
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 10 January 2023
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.1032541
This article is part of the Research Topic Lipid-Protein Interactions and Regulation in Neurodegenerative Disorders View all 6 articles
 Cyntia Alves Conceição1,2
Cyntia Alves Conceição1,2 Gabriela Assis de Lemos1,2
Gabriela Assis de Lemos1,2 Caroline Augusto Barros1,2
Caroline Augusto Barros1,2 Tuane C. R. G. Vieira1,2*
Tuane C. R. G. Vieira1,2*The molecular cause of transmissible spongiform encephalopathies (TSEs) involves the conversion of the cellular prion protein (PrPC) into its pathogenic form, called prion scrapie (PrPSc), which is prone to the formation of amorphous and amyloid aggregates found in TSE patients. Although the mechanisms of conversion of PrPC into PrPSc are not entirely understood, two key points are currently accepted: (i) PrPSc acts as a seed for the recruitment of native PrPC, inducing the latter’s conversion to PrPSc; and (ii) other biomolecules, such as DNA, RNA, or lipids, can act as cofactors, mediating the conversion from PrPC to PrPSc. Interestingly, PrPC is anchored by a glycosylphosphatidylinositol molecule in the outer cell membrane. Therefore, interactions with lipid membranes or alterations in the membranes themselves have been widely investigated as possible factors for conversion. Alone or in combination with RNA molecules, lipids can induce the formation of PrP in vitro-produced aggregates capable of infecting animal models. Here, we discuss the role of lipids in prion conversion and infectivity, highlighting the structural and cytotoxic aspects of lipid-prion interactions. Strikingly, disorders like Alzheimer’s and Parkinson’s disease also seem to be caused by changes in protein structure and share pathogenic mechanisms with TSEs. Thus, we posit that comprehending the process of PrP conversion is relevant to understanding critical events involved in a variety of neurodegenerative disorders and will contribute to developing future therapeutic strategies for these devastating conditions.
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, are a group of fatal neurodegenerative disorders that directly affect the central nervous system (CNS), causing loss of neuronal cells and, consequently, neurological symptoms (Greenlee and Greenlee, 2015; Asher and Gregori, 2018). The TSEs that affect humans are Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), kuru, Gerstmann-Sträussler-Scheinker (GSS) syndrome, and variably protease-sensitive prionopathy (VPSPr; Collins et al., 2004; Gambetti et al., 2011; Baldwin and Correll, 2019; Liberski et al., 2019). The structural conversion and accumulation of prion protein (PrP) play a significant role in the development of TSEs (Gill and Castle, 2018).
The cellular prion protein (PrPC) is encoded by PRNP, a highly conserved gene in mammals located on chromosome 20 in humans (Sarnataro et al., 2017). PRNP encodes a sequence of 253 amino acids that undergo post-translational modifications in the endoplasmic reticulum. These modifications include the removal of a signal peptide in the N-terminal domain (1-22) that directs PrPC to the plasma membrane; the removal of a signal peptide in the C-terminal domain (232-253) in the endoplasmic reticulum, and the attachment of a glycosylphosphatidylinositol (GPI) anchor; addition of glycans (N-181 and N-197), and the formation of disulfide bonds (C-179 and C-214; Stahl et al., 1987; Haraguchi et al., 1989; Harris, 2003). The GPI anchor is added to PrPC via the GPI-transamidase enzyme (Puig et al., 2014; Sarnataro et al., 2017). PrPC is widely found in CNS cells and is also expressed in other tissues, such as the heart and lungs (Bendheim et al., 1992; Wulf et al., 2017; Gill and Castle, 2018); it is found in regions of lipid rafts anchored by the GPI in the extracellular membrane (Prado et al., 2004).
The mature prion protein structure (PrP23-231) can be structurally divided into N- and C-terminal domains. The N-terminal domain is intrinsically disordered, with a repeated octapeptide sequence (PHGGGWGQ), containing histidine residues that are important for interaction with metallic ligands such as copper (II) (Brown et al., 1997; Salzano et al., 2019). The C-terminal domain is structured and globular, with three α-helices and a small antiparallel β-sheet (Heske et al., 2004; Acevedo-Morantes and Wille, 2014).
PrPC plays many different roles in cells since it interacts with many other partners. These functions are related to metal ion metabolism, neurotransmission, neurogenesis, neuroprotection by acting as an antioxidant, cell–cell adhesion, and memory, among others (Das and Zou, 2016; Linden, 2017; Wulf et al., 2017). The cellular location of PrPC at the plasma membrane may be related to its cell signaling function, as discussed below. PrPC may interact with membrane lipids and associate with other transmembrane proteins, thereby transmitting signals into the intracellular compartment (Legname, 2017; Sarnataro et al., 2017).
Conformational changes in PrPC cause TSEs; it has its structure rich in α-helices (about 40%) and a small percentage of β-sheets (about 3%), which transforms into a structure enriched in β-sheets (about 45% β-sheets and 30% α-helices) called prion scrapie (PrPSc; Pan et al., 1993; Prusiner, 1998; Wulf et al., 2017). The conversion of PrPC to PrPSc leads to biochemical changes in the physicochemical properties, increasing the tendency to aggregation, resistance to protease digestion, and partial resistance to heat and denaturing agents (Prusiner, 1998; van Rheede et al., 2003). The diseases caused by PrPSc can have an infectious and genetic origin and have been classified as hereditary, acquired, or sporadic. Mutations in the PRNP gene are associated with the hereditary form, while there is a spontaneous conversion of PrPC to PrPSc in the sporadic form. In the acquired form, transmission can occur in several ways, such as using surgical instruments and ingesting contaminated food (Will, 2003; Geschwind, 2015).
Explanations still need to be made available for how exactly PrPC structural changes initiate and propagate in the misfolded form. According to the protein-only hypothesis, the presence of PrPSc alone is sufficient to induce PrPC conversion, as the former acts as a template, recruits PrPC, and causes conformational changes for more PrPSc formation and subsequent aggregation (Prusiner, 1998; Baskakov and Bocharova, 2005). Some studies reproduced this conversion hypothesis in vitro but with low efficiency (Deleault et al., 2005; Saá et al., 2006; Zhang et al., 2013). In vitro conversion in the presence of other molecules was more efficient (Deleault et al., 2003; Geoghegan et al., 2007; Kovachev et al., 2019). Therefore, it is hypothesized that the presence of cofactors benefits PrP conversion (Cohen and Prusiner, 1998; Silva et al., 2010a; Wang et al., 2010a,b). Other biomolecules, like lipids, RNA, DNA, and other proteins at the cell membrane and in the cytoplasm, could act as cofactors that accelerate prion structural conversion and subsequently modulate infectivity and toxicity.
In this review, we provide information about membrane lipid interaction with prion protein and the role of this interaction in PrP function and the conversion process. First, we discuss prion protein attachment to cell membranes and the effects of this interaction on prion structure and stability. Then, we discuss the roles of PrP-lipid interactions in physiology. Finally, we relate studies that investigate lipid involvement in RNA- and lipid-mediated PrP conversion and the toxic effects of this process.
Cells and some cell organelles are delimited by a lipid membrane organized in bilayers of two lamellae, where the hydrophobic portion of the lipids hides from the water, and the hydrophilic portion interacts with the outer and inner cellular spaces. This lipid bilayer is responsible for cell protection and cell–cell communication and selectively internalizes some molecules to the cytoplasm (Simons and Sampaio, 2011). A vast repertoire of lipid species can participate in the structure of membrane bilayers. Most lipids in the mammalian cell membrane are glycerophospholipids (GLPs), sphingolipids (SPs), and cholesterol. GLPs, the major lipid components of the membranes, comprise a glycerol backbone linked to a hydrophobic portion of two acyl chains and a hydrophilic headgroup with phosphoric acid. This basic structure of a GLP is named phosphatidic acid (PA). The interaction of the PA headgroup with alcohol molecules leads to the formation of a diversity of GLPs, namely phosphatidylinositol (PI), phosphatidylethanolamine (PE), phosphatidylcholine (PC), and other GLPs. SPs comprise a sphingosine backbone linked to an acyl chain on its amine group, and a headgroup, such as phosphocholine, phosphoethanolamine and others, on its hydroxyl group.
The sphingosine backbone also forms the core of glycolipids, which may interact with lipid bilayers. In the case of glycolipids, the hydroxyl group of the sphingosine binds to glycan units (Harayama and Riezman, 2018). Each cell organelle has different membrane lipid distribution, influencing its fluidity and function. High amounts of cholesterol contribute to the formation of less impermeable bilayers; for instance, this lipid is found in higher proportions in the plasma membrane (PM), which is essential for controlling molecule exchanges with the external environment. Cholesterol and SPs are associated with more organized regions of the membrane bilayers, such as caveolae and lipid rafts (Simons and Sampaio, 2011; Egawa et al., 2016).
The lipid bilayer also comprises transmembrane proteins and proteins that interact with the membrane through glycolipids, allowing communication between cells and activating various physiological mechanisms (Egawa et al., 2016). PrPC is attached to the outer leaflet of cellular membranes through a GPI anchor, bound to lipid raft regions via the amino acid residue S231 (Rudd et al., 2001; Taylor and Hooper, 2006). GPI is composed of a core made of three mannose and one glucosamine residue. A PI headgroup with a saturated acyl chain connects to the GPI glucosamine residue, and during the traffic to the cell membrane, sialic acid can be linked to one of the mannose residues (Taylor and Hooper, 2006). Lipid rafts are lipid membrane microdomains formed by the ordered assembling of cholesterol and SPs that create a liquid domain resistant to detergent solubilization, unsaturated GLPs, and proteins (Egawa et al., 2016). Changes in GPI composition and PrPC attachment to lipid raft influence PrP’s physiological and pathological roles (Bate et al., 2016a,b).
PrPC internalization is required for it to be trafficked to the secretory and recycling pathways. Two principal mechanisms of PrP internalization have been described: caveolae-dependent and clathrin-coated pit internalization (Shyng et al., 1994; Peters et al., 2003; Fehlinger et al., 2017). Caveolae are membrane invagination regions, considered a special class of rafts, which are rich in cholesterol and glycosphingolipids, and are coated by the protein caveolin-1; Caveolin-1 mediates signal transduction mechanisms and other physiological processes (Anderson, 1998; Peters et al., 2003; Echarri et al., 2007). PrPC was found in the caveolae regions of the PM and trans-Golgi network (Peters et al., 2003). It was found that the depletion of cholesterol from these regions is associated with the impairment of PrPC endocytosis, suggesting that the lipid is of great importance for caveolae-mediated PrP internalization and that PrP must be present in lipid raft-like domains to be internalized via caveolae (Marella et al., 2002; Bate et al., 2004; Cashion et al., 2022). Nonetheless, some neuronal cells do not have the machinery for caveolae internalization of proteins. Thus, other internalization pathways, like clathrin-mediated endocytosis, are important in these cells’ PrP internalization mechanism.
In contrast to the caveolae mechanism, PrP internalization mediated by clathrin-coated pits occurs out of the lipid rafts in non-raft membrane domains (Taylor and Hooper, 2006). Since the clathrin pathway is only related to the endocytosis of transmembrane proteins (Trowbridge, 1991), PrP internalization mediated by clathrin depends on its interaction with a transmembrane adaptor (Shyng et al., 1994, 1995; Taylor and Hooper, 2006). Moreover, deletions of N-terminal amino acid residues, or point mutations in its polybasic region, impair PrP endocytosis (Nunziante et al., 2003; Sunyach et al., 2003). Cu2+ binding to the octarepeat region at the N-terminal destabilizes the PrP structure, facilitating its translocation from lipid rafts (Taylor et al., 2005). Thus, the PrP N-terminal domain and the lipid membrane composition, together with anchoring through GPI, are critical for PrP endocytosis.
During endocytosis processing, PrP loses its stability and may detach from the GPI anchor, leading to the conversion and loss of its physiological functions (Chesebro et al., 2005; Bate et al., 2016c). Moreover, PrPC localization in lipid rafts and the maturation or degradation process facilitate its interaction with various molecules in the membrane traffic. These interactions influence its physiological effects and the conversion process to PrPSc.
Although the precise function of PrPC remains elusive, its physiological participation in multiple cell-signaling pathways may be determined by its interaction with different lipids in the cell membrane (Figure 1; Godsave et al., 2015; Wulf et al., 2017). Structural and functional studies of PrP have revealed the presence of multiple membrane interaction motifs. At the N-terminus, an unstructured basic-hydrophobic region is predicted to be membrane-interactive since it inserts into micelles and may have a role in cell penetration (Magzoub et al., 2006). Multiple tryptophan residues in the octapeptide repeats between residues 60 and 91 interact with dodecyl phosphocholine (DPC) micelles regardless of the presence or absence of Cu2+ and insert into membranes (Dong et al., 2007). The protonation of the histidine residues in these repeats within low pH environments of endocytic compartments could favor interactions with acidic lipids (Morillas et al., 1999).
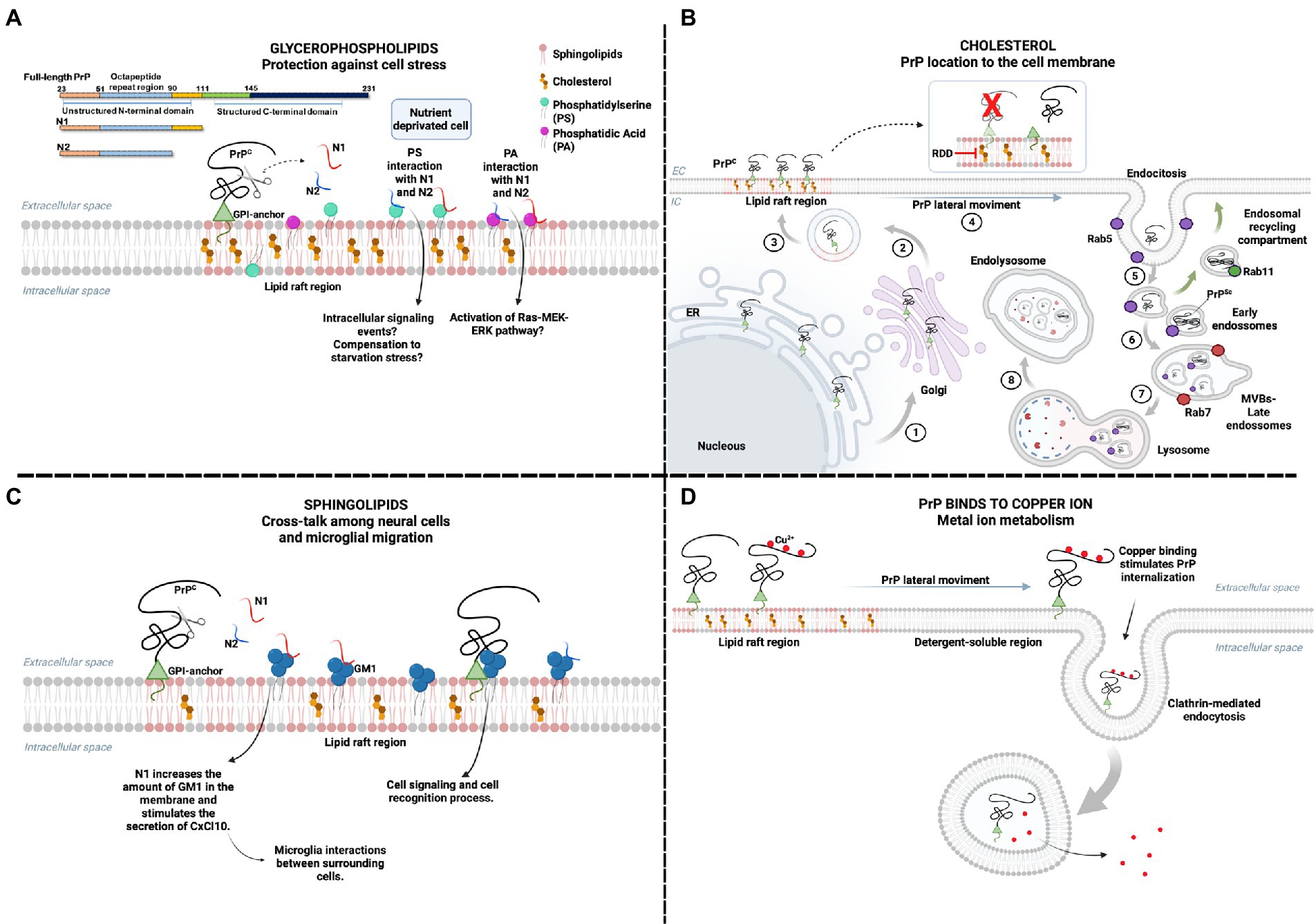
Figure 1. The physiological effects of PrP interaction with membrane lipids. Location on lipid rafts enables PrPC interaction with various ligands, including membrane lipids. (A) Cellular starvation can alter the plasma membrane order, leading to PS exposure to the extracellular medium and increased PA. The interaction of N1 and N2 fragments, peptides derived from the proteolytic cleavage of PrP, may have a regulatory effect on cellular stress by binding to PS and PA. The interaction of N1 and N2 with PA can activate the Ras–MEK–ERK pathway and promote cell survival. At the same time, the interaction with PS can also activate pathways related to the same function. (B) PrPC is formed in the endoplasmic reticulum, which undergoes post-translational modifications, such as binding to the GPI anchor. Subsequently, it is taken to the Golgi complex (1) and forwarded to the plasma membrane (2), mainly in lipid rafts (3). The importance of cholesterol in lipid rafts for the physiological location of PrPC has already been reported. The process of internalization of PrP requires a lateral movement (4) to areas where the membrane is more soluble, outside the lipid rafts. Endosome motility is related to the function of Rab proteins, such as Rab 5 for early endosomes (5), Rab 7 for the multivesicular body (MVB)/late endosomes (6), and Rab11 for the endosomal recycling compartment (ERC) (green arrows). Conversion of PrPC to PrPSc can occur within ERC and MVBs. MVBs fuse to lysosomes (7) and proteins are degraded in endolysosomes (8). The cleavage of PrPC from the GPI anchor may favor PrP-lipid interaction and PrP conversion, but cells expressing PrP without GPI are not infected. Drugs that sequester cholesterol are raft dissociation drugs (RDD), known to inhibit PrPSc formation. (C) The interaction of GM1 ganglioside and N1 fragment promotes the increase of GM1 in the plasma membrane. It stimulates the secretion of the cytokine CxCl10, enabling the interaction between microglia and other surrounding cells. The interaction between PrPC and GM1 has been reported, but the mechanisms triggered by this interaction have not been elucidated but are possibly related to cell signaling and cell recognition process. (D) PrPC binds to copper ions and is involved in ion metabolism. It has already been reported that the interaction between copper and PrPC stimulates the endocytosis of PrP. In the presence of copper, PrPC leaves the lipid raft region and internalizes mainly via clathrin-mediated endocytosis, balancing the amount of copper inside and outside the cell. Created with BioRender.com.
These membrane-interacting elements are disordered regions and may mediate pH-dependent associations with acidic phospholipid bilayers via relatively non-stereospecific electrostatic and hydrophobic interactions. The investigation of the interaction between full-length PrP and Supported lipid bilayers, a membrane mimetic surface, by single-molecule force spectroscopy revealed three lipid-binding regions at the PrP N-terminal: PrP95-110; PrP23-51 and the octapeptide repeat PrP51-90 (Pan et al., 2019). Motifs in the C-terminal domain of PrP, which shows structured elements, may mediate more specific lipid interactions (Overduin et al., 2021). Three proximal membrane interacting motifs, including V122GGL, which precedes the β1 strand, Y169SN between β2 and α2, and Y225YQR in α3, were implicated in the association with lipids, forming a single continuous binding surface with lipid membranes (Overduin et al., 2021).
The N-terminal fragments resulting from the physiological post-translational endoproteolytic cleavage of PrPC – the N1 and N2 fragments – were shown to bind in vitro to phosphatidylserine (PS) and PA, which are components of cell membranes responsive to cellular stress. In a model of serum deprivation, the N2 fragment protected neural cells from disturbance in the cellular lipid environment, including externalization of membrane PS and increased PA levels (Haigh et al., 2015). This suggests that PrP’s protective role in cellular stress conditions may involve interaction with PS and PA (Figure 1A).
The N1 fragment, which also interacts with PS and PA, was shown to enhance cell viability in a co-culture of neuronal and microglia cells and, significantly, to modulate the interaction and cross-talk among the cells through an increase of the sphingolipid GM1 at sites of interaction in the membrane of microglial cells (Carroll et al., 2020; Figure 1C). GM1 is a ganglioside that resides in cholesterol-rich domains of the cell membrane, whose disruption has been related to the reduced migratory capacity of microglial cells (Kuipers et al., 2006), and PrP was shown to bind to GM1 directly (Sanghera et al., 2011). Since PrP is necessary for inducing cell migration by microglial stress-inducible protein 1 (STI1; da Fonseca et al., 2012), also a PrP ligand, the impairment in the migration of microglia by the disruption of cholesterol-rich domains may be related to the loss of interaction of PrP with GM1.
The physiological interaction between gangliosides and PrP was observed in neural and immune system cells like T lymphocytes. In human T cells, PrP was shown by immunoprecipitation to interact with the ganglioside GM3 specifically, the main component of glycosphingolipid-enriched microdomains (GEM) in the cell membrane (Mattei et al., 2002, 2004), which is involved in T-cell activation signaling by assembly with signal transducer molecules, such as Fyn and phosphorylated ZAP-70 (P-ZAP-70). Since PrP interacts with GM3, Fyn, and P-ZAP-70, it is proposed to be a component of the multimolecular signaling complex involved in ligand-specific T-cell activation, suggesting a role for PrP in this context. In a human T-cell line, PrP colocalized with GM1 and CD3, also components of the multimolecular T-cell receptor (TCR) complex, in response to hypothermic stress, which led to lymphocyte activation (Wurm et al., 2004). The depletion of cholesterol by methyl-β-cyclodextrin, which interferes with the interaction of GPI-anchored proteins such as PrP, impaired the hypothermal activation of T-cells (Wurm et al., 2004), reinforcing a role for PrP.
Cholesterol, also abundant in lipid rafts domains, was essential to the cell membrane localization of PrP in neurons since inhibiting cholesterol synthesis led to an accumulation of PrP in the Golgi compartment (Gilch et al., 2006). Therefore, cholesterol determines PrP roles depending on its cell membrane location (Figure 1B).
Nonetheless, the characteristic structural assemblies between lipids and proteins in lipid rafts are not confined to the plasma membrane. Raft-like microdomains are found in membranes of subcellular compartments like the endoplasmic reticulum, Golgi, and mitochondria (Hayashi and Su, 2003; Garofalo et al., 2005). Mitochondrial lipid raft-like microdomains have been proposed to regulate cell apoptosis in different cell types (Garofalo et al., 2015). Interestingly, PrP is present in the inner mitochondrial membrane of healthy brain tissue, suggesting a role for PrP in mitochondrial function (Faris et al., 2017). It is well known that mitochondria are an important modulator of cell apoptosis through the release of cytochrome C and the resulting activation of the caspase signaling pathway (Wang and Youle, 2009). PrP was also shown to modulate apoptosis in multiple conditions, exerting an anti-apoptotic role in neuronal and cancer cells (Kim et al., 2004; Roucou et al., 2005; Gao et al., 2019; Adhikari et al., 2021). Therefore, it is plausible to suggest that the anti-apoptotic role of PrP may involve its interaction with mitochondrial lipid raft-like microdomains.
Among the multiple roles of PrP in cellular homeostasis, its role as a copper-binding protein is the most well-accepted (Kawahara et al., 2021). Importantly, it may function as an antioxidant as it quenches free radicals generated by Cu2+ redox cycling (Haigh and Brown, 2006; Viles et al., 2008). The excess of free Cu2+ ions is toxic due to producing reactive oxygen species (ROS), such as superoxide and nitric oxide (Wong et al., 2008; Kodama et al., 2012). Studies show that enzyme superoxide dismutase (SOD) activity, crucial for controlling ROS homeostasis, is regulated by the presence of PrPC in cell membranes (Brown et al., 1997, 1999; Sakudo et al., 2005). Brown et al. showed that adding Cu2+ to PrP induces SOD activity. This study compared the SOD activity of PrPC extracted from wild-type and Prnp knockout mice (Prnp-/-) and showed that SOD activity is abolished in Prnp-/- mice, suggesting that PrPC is vital for SOD activity. Also, when Cu2+ was chelated in wild-type mice using diethyldithiocarbamate (DDC), PrPC-SOD activity was abolished (Brown et al., 1999).
The leading site for Cu2+ binding is the PrPC N-terminal region, with four Cu2+ binding to four octarepeats (PHGGGWGQ), one to His96 and another to His111 (Sánchez-López et al., 2018). Binding to Cu2+ drives PrPC lateral movement outside lipid rafts, stimulating endocytosis (Pauly and Harris, 1998; Taylor et al., 2005). It regulates Cu2+ levels and the activity of Cu-dependent enzymes (Figure 1D; Brown et al., 1997). It is also suggested to regulate PrPC interaction with membrane partners, affecting PrPC’s physiological role (Posadas et al., 2022).
Alpha-cleavage, the main proteolytic event of PrPC, yields fragments N1, including residues 23-110, and C1, residues 111-231 with a free NH2-terminus, which remains membrane-bound and retains the copper binding site at His111 (Sánchez-López et al., 2018). Although it has only one coordination site for Cu+2 precisely at the alpha-cleavage site, PrP(111-115) peptide was proposed to bind Cu2+ depending on proton (pH) and copper-peptide ratios (Sánchez-López et al., 2018). Since the C1 fragment can represent up to 50% of total PrPC at the PM (Altmeppen et al., 2012) and is exposed to fluctuations in copper concentration during synaptic transmission (D’Ambrosi and Rossi, 2015; Gromadzka et al., 2020), Cu2+ trafficking may be physiologically modulated by the membrane-bound C1 fragment.
Interestingly, a disturbance in lipid rafts composition by exposure to exogenous gangliosides GM1, GM3, and GD1a in cell culture, did not impact PrP cleavage and consequent generation of C1 and N1 fragments. However, it led to the structural rearrangement of PrPC (Botto et al., 2014). In accordance, neither lipid raft location nor membrane anchorage of PrPC was central for the generation of C1, since cells expressing (i) PrP-CTM, a PrP construct known to not localize in lipid rafts or (ii) a GPI-anchorless mutant PrP, produced a fragment analogous to C1 in cell lysates (Walmsley et al., 2009). However, the C1 fragment strongly colocalized with the lipid raft marker Cholerae Toxin B subunit, showing a preferential enrichment in raft regions (Botto et al., 2014), suggesting cholesterol and sphingolipids may be necessary for the Cu2+-binding property of this fragment, although in a manner not related to its alpha-cleavage.
PrPC anchoring to lipid rafts was shown to increase its stability; thus, the absence of the GPI anchor may influence PrP susceptibility to conversion and aggregation (Figure 1B; Baron et al., 2002; Chu et al., 2014). Moreover, it has been reported that characteristics related to prion diseases, such as neuropathology and disease incubation time, are modified depending on the presence and composition of the GPI anchor (Chesebro et al., 2005; Bate et al., 2010; Bate and Williams, 2011). The absence of sialic acid in the GPI composition may change the lipid environment and be related to the reduction of PrPSc neurotoxicity (Bate and Williams, 2011; Bate et al., 2016d). A recent study showed that changes in the GPI signaling sequence of the PrP C-terminal domain generate a GPI anchor lacking sialic acid. The new composition was associated with increased prion disease incubation time and reduced PrPSc levels (Puig et al., 2019). Therefore, changes in the polysaccharide composition of the GPI anchor directly interfere with PrP conversion, becoming an attractive target to modulate PrP aggregation.
Cholesterol lipid is essential for PrP localization in lipid rafts; however, high concentrations of this lipid are cytotoxic as it leads to decreased membrane fluidity and membrane disruption, besides other toxic effects related to cholesterol oxidation. These changes may cause defects in integral membrane activity, cell signaling, and death (Tabas, 2002). Upon prion infection, enzymes involved in cholesterol synthesis are upregulated in neuronal cell lines and infected neurons (Bach et al., 2009). Also, cholesterol efflux from the brain to the circulation system is affected by reduced levels of the enzyme cholesterol 24-hydroxylase [Cytochrome P450 46A1 (CYP46A1)] in mice brains infected with PrPSc (Ali et al., 2021). Consequently, cholesterol levels are increased in neuronal cells and sequestered to membranes during prion disease progression, contributing to PrPSc pathogenic mechanism (Bate et al., 2008a,b,c; Cashion et al., 2022).
Inhibition of cholesterol synthesis using lovastatin and squalestatin reduced PrPSc formation in prion-infected cells (Taraboulos et al., 1995; Bate et al., 2004). The administration of efavirenz to N2a-infected cells, an allosteric activator of CYP46A1 enzyme, reduced PrPSc levels without affecting cell membrane stability nor PrPC levels. Moreover, an increase in survival time and a decrease in the disease progression were observed in prion-infected mice treated with this drug (Taraboulos et al., 1995; Ali et al., 2021; Cashion et al., 2022). Drugs affecting lipid raft formation by biding cholesterol, such as filipin and amphotericin B, showed the same effect over PrPSc levels (Mangé et al., 2000; Marella et al., 2002). A drug affecting cholesterol transport named U18666A caused a redistribution of cholesterol from the plasma membrane to the intracellular space, reducing PrPSc in N2a cells, although it failed when administrated to infected mice (Klingenstein et al., 2006; Hagiwara et al., 2007). Thus, the stability of the PrPC and its conversion must be significantly affected not only by the GPI anchor but also by the composition of the lipid raft itself. In this context, drugs that affect cholesterol metabolism are promising therapeutic candidates for prion diseases.
In addition to its importance in modulating lipid metabolism, the presence of PrPC or its scrapie form is also essential for modulating endosomal trafficking processes and being modulated by it. PrP trafficking during the endocytic pathway favors the formation of resistant PrP (PrPres) in cell cultures infected with PrPSc (Caughey and Raymond, 1991; Marijanovic et al., 2009; Priola and McNally, 2009). Thus, PrP internalization is an essential step for the conversion into PrPSc and the associated aggregation.
PrP trafficking is essential for conversion and especially for prion transmission and propagation. The lateral translocation of PrP from the lipid raft region for clathrin-coated pits makes PrP more susceptible to conversion since the protein is less stable in non-raft regions (Taylor et al., 2005). Furthermore, once endocytosed, PrP traffics into early endosomes to be either directed to recycling and returns to the plasma membrane or to late endosomes to be degraded in lysosomes (Campana et al., 2005; Figure 1B). Studies suggest that PrP conversion occurs in the endosomal recycling compartment (Marijanovic et al., 2009) and in late endosomes (or multivesicular endosomes; Yim et al., 2015), which may propagate through secreted exosomes (Fevrier et al., 2004; Yim et al., 2015).
PrPSc alters Rab GTPases profile (Kovács et al., 2007; Shim et al., 2016), known to regulate intracellular transport and vesicle fusion, interfering with the endo-lysosomal pathway, and it may enhance conversion and toxicity. PrPSc infection also affects post-Golgi vesicle transportation of membrane proteins such as PrPC, which accumulates in the Golgi apparatus. Other membrane protein distributions are also affected by PrPSc, such as insulin receptor, which is essential for neuroprotection, and attractin, which absence may have implications in spongiform degeneration since it plays a role in the myelination process. Thus, prion toxicity involves not only PrPSc activity but also the impairment of other membrane protein functions (Kuramoto et al., 2001; Barmada, 2005; Nelson and Alkon, 2005; Uchiyama et al., 2013). Moreover, PrPSc amyloid aggregates may accumulate on cell membranes, forming amyloid deposits. In neurons, PrPSc deposition is associated with dendritic degeneration in the early steps of prion disease, leading to severe synapses dysfunction (DeArmond and Bajsarowicz, 2010).
Other cells’ subcompartments may influence PrP aggregation (Campana et al., 2005). Genetic prion diseases, such as FFI and GSS, are characterized by point mutations in PrP amino acid sequence (Baldwin and Correll, 2019). Mutation in the PrP residue 117, associated with the GSS pathology, generates the accumulation of PrP with its C-terminal inside the ER lumen, which is associated with increased ER stress and PrPSc accumulation (Hegde et al., 1998). PrP is also found in the cytosol (Mironov et al., 2003; Levine et al., 2005), and the overexpression of PrP without its signal sequence for GPI interaction is associated with the formation of PK-resistant cytosolic aggregates (Godsave et al., 2015).
The pathophysiology of prion infection may be related to PrPC loss of function or PrPSc gain of cytotoxic function (Winklhofer et al., 2008). A well-established characteristic of PrPC is its N-terminal octapeptide repeat region binding affinity for Cu2+, which is required for some of the PrPC physiological roles. PrPC binding to Cu2+ regulates Cu2+ levels in neuronal cells, playing a neuroprotective role. Thus, PrPC conversion to PrPSc causes perturbations in neuronal antioxidant activity (Figure 2D).
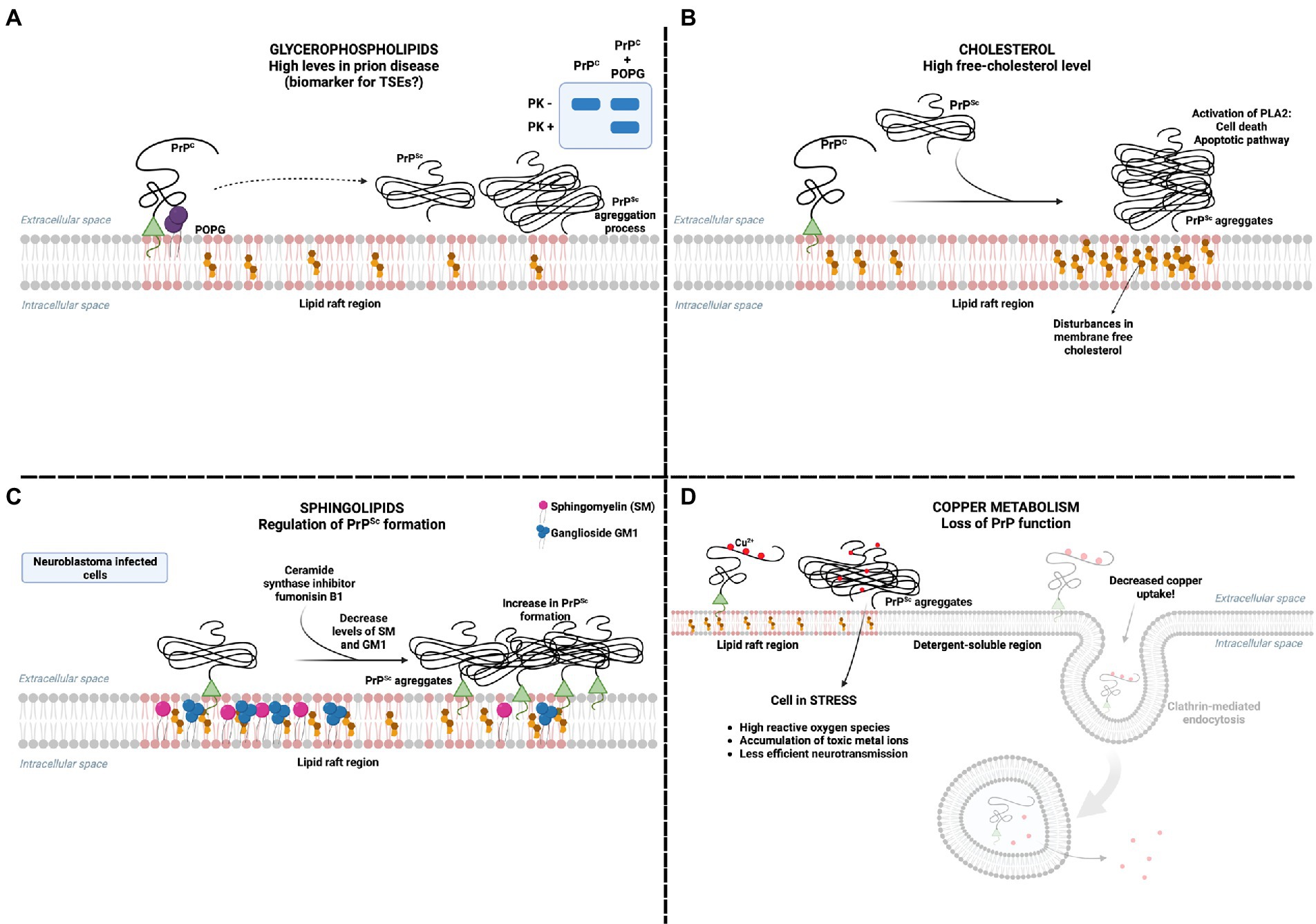
Figure 2. PrPC interaction with lipids in pathology (A) Glycerophospholipids are proposed as potential biomarkers for TSEs as their concentration increases in prion disease models. The interaction of PrP with POPG can modulate the formation of proteinase K-resistant PrPSc aggregates. (B) It has already been reported that the presence of PrPSc increases free cholesterol and activates cell death mechanisms by activating phospholipase A (PLA). (C) The presence of sphingolipids in the membrane is essential in inhibiting the presence of PrPSc. Depletion of sphingolipids SM and GM1, using the ceramide synthase inhibitor fumonisin B(1), has been reported to increase the presence of PrPSc in neuroblastoma cells infected with PrPSc. (D) The presence of PrPSc in the plasma membrane prevents the internalization of copper, promoting a series of harmful consequences to the cell, mainly related to oxidative stress. Created with BioRender.com.
Cu2+ may be directly involved with PrP conversion and transmission. Cu2+ binding to octarepeats leads to PrP conformational change to a beta-sheet-rich structure (Salzano et al., 2019). Chelation of Cu2+ delayed prion disease in infected mice model (Sigurdsson et al., 2003). The presence of extra octapeptide regions (Krasemann et al., 1995) or the deletion of this region (Flechsig et al., 2000) impacts the formation of PrPSc, being favored when this region is available, probably binding Cu2+. On the other hand, Cu2+ was also beneficial for prion diseases once its supplementation prolonged survival time in an infected animal model and protected N2A cells from infection (Hijazi et al., 2003). Once it enhances PrPC internalization, it should reduce the encounter with PrPSc and, consequently, its conversion. The controversial results observed for Cu2+ must be further investigated for complete understanding. The findings probably result from a delicate balance between Cu2+ and PrP metabolism and its beneficial and deleterious roles.
Two molecular hypotheses for PrP conversion have been investigated in recent decades. The protein-only hypotheses suggest that the presence of PrPSc alone can induce PrPC structural changes and conversion (Prusiner, 1998; Baskakov and Bocharova, 2005). However, the energy barrier between both PrP structures could not be transposed only by the influence of PrPSc (Cohen and Prusiner, 1998; Silva et al., 2010b). Therefore, other macromolecules co-purified with PrPSc from brain tissues were suspected to be involved in PrP conversion. One of the main findings was that polyanionic compounds, such as RNA and proteoglycans, interact and convert endogenous and bacterially expressed PrPC (recombinant PrP-rPrP; Deleault et al., 2003, 2007; Kovachev et al., 2019).
Following the finding that RNA was an essential molecule for PrP conversion, and since the RNA molecule is negatively charged and PrP is found at lipid membranes, it was suggested that negatively charged lipids could interact and induce PrP conversion (Table 1). PMCA (protein misfolding cyclic amplification) is a methodology that uses recombinant PrP as substrate and infected brain homogenates as seed. Preparations containing supposedly converting molecules can also be used in place of PrPSc seeds. The sample goes through sonication cycles, and PK-resistant PrP formation is observed if you have conversion and aggregation. Using this technique, Wang et al. (2010a) showed that both RNA and synthetic palmitoyl oleoyl-phosphoglycerol (POPG) caused recombinant PrP conversion to a PK-resistant form capable of propagating its conformation to endogenous PrPC and causing clinical signs of prion disease when inoculated in wild-type mice. POPG vesicles induced the exposure of PrP-RNA binding sites leading to RNA direct interaction and PrP aggregation (Miller et al., 2013; Zurawel et al., 2014). In contrast, another study suggested that POPG and RNA may refold PrPC to its PK-resistant form, PrPSc-like, but lacking infectivity (Timmes et al., 2013). This contrasting result is probably a consequence of the depletion of cofactors necessary for maintaining PrPSc infectious conformation (Gomes et al., 2008; Deleault et al., 2012a,b). In addition, spectroscopic methods showed that POPG vesicles alone induce recombinant PrP conversion to a β-sheet enriched form, resistant to PK digestion (Wang et al., 2007; Sanghera et al., 2011), suggesting that membrane lipids could interact and convert PrP in the absence of any other molecule.
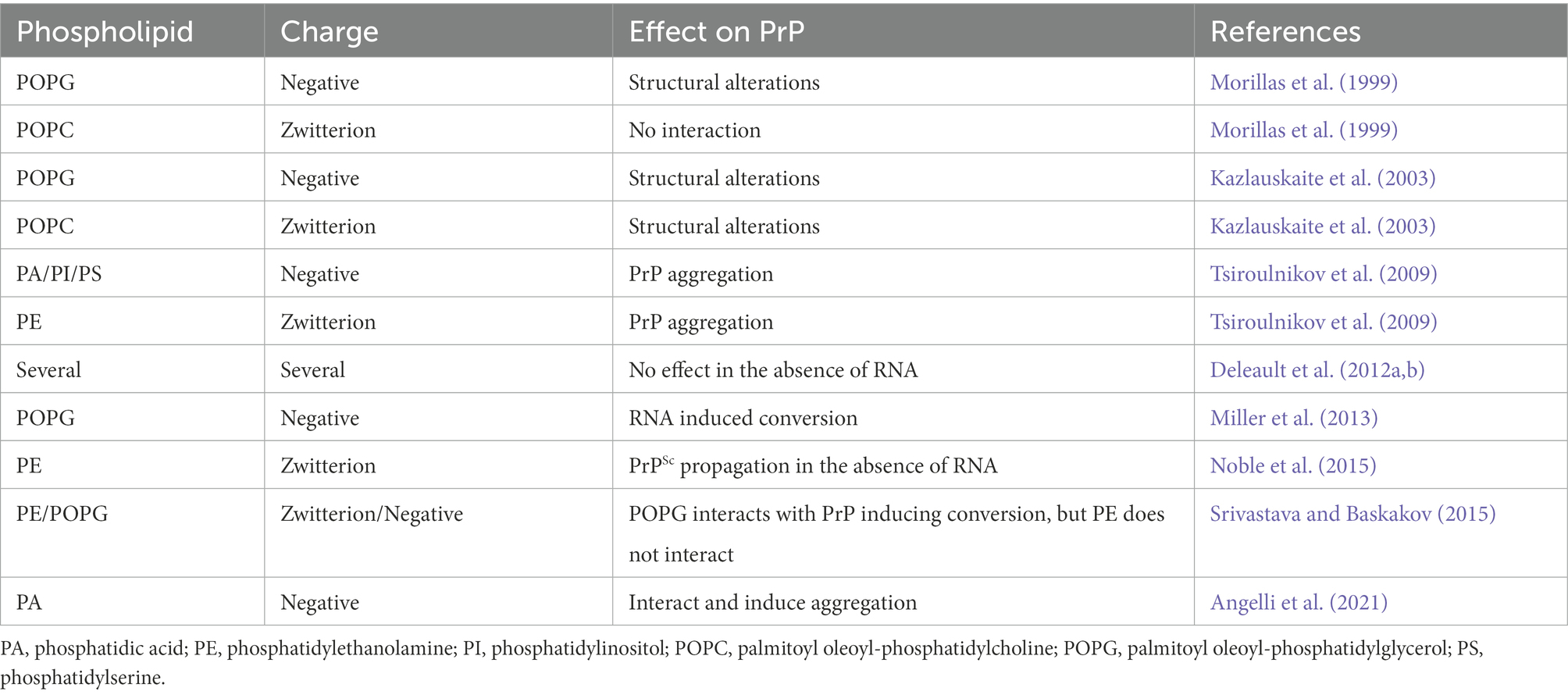
Table 1. Described effects of phospholipids with different charges on prion protein (PrP).
Different negative and zwitterionic phospholipids can interact and induce PrP aggregation, such as PI, PS, PA, PE, and PC (Tsiroulnikov et al., 2009; Table 1 and Figure 3). Studies suggest that the interaction of phospholipids with PrP is linked to the charge of the lipid and other types of interactions, like van der Waals and hydrophobic forces, and may be determinants for phospholipid-induced PrP aggregation. PA vesicles were shown to interact and induce aggregation of either murine (MuPrP) and rabbit PrP (RbPrP), with greater affinity for RbPrP, despite leading to a more significant aggregation of MuPrP (Angelli et al., 2021). The higher affinity of RbPrP for PA is probably due to its more positively charged surface (Wen et al., 2010). Positively charged residues 100-110 at the PrP N-terminus and the hydrophobic region were necessary for PrP-POPG vesicle interaction, where the positively charged residues would be responsible for the first contact with POPG by electrostatic interactions. Together with the hydrophobic region (residues 111-134), these residues were critical for POPG-induced PrP conversion (Wang et al., 2010b).
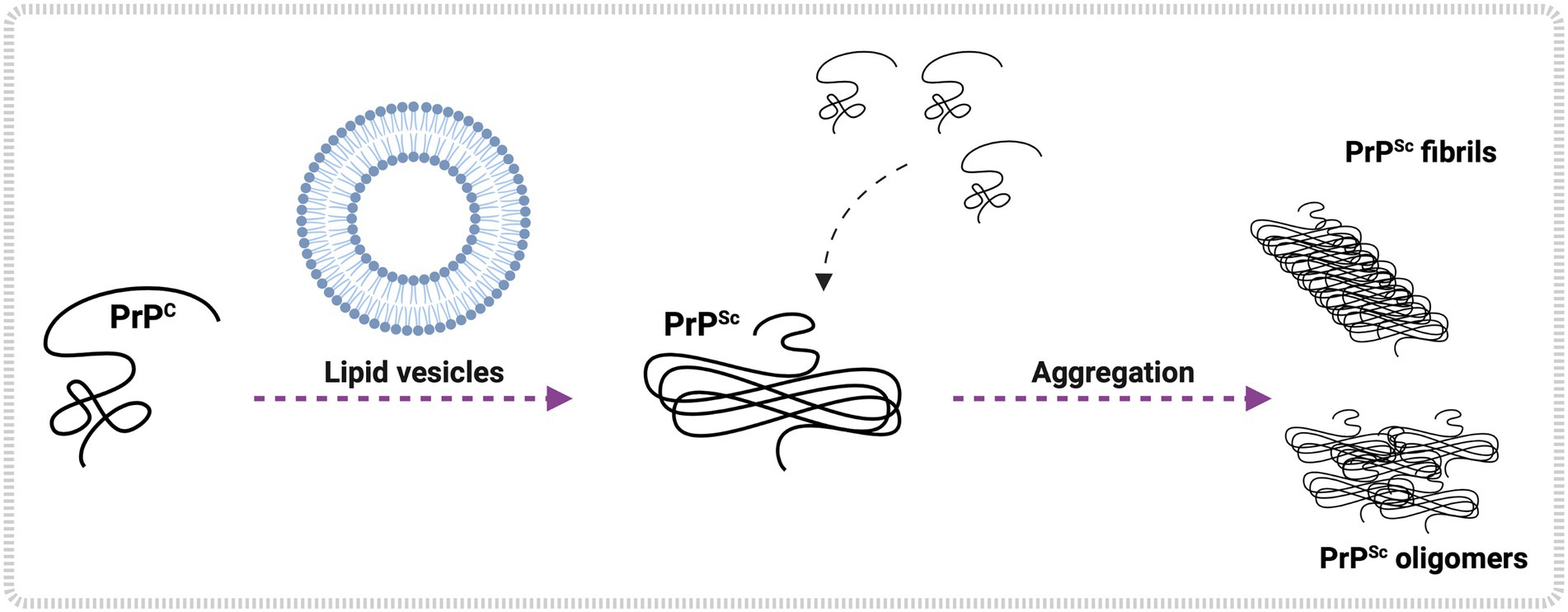
Figure 3. Lipid-mediated PrPC conversion and aggregation in vitro. In vitro studies show lipid vesicle interaction with PrPC, leading to PrPSc formation. Examples of glycerophospholipids are POPG, PE, PA, and others. Once formed, PrPSc converts more PrPC to the pathological form, initiating an aggregation process that may culminate in the shape of PrPSc oligomers and PrPSc fibrils. Created with BioRender.com.
It is still unclear how PrP-lipid interactions occur, and more studies are needed to explain the pathways in which these interactions may take place. Contrasting results from the literature suggest that differences in sample preparation, lipid systems, and methodologies can generate different effects on PrP conversion and aggregation. Deleault et al. (2012a) isolated PE as a lipid cofactor capable of inducing PrP conversion to infectious PrP without any other molecules. In contrast, Srivastava and Baskakov (2015) showed that PE did not significantly alter PrPC structure nor lead to aggregation. Both studies worked with very similar buffer conditions but employed different lipid systems and methodologies. Deleault et al. (2012a) dissolved lipid powder in Triton and evaluated conversion after PMCA cycles. Srivastava and Baskakov (2015) prepared PE vesicles and evaluated the direct interaction with rPrP through different biophysical techniques without employing any amplification procedure.
Another study by Hoover et al. (2017) dissolved lipid powder in a chloroform-methanol solvent and performed the RT-QuIC (real-time quaking-induced conversion) assay. RT-QuIC, like PMCA, is an amplification assay that uses recombinant PrP as substrate and brain homogenates as seeds. Still, in specific buffer conditions, agitation cycles under temperatures around 55°C allow conversion only by PrPSc seeds. This work evaluated the effect of PI, PC, and PE and showed that these lipids did not induce PrP conversion when used as seed. When added together with Chronic wasting disease brain homogenates, it inhibited prion-seeded amyloid formation, suggesting an inhibitory effect for these lipids.
When comparing results from different cell models, different recombinant proteins used in vitro, and different lipid preparations and methodologies, it is difficult to define specific and clear roles for lipid cofactors, resulting in paradoxical results. At the same time, various approaches explore different characteristics of the PrP-lipid system, generating significant contributions. More studies on this topic will be essential to reveal the different paths that this interaction can take.
Although the interaction of PrP with membrane lipids determines its function in cell physiology, in models of prion pathology, lipids have also been identified as mediators of prion toxicity. PrP aggregation involves the formation of oligomers, which are thought to be the neurotoxic forms rather than monomers or fibrils, with hydrophobic regions that may insert into lipid membranes and lead to destabilization (Simoneau et al., 2007). Since interactions of PrP with membrane lipids, such as POPG, were believed to induce a conformational change of PrPC to a PrPSc-like structure (Wang et al., 2007; Figure 2A), lipids emerged as potential cofactors in the formation of PrP neurotoxic aggregates in TSEs.
The infection of neuronal cell lines with PrPSc leads to an alteration in cell membrane composition, increasing free-cholesterol levels (Figure 2B), an effect not generated by PrPC or by increasing overall cholesterol synthesis (Bate et al., 2008a,b,c), suggesting that the disturbance on membrane composition culminating in high levels of free cholesterol may be part of the mechanism of neurotoxicity in prion pathologies. Indeed, cholesterol was shown to stabilize prion multimers and may be required for the efficient formation of PrPSc (Taraboulos et al., 1995). Also, inhibiting the esterification of cholesterol by acyl-coenzyme A:cholesterol acyltransferase in neuronal cell lines was significantly more toxic for prion-infected cells (Bate et al., 2008a,b,c), suggesting that esterification of free cholesterol may be an important protective mechanism against PrP neurotoxicity. Inhibiting cholesterol synthesis protected primary neurons from cell death induced by platelet-activating factor (PAF; Bate et al., 2007), a phospholipid implicated in neuronal damage in different brain diseases, including TSEs. Therefore, there is strong evidence for cholesterol’s role as a mediator of PrP toxicity.
The apparent importance of the localization of PrP on lipid rafts during the conversion of PrPC to PrPSc (Taylor and Hooper, 2006; Wälzlein et al., 2021) may be related to lipid-mediated toxicity in prion diseases. Indeed, the composition of the bilayer membrane may be a determinant of the PrP oligomer’s cytotoxicity. As in a mimetic model of the bacterial anionic membrane, oligomers led to membrane disruption through the detergent model (as proposed for other antimicrobial peptides). In contrast, in a model of the mammalian cell membrane, which is zwitterionic and contains cholesterol-rich domains, it induced a loss of domain separation and has been associated with the activation of apoptotic pathways (Walsh et al., 2014).
Two polyunsaturated fatty acids, docosahexaenoic and eicosapentaenoic acids, even with lower cholesterol levels, increased PrPC expression and PrPSc formation in prion-infected neuronal cell lines. This was followed by increased activation of cytoplasmic phospholipase A2 (Bate et al., 2008a,b,c), whose inhibition was shown to prevent prion-induced neuronal damage (Last et al., 2012). These studies suggest that fatty acids may also mediate the toxicity of PrP aggregates.
Depletion of sphingolipids in prion-infected neuroblastoma cells, using the ceramide synthase inhibitor fumonisin B(1), led to a 4-fold increase in PrPSc formation, which seemed to inversely correlate specifically with sphingomyelin levels (Naslavsky et al., 1999; Figure 2C). Keeping in mind the importance of sphingolipid-rich rafts in the formation of PrPSc and the possible relation with its cytotoxicity (Taylor and Hooper, 2006; Walsh et al., 2014), alterations in the levels of membrane sphingolipids may be involved in neuronal damage in TSEs.
To date, the precise way PrPSc causes neurodegeneration is still unclear; studies suggest that it is related either to the deposition of PrPSc fibrils on cellular membranes, to PrPC loss of function after its conversion, or to the conversion process itself (Westergard et al., 2007). Moreover, the clinical signs of prion disorders appear only in the late stages of the disease, hindering the possibility of early diagnosis (Connor et al., 2019). This, in turn, hinders the development of effective therapeutic strategies for blocking prion diseases. Although many polymers capable of impairing PrPSc accumulation or diminishing the incubation time of TSEs in neuronal cells have been developed in the last decades, none of them has been effective in humans (Teruya and Doh-ura, 2022). Interestingly, in a mouse model of prion disease, global changes in lipidomic profiling were demonstrated in the disease early stage; 75% of the alterations were on GLPs, upregulated in prion-infected animals, suggesting GLPs as potential lipid biomarkers for TSEs (Kim et al., 2021). Although more studies are needed, lipid alterations may allow an early diagnosis of prion diseases before clinical signs appear, representing a significant therapeutical potential in TSEs.
Over the last decade, many studies have revealed mechanistic similarities between prion diseases and other diseases that involve protein aggregation. For this reason, these proteins have been termed prion-like or prionoids (Ritchie and Barria, 2021). The sporadic nature of these diseases reveals the intricate role of factors that can be altered throughout the turnover of these proteins to favor the establishment of pathological pathways. Protein interaction with macromolecules is a factor in this process (Silva et al., 2010a; Burke et al., 2020); as such, their interaction with membranes and their lipid repertoire are relevant.
In this review, we showed the relationship between lipid membranes and PrP physiology, highlighting its importance in copper metabolism, remedying cellular stress conditions, and contributing to cell viability and migration. We also address the importance of PrP-lipid interaction in PrPSc conversion and pathology. Since PrP is a GPI-anchored membrane protein, it traffics from its synthesis to its recycling and degradation, facing different lipid repertoires. These different environments directly interfere with its stability and its propensity for conversion. Many phospholipids interact with PrP and recapitulate their conversion to toxic and infectious aggregates in vitro. PrPSc interaction with lipid membranes is also responsible for neuronal damage.
While in vitro models are vital for understanding details of more complex processes at the cellular and organismal level, recapitulating the complexity of the cellular environment, especially of the plasma membrane, is a significant challenge. Most studies have used vesicle models to assess the specific importance of certain lipids and to understand the physicochemical details of these interactions (Wang et al., 2007; Sanghera et al., 2011; Angelli et al., 2021). However, these lipid systems are poor in membrane composition and organization regarding lipids and the other molecules that compose these structures. Nanodiscs isolated from cell membranes using SMALPs (styrene maleic acid lipid particles) show lipid and protein profiles biologically relevant (Overduin et al., 2021). SMALPs can fragment membranes maintaining protein and lipid integrity. Studies with native nanodiscs are increasing and may bring important information about new membrane complex structures, improving the field of structural membrane biology. It will enhance the knowledge about the prion environment and partners, which is important for understanding physiology and pathology.
Understanding the role of lipids in prion physiology and pathology will lead to the development of therapeutic agents for prion diseases. Cerebral cholesterol originates most from de novo synthesis (Dai et al., 2021). So, modulation of its metabolism is an attractive therapeutic approach. Interestingly, prion-infected neurons show increased unesterified cholesterol levels due to the up-regulation of cholesterol synthesis enzymes (Bach et al., 2009) and inhibition of cholesterol export (Sodero, 2021). Some studies about the administration of cholesterol synthesis inhibitors, such as simvastatin and pravastatin, showed increased survival and delayed clinical signs in animal models (Kempster et al., 2007; Haviv et al., 2008; Vetrugno et al., 2009), but another study found no effect (Carroll et al., 2017). These findings demonstrate the need to carry out well-controlled experiments to exclude experimental and analysis variables among the research carried out and for more studies to understand the real benefit of these strategies better.
The imbalance of cholesterol levels occurs together with changes in sphingolipid and glycerophospholipid metabolism (Kim et al., 2021), including increased levels of PE that showed to be an in vitro conversion cofactor (Deleault et al., 2012a). Changes observed in lipid profile in the early stage of the disease in infected animal models (Kim et al., 2021) suggest that they are consequences of the infection but do not exclude the possibility that they are also involved with the cause of the disease. Targeting the conversion process and understanding the effect of lipids through direct conversion or modulation of PrP localization can be interesting therapeutically.
PrPSc propagates through four main mechanisms: direct cell-to-cell contact, tunneling nanotubes, GPI painting (spontaneous incorporation of GPI-protein into the cell surface membrane), and extracellular vesicles (Heumüller et al., 2022). Alterations of the lipid membrane profile may interfere with many of these processes: (i) Changes in GPI anchor or even detachment of PrP from the cell membrane will directly interfere with cell-to-cell propagation and GPI painting. (ii) Changes in cholesterol and sphingolipids perturb lipid raft domains and PrP localization, also interfering with cell-to-cell propagation and GPI painting. (iii) Modulating the endocytosis process will affect the intercellular transmission of endocytic compartments through tunneling nanotubes and extracellular vesicles. All these approaches are exciting to be therapeutically investigated.
Although we know a lot about the importance of lipids for prion diseases, many questions remain unanswered about their role in the conversion and propagation of PrPSc. The extent to which changes in the cellular lipid profile are a cause or consequence of the conversion and propagation of PrP remains to be elucidated. The aggregation pathways involved in the interaction with lipids also need to be explained to understand better the molecular mechanism involved in establishing prion diseases, thus enabling the development of relevant therapeutic strategies. The mechanisms are still quite elusive, bringing an exciting perspective for developing new studies in this area. Since the membrane environment is essential for the aggregation and toxicity of other prion-like proteins, the findings relating to PrP are of considerable importance for prion diseases and many other diseases.
TV and CA contributed to the conception and design of the study. TV, CA, GL, and CB wrote sections of the manuscript. All authors contributed to the manuscript revision and read and approved the submitted version.
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001; Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ); and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acevedo-Morantes, C., and Wille, H. (2014). The structure of human prions: from biology to structural models—considerations and pitfalls. Viruses 6, 3875–3892. doi: 10.3390/v6103875
Adhikari, U. K., Sakiz, E., Habiba, U., Mikhael, M., Senesi, M., David, M. A., et al. (2021). Treatment of microglia with anti-PrP monoclonal antibodies induces neuronal apoptosis in vitro. Heliyon 7:e08644. doi: 10.1016/j.heliyon.2021.e08644
Ali, T., Hannaoui, S., Nemani, S., Tahir, W., Zemlyankina, I., Cherry, P., et al. (2021). Oral administration of repurposed drug targeting Cyp46A1 increases survival times of prion infected mice. Acta Neuropathol. Commun. 9:58. doi: 10.1186/s40478-021-01162-1
Altmeppen, H. C., Puig, B., Dohler, F., Thurm, D. K., Falker, C., Krasemann, S., et al. (2012). Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 1, 15–31.
Anderson, R. G. W. (1998). The caveolae membrane system. Annu. Rev. Biochem. 67, 199–225. doi: 10.1146/annurev.biochem.67.1.199
Angelli, J. N., Passos, Y. M., Brito, J. M. A., Silva, J. L., Cordeiro, Y., and Vieira, T. C. R. G. (2021). Rabbit PrP is partially resistant to in vitro aggregation induced by different biological cofactors. Front. Neurosci. 15:689315. doi: 10.3389/fnins.2021.689315
Asher, D. M., and Gregori, L. (2018). Human transmissible spongiform encephalopathies: historic view. Handb. Clin. Neurol. 153, 1–17. doi: 10.1016/B978-0-444-63945-5.00001-5
Bach, C., Gilch, S., Rost, R., Greenwood, A. D., Horsch, M., Hajj, G. N. M., et al. (2009). Prion-induced activation of cholesterogenic gene expression by Srebp2 in neuronal cells. J. Biol. Chem. 284, 31260–31269. doi: 10.1074/jbc.M109.004382
Baldwin, K. J., and Correll, C. M. (2019). Prion disease. Semin. Neurol. 39, 428–439. doi: 10.1055/s-0039-1687841
Barmada, S. J. (2005). Visualization of prion infection in transgenic mice expressing green fluorescent protein-tagged prion protein. J. Neurosci. 25, 5824–5832. doi: 10.1523/JNEUROSCI.1192-05.2005
Baron, G. S., Wehrly, K., Dorward, D. W., Chesebro, B., and Caughey, B. (2002). Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrPSc) into contiguous membranes. EMBO J. 21, 1031–1040. doi: 10.1093/emboj/21.5.1031
Baskakov, I. V., and Bocharova, O. V. (2005). In vitro conversion of mammalian prion protein into amyloid fibrils displays unusual features. Biochemistry 44, 2339–2348. doi: 10.1021/bi048322t
Bate, C., Nolan, W., McHale-Owen, H., and Williams, A. (2016a). Sialic acid within the glycosylphosphatidylinositol anchor targets the cellular prion protein to synapses. J. Biol. Chem. 291, 17093–17101. doi: 10.1074/jbc.M116.731117
Bate, C., Nolan, W., and Williams, A. (2016b). Does the tail wag the dog? How the structure of a glycosylphosphatidylinositol anchor affects prion formation. Prion 10, 127–130. doi: 10.1080/19336896.2016.1148237
Bate, C., Nolan, W., and Williams, A. (2016c). Glycosylphosphatidylinositols: more than just an anchor? J. Biol. Chem. 291, 160–170. doi: 10.1074/jbc.M115.672394
Bate, C., Nolan, W., and Williams, A. (2016d). Sialic acid on the glycosylphosphatidylinositol anchor regulates PrP-mediated cell signaling and prion formation. J. Biol. Chem. 291, 160–170. doi: 10.1074/jbc.M115.672394
Bate, C., Rumbold, L., and Williams, A. (2007). Cholesterol synthesis inhibitors protect against platelet-activating factor-induced neuronal damage. J. Neuroinflammation 4:5. doi: 10.1186/1742-2094-4-5
Bate, C., Salmona, M., Diomede, L., and Williams, A. (2004). Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J. Biol. Chem. 279, 14983–14990. doi: 10.1074/jbc.M313061200
Bate, C., Tayebi, M., Diomede, L., Salmona, M., and Williams, A. (2008a). Docosahexaenoic and eicosapentaenoic acids increase prion formation in neuronal cells. BMC Biol. 6:39. doi: 10.1186/1741-7007-6-39
Bate, C., Tayebi, M., and Williams, A. (2008b). Cholesterol esterification reduces the neurotoxicity of prions. Neuropharmacology 54, 1247–1253. doi: 10.1016/j.neuropharm.2008.04.002
Bate, C., Tayebi, M., and Williams, A. (2008c). Sequestration of free cholesterol in cell membranes by prions correlates with cytoplasmic phospholipase A2activation. BMC Biol. 6:8. doi: 10.1186/1741-7007-6-8
Bate, C., Tayebi, M., and Williams, A. (2010). A glycosylphosphatidylinositol analogue reduced prion-derived peptide mediated activation of cytoplasmic phospholipase A2, synapse degeneration and neuronal death. Neuropharmacology 59, 93–99. doi: 10.1016/j.neuropharm.2010.04.002
Bate, C., and Williams, A. (2011). The cellular prion protein with a monoacylated glycosylphosphatidylinositol anchor modifies cell membranes, inhibits cell signaling and reduces prion formation. Prion 5, 65–68. doi: 10.4161/pri.5.2.16095
Bendheim, P. E., Brown, H. R., Rudelli, R. D., Scala, L. J., Goller, N. L., Wen, G. Y., et al. (1992). Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 42, 149–156. doi: 10.1212/WNL.42.1.149
Botto, L., Cunati, D., Coco, S., Sesana, S., Bulbarelli, A., Biasini, E., et al. (2014). Role of lipid rafts and GM1 in the segregation and processing of prion protein. PLoS One 9:e98344. doi: 10.1371/journal.pone.0098344
Brown, D. R., Qin, K., Herms, J. W., Madlung, A., Manson, J., Strome, R., et al. (1997). The cellular prion protein binds copper in vivo. Nature 390, 684–687. doi: 10.1038/37783
Brown, D. R., Wong, B. S., Hafiz, F., Clive, C., Haswell, S. J., and Jones, I. M. (1999). Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 344, 1–5.
Burke, C. M., Walsh, D. J., Mark, K. M. K., Deleault, N. R., Nishina, K. A., Agrimi, U., et al. (2020). Cofactor and glycosylation preferences for in vitro prion conversion are predominantly determined by strain conformation. PLoS Pathog. 16:e1008495. doi: 10.1371/journal.ppat.1008495
Campana, V., Sarnataro, D., and Zurzolo, C. (2005). The highways and byways of prion protein trafficking. Trends Cell Biol. 15, 102–111. doi: 10.1016/j.tcb.2004.12.002
Carroll, J. A., Groveman, B. R., Williams, K., Moore, R., Race, B., and Haigh, C. L. (2020). Prion protein N1 cleavage peptides stimulate microglial interaction with surrounding cells. Sci. Rep. 10:6654. doi: 10.1038/s41598-020-63472-z
Carroll, J. A., Race, B., Phillips, K., Striebel, J. F., and Chesebro, B. (2017). Statins are ineffective at reducing neuroinflammation or prolonging survival in scrapie-infected mice. J. Gen. Virol. 98, 2190–2199. doi: 10.1099/jgv.0.000876
Cashion, J., Zhang, W., Ali, T., and Gilch, S. (2022). Cholesterol and its reciprocal association with prion infection. Cell Tissue Res. doi: 10.1007/s00441-022-03669-y
Caughey, B., and Raymond, G. J. (1991). The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 266, 18217–18223. doi: 10.1016/s0021-9258(18)55257-1
Chesebro, B., Trifilo, M., Race, R., Meade-White, K., Teng, C., LaCasse, R., et al. (2005). Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 1979:308. doi: 10.1126/science.1110252
Chu, N. K., Shabbir, W., Bove-Fenderson, E., Araman, C., Lemmens-Gruber, R., Harris, D. A., et al. (2014). A C-terminal membrane anchor affects the interactions of prion proteins with lipid membranes. J. Biol. Chem. 289, 30144–30160. doi: 10.1074/jbc.M114.587345
Cohen, F. E., and Prusiner, S. B. (1998). Pathologic conformations of prion proteins. Available at: https://www.annualreviews.org/
Collins, S. J., Lawson, V. A., and Masters, C. L. (2004). Transmissible spongiform encephalopathies. Lancet 363, 51–61. doi: 10.1016/S0140-6736(03)15171-9
Connor, A., Wang, H., Appleby, B. S., and Rhoads, D. D. (2019). Clinical laboratory tests used to aid in diagnosis of human prion disease. J. Clin. Microbiol. 57:e00769-19. doi: 10.1128/JCM.00769-19
D’Ambrosi, N., and Rossi, L. (2015). Copper at synapse: release, binding and modulation of neurotransmission. Neurochem. Int. 90, 36–45. doi: 10.1016/j.neuint.2015.07.006
da Fonseca, A. C. C., Romão, L., Amaral, R. F., Kahn, S. A., Lobo, D., Martins, S., et al. (2012). Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience 200, 130–141. doi: 10.1016/j.neuroscience.2011.10.025
Dai, L., Zou, L., Meng, L., Qiang, G., Yan, M., and Zhang, Z. (2021). Cholesterol metabolism in neurodegenerative diseases: molecular mechanisms and therapeutic targets. Mol. Neurobiol. 58, 2183–2201. doi: 10.1007/s12035-020-02232-6
Das, A. S., and Zou, W.-Q. (2016). Prions: beyond a single protein. Clin. Microbiol. Rev. 29, 633–658. doi: 10.1128/CMR.00046-15
DeArmond, S. J., and Bajsarowicz, K. (2010). PrPSc accumulation in neuronal plasma membranes links Notch-1 activation to dendritic degeneration in prion diseases. Mol. Neurodegener. 5:6. doi: 10.1186/1750-1326-5-6
Deleault, N. R., Geoghegan, J. C., Nishina, K., Kascsak, R., Williamson, R. A., and Supattapone, S. (2005). Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J. Biol. Chem. 280, 26873–26879. doi: 10.1074/jbc.M503973200
Deleault, N. R., Harris, B. T., Rees, J. R., and Supattapone, S. (2007). Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U. S. A. 104, 9741–9746. doi: 10.1073/pnas.0702662104
Deleault, N. R., Lucassen, R. W., and Supattapone, S. (2003). RNA molecules stimulate prion protein conversion. Nature 425, 717–720. doi: 10.1038/nature01979
Deleault, N. R., Piro, J. R., Walsh, D. J., Wang, F., Jiyan, M., Geoghegan, J. C., et al. (2012a). Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U. S. A. 109, 8546–8551. doi: 10.1073/pnas.1204498109
Deleault, N. R., Walsh, D. J., Piro, J. R., Wang, F., Wang, X., Ma, J., et al. (2012b). Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U. S. A. 109, E1938–E1946. doi: 10.1073/pnas.1206999109
Dong, S.-L., Cadamuro, S. A., Fiorino, F., Bertsch, U., Moroder, L., and Renner, C. (2007). Copper binding and conformation of the N-terminal octarepeats of the prion protein in the presence of DPC micelles as membrane mimetic. Biopolymers 88, 840–847. doi: 10.1002/bip.20860
Echarri, A., Muriel, O., and del Pozo, M. A. (2007). Intracellular trafficking of raft/caveolae domains: insights from integrin signaling. Semin. Cell Dev. Biol. 18, 627–637. doi: 10.1016/j.semcdb.2007.08.004
Egawa, J., Pearn, M. L., Lemkuil, B. P., Patel, P. M., and Head, B. P. (2016). Membrane lipid rafts and neurobiology: age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 594, 4565–4579. doi: 10.1113/JP270590
Faris, R., Moore, R. A., Ward, A., Race, B., Dorward, D. W., Hollister, J. R., et al. (2017). Cellular prion protein is present in mitochondria of healthy mice. Sci. Rep. 7:41556. doi: 10.1038/srep41556
Fehlinger, A., Wolf, H., Hossinger, A., Duernberger, Y., Pleschka, C., Riemschoss, K., et al. (2017). Prion strains depend on different endocytic routes for productive infection. Sci. Rep. 7:6923. doi: 10.1038/s41598-017-07260-2
Fevrier, B., Vilette, D., Archer, F., Loew, D., Faigle, W., Vidal, M., et al. (2004). Cells release prions in association with exosomes. Proc Natl. Acad. Sci. U. S. A 101, 9683–9688. doi: 10.1126/science.279.5352.827
Flechsig, E., Shmerling, D., Hegyi, I., Raeber, A. J., Fischer, M., Cozzio, A., et al. (2000). Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 27, 399–408. doi: 10.1016/S0896-6273(00)00046-5
Gambetti, P., Puoti, G., and Zou, W.-Q. (2011). Variably protease-sensitive prionopathy: a novel disease of the prion protein. J. Mol. Neurosci. 45, 422–424. doi: 10.1007/s12031-011-9543-1
Gao, Z., Peng, M., Chen, L., Yang, X., Li, H., Shi, R., et al. (2019). Prion protein protects cancer cells against endoplasmic reticulum stress induced apoptosis. Virol. Sin. 34, 222–234. doi: 10.1007/s12250-019-00107-2
Garofalo, T., Giammarioli, A. M., Misasi, R., Tinari, A., Manganelli, V., Gambardella, L., et al. (2005). Lipid microdomains contribute to apoptosis-associated modifications of mitochondria in T cells. Cell Death Differ. 12, 1378–1389. doi: 10.1038/sj.cdd.4401672
Garofalo, T., Manganelli, V., Grasso, M., Mattei, V., Ferri, A., Misasi, R., et al. (2015). Role of mitochondrial raft-like microdomains in the regulation of cell apoptosis. Apoptosis 20, 621–634. doi: 10.1007/s10495-015-1100-x
Geoghegan, J. C., Valdes, P. A., Orem, N. R., Deleault, N. R., Anthony Williamson, R., Harris, B. T., et al. (2007). Selective incorporation of polyanionic molecules into hamster prions. J. Biol. Chem. 282, 36341–36353. doi: 10.1074/jbc.M704447200
Geschwind, M. D. (2015). Prion diseases. Contin. Lifelong Learn. Neurol 21, 1612–1638. doi: 10.1212/CON.0000000000000251
Gilch, S., Kehler, C., and Schätzl, H. M. (2006). The prion protein requires cholesterol for cell surface localization. Mol. Cell. Neurosci. 31, 346–353. doi: 10.1016/j.mcn.2005.10.008
Gill, A. C., and Castle, A. R. (2018). The cellular and pathologic prion protein. Handb. Clin. Neurol. 151, 21–44. doi: 10.1016/B978-0-444-63945-5.00002-7
Godsave, S. F., Peters, P. J., and Wille, H. (2015). Subcellular distribution of the prion protein in sickness and in health. Virus Res. 207, 136–145. doi: 10.1016/j.virusres.2015.02.004
Gomes, M. P. B., Millen, T. A., Ferreira, P. S., Cunha e Silva, N. L., Vieira, T. C. R. G., Almeida, M. S., et al. (2008). Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J. Biol. Chem. 283, 19616–19625. doi: 10.1074/jbc.M802102200
Greenlee, J. J., and Greenlee, M. H. W. (2015). The transmissible spongiform encephalopathies of livestock. ILAR J. 56, 7–25. doi: 10.1093/ilar/ilv008
Gromadzka, G., Tarnacka, B., Flaga, A., and Adamczyk, A. (2020). Copper dyshomeostasis in neurodegenerative diseases—therapeutic implications. Int. J. Mol. Sci. 21:9259. doi: 10.3390/ijms21239259
Hagiwara, K., Nakamura, Y., Nishijima, M., and Yamakawa, Y. (2007). Prevention of prion propagation by dehydrocholesterol reductase inhibitors in cultured cells and a therapeutic trial in mice. Biol. Pharm. Bull. 30, 835–838. doi: 10.1248/bpb.30.835
Haigh, C. L., and Brown, D. R. (2006). Prion protein reduces both oxidative and non-oxidative copper toxicity. J. Neurochem. 98, 677–689. doi: 10.1111/j.1471-4159.2006.03906.x
Haigh, C. L., Tumpach, C., Drew, S. C., and Collins, S. J. (2015). The prion protein N1 and N2 cleavage fragments bind to phosphatidylserine and phosphatidic acid; relevance to stress-protection responses. PLoS One 10, –e0134680. doi: 10.1371/journal.pone.0134680
Haraguchi, T., Fisher, S., Olofsson, S., Endo, T., Groth, D., Tarentino, A., et al. (1989). Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 274, 1–13. doi: 10.1016/0003-9861(89)90409-8
Harayama, T., and Riezman, H. (2018). Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 19, 281–296. doi: 10.1038/nrm.2017.138
Harris, D. A. (2003). Trafficking, turnover and membrane topology of PrP. Br. Med. Bull. 66, 71–85. doi: 10.1093/bmb/66.1.71
Haviv, Y., Avrahami, D., Ovadia, H., Ben-Hur, T., Gabizon, R., and Sharon, R. (2008). Induced neuroprotection independently from PrPSc accumulation in a mouse model for prion disease treated with simvastatin. Arch. Neurol. 65, 762–775. doi: 10.1001/archneur.65.6.762
Hayashi, T., and Su, T.-P. (2003). σ-1 receptors (σ 1 binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 306, 718–725. doi: 10.1124/jpet.103.051284
Hegde, R. S., Mastrianni, J. A., Scott, M. R., Defea, K. A., Tremblay, P., Torchia, M., et al. (1998). A transmembrane form of the prion protein in neurodegenerative disease. Science 279, 827–834. doi: 10.1126/science.279.5352.827
Heske, J., Heller, U., Winklhofer, K. F., and Tatzelt, J. (2004). The C-terminal globular domain of the prion protein is necessary and sufficient for import into the endoplasmic reticulum. J. Biol. Chem. 279, 5435–5443. doi: 10.1074/jbc.M309570200
Heumüller, S.-E., Hornberger, A. C., Hebestreit, A. S., Hossinger, A., and Vorberg, I. M. (2022). Propagation and dissemination strategies of transmissible spongiform encephalopathy agents in mammalian cells. Int. J. Mol. Sci. 23:2909. doi: 10.3390/ijms23062909
Hijazi, N., Shaked, Y., Rosenmann, H., Ben-Hur, T., and Gabizon, R. (2003). Copper binding to PrPC may inhibit prion disease propagation. Brain Res. 993, 192–200. doi: 10.1016/j.brainres.2003.09.014
Hoover, C. E., Davenport, K. A., Henderson, D. M., Zabel, M. D., and Hoover, E. A. (2017). Endogenous brain lipids inhibit prion amyloid formation in vitro. J. Virol. 91, e02162–e02166. doi: 10.1128/JVI.02162-16
Kawahara, M., Kato-Negishi, M., and Tanaka, K. (2021). Neurometals in the pathogenesis of prion diseases. Int. J. Mol. Sci. 22:1267. doi: 10.3390/ijms22031267
Kazlauskaite, J., Sanghera, N., Sylvester, I., Vénien-Bryan, C., and Pinheiro, T. J. T. (2003). Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 42, 3295–3304. doi: 10.1021/bi026872q
Kempster, S., Bate, C., and Williams, A. (2007). Simvastatin treatment prolongs the survival of scrapie-infected mice. Neuroreport 18, 479–482. doi: 10.1097/WNR.0b013e328058678d
Kim, B.-H., Lee, H.-G., Choi, J.-K., Kim, J.-I., Choi, E.-K., Carp, R. I., et al. (2004). The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Mol. Brain Res. 124, 40–50. doi: 10.1016/j.molbrainres.2004.02.005
Kim, Y.-C., Lee, J., Lee, D.-W., and Jeong, B.-H. (2021). Large-scale lipidomic profiling identifies novel potential biomarkers for prion diseases and highlights lipid raft-related pathways. Vet. Res. 52:105. doi: 10.1186/s13567-021-00975-1
Klingenstein, R., Lober, S., Kujala, P., Godsave, S., Leliveld, S. R., Gmeiner, P., et al. (2006). Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J. Neurochem. 98, 748–759. doi: 10.1111/j.1471-4159.2006.03889.x
Kodama, H., Fujisawa, C., and Bhadhprasit, W. (2012). Inherited copper transport disorders: biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 13, 237–250. doi: 10.2174/138920012799320455
Kovachev, P. S., Gomes, M. P. B., Cordeiro, Y., Ferreira, N. C., Valadão, L. P. F., Ascari, L. M., et al. (2019). RNA modulates aggregation of the recombinant mammalian prion protein by direct interaction. Sci. Rep. 9:12406. doi: 10.1038/s41598-019-48883-x
Kovács, G. G., Gelpi, E., Ströbel, T., Ricken, G., Nyengaard, J. R., Bernheimer, H., et al. (2007). Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 66, 628–636. doi: 10.1097/nen.0b013e318093ecc7
Krasemann, S., Zerr, I., Weber, T., Poser, S., Kretzschmar, H., Hunsmann, G., et al. (1995). Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Mol. Brain Res. 34, 173–176. doi: 10.1016/0169-328X(95)00175-R
Kuipers, H. F., Rappert, A. A. C., Mommaas, A. M., Van Haastert, E. S., Van Valk, P., Boddeke, H. W. G. M., et al. (2006). Simvastatin affects cell motility and actin cytoskeleton distribution of microglia. Glia 53, 115–123. doi: 10.1002/glia.20269
Kuramoto, T., Kitada, K., Inui, T., Sasaki, Y., Ito, K., Hase, T., et al. (2001). Attractin/mahogany/Zitter plays a critical role in myelination of the central nervous system. Proc. Natl. Acad. Sci. 98, 559–564. doi: 10.1073/pnas.98.2.559
Last, V., Williams, A., and Werling, D. (2012). Inhibition of cytosolic phospholipase A2prevents prion peptide-induced neuronal damage and co-localisation with Beta III tubulin. BMC Neurosci. 13:106. doi: 10.1186/1471-2202-13-106
Legname, G. (2017). Elucidating the function of the prion protein. PLoS Pathog. 13:e1006458. doi: 10.1371/journal.ppat.1006458
Levine, C. G., Mitra, D., Sharma, A., Smith, C. L., and Hegde, R. S. (2005). The efficiency of protein compartmentalization into the secretory pathway. Mol. Biol. Cell 16, 279–291. doi: 10.1091/mbc.E04
Liberski, P., Gajos, A., Sikorska, B., and Lindenbaum, S. (2019). Kuru, the first human prion disease. Viruses 11:232. doi: 10.3390/v11030232
Linden, R. (2017). The biological function of the prion protein: a cell surface scaffold of signaling modules. Front. Mol. Neurosci. 10:77. doi: 10.3389/fnmol.2017.00077
Magzoub, M., Sandgren, S., Lundberg, P., Oglęcka, K., Lilja, J., Wittrup, A., et al. (2006). N-terminal peptides from unprocessed prion proteins enter cells by macropinocytosis. Biochem. Biophys. Res. Commun. 348, 379–385. doi: 10.1016/j.bbrc.2006.07.065
Mangé, A., Nishida, N., Milhavet, O., McMahon, H. E. M., Casanova, D., and Lehmann, S. (2000). Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J. Virol. 74, 3135–3140. doi: 10.1128/JVI.74.7.3135-3140.2000
Marella, M., Lehmann, S., Grassi, J., and Chabry, J. (2002). Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J. Biol. Chem. 277, 25457–25464. doi: 10.1074/jbc.M203248200
Marijanovic, Z., Caputo, A., Campana, V., and Zurzolo, C. (2009). Identification of an intracellular site of prion conversion. PLoS Pathog. 5:e1000426. doi: 10.1371/journal.ppat.1000426
Mattei, V., Garofalo, T., Misasi, R., Circella, A., Manganelli, V., Lucania, G., et al. (2004). Prion protein is a component of the multimolecular signaling complex involved in T cell activation. FEBS Lett. 560, 14–18. doi: 10.1016/S0014-5793(04)00029-8
Mattei, V., Garofalo, T., Misasi, R., Gizzi, C., Mascellino, M. T., Dolo, V., et al. (2002). Association of cellular prion protein with gangliosides in plasma membrane microdomains of neural and lymphocytic cells. Neurochem. Res. 27, 743–749. doi: 10.1023/A:1020244621373
Miller, M. B., Wang, D. W., Wang, F., Noble, G. P., Ma, J., Woods, V. L., et al. (2013). Cofactor molecules induce structural transformation during infectious prion formation. Structure 21, 2061–2068. doi: 10.1016/j.str.2013.08.025
Mironov, A., Latawiec, D., Wille, H., Bouzamondo-Bernstein, E., Legname, G., Williamson, R. A., et al. (2003). Cytosolic prion protein in neurons. J. Neurosci. 23, 7183–7193. doi: 10.1523/JNEUROSCI.23-18-07183.2003
Morillas, M., Swietnicki, W., Gambetti, P., and Surewicz, W. K. (1999). Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 274, 36859–36865. doi: 10.1074/jbc.274.52.36859
Naslavsky, N., Shmeeda, H., Friedlander, G., Yanai, A., Futerman, A. H., Barenholz, Y., et al. (1999). Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J. Biol. Chem. 274, 20763–20771. doi: 10.1074/jbc.274.30.20763
Nelson, T. J., and Alkon, D. L. (2005). Insulin and cholesterol pathways in neuronal function, memory and neurodegeneration. Biochem. Soc. Trans. 33:1033. doi: 10.1042/BST20051033
Noble, G., Walsh, D., Miller, M., Jackson, W., and Supattapone, S. (2015). Requirements for mutant and wild-type prion protein misfolding in vitro. Biochemistry 54, 1180–1187. doi: 10.1021/bi501495j
Nunziante, M., Gilch, S., and Schätzl, H. M. (2003). Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J. Biol. Chem. 278, 3726–3734. doi: 10.1074/jbc.M206313200
Overduin, M., Wille, H., and Westaway, D. (2021). Multisite interactions of prions with membranes and native nanodiscs. Chem. Phys. Lipids 236:105063. doi: 10.1016/j.chemphyslip.2021.105063
Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., et al. (1993). Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. 90, 10962–10966. doi: 10.1073/pnas.90.23.10962
Pan, Y., Wang, B., Reese, R. A., and Xu, B. (2019). The molecular basis of interaction domains of full-length PrP with lipid membranes. Nanoscale 11, 12087–12091. doi: 10.1039/C9NR02735A
Pauly, P. C., and Harris, D. A. (1998). Copper stimulates endocytosis of the prion protein. J. Biol. Chem. 273, 33107–33110. doi: 10.1074/jbc.273.50.33107
Peters, P. J., Mironov, A., Peretz, D., van Donselaar, E., Leclerc, E., Erpel, S., et al. (2003). Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J. Cell Biol. 162, 703–717. doi: 10.1083/jcb.200304140
Posadas, Y., López-Guerrero, V. E., Segovia, J., Perez-Cruz, C., and Quintanar, L. (2022). Dissecting the copper bioinorganic chemistry of the functional and pathological roles of the prion protein: relevance in Alzheimer’s disease and cancer. Curr. Opin. Chem. Biol. 66:102098. doi: 10.1016/j.cbpa.2021.102098
Prado, M., Alves-Silva, J., Magalhães, A., Prado, V., Linden, R., Martins, V., et al. (2004). PrPc on the road: trafficking of the cellular prion protein. J. Neurochem. 88, 769–781. doi: 10.1046/j.1471-4159.2003.02199.x
Priola, S. A., and McNally, K. L. (2009). The role of the prion protein membrane anchor in prion infection. Prion 3, 134–138. doi: 10.4161/pri.3.3.9771
Prusiner, S. B. (1998). Prions [review]. Proc. Natl. Acad. Sci. 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Puig, B., Altmeppen, H., and Glatzel, M. (2014). The GPI-anchoring of PrP: implications in sorting and pathogenesis. Prion 8, 11–18. doi: 10.4161/pri.27892
Puig, B., Altmeppen, H. C., Linsenmeier, L., Chakroun, K., Wegwitz, F., Piontek, U. K., et al. (2019). GPI-anchor signal sequence influences PrP C sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog. 15:e1007520. doi: 10.1371/journal.ppat.1007520
Ritchie, D. L., and Barria, M. A. (2021). Prion diseases: a unique transmissible agent or a model for neurodegenerative diseases? Biomol. Ther. 11:207. doi: 10.3390/biom11020207
Roucou, X., Giannopoulos, P. N., Zhang, Y., Jodoin, J., Goodyer, C. G., and LeBlanc, A. (2005). Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 12, 783–795. doi: 10.1038/sj.cdd.4401629
Rudd, P. M., Wormald, M. R., Wing, D. R., Prusiner, S. B., and Dwek, R. A. (2001). Prion glycoprotein: structure, dynamics, and roles for the sugars. Biochemistry 40, 3759–3766. doi: 10.1021/bi002625f
Saá, P., Castilla, J., and Soto, C. (2006). Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252. doi: 10.1074/jbc.M603964200
Sakudo, A., Lee, D., Nishimura, T., Li, S., Tsuji, S., Nakamura, T., et al. (2005). Octapeptide repeat region and N-terminal half of hydrophobic region of prion protein mediate PrP-dependent activation of superoxide dismutase. Biochem. Biophys. Res. Commun. 326, 600–606. doi: 10.1016/j.bbrc.2004.11.092
Salzano, G., Giachin, G., and Legname, G. (2019). Structural consequences of copper binding to the prion protein. Cells 8:770. doi: 10.3390/cells8080770
Sánchez-López, C., Rossetti, G., Quintanar, L., and Carloni, P. (2018). Structural determinants of the prion protein N-terminus and its adducts with copper ions. Int. J. Mol. Sci. 20:18. doi: 10.3390/ijms20010018
Sanghera, N., Correia, B. E. F. S., Correia, J. R. S., Ludwig, C., Agarwal, S., Nakamura, H. K., et al. (2011). Deciphering the molecular details for the binding of the prion protein to main ganglioside GM1 of neuronal membranes. Chem. Biol. 18, 1422–1431. doi: 10.1016/j.chembiol.2011.08.016
Sarnataro, D., Pepe, A., and Zurzolo, C. (2017). Cell biology of prion protein. Prog. Mol. Biol. Transl. Sci. 150, 57–82. doi: 10.1016/bs.pmbts.2017.06.018
Shim, S. Y., Karri, S., Law, S., Schatzl, H. M., and Gilch, S. (2016). Prion infection impairs lysosomal degradation capacity by interfering with Rab 7 membrane attachment in neuronal cells. Sci. Rep. 6:21658. doi: 10.1038/srep21658
Shyng, L., Heuser, J. E., and Harris, D. A. (1994). A Glycolipid-anchored Prion Protein Is Endocytosed via Clathrin-coated Pits. J. Cell Biol. 125, 1239–1250. doi: 10.1083/jcb.125.6.1239
Shyng, S. L., Moulder, K. L., Lesko, A., and Harris, D. A. (1995). The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J. Biol. Chem. 270, 14793–14800. doi: 10.1074/jbc.270.24.14793
Sigurdsson, E. M., Brown, D. R., Alim, M. A., Scholtzova, H., Carp, R., Meeker, H. C., et al. (2003). Copper chelation delays the onset of prion disease. J. Biol. Chem. 278, 46199–46202. doi: 10.1074/jbc.C300303200
Silva, J. L., Gomes, M. P. B., Vieira, T. C. R. G., and Cordeiro, Y. (2010a). PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front. Biosci. 15, 132–150. doi: 10.2741/3611
Silva, J. L., Vieira, T. C. R. G., Gomes, M. P. B., Ano Bom, A. P., Lima, L. M. T. R., Freitas, M. S., et al. (2010b). Ligand binding and hydration in protein misfolding: insights from studies of prion and p 53 tumor suppressor proteins. Acc. Chem. Res. 43, 271–279. doi: 10.1021/ar900179t
Simoneau, S., Rezaei, H., Salès, N., Kaiser-Schulz, G., Lefebvre-Roque, M., Vidal, C., et al. (2007). In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 3:e125. doi: 10.1371/journal.ppat.0030125
Simons, K., and Sampaio, J. L. (2011). Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 3, 1–17. doi: 10.1101/cshperspect.a004697
Sodero, A. O. (2021). 24S-hydroxycholesterol: cellular effects and variations in brain diseases. J. Neurochem. 157, 899–918. doi: 10.1111/jnc.15228
Srivastava, S., and Baskakov, I. (2015). Contrasting effects of two lipid cofactors of prion replication on the conformation of the prion protein. PLoS One 10:e0130283. doi: 10.1371/journal.pone.0130283
Stahl, N., Borchelt, D. R., Hsiao, K., and Prusiner, S. B. (1987). Scrapie prion protein contains a phosphatidylinositol glycolipid. Cells 51, 229–240.
Sunyach, C., Jen, A., Deng, J., Fitzgerald, K. T., Frobert, Y., Grassi, J., et al. (2003). The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 22, 3591–3601. doi: 10.1093/emboj/cdg344
Tabas, I. (2002). Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Investig. 110, 905–911. doi: 10.1172/JCI16452
Taraboulos, A., Scott, M., Semenov, A., Avraham, D., Laszlo, L., and Prusiner, S. B. (1995). Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 129, 121–132. doi: 10.1083/jcb.129.1.121
Taylor, D. R., and Hooper, N. M. (2006). The prion protein and lipid rafts (review). Mol. Membr. Biol. 23, 89–99. doi: 10.1080/09687860500449994
Taylor, D. R., Watt, N. T., Perera, W. S. S., and Hooper, N. M. (2005). Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 118, 5141–5153. doi: 10.1242/jcs.02627
Teruya, K., and Doh-ura, K. (2022). Therapeutic development of polymers for prion disease. Cell Tissue Res. doi: 10.1007/s00441-022-03604-1
Timmes, A. G., Moore, R. A., Fischer, E. R., and Priola, S. A. (2013). Recombinant prion protein refolded with lipid and RNA has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS One 8:e71081. doi: 10.1371/journal.pone.0071081
Trowbridge, I. S. (1991). Endocytosis and signals for internalization. Curr. Opin. Cell Biol. 3, 634–641. doi: 10.1016/0955-0674(91)90034-v
Tsiroulnikov, K., Shchutskaya, Y., Muronetz, V., Chobert, J. M., and Haertlé, T. (2009). Phospholipids influence the aggregation of recombinant ovine prions. From rapid extensive aggregation to amyloidogenic conversion. Biochim. Biophys. Acta 1794, 506–511. doi: 10.1016/j.bbapap.2008.12.002
Uchiyama, K., Muramatsu, N., Yano, M., Usui, T., Miyata, H., and Sakaguchi, S. (2013). Prions disturb post-Golgi trafficking of membrane proteins. Nat. Commun. 4:1846. doi: 10.1038/ncomms2873
van Rheede, T., Smolenaars, M. M. W., Madsen, O., and de Jong, W. W. (2003). Molecular evolution of the mammalian prion protein. Mol. Biol. Evol. 20, 111–121. doi: 10.1093/molbev/msg014
Vetrugno, V., di Bari, M. A., Nonno, R., Puopolo, M., D’Agostino, C., Pirisinu, L., et al. (2009). Oral pravastatin prolongs survival time of scrapie-infected mice. J. Gen. Virol. 90, 1775–1780. doi: 10.1099/vir.0.009936-0
Viles, J. H., Klewpatinond, M., and Nadal, R. C. (2008). Copper and the structural biology of the prion protein. Biochem. Soc. Trans. 36, 1288–1292. doi: 10.1042/BST0361288
Walmsley, A. R., Watt, N. T., Taylor, D. R., Perera, W. S. S., and Hooper, N. M. (2009). α-Cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol. Cell. Neurosci. 40, 242–248. doi: 10.1016/j.mcn.2008.10.012
Walsh, P., Vanderlee, G., Yau, J., Campeau, J., Sim, V. L., Yip, C. M., et al. (2014). The mechanism of membrane disruption by cytotoxic amyloid oligomers formed by prion protein (106–126) is dependent on bilayer composition. J. Biol. Chem. 289, 10419–10430. doi: 10.1074/jbc.M113.515866
Wälzlein, J.-H., Schwenke, K. A., and Beekes, M. (2021). Propagation of CJD prions in primary murine glia cells expressing human PrPc. Pathogens 10:1060. doi: 10.3390/pathogens10081060
Wang, F., Wang, X., Yuan, C. G., and Ma, J. (2010a). Generating a prion with bacterially expressed recombinant prion protein. Science 1979, 1132–1135. doi: 10.1126/science.1183748
Wang, F., Yang, F., Hu, Y., Wang, X., Wang, X., Jin, C., et al. (2007). Lipid interaction converts prion protein to a PrPSc-like proteinase k-resistant conformation under physiological conditions. Biochemistry 46, 7045–7053. doi: 10.1021/bi700299h
Wang, F., Yin, S., Wang, X., Zha, L., Sy, M. S., and Ma, J. (2010b). Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry 49, 8169–8176. doi: 10.1021/bi101146v
Wang, C., and Youle, R. J. (2009). The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118. doi: 10.1146/annurev-genet-102108-134850
Wen, Y., Li, J., Xiong, M., Peng, Y., Yao, W., Hong, J., et al. (2010). Solution structure and dynamics of the I214V mutant of the rabbit prion protein. PLoS One 5:e13273. doi: 10.1371/journal.pone.0013273
Westergard, L., Christensen, H. M., and Harris, D. A. (2007). The cellular prion protein (PrP C): its physiological function and role in disease. Biochim. Biophys. Acta 1772, 629–644. doi: 10.1016/j.bbadis.2007.02.011
Will, R. G. (2003). Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br. Med. Bull. 66, 255–265. doi: 10.1093/bmb/66.1.255
Winklhofer, K. F., Tatzelt, J., and Haass, C. (2008). The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336–349. doi: 10.1038/sj.emboj.7601930
Wong, B.-S., Brown, D. R., Pan, T., Whiteman, M., Liu, T., Bu, X., et al. (2008). Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations and altered antioxidant activities. J. Neurochem. 79, 689–698. doi: 10.1046/j.1471-4159.2001.00625.x
Wulf, M. A., Senatore, A., and Aguzzi, A. (2017). The biological function of the cellular prion protein: an update. BMC Biol. 15:34. doi: 10.1186/s12915-017-0375-5
Wurm, S., Paar, C., Sonnleitner, A., Sonnleitner, M., Höglinger, O., Romanin, C., et al. (2004). Co-localization of CD3 and prion protein in Jurkat lymphocytes after hypothermal stimulation. FEBS Lett. 566, 121–125. doi: 10.1016/j.febslet.2004.03.114
Yim, Y.-I., Park, B.-C., Yadavalli, R., Zhao, X., Eisenberg, E., and Greene, L. E. (2015). The multivesicular body is the major internal site of prion conversion. J. Cell Sci. 128, 1434–1443. doi: 10.1242/jcs.165472
Zhang, Z., Zhang, Y., Wang, F., Wang, X., Xu, Y., Yang, H., et al. (2013). De novo generation of infectious prions with bacterially expressed recombinant prion protein. FASEB J. 27, 4768–4775. doi: 10.1096/fj.13-233965
Keywords: prion diseases, prion protein, protein-lipid interaction, aggregation, neurodegenerative disease
Citation: Alves Conceição C, Assis de Lemos G, Barros CA and Vieira TCRG (2023) What is the role of lipids in prion conversion and disease? Front. Mol. Neurosci. 15:1032541. doi: 10.3389/fnmol.2022.1032541
Edited by:
Mohammad Haddadi, University of Zabol, IranReviewed by:
Julie Ann Moreno, Colorado State University, United StatesCopyright © 2023 Alves Conceição, Assis de Lemos, Barros and Vieira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tuane C. R. G. Vieira, ✉ dHVhbmVAYmlvcW1lZC51ZnJqLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.