 Qi Tian1,2,3†
Qi Tian1,2,3† Li Shu1,2†
Li Shu1,2† Pu Zhang3†Ting Zeng4Yang Cao5
Pu Zhang3†Ting Zeng4Yang Cao5 Hui Xi1Ying Peng1
Hui Xi1Ying Peng1 Yaqin Wang6*
Yaqin Wang6* Xiao Mao1,2*
Xiao Mao1,2* Hua Wang1,2*
Hua Wang1,2*
- 1Department of Medical Genetics, Maternal and Child Health Hospital of Hunan Province, Changsha, China
- 2National Health Commission Key Laboratory of Birth Defects Research, Prevention and Treatment, Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
- 3Department of Obstetrics and Gynecology, Maternal and Child Health Hospital of Hunan Province, Changsha, China
- 4The Ministry of Education and Science, Maternal and Child Health Hospital of Hunan Province, Changsha, China
- 5Department of Radiology, Chenzhou First People’s Hospital, Chenzhou, China
- 6Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China
Background: MN1 C-terminal truncation (MCTT) syndrome is caused by variants in the C-terminal region of MN1, which were first described in 2020. The clinical features of MCTT syndrome includes severe neurodevelopmental and brain abnormalities. We reported on a patient who carried the MN1 variant in the C-terminal region with mild developmental delay and normal brain magnetic resonance image (MRI).
Methods: Detailed clinical information was collected in the pedigree. Whole-exome sequencing (WES) accompanied with Sanger sequencing validation were performed. A functional study based on HEK239T cells was performed.
Results: A de novo heterozygous c.3734delT: p.L1245fs variant was detected. HEK239T cells transinfected with the de novo variant showed decreased proliferation, enhanced apoptotic rate, and MN1 nuclear aggregation.
Conclusion: Our study expended the clinical and genetic spectrum of MCTT which contributes to the genetic counseling of the MN1 gene.
Introduction
MN1 (MIM 156100) gene was initially reported to be a tumor suppressor gene associated with meningioma and myeloproliferative diseases. Mak et al. (2020) first described the MN1 C-terminal truncation (MCTT) syndrome as causing craniofacial symptoms and severe neurodevelopmental abnormalities and brain abnormalities (Buijs et al., 1995; Lekanne Deprez et al., 1995). Another research group in the same year reported three probands with MN1 C-terminal variants who showed consistent clinical features (Vegas et al., 2021).
There is growing evidence of genotype–phenotype correlations of MN1-related clinical syndrome. Different from MN1 C-terminal variants, MN1 N-terminal variants were reported to cause less severe clinical syndromes. Patients with variants located in the N-terminal region of MN1 showed speech defects without significant intellectual disability, mild conductive hearing loss, and non-specific facial features (Shu et al., 2021).
Till now, atypical clinical presentations of MN1-related clinical syndrome caused by variants in C-terminal region have never been described. In our study, we first presented a MN1 C-terminal frameshift deletion variant that caused mild global developmental delay, cleft palate, and dysmorphic facial features but with no hearing loss or brain magnetic resonance image (MRI) abnormalities.
Materials and Methods
Genetic Investigation
Genomic DNA from peripheral blood leukocytes of the trio were extracted by Qiagen DNA Blood Midi/Mini Kit (Qiagen GmbH, Hilden, Germany). Data were processed preliminarily according to the protocols of whole-exome sequencing (WES) (Ulintz et al., 2019). In detail, DNA was sheared by sonication (Biorupter UCD-200, Diagenode) to approximately 200 bp. DNA fragments were repaired at the end. The sequencing adaptors were used to collect DNA fragments and the fragments (approximately 320 bp) were collected by XP beads. After amplification, the DNA fragments were captured by IDT’s xGen Exome Research Panel (Integrated DNA Technologies, San Diego, CA, United States) according to the protocol. The products were eluted and collected. DNA was then amplified and purified by PCR. The enrichment of libraries was tested by qPCR, and size distribution and concentration were determined by Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, United States). To sequence the genomic DNA of the family, WES was performed on the Illumina HiSeq 2500 system with an average coverage depth of 100× of the variants. Raw image files were processed using CASAVA v1.82 for base calling and raw data generating (Markus et al., 2020). Variants were then annotated using ANNOVAR (Wang et al., 2010).
The variants were initially filtered following HGMD and ACMG guidelines. Disease-causing mutations (DMs) and probable/possible pathological mutation (DM) in the HGMD database (Prof. version 2019.1), and pathogenic (P) and likely pathogenic (LP) variants were interpreted by ACMG guidelines. The variants were then filtered according to allele frequency, variant type, and mode of inheritance. Variants with minor allele frequencies (MAFs) <0.1%, variant depth of coverage ≥20, and alteration base depth of coverage ≥4 were chosen for further analyses. The remaining variants were further filtered according to variant type and inheritance model of the associated disease. Sanger sequencing was performed on the DNA of the proband’s parents to validate the mutation found in WES.
MN1 Subcellular Localization and Aggregation in HEK293T Cells
To create N-terminal GFP-fused human MN1 expression vector, the MN1 (GenBank: NM_002430.3) open-reading frame (ORF) was incorporated into DEST53 via the Gateway cloning system (Thermo Fisher). The mutant ORF and MN1 was amplified with a human cDNA library (Clontech). The mutant MN1 (M-MN1) was created by a KOD-plus-Mutagenesis Kit (TOYOBO). ViaFect Transfection Reagent (Promega) was used to transfect construct (500 ng each) into HEK239T cells. After 48 h of transfection, the cells were fixed with 2% paraformaldehyde, washed with PBS, stained with DAPI (Vector Laboratories), and then mounted onto slides. The sub-cellular localization and aggregation of MN1/M-MN1 were observed under confocal microscopy.
Cell Proliferation and Apoptosis Assay
Cell Proliferation Assay was carried out with Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s protocol (Vegas et al., 2021). Cell Apoptosis Assay Kit (Solarbio, CA1020) was used to detect apoptotic rate (Xiao et al., 2019).
Results
Case Description
Our patient was the first child born to the non-consanguineous Chinese parents. The proband was a 3 year and 5-month-old male born at full term from a normal pregnancy. The birth weight was 2800 g and Apgar score was 9/10. The patient presented difficulties in breast feeding. He lifted his head at 7 months old and could sit with support at the age of 8 months. He stared to walk when he was 1 year and 7 months old. He had no seizures and language development delay was observed. Physical examination showed that he had facial dysmorphism, hypertelorism, auricle deformation, upper palate cleft, plagiocephaly, protruding occipital bone, and hypotonia (Figure 1A). Behavioral observation audiometry and auditory brainstem response were normal. The brain MRI was normal (Figure 1B). The developmental milestones were presented in weight/length-for-age percentiles (Supplementary Figure 1). A de novo MN1 gene frameshift variant NM_002430.2: c.3734delT: p.L1245fs (chr22:28192798-28192798) was identified and proved by sanger sequencing in the pedigree (Figures 1C,D). The table showing the follow-up timeline is available in Supplementary Table 1.
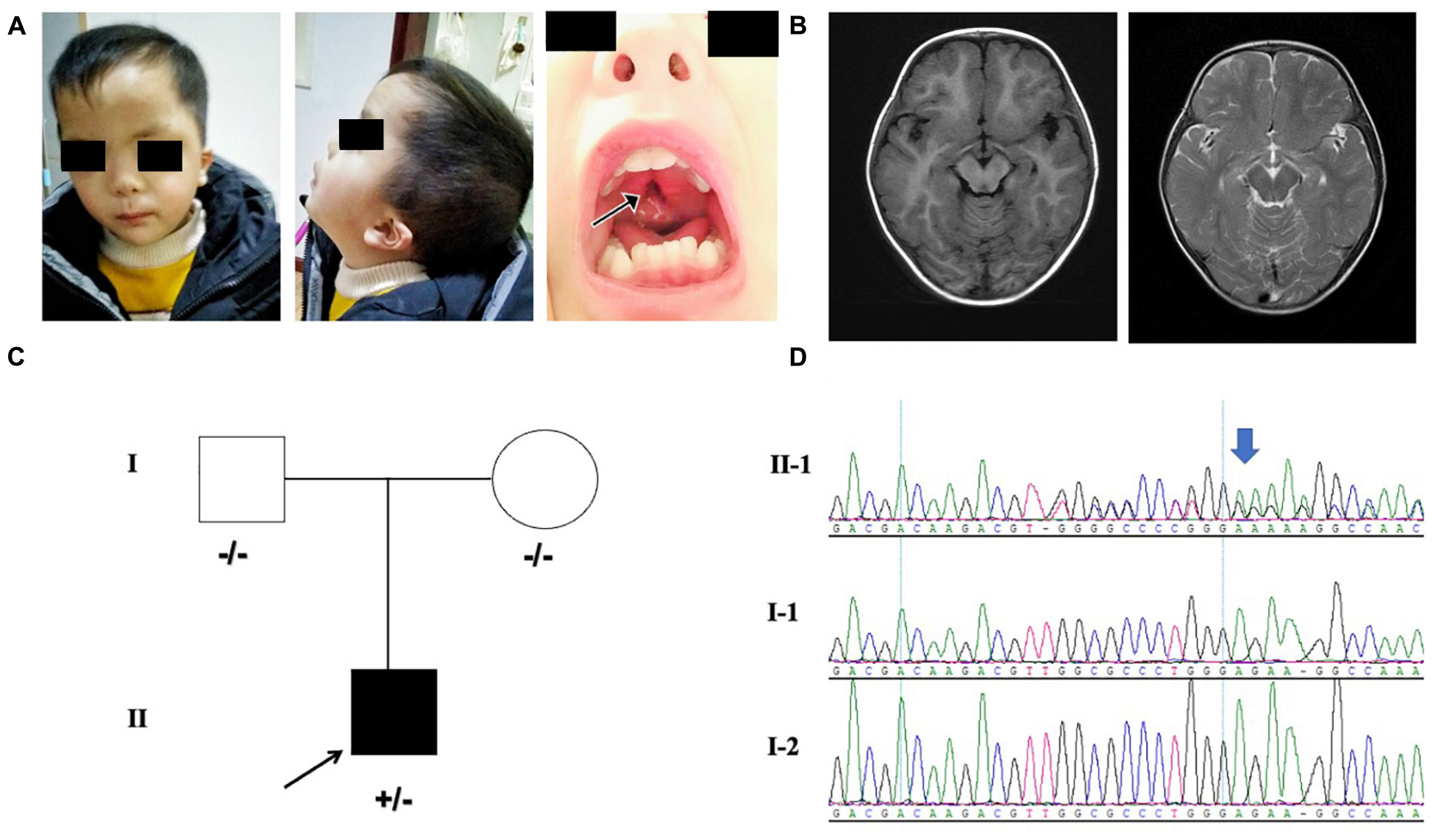
Figure 1. (A) Facial features of the proband. The arrows point to the cleft palate. (B) MRI indicating the normal brain tissue and the protruding occipital bone (image on the left is axial T1-weighted; image on the right is axial T2-weighted) of the proband at 18-months-old. (C) Pedigree with MN1 variant. Individuals with heterozygous variants are indicated by plus/minus (+/–) symbols and individuals without the variant are labeled as minus/minus (–/–) symbols. (D) Sanger sequencing results of MN1 frameshift variant in family members is presented on the right.
Functional Study for MN1 Variant in HEK239T Cells
GFP-fused wild-type and mutated-MN1 proteins were expressed in HEK239T cells. Both wild-type MN1 and M-MN1 were found to be aggregated and localized in the nuclear of HEK239T cells (Figure 2A). Intensity of M-MN1 aggregates were significantly higher than the wild-type MN1 (t-test, p = 0.017) (Figure 2B). Cell apoptotic rate were statistically higher in M-MN1 group compared with HEK239T and wild-type group (t-test, p = 0.009, 0.004, respectively) (Figure 2C).
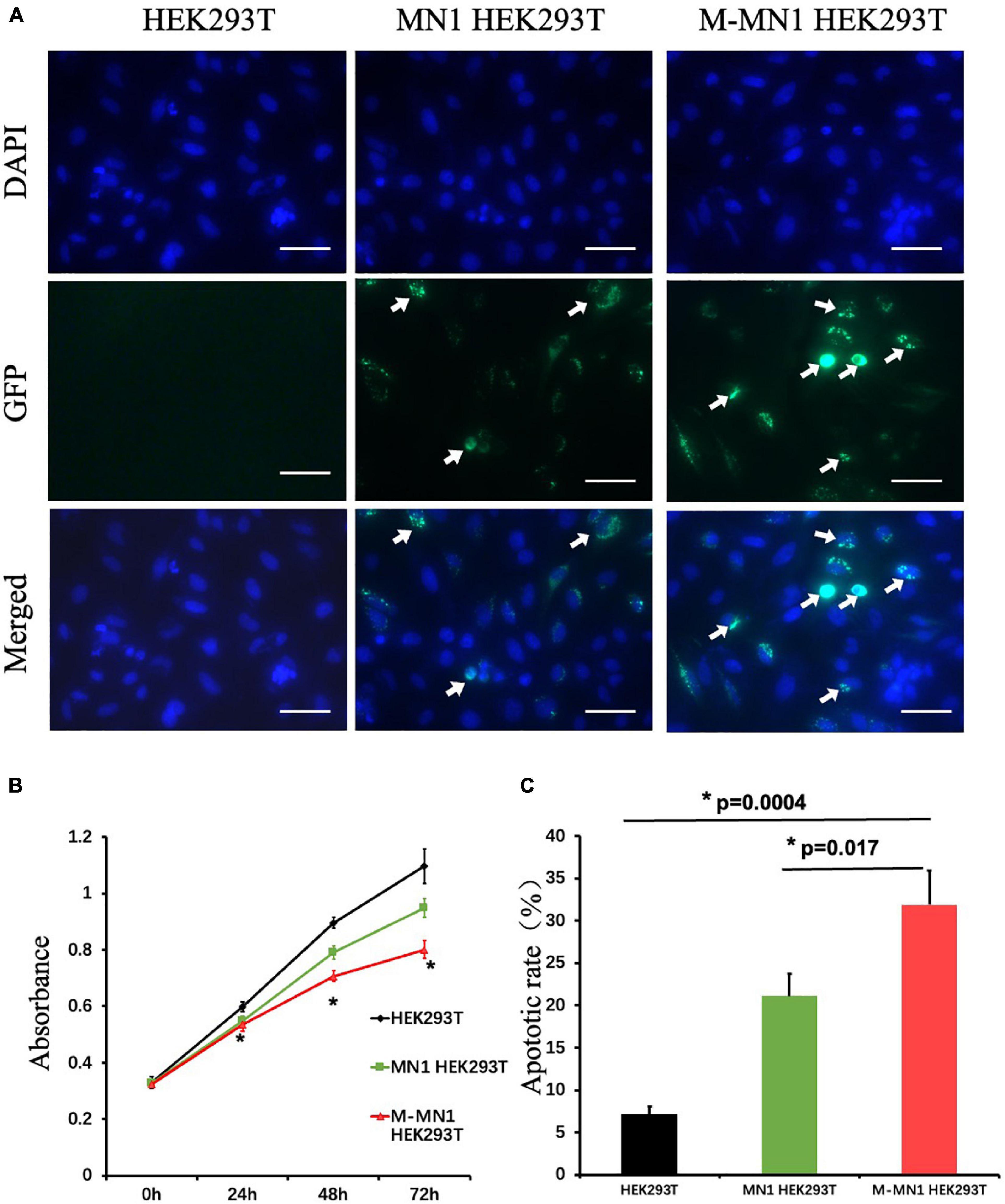
Figure 2. (A) Immunofluorescence of GFP-fused MN1 and M-MN1 showing the subcellular localization and aggregation of MN1 proteins in HEK239T cells. The arrows pointed out the represented MN1 fluorescence. Scale bars represent 50 mm. (B) Cell proliferation at 0, 24, 48, and 72 h after transinfection. The cell numbers indicated by the absorbance at 450 nm were significantly lower in transinfected groups (t-test, p = 0.017). And the M-MN1 group had the lowest absorbance reading across all time points; at 48 and 72 h the readings of M-MN1 group were significantly lower compared to MN1 group (t-test, p = 0.009, 0.004, respectively). (C) Cell apoptotic rate was significantly higher in M-MN1 group compared with HEK239T and MN1 group. *p ≤ 0.05.
Discussion
The clinical characteristics of MCTT syndrome included craniofacial features, hearing loss, severe neurodevelopmental abnormalities, and abnormal brain MRI (Vegas et al., 2021). In our study, we identified a de novo frameshift deletion variant located in C-terminal of MN1 gene in a pedigree. Our proband presented a few atypical clinical manifestations different from the reported ones (Mak et al., 2020; Miyake et al., 2020). Our patients did not show cranial shape defects, hearing loss, or brain MRI abnormalities (Table 1). Our patient provided evidence for MN1 MCTT syndrome and the different clinical features of our patient may help to refine the clinical spectrum of MN1 C-terminal variants’ related syndromes.
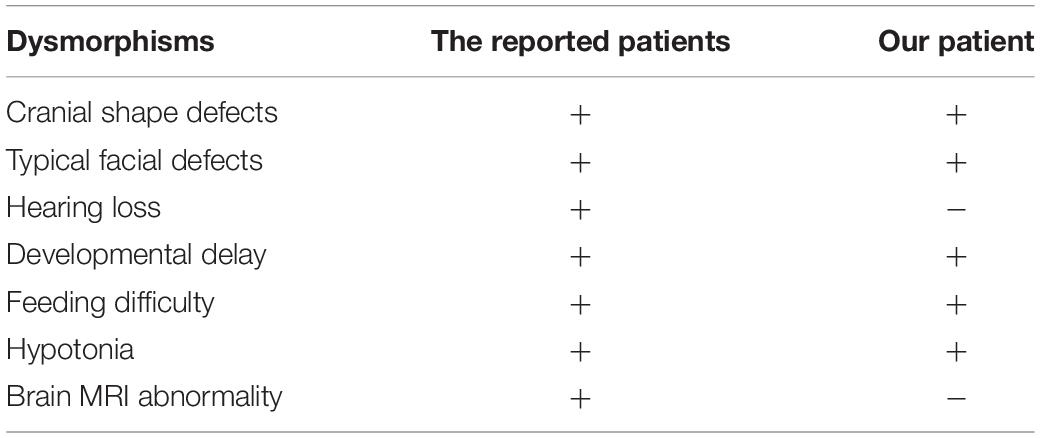
Table 1. The clinical comparison of our patient and the patients reported previously.
MN1 C-terminal heterozygous variants exert a dominant-negative or gain-of-function effect on the MN1 protein (Mak et al., 2020). The variant led to increased protein MN1 stability and enhanced MN1 nuclear aggregation, which were related to the MCTT syndrome (Miyake et al., 2020). In our study, the variant was tested by functional study and the abnormal cellular functions were detected in M-MN1 group. Combining the functional studies with the genetic findings, we proved that our MN1 variant was the cause of the diseases. Further research on how different MN1 variants lead to various clinical manifestations is needed.
Some research has revealed the molecular functions of MN1 protein. MN1 encodes a developmentally expressed transcriptional co-regulator (Liu et al., 2008). MN1 protein may act as a transcriptional cofactor and the mutant protein could impair downstream binding transcription factors, such as Cbf-β and Runx2 (Meester-Smoor et al., 2005; Miyake et al., 2020). The clinical heterogeneities of MN1 C-terminal variants may be due to the regulation of various corresponding downstream target genes (Lai et al., 2014). Our present study contributes to expanding the genetic and clinical spectrum of MN1 and aid in precise genetic counseling in the future.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author (XM), upon reasonable request. Requests to access these datasets should be directed to XM (Z2J0ZWNoaWVzQG91dGxvb2suY29t).
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Maternal and Child Health Hospital of Hunan Province (2020-S003). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XM, HW, and YW designed the research. QT, LS, and TZ interpreted the data and wrote the manuscript. QT, LS, PZ, YC, HX, and YP did the follow-up study and collected, evaluated the clinical, and genetic evidence. TZ revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81801136 and 32100162), Major Scientific and Technological Projects for Collaborative Prevention and Control of Birth Defects in Hunan Province (Nos. 2019SK1010, 2019SK1012, and 2019SK1014), the National Key R&D Program of China (No. 2019YFC1005100), the China Postdoctoral Science Foundation (No. 2019M662804), the Changsha Municipal Natural Science Foundation (No. kq2007048), Rui Xin Project of Maternal and Child Health Hospital of Hunan Province, and the Natural Science Foundation of Hunan Province (No. 2020JJ4854).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the families and clinical staff for participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2021.789778/full#supplementary-material
References
Buijs, A., Sherr, S., van Baal, S., van Bezouw, S., van der Plas, D., Geurts, et al. (1995). Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS-like TEL gene on 12p13 to the MN1 gene on 22q11. Oncogene 10, 1511–1519.
Lai, C. K., Moon, Y., Kuchenbauer, F., Starzcynowski, D. T., Argiropoulos, B., Yung, E., et al. (2014). Cell fate decisions in malignant hematopoiesis: leukemia phenotype is determined by distinct functional domains of the MN1 oncogene. PLoS One 9:e112671. doi: 10.1371/journal.pone.0112671
Lekanne Deprez, R. H., Riegman, P. H., Groen, N. A., Warringa, U. L., van Biezen, N. A., Molijn, A. C., et al. (1995). Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene 10, 1521–1528.
Liu, W., Lan, Y., Pauws, E., Meester-Smoor, M. A., Stanier, P., Zwarthoff, E. C., et al. (2008). The Mn1 transcription factor acts upstream of Tbx22 and preferentially regulates posterior palate growth in mice. Development 135, 3959–3968. doi: 10.1242/dev.025304
Mak, C. C. Y., Doherty, D., Lin, A. E., Vegas, N., Cho, M. T., Viot, G., et al. (2020). MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain 143, 55–68. doi: 10.1093/brain/awz379
Markus, F., Angelini, C., Trimouille, A., Rudolf, G., Lesca, G., Goizet, C., et al. (2020). Rare variants in the GABAA receptor subunit epsilon identified in patients with a wide spectrum of epileptic phenotypes. Mol. Genet. Genomic Med. 8:e1388. doi: 10.1002/mgg3.1388
Meester-Smoor, M. A., Vermeij, M., van Helmond, M. J., Molijn, A. C., van Wely, K. H., Hekman, A. C., et al. (2005). Targeted disruption of the Mn1 oncogene results in severe defects in development of membranous bones of the cranial skeleton. Mol. Cell Biol. 25, 4229–4236. doi: 10.1128/MCB.25.10.4229-4236.2005
Miyake, N., Takahashi, H., Nakamura, K., Isidor, B., Hiraki, Y., Koshimizu, E., et al. (2020). Gain-of-Function MN1 truncation variants cause a recognizable syndrome with craniofacial and brain abnormalities. Am. J. Hum. Genet. 106, 13–25. doi: 10.1016/j.ajhg.2019.11.011
Shu, L., He, D., Wu, D., Peng, Y., Xi, H., Mao, X., et al. (2021). MN1 gene loss-of-function mutation causes cleft palate in a pedigree. Brain 144:e18.
Ulintz, P. J., Wu, W., and Gates, C. M. (2019). Bioinformatics analysis of whole exome sequencing data. Methods Mol. Biol. 1881, 277–318.
Vegas, N., Low, K., Mak, C. C. Y., Fung, J. L. F., Hing, A. V., Chung, B. H. Y., et al. (2021). Reply: MN1 gene loss-of-function mutation causes cleft palate in a pedigree. Brain 144:e19. doi: 10.1093/brain/awaa432
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Keywords: MN1, MN1 C-terminal truncation (MCTT) syndrome, neurodevelopmental outcome, developmental delay, whole-exome sequencing
Citation: Tian Q, Shu L, Zhang P, Zeng T, Cao Y, Xi H, Peng Y, Wang Y, Mao X and Wang H (2021) MN1 Neurodevelopmental Disease-Atypical Phenotype Due to a Novel Frameshift Variant in the MN1 Gene. Front. Mol. Neurosci. 14:789778. doi: 10.3389/fnmol.2021.789778
Received: 05 October 2021; Accepted: 19 November 2021;
Published: 16 December 2021.
Edited by:
Weiping Liao, Second Affiliated Hospital of Guangzhou Medical University, ChinaCopyright © 2021 Tian, Shu, Zhang, Zeng, Cao, Xi, Peng, Wang, Mao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaqin Wang, d2FuZ3kxMUBjc3UuZWR1LmNu; Xiao Mao, Z2J0ZWNoaWVzQG91dGxvb2suY29t; Hua Wang, d2FuZ2h1YV8yMTNAaG90bWFpbC5jb20=
†These authors share first authorship