 Shuang Chen
Shuang Chen Da Xu
Da Xu Liu Fan†
Liu Fan† Zhi Fang
Zhi Fang Man Li
Man Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 07 January 2022
Sec. Brain Disease Mechanisms
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.797253
This article is part of the Research Topic The Molecular Mechanisms of Epilepsy and Potential Therapeutics View all 12 articles
Epilepsy is one of the most common neurological disorders characterized by recurrent seizures. The mechanism of epilepsy remains unclear and previous studies suggest that N-methyl-D-aspartate receptors (NMDARs) play an important role in abnormal discharges, nerve conduction, neuron injury and inflammation, thereby they may participate in epileptogenesis. NMDARs belong to a family of ionotropic glutamate receptors that play essential roles in excitatory neurotransmission and synaptic plasticity in the mammalian CNS. Despite numerous studies focusing on the role of NMDAR in epilepsy, the relationship appeared to be elusive. In this article, we reviewed the regulation of NMDAR and possible mechanisms of NMDAR in epilepsy and in respect of onset, development, and treatment, trying to provide more evidence for future studies.
Epilepsy is one of the most common neurological disorders characterized by recurrent seizures. Long-term recurrent seizures could lead to cognitive impairment and mental disorders, which severely affect the social interaction level and employment ability of epileptic patients, and result in a decline in the quality of life (Chen et al., 2020a). At present, antiepileptic drugs (AEDs) remain the main therapy of epilepsy, despite no response in about 1/3 patients with epilepsy (Moshé et al., 2015). The mechanism of epilepsy remains unclear but it is generally regarded as a self-facilitated pathological process triggered by brain injury, ultimately resulting in nerve damage, mossin fibrosis, synaptic plasticity, inflammatory response, and ionic pathway dysfunction (Gan et al., 2015). It is widely acknowledged that abnormal excessive synchronous discharge, i.e., the imbalance between excitation and inhibition of neurons, plays an essential role in epileptogenesis.
However, the factors affecting this imbalance were sophisticated, and excitatory amino acids were supposed to participate in this imbalance (Bonansco and Fuenzalida, 2016).
In the central nervous system (CNS), N-methyl-D-aspartate receptor (NMDAR) is one of the main excitatory receptors on the synapses of neurons including glutamatergic neurons and GABAergic interneurons, which regulate the balance between neuronal excitation and inhibition (Hendry et al., 1987; Hanada, 2020). Meanwhile, NMDARs, the ionotropic glutamate receptors in the brain, are involved in neuroplasticity, excitatory neurotransmission, and neurotoxicity (Fricker et al., 2018; Horak et al., 2021). Related studies have shown that overexcitation of NMDAR leads to neuronal death in neurological diseases such as epilepsy, stroke, Alzheimer’s disease (AD), and Parkinson’s disease (PD; Essiz et al., 2021). In the brain, NMDARs are hetero-tetramers generally composed of two GluN1 subunits and four distinct GluN2 (GluN2A-D) or a mixture of GluN2 with two different GluN3 (GluN3A and 3B) subunits. The GluN1 subunit is required for NMDAR activation and binds to the necessary co-agonists through the amino-terminal domain of the extracellular region. GluN2 subunits are able to bind glutamate specifically and these subunits are different from each other by providing different functional properties of NMDAR. In the NMDAR, GRIN1 codes for GluN1 subunit, GRIN2 codes for GluN2 subunit, and GRIN3 codes for GluN3 subunit (Beesley et al., 2020). Meanwhile, the triheteromer (GluN1/GluN2A/GluN2B) is the main subtype of NMDAR and is widely expressed in the cortex and hippocampus (Luo et al., 1997; Tovar et al., 2013).
Previous studies have shown that glutamate levels increase in the extracellular fluid during seizures in temporal lobe epilepsy (TLE) and glutamate can directly activate NMDAR and induce neuroexcitatory toxicity (Albrecht and Zielińska, 2017). Meanwhile, it has been reported that NMDA, AMPA, and kainite (KA) can induce seizures in animal models, and glutamate receptor antagonists inhibit seizures in animals (Celli and Fornai, 2020). In the PTZ-induced status epilepticus (SE), GluN1, GluN2A, and GluN2B subunits are increased and synaptic plasticity impairs in the hippocampus of rats. Meanwhile, the increase in the GluN2B subunit may result in the decrease of long-term potentiation (LTP; Postnikova et al., 2017). Related studies have shown that GRIN1, GRIN2A, and GRIN2B mutations can lead to epilepsy. In all mutations, GRIN2A variants are associated with neurological diseases including developmental and epileptic encephalopathy, which may be manifested as seizures, mild speech and language delay, and cognitive impairment (Lemke et al., 2013, 2014; Fry et al., 2018). In addition, anti-NMDAR encephalitis, a major type of autoimmune encephalitis (AE), has been reported to be an entity of epilepsy (Leypoldt et al., 2015). In addition to neurotoxicity, NMDARs can also participate in neuroprotection by activating cAMP response element-binding protein (CREB) signals in epilepsy (Wang et al., 2020b).
Owing to its role in brain functional plasticity and neuroexcitatory, the regulation of NMDAR in epilepsy has attracted extensive attention. As noted above, NMDARs have been shown to involve in seizures, but functions and mechanisms of NMDARs in epilepsy appear to be elusive. In clinical study, understanding the function of NMDAR is of great significance for the treatment of epilepsy and AEDs selection. This article reviews the regulatory mechanism of NMDAR and the progress of NMDAR in the occurrence, development, and treatment of epilepsy from various points of view (Figure 1).
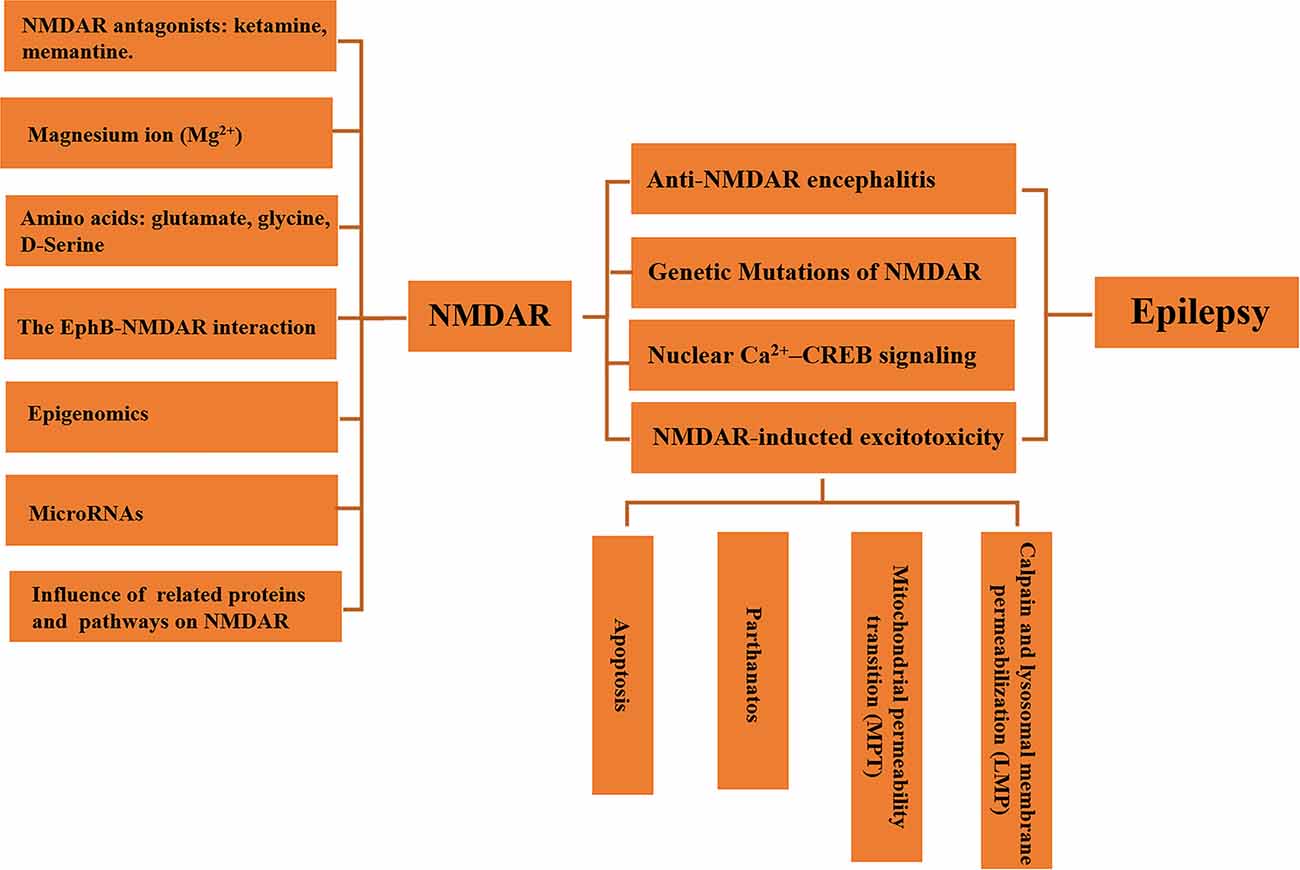
Figure 1. Regulation of neuroexcitatory receptor N-Methyl-D-Aspartate Receptor (NMDAR) in epilepsy.
Anti-NMDAR encephalitis is a major type of AE in which the over-production of NMDAR autoantibodies results in profound neurotransmitter dysregulation, causing seizures and other symptoms such as dysautonomia, orofacial dyskinesia, psychosis, mental status changes, hallucinations, and headaches (Tong et al., 2020). Related studies have shown that approximately 75% of patients with anti-NMDAR encephalitis develop seizures, and refractory status epilepticus (RSE) can lead to neuronal death (Geis et al., 2019). However, the mechanism of anti-NMDAR encephalitis leading to seizures is not fully understood.
NMDARs have been reported to play a role in synaptic homeostasis. Related studies have found that NMDAR autoantibodies could increase extracellular glutamate levels in the brain (Manto et al., 2010). Some findings also have confirmed that NMDAR autoantibodies can result in the internalization of surface NMDARs and the decreasing of receptor density in the patients with anti-NMDAR encephalitis (Hughes et al., 2010; Takahashi et al., 2020). In the patients, NMDAR autoantibodies bind and cross-link to a specific region of NMDARs GluN1, then internalize NMDARs (Gleichman et al., 2012). NMDAR-EphB2 interaction plays a key role in the NMDAR autoantibody-mediated NMDAR internalization. When autoantibodies bind to endogenous NMDARs, the interaction between NMDAR and EphB2 is disrupted, thereby leading to NMDAR internalization and dysfunction, and the reduction of NMDAR-mediated synaptic currents (Hughes et al., 2010; Mikasova et al., 2012). After activation of EphB2, the extracellular domain of EphB2 interacts directly with the GluN1 subunit, thereby stabilizing NMDARs in the synapse (Dalva et al., 2000; Washburn et al., 2020). Meanwhile, a related report has indicated that NMDAR autoantibody interferes with the interaction between NMDARs and EphB2 in cultured hippocampal neurons (Mikasova et al., 2012). In addition, cerebrospinal fluid (CSF) of the patient with anti-NMDAR encephalitis can also result in the reduction of both GluN2A and GluN2B on the synaptic surface and prevent a chemically induced LTP of glutamate synapses (Mikasova et al., 2012). Similar to the anti-NMDAR antibodies-mediated effect, the amino terminal domain (ATD) peptide of GluN1 subunit can also actively immunize against NMDARs and induce anti-NMDAR encephalitis in a mouse model (Ding et al., 2021). A study shows that a single injection of anti-NMDAR antibodies from the patient with anti-NMDAR encephalitis into mice does not induce seizures (Wright et al., 2015). However, injection of anti-NMDAR antibodies in vivo can increase the number of seizures in the PTZ induced-mice model. Moreover, about 75–93% of mice developed epilepsy after long-term infusion of CSF or purified anti-NMDAR antibodies from patients with anti-NMDAR encephalitis (Wright et al., 2015). It is puzzling that the function reduction of NMDAR is more likely to activate the persistent abnormal discharge of neurons, but a specific inhibition of the NMDARs in the GABAergic interneurons can also explain this phenomenon (Manto et al., 2010). NMDAR autoantibodies reduce the excitability of GABAergic interneurons through the interaction with NMDAR, thereby weakening the inhibitory effect on excitatory transduction of glutamatergic neurons (Geis et al., 2019). The disinhibition of excitatory glutamatergic neurons may also account for seizures in anti-NMDAR encephalitis. In addition, it has been found that ketamine, an NMDAR antagonist, may be useful in super-refractory status epilepticus (SRSE) developed in patients with anti-NMDAR encephalitis (Santoro et al., 2019). Despite the profound effect on NMDARs, there is no evidence that NMDAR autoantibodies can alter the localization and expression of other glutamate receptors such as AMPARs, the synaptic protein PSD-95, as well as the number of synapses, or affect the survival of nerve cells in vitro or in vivo models (Huang and Xiong, 2021). In a word, both the activation of NMDARs in glutamatergic neurons and the inhibition of NMDARs in GABAergic interneurons may participate in epileptogenesis and more researches are urgently needed.
In recent years, NMDAR subunit-encoding genes have been confirmed to be involved in epilepsy and the genetic mutations in NMDARs may cause epilepsy in humans, suggesting that NMDAR is closely related to epilepsy (Xu and Luo, 2018). Besides, impairment of NMDAR signals as a result of genetic or environmental insults leads to a variety of neurodevelopmental disorders, including epilepsy, schizophrenia, intellectual disability, or autism (Mielnik et al., 2021). Meanwhile, NMDAR-mediated excitotoxicity was supposed to participate in neuronal death induced by high levels of glutamate and aspartate in neurological diseases such as epilepsy, stroke, AD, and PD (Fricker et al., 2018; Essiz et al., 2021). This article will review genetic mutations of NMDAR, signaling pathways of NMDAR-mediated excitotoxicity, and NMDAR-dependent neuroprotection in epilepsy.
GluN1 subunit, the essential subunit of functional NMDAR is encoded by GRIN1, and GRIN1 mutations have a significant effect on neuronal activity, causing various types of epilepsy, including SE, focal dyscognitive seizures, myoclonic seizures, febrile seizures, spasms, hypermotor seizures, tonic and atonic seizures, generalized seizures, etc (Wyllie et al., 2013; Fry et al., 2018). Besides, the epileptic phenotype may contribute to the p. Met641Leu de novo variant in GRIN1 gene, and de novo GRIN1 mutations were gradually recognized to be in association with severe early infantile encephalopathy (Pironti et al., 2018). The common characteristics are extensive bilateral polymicrogyria with intractable epilepsy, cortical visual impairment, postnatal microcephaly, and severe developmental delay in patients with de novo GRIN1 mutations (Fry et al., 2018). Extensive bilateral polymicrogyria is associated with severe developmental delay and intractable epilepsy. At present, a variety of polymicrogyria-associated mutations have been found, including p.Asn674Ile, p.Arg794Gln, p.Arg659Trp, p.Asp789Asn, p.Tyr647Cys, p.Asn650Ile, p.Ala653Gly, p.Leu551Pro, p.Ser553Leu, etc (Fry et al., 2018). In GRIN1 mutations, the mechanisms remained unclear but disrupted gating of the ion channel by p.Gly827Arg mutation and disruption of NMDAR ligand binding by p.Ser688Tyr mutation may be concerned (Zehavi et al., 2017).
Current studies have found that GluN2 subunits may control epileptiform events in the hippocampus (Punnakkal and Dominic, 2018). GRIN2A, which encodes the GluN2A subunits, is widely considered to be epileptogenic. The most common types of seizure caused by GRIN2A mutations include atypical benign partial epilepsy, Landau–Kleffner syndrome (LKS), and benign epilepsy with centro-temporal spikes (BECT; Hanada, 2020). GluN2 subunits mainly regulate the open/close of the NMDARs. GluN2A-containing receptors have a reversible calcium-dependent inactivation, whereas GluN2B does not (Franchini et al., 2020). Meanwhile, GluN2A subunits can regulate neuronal NMDAR-induced microglia-neuron physical interactions (Eyo et al., 2018). Related studies have shown that voltage-independent GluN2A-related NMDAR-Ca2+ signaling is related to audiogenic seizures, attentional and cognitive deficits in mice (Bertocchi et al., 2021). A rare variant of GRIN2A associated with epilepsy disrupts CaMKIIα phosphorylation of GluN2A and NMDAR trafficking, which demonstrates a role of GluN2A for CaMKIIα phosphorylation in receptor targeting and suggests that the defects of NMDAR trafficking are related to epilepsy (Mota Vieira et al., 2020). There were defects of GRIN2A related to epileptiform discharges and transient microstructural brain abnormalities in mice with epilepsy (Salmi et al., 2018). The mutant GluN2A (p.Met817Val)-containing receptors decreased sensitivity to endogenous negative inhibitors (Mg, zinc), prolonged the time of synaptic response, increased the time of single-channel mean open, and the probability of channel open. These acquired GRIN2A mutations lead to overactivation of NMDAR and increase neuronal excitability, which may be related to epileptogenesis observed in patients (Chen et al., 2017). A de novo GRIN2A missense mutation (p.Asp731Asn) in a child with focal epilepsy and acquired epileptic aphasia was reported. However, this mutant reduced NMDAR activation, suggesting that NMDAR hypofunction may also be related to epilepsy pathogenesis (Gao et al., 2017).
GRIN2B mutation is a rare cause of severe epileptic encephalopathy (Sharawat et al., 2019). A related study demonstrated that GRIN2B, BDNF, and IL-1β gene significantly were upregulated and GRIN2B was positively correlated with the expressions of BDNF and IL-1β gene in people with epilepsy (Zhand et al., 2018). Some GRIN2B mutations (p.Val618Gly and p.Asn615Ile) were found in patients with early-onset epilepsy and epileptic encephalopathy (Lemke et al., 2014; Smigiel et al., 2016). Those GluN2B heteromers showed a significant loss of ion-channel block by extracellular Mg2+ and a significant increase of Ca2+ permeability (Lemke et al., 2014). Meanwhile, blocking GluN2B-containing NMDARs can reduce short-term brain injury caused by early-life SE (Loss et al., 2019).
In addition, GRIN2C expression is limited to astrocytes whereas GRIN2D are expressed at high levels in GABAergic interneurons in the hippocampus (Shelkar et al., 2019; Dubois and Liu, 2021). These specific distributions are likely to be highly relevant to mechanisms of epilepsy and dysregulation of glutamatergic signaling. Some GRIN2D variants (p. Thr674Lys, p.Met681Ile, p.Ser694Arg, p, Asp449Asn, p.Val667Ile, p.Ser573Phe, p.Leu670Phe, p.Ala675Thr, p.Ala678Asp, p.Ser1271Leu, and p.Arg1313Trp) have been found in developmental and epileptic encephalopathy (Tsuchida et al., 2018; Jiao et al., 2021). In a novel GRIN2D variant with epileptic encephalopathy, GRIN2D mutation-related epilepsy is found to be refractory to conventional AEDs (Camp and Yuan, 2020; Jiao et al., 2021). However, GRIN2D dominant mutations can cause severe epileptic encephalopathy, which can be treated with NMDAR channel blockers (Li et al., 2016). In epilepsy, we need to further understand the unique characteristics of GRIN2D mutations in neurological function and pathology, which is conducive to the treatment of refractory epilepsy.
In the CNS, high levels of glutamate induce neuronal death by NMDAR-mediated excitotoxicity (Olney, 1971). Glutamate-induced excitotoxicity is mainly attributed to apoptosis, autophagy, parthanatos, phagocytosis, ferroptosis, apoptosis- inducing factor (AIF), calpain I, mitochondrial permeability transformation (MPT), lysosomal membrane permeability (LMP), and RNS and ROS production (Figure 2; Fricker et al., 2018). Activated NMDARs lead to neuronal depolarization and calcium (Ca2+) loading. The increasing of cytoplasmic Ca2+ can cause the activation of nNOS, calpain I, and MPT pore, eventually leading to neuronal death (Figure 2; Fricker et al., 2018). Overexcitation of NMDAR leads to neuronal death in neurological diseases such as epilepsy, stroke, AD, and PD, and blockade of NMDARs can reduce neuronal death in the brain (Essiz et al., 2021).
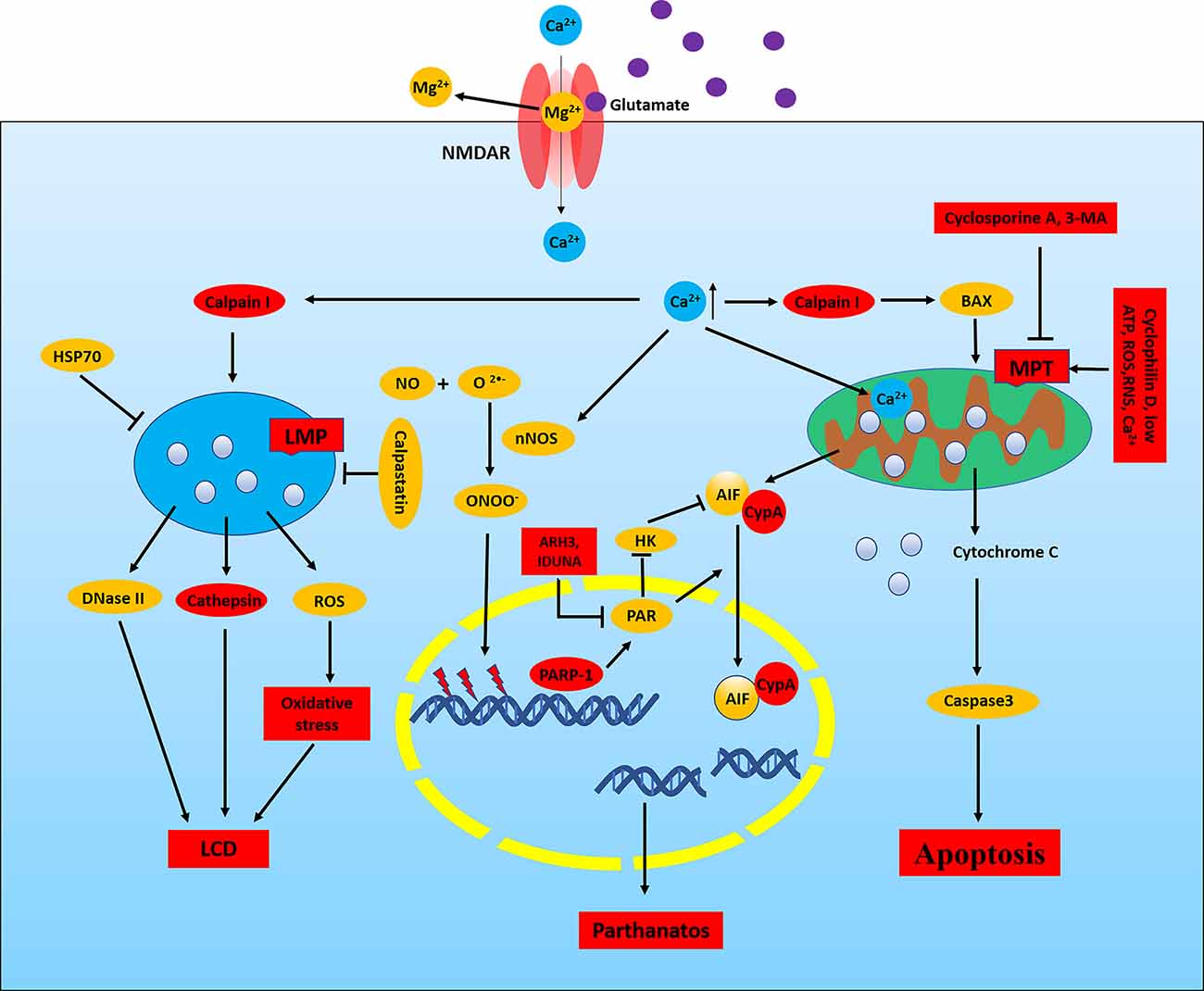
Figure 2. NMDAR-mediated excitotoxicity in epilepsy. In neurons, the NMDAR channel is blocked by Mg2+ at neuronal resting membrane potential, and Mg2+ is removed when the membrane is depolarized. Activated NMDAR leads to calcium loading which will cause the activation of nNOS, calpain I, and mitochondrial permeability transformation (MPT) pore and eventually lead to neuronal death. Calpain I can cleave Bid and Bax, leading to the release of apoptosis-inducing factor (AIF) and cytochrome C from the mitochondria. Meanwhile, cytochrome C can induce the activation of caspase, and calpain I can also directly cleave and activate caspases, thus resulting in apoptosis. In addition, AIF is cleaved by calpain I to a tAIF, which translocates to the nucleus and induces DNA cleavage, thereby leading to apoptosis and parthanatos. Activation of calpain can cause lysosomal membrane permeability (LMP), which releases the toxic cathepsin, DNase II, and ROS, thereby resulting in LCD. Meanwhile, HSP70 and calpastatin can resist LMP. Increased Ca2+, ROS, RNS, and low ATP in mitochondrial matrix results in MPT which depends on the opening of mPTP. Cyclosporine A and 3-MA can block MPT. Ca2+ directly activates nNOS, which can catalyze NO and O2− to form ONOO−. ONOO− damages DNA, thereby activating PARP1, resulting in parthanatos. PARP1 is involved in chromosomal stability, DNA repair, and inflammatory responses. PAR, the product of PARP1 activity, induces nuclear translocation of AIF and inhibits HK. Nuclear translocation of AIF requires the involvement of CypA, which binds to AIF and forms CypA-AIF complex after the release from mitochondria, thereby participating in DNA degradation and leading to parthanatos. ARH3 reduces PAR levels in the nucleus and cytoplasm and IDUNA reduces the release of AIF by binding to the PAR polymers and prevents PARP1-induced cell death. LCD, lysosomal cell death; HSP70, heat shock protein 70; HK, hexokinase.
Calpain and Lysosomal Membrane Permeabilization (LMP). Activated NMDAR leads to Ca2+ influx, which activates calpain. Calpain I is involved in the late phase of neuronal death caused by mitochondrial dysfunction. Calpain I, a cysteine protease highly expressed in neurons, is activated by high levels of Ca2+ in the cytoplasm and can cleave Bid and Bax, leading to the release of AIF and cytochrome C from the mitochondria (Wang, 2000; D’Orsi et al., 2012). Meanwhile, the release of cytochrome C can induce the activation of caspases, and activated calpain I can also directly cleave and activate caspases, thus resulting in apoptosis (Wang, 2000). However, AIF is cleaved by calpain I to a truncated AIF (tAIF), which translocates to the nucleus and induces DNA cleavage, thereby leading to apoptosis and parthanatos (Fricker et al., 2018). In addition, GluN2A subunit-containing synaptic NMDARs preferentially activates calpain I, which is conducive to neuronal survival by selectively degrading the protein phosphatase PHLPP1α and PHLPP1β (Wang et al., 2013). On the contrary, calpain II is selectively activated by GluN2B subunit-containing extrasynaptic NMDARs and calpain II participates in neuronal death by degrading the protein tyrosine phosphatase STEP (Hoque et al., 2016). Further studies are needed to clarify the role of these two isoforms of calpains in excitotoxic neuronal death.
Activated calpain can also cause LMP, thereby releasing the toxic cathepsin into the cytoplasm and leading to lysosomal cell death (LCD), also known as autolysis in neurodegenerative diseases (Fricker et al., 2018). It has been found that ischemia can induce calpain I to be localized in lysosomes and cause neuronal LMP (Yamashima et al., 1998; Windelborn and Lipton, 2008). Meanwhile, multiple stimuli can also induce LMP, release cathepsin, and induce cell death through a variety of pathways. However, activated NMDAR may also lead to LMP, which is dependent on the activation of calpain (Yan et al., 2016). Activated calpain I can also cleave and inactivate Na+/Ca2+ exchangers in the plasma membrane of the nerve cells during neuroexcitatory toxicity, thereby leading to calcium overload and necrosis (Bano et al., 2005). Thus, calcium overload and neuronal death can be effectively inhibited by inhibiting calpain. In addition, the absence of calpastatin, a natural calpain inhibitor expressed in neurons, makes neurons more susceptible to excitotoxicity, and its overexpression inhibits neuronal death attributed to excitotoxicity (Descloux et al., 2015). Meanwhile, heat shock protein 70 (HSP70) also stabilizes lysosomes to resist LMP (Aits and Jäättelä, 2013). In conclusion, NMDAR can promote activation of calpain and LMP, while inhibition of calpain and LMP may be an effective method to reduce neuronal death caused by NMDAR-mediated excitotoxicity in epilepsy.
Mitochondrial Permeability Transition (MPT). Activated NMDARs can also result in MPT by Ca2+ influx, involved in neuronal death. MPT is characterized by a significant increase in the permeability of the inner mitochondrial membrane with increased Ca2+ concentration, which eventually leads to oxidative phosphorylation decoupling, depletion of cell energy, and necrotic cell death. MPT largely depends on the opening of mitochondrial permeability transition pore (mPTP; Fricker et al., 2018). However, increased Ca2+ concentration in the mitochondrial matrix is an important factor leading to the opening of mPTP. In addition, it is also closely associated with ROS and RNS, the decreasing of ATP, the decreasing of mitochondrial membrane potential, and intracellular acidification (Fricker et al., 2018). The opening of mPTP leads to the depolarization of the inner mitochondrial membrane, the reduction of ATP production and the increasing of ATP consumption, the rupture of the mitochondrial membrane and the release of cytochrome C and cytochrome G, eventually causing irreversible cellular respiratory arrest and cell death (El-Mir et al., 2008; Fricker et al., 2018). Activated NMDARs can induce MPT by increasing Ca2+, ROS, and RNS in neurons, while cyclosporine A can reduce neuronal death by blocking MPT (Schinder et al., 1996). Meanwhile, MPT is also inhibited by 3-methyladenine (3-MA), an inhibitor of autophagosome formation, which can inhibit kinases to regulate neuronal survival and death (Xue et al., 2002). Correlative experimental data showed that cyclosporine A had different protective effects on excitotoxicity induced hippocampal nerve cell death (Santos and Schauwecker, 2003). Thus, blocking the opening of mPTP by cyclosporine A or the gene knockout of cyclophilin D can partially prevent neuronal death caused by excitotoxicity. In addition, studies have shown that high levels of Ca2+, ROS, and low levels of ATP in the cytoplasm can promote MPT in the brain of epilepsy, and ketone bodies also mediate antiepileptic effects through MPT (Kim et al., 2015). These studies suggest that MPT may play an important role in the occurrence and treatment of epilepsy.
Parthanatos. Ca2+ enters the cytoplasm by activating NMDARs and directly activates nNOS which is significantly expressed in the cytoplasm of some GABAergic neurons of the hippocampus and cortex. Activated nNOS can catalyze the reaction of NO with O2−, thereby producing peroxynitrite (ONOO−), which interacts with DNA, lipids, and proteins through a direct oxidative stress response, leading to parthanatos or apoptosis (Figure 2; Conrad et al., 2016; Ivanova V. et al., 2020). Parthanatos is an important form of cell death, characterized by dependence on the overactivation of the nuclear protein PARP1 after DNA damage and ROS production (Virág et al., 2013).
Related studies have shown that glutamate is involved in inducing neuronal injury through the activation of PARP-1 and generation of poly-ADP-ribose (PAR) polymer, thereby participating in parthanatos (Andrabi et al., 2011). In brief, activated NMDARs can promote ONOO− production by activating nNOS, which damages DNA and activates PARP, eventually resulting in parthanatos.
As is well-known, PARP1 and PARP2 are involved in chromosomal stability, DNA repair, and inflammatory responses (Curtin and Szabo, 2013). In PARP-dependent death, activated PARP1 results in production and NAD+ depletion. The direct interaction between AIF and PAR promotes the nuclear translocation of AIF, which leads to chromatin degradation (Andrabi et al., 2008; Wang et al., 2011). Meanwhile, PAR is the product of PARP1 activity and also induces nuclear translocation of AIF by inhibiting hexokinase (HK; Wang et al., 2009). Overactivated PARP1 leads to NAD+ depletion that further disrupts cellular metabolic processes and promotes cell death (Alano et al., 2010). Nuclear translocation of AIF also requires the involvement of cyclophilin A (CypA), which binds to AIF after the release from mitochondria and forms CypA-AIF complex, thereby participating in DNA degradation under various cellular stress conditions, such as cerebral hypoxia-ischemia and traumatic brain injury (TBI; Zhu et al., 2007; Farina et al., 2017). Related studies have shown that inhibiting the formation of the CypA-AIF complex can reduce glutamate-induced HT22 hippocampal cell death (Doti et al., 2014). The release of AIF in mitochondria may also be associated with calpain, BH3-only protein Bid, and BNIP3 (Fricker et al., 2018). In addition, PAR levels are also regulated by the ADP-ribosyl-acceptor hydrolase 3 (ARH3), which reduces PAR levels in the nucleus and cytoplasm (Mashimo et al., 2013). Meanwhile, the protein IDUNA, also known as E3 ubiquitin protein ligase RNF146, binds to the PAR polymers, thereby reducing the release of AIF and preventing PARP1-induced cell death (Andrabi et al., 2011).
Glutamate acting on NMDAR induces neuronal injury through activation of PARP-1 and generation of PAR polymer (Andrabi et al., 2011). There is currently considerable evidence supporting the role of parthanatos in a variety of neurological disorders including epilepsy, stroke, PD, and TBI, and the inhibition of PARP-1 and PARP-2 can reduce nuclear translocation of AIF and increase neuroprotection (D’Orsi et al., 2016; Xu H. et al., 2019; Dionísio et al., 2021; Koehler et al., 2021). Related studies have found that the formation of the neuronal AIF-CypA complex is considered to be the main target for the recovery of ischemia-stroke injury (Farina et al., 2018). However, in the hippocampal neuronal culture (HNC) model of acute acquired epilepsy, activation of PARP-1 is thought to be a major cause of caspase-independent cell death (Wang S. et al., 2013). Meanwhile, PARP-1-mediated mitochondrial dysfunction promotes neuronal damage in the hippocampus after SE (Lai et al., 2017). In addition, inhibition of PARP-dependent cell death pathways has been shown to prevent seizure-induced neuronal damage (D’Orsi et al., 2016). In epilepsy, activated NMDAR may damage DNA and activate PARP. Thus, blocking PARP-dependent cell death pathways may be a way to mitigate NMDAR-mediated excitotoxicity in epilepsy.
Other Signaling Pathways in NMDAR-Mediated Excitotoxicity. In addition to the signaling pathways described above, NMDAR regulates nerve cell death through other pathways. Activated NMDAR can promote the NADPH oxidase (NOX) to produce O2− in neurons, leading to neuronal death. A related study has shown that seizures are induced by NMDAR-mediated activation of NOX-induced oxidative stress and can be arrested by NOX inhibition (Malkov et al., 2019). Meanwhile, activated NMDAR increases c-Jun abundance in several neurodegenerative disorders and following ischemia and SE. Phosphorylated c-Jun is transcriptionally active and can induce apoptosis by upregulation of cell death-inducing genes or by downregulating anti-apoptotic genes (Kravchick et al., 2016). In addition, NMDAR overactivation activates NF-κB signaling to promote IL-1β and IL-6 macrophage marker expression. NMDAR silencing and calpain inhibition reduce inflammatory responses (Cheng et al., 2020). It has been reported that glutamate can reduce the insulin-like growth factor-1 (IGF-1) signal through GluN2B-containing NMDAR in cultured cortical neurons, which is considered to be a new mechanism of glutamate-induced neurotoxicity (Zhao et al., 2020). In epilepsy, increased IGF-1 levels after recurrent hippocampal neuronal discharges might promote seizure by IGF-1R-dependent signaling pathways (Jiang et al., 2015). However, the previous role of the NMDAR-IGF-1 signal is unappreciated in the development of seizure activity.
Ca2+ influx could stimulate and induce cell death, but it is also in association with NMDAR-dependent neuroprotection (Wang et al., 2020b). Ca2+ entering the cytoplasm via synaptic NMDAR causes an increase of nuclear Ca2+ (Wang et al., 2020b). Nuclear Ca2+ is one of the most effective activators of neuronal gene expression, and nuclear Ca2+ can regulate about 200 neuronal genes in hippocampal neurons (Zhang et al., 2007, 2009). The CREB is a signal-regulating transcription factor that plays a critical role in neuronal survival, synaptic plasticity, neurogenesis, learning, and memory. Activated NMDAR results in translocation of CREB regulators from synapse to nucleus (Hardingham and Bading, 2010). In hippocampal neurons, CREB-dependent gene expression was associated with neuroprotection against apoptosis and excitatory damage, which depends on the nuclear Ca2+ signaling (Papadia et al., 2005). However, some studies have also shown that activated extrasynaptic NMDARs can promote the shutdown of CREB, thereby causing mitochondrial membrane potential loss and cell death. Meanwhile, activated synaptic NMDARs activate only the CREB pathway and do not activate apoptosis (Hardingham and Bading, 2010; Franchini et al., 2020).
Synaptic NMDAR activity can activate CREB-dependent gene expression by a variety of signal pathways (Figure 3). CREB phosphorylates at serine-133 in order to recruit its co-activator CREB binding protein (CBP). Phosphorylation of CREB is mediated by the fast-acting nuclear Ca2+/CaMK pathway and the slower acting, longer lasting Ras-ERK1/2 pathway, both of them are promoted by activation of synaptic NMDARs (Hardingham and Bading, 2010). Nuclear Ca2+-dependent CaMKIV/CaMKII phosphorylates CBP at serine-301. Meanwhile, CBP is also phosphorylated by the Ras-MEK-ERK1/2 pathway or the CaMKII/PKC/PKA-ERK1/2 pathway (Cortés-Mendoza et al., 2013; Lyu et al., 2020). Nuclear translocation of the transducer of regulated CREB (TORC) activity is a key step in CREB activation. Synaptic NMDAR-induced Ca2+ signals promote TORC import into the nucleus by calcineurin (CaN)-dependent dephosphorylation (Screaton et al., 2004; Kovács et al., 2007). TORC also acts at least in part by assisting in the recruitment of CBP to CREB. Meanwhile, CREB-regulated transcription coactivator 1(CRTC1) can also dephosphorylate at Ser-151 and is recruited from cytoplasm to the nucleus, where it competes with FXR (fed-state sensing nuclear receptor) for binding to CREB and drives autophagy gene expression (Pan et al., 2021). Some studies have shown that Ca2+ influx activates CREB through TRPC6, which is an important transcription factor linked to neuronal survival. Activated TRPC6 may inhibit neuronal NMDAR activity through the post-translational means to combat glutamate-induced excitotoxic damage (Shekhar et al., 2021). Finally, CREB can also be activated through the PI3K-AKT-GSK3β pathway and play a neuroprotective role in the hippocampus. GSK-3β deletion also inhibits the activity-dependent neural activation and Ca2+/CaMKIV/CaMKII-CREB signaling (Liu et al., 2017; Srivastava et al., 2018). Related studies have shown that the epileptogenesis of pilocarpine-induced medial temporal lobe epilepsy (MTLE) is associated with abnormal regulation of NMDAR-mediated excitatory neuronal mechanisms and neuronal activity regulated by Ca2+/CaMK signaling (Canto et al., 2021).
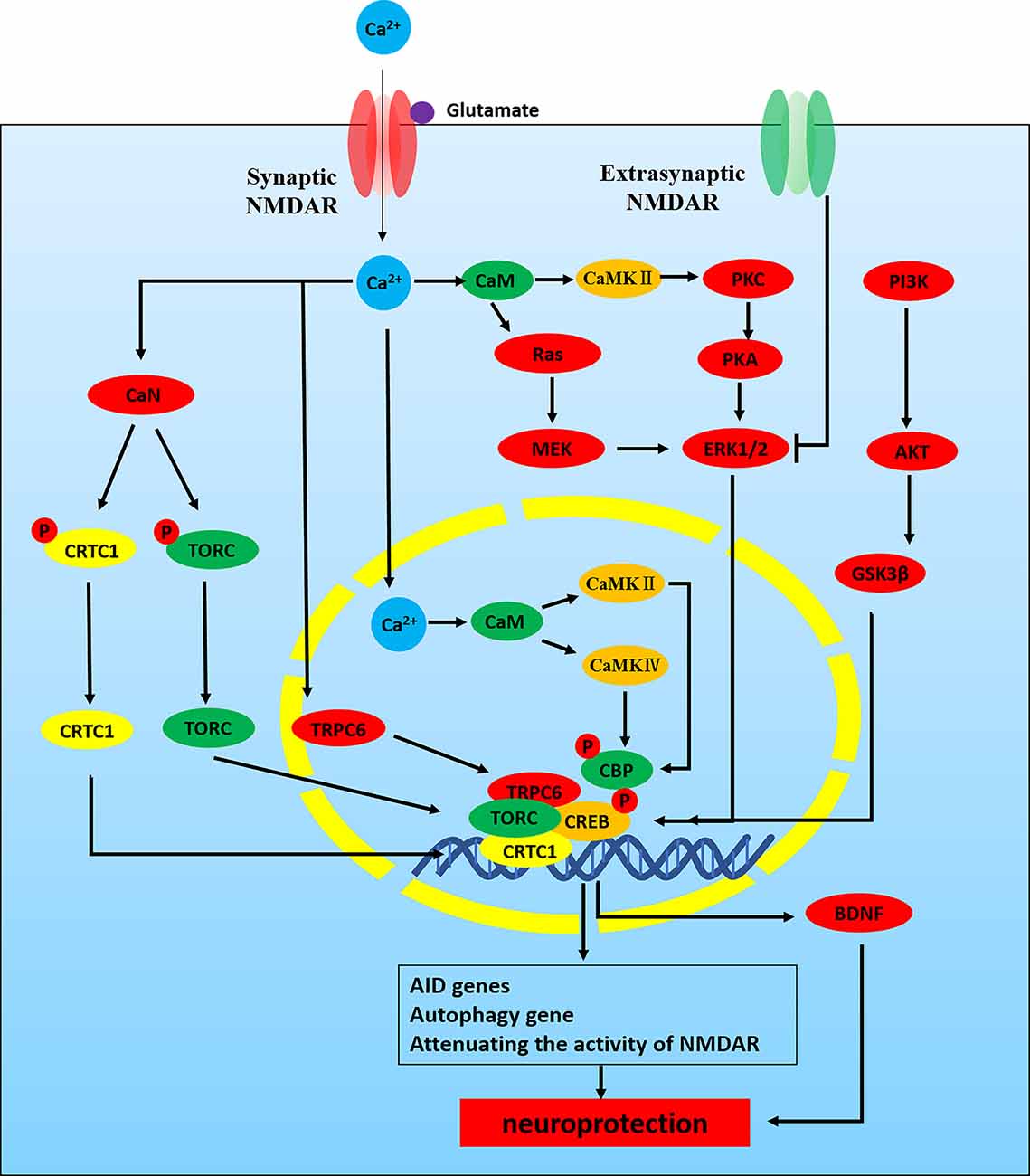
Figure 3. NMDAR -Ca2+-CREB signaling pathways in neuroprotection. NMDAR activity can activate CREB-dependent gene expression. CREB must be phosphorylated at serine-133 in order to recruit its co-activator CREB binding protein (CBP). Phosphorylation of CREB is mediated by the fast-acting nuclear Ca2+/CaMK pathway and the slower acting, longer lasting Ras-ERK1/2 pathway, both of which are promoted by activation of synaptic NMDARs. (1) Nuclear Ca2+-CaM-CaMKIV/CaMKII-CREB: nuclear Ca2+-dependent CaMKIV/CaMKII phosphorylates CBP at serine-301. (2) ERK1/2-CREB: CBP is also phosphorylated by Ras-MEK-ERK1/2 pathway or CaMKII/PKC/PKA-ERK1/2 pathway. CREB phosphorylated at serine-133 recruits its CBP. In addition, nuclear translocation of TORC activity is a key step in CREB activation. (3) Ca2+-TORC-CREB: synaptic NMDAR-induced Ca2+ signals promote TORC import into the nucleus by CaN-dependent dephosphorylation. TORC acts at least in part by assisting in the recruitment of CBP to CREB. (4) Ca2+-CRTC1-CREB: CRTC1 dephosphorylates at Ser-151 and is recruited from cytoplasm to the nucleus, where it competes with FXR for binding to CREB and drives autophagy gene expression. (5) Ca2+-TRPC6-CREB: Ca2+ influx through TRPC6 activates CREB, an important transcription factor linked to neuronal survival. (6) PI3K-AKT-GSK3β-CREB.
The gene of neurotrophin BDNF is regulated by nuclear Ca2+-CREB signaling (Favaron et al., 1993; Hardingham and Bading, 2010). NMDAR activation increases the release of BDNF, which protects neurons from damage caused by NMDAR blockade (Fabbrin et al., 2020; Lian et al., 2021). Some studies have shown that improving mitochondrial dynamics and increasing the activity of the NMDAR-CREB-BDNF pathway could ameliorate synaptic function and neuronal survival in SAMP8 mice (Lian et al., 2021). Synaptic NMDARs and extrasynaptic NMDARs have different physiological functions. Activated synaptic NMDARs lead to phosphorylation and activation of CREB, while activated extrasynaptic NMDARs inhibit CREB pathway (Hardingham and Bading, 2010). ERK1/2 pathway also promotes CREB activation and inactivates the pre-death protein BAD, which is associated with NMDAR-dependent neuroprotection (Hetman and Kharebava, 2006). In addition, nuclear factor I subtype A (NFIA), an NMDAR-dependent activation of other neuroprotective factors, may not be associated with the increase of nuclear Ca2+, but its activation depends on the ERK1/2 pathway and nNOS (Zheng et al., 2010).
The NMDAR-CREB-BDNF pathway plays an important role in inhibiting epileptic seizures (Yu et al., 2019; Sharma et al., 2021). Recent studies have indicated that CREB is involved in the etiology of epilepsy (Wang G. et al., 2020). In the KA-induced epilepsy model, CREB is considered to be one of the main upstream transcription factors regulating gene expression and is closely related to the severity of epilepsy (Conte et al., 2020). Meanwhile, recent studies have shown that CREB reduces oxidative neuronal damage in TLE associated with cognitive impairment (Xing et al., 2019). In addition, microRNA-204 also inhibits the epileptiform discharge of hippocampal neurons in vitro by regulating TrkB-ERK1/2-CREB signaling (Xiang et al., 2016). NMDAR mediates CREB-dependent gene expression, which is closely associated with neuroprotection against apoptosis and excitatory damage, so the study of the regulatory mechanism of the NMDAR-CREB pathway in epilepsy is conducive to the neuroprotection of the epileptic brain. However, the regulatory mechanism of the NMDAR-CREB pathway in epilepsy is not entirely clear and needs further exploration.
Although NMDAR affects the occurrence of epilepsy through a variety of mechanisms, it is also regulated by a variety of factors. Activation of the NMDAR is a cooperative process, which depends on: (1) the relief of Mg2+ block of the ion channel pore; (2) depolarization of the postsynaptic membrane; and (3) the agonist glutamate, and co-agonists (glycine, D-serine; Jorratt et al., 2021). In addition, the expression and function of NMDAR are also affected by the expression and transcription process of related NMDAR genes, microRNAs, related proteins, and signaling pathways. On this basis, we will discuss a variety of factors for the regulation of NMDARs in the occurrence, development, and treatment of epilepsy (Tables 1, 2).

Table 1. The regulation of NMDAR in epilepsy.
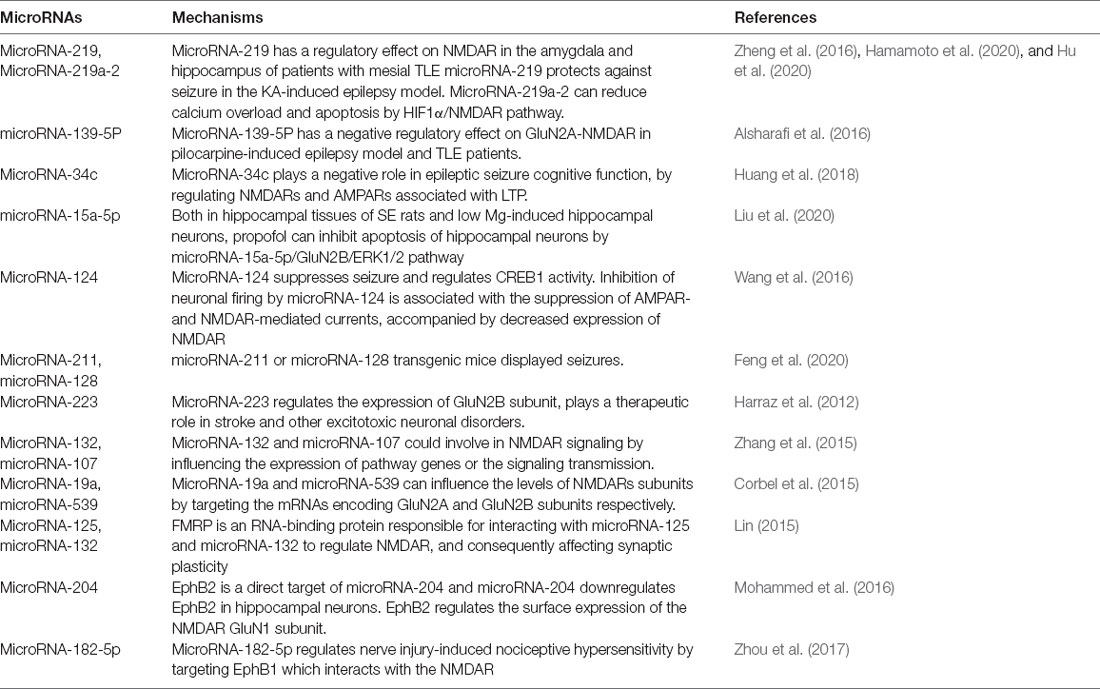
Table 2. Regulation of microRNAs on NMDARs in nervous system.
NMDAR antagonists play an important role in the treatment of epilepsy. NMDAR antagonists enhance the anticonvulsant effect of lithium chloride on PTZ-induced clonic seizures in mouse (Ghasemi et al., 2010). Animal studies have suggested NMDAR antagonists may become more effective with seizures lasting longer after the failure of the first line therapies (Sánchez Fernández et al., 2019). In addition, the latter epilepsy patients might respond to positive allosteric modulators of the NMDARs (Zhu and Paoletti, 2015).
Ketamine is a noncompetitive NMDAR antagonist and blocks Ca2+ influx by binding to phencyclidine-binding sites of NMDAR. Meanwhile, it is used for evidence of clinically RSE and SRSE (Borsato et al., 2020). Ketamine induces developmental neurotoxicity by inhibiting the expression of NMDAR and increasing the sensitivity of neurons to glutamate excitotoxicity, thereby leading to deregulation of Ca2+ signaling and triggering oxidative stress and even mitochondrial apoptosis pathways in neurons. Ketamine significantly upregulates the GluN1 subunit of NMDAR in the frontal cortex, thereby triggering neuronal apoptosis (Liu et al., 2011). Meanwhile, mitochondrial dysfunction and oxidative stress in the hippocampus of rats exposed to ketamine are associated with down-regulation of the ERK signaling cascade (Huang et al., 2012; Li et al., 2017). In addition, ketamine induces apoptosis through the mechanism associated with caspase-1-dependent pyroptosis in the hippocampus (Ye et al., 2018). Ketamine also aggravated cognitive impairment and hippocampal neurodegeneration through the ROS/HIF-1α pathway (Yan et al., 2014). However, ketamine inhibits lipopolysaccharide-mediated BV2 microglia inflammation by blocking NMDARs (Lu et al., 2020). It has been shown that ketamine inhibits the NOX2 activation to produce ROS of the mice brain in pilocarpine-induced epilepsy (Tannich et al., 2021). Ketamine-midazolam therapy can reduce the severity of seizures and improve brain pathology in plasma carboxylesterase knockout mice (Marrero-Rosado et al., 2020). A low dose of ketamine can reduce the behavioral changes in pilocarpine-induced epilepsy mice (Tannich et al., 2020).
In clinical studies, ketamine was used in the treatment of RSE and SRSE (Samanta, 2020). Ketamine treatment is associated with a decrease in seizure burden in patients with SRSE (Alkhachroum et al., 2020). Midazolam-ketamine-valproate therapy is significantly more effective than midazolam-fosphenytoin-valproate therapy in seizure reduction (Niquet et al., 2017). Meanwhile, the combination of ketamine-midazolam can reduce the severity of epilepsy, epileptogenesis, and neuropathology in cholinergic-induced SE (Lumley et al., 2021). And ketamine is also used in anti-NMDAR encephalitis-associated RSE (Santoro et al., 2019). Severe epileptic encephalopathy caused by GRIN2D mutations can be treated with NMDAR channel blockers (ketamine, magnesium; Li et al., 2016).
Different NMDAR subunit gene mutations also have different responses to NMDAR antagonists. it was reported that a 9-year-old boy with severe early-onset epileptic encephalopathy caused by a GRIN2A missense mutation was trialed on memantine and a significant reduction in seizure frequency was revealed (Pierson et al., 2014). Whereas, another study showed that no seizure reduction was found in patients with GRIN2B mutation-related encephalopathy treated by memantine despite improved consciousness, behavior, and sleep (Platzer et al., 2017). In addition, a combination of memantine and cathodal direct current stimulation (cDCS) suppressed KA-induced seizures (Sun et al., 2020). Thus, early identifying the location and type of NMDAR subunit gene mutation in epilepsy has guiding significance for the selection of AEDs.
The NMDAR ion channel pores are permeable to Ca2+ but can be blocked by Magnesium ion (Mg2+) in a strongly voltage-dependent manner, which makes them largely inactive at resting voltages, even in the presence of agonists (Mayer et al., 1984; Nikolaev et al., 2021). Activation of the NMDAR is a cooperative process, which depends on the relief of the Mg2+ block of the ion channel pore (Mg2+ is removed into the extracellular compartment from the channel pore; Hou et al., 2020; Jorratt et al., 2021). The NMDAR channel is blocked by Mg2+ at neuronal resting membrane potential, and Mg2+ is removed when the membrane is depolarized (Jorratt et al., 2021; Li et al., 2021). In addition, non-competitive NMDAR ion channel blockers such as MK-801 mainly bind to ion channels of TMD. Since Mg2+ normally blocks this channel, the binding of MK-801 requires NMDAR activation and depolarization to release Mg2+ (Wong et al., 2021). A relevant study has shown that Mg2+ influx, dependent on NMDAR opening, can transduce a signaling pathway to activate CREB in neurons (Hou et al., 2020). Magnesium sulfate is a neuroprotective agent in clinical practice. By noncompetitively blocking NMDARs, magnesium sulfate can inhibit glutamatergic signaling, thereby altering Ca2+ influx, leading to reduced excitotoxicity (Elsayed et al., 2021). In vitro, TLE cell model is often established by treating primary hippocampal cells with magnesium-free extracellular fluid. Transient culture of hippocampal neurons in magnesium-free induces rhythmic and synchronous epileptiform-like activity (Mele et al., 2021).
The low-affinity binding site of Mg2+ is located deep in the ion channel and is modulated by the NMDAR subunit. Related studies have shown that NMDAR complexes formed by GluN2A or GluN2B subunits have a higher affinity for Mg2+ than those containing GluN2C or GluN2D (Monyer et al., 1994). Due to different GluN2 subunits, NMDAR has different sensitivity to Mg2+ (Valdivielso et al., 2020). An important feature of the GluN2 subunits is that GluN2A and GluN2B subunits are more sensitive to voltage-dependent Mg2+ blocking than GluN2C and GluN2D subunits (Qian et al., 2005). Meanwhile, the GluN2C subunit contributes to a lower threshold for Mg2+ block and influences NMDAR agonist activity (Intson et al., 2020). Compared to GluN1/GluN2D receptors or other NMDAR subtypes, GluN1/GluN2C receptors exhibit higher blockade with ketamine in the presence of Mg2+(Shelkar et al., 2019). The human NMDAR GluN2A variant influences channel blocker potency. A novel genetic variant of GRIN2A has been identified in patients with epileptic encephalopathy altering residues located in the NMDAR ion channel pore and significantly reducing Mg2+ blockade and channel conductance (Marwick et al., 2019). In functional studies, the GRIN2A mutation decreased the potency of endogenous negative modulators, including magnesium and zinc (Fernández-Marmiesse et al., 2018). In addition, missense mutations of GRIN2B also alter NMDAR ligand binding and ion channel properties. GRIN2B mutants showed decreased glutamate potency, increased NMDAR desensitization, and disappearance of voltage-dependent Mg2+ block (Fedele et al., 2018). Meanwhile, Mg2+ deficiency down-regulated GluN2B subunits expression in cultured hippocampal slices (Zhou et al., 2021). In addition, presynaptic release and postsynaptic transporter transport zinc (Zn) to different microdomains to regulate NMDAR neurotransmission. Meanwhile, zinc inhibits synaptic NMDARs, which depend on the binding of GluN2A to zinc transporter ZnT1(Krall et al., 2020). In conclusion, Mg2+ plays an important role in the pathogenesis of epilepsy. The most prominent of these is voltage-dependent block of the NMDAR channel by Mg2+.
It is widely believed that the imbalance between excitatory and inhibitory neurotransmission leads to hyperexcitability of neuronal circuits, which is the basis of the process of epileptogenesis (Alcoreza et al., 2021). Glutamate is an excitatory neurotransmitter in the brain involved in various neural functions and metabolic processes of the CNS (Wang et al., 2020b). NMDAR is one of the excitatory receptors that glutamate acts on directly and may lead to diseases such as epilepsy, stroke, AD, and PD (Essiz et al., 2021). On the one hand, glutamate can be directly synthesized de novo by astrocytes in the brain. On the other hand, glutamate can also be produced indirectly with glucose molecules through the action of pyruvate dehydrogenase and pyruvate carboxylase in astrocytes (Schousboe et al., 2014). Meanwhile, extracellular glutamate can be transferred to astrocytes by excitatory amino acid transporter 2 (EAAT2) and then converted to glutamine by glutamine synthetase (GS). Glutamine is transported by astrocytic glutamine transporter-5 (SNAT-5) to the extracellular environment, where it can then be transferred to neurons by astrocytic glutamine transporter-1 (SNAT-1; Danbolt, 2001). In the pre-synaptic neurons, phosphate-activated glutaminase (PAG/GLS-1) converts inactive glutamine to glutamate, which is repackaged into synaptic vesicles and released into the synaptic cleft and directly acts on the NMDAR in the post-synaptic neurons, thus activating NMDAR (Limón et al., 2021). In a word, the glutamate and glutamine cycle in astrocytes and neurons is called the glutamate-glutamine cycle (Figure 4).
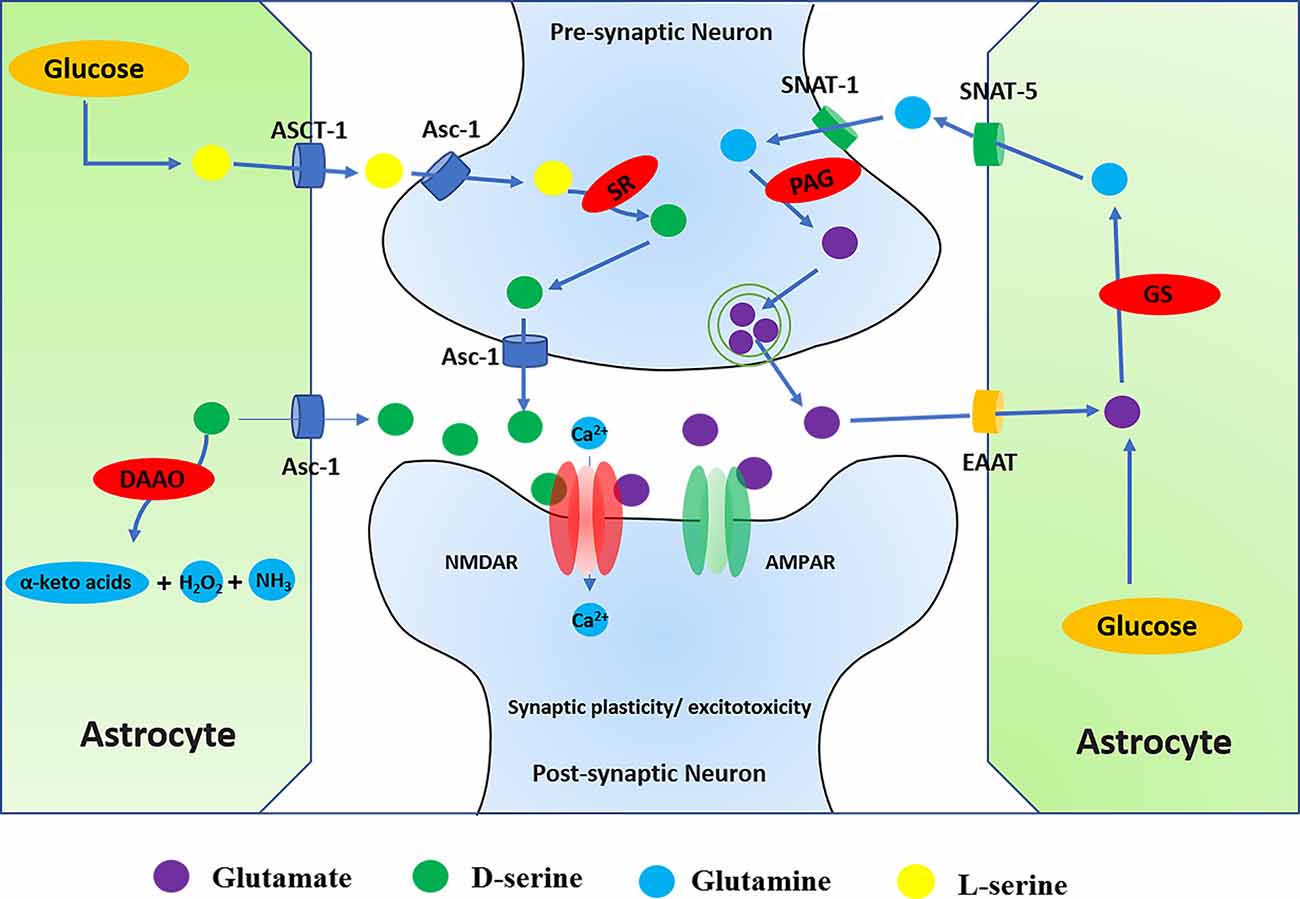
Figure 4. Regulation of NMDARs by D-serine and glutamate. (1) Glutamate can directly act on NMDAR, and the glutamate-glutamine cycle is involved in the regulation of NMDAR. Glutamate-glutamine cycle: glutamate can be directly synthesized de novo by astrocytes or indirectly produced from glucose molecules through the actions of pyruvate dehydrogenase and astrocyte-specific enzyme pyruvate carboxylase in the brain. Meanwhile, extracellular glutamate can be transferred to astrocytes by ETTA2 (GLT-1) and then converted to glutamine by glutamine synthetase (GS). Glutamine is transported by SNAT-5 to the extracellular environment, where it can then be transferred to neurons by SNAT-1. In the neuron, glutamine is degraded by PAG into glutamate and ammonia. Glutamate enters the synaptic vesicles in the pre-synaptic neurons and then is released from the pre-synaptic membrane into the synaptic cleave. It directly acts on the NMDAR in the post-synaptic neurons, thus activating NMDAR. (2) In addition to glutamate, activation of NMDAR also requires the binding of D-serine at the glycine binding site. SR converts L-serine to D-serine in the neuron, while DAAO catalyzes the breakdown of d-serine in the astrocyte. D-serine is released from neurons by Asc-1, which mediates D-serine efflux in exchange for external amino acid substrates. L-serine can be directly synthesized de novo in astrocytes. Through orchestrated Asc-1 and ASCT1 subtypes, L-serine from astrocytes enters the neuron and is catalyzed by SR to produce D-serine.
Astrocyte dysfunction can alter glutamate homeostasis, leading to neuroexcitatory toxicity (Niciu et al., 2012). Excessive glutamate can cause neuroexcitatory toxicity after being released into the synaptic cleft, and excess glutamate needs to be cleared quickly in the brain (Schousboe et al., 2014). However, due to the absence of extracellular enzymes, the uptake of extracellular glutamate mainly relies on EAATs which are located in the plasma membranes of neurons and glia (Zhang et al., 2016). Meanwhile, astrocytes can also completely enclose the glutamate synapse to quickly clear glutamate from the synaptic cleft (Alcoreza et al., 2021). Once glutamate enters the astrocyte, it is converted to glutamine and returned to the neuron by the glutamate-glutamine cycle (Alcoreza et al., 2021). In addition, over-activated astrocytic NMDARs could lead to the release of many other molecules that are likely to be relevant. Astrocyte GluN2A regulates nerve growth factor β (β-NGF) synthesis, maturation, and secretion by regulating pNF-κB, Furin, and VAMP3.
It is found that the neuroprotective role of astrocytic GluN2A in the promotion of synapse survival is by regulating these molecules (Du et al., 2021). Both glutamate and quinolinic acid (QUIN) could activate astrocytic NMDARs, which stimulate Ca2+ influx into the cell and can result in dysfunction and death of astrocytes (Lee et al., 2010).
In addition to glutamate, the activation of NMDAR also requires the binding of a co-agonist at the glycine binding site. Originally similar to glycine, D-serine can also control the activation of NMDAR by binding to the receptor’s glycine binding site in the brain (Figure 4; Mothet et al., 2015). Recent studies have indicated that the expression of D-serine and NMDAR is closely related to intractable epilepsy (Zhu and Paoletti, 2015). The review investigated the regulation of NMDAR by D-serine in CNS diseases, including epilepsy.
D-serine is the main endogenous co-agonist of NMDARs, which is significantly dependent on the activity of the metabolic enzyme d-amino acid oxidase (DAAO) and serine racemase (SR). In the brain, DAAO catalyzes the decomposition of D-serine, while the cytoplasmic enzyme SR converts L-serine to D-serine (Takagi et al., 2021). SR is mainly found in neurons, that catalyzes the reversible racemization of L-serine and D-serine (Raboni et al., 2019). On the one hand, D-serine is released from the neuron via a plasma membrane transporter (ASC-1/SLC7A10), which mediates D-serine efflux. On the other hand, astrocytes synthesize SR substrate L-serine, which is transferred to neurons through a mechanism of serine shuttle to participate in neuronal D-serine synthesis. L-serine synthesized by astrocytes is dependent on the activity of 3-phosphoglycerate dehydrogenase (PHGDH), which is the key to de novo synthesis of L-serine and activation of NMDAR (Neame et al., 2019). Clinically, serine deficiency patients present with severe neurological symptoms, including intractable epilepsy, which suggests the relevance of serine to brain development and morphogenesis (Murtas et al., 2020). Although the activity of SR and DAAO is important for the regulation of D-serine levels, the regulatory mechanisms of SR and DAAO are not fully understood in epilepsy.
DAAO selectively catalyzes the oxidative deamination of natural D-serine to produce imino acid, which is naturally hydrolyzed to the corresponding α-keto acids and ammonia (Pollegioni et al., 2018). Related research has confirmed that inhibition of DAAO can lead to increased D-serine in the brain, thereby regulating a variety of neurophysiological functions including cognitive behavior (Nagy et al., 2020). Meanwhile, it was also found that NMDAR antagonists (MK801 and cocaine) could increase the release of glutamate and decrease the expression of SR and DAAO. However, D-serine and antipsychotics did not modulate the levels of SR and DAAO (Takagi et al., 2021). In addition, the neuroprotective effect of DAAO is also mediated by the ERK1/2 signal pathway (Zhang X. et al., 2020).
In the brain, SR is mainly in excitatory neurons and GABAergic inhibitory interneurons, and is only weakly expressed in astrocytes (Billard, 2018). Through the coordinated activities of ASC-1 and ASCT1 subtypes, D-serine is released and binds with NMDAR to perform neurophysiological functions (Sason et al., 2017; Billard, 2018; Kaplan et al., 2018). The deletion of SR affects the balance of excitatory and inhibitory in the hippocampal CA1 network (Ploux et al., 2020). In addition, related research has found that PKC phosphorylates SR on serine residues and reduces the activity of SR in vitro. Similarly, activated PKC also increases SR phosphorylation and decreases the levels of D-serine in the rat frontal cortex (Vargas-Lopes et al., 2011). Therefore, PKC-mediated SR phosphorylation may be important for the activation of NMDARs.
In fact, the specific degradation of D-serine by the enzyme DAAO and the genetic deletion of SR significantly altered the activation of NMDAR. D-serine plays a key role in regulating the functional plasticity of many synapses in the brain (Billard, 2018). Related studies have found that the beneficial effects of D-serine supplementation may reflect that D-serine levels decreased significantly with age, as supported in the hippocampal trial. Interestingly, this decline of D-serine is also found in human plasma levels (Potier et al., 2010). In addition, in addition to affecting synaptic plasticity and synaptogenesis, dysregulation of D-serine metabolism may also enhance NMDAR-dependent excitotoxicity and promote cognitive impairment and neurodegeneration (Beltrán-Castillo et al., 2018). Therefore, the synaptic availability of D-serine and the preservation of SR activity are critical for maintaining strong cognitive abilities in the brain.
D-serine and NMDAR were found to be significantly upregulated in patients with intractable epilepsy (Zhang et al., 2021). Therefore, the D-serine signal pathway may be a potential target for epilepsy therapy. Endogenous D-serine deficiency may lead to decreased inhibition of the hippocampal CA1 network and altered excitatory/inhibitory balance. besides, D-serine contributes to maintaining cognitive abilities and functional plasticity of synapses (Ploux et al., 2020). Related studies have shown that intracranial injection of D-serine into the medial entorhinal area (MEA) in the TLE is beneficial to prevent neuronal loss and epileptogenesis by rescuing hippocampal CA1 neurons in the epileptic brain and reducing the number of astrocytes and microglia, thus alleviating the effect of neuroinflammation (Beesley et al., 2021). Therefore, D-serine might be a potential therapy target via regulating NMDAR in epilepsy, and more studies are needed in the future.
DNA methylation is a crucial epigenetic mark for activity-dependent gene expression in neurons. It has been shown that the levels of DNA methyltransferase 3A1 (DNMT3A1) in neurons are closely controlled by the activation of NMDAR-containing GluN2A subunits (Bayraktar et al., 2020). Interestingly, synaptic NMDARs drive methyltransferase degradation in a ubiquitin-like dependent manner. The binding of NEDD8 ubiquitin-like protein to lysine residues inhibits ubiquitination, thereby blocking DNMT3A1 degradation (Bayraktar et al., 2020). Defects in promoter methylation of these activity-dependent genes may be related to synaptic plasticity and memory formation (Bayraktar et al., 2020). Overall, the activity-dependent DNA methylation is regulated by GluN2A-containing NMDAR signals to participate in memory formation. Meanwhile, NO produced by NMDAR activation can also up-regulated DNA methyltransferase 3B (DNMT3B) in the hippocampus (de Sousa Maciel et al., 2020). A related analysis showed that both GluN2B expression levels and histone H3K9 acetylation of GRIN2B gene promoter were positively correlated with ethanol withdrawal syndrome (EWS; Li et al., 2019). In addition, the changes of GRIN2B promoter methylation may be associated with cognition reduction and glutamatergic dysfunction in schizophrenia (Fachim et al., 2019). Meanwhile, this epigenetic change leads to upregulation of functional NMDAR and abnormal neuronal differentiation (D’Aiuto et al., 2011). In addition, NMDAR expression is also regulated by histone methylation in the brain. Histone methyltransferases targeting histone H3-lysine 9 residues, including Setdb1 (Set domain, bifurcated1)/Eset/Kmt1e, are closely related to inhibition of chromatin remodeling in the adult brain. Meanwhile, the inhibition of Setdb1-mediated histone methylation at GRIN2B is associated with decreased expression of GluN2B (Jiang et al., 2010).
In epilepsy, control of epigenetic of epilepsy target genes contributes to cellular memory of epileptogenesis in cultured rat hippocampal neurons (Kiese et al., 2017). In cultured neurons, the altered gene expression and epigenetic modifications can be rescued by blocking action potential propagation or inhibiting glutamatergic activation (Kiese et al., 2017). The epigenetic modification of epileptic target genes and cellular memory of epileptogenesis, which can transform normal neurons and circuits into pro-epileptic neurons and neural circuits (Kiese et al., 2017). SE can also lead to abnormal expression of GRIN2B and BDNF genes in the hippocampus in TLE (Ryley Parrish et al., 2013). In the epileptic hippocampus, GRIN2B DNA methylation levels were increased and BDNF DNA methylation levels were decreased, which led to decreased mRNA and protein expression of GluN2B and increased mRNA and protein expression of BDNF. Meanwhile, suppressive DNMT can increase excitatory postsynaptic potential in hippocampal slices of epileptic rats (Ryley Parrish et al., 2013). Therefore, GluN2B DNA methylation may be an early SE-induced event that persists into late epilepsy in the hippocampus and promotes changes of gene expression in TLE. In addition, leukemia-related AF9/MLLT3 mutations are involved in neurodevelopmental disorders such as epilepsy and ataxia (Büttner et al., 2010). Meanwhile, AF9 is found to be an active epigenetic modifier by increasing the expression of GluN1 subunits during the generation of cortical projection neurons (Büttner et al., 2010).
In conclusion, the epigenetic regulation of NMDAR and epilepsy target genes by NMDAR play an important role in the onset and development of seizures. Whereas, the mechanism remains unclear and more studies are urgently needed.
In recent years, researchers have found a link between microRNAs and NMDARs-mediated neurological diseases (Table 2; Alsharafi et al., 2017). A growing body of evidence indicates that microRNAs regulate synaptic homeostasis and plasticity processes, suggesting that they may be involved in synaptic dysfunction during epilepsy, stroke, and AD (Alsharafi et al., 2017). MicroRNA-34a deficiency promotes cognitive function by regulating AMPARs and NMDARs to increase synaptic plasticity (Xu et al., 2018). MicroRNA-219-5p alleviates morphine tolerance by inhibiting the CaMKII/NMDAR pathway (Wang J. et al., 2017). Meanwhile, microRNA-219a-2 has been reported to reduce calcium overload and apoptosis through HIF1α/NMDAR pathway, thus alleviating myocardial ischemia-reperfusion injury (Hu et al., 2020). MicroRNA-182-5p regulates nerve injury-induced nociceptive hypersensitivity by targeting Ephrin type-b receptor 1 (EphB1) which interacts with the NMDAR (Zhou et al., 2017). EphB2 is a direct target of microRNA-204 and microRNA-204 downregulates EphB2 in hippocampal neurons. EphB2 is a known regulator of synaptic plasticity and regulates the surface expression of the NMDAR GluN1 subunit (Mohammed et al., 2016). Thus, microRNA-204 may play an important role in anti-NMDAR encephalitis by regulating EphB2-NMDAR, which remains to be explored. In normal neuronal development, FMRP is an RNA-binding protein responsible for interacting with microRNA-125 and microRNA-132 to regulate NMDAR and consequently affecting synaptic plasticity (Lin, 2015). MicroRNA-19a and microRNA-539 can influence the levels of NMDARs subunits by targeting the mRNAs encoding GluN2A and GluN2B subunits respectively (Corbel et al., 2015). MicroRNA-219, microRNA-132, and microRNA-107 could be involved in NMDAR signaling by influencing the expression of pathway genes or the signaling transmission (Zhang et al., 2015). MicroRNA-223 as a major regulator of the expression of the GluN2B subunit, plays a therapeutic role in stroke and other excitotoxic neuronal disorders (Harraz et al., 2012). These microRNAs provide an entry point for affecting neural plasticity and abnormal nerve firing and provide a new approach for the treatment of NMDAR-related neurological diseases.
In epilepsy, some microRNAs (microRNA-34, microRNA-124, microRNA-146a, microRNA-135a, microRNA-23a, microRNA-132, microRNA-234-5p, microRNA-203, microRNA-181b, microRNA-155 microRNA-219, microRNA-211, microRNA-128, microRNA-23) have been reported and each microRNA has limitations as a potential epilepsy target (Feng et al., 2020). Importantly, microRNA-211 or microRNA-128 transgenic mice displayed seizures (Feng et al., 2020). However, some microRNAs play an important role in epilepsy by regulating NMDARs. MicroRNA-219 had a regulatory effect on NMDAR in the amygdala and hippocampus of patients with mesial TLE and microRNA-219 protects against seizure in the KA-induced epilepsy model (Zheng et al., 2016; Hamamoto et al., 2020). Meanwhile, microRNA-139-5P has a negative regulatory effect on GluN2A-NMDAR in pilocarpine-induced epileptic rat models and TLE patients (Alsharafi et al., 2016). MicroRNA-34c has also been found to play a negative role in seizure and cognitive function, possibly by regulating NMDARs and AMPARs associated with LTP (Huang et al., 2018). Both in hippocampal tissues of SE rats and low Mg-induced hippocampal neurons, propofol can inhibit apoptosis of hippocampal neurons by microRNA-15a-5p/GluN2B/ERK1/2 pathway, which provides theoretical support for propofol treatment of SE (Liu et al., 2020). MicroRNA-124 suppresses seizure and regulates CREB1 activity. Inhibition of neuronal firing by microRNA-124 is associated with the suppression of mEPSC, AMPAR- and NMDAR-mediated currents, which are accompanied by decreased surface expression of NMDAR (Wang et al., 2016). However, many microRNAs have not been confirmed to function in epilepsy by regulating NMDAR. The discovery of various microRNA is also beneficial for the treatment of epilepsy and reducing the occurrence of epilepsy.
The interaction between NMDAR and EphB2 was found in anti-NMDAR encephalitis (Hughes et al., 2010; Mikasova et al., 2012). It is reported that transcranial direct current stimulation promotes hippocampal neurogenesis in mice with cerebral ischemia by activation of Ephrinb1/EphB2/MAP-2/NMDAR pathway (Ma et al., 2021). Meanwhile, activated EphB receptors promote the excitability of primary sensory neurons either directly through Ca2+ influx or by phosphorylation of Src kinase-mediated NMDAR (Washburn et al., 2020). In the acute phase of ischemic stroke, EphB2-dependent signal pathways are found to promote neuronal NMDAR-induced excitotoxicity and inflammation (Ernst et al., 2019). In AD models, overexpression of EphB2 in hippocampal neurons improved impaired NMDAR and cognitive dysfunction (Hu et al., 2017). In addition, EphB2 has a positive protective effect on Aβ1-42 oligomer-induced neurotoxicity by synaptic NMDAR signal pathway in hippocampal neurons (Geng et al., 2013). EphB2 can also prevent the effects of NMDAR antibodies on memory and neuroplasticity (Planagumà et al., 2016). EphB2 regulates NMDAR function and synaptic targeting. In mature neurons, EphB2 regulates the number of synaptic NMDAR, while activated EphB2 reduces desensitization of Ca2+-dependent NMDAR and is required for enhanced synaptic localization of GluN2B-containing NMDAR. EphB knockout mice showed the homeostatic upregulation of NMDAR expression (Nolt et al., 2011). Synaptic plasticity is regulated by the EphB2-GluN2A-AKT cascade, which might be a potential pathogenesis of depression and potential therapeutic target of glutamatergic transmission dysfunction (Wu et al., 2019). Meanwhile, EphrinB/EphB signaling is conducive to synaptic plasticity by GluN2B phosphorylation in chronic migraine (Wang et al., 2020a). In dentate granular neurons of EphB2-deficient mouse, synaptic NMDAR-mediated current was reduced (Henderson et al., 2001). These findings suggest that EphB is a key regulator of NMDAR synaptic localization and NMDAR-dependent synaptic function in the CNS. Together, the regulation of synaptic function may be closely related to EphB2-NMDAR interaction in epilepsy.
In addition to the above regulation mechanisms, NMDAR is also modulated by other pathways. Related studies have demonstrated that both purinergic P2X receptors (P2X2) and P2X4 interact with NMDAR in an inhibitory manner (Rodriguez et al., 2020). Meanwhile, SULT4A1 promotes the formation of the PSD-95/NMDAR complex to modulate synaptic development and function (Culotta et al., 2020). It is also found that S-PrP interacts with LRP1/NMDAR system to activate ERK1/2, thereby promoting cell migration in Schwann cells (Mantuano et al., 2020). Neuronal surface P antigen (NSPA) regulates the postsynaptic stability of NMDAR by ubiquitination of tyrosine phosphatase PTPMEG (Espinoza et al., 2020). In addition, Cyclin B/CDK1 mediates NMDAR phosphorylation and regulates calcium kinetics and mitosis (Rosendo-Pineda et al., 2020). Neuroinflammation modulation is known to be controlled by cholinergic signals (Mizrachi et al., 2021). However, ACh potentiates NMDARs through muscarinic receptors in CA1 neurons of the hippocampus (Markram and Segal, 1990). Nicotinic α7-nAChR is enriched in the glutamate network synapses in the dorsolateral PFC (dlPFC) and is required for NMDAR action (Yang et al., 2013). LTP can be induced by exposure to the cholinergic receptor agonist carbachol in the hippocampus, which depends on NMDAR activation (Flores-Hernandez et al., 2009).
In addition, NMDAR is also regulated by ERK signaling pathway. In the spinal cord, CCL2 rapidly enhances NMDAR-induced neuronal electrical currents through the ERK-Glun2B pathway, thereby promoting pain sensitivity (Zhang H. et al., 2020). Related studies have found that CXCR7 can control the synaptic activity of hippocampal granular cells to regulate seizures. CXCR7 regulates GluN2A expression on the cell membrane by activating ERK1/2, thereby selectively modulating NMDAR-mediated synaptic neurotransmission in hippocampal granular cells (Xu T. et al., 2019). Therefore, CXCR7 may regulate seizures and become a new target for antiepileptic therapy by regulating the cell membrane expression of NMDAR. Some studies find that icaritin (ICT) has a neuroprotective effect on glutamate-induced neuronal damage and its mechanism may be associated with inactivating GluN2B-containing NMDAR by ERK/DAPK1 pathway (Liu et al., 2021). Meanwhile, DAPK1 interacts with NMDAR involved in glutamate-induced neurological events during sudden physiopathologic conditions in the brain (DeGregorio-Rocasolano et al., 2020). Inhibition of DAPK1 results in the phosphorylation and surface normalization of GluN2B expression outside the synapse (Schmidt et al., 2020).
Redox modulation of cysteine residues is one of the post-translational modifications of NMDAR. HCY accumulation in the human plasma, known as hyperhomocysteinemia, can exacerbate neurodegenerative diseases and act as a persistent NMDAR agonist (Ivanova M. et al., 2020). Meanwhile, HCY activates GluN2 subunit-dependent redox regulation of NMDAR by the reduction of NMDAR disulfide (Sibarov et al., 2020). The protein disulfide isomerase (PDI) binds to NMDAR in chronic epileptic rats and increases the mercaptan content on recombinant GluN1 protein (Kim et al., 2017). In fact, PDI plays a crucial role in catalyzing disulfide bond formation, reduction, and isomerization (Kim et al., 2017). Besides, H2S blocks the enhancement of neuronal excitability in the early hippocampal network by inhibiting voltage-gated sodium channels and NMDARs (Yakovlev et al., 2017). Thus, redox regulation of NMDAR may affect the occurrence and development of epilepsy and provide a new way for reducing the occurrence of epilepsy.
In epilepsy, some proteins and organisms can also affect NMDAR activity. As shown in the treatment of epilepsy, the inhibitory effect of β-hydroxybutyrate and acetone on NMDARs may underlie the therapeutic effects of the ketogenic diet in epilepsy (Pflanz et al., 2019). The interaction between the PCDH7 and the GluN1 subunit regulates the dendritic spine morphology and synaptic function, and it is associated with several CNS diseases including epilepsy (Wang Y. et al., 2020). In acute and chronic epileptic models, SPDI knockdown can inhibit seizure activity by nitrosylation-independent thiolation on NMDAR (Jeon and Kim, 2018). Inhibition of Nwd1 activity can reduce the hyperexcitability and phosphorylation of GluN2B in hippocampal neurons (Yang et al., 2019). Meanwhile, TMEM25 can also modulate the degradation of the GluN2B subunit and neuronal excitability (Zhang et al., 2019). Inhibition of acid-sensing ion channel 3 can regulate NMDAR function, thereby aggravating seizure severity (Cao et al., 2018). POSH could be a potential therapeutic target for epilepsy via increasing surface expression of NMDAR (Wang X. et al., 2017). Previous studies have shown that neuregulin1(NRG1)-ErbB4 signaling pathway may regulate the excitability of neurons and participate in primary epilepsy. NRG1-ErbB4 signaling can inhibit the phosphorylation of GluN2B, which has been detected in symptomatic human epileptic tissue (Zhu et al., 2017). In addition, the extracellular matrix protein SPARCL-1 also directly promotes synapse formation and NMDAR recruitment. In addition, SPARCL-1 might directly increase branches of dendrites, augment the numbers of synapse, and induce the formation of NMDARs, thereby increasing synaptic connectivity and reducing the risk for neurodegenerative disease (Chen et al., 2020b; Gan and Südhof, 2020). Leptin resists glutamate-induced excitotoxicity in HT22 hippocampal neurons and leptin also increases postsynaptic NMDAR currents to sensitize the nucleus of the solitary tract (NTS) neurons to vagal input (Jin et al., 2018; Neyens et al., 2020).
In this review, we reviewed and elucidated the regulatory mechanisms of NMDAR and its role in the onset, development, and treatment of epilepsy. Increasing evidence suggests that NMDAR is closely related to epilepsy and the autoimmune encephalopathy. Synaptic NMDARs mainly mediate pro-survival and synaptic plasticity pathways, whereas extrasynaptic NMDARs are mostly responsible for glutamate-induced excitotoxicity. Meanwhile, different NMDAR subunit also has different physiological functions in epilepsy. Studying the role of various NMDAR subunits in epilepsy may be beneficial to understand epileptogenesis. At present, there are many ways to regulate NMDAR, but the regulatory mechanism of NMDAR in the onset and development of epilepsy is not fully understood. Therefore, targeting upstream and downstream signal pathways of NMDAR may be a new approach to inhibit seizures and slow the progression of epilepsy. This type of treatment is yet to be discovered and explored.
SC participated in experimental studies. SC and LF searched and sorted out the references and participated in drafting the manuscript. DX and ML coordinated and supervised the work, provided research direction, designed research plans, and modified the final drafts. All authors have carefully read and confirmed the final manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Natural Science Foundation of China (No. 82171315).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank for the support of Department of Neurology, Wuhan Union hospital.
Aits, S., and Jäättelä, M. (2013). Lysosomal cell death at a glance. J. Cell Sci. 126, 1905–1912. doi: 10.1242/jcs.091181
Alano, C., Garnier, P., Ying, W., Higashi, Y., Kauppinen, T., and Swanson, R. (2010). NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 30, 2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010
Albrecht, J., and Zielińska, M. (2017). Mechanisms of excessive extracellular glutamate accumulation in temporal lobe epilepsy. Neurochem. Res. 42, 1724–1734. doi: 10.1007/s11064-016-2105-8
Alcoreza, O., Patel, D., Tewari, B., and Sontheimer, H. (2021). Dysregulation of ambient glutamate and glutamate receptors in epilepsy: an astrocytic perspective. Front. Neurol. 12:652159. doi: 10.3389/fneur.2021.652159
Alkhachroum, A., Der-Nigoghossian, C., Mathews, E., Massad, N., Letchinger, R., Doyle, K., et al. (2020). Ketamine to treat super-refractory status epilepticus. Neurology 95, e2286–e2294. doi: 10.1212/WNL.0000000000010611
Alsharafi, W., Luo, Z., Long, X., Xie, Y., and Xiao, B. (2017). MicroRNA in glutamate receptor-dependent neurological diseases. Clin. Sci. (London) 131, 1591–1604. doi: 10.1042/CS20170964
Alsharafi, W., Xiao, B., and Li, J. (2016). MicroRNA-139–5p negatively regulates NR2A-containing NMDA receptor in the rat pilocarpine model and patients with temporal lobe epilepsy. Epilepsia 57, 1931–1940. doi: 10.1111/epi.13568
Andrabi, S., Dawson, T., and Dawson, V. (2008). Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N Y Acad. Sci. 1147, 233–241. doi: 10.1196/annals.1427.014
Andrabi, S., Kang, H., Haince, J., Lee, Y., Zhang, J., Chi, Z., et al. (2011). Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat. Med. 17, 692–699. doi: 10.1038/nm.2387
Bano, D., Young, K., Guerin, C., Lefeuvre, R., Rothwell, N., Naldini, L., et al. (2005). Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120, 275–285. doi: 10.1016/j.cell.2004.11.049
Bayraktar, G., Yuanxiang, P., Confettura, A., Gomes, G., Raza, S., Stork, O., et al. (2020). Synaptic control of DNA methylation involves activity-dependent degradation of DNMT3A1 in the nucleus. Neuropsychopharmacology 45, 2120–2130. doi: 10.1038/s41386-020-0780-2
Beesley, S., Sullenberger, T., Ailani, R., D’Orio, C., Crockett, M., and Kumar, S. (2021). d-Serine intervention in the medial entorhinal area alters TLE-related pathology in CA1 hippocampus via the temporoammonic pathway. Neuroscience 453, 168–186. doi: 10.1016/j.neuroscience.2020.10.025
Beesley, S., Sullenberger, T., and Kumar, S. (2020). The GluN3 subunit regulates ion selectivity within native N-methyl-d-aspartate receptors. IBRO Rep. 9, 147–156. doi: 10.1016/j.ibror.2020.07.009
Beltrán-Castillo, S., Eugenín, J., and von Bernhardi, R. (2018). Impact of aging in microglia-mediated D-serine balance in the CNS. Mediators Inflamm. 2018:7219732. doi: 10.1155/2018/7219732
Bertocchi, I., Eltokhi, A., Rozov, A., Chi, V., Jensen, V., Bus, T., et al. (2021). Voltage-independent GluN2A-type NMDA receptor Ca signaling promotes audiogenic seizures, attentional and cognitive deficits in mice. Commun. Biol. 4:59. doi: 10.1038/s42003-020-01538-4
Billard, J. (2018). Changes in serine racemase-dependent modulation of NMDA receptor: impact on physiological and pathological brain aging. Front. Mol. Biosci. 5:106. doi: 10.3389/fmolb.2018.00106
Bonansco, C., and Fuenzalida, M. (2016). Plasticity of hippocampal excitatory-inhibitory balance: missing the synaptic control in the epileptic brain. Neural Plast. 2016:8607038. doi: 10.1155/2016/8607038
Borsato, G., Siegel, J., Rose, M., Ojard, M., Feyissa, A., Quinones-Hinojosa, A., et al. (2020). Ketamine in seizure management and future pharmacogenomic considerations. Pharmacogenomics J. 20, 351–354. doi: 10.1038/s41397-019-0120-2
Büttner, N., Johnsen, S., Kügler, S., and Vogel, T. (2010). Af9/Mllt3 interferes with Tbr1 expression through epigenetic modification of histone H3K79 during development of the cerebral cortex. Proc. Natl. Acad. Sci. U S A 107, 7042–7047. doi: 10.1055/a-1651-7450
Camp, C., and Yuan, H. (2020). GRIN2D/GluN2D NMDA receptor: unique features and its contribution to pediatric developmental and epileptic encephalopathy. Eur. J. Paediatr. Neurol. 24, 89–99. doi: 10.1016/j.ejpn.2019.12.007
Canto, A., Matos, A., Godoi, A., Vieira, A., Aoyama, B., Rocha, C., et al. (2021). Multi-omics analysis suggests enhanced epileptogenesis in the cornu ammonis 3 of the pilocarpine model of mesial temporal lobe epilepsy. Hippocampus 31, 122–139. doi: 10.1002/hipo.23268
Cao, Q., Xiao, Z., Wang, X., Weng, C., Ding, M., Zhu, F., et al. (2018). Inhibition of acid sensing ion channel 3 aggravates seizures by regulating NMDAR function. Neurochem. Res. 43, 1227–1241. doi: 10.1007/s11064-018-2540-9
Celli, R., and Fornai, F. (2020). Targeting ionotropic glutamate receptors in the treatment of epilepsy. Curr. Neuropharmacol. 19, 747–765. doi: 10.2174/1570159X18666200831154658
Chen, S., Chen, Y., Zhang, Y., Kuang, X., Liu, Y., Guo, M., et al. (2020a). Iron Metabolism and ferroptosis in epilepsy. Front. Neurosci. 14:601193. doi: 10.3389/fnins.2020.601193
Chen, W., Tankovic, A., Burger, P., Kusumoto, H., Traynelis, S., and Yuan, H. (2017). GRIN2AFunctional evaluation of a de novo mutation identified in a patient with profound global developmental delay and refractory epilepsy. Mol. Pharmacol. 91, 317–330. doi: 10.1124/mol.116.106781
Chen, S., Zou, Q., Chen, Y., Kuang, X., Wu, W., Guo, M., et al. (2020b). Regulation of SPARC family proteins in disorders of the central nervous system. Brain Res. Bull. 163, 178–189. doi: 10.1016/j.brainresbull.2020.05.005
Cheng, H., Cheng, Q., Bao, X., Luo, Y., Zhou, Y., Li, Y., et al. (2020). Over-activation of NMDA receptors promotes ABCA1 degradation and foam cell formation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1865:158778. doi: 10.1016/j.bbalip.2020.158778
Conrad, M., Angeli, J., Vandenabeele, P., and Stockwell, B. (2016). Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 15, 348–366. doi: 10.1038/nrd.2015.6
Conte, G., Parras, A., Alves, M., Ollà, I., De Diego-Garcia, L., Beamer, E., et al. (2020). High concordance between hippocampal transcriptome of the mouse intra-amygdala kainic acid model and human temporal lobe epilepsy. Epilepsia 61, 2795–2810. doi: 10.1111/epi.16714
Corbel, C., Hernandez, I., Wu, B., and Kosik, K. (2015). Developmental attenuation of N-methyl-D-aspartate receptor subunit expression by microRNAs. Neural Dev. 10:20. doi: 10.1186/s13064-015-0047-5
Cortés-Mendoza, J., Díaz de León-Guerrero, S., Pedraza-Alva, G., and Pérez-Martínez, L. (2013). Shaping synaptic plasticity: the role of activity-mediated epigenetic regulation on gene transcription. Int J. Dev. Neurosci. 31, 359–369. doi: 10.1016/j.steroids.2021.108947
Culotta, L., Scalmani, P., Vinci, E., Terragni, B., Sessa, A., Broccoli, V., et al. (2020). SULT4A1 modulates synaptic development and function by promoting the formation of PSD-95/NMDAR complex. J. Neurosci. 40, 7013–7026. doi: 10.1523/JNEUROSCI.2194-19.2020
Curtin, N., and Szabo, C. (2013). Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol. Aspects Med. 34, 1217–1256. doi: 10.1016/j.mam.2013.01.006
D’Aiuto, L., Di Maio, R., Mohan, K., Minervini, C., Saporiti, F., Soreca, I., et al. (2011). Mouse ES cells overexpressing DNMT1 produce abnormal neurons with upregulated NMDA/NR1 subunit. Differentiation 82, 9–17. doi: 10.1016/j.diff.2011.03.003
Dalva, M., Takasu, M., Lin, M., Shamah, S., Hu, L., Gale, N., et al. (2000). EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 103, 945–956. doi: 10.1016/s0092-8674(00)00197-5
Danbolt, N. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/s0301-0082(00)00067-8
de Sousa Maciel, I., Sales, A., Casarotto, P., Castrén, E., Biojone, C., and Joca, S. (2020). Nitric Oxide Synthase inhibition counteracts the stress-induced DNA methyltransferase 3b expression in the hippocampus of rats. Eur. J. Neurosci. [Online ahead of print]. doi: 10.1111/ejn.15042
DeGregorio-Rocasolano, N., Guirao, V., Ponce, J., Melià-Sorolla, M., Aliena-Valero, A., García-Serran, A., et al. (2020). Comparative proteomics unveils LRRFIP1 as a new player in the DAPK1 interactome of neurons exposed to oxygen and glucose deprivation. Antioxidants (Basel) 9:1202. doi: 10.3390/antiox9121202
Descloux, C., Ginet, V., Clarke, P., Puyal, J., and Truttmann, A. (2015). Neuronal death after perinatal cerebral hypoxia-ischemia: focus on autophagy-mediated cell death. Int. J. Dev. Neurosci. 45, 75–85. doi: 10.1016/j.ijdevneu.2015.06.008
Ding, Y., Zhou, Z., Chen, J., Peng, Y., Wang, H., Qiu, W., et al. (2021). Anti-NMDAR encephalitis induced in mice by active immunization with a peptide from the amino-terminal domain of the GluN1 subunit. J. Neuroinflammation 18:53. doi: 10.1186/s12974-021-02107-0
Dionísio, P., Amaral, J., and Rodrigues, C. (2021). Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res. Rev. 67:101263. doi: 10.1016/j.arr.2021.101263
D’Orsi, B., Bonner, H., Tuffy, L., Düssmann, H., Woods, I., Courtney, M., et al. (2012). Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J. Neurosci. 32, 1847–1858. doi: 10.1523/JNEUROSCI.2345-11.2012
D’Orsi, B., Engel, T., Pfeiffer, S., Nandi, S., Kaufmann, T., Henshall, D., et al. (2016). Bok is not pro-apoptotic but suppresses poly ADP-ribose polymerase-dependent cell death pathways and protects against excitotoxic and seizure-induced neuronal injury. J. Neurosci. 36, 4564–4578. doi: 10.1523/JNEUROSCI.3780-15.2016
Doti, N., Reuther, C., Scognamiglio, P., Dolga, A., Plesnila, N., Ruvo, M., et al. (2014). Inhibition of the AIF/CypA complex protects against intrinsic death pathways induced by oxidative stress. Cell Death Dis. 5:e993. doi: 10.1038/cddis.2013.518
Du, Z., Song, Y., Chen, X., Zhang, W., Zhang, G., Li, H., et al. (2021). Knockdown of astrocytic Grin2a aggravates β-amyloid-induced memory and cognitive deficits through regulating nerve growth factor. Aging Cell 20:e13437. doi: 10.1111/acel.13437
Dubois, C., and Liu, S. (2021). GluN2D NMDA receptors gate fear extinction learning and interneuron plasticity. Front. Synaptic Neurosci. 13:681068. doi: 10.3389/fnsyn.2021.681068
El-Mir, M., Detaille, D., R-Villanueva, G., Delgado-Esteban, M., Guigas, B., Attia, S., et al. (2008). Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J. Mol. Neurosci. 34, 77–87. doi: 10.1007/s12031-007-9002-1
Elsayed, N., Boyer, T., and Burd, I. (2021). Fetal neuroprotective strategies: therapeutic agents and their underlying synaptic pathways. Front. Synaptic Neurosci. 13:680899. doi: 10.3389/fnsyn.2021.680899
Ernst, A., Böhler, L., Hagenston, A., Hoffmann, A., Heiland, S., Sticht, C., et al. (2019). EphB2-dependent signaling promotes neuronal excitotoxicity and inflammation in the acute phase of ischemic stroke. Acta Neuropathol. Commun. 7:15. doi: 10.1186/s40478-019-0669-7
Espinoza, S., Arredondo, S., Barake, F., Carvajal, F., Guerrero, F., Segovia-Miranda, F., et al. (2020). Neuronal surface P antigen (NSPA) modulates postsynaptic NMDAR stability through ubiquitination of tyrosine phosphatase PTPMEG. BMC Biol. 18:164. doi: 10.1186/s12915-020-00877-2
Essiz, S., Gencel, M., Aktolun, M., Demir, A., Carpenter, T., and Servili, B. (2021). Correlated conformational dynamics of the human GluN1-GluN2A type N-methyl-D-aspartate (NMDA) receptor. J. Mol. Model. 27:162. doi: 10.1007/s00894-021-04755-8
Eyo, U., Bispo, A., Liu, J., Sabu, S., Wu, R., DiBona, V., et al. (2018). The GluN2A subunit regulates neuronal NMDA receptor-induced microglia-neuron physical interactions. Sci. Rep. 8:828. doi: 10.1038/s41598-018-19205-4
Fabbrin, S., Girardi, B., de Lorena Wendel, A., Coelho Ilha Valin, C., Pillat, M., Viero, F., et al. (2020). Spermidine-induced improvement of memory consolidation involves PI3K/Akt signaling pathway. Brain Res. Bull. 164, 208–213. doi: 10.1016/j.brainresbull.2020.08.018
Fachim, H., Loureiro, C., Corsi-Zuelli, F., Shuhama, R., Louzada-Junior, P., Menezes, P., et al. (2019). GRIN2B promoter methylation deficits in early-onset schizophrenia and its association with cognitive function. Epigenomics 11, 401–410. doi: 10.2217/epi-2018-0127
Farina, B., Di Sorbo, G., Chambery, A., Caporale, A., Leoni, G., Russo, R., et al. (2017). Structural and biochemical insights of CypA and AIF interaction. Sci. Rep. 7:1138. doi: 10.1038/s41598-017-01337-8
Farina, B., Sturlese, M., Mascanzoni, F., Caporale, A., Monti, A., Di Sorbo, G., et al. (2018). Binding mode of AIF(370–394) peptide to CypA: insights from NMR, label-free and molecular docking studies. Biochem. J. 475, 2377–2393. doi: 10.1042/BCJ20180177
Favaron, M., Manev, R., Rimland, J., Candeo, P., Beccaro, M., and Manev, H. (1993). NMDA-stimulated expression of BDNF mRNA in cultured cerebellar granule neurones. Neuroreport 4, 1171–1174.
Fedele, L., Newcombe, J., Topf, M., Gibb, A., Harvey, R., and Smart, T. (2018). Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat. Commun. 9:957. doi: 10.1038/s41467-018-02927-4
Feng, Y., Yang, H., Yue, Y., and Tian, F. (2020). MicroRNAs and target genes in epileptogenesis. Epilepsia 61, 2086–2096. doi: 10.1111/epi.16687
Fernández-Marmiesse, A., Kusumoto, H., Rekarte, S., Roca, I., Zhang, J., Myers, S., et al. (2018). A novel missense mutation in GRIN2A causes a nonepileptic neurodevelopmental disorder. Mov. Disord. 33, 992–999. doi: 10.1111/cla.12461
Flores-Hernandez, J., Salgado, H., De La Rosa, V., Avila-Ruiz, T., Torres-Ramirez, O., Lopez-Lopez, G., et al. (2009). Cholinergic direct inhibition of N-methyl-D aspartate receptor-mediated currents in the rat neocortex. Synapse 63, 308–318. doi: 10.1002/syn.20609
Franchini, L., Carrano, N., Di Luca, M., and Gardoni, F. (2020). Synaptic GluN2A-containing NMDA receptors: from physiology to pathological synaptic plasticity. Int. J. Mol. Sci. 21:1538. doi: 10.3390/ijms21041538
Fricker, M., Tolkovsky, A., Borutaite, V., Coleman, M., and Brown, G. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Fry, A., Fawcett, K., Zelnik, N., Yuan, H., Thompson, B., Shemer-Meiri, L., et al. (2018). De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain 141, 698–712. doi: 10.1093/brain/awx358
Gan, J., Qu, Y., Li, J., Zhao, F., and Mu, D. (2015). An evaluation of the links between microRNA, autophagy and epilepsy. Rev. Neurosci. 26, 225–237. doi: 10.1515/revneuro-2014-0062
Gan, K., and Südhof, T. (2020). SPARCL1 promotes excitatory but not inhibitory synapse formation and function independent of neurexins and neuroligins. J. Neurosci. 40, 8088–8102. doi: 10.1523/JNEUROSCI.0454-20.2020
Gao, K., Tankovic, A., Zhang, Y., Kusumoto, H., Zhang, J., Chen, W., et al. (2017). A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS One 12:e0170818. doi: 10.1371/journal.pone.0170818
Geis, C., Planagumà, J., Carreño, M., Graus, F., and Dalmau, J. (2019). Autoimmune seizures and epilepsy. J. Clin. Invest. 129, 926–940. doi: 10.1172/JCI125178
Geng, D., Kang, L., Su, Y., Jia, J., Ma, J., Li, S., et al. (2013). Protective effects of EphB2 on Aβ1–42 oligomer-induced neurotoxicity and synaptic NMDA receptor signaling in hippocampal neurons. Neurochem. Int. 63, 283–290. doi: 10.1016/j.neuint.2013.06.016
Ghasemi, M., Shafaroodi, H., Nazarbeiki, S., Meskar, H., Heydarpour, P., Ghasemi, A., et al. (2010). Voltage-dependent calcium channel and NMDA receptor antagonists augment anticonvulsant effects of lithium chloride on pentylenetetrazole-induced clonic seizures in mice. Epilepsy Behav. 18, 171–178. doi: 10.1016/j.yebeh.2010.04.002
Gleichman, A., Spruce, L., Dalmau, J., Seeholzer, S., and Lynch, D. (2012). Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J. Neurosci. 32, 11082–11094. doi: 10.1523/JNEUROSCI.0064-12.2012
Hamamoto, O., Tirapelli, D., Lizarte Neto, F., Freitas-Lima, P., Saggioro, F., Cirino, M., et al. (2020). Modulation of NMDA receptor by miR-219 in the amygdala and hippocampus of patients with mesial temporal lobe epilepsy. J. Clin. Neurosci. 74, 180–186. doi: 10.1016/j.jocn.2020.02.024
Hanada, T. (2020). Ionotropic glutamate receptors in epilepsy: a review focusing on AMPA and NMDA receptors. Biomolecules 10:464. doi: 10.3390/biom10030464
Hardingham, G., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Harraz, M., Eacker, S., Wang, X., Dawson, T., and Dawson, V. (2012). MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc. Natl. Acad. Sci. U S A 109, 18962–18967. doi: 10.1073/pnas.1121288109
Henderson, J., Georgiou, J., Jia, Z., Robertson, J., Elowe, S., Roder, J., et al. (2001). The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron 32, 1041–1056. doi: 10.1016/s0896-6273(01)00553-0
Hendry, S., Schwark, H., Jones, E., and Yan, J. (1987). Numbers and proportions of GABA-immunoreactive neurons in different areas of monkey cerebral cortex. J. Neurosci. 7, 1503–1519. doi: 10.1523/JNEUROSCI.07-05-01503.1987
Hetman, M., and Kharebava, G. (2006). Survival signaling pathways activated by NMDA receptors. Curr. Top. Med. Chem. 6, 787–799. doi: 10.2174/156802606777057553
Hoque, A., Hossain, M., Ameen, S., Ang, C., Williamson, N., Ng, D., et al. (2016). A beacon of hope in stroke therapy-Blockade of pathologically activated cellular events in excitotoxic neuronal death as potential neuroprotective strategies. Pharmacol. Ther. 160, 159–179. doi: 10.1016/j.pharmthera.2016.02.009
Horak, M., Barackova, P., Langore, E., Netolicky, J., Rivas-Ramirez, P., and Rehakova, K. (2021). The extracellular domains of GluN subunits play an essential role in processing NMDA receptors in the ER. Front. Neurosci. 15:603715. doi: 10.3389/fnins.2021.603715
Hou, H., Wang, L., Fu, T., Papasergi, M., Yule, D., and Xia, H. (2020). Magnesium acts as a second messenger in the regulation of NMDA receptor-mediated CREB signaling in neurons. Mol. Neurobiol. 57, 2539–2550. doi: 10.1007/s12035-020-01871-z
Hu, R., Wei, P., Jin, L., Zheng, T., Chen, W., Liu, X., et al. (2017). Overexpression of EphB2 in hippocampus rescues impaired NMDA receptors trafficking and cognitive dysfunction in Alzheimer model. Cell Death Dis. 8:e2717. doi: 10.1038/cddis.2017.140
Hu, F., Zhang, S., Chen, X., Fu, X., Guo, S., Jiang, Z., et al. (2020). MiR-219a-2 relieves myocardial ischemia-reperfusion injury by reducing calcium overload and cell apoptosis through HIF1α/ NMDAR pathway. Exp. Cell Res. 395:112172. doi: 10.1016/j.yexcr.2020.112172
Huang, L., Liu, Y., Jin, W., Ji, X., and Dong, Z. (2012). Ketamine potentiates hippocampal neurodegeneration and persistent learning and memory impairment through the PKCγ-ERK signaling pathway in the developing brain. Brain Res. 1476, 164–171. doi: 10.1016/j.brainres.2012.07.059
Huang, Y., Liu, X., Liao, Y., Liao, Y., Zou, D., Wei, X., et al. (2018). Role of miR-34c in the cognitive function of epileptic rats induced by pentylenetetrazol. Mol. Med. Rep. 17, 4173–4180. doi: 10.3892/mmr.2018.8441
Huang, Y., and Xiong, H. (2021). Anti-NMDA receptor encephalitis: a review of mechanistic studies. Int. J. Physiol. Pathophysiol. Pharmacol. 13, 1–11.
Hughes, E., Peng, X., Gleichman, A., Lai, M., Zhou, L., Tsou, R., et al. (2010). Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 30, 5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010
Intson, K., Geissah, S., McCullumsmith, R., and Ramsey, A. (2020). A role for endothelial NMDA receptors in the pathophysiology of schizophrenia. Schizophr. Res. [Online ahead of print]. doi: 10.1016/j.schres.2020.10.004
Ivanova, V., Balaban, P., and Bal, N. (2020). Modulation of AMPA receptors by nitric oxide in nerve cells. Int. J. Mol. Sci. 21:981. doi: 10.3390/ijms21030981
Ivanova, M., Kokorina, A., Timofeeva, P., Karelina, T., Abushik, P., Stepanenko, J., et al. (2020). Calcium export from neurons and multi-kinase signaling cascades contribute to ouabain neuroprotection in hyperhomocysteinemia. Biomolecules 10:1104. doi: 10.3390/biom10081104
Jeon, A., and Kim, J. (2018). SPDI knockdown inhibits seizure activity in acute seizure and chronic epilepsy rat models via S-nitrosylation-independent thiolation on NMDA receptor. Front. Cell. Neurosci. 12:438. doi: 10.3389/fncel.2018.00438
Jiang, Y., Jakovcevski, M., Bharadwaj, R., Connor, C., Schroeder, F., Lin, C., et al. (2010). Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J. Neurosci. 30, 7152–7167. doi: 10.1523/JNEUROSCI.1314-10.2010
Jiang, G., Wang, W., Cao, Q., Gu, J., Mi, X., Wang, K., et al. (2015). Insulin growth factor-1 (IGF-1) enhances hippocampal excitatory and seizure activity through IGF-1 receptor-mediated mechanisms in the epileptic brain. Clin. Sci. (London) 129, 1047–1060. doi: 10.1042/CS20150312
Jiao, J., Li, L., Sun, M., Fang, J., Meng, L., Zhang, Y., et al. (2021). Identification of a novel GRIN2D variant in a neonate with intractable epileptic encephalopathy-a case report. BMC Pediatr. 21:5. doi: 10.1186/s12887-020-02462-6
Jin, M., Ni, H., and Li, L. (2018). Leptin maintained zinc homeostasis against glutamate-induced excitotoxicity by preventing mitophagy-mediated mitochondrial activation in HT22 hippocampal neuronal cells. Front. Neurol. 9:322. doi: 10.3389/fneur.2018.00322
Jorratt, P., Hoschl, C., and Ovsepian, S. (2021). Endogenous antagonists of N-methyl-d-aspartate receptor in schizophrenia. Alzheimer’s Dement. 17, 888–905. doi: 10.1002/alz.12244
Kaplan, E., Zubedat, S., Radzishevsky, I., Valenta, A., Rechnitz, O., Sason, H., et al. (2018). ASCT1 (Slc1a4) transporter is a physiologic regulator of brain d-serine and neurodevelopment. Proc. Natl. Acad. Sci. U S A 115, 9628–9633. doi: 10.1073/pnas.1722677115
Kiese, K., Jablonski, J., Hackenbracht, J., Wrosch, J., Groemer, T., Kornhuber, J., et al. (2017). Epigenetic control of epilepsy target genes contributes to a cellular memory of epileptogenesis in cultured rat hippocampal neurons. Acta Neuropathol. commun. 5:79. doi: 10.1186/s40478-017-0485-x
Kim, J., Ko, A., Hyun, H., Min, S., and Kim, J. (2017). PDI regulates seizure activity via NMDA receptor redox in rats. Sci. Rep. 7:42491. doi: 10.1038/srep42491
Kim, D., Simeone, K., Simeone, T., Pandya, J., Wilke, J., Ahn, Y., et al. (2015). Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 78, 77–87. doi: 10.1002/ana.24424
Koehler, R., Dawson, V., and Dawson, T. (2021). Targeting parthanatos in ischemic stroke. Front. Neurol. 12:662034. doi: 10.3389/fneur.2021.662034
Kovács, K., Steullet, P., Steinmann, M., Do, K., Magistretti, P., Halfon, O., et al. (2007). TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. U S A 104, 4700–4705. doi: 10.1073/pnas.0607524104
Krall, R., Moutal, A., Phillips, M., Asraf, H., Johnson, J., Khanna, R., et al. (2020). Synaptic zinc inhibition of NMDA receptors depends on the association of GluN2A with the zinc transporter ZnT1. Sci. Adv. 6:eabb1515. doi: 10.1126/sciadv.abb1515
Kravchick, D., Karpova, A., Hrdinka, M., Lopez-Rojas, J., Iacobas, S., Carbonell, A., et al. (2016). Synaptonuclear messenger PRR7 inhibits c-Jun ubiquitination and regulates NMDA-mediated excitotoxicity. EMBO J. 35, 1923–1934. doi: 10.15252/embj.201593070
Lai, Y., Baker, J., Donti, T., Graham, B., Craigen, W., and Anderson, A. (2017). Mitochondrial dysfunction mediated by poly(ADP-Ribose) polymerase-1 activation contributes to hippocampal neuronal damage following status epilepticus. Int. J. Mol. Sci. 18:1502. doi: 10.3390/ijms18071502
Lee, M., Ting, K., Adams, S., Brew, B., Chung, R., and Guillemin, G. (2010). Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One 5:e14123. doi: 10.1371/journal.pone.0014123
Lemke, J., Hendrickx, R., Geider, K., Laube, B., Schwake, M., Harvey, R., et al. (2014). GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 75, 147–154. doi: 10.1002/ana.24073
Lemke, J., Lal, D., Reinthaler, E., Steiner, I., Nothnagel, M., Alber, M., et al. (2013). Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 45, 1067–1072. doi: 10.1038/ng.2728
Leypoldt, F., Armangue, T., and Dalmau, J. (2015). Autoimmune encephalopathies. Ann. N Y Acad. Sci. 1338, 94–114. doi: 10.1111/nyas.12553
Li, X., Guo, C., Li, Y., Li, L., Wang, Y., Zhang, Y., et al. (2017). Ketamine administered pregnant rats impair learning and memory in offspring via the CREB pathway. Oncotarget 8, 32433–32449. doi: 10.18632/oncotarget.15405
Li, S., Raychaudhuri, S., Lee, S., Brockmann, M., Wang, J., Kusick, G., et al. (2021). Asynchronous release sites align with NMDA receptors in mouse hippocampal synapses. Nat. Commun. 12:677. doi: 10.1038/s41467-021-21004-x
Li, D., Yuan, H., Ortiz-Gonzalez, X., Marsh, E., Tian, L., McCormick, E., et al. (2016). GRIN2D recurrent de novo dominant mutation causes a severe epileptic encephalopathy treatable with NMDA receptor channel blockers. Am. J. Hum. Genet. 99, 802–816. doi: 10.1016/j.ajhg.2016.07.013
Li, D., Zhang, Y., Zhang, Y., Wang, Q., Miao, Q., Xu, Y., et al. (2019). Correlation between the epigenetic modification of histone H3K9 acetylation of NR2B gene promoter in rat hippocampus and ethanol withdrawal syndrome. Mol. Biol. Rep. 46, 2867–2875. doi: 10.1007/s11033-019-04733-7
Lian, W., Zhou, W., Zhang, B., Jia, H., Xu, L., Liu, A., et al. (2021). DL0410 ameliorates cognitive disorder in SAMP8 mice by promoting mitochondrial dynamics and the NMDAR-CREB-BDNF pathway. Acta Pharmacol. Sin. 42, 1055–1068. doi: 10.1038/s41401-020-00506-2
Limón, I., Angulo-Cruz, I., Sánchez-Abdon, L., and Patricio-Martínez, A. (2021). Disturbance of the glutamate-glutamine cycle, secondary to hepatic damage, compromises memory function. Front. Neurosci. 15:578922. doi: 10.3389/fnins.2021.578922
Lin, S. (2015). microRNAs and fragile X syndrome. Adv. Exp. Med. Biol. 888, 107–121. doi: 10.1007/978-3-319-22671-2_7
Liu, X., Geng, J., Guo, H., Zhao, H., and Ai, Y. (2020). Propofol inhibited apoptosis of hippocampal neurons in status epilepticus through miR-15a-5p/NR2B/ERK1/2 pathway. Cell Cycle 19, 1000–1011. doi: 10.1080/15384101.2020.1743909
Liu, S., Liu, C., Xiong, L., Xie, J., Huang, C., Pi, R., et al. (2021). Icaritin alleviates glutamate-induced neuronal damage by inactivating GluN2B-containing NMDARs through the ERK/DAPK1 pathway. Front. Neurosci. 15:525615. doi: 10.3389/fnins.2021.525615
Liu, F., Paule, M., Ali, S., and Wang, C. (2011). Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr. Neuropharmacol. 9, 256–261. doi: 10.2174/157015911795017155
Liu, E., Xie, A., Zhou, Q., Li, M., Zhang, S., Li, S., et al. (2017). GSK-3β deletion in dentate gyrus excitatory neuron impairs synaptic plasticity and memory. Sci. Rep. 7:5781. doi: 10.1038/s41598-017-06173-4
Loss, C., da Rosa, N., Mestriner, R., Xavier, L., and Oliveira, D. (2019). Blockade of GluN2B-containing NMDA receptors reduces short-term brain damage induced by early-life status epilepticus. Neurotoxicology 71, 138–149. doi: 10.1016/j.neuro.2019.01.002
Lu, Y., Ding, X., Wu, X., and Huang, S. (2020). Ketamine inhibits LPS-mediated BV2 microglial inflammation via NMDA receptor blockage. Fundam. Clin. Pharmacol. 34, 229–237. doi: 10.1111/fcp.12508
Lumley, L., Niquet, J., Marrero-Rosado, B., Schultz, M., Rossetti, F., de Araujo Furtado, M., et al. (2021). Treatment of acetylcholinesterase inhibitor-induced seizures with polytherapy targeting GABA and glutamate receptors. Neuropharmacology 185:108444. doi: 10.1016/j.neuropharm.2020.108444
Luo, J., Wang, Y., Yasuda, R., Dunah, A., and Wolfe, B. (1997). The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol. Pharmacol. 51, 79–86. doi: 10.1124/mol.51.1.79
Lyu, Y., Ren, X., Zhang, H., Tian, F., Mu, J., and Zheng, J. (2020). Sub-chronic administration of benzo[a]pyrene disrupts hippocampal long-term potentiation via inhibiting CaMK II/PKC/PKA-ERK-CREB signaling in rats. Environ. Toxicol. 35, 961–970. doi: 10.1002/tox.22932
Ma, X., Cheng, O., Jiang, Q., Yang, J., Xiao, H., and Qiu, H. (2021). Activation of ephrinb1/EPHB2/MAP-2/NMDAR mediates hippocampal neurogenesis promoted by transcranial direct current stimulation in cerebral-ischemic mice. Neuromol. Med. 23, 521–530. doi: 10.1007/s12017-021-08654-2
Malkov, A., Ivanov, A., Latyshkova, A., Bregestovski, P., Zilberter, M., and Zilberter, Y. (2019). Activation of nicotinamide adenine dinucleotide phosphate oxidase is the primary trigger of epileptic seizures in rodent models. Ann. Neurol. 85, 907–920. doi: 10.1002/ana.25474
Manto, M., Dalmau, J., Didelot, A., Rogemond, V., and Honnorat, J. (2010). in vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J. Rare Dis. 5:31. doi: 10.1186/1750-1172-5-31
Mantuano, E., Azmoon, P., Banki, M., Lam, M., Sigurdson, C., and Gonias, S. (2020). A soluble derivative of PrP activates cell-signaling and regulates cell physiology through LRP1 and the NMDA receptor. J. Biol. Chem. 295, 14178–14188. doi: 10.1074/jbc.RA120.013779
Markram, H., and Segal, M. (1990). Acetylcholine potentiates responses to N-methyl-D-aspartate in the rat hippocampus. Neurosci. Lett. 113, 62–65. doi: 10.1016/0304-3940(90)90495-u
Marrero-Rosado, B., de Araujo Furtado, M., Kundrick, E., Walker, K., Stone, M., Schultz, C., et al. (2020). Ketamine as adjunct to midazolam treatment following soman-induced status epilepticus reduces seizure severity, epileptogenesis and brain pathology in plasma carboxylesterase knockout mice. Epilepsy Behav. 111:107229. doi: 10.1016/j.yebeh.2020.107229
Marwick, K., Hansen, K., Skehel, P., Hardingham, G., and Wyllie, D. (2019). Functional assessment of triheteromeric NMDA receptors containing a human variant associated with epilepsy. J. Physiol. 597, 1691–1704. doi: 10.1113/JP277292
Mashimo, M., Kato, J., and Moss, J. (2013). ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl. Acad. Sci. U S A 110, 18964–18969. doi: 10.1073/pnas.1312783110
Mayer, M., Westbrook, G., and Guthrie, P. (1984). Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309, 261–263. doi: 10.1038/309261a0
Mele, M., Vieira, R., Correia, B., De Luca, P., Duarte, F., Pinheiro, P., et al. (2021). Transient incubation of cultured hippocampal neurons in the absence of magnesium induces rhythmic and synchronized epileptiform-like activity. Sci. Rep. 11:11374. doi: 10.1038/s41598-021-90486-y
Mielnik, C., Binko, M., Chen, Y., Funk, A., Johansson, E., Intson, K., et al. (2021). Consequences of NMDA receptor deficiency can be rescued in the adult brain. Mol. Psychiatry 26, 2929–2942. doi: 10.1038/s41380-020-00859-4
Mikasova, L., De Rossi, P., Bouchet, D., Georges, F., Rogemond, V., Didelot, A., et al. (2012). Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 135, 1606–1621. doi: 10.1093/brain/aws092
Mizrachi, T., Vaknin-Dembinsky, A., Brenner, T., and Treinin, M. (2021). Neuroinflammation modulation via α7 nicotinic acetylcholine receptor and its chaperone, RIC-3. Molecules 26:6139. doi: 10.3390/molecules26206139
Mohammed, C., Rhee, H., Phee, B., Kim, K., Kim, H., Lee, H., et al. (2016). miR-204 downregulates EphB2 in aging mouse hippocampal neurons. Aging cell 15, 380–388. doi: 10.1111/acel.12444
Monyer, H., Burnashev, N., Laurie, D., Sakmann, B., and Seeburg, P. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. doi: 10.1016/0896-6273(94)90210-0
Moshé, S., Perucca, E., Ryvlin, P., and Tomson, T. (2015). Epilepsy: new advances. Lancet 385, 884–898. doi: 10.1016/S0140-6736(14)60456-6
Mota Vieira, M., Nguyen, T., Wu, K., Badger, J., Collins, B., Anggono, V., et al. (2020). An epilepsy-associated GRIN2A rare variant disrupts CaMKIIα phosphorylation of GluN2A and NMDA receptor trafficking. Cell Rep. 32:108104. doi: 10.1016/j.celrep.2020.108104
Mothet, J. P., Le Bail, M., and Billard, J. (2015). Time and space profiling of NMDA receptor co-agonist functions. J. Neurochem. 135, 210–225. doi: 10.1111/jnc.13204
Murtas, G., Marcone, G., Sacchi, S., and Pollegioni, L. (2020). L-serine synthesis via the phosphorylated pathway in humans. Cell. Mol. Life Sci. 77, 5131–5148. doi: 10.1007/s00018-020-03574-z
Nagy, L. V., Bali, Z., Kapus, G., Pelsöczi, P., Farkas, B., Lendvai, B., et al. (2020). Converging evidence on D-amino acid oxidase-dependent enhancement of hippocampal firing activity and passive avoidance learning in rats. Int. J. Neuropsychopharmacol. 24, 434–445. doi: 10.1093/ijnp/pyaa095
Neame, S., Safory, H., Radzishevsky, I., Touitou, A., Marchesani, F., Marchetti, M., et al. (2019). The NMDA receptor activation by d-serine and glycine is controlled by an astrocytic Phgdh-dependent serine shuttle. Proc. Natl. Acad. Sci. U S A 116, 20736–20742. doi: 10.1073/pnas.1909458116
Neyens, D., Zhao, H., Huston, N., Wayman, G., Ritter, R., and Appleyard, S. (2020). Leptin sensitizes NTS neurons to vagal input by increasing postsynaptic NMDA receptor currents. J. Neurosci. 40, 7054–7064. doi: 10.1523/JNEUROSCI.1865-19.2020
Niciu, M., Kelmendi, B., and Sanacora, G. (2012). Overview of glutamatergic neurotransmission in the nervous system. Pharmacol. Biochem. Behav. 100, 656–664. doi: 10.1016/j.pbb.2011.08.008
Nikolaev, M., Chizhov, A., and Tikhonov, D. (2021). Molecular mechanisms of action determine inhibition of paroxysmal depolarizing shifts by NMDA receptor antagonists in rat cortical neurons. Neuropharmacology 184:108443. doi: 10.1016/j.neuropharm.2020.108443
Niquet, J., Baldwin, R., Norman, K., Suchomelova, L., Lumley, L., and Wasterlain, C. (2017). Simultaneous triple therapy for the treatment of status epilepticus. Neurobiol. Dis. 104, 41–49. doi: 10.1016/j.nbd.2017.04.019
Nolt, M., Lin, Y., Hruska, M., Murphy, J., Sheffler-Colins, S., Kayser, M., et al. (2011). EphB controls NMDA receptor function and synaptic targeting in a subunit-specific manner. J. Neurosci. 31, 5353–5364. doi: 10.1523/JNEUROSCI.0282-11.2011
Olney, J. (1971). Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J. Neuropathol. Exp. Neurol. 30, 75–90. doi: 10.1097/00005072-197101000-00008
Pan, Y., He, X., Li, C., Li, Y., Li, W., Zhang, H., et al. (2021). Neuronal activity recruits the CRTC1/CREB axis to drive transcription-dependent autophagy for maintaining late-phase LTD. Cell Rep. 36:109398. doi: 10.1016/j.celrep.2021.109398
Papadia, S., Stevenson, P., Hardingham, N., Bading, H., and Hardingham, G. (2005). Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J. Neurosci. 25, 4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005
Pflanz, N., Daszkowski, A., James, K., and Mihic, S. (2019). Ketone body modulation of ligand-gated ion channels. Neuropharmacology 148, 21–30. doi: 10.1016/j.neuropharm.2018.12.013
Pierson, T., Yuan, H., Marsh, E., Fuentes-Fajardo, K., Adams, D., Markello, T., et al. (2014). GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann. Clin. Transl. Neurol. 1, 190–198. doi: 10.1002/acn3.39
Pironti, E., Granata, F., Cucinotta, F., Gagliano, A., Efthymiou, S., Houlden, H., et al. (2018). Electroclinical history of a five-year-old girl with GRIN1-related early-onset epileptic encephalopathy: a video-case study. Epileptic Disord. 20, 423–427. doi: 10.1684/epd.2018.0992
Planagumà, J., Haselmann, H., Mannara, F., Petit-Pedrol, M., Grünewald, B., Aguilar, E., et al. (2016). Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann. Neurol. 80, 388–400. doi: 10.1002/ana.24721
Platzer, K., Yuan, H., Schütz, H., Winschel, A., Chen, W., Hu, C., et al. (2017). GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 54, 460–470. doi: 10.1136/jmedgenet-2016-104509
Ploux, E., Bouet, V., Radzishevsky, I., Wolosker, H., Freret, T., and Billard, J. (2020). Serine racemase deletion affects the excitatory/inhibitory balance of the hippocampal CA1 network. Int. J. Mol. Sci. 21:9447. doi: 10.3390/ijms21249447
Pollegioni, L., Sacchi, S., and Murtas, G. (2018). Human D-amino acid oxidase: structure, function and regulation. Front. Mol. Biosci. 5:107. doi: 10.3389/fmolb.2018.00107
Postnikova, T., Zubareva, O., Kovalenko, A., Kim, K., Magazanik, L., and Zaitsev, A. (2017). Status epilepticus impairs synaptic plasticity in rat hippocampus and is followed by changes in expression of NMDA receptors. Biochemistry (Mosc) 82, 282–290. doi: 10.1134/S0006297917030063
Potier, B., Turpin, F., Sinet, P., Rouaud, E., Mothet, J., Videau, C., et al. (2010). Contribution of the d-serine-dependent pathway to the cellular mechanisms underlying cognitive aging. Front. Aging Neurosci. 2:1. doi: 10.3389/neuro.24.001.2010
Punnakkal, P., and Dominic, D. (2018). NMDA receptor GluN2 subtypes control epileptiform events in the hippocampus. Neuromol. Med. 20, 90–96. doi: 10.1007/s12017-018-8477-y
Qian, A., Buller, A., and Johnson, J. (2005). NR2 subunit-dependence of NMDA receptor channel block by external Mg2+. J. Physiol. 562, 319–331. doi: 10.1113/jphysiol.2004.076737
Raboni, S., Marchetti, M., Faggiano, S., Campanini, B., Bruno, S., Marchesani, F., et al. (2019). The energy landscape of human serine racemase. Front. Mol. Biosci. 5:112. doi: 10.3389/fmolb.2018.00112
Rodriguez, L., Yi, C., Chu, C., Duriez, Q., Watanabe, S., Ryu, M., et al. (2020). Cross-talk between P2X and NMDA receptors. Int. J. Mol. Sci. 21:7187. doi: 10.3390/ijms21197187
Rosendo-Pineda, M., Vicente, J., Vivas, O., Pacheco, J., Loza-Huerta, A., Sampieri, A., et al. (2020). Phosphorylation of NMDA receptors by cyclin B/CDK1 modulates calcium dynamics and mitosis. Commun. Biol. 3:665. doi: 10.1038/s42003-020-01393-3
Ryley Parrish, R., Albertson, A., Buckingham, S., Hablitz, J., Mascia, K., Davis Haselden, W., et al. (2013). Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus. Neuroscience 248, 602–619. doi: 10.1016/j.neuroscience.2013.06.029
Salmi, M., Bolbos, R., Bauer, S., Minlebaev, M., Burnashev, N., and Szepetowski, P. (2018). Transient microstructural brain anomalies and epileptiform discharges in mice defective for epilepsy and language-related NMDA receptor subunit gene Grin2a. Epilepsia 59, 1919–1930. doi: 10.1111/epi.14543
Samanta, D. (2020). Ketamine infusion for super refractory status epilepticus in alternating hemiplegia of childhood. Neuropediatrics 51, 225–228. doi: 10.1055/s-0039-3402005
Sánchez Fernández, I., Goodkin, H., and Scott, R. (2019). Pathophysiology of convulsive status epilepticus. Seizure 68, 16–21. doi: 10.1016/j.seizure.2018.08.002
Santoro, J., Filippakis, A., and Chitnis, T. (2019). Ketamine use in refractory status epilepticus associated with anti-NMDA receptor antibody encephalitis. Epilepsy Behav. Rep. 12:100326. doi: 10.1016/j.ebr.2019.100326
Santos, J., and Schauwecker, P. (2003). Protection provided by cyclosporin A against excitotoxic neuronal death is genotype dependent. Epilepsia 44, 995–1002. doi: 10.1046/j.1528-1157.2003.66302.x
Sason, H., Billard, J., Smith, G., Safory, H., Neame, S., Kaplan, E., et al. (2017). Asc-1 transporter regulation of synaptic activity via the tonic release of d-serine in the forebrain. Cereb. Cortex 27, 1573–1587. doi: 10.1093/cercor/bhv350
Schinder, A., Olson, E., Spitzer, N., and Montal, M. (1996). Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16, 6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996
Schmidt, M., Caron, N., Aly, A., Lemarié, F., Dal Cengio, L., Ko, Y., et al. (2020). DAPK1 promotes extrasynaptic glun2b phosphorylation and striatal spine instability in the YAC128 mouse model of huntington disease. Front. Cell. Neurosci. 14:590569. doi: 10.3389/fncel.2020.590569
Schousboe, A., Scafidi, S., Bak, L., Waagepetersen, H., and McKenna, M. (2014). Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 11, 13–30. doi: 10.1007/978-3-319-08894-5_2
Screaton, R., Conkright, M., Katoh, Y., Best, J., Canettieri, G., Jeffries, S., et al. (2004). The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119, 61–74. doi: 10.1016/j.cell.2004.09.015
Sharawat, I., Yadav, J., and Saini, L. (2019). GRIN2BNovel mutation: a rare cause of severe epileptic encephalopathy. Neurol. India 67, 562–563. doi: 10.4103/0028-3886.257986
Sharma, P., Kumari, S., Sharma, J., Purohit, R., and Singh, D. (2021). Hesperidin interacts With CREB-BDNF signaling pathway to suppress pentylenetetrazole-induced convulsions in zebrafish. Front. Pharmacol. 11:607797. doi: 10.3389/fphar.2020.607797
Shekhar, S., Liu, Y., Wang, S., Zhang, H., Fang, X., Zhang, J., et al. (2021). Novel mechanistic insights and potential therapeutic impact of TRPC6 in neurovascular coupling and ischemic stroke. Int. J. Mol. Sci. 22:2074. doi: 10.3390/ijms22042074
Shelkar, G., Pavuluri, R., Gandhi, P., Ravikrishnan, A., Gawande, D., Liu, J., et al. (2019). Differential effect of NMDA receptor GluN2C and GluN2D subunit ablation on behavior and channel blocker-induced schizophrenia phenotypes. Sci. Rep. 9:7572. doi: 10.1038/s41598-019-43957-2
Sibarov, D., Boikov, S., Karelina, T., and Antonov, S. (2020). GluN2 subunit-dependent redox modulation of nmda receptor activation by homocysteine. Biomolecules 10:1441. doi: 10.3390/biom10101441
Smigiel, R., Kostrzewa, G., Kosinska, J., Pollak, A., Stawinski, P., Szmida, E., et al. (2016). Further evidence for GRIN2B mutation as the cause of severe epileptic encephalopathy. Am. J. Med. Genet. A 170, 3265–3270. doi: 10.1002/ajmg.a.37887
Srivastava, P., Dhuriya, Y., Kumar, V., Srivastava, A., Gupta, R., Shukla, R., et al. (2018). PI3K/Akt/GSK3β induced CREB activation ameliorates arsenic mediated alterations in NMDA receptors and associated signaling in rat hippocampus: Neuroprotective role of curcumin. Neurotoxicology 67, 190–205. doi: 10.1016/j.neuro.2018.04.018
Sun, Y., Dhamne, S., Carretero-Guillén, A., Salvador, R., Goldenberg, M., Godlewski, B., et al. (2020). Drug-responsive inhomogeneous cortical modulation by direct current stimulation. Ann. Neurol. 88, 489–502. doi: 10.1002/ana.25822
Takagi, S., Balu, D., and Coyle, J. (2021). Factors regulating serine racemase and d-amino acid oxidase expression in the mouse striatum. Brain Res. 1751:147202. doi: 10.1016/j.brainres.2020.147202
Takahashi, Y., Nishimura, S., Takao, E., Kasai, R., Enokida, K., Ida, K., et al. (2020). Characteristics of internalization of NMDA-type GluRs with antibodies to GluN1 and GluN2B. J. Neuroimmunol. 349:577427. doi: 10.1016/j.jneuroim.2020.577427
Tannich, F., Barhoumi, K., Rejeb, A., Aouichri, M., and Souilem, O. (2020). Ketamine, at low dose, decrease behavioural alterations in epileptic diseases induced by pilocarpine in mice. Int. J. Neurosci. 130, 1118–1124. doi: 10.1080/00207454.2020.1730363
Tannich, F., Tlili, A., Pintard, C., Chniguir, A., Eto, B., Dang, P., et al. (2021). Correction to: activation of the phagocyte NADPH oxidase/NOX2 and myeloperoxidase in the mouse brain during pilocarpine-induced temporal lobe epilepsy and inhibition by ketamine. Inflammopharmacology 29, 333–334. doi: 10.1007/s10787-020-00779-3
Tong, L., Yang, X., Zhang, S., Zhang, D., Yang, X., Li, B., et al. (2020). Clinical and EEG characteristics analysis of autoimmune encephalitis in children with positive and negative anti-N-methyl- D-aspartate receptor antibodies. Ann. Palliat. Med. 9, 2575–2585. doi: 10.21037/apm-19-484
Tovar, K., McGinley, M., and Westbrook, G. (2013). Triheteromeric NMDA receptors at hippocampal synapses. J. Neurosci. 33, 9150–9160. doi: 10.1523/JNEUROSCI.0829-13.2013
Tsuchida, N., Hamada, K., Shiina, M., Kato, M., Kobayashi, Y., Tohyama, J., et al. (2018). GRIN2D variants in three cases of developmental and epileptic encephalopathy. Clin. Genet. 94, 538–547. doi: 10.1111/cge.13454
Valdivielso, J., Eritja, à., Caus, M., and Bozic, M. (2020). Glutamate-gated NMDA receptors: insights into the function and signaling in the kidney. Biomolecules 10:1051. doi: 10.3390/biom10071051
Vargas-Lopes, C., Madeira, C., Kahn, S., Albino do Couto, I., Bado, P., Houzel, J., et al. (2011). Protein kinase C activity regulates D-serine availability in the brain. J. Neurochem. 116, 281–290. doi: 10.1111/j.1471-4159.2010.07102.x
Virág, L., Robaszkiewicz, A., Rodriguez-Vargas, J., and Oliver, F. (2013). Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 34, 1153–1167. doi: 10.1016/j.mam.2013.01.007
Wang, K. (2000). Calpain and caspase: can you tell the difference? Trends Neurosci. 23, 20–26. doi: 10.1016/s0166-2236(99)01479-4
Wang, Y., Briz, V., Chishti, A., Bi, X., and Baudry, M. (2013). Distinct roles for μ-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J. Neurosci. 33, 18880–18892. doi: 10.1523/JNEUROSCI.3293-13.2013
Wang, Y., Dawson, V., and Dawson, T. (2009). Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol. 218, 193–202. doi: 10.1016/j.expneurol.2009.03.020
Wang, Y., Kerrisk Campbell, M., Tom, I., Foreman, O., Hanson, J., and Sheng, M. (2020). PCDH7 interacts with GluN1 and regulates dendritic spine morphology and synaptic function. Sci. Rep. 10:10951. doi: 10.1038/s41598-020-67831-8
Wang, Y., Kim, N., Haince, J., Kang, H., David, K., Andrabi, S., et al. (2011). Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 4:ra20. doi: 10.1126/scisignal.2000902
Wang, X., Tian, X., Yang, Y., Lu, X., Li, Y., Ma, Y., et al. (2017). POSH participates in epileptogenesis by increasing the surface expression of the NMDA receptor: a promising therapeutic target for epilepsy. Expert Opin. Ther. Targets 21, 1083–1094. doi: 10.1080/14728222.2017.1394456
Wang, W., Wang, X., Chen, L., Zhang, Y., Xu, Z., Liu, J., et al. (2016). The microRNA miR-124 suppresses seizure activity and regulates CREB1 activity. Expert Rev. Mol. Med. 18:e4. doi: 10.1017/erm.2016.3
Wang, J., Xu, W., Shao, J., He, Z., Ding, Z., Huang, J., et al. (2017). miR-219-5p targets CaMKIIγ to attenuate morphine tolerance in rats. Oncotarget 8, 28203–28214. doi: 10.18632/oncotarget.15997
Wang, S., Yang, X., Lin, Y., Qiu, X., Li, H., Zhao, X., et al. (2013). Cellular NAD depletion and decline of SIRT1 activity play critical roles in PARP-1-mediated acute epileptic neuronal death in vitro. Brain Res. 1535, 14–23. doi: 10.1016/j.brainres.2013.08.038
Wang, G., Zhu, Z., Xu, D., and Sun, L. (2020). Advances in understanding CREB signaling-mediated regulation of the pathogenesis and progression of epilepsy. Clin. Neurol. Neurosurg. 196:106018. doi: 10.1016/j.clineuro.2020.106018
Wang, J., Wang, F., Mai, D., and Qu, S. (2020a). Molecular mechanisms of glutamate toxicity in Parkinson’s disease. Front. Neurosci. 14:585584. doi: 10.3389/fnins.2020.585584
Wang, J., Fei, Z., Liang, J., Zhou, X., Qin, G., Zhang, D., et al. (2020b). EphrinB/EphB signaling contributes to the synaptic plasticity of chronic migraine through NR2B phosphorylation. Neuroscience 428, 178–191. doi: 10.1016/j.neuroscience.2019.12.038
Washburn, H., Xia, N., Zhou, W., Mao, Y., and Dalva, M. (2020). Positive surface charge of GluN1 N-terminus mediates the direct interaction with EphB2 and NMDAR mobility. Nat. Commun. 11:570. doi: 10.1038/s41467-020-14345-6
Windelborn, J., and Lipton, P. (2008). Lysosomal release of cathepsins causes ischemic damage in the rat hippocampal slice and depends on NMDA-mediated calcium influx, arachidonic acid metabolism and free radical production. J. Neurochem. 106, 56–69. doi: 10.1111/j.1471-4159.2008.05349.x
Wong, H. H.-W., Rannio, S., Jones, V., Thomazeau, A., and Sjöström, P. (2021). NMDA receptors in axons: there’s no coincidence. J. Physiol. 599, 367–387. doi: 10.1113/JP280059
Wright, S., Hashemi, K., Stasiak, L., Bartram, J., Lang, B., Vincent, A., et al. (2015). Epileptogenic effects of NMDAR antibodies in a passive transfer mouse model. Brain 138, 3159–3167. doi: 10.1093/brain/awv257
Wu, Y., Wei, Z., Li, Y., Wei, C., Li, Y., Cheng, P., et al. (2019). Perturbation of ephrin receptor signaling and glutamatergic transmission in the hypothalamus in depression using proteomics integrated with metabolomics. Front. Neurosci. 13:1359. doi: 10.3389/fnins.2019.01359
Wyllie, D., Livesey, M., and Hardingham, G. (2013). Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74, 4–17. doi: 10.1016/j.neuropharm.2013.01.016
Xiang, L., Ren, Y., Li, X., Zhao, W., and Song, Y. (2016). MicroRNA-204 suppresses epileptiform discharges through regulating TrkB-ERK1/2-CREB signaling in cultured hippocampal neurons. Brain Res. 1639, 99–107. doi: 10.1016/j.brainres.2016.02.045
Xing, J., Han, D., Xu, D., Li, X., and Sun, L. (2019). CREB protects against temporal lobe epilepsy associated with cognitive impairment by controlling oxidative neuronal damage. Neurodegener. Dis. 19, 225–237. doi: 10.1159/000507023
Xu, Y., Chen, P., Wang, X., Yao, J., and Zhuang, S. (2018). miR-34a deficiency in APP/PS1 mice promotes cognitive function by increasing synaptic plasticity via AMPA and NMDA receptors. Neurosci. Lett. 670, 94–104. doi: 10.1016/j.neulet.2018.01.045
Xu, H., Li, X., Wu, X., Yang, Y., Dai, S., Lei, T., et al. (2019). Iduna protects HT22 cells by inhibiting parthanatos: the role of the p53-MDM2 pathway. Exp. Cell Res. 384:111547. doi: 10.1016/j.yexcr.2019.111547
Xu, X., and Luo, J. (2018). Mutations of N-methyl-D-aspartate receptor subunits in epilepsy. Neurosci. Bull. 34, 549–565. doi: 10.1007/s12264-017-0191-5
Xu, T., Yu, X., Deng, J., Ou, S., Liu, X., Wang, T., et al. (2019). CXCR7 regulates epileptic seizures by controlling the synaptic activity of hippocampal granule cells. Cell Death Dis. 10:825. doi: 10.1038/s41419-019-2052-9
Xue, L., Borutaite, V., and Tolkovsky, A. (2002). Inhibition of mitochondrial permeability transition and release of cytochrome c by anti-apoptotic nucleoside analogues. Biochem. Pharmacol. 64, 441–449. doi: 10.1016/s0006-2952(02)01181-4
Yakovlev, A., Kurmasheva, E., Giniatullin, R., Khalilov, I., and Sitdikova, G. (2017). Hydrogen sulfide inhibits giant depolarizing potentials and abolishes epileptiform activity of neonatal rat hippocampal slices. Neuroscience 340, 153–165. doi: 10.1016/j.neuroscience.2016.10.051
Yamashima, T., Kohda, Y., Tsuchiya, K., Ueno, T., Yamashita, J., Yoshioka, T., et al. (1998). Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ’calpain-cathepsin hypothesis’. Eur. J. Neurosci. 10, 1723–1733. doi: 10.1046/j.1460-9568.1998.00184.x
Yan, J., Huang, Y., Lu, Y., Chen, J., and Jiang, H. (2014). Repeated administration of ketamine can induce hippocampal neurodegeneration and long-term cognitive impairment via the ROS/HIF-1α pathway in developing rats. Cell. Physiol. Biochem. 33, 1715–1732. doi: 10.1159/000362953
Yan, M., Zhu, W., Zheng, X., Li, Y., Tang, L., Lu, B., et al. (2016). Effect of glutamate on lysosomal membrane permeabilization in primary cultured cortical neurons. Mol. Med. Rep. 13, 2499–2505. doi: 10.3892/mmr.2016.4819
Yang, Q., Huang, Z., Luo, Y., Zheng, F., Hu, Y., Liu, H., et al. (2019). Inhibition of Nwd1 activity attenuates neuronal hyperexcitability and GluN2B phosphorylation in the hippocampus. EBioMedicine 47, 470–483. doi: 10.1016/j.ebiom.2019.08.050
Yang, Y., Paspalas, C., Jin, L., Picciotto, M., Arnsten, A., and Wang, M. (2013). Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc. Natl. Acad. Sci. U S A 110, 12078–12083. doi: 10.1073/pnas.1307849110
Ye, Z., Li, Q., Guo, Q., Xiong, Y., Guo, D., Yang, H., et al. (2018). Ketamine induces hippocampal apoptosis through a mechanism associated with the caspase-1 dependent pyroptosis. Neuropharmacology 128, 63–75. doi: 10.1016/j.neuropharm.2017.09.035
Yu, X., Guan, Q., Wang, Y., Shen, H., Zhai, L., Lu, X., et al. (2019). Anticonvulsant and anti-apoptosis effects of salvianolic acid B on pentylenetetrazole-kindled rats via AKT/CREB/BDNF signaling. Epilepsy Res. 154, 90–96. doi: 10.1016/j.eplepsyres.2019.05.007
Zehavi, Y., Mandel, H., Zehavi, A., Rashid, M., Straussberg, R., Jabur, B., et al. (2017). De novo GRIN1 mutations: an emerging cause of severe early infantile encephalopathy. Eur. J. Med. Genet. 60, 317–320. doi: 10.1016/j.ejmg.2017.04.001
Zhand, A., Sayad, A., Ghafouri-Fard, S., Arsang-Jang, S., Mazdeh, M., and Taheri, M. (2018). Expression analysis of GRIN2B, BDNF and IL-1β genes in the whole blood of epileptic patients. Neurol. Sci. 39, 1945–1953. doi: 10.1007/s10072-018-3533-9
Zhang, Y., Fan, M., Wang, Q., He, G., Fu, Y., Li, H., et al. (2015). Polymorphisms in microRNA genes and genes involving in NMDAR signaling and schizophrenia: a case-control study in chinese han population. Sci. Rep. 5:12984. doi: 10.1038/srep12984
Zhang, X., Hu, B., Lu, L., Xu, D., Sun, L., and Lin, W. (2021). D-serine and NMDA receptor 1 expression in patients with intractable epilepsy. Turk. Neurosurg. 31, 76–82. doi: 10.5137/1019-5149.JTN.28138-19.2
Zhang, H., Ma, S., Gao, Y., Xing, J., Xian, H., Li, Z., et al. (2020). Spinal CCL2 promotes pain sensitization by rapid enhancement of NMDA-induced currents through the ERK-GluN2B pathway in mouse lamina II neurons. Neurosci. Bull. 36, 1344–1354. doi: 10.1007/s12264-020-00557-9
Zhang, S., Steijaert, M., Lau, D., Schütz, G., Delucinge-Vivier, C., Descombes, P., et al. (2007). Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron 53, 549–562. doi: 10.1016/j.neuron.2007.01.025
Zhang, Y., Tan, F., Xu, P., and Qu, S. (2016). Recent advance in the relationship between excitatory amino acid transporters and Parkinson’s disease. Neural Plast. 2016:8941327. doi: 10.1155/2016/8941327
Zhang, H., Tian, X., Lu, X., Xu, D., Guo, Y., Dong, Z., et al. (2019). TMEM25 modulates neuronal excitability and NMDA receptor subunit NR2B degradation. J. Clin. Invest. 129, 3864–3876. doi: 10.1172/JCI122599
Zhang, X., Zhang, R., Chen, J., and Wu, J. (2020). Neuroprotective effects of DAAO are mediated via the ERK1/2 signaling pathway in a glaucomatous animal model. Exp. Eye Res. 190:107892. doi: 10.1016/j.exer.2019.107892
Zhang, S., Zou, M., Lu, L., Lau, D., Ditzel, D., Delucinge-Vivier, C., et al. (2009). Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 5:e1000604. doi: 10.1371/journal.pgen.1000604
Zhao, X., Han, C., Zeng, Z., Liu, L., Wang, H., Xu, J., et al. (2020). Glutamate attenuates the survival property of IGFR through NR2B containing N-methyl-D-aspartate receptors in cortical neurons. Oxid. Med. Cell. Longev. 2020:5173184. doi: 10.1155/2020/5173184
Zheng, S., Eacker, S., Hong, S., Gronostajski, R., Dawson, T., and Dawson, V. (2010). NMDA-induced neuronal survival is mediated through nuclear factor I-A in mice. J. Clin. Invest. 120, 2446–2456. doi: 10.1172/JCI33144
Zheng, H., Tang, R., Yao, Y., Ji, Z., Cao, Y., Liu, Z., et al. (2016). MiR-219 protects against seizure in the kainic acid model of epilepsy. Mol. Neurobiol. 53, 1–7. doi: 10.1007/s12035-014-8981-5
Zhou, X., Huang, Z., Zhang, J., Chen, J., Yao, P., Mai, C., et al. (2021). Chronic oral administration of magnesium-L-threonate prevents oxaliplatin-induced memory and emotional deficits by normalization of TNF-α/NF-κB signaling in rats. Neurosci. Bull. 37, 55–69. doi: 10.1007/s12264-020-00563-x
Zhou, X., Zhang, C., Zhang, C., Peng, Y., Wang, Y., and Xu, H. (2017). MicroRNA-182-5p regulates nerve injury-induced nociceptive hypersensitivity by targeting ephrin type-b receptor 1. Anesthesiology 126, 967–977. doi: 10.1097/ALN.0000000000001588
Zhu, J., Li, K., Cao, S., Chen, X., Shen, C., Zhang, Y., et al. (2017). Increased NRG1-ErbB4 signaling in human symptomatic epilepsy. Sci. Rep. 7:141. doi: 10.1038/s41598-017-00207-7
Zhu, S., and Paoletti, P. (2015). Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr. Opin. Pharmacol. 20, 14–23. doi: 10.1016/j.coph.2014.10.009
Keywords: N-methyl-D-aspartate receptor, epilepsy, anti-NMDAR encephalitis, D-serine, glutamate, excitotoxicity, CREB, epigenomics
Citation: Chen S, Xu D, Fan L, Fang Z, Wang X and Li M (2022) Roles of N-Methyl-D-Aspartate Receptors (NMDARs) in Epilepsy. Front. Mol. Neurosci. 14:797253. doi: 10.3389/fnmol.2021.797253
Received: 18 October 2021; Accepted: 29 November 2021;
Published: 07 January 2022.
Edited by:
Tobias Engel, Royal College of Surgeons in Ireland, IrelandReviewed by:
Hermona Soreq, Hebrew University of Jerusalem, IsraelCopyright © 2022 Chen, Xu, Fan, Fang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man Li, bW1sbHN5QDEyNi5jb20=
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.