 Frances St. George-Hyslop
Frances St. George-Hyslop Toomas Kivisild
Toomas Kivisild Frederick J. Livesey1
Frederick J. Livesey1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 20 October 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.1017144
This article is part of the Research TopicMolecular Advances and Applications of Machine Learning in Understanding Autism and Comorbid Psychiatric DisordersView all 16 articles
The contactin-associated protein-like 2 (CNTNAP2) gene is associated with multiple neurodevelopmental disorders, including autism spectrum disorder (ASD), intellectual disability (ID), and specific language impairment (SLI). Experimental work has shown that CNTNAP2 is important for neuronal development and synapse formation. There is also accumulating evidence for the differential use of CNTNAP2 in the human cerebral cortex compared with other primates. Here, we review the current literature on CNTNAP2, including what is known about its expression, disease associations, and molecular/cellular functions. We also review the evidence for its role in human brain evolution, such as the presence of eight human accelerated regions (HARs) within the introns of the gene. While progress has been made in understanding the function(s) of CNTNAP2, more work is needed to clarify the precise mechanisms through which CNTNAP2 acts. Such information will be crucial for developing effective treatments for CNTNAP2 patients. It may also shed light on the longstanding question of what makes us human.
The contactin-associated protein-like 2 gene (CNTNAP2) is located at chromosome 7q35. CNTNAP2 spans 2.3 Mb across 24 exons and is the one of the largest genes in the genome (Figures 1A,B; Rodenas-Cuadrado et al., 2014). Mutations in CNTNAP2 have been linked to neurodevelopmental disorders like autism spectrum disorder (ASD), intellectual disability (ID), and specific language impairment (SLI). There is also growing evidence that the temporal and spatial expression of CNTNAP2 during brain development in humans is different from other primates. The potential evolutionary significance of CNTNAP2 is further highlighted by the presence of eight human accelerated regions (HARs) in its introns. HARs are DNA sequences that are highly conserved across primates or mammals, but have an unexpectedly large number of human-specific nucleotide changes (see section “What are HARs?” for a full description) (Pollard et al., 2006; Prabhakar et al., 2006; Bird et al., 2007; Bush and Lahn, 2008; Lindblad-Toh et al., 2011). Given the association between CNTNAP2 and human cognitive disorders, and the evidence for its potential role in human evolution, this review aims to synthesize clinical, experimental, and evolutionary data to understand CNTNAP2 function and the biological consequences of its disruption.
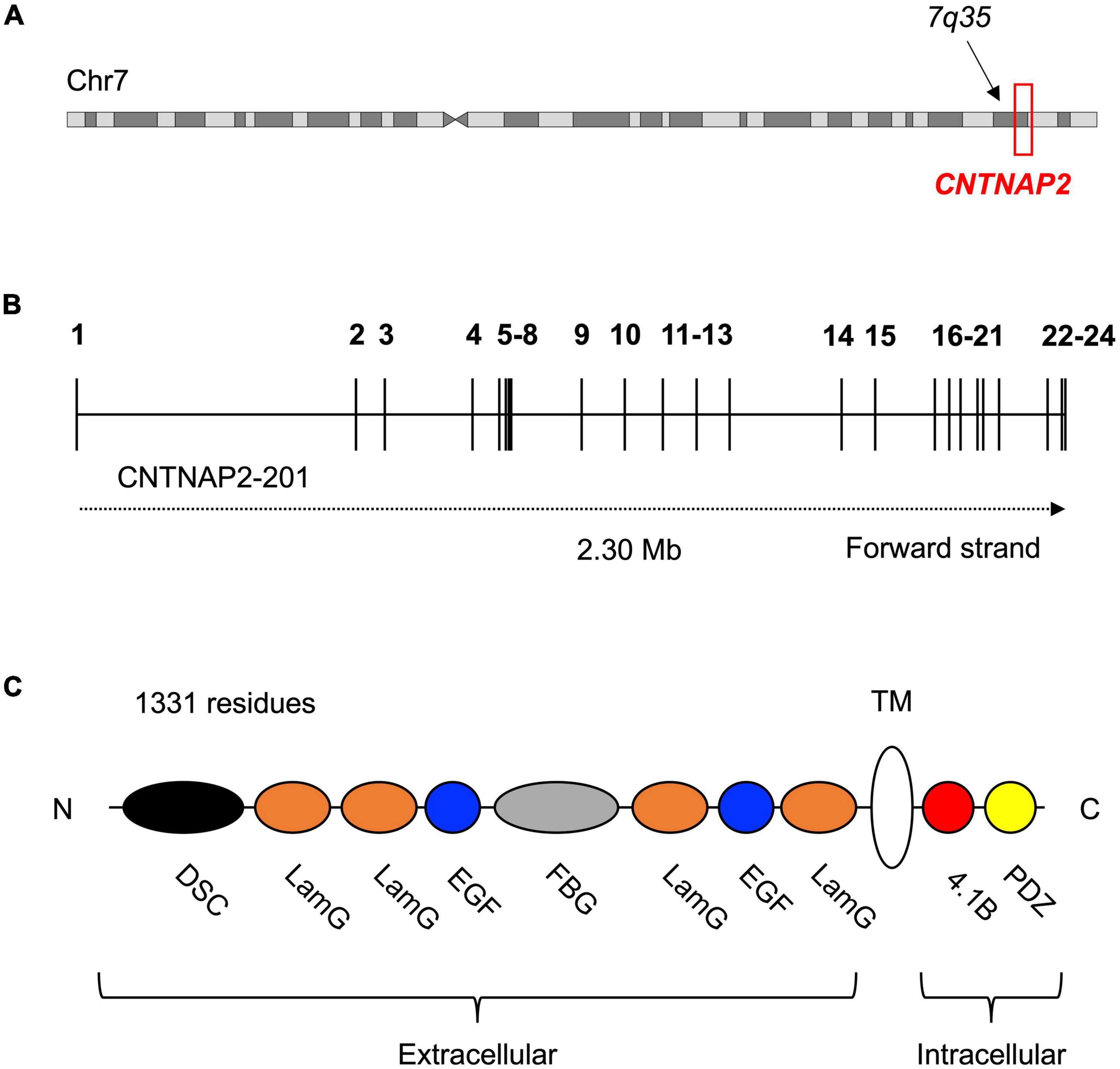
Figure 1. Location and structure of the contactin-associated protein-like 2 (CNTNAP2) gene and protein. (A) Contactin-associated protein-like 2 is located at the distal end of the long arm (q) of chromosome 7 (GRCh38 chr7: 146,116,002–148,420,998). (B) Schematic of the 24 exons (numbered) and 23 introns of the CNTNAP2 gene. Transcript CNTNAP2-201 is the canonical transcript. (C) Structure of the contactin-associated protein-like 2 (CASPR2) protein and its eight extracellular, one transmembrane, and two intracellular subdomains. The extracellular subdomains include a discoidin domain (DSC) and a fibrinogen-like domain (FBG), both of which are known to facilitate cell-cell adhesion and interactions with the extracellular matrix. The remaining extracellular subdomains are four laminin-G domains (LamG) and two epidermal growth factor-like domains (EGF). These are predicted to mediate receptor-ligand interactions and cell adhesion, migration, and differentiation. The intracellular region of CASPR2 is mostly involved in protein-protein interactions, as it contains a type II PDZ domain (PDZ) and a protein 4.1B binding site (4.1B). N-terminus (N); C-terminus (C).
Contactin-associated protein-like 2 encodes the contactin-associated protein-like 2 (CASPR2) protein. CASPR2 is a single-pass transmembrane protein composed of 1,331 residues and with a mass of 138 kDa (Rodenas-Cuadrado et al., 2014). The protein contains eight extracellular, one transmembrane, and two intracellular subdomains (Figure 1C). CASPR2 belongs to the neurexin superfamily of transmembrane proteins, which are cell adhesion molecules involved in synapse formation and function. Several other CASPR proteins exist (CASPR1 – 5), however, these proteins have fewer disease associations than CASPR2 and there is less evidence for their involvement in human evolution. They also all share less than 50% sequence identity to CASPR2, implying that CASPR2 serves differing functions.
CASPR2 is highly conserved amongst mammals – for example, human and mouse amino acid sequences are 94% identical (Rodenas-Cuadrado et al., 2014). This conservation is even greater between humans and chimpanzees, with only 6/1,331 residues differing (99.5% identity). Comparisons with archaic humans show yet further conservation. Between both Denisovans and Neanderthals, only one amino acid difference is noted with modern humans. Residue 345 is a valine in Homo sapiens but isoleucine in the two other species. One other noteworthy difference exists: position 215 is an asparagine in humans and Denisovans, but in all other species (including Neanderthals) it is a Lysine. It is currently unclear whether either of these amino acid changes have had functional consequences. However, PolyPhen-2 predicts both 345-I and 215-N to be functionally neutral (Adzhubei et al., 2010).
Although the molecular function(s) of CASPR2 are not completely understood, it was first characterized in the axon initial segment (AIS) and juxtaparanodal regions of myelinated neurons in the peripheral nervous system (Poliak et al., 1999). At juxtaparanodes, CASPR2 forms a complex with the contactin-2 protein (CNTN2). CASPR2 also binds to contactin-1 (CNTN1), an interaction which may play a role in nerve myelination, but this is currently not well characterized (Rubio-Marrero et al., 2016). Conversely, the CASPR2-CNTN2 complex has been robustly demonstrated to be required for the clustering of voltage-gated K+ channels, which function in the conduction of nerve impulses (Strauss et al., 2006). Several studies have reported that Cntnap2 knockout mice have a significant reduction in the density of Kv1.2 potassium channels in cortical myelinated axons (Poliak et al., 2003; Scott et al., 2019). Scott et al. (2019) observed these changes correlated with an increase in excitatory transmission and increased probability of neurotransmitter release. However, given that CNTNAP2 expression is high in development - at timepoints prior to nerve myelination - other developmental functions are also likely to exist. These include proposed roles in neurite outgrowth and in the formation of synaptic connections (both discussed later in this review).
Lastly, recent work has identified that synaptic potentiation triggers the N-terminal region of CASPR2 to be cleaved (Martín-de-Saavedra et al., 2022). This ecto-domain localizes extracellularly near synapses, and binds and activates the PMCA calcium extrusion pump. In other words, synaptic activity causes CASPR2 to promote calcium export from neurons, thereby suppressing excitability and network activity. A loss of CASPR2 could therefore lead to hyperexcitability by increasing intracellular calcium. This finding is especially noteworthy in light of the neuronal over-excitation frequently reported in the brains of autistic individuals (Rodenas-Cuadrado et al., 2016), and could relate to the seizures that commonly occur in individuals with CNTNAP2 loss-of-function mutations. Accordingly, the levels of the cleaved CASPR2 N-terminus are reduced in the cerebral spinal fluid of individuals with ASD, which further supports this hypothesis (Martín-de-Saavedra et al., 2022).
Other proteins that interact with CASPR2 include the scaffolding protein PAR3 (Gao et al., 2020) and the serine protein kinase CASK (Gao et al., 2019). These interactions may be required for the correct localization of CASPR2 within developing neurons – either intracellularly or on the plasma membrane, respectively – as mis-localization of CASPR2 occurs with a loss of their binding (Gao et al., 2019, 2020). Immunoprecipitation experiments have also suggested CASPR2 binds intracellularly to ADAM22, LGI1, GPR37, and subunits of the Kv1.1 channel (KCNA1) (Poot, 2017). Extracellularly, CASPR2 binds to the scaffolding proteins DLG1 and DLG4. However, the precise functions of these interactions are not currently known.
Although CNTNAP2 was first described in the peripheral nervous system (Poliak et al., 1999), it is primarily expressed in the cerebral cortex (Bakkaloglu et al., 2008; Vernes et al., 2008). In adult humans, the highest expression is observed in layers II–V of the frontal and temporal cortices. Sub-cortically, CNTNAP2 is also present in the thalamus, amygdala, and striatum. Interestingly, this pattern of expression is dramatically restricted to the cortico-striato-thalamic circuitry that mediates higher cognitive functions (Abrahams et al., 2007).
In the human fetal brain, CNTNAP2 is expressed in the frontal and anterior temporal lobes, medial ganglionic eminence, striatum, and dorsal thalamus (Alarcon et al., 2008). This anterior cortical enrichment is not observed in rodents. In the developing mouse and rat cortex, Cntnap2 is broadly expressed throughout the brain and is low or absent in the cortical plate (with highest expression – when present – located posteriorly) (Abrahams et al., 2007). Even in adulthood, Cntnap2 is never enriched in the rodent frontal cortex, unlike in humans.
In addition to the differences in CNTNAP2 expression between humans and rodents, differential expression has also been observed between humans and non-human primates. Recent single cell RNA-Seq (scRNA-Seq) data revealed CNTNAP2 is significantly increased in the excitatory neurons of 2 month old cortical organoids derived from humans versus chimpanzees (Kanton et al., 2019). Increased expression in human organoids was also identified relative to macaque for both excitatory neurons and interneurons. CNTNAP2 expression was also significantly higher in chimpanzee excitatory neurons than in macaque. These findings suggest that cortical CNTNAP2 expression has increased along the lineage leading to humans.
Due to the limits on accessing primate fetal cortex, only one scRNA-Seq dataset of primary human and non-human primate developing brain currently exists (Pollen et al., 2019). This study includes matched samples from macaque (post-conception weeks 8–24) and human (post-conception weeks 4–40). In both deep and upper layer excitatory neurons, CNTNAP2 is increased in human relative to macaque. However, neither remain significant after correction for multiple testing. As additional transcriptomic studies of human and non-human primate fetal brain become available, this relationship can be tested further.
In primary adult cortex, scRNA-Seq of human, chimpanzee, and macaque does not show evidence of increased expression in humans. Indeed, in a study by Kanton et al. (2019) the only significant differences identified were decreases in human CNTNAP2 expression. In chimpanzees, excitatory layer 6a neurons showed significantly increased CNTNAP2 compared to human. In macaques, excitatory layers 1, 3e, and 6b as well as inhibitory layers 4b and 6b all had increased expression relative to human. The lack of increased human CNTNAP2 expression in adulthood reinforces that a human-specific role for CNTNAP2 is most likely occurring during cortical development.
It is important to note, however, that CNTNAP2 has multiple protein-coding isoforms that cluster at the 3’ end of the locus. As most scRNA-Seq technology only detects the 3’ end of a transcript, the different isoforms cannot be distinguished. Thus, the above scRNA-seq expression data of CNTNAP2 should be interpreted with this in mind.
Individuals with CNTNAP2 loss-of-function mutations typically display four core phenotypes: (1) ID (Lu et al., 2021), (2) ASD (O’Roak et al., 2011), (3) SLI (Centanni et al., 2015), and (4) epilepsy (Friedman et al., 2008). Other disorders associated with CNTNAP2 include schizophrenia (Lee et al., 2015), attention deficit hyperactivity disorder (ADHD) (Elia et al., 2010), Tourette syndrome (Verkerk et al., 2003), dyslexia (Veerappa et al., 2013), and major depression (Rodenas-Cuadrado et al., 2014). These illnesses have been identified in individuals with microdeletions or point mutations affecting only the CNTNAP2 locus.
Homozygous loss-of-function mutations usually lead to a diagnosis of either cortical dysplasia focal epilepsy (CDFE) or Pitt-Hopkins syndrome (PTHS) (Zweier et al., 2009; Freri et al., 2021). Both disorders are characterized by uncontrollable seizures, language regression, social/behavioral disturbances, and intellectual disability (Figure 2 and Table 1). Most CNTNAP2 mutations are heterozygous, suggesting two functional copies of the gene are required for normal cognitive function (Figure 3). Homozygous mutations cause the most severe phenotypes and are often found in children of unaffected carrier parents (Strauss et al., 2006). This implies certain CNTNAP2 mutations are fully penetrant while others are not.
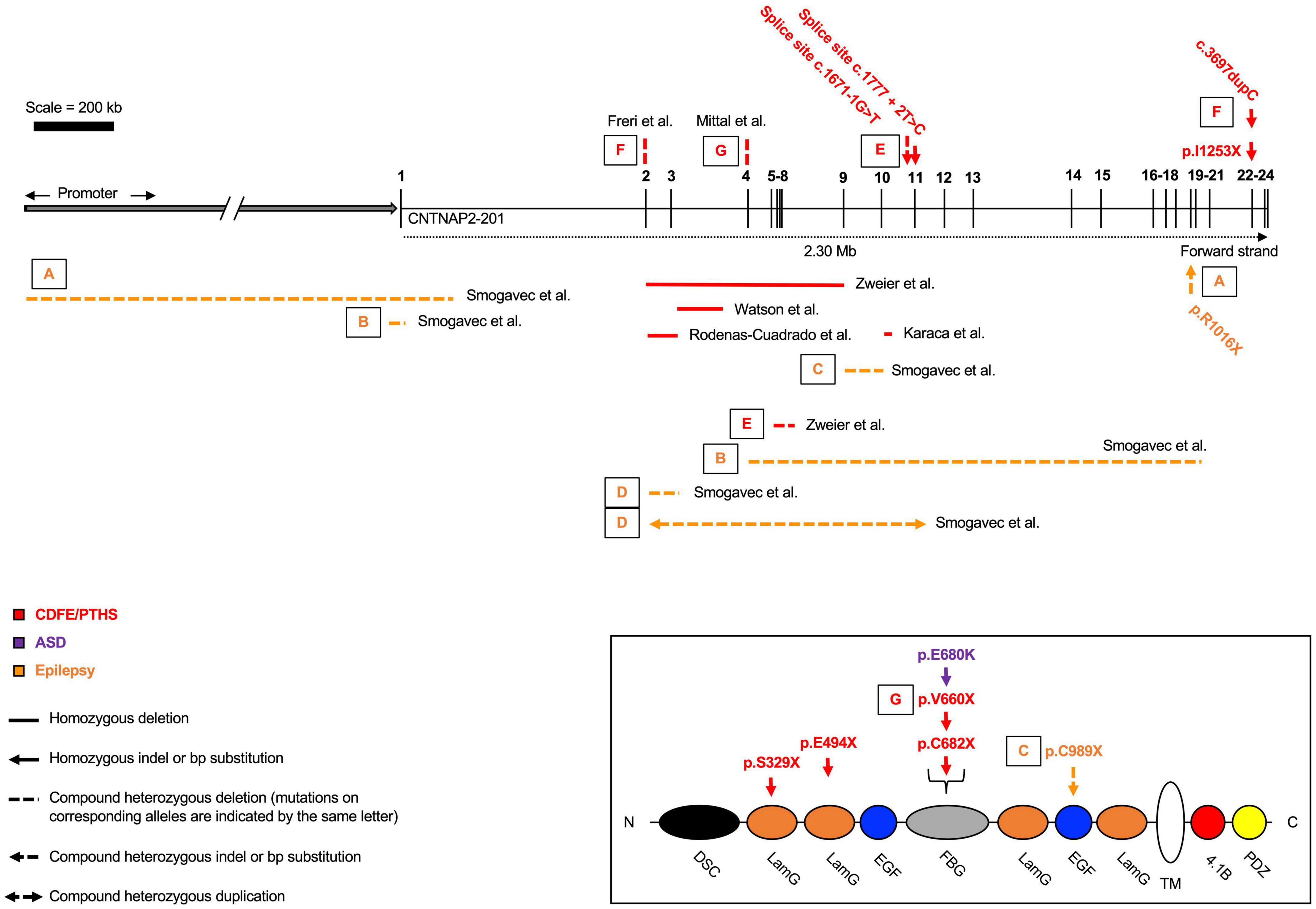
Figure 2. Homozygous and compound heterozygous contactin-associated protein-like 2 (CNTNAP2) mutations. Mutations affecting the CNTNAP2 locus are shown with their associated disorders. Mutations are commonly found in the middle of the gene, between exons 2 – 13. Cortical dysplasia focal epilepsy/Pitt-Hopkins syndrome (CDFE/PTHS) are the most common diagnoses, though patients typically fall into multiple categories. Mutations that map to a subdomain of the contactin-associated protein-like 2 (CASPR2) protein are shown separately. Diagram not perfectly to scale. Works cited are listed in Supplementary Material 1. ASD, autism spectrum disorder.
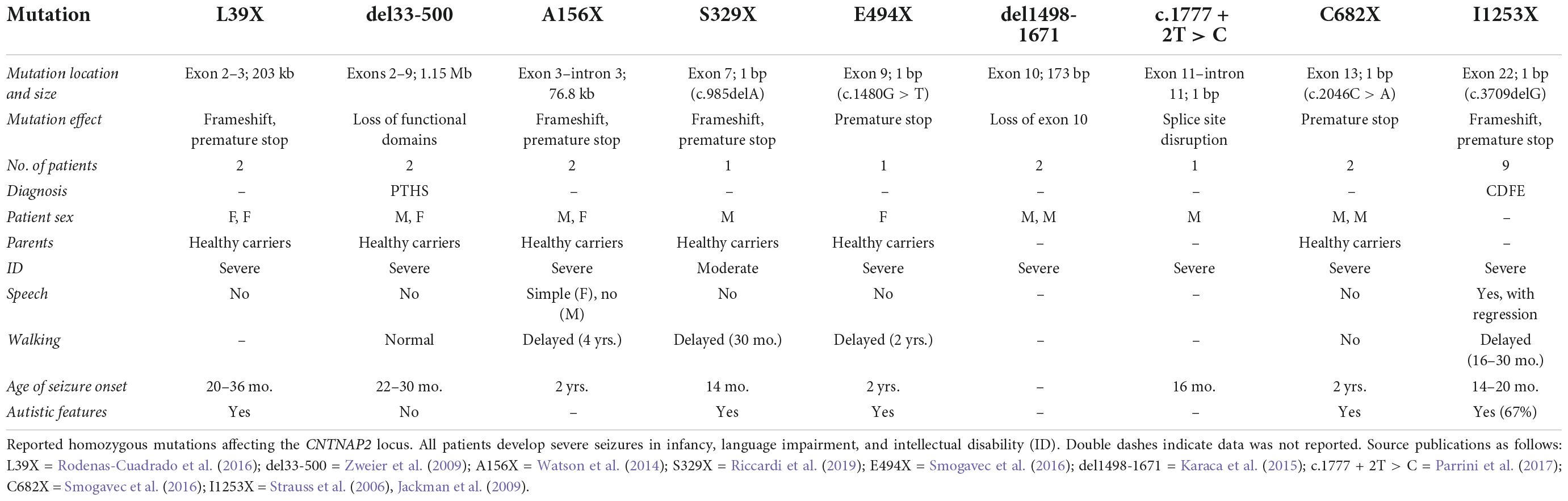
Table 1. Homozygous contactin-associated protein-like 2 (CNTNAP2) mutations.
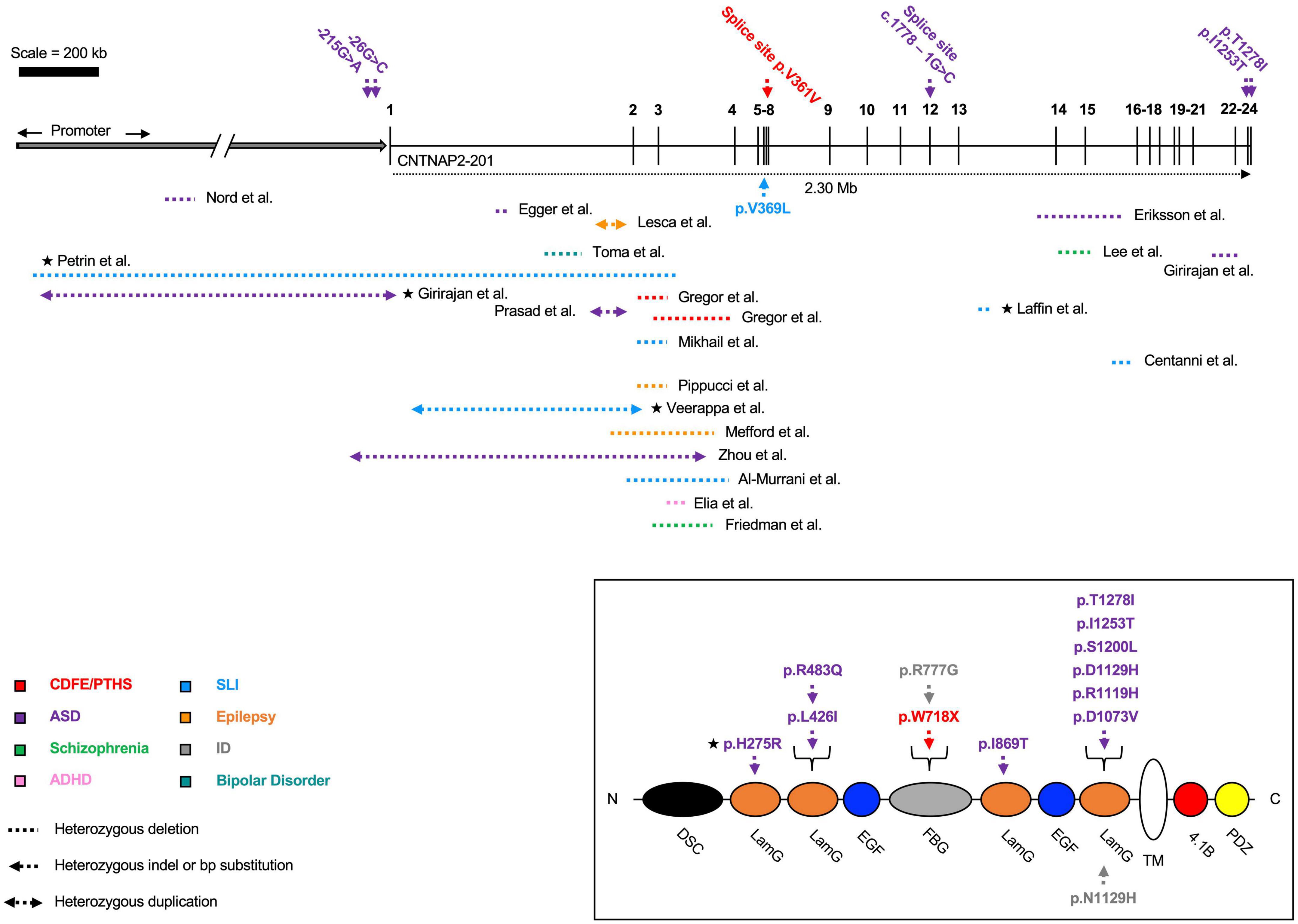
Figure 3. Heterozygous contactin-associated protein-like 2 (CNTNAP2) mutations. Mutations affecting the CNTNAP2 locus are shown with their associated disorders. Mutations in patients with multiple affected genes are marked by a star (★). Autism spectrum disorder (ASD) is the most common diagnosis, though patients typically fall into multiple categories. The majority of deletions are found before exon 8. Mutations that map to a subdomain of the contactin-associated protein-like 2 (CASPR2) protein are shown separately. Diagram not perfectly to scale. Works cited are listed in Supplementary Material 2. CDFE/PTHS (cortical dysplasia focal epilepsy); ADHD (attention deficit hyperactivity disorder); SLI (specific language impairment); ID (intellectual disability).
In addition to the association between mutation homozygosity and disease severity, there are other lines of evidence suggesting CNTNAP2 dosage is important for determining phenotype. Specifically, Nord et al. (2011) identified an autism patient with a 62 kb deletion in the CNTNAP2 promoter region. This deletion was confirmed to reduce CNTNAP2 gene expression in patient-derived lymphoblasts (versus those from healthy controls). A separate study from Chiocchetti et al. (2015) found a novel variant, g.-215G > A, associated with ASD and delayed age of first word. The variant also lies within the CNTNAP2 promoter and was predicted to disrupt transcription factor binding sites. In a luciferase assay, the g.-215G > A variant was shown to significantly decrease enhancer potential in SH-SY5Y cells.
Intronic disease-causing mutations in CNTNAP2 have also been discovered. Three unrelated patients with deletions in intron 1, which contains a regulatory element that binds with the FOXP2 transcription factor, displayed dysarthric language, autism, intellectual disability, bipolar disorder and/or ADHD (Rodenas-Cuadrado et al., 2014; Scala et al., 2021). Thus, changes in the expression level of the gene itself are sufficient to cause disorder and may explain the emergence of specific brain phenotypes.
Dendritic abnormalities have been noted in humans harboring CNTNAP2 mutations. Post-mortem brain analyses of CNTNAP2 patients with CDFE have identified neurons in the temporal cortex with irregularly oriented dendritic processes (Strauss et al., 2006). While no additional studies in human patients are available, there is strong evidence in mice to suggest Cntnap2 is involved in neurite development and/or synaptic transmission.
Anderson et al. (2012) showed shRNA-mediated knockdown of CASPR2 in mouse cortical cultures decreased the length and branching of neurites in excitatory neurons. These effects caused a reduction in the amplitude and frequency of excitatory and inhibitory mini post-synaptic currents. Delivery of the CASPR2 shRNA with either lentivirus or calcium phosphate transfection – targeting all neurons or individual neurons, respectively – both produced the same results, suggesting the observed effects were cell-autonomous. No changes to dendritic spine density or synapse density were observed, however, the width of spine heads was significantly reduced.
A subsequent study by Canali et al. (2018) examined neuronal cultures from both homozygous and heterozygous knockout mice. Homozygous knockout neurons had significantly reduced axon lengths, while heterozygous neurons were intermediate to wild type and homozygous. Recent work by Elia et al. (2010) came to similar findings. They over-expressed CNTNAP2 p.R777G, a mutation associated with intellectual disability and epilepsy, in mouse cortical neurons. Both neurite branching and neurite outgrowth were decreased in the mutant cultures. These neurons had reduced amplitude of spontaneous excitatory post-synaptic currents (EPSCs), in addition to decreased action potential firing. Such reductions in EPSC amplitude have also been reported by other groups (Fernandes et al., 2019; Kim et al., 2019; Lazaro et al., 2019; Sacai et al., 2020).
Finally, Gao et al. (2018) reported that Cntnap2 is involved in neurite outgrowth, but only in cortical interneurons. The authors showed mature interneurons from Cntnap2 knockout mice (in vitro and in vivo) had reduced dendritic branching and dendritic length. No phenotype was observed in excitatory neurons from the same knockout mice or cell cultures. Moreover, as no difference in branching or length was observed in immature neurons (in either inhibitory or excitatory cells), the authors concluded the reduction in branching was due to decreased neurite stabilization and not impaired outgrowth.
As well as the proposed role for Cntnap2 in neurite branching and stabilization, several studies have suggested the gene may regulate dendritic spine dynamics. Gdalyahu et al. (2015) were the first to report a reduction in dendritic spine density in an in vivo study of Thy1-GFP/Cntnap2 null mice. Using 2-photon microscopy, the authors showed knockout mice had significantly decreased spine density in cortical layer Vb. The reduction was caused by decreased stability of newly formed spines (i.e., loss of spines shortly after they form). No reduction in the formation of new spines was observed, nor was any effect on the maintenance or pruning of already-formed/stable spines. This suggests CNTNAP2 may be required for the stabilization of new synaptic contacts, a process thought to be critical for brain plasticity (Gdalyahu et al., 2015).
Lazaro et al. (2019) added further evidence that loss of Cntnap2 reduces dendritic spine density in vivo. The authors reported knockout mice had significantly decreased spine densities and synaptic inputs in layer II–III excitatory neurons. This resulted in a 2-fold decrease in the frequency and amplitude of mEPSCs. No differences in intrinsic neuronal excitability, neurotransmitter release probability, or synapse maturity were observed between genotypes. The knockout neurons did, however, have reduced network synchrony and less precise firing patterns.
Lastly, Varea et al. (2015) have also reported reductions in dendritic spine density in in vitro knockout cultures. The authors additionally observed reduced GluA1 AMPA receptor subunit expression in spines, and GluA1 cytoplasmic aggregates in cell bodies. These aggregates were found to contain trafficking proteins (e.g., clathrin and rab5), suggesting loss of Cntnap2 could affect intracellular GluA1 transport. Two other papers reported similar reductions in glutamate receptor expression (Fernandes et al., 2019; Kim et al., 2019). More recent work identified that CASPR2 binds GluA1 through complexing with the protein CASK (Gao et al., 2019). Mutations in the gene encoding CASK have been linked to intellectual disability and ASD, which implies the pathway downstream of the CASPR2-CASK complex is important for disease pathophysiology (Becker et al., 2020).
Contactin-associated protein-like 2 is robustly expressed in interneurons, and in the ganglionic eminence where interneurons derive from Peñagarikano et al. (2011); Gordon et al. (2016). Peñagarikano et al. (2011) described a loss of GABAergic interneurons in Cntnap2 null mice. The authors noted a significant reduction in all cortical layers. Parvalbumin positive (PV+) interneurons were the most affected (20% loss), while calretinin- (CALB2) and neuropeptide Y- (NPY) positive neurons were also significantly reduced. The loss of interneurons was hypothesized to underly the frequent seizures observed in Cntnap2 null mice (as reported by others, see Hoffman et al., 2016; Thomas et al., 2017). In vivo 2-photon calcium imaging of layer II–III neurons revealed firing was highly asynchronous relative to wild type. The authors did not detect any changes to firing amplitude or frequency, suggesting the asynchronicity was not due to abnormal neuronal activity/conduction, but to defects in synaptic networks.
These findings were followed up by a study from Selimbeyoglu et al. (2017), who found that the PV+ interneurons of Cntnap2 knockout mice had significantly decreased activity in vivo. Activating PV+ interneurons or inhibiting excitatory neurons rescued the observed excitation: inhibition imbalance. Finally, a recent study by Hali et al. (2020) reported significant reductions in the number of interneurons in cortical organoids derived from Cntnap2 knockout mice. No differences in glutamatergic neurons were observed. The authors also noted knockout organoids had dramatically reduced expression of transcription factors expressed in ventral telencephalic (interneuron) progenitor cells (e.g., Dlx2, Nkx2.1, and Ascl1). Similar results have also been observed in a zebrafish Cntnap2 knockout model (Hoffman et al., 2016) and in Cntnap2 knockout mouse hippocampal neurons (Paterno et al., 2021).
The sheer size of the human genome (approximately 3 billion nucleotides) poses a major challenge for identifying the genomic regions important for human evolution. In order to prioritize sequences for further study, Pollard et al. (2006) published a statistical test to find “HARs.” As mentioned in the introduction of this paper, HARs are DNA sequences that fulfill two key criteria (see Figure 4; Pollard et al., 2006; Prabhakar et al., 2006; Bird et al., 2007; Bush and Lahn, 2008; Lindblad-Toh et al., 2011):

Figure 4. Multiple species alignment of human accelerated conserved non-coding sequence 97 (HACNS_97) [109 bp HAR located within intron 1 of contactin-associated protein-like 2 (CNTNAP2)]. Human accelerated regions (HARs) are sequences of DNA that are highly conserved in non-human primates but have an unexpected number of unique nucleotide substitutions in humans. On average, a 100 bp-long HAR will contain ∼1.7 human-specific substitutions (HARs have a mean length of 266 bp). In contrast, chimpanzees (who carry the highly conserved ortholog), will have ∼0.2 unique substitutions. Even if a HAR gains only a small number of human-specific changes, this rate is significantly higher than observed in other conserved elements. Moreover, the regions surrounding HARs are usually still conserved, suggesting HARs may be part of a larger functional structure. Human Accelerated Conserved Non-coding Sequence 97 (HACNS_97) is shown as an example. HACNS_97 contains four nucleotides that are fixed in all humans but are absent in other primates (nucleotides highlighted in red and blue, respectively). Pan troglodytes = chimpanzee; Gorilla gorilla = western gorilla; Macaca fascicularis = crab-eating macaque.
(i) They are highly conserved across a wider clade (e.g., primates, mammals, or vertebrates) – this suggests the region may be functional;
(ii) They have an unexpectedly large number of human-specific nucleotide changes – this suggests the sequence may be important for human evolution.
Approximately 4,000 HARs have been identified to date – ∼97% of them in non-coding portions of the genome (Capra et al., 2013; Uebbing et al., 2021). This distribution has made it difficult to assign HARs a function, because most of the non-coding genome is uncharacterized. Even so, there is growing evidence that many HARs are gene enhancers. For example, Capra et al. (2013) used existing functional genomics data, in combination with machine learning algorithms, to show that 60% of non-coding HARs overlap epigenetic enhancer marks like H3K4me1, H3K27ac, or p300. Half of these HARs were predicted to target genes active during development, and one third to act in the brain.
These predictions were supported by the finding that HARs are highly enriched in transcription factor binding motifs (Capra et al., 2013; Doan et al., 2016). ∼60% of HARs are also located within 1 Mb of a gene that is differentially expressed between humans and chimpanzees (Levchenko et al., 2018). While only a few HARs have been experimentally characterized (most using mouse enhancer assays), these studies have generally validated the predictions made by Capra et al. (2013) (Ryu et al., 2018; Girskis et al., 2021; Uebbing et al., 2021).
Contactin-associated protein-like 2 is unusual for containing eight HARs in its introns (Table 2):
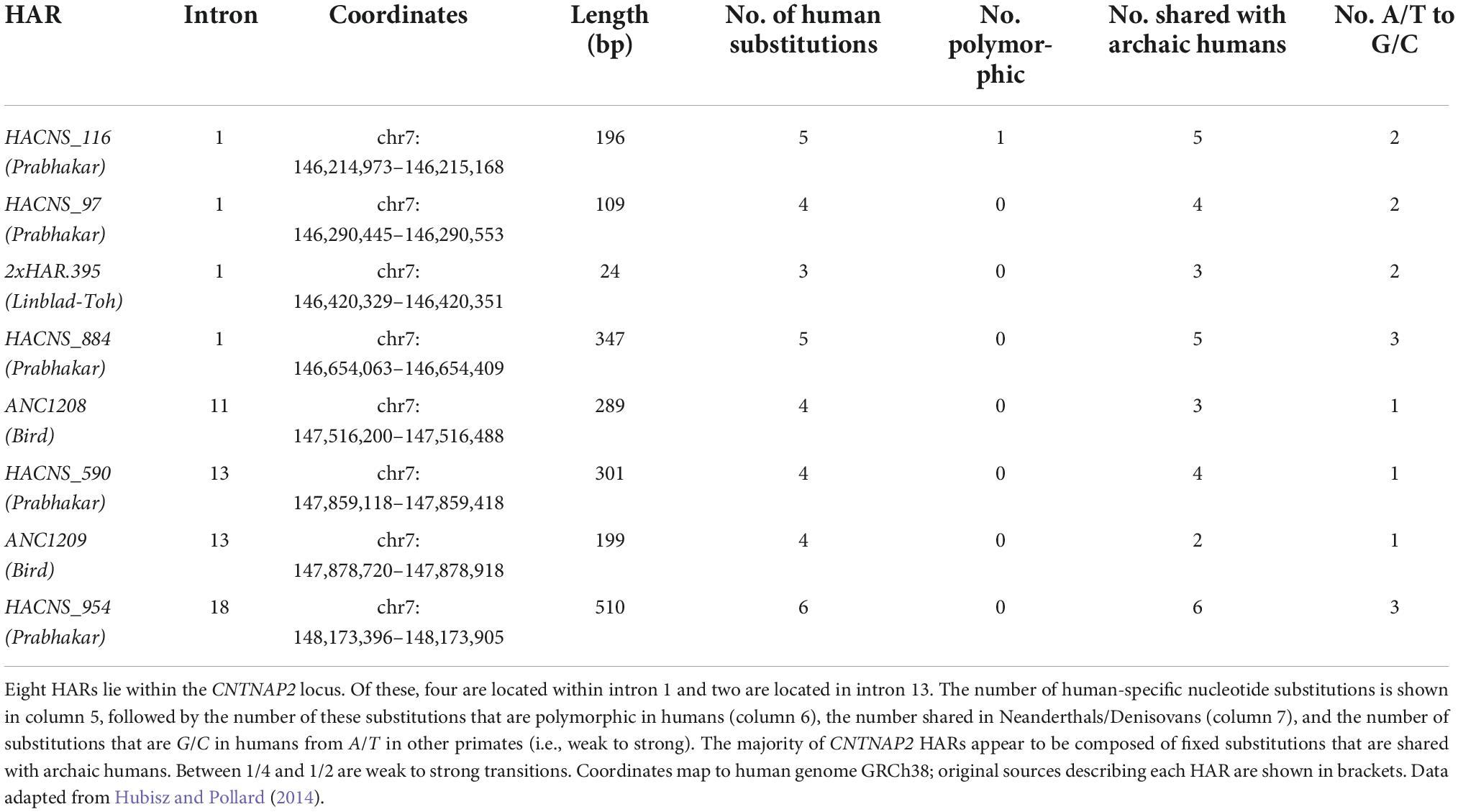
Table 2. Contactin-associated protein-like 2 (CNTNAP2) human accelerated regions (HARs).
1. HACNS_1161 (Prabhakar et al., 2006);
2. HACNS_97 (Prabhakar et al., 2006);
3. 2xHAR.395 (Lindblad-Toh et al., 2011);
4. HACNS_884 (Prabhakar et al., 2006);
5. ANC12082 (Bird et al., 2007);
6. HACNS_590 (Prabhakar et al., 2006);
7. ANC1209 (Bird et al., 2007);
8. HACNS_954 (Prabhakar et al., 2006).
While this is likely related to the sheer size of the gene, the density of HARs is still higher than expected. Since CNTNAP2 is 2.3 Mb long, one would expect only two to three HARs to fall within the gene (using the 4,000 HARs identified, and assuming that HARs are evenly distributed across the genome). From this perspective, eight HARs would be unexpectedly high.
Half of the CNTNAP2 HARs are located in intron 1 – a common location for gene regulatory elements (Chorev and Carmel, 2012). The HARs range in length from 24 bp (2xHAR.395) to 510 bp (HACNS_954), and from 3 to 6 human-specific substitutions. Most of these human-specific changes are shared with Neanderthals and Denisovans, indicating they arose before the emergence of Homo sapiens. However, HACNS_97, ANC1209, ANC1209, and HACNS_954 each contain one substitution that is unique to modern humans alone (Burbano et al., 2012). Multiple species alignments for each CNTNAP2 HAR can be found in Figure 4 and Supplementary Figures 1–7.
Although HARs are a highly powerful tool, a number of caveats apply to their interpretation. These include the low reproducibility between studies that have used different methods to detect HARs, and the dependence on the size of human reference panels in defining which mutations are likely to be fixed in humans species-wide. Each of the CNTNAP2 HARs was identified by only one of the six major HAR publications [excluding HACNS_884, which was identified by both Gittelman et al. (2015) and Prabhakar et al. (2006)]. Secondly, one of the HARs, HACNS_116, has a human-specific substitution that was subsequently found to be polymorphic (Hubisz and Pollard, 2014). All remaining HAR substitutions appear – according to currently available evidence – to be fixed in humans.
That said, there is some indication that one or more of the CNTNAP2 HARs could be enhancers. HACNS_884 was shown by Gittelman et al. (2015) to overlap a human-specific DNase I hypersensitive site (DHS). DNase I selectively cleaves regions of open/active DNA, which is the expected chromatin state of regulatory elements (Dorschner et al., 2004). Won et al. (2019) further identified six of the eight HARs as overlapping DHSs in fetal brain (all except 2xHAR.395 and ANC1208). Similarly, Capra et al. (2013) detected HACNS_884 and HACNS_954 as putative enhancers using their enhancer finding pipeline. They were also able to bioinformatically predict that HACNS_884 is active in fetal brain but could not provide a prediction for HACNS_954. More recently, Girskis et al. (2021) identified HACNS_116, HACNS_590, and ANC1209 as potential enhancers using a massively parallel reporter assay. Interestingly, HACNS_116 was found to have over 2x the enhancer activity as its chimpanzee ortholog.
Also suggestive of their functional role is the fact that the CNTNAP2 HARs overlap deletions associated with neurodevelopmental disorders, including ASD, SLI, ID, and epilepsy (Figure 5). Curiously, all of these disease-linked mutations are heterozygous (or compound heterozygous), which suggests that the loss of even a single copy of a HAR may be enough to affect brain function. However, there is no evidence to suggest directly that these mutations are pathogenic due to the loss of the HAR. This cannot be assumed, particularly because many of the mutations are large, and therefore not specific. Moreover, most of the mutations encompass multiple HARs, suggesting it could be a combinatorial loss of HAR function(s) that leads to disease. HACNS_116 overlaps the most pathogenic mutations (five in number), followed by HACNS_884 (four in number), and then HACNS_97 and 2xHAR.395 each with three.
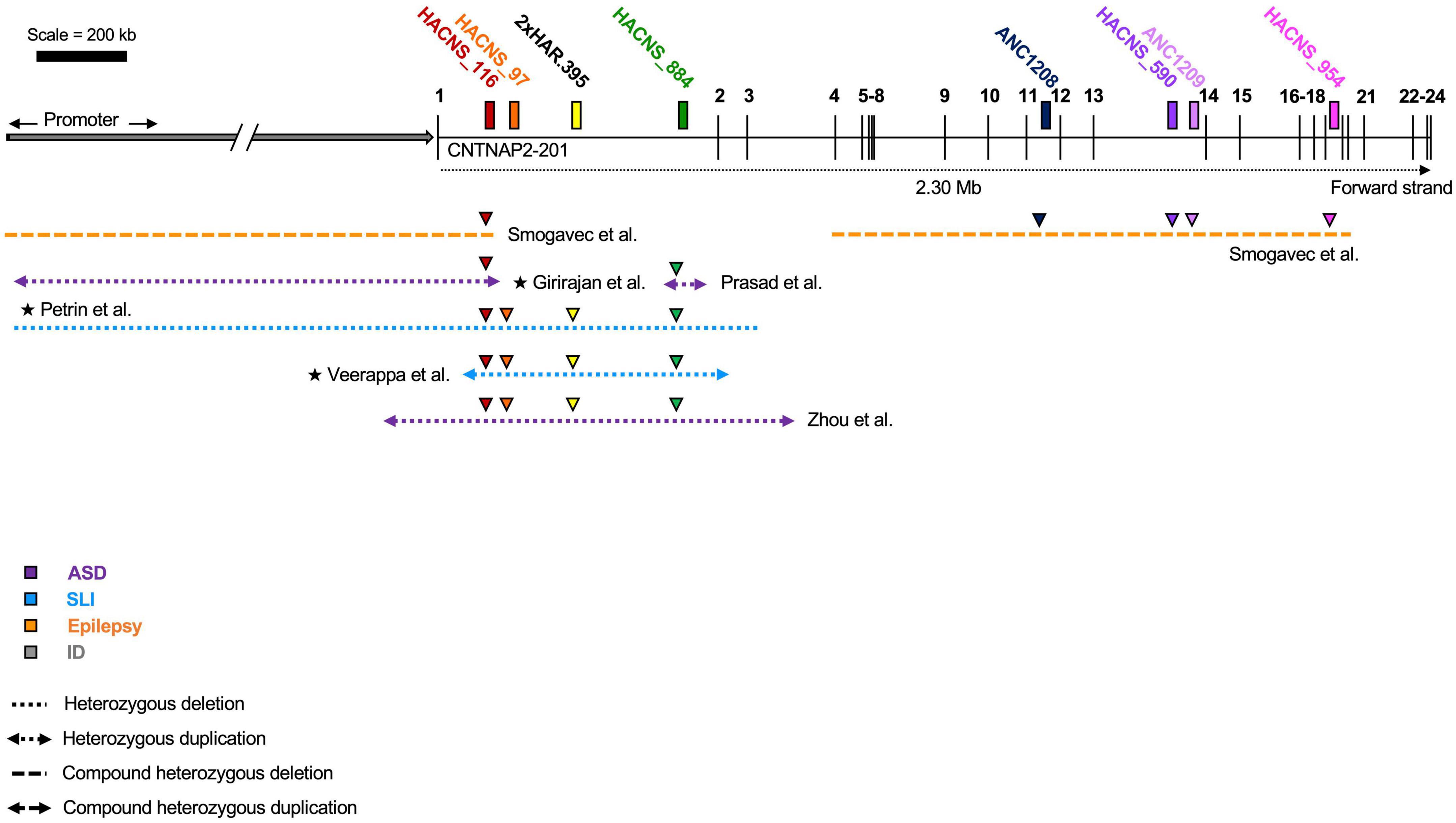
Figure 5. Overlap of human accelerated regions (HARs) and contactin-associated protein-like 2 (CNTNAP2) mutations. Disease-associated mutations that overlap one or more human accelerated region. Colored triangles indicate overlap: the color represents the specific HAR involved. Mutations in patients with multiple affected genes are marked by a star (★). HACNS_116 in intron 1 intersects the most mutations (five), while ANC1208, HACNS_590, ANC1209, and HACNS_954 all intersect the fewest (one mutation). ASD (autism spectrum disorder); SLI (specific language impairment); ID (intellectual disability). Diagram not perfectly to scale.
A “selective sweep” occurs when a positively selected variant rapidly increases in frequency, and nearby linked variants rise in frequency along with it (Jobling, 2014). This occurs as there is no time for recombination to break down the linkage between the selected and non-selected loci. In other words, sweeps cause genetic diversity in the region around the selected variant to decrease (Sabeti et al., 2006).
Evidence for selective sweeps have been identified at the CNTNAP2 locus in humans (Ayub et al., 2013; Mozzi et al., 2016). Specifically, Ayub et al. (2013) detected sweep signatures in CNTNAP2 introns 1 and 13. Since Neanderthals and Denisovans carry the ancestral sequences at these loci, this suggests selection began after the split of modern humans from archaic hominins. The sweep signatures were identified in some but not all human populations, implying that selection occurred after the Out-of-Africa (OOA) dispersals. Introns 1 and 13 each contain several HARs: HACNS_116, HACNS_97, 2xHAR.395, and HACNS_884 in intron 1, and HACNS_590 and ANC1208 in intron 13. Notably, HACNS_97 coincides directly with one of the sweeps and may therefore be a target for further study.
Taken altogether, there are initial grounds to suggest CNTNAP2 has played a role in the evolution of the human brain. There is also a strong body of literature linking mutations in CNTNAP2 to neurodevelopmental disorders. Despite these advances, a number of unanswered questions remain about CNTNAP2 function, and in particular, the mechanism(s) through which CNTNAP2 acts.
One attractive hypothesis is that one or more of the CNTNAP2 HARs are gene enhancers. These may be driving human-specific patterns of CNTNAP2 expression in the developing cortex. In turn, this human-specific increase in CNTNAP2 expression may have contributed to human brain function and increased human susceptibility to cognitive disorders. The studies summarized in this review suggest five mechanisms by which this may have occurred: increases in (1) potassium channel expression, (2) neurite development, (3) dendritic spine formation, (4) glutamate receptor expression, and/or (5) cortical interneuron abundance (Figure 6). Further experimentation is needed to clarify which of these mechanisms (if any) are at play, and which genes/pathways they involve.
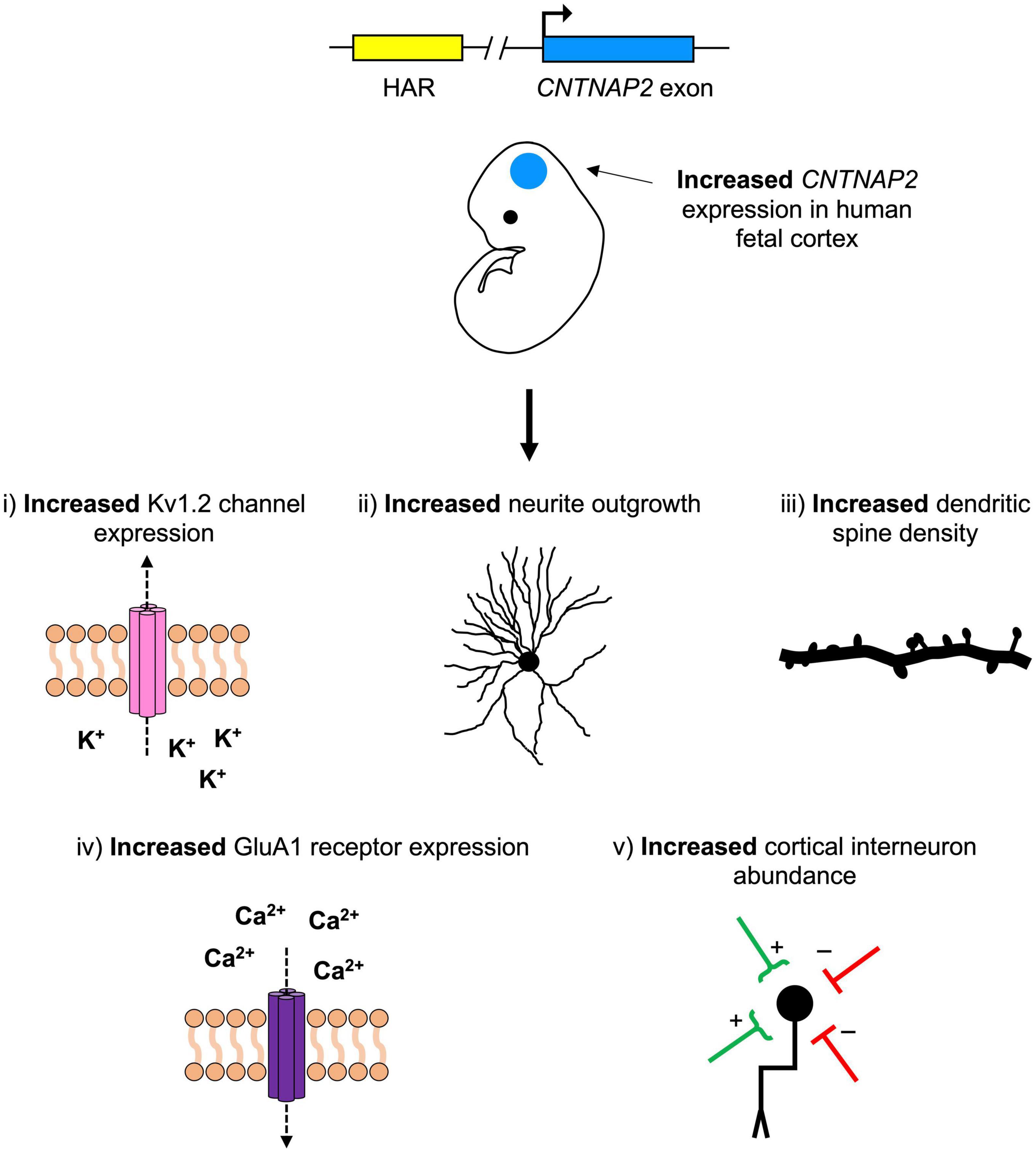
Figure 6. A hypothetical model for the role of contactin-associated protein-like 2 (CNTNAP2) in human neurodevelopmental disease and cerebral cortex evolution. Mutations that decrease CNTNAP2 expression [e.g., nonsense mutations or those disrupting a human accelerated region (HAR)] cause reductions in (1) potassium channel (Kv1.2) expression, (2) neurite branching, (3) dendritic spine density, (4) glutamate receptor (GluA1) expression, and (5) cortical interneuron abundance. Since CNTNAP2 has higher expression in the cortex of humans than in non-human primates, the opposite (i.e., increases in these features) may have contributed to differences in human versus non-human primate brain function. One (or more) of the CNTNAP2 HARs may be driving this increase in expression by functioning as an enhancer with human-specific properties.
Additionally, little experimental work has been conducted in human models, with most studies limited to either rodents or zebrafish. Human systems should be adopted for investigations into CNTNAP2 function, including the use of stem cell-derived neurons and primary patient samples, where possible. Such studies will be invaluable for understanding the role of CNTNAP2 in human-specific neurobiology, and in deciphering the downstream molecular events caused by human CNTNAP2 mutations.
FS researched and wrote the manuscript under the supervision of FL and TK. All authors contributed to the article and approved the submitted version.
FL’s group was supported by a Wellcome Trust Senior Investigator Award WT101052MA, Great Ormond Street Children’s Charity (Stem Cell Professorship) and Alzheimer’s Research UK (Stem Cell Research Center). FS was funded by the Canadian Centennial Scholarship Fund. This work was supported by the NIHR Great Ormond Street Biomedical Research Center.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.1017144/full#supplementary-material
Abrahams, B. S., Tentler, D., Perederiy, J. V., Oldham, M. C., Coppola, G., and Geschwind, D. H. (2007). Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc. Natl. Acad. Sci. U.S.A. 104, 17849–17854. doi: 10.1073/pnas.0706128104
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249.
Alarcon, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Anderson, G. R., Galfin, T., Xu, W., Aoto, J., Malenka, R. C., and Sudhof, T. C. (2012). Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. U.S.A. 109, 18120–18125. doi: 10.1073/pnas.1216398109
Ayub, Q., Yngvadottir, B., Chen, Y., Xue, Y., Hu, M., Vernes, S. C., et al. (2013). FOXP2 targets show evidence of positive selection in European populations. Am. J. Hum. Genet. 92, 696–706. doi: 10.1016/j.ajhg.2013.03.019
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Becker, M., Mastropasqua, F., Reising, J. P., Maier, S., Ho, M. L., Rabkina, I., et al. (2020). Presynaptic dysfunction in CASK-related neurodevelopmental disorders. Transl. Psychiatry 10:312. doi: 10.1038/s41398-020-00994-0
Bird, C. P., Stranger, B. E., Liu, M., Thomas, D. J., Ingle, C. E., Beazley, C., et al. (2007). Fast-evolving noncoding sequences in the human genome. Genome Biol. 8:R118.
Burbano, H. A., Green, R. E., Maricic, T., Lalueza-Fox, C., de la Rasilla, M., Rosas, A., et al. (2012). Analysis of human accelerated DNA regions using archaic hominin genomes. PLoS One 7:e32877. doi: 10.1371/journal.pone.0032877
Bush, E. C., and Lahn, B. T. (2008). A genome-wide screen for noncoding elements important in primate evolution. BMC Evol. Biol. 8:17. doi: 10.1186/1471-2148-8-17
Canali, G., Garcia, M., Hivert, B., Pinatel, D., Goullancourt, A., Oguievetskaia, K., et al. (2018). Genetic variants in autism-related CNTNAP2 impair axonal growth of cortical neurons. Hum. Mol. Genet. 27, 1941–1954. doi: 10.1093/hmg/ddy102
Capra, J. A., Erwin, G. D., McKinsey, G., Rubenstein, J. L., and Pollard, K. S. (2013). Many human accelerated regions are developmental enhancers. Philos. Trans. R Soc. Lond. B Biol. Sci. 368:20130025.
Centanni, T. M., Sanmann, J. N., Green, J. R., Iuzzini-Seigel, J., Bartlett, C., Sanger, W. G., et al. (2015). The role of candidate-gene CNTNAP2 in childhood apraxia of speech and specific language impairment. Am. J. Med. Genet. B Neuropsychiatr. Genet. 168, 536–543. doi: 10.1002/ajmg.b.32325
Chiocchetti, A. G., Kopp, M., Waltes, R., Haslinger, D., Duketis, E., Jarczok, T. A., et al. (2015). Variants of the CNTNAP2 5’ promoter as risk factors for autism spectrum disorders: A genetic and functional approach. Mol. Psychiatry 20, 839–849. doi: 10.1038/mp.2014.103
Chorev, M., and Carmel, L. (2012). The function of introns. Front. Genet. 3:55. doi: 10.3389/fgene.2012.00055
Doan, R. N., Bae, B. I., Cubelos, B., Chang, C., Hossain, A. A., Al-Saad, S., et al. (2016). Mutations in human accelerated regions disrupt cognition and social behavior. Cell 167, 341.e–354.e. doi: 10.1016/j.cell.2016.08.071
Dorschner, M. O., Hawrylycz, M., Humbert, R., Wallace, J. C., Shafer, A., Kawamoto, J., et al. (2004). High-throughput localization of functional elements by quantitative chromatin profiling. Nat. Methods 1, 219–225.
Elia, J., Gai, X., Xie, H. M., Perin, J. C., Geiger, E., Glessner, J. T., et al. (2010). Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 15, 637–646. doi: 10.1038/mp.2009.57
Fernandes, D., Santos, S. D., Coutinho, E., Whitt, J. L., Beltrao, N., Rondao, T., et al. (2019). Disrupted AMPA receptor function upon genetic- or antibody-mediated loss of autism-associated CASPR2. Cereb. Cortex 29, 4919–4931.
Freri, E., Castellotti, B., Canafoglia, L., Ragona, F., Solazzi, R., Vannicola, C., et al. (2021). Severe epilepsy in CNTNAP2-related Pitt-Hopkins-like syndrome successfully treated with stiripentol. Seizure 88, 143–145. doi: 10.1016/j.seizure.2021.04.012
Friedman, J. I., Vrijenhoek, T., Markx, S., Janssen, I. M., van der Vliet, W. A., Faas, B. H., et al. (2008). CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol. Psychiatry 13, 261–266.
Gao, R., Piguel, N. H., Melendez-Zaidi, A. E., Martin-de-Saavedra, M. D., Yoon, S., Forrest, M. P., et al. (2018). CNTNAP2 stabilizes interneuron dendritic arbors through CASK. Mol. Psychiatry 23, 1832–1850.
Gao, R., Pratt, C. P., Yoon, S., Martin-de-Saavedra, M. D., Forrest, M. P., and Penzes, P. (2020). CNTNAP2 is targeted to endosomes by the polarity protein PAR3. Eur. J. Neurosci. 51, 1074–1086. doi: 10.1111/ejn.14620
Gao, R., Zaccard, C. R., Shapiro, L. P., Dionisio, L. E., Martin-de-Saavedra, M. D., Piguel, N. H., et al. (2019). The CNTNAP2-CASK complex modulates GluA1 subcellular distribution in interneurons. Neurosci. Lett. 701, 92–99. doi: 10.1016/j.neulet.2019.02.025
Gdalyahu, A., Lazaro, M., Penagarikano, O., Golshani, P., Trachtenberg, J. T., and Geschwind, D. H. (2015). The autism related protein contactin-associated protein-like 2 (CNTNAP2) stabilizes new spines: An in vivo mouse study. PLoS One 10:e0125633. doi: 10.1371/journal.pone.0125633
Girskis, K. M., Stergachis, A. B., DeGennaro, E. M., Doan, R. N., Qian, X., Johnson, M. B., et al. (2021). Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron 109, 3239.e–3251.e. doi: 10.1016/j.neuron.2021.08.005
Gittelman, R. M., Hun, E., Ay, F., Madeoy, J., Pennacchio, L., Noble, W. S., et al. (2015). Comprehensive identification and analysis of human accelerated regulatory DNA. Genome Res. 25, 1245–1255.
Gordon, A., Salomon, D., Barak, N., Pen, Y., Tsoory, M., Kimchi, T., et al. (2016). Expression of Cntnap2 (Caspr2) in multiple levels of sensory systems. Mol. Cell. Neurosci. 70, 42–53. doi: 10.1016/j.mcn.2015.11.012
Hali, S., Kim, J., Kwak, T. H., Lee, H., Shin, C. Y., and Han, D. W. (2020). Modelling monogenic autism spectrum disorder using mouse cortical organoids. Biochem. Biophys. Res. Commun. 521, 164–171. doi: 10.1016/j.bbrc.2019.10.097
Hoffman, E. J., Turner, K. J., Fernandez, J. M., Cifuentes, D., Ghosh, M., Ijaz, S., et al. (2016). Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene. CNTNAP2. Neuron 89, 725–733. doi: 10.1016/j.neuron.2015.12.039
Hubisz, M. J., and Pollard, K. S. (2014). Exploring the genesis and functions of human accelerated regions sheds light on their role in human evolution. Curr. Opin. Genet. Dev. 29, 15–21. doi: 10.1016/j.gde.2014.07.005
Jackman, C., Horn, N. D., Molleston, J. P., and Sokol, D. K. (2009). Gene associated with seizures, autism, and hepatomegaly in an Amish girl. Pediatr. Neurol. 40, 310–313.
Kanton, S., Boyle, M. J., He, Z., Santel, M., Weigert, A., Sanchís-Calleja, F., et al. (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. doi: 10.1038/s41586-019-1654-9
Karaca, E., Harel, T., Pehlivan, D., Jhangiani, S. N., Gambin, T., Coban Akdemir, Z., et al. (2015). Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron 88, 499–513.
Kim, J. W., Park, K., Kang, R. J., Gonzales, E. L. T., Kim, D. G., Oh, H. A., et al. (2019). Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology 44, 314–323.
Lazaro, M. T., Taxidis, J., Shuman, T., Bachmutsky, I., Ikrar, T., Santos, R., et al. (2019). Reduced prefrontal synaptic connectivity and disturbed oscillatory population dynamics in the CNTNAP2 model of autism. Cell Rep. 27, 2567.e–2578.e. doi: 10.1016/j.celrep.2019.05.006
Lee, I. S., Carvalho, C. M., Douvaras, P., Ho, S. M., Hartley, B. J., Zuccherato, L. W., et al. (2015). Characterization of molecular and cellular phenotypes associated with a heterozygous CNTNAP2 deletion using patient-derived hiPSC neural cells. NPJ Schizophr. 1:15019. doi: 10.1038/npjschz.2015.19
Levchenko, A., Kanapin, A., Samsonova, A., and Gainetdinov, R. R. (2018). Human accelerated regions and other human-specific sequence variations in the context of evolution and their relevance for brain development. Genome Biol. Evol. 10, 166–188. doi: 10.1093/gbe/evx240
Lindblad-Toh, K., Garber, M., Zuk, O., Lin, M. F., Parker, B. J., Washietl, S., et al. (2011). A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482. doi: 10.1038/nature10530
Lu, P., Wang, F., Zhou, S., Huang, X., Sun, H., Zhang, Y. W., et al. (2021). A Novel CNTNAP2 mutation results in abnormal neuronal E/I balance. Front. Neurol. 12:712773. doi: 10.3389/fneur.2021.712773
Martín-de-Saavedra, M. D., Dos Santos, M., Culotta, L., Varea, O., Spielman, B. P., Parnell, E., et al. (2022). Shed CNTNAP2 ectodomain is detectable in CSF and regulates Ca(2+) homeostasis and network synchrony via PMCA2/ATP2B2. Neuron 110, 627.e–643.e. doi: 10.1016/j.neuron.2021.11.025
Mozzi, A., Forni, D., Clerici, M., Pozzoli, U., Mascheretti, S., Guerini, F. R., et al. (2016). The evolutionary history of genes involved in spoken and written language: Beyond FOXP2. Sci Rep. 6:22157. doi: 10.1038/srep22157
Nord, A. S., Roeb, W., Dickel, D. E., Walsh, T., Kusenda, M., O’Connor, K. L., et al. (2011). Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 19, 727–731. doi: 10.1038/ejhg.2011.24
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 43, 585–589.
Parrini, E., Marini, C., Mei, D., Galuppi, A., Cellini, E., Pucatti, D., et al. (2017). Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum. Mutat. 38, 216–225.
Paterno, R., Marafiga, J. R., Ramsay, H., Li, T., Salvati, K. A., and Baraban, S. C. (2021). Hippocampal gamma and sharp-wave ripple oscillations are altered in a Cntnap2 mouse model of autism spectrum disorder. Cell Rep. 37:109970. doi: 10.1016/j.celrep.2021.109970
Peñagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Poliak, S., Gollan, L., Martinez, R., Custer, A., Einheber, S., Salzer, J. L., et al. (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24, 1037–1047. doi: 10.1016/s0896-6273(00)81049-1
Poliak, S., Salomon, D., Elhanany, H., Sabanay, H., Kiernan, B., Pevny, L., et al. (2003). Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 162, 1149–1160. doi: 10.1083/jcb.200305018
Pollard, K. S., Salama, S. R., King, B., Kern, A. D., Dreszer, T., Katzman, S., et al. (2006). Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2:e168. doi: 10.1371/journal.pgen.0020168
Pollen, A. A., Bhaduri, A., Andrews, M. G., Nowakowski, T. J., Meyerson, O. S., Mostajo-Radji, M. A., et al. (2019). Establishing cerebral organoids as models of human-specific brain evolution. Cell 176, 743.e–756.e.
Poot, M. (2017). Intragenic CNTNAP2 deletions: A bridge too far? Mol. Syndromol. 8, 118–130. doi: 10.1159/000456021
Prabhakar, S., Noonan, J. P., Paabo, S., and Rubin, E. M. (2006). Accelerated evolution of conserved noncoding sequences in humans. Science 314:786.
Riccardi, F., Urquhart, J., McCullagh, G., Lawrence, P., and Douzgou, S. A. (2019). patient with a novel CNTNAP2 homozygous variant: Further delineation of the CASPR2 deficiency syndrome and review of the literature. Clin. Dysmorphol. 28, 66–70.
Rodenas-Cuadrado, P., Ho, J., and Vernes, S. C. (2014). Shining a light on CNTNAP2: Complex functions to complex disorders. Eur. J. Hum. Genet. 22, 171–178. doi: 10.1038/ejhg.2013.100
Rodenas-Cuadrado, P., Pietrafusa, N., Francavilla, T., La Neve, A., Striano, P., and Vernes, S. C. (2016). Characterisation of CASPR2 deficiency disorder–a syndrome involving autism, epilepsy and language impairment. BMC Med. Genet. 17:8. doi: 10.1186/s12881-016-0272-8
Rubio-Marrero, E. N., Vincelli, G., Jeffries, C. M., Shaikh, T. R., Pakos, I. S., Ranaivoson, F. M., et al. (2016). Structural characterization of the extracellular domain of CASPR2 and insights into its association with the novel ligand contactin1. J. Biol. Chem. 291, 5788–5802. doi: 10.1074/jbc.M115.705681
Ryu, H., Inoue, F., Whalen, S., Williams, A., Kircher, M., Martin, B., et al. (2018). Massively parallel dissection of human accelerated regions in human and chimpanzee neural progenitors. bioRxiv [Preprint]. doi: 10.1101/256313
Sabeti, P. C., Schaffner, S. F., Fry, B., Lohmueller, J., Varilly, P., Shamovsky, O., et al. (2006). Positive natural selection in the human lineage. Science 312, 1614–1620.
Sacai, H., Sakoori, K., Konno, K., Nagahama, K., Suzuki, H., Watanabe, T., et al. (2020). Autism spectrum disorder-like behavior caused by reduced excitatory synaptic transmission in pyramidal neurons of mouse prefrontal cortex. Nat. Commun. 11:5140. doi: 10.1038/s41467-020-18861-3
Scala, M., Anijs, M., Battini, R., Madia, F., Capra, V., Scudieri, P., et al. (2021). Hyperkinetic stereotyped movements in a boy with biallelic CNTNAP2 variants. Ital. J. Pediatr. 47:208. doi: 10.1186/s13052-021-01162-w
Scott, R., Sanchez-Aguilera, A., van Elst, K., Lim, L., Dehorter, N., Bae, S. E., et al. (2019). Loss of Cntnap2 causes axonal excitability deficits. Developmental Delay in cortical myelination, and abnormal stereotyped motor behavior. Cereb Cortex 29, 586–597. doi: 10.1093/cercor/bhx341
Selimbeyoglu, A., Kim, C. K., Inoue, M., Lee, S. Y., Hong, A. S. O., Kauvar, I., et al. (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci. Transl. Med. 9:eaah6733. doi: 10.1126/scitranslmed.aah6733
Smogavec, M., Cleall, A., Hoyer, J., Lederer, D., Nassogne, M. C., Palmer, E. E., et al. (2016). Eight further individuals with intellectual disability and epilepsy carrying bi-allelic CNTNAP2 aberrations allow delineation of the mutational and phenotypic spectrum. J. Med. Genet. 53, 820–827.
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M., et al. (2006). Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 354, 1370–1377. doi: 10.1056/NEJMoa052773
Thomas, A. M., Schwartz, M. D., Saxe, M. D., and Kilduff, T. S. (2017). Cntnap2 knockout rats and mice exhibit epileptiform activity and abnormal sleep-wake physiology. Sleep 40. *P. doi: 10.1093/sleep/zsw026
Uebbing, S., Gockley, J., Reilly, S. K., Kocher, A. A., Geller, E., Gandotra, N., et al. (2021). Massively parallel discovery of human-specific substitutions that alter enhancer activity. Proc. Natl. Acad. Sci. U.S.A. 118:e2007049118.
Varea, O., Martin-de-Saavedra, M. D., Kopeikina, K. J., Schurmann, B., Fleming, H. J., Fawcett-Patel, J. M., et al. (2015). Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc. Natl. Acad. Sci. U.S.A. 112, 6176–6181. doi: 10.1073/pnas.1423205112
Veerappa, A. M., Saldanha, M., Padakannaya, P., and Ramachandra, N. B. (2013). Family-based genome-wide copy number scan identifies five new genes of dyslexia involved in dendritic spinal plasticity. J. Hum. Genet. 58, 539–547. doi: 10.1038/jhg.2013.47
Verkerk, A. J., Mathews, C. A., Joosse, M., Eussen, B. H., Heutink, P., and Oostra, B. A. (2003). CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 82, 1–9. doi: 10.1016/s0888-7543(03)00097-1
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., et al. (2008). A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 359, 2337–2345.
Watson, C. M., Crinnion, L. A., Tzika, A., Mills, A., Coates, A., Pendlebury, M., et al. (2014). Diagnostic whole genome sequencing and split-read mapping for nucleotide resolution breakpoint identification in CNTNAP2 deficiency syndrome. Am. J. Med. Genet. A 164a, 2649–2655.
Won, H., Huang, J., Opland, C. K., Hartl, C. L., and Geschwind, D. H. (2019). Human evolved regulatory elements modulate genes involved in cortical expansion and neurodevelopmental disease susceptibility. Nat. Commun. 10:2396. doi: 10.1038/s41467-019-10248-3
Zweier, C., de Jong, E. K., Zweier, M., Orrico, A., Ousager, L. B., Collins, A. L., et al. (2009). CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 85, 655–666. doi: 10.1016/j.ajhg.2009.10.004
Keywords: autism (ASD), neurodevelopmental disorders, cerebral cortex, genetics, CNTNAP2
Citation: St. George-Hyslop F, Kivisild T and Livesey FJ (2022) The role of contactin-associated protein-like 2 in neurodevelopmental disease and human cerebral cortex evolution. Front. Mol. Neurosci. 15:1017144. doi: 10.3389/fnmol.2022.1017144
Received: 11 August 2022; Accepted: 20 September 2022;
Published: 20 October 2022.
Edited by:
Michel J. Simonneau, École Normale Supérieure Paris-Saclay, FranceReviewed by:
J. Peter H. Burbach, University Medical Center Utrecht, NetherlandsCopyright © 2022 St. George-Hyslop, Kivisild and Livesey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frances St. George-Hyslop, Zi5zdGdlb3JnZS5oeXNsb3BAbWFpbC51dG9yb250by5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.