 Xin Yi Yeo
Xin Yi Yeo Yi Tang Lim1
Yi Tang Lim1 Hyokeun Park
Hyokeun Park Sangyong Jung
Sangyong Jung- 1Institute of Molecular and Cell Biology, Agency for Science, Technology and Research, Singapore, Singapore
- 2Department of Psychological Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 3Department of BioNano Technology, Gachon University, Seongnam, South Korea
- 4Division of Life Science, The Hong Kong University of Science and Technology, Kowloon, Hong Kong SAR, China
- 5Department of Physics, The Hong Kong University of Science and Technology, Kowloon, Hong Kong SAR, China
- 6State Key Laboratory of Molecular Neuroscience, The Hong Kong University of Science and Technology, Kowloon, Hong Kong SAR, China
- 7Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
The expanded use of hypothesis-free gene analysis methods in autism research has significantly increased the number of genetic risk factors associated with the pathogenesis of autism. A further examination of the implicated genes directly revealed the involvement in processes pertinent to neuronal differentiation, development, and function, with a predominant contribution from the regulators of synaptic function. Despite the importance of presynaptic function in synaptic transmission, the regulation of neuronal network activity, and the final behavioral output, there is a relative lack of understanding of the presynaptic contribution to the pathology of autism. Here, we will review the close association among autism-related mutations, autism spectrum disorders (ASD) phenotypes, and the altered presynaptic protein functions through a systematic examination of the presynaptic risk genes relating to the critical stages of synaptogenesis and neurotransmission.
Introduction
When first described, autism was regarded as a neuropsychiatric condition rooted in psychosocial distress and family burden and characterized by a range of emotional and communication defects from the early stages of life (Kanner, 1968; MacCULLOCH and Sambrooks, 1973). Subsequently, patients with metabolic defects such as phenylketonuria or creatine deficiency syndromes (Manzi et al., 2008) and mitochondrial disorders were observed to suffer similar autistic symptoms (Frye, 2020). Autistic patients often have problems with social communication, interactions, and attention and consequently present with abnormal behaviors (Fakhoury, 2015). Due to the lack of biological understanding of the development of autism and the wide variation in severity of symptoms, autism has been considered a spectrum disorder (ASD), with diagnosis hinged on the clinical phenotypes observed in autistic individuals reported in the Diagnostic and Statistical Manual of Mental Disorders (DSM) (Rosen et al., 2021).
Follow-up epidemiological studies further revealed the involvement of intellectual disability in autistic individuals (Kerbeshian et al., 2008), high adjusted concordance rate of autism among monozygotic twins (60%) and siblings (2%) compared to general population prevalence rates of 0.04% (Folstein and Rutter, 1977; Bolton et al., 1994; Bailey et al., 1995), and a skewed male preference for the development of ASD (Smalley, 1988), suggesting a genetic involvement in etiology of ASD. In an attempt to pinpoint the etiology of ASD, karyotypic and linkage studies have presented the debatable association of susceptibility loci on chromosome 2q (Buxbaum et al., 2001), 7q (International Molecular Genetic Study of Autism Consortium, 1998), 15q (Shao et al., 2003), 16p, and 19 (Liu et al., 2001) with ASD development. Though not all candidates identified in these first genetic studies are convincingly associated with the core symptoms of ASD (Talebizadeh, 2002; Zhang et al., 2002), genes with validated links with ASD phenotypes, such as the contactin-associated protein-like 2 (CNTNAP2) (Alarcón et al., 2008), gamma-aminobutyric acid type A receptor gamma 3 subunit (GABRG3) (Buxbaum et al., 2002), methyl-CpG binding protein 2 (MeCP2) (Lam, 2000; Carney et al., 2003), ubiquitin-protein ligase E3A (UBE3A) (Nurmi et al., 2001), and neuroligin 3 (NLGN3) (Paris Autism Research International Sibpair Study et al., 2003) point to the neurodevelopmental and multifactorial nature of autism.
More importantly, it is clear that autistic phenotypes likely stem from the disruption of processes critical for neuronal differentiation, development, and function. Interestingly, the re-examination of the genetic landscape of autistic individuals with new hypothesis-free whole genome sequencing and specific single nucleotide polymorphisms (SNP) identification methods have resulted in the confirmation and preferential pick-up of a myriad of highly penetrant mutations and variations in regulators of synaptic function (Buxbaum et al., 2007; Zoghbi and Bear, 2012; Giovedà et al., 2014; Leblond et al., 2014). Synapses are the basic computational unit of the nervous system responsible for the organized transmission and processing of information in the central nervous system (Juusola et al., 1996), which inevitably modulates an individual’s behavioral output and cognitive abilities (Woodburn et al., 2021). As the synaptic function is dependent on a series of coordinated and organized processes that drives synapse formation (Naskar et al., 2019), maintenance, and activity-dependent neurotransmitter release (Südhof and Malenka, 2008), it is not surprising that mutations in synaptic regulators results in a predisposition to the development of cognitive deficits.
There has been a preferential focus on the factors involved in the organization of synaptic structure and functional outcome that makes up a significant proportion of the risk genes identified in the autistic population (Guang et al., 2018). The extensive investigation of the contribution of the neuroligin (NLGN)/neurexin (NRXN) family of cell adhesion molecule and SRC Homology 3 Domain (SH3) and multiple ankyrin repeat domains (SHANK) family of glutamatergic postsynaptic density protein and their autism-associated mutations (Jiang and Ehlers, 2013; Trobiani et al., 2020), which cumulate into the excitation-inhibition balance model of autism development (Sohal and Rubenstein, 2019). Despite the potential involvement of presynaptic active zone proteins in neuronal circuit regulation throughout various stages of development and the importance of presynaptic neurotransmitter release machinery in basic neuronal signal propagation, there has been a lack of understanding of the presynaptic contribution to the development of ASD. In this review, we summarize the reported autism-related mutations related to presynaptic functionality and their potential impact on the development of autistic phenotypes (Table 1). We also recognize the previous attempt to understand the state of involvement of the presynaptic vesicle release machinery in general neurodevelopmental disorders (Bonnycastle et al., 2021). Nevertheless, by examining the novel presynaptic autism risk factors, we aimed to provide a renewed and balanced perspective of the current understanding of the presynaptic involvement in cognitive defects (Figure 1), which are characteristic of autism pathogenesis.
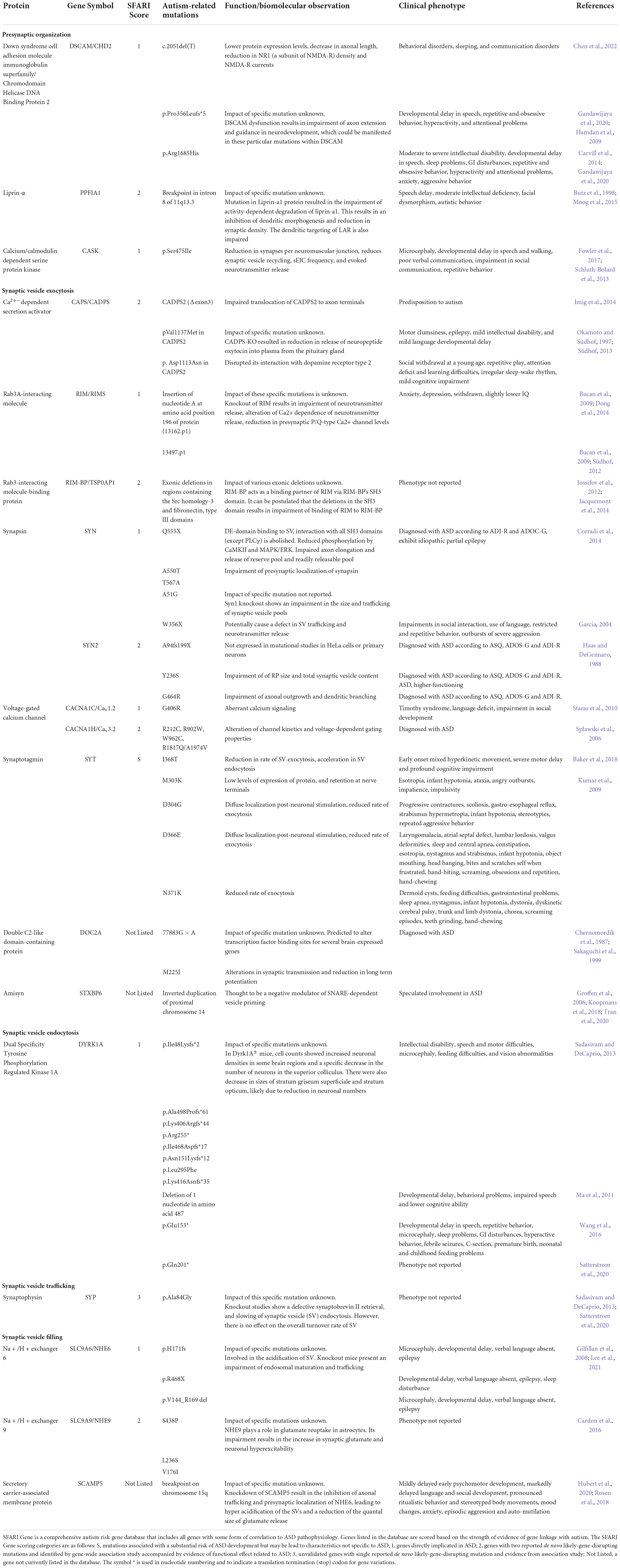
Table 1. Autism-associated mutations in presynaptic proteins.
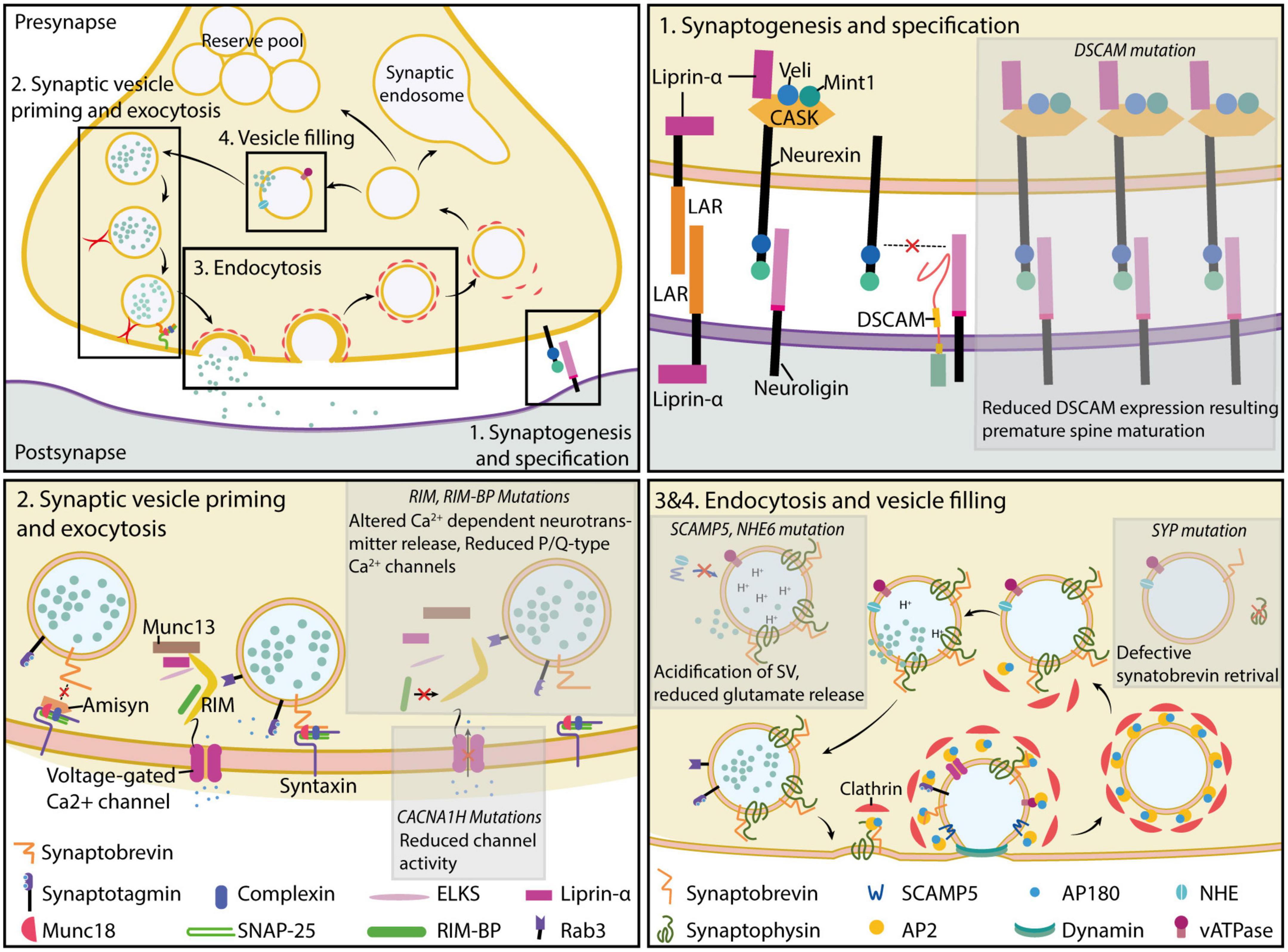
Figure 1. Molecular players of presynaptic function. Presynaptic formation and function are critically dependent on the expression and localization of proteins involved in the process of synaptogenesis and specification (top right) (section “Synaptogenesis and specification”), synaptic vesicle priming and exocytosis (bottom left) (sections “Synaptic vesicle priming” and “Synaptic vesicle exocytosis”), retrieval of synaptic vesicles (bottom right) (section “Retrieval of vesicle from plasma membrane and endocytosis”), and filling of empty synaptic vesicles (bottom left) (section “Regulation of synaptic vesicle filling and synaptic vesicle pools”). Mutations disrupting presynaptic protein function impair normal synaptic formation and neurotransmission, resulting in a loss of synaptic function. Further discussion of the mutations and their potential implications in autism etiology can be found in the main text.
Developmental profile of presynaptic terminals and their involvement in neuronal function
Synaptogenesis and specification
Synapses are specialized and asymmetrical structures formed through the initial formation of dendritic or axonal filopodia to bring nascent presynaptic sites closer to targeted postsynaptic specializations. In the neuron, the actin-capping protein (CP) works in concert with actin-related protein 2/3 (Arp2/3) to prevent further elongation of filopodia structures (Akin and Mullins, 2008; Fan et al., 2011), likely promoting the formation of more stable, putative synaptic structures. Subsequently, the anti-capping protein-enabled/vasodilator-stimulated phosphoprotein (Ena/VASP) regulates presynaptic actin polymerization, which affects synaptic protein anchoring and the eventual bouton size (Lin et al., 2010). The process is followed by the recruitment of cell adhesion molecules (CAMs) to stabilize axo-dendritic contact. The importance of CAMs in synapse formation, specialization, and function is well understood and has been extensively examined in other reviews (Dalva et al., 2007; Kilinc, 2018; Südhof, 2018). Previous reviews on the CAMs’ ASD-associated mutations and their link to autism development have provided many insights into their roles in ASD pathology (Ye et al., 2010; Redies et al., 2012; Gandawijaya et al., 2020; Tromp et al., 2021).
The DS Cell Adhesion Molecule (DSCAM), also known as chromodomain helicase DNA binding protein 2 (CHD2), is a member of the neuronal immunoglobulin superfamily of molecules broadly expressed in the nervous system and implicated in the process of pathfinding (Liu et al., 2009), axon branching (Wang et al., 2002), and dendritic arborization (Maynard and Stein, 2012). The gene coding for DSCAM has been mapped to chromosome 21q22.12 → q22.3, commonly duplicated in Down syndrome patients (Yamakawa, 1998), and its upregulation has been linked to altered neuronal circuits and learning and behavioral abnormalities in early studies on the animal models of Down syndrome (Sago et al., 1998). In addition to its link with Down syndrome, DSCAM is a strong ASD risk gene recurringly identified in various genome association studies (Iossifov et al., 2014; Sanders et al., 2015; Wang et al., 2016). Mutations that result in the premature termination of DSCAM expression have also been identified within the human autistic population, supporting the importance of DSCAM function in general neurological function.
DSCAM exhibits prominent synaptogenic capabilities. In Drosophila and Aplysia, DSCAM is involved in the regulation of synapse targeting (Millard et al., 2010) and the activity-dependent clustering of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Li et al., 2009), leading to an alteration in presynaptic size (Kim et al., 2013). This observation is likely due to the preferential interaction with, and aggregation of, postsynaptic proteins associated with nanoclusters of protein determinants of synapse maturity and functionality, similar to effects observed with neurexin ligands (Nozawa et al., 2022). Furthermore, DSCAM can negatively regulate synaptic maturation through competitive interaction with neuroligin 1 (NLGN1) and the inhibition of NLGN1-NRXN1β-mediated synaptic specification (Chen et al., 2022) in the developing mouse cortex. Using an induced pluripotent stem cell (iPSC)-derived model of telencephalic neurons, an early truncated variant of DCAM (amino acid 684) resulted in a reduced DSCAM/N-methyl-D-aspartate receptor subunit 1 (NR1) colocalization. As a result, the synaptic N-methyl-D-aspartate (NMDA) receptor response was compromised (Lim et al., 2021), suggesting DSCAM’s role in synaptic maintenance.
CASK is an alternative presynaptic scaffold protein encoded by chromosome Xp11.4. Due to its localization on the X chromosome, pathogenic variants are disproportionately identified in the female-dominant X-linked microcephaly with pontine and cerebellar hypoplasia (OMIM 300749) and FG syndrome 4 (OMIM 300422) (Piluso et al., 2009; Moog et al., 2015), with a comparatively rare occurrence in general autistic patients (Gupta et al., 2014; Seto et al., 2017). Identified mutations are widely distributed along the gene without preferential mutation spots (Moog et al., 2015), while observed phenotypes vary in individuals. Hence, little can be deduced regarding the dominance of mutations and the key impact of CASK on the development of intellectual disorders and ASD. In normal physiology, CASK is a neurexin-binding protein (Hata et al., 1996) with a suspected role as a synaptic organizer that links cell adhesion and transsynaptic signaling with vesicle exocytosis (Butz et al., 1998). Despite an absence of effect on neuron excitability, and microstructure of synapses when CASK is ablated (Atasoy et al., 2007), there is a reduction in spontaneous synaptic events and synaptic vesicle cycling in the Drosophila neuromuscular junction (Chen and Featherstone, 2011) that corroborate the potential function of CASK in presynaptic vesicle exocytosis. Further work is required to pinpoint the precise neurological function of CASK and its correlation with the observed neurodevelopmental conditions.
The ability of non-neuronal HEK293 cells artificially expressing postsynaptic-specific CAM to induce the formation of presynaptic specializations in cocultured-primary neurons exemplifies the synaptogenic capability of CAMs (Scheiffele et al., 2000). Conversely, the reverse applies to neurexin—its presynaptic expression is sufficient to induce the formation of postsynaptic compartments (Graf et al., 2004). Despite their distinct role in synaptogenesis, the deletion of individual synaptic organizers does not adversely disrupt neurological development. Given the frequent presence of alternative genes, promoters, splice site in CAM coding sequences (Chih et al., 2006; Schor et al., 2013; Treutlein et al., 2014; Li et al., 2020), and preferential binding partners, the possible permutations of the interactions (Fowler et al., 2017; Nozawa et al., 2022), revealing the precise nanoscopic architecture of CAM interactions, is a monumental task. The importance of CAMs in neurological development can only be addressed with the resolution of the relative contribution of each type of CAMs in neurological development and function.
Presynaptic proteins that support the function of CAMs and synaptic organization complicate the process of synaptogenesis and specification. Liprin was identified as an interacting partner and regulator of the localization of the adhesion molecule leukocyte common antigen-related receptor protein tyrosine phosphatase (LAR-RPTP) (Serra-Pagès et al., 1995). As LAR-RPTPs were required for the synaptogenic function of presynaptic neurexin (Han et al., 2020) and postsynaptic Slit- and Trk-like proteins (Slitrks) (Yim et al., 2013), presynaptic Liprin can modulate synapse specification. The vertebrate Liprin family of proteins consists of four different isoforms of Liprin-α (α1–4), two Liprin-β (β1, 2), and a single KazrinE (Sakamoto et al., 2012). In mammals, liprin-α1 is ubiquitously expressed throughout the body, while Liprin-α2 and 3 are predominant in the central nervous system (CNS). Rare de novo missense mutations and chromosomal rearrangement that disrupt the intronic sequence of the Liprin-α1 coding gene PPFIA1 were found in autism proband and patients (Schluth-Bolard et al., 2013; Iossifov et al., 2014).
Liprin-α is also involved in the interaction and recruitment of several components of the presynaptic vesicle release machinery, such as the calcium/calmodulin-dependent protein kinase II (CaMKII), calcium/calmodulin-dependent serine protein kinase (CASK), and regulating synaptic membrane exocytosis protein 1 (RIM1) (Spangler and Hoogenraad, 2007; Spangler et al., 2013), to facilitate synaptic transmission. Interestingly, activity-dependent phosphorylation of neurexin by CASK destabilizes the CASK-Liprin-Neurexin complex, leading to dissociation and an increase in turnover of neurexin (LaConte et al., 2016). Consistent with the observation, mutations in the Caenorhabditis elegans homolog of Liprin-α syd-2 caused the a decrease in active site electron density, mislocation of synaptobrevin-GFP labeling in the active zone, and an increase in the overall size of the presynaptic active site, without affecting synaptic density (Zhen and Jin, 1999). A similar observation was made in the mammalian system—the depletion of Liprin-α2/3 in mice results in the disruption of the active zone ultrastructure, synaptic vesicle tethering, and vesicle exocytosis (Spangler et al., 2013; Wong et al., 2018). Liprin-α is further involved in postsynaptic Slitrk6 and neuroligin 2 (NLGN2)-mediated presynaptic differentiation in cultured rat hippocampal neuron culture (Han et al., 2018), likely via its function as a presynaptic anchor and organizer.
Synaptic vesicle priming
Upon targeted interactions between CAMs, there is a rapid and bulk recruitment of preassembled active zone proteins in the form of piccolo-bassoon transport vesicles (PTVs) and synaptic vesicle protein transport vesicles (STVs) (Sabo and McAllister, 2003; Shapira et al., 2003; Sabo et al., 2006; Tao-Cheng, 2007). These are thought to be readily integrated into the nascent presynaptic structure, and form the basis for further defined recruitment and localization of the functional subunits of the presynaptic release machinery. Regulating synaptic membrane exocytosis (RIM), RIM-binding protein (RIM-BP), protein-rich in the amino acids glutamic acid (E), leucine (L), lysine (K), and serine (S) (ELKS), and Liprins interact with each other to form a large protein network that recruits the necessary molecular components for effective vesicle exocytosis (Südhof, 2012). Rare inherited mutations in RIM and de novo RIM-BP variants have been identified in the human ASD population (Bucan et al., 2009; Iossifov et al., 2012; Dong et al., 2014). The localization of voltage-gated calcium channels (VGCC) in the presynaptic compartment is directly dependent on the interaction between their cytoplasmic domain and the PDZ domain of RIM and RIM-BP (Kaeser et al., 2011), while the interaction of RIM with Munc13 places Munc13 near the soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE) machinery for the efficient coupling of vesicle priming and docking events (Betz et al., 2001; Dulubova et al., 2005; Ma et al., 2011). Indeed, the deletion of RIM or the RIM-BP results in the loss of presynaptic Ca2+ channels, reduced synaptic release probability in the immediate proximity of the presynaptic active zone, and impairment in activity-dependent neurotransmitter release (Kaeser et al., 2011; Liu et al., 2011; Zarebidaki et al., 2020). When combined with mutations in presynaptic factors associated with RIM or RIM-BP function, basic presynaptic homeostatic plasticity fails in Drosophila (Genç et al., 2020), suggesting a potential convergence of seemingly unrelated autism risk genes to the susceptibility of disturbed presynaptic function.
Distinct from the neurotransmitter-containing synaptic vesicles, large dense core vesicle (LDCV) exocytosis is regulated by the Ca2+-dependent secretion activator (CAPS), a two-member multidomain protein (Berwin et al., 1998; Renden et al., 2001). However, the enrichment of CAPS proteins in the neuron presynaptic compartments where there is very little LDCV (Sadakata et al., 2006) raised questions surrounding their possible involvement in synaptic vesicle exocytosis. Subsequent studies revealed their fundamental importance in priming synaptic vesicles for fusion and neurotransmitter release. CAPS-1 ablation in mouse hippocampal neurons drastically reduced the readily releasable pool (RRP) size and halved the evoked excitatory postsynaptic current (EPSC) amplitude, despite comparable release probability during RRP release and EPSC kinetics (Jockusch et al., 2007). Furthermore, the double knockout of CAPS renders a significant proportion of the analyzed neurons (39%) incapable of eliciting an EPSC response even with hypertonic buffer treatment. The functional defects observed are due to the loss of docked vesicles with CAPS-1 knockout (Shinoda et al., 2016). CAPS mutations are further linked to the disrupted release of neuropeptides involved in the modulation of social behavior (Fujima et al., 2021). This is in line with the observation that ASD patients have lower blood oxytocin concentration (Zhang et al., 2016), and the oxytocin-treatment-dependent improvement in social skills is only present in individuals with lowered oxytocin levels (Parker et al., 2017). Nonetheless, there have been mixed results in clinical trials involving oxytocin-related treatments, and the efficacy of oxytocin in enhancing the social function of ASD patients remains unclear. Although CAPS mutations are rare in human autism patients (Sadakata et al., 2007; Bonora et al., 2014), likely due to CAPS’ functional importance in the nervous system, gene mutations directly impacting the priming function of CAPS-1/2 would be easily translated into behavioral changes.
Synaptic vesicle exocytosis
The process of synaptic vesicle recruitment, priming, fusion, and recycling during chemical neurotransmission has been extensively examined (Okamoto and Südhof, 1997; Rizo and Rosenmund, 2008; Südhof, 2013; Imig et al., 2014; Chanaday et al., 2019). Due to the ubiquitous nature of Ca2+ in excitable cell function, function-modifying mutations in the VGCC have been identified in several neurological and neuromuscular conditions, including ASD (Bidaud et al., 2006; Lu et al., 2012). In the neuron, activity-dependent activation of the VGCC and influx of Ca2+ in the presynaptic terminal is a critical step in the release of neurotransmitters. The formation and assembly of the SNARE complex are heavily dependent on the presence of Ca2+ (Chen et al., 1999) whereby the forebrain-specific ablation of calcium voltage-gated channel subunit α1 A (CACNA1A) results in deficits in a variety of cognitive functions related to learning and memory and circadian rhythms (Mallmann et al., 2013). Depending on the site of impact, mutations in calcium voltage-gated channel subunit α (CACNA) which encodes for the main pore-forming subunit of the VGCC likely have an impact on the activation profile and kinetics of VGCC function.
Synaptic vesicles containing neurotransmitters can translocate between presynaptic terminals (Staras et al., 2010), and the vesicle distribution can be affected by synapsin (SYN) and β-catenin (Bamji et al., 2003; Pechstein et al., 2020), which sequester mobile vesicles. SYN is a family of evolutionarily conserved presynaptic proteins (Candiani et al., 2010) that plays a critical role in the structural and functional organization of the presynaptic terminal. Particularly, it reversibly tethers synaptic vesicles to the actin cytoskeleton which is crucial for the establishment of the vesicle pool for efficient neurotransmission (Baldelli et al., 2007; Cesca et al., 2010). Nonsense (Q555X) and missense (A550T and T567A) mutations, and maternally-inherited frameshift (A94fs199X) and missense (Y236S and G464) mutations have been identified in SYN1 and SYN2, respectively (Fassio et al., 2011; Corradi et al., 2014). Defects in directed targeting of the A550T and T567A SYN1 mutants, the lack of expression of the nonsense A94fs199X variant of SYN2, and the ablation of SYN2 function with Y236S and G464R revealed the importance of the SYN family activity in the regulation of synaptic function. The onset of SYN expression coincides with neuronal differentiation and peaks during synaptogenesis (Haas and DeGennaro, 1988; Melloni and Degennaro, 1994). Beyond this, SYN knockout in mice results in a differential impact on the populations of synaptic vesicles within the excitatory and inhibitor terminals (Gitler, 2004; Chiappalone et al., 2009), alongside epileptic phenotypes and ASD-like behaviors (Li et al., 1995).
It is recognized that spontaneous neurotransmitter release is important in the regulation of presynaptic maturation in the developing nervous system (Choi et al., 2014), as well as the regulation of postsynaptic receptor clustering and synaptic strength (Saitoe et al., 2001). Spontaneous release is dependent on the function of the putative presynaptic calcium sensors synaptotagmin (SYT), double C2-like domain-containing protein alpha (DOC2α), and double C2-like domain-containing protein beta (DOC2β) (Xu et al., 2009; Groffen et al., 2010), which differs in their cell-type specific expression profile (Courtney et al., 2018), sensitivity toward Ca2+, kinetics, and preference for phospholipid binding (Kojima et al., 1996; Groffen et al., 2006). Calcium binding changes the conformation of C2 domains, allowing them to insert into, buckle, and bring the presynaptic membrane closer to the synaptic vesicle to promote membrane fusion and neurotransmitter release (Martens et al., 2007). Mutations of SYT1 clustered mainly within the C2B domain which affected exocytosis rates following sustained action potential stimulation (Baker et al., 2015, 2018). SYT with M303K, D304G, and D366E mutations specifically failed to localize or relocalize to the presynaptic terminals following exocytosis. Comparatively, the DOC2A M225I variant likely affects its interaction with Munc13 (Kumar et al., 2009) and consequently SNARE-dependent fusion of synaptic vesicles. Hence, phenotypes associated with autism-associated SYT and DOC2 mutations originate from the disturbed neuronal transmission.
The importance of the SNARE complex in vesicle exocytosis cannot be overstated. Spontaneous membrane fusion in living organisms is energetically unfavorable—a large amount of directed force is required to overcome the repulsive forces between the lipid structures and lateral tension on the membrane surface (Chernomordik et al., 1987; Kozlovsky et al., 2002). Cooperative interactions between the vesicle SNARE (v-SNARE) synaptobrevin, and target-localized SNAREs (t-SNAREs) syntaxin-1 and synaptosomal-Associated Protein, 25kDa (SNAP-25) complex provide sufficient mechanical force for the fusion of opposing bilayers (McNew et al., 2000). Amisyn is a brain-enriched protein with a tomosyn- and synaptobrevin (VAMP)-liked coiled-coil-forming domain that competes with synaptobrevin-2 (VAMP2) for the assembly of the SNARE complex and inhibition of SNARE-dependent vesicle priming (Kondratiuk et al., 2020). Amisyn-containing SNARE complexes are more thermally stable than conventional VAMP2-containing SNARE complexes (Scales et al., 2002), but fusion-incompetent due to the absence of a transmembrane anchor in Amisyn. Interestingly, amisyn or syntaxin binding protein 6 (STXBP6) knockout in mice does not lead to any behavioral abnormalities (Liu et al., 2021), suggesting that Amisyn is not critical for neuronal development but may be involved in the regulation of activity-dependent of synaptic vesicular release. To date, further information regarding the regulatory mechanism of Amisyn function is absent, with only speculations of the full implication of STXBP6 mutation in ASD.
Retrieval of vesicle from plasma membrane and endocytosis
Following activity-dependent exocytosis, synaptic vesicles can be regenerated through the reuse of synaptic vesicles that have transiently docked and fused with the synaptic membrane (kiss-and-run fusion) (Alabi and Tsien, 2013), clathrin-mediated endocytosis of fully collapsed lipid structures from the plasma membrane, activity-dependent bulk endocytosis (Cheung et al., 2010) or fast endophilin-mediated endocytosis (Watanabe and Boucrot, 2017). Dual Specificity Tyrosine Phosphorylation Regulated Kinase 1A (DYRK1A) encodes a chromosome 21-associated proline-directed serine/threonine kinase with dual function in the regulation of gene transcription and clathrin-mediated endocytosis. Recurring DYRK1A haploinsufficiency has been observed in ASD and is associated with microcephaly, intellectual disability, and epileptic seizures (Earl et al., 2017).
At the early stages of neurodevelopment, DYRK1A phosphorylation of Lin-52 dREAM MuvB core complex component (LIN52) is required for the assembly of the dimerization partner (DP), retinoblastoma (RB)-like, E2F, and multi-vulval class B (MuvB) (DREAM) complex which coordinates cell cycle-dependent gene expression (Litovchick et al., 2011; Sadasivam and DeCaprio, 2013). Despite the presence of a nuclear localization sequence, the majority of the DYRK1A remained localized to the cytoplasm of neurons (Hämmerle et al., 2003; Martí et al., 2003). DYRK1A was detected in isolated latherin-coated vesicles and colocalized with latherin in mouse neurons. Mass spectrometry analysis revealed the phosphorylation of adaptins, dymain 1 (DYN1), amphiphysin 1 (AMPH1), and synaptojanin 1 (SYNJ1) by DYRKA1 which inhibits the onset of latherin-mediated endocytosis and promotes the dissociation of clathrin structures on vesicles (Murakami et al., 2012). While the autism-associated R205X and E239X truncations are linked to defects in dendritic growth and spine development in rodents (Dang et al., 2018), the normalization of DYRK1A expression postnatal is sufficient to ameliorate synaptic and the functional changes linked to altered synaptic plasticity (Duchon and Herault, 2016). Similarly, the multimodular intersectin 1 (ITSN1) has been shown to interact with endocytic proteins and control endocytosis in various cell types and organisms, with contradictory effects observed for mammalian synaptic vesicle endocytosis (Gubar et al., 2013).
Although synaptophysin (SYP) is the most abundant synaptic vesicle membrane protein, its function is enigmatic. The existing report suggests the diverse roles of SYP function, including synaptogenesis (Tarsa and Goda, 2002), and the biogenesis of synaptic vesicles (Leube et al., 1989; Cameron et al., 1991; Kwon and Chapman, 2011). Mutant mice lacking SYP are viable and does not exhibit changes in neuronal structure and behavior (McMahon et al., 1996). The lack of phenotypes is accounted for by the redundancy of function between the synaptophysin and synaptogyrin family of proteins (Raja et al., 2019). Interestingly, SYP knockout results in the mislocalization of VAMP2 with no impact on vesicle turnover, likely reflecting a defect in SYP-dependent VAMP2 retrieval during synaptic vesicle endocytosis (Gordon et al., 2011). As such, while SYP variants have been identified in a population of Vietnamese autistic patients (Tran et al., 2020) and subjects recruited by the Autism Sequencing Consortium (Satterstrom et al., 2020), their role in ASD is not known.
Regulation of synaptic vesicle filling and synaptic vesicle pools
Regardless of the mechanism adopted for the replenishment of synaptic vesicles, empty vesicles need to be rapidly refilled in the anticipation of a new round of neuronal activity and neurotransmission. Partially filled synaptic vesicles have a lower release probability (Rost et al., 2015) and can affect signal transmission. Vacuolar H+-ATPase (vATPase) is first required to establish a proton gradient across the vesicle membrane before neurotransmitters can be efficiently loaded into the vesicle lumen through the established electrochemical gradient (Farsi et al., 2017). The precise molecular composition and arrangement of synaptic-vesicle-associated vATPase have not been fully resolved.
Unlike conventional ASD risk genes, the correlation of nuclear receptor coactivator (NCOA7) with autism pathogenesis was revealed through the multidimensional examination of shared co-expression relationships of previously identified autism candidate genes with normal neurodevelopmental processes (Mahfouz et al., 2015). Up to date, there is only a single recessive case of autism known to be due to mutation in nuclear receptor coactivator 7 (NCOA7) (Autism Sequencing Consortium et al., 2019), and very little is known about NCOA7 function in neuronal physiology. NCOA7 is widely expressed throughout all developmental time frames of the mouse brain and interacts with various cytosolic V1 subunits of the vATPase in vivo. The loss of NCOA7 results in the increase in the number of proximal neurites of cultured primary neurons and a reduction in inhibitory synapses in layer 2/3 of the somatosensory cortex, which is related to the impaired social behavior observed (Castroflorio et al., 2021). A closer examination of potential ASD risk factors linked to vATPase function could reveal novel mechanisms for the development of neurodevelopmental and neuropsychiatric disorders.
The Na+/H+ exchanger 6 and 9 (NHE6/9 or SLC9A6/9) has been identified as candidate gene of interest for attention deficit hyperactivity disorder (ADHD) (Lasky-Su et al., 2008) and ASD (Morrow et al., 2008). Nonsense mutations that result in a similar premature termination of the last transmembrane segment of NHE9 have been observed in the closely related NHE1 and NHE6, associated with lower cognitive ability and epilepsy (Cox et al., 1997; Gilfillan et al., 2008). The sensitivity of Na uptake by glutamatergic synaptic vesicles toward a low micromolar amount of 5-(N-ethyl-N-isopropyl)amiloride, an inhibitor of NHEs suggests the involvement of NHEs in the filling of synaptic vesicles (Goh et al., 2011). Furthermore, mutations in the secretory carrier-associated membrane protein 5 (SCAMP5), a synaptic vesicle enriched protein responsible for the trafficking and synaptic localization of NHE6, were reported in idiopathic ASD and autism-like neurodevelopmental disorder with the manifestation of epilepsy (Castermans et al., 2010; Hubert et al., 2020). The knockdown of SCAMP5 in rat hippocampal neurons mislocalized NHE6 and hyperacidified the synaptic vesicles within the neuron (Lee et al., 2021). Given the prominent endosomal function of NHE, a careful evaluation of the contribution of NHEs is warranted before any conclusion can be made regarding its role in ASD development.
Discussion
ASD diagnosis and treatment remain difficult despite increasing research and clinical efforts to tackle the condition. Due to the essentiality of synaptic genes in proper neurological function, it is not surprising that many of the reported ASD risk genes were associated with alternate neuropsychiatric conditions (Zhu et al., 2014). However, varying neurological or non-neurological-related developmental trajectories can result in a similar cognitive outcome (Sala et al., 2020). Furthermore, patients with ASD often suffer from other comorbid conditions (Rosen et al., 2018) that may mask proper ASD symptoms, delay diagnosis (Mazefsky et al., 2012), and affect treatment efficacy (McDougle et al., 2003). There is currently no efficient method for identifying the causal factor and linking it directly to potential neurological defects in ASD patient, and neither is there a consensus on the optimal instrument for measuring the co-occurrence of other psychiatric disorders in ASD.
In addition to the above reported presynaptic targets, there was a pickup of mutations in alternate presynaptic factors with unknown significance in ASD. Individuals diagnosed with delayed development and amyotrophic lateral sclerosis carry UNC13A (coding for Munc13-1) (Engel et al., 2016; Tan et al., 2020), syntaxin-binding protein (STXBP1, coding for Munc18-1) in individuals with intellectual disorder and epilepsy (Hamdan et al., 2009; Carvill et al., 2014), and dynamin 1 (DMN1) in epilepsy (Appenzeller et al., 2014). Despite their equal importance in presynaptic function and neurotransmission, the restricted clinical outcome and lack of association with autism are surprising. We recognize that a multifactorial model may be a better representative of the development of ASD (Guo et al., 2018). Even among closely related genes, the skewed prevalence of mutations in specific targets is particularly perplexing. Although current genetic sequencing methods have been efficient in covering a significant proportion of the protein-coding region of the human genome, annotations of GC-rich sequences (Kim et al., 2022), the consensus in heterogenous locus, low-level genetic mosaics due to somatic de novo mutations (Rodríguez-Santiago et al., 2010), and gender biases have not been adequately examined (Jacquemont et al., 2014). Unless validated, caution is necessary for implicating single instances of mutations with ASD pathogenesis.
Experimental animal models are actively employed to understand the biological relevance of human mutations in neurological functions. Genetically modified rodent models have provided invaluable insights into the precise role of several presynaptic factors in specific neurological functions (Annamneedi et al., 2021; Castroflorio et al., 2021; Mitsogiannis et al., 2021). Nonetheless, the generation of animal models that would fully recapitulate the autistic human behavioral phenotype is difficult, if not currently impossible. ASD diagnosis is based purely on the characteristic behavioral defects in social interactions, communication, and motor stereotypes, which cannot be recapitulated satisfactorily with stereotypic rodent behavior and existing behavioral tests (Silverman et al., 2010). Although behavioral studies are often supplemented with biochemical and electrophysiological assays to provide a comprehensive understanding for the analysis of target function, inherent differences in the structural and molecular organization of synapses between humans and rodents (Testa-Silva, 2010; Testa-Silva et al., 2014; Koopmans et al., 2018; Wildenberg et al., 2021) reduce the relevance of phenotypes observed to human conditions. Cell reprogramming methods combined with pre-existing animal models, such as the human neuron xenograft models (Real et al., 2018; Linaro et al., 2019), and advanced analysis tools are valuable for understanding the role of presynaptic genes in actual human neurological function.
While ASD patients often suffer from the cognitive impact of irreversible neurodevelopmental defects (Xiao et al., 2014; Kumar et al., 2019), the sensitivity of human behavior and cognitive function to regulation from multiple molecular levels (Ganguly and Poo, 2013; Juárez-Portilla et al., 2018) provides opportunities to reverse autistic symptoms even later in life. Synaptic remodeling continues through adulthood after the conclusion of the critical stages of neurogenesis, cell migration, and maturation in the nervous system (Zito and Svoboda, 2002; Kelsch et al., 2008). Interestingly, the adult restoration of synaptic protein expression can reverse part of the autism-like phenotypes observed in mice (Mei et al., 2016). Furthermore, the neuronal nicotinic acetylcholine receptor agonists Nefiracetam and PHA 543613 developed for Alzheimer’s disease treatment can reverse synaptic defects observed in an induced human pluripotent stem cell model of MECP2 knockout cortical organoid (Trujillo et al., 2021). Although FDA-approved modifiers of synaptic function such as memantine and ketamine have met with a certain level of success in clinical use (Blanco-Silvente et al., 2018; Nuñez et al., 2020), their effect is mediocre likely due to the difference in etiology between patients. There are also no existing drugs targeting specifically presynaptic proteins and functions. Further considerations are essential for effective novel presynapse-targeting therapeutics development against the greatly heterogenous ASD etiology and symptoms.
Author contributions
XY and SJ: conceptualization. XY, YL, CP, HP, and SJ: writing—original draft preparation. XY, WC, HP, and SJ: writing—review and editing. SJ: supervision. HP and SJ: funding acquisition and critical revision of manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Joint Council Office grant (BMSI/15- 800003-SBIC-00E) from Agency for Science, technology and Research (A*STAR), Singapore (SJ), grants from the Research Grants Council of Hong Kong (16101518, 16102322, N_HKUST613/17, and A-HKUST603/17 to HP) and the Innovation and Technology Commission (ITCPD/17-9 to HP).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akin, O., and Mullins, R. D. (2008). Capping protein increases the rate of actin-based motility by promoting filament nucleation by the Arp2/3 complex. Cell 133, 841–851. doi: 10.1016/j.cell.2008.04.011
Alabi, A. A., and Tsien, R. W. (2013). Perspectives on kiss-and-run: Role in exocytosis, endocytosis, and neurotransmission. Annu. Rev. Physiol. 75, 393–422. doi: 10.1146/annurev-physiol-020911-153305
Alarcón, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Annamneedi, A., Del Angel, M., Gundelfinger, E. D., Stork, O., and Çalışkan, G. (2021). The Presynaptic scaffold protein bassoon in forebrain excitatory neurons mediates hippocampal circuit maturation: Potential involvement of TrkB signalling. Int. J. Mol. Sci. 22:7944. doi: 10.3390/ijms22157944
Appenzeller, S., Balling, R., Barisic, N., Baulac, S., Caglayan, H., Craiu, D., et al. (2014). De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370. doi: 10.1016/j.ajhg.2014.08.013
Atasoy, D., Schoch, S., Ho, A., Nadasy, K. A., Liu, X., Zhang, W., et al. (2007). Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. U.S.A. 104, 2525–2530. doi: 10.1073/pnas.0611003104
Autism Sequencing Consortium, Doan, R. N., Lim, E. T., De Rubeis, S., Betancur, C., Cutler, D. J., et al. (2019). Recessive gene disruptions in autism spectrum disorder. Nat. Genet. 51, 1092–1098. doi: 10.1038/s41588-019-0433-8
Bailey, A., Le Couteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., et al. (1995). Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 25, 63–77. doi: 10.1017/S0033291700028099
Baker, K., Gordon, S. L., Grozeva, D., van Kogelenberg, M., Roberts, N. Y., Pike, M., et al. (2015). Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J. Clin. Invest. 125, 1670–1678. doi: 10.1172/JCI79765
Baker, K., Gordon, S. L., Melland, H., Bumbak, F., Scott, D. J., Jiang, T. J., et al. (2018). SYT1-associated neurodevelopmental disorder: A case series. Brain J. Neurol. 141, 2576–2591. doi: 10.1093/brain/awy209
Baldelli, P., Fassio, A., Valtorta, F., and Benfenati, F. (2007). Lack of synapsin i reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J. Neurosci. 27, 13520–13531. doi: 10.1523/JNEUROSCI.3151-07.2007
Bamji, S. X., Shimazu, K., Kimes, N., Huelsken, J., Birchmeier, W., Lu, B., et al. (2003). Role of β-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40, 719–731. doi: 10.1016/S0896-6273(03)00718-9
Berwin, B., Floor, E., and Martin, T. F. J. (1998). CAPS (Mammalian UNC-31) protein localizes to membranes involved in dense-core vesicle exocytosis. Neuron 21, 137–145. doi: 10.1016/S0896-6273(00)80521-8
Betz, A., Thakur, P., Junge, H. J., Ashery, U., Rhee, J. S., Scheuss, V., et al. (2001). Functional Interaction of the Active Zone Proteins Munc13-1 and RIM1 in Synaptic Vesicle Priming. Neuron. 30, 183–196. doi: 10.1016/S0896-6273(01)00272-0
Bidaud, I., Mezghrani, A., Swayne, L. A., Monteil, A., and Lory, P. (2006). Voltage-gated calcium channels in genetic diseases. Biochim. Biophys. Acta BBA Mol. Cell Res. 1763, 1169–1174. doi: 10.1016/j.bbamcr.2006.08.049
Blanco-Silvente, L., Capellà, D., Garre-Olmo, J., Vilalta-Franch, J., and Castells, X. (2018). Predictors of discontinuation, efficacy, and safety of memantine treatment for Alzheimer’s disease: Meta-analysis and meta-regression of 18 randomized clinical trials involving 5004 patients. BMC Geriatr. 18:168. doi: 10.1186/s12877-018-0857-5
Bolton, P., Macdonald, H., Pickles, A., Rios, P., Goode, S., Crowson, M., et al. (1994). A Case-control family history study of Autism. J. Child Psychol. Psychiatry 35, 877–900. doi: 10.1111/j.1469-7610.1994.tb02300.x
Bonnycastle, K., Davenport, E. C., and Cousin, M. A. (2021). Presynaptic dysfunction in neurodevelopmental disorders: Insights from the synaptic vesicle life cycle. J. Neurochem. 157, 179–207. doi: 10.1111/jnc.15035
Bonora, E., Graziano, C., Minopoli, F., Bacchelli, E., Magini, P., Diquigiovanni, C., et al. (2014). Maternally inherited genetic variants of CADPS 2 are present in autism spectrum disorders and intellectual disability patients. EMBO Mol. Med. 6, 795–809. doi: 10.1002/emmm.201303235
Bucan, M., Abrahams, B. S., Wang, K., Glessner, J. T., Herman, E. I., Sonnenblick, L. I., et al. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5e1000536. doi: 10.1371/journal.pgen.1000536
Butz, S., Okamoto, M., and Südhof, T. C. A. (1998). Tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94, 773–782. doi: 10.1016/S0092-8674(00)81736-5
Buxbaum, J. D., Cai, G., Chaste, P., Nygren, G., Goldsmith, J., Reichert, J., et al. (2007). Mutation screening of thePTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 484–491. doi: 10.1002/ajmg.b.30493
Buxbaum, J. D., Silverman, J. M., Smith, C. J., Greenberg, D. A., Kilifarski, M., Reichert, J., et al. (2002). Association between a GABRB3 polymorphism and autism. Mol. Psychiatry 7, 311–316. doi: 10.1038/sj.mp.4001011
Buxbaum, J. D., Silverman, J. M., Smith, C. J., Kilifarski, M., Reichert, J., Hollander, E., et al. (2001). Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am. J. Hum. Genet. 68, 1514–1520. doi: 10.1086/320588
Cameron, P. L., Südhof, T. C., Jahn, R., and De Camilli, P. (1991). Colocalization of synaptophysin with transferrin receptors: Implications for synaptic vesicle biogenesis. J. Cell Biol. 115, 151–164. doi: 10.1083/jcb.115.1.151
Candiani, S., Moronti, L., Pennati, R., De Bernardi, F., Benfenati, F., and Pestarino, M. (2010). The synapsin gene family in basal chordates: Evolutionary perspectives in metazoans. BMC Evol. Biol. 10:32. doi: 10.1186/1471-2148-10-32
Cardon, M., Evankovich, K. D., and Holder, J. L. (2016). Exonic deletion of SLC9A9 in autism with epilepsy. Neurol Genet. 2:e62. doi: 10.1212/NXG.0000000000000062
Carney, R. M., Wolpert, C. M., Ravan, S. A., Shahbazian, M., Ashley-Koch, A., Cuccaro, M. L., et al. (2003). Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol. 28, 205–211. doi: 10.1016/S0887-8994(02)00624-0
Carvill, G. L., Weckhuysen, S., McMahon, J. M., Hartmann, C., Moller, R. S., Hjalgrim, H., et al. (2014). GABRA1 and STXBP1: Novel genetic causes of Dravet syndrome. Neurology 82, 1245–1253. doi: 10.1212/WNL.0000000000000291
Castermans, D., Volders, K., Crepel, A., Backx, L., De Vos, R., Freson, K., et al. (2010). SCAMP5, NBEA and AMISYN: Three candidate genes for autism involved in secretion of large dense-core vesicles. Hum. Mol. Genet. 19, 1368–1378. doi: 10.1093/hmg/ddq013
Castroflorio, E., den Hoed, J., Svistunova, D., Finelli, M. J., Cebrian-Serrano, A., Corrochano, S., et al. (2021). The Ncoa7 locus regulates V-ATPase formation and function, neurodevelopment and behaviour. Cell. Mol. Life Sci. CMLS 78, 3503–3524. doi: 10.1007/s00018-020-03721-6
Cesca, F., Baldelli, P., Valtorta, F., and Benfenati, F. (2010). The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 91, 313–348. doi: 10.1016/j.pneurobio.2010.04.006
Chanaday, N. L., Cousin, M. A., Milosevic, I., Watanabe, S., and Morgan, J. R. (2019). The synaptic vesicle cycle revisited: New insights into the modes and mechanisms. J. Neurosci. 39, 8209–8216. doi: 10.1523/JNEUROSCI.1158-19.2019
Chen, K., and Featherstone, D. E. (2011). Pre and postsynaptic roles for Drosophila CASK. Mol. Cell Neurosci. 48, 171–182. doi: 10.1016/j.mcn.2011.07.009
Chen, P., Liu, Z., Zhang, Q., Lin, D., Song, L., Liu, J., et al. (2022). DSCAM deficiency leads to premature spine maturation and autism-like behaviors. J. Neurosci. 42, 532–551. doi: 10.1523/JNEUROSCI.1003-21.2021
Chen, Y. A., Scales, S. J., Patel, S. M., Doung, Y. C., and Scheller, R. H. (1999). SNARE complex formation is triggered by Ca 2+ and drives membrane fusion. Cell 97, 165–174. doi: 10.1016/S0092-8674(00)80727-8
Chernomordik, L. V., Melikyan, G. B., and Chizmadzhev, Y. A. (1987). Biomembrane fusion: A new concept derived from model studies using two interacting planar lipid bilayers. Biochim. Biophys. Acta BBA Rev. Biomembr. 906, 309–352. doi: 10.1016/0304-4157(87)90016-5
Cheung, G., Jupp, O. J., and Cousin, M. A. (2010). Activity-dependent bulk endocytosis and clathrin-dependent endocytosis replenish specific synaptic vesicle pools in central nerve terminals. J. Neurosci. 30, 8151–8161. doi: 10.1523/JNEUROSCI.0293-10.2010
Chiappalone, M., Casagrande, S., Tedesco, M., Valtorta, F., Baldelli, P., Martinoia, S., et al. (2009). Opposite changes in glutamatergic and GABAergic transmission underlie the diffuse hyperexcitability of synapsin I–deficient cortical networks. Cereb. Cortex 19, 1422–1439. doi: 10.1093/cercor/bhn182
Chih, B., Gollan, L., and Scheiffele, P. (2006). Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron 51, 171–178. doi: 10.1016/j.neuron.2006.06.005
Choi, B. J., Imlach, W. L., Jiao, W., Wolfram, V., Wu, Y., Grbic, M., et al. (2014). Miniature neurotransmission regulates drosophila synaptic structural maturation. Neuron 82, 618–634. doi: 10.1016/j.neuron.2014.03.012
Corradi, A., Fadda, M., Piton, A., Patry, L., Marte, A., Rossi, P., et al. (2014). SYN2 is an autism predisposing gene: Loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum. Mol. Genet. 23, 90–103. doi: 10.1093/hmg/ddt401
Courtney, N. A., Briguglio, J. S., Bradberry, M. M., Greer, C., and Chapman, E. R. (2018). Excitatory and inhibitory neurons utilize different Ca2+ sensors and sources to regulate spontaneous release. Neuron 98, 977.e–991.e. doi: 10.1016/j.neuron.2018.04.022
Cox, G. A., Lutz, C. M., Yang, C. L., Biemesderfer, D., Bronson, R. T., Fu, A., et al. (1997). Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell 91, 139–148. doi: 10.1016/S0092-8674(01)80016-7
Dalva, M. B., McClelland, A. C., and Kayser, M. S. (2007). Cell adhesion molecules: Signalling functions at the synapse. Nat. Rev. Neurosci. 8, 206–220. doi: 10.1038/nrn2075
Dang, T., Duan, W. Y., Yu, B., Tong, D. L., Cheng, C., Zhang, Y. F., et al. (2018). Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol. Psychiatry 23, 747–758. doi: 10.1038/mp.2016.253
Dong, S., Walker, M. F., Carriero, N. J., DiCola, M., Willsey, A. J., Ye, A. Y., et al. (2014). De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 9, 16–23. doi: 10.1016/j.celrep.2014.08.068
Duchon, A., and Herault, Y. (2016). DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in down syndrome. Front. Behav. Neurosci. 10:104. doi: 10.3389/fnbeh.2016.00104
Dulubova, I., Lou, X., Lu, J., Huryeva, I., Alam, A., Schneggenburger, R., et al. (2005). A Munc13/RIM/Rab3 tripartite complex: From priming to plasticity? EMBO J. 24, 2839–2850. doi: 10.1038/sj.emboj.7600753
Earl, R. K., Turner, T. N., Mefford, H. C., Hudac, C. M., Gerdts, J., Eichler, E. E., et al. (2017). Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol. Autism 8:54. doi: 10.1186/s13229-017-0173-5
Engel, A. G., Selcen, D., Shen, X. M., Milone, M., and Harper, C. M. (2016). Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol. Genet. 2:e105. doi: 10.1212/NXG.0000000000000105
Fakhoury, M. (2015). Autistic spectrum disorders: A review of clinical features, theories and diagnosis. Int. J. Dev. Neurosci. 43, 70–77. doi: 10.1016/j.ijdevneu.2015.04.003
Fan, Y., Tang, X., Vitriol, E., Chen, G., and Zheng, J. Q. (2011). Actin capping protein is required for dendritic spine development and synapse formation. J. Neurosci. 31, 10228–10233. doi: 10.1523/JNEUROSCI.0115-11.2011
Farsi, Z., Jahn, R., and Woehler, A. (2017). Proton electrochemical gradient: Driving and regulating neurotransmitter uptake. BioEssays 39:1600240. doi: 10.1002/bies.201600240
Fassio, A., Patry, L., Congia, S., Onofri, F., Piton, A., Gauthier, J., et al. (2011). SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 20, 2297–2307. doi: 10.1093/hmg/ddr122
Folstein, S., and Rutter, M. (1977). Genetic influences and infantile autism. Nature 265, 726–728. doi: 10.1038/265726a0
Fowler, D. K., Peters, J. H., Williams, C., and Washbourne, P. (2017). Redundant postsynaptic functions of SynCAMs 1-3 during synapse formation. Front. Mol. Neurosci. 10:24. doi: 10.3389/fnmol.2017.00024
Frye, R. E. (2020). Mitochondrial dysfunction in autism spectrum disorder: Unique abnormalities and targeted treatments. Semin. Pediatr. Neurol. 35:100829. doi: 10.1016/j.spen.2020.100829
Fujima, S., Yamaga, R., Minami, H., Mizuno, S., Shinoda, Y., Sadakata, T., et al. (2021). CAPS2 deficiency impairs the release of the social peptide oxytocin, as well as oxytocin-associated social behavior. J. Neurosci. 41, 4524–4535. doi: 10.1523/JNEUROSCI.3240-20.2021
Gandawijaya, J., Bamford, R. A., Burbach, J. P. H., and Oguro-Ando, A. (2020). Cell Adhesion molecules involved in neurodevelopmental pathways implicated in 3p-deletion syndrome and autism spectrum disorder. Front. Cell Neurosci. 14:611379. doi: 10.3389/fncel.2020.611379
Ganguly, K., and Poo, M. M. (2013). Activity-dependent neural plasticity from bench to bedside. Neuron 80, 729–741. doi: 10.1016/j.neuron.2013.10.028
Garcia, C. C. (2004). Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J. Med. Genet. 41, 183–186. doi: 10.1136/jmg.2003.013680
Genç, Ö, An, J. Y., Fetter, R. D., Kulik, Y., Zunino, G., Sanders, S. J., et al. (2020). Homeostatic plasticity fails at the intersection of autism-gene mutations and a novel class of common genetic modifiers. Elife 9:e55775. doi: 10.7554/eLife.55775
Gilfillan, G. D., Selmer, K. K., Roxrud, I., Smith, R., Kyllerman, M., Eiklid, K., et al. (2008). SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking angelman syndrome. Am. J. Hum. Genet. 82, 1003–1010. doi: 10.1016/j.ajhg.2008.01.013
GiovedÃ, S., Corradi, A., Fassio, A., and Benfenati, F. (2014). Involvement of synaptic genes in the pathogenesis of autism spectrum disorders: The case of synapsins. Front. Pediatr. 2:94. doi: 10.3389/fped.2014.00094
Gitler, D. (2004). Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J. Neurosci. 24, 11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004
Goh, G. Y., Huang, H., Ullman, J., Borre, L., Hnasko, T. S., Trussell, L. O., et al. (2011). Presynaptic regulation of quantal size: K+/H+ exchange stimulates vesicular glutamate transport. Nat. Neurosci. 14, 1285–1292. doi: 10.1038/nn.2898
Gordon, S. L., Leube, R. E., and Cousin, M. A. (2011). Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J. Neurosci. 31, 14032–14036. doi: 10.1523/JNEUROSCI.3162-11.2011
Graf, E. R., Zhang, X., Jin, S. X., Linhoff, M. W., and Craig, A. M. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026. doi: 10.1016/j.cell.2004.11.035
Groffen, A. J. A., Friedrich, R., Brian, E. C., Ashery, U., and Verhage, M. (2006). DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties: DOC2A and DOC2B are sensors for neuronal activity. J. Neurochem. 97, 818–833. doi: 10.1111/j.1471-4159.2006.03755.x
Groffen, A. J., Martens, S., Arazola, R. D., Cornelisse, L. N., Lozovaya, N., de Jong, A. P. H., et al. (2010). Doc2b is a high-affinity Ca 2+ sensor for spontaneous neurotransmitter release. Science 327, 1614–1618. doi: 10.1126/science.1183765
Guang, S., Pang, N., Deng, X., Yang, L., He, F., Wu, L., et al. (2018). Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell Neurosci. 12:470. doi: 10.3389/fncel.2018.00470
Gubar, O., Morderer, D., Tsyba, L., Croisé, P., Houy, S., Ory, S., et al. (2013). Intersectin: The crossroad between vesicle exocytosis and endocytosis. Front. Endocrinol. 4:109. doi: 10.3389/fendo.2013.00109
Guo, H., Wang, T., Wu, H., Long, M., Coe, B. P., Li, H., et al. (2018). Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism. 9:64. doi: 10.1186/s13229-018-0247-z
Gupta, A. R., Pirruccello, M., Cheng, F., Kang, H., Fernandez, T. V., Baskin, J. M., et al. (2014). Rare deleterious mutations of the gene EFR3A in autism spectrum disorders. Mol. Autism 5:31. doi: 10.1186/2040-2392-5-31
Haas, C. A., and DeGennaro, L. J. (1988). Multiple synapsin I messenger RNAs are differentially regulated during neuronal development. J. Cell Biol. 106, 195–203. doi: 10.1083/jcb.106.1.195
Hamdan, F. F., Piton, A., Gauthier, J., Lortie, A., Dubeau, F., Dobrzeniecka, S., et al. (2009). De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann. Neurol. 65, 748–753. doi: 10.1002/ana.21625
Hämmerle, B., Carnicero, A., Elizalde, C., Ceron, J., Martínez, S., and Tejedor, F. J. (2003). Expression patterns and subcellular localization of the Down syndrome candidate protein MNB/DYRK1A suggest a role in late neuronal differentiation: Mnb expression during neuronal differentiation. Eur. J. Neurosci. 17, 2277–2286. doi: 10.1046/j.1460-9568.2003.02665.x
Han, K. A., Kim, Y. J., Yoon, T. H., Kim, H., Bae, S., Um, J. W., et al. (2020). LAR-RPTPs directly interact with neurexins to coordinate bidirectional assembly of molecular machineries. J. Neurosci. 40, 8438–8462. doi: 10.1523/JNEUROSCI.1091-20.2020
Han, K. A., Ko, J. S., Pramanik, G., Kim, J. Y., Tabuchi, K., Um, J. W., et al. (2018). PTPσ drives excitatory presynaptic assembly via various extracellular and intracellular mechanisms. J. Neurosci. 38, 6700–6721. doi: 10.1523/JNEUROSCI.0672-18.2018
Hata, Y., Butz, S., and Sudhof, T. (1996). CASK: A novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 16, 2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996
Hubert, L., Cannata Serio, M., Villoing-Gaudé, L., Boddaert, N., Kaminska, A., Rio, M., et al. (2020). De novo SCAMP5 mutation causes a neurodevelopmental disorder with autistic features and seizures. J. Med. Genet. 57, 138–144. doi: 10.1136/jmedgenet-2018-105927
Imig, C., Min, S. W., Krinner, S., Arancillo, M., Rosenmund, C., Südhof, T. C., et al. (2014). The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84, 416–431. doi: 10.1016/j.neuron.2014.10.009
International Molecular Genetic Study of Autism Consortium (1998). A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum. Mol. Genet. 7, 571–578. doi: 10.1093/hmg/7.3.571
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Jacquemont, S., Coe, B. P., Hersch, M., Duyzend, M. H., Krumm, N., Bergmann, S., et al. (2014). A higher mutational burden in females supports a “Female Protective Model” in neurodevelopmental disorders. Am. J. Hum. Genet. 94, 415–425. doi: 10.1016/j.ajhg.2014.02.001
Jiang, Y. H., and Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. doi: 10.1016/j.neuron.2013.03.016
Jockusch, W. J., Speidel, D., Sigler, A., Sørensen, J. B., Varoqueaux, F., Rhee, J. S., et al. (2007). CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell 131, 796–808. doi: 10.1016/j.cell.2007.11.002
Juárez-Portilla, C., Molina-Jiménez, T., Morin, J. P., Roldán-Roldán, G., and Zepeda, R. C. (2018). “Influence of Drugs on Cognitive Functions,” in Health and Academic Achievement, ed. B. Bernal-Morales (London: IntechOpen). doi: 10.5772/intechopen.71842
Juusola, M., French, A. S., Uusitalo, R. O., and Weckström, M. (1996). Information processing by graded-potential transmission through tonically active synapses. Trends Neurosci. 19, 292–297. doi: 10.1016/S0166-2236(96)10028-X
Kaeser, P. S., Deng, L., Wang, Y., Dulubova, I., Liu, X., Rizo, J., et al. (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295. doi: 10.1016/j.cell.2010.12.029
Kelsch, W., Lin, C. W., and Lois, C. (2008). Sequential development of synapses in dendritic domains during adult neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 16803–16808. doi: 10.1073/pnas.0807970105
Kerbeshian, J., Burd, L., Randall, T., Martsolf, J., and Jalal, S. (2008). Autism, profound mental retardation and atypical bipolar disorder in a 33-year-old female with a deletion of 15q12. J. Intellect Disabil. Res. 34, 205–210. doi: 10.1111/j.1365-2788.1990.tb01530.x
Kilinc, D. (2018). The emerging role of mechanics in synapse formation and plasticity. Front. Cell Neurosci. 12:483. doi: 10.3389/fncel.2018.00483
Kim, J. H., Wang, X., Coolon, R., and Ye, B. (2013). Dscam expression levels determine presynaptic arbor sizes in drosophila sensory neurons. Neuron 78, 827–838. doi: 10.1016/j.neuron.2013.05.020
Kim, J., Lee, C., Ko, B. J., Yoo, D. A., Won, S., Phillippy, A. M., et al. (2022). False gene and chromosome losses in genome assemblies caused by GC content variation and repeats. Genome Biol. 23:204. doi: 10.1186/s13059-022-02765-0
Kojima, T., Fukuda, M., Aruga, J., and Mikoshiba, K. (1996). Calcium-dependent phospholipid binding to the C2A domain of a ubiquitous form of double C2 protein (Doc2). J. Biochem (Tokyo). 120, 671–676. doi: 10.1093/oxfordjournals.jbchem.a021464
Kondratiuk, I., Jakhanwal, S., Jin, J., Sathyanarayanan, U., Kroppen, B., Pobbati, A. V., et al. (2020). PI(4,5)P2-dependent regulation of exocytosis by amisyn, the vertebrate-specific competitor of synaptobrevin 2. Proc. Natl. Acad. Sci. U.S.A. 117, 13468–13479. doi: 10.1073/pnas.1908232117
Koopmans, F., Pandya, N. J., Franke, S. K., Phillippens, I. H. C. M. H., Paliukhovich, I., Li, K. W., et al. (2018). Comparative hippocampal synaptic proteomes of rodents and primates: Differences in neuroplasticity-related proteins. Front. Mol. Neurosci. 11:364. doi: 10.3389/fnmol.2018.00364
Kozlovsky, Y., Chernomordik, L. V., and Kozlov, M. M. (2002). Lipid intermediates in membrane fusion: Formation, structure, and decay of hemifusion diaphragm. Biophys. J. 83, 2634–2651. doi: 10.1016/S0006-3495(02)75274-0
Kumar, R. A., Marshall, C. R., Badner, J. A., Babatz, T. D., Mukamel, Z., Aldinger, K. A., et al. (2009). Association and mutation analyses of 16p11.2 autism candidate genes. PLoS One 4:e4582. doi: 10.1371/journal.pone.0004582
Kumar, S., Reynolds, K., Ji, Y., Gu, R., Rai, S., and Zhou, C. J. (2019). Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 11:10. doi: 10.1186/s11689-019-9268-y
Kwon, S. E., and Chapman, E. R. (2011). Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70, 847–854. doi: 10.1016/j.neuron.2011.04.001
LaConte, L. E. W., Chavan, V., Liang, C., Willis, J., Schönhense, E. M., Schoch, S., et al. (2016). CASK stabilizes neurexin and links it to liprin-α in a neuronal activity-dependent manner. Cell. Mol. Life Sci. 73, 3599–3621. doi: 10.1007/s00018-016-2183-4
Lam, C. W. (2000). Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J. Med. Genet. 37:E41. doi: 10.1136/jmg.37.12.e41
Lasky-Su, J., Neale, B. M., Franke, B., Anney, R. J. L., Zhou, K., Maller, J. B., et al. (2008). Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1345–1354. doi: 10.1002/ajmg.b.30867
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: A gradient of severity in cognitive impairments. PLoS Genet. 10:e1004580. doi: 10.1371/journal.pgen.1004580
Lee, U., Choi, C., Ryu, S. H., Park, D., Lee, S. E., Kim, K., et al. (2021). SCAMP5 plays a critical role in axonal trafficking and synaptic localization of NHE6 to adjust quantal size at glutamatergic synapses. Proc. Natl. Acad. Sci. U.S.A. 118:e2011371118. doi: 10.1073/pnas.2011371118
Leube, R. E., Wiedenmann, B., and Franke, W. W. (1989). Topogenesis and sorting of synaptophysin: Synthesis of a synaptic vesicle protein from a gene transfected into nonneuroendocrine cells. Cell 59, 433–446. doi: 10.1016/0092-8674(89)90028-7
Li, H. L., Huang, B. S., Vishwasrao, H., Sutedja, N., Chen, W., Jin, I., et al. (2009). Dscam mediates remodeling of glutamate receptors in aplysia during de novo and learning-related synapse formation. Neuron 61, 527–540. doi: 10.1016/j.neuron.2009.01.010
Li, J., Xie, Y., Cornelius, S., Jiang, X., Sando, R., Kordon, S. P., et al. (2020). Alternative splicing controls teneurin-latrophilin interaction and synapse specificity by a shape-shifting mechanism. Nat. Commun. 11:2140. doi: 10.1038/s41467-020-16029-7
Li, L., Chin, L. S., Shupliakov, O., Brodin, L., Sihra, T. S., Hvalby, O., et al. (1995). Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 92, 9235–9239. doi: 10.1073/pnas.92.20.9235
Lim, C. S., Kim, M. J., Choi, J. E., Islam, M. A., Lee, Y. K., Xiong, Y., et al. (2021). Dysfunction of NMDA receptors in neuronal models of an autism spectrum disorder patient with a DSCAM mutation and in Dscam-knockout mice. Mol. Psychiatry 26, 7538–7549. doi: 10.1038/s41380-021-01216-9
Lin, W. H., Nebhan, C. A., Anderson, B. R., and Webb, D. J. (2010). Vasodilator-stimulated Phosphoprotein (VASP) induces actin assembly in dendritic spines to promote their development and potentiate synaptic strength. J. Biol. Chem. 285, 36010–36020. doi: 10.1074/jbc.M110.129841
Linaro, D., Vermaercke, B., Iwata, R., Ramaswamy, A., Libé-Philippot, B., Boubakar, L., et al. (2019). Xenotransplanted human cortical neurons reveal species-specific development and functional integration into mouse visual circuits. Neuron 104, 972.e–986.e. doi: 10.1016/j.neuron.2019.10.002
Litovchick, L., Florens, L. A., Swanson, S. K., Washburn, M. P., and DeCaprio, J. A. (2011). DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 25, 801–813. doi: 10.1101/gad.2034211
Liu, C., Hu, Q., Chen, Y., Wu, L., Liu, X., and Liang, D. (2021). Behavioral and gene expression analysis of Stxbp6-knockout mice. Brain Sci. 11:436. doi: 10.3390/brainsci11040436
Liu, G., Li, W., Wang, L., Kar, A., Guan, K. L., Rao, Y., et al. (2009). DSCAM functions as a netrin receptor in commissural axon pathfinding. Proc. Natl. Acad. Sci. U.S.A. 106, 2951–2956. doi: 10.1073/pnas.0811083106
Liu, J., Nyholt, D. R., Magnussen, P., Parano, E., Pavone, P., Geschwind, D., et al. (2001). A genomewide screen for autism susceptibility loci. Am. J. Hum. Genet. 69, 327–340. doi: 10.1086/321980
Liu, K. S. Y., Siebert, M., Mertel, S., Knoche, E., Wegener, S., Wichmann, C., et al. (2011). RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 334, 1565–1569. doi: 10.1126/science.1212991
Lu, A. T. H., Dai, X., Martinez-Agosto, J. A., and Cantor, R. M. (2012). Support for calcium channel gene defects in autism spectrum disorders. Mol Autism 3:18. doi: 10.1186/2040-2392-3-18
Ma, C., Li, W., Xu, Y., and Rizo, J. (2011). Munc13 mediates the transition from the closed syntaxin–Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol. 18, 542–549. doi: 10.1038/nsmb.2047
MacCULLOCH, M. J., and Sambrooks, J. E. (1973). Concepts of autisma: Review. J. Paediatr. Child Health 9, 237–245. doi: 10.1111/j.1440-1754.1973.tb02226.x
Mahfouz, A., Ziats, M. N., Rennert, O. M., Lelieveldt, B. P. F., and Reinders, M. J. T. (2015). Shared pathways among autism candidate genes determined by co-expression network analysis of the developing human brain transcriptome. J. Mol. Neurosci. 57, 580–594. doi: 10.1007/s12031-015-0641-3
Mallmann, R. T., Elgueta, C., Sleman, F., Castonguay, J., Wilmes, T., van den Maagdenberg, A., et al. (2013). Ablation of CaV2.1 Voltage-Gated Ca2+ channels in mouse forebrain generates multiple cognitive impairments. PLoS One 8:e78598. doi: 10.1371/journal.pone.0078598
Manzi, B., Loizzo, A. L., Giana, G., and Curatolo, P. (2008). Autism and Metabolic Diseases. J. Child Neurol. 23, 307–314. doi: 10.1177/0883073807308698
Martens, S., Kozlov, M. M., and McMahon, H. T. (2007). How synaptotagmin promotes membrane fusion. Science 316, 1205–1208. doi: 10.1126/science.1142614
Martí, E., Altafaj, X., Dierssen, M., de la Luna, S., Fotaki, V., Alvarez, M., et al. (2003). Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res. 964, 250–263. doi: 10.1016/S0006-8993(02)04069-6
Maynard, K. R., and Stein, E. (2012). Contributes to dendrite arborization and spine formation in the developing cerebral cortex. J. Neurosci. 32, 16637–16650. doi: 10.1523/JNEUROSCI.2811-12.2012
Mazefsky, C. A., Oswald, D. P., Day, T. N., Eack, S. M., Minshew, N. J., and Lainhart, J. E. (2012). ASD, a Psychiatric disorder, or both? Psychiatric diagnoses in adolescents with high-functioning ASD. J. Clin. Child Adolesc. Psychol. 41, 516–523. doi: 10.1080/15374416.2012.686102
McDougle, C. J., Stigler, K. A., and Posey, D. J. (2003). Treatment of aggression in children and adolescents with autism and conduct disorder. J. Clin. Psychiatry 64 Suppl 4, 16–25.
McMahon, H. T., Bolshakov, V. Y., Janz, R., Hammer, R. E., Siegelbaum, S. A., and Südhof, T. C. (1996). Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 93, 4760–4764. doi: 10.1073/pnas.93.10.4760
McNew, J. A., Weber, T., Parlati, F., Johnston, R. J., Melia, T. J., Söllner, T. H., et al. (2000). Close is not enough. J. Cell Biol. 150, 105–118. doi: 10.1083/jcb.150.1.105
Mei, Y., Monteiro, P., Zhou, Y., Kim, J. A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Melloni, R. H., and Degennaro, L. J. (1994). Temporal onset of synapsin I gene expression coincides with neuronal differentiation during the development of the nervous system. J. Comp. Neurol. 342, 449–462. doi: 10.1002/cne.903420311
Millard, S. S., Lu, Z., Zipursky, S. L., and Meinertzhagen, I. A. (2010). Drosophila dscam proteins regulate postsynaptic specificity at multiple-contact synapses. Neuron 67, 761–768. doi: 10.1016/j.neuron.2010.08.030
Mitsogiannis, M. D., Pancho, A., Aerts, T., Sachse, S. M., Vanlaer, R., Noterdaeme, L., et al. (2021). Subtle roles of down syndrome cell adhesion molecules in embryonic forebrain development and neuronal migration. Front. Cell Dev. Biol. 8:624181. doi: 10.3389/fcell.2020.624181
Moog, U., Bierhals, T., Brand, K., Bautsch, J., Biskup, S., Brune, T., et al. (2015). Phenotypic and molecular insights into CASK-related disorders in males. Orphanet J. Rare Dis. 10:44. doi: 10.1186/s13023-015-0256-3
Morrow, E. M., Yoo, S. Y., Flavell, S. W., Kim, T. K., Lin, Y., Hill, R. S., et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. doi: 10.1126/science.1157657
Murakami, N., Bolton, D. C., Kida, E., Xie, W., and Hwang, Y. W. (2012). Phosphorylation by Dyrk1A of clathrin coated vesicle-associated proteins: Identification of the substrate proteins and the effects of phosphorylation. PLoS One 7:e34845. doi: 10.1371/journal.pone.0034845
Naskar, S., Narducci, R., Balzani, E., Cwetsch, A. W., Tucci, V., and Cancedda, L. (2019). The development of synaptic transmission is time-locked to early social behaviors in rats. Nat. Commun. 10:1195. doi: 10.1038/s41467-019-09156-3
Nozawa, K., Sogabe, T., Hayashi, A., Motohashi, J., Miura, E., Arai, I., et al. (2022). In vivo nanoscopic landscape of neurexin ligands underlying anterograde synapse specification. Neuron 110, 3168.e–3185.e. doi: 10.1016/j.neuron.2022.07.027
Nuñez, N. A., Joseph, B., Pahwa, M., Seshadri, A., Prokop, L. J., Kung, S., et al. (2020). An Update on the efficacy and tolerability of oral ketamine for major depression: A systematic review and meta-analysis. Psychopharmacol. Bull. 50, 137–163.
Nurmi, E. L., Bradford, Y., Chen, Y., Hall, J., Arnone, B., Gardiner, M. B., et al. (2001). Linkage disequilibrium at the angelman syndrome gene UBE3A in autism families. Genomics 77, 105–113. doi: 10.1006/geno.2001.6617
Okamoto, M., and Südhof, T. C. (1997). Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J. Biol. Chem. 272, 31459–31464. doi: 10.1074/jbc.272.50.31459
Paris Autism Research International Sibpair Study, Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Parker, K. J., Oztan, O., Libove, R. A., Sumiyoshi, R. D., Jackson, L. P., Karhson, D. S., et al. (2017). Intranasal oxytocin treatment for social deficits and biomarkers of response in children with autism. Proc. Natl. Acad. Sci. U.S.A. 114, 8119–8124. doi: 10.1073/pnas.1705521114
Pechstein, A., Tomilin, N., Fredrich, K., Vorontsova, O., Sopova, E., Evergren, E., et al. (2020). Vesicle clustering in a living synapse depends on a synapsin region that mediates phase separation. Cell Rep. 30, 2594.e–2602.e. doi: 10.1016/j.celrep.2020.01.092
Piluso, G., D’Amico, F., Saccone, V., Bismuto, E., Rotundo, I. L., Di Domenico, M., et al. (2009). A missense mutation in CASK causes FG syndrome in an italian family. Am. J. Hum. Genet. 84, 162–177. doi: 10.1016/j.ajhg.2008.12.018
Raja, M. K., Preobraschenski, J., Del Olmo-Cabrera, S., Martinez-Turrillas, R., Jahn, R., Perez-Otano, I., et al. (2019). Elevated synaptic vesicle release probability in synaptophysin/gyrin family quadruple knockouts. Elife 8:e40744. doi: 10.7554/eLife.40744
Real, R., Peter, M., Trabalza, A., Khan, S., Smith, M. A., Dopp, J., et al. (2018). In vivo modeling of human neuron dynamics and Down syndrome. Science 362:eaau1810. doi: 10.1126/science.aau1810
Redies, C., Hertel, N., and Hübner, C. A. (2012). Cadherins and neuropsychiatric disorders. Brain Res. 1470, 130–144. doi: 10.1016/j.brainres.2012.06.020
Renden, R., Berwin, B., Davis, W., Ann, K., Chin, C. T., Kreber, R., et al. (2001). Drosophila CAPS is an essential gene that regulates dense-core vesicle release and synaptic vesicle fusion. Neuron 31, 421–437. doi: 10.1016/S0896-6273(01)00382-8
Rizo, J., and Rosenmund, C. (2008). Synaptic vesicle fusion. Nat. Struct. Mol. Biol. 15, 665–674. doi: 10.1038/nsmb.1450
Rodríguez-Santiago, B., Malats, N., Rothman, N., Armengol, L., Garcia-Closas, M., Kogevinas, M., et al. (2010). Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 87, 129–138. doi: 10.1016/j.ajhg.2010.06.002
Rosen, N. E., Lord, C., and Volkmar, F. R. (2021). The diagnosis of autism: From kanner to DSM-III to DSM-5 and Beyond. J. Autism Dev. Disord. 51, 4253–4270. doi: 10.1007/s10803-021-04904-1
Rosen, T. E., Mazefsky, C. A., Vasa, R. A., and Lerner, M. D. (2018). Co-occurring psychiatric conditions in autism spectrum disorder. Int. Rev. Psychiatry 30, 40–61. doi: 10.1080/09540261.2018.1450229
Rost, B. R., Schneider, F., Grauel, M. K., Wozny, C., Bentz, C., Blessing, A., et al. (2015). Optogenetic acidification of synaptic vesicles and lysosomes. Nat. Neurosci. 18, 1845–1852. doi: 10.1038/nn.4161
Sabo, S. L., and McAllister, A. K. (2003). Mobility and cycling of synaptic protein–containing vesicles in axonal growth cone filopodia. Nat. Neurosci. 6, 1264–1269. doi: 10.1038/nn1149
Sabo, S. L., Gomes, R. A., and McAllister, A. K. (2006). Formation of presynaptic terminals at predefined sites along axons. J. Neurosci. 26, 10813–10825. doi: 10.1523/JNEUROSCI.2052-06.2006
Sadakata, T., Itakura, M., Kozaki, S., Sekine, Y., Takahashi, M., and Furuichi, T. (2006). Differential distributions of the Ca2+-dependent activator protein for secretion family proteins (CAPS2 and CAPS1) in the mouse brain. J. Comp. Neurol. 495, 735–753. doi: 10.1002/cne.20947
Sadakata, T., Washida, M., Iwayama, Y., Shoji, S., Sato, Y., Ohkura, T., et al. (2007). Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J. Clin. Invest. 117, 931–943. doi: 10.1172/JCI29031
Sadasivam, S., and DeCaprio, J. A. (2013). The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 13, 585–595. doi: 10.1038/nrc3556
Sago, H., Carlson, E. J., Smith, D. J., Kilbridge, J., Rubin, E. M., Mobley, W. C., et al. (1998). Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. 95, 6256–6261. doi: 10.1073/pnas.95.11.6256
Saitoe, M., Schwarz, T. L., Umbach, J. A., Gundersen, C. B., and Kidokoro, Y. (2001). Absence of junctional glutamate receptor clusters in drosophila mutants lacking spontaneous transmitter release. Science 293, 514–517. doi: 10.1126/science.1061270
Sakaguchi, G., Manabe, T., Kobayashi, K., Orita, S., Sasaki, T., Naito, A., et al. (1999). Doc2α is an activity-dependent modulator of excitatory synaptic transmission: Role of Doc2α in activity-dependent synaptic modulation. Eur. J. Neurosci. 11, 4262–4268. doi: 10.1046/j.1460-9568.1999.00855.x
Sakamoto, S., Narumiya, S., and Ishizaki, T. (2012). A new role of multi scaffold protein Liprin-α: Liprin-α suppresses Rho-mDia mediated stress fiber formation. Bioarchitecture 2, 43–49. doi: 10.4161/bioa.20442
Sala, R., Amet, L., Blagojevic-Stokic, N., Shattock, P., and Whiteley, P. (2020). Bridging the Gap between physical health and autism spectrum disorder. Neuropsychiatr. Dis. Treat. 16, 1605–1618. doi: 10.2147/NDT.S251394
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233. doi: 10.1016/j.neuron.2015.09.016
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568.e–584.e. doi: 10.1016/j.cell.2019.12.036
Scales, S. J., Hesser, B. A., Masuda, E. S., and Scheller, R. H. (2002). Amisyn, a novel syntaxin-binding protein that may regulate SNARE complex assembly. J. Biol. Chem. 277, 28271–28279. doi: 10.1074/jbc.M204929200
Scheiffele, P., Fan, J., Choih, J., Fetter, R., and Serafini, T. (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. doi: 10.1016/S0092-8674(00)80877-6
Schluth-Bolard, C., Labalme, A., Cordier, M. P., Till, M., Nadeau, G., Tevissen, H., et al. (2013). Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J. Med. Genet. 50, 144–150. doi: 10.1136/jmedgenet-2012-101351
Schor, I. E., Fiszbein, A., Petrillo, E., and Kornblihtt, A. R. (2013). Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. EMBO J. 32, 2264–2274. doi: 10.1038/emboj.2013.167
Serra-Pagès, C., Kedersha, N. L., Fazikas, L., Medley, Q., Debant, A., and Streuli, M. (1995). The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO J. 14, 2827–2838. doi: 10.1002/j.1460-2075.1995.tb07282.x
Seto, T., Hamazaki, T., Nishigaki, S., Kudo, S., Shintaku, H., Ondo, Y., et al. (2017). A novel CASK mutation identified in siblings exhibiting developmental disorders with/without microcephaly. Intractable Rare Dis. Res. 6, 177–182. doi: 10.5582/irdr.2017.01031
Shao, Y., Cuccaro, M. L., Hauser, E. R., Raiford, K. L., Menold, M. M., Wolpert, C. M., et al. (2003). Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am. J. Hum. Genet. 72, 539–548. doi: 10.1086/367846
Shapira, M., Zhai, R. G., Dresbach, T., Bresler, T., Torres, V. I., Gundelfinger, E. D., et al. (2003). Unitary assembly of presynaptic active zones from piccolo-bassoon transport vesicles. Neuron 38, 237–252. doi: 10.1016/S0896-6273(03)00207-1
Shinoda, Y., Ishii, C., Fukazawa, Y., Sadakata, T., Ishii, Y., Sano, Y., et al. (2016). CAPS1 stabilizes the state of readily releasable synaptic vesicles to fusion competence at CA3–CA1 synapses in adult hippocampus. Sci. Rep. 6:31540. doi: 10.1038/srep31540
Silverman, J. L., Yang, M., Lord, C., and Crawley, J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502. doi: 10.1038/nrn2851
Smalley, S. L. (1988). Autism and genetics: A decade of research. Arch. Gen. Psychiatry 45:953. doi: 10.1001/archpsyc.1988.01800340081013
Sohal, V. S., and Rubenstein, J. L. R. (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. doi: 10.1038/s41380-019-0426-0
Spangler, S. A., and Hoogenraad, C. C. (2007). Liprin-α proteins: Scaffold molecules for synapse maturation. Biochem. Soc. Trans. 35, 1278–1282. doi: 10.1042/BST0351278
Spangler, S. A., Schmitz, S. K., Kevenaar, J. T., de Graaff, E., de Wit, H., Demmers, J., et al. (2013). Liprin-α2 promotes the presynaptic recruitment and turnover of RIM1/CASK to facilitate synaptic transmission. J. Cell Biol. 201, 915–928. doi: 10.1083/jcb.201301011
Splawski, I., Yoo, D. S., Stotz, S. C., Cherry, A., Clapham, D. E., and Keating, M. T. (2006). CACNA1H Mutations in Autism Spectrum Disorders. J. Biol. Chem. 281, 22085–22091. doi: 10.1074/jbc.M603316200
Staras, K., Branco, T., Burden, J. J., Pozo, K., Darcy, K., Marra, V., et al. (2010). A vesicle superpool spans multiple presynaptic terminals in hippocampal neurons. Neuron 66, 37–44. doi: 10.1016/j.neuron.2010.03.020
Südhof, T. C. (2012). The presynaptic active zone. Neuron 75, 11–25. doi: 10.1016/j.neuron.2012.06.012
Südhof, T. C. (2013). Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. doi: 10.1016/j.neuron.2013.10.022
Südhof, T. C. (2018). Towards an understanding of synapse formation. Neuron 100, 276–293. doi: 10.1016/j.neuron.2018.09.040
Südhof, T. C., and Malenka, R. C. (2008). Understanding Synapses: Past. Present, and Future. Neuron 60, 469–476. doi: 10.1016/j.neuron.2008.10.011
Talebizadeh, Z. (2002). No association between HOXA1 and HOXB1 genes and autism spectrum disorders (ASD). J. Med. Genet. 39:70e. doi: 10.1136/jmg.39.11.e70
Tan, H. H. G., Westeneng, H., Burgh, H. K., Es, M. A., Bakker, L. A., Veenhuijzen, K., et al. (2020). The Distinct Traits Of the UNC13A polymorphism in amyotrophic lateral sclerosis. Ann. Neurol. 88, 796–806. doi: 10.1002/ana.25841
Tao-Cheng, J. H. (2007). Ultrastructural localization of active zone and synaptic vesicle proteins in a preassembled multi-vesicle transport aggregate. Neuroscience 150, 575–584. doi: 10.1016/j.neuroscience.2007.09.031
Tarsa, L., and Goda, Y. (2002). Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 1012–1016. doi: 10.1073/pnas.022575999
Testa-Silva, G. (2010). Human synapses show a wide temporal window for spike-timing-dependent plasticity. Front. Synaptic Neurosci. 2:12. doi: 10.3389/fnsyn.2010.00012
Testa-Silva, G., Verhoog, M. B., Linaro, D., de Kock, C. P. J., Baayen, J. C., Meredith, R. M., et al. (2014). High bandwidth synaptic communication and frequency tracking in human neocortex. PLoS Biol. 12:e1002007. doi: 10.1371/journal.pbio.1002007
Tran, K. T., Le, V. S., Bui, H. T. P., Do, D. H., Ly, H. T. T., Nguyen, H. T., et al. (2020). Genetic landscape of autism spectrum disorder in Vietnamese children. Sci. Rep. 10:5034. doi: 10.1038/s41598-020-61695-8
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. 111, E1291–E1299. doi: 10.1073/pnas.1403244111
Trobiani, L., Meringolo, M., Diamanti, T., Bourne, Y., Marchot, P., Martella, G., et al. (2020). The neuroligins and the synaptic pathway in autism spectrum disorder. Neurosci. Biobehav. Rev. 119, 37–51. doi: 10.1016/j.neubiorev.2020.09.017
Tromp, A., Mowry, B., and Giacomotto, J. (2021). Neurexins in autism and schizophrenia-a review of patient mutations, mouse models and potential future directions. Mol. Psychiatry 26, 747–760. doi: 10.1038/s41380-020-00944-8
Trujillo, C. A., Adams, J. W., Negraes, P. D., Carromeu, C., Tejwani, L., Acab, A., et al. (2021). Pharmacological reversal of synaptic and network pathology in human MECP2 -KO neurons and cortical organoids. EMBO Mol. Med. 13:e12523. doi: 10.15252/emmm.202012523
Wang, J., Zugates, C. T., Liang, I. H., Lee, C. H. J., and Lee, T. (2002). Drosophila dscam is required for divergent segregation of sister branches and suppresses ectopic bifurcation of axons. Neuron 33, 559–571. doi: 10.1016/S0896-6273(02)00570-6
Wang, T., Guo, H., Xiong, B., Stessman, H. A. F., Wu, H., Coe, B. P., et al. (2016). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 7:13316. doi: 10.1038/ncomms13316
Watanabe, S., and Boucrot, E. (2017). Fast and ultrafast endocytosis. Curr. Opin. Cell Biol. 47, 64–71. doi: 10.1016/j.ceb.2017.02.013
Wildenberg, G. A., Rosen, M. R., Lundell, J., Paukner, D., Freedman, D. J., and Kasthuri, N. (2021). Primate neuronal connections are sparse in cortex as compared to mouse. Cell Rep. 36:109709. doi: 10.1016/j.celrep.2021.109709
Wong, M. Y., Liu, C., Wang, S. S. H., Roquas, A. C. F., Fowler, S. C., and Kaeser, P. S. (2018). Liprin-α3 controls vesicle docking and exocytosis at the active zone of hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 115, 2234–2239. doi: 10.1073/pnas.1719012115
Woodburn, S. C., Bollinger, J. L., and Wohleb, E. S. (2021). Synaptic and behavioral effects of chronic stress are linked to dynamic and sex-specific changes in microglia function and astrocyte dystrophy. Neurobiol. Stress 14:100312. doi: 10.1016/j.ynstr.2021.100312
Xiao, Z., Qiu, T., Ke, X., Xiao, X., Xiao, T., Liang, F., et al. (2014). Autism spectrum disorder as early neurodevelopmental disorder: Evidence from the brain imaging abnormalities in 2-3 years old toddlers. J. Autism Dev. Disord. 44, 1633–1640. doi: 10.1007/s10803-014-2033-x
Xu, J., Pang, Z. P., Shin, O. H., and Südhof, T. C. (2009). Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat. Neurosci. 12, 759–766. doi: 10.1038/nn.2320
Yamakawa, K. (1998). DSCAM: A novel member of the immunoglobulin superfamily maps in a Down syndrome region and is involved in the development of the nervous system. Hum. Mol. Genet. 7, 227–237. doi: 10.1093/hmg/7.2.227
Ye, H., Liu, J., and Wu, J. Y. (2010). Cell adhesion molecules and their involvement in autism spectrum disorder. Neurosignals 18, 62–71. doi: 10.1159/000322543
Yim, Y. S., Kwon, Y., Nam, J., Yoon, H. I., Lee, K., Kim, D. G., et al. (2013). Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 110, 4057–4062. doi: 10.1073/pnas.1209881110
Zarebidaki, F., Camacho, M., Brockmann, M. M., Trimbuch, T., Herman, M. A., and Rosenmund, C. (2020). Disentangling the Roles of RIM and Munc13 in synaptic vesicle localization and neurotransmission. J. Neurosci. 40, 9372–9385. doi: 10.1523/JNEUROSCI.1922-20.2020
Zhang, H. F., Dai, Y. C., Wu, J., Jia, M. X., Zhang, J. S., Shou, X. J., et al. (2016). Plasma Oxytocin and arginine-vasopressin levels in children with autism spectrum disorder in china: Associations with symptoms. Neurosci. Bull. 32, 423–432. doi: 10.1007/s12264-016-0046-5
Zhang, H., Liu, X., Zhang, C., Mundo, E., Macciardi, F., Grayson, D. R., et al. (2002). Reelin gene alleles and susceptibility to autism spectrum disorders. Mol. Psychiatry 7, 1012–1017. doi: 10.1038/sj.mp.4001124
Zhen, M., and Jin, Y. (1999). The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature 401, 371–375. doi: 10.1038/43886
Zhu, X., Need, A. C., Petrovski, S., and Goldstein, D. B. (2014). One gene, many neuropsychiatric disorders: Lessons from Mendelian diseases. Nat. Neurosci. 17, 773–781. doi: 10.1038/nn.3713
Zito, K., and Svoboda, K. (2002). Activity-dependent synaptogenesis in the adult mammalian cortex. Neuron 35, 1015–1017. doi: 10.1016/S0896-6273(02)00903-0
Keywords: presynaptic proteins, synaptopathy, presynaptic vesicle dynamics, vesicle release machinery, synaptogenesis, autism spectrum disorders (ASD)
Citation: Yeo XY, Lim YT, Chae WR, Park C, Park H and Jung S (2022) Alterations of presynaptic proteins in autism spectrum disorder. Front. Mol. Neurosci. 15:1062878. doi: 10.3389/fnmol.2022.1062878
Received: 06 October 2022; Accepted: 31 October 2022;
Published: 17 November 2022.
Edited by:
Katsuhiko Tabuchi, Shinshu University, JapanReviewed by:
Paola Bonsi, Santa Lucia Foundation (IRCCS), ItalySudeep Karki, University of Helsinki, Finland
Copyright © 2022 Yeo, Lim, Chae, Park, Park and Jung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyokeun Park, aGtwYXJrQHVzdC5oaw==; Sangyong Jung, SnVuZ19TYW5neW9uZ0BpbWNiLmEtc3Rhci5lZHUuc2c=