 Werner Kilb
Werner Kilb- Institute of Physiology, University Medical Center of the Johannes Gutenberg-University Mainz, Mainz, Germany
The membrane responses upon activation of GABA(A) receptors critically depend on the intracellular Cl− concentration ([Cl−]i), which is maintained by a set of transmembrane transporters for Cl−. During neuronal development, but also under several pathophysiological conditions, the prevailing expression of the Cl− loader NKCC1 and the low expression of the Cl− extruder KCC2 causes elevated [Cl−]i, which result in depolarizing GABAergic membrane responses. However, depolarizing GABAergic responses are not necessarily excitatory, as GABA(A) receptors also reduces the input resistance of neurons and thereby shunt excitatory inputs. To summarize our knowledge on the effect of depolarizing GABA responses on neuronal excitability, this review discusses theoretical considerations and experimental studies illustrating the relation between GABA conductances, GABA reversal potential and neuronal excitability. In addition, evidences for the complex spatiotemporal interaction between depolarizing GABAergic and glutamatergic inputs are described. Moreover, mechanisms that influence [Cl−]i beyond the expression of Cl− transporters are presented. And finally, several in vitro and in vivo studies that directly investigated whether GABA mediates excitation or inhibition during early developmental stages are summarized. In summary, these theoretical considerations and experimental evidences suggest that GABA can act as inhibitory neurotransmitter even under conditions that maintain substantial depolarizing membrane responses.
Introduction
About 30–40 years ago it was first published that GABAA receptors can mediate depolarizing and even excitatory membrane responses in the immature brain (Mueller et al., 1983; Ben-Ari et al., 1989; Luhmann and Prince, 1991), in contrast to the general textbook knowledge that GABA mediates inhibitory and mostly hyperpolarizing neurotransmission in the CNS. In the following decades, it has been shown that depolarizing GABAergic membrane responses play an essential role for cortical development (Ben-Ari, 2002; Owens and Kriegstein, 2002; Kirmse and Holthoff, 2020) and that such depolarizing GABAergic responses can be re-attained under pathophysiological conditions like trauma, stroke or epilepsy (Jaenisch et al., 2010; Dzhala et al., 2012; Kaila et al., 2014; Liu et al., 2020).
The main molecular mechanisms underlying these depolarizing membrane responses have in the meantime been unraveled (Blaesse et al., 2009; Loscher et al., 2013; Watanabe and Fukuda, 2015; Virtanen et al., 2020) and the existence of depolarizing GABAergic responses had been demonstrated in vivo (Kirmse et al., 2015; Valeeva et al., 2016; Murata and Colonnese, 2020). Thus evidences indicate that the direction of GABAergic membrane responses shows a striking modification during neurodevelopment and under pathophysiological conditions, a process termed “GABA-shift”. Regarding the physiological consequences of this “GABA-shift” it is, however, important to consider that depolarizing GABAergic responses can mediate inhibition already during early postnatal development (Khalilov et al., 1999; Kolbaev et al., 2011a; Kirmse et al., 2015; Valeeva et al., 2016) and that inhibitory responses in the adult CNS can be accompanied by GABAergic depolarizations (Andersen et al., 1980; Misgeld et al., 1982; Staley and Mody, 1992). To provide a current concept of the functional impact of depolarizing GABAergic responses, I summarize in this review theoretical considerations and experimental studies that illustrate how the relation between GABA conductances, GABA reversal potential and membrane potential changes determines the impact of GABA on neuronal excitability. In addition, I review studies that directly investigated whether GABA mediates excitation during early developmental stages.
Cl− and Hco3− Set The Pace for The Effects of GABAA Receptors
The flux of the hydrophilic Cl− ions across the hydrophobic plasma membrane occurs exclusively via integral membrane proteins that mediate Cl− transport. Passive Cl− fluxes are mediated by a heterogeneous set of anion channels that are more or less specific for Cl− ions (Duran et al., 2010; Jentsch and Pusch, 2018), including the GABAA receptor (Farrant and Kaila, 2007). The passive Cl− fluxes through these anion channels follow the electromotive force for Cl− ions (EMFCl), which depends on the difference between the Cl− equilibrium potential (ECl) and the membrane potential (Em). In consequence, hyperpolarizing or depolarizing GABAergic responses require that the intracellular Cl− concentration ([Cl−]i) is not in an equilibrium state. Active transmembrane transport is required for accumulation or depletion of Cl− from cells (Huebner and Holthoff, 2013; Kaila et al., 2014). In the absence of such active transport processes [Cl−]i follows a passive distribution, which due to the negative Em is set at low millimolar concentrations under physiological conditions (given by the Nernst equation). Either primary active Cl− transport, via an ATP-dependent Cl− pump, or secondary active transport, coupling Cl− transport to the transport of another ions along their gradient, is required to obtain [Cl−]i below or above this passive distribution. The major proteins that mediate secondary active transmembrane Cl− transport are Na+-dependent K+/Cl−-cotransporters (NKCC), K+/Cl−-cotransporters (KCC) and Cl−/HCO3−-antiporters (Payne et al., 2003; Blaesse et al., 2009; Figure 1), whereas there is currently little evidence for a neuronal Cl−-dependent ATPase or Cl− pump (Gerencser and Zhang, 2003). The most important Cl− loader in neurons is NKCC1 (SLC12A2), an ubiquitously expressed Cl− transporter that utilizes the inwardly directed Na+ gradient to mediate an electroneutral import of Cl− ions (and K+ ions) into cells (Russell, 2000; Virtanen et al., 2020). The main transporter responsible for the low [Cl−]i of mature neurons is KCC2 (SLC12A5; Rivera et al., 1999; Lee et al., 2005), which uses the outwardly directed K+ gradient to extrude Cl−. In addition, the isoforms KCC1, KCC3, and KCC4 were expressed in some neuron populations, but these isoforms can also be found in non-neuronal tissue (Becker et al., 2003). The anion exchanger (AE3) mediates the counter-transport of one Cl− with one HCO3−, thus leading to Cl− accumulation at physiological pH values (Gonzalez-Islas et al., 2009; Pfeffer et al., 2009). The contribution of Na+-dependent Cl−/HCO3−-antiporters, which mediate Cl− extrusion, to neuronal [Cl−]i homeostasis is less clear (Huebner and Holthoff, 2013). And finally, Misgeld and coworkers demonstrated that the combination of voltage-dependent Cl− channels with depolarizing Em transients can also lead to an elevated [Cl−]i (Titz et al., 2003).
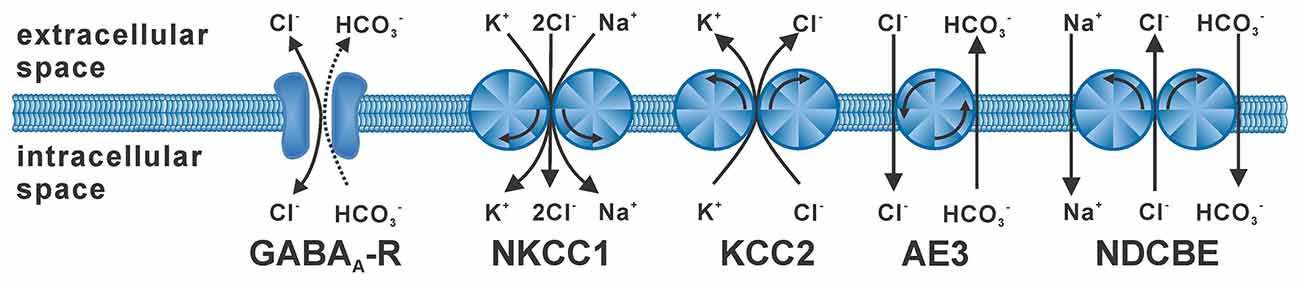
Figure 1. Stoichiometry and typical operation of secondary active Cl− transporters. The GABAA receptor (GABAA-R) mediates mainly Cl− fluxes and to a lesser extent HCO3− fluxes. NKCC1 mediates uptake of two Cl− ions with of one K+ and one Na+ ion. KCC2 mediates extrusion of one Cl− with one K+ ion. The anion-exchanger (AE3) is supposed to mediate uptake of one Cl− ion in antiport with one HCO3− ion. The Na+-dependent Cl−/HCO3− exchanger (NDCBE) utilizes the Na+ gradient to extrude Cl− ions.
GABAA receptors also have a considerable HCO3− permeability (Farrant and Kaila, 2007; Blaesse et al., 2009). The relative HCO3− permeability of GABAA receptors is between 0.18 and 0.44 of the Cl− permeability (Bormann et al., 1987; Fatima-Shad and Barry, 1993). Due to the rather positive equilibrium potential for HCO3− (EHCO3), which is around −10 mV, the HCO3− fluxes always add a depolarizing component to the GABAergic current (Rivera et al., 2005; Huebner and Holthoff, 2013). The high EHCO3 is a consequence of the low intracellular HCO3− concentration ([HCO3−]i), which is on one hand maintained by secondary active HCO3− uptake via electroneutral and electrogenic Na+/HCO3− symporters (Sinning et al., 2011; Huebner and Holthoff, 2013). On the other hand, the [HCO3−]i is directly linked to the intracellular pH (pHi) via the carbonic anhydrase (Sinning and Hübner, 2013). Thus at physiological pH values between 7.0 and 7.4 (Ruffin et al., 2014), which is maintained by aforementioned Na+/HCO3− symporters and the Na+/H+ exchanger (Ruffin et al., 2014), [HCO3−]i values of ca. 14 mM can be estimated (Lombardi et al., 2019). In consequence, EGABA is typically positive to ECl, albeit the contribution of EHCO3 to EGABA becomes smaller with higher [Cl−]i (Farrant and Kaila, 2007). During massive GABAergic stimulation, e.g., during epileptic seizures, the stable depolarizing drive of the GABAergic HCO3− currents will enhance activity-dependent Cl− uptake and thus directly contributes to the generation of GABAergic excitation under this conditions (Kaila et al., 1997; Ruusuvuori et al., 2004).
Expression Profile of Cl− Loaders and Cl− Extruders (And Why This Does Not Tell Everything About GABA Actions)
During development the different Cl−-transporters are differentially expressed in the nervous system (Blaesse et al., 2009; Huebner and Holthoff, 2013; Kaila et al., 2014; Kirmse et al., 2018). The expression levels of NKCC1 vary significantly between neuronal cell types and between different developmental or functional states of individual neurons (Watanabe and Fukuda, 2015; Virtanen et al., 2020).
Experimental studies show that NKCC1 expression either declines (Plotkin et al., 1997) or increases (Clayton et al., 1998) during development. Therefore, no general statements about the trend of NKCC1 expression level during development could be made (Virtanen et al., 2020). In contrast, the expression of KCC2 has been tightly correlated to the developmental downregulation of [Cl−]i. Suppression of functional KCC2 expression increases neuronal [Cl−]i (Rivera et al., 1999; Pellegrino et al., 2011), whereas enhancing the functional expression of KCC2 during early developmental stages leads to a reduced neuronal [Cl−]i (Lee et al., 2005). The expression of KCC2 occurs typically delayed to the expression of NKCC1 (Lu et al., 1999; Rivera et al., 1999; Stein et al., 2004), with the temporal profile of KCC2 expression depending on brain structures (Watanabe and Fukuda, 2015), cortical layers (Li et al., 2002; Shimizu-Okabe et al., 2002), and neuronal cell types (Ikeda et al., 2003; Clarkson and Herbison, 2006). In particular, GABAergic interneurons seem to display systematically more depolarized GABA reversal potentials (EGABA) than glutamatergic or principle neurons in the amygdala and neocortex (Martina et al., 2001), the cerebellum (Chavas and Marty, 2003), and in the hippocampus (Elgueta and Bartos, 2019; Otsu et al., 2020). For the hippocampus this has directly been related to a lower KCC2 expression in the GABAergic interneurons (Elgueta and Bartos, 2019). There are also evidences that the expression ratio between NKCC1 and KCC2 can be different in distinct compartments of the same cell (Virtanen et al., 2020). The most striking example is the axon initial segment, in which GABAergic synapses mediate depolarizing responses with putative excitatory effect (Szabadics et al., 2006; Khirug et al., 2008), which has been linked to the delayed shift in the expression ration of Cl− transporter expression in this compartment (Rinetti-Vargas et al., 2017). The observation of a somatodencritic [Cl−]i gradient (Kuner and Augustine, 2000; Elgueta and Bartos, 2019) suggest a variable NKCC1/KCC2 expression ratio within dendritic membranes.
A variety of pathophysiological conditions are also linked to massive changes in the [Cl−]i -homeostasis (Kaila et al., 2014). It has been shown that depolarizing GABAergic responses and/or an high NKCC1/KCC2 expression ration can be re-attained for example after traumatic (Toyoda et al., 2003; Dzhala et al., 2012) or ischemic (Jaenisch et al., 2010) insults, in peri-tumor regions (Pallud et al., 2014; Campbell et al., 2015) and as cause or consequence of epilepsy (Fujiwara-Tsukamoto et al., 2003; Aronica et al., 2007; Huberfeld et al., 2007; Buchin et al., 2016; Burman et al., 2019; Liu et al., 2020). The putative switch in GABAergic responses from excitation to inhibition related to this alterations can aggravate the clinical consequences of these neuropathologies (Kaila et al., 2014).
However, it should always be kept in mind that both KCC2 and NKCC1 are highly regulated proteins (Russell, 2000; Blaesse et al., 2009; Kaila et al., 2014). For example it has been demonstrated that membrane trafficking and functional expression of KCC2 is tightly controlled by the sonic hedgehog pathway, neuronal-restricted silencing element or the neurotrophin BDNF (Karadsheh and Delpire, 2001; Rivera et al., 2002; Delmotte et al., 2020). In addition, the activity of KCC2 can be modulated by phosphorylation (Wake et al., 2007; Banke and Gegelashvili, 2008) and thereby neurotransmitters can directly interfere with the [Cl−]i homeostasis (Banke and Gegelashvili, 2008; Inoue et al., 2012; Yang et al., 2015). Thus the [Cl−]i is clearly regulated beyond the expression ratio of Cl− transporters.
The situation is made even more complicated by the fact that [Cl−]i, is also directly influenced by GABAergic activity via Cl− fluxes across GABAA receptors (Wright et al., 2011; Raimondo et al., 2012; Branchereau et al., 2016). This is not only relevant for high, pathophysiological activity patterns, but also for physiological levels of neuronal activity (Gonzalez-Islas et al., 2010; Kolbaev et al., 2011b; Currin et al., 2020). In-silico experiments show that these activity-dependent [Cl−]i changes are influenced by the dendritic morphology, membrane properties as well as the kinetics of GABAergic inputs, [Cl−]i homeostasis and [HCO3−]i homeostasis (Doyon et al., 2016a; Mohapatra et al., 2016; Düsterwald et al., 2018; Lombardi et al., 2019). In addition, such activity-dependent [Cl−]i transients are augmented by coincident glutamatergic inputs (Halbhuber et al., 2019; Lombardi et al., 2021). Thus the impact of neuronal activity on [Cl−]i is probably particularly relevant under in vivo situations, where an ongoing “bombardment” with GABAergic and glutamatergic synaptic inputs has been suggested (Steriade, 2001).
In summary, these facts demonstrate that, although an increasing KCC2 expression is correlated to a [Cl−]i decline during neuronal development, the precise [Cl−]i, and thus the GABAergic effects cannot be directly estimated from the general expression level of KCC2. In particular, results from in vitro experiments may overestimate the in vivo levels of [Cl−]i in immature neurons (and underestimate them in adult neurons), because the influence of neuronal activity on [Cl−]i is negligible due to the limited activity in the in vitro preparations.
Relation Between [Cl−]I, GABAergic Membrane Responses and Excitation/Inhibition
The [Cl−]i is a main factor that determines EMFCl and thus the direction and size of Cl− fluxes through the anion-pore of GABAA receptors (Farrant and Kaila, 2007). As mentioned before, HCO3− fluxes contribute to GABAergic membrane responses, because GABAA receptors have a considerable HCO3− permeability (Farrant and Kaila, 2007). EMFHCO3 is directed outwards and therefore HCO3− efflux adds a depolarizing component to GABAergic responses. Whereas the HCO3− fluxes shift GABA responses in depolarizing direction for low [Cl−]i, their contribution is relatively small at higher [Cl−]i (Farrant and Kaila, 2007). Because of its mostly minor contribution and in order to make the following considerations more concise, HCO3−-fluxes will not be taken into account in the remainder of this review. Nevertheless, the (small) depolarizing HCO3−-currents via GABAA receptors will slightly enhance the excitatory potency of GABAergic effects.
When considering the effect of GABA on neuronal excitability, one should keep in mind that GABA can mediate inhibition by two mechanisms (Farrant and Kaila, 2007): first, by hyperpolarization, which increases the difference between Em and the AP threshold (Figure 2A, left traces), and second by a decreased membrane resistivity upon GABAA receptor activation (Figure 2A, right traces), which shunts excitatory synaptic inputs (Edwards, 1990; Staley and Mody, 1992; Farrant and Kaila, 2007). However, in reality both effects act in parallel (Figure 2B). To predict the effects of GABA on the excitability, it is necessary to delineate how both mechanisms are related to [Cl−]i. If EMFCl− is negative (i.e., [Cl−]i is below the passive distribution) activation of GABAA receptors will induce a Cl− influx and thus hyperpolarize the membrane. Such a membrane hyperpolarization, together with the membrane shunting, will increases the amount of excitatory synaptic currents required to cross the AP threshold. Thus it is obvious that under low [Cl−]i conditions GABAA receptors mediate an inhibitory effect on neuronal membranes. It is sometimes considered that at higher [Cl−]i GABA receptors mediate an opposite effect, because the depolarizing GABAergic responses shift Em towards AP threshold. However, under this condition the excitatory influence of this depolarizing effect is opposed by the reduction in the membrane resistivity, which in parallel shunts excitatory postsynaptic potentials (Edwards, 1990; Staley and Mody, 1992; Egawa and Fukuda, 2013). Thereby depolarizing GABAergic responses can also reduce excitatory influence and thus mediate inhibition (Figure 2B). The central question arising from these considerations is: Which GABAergic membrane depolarization is required to mediate an excitatory response, i.e., to increase the probability to trigger an AP?
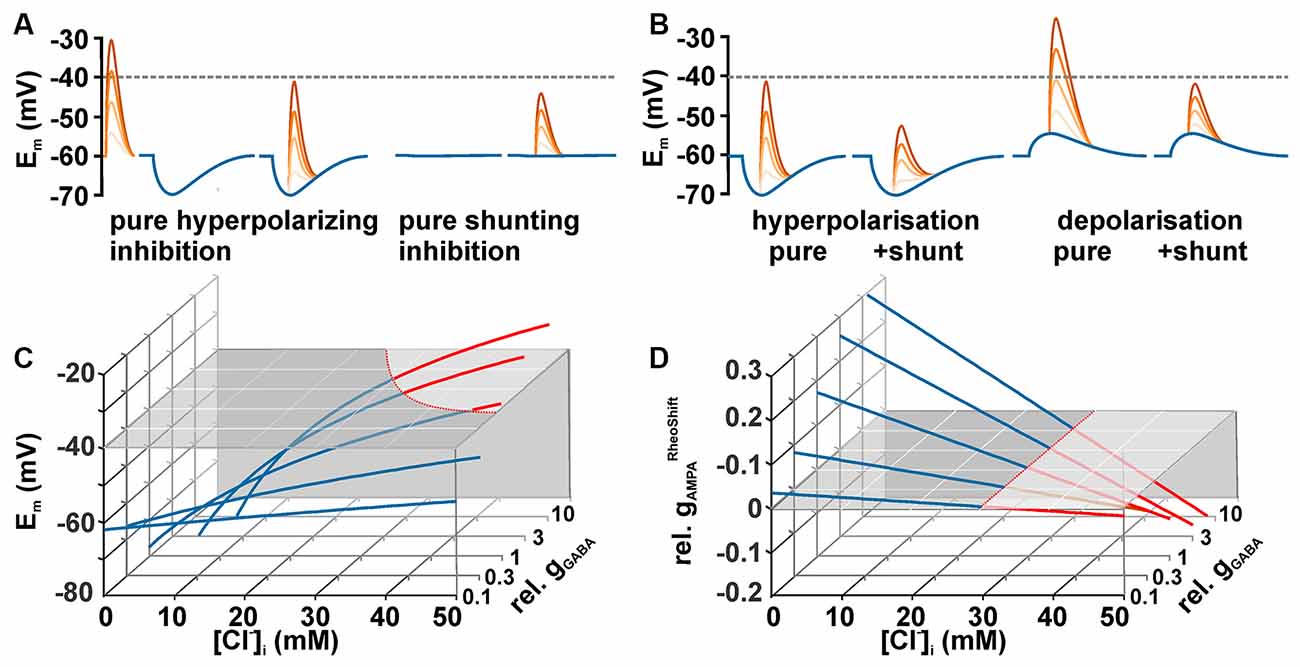
Figure 2. Dependency between [Cl−]i and GABAergic actions. (A) Schematic diagrams illustrating the two exemplary effects of GABAergic inputs (blue traces) on glutamatergic inputs (red traces) of different intensities. A GABAergic hyperpolarization augments the distance between peak glutamate depolarization and the AP threshold (hyperpolarizing inhibition). At a passive Cl−-distribution GABA does not affect Em, but the decreased membrane resistivity induced by GABA reduced the peak glutamate depolarization (shunting inhibition). The dashed line represents a hypothetical action potential threshold. (B) Schematic diagrams illustrating that the combination of the membrane potential shift with the shunting effect caused by the decreased membrane resistivity augments the effect of a hyperpolarization inhibition (left traces) and can lead to inhibition even at depolarizing GABAergic membrane responses (right traces). (C) [Cl−]i-dependency of the membrane potential (Em) calculated for five different GABAergic conductances (gGABA, normalized to gInput) under stationary conditions (see main text for details). The gray plane represents AP threshold. Note that considerable gGABA in combination with high [Cl−]i is needed for a suprathreshold GABAergic depolarization. (D) Dependency of gAMPARheoshift (normalized to gInput, ; gAMPARheoshift = gAMPA in presence of GABA minus gAMPA in absence of GABA) on [Cl−]i and gGABA. Red traces indicate excitatory and blue traces inhibitory GABAergic effects. Note that gGABARheoshift becomes negative at identical [Cl−]i independent of gGABA.
Theoretically, this question can be easily addressed by calculating the membrane depolarization and the shunting effect that are caused by a given GABAergic conductance. The maximal GABAergic depolarization is, in accordance with Ohm’s law, given by the product of the GABAergic current (IGABA) and the input resistance (RInput). IGABA is given by the product of EMFCl (when the GABAergic HCO3− permeability is neglected) and gGABA. A combination of these functions define the relation between gGABA and EMFCl and can be used to estimate under which conditions GABA responses itself can reach AP threshold and trigger APs (Figure 2C).
However, this situation does not reflect the physiologically relevant situation. More relevant is the question whether subtreshold GABAergic depolarizations can attenuate or augment the excitatory effect of additional excitatory synaptic inputs. In this respect it must be considered that the increase in gGABA, which is necessarily linked to the depolarization, also reduces the membrane resistivity and thus shunts excitatory synaptic inputs. The impact of both effects on the excitability can be estimated in a simplified model for Em (which neglects the HCO3− conductance of GABAA receptors) as follows:
(with and as passive membrane conductance for Na+ and K+, gGABA and gAMPA as conductance of GABA and AMPA receptors, and [X+]i and [X+]e as intra- and extracellular concentration of the ion X+, respectively). Note that at this moment we use a rather simple model that neglects capacitive currents, the time course of GABAergic and glutamatergic conductances, as well as spatial integration (Gidon and Segev, 2012). Thus this formula describes only the interaction of stationary GABAergic and glutamatergic conductances.
Using this formula, the gGlu value leading to a depolarization that equals the AP threshold (EThr) can be calculated as follows:
These threshold gGlu values () describe the excitatory conductance required to just reach AP threshold. To quantify the GABAergic effect on the excitability, determined in the absence of GABA (i.e., gGABA = 0) is subtracted from determined in the presence of GABA. This value is termed GABAergic rheobase shift (). Negative values characterize an excitatory GABAergic action (less gGlu is required to induce APs). Intriguingly, GABA mediates an excitatory action, independent of the gGABA values, for all [Cl−]i ≥ 29.5 mM in the exemplary simulated neuron used for Figure 2D. Note that this [Cl−]i corresponds to an EGABA that is identical to the AP threshold of −40 mV used in this model. This relation suggests that GABA mediate an excitation whenever EGABA is positive to AP threshold. This theoretical suggestion is in line with previous assumptions that GABA mediate an excitatory effect as long as EGABA is above AP threshold (Ben-Ari, 2002; Owens and Kriegstein, 2002) and it was replicated in patch-clamp experiments (Kolbaev et al., 2011a).
However, as already mentioned the previous considerations are clearly an oversimplification as they: (i) neglect additional voltage gated conductances that contribute to excitability (Valeeva et al., 2010); and (ii) represent only stationary conductances in a quasi one-dimensional situation ignoring the consequences of the temporal relation between GABA and glutamatergic inputs (Gao et al., 1998; Gulledge and Stuart, 2003) and the complex neuronal topologies (Jadi et al., 2012; Spruston et al., 2016) on spatiotemporal properties of GABAergic inhibition/excitation.
To understand the influence of temporal relation between GABA and glutamatergic inputs on the excitability, it is important to consider that the GABAergic membrane depolarization outlasts the GABAergic conductance increase (Figure 3A). The amplitude of a glutamatergic excitatory postsynaptic potential (ePSP) drops by shunting effects when it was evoked in synchrony to the GABAergic input. However, when the glutamatergic input was stimulated during the late phase of the GABAergic depolarization (when the GABAergic conductance ceases, but the GABAergic depolarization is still present), temporal summation lead to an increased peak voltage of the compound postsynaptic potential (PSP; Figure 3B). Thereby, GABA can mediate a substantial inhibitory effect on synchronously occurring glutamatergic inputs, while the longer lasting depolarization can enhance the amplitude of delayed ePSPs and thus mediate excitation (Figure 3C). Such a temporal shunting-to-excitation sequence has already been shown in vitro (Gao et al., 1998; Gulledge and Stuart, 2003; Bracci and Panzeri, 2006). This finding also implies that for excitatory inputs that occur with a substantial delay after the GABA input mainly the GABAergic depolarization is effective and thus an excitatory effect can be imposed whenever EGABA is positive to RMP. In consequence, GABA may more probably have an excitatory effect when GABA inputs are not temporally correlated to glutamatergic inputs, e.g., for tonic GABAergic inhibition (Song et al., 2011; Kolbaev et al., 2012). On the other hand, when GABA and glutamate inputs are temporally highly correlated, e.g., at feedback, feedforward, or lateral inhibition, GABA mediates a reliable inhibition as long as EGABA is below AP threshold.
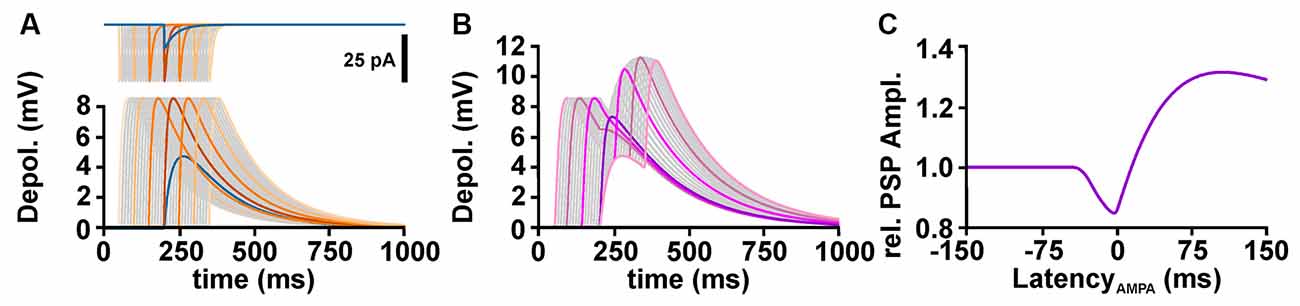
Figure 3. Temporal profile of GABAergic shunting and GABAergic depolarizing effects on excitatory glutamatergic inputs. (A) The upper traces illustrate GABAergic (blue line) and glutamatergic (orange and gray lines) currents provided at latencies between −150 and +150 ms. The lower traces illustrate the postsynaptic potentials (PSPs) evoked by these currents. Note that the PSPs outlast the synaptic currents. (B) Compound PSPs induced by the co-stimulation of GABA and glutamate synapses, with the glutamatergic inputs provided at latencies between −150 and +150 ms. (C) Peak amplitude of compound PSP, normalized to the glutamatergic PSP in the absence of GABA, plotted against the latency between AMPA and GABA stimuli, as shown in (B). Note that the compound PSP amplitude drops if glutamatergic synapses are activated within a narrow interval around coincident stimulation, but increases when AMPA receptors are stimulated several ms after the GABA input.
To address the role of complex neuronal topologies on the GABAergic effect, it must be considered: (i) that GABAA receptor activation influences the length and time constant of membranes and (ii) that the GABAergic effect depends on the spatial relation between the GABAergic and the glutamatergic inputs. A simple NEURON-based in-silico simulation demonstrate that a GABAergic PSPs showed the typical decline in the amplitude with increasing dendritic distance (Figure 4A), which reflects the length constant within a linear neuronal structure (Rall, 1989). On the other hand, the membrane shunting effect demonstrated a more complex behavior (Gulledge and Stuart, 2003; Gidon and Segev, 2012). If the GABA synapse is located between the soma and the excitatory glutamatergic input (“on-path”, Gidon and Segev, 2012) a stable attenuation of the ePSP amplitude occurs (Figure 4B). But when the GABA synapse is distal to the AMPA synapse (“off-path”), the shunting declines rather fast (Figure 4B). To estimate whether GABA has an excitatory or inhibitory influence, the interaction between the wide spreading depolarizing effect (Figure 4A) and the complex spatial profile of shunting inhibition (Figure 4B) is fundamental. The co-stimulation of a depolarizing GABA synapse which is co-localized to an AMPA synapse mediate an inhibitory effect (Figure 4C), suggesting a dominance of the shunting effect. With increasing distance between the GABA and the AMPA synapse the shunting effect was attenuated and the excitatory potential of the GABAergic depolarization dominates, leading to an excitatory effect of the co-stimulation (Figure 4C). Thus even under mild depolarizing conditions found in dendrites of mature cortical pyramidal cells (Kuner and Augustine, 2000) GABAergic stimulation in the remote dendritic compartment can mediate an excitatory response (Gulledge and Stuart, 2003).
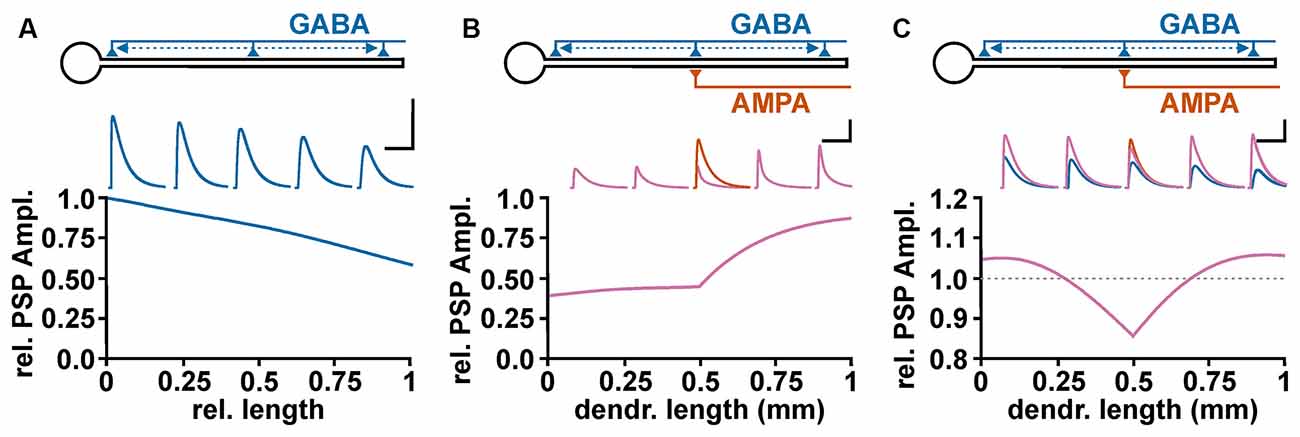
Figure 4. Spatial profile of GABAergic depolarization and GABAergic shunting effects on excitatory glutamatergic inputs. (A) Relative amplitude of GABAergic PSPs, as measured at the soma, upon activation of a depolarizing GABA synapse (EGABA = −52 mV) at different dendritic positions. The voltage traces above graph illustrate GABAergic PSPs at 0%, 25%, 50%, 75%, and 100% of the dendritic length. Scale bar in (A–C) is 5 mV/500 ms. (B) Profile of the GABAergic shunting effect on glutamatergic inputs, calculated by normalizing the amplitude of the compound PSPs obtained in the presence of GABA (purple traces) to the EPSC amplitude obtained in the absence of GABA inputs (orange trace). In these experiments the shunting effect was isolated by maintaining EGABA at resting membrane potential. Note that GABA synapses located proximally to the AMPA synapse (“on-path”) mediate a stable shunting effect, while for GABA synapses distal to the AMPA synapse (“off-path”) the shunting effect declines rather fast. (C) Effect of a depolarizing GABAergic input (EGABA = −52 mV) at different positions along the dendrite on the peak compound PSP amplitude during co-stimulation The blue traces represent purely GABAergic PSPs, the orange trace the glutamatergic PSP, and the purple traces the compound PSPs upon co-activation of AMPA and GABA synapses. Note that GABA inputs mediate an inhibitory effect when co-localized with the AMPA synapse, while at more distant on-path and off-path synapses an excitatory effect is observed.
Of note, these findings have some implication for the inhibition mediated by GABA receptors. The typical perisomatic GABAergic inputs of parvalbumin-positive basket interneurons (Freund, 2003; Elgueta and Bartos, 2019) will mediate a stable inhibitory effect, even at depolarizing GABAergic responses, since they can effectively shunt ePSPs. In contrast, for GABAergic synapses located in the dendritic periphery, e.g., from hippocampal O-LM interneurons (Somogyi and Klausberger, 2005) or neocortical Martinotti interneurons (Ascoli et al., 2008; Gidon and Segev, 2012), depolarizing GABA responses can more easily mediate an excitatory effect on glutamatergic inputs from distant sites in the dendrite.
In summary, the action of GABA did not only depend on the ratio between EGABA and the AP threshold, but also on the spatiotemporal relation between the GABAergic and glutamatergic inputs. Thus, under physiological conditions depolarizing GABAergic inputs can mediate in the same cell excitation as well as inhibition, depending on the exact spatiotemporal relation between both inputs. Thus it is difficult or even impossible to make general predictions for a global effect of depolarizing GABAergic responses. However, from the published observations one can presume: (i) that at sufficiently high EGABA above the AP threshold reliable excitation is mediated by GABAA receptors; (ii) that at intermediate EGABA levels GABA mediates a dominant inhibitory effect for spatially and temporally correlated inputs; and (iii) that the effect of GABA on delayed or spatially separated inputs can be excitatory under this conditions. Thus the typical GABAergic feedforward or feedback loops with perisomatic terminals will already mediate inhibition, even at higher [Cl−]I that are typical during development (Farrant and Kaila, 2007; Blaesse et al., 2009). In addition, these synapses will be rather resistant to activity-dependent [Cl−]i increases (Wright et al., 2011; Doyon et al., 2016b; Lombardi et al., 2018). In contrast, other modes of GABAergic mechanisms are more prone to mediate an excitatory influence at rather moderate [Cl−]i increases.
Examples for Excitatory and Inhibitory GABAergic Effects in The Immature CNS
With all of the information provided above, one of the major questions remaining is, of course, whether the depolarizing GABAA receptor-mediated responses in the immature CNS (Ben Ari et al., 2012) have a net excitatory or inhibitory effect.
Several in vitro studies demonstrate that activation of GABAA receptors can mediate excitatory inputs in immature neurons. For example, it was demonstrated that hippocampal giant depolarizations critically depend on tonic depolarizing GABAergic currents (Ben-Ari et al., 1989; Sipila et al., 2005). Gramicidin-perforated or cell-attached patch-clamp experiments, which both did not artificially alter [Cl−]i and thus allow estimating the physiological GABA responses, demonstrated suprathreshold GABAergic responses in neocortical (Dammerman et al., 2000; Hanganu et al., 2002; Rheims et al., 2008; Sava et al., 2014), hippocampal (Khazipov et al., 1997; Leinekugel et al., 1997; Sauer and Bartos, 2010; Valeeva et al., 2013), and hypothalamic neurons (Wang et al., 2001). And finally, optogenetic activation of GABAergic interneurons in vitro increases the frequency of neocortical and hippocampal EPSCs (Valeeva et al., 2016), as well as synchronous network activity (Flossmann et al., 2019), demonstrating a direct excitatory effect of GABA in neuronal networks. Excitatory GABAergic actions have also been found in mature neurons for distant off-path GABAergic inputs (Gulledge and Stuart, 2003).
In contrast to these reports of excitatory GABAergic actions, several in vitro studies also demonstrate that GABAergic stimulation can mediate inhibition already in early postnatal neurons (Agmon et al., 1996; Khalilov et al., 1999; Lamsa et al., 2000). The frequent observations that inhibition of GABAA receptors provoke epileptiform discharges in the immature CNS (Khalilov et al., 1999; Wells et al., 2000; Richter et al., 2010; Kolbaev et al., 2012; Sharopov et al., 2019) also suggest that GABA may mediate a net inhibitory effect in immature hippocampal and neocortical networks. However, depolarizing GABAergic responses has been suggested to significantly contribute to epilepsy in the immature CNS (Dzhala and Staley, 2003; Dzhala et al., 2005; Khalilov et al., 2005; Nardou et al., 2009, 2013). This discrepancy most probably reflects the complexity of functional consequences of depolarizing GABAergic responses. Notably, in vitro experiments demonstrated that weak GABAergic stimulation can promote excitation, whereas stronger GABAergic currents mediate inhibition (Khalilov et al., 1999; Winkler et al., 2019), indicating that the balance between GABAergic depolarization and shunt determines the net effect. In line with the aforementioned spatiotemporal dependency of GABAergic effects, it has been observed in the immature hippocampus that synaptic GABAA receptors mediate an anticonvulsive and tonic GABAA receptors a proconvulsive effect (Kolbaev et al., 2012). In summary, these experiments promote the view that it depends, in addition to the [Cl−]i, on the properties and mode of GABAergic stimulation whether GABA has a pro- or anticonvulsive effect.
However, the in vitro experiments summarized above represent fairly artificial conditions that may severely interfere with [Cl−]i homeostasis. The slicing procedures used for the generation of most in vitro preparation represent a traumatic insult, which alters the expression and function of NKCC1 and/or KCC2 and led to an increased [Cl−]i in many neurons within such preparations (Dzhala et al., 2012). In addition, neuronal activity, and thus most probably also the frequency of GABAergic inputs, is massively reduced in most in vitro preparations (Steriade, 2001). However, the frequency of GABAergic inputs massively influences [Cl−]i and lead in immature neurons to a reduction of their high [Cl−]i (Kolbaev et al., 2011b; Wright et al., 2011; Lombardi et al., 2018). Thus, in vitro condition may systematically overestimate the excitatory capacity of GABAA receptors. Thus it is essential that the effect of GABA in immature nervous systems must also be investigated under in vivo conditions.
Seminal in vivo experiments addressing the functional responses of GABA on cortical neurons during early developmental stages demonstrated that exogenously applied GABA indeed mediates depolarizing membrane responses, but that these responses reduce neuronal activity in the developing neocortex (Kirmse et al., 2015). In line with these results, optogenetic activation of GABAergic interneurons in vivo decreases the frequency of neocortical and hippocampal EPSCs already at early developmental stages (Valeeva et al., 2016), demonstrating a direct inhibitory effect of GABA on neuronal networks. Interestingly, this in vivo result is opposing the observation made in the same study under in vitro conditions, emphasizing the limitations of conclusions drawn from in vitro experiments. Also the observations that the GABA antagonist gabazine enhances the frequency of spindle bust oscillation in the early postnatal neocortex in vivo (Minlebaev et al., 2007), and that GABAergic agonists attenuate epileptiform activity in vivo (Isaev et al., 2005) already suggest a putative inhibitory role of GABA at this developmental stage.
On the other hand, recent studies demonstrated that activation of GABAergic interneurons in vivo can also enhance network activity in the immature hippocampus, suggesting that also under in vivo conditions GABA may exert an excitatory effect in this region. Using DREADD as well as optogenetic approaches, it was demonstrated that activation of GABAergic interneurons enhances and inhibition of GABAergic interneurons suppresses network activity in hippocampus of non-anesthetized 3 day old mice pups (Murata and Colonnese, 2020). This effect reversed to GABAergic inhibition already at the 7th postnatal day (Murata and Colonnese, 2020). Comparable results are observed when depolarizing GABAergic responses during early development are minimized by a conditional NKCC1 knockout in pyramidal neurons. In these animals the spontaneous correlated network activity in the hippocampus was attenuated (Graf et al., 2021), suggesting a putative excitatory effect of depolarizing GABAergic responses in the immature hippocampus. However, in line with the previous in vivo studies on neocortical areas these two in vivo study demonstrated for the visual cortex that activation of GABAergic interneurons mediates inhibition already at the 3rd postnatal day (Murata and Colonnese, 2020) and that a conditional NKCC1 knockout in pyramidal neurons of the visual cortex has no effect on the typical network activity (Graf et al., 2021).
Conclusion
While it is obvious that the effect of GABAA receptor activation critically depends on the [Cl−]i, and thus on the expression and function of Cl− transporters, theoretical consideration and many experimental findings indicate that the effect of GABA on the excitability cannot reliably be predicted only from the expression ratio of Cl− transporters or the [Cl−]i. Several additional parameters determine whether GABA mediate excitation or inhibition at a given [Cl−]i. Recent experimental evidences suggest that GABA probably mediates inhibition already in the immature cortex, whereas it may contribute to excitation in the immature hippocampus. However, these experiments can, of course, not predict GABAergic effects during fetal stages or in brain structures that have not been investigated yet.
Author Contributions
WK conceptualized and wrote this manuscript.
Funding
WK received funding from the Deutsche Forschungsgemeinschaft (DFG-grant Ki835/3).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
WK thanks Heiko J. Luhmann, Anne Sinning, Peter Jedlicka, and Atsuo Fukuda for intense discussion on this topic. The valuable contribution of Aniello Lombardi, Sergey Kolbaev, Katharina Achilles, Lisa Halbhuber, Akihito Okabe, Chigusa Shimizu-Okabe, Ileana Hanganu-Opatz, Junko Yamada, Salim Sharopov, Daniel Richter, Bogdan Sava, Haiyan Sun, Sigrid Stroh-Kaffei, Violetta Steinbrecher, and Beate Krumm to the experimental evaluation of the complex relation between GABAA receptors and the [Cl−]i is highly acknowledged.
References
Agmon, A., Hollrigel, G., and O’Dowd, D. K. (1996). Functional GABAergic synaptic connection in neonatal mouse barrel cortex. J. Neurosci. 16, 4684–4695. doi: 10.1523/JNEUROSCI.16-15-04684.1996
Andersen, P., Dingledine, R., Gjerstad, L., Langmoen, I. A., and Laursen, A. M. (1980). Two different responses of hippocampal pyramidal cells to application of gamma-amino butyric acid. J. Physiol. 305, 279–296. doi: 10.1113/jphysiol.1980.sp013363
Aronica, E., Boer, K., Redeker, S., Spliet, W. G. M., van Rijen, P. C., Troost, D., et al. (2007). Differential expression patterns of chloride transporters, Na+-K+-2Cl−-cotransporter and K+-Cl−-cotransporter, in epilepsy-associated malformations of cortical development. Neuroscience 145, 185–196. doi: 10.1016/j.neuroscience.2006.11.041
Ascoli, G. A., Alonso-Nanclares, L., Anderson, S. A., Barrionuevo, G., Benavides-Piccione, R., Burkhalter, A., et al. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568. doi: 10.1038/nrn2402
Banke, T. G., and Gegelashvili, G. (2008). Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J. Physiol. 586, 4925–4934. doi: 10.1113/jphysiol.2008.157024
Becker, M., Nothwang, H. G., and Friauf, E. (2003). Differential expression pattern of chloride transporters NCC, NKCC2, KCC1, KCC3, KCC4 and AE3 in the developing rat auditory brainstem. Cell Tissue Res. 312, 155–165. doi: 10.1007/s00441-003-0713-5
Ben-Ari, Y. (2002). Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J.-L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325. doi: 10.1113/jphysiol.1989.sp017762
Ben Ari, Y., Woodin, M. A., Sernagor, E., Cancedda, L., Vinay, L., Rivera, C., et al. (2012). Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever!. Front. Cell. Neurosci. 6:35. doi: 10.3389/fncel.2012.00035
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. doi: 10.1016/j.neuron.2009.03.003
Bormann, J., Hamill, O. P., and Sakmann, B. (1987). Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J. Physiol. 385, 243–286. doi: 10.1113/jphysiol.1987.sp016493
Bracci, E., and Panzeri, S. (2006). Excitatory GABAergic effects in striatal projection neurons. J. Neurophysiol. 95, 1285–1290. doi: 10.1152/jn.00598.2005
Branchereau, P., Cattaert, D., Delpy, A., and Allain, A. (2016). Depolarizing GABA/glycine synaptic events switch from excitation to inhibition during frequency increases. Sci. Rep. 6:21753. doi: 10.1038/srep21753
Buchin, A., Chizhov, A., Huberfeld, G., Miles, R., and Gutkin, B. S. (2016). Reduced efficacy of the KCC2 cotransporter promotes epileptic oscillations in a subiculum network model. J. Neurosci. 36, 11619–11633. doi: 10.1523/JNEUROSCI.4228-15.2016
Burman, R. J., Selfe, J. S., Lee, J. H., Van Den Berg, M., Calin, A., Codadu, N. K., et al. (2019). Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain 142, 3482–3501. doi: 10.1093/brain/awz283
Campbell, S. L., Robel, S., Cuddapah, V. A., Robert, S., Buckingham, S. C., Kahle, K. T., et al. (2015). GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 63, 23–36. doi: 10.1002/glia.22730
Chavas, J., and Marty, A. (2003). Coexistence of excitatory and inhibitory GABA synapses in the cerebellar interneuron network. J. Neurosci. 23, 2019–2031. doi: 10.1523/JNEUROSCI.23-06-02019.2003
Clarkson, J., and Herbison, A. E. (2006). Development of GABA and glutamate signaling at the GnRH neuron in relation to puberty. Mol. Cell. Endocrinol. 254–255, 32–38. doi: 10.1016/j.mce.2006.04.036
Clayton, G. H., Owens, G. C., Wolff, J. S., and Smith, R. L. (1998). Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Brain Res. Dev. Brain Res. 109, 281–292. doi: 10.1016/s0165-3806(98)00078-9
Currin, C. B., Trevelyan, A. J., Akerman, C. J., and Raimondo, J. V. (2020). Chloride dynamics alter the input-output properties of neurons. PLoS Comput. Biol. 16:e1007932. doi: 10.1371/journal.pcbi.1007932
Dammerman, R. S., Flint, A. C., Noctor, S., and Kriegstein, A. R. (2000). An excitatory GABAergic plexus in developing neocortical layer 1. J. Neurophysiol. 84, 428–434. doi: 10.1152/jn.2000.84.1.428
Delmotte, Q., Hamze, M., Medina, I., Buhler, E., Zhang, J., Belgacem, Y. H., et al. (2020). Smoothened receptor signaling regulates the developmental shift of GABA polarity in rat somatosensory cortex. J. Cell Sci. 133:jcs247700. doi: 10.1242/jcs.247700
Doyon, N., Prescott, S. A., and De Koninck, Y. (2016a). Mild KCC2 hypofunction causes inconspicuous chloride dysregulation that degrades neural coding. Front. Cell. Neurosci. 9:516. doi: 10.3389/fncel.2015.00516
Doyon, N., Vinay, L., Prescott, S. A., and De Koninck, Y. (2016b). Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron 89, 1157–1172. doi: 10.1016/j.neuron.2016.02.030
Duran, C., Thompson, C. H., Xiao, Q., and Hartzell, H. C. (2010). Chloride channels: often enigmatic, rarely predictable. Annu. Rev. Physiol. 72, 95–121. doi: 10.1146/annurev-physiol-021909-135811
Düsterwald, K. M., Currin, C. B., Burman, R. J., Akerman, C. J., Kay, A. R., and Raimondo, J. V. (2018). Biophysical models reveal the relative importance of transporter proteins and impermeant anions in chloride homeostasis. eLife 7:e39575. doi: 10.7554/eLife.39575
Dzhala, V. I., and Staley, K. J. (2003). Excitatory actions of endogenously released GABA contribute to initiation of ictal epileptiform activity in the developing hippocampus. J. Neurosci. 23, 1840–1846. doi: 10.1523/JNEUROSCI.23-05-01840.2003
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., et al. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213. doi: 10.1038/nm1301
Dzhala, V., Valeeva, G., Glykys, J., Khazipov, R., and Staley, K. (2012). Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J. Neurosci. 32, 4017–4031. doi: 10.1523/JNEUROSCI.5139-11.2012
Edwards, D. H. (1990). Mechanisms of depolarizing inhibition at the crayfish giant motor synapse. I. Electrophysiology. J. Neurophysiol. 64, 532–540. doi: 10.1152/jn.1990.64.2.532
Egawa, K., and Fukuda, A. (2013). Pathophysiological power of improper tonic GABAAconductances in mature and immature models. Front. Neural Circuits 7:170. doi: 10.3389/fncir.2013.00170
Elgueta, C., and Bartos, M. (2019). Dendritic inhibition differentially regulates excitability of dentate gyrus parvalbumin-expressing interneurons and granule cells. Nat. Commun. 10:5561. doi: 10.1038/s41467-019-13533-3
Farrant, M., and Kaila, K. (2007). The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog. Brain Res. 160, 59–87. doi: 10.1016/S0079-6123(06)60005-8
Fatima-Shad, K., and Barry, P. H. (1993). Anion permeation in GABA- and glycine-gated channels of mammalian cultured hippocampal neurons. Proc. Biol. Sci. 253, 69–75. doi: 10.1098/rspb.1993.0083
Flossmann, T., Kaas, T., Rahmati, V., Kiebel, S. J., Witte, O. W., Holthoff, K., et al. (2019). Somatostatin interneurons promote neuronal synchrony in the neonatal hippocampus. Cell Rep. 26, 3173–3182.e5. doi: 10.1016/j.celrep.2019.02.061
Freund, T. F. (2003). Interneuron Diversity series: Rhythm and mood in perisomatic inhibition. Trends Neurosci. 26, 489–495. doi: 10.1016/S0166-2236(03)00227-3
Fujiwara-Tsukamoto, Y., Isomura, Y., Nambu, A., and Takada, M. (2003). Excitatory gaba input directly drives seizure-like rhythmic synchronization in mature hippocampal CA1 pyramidal cells. Neuroscience 119, 265–275. doi: 10.1016/s0306-4522(03)00102-7
Gao, X. B., Chen, G., and Van Den Pol, A. N. (1998). GABA-dependent firing of glutamate-evoked action potentials at AMPA/kainate receptors in developing hypothalamic neurons. J. Neurophysiol. 79, 716–726. doi: 10.1152/jn.1998.79.2.716
Gerencser, G. A., and Zhang, J. L. (2003). Existence and nature of the chloride pump. Biochim. Biophys. Acta 1618, 133–139. doi: 10.1016/j.bbamem.2003.09.013
Gidon, A., and Segev, I. (2012). Principles governing the operation of synaptic inhibition in dendrites. Neuron 75, 330–341. doi: 10.1016/j.neuron.2012.05.015
Gonzalez-Islas, C., Chub, N., Garcia-Bereguiain, M. A., and Wenner, P. (2010). GABAergic synaptic scaling in embryonic motoneurons is mediated by a shift in the chloride reversal potential. J. Neurosci. 30, 13016–13020. doi: 10.1523/JNEUROSCI.1659-10.2010
Gonzalez-Islas, C., Chub, N., and Wenner, P. (2009). NKCC1 and AE3 appear to accumulate chloride in embryonic motoneurons. J. Neurophysiol. 101, 507–518. doi: 10.1152/jn.90986.2008
Graf, J., Zhang, C., Marguet, S. L., Herrmann, T., Flossmann, T., Hinsch, R., et al. (2021). A limited role of NKCC1 in telencephalic glutamatergic neurons for developing hippocampal network dynamics and behavior. Proc. Natl. Acad. Sci. U S A 118:e2014784118. doi: 10.1073/pnas.2014784118
Gulledge, A. T., and Stuart, G. J. (2003). Excitatory actions of GABA in the cortex. Neuron 37, 299–309. doi: 10.1016/s0896-6273(02)01146-7
Halbhuber, L., Achtner, C., Luhmann, H. J., Sinning, A., and Kilb, W. (2019). Coincident activation of glutamate receptors enhances GABAA receptor-induced ionic plasticity of the intracellular Cl−-concentration in dissociated neuronal cultures. Front. Cell. Neurosci. 13:497. doi: 10.3389/fncel.2019.00497
Hanganu, I. L., Kilb, W., and Luhmann, H. J. (2002). Functional synaptic projections onto subplate neurons in neonatal rat somatosensory cortex. J. Neurosci. 22, 7165–7176. doi: 10.1523/JNEUROSCI.22-16-07165.2002
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., et al. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007
Huebner, C. A., and Holthoff, K. (2013). Anion transport and GABA signaling. Front. Cell. Neurosci. 7:177. doi: 10.3389/fncel.2013.00177
Ikeda, M., Toyoda, H., Yamada, J., Okabe, A., Sato, K., Hotta, Y., et al. (2003). Differential development of cation-chloride cotransporters and Cl- homeostasis contributes to differential GABAergic actions between developing rat visual cortex and dorsal lateral geniculate nucleus. Brain Res. 984, 149–159. doi: 10.1016/s0006-8993(03)03126-3
Inoue, K., Furukawa, T., Kumada, T., Yamada, J., Wang, T., Inoue, R., et al. (2012). Taurine inhibits K+-Cl− cotransporter KCC2 to regulate embryonic Cl− homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem. 287, 20839–20850. doi: 10.1074/jbc.M111.319418
Isaev, D., Isaeva, E., Khazipov, R., and Holmes, G. L. (2005). Anticonvulsant action of GABA in the high potassium-low magnesium model of ictogenesis in the neonatal rat hippocampus in vivo and in vitro. J. Neurophysiol. 94, 2987–2992. doi: 10.1152/jn.00138.2005
Jadi, M., Polsky, A., Schiller, J., and Mel, B. W. (2012). Location-dependent effects of inhibition on local spiking in pyramidal neuron dendrites. PLoS Comput. Biol. 8:e1002550. doi: 10.1371/journal.pcbi.1002550
Jaenisch, N., Witte, O. W., and Frahm, C. (2010). Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 41, e151–e159. doi: 10.1161/STROKEAHA.109.570424
Jentsch, T. J., and Pusch, M. (2018). CLC chloride channels and transporters: Structure, function, physiology and disease. Physiol. Rev. 98, 1493–1590. doi: 10.1152/physrev.00047.2017
Kaila, K., Lamsa, K., Smirnov, S., Taira, T., and Voipio, J. (1997). Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J. Neurosci. 17, 7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M., and Voipio, J. (2014). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. doi: 10.1038/nrn3819
Karadsheh, M. F., and Delpire, E. (2001). Neuronal restrictive silencing element is found in the KCC2 gene: molecular basis for KCC2-specific expression in neurons. J. Neurophysiol. 85, 995–997. doi: 10.1152/jn.2001.85.2.995
Khalilov, I., Dzhala, V., Ben-Ari, Y., and Khazipov, R. (1999). Dual role of GABA in the neonatal rat hippocampus. Dev. Neurosci. 21, 310–319. doi: 10.1159/000017380
Khalilov, I., Le Van Quyen, M., Gozlan, H., and Ben-Ari, Y. (2005). Epileptogenic actions of GABA and fast oscillations in the developing hippocampus. Neuron 48, 787–796. doi: 10.1016/j.neuron.2005.09.026
Khazipov, R., Leinekugel, X., Khalilov, I., Gaiarsa, J. L., and Ben-Ari, Y. (1997). Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J. Physiol. 498, 763–772. doi: 10.1113/jphysiol.1997.sp021900
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., and Kaila, K. (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci. 28, 4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008
Kirmse, K., and Holthoff, K. (2020). “Chloride transporter activities shape early brain circuit development,” in Neuronal Chloride Transporters in Health and Disease, ed X. Tang (London, UK: Academic Press), 59–88.
Kirmse, K., Hübner, C. A., Isbrandt, D., Witte, O. W., and Holthoff, K. (2018). GABAergic transmission during brain development: multiple effects at multiple stages. Neuroscientist 24, 36–53. doi: 10.1177/1073858417701382
Kirmse, K., Kummer, M., Kovalchuk, Y., Witte, O. W., Garaschuk, O., and Holthoff, K. (2015). GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 6:7750. doi: 10.1038/ncomms8750
Kolbaev, S. N., Achilles, K., Luhmann, H. J., and Kilb, W. (2011a). Effect of depolarizing GABA A-mediated membrane responses on excitability of cajal-retzius cells in the immature rat neocortex. J. Neurophysiol. 106, 2034–2044. doi: 10.1152/jn.00699.2010
Kolbaev, S. N., Luhmann, H. J., and Kilb, W. (2011b). Activity-dependent scaling of GABAergic excitation by dynamic Cl− changes in Cajal-Retzius cells. Pflugers Arch. 461, 557–565. doi: 10.1007/s00424-011-0935-4
Kolbaev, S. N., Sharopov, S., Dierkes, P. W., Luhmann, H. J., and Kilb, W. (2012). Phasic GABAA-receptor activation is required to suppress epileptiform activity in the CA3 region of the immature rat hippocampus. Epilepsia 53, 888–896. doi: 10.1111/j.1528-1167.2012.03442.x
Kuner, T., and Augustine, G. J. (2000). A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27, 447–459. doi: 10.1016/s0896-6273(00)00056-8
Lamsa, K., Palva, J. M., Ruusuvuori, E., Kaila, K., and Taira, T. (2000). Synaptic GABA(A) activation inhibits AMPA-kainate receptor-mediated bursting in the newborn (P0–P2) rat hippocampus. J. Neurophysiol. 83, 359–366. doi: 10.1152/jn.2000.83.1.359
Lee, H., Chen, C. X., Liu, Y. J., Aizenman, E., and Kandler, K. (2005). KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur. J. Neurosci. 21, 2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x
Leinekugel, X., Medina, I., Khalilov, I., Ben-Ari, Y., and Khazipov, R. (1997). Ca2+ oscillations mediated by the synergistic excitatory actions of GABA(A) and NMDA receptors in the neonatal hippocampus. Neuron 18, 243–255. doi: 10.1016/s0896-6273(00)80265-2
Li, H., Tornberg, J., Kaila, K., Airaksinen, M. S., and Rivera, C. (2002). Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 16, 2358–2370. doi: 10.1046/j.1460-9568.2002.02419.x
Liu, R., Wang, J., Liang, S., Zhang, G., and Yang, X. (2020). Role of NKCC1 and KCC2 in epilepsy: from expression to function. Front. Neurol. 10:1407. doi: 10.3389/fneur.2019.01407
Lombardi, A., Jedlicka, P., Luhmann, H. J., and Kilb, W. (2018). Giant depolarizing potentials trigger transient changes in the intracellular Cl− concentration in CA3 pyramidal neurons of the immature mouse hippocampus. Front. Cell. Neurosci. 12:420. doi: 10.3389/fncel.2018.00420
Lombardi, A., Jedlicka, P., Luhmann, H. J., and Kilb, W. (2019). Interactions between membrane resistance, GABA-A receptor properties, bicarbonate dynamics and Cl−-transport shape activity-dependent changes of intracellular Cl− concentration. Int. J. Mol. Sci. 20:1416. doi: 10.3390/ijms20061416
Lombardi, A., Jedlicka, P., Luhmann, H. J., and Kilb, W. (2021). Coincident glutamatergic depolarizations enhance GABAA receptor-dependent Cl− influx in mature and suppress Cl− efflux in immature neurons. PLoS Comput. Biol. 17:e1008573. doi: 10.1371/journal.pcbi.1008573
Loscher, W., Puskarjov, M., and Kaila, K. (2013). Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69, 62–74. doi: 10.1016/j.neuropharm.2012.05.045
Lu, J., Karadsheh, M., and Delpire, E. (1999). Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J. Neurobiol. 39, 558–568.
Luhmann, H. J., and Prince, D. A. (1991). Postnatal maturation of the GABAergic system in rat neocortex. J. Neurophysiol. 65, 247–263. doi: 10.1152/jn.1991.65.2.247
Martina, M., Royer, B., and Pare, D. (2001). Cell-type-specific GABA responses and chloride homeostasis in the cortex and amygdala. J. Neurophysiol. 86, 2887–2895. doi: 10.1152/jn.2001.86.6.2887
Minlebaev, M., Ben-Ari, Y., and Khazipov, R. (2007). Network mechanisms of spindle-burst oscillations in the neonatal rat barrel cortex in vivo. J. Neurophysiol. 97, 692–700. doi: 10.1152/jn.00759.2006
Misgeld, U., Wagner, A., and Ohno, T. (1982). Depolarizing IPSPs and depolarization by GABA of rat neostriatum cells in vitro. Exp. Brain Res. 45, 108–114. doi: 10.1007/BF00235769
Mohapatra, N., Tonnesen, J., Vlachos, A., Kuner, T., Deller, T., Nagerl, U. V., et al. (2016). Spines slow down dendritic chloride diffusion and affect short-term ionic plasticity of GABAergic inhibition. Sci. Rep. 6:23196. doi: 10.1038/srep23196
Mueller, A. L., Chesnut, R. M., and Schwartzkroin, P. A. (1983). Actions of GABA in developing rabbit hippocampus: an in vitro study. Neurosci. Lett. 39, 193–198. doi: 10.1016/0304-3940(83)90076-9
Murata, Y., and Colonnese, M. T. (2020). GABAergic interneurons excite neonatal hippocampus in vivo. Sci. Adv. 6:eaba1430. doi: 10.1126/sciadv.aba1430
Nardou, R., Ben-Ari, Y., and Khalilov, I. (2009). Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J. Neurophysiol. 101, 2878–2888. doi: 10.1152/jn.90761.2008
Nardou, R., Ferrari, D. C., and Ben-Ari, Y. (2013). Mechanisms and effects of seizures in the immature brain. Semin. Fetal Neonatal Med. 18, 175–184. doi: 10.1016/j.siny.2013.02.003
Otsu, Y., Donneger, F., Schwartz, E. J., and Poncer, J. C. (2020). Cation-chloride cotransporters and the polarity of GABA signalling in mouse hippocampal parvalbumin interneurons. J. Physiol. 598, 1865–1880. doi: 10.1113/JP279221
Owens, D. F., and Kriegstein, A. R. (2002). Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 3, 715–727. doi: 10.1038/nrn919
Pallud, J., Le Van Quyen, M., Bielle, F., Pellegrino, C., Varlet, P., Labussiere, M., et al. (2014). Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 6:244ra89. doi: 10.1126/scitranslmed.3008065
Payne, J. A., Rivera, C., Voipio, J., and Kaila, K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206. doi: 10.1016/S0166-2236(03)00068-7
Pellegrino, C., Gubkina, O., Schaefer, M., Becq, H., Ludwig, A., Mukhtarov, M., et al. (2011). Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J. Physiol. 589, 2475–2496. doi: 10.1113/jphysiol.2010.203703
Pfeffer, C. K., Stein, V., Keating, D. J., Maier, H., Rinke, I., Rudhard, Y., et al. (2009). NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J. Neurosci. 29, 3419–3430. doi: 10.1523/JNEUROSCI.1377-08.2009
Plotkin, M. D., Snyder, E. Y., Hebert, S. C., and Delpire, E. (1997). Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: A possible mechanism underlying GABA’s excitatory role in immature brain. J. Neurobiol. 33, 781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5
Raimondo, J. V., Markram, H., and Akerman, C. J. (2012). Short-term ionic plasticity at GABAergic synapses. Front. Synaptic Neurosci. 4:5. doi: 10.3389/fnsyn.2012.00005
Rall, W. (1989). “Cable theory for dendritic neurons,” in Methods in Neuronal Modeling, eds K. Koch and I. Segev (Cambridge, MA: MIT Press), 9–92.
Rheims, S., Minlebaev, M., Ivanov, A., Represa, A., Khazipov, R., Holmes, G. L., et al. (2008). Excitatory GABA in rodent developing neocortex in vitro. J. Neurophysiol. 100, 609–619. doi: 10.1152/jn.90402.2008
Richter, D., Luhmann, H. J., and Kilb, W. (2010). Intrinsic activation of GABAA receptors suppresses epileptiform activity in the cerebral cortex of immature mice. Epilepsia 51, 1483–1492. doi: 10.1111/j.1528-1167.2010.02591.x
Rinetti-Vargas, G., Phamluong, K., Ron, D., and Bender, K. J. (2017). Periadolescent maturation of GABAergic hyperpolarization at the axon initial segment. Cell Rep. 20, 21–29. doi: 10.1016/j.celrep.2017.06.030
Rivera, C., Li, H., Thomas-Crusells, J., Lahtinen, H., Viitanen, T., Nanobashvili, A., et al. (2002). BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 159, 747–752. doi: 10.1083/jcb.200209011
Rivera, C., Voipio, J., and Kaila, K. (2005). Two developmental switches in GABAergic signalling: the K+-Cl− cotransporter KCC2 and carbonic anhydrase CAVII. J. Physiol. 562, 27–36. doi: 10.1113/jphysiol.2004.077495
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Ruffin, V. A., Salameh, A. I., Boron, W. F., and Parker, M. D. (2014). Intracellular pH regulation by acid-base transporters in mammalian neurons. Front. Physiol. 5:43. doi: 10.3389/fphys.2014.00043
Russell, J. M. (2000). Sodium-potassium-chloride cotransport. Physiol. Rev. 80, 211–276. doi: 10.1152/physrev.2000.80.1.211
Ruusuvuori, E., Li, H., Huttu, K., Palva, J. M., Smirnov, S., Rivera, C., et al. (2004). Carbonic anhydrase isoform VII acts as a molecular switch in the development of synchronous gamma-frequency firing of hippocampal CA1 pyramidal cells. J. Neurosci. 24, 2699–2707. doi: 10.1523/JNEUROSCI.5176-03.2004
Sauer, J. F., and Bartos, M. (2010). Recruitment of early postnatal parvalbumin-positive hippocampal interneurons by GABAergic excitation. J. Neurosci. 30, 110–115. doi: 10.1523/JNEUROSCI.4125-09.2010
Sava, B. A., Chen, R., Sun, H., Luhmann, H. J., and Kilb, W. (2014). Taurine activates GABAergic networks in the neocortex of immature mice. Front. Cell. Neurosci. 8:26. doi: 10.3389/fncel.2014.00026
Sharopov, S., Winkler, P., Uehara, R., Lombardi, A., Halbhuber, L., Okabe, A., et al. (2019). Allopregnanolone augments epileptiform activity of an in vitro mouse hippocampal preparation in the first postnatal week. Epilepsy Res. 157:106196. doi: 10.1016/j.eplepsyres.2019.106196
Shimizu-Okabe, C., Yokokura, M., Okabe, A., Ikeda, M., Sato, K., Kilb, W., et al. (2002). Layer-specific expression of Cl− transporters and differential [Cl−]i in newborn rat cortex. Neuroreport 13, 2433–2437. doi: 10.1097/00001756-200212200-00012
Sinning, A., and Hübner, C. A. (2013). Minireview: PH and synaptic transmission. FEBS Lett. 587, 1923–1928. doi: 10.1016/j.febslet.2013.04.045
Sinning, A., Liebmann, L., Kougioumtzes, A., Westermann, M., Bruehl, C., and Hübner, C. A. (2011). Synaptic glutamate release is modulated by the Na+-driven Cl−/HCO−3 exchanger Slc4a8. J. Neurosci. 31, 7300–7311. doi: 10.1523/JNEUROSCI.0269-11.2011
Sipila, S. T., Huttu, K., Soltesz, I., Voipio, J., and Kaila, K. (2005). Depolarizing GABA acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J. Neurosci. 25, 5280–5289. doi: 10.1523/JNEUROSCI.0378-05.2005
Somogyi, P., and Klausberger, T. (2005). Defined types of cortical interneurone structure space and spike timing in the hippocampus. J. Physiol. 562, 9–26. doi: 10.1113/jphysiol.2004.078915
Song, I., Savtchenko, L., and Semyanov, A. (2011). Tonic excitation or inhibition is set by GABAA conductance in hippocampal interneurons. Nat. Commun. 2:376. doi: 10.1038/ncomms1377
Spruston, N., Stuart, G., and Häusser, M. (2016). “Principles of dendritic integration,” in Dendrites, eds G. Stuart, N. Spruston and M. Häusser (Oxford, UK: Oxford Scholarship), 351–398.
Staley, K. J., and Mody, I. (1992). Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABA(A) receptor-mediated postsynaptic conductance. J. Neurophysiol. 68, 197–212. doi: 10.1152/jn.1992.68.1.197
Stein, V., Hermans-Borgmeyer, I., Jentsch, T. J., and Hubner, C. A. (2004). Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J. Comp. Neurol. 468, 57–64. doi: 10.1002/cne.10983
Szabadics, J., Varga, C., Molnár, G., Oláh, S., Barzó, P., and Tamás, G. (2006). Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311, 233–235. doi: 10.1126/science.1121325
Titz, S., Hans, M., Kelsch, W., Lewen, A., Swandulla, D., and Misgeld, U. (2003). Hyperpolarizing inhibition develops without trophic support by GABA in cultured rat midbrain neurons. J. Physiol. 550, 719–730. doi: 10.1113/jphysiol.2003.041863
Toyoda, H., Ohno, K., Yamada, J., Ikeda, M., Okabe, A., Sato, K., et al. (2003). Induction of NMDA and GABAA receptor-mediated Ca2+ oscillations with KCC2 mRNA downregulation in injured facial motoneurons. J. Neurophysiol. 89, 1353–1362. doi: 10.1152/jn.00721.2002
Valeeva, G., Abdullin, A., Tyzio, R., Skorinkin, A., Nikolski, E., Ben-Ari, Y., et al. (2010). Temporal coding at the immature depolarizing gabaergic synapse. Front. Cell. Neurosci. 4:17. doi: 10.3389/fncel.2010.00017
Valeeva, G., Tressard, T., Mukhtarov, M., Baude, A., and Khazipov, R. (2016). An optogenetic approach for investigation of excitatory and inhibitory network GABA actions in mice expressing channelrhodopsin-2 in GABAergic neurons. J. Neurosci. 36, 5961–5973. doi: 10.1523/JNEUROSCI.3482-15.2016
Valeeva, G., Valiullina, F., and Khazipov, R. (2013). Excitatory actions of GABA in the intact neonatal rodent hippocampus in vitro. Front. Cell. Neurosci. 7:20. doi: 10.3389/fncel.2013.00020
Virtanen, M. A., Uvarov, P., Hübner, C. A., and Kaila, K. (2020). NKCC1, an elusive molecular target in brain development: making sense of the existing data. Cells 9:2607. doi: 10.3390/cells9122607
Wake, H., Watanabe, M., Moorhouse, A. J., Kanematsu, T., Horibe, S., Matsukawa, N., et al. (2007). Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 27, 1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007
Wang, Y. F., Gao, X. B., and Van Den Pol, A. N. (2001). Membrane properties underlying patterns of GABA-dependent action potentials in developing mouse hypothalamic neurons. J. Neurophysiol. 86, 1252–1265. doi: 10.1152/jn.2001.86.3.1252
Watanabe, M., and Fukuda, A. (2015). Development and regulation of chloride homeostasis in the central nervous system. Front. Cell. Neurosci. 9:371. doi: 10.3389/fncel.2015.00371
Wells, J. E., Porter, J. T., and Agmon, A. (2000). GABAergic inhibition suppresses paroxysmal network activity in the neonatal rodent hippocampus and neocortex. J. Neurosci. 20, 8822–8830. doi: 10.1523/JNEUROSCI.20-23-08822.2000
Winkler, P., Luhmann, H. J., and Kilb, W. (2019). Taurine potentiates the anticonvulsive effect of the GABA A agonist muscimol and pentobarbital in the immature mouse hippocampus. Epilepsia 60, 464–474. doi: 10.1111/epi.14651
Wright, R., Raimondo, J. V., and Akerman, C. J. (2011). Spatial and Temporal Dynamics in the Ionic Driving Force for GABA(A) Receptors. Neural Plast. 2011:728395. doi: 10.1155/2011/728395
Keywords: chloride homeostasis, NKCC1, KCC2, SLC12A2, SLC12A5, gaba receptor, neuronal development
Citation: Kilb W (2021) When Are Depolarizing GABAergic Responses Excitatory? Front. Mol. Neurosci. 14:747835. doi: 10.3389/fnmol.2021.747835
Received: 26 July 2021; Accepted: 28 October 2021;
Published: 24 November 2021.
Edited by:
Andreas Vlachos, University of Freiburg, GermanyReviewed by:
Imre Vida, Charité University Medicine Berlin, GermanyCorette J. Wierenga, Utrecht University, Netherlands
Copyright © 2021 Kilb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Werner Kilb, d2tpbGJAdW5pLW1haW56LmRl