, R. L., Gerardy-Schahn, R., and Hildebrandt, H. (2014). Sialic acids in the brain: Gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol. Rev. 94, 461–518. doi: 10.1152/physrev.00033.2013
Schneider, J. S. (2018). Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson’s disease. PLoS One 13:e0199189. doi: 10.1371/journal.pone.0199189
Schneider, J. S. (2022). A critical role for GM1 ganglioside in the pathophysiology and potential treatment of Parkinson’s disease. Glycoconj. J. 39, 13–26. doi: 10.1007/s10719-021-10002-2
Schneider, J. S., and Singh, V. (2019). Dysregulation of sphingolipid expression and metabolism and its role in the pathogenesis of Parkinson’s disease. Society for neuroscience annual meeting. Chicago, IL: Society for Neuroscience.
Seyfried, T. N., Choi, H., Chevalier, A., Hogan, D., Akgoc, Z., and Schneider, J. S. (2018). Sex-related abnormalities in substantia nigra lipids in Parkinson’s disease. ASN Neuro 10:1759091418781889. doi: 10.1177/1759091418781889
Seyrantepe, V., Canuel, M., Carpentier, S., Landry, K., Durand, S., Liang, F., et al. (2008). Mice deficient in Neu4 sialidase exhibit abnormal ganglioside catabolism and lysosomal storage. Hum. Mol. Genet. 17, 1556–1568. doi: 10.1093/hmg/ddn043
Sivasubramanian, M., Kanagaraj, N., Dheen, S. T., and Tay, S. S. (2015). Sphingosine kinase 2 and sphingosine-1-phosphate promotes mitochondrial function in dopaminergic neurons of mouse model of Parkinson’s disease and in MPP+ –treated MN9D cells in vitro. Neuroscience 290, 636–648. doi: 10.1016/j.neuroscience.2015.01.032
Szewczyk, L. M., Brozko, N., Nagalski, A., Rockle, I., Werneburg, S., Hildebrandt, H., et al. (2017). ST8SIA2 promotes oligodendrocyte differentiation and the integrity of myelin and axons. Glia 65, 34–49. doi: 10.1002/glia.23048
Takahashi, K., Mitoma, J., Hosono, M., Shiozaki, K., Sato, C., Yamaguchi, K., et al. (2012). Sialidase NEU4 hydrolyzes polysialic acids of neural cell adhesion molecules and negatively regulates neurite formation by hippocampal neurons. J. Biol. Chem. 287, 14816–14826. doi: 10.1074/jbc.M111.324186
Tsujii, S., Ishisaka, M., and Hara, H. (2015). Modulation of endoplasmic reticulum stress in Parkinson’s disease. Eur. J. Pharmacol. 765, 154–156. doi: 10.1016/j.ejphar.2015.08.033
Varadi, C., Nehez, K., Hornyak, O., Viskolcz, B., and Bones, J. (2019). Serum N-glycosylation in Parkinson’s disease: A novel approach for potential alterations. Molecules 24:2220. doi: 10.3390/molecules24122220
Videira, P. A. Q., and Castro-Caldas, M. (2018). Linking glycation and glycosylation with inflammation and mitochondrial dysfunction in Parkinson’s disease. Front. Neurosci. 12:381. doi: 10.3389/fnins.2018.00381
Wang, P., Li, X., and Xie, Y. (2020). B4GalT1 regulates apoptosis and autophagy of glioblastoma in vitro and in vivo. Technol. Cancer Res. Treat. 19:1533033820980104. doi: 10.1177/1533033820980104
Wilkinson, H., Thomsson, K. A., Rebelo, A. L., Hilliard, M., Pandit, A., Rudd, P. M., et al. (2021). The O-glycome of human nigrostriatal tissue and its alteration in Parkinson’s disease. J. Proteome Res. 20, 3913–3924. doi: 10.1021/acs.jproteome.1c00219
Yang, X., Li, Z. Y., Yuan, C. L., Tan, Y. F., Zhang, N. D., Liu, L. H., et al. (2020). Study on the role and mechanism of beta4GalT1 both in vivo and in vitro glioma. Eur. Rev. Med. Pharmacol. Sci. 24, 4368–4381. doi: 10.26355/eurrev_202004_21018
Zhang, L., Okada, T., Badawy, S. M. M., Hirai, C., Kajimoto, T., and Nakamura, S. I. (2018). Erratum: Extracellular alpha-synuclein induces sphingosine 1-phosphate receptor subtype 1 uncoupled from inhibitory G-protein leaving beta-arrestin signal intact. Sci. Rep. 8:46964. doi: 10.1038/srep46964
Keywords: Parkinson’s disease, glycolipid, sphingolipid, substantia nigra, putamen, gene expression
Citation: Schneider JS and Singh G (2022) Altered expression of glycobiology-related genes in Parkinson’s disease brain. Front. Mol. Neurosci. 15:1078854. doi: 10.3389/fnmol.2022.1078854
Received: 24 October 2022; Accepted: 11 November 2022;
Published: 24 November 2022.
Edited by:
Andrei Surguchov, University of Kansas Medical Center, United StatesReviewed by:
Irina G. Sourgoutcheva, University of Kansas Medical Center, United StatesVittorio Maglione, Mediterranean Neurological Institute Neuromed (IRCCS), Italy
Copyright © 2022 Schneider and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jay S. Schneider, amF5LnNjaG5laWRlckBqZWZmZXJzb24uZWR1