 Yue Hu1
Yue Hu1 Weiwei Tao1,2*
Weiwei Tao1,2*- 1School of Chinese Medicine, School of Integrated Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing, China
- 2Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization, National and Local Collaborative Engineering Center of Chinese Medicinal Resources Industrialization and Formulae Innovative Medicine, Nanjing University of Chinese Medicine, Nanjing, China
Traumatic brain injury (TBI) is linked to several pathologies. The blood-brain barrier (BBB) breakdown is considered to be one of the initial changes. Further, the microenvironmental alteration following TBI-induced BBB breakdown can be multi-scaled, constant, and dramatic. The microenvironmental variations after disruption of BBB includes several pathological changes, such as cerebral blood flow (CBF) alteration, brain edema, cerebral metabolism imbalances, and accumulation of inflammatory molecules. The modulation of the microenvironment presents attractive targets for TBI recovery, such as reducing toxic substances, inhibiting inflammation, and promoting neurogenesis. Herein, we briefly review the pathological alterations of the microenvironmental changes following BBB breakdown and outline potential interventions for TBI recovery based on microenvironmental modulation.
Introduction
Traumatic brain injury (TBI) is a critical public health problem in many areas worldwide, especially in the developed countries (Hydera et al., 2007; Corrigan et al., 2010; Roozenbeek et al., 2013). This injury has both short- and long-term effects on prognosis, such as TBI-associated disabilities, amnesia, depression, and other related physical or mental disorders (Dixon, 2017). The studies have found out that not only severe TBI, but also mild TBI result in long-term sequelae and psychological morbidity (Levin and Diaz-Arrastia, 2015). Despite the well-developed medical management of TBI in the United States and other countries (Stonesifer, 2008; Coronado et al., 2012), many of the survivors of TBI do not fully recover and left permanent sequela. Thus, novel perspective of pathophysiologic mechanism for TBI and the therapeutic targets are desperately needed.
The microenvironment around neurons and other cells in brain parenchyma consists of elements that greatly influence the conditions around a cell or a cell cluster, and these elements may play a direct or indirect role in affecting cell behavior biophysically or biochemically (Charles et al., 2011). Since TBI is a complex and heterogeneous disease, microenvironment in the lesion areas following TBI may changes multi-scaled, constantly and dramatically (Hemphill et al., 2015). The cell–cell and cell–matrix interactions are greatly regulated by the molecules or factors which consist in microenvironment, suggesting that the microenvironmental changes in brain play an essential role in brain injury and remodeling after TBI (Kan et al., 2012; Teschemacher et al., 2015).
Because of blood-brain barrier (BBB), most compounds from blood to brain were impeded (Daneman, 2012; Zhao et al., 2015). Thus, BBB is one of the most important sites for the control of the central nerve system (CNS) microenvironment and homeostasis (Ballabh et al., 2004; Lampron et al., 2013). At present, many researchers show great interest in the association of brain microvessels, pericytes, astrocytes, and neurons to form functional “neurovascular units” (NVU), which contribute to neurovascular coupling (McCarty, 2009; Chen et al., 2014). In addition, the BBB is the most important structure of NVU not only anatomically but also physiologically (Muoio et al., 2014; Price et al., 2016). When TBI occurred, the BBB breakdown frequently follows, and might lead to the signaling cascades and complex interactions between the pathological processes within the NVU (Korn et al., 2005; Tomkins et al., 2008), such as edema, neuroinflammation, and cell death (Shlosberg et al., 2010). These processes are closely associated with the microenvironmental changes in the damaged brain (Kan et al., 2012).
In this review, we briefly discussed the pathological alteration of TBI after BBB breakdown and the microenvironmental changes related to BBB dysfunction, e.g., the cerebral metabolic changes, cerebral blood flow (CBF), toxic molecules accumulation, inflammation, and edema. In addition, we outlined the potential intervention schemes that target BBB-related microenvironment balance, homeostasis, and improvement for post-TBI recovery.
Traumatic Brain Injury and Blood-Brain Barrier Dysfunction
The Structure and Function of Blood-Brain Barrier
Since first observed by Paul Ehrlich in 1885, until recent decades, basically, the BBB has well-known as a complex, dynamic, adaptable structure to prevent the uncontrolled leakage of substances from the blood into the brain. Herein, we briefly overview the structure and function of BBB.
Anatomically, the elements compose the BBB are the endothelial cells, astrocyte end-feet, pericytes, and the basement membranes (BM) (Figure 1, left panel): (1) For endothelial cells, they are the central component of the BBB, connected with each other through the tight junction (TJ), adheres junction (AJ), and gap junction (GJ) proteins (Liebner et al., 2018; Sharif et al., 2018). TJs composed of at least three major transmembrane proteins, such as claudin, occludin, and junctional adhesion molecules (JAMs) (Furuse et al., 1999; Balda et al., 2000; Mankertz et al., 2002; Wolburg and Lippoldt, 2002). These proteins form an impermeable barrier to fluid. In addition, many cytoplasmic proteins involved in TJ formation include zonula occludens proteins (ZO-1, ZO-2, and ZO-3), cingulin, 7H6, and so on (Matter and Balda, 2003; Tepass and Harris, 2007; Peglion et al., 2014). (2) The end-feet of astrocyte tightly sheath the vessel wall and the loss of contact between the end-feet and blood vessels also leads to a loss of TJ (Willis et al., 2004; Watkins et al., 2014). The astrocytes promote the BBB creation and maintenance by the release of various secreted factors which may be important to contribute to vessel stabilization and junctional proteins regulation (Janzer and Raff, 1987; Wolburg and Lippoldt, 2002; Lee et al., 2003; Alvarez et al., 2013; Broux et al., 2015). In addition, the astrocytes produce the biochemical enzymes and regulate blood flow which is important for BBB maintenance (Wolburg-Buchholz et al., 2009; MacVicar and Newman, 2015). (3) The pericytes share a basement membrane with endothelial cell (Attwell et al., 2016), and anchored to the basement membrane via integrins (Armulik et al., 2010). They confirmed to play the essential roles in maintaining BBB integrity (Daneman et al., 2010; Armulik et al., 2011), regulating capillary diameter, and CBF (Yemisci et al., 2009; Fernández-Kletta et al., 2010; Hall et al., 2014; Sweeney et al., 2016), promoting angiogenesis (Winkler et al., 2011) and phagocytosing toxic metabolites (Hartmann et al., 2015). Moreover, signaling between the astrocytes and pericytes exerts significant impact on BBB integrity (Yao et al., 2014; Mishra et al., 2016). (4) The BM abound all the kinds of cells mainly consist of type IV collagens, laminins, nidogen, and HSPGs also vital for BBB structural integrity. Because access of the molecules and cells to the CNS parenchyma requires not only crossing the endothelial cell, but traversing both the layers of BM (Banerjee et al., 2016).
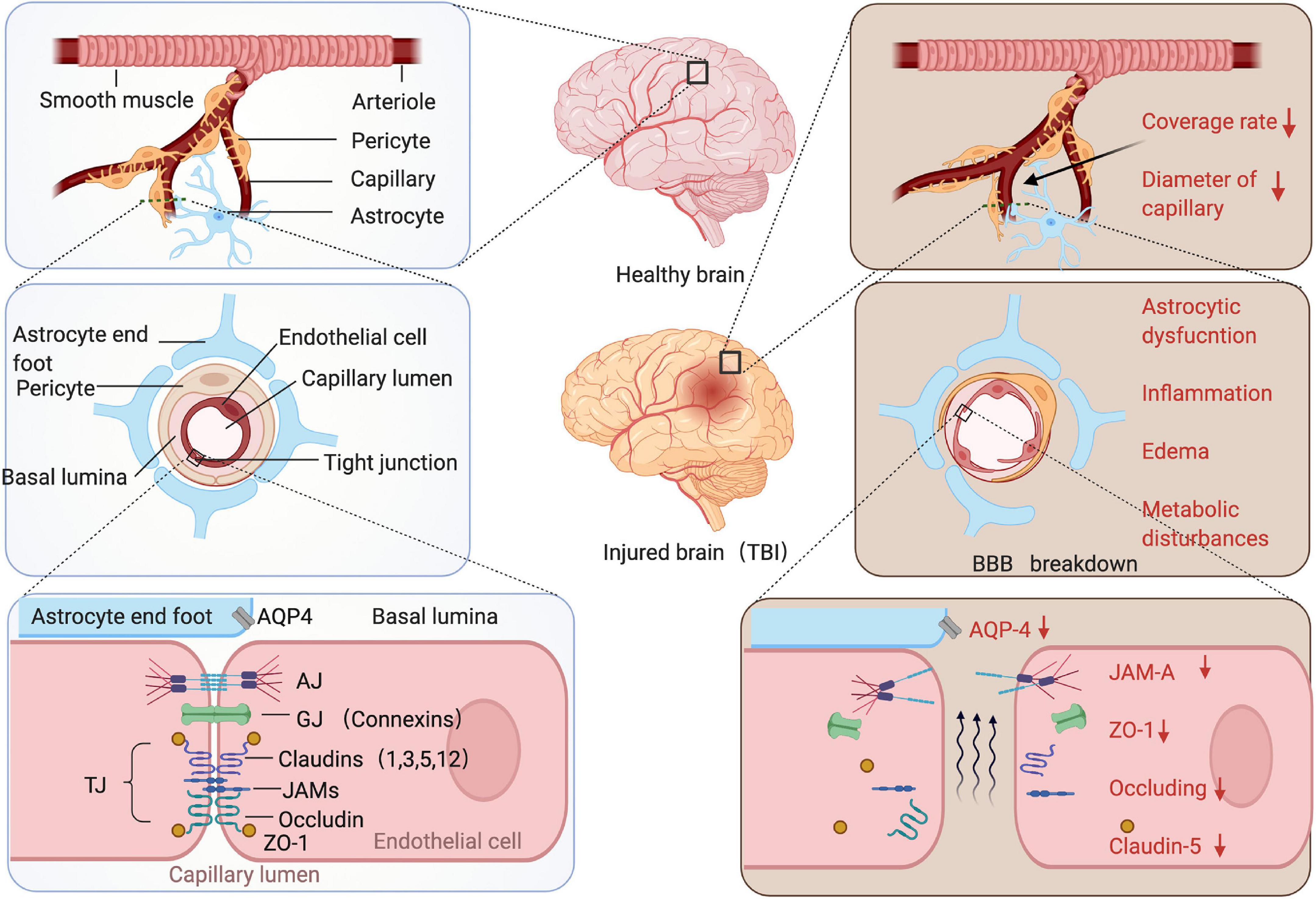
Figure 1. The stepwise amplified structure of the BBB of the healthy or injured brain. Arterioles branch off into capillaries, and capillaries are covered by pericytes and astrocytes end-feet. The pericytes and endothelium share a common basement membrane and connect with each other with several transmembrane junctional proteins. After traumatic brain injury (TBI), coverage rate of the pericytes dramatically decreased and diameter of capillary reduced, junction proteins were downregulated. There are several pathological changes occur following TBI, e.g., astrocytic dysfunction, inflammation, edema, and metabolic disturbance.
For function of BBB, in the physical condition, BBB are permeable to O2 and CO2 as well as other gaseous molecules, such as helium, N2, and many gaseous anesthetics. In addition, BBB is also permeable to water and lipid soluble. However, transfer of some molecules, especially the macromolecules through BBB are limited, it seems that the regulation of macromolecules is more complicated and usually mediated with transporters (Pardridge, 2005; Obermeier et al., 2013; Serlin et al., 2015). BBB permeability contains two aspects: (1) the ions and other small molecules cross the BBB by paracellular diffusion through the junctional complex or by the transcellular pathway across the cells. However, in some circumstances, the tight junctions may limit the paracellular flux of hydrophilic molecules across the BBB (Cancilla and DeBault, 1983; Simard and Nedergaard, 2004; Jeong et al., 2006). (2) For the macromolecules, accumulating evidence suggests that the large molecular weight serum proteins infiltration though a dysfunctional BBB carries a potential risk for pathological outcomes (Tajes et al., 2014). Thus, nearly 98% of all these molecules are not freely transported across the BBB (Pardridge, 2005). The delivery of large molecules, such as the proteins and peptides are mainly regulated by adsorptive-mediated transcytosis (AMT) and receptor-mediated transcytosis (RMT) (Dogrukol-Ak et al., 2009). Both of these processes result in passage across the BBB.
A new concept is that the BBB changes from “barrier” to “interface,” which means this structure is not only a substantial barrier for drug delivery to the brain but also a complex, dynamic interface that adapts to the needs of the CNS (Banks, 2016). BBB itself is now considered to be a therapeutic target for CNS disease and is often more accessible to the manipulation than the cells that it protects (Cho et al., 2017).
Blood-Brain Barrier Breakdown Following Traumatic Brain Injury
Under the physiological conditions, the BBB acts as a barrier that impairs the access of molecules and immune cells, such as monocytes, lymphocytes, and other leukocytes. However, BBB can easily breakdown in many neurological diseases, such as brain trauma, stroke, as well as other neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease (Kortekaas et al., 2005; Bowman et al., 2007; Zlokovic, 2008).
In general, TBI can be divided into two phases: primary and secondary injury (Hawryluk and Manley, 2015; Hay et al., 2015). The primary injuries are the result of mechanical forces causing compressive and shearing injuries, the secondary injuries are the consequence of subsequent damages, such as hypoxia, inflammation, and metabolic disturbances (Sahuquillo et al., 2001; Shlosberg et al., 2010; Lozano et al., 2015). Both the animal model and substantial clinical data indicated that BBB disruption frequently follows brain trauma and can last from several days to weeks (Tomkins et al., 2001; Korn et al., 2005). In the focal controlled cortex impact CCI animal model, the severe force delivered to the brain directly cause BBB disruption (Barzo et al., 1996; Esen et al., 2003), which is called as primary BBB damage. Following the infliction of a focal head impact, the small blood vessels often incur a concomitant shear injury, which lead to the impairments in the regulation of the BBB, CBF, and metabolic processes (Rodriguez-Baeza et al., 2003; Akbik et al., 2016). During the secondary phase, the abnormalities in the BBB can arise the abnormal brain activity, astrocytic dysfunction (Wolburg-Buchholz et al., 2009; Heinemann et al., 2012), inflammatory responses (Harting et al., 2008; Plesnila, 2016), brain edema (Unterberg et al., 2004), and metabolic disturbances (Alluri et al., 2015).
For BBB structures damage, an inevitable consequence of BBB breakdown is an increase in the permeability of the damaged endothelium (Bhowmick et al., 2019). Following TBI, the endothelium-associated tight junction proteins JAM-A, ZO-1, occludin, and claudin-5 were down-regulated indicating acute TBI-associated tight junction protein disruption (Evran et al., 2020; Sivandzade et al., 2020; Kempuraj et al., 2021). The studies showed that after animal TBI model, as many as 40% of the pericytes loss the contact of basement membrane within the first hours of the injury (Dore-Duffy et al., 2000). Then, the diameter of the arteriolar and capillary was reduced at a later time point following TBI (Prager et al., 2019). For astrocyte end-feet, AQP4 proteins are expressed abundantly on the perivascular end-foot membranes and astrocytic membranes in a polarized pattern, which mainly contribute to edema that evolves after TBI. The studies demonstrated that expression of AQP4 on the perivascular end-foot membrane reduced rapidly following TBI (Lu et al., 2020; Ma et al., 2021; Figure 1, right panel). In chronic phase, the mural cells (pericytes and smooth muscle cells) can be degenerated up to 12 months post injury, causing the alterations in tau uptake may further contribute to tau deposition in the brain (Ojo et al., 2021).
It seems that following TBI-induced BBB breakdown, together with the damage of BBB structure, microenvironmental homeostasis is quickly destructed. The imbalance of microenvironment may lead to further damage to BBB, on the other side, targeting some novel factors to improve the brain microenvironment may provide a potential approach to TBI recovery.
Microenvironmental Changes Following Traumatic Brain Injury-Induced Blood-Brain Barrier Breakdown
Although the underlying molecular changes in the microenvironment following TBI are not completely clear, with the development of microdialysis, angiography, imaging, and other techniques, our understanding of the microenvironmental changes after TBI become deeper. This section discusses the new perspective on the microenvironmental changes following TBI-induced BBB breakdown (Figure 2).
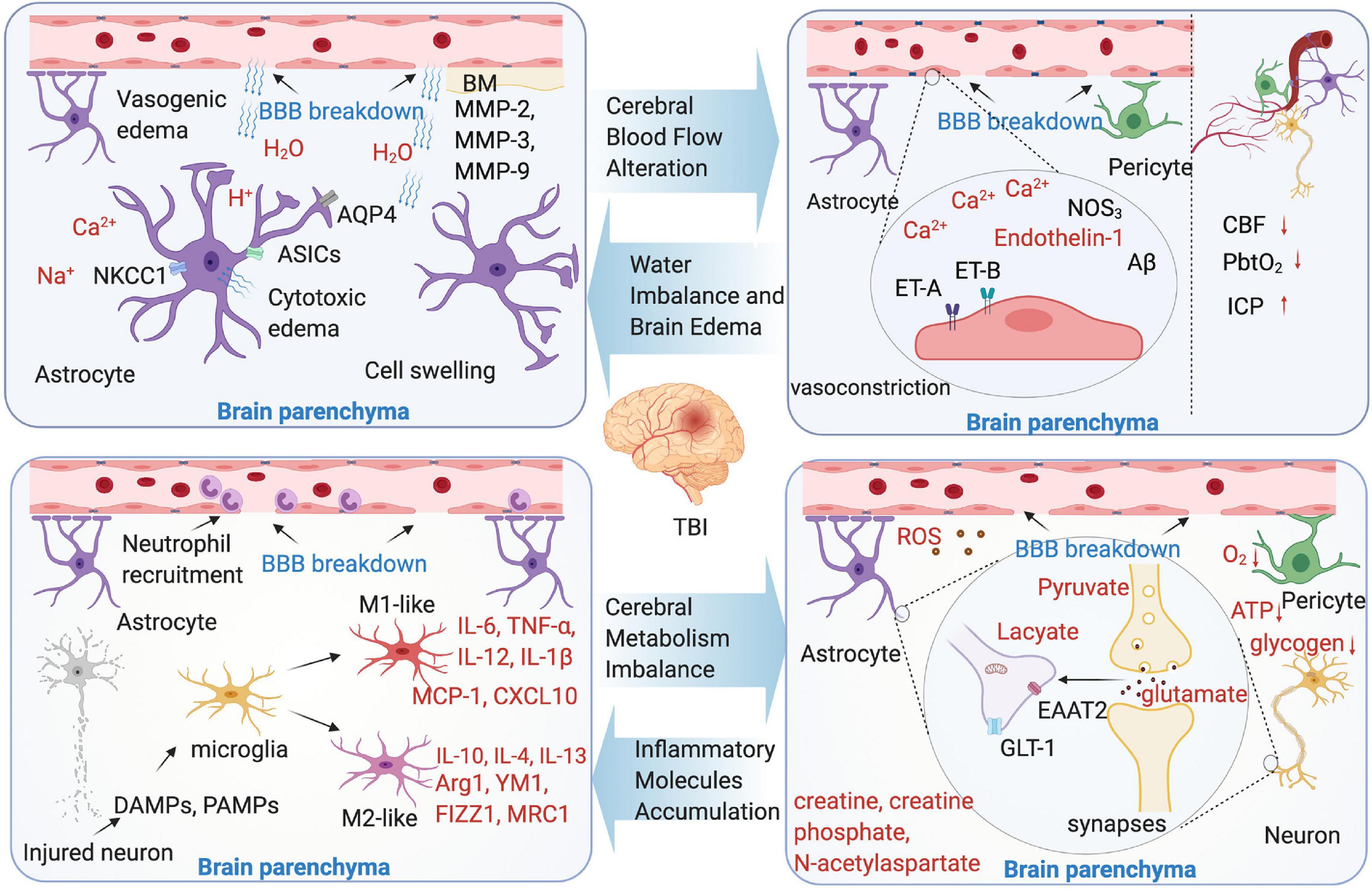
Figure 2. Microenvironment changes following TBI-induced blood-brain barrier (BBB) breakdown. Four aspects were shown as cerebral blood flow (CBF) alteration, water imbalance and brain edema, cerebral metabolism imbalance, and inflammatory molecules accumulation. The text marked red in the picture indicate the substance in brain parenchyma microenvironment.
Cerebral Blood Flow Alteration
It is already clear that both O2 and glucose are delivered to the neurons by CBF and are transported across the BBB (Moskowitz et al., 2010). CBF regulation involves complicated mechanism and contains many types of cells, such as pericyte and astrocyte (Hall et al., 2014; Hill et al., 2015; MacVicar and Newman, 2015; Kisler et al., 2017). Proper structural and functional BBB connectivity, synaptic activity, and information processing all requires precise regulation of CBF (Attwell et al., 2010). In TBI, the measurement of CBF can be invasive or non-invasive (Rostami et al., 2014), the markers of CBF, such as brain tissue oxygenation (PbtO2), Jugular venous bulb oximetry (SjvO2), ICP, and CPP, each has inherent limitations (Akbik et al., 2016).
Numerous findings from the animal TBI models have linked the endothelium cells to decreased CBF and poor outcome following brain injury. In brain vascular system, the endothelium cells, which is the main structure of BBB, play a key role to maintain vascular integrity and microenvironmental homeostasis (Graves and Kreipke, 2015). Endothelin-mediated vasoconstriction that decreases arterial luminal areas is the main reason of CBF reduction in TBI. The main mechanism is that vasoconstriction through the synthesis of endothelin-1 or upregulate endothelin receptors A and B (Faraci and Breese, 1993; Steiner et al., 2004; Kallakuri et al., 2010; Schwarzmaier et al., 2015b). In addition, in the mild to moderate TBI model, mitochondrial Ca2+ uptake improves CBF, and the intervention of this pathway may reduce behavioral deficit (Murugan et al., 2016). The pericytes and astrocyte end-feet swelling are found to contribute to CBF regulation (Ostergaard et al., 2014). Astrocytic end-feet swelling has been observed as early as 1 h after TBI (Dietrich et al., 1994), and lasts until 11 days after the initial injury (Bullock et al., 1991), which cause compression of the capillary lumen that negatively affect CBF in the injured brain. The pericytes are involved in the regulation of capillary diameter to affect CBF. After brain insult, the pericytes leave their pericapillary location within the first hour (Dore-Duffy et al., 2000), and decline in the acute phase. However, in the trauma zone, the pericytes increase days after the initial injury (Zehendner et al., 2015). It seems that the brain trauma causes a biphasic response of pericytes in the early phase of brain trauma. Loss of pericytes or the impairment of pericyte-endothelium interaction increases the BBB permeability, facilitates the formation of brain edema, and decreases the CBF in the surrounding parenchyma (Bhowmick et al., 2019). Additionally, the variants of some genes are confirmed to be related with CBF alteration in an animal TBI model. These genes include NOS3 and Aβ (Abrahamson et al., 2013).
Water Imbalance and Brain Edema
Following the primary injury of TBI, the structural and functional integrity of the BBB is disrupted, the alterations in blood flow lead to the hypoxic conditions in tissue with the activation of proteases, initiation of inflammatory pathways, generation of toxic substance, and production of reactive oxygen species (ROS), which are described previously, leading to brain edema. This edema is the result of BBB injury and can further cause tissue damage, it can be mainly classified into two types: vasogenic and cytotoxic (Unterberg et al., 2004; Lukaszewicz et al., 2011; Jha et al., 2019).
Briefly, the definition of vasogenic edema is that the water moves from the vasculature to the extracellular space, results in brain water content increase, tissue swelling, and ICP increase. Thus, the vasogenic edema from BBB opening considered to be the main contributor of the injury (Reulen et al., 1977). By using a two-photon microscopy and in vivo 3D deep-brain imaging, TBI induces vasogenic brain edema that is identified from capillaries, venules, and arterioles (Schwarzmaier et al., 2015a). Moreover, the development of vasogenic edema showed a biphasic pattern, peaking 4 and 48–72 h after TBI (Hu et al., 2021). Cytotoxic edema is characterized by the sustained intracellular water accumulation, this type of edema usually associated with a failure of the ATP-dependent Na+/K+-pumps, which further lead to the cellular ionic content increase and influx of water into the neuronal and other cells (Shapira et al., 1993). In contrast to vasogenic brain edema, cytotoxic edema with no change in tissue water content or volume and independently of the BBB integrity. Osmotic brain edema develops with osmotic gradient, and the imbalances between the blood and tissue cause cell swelling as cytotoxic edema does (Katayama and Kawamata, 2003; Unterberg et al., 2004). Additionally, numerous mediators are identified that are involved in the process of brain edema, for instance, aquaporins (AQPs), matrix metalloproteinases (MMPs), and vasoactive agents following BBB breakdown (Ke et al., 2002; Higashida et al., 2011; Blixt et al., 2015). The AQP4 is associated with the cytotoxic edema (Haj-Yasein et al., 2011), however, the opinions are controversial: the inhibition of AQP4 expression is identified associated with the brain edema reduction (Fazzina et al., 2010; Keisuke et al., 2010), however, conversely, in the AQP4 knockout animals, vasogenic edema was exacerbated after cold lesion injury, identified that AQP4 may have the function to reduce vasogenic edema (Papadopoulos et al., 2004). Other studies focus on target AQP4 to treat brain edema following TBI-induced BBB breakdown, such as oloxamer-188, edaravone, and nerve growth factor (Kikuchi et al., 2009; Bao et al., 2012; Lv et al., 2013). The MMPs are zinc-dependent endopeptidases involved in the formation of BBB. The MMPs, mainly include MMP-2, MMP-3, and MMP-9, all upregulated in the TBI animal models (Asahi et al., 2001; Falo et al., 2006; Alluri et al., 2016). The MMPs can cause BBB breakdown and further vasogenic edema, especially MMP-9. The result of a recent study shows that, in MMP-9 knock-out mice, BBB disruption was attenuated compared with the wild type mice (Asahi et al., 2001).
Cerebral Metabolism Imbalance
It is well-known that the brain undergoes a metabolic crisis after TBI, especially after BBB breakdown. As a consequence of extracellular and intracellular ionic imbalance following neuronal activation, energy production has to take place (Lin et al., 2010; Mishra et al., 2011; Lovatt et al., 2012). Usually, BBB breakdown causes a mismatch between energy demand and supply, and the tissue metabolism is regionally heterogeneous following TBI (Buxton, 2010; O’Phelan et al., 2013; Brooks and Martin, 2014). With the CBF breakdown and limited oxygen delivery, the ionic and cellular homeostasis are destroyed, resulting in intracellular calcium flux, further mitochondrial dysfunction (Giza and Hovda, 2014). In the very early phases, the oxidative metabolism may occur, it can be measured by microdialysis and MR spectroscopy imaging (Alves et al., 2003; Belli et al., 2008). The initial oxidative metabolism increases the glucose uptake in a very short period of time, however, in long term, it worsens the energy crisis of TBI. With the metabolic pathways change, the glucose metabolic rates reduce due to the breakdown of ATP-dependent pumps/transporters, at the same time, other metabolic product changes as well, such as creatine, creatine phosphate, and N-acetylaspartate (Signoretti et al., 2009). Increasingly, the lactate pyruvate ratio, which reflects impairment of hypoxic episode or cellular respiration is dramatically changed. As consequence of anaerobic metabolism and glycolysis, the amounts of lactate increased, a study by Bouzat et al. (2014) showed that exogenous systemic lactate was utilized by the injured human brain as a preferential energy substrate in TBI. This study suggests that hypertonic lactate therapy has beneficial cerebral metabolic and hemodynamic effects after TBI.
The cytotoxic molecules are released, such as excitatory amino acids which can cause damage to the brain. In general, glutamate, which is taken up by the astrocytes, largely by excitatory amino acid transporter 2 (EAAT2) or glutamate transporter-1 (GLT-1), is considered to be a main contributor to cellular apoptosis (Jansson and Akerman, 2014; Guerriero et al., 2015). In TBI, glutamate increase is among the first events to occur post-injury, and results in destroying the astrocyte function and increase BBB permeability (Obrenovitch and Urenjak, 1997; Guerriero et al., 2015). Measured by cerebral microdialysis, the glutamate levels, not only in brain, but also in blood, are confirmed to correlated with the mortality rate and long-term functional outcome in TBI clinical practice (Chamoun et al., 2010; Quintard et al., 2015). In an animal CCI model, glutamate signaling is significantly increased in the injured cortex (Cantu et al., 2015), another study by Goodrich demonstrated that GLT-1 expression is depressed, which means more glutamate gathered (Goodrich et al., 2013).
By using two-photon microscopy, tissue oxygenation, the diameters of single arterioles and capillaries at different depths in the brain cortex are measured (Tiana et al., 2010; Devor et al., 2011; Kasischke et al., 2011; Schwarzmaier et al., 2016). As for calcium flux, the astrocytes play a key role, when oxygen is limited after BBB breakdown, the astrocyte glycolysis and lactate release are maximized. The astrocytes induce vasodilation relies on the metabolic state (Gordon et al., 2008). The other cell type is pericyte, by using pericyte-deficient mice, Kisler et al. (2017) show that the pericyte degeneration diminishes capillary CBF responses, resulting in oxygen supply reduction to the brain and metabolic stress.
During primary injuries phase of TBI, the immediate cell death can cause noxious substances release and BBB breakdown. It is confirmed that ROS, mainly generated in the neurons under the pathological conditions, are the key mediators of BBB breakdown, and overproduced after BBB dysfunction (Gilgun-Sherki et al., 2002; Pun et al., 2009). ROS directly downregulate the proteins of tight junctions and indirectly activate MMPs, which lead to leakiness of the BBB and progression of neuroinflammation (Abdul-Muneer et al., 2015). In addition, ROS contribute to active Src family kinases, resulting in further dysfunction of BBB and brain edema (Liu et al., 2016). In addition, Lutton et al. (2017) reported that following TBI, with the BBB hyperpermeability, endothelial activation results in an increase expression of ICAM-1, which induce more ROS generation. Moreover, the excessive glutamate facilitates the excessive calcium influx further results in the generation of ROS, mitochondrial dysfunction, and cell death (Khatri et al., 2018).
Inflammatory Molecules Accumulation
The inflammatory response starts within hours after initial insult, corresponding with BBB disruption. The animal studies showed that the peripheral neutrophils, macrophages, T cells, and natural killer cells present in the brain within few hours after TBI (Holmin et al., 1998; Holmin and Mathiesen, 2000; Lin et al., 2017). Then, the leukocytes release pro-inflammatory cytokines and then active resident microglia (Schwarzmaier et al., 2013; Cunningham et al., 2014; Schwarzmaier and Plesnila, 2014; Corps et al., 2015; Salvador et al., 2015; Corrigan et al., 2016). Microglia sense a large repertoire of exogenous and endogenous signals and express certain surface and cytoplasmic receptors as a result of activation (Loane and Kumar, 2016).
In the acute phase following TBI, the damaged neurons and other cells release danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) into the brain (Hanisch and Kettenmann, 2007). Microglia response to these environmental signals and change their phenotypes into M1 or M2 (Xu et al., 2017). M1-like phenotype causes neuroinflammation by releasing the high level of pro-inflammatory molecules [tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), IL-12, and IL-1β], chemokines (monocyte chemoattractant protein-1 (MCP-1), CXCL10) into the microenvironment of the brain (Semple et al., 2010; Clausen et al., 2011; Tian et al., 2016; Sanchis et al., 2020; Sen et al., 2020; Zhao et al., 2020). In the lipopolysaccharide-stimulated (LPS) model, microglia are activated and release TNF-α contributed to BBB dysfunction (Nishioku et al., 2010; Semple et al., 2010; Willis et al., 2020). In addition, another study from Schlegel and Waschke (2009) suggested that TNF-α can induce microvascular endothelial barrier breakdown and reduce BBB stabilization by inhibiting cAMP level and Rac1 signaling (Baumer et al., 2009). For M2-like phenotype microglia, it is associated with the memory immune responses and may have either pro- or anti-inflammatory function. They not only produce anti-inflammatory cytokines, such as IL-10, IL-4, and IL-13, but also upregulate several factors, such as Arg1, YM1, FIZZ1, and MRC1 (Ansari, 2015). In chronic phase, inflammation following BBB dysfunction in TBI can be simultaneously helpful and deleterious (Simon et al., 2017). The experiments in the TBI animal models have shown that the levels of IL-1β, IL-6, CXCL8, IL-10, and TNFα are chronically increased together with chronic microglial activation which link to neurodegeneration and dementia, suggesting that the inflammatory molecules accumulation in brain microenvironment following TBI may last for a long time. For apoptotic factors, a study indicated that, following TBI and BBB breakdown, accumulation of caspase-3, an apoptotic factor, and its cleaved tau may contribute to microvascular disruption and cause further chronic BBB damage. This process may also accompanied by the chronic inflammatory responses, such as astrocytes and microglia activation (Glushakova et al., 2017).
Modulation of Microenvironment for Post-Traumatic Brain Injury Recovery
This section discusses the interventions that have been recently reported to modulate the microenvironment for post TBI recovery. In a neuropathological condition, the microenvironment in the brain can be toxic, which may prohibit the neural recovery. Thus, creating an optimal microenvironment in toxic “soil,” is capable of executing neural repair to promote the post-TBI recovery.
Eliminate the Toxic Substances and Excessive Water in Microenvironment
The acute microenvironmental changes post-TBI present an attractive target for modulation of the TBI symptoms and the development of cognitive changes later in life. For toxic substances eliminate, the methods should be use of specific receptor inhibitors or prevent the entry of ions, such as sodium and calcium, or reduce the content of toxic substance, e.g., ROS, malondialdehyde (MDA), or glutamate. The water elimination, the widely used mannitol is an osmotic agent, however, only for symptomatic treatment but not causal treatment. More strategies are urgently needed to point at causal treatment of edema to enhance brain microenvironment for recovery.
The administration of many drugs targets different type of toxic substances to enhance the microenvironment for neurological function improvement. The accumulating studies have shown that by inhibiting specific receptors which abundantly expressed in CNS, e.g., arginine-vasopressin (AVP) receptor, bradykinin 2 receptor, β2 adrenergic receptor, endothelin receptors B (ETB), myosin light-chain kinase (MLCK), and peroxisome proliferator-activated receptor γ (PPARγ), brain edema can be reduced (Marmarou et al., 2005; Zweckberger and Plesnila, 2009; Zlotnik et al., 2012; Rossi et al., 2013; Krieg et al., 2015, 2016; Michinaga et al., 2018, 2020; Deng et al., 2020; Table 1). The studies have reported that by using AVP V1 and V2 receptor antagonist, brain water content, and intracranial pressure of CCI model were reduced (Krieg et al., 2015, 2016). Additionally, the bradykinin and its B2 receptors play key roles in TBI recovery (Marmarou et al., 2005; Zweckberger and Plesnila, 2009; Trabold et al., 2010). The other study demonstrated that propranolol and metoprolol, β2 adrenergic receptor inhibitors, reduce excess brain glutamate levels in the microenvironment after TBI (Zlotnik et al., 2012). The highly expressed endothelin-1 (ET-1) in brain after TBI usually links with the BBB dysfunction and increases the inflammatory cytokines and chemokines. It is demonstrated that inhibitory of ETB receptor could reduce the brain edema by decreasing the level of claudin-5, occludin, and zonula occludens-1 proteins (Michinaga et al., 2018). In addition, using a MLCK inhibitor ML-7, cerebral edema can be attenuated in a close head injury model (Rossi et al., 2013). Several drugs which already approved in clinical practice show curative effect in TBI treatment, e.g., pioglitazone, bumetanide, and glibenclamide (Deng et al., 2020; Sawant-Pokam et al., 2020; Jha et al., 2021). However, the mechanism of these drugs for treating TBI only explored in the animal models: Deng et al. (2020) demonstrated that pioglitazone increased the expression of PPARγ after TBI, thus, to alleviate TBI-caused brain edema. To block the water or ion channels is also an option to reduce the brain edema. Inhibition of NKCC1/KCC2 channel (Sawant-Pokam et al., 2020), Sur1-Trpm4 channel (Jha et al., 2021), AQP4 transporter (Farr et al., 2019; Glober et al., 2019), ASIC (Yin et al., 2013), NHE-1 (Zhao et al., 2008).
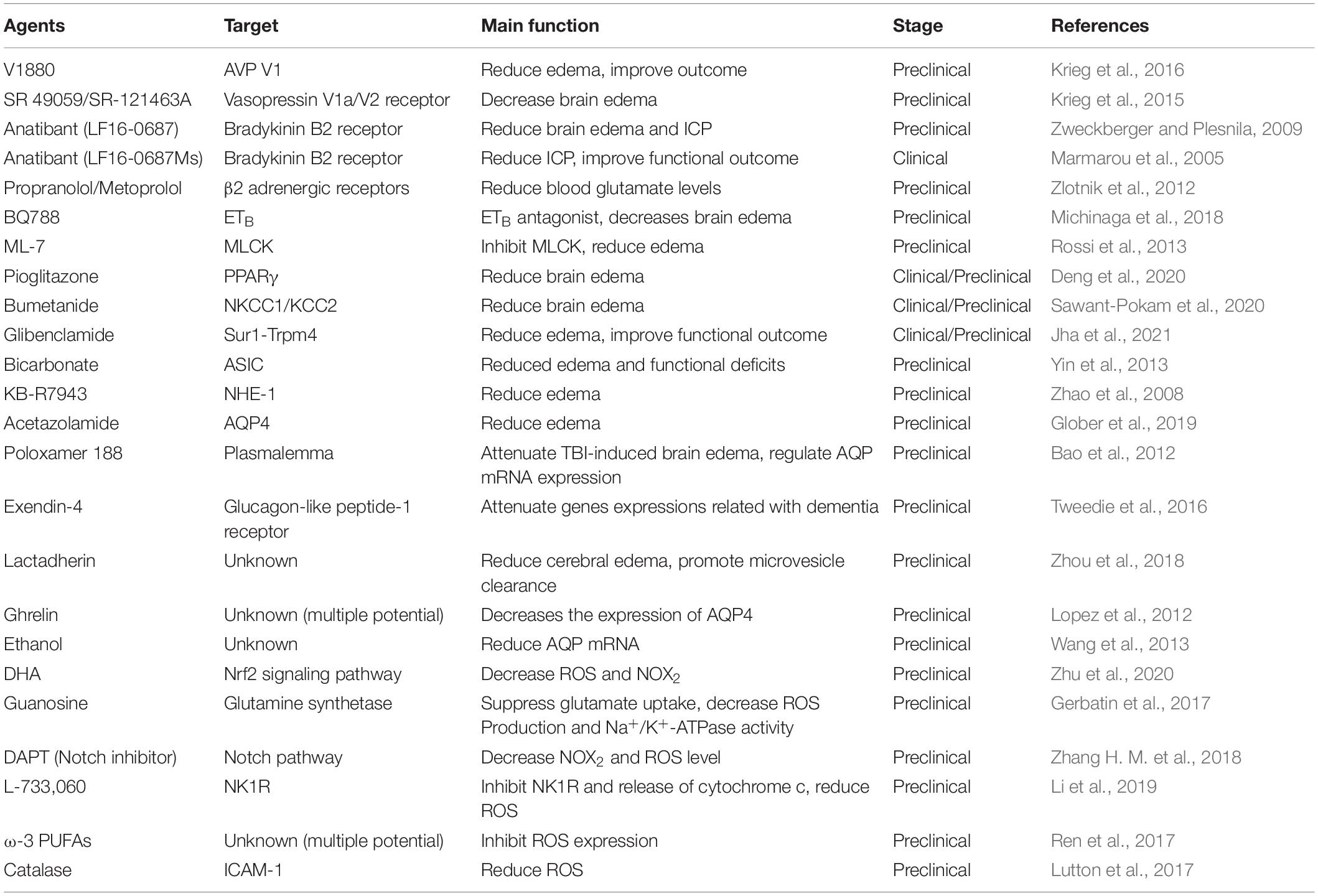
Table 1. The pharmacologic agents targeting toxic substances and edema in the microenvironment.
Besides applying the specific receptor inhibitors, some agents may have effects on regulating the essential gene expressions to help eliminate excess water, although the particular target of some agents remains unclear. For instance, poloxamer 188 could attenuate TBI-induced brain edema by regulating AQP mRNA expression (Bao et al., 2012). As an agonist of G-protein coupled receptor (GLP-1R), exendin-4 was confirmed beneficial to both type 2 diabetes mellitus (T2DM) and TBI (Tweedie et al., 2016). The studies report that exendin-4 is able to regulate the gene expression which is associated with TBI-caused dementia (Tweedie et al., 2016). Although there is no evidence that shows the specific target of lactadherin, ghrelin, and ethanol in treating TBI, these agents could influence the brain edema or the expression of AQP4 post TBI (Lopez et al., 2012; Wang et al., 2013; Zhou et al., 2018). To eliminate the toxic substance in brain parenchyma following TBI, the main option is to reduce the content of ROS. There are several agents or molecules that have confirmed to decrease the level of ROS after TBI, e.g., docosahexaenoic acid (DHA), guanosine, dual antiplatelet therapy (DAPT), omega-3 polyunsaturated fatty acids (ω-3 PUFAs), L-733,060, and catalase (Gerbatin et al., 2017; Lutton et al., 2017; Ren et al., 2017; Zhang H. M. et al., 2018; Li et al., 2019; Zhu et al., 2020). Some of these factors may have other functions. For instance, guanosine could suppress the glutamate uptake and decrease Na+/K+-ATPase activity. By inhibiting tachykinin neurokinin-1 receptor (NK1R), L-733,060 could reduce the release of cytochrome c (Li et al., 2019; Table 1).
Anti-inflammation to Enhance the Microenvironment
As mentioned previously, inflammatory response after TBI occurs within minutes and may last for days, weeks, months, or years. Due to the complexity of neural inflammatory response after TBI, certain anti-inflammatory agents are failed to improve the TBI outcomes in some clinical trials (Gaab et al., 1994; Marshall et al., 1998; Asehnoune et al., 2014). For instance, treatment with dexamethasone is failed to improve the Modified Glasgow Coma Scale for the patients with TBI (Gaab et al., 1994). A low-dose of hydrocortisone and fludrocortisone have no effect on the outcome of patients with severe TBI (Asehnoune et al., 2014). However, the emerging pre-clinical studies have been focused on the agents and drugs that can directly target the environmental inflammasome, cytokines, or chemokines, some of them may also alternatively change the macrophage/microglia polarization or regulate classical NF-κB pathway (Table 2 and Figure 3).
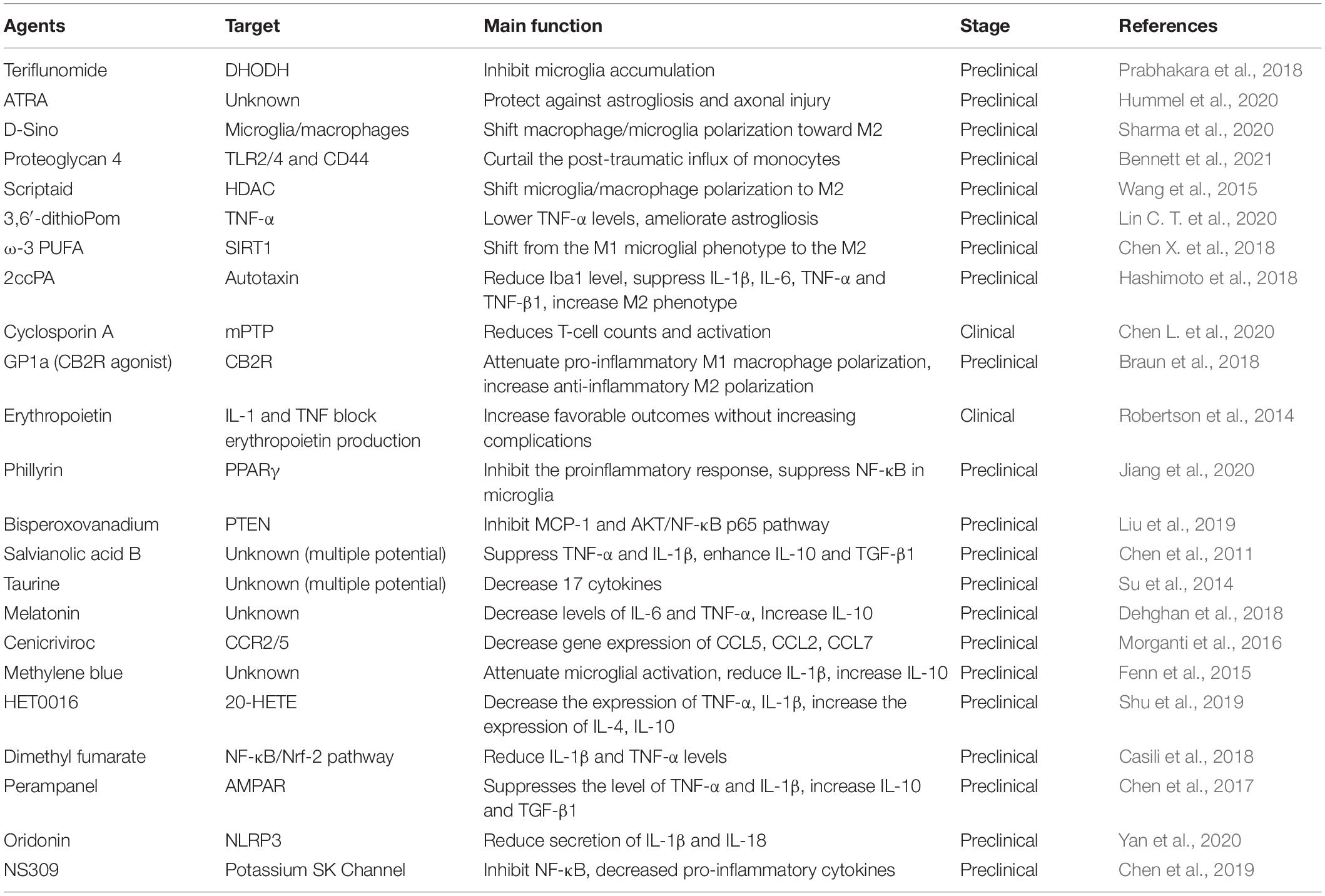
Table 2. The pharmacologic agents with anti-inflammatory effect in the microenvironment.
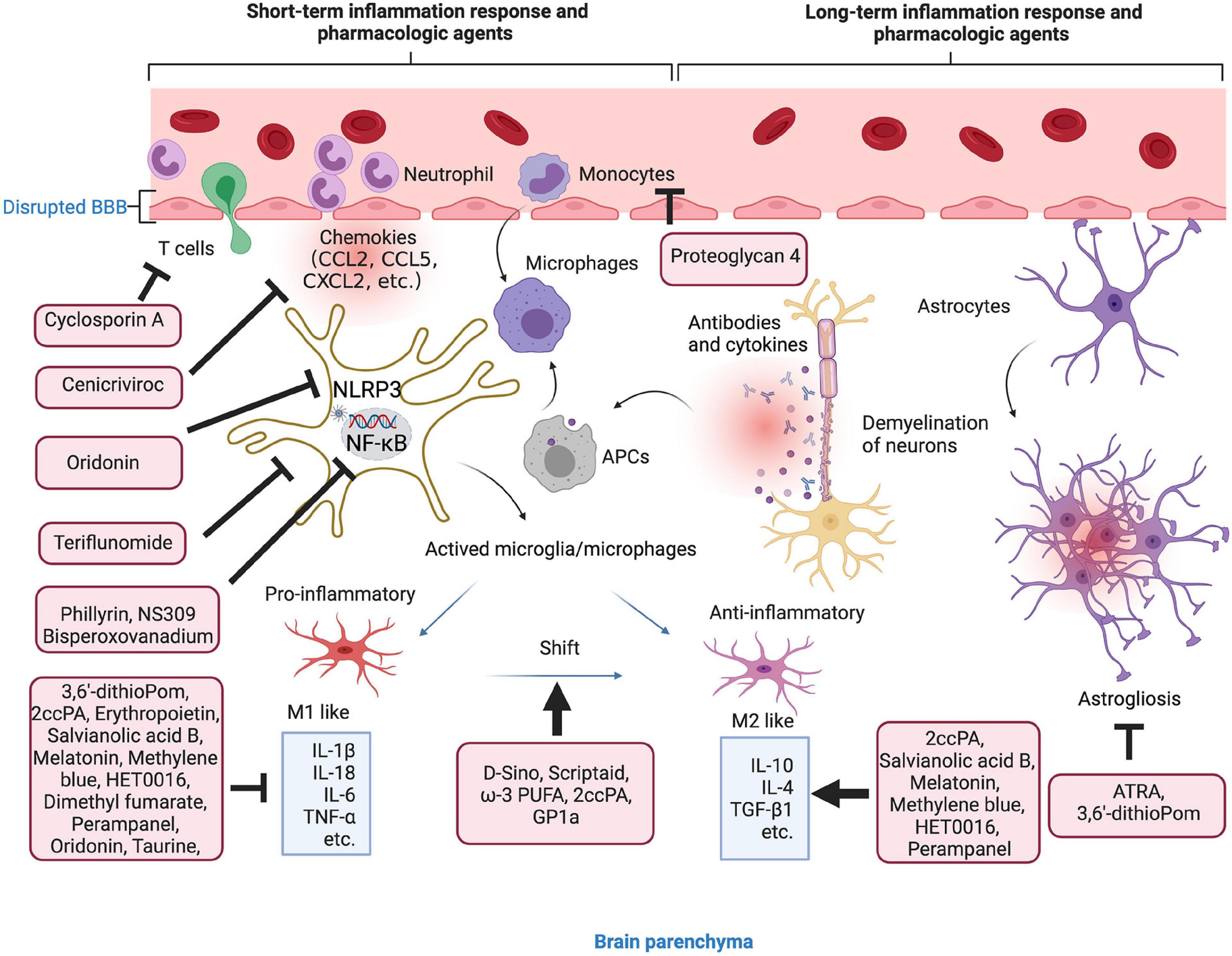
Figure 3. Anti-inflammation strategies in microenvironment. Short- and long-term inflammation response and pharmacologic agents in TBI. The agents in the red boxes showed anti-inflammatory effect in the different stages of inflammatory response.
To exert the anti-inflammatory effect, the agents or molecule may target certain type of immune cells to enhance their function, change the phenotypes, inhibit the secretion of pro-inflammatory factors, or enhance the secretion of anti-inflammatory factors (Table 2). There are several agents attenuate inflammation by inhibiting the accumulation and activation of immune cells, such as microglia, T cells, astrocytes, and monocytes (Prabhakara et al., 2018; Chen Y. et al., 2020; Hummel et al., 2020; Bennett et al., 2021). More studies have focused on the process of shifting from M1 microglial phenotype to the M2. For instance, scriptaid, a HDAC inhibitor has been found to play a critical role in shifting microglia/macrophage polarization by upregulating glycogen synthase kinase 3 beta (GSK3β) (Wang et al., 2015). The experimental studies demonstrate that small molecule, such as ω-3 PUFA, GP1a (cannabinoid receptor-2 agonist), attenuate pro-inflammatory M1 macrophage polarization, and increased anti-inflammatory M2 polarization via virous pathways (Chen et al., 2017; Lin et al., 2017; Braun et al., 2018; Chen X. et al., 2018).
Genes associated with chemotaxis (CCL2, CCL5, and CCL7), cytokine signaling (IL-6, IL-1β, TNF-β1, TNF-α, and IL-10) can be regulated or specifically inhibited by several agents or drugs, such as 3,6′-dithioPom/Pom (Lin C. T. et al., 2020), 2ccPA (Hashimoto et al., 2018), erythropoietin (Robertson et al., 2014), salvianolic acid B (Chen et al., 2011), taurine (Su et al., 2014), melatonin (Dehghan et al., 2018), cenicriviroc (Morganti et al., 2016), methylene blue (Fenn et al., 2015), HET0016 (Shu et al., 2019), dimethyl fumarate (Casili et al., 2018), and perampanel (Chen et al., 2017). The agents exert anti-inflammatory effect mainly by suppressing the pro-inflammatory factors, e.g., TNF-α, IL-1β, and IL-6, while promoting anti-inflammatory factors, e.g., IL-10 and TGF-β1. For mechanisms, NLRP3 inflammasome attracted much attention in recent years. For instance, oridonin suppresses the expression of NLRP3 inflammasome to decrease the secretion of IL-1β and IL-18 (Yan et al., 2020). In addition, small-molecule NLRP3 inflammasome inhibitor, MCC950, reduces neuroinflammation, preserves BBB integrity, alleviates TBI-induced loss of tight junction proteins, and attenuate cell death in a CCI mice model (Xu et al., 2018). Potassium SK Channel Activator NS309 inhibit NF-κB activation and further decreased the levels of pro-inflammatory cytokines and chemokines (Chen et al., 2019).
Agents in Microenvironment Targeting Blood-Brain Barrier
As we have discussed, BBB breakdown and the associated microvascular hyperpermeability are hallmark features of TBI pathological change. Thus, the agents contributing to the maintenance of BBB integrity may enhance the microenvironment and further exert brain protective function in TBI.
The efforts aimed at modification of molecular components of the BBB, e.g., TJ, AJ, and BM have shown promising therapeutic effect in treating TBI (Table 3). In recent years, various mediators targeting TJ, AJ, and BM proteins has been confirmed to play important roles in BBB repairment following TBI. Cyclosporin A antagonist CsA has been found to attenuate MMP-9 responses and enhances BBB repair in TBI animal model (Main et al., 2018). Other compounds or molecules, such as microRNA-9-5p agomir (Wu et al., 2020), FABP7 (Rui et al., 2019), mdivi-1 (Wu et al., 2018), bosentan (Michinaga et al., 2020), SB-3CT (Jia et al., 2014), also have effect on expression of the BM proteins (mainly MMP-2 and MMP-9) (Table 3). These agents could inhibit the expression of BM proteins to protect against BBB disruption through different signaling pathways. For instance, by targeting Ptch-1, microRNA-9-5p could alleviate BBB disruption though activating the Hedgehog pathway and inhibiting NF-kB/MMP-9 pathway, and further promote the recovery of neurological dysfunction in TBI (Wu et al., 2020). Wang et al. (2016) demonstrated that rhubarb, a traditional Chinese herbal medicine, prevented activation of gp91phox subunit and protect the BBB via modulating NADPH oxidase/ROS/ERK/MMP-9 signaling pathway.
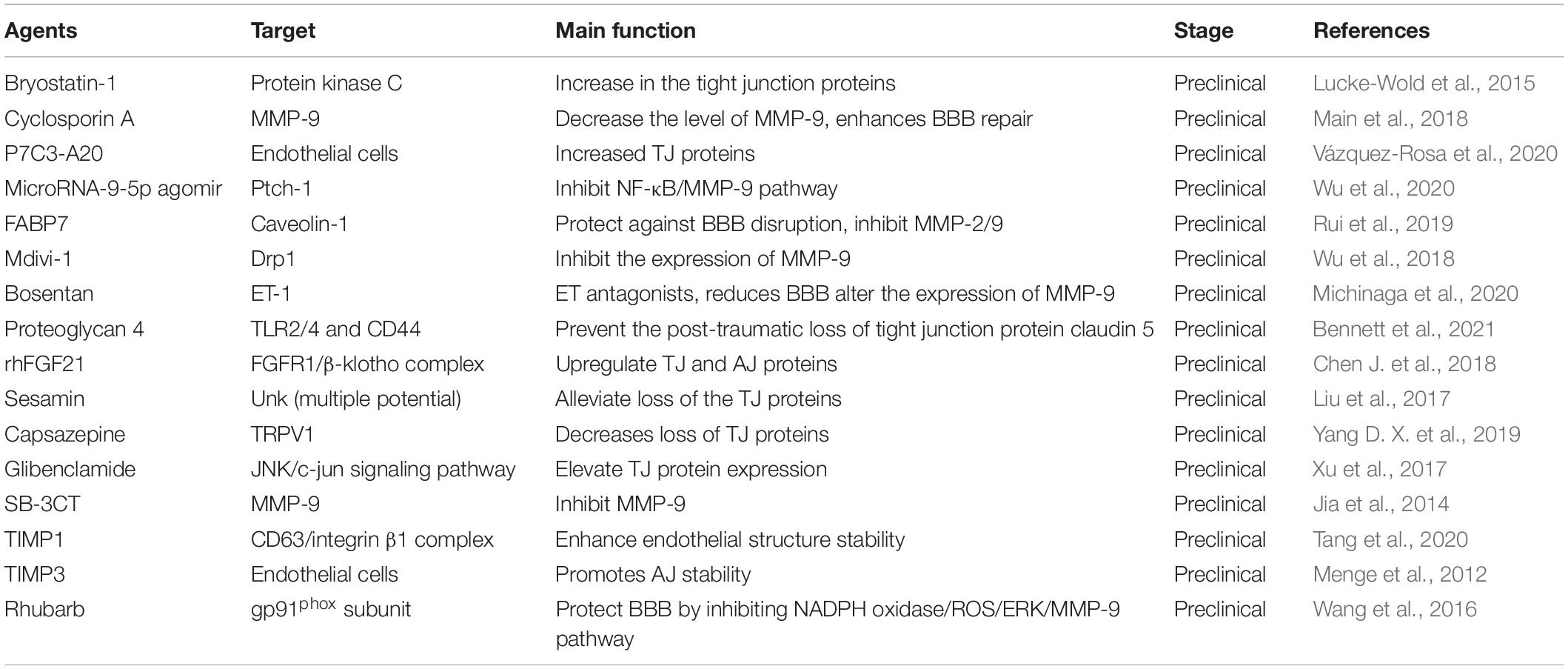
Table 3. The agents in microenvironment targeting BBB components.
Besides to suppress the BM proteins, TJ and AJ proteins are also considered as main targets for BBB protection following TBI. A study has demonstrated that P7C3-A20, a compound that stabilizes the cellular energy levels, could increase the expression of TJ proteins in different region of the brain, e.g., claudin-5 in the cortex and hippocampus, and zona occludens-1 in the cortex (Vázquez-Rosa et al., 2020). Other agents or drugs, such as proteoglycan 4, rhFGF21 (Bennett et al., 2021), sesamin (Liu et al., 2017), capsazepine (TRPV1 inhibitor) (Yang D. X. et al., 2019), glibenclamide (Xu et al., 2017), TIMP1 (Tang et al., 2020), and TIMP3 (Menge et al., 2012) also have the effect on the expression of TJ and AJ proteins, such as claudin 5, occludens-1, and ZO-1.
Molecules and Factors in Microenvironment for Neurogenesis
In a neuropathological condition, the damaged brain can activate a system of self-repair by promoting neurogenesis. Although brain tissue is poor at self-regeneration, in some cases, the quiescent cells can be mitotically activated by the vinous factors in the microenvironment. Recently, the emerging pre-clinical studies have investigated that stem cell transplantation is a novel method for treatment of TBI (Richardson et al., 2010; Koliatsos et al., 2015). However, this therapy has very low rates of cell survival due to the unbefitting microenvironment (Riess et al., 2002). Thus, targeting the specific molecules and factors to enhance the neuro-microenvironment considered to be the strategy. Recent studies show that numerous secrete factors can promote the endogenous repair response, i.e., chemokine stromal cell-derived factor 1α (SDF-1α) (Addington et al., 2015), cytokine signaling-2 (SOCS2) (Basrai et al., 2016), carbon monoxide (Choi et al., 2016), brain-derived neurotrophic factor (BDNF) (Failla et al., 2015; Shi et al., 2016), fibroblast growth factor (FGF2) (Nichols et al., 2013), and Wnt3a (Zhao Y. et al., 2016; Table 4). A new study reported that repopulating microglia can promote brain repair after TBI by regulating IL-6 and IL-6 receptor to support neurogenesis (Willis et al., 2020). In addition, mild hypothermia (MHT) therapy mitigates the degree of microenvironment and benefit for neurogenesis (Chen et al., 2016).
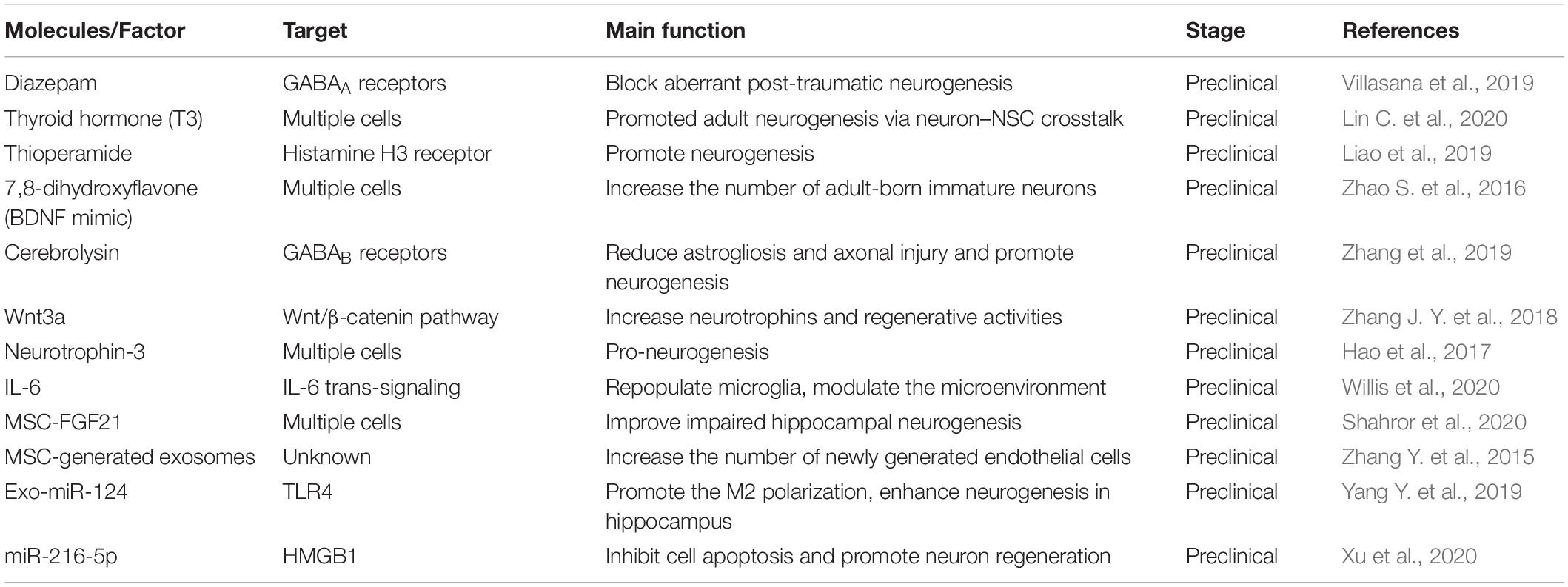
Table 4. The molecules and factors in the microenvironment for neurogenesis.
Other pathways to enhance the microenvironment for neurogenesis is exosomes delivery (Lai et al., 2013; Zhang et al., 2016). The exosomes are kind of vesicles that carry proteins and RNAs for intercellular communication, and usually have ability to cross the BBB and reach the brain parenchyma. Among them, MSCs-derived exosomes might play an essential role in neurogenesis following TBI and promise to be a novel and valuable therapeutic strategy (Xiong et al., 2017; Yang et al., 2017; Chen Y. et al., 2020). The injection of exosomes derived from the MSCs effectively improve functional recovery after TBI (Zhang Y. et al., 2015). However, the cellular and molecular mechanism of this neurogenic process remains unclear. The majority of the studies are inclined to believe that the MSCs participate in neurogenesis after TBI is not their cell replacement effects but their secretion-based paracrine effect (Zhang et al., 2016). The exosomes-induced microenvironment acts as a crucial role in the regulation of plasticity and homeostasis in the neurogenesis process. The injection of exosomes derived from the MSCs effectively improve functional recovery after TBI. In the recent years, exosomes related studies of TBI focused on miRNAs in exosomes, such as miR-124 and miR-216a-5p (Zhang L. et al., 2015; Yang Y. et al., 2019; Long et al., 2020; Table 4). Moreover, in clinical study, the exosomes can be used as the injury-specific biomarkers for TBI diagnose and considered to be potential therapeutic target (Moyron et al., 2017). Additional emphasis may be placed on promoting endogenous neurogenesis to limit cognitive impairment and to promote repair of the injured brain.
Conclusion
Traumatic brain injury is a complex, heterogeneous, and mechanobiology problem with the dynamic changes of the microenvironment following BBB disruption (Logsdon et al., 2015; Cash and Theus, 2020). Not only cells and vascular conditions are dramatically changed (Logsdon et al., 2017; Johnson et al., 2018), but also the microenvironment around neurons and other cells. Thus, understanding the underlying mechanisms of these variations after TBI are necessary in appropriate patient management (Lucke-Wold et al., 2015). Abundant studies of brain microenvironment have emerged in the areas of brain tumors and cancers (Subramani et al., 2013; Batista et al., 2015; Placone et al., 2016). However, the evidence of microenvironmental changes following TBI is inadequate. In this review, we briefly overviewed the structure and function of BBB, the pathophysiologic process of microenvironmental changes following TBI-induced BBB breakdown, such as CBF alteration, water imbalance, cerebral metabolism imbalance, and the accumulation of inflammatory molecules. By summarizing the current literature, we also listed the potential intervention to target BBB-disruption-related microenvironment for post TBI recovery. The key aspects included are reducing toxic substances and in the intercellular matrix, eliminating excessive water, inhibiting inflammation, protecting BBB components, and promoting neurogenesis. Over the up-coming years, more emerging information on the mechanism of microenvironmental changes following TBI-induced BBB disruption may help in formulating the novel strategies for post-TBI treatment.
Author Contributions
YH did major work of writing the manuscript. WT made the outline of this review. Both authors agreed to be accountable for the content of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81873096), the Priority Academic Program Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine), the Key Research and Development Projects of Ningxia (Grant No. 2021BEG02040), the Jiangsu Chinese Medicine Science and Technology Development Project (Grant No. QN202001), and Innovative and Entrepreneurial Doctor Program of Jiangsu Province (Grant No. JSSCBS20210326).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The figures were created with BioRender.com.
Abbreviations
2ccPA, 2-carba-cyclic phosphatidic acid; 20-HETE, 20-hydroxyeicosatetraenoic acid; ω -3 PUFAs, omega-3 polyunsaturated fatty acids; AJ, adherens junction; AMPAR, α-amino -3-hydroxy-5-methyl-4-isoxazole propionate receptor; APCs, antigen-presenting cell; AQPs, aquaporins, Arg1, arginase 1; ASICs, Acid sensing ion channels; ATRA, All-trans retinoic acid; ATP, adenosine triphosphate; AVP V1, arginine-vasopressin V1; BBB, blood-brain barrier; BDNF, brain-derived neurotrophic factor; BM, basement membranes; CB2R, cannabinoid B2 receptors; CBF, cerebral blood flow; controlled cortex impact, CCI; CCL2, chemokine (C-C motif) ligand 2; CCL5, chemokine (C-C motif) ligand 5; CD44, cluster of differentiation 44; CNS, central nerve systems; CXCL2, chemokine (C-X-C motif) ligand 2; DAMPs, danger-associated molecular pattern; DHA, Docosahexaenoic acid; DHODH, dihydroorotate-dehydrogenase; Drp1, dynamin-related protein 1; D-Sino, dendrimer sinomenine; EAAT2, excitatory amino acid transporter 2; ET-1, endothelin-1; ET-A, endothelin receptors A; ET-B, endothelin receptors B; Exo-miR-124, miR-124 enriched exosomes; FGFR1, Fibroblast growth factor receptor; FIZZ1, resistin-like- α; FGF21, Fibroblast growth factor 21; GABA, gamma-aminobutyric acid; GJ, gap junction; GLT-1, glutamate transporter-1; HET0016, N-hydroxy -N-4-butyl-2-methylphenylformamidine; HDACs, histone deacetylases; HMGB1, high-mobility group box 1; ICAM-1, intercellular adhesion molecule 1; ICP, intracranial pressure; IL, interleukin; JAMs, junctional adhesion molecules; JNK, stress activated protein kinase; Mdivi-1, mitochondrial division inhibitor 1; MCP-1, monocyte chemoattractant protein-1; MLCK, myosin light-chain kinase; MMP, matrix metalloproteinases; mPTP, mitochondrial permeability transition pore; MRC1, mannose receptor C-1; MSC, mesenchymal stem cells; NK1R, tachykinin neurokinin-1 receptor; NKCC1/KCC2, Na+-K+-Cl– cotransporter 1/K+-Cl– cotransporter 2; NF- κ B, transcription factors of the nuclear factor kappa B; Nrf2, nuclear factor erythroid-2 related factor 2; PAMPs, pathogen-associated molecular patterns; PbtO2, trial pressure of brain tissue oxygen; PPAR γ, peroxisome proliferator-activated receptor γ; PTEN, Phosphatase and tensin homolog; Ptch-1, Patched 1; ROS, reactive oxygen species; SEMA3A, Semaphorin 3A; SIRT1, sirtuin1; TBI, Traumatic brain injury; TJ, tight junction; TIMP1, Tissue inhibitor of metalloproteinase-1; TIMP3, tissue inhibitor of matrix metalloproteinase-3; TLR2/4, Toll-like receptor 2/4; TNF- α, tumor necrosis factor-alpha; TRPV1, Transient receptor potential vanilloid 1; NVU, neurovascular units; YM1, chitinase 3-like 3; ZO-1, zonula occludens-1.
References
Abdul-Muneer, P. M., Chandra, N., and Haorah, J. (2015). Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 51, 966–979. doi: 10.1007/s12035-014-8752-3
Abrahamson, E. E., Foley, L. M., Dekosky, S. T., Hitchens, T. K., Ho, C., Kochanek, P. M., et al. (2013). Cerebral blood flow changes after brain injury in human amyloid-beta knock-in mice. J. Cereb. Blood Flow. Metab. 33, 826–833. doi: 10.1038/jcbfm.2013.24
Addington, C. P., Heffernan, J. M., Millar-Haskell, C. S., Tucker, E. W., Sirianni, R. W., and Stabenfeldt, S. E. (2015). Enhancing neural stem cell response to SDF-1alpha gradients through hyaluronic acid-laminin hydrogels. Biomaterials 72, 11–19. doi: 10.1016/j.biomaterials.2015.08.041
Akbik, O. S., Carlson, A. P., Krasberg, M., and Yonas, H. (2016). The Utility of Cerebral Blood Flow Assessment in TBI. Curr. Neurol. Neurosci. Rep. 16:72. doi: 10.1007/s11910-016-0672-3
Alluri, H., Wiggins-Dohlvik, K., Davis, M. L., Huang, J. H., and Tharakan, B. (2015). Blood-brain barrier dysfunction following traumatic brain injury. Metab. Brain Dis. 30, 1093–1104. doi: 10.1007/s11011-015-9651-7
Alluri, H., Wilson, R. L., Anasooya Shaji, C., Wiggins-Dohlvik, K., Patel, S., Liu, Y., et al. (2016). Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS One 11:e0154427. doi: 10.1371/journal.pone.0154427
Alvarez, J. I., Katayama, T., and Prat, A. (2013). Glial influence on the blood brain barrier. Glia 61, 1939–1958. doi: 10.1002/glia.22575
Alves, O. L., Doyle, J., Tobias, C., Gilman, C. I., and Bullock, R. (2003). Evaluation of Topiramate Neuroprotective Effect in Severe TBI Using Microdialysis. Ann. N.Y. Acad. Sci. 993, 25–34.
Ansari, M. A. (2015). Temporal profile of M1 and M2 responses in the hippocampus following early 24h of neurotrauma. J. Neurol. Sci. 357, 41–49. doi: 10.1016/j.jns.2015.06.062
Armulik, A., Genove, G., and Betsholtz, C. (2011). Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 21, 193–215. doi: 10.1016/j.devcel.2011.07.001
Armulik, A., Genove, G., Mae, M., Nisancioglu, M. H., Wallgard, E., Niaudet, C., et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468, 557–561. doi: 10.1038/nature09522
Asahi, M., Wang, X., Mori, T., Sumii, T., Jung, J. C., Moskowitz, M. A., et al. (2001). Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 21, 7724–7732.
Asehnoune, K., Seguin, P., Allary, J., Feuillet, F., Lasocki, S., Cook, F., et al. (2014). Hydrocortisone and fludrocortisone for prevention of hospital-acquired pneumonia in patients with severe traumatic brain injury (Corti-TC): a double-blind, multicentre phase 3, randomised placebo-controlled trial. Lancet Respir. Med. 2, 706–716. doi: 10.1016/s2213-2600(14)70144-4
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., Macvicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi: 10.1038/nature09613
Attwell, D., Mishra, A., Hall, C. N., O’Farrell, F. M., and Dalkara, T. (2016). What is a pericyte? J. Cereb. Blood Flow Metab. 36, 451–455. doi: 10.1177/0271678X15610340
Balda, M. S., Flores-Maldonado, C., Cereijido, M., and Matter, K. (2000). Multiple domains of occludin are involved in the regulation of paracellular permeability. J. Cell Biochem. 78, 85–96.
Ballabh, P., Braun, A., and Nedergaard, M. (2004). The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol. Dis. 16, 1–13. doi: 10.1016/j.nbd.2003.12.016
Banerjee, J., Shi, Y., and Azevedo, H. S. (2016). In vitro blood-brain barrier models for drug research: state-of-the-art and new perspectives on reconstituting these models on artificial basement membrane platforms. Drug Discov. Today 21, 1367–1386. doi: 10.1016/j.drudis.2016.05.020
Banks, W. A. (2016). From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 15, 275–292. doi: 10.1038/nrd.2015.21
Bao, H. J., Wang, T., Zhang, M. Y., Liu, R., Dai, D. K., Wang, Y. Q., et al. (2012). Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem. Res. 37, 2856–2867. doi: 10.1007/s11064-012-0880-4
Barzo, P., Marmarou, A., Fatouros, P., Corwin, F., and Dunbar, J. (1996). Magnetic resonance imaging-monitored acute blood-brain barrier changes in experimental traumatic brain injury. J. Neurosurg. 85, 1113–1121. doi: 10.3171/jns.1996.85.6.1113
Basrai, H. S., Christie, K. J., Turbic, A., Bye, N., and Turnley, A. M. (2016). Suppressor of Cytokine Signaling-2 (SOCS2) Regulates the Microglial Response and Improves Functional Outcome after Traumatic Brain Injury in Mice. PLoS One 11:e0153418. doi: 10.1371/journal.pone.0153418
Batista, A., Riedemann, L., Vardam, T., and Jain, R. K. (2015). Targeting the Tumor Microenvironment to Enhance Pediatric Brain Cancer Treatment. Cancer J. 21, 307–313. doi: 10.1097/ppo.0000000000000125
Baumer, Y., Spindler, V., Werthmann, R. C., Bunemann, M., and Waschke, J. (2009). Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J. Cell Physiol. 220, 716–726. doi: 10.1002/jcp.21819
Belli, A., Sen, J., Petzold, A., Russo, S., Kitchen, N., and Smith, M. (2008). Metabolic failure precedes intracranial pressure rises in traumatic brain injury: a microdialysis study. Acta Neurochir. 150, 461–469. doi: 10.1007/s00701-008-1580-3
Bennett, M., Chin, A., Lee, H. J., Morales Cestero, E., Strazielle, N., Ghersi-Egea, J. F., et al. (2021). Proteoglycan 4 Reduces Neuroinflammation and Protects the Blood-Brain Barrier after Traumatic Brain Injury. J. Neurotrauma 38, 385–398. doi: 10.1089/neu.2020.7229
Bhowmick, S., D’Mello, V., Caruso, D., Wallerstein, A., and Abdul-Muneer, P. M. (2019). Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 317, 260–270. doi: 10.1016/j.expneurol.2019.03.014
Blixt, J., Svensson, M., Gunnarson, E., and Wanecek, M. (2015). Aquaporins and blood-brain barrier permeability in early edema development after traumatic brain injury. Brain Res. 1611, 18–28. doi: 10.1016/j.brainres.2015.03.004
Bouzat, P., Sala, N., Suys, T., Zerlauth, J. B., Marques-Vidal, P., Feihl, F., et al. (2014). Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 40, 412–421. doi: 10.1007/s00134-013-3203-6
Bowman, G. L., Kaye, J. A., Moore, M., Waichunas, D., Carlson, N. E., and Quinn, J. F. (2007). Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology 68, 1809–1814. doi: 10.1212/01.wnl.0000262031.18018.1a
Braun, M., Khan, Z. T., Khan, M. B., Kumar, M., Ward, A., Achyut, B. R., et al. (2018). Selective activation of cannabinoid receptor-2 reduces neuroinflammation after traumatic brain injury via alternative macrophage polarization. Brain Behav. Immun. 68, 224–237. doi: 10.1016/j.bbi.2017.10.021
Brooks, G. A., and Martin, N. A. (2014). Cerebral metabolism following traumatic brain injury: new discoveries with implications for treatment. Front. Neurosci. 8:408. doi: 10.3389/fnins.2014.00408
Broux, B., Gowing, E., and Prat, A. (2015). Glial regulation of the blood-brain barrier in health and disease. Semin. Immunopathol. 37, 577–590. doi: 10.1007/s00281-015-0516-2
Bullock, R., Maxwell, W. L., Graham, D. I., Teasdale, G. M., and Adams, J. H. (1991). Glial swelling following human cerebral contusion: an ultrastructural study. J. Neurol. Neurosurg. Psychiatry 54, 427–434. doi: 10.1136/jnnp.54.5.427
Buxton, R. B. (2010). Interpreting oxygenation-based neuroimaging signals: the importance and the challenge of understanding brain oxygen metabolism. Front. Neuroenergetics 2:8. doi: 10.3389/fnene.2010.00008
Cancilla, P. A., and DeBault, L. E. (1983). Neutral amino acid transport properties of cerebral endothelial cells in vitro. J. Neuropathol. Exp. Neurol. 42, 191–199.
Cantu, D., Walker, K., Andresen, L., Taylor-Weiner, A., Hampton, D., Tesco, G., et al. (2015). Traumatic Brain Injury Increases Cortical Glutamate Network Activity by Compromising GABAergic Control. Cereb Cortex 25, 2306–2320. doi: 10.1093/cercor/bhu041
Cash, A., and Theus, M. H. (2020). Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 21:9. doi: 10.3390/ijms21093344
Casili, G., Campolo, M., Paterniti, I., Lanza, M., Filippone, A., Cuzzocrea, S., et al. (2018). Dimethyl Fumarate Attenuates Neuroinflammation and Neurobehavioral Deficits Induced by Experimental Traumatic Brain Injury. J. Neurotrauma 35, 1437–1451. doi: 10.1089/neu.2017.5260
Chamoun, R., Suki, D., Gopinath, S. P., Goodman, J. C., and Robertson, C. (2010). Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J. Neurosurg. Anesthesiol. 113, 564–570. doi: 10.3171/2009.12.JNS09689)
Charles, N. A., Holland, E. C., Gilbertson, R., Glass, R., and Kettenmann, H. (2011). The brain tumor microenvironment. Glia 59, 1169–1180. doi: 10.1002/glia.21136
Chen, B. R., Kozberg, M. G., Bouchard, M. B., Shaik, M. A., and Hillman, E. M. (2014). A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J. Am. Heart Assoc. 3:e000787. doi: 10.1161/JAHA.114.000787
Chen, C., Ma, T. Z., Wang, L. N., Wang, J. J., Tu, Y., Zhao, M. L., et al. (2016). Mild hypothermia facilitates the long-term survival of newborn cells in the dentate gyrus after traumatic brain injury by diminishing a pro-apoptotic microenvironment. Neuroscience 335, 114–121. doi: 10.1016/j.neuroscience.2016.08.038
Chen, J., Hu, J., Liu, H., Xiong, Y., Zou, Y., Huang, W., et al. (2018). FGF21 Protects the Blood-Brain Barrier by Upregulating PPARγ via FGFR1/β-klotho after Traumatic Brain Injury. J. Neurotrauma 35, 2091–2103. doi: 10.1089/neu.2017.5271
Chen, L., Song, Q., Chen, Y., Meng, S., Zheng, M., Huang, J., et al. (2020). Tailored Reconstituted Lipoprotein for Site-Specific and Mitochondria-Targeted Cyclosporine A Delivery to Treat Traumatic Brain Injury. ACS Nano 14, 6636–6648. doi: 10.1021/acsnano.9b09186
Chen, T., Dai, S. H., Jiang, Z. Q., Luo, P., Jiang, X. F., Fei, Z., et al. (2017). The AMPAR Antagonist Perampanel Attenuates Traumatic Brain Injury Through Anti-Oxidative and Anti-Inflammatory Activity. Cell Mol. Neurobiol. 37, 43–52. doi: 10.1007/s10571-016-0341-8
Chen, T., Liu, W., Chao, X., Zhang, L., Qu, Y., Huo, J., et al. (2011). Salvianolic acid B attenuates brain damage and inflammation after traumatic brain injury in mice. Brain Res. Bull 84, 163–168. doi: 10.1016/j.brainresbull.2010.11.015
Chen, T., Zhu, J., Hang, C. H., and Wang, Y. H. (2019). The Potassium SK Channel Activator NS309 Protects Against Experimental Traumatic Brain Injury Through Anti-Inflammatory and Immunomodulatory Mechanisms. Front. Pharmacol. 10:1432. doi: 10.3389/fphar.2019.01432
Chen, X., Chen, C., Fan, S., Wu, S., Yang, F., Fang, Z., et al. (2018). Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflammation 15:116. doi: 10.1186/s12974-018-1151-3
Chen, Y., Li, J., Ma, B., Li, N., Wang, S., Sun, Z., et al. (2020). MSC-derived exosomes promote recovery from traumatic brain injury via microglia/macrophages in rat. Aging 12, 18274–18296. doi: 10.18632/aging.103692
Cho, C. F., Wolfe, J. M., Fadzen, C. M., Calligaris, D., Hornburg, K., Chiocca, E. A., et al. (2017). Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 8:15623. doi: 10.1038/ncomms15623
Choi, Y. K., Maki, T., Mandeville, E. T., Koh, S. H., Hayakawa, K., Arai, K., et al. (2016). Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 22, 1335–1341. doi: 10.1038/nm.4188
Clausen, F., Hånell, A., Israelsson, C., Hedin, J., Ebendal, T., Mir, A. K., et al. (2011). Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 34, 110–123. doi: 10.1111/j.1460-9568.2011.07723.x
Coronado, V. G., McGuire, L. C., Sarmiento, K., Bell, J., Lionbarger, M. R., Jones, C. D., et al. (2012). Trends in Traumatic Brain Injury in the U.S. and the public health response: 1995-2009. J. Safety Res. 43, 299–307. doi: 10.1016/j.jsr.2012.08.011
Corps, K. N., Roth, T. L., and McGavern, D. B. (2015). Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 72, 355–362. doi: 10.1001/jamaneurol.2014.3558
Corrigan, F., Mander, K. A., Leonard, A. V., and Vink, R. (2016). Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflammation 13:264. doi: 10.1186/s12974-016-0738-9
Corrigan, J. D., Selassie, A. W., Orman, J. A., and Corrigan, J. D. (2010). The Epidemiology of Traumatic Brain Injury. J. Head Trauma Rehabil. 25, 72–80.
Cunningham, T. L., Cartagena, C. M., Lu, X. C., Konopko, M., Dave, J. R., Tortella, F. C., et al. (2014). Correlations between blood-brain barrier disruption and neuroinflammation in an experimental model of penetrating ballistic-like brain injury. J. Neurotrauma 31, 505–514. doi: 10.1089/neu.2013.2965
Daneman, R. (2012). The blood-brain barrier in health and disease. Ann. Neurol. 72, 648–672. doi: 10.1002/ana.23648
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Dehghan, F., Shahrokhi, N., Khaksari, M., Soltani, Z., Asadikorom, G., Najafi, A., et al. (2018). Does the administration of melatonin during post-traumatic brain injury affect cytokine levels? Inflammopharmacology 26, 1017–1023. doi: 10.1007/s10787-017-0417-1
Deng, Y., Jiang, X., Deng, X., Chen, H., Xu, J., Zhang, Z., et al. (2020). Pioglitazone ameliorates neuronal damage after traumatic brain injury via the PPARγ/NF-κB/IL-6 signaling pathway. Genes Dis. 7, 253–265. doi: 10.1016/j.gendis.2019.05.002
Devor, A., Sakadzic, S., Saisan, P. A., Yaseen, M. A., Roussakis, E., Srinivasan, V. J., et al. (2011). “Overshoot” of O(2) is required to maintain baseline tissue oxygenation at locations distal to blood vessels. J. Neurosci. 31, 13676–13681. doi: 10.1523/JNEUROSCI.1968-11.2011
Dietrich, W. D., Alonso, O., and Halley, M. (1994). Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J. Neurotrauma 11, 289–301. doi: 10.1089/neu.1994.11.289
Dixon, K. J. (2017). Pathophysiology of Traumatic Brain Injury. Phys. Med. Rehabil. Clin. N. Am. 28, 215–225. doi: 10.1016/j.pmr.2016.12.001
Dogrukol-Ak, D., Kumar, V. B., Ryerse, J. S., Farr, S. A., Verma, S., Nonaka, N., et al. (2009). Isolation of peptide transport system-6 from brain endothelial cells: therapeutic effects with antisense inhibition in Alzheimer and stroke models. J. Cereb. Blood Flow Metab. 29, 411–422. doi: 10.1038/jcbfm.2008.131
Dore-Duffy, P., Owen, C., Balabanov, R., Murphy, S., Beaumont, T., and Rafols, J. A. (2000). Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc. Res. 60, 55–69. doi: 10.1006/mvre.2000.2244
Esen, F., Erdem, T., Aktan, D., Kalayci, R., Cakar, N., Kaya, M., et al. (2003). Effects of magnesium administration on brain edema and blood-brain barrier breakdown after experimental traumatic brain injury in rats. J. Neurosurg. Anesthesiol. 15, 119–125.
Evran, S., Calis, F., Akkaya, E., Baran, O., Cevik, S., Katar, S., et al. (2020). The effect of high mobility group box-1 protein on cerebral edema, blood-brain barrier, oxidative stress and apoptosis in an experimental traumatic brain injury model. Brain Res. Bull. 154, 68–80. doi: 10.1016/j.brainresbull.2019.10.013
Failla, M. D., Kumar, R. G., Peitzman, A. B., Conley, Y. P., Ferrell, R. E., and Wagner, A. K. (2015). Variation in the BDNF gene interacts with age to predict mortality in a prospective, longitudinal cohort with severe TBI. Neurorehabil. Neural. Repair. 29, 234–246. doi: 10.1177/1545968314542617
Falo, M. C., Fillmore, H. L., Reeves, T. M., and Phillips, L. L. (2006). Matrix metalloproteinase-3 expression profile differentiates adaptive and maladaptive synaptic plasticity induced by traumatic brain injury. J. Neurosci. Res. 84, 768–781. doi: 10.1002/jnr.20986
Faraci, F. M., and Breese, K. R. (1993). Nitric Oxide Mediate Vasodilatation in Response to Activation of N-Methyl-D-Aspartate Receptors in Brain. Circulat. Res. 72, 476–480.
Farr, G. W., Hall, C. H., Farr, S. M., Wade, R., Detzel, J. M., Adams, A. G., et al. (2019). Functionalized Phenylbenzamides Inhibit Aquaporin-4 Reducing Cerebral Edema and Improving Outcome in Two Models of CNS Injury. Neuroscience 404, 484–498. doi: 10.1016/j.neuroscience.2019.01.034
Fazzina, G., Amorini, A. M., Marmarou, C. R., Fukui, S., Okuno, K., Dunbar, J. G., et al. (2010). The protein kinase C activator phorbol myristate acetate decreases brain edema by aquaporin 4 downregulation after middle cerebral artery occlusion in the rat. J. Neurotrauma 27, 453–461. doi: 10.1089/neu.2008.0782
Fenn, A. M., Skendelas, J. P., Moussa, D. N., Muccigrosso, M. M., Popovich, P. G., Lifshitz, J., et al. (2015). Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J. Neurotrauma 32, 127–138. doi: 10.1089/neu.2014.3514
Fernández-Kletta, F., Offenhauserb, N., Dirnaglb, U., and Prillera, J. and Lindauerb, U. (2010). Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. PNAS 107, 22290–22295.
Furuse, M., Sasaki, H., and Tsukita, S. (1999). Manner of interaction of heterogeneous claudin species within and between tight junction strands. J. Cell Biol. 147, 891–903.
Gaab, M. R., Trost, H. A., Alcantara, A., Karimi-Nejad, A., Moskopp, D., Schultheiss, R., et al. (1994). “Ultrahigh” dexamethasone in acute brain injury. Results from a prospective randomized double-blind multicenter trial (GUDHIS). German Ultrahigh Dexamethasone Head Injury Study Group. Zentralbl. Neurochir. 55, 135–143.
Gerbatin, R. D. R., Cassol, G., Dobrachinski, F., Ferreira, A. P. O., Quines, C. B., Pace, I. D. D., et al. (2017). Guanosine Protects Against Traumatic Brain Injury-Induced Functional Impairments and Neuronal Loss by Modulating Excitotoxicity, Mitochondrial Dysfunction, and Inflammation. Mol. Neurobiol. 54, 7585–7596. doi: 10.1007/s12035-016-0238-z
Gilgun-Sherki, Y., Rosenbaum, Z., Melamed, E., and Offen, D. (2002). Antioxidant therapy in acute central nervous system injury: current state. Pharmacol. Rev. 54, 271–284. doi: 10.1124/pr.54.2.271
Giza, C. C., and Hovda, D. A. (2014). The new neurometabolic cascade of concussion. Neurosurgery 75, (Suppl. 4), S24–S33. doi: 10.1227/NEU.0000000000000505
Glober, N. K., Sprague, S., Ahmad, S., Mayfield, K. G., Fletcher, L. M., Digicaylioglu, M. H., et al. (2019). Acetazolamide Treatment Prevents Redistribution of Astrocyte Aquaporin 4 after Murine Traumatic Brain Injury. Neurosci. J. 2019:2831501. doi: 10.1155/2019/2831501
Glushakova, O. Y., Glushakov, A. O., Cesar, V., Borlongan, Alex, B., and Valadka, et al. (2017). Role of Caspase-3-mediated Apoptosis in Chronic Caspase-3-cleaved Tau Accumulation and Blood-brain Barrier Damage in the Corpus Callosum after Traumatic Brain Injury in Rats. J. Neurotr. 1, 1–47. doi: 10.1089/neu.20110.1089/neu.2017.4999
Goodrich, G. S., Kabakov, A. Y., Hameed, M. Q., Dhamne, S. C., Rosenberg, P. A., and Rotenberg, A. (2013). Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J. Neurotrauma 30, 1434–1441. doi: 10.1089/neu.2012.2712
Gordon, G. R., Choi, H. B., Rungta, R. L., Ellis-Davies, G. C., and MacVicar, B. A. (2008). Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749. doi: 10.1038/nature07525
Graves, J. C., and Kreipke, C. W. (2015). “Frontiers in Neuroengineering Endothelin, Cerebral Blood Flow, and Traumatic Brain Injury: Implications for a Future Therapeutic Target,” in Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects, ed. F. H. Kobeissy (Boca Raton, FL: CRC Press/Taylor & Francis (c) 2015 by Taylor & Francis Group, LLC).
Guerriero, R. M., Giza, C. C., and Rotenberg, A. (2015). Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 15:27. doi: 10.1007/s11910-015-0545-1
Haj-Yasein, N. N., Vindedal, G. F., Eilert-Olsen, M., Gundersen, G. A., Skare, O., Laake, P., et al. (2011). Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. U S A 108, 17815–17820. doi: 10.1073/pnas.1110655108
Hall, C. N., Reynell, C., Gesslein, B., Hamilton, N. B., Mishra, A., Sutherland, B. A., et al. (2014). Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. doi: 10.1038/nature13165
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hao, P., Duan, H., Hao, F., Chen, L., Sun, M., Fan, K. S., et al. (2017). Neural repair by NT3-chitosan via enhancement of endogenous neurogenesis after adult focal aspiration brain injury. Biomaterials 140, 88–102. doi: 10.1016/j.biomaterials.2017.04.014
Harting, M. T., Jimenez, F., Adams, S. D., Mercer, D. W., and Cox, C. S. Jr. (2008). Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery 144, 803–813. doi: 10.1016/j.surg.2008.05.017
Hartmann, D. A., Underly, R. G., Grant, R. I., Watson, A. N., Lindner, V., and Shih, A. Y. (2015). Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics 2:041402. doi: 10.1117/1.NPh.2.4.041402
Hashimoto, K., Nakashima, M., Hamano, A., Gotoh, M., Ikeshima-Kataoka, H., Murakami-Murofushi, K., et al. (2018). 2-carba cyclic phosphatidic acid suppresses inflammation via regulation of microglial polarisation in the stab-wounded mouse cerebral cortex. Sci. Rep. 8:9715. doi: 10.1038/s41598-018-27990-1
Hawryluk, G. W. J., and Manley, G. T. (2015). Classification of traumatic brain injury. Handbook Clin. Neurol. 127, 15–21. doi: 10.1016/B978-0-444-52892-6.00002-7
Hay, J. R., Johnson, V. E., Young, A. M., Smith, D. H., and Stewart, W. (2015). Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years After Traumatic Brain Injury in Humans. J. Neuropathol. Exp. Neurol. 74, 1147–1157. doi: 10.1097/nen.0000000000000261
Heinemann, U., Kaufer, D., and Friedman, A. (2012). Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 60, 1251–1257. doi: 10.1002/glia.22311
Hemphill, M. A., Dauth, S., Yu, C. J., Dabiri, B. E., and Parker, K. K. (2015). Traumatic brain injury and the neuronal microenvironment: a potential role for neuropathological mechanotransduction. Neuron 85, 1177–1192. doi: 10.1016/j.neuron.2015.02.041
Higashida, T., Kreipke, C. W., Rafols, J. A., Peng, C., Schafer, S., Schafer, P., et al. (2011). The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J. Neurosurg. 114, 92–101. doi: 10.3171/2010.6.jns10207
Hill, R. A., Tong, L., Yuan, P., Murikinati, S., Gupta, S., and Grutzendler, J. (2015). Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron 87, 95–110. doi: 10.1016/j.neuron.2015.06.001
Holmin, S., and Mathiesen, T. (2000). Intracerebral administration of interleukin-1beta and induction of inflammation, apoptosis, and vasogenic edema. J. Neurosurg. 92, 108–120. doi: 10.3171/jns.2000.92.1.0108
Holmin, S., Söderlund, J., Biberfeld, P., and Mathiesen, T. (1998). Intracerebral inflammation after human brain contusion. Neurosurgery 42, 291–298. doi: 10.1097/00006123-199802000-00047
Hu, Y., Seker, B., Exner, C., Zhang, J., Plesnila, N., and Schwarzmaier, S. M. (2021). Longitudinal Characterization of Blood-Brain Barrier Permeability after Experimental Traumatic Brain Injury by In Vivo 2-Photon Microscopy. J. Neurotrauma 38, 399–410. doi: 10.1089/neu.2020.7271
Hummel, R., Ulbrich, S., Appel, D., Li, S., Hirnet, T., Zander, S., et al. (2020). Administration of all-trans retinoic acid after experimental traumatic brain injury is brain protective. Br. J. Pharmacol. 177, 5208–5223. doi: 10.1111/bph.15259
Hydera, A. A., Wunderlichb, C., Puvanachandraa, P., Gururajc, G., and Kobusingye, O. C. (2007). The impact of traumatic brain injuries: A global perspective. NeuroRehabilitation 22, 341–353.
Jansson, L. C., and Akerman, K. E. (2014). The role of glutamate and its receptors in the proliferation, migration, differentiation and survival of neural progenitor cells. J. Neural. Transm. 121, 819–836. doi: 10.1007/s00702-014-1174-6
Janzer, R. C., and Raff, M. C. (1987). Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 325, 253–257. doi: 10.1038/325253a0
Jeong, S. M., Hahm, K. D., Shin, J. W., Leem, J. G., Lee, C., and Han, S. M. (2006). Changes in magnesium concentration in the serum and cerebrospinal fluid of neuropathic rats. Acta Anaesthesiolog. Scand. 50, 211–216. doi: 10.1111/j.1399-6576.2006.00925.x
Jha, R. M., Kochanek, P. M., and Simard, J. M. (2019). Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 145, 230–246. doi: 10.1016/j.neuropharm.2018.08.004
Jha, R. M., Mondello, S., Bramlett, H. M., Dixon, C. E., Shear, D. A., Dietrich, W. D., et al. (2021). Glibenclamide Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 38, 628–645. doi: 10.1089/neu.2020.7421
Jia, F., Yin, Y. H., Gao, G. Y., Wang, Y., Cen, L., and Jiang, J. Y. (2014). MMP-9 inhibitor SB-3CT attenuates behavioral impairments and hippocampal loss after traumatic brain injury in rat. J. Neurotrauma 31, 1225–1234. doi: 10.1089/neu.2013.3230
Jiang, Q., Chen, J., Long, X., Yao, X., Zou, X., Yang, Y., et al. (2020). Phillyrin protects mice from traumatic brain injury by inhibiting the inflammation of microglia via PPARγ signaling pathway. Int. Immunopharmacol. 79:106083. doi: 10.1016/j.intimp.2019.106083
Johnson, V. E., Weber, M. T., Xiao, R., Cullen, D. K., Meaney, D. F., Stewart, W., et al. (2018). Mechanical disruption of the blood-brain barrier following experimental concussion. Acta Neuropathol. 135, 711–726. doi: 10.1007/s00401-018-1824-0
Kallakuri, S., Kreipke, C. W., Schafer, P. C., Schafer, S. M., and Rafols, J. A. (2010). Brain cellular localization of endothelin receptors A and B in a rodent model of diffuse traumatic brain injury. Neuroscience 168, 820–830. doi: 10.1016/j.neuroscience.2010.01.018
Kan, E. M., Ling, E. A., and Lu, J. (2012). Microenvironment changes in mild traumatic brain injury. Brain Res. Bull. 87, 359–372. doi: 10.1016/j.brainresbull.2012.01.007
Kasischke, K. A., Lambert, E. M., Panepento, B., Sun, A., Gelbard, H. A., Burgess, R. W., et al. (2011). Two-photon NADH imaging exposes boundaries of oxygen diffusion in cortical vascular supply regions. J. Cereb. Blood Flow Metab. 31, 68–81. doi: 10.1038/jcbfm.2010.158
Katayama, Y., and Kawamata, T. (2003). Edema fluid accumulation within necrotic brain tissue as a cause of the mass effect of cerebral contusion in head trauma patients. Acta Neurochir. Suppl. 86, 323–327.
Ke, C., Poon, W. S., Ng, H. K., Lai, F. M., Tang, N. L., and Pang, J. C. (2002). Impact of experimental acute hyponatremia on severe traumatic brain injury in rats: influences on injuries, permeability of blood-brain barrier, ultrastructural features, and aquaporin-4 expression. Exp. Neurol. 178, 194–206.
Keisuke, T., Marmarou, C. R., Kenji, O., Ruth, P., and Marmarou, A. (2010). Effect of Secondary Insults upon Aquaporin-4 Water Channels following Experimental Cortical Contusion in Rats. J. Neurotr. 27, 229–239.
Kempuraj, D., Ahmed, M. E., Selvakumar, G. P., Thangavel, R., Raikwar, S. P., Zaheer, S. A., et al. (2021). Acute Traumatic Brain Injury-Induced Neuroinflammatory Response and Neurovascular Disorders in the Brain. Neurotox Res. 39, 359–368. doi: 10.1007/s12640-020-00288-9
Khatri, N., Thakur, M., Pareek, V., Kumar, S., Sharma, S., and Datusalia, A. K. (2018). Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord Drug Targets 17, 689–695. doi: 10.2174/1871527317666180627120501
Kikuchi, K., Tancharoen, S., Matsuda, F., Biswas, K. K., Ito, T., Morimoto, Y., et al. (2009). Edaravone attenuates cerebral ischemic injury by suppressing aquaporin-4. Biochem. Biophys. Res. Commun. 390, 1121–1125. doi: 10.1016/j.bbrc.2009.09.015
Kisler, K., Nelson, A. R., Montagne, A., and Zlokovic, B. V. (2017). Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017:48. doi: 10.1038/nrn.2017.48
Koliatsos, V. E., Xu, L., and Cummings, B. J. (2015). Stem cell therapies for traumatic brain injury. Regen Med. 10, 917–920. doi: 10.2217/rme.15.62
Korn, A., Golan, H., Melamed, I., Pascual-Marqui, R., and Friedman, A. (2005). Focal cortical dysfunction and blood-brain barrier disruption in patients with Postconcussion syndrome. J. Clin. Neurophysiol. 22, 1–9.
Kortekaas, R., Leenders, K. L., van Oostrom, J. C., Vaalburg, W., Bart, J., Willemsen, A. T., et al. (2005). Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 57, 176–179. doi: 10.1002/ana.20369
Krieg, S. M., Sonanini, S., Plesnila, N., and Trabold, R. (2015). Effect of small molecule vasopressin V1a and V2 receptor antagonists on brain edema formation and secondary brain damage following traumatic brain injury in mice. J. Neurotrauma 32, 221–227. doi: 10.1089/neu.2013.3274
Krieg, S. M., Trabold, R., and Plesnila, N. (2016). Time-dependent effects of arginine-vasopressin V1 receptor inhibition on secondary brain damage after traumatic brain injury. J. Neurotrauma 2016:4514. doi: 10.1089/neu.2016.4514
Lai, R. C., Yeo, R. W., Tan, K. H., and Lim, S. K. (2013). Exosomes for drug delivery - a novel application for the mesenchymal stem cell. Biotechnol. Adv. 31, 543–551. doi: 10.1016/j.biotechadv.2012.08.008
Lampron, A., Elali, A., and Rivest, S. (2013). Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron 78, 214–232. doi: 10.1016/j.neuron.2013.04.005
Lee, S.-W., Kim, W. J., Choi, Y. K., Song, H. S., Son, M. J., Irwin, H., et al. (2003). SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 9, 900–906.
Levin, H. S., and Diaz-Arrastia, R. R. (2015). Diagnosis, prognosis, and clinical management of mild traumatic brain injury. Lancet Neurol. 14, 506–517. doi: 10.1016/s1474-4422(15)00002-2
Li, Q., Wu, X., Yang, Y., Zhang, Y., He, F., Xu, X., et al. (2019). Tachykinin NK1 receptor antagonist L-733,060 and substance P deletion exert neuroprotection through inhibiting oxidative stress and cell death after traumatic brain injury in mice. Int. J. Biochem. Cell Biol. 107, 154–165. doi: 10.1016/j.biocel.2018.12.018
Liao, R., Chen, Y., Cheng, L., Fan, L., Chen, H., Wan, Y., et al. (2019). Histamine H1 Receptors in Neural Stem Cells Are Required for the Promotion of Neurogenesis Conferred by H3 Receptor Antagonism following Traumatic Brain Injury. Stem Cell Rep. 12, 532–544. doi: 10.1016/j.stemcr.2019.01.004
Liebner, S., Dijkhuizen, R. M., Reiss, Y., Plate, K. H., Agalliu, D., and Constantin, G. (2018). Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 135, 311–336. doi: 10.1007/s00401-018-1815-1
Lin, A. L., Fox, P. T., Hardies, J., Duong, T. Q., and Gao, J. H. (2010). Nonlinear coupling between cerebral blood flow, oxygen consumption, and ATP production in human visual cortex. Proc. Natl. Acad. Sci. U S A 107, 8446–8451. doi: 10.1073/pnas.0909711107
Lin, C., Chao, H., Li, Z., Xu, X., Liu, Y., Bao, Z., et al. (2017). Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp. Neurol. 290, 115–122. doi: 10.1016/j.expneurol.2017.01.005
Lin, C., Li, N., Chang, H., Shen, Y., Li, Z., Wei, W., et al. (2020). Dual effects of thyroid hormone on neurons and neurogenesis in traumatic brain injury. Cell Death Dis. 11:671. doi: 10.1038/s41419-020-02836-9
Lin, C. T., Lecca, D., Yang, L. Y., Luo, W., Scerba, M. T., Tweedie, D., et al. (2020). 3,6’-dithiopomalidomide reduces neural loss, inflammation, behavioral deficits in brain injury and microglial activation. Elife 9:54726. doi: 10.7554/eLife.54726
Liu, D., Zhang, X., Hu, B., and Ander, B. P. (2016). Src Family Kinases in Brain Edema After Acute Brain Injury. Acta Neurochir. Suppl. 121, 185–190. doi: 10.1007/978-3-319-18497-5_33
Liu, R., Liao, X. Y., Tang, J. C., Pan, M. X., Chen, S. F., Lu, P. X., et al. (2019). BpV(pic) confers neuroprotection by inhibiting M1 microglial polarization and MCP-1 expression in rat traumatic brain injury. Mol. Immunol. 112, 30–39. doi: 10.1016/j.molimm.2019.04.010
Liu, Y. L., Xu, Z. M., Yang, G. Y., Yang, D. X., Ding, J., Chen, H., et al. (2017). Sesamin alleviates blood-brain barrier disruption in mice with experimental traumatic brain injury. Acta Pharmacol. Sin. 38, 1445–1455. doi: 10.1038/aps.2017.103
Loane, D. J., and Kumar, A. (2016). Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 0, 316–327. doi: 10.1016/j.expneurol.2015.08.018
Logsdon, A. F., Lucke-Wold, B. P., Turner, R. C., Huber, J. D., Rosen, C. L., and Simpkins, J. W. (2015). Role of Microvascular Disruption in Brain Damage from Traumatic Brain Injury. Compr. Physiol. 5, 1147–1160. doi: 10.1002/cphy.c140057
Logsdon, A. F., Lucke-Wold, B. P., Turner, R. C., Li, X., Adkins, C. E., Mohammad, A. S., et al. (2017). A mouse Model of Focal Vascular Injury Induces Astrocyte Reactivity, Tau Oligomers, and Aberrant Behavior. Arch. Neurosci. 4:2. doi: 10.5812/archneurosci.44254
Long, X., Yao, X., Jiang, Q., Yang, Y., He, X., Tian, W., et al. (2020). Astrocyte-derived exosomes enriched with miR-873a-5p inhibit neuroinflammation via microglia phenotype modulation after traumatic brain injury. J. Neuroinflammation 17:89. doi: 10.1186/s12974-020-01761-0
Lopez, N. E., Krzyzaniak, M. J., Blow, C., Putnam, J., Ortiz-Pomales, Y., Hageny, A. M., et al. (2012). Ghrelin prevents disruption of the blood-brain barrier after traumatic brain injury. J. Neurotrauma 29, 385–393. doi: 10.1089/neu.2011.2053
Lovatt, D., Xu, Q., Liu, W., Takano, T., Smith, N. A., Schnermann, J., et al. (2012). Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc. Natl. Acad. Sci. U S A 109, 6265–6270. doi: 10.1073/pnas.1120997109
Lozano, D., Gonzales-Portillo, G. S., Acosta, S., de la Pena, I., Tajiri, N., Kaneko, Y., et al. (2015). Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat 11, 97–106. doi: 10.2147/NDT.S65815
Lu, H., Zhan, Y., Ai, L., Chen, H., and Chen, J. (2020). AQP4-siRNA alleviates traumatic brain edema by altering post-traumatic AQP4 polarity reversal in TBI rats. J. Clin. Neurosci. 81, 113–119. doi: 10.1016/j.jocn.2020.09.015
Lucke-Wold, B. P., Logsdon, A. F., Smith, K. E., Turner, R. C., Alkon, D. L., Tan, Z., et al. (2015). Bryostatin-1 Restores Blood Brain Barrier Integrity following Blast-Induced Traumatic Brain Injury. Mol. Neurobiol. 52, 1119–1134. doi: 10.1007/s12035-014-8902-7
Lukaszewicz, A. C., Soyer, B., and Payen, D. (2011). Water, water, everywhere: sodium and water balance and the injured brain. Curr. Opin. Anaesthesiol. 24, 138–143. doi: 10.1097/ACO.0b013e32834458af
Lutton, E. M., Razmpour, R., Andrews, A. M., Cannella, L. A., Son, Y. J., Shuvaev, V. V., et al. (2017). Acute administration of catalase targeted to ICAM-1 attenuates neuropathology in experimental traumatic brain injury. Sci. Rep. 7:3846. doi: 10.1038/s41598-017-03309-4
Lv, Q., Fan, X., Xu, G., Liu, Q., Tian, L., Cai, X., et al. (2013). Intranasal delivery of nerve growth factor attenuates aquaporins-4-induced edema following traumatic brain injury in rats. Brain Res. 1493, 80–89. doi: 10.1016/j.brainres.2012.11.028
Ma, H., Cui, L. X., Lam, P. K., Tong, C. S. W., Lo, K. K. Y., Wong, G. K. C., et al. (2021). Topical application of adipose tissue-derived mesenchymal stem cells (ADMSCs) reduced cerebral edema in experimental traumatic brain injury (TBI)-a preliminary study. Chin. Neurosurg. J. 7:2. doi: 10.1186/s41016-020-00219-9
MacVicar, B. A., and Newman, E. A. (2015). Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 7:5. doi: 10.1101/cshperspect.a020388
Main, B. S., Villapol, S., Sloley, S. S., Barton, D. J., Parsadanian, M., Agbaegbu, C., et al. (2018). Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol. Neurodegener. 13:17. doi: 10.1186/s13024-018-0249-5
Mankertz, J., Waller, J. S., Hillenbrand, B., Tavalali, S., Florian, P., Schoneberg, T., et al. (2002). Gene expression of the tight junction protein occludin includes differential splicing and alternative promoter usage. Biochem. Biophys. Res. Commun. 298, 657–666.
Marmarou, A., Guy, M., Murphey, L., Roy, F., Layani, L., Combal, J. P., et al. (2005). A single dose, three-arm, placebo-controlled, phase I study of the bradykinin B2 receptor antagonist Anatibant (LF16-0687Ms) in patients with severe traumatic brain injury. J. Neurotrauma 22, 1444–1455. doi: 10.1089/neu.2005.22.1444
Marshall, L. F., Maas, A. I., Marshall, S. B., Bricolo, A., Fearnside, M., Iannotti, F., et al. (1998). A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J. Neurosurg. 89, 519–525. doi: 10.3171/jns.1998.89.4.0519
Matter, K., and Balda, M. S. (2003). Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 4, 225–236. doi: 10.1038/nrm1055
McCarty, J. H. (2009). Cell adhesion and signaling networks in brain neurovascular units. Curr. Opin. Hematol. 16, 209–214. doi: 10.1097/MOH.0b013e32832a07eb
Menge, T., Zhao, Y., Zhao, J., Wataha, K., Gerber, M., Zhang, J., et al. (2012). Mesenchymal stem cells regulate blood-brain barrier integrity through TIMP3 release after traumatic brain injury. Sci. Transl. Med. 4:161ra150. doi: 10.1126/scitranslmed.3004660
Michinaga, S., Inoue, A., Yamamoto, H., Ryu, R., Inoue, A., Mizuguchi, H., et al. (2020). Endothelin receptor antagonists alleviate blood-brain barrier disruption and cerebral edema in a mouse model of traumatic brain injury: A comparison between bosentan and ambrisentan. Neuropharmacology 175:108182. doi: 10.1016/j.neuropharm.2020.108182
Michinaga, S., Kimura, A., Hatanaka, S., Minami, S., Asano, A., Ikushima, Y., et al. (2018). Delayed Administration of BQ788, an ET(B) Antagonist, after Experimental Traumatic Brain Injury Promotes Recovery of Blood-Brain Barrier Function and a Reduction of Cerebral Edema in Mice. J. Neurotrauma 35, 1481–1494. doi: 10.1089/neu.2017.5421
Mishra, A., Hamid, A., and Newman, E. A. (2011). Oxygen modulation of neurovascular coupling in the retina. Proc. Natl. Acad. Sci. U S A 108, 17827–17831. doi: 10.1073/pnas.1110533108
Mishra, A., Reynolds, J. P., Chen, Y., Gourine, A. V., Rusakov, D. A., and Attwell, D. (2016). Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 19, 1619–1627. doi: 10.1038/nn.4428
Morganti, J. M., Riparip, L. K., Chou, A., Liu, S., Gupta, N., and Rosi, S. (2016). Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. J. Neuroinflammation 13:80. doi: 10.1186/s12974-016-0547-1
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The Science of Stroke: Mechanisms in Search of Treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Moyron, R. B., Gonda, A., Selleck, M. J., Luo-Owen, X., Catalano, R. D., O’Callahan, T., et al. (2017). Differential protein expression in exosomal samples taken from trauma patients. Proteomics Clin. Appl. 2017:201700061. doi: 10.1002/prca.201700061
Muoio, V., Persson, P. B., and Sendeski, M. M. (2014). The neurovascular unit - concept review. Acta Physiol. 210, 790–798. doi: 10.1111/apha.12250
Murugan, M., Santhakumar, V., and Kannurpatti, S. S. (2016). Facilitating Mitochondrial Calcium Uptake Improves Activation-Induced Cerebral Blood Flow and Behavior after mTBI. Front. Syst. Neurosci. 10:19. doi: 10.3389/fnsys.2016.00019
Nichols, J. E., Niles, J. A., DeWitt, D., Prough, D., Parsley, M., Vega, S., et al. (2013). Neurogenic and neuro-protective potential of a novel subpopulation of peripheral blood-derived CD133+ ABCG2+CXCR4+ mesenchymal stem cells: development of autologous cell-based therapeutics for traumatic brain injury. Stem Cell Res. Ther. 4:3. doi: 10.1186/scrt151
Nishioku, T., Matsumoto, J., Dohgu, S., Sumi, N., Miyao, K., Takata, F., et al. (2010). Tumor Necrosis Factor-α Mediates the Blood–Brain Barrier Dysfunction Induced by Activated Microglia in Mouse Brain Microvascular Endothelial Cells. J. Pharm. Sci. 112, 251–254. doi: 10.1254/jphs.09292SC
Obermeier, B., Daneman, R., and Ransohoff, R. M. (2013). Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 19, 1584–1596. doi: 10.1038/nm.3407
Obrenovitch, T. P., and Urenjak, J. (1997). Is High Extracellular Glutamate the Key to Excitotoxicity in Traumatic Brain Injury? J. Neurotr. 14, 677–697.
Ojo, J., Eisenbaum, M., Shackleton, B., Lynch, C., Joshi, U., Saltiel, N., et al. (2021). Mural cell dysfunction leads to altered cerebrovascular tau uptake following repetitive head trauma. Neurobiol. Dis. 150:105237. doi: 10.1016/j.nbd.2020.105237
O’Phelan, K., Denny, K., Ernst, T., Green, D., Park, D., Chang, C., et al. (2013). Impact of Methamphetamine on Regional Metabolism and Cerebral Blood Flow After Traumatic Brain Injury. Neurocrit. Care 19, 183–191. doi: 10.1007/s12028-013-9871-9)
Ostergaard, L., Engedal, T. S., Aamand, R., Mikkelsen, R., Iversen, N. K., Anzabi, M., et al. (2014). Capillary transit time heterogeneity and flow-metabolism coupling after traumatic brain injury. J. Cereb. Blood Flow Metab. 34, 1585–1598. doi: 10.1038/jcbfm.2014.131
Papadopoulos, M. C., Manley, G. T., Krishna, S., and Verkman, A. S. (2004). Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. Faseb J. 18, 1291–1293. doi: 10.1096/fj.04-1723fje
Pardridge, W. M. (2005). The blood-brain barrier: Bottleneck in brain drug development. NeuroRX 2, 3–14. doi: 10.1602/neurorx.2.1.3
Peglion, F., Llense, F., and Etienne-Manneville, S. (2014). Adherens junction treadmilling during collective migration. Nat. Cell Biol. 16, 639–651. doi: 10.1038/ncb2985
Placone, A. L., Quinones-Hinojosa, A., and Searson, P. C. (2016). The role of astrocytes in the progression of brain cancer: complicating the picture of the tumor microenvironment. Tumour. Biol. 37, 61–69. doi: 10.1007/s13277-015-4242-0
Plesnila, N. (2016). The immune system in traumatic brain injury. Curr. Opin. Pharmacol. 26, 110–117. doi: 10.1016/j.coph.2015.10.008
Prabhakara, K. S., Kota, D. J., Jones, G. H., Srivastava, A. K., Cox, C. S. Jr., and Olson, S. D. (2018). Teriflunomide Modulates Vascular Permeability and Microglial Activation after Experimental Traumatic Brain Injury. Mol. Ther. 26, 2152–2162. doi: 10.1016/j.ymthe.2018.06.022
Prager, O., Kamintsky, L., Hasam-Henderson, L. A., Schoknecht, K., Wuntke, V., Papageorgiou, I., et al. (2019). Seizure-induced microvascular injury is associated with impaired neurovascular coupling and blood-brain barrier dysfunction. Epilepsia 60, 322–336. doi: 10.1111/epi.14631
Price, L., Wilson, C., and Grant, G. (2016). ““Blood-Brain Barrier Pathophysiology following Traumatic Brain Injury,”,” in Translational Research in Traumatic Brain Injury, eds D. Laskowitz and G. Grant (Boca Raton (FL): CRC Press/Taylor and Francis Group (c) 2016 by Taylor & Francis Group, LLC).
Pun, P. B., Lu, J., and Moochhala, S. (2009). Involvement of ROS in BBB dysfunction. Free Radic. Res. 43, 348–364. doi: 10.1080/10715760902751902
Quintard, H., Patet, C., Marques-Vidal, P., Suys, T., and Oddo, M. (2015). Normobaric Hyperoxia is Associated with Increased Cerebral Excitotoxicity After Severe Traumatic Brain Injury. Neurocrit. Care 22, 243–250.
Ren, H., Yang, Z., Luo, C., Zeng, H., Li, P., Kang, J. X., et al. (2017). Enriched Endogenous Omega-3 Fatty Acids in Mice Ameliorate Parenchymal Cell Death After Traumatic Brain Injury. Mol. Neurobiol. 54, 3317–3326. doi: 10.1007/s12035-016-9931-1
Reulen, H. J., Graham, R., Spatz, M., and Klatzo, I. (1977). Role of pressure gradients and bulk flow in dynamics of vasogenic brain edema. J. Neurosurg. 46, 24–35. doi: 10.3171/jns.1977.46.1.0024
Richardson, R. M., Singh, A., Sun, D., Fillmore, H. L., Dietrich, D. W. III, and Bullock, M. R. (2010). Stem cell biology in traumatic brain injury: effects of injury and strategies for repair. J. Neurosurg. 112, 1125–1138. doi: 10.3171/2009.4.jns081087
Riess, P., Zhang, C., Saatman, K. E., Laurer, H. L., Longhi, L. G., Raghupathi, R., et al. (2002). Transplanted neural stem cells survive, differentiate, and improve neurological motor function after experimental traumatic brain injury. Neurosurgery 51, 1043–1052.
Robertson, C. S., Hannay, H. J., Yamal, J. M., Gopinath, S., Goodman, J. C., Tilley, B. C., et al. (2014). Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. Jama 312, 36–47. doi: 10.1001/jama.2014.6490
Rodriguez-Baeza, A., Reina-de la Torre, F., Poca, A., Marti, M., and Garnacho, A. (2003). Morphological features in human cortical brain microvessels after head injury: a three-dimensional and immunocytochemical study. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 273, 583–593. doi: 10.1002/ar.a.10069
Roozenbeek, B., Maas, A. I. R., and Menon, D. K. (2013). Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 9, 231–236. doi: 10.1038/nrneurol.2013.22
Rossi, J. L., Todd, T., Bazan, N. G., and Belayev, L. (2013). Inhibition of Myosin light-chain kinase attenuates cerebral edema after traumatic brain injury in postnatal mice. J. Neurotrauma 30, 1672–1679. doi: 10.1089/neu.2013.2898
Rostami, E., Engquist, H., and Enblad, P. (2014). Imaging of cerebral blood flow in patients with severe traumatic brain injury in the neurointensive care. Front. Neurol. 5:114. doi: 10.3389/fneur.2014.00114
Rui, Q., Ni, H., Lin, X., Zhu, X., Li, D., Liu, H., et al. (2019). Astrocyte-derived fatty acid-binding protein 7 protects blood-brain barrier integrity through a caveolin-1/MMP signaling pathway following traumatic brain injury. Exp. Neurol. 322:113044. doi: 10.1016/j.expneurol.2019.113044
Sahuquillo, J., Poca, M. A., and Amorós, S. (2001). current aspects of pph and cell dysfunction after severe head injury. Curr. Pharm. Design 7, 1475–1503.
Salvador, E., Burek, M., and Forster, C. Y. (2015). Stretch and/or oxygen glucose deprivation (OGD) in an in vitro traumatic brain injury (TBI) model induces calcium alteration and inflammatory cascade. Front. Cell Neurosci. 9:323. doi: 10.3389/fncel.2015.00323
Sanchis, P., Fernández-Gayol, O., Vizueta, J., Comes, G., Canal, C., Escrig, A., et al. (2020). Microglial cell-derived interleukin-6 influences behavior and inflammatory response in the brain following traumatic brain injury. Glia 68, 999–1016. doi: 10.1002/glia.23758
Sawant-Pokam, P. A., Vail, T. J., Metcalf, C. S., Maguire, J. L., McKean, T. O., McKean, N. O., et al. (2020). Preventing neuronal edema increases network excitability after traumatic brain injury. J. Clin. Invest. 130, 6005–6020. doi: 10.1172/jci134793
Schlegel, N., and Waschke, J. (2009). Impaired cAMP and Rac 1 signaling contribute to TNF-alpha-induced endothelial barrier breakdown in microvascular endothelium. Microcirculation 16, 521–533. doi: 10.1080/10739680902967427
Schwarzmaier, S. M., de Chaumont, C., Balbi, M., Terpolilli, N. A., Kleinschnitz, C., Gruber, A., et al. (2016). The Formation of Microthrombi in Parenchymal Microvessels after Traumatic Brain Injury Is Independent of Coagulation Factor XI. J. Neurotrauma 33, 1634–1644. doi: 10.1089/neu.2015.4173
Schwarzmaier, S. M., Terpolilli, N. A., Dienel, A., Gallozzi, M., Schinzel, R., Tegtmeier, F., et al. (2015b). Endothelial nitric oxide synthase mediates arteriolar vasodilatation after traumatic brain injury in mice. J. Neurotrauma 32, 731–738. doi: 10.1089/neu.2014.3650
Schwarzmaier, S. M., Gallozzi, M., and Plesnila, N. (2015a). Identification of the Vascular Source of Vasogenic Brain Edema following Traumatic Brain Injury Using In Vivo 2-Photon Microscopy in Mice. J. Neurotrauma 32, 990–1000. doi: 10.1089/neu.2014.3775
Schwarzmaier, S. M., and Plesnila, N. (2014). Contributions of the immune system to the pathophysiology of traumatic brain injury - evidence by intravital microscopy. Front. Cell Neurosci. 8:358. doi: 10.3389/fncel.2014.00358
Schwarzmaier, S. M., Zimmermann, R., McGarry, N. B., Trabold, R., Kim, S.-W., and Plesnila, N. (2013). In vivo temporal and spatial profile of leukocyte adhesion and migration after experimental traumatic brain injury in mice. J. Neuroinflam. 10, 1–17.
Semple, B. D., Bye, N., Rancan, M., Ziebell, J. M., and Morganti-Kossmann, M. C. (2010). Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2-/- mice. J. Cereb. Blood Flow Metab. 30, 769–782. doi: 10.1038/jcbfm.2009.262
Sen, T., Saha, P., Gupta, R., Foley, L. M., Jiang, T., Abakumova, O. S., et al. (2020). Aberrant ER Stress Induced Neuronal-IFNβ Elicits White Matter Injury Due to Microglial Activation and T-Cell Infiltration after TBI. J. Neurosci. 40, 424–446. doi: 10.1523/jneurosci.0718-19.2019
Serlin, Y., Shelef, I., Knyazer, B., and Friedman, A. (2015). Anatomy and physiology of the blood-brain barrier. Semin Cell Dev. Biol. 38, 2–6. doi: 10.1016/j.semcdb.2015.01.002
Shahror, R. A., Linares, G. R., Wang, Y., Hsueh, S. C., Wu, C. C., Chuang, D. M., et al. (2020). Transplantation of Mesenchymal Stem Cells Overexpressing Fibroblast Growth Factor 21 Facilitates Cognitive Recovery and Enhances Neurogenesis in a Mouse Model of Traumatic Brain Injury. J. Neurotrauma 37, 14–26. doi: 10.1089/neu.2019.6422
Shapira, Y., Setton, D., Artru, A. A., and Shohami, E. (1993). Blood-brain barrier permeability, cerebral edema, and neurologic function after closed head injury in rats. Anesth. Analg. 77, 141–148.
Sharif, Y., Jumah, F., Coplan, L., Krosser, A., Sharif, K., and Tubbs, R. S. (2018). Blood brain barrier: A review of its anatomy and physiology in health and disease. Clin. Anat. 31, 812–823. doi: 10.1002/ca.23083
Sharma, R., Kambhampati, S. P., Zhang, Z., Sharma, A., Chen, S., Duh, E. I., et al. (2020). Dendrimer mediated targeted delivery of sinomenine for the treatment of acute neuroinflammation in traumatic brain injury. J. Control Release 323, 361–375. doi: 10.1016/j.jconrel.2020.04.036
Shi, W., Huang, C. J., Xu, X. D., Jin, G. H., Huang, R. Q., Huang, J. F., et al. (2016). Transplantation of RADA16-BDNF peptide scaffold with human umbilical cord mesenchymal stem cells forced with CXCR4 and activated astrocytes for repair of traumatic brain injury. Acta Biomater. 45, 247–261. doi: 10.1016/j.actbio.2016.09.001
Shlosberg, D., Benifla, M., Kaufer, D., and Friedman, A. (2010). Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 6, 393–403. doi: 10.1038/nrneurol.2010.74
Shu, S., Zhang, Z., Spicer, D., Kulikowicz, E., Hu, K., Babapoor-Farrokhran, S., et al. (2019). Administration of a 20-Hydroxyeicosatetraenoic Acid Synthesis Inhibitor Improves Outcome in a Rat Model of Pediatric Traumatic Brain Injury. Dev. Neurosci. 41, 166–176. doi: 10.1159/000500895
Signoretti, S., Di Pietro, V., Vagnozzi, R., Lazzarino, G., Amorini, A. M., Belli, A., et al. (2009). Transient alterations of creatine, creatine phosphate, N-acetylaspartate and high-energy phosphates after mild traumatic brain injury in the rat. Mole. Cell. Biochem. 333:269. doi: 10.1007/s11010-009-0228-9
Simard, M., and Nedergaard, M. (2004). The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 129, 877–896. doi: 10.1016/j.neuroscience.2004.09.053
Simon, D. W., McGeachy, M. J., Bayır, H., Clark, R. S., Loane, D. J., and Kochanek, P. M. (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 13, 171–191. doi: 10.1038/nrneurol.2017.13
Sivandzade, F., Alqahtani, F., Sifat, A., and Cucullo, L. (2020). The cerebrovascular and neurological impact of chronic smoking on post-traumatic brain injury outcome and recovery: an in vivo study. J. Neuroinflam. 17:133. doi: 10.1186/s12974-020-01818-0
Steiner, J., Rafols, D., Park, H. K., Katar, M. S., Rafols, J. A., and Petrov, T. (2004). Attenuation of iNOS mRNA exacerbates hypoperfusion and upregulates endothelin-1 expression in hippocampus and cortex after brain trauma. Nitric Oxide 10, 162–169. doi: 10.1016/j.niox.2004.03.005
Stonesifer, L. D. (2008). Mild traumatic brain injury in U.S. soldiers returning from Iraq. N. Engl. J. Med. 358:2178.
Su, Y., Fan, W., Ma, Z., Wen, X., Wang, W., Wu, Q., et al. (2014). Taurine improves functional and histological outcomes and reduces inflammation in traumatic brain injury. Neuroscience 266, 56–65. doi: 10.1016/j.neuroscience.2014.02.006
Subramani, A., Alsidawi, S., Jagannathan, S., Sumita, K., Sasaki, A. T., Aronow, B., et al. (2013). The brain microenvironment negatively regulates miRNA-768-3p to promote K-ras expression and lung cancer metastasis. Sci. Rep. 3:2392. doi: 10.1038/srep02392
Sweeney, M. D., Ayyadurai, S., and Zlokovic, B. V. (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 19, 771–783. doi: 10.1038/nn.4288
Tajes, M., Ramos-Fernandez, E., Weng-Jiang, X., Bosch-Morato, M., Guivernau, B., Eraso-Pichot, A., et al. (2014). The blood-brain barrier: structure, function and therapeutic approaches to cross it. Mol. Membr. Biol. 31, 152–167. doi: 10.3109/09687688.2014.937468
Tang, J., Kang, Y., Huang, L., Wu, L., and Peng, Y. (2020). TIMP1 preserves the blood-brain barrier through interacting with CD63/integrin β 1 complex and regulating downstream FAK/RhoA signaling. Acta Pharm. Sin. B 10, 987–1003. doi: 10.1016/j.apsb.2020.02.015
Tepass, U., and Harris, K. P. (2007). Adherens junctions in Drosophila retinal morphogenesis. Trends Cell Biol. 17, 26–35. doi: 10.1016/j.tcb.2006.11.006
Teschemacher, A. G., Gourine, A. V., and Kasparov, S. (2015). A Role for Astrocytes in Sensing the Brain Microenvironment and Neuro-Metabolic Integration. Neurochem. Res. 40, 2386–2393. doi: 10.1007/s11064-015-1562-9
Tian, R., Hou, Z., Hao, S., Wu, W., Mao, X., Tao, X., et al. (2016). Hydrogen-rich water attenuates brain damage and inflammation after traumatic brain injury in rats. Brain Res. 1637, 1–13. doi: 10.1016/j.brainres.2016.01.029
Tiana, P., Tenga, I. C., Mayb, L. D., Kurzb, R., Lub, K., Scadengb, M., et al. (2010). Cortical depth-specific microvascular dilation underlies laminar differences in blood oxygenation level-dependent functional MRI signal. PNAS 107, 15246–15251.
Tomkins, O., Kaufer, D., Korn, A., Shelef, I., Golan, H., Reichenthal, E., et al. (2001). Frequent blood-brain barrier disruption in the human cerebral cortex. Cell Mol. Neurobiol. 21, 675–691.
Tomkins, O., Shelef, I., Kaizerman, I., Eliushin, A., Afawi, Z., Misk, A., et al. (2008). Blood-brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry 79, 774–777. doi: 10.1136/jnnp.2007.126425
Trabold, R., Eros, C., Zweckberger, K., Relton, J., Beck, H., Nussberger, J., et al. (2010). The role of bradykinin B(1) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 30, 130–139. doi: 10.1038/jcbfm.2009.196
Tweedie, D., Rachmany, L., Rubovitch, V., Li, Y., Holloway, H. W., Lehrmann, E., et al. (2016). Blast traumatic brain injury-induced cognitive deficits are attenuated by preinjury or postinjury treatment with the glucagon-like peptide-1 receptor agonist, exendin-4. Alzheimers Dement 12, 34–48. doi: 10.1016/j.jalz.2015.07.489
Unterberg, A. W., Stover, J., Kress, B., and Kiening, K. L. (2004). Edema and brain trauma. Neuroscience 129, 1021–1029. doi: 10.1016/j.neuroscience.2004.06.046
Vázquez-Rosa, E., Shin, M. K., Dhar, M., Chaubey, K., Cintrón-Pérez, C. J., Tang, X., et al. (2020). P7C3-A20 treatment one year after TBI in mice repairs the blood-brain barrier, arrests chronic neurodegeneration, and restores cognition. Proc. Natl. Acad. Sci. U S A 117, 27667–27675. doi: 10.1073/pnas.2010430117
Villasana, L. E., Peters, A., McCallum, R., Liu, C., and Schnell, E. (2019). Diazepam Inhibits Post-Traumatic Neurogenesis and Blocks Aberrant Dendritic Development. J. Neurotrauma 36, 2454–2467. doi: 10.1089/neu.2018.6162
Wang, G., Shi, Y., Jiang, X., Leak, R. K., Hu, X., Wu, Y., et al. (2015). HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc. Natl. Acad. Sci. U S A 112, 2853–2858. doi: 10.1073/pnas.1501441112
Wang, T., Chou, D. Y., Ding, J. Y., Fredrickson, V., Peng, C., Schafer, S., et al. (2013). Reduction of brain edema and expression of aquaporins with acute ethanol treatment after traumatic brain injury. J. Neurosurg. 118, 390–396. doi: 10.3171/2012.8.Jns12736
Wang, Y., Fan, X., Tang, T., Fan, R., Zhang, C., Huang, Z., et al. (2016). Rhein and rhubarb similarly protect the blood-brain barrier after experimental traumatic brain injury via gp91(phox) subunit of NADPH oxidase/ROS/ERK/MMP-9 signaling pathway. Sci. Rep. 6:37098. doi: 10.1038/srep37098
Watkins, S., Robel, S., Kimbrough, I. F., Robert, S. M., Ellis-Davies, G., and Sontheimer, H. (2014). Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 5:4196. doi: 10.1038/ncomms5196
Willis, C. L., Nolan, C. C., Reith, S. N., Lister, T., Prior, M. J., Guerin, C. J., et al. (2004). Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia 45, 325–337. doi: 10.1002/glia.10333
Willis, E. F., MacDonald, K. P. A., Nguyen, Q. H., Garrido, A. L., Gillespie, E. R., Harley, S. B. R., et al. (2020). Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell 180, 833.e–846.e. doi: 10.1016/j.cell.2020.02.013
Winkler, E. A., Bell, R. D., and Zlokovic, B. V. (2011). Central nervous system pericytes in health and disease. Nat. Neurosci. 14, 1398–1405. doi: 10.1038/nn.2946
Wolburg, H., and Lippoldt, A. (2002). Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul. Pharmacol. 38, 323–337.
Wolburg-Buchholz, K., Mack, A. F., Steiner, E., Pfeiffer, F., Engelhardt, B., and Wolburg, H. (2009). Loss of astrocyte polarity marks blood-brain barrier impairment during experimental autoimmune encephalomyelitis. Acta Neuropathol. 118, 219–233. doi: 10.1007/s00401-009-0558-4
Wu, J., He, J., Tian, X., Luo, Y., Zhong, J., Zhang, H., et al. (2020). microRNA-9-5p alleviates blood-brain barrier damage and neuroinflammation after traumatic brain injury. J. Neurochem. 153, 710–726. doi: 10.1111/jnc.14963
Wu, Q., Gao, C., Wang, H., Zhang, X., Li, Q., Gu, Z., et al. (2018). Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int. J. Biochem. Cell Biol. 94, 44–55. doi: 10.1016/j.biocel.2017.11.007
Xiong, Y., Mahmood, A., and Chopp, M. (2017). Emerging potential of exosomes for treatment of traumatic brain injury. Neural. Regen Res. 12, 19–22. doi: 10.4103/1673-5374.198966
Xu, H., Jia, Z., Ma, K., Zhang, J., Dai, C., Yao, Z., et al. (2020). Protective effect of BMSCs-derived exosomes mediated by BDNF on TBI via miR-216a-5p. Med. Sci. Monit. 26:e920855. doi: 10.12659/msm.920855
Xu, X., Yin, D., Ren, H., Gao, W., Li, F., Sun, D., et al. (2018). Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol. Dis. 117, 15–27. doi: 10.1016/j.nbd.2018.05.016
Xu, Z. M., Yuan, F., Liu, Y. L., Ding, J., and Tian, H. L. (2017). Glibenclamide Attenuates Blood-Brain Barrier Disruption in Adult Mice after Traumatic Brain Injury. J. Neurotrauma 34, 925–933. doi: 10.1089/neu.2016.4491
Yan, C., Yan, H., Mao, J., Liu, Y., Xu, L., Zhao, H., et al. (2020). Neuroprotective Effect of Oridonin on Traumatic Brain Injury via Inhibiting NLRP3 Inflammasome in Experimental Mice. Front. Neurosci. 14:557170. doi: 10.3389/fnins.2020.557170
Yang, D. X., Jing, Y., Liu, Y. L., Xu, Z. M., Yuan, F., Wang, M. L., et al. (2019). Inhibition of Transient Receptor Potential Vanilloid 1 Attenuates Blood-Brain Barrier Disruption after Traumatic Brain Injury in Mice. J. Neurotrauma 36, 1279–1290. doi: 10.1089/neu.2018.5942
Yang, Y., Ye, Y., Kong, C., Su, X., Zhang, X., Bai, W., et al. (2019). MiR-124 Enriched Exosomes Promoted the M2 Polarization of Microglia and Enhanced Hippocampus Neurogenesis After Traumatic Brain Injury by Inhibiting TLR4 Pathway. Neurochem. Res. 44, 811–828. doi: 10.1007/s11064-018-02714-z
Yang, Y., Ye, Y., Su, X., He, J., Bai, W., and He, X. (2017). MSCs-Derived Exosomes and Neuroinflammation, Neurogenesis and Therapy of Traumatic Brain Injury. Front. Cell Neurosci. 11:55. doi: 10.3389/fncel.2017.00055
Yao, Y., Chen, Z. L., Norris, E. H., and Strickland, S. (2014). Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat. Commun. 5:3413. doi: 10.1038/ncomms4413
Yemisci, M., Gursoy-Ozdemir, Y., Vural, A., Can, A., Topalkara, K., and Dalkara, T. (2009). Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 15, 1031–1037. doi: 10.1038/nm.2022
Yin, T., Lindley, T. E., Albert, G. W., Ahmed, R., Schmeiser, P. B., Grady, M. S., et al. (2013). Loss of Acid sensing ion channel-1a and bicarbonate administration attenuate the severity of traumatic brain injury. PLoS One 8:e72379. doi: 10.1371/journal.pone.0072379
Zehendner, C. M., Sebastiani, A., Hugonnet, A., Bischoff, F., Luhmann, H. J., and Thal, S. C. (2015). Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci. Rep. 5:13497. doi: 10.1038/srep13497
Zhang, H. M., Chen, W., Liu, R. N., and Zhao, Y. (2018). Notch inhibitor can attenuate apparent diffusion coefficient and improve neurological function through downregulating NOX2-ROS in severe traumatic brain injury. Drug Des. Devel. Ther. 12, 3847–3854. doi: 10.2147/dddt.S174037
Zhang, J. Y., Lee, J. H., Gu, X., Wei, Z. Z., Harris, M. J., Yu, S. P., et al. (2018). Intranasally Delivered Wnt3a Improves Functional Recovery after Traumatic Brain Injury by Modulating Autophagic, Apoptotic, and Regenerative Pathways in the Mouse Brain. J. Neurotrauma 35, 802–813. doi: 10.1089/neu.2016.4871
Zhang, L., Zhang, S., Yao, J., Lowery, F. J., Zhang, Q., Huang, W. C., et al. (2015). Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104. doi: 10.1038/nature15376
Zhang, Y., Chopp, M., Meng, Y., Katakowski, M., Xin, H., Mahmood, A., et al. (2015). Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J. Neurosurg 122, 856–867. doi: 10.3171/2014.11.Jns14770
Zhang, Y., Chopp, M., Zhang, Z. G., Katakowski, M., Xin, H., Qu, C., et al. (2016). Systemic administration of cell-free exosomes generated by human bone marrow derived mesenchymal stem cells cultured under 2D and 3D conditions improves functional recovery in rats after traumatic brain injury. Neurochem. Int. 2016:003. doi: 10.1016/j.neuint.2016.08.003
Zhang, Y., Chopp, M., Zhang, Z. G., Zhang, Y., Zhang, L., Lu, M., et al. (2019). Cerebrolysin Reduces Astrogliosis and Axonal Injury and Enhances Neurogenesis in Rats After Closed Head Injury. Neurorehabil. Neural. Repair. 33, 15–26. doi: 10.1177/1545968318809916
Zhao, Q. H., Xie, F., Guo, D. Z., Ju, F. D., He, J., Yao, T. T., et al. (2020). Hydrogen inhalation inhibits microglia activation and neuroinflammation in a rat model of traumatic brain injury. Brain Res. 1748:147053. doi: 10.1016/j.brainres.2020.147053
Zhao, S., Yu, A., Wang, X., Gao, X., and Chen, J. (2016). Post-Injury Treatment of 7,8-Dihydroxyflavone Promotes Neurogenesis in the Hippocampus of the Adult Mouse. J. Neurotrauma 33, 2055–2064. doi: 10.1089/neu.2015.4036
Zhao, X., Gorin, F. A., Berman, R. F., and Lyeth, B. G. (2008). Differential hippocampal protection when blocking intracellular sodium and calcium entry during traumatic brain injury in rats. J. Neurotrauma 25, 1195–1205. doi: 10.1089/neu.2008.0635
Zhao, Y., Gibb, S. L., Zhao, J., Moore, A. N., Hylin, M. J., Menge, T., et al. (2016). Wnt3a, a Protein Secreted by Mesenchymal Stem Cells Is Neuroprotective and Promotes Neurocognitive Recovery Following Traumatic Brain Injury. Stem Cells 34, 1263–1272. doi: 10.1002/stem.2310
Zhao, Z., Nelson, A. R., Betsholtz, C., and Zlokovic, B. V. (2015). Establishment and Dysfunction of the Blood-Brain Barrier. Cell 163, 1064–1078. doi: 10.1016/j.cell.2015.10.067
Zhou, Y., Cai, W., Zhao, Z., Hilton, T., Wang, M., Yeon, J., et al. (2018). Lactadherin promotes microvesicle clearance to prevent coagulopathy and improves survival of severe TBI mice. Blood 131, 563–572. doi: 10.1182/blood-2017-08-801738
Zhu, W., Cui, G., Li, T., Chen, H., Zhu, J., Ding, Y., et al. (2020). Docosahexaenoic Acid Protects Traumatic Brain Injury by Regulating NOX(2) Generation via Nrf2 Signaling Pathway. Neurochem. Res. 45, 1839–1850. doi: 10.1007/s11064-020-03078-z
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Zlotnik, A., Klin, Y., Gruenbaum, B. F., Gruenbaum, S. E., Ohayon, S., Leibowitz, A., et al. (2012). β2 adrenergic-mediated reduction of blood glutamate levels and improved neurological outcome after traumatic brain injury in rats. J. Neurosurg. Anesthesiol. 24, 30–38. doi: 10.1097/ANA.0b013e318232deaa
Keywords: traumatic brain injury, blood-brain barrier, microenvironment, edema, inflammation, toxic substances, recovery
Citation: Hu Y and Tao W (2021) Microenvironmental Variations After Blood-Brain Barrier Breakdown in Traumatic Brain Injury. Front. Mol. Neurosci. 14:750810. doi: 10.3389/fnmol.2021.750810
Received: 31 July 2021; Accepted: 18 October 2021;
Published: 26 November 2021.
Edited by:
Michele Papa, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Ping Liao, National Neuroscience Institute (NNI), SingaporeAruna Sharma, Uppsala University, Sweden
Brandon Peter Lucke-Wold, University of Florida, United States
Copyright © 2021 Hu and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiwei Tao, dGFvd3dAbmp1Y20uZWR1LmNu