 Joke Terryn
Joke Terryn Catherine M. Verfaillie
Catherine M. Verfaillie Philip Van Damme1,2,4*
Philip Van Damme1,2,4*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 23 July 2021
Sec. Brain Disease Mechanisms
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.713031
This article is part of the Research Topic Current Concept and Translational Study in ALS–FTD Spectrum: from Genetics, Neuroinflammation to Neurodegeneration View all 9 articles
Frontotemporal dementia (FTD) is a neurodegenerative disease, leading to behavioral changes and language difficulties. Heterozygous loss-of-function mutations in progranulin (GRN) induce haploinsufficiency of the protein and are associated with up to one-third of all genetic FTD cases worldwide. While the loss of GRN is primarily associated with neurodegeneration, the biological functions of the secreted growth factor-like protein are more diverse, ranging from wound healing, inflammation, vasculogenesis, and metabolic regulation to tumor cell growth and metastasis. To date, no disease-modifying treatments exist for FTD, but different therapeutic approaches to boost GRN levels in the central nervous system are currently being developed (including AAV-mediated GRN gene delivery as well as anti-SORT1 antibody therapy). In this review, we provide an overview of the multifaceted regulation of GRN levels and the corresponding therapeutic avenues. We discuss the opportunities, advantages, and potential drawbacks of the diverse approaches. Additionally, we highlight the therapeutic potential of elevating GRN levels beyond patients with loss-of-function mutations in GRN.
Frontotemporal dementia (FTD) is a neurodegenerative disease, leading to behavioral changes and language difficulties. Previously considered a rare disease entity, FTD is in fact the third leading cause of all dementia cases (Rabinovici and Miller, 2010). FTD typically presents before the age of 65 and is therefore considered a presenile dementia. In this age group (<65), FTD is as common as Alzheimer’s dementia (Rabinovici and Miller, 2010; Ferrari et al., 2014). The prevalence of FTD varies regionally (ranging from 3 to 15 per 100,000 in the 45- to 64-year-old population; Rabinovici and Miller, 2010). In contrast to other types of dementia, FTD patients have a positive family history in 30–40 percent of all cases, with mutations in three genes (GRN, MAPT, and C9orf72) explaining the majority of the genetic cases (Moore et al., 2020).
The term FTD is used to bundle the spectrum of clinical manifestations, while frontotemporal lobar degeneration (FTLD) refers to the underlying neuropathology. FTLD neuropathological subtypes were established based on the proteins found in the neuropathological inclusions. The two main subtypes are FTLD-tau for microtubule-associated protein tau (MAPT) positive inclusions, and FTLD-TDP/FTD-U for ubiquitin and transactive response DNA-binding protein 43 (TDP-43) positive inclusions (Mackenzie et al., 2006).
The first FTD gene was discovered in 1998, when mutations in MAPT were identified in some families with FTD and Parkinsonism. Surprisingly, some of the cases linked to the same chromosomal locus (17q21) were tau-negative. In 2006, mutations in the neighboring GRN gene were discovered, explaining the genetic linkage to the same chromosomal locus (Baker et al., 2006; Cruts et al., 2006). The neuronal inclusions found in these patients were not GRN immunoreactive and GRN mutations were shown to lead to reduced GRN expression (Baker et al., 2006; Cruts et al., 2006). Shortly after the discovery of the GRN gene, TDP-43 was identified as the key constituent of the tau-negative neuronal inclusions (Neumann et al., 2006). As FTLD-TDP pathology is responsible for 45% of FTD cases and the majority of amyotrophic lateral sclerosis (ALS) cases, the finding consolidated the link between FTD and ALS (Ling et al., 2013; Tziortzouda et al., 2021). In 2011, the discovery of a hexanucleotide (GGGGCC) repeat expansion mutation in C9orf72 finally explained the genetic link to the ninth chromosome in cases with ALS and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Repeat expansions in C9orf72 are the most common genetic cause of FTD and ALS. Other, more rare mutations, such as mutations in charged multivesicular body protein 2B (CHMP2B), Valosin-Containing Protein (VCP), ubiquilin 2 (UBQLN2), TANK-Binding Kinase 1 (TBK1), sequestosome 1 (SQSTM1), and T cell–restricted intracellular antigen 1 (TIA1) explain residual cases of genetic FTD (Pottier et al., 2016).
Behavioral variant frontotemporal dementia (bvFTD) is the most common clinical manifestation of frontotemporal lobar degeneration, occurring in 60 percent of cases. When language is primarily impaired the term primary progressive aphasia is used (PPA) (Rascovsky and Grossman, 2013). Generally, three variants of PPA are described—the semantic, logopenic and nonfluent/agrammatic variant—but mixed variants exist (Gorno-Tempini et al., 2011; Vandenberghe, 2016). In addition to the typical manifestations (bvFTD and PPA), other disorders have been linked to the FTD spectrum: Frontotemporal Dementia with Motor Neuron Disease (FTD-MND), corticobasal degeneration, and progressive supranuclear palsy (Olney et al., 2017).
In humans, the progranulin (GRN) gene is located on chromosome 17q21, 1.7 Mb centromeric of MAPT, and consists of 12 protein-coding exons and a 5′ non-coding exon. The mammalian GRN gene codes for a repetition of seven and a half granulin domains. Each granulin domain is encoded by two neighboring exons, contributing either the N- or C-terminal half of the domain. The unique structure of the evolutionary conserved granulin motif is reviewed in Palfree et al. (2015).
Since the discovery in 2006 of GRN mutations linked to FTD (Baker et al., 2006; Cruts et al., 2006), over 130 pathological mutations in the GRN gene have been described (Moore et al., 2020). The majority of GRN mutations are nonsense and frameshift mutations, that introduce premature stop codons and result in nuclear degradation of the mutant mRNA, suggesting GRN haploinsufficiency underlying GRN-linked neurodegeneration (Yu et al., 2010; Kleinberger et al., 2013). Other mutations in the growing list of GRN mutations, result in deletion of the gene (Gijselinck et al., 2008), affect the initiation of translation (Le Ber et al., 2008), processing (Shankaran et al., 2008), and secretion (Mukherjee et al., 2006). Several studies showed that patients with a GRN mutation have reduced GRN protein levels in the cerebrospinal fluid (Ghidoni et al., 2008; Van Damme et al., 2008). Similarly, a reduction of blood GRN levels can be seen, which can be used to predict the presence of a pathogenic GRN mutation (Finch et al., 2009).
The phenotypic variability seen in patients with GRN mutation is astonishing (Moore et al., 2020). Behavioral variant frontotemporal dementia and nonfluent/agrammatic variant PPA are the most common diagnoses in this genetic group (Kim et al., 2013). Mild Parkinsonism is a common clinical finding in GRN mutation carriers (Le Ber et al., 2008). Clinical presentations indistinguishable from typical Alzheimer’s or Parkinson’s disease are occasionally seen (Brouwers et al., 2007). Rarely, GRN mutations present as motor neuron disease (Benussi et al., 2009), corticobasal syndrome or progressive supranuclear palsy (Baizabal-Carvallo and Jankovic, 2016).
When GRN entered the neurodegenerative scene, the protein was already known as a widely expressed growth factor. The GRN protein had been identified by a number of independent research groups in different biological contexts and is therefore known by many names. Sequencing revealed the shared genetic origin of proepithelin (Plowman et al., 1992), granulin-epithelin precursor (GEP) (Zanocco-Marani et al., 1999), PC cell derived growth factor (PCCDGF) (Zhou et al., 1993), and acrogranin (Baba et al., 1993). A multitude of names underscore the pleiotrophy of the GRN protein.
Initial work was focused on the mitogenic and tumor-promoting effects of GRN. High GRN expression has been detected in many types of cancer and elevated serum GRN levels could be used as a potential prognostic biomarker in for example breast cancer, chronic lymphocytic leukemia, and non-small-cell lung carcinoma (Arechavaleta-Velasco et al., 2017). GRN seems to stimulate tumor growth and promotes tumor cell migration, invasiveness, anchorage independence, and chemo-resistance (He and Bateman, 1999; Bandey et al., 2015; Tanimoto et al., 2016). Important to note, however, is that while GRN stimulates tumor progression, it does not induce malignant transformation on its own. GRN however, can influence the transition from a precancerous state to a highly tumorigenic state (Matsumura et al., 2006). As GRN was shown to be an important therapeutic and diagnostic target in breast cancer, GRN diagnostic kits and neutralizing antibodies are being developed (Guha et al., 2021).
Endogenous GRN expression is found throughout the body, with high expression levels in neural tissue, immune cells and epithelia lining the reproductive organs, the gastrointestinal tract, and the skin (Daniel et al., 2000). GRN is closely involved in embryogenesis, but its expression becomes more restricted later in development (Bateman and Bennett, 2009). At physiological levels, GRN is typically expressed under conditions of tissue remodeling and repair (Bateman and Bennett, 2009), where it exerts multifaceted roles. For example in wound healing, GRN stimulates migration and mitosis of fibroblast and endothelial cells, promotes blood vessel formation, and controls the recruitment and activity of leukocytes (He et al., 2003). In adipose tissue, GRN secretion correlates with macrophage infiltration (Youn et al., 2009) and GRN was found to be a key regulator of insulin sensitivity (Matsubara et al., 2012; Zhou B. et al., 2015). Because GRN expression is increased in acute and chronic inflammatory states, GRN has been proposed as a biomarker for diseases with a strong inflammatory component, including metabolic, cardiovascular, and auto-immune diseases (Abella et al., 2017). Spontaneously occurring neutralizing GRN antibodies have been detected in patient’s sera in association with multiple auto-immune diseases (Thurner et al., 2013), suggesting an important function of GRN as a modulator of the immune response. Figure 1 summarizes the diverse biological roles of GRN.
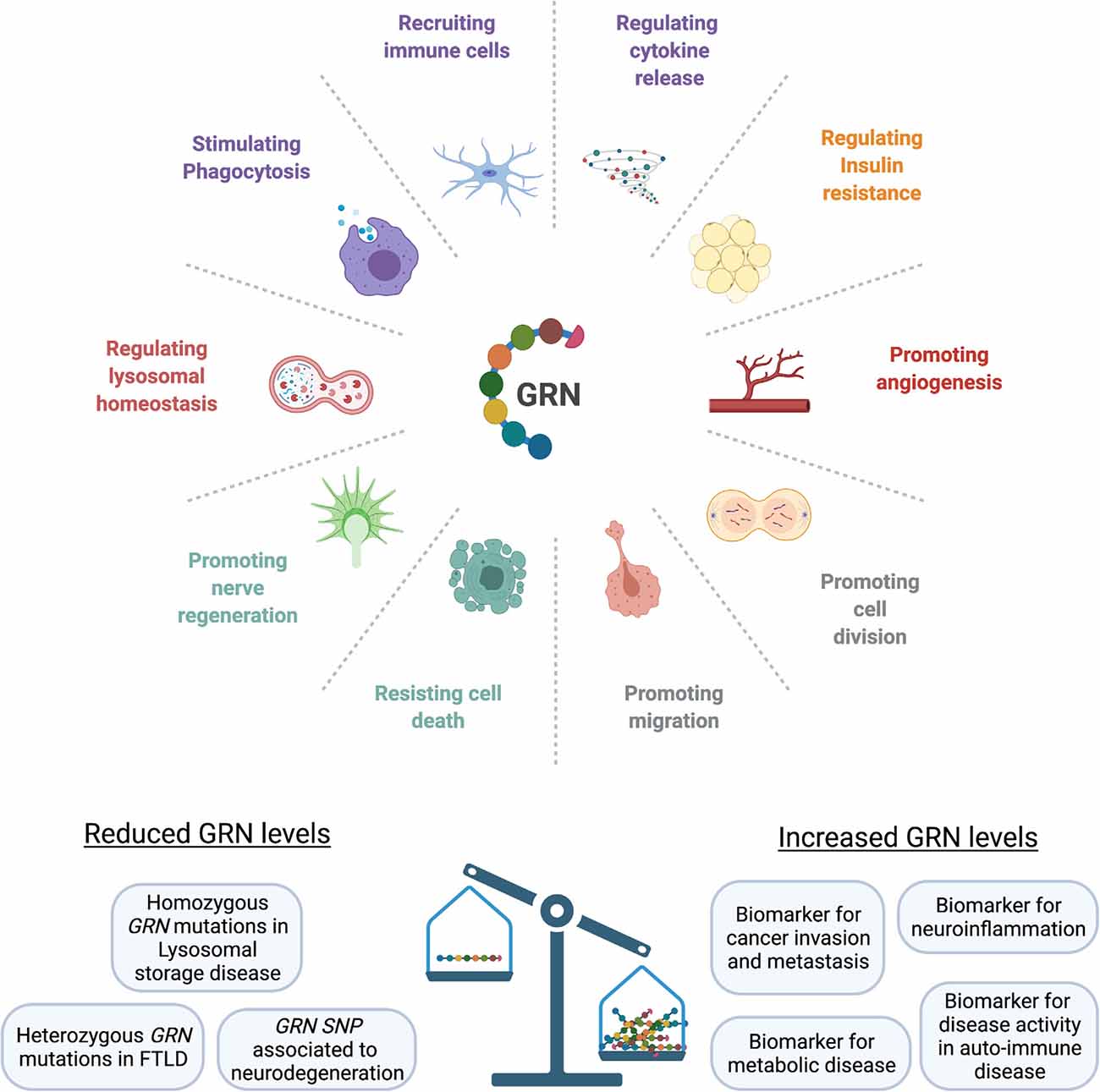
Figure 1. GRN affects multiple biological functions. At physiological levels, GRN is involved in development, fine-tuning the immune response, orchestrating tissue repair, and providing neurotrophic support. Loss of progranulin can lead to an uncontrolled inflammatory response, loss of lysosomal homeostasis, and neuronal death. GRN levels are elevated in cancer, metabolic diseases, and chronic inflammatory states (SNP, single-nucleotide polymorphism; FTLD, Frontotemporal lobar degeneration).
Because GRN had been known as a widely expressed growth factor with important roles in development, inflammation, and tumorigenesis (He and Bateman, 2003), the discovery of GRN mutations in an adult onset neurodegenerative disease was surprising. GRN haploinsufficiency in the brain, as seen in FTD cases with GRN mutations, leads to chronic degenerative changes of neuronal loss, gliosis, and microglial activation (Mackenzie et al., 2006). Because GRN expression is restricted to neurons and microglia in the central nervous system (CNS) (Daniel et al., 2000; Petkau et al., 2010), both celltypes have been postulated as drivers of neuropathology in GRN-FTLD cases.
Proof for the direct neurotrophic effect of GRN was provided when extracellular administration of GRN was shown to stimulate neurite outgrowth and survival in cultured primary cortical and motor neurons (Van Damme et al., 2008; Gao et al., 2010). Likewise, neurons derived from Grn null mice were more susceptible to toxic insults (Kleinberger et al., 2010) and showed reduced neurite outgrowth and branching (Gass et al., 2012; Beel et al., 2017). While studies in multiple model systems have supported a direct role for GRN in neuronal development and survival (Kleinberger et al., 2013), the mechanisms by which GRN functions and how the shortage of GRN causes TDP-43 pathology and neurodegeneration, is only partially understood.
In addition to the growth factor properties, the immune modulatory role of GRN has been explored in the CNS as well. A prominent feature of Grn null mice is the consistent age-dependent microgliosis and astrogliosis (Ahmed et al., 2010; Yin et al., 2010). Mouse Grn-deficient microglia were shown to express higher levels of proinflammatory cytokines and contributed to neuronal death (Martens et al., 2012). Enhanced phagocytosis in response to the loss of GRN has been demonstrated in multiple animal models. Nematodes lacking pgrn-1 appeared normal, but exhibit fewer apoptotic cell corpses during development, due to rapid clearance of apoptotic cells (Kao et al., 2011). An increased appetite for inhibitory synapses was demonstrated in Grn null microglia and was shown to be associated with compulsive grooming behavior (Lui et al., 2016). These findings could indicate that GRN normally suppresses microglial activity and that excessive clearance upon loss of GRN could prevent normal neuronal recovery.
While many studies point to microglia as drivers of pathology, a number of conditional knockout studies nuance this hypothesis. Selective depletion of either neuronal or microglial Grn was shown to produce different behavioral deficits. While depletion of neuronal Grn provoked social dominance defects (Arrant et al., 2017, 2019), depletion of microglial Grn triggered compulsive behaviors (e.g., excessive grooming) (Krabbe et al., 2017; Arrant et al., 2019). Interestingly, neither the combined nor isolated depletion of neuronal or microglial Grn was sufficient to produce gliosis and lipofuscinosis in mice (Petkau et al., 2017; Arrant et al., 2019). Seemingly, low residual doses of Grn are sufficient to prevent gliosis and lipofuscinosis. This is in analogy with the lack of obvious neuropathological changes in heterozygous Grn mutant mice (Ahmed et al., 2010). In addition to the selective depletion studies focusing on the CNS, a study examined the relative contribution of microglial and neuronal Grn on the recovery from facial nerve injury (Beel et al., 2017). The study showed that neuronal Grn and not microglial Grn is essential for nerve regeneration.
Given that different cell types provoke different behavioral deficits (Krabbe et al., 2017; Arrant et al., 2019) and different mechanisms seem to be at play in neurons and microglia (Lui et al., 2016; Beel et al., 2017; Krabbe et al., 2017), combined targeting of both neurons and microglia might be needed to tackle different aspects of GRN-linked neurodegeneration.
A very curious case report in 2012 redirected the GRN field towards the lysome (Smith et al., 2012). Exome sequencing of two Italian siblings with neuronal ceroid lipofuscinosis (NCL) revealed that homozygous loss of GRN caused the lysosomal storage disease (Smith et al., 2012). The clinical picture of the siblings resembled in no apparent way a more severe manifestation of FTD. The sisters had developed generalized epilepsy in their early twenties, combined with retinal dystrophy, cerebellar ataxia, and cognitive deterioration. Brain MRI showed focal atrophy of the cerebellum, not of the frontal and temporal lobes. Skin biopsies were taken as the clinical picture of progressive dementia, visual loss, epilepsy, and motor deterioration was suggestive for neuronal ceroid lipofuscinosis (NCL). Electron microscopic examination confirmed the presence of typical fingerprint profiles in endothelium and secretory cells, diagnostic of NCL. Consequently, the GRN gene joined the list of NCL genes as CLN11 (Smith et al., 2012). The neuronal ceroid lipofucinoses are, as a group, defined by selective neurodegeneration and the deposition of autofluorescent storage material (either subunit c of mitochondrial ATP synthase or saposins in lysosome-derived organelles; Palmer et al., 2013).
While only a few patients have been described with homozygous loss of GRN (Huin et al., 2020), the genetic and pathological link to NCL confirmed a previously suspected role for GRN in the lysosome. Earlier studies had already reported the transcriptional co-regulation of GRN and other lysosomal genes (Belcastro et al., 2011), and GRN was known to be rapidly endocytosed and delivered to the lysosome by the sorting receptor sortilin (SORT1) (Hu et al., 2010). The connection to NCL urged a reevaluation of brain tissue from previously described homozygous Grn knockout mice, revealing lipofuscin deposits and enlarged lysosomes, consistent with NCL (Ahmed et al., 2010; Smith et al., 2012; Wils et al., 2012). Consecutive studies demonstrated that GRN haploinsufficiency in humans similarly leads to features of NCL. Heterozygous GRN mutation carriers were shown to display preclinical retinal lipofuscinosis (Ward et al., 2017). At the neuropathological level, FTD brains demonstrate, in addition to TDP-43 proteinopathy, subtle features of NCL such as increased lipofuscinosis, intracellular NCL-like storage material as well as elevated expression of lysosomal proteins (for example CTSD and LAMP1/2) (Gotzl et al., 2014; Ward et al., 2017). Unexpectedly, the brain tissue of a homozygous GRN mutation carrier was devoid of TDP-43 cytoplasmic inclusions, although the loss of normal TDP-43 staining was seen in the nucleus of some neurons (Huin et al., 2020). Consistent with these findings, some NCL patients demonstrated increased phosphorylated TDP-43, although it remained largely soluble (Gotzl et al., 2014). This rare and surprising association of GRN mutations in NCL provided new insights into the pathological mechanisms underlying GRN-linked neurodegeneration and further broadened the therapeutic window.
The neuroprotective and immune modulatory roles of GRN have encouraged the therapeutic overexpression of GRN in a number of preclinical models for neurodegeneration.
In Alzheimer’s disease (AD), GRN cerebrospinal fluid (CSF) levels rise with disease progression and correlate with cognitive impairment and with the expression of the microglia derived soluble TREM2 (triggering receptor expressed on myeloid cells 2) (Suárez-Calvet et al., 2018). The rise in GRN is believed to reflect microglial activation in response to neurodegeneration. In AD mouse models, loss of GRN exacerbated plaque load and cognitive deficits, while lentiviral overexpression improved spatial memory and prevented hippocampal neuronal loss (Minami et al., 2014).
In a mouse model of motor neuron disease, increased microglial GRN expression marked the degenerative process (Philips et al., 2010). Human GRN overexpression was shown to rescue motor neuron abnormalities in zebrafish caused by mutant TDP-43 (Laird et al., 2010), but failed to affect a mutant SOD1 phenotype in zebrafish and mice (Laird et al., 2010). In mice, similar effects were observed: GRN overexpression could reduce the accumulation of insoluble TDP-43, slow down the axonal loss and improve survival of mutant TDP-43 mice, but was ineffective in mutant SOD1 mice (Herdewyn et al., 2013; Beel et al., 2018). Motor neuron defects in zebrafish caused by FUS mutants or by loss of SMN1, could be rescued by GRN as well (Chitramuthu et al., 2010, 2017).
The striatum is typically affected in GRN mutation carriers (Mackenzie, 2007), consistent with the high prevalence of Parkinsonism in these patients (Le Ber et al., 2008). In accordance, GRN has been shown to support the health of striatal neurons. In a mouse model of Parkinson’s disease, lentiviral Grn overexpression bolstered the resilience of nigrostriatal neurons to acute MTPT toxicity (Van Kampen et al., 2014), while loss of Grn rendered neurons vulnerable to toxicity (Martens et al., 2012). Additionally, neuroprotective effects of GRN have been demonstrated in ischemic brain injury. Intracerebroventricular administration of recombinant Grn two hours after ischemic brain injury was shown to reduce infarction volume and brain edema in a mouse stroke model (Egashira et al., 2013).
These studies demonstrate the therapeutic potential of GRN as a potent modulator of neuroinflammation and neurotrophic factor in acute and chronic neurological disorders. The protective effects of GRN extend far beyond the neuropathology associated with GRN mutations. In the next paragraphs, we will discuss the complex cellular regulation of GRN expression and the associated therapeutic interventions. An overview of the life-cycle of GRN is provided in Figure 2.
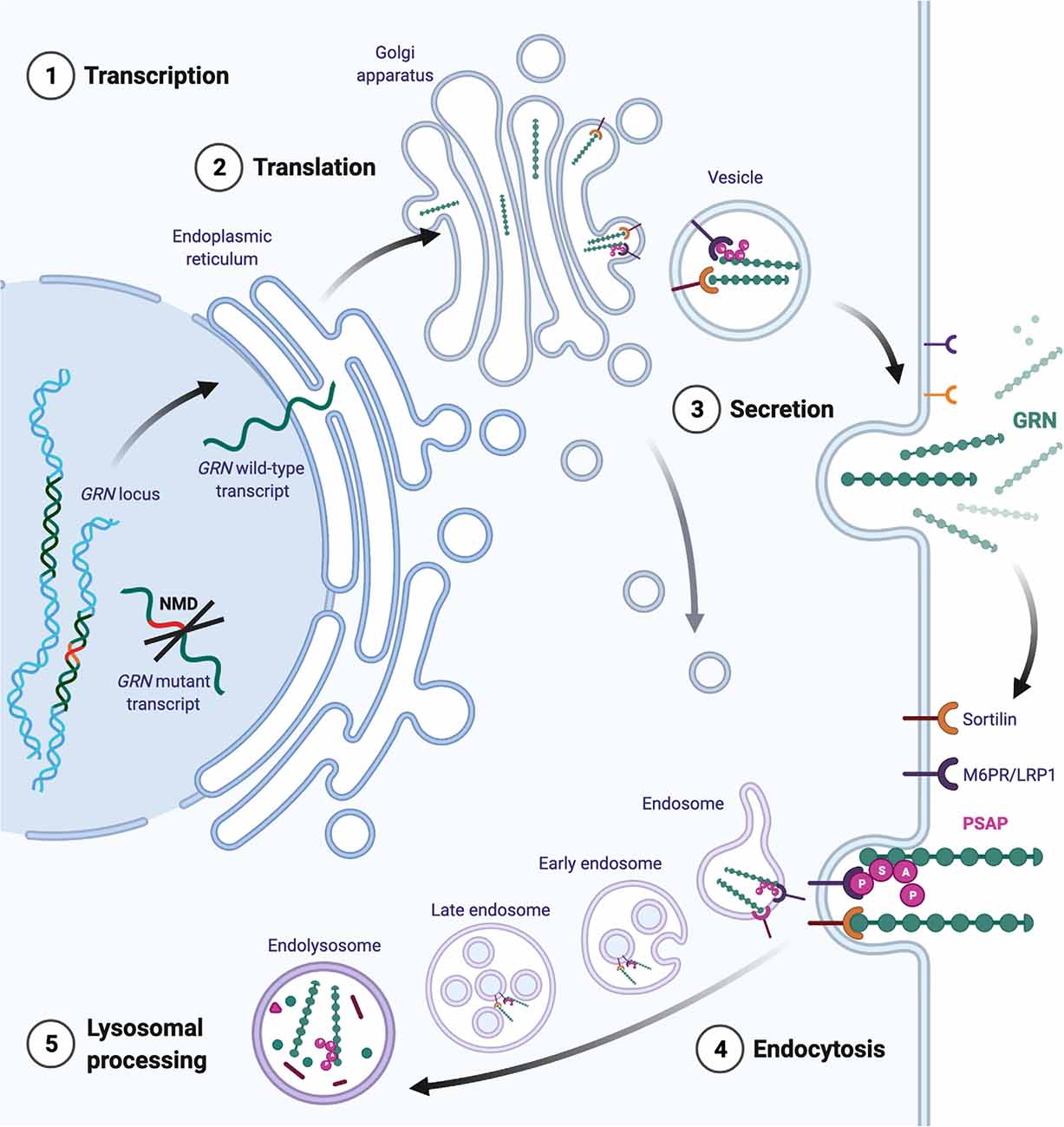
Figure 2. Intracellular trafficking of GRN. Transcripts from the mutated GRN allele are generally removed by nonsense-mediated mRNA decay (NMD). Transcripts from the healthy allele are translated directly into the endoplasmatic reticulum, where chaperones ensure proper folding. Mature GRN can be secreted constitutively or in an activity- dependent manner (for example in neurons). Extracellularly, GRN initiates tissue- and cell-type-dependent autocrine and paracrine signaling cascades. GRN binding partners are omitted for simplicity. Extracellular GRN levels are regulated through proteolytic degradation and by endocytosis. The binding of GRN to sortilin facilitates the uptake of GRN to endosomes. Alternatively, GRN can traffic to the endosome by indirect binding of the mannose-6-phosphate receptor (M6P) via prosaposin (PSAP). Additionally, GRN is thought to travel directly to the endosomal system via the trans-Golgi network.
In patients with GRN mutations, transcripts from the mutated allele are generally removed by nonsense-mediated mRNA decay (NMD), as the majority of GRN mutations are nonsense and frameshift mutations that introduce premature termination codons (PTC). One therapeutic option could therefore be to block NMD in order to salvage truncated transcripts or to promote read-through of nonsense codons. The efficacy of these approaches hinges on whether the proteins resulting from nonsense suppression retain substantial function. A study in rodents, focusing on the most prevalent nonsense mutation (GRNR493X), provided proof of principle for the inhibition of NMD by use of antisense oligonucleotides (ASO) as well as by genetic and pharmacological intervention (Nguyen et al., 2018). The GRN R493X truncation mutant, lacking 17 percent of the protein, retained the ability to suppress the expression of inflammatory markers and influenced gene expression (Nguyen et al., 2018). In human GRN mutant iPSC-derived cortical neurons and astrocytes, a novel class of aminoglycoside PTC readthrough enhancer compounds increased GRN expression and improved lysosomal function (Frew et al., 2020). As the compounds did not readily cross the blood-brain barrier, the authors used intracerebroventricular delivery in a rodent model, to provide in vivo efficacy. Preventing NMD via antisense oligonucleotide-based approaches or pharmacological compounds thus seems feasible, albeit tailored to a selective number of pathogenic GRN mutations. Figure 3 illustrates the mechanisms to increase GRN gene expression.
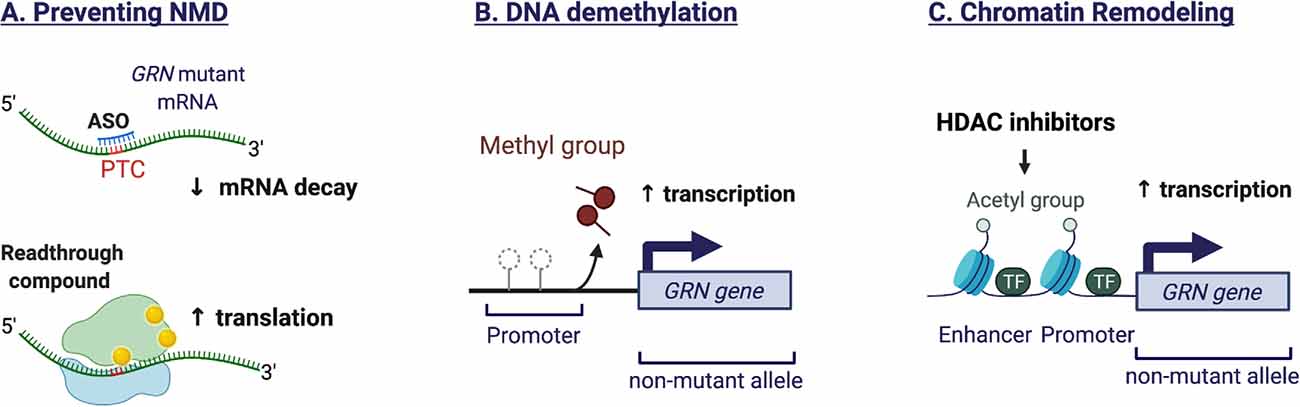
Figure 3. Mechanisms to increase endogenous GRN expression. (A) The majority of GRN mutations are nonsense and frameshift mutations that introduce premature termination codons (PTC). The presence of a PTC triggers the nonsense-mediated mRNA decay (NMD) machinery, resulting in degradation of the aberrant transcript and preventing protein translation. Antisense oligonucleotides (ASO) can stabilize the mutant mRNA by preventing the recruitment of NMD proteins. Alternatively, readthrough compounds can suppress nonsense mutations, leading to increased mRNA stability and protein translation. The efficacy of these approaches is mutation-dependent. (B) Epigenetic mechanisms regulate the expression of GRN from the healthy allele. GRN promotor demethylation promotes transcription. (C) Histone acetylation favors an open chromatin structure that is more permissive to gene transcription. HDAC inhibitors can prevent histone deacetylation and therefore increase GRN expression.
Very little is known about the physiological regulation of GRN expression and the differences between cell types. In the brain, GRN is primarily expressed by neurons and microglia. While microglia can upregulate GRN expression when activated, neurons maintain a stable GRN expression, only to increase with age (Petkau et al., 2010). Candidate epigenetic mechanisms are gene inactivation by methylation of the GRN promoter region or by limiting the accessibility of chromatin by histone deacetylation.
Evidence for the epigenetic regulation of GRN expression was found in sporadic patients with FTD. Compared to controls as well as patients with Alzheimer’s and Parkinson’s disease, sporadic FTLD brains exhibited altered expression of the DNA methyltransferase 3a as well as hypermethylation of the GRN gene promoter. Chronic treatment of GRN mutant lymphoblast lines with a methyltransferase inhibitor could rescue GRN transcript and protein levels in the study (Banzhaf-Strathmann et al., 2013).
In another attempt to influence the transcription of the healthy GRN allele in patients, Cenik and colleagues assessed GRN promotor activity in a luciferase-based screen. They identified the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) as an enhancer of GRN expression and validated their results in cells from patients with frontotemporal dementia (Cenik et al., 2011). SAHA is currently in use as a treatment for cutaneous T-cell lymphoma but has significant side effects and limited brain penetrance (Khan and La Thangue, 2008). In December 2014 a phase 2a clinical trial was launched to assess the safety, tolerability, and pharmacodynamics of the highly brain penetrant HDAC inhibitor FRM-0334 (ClinicalTrials.gov identifier: NCT02149160). The compound induced GRN in rodent brain and in FTD-GRN patient-derived lymphoblasts but failed to increase GRN levels in patients at the doses tested (Boxer et al., 2019). Of note, the HDAC inhibitor valproic acid, frequently used as a mood stabilizer and for the treatment of epilepsy, was shown to increase GRN expression as well, notwithstanding the compound is less potent than SAHA (She et al., 2017). While HDAC inhibitors remain an interesting therapeutic option to increase GRN expression, their lack of specificity as general transcriptional enhancers is potentially problematic. An in-depth analysis of HDAC inhibitor binding kinetics and structural features required for the upregulation of GRN expression is provided by Moreno-Yruela et al. (2019).
Focusing on known autophagy-lysosome modulators, the compound trehalose was identified as a potential therapeutic target. Trehalose, a natural disaccharide, increased GRN mRNA as well as intracellular and secreted GRN in GRN mutant iPSC-derived cortical neurons (Holler et al., 2016). To improve blood–brain-barrier penetrance, novel trehalose derivatives are currently in development (Holler et al., 2016). The importance of using human model systems in the identification of potential therapeutic agents was demonstrated in a trial investigating the calcium channel blocker Nimodipine. In a rodent model system, the compound was shown to increase GRN levels but in GRN mutation carriers, Nimodipine failed to significantly raise GRN CSF levels (Sha et al., 2017).
Gene therapy, the in vivo delivery of exogenous DNA, could be an alternative to influencing endogenous gene expression. Recombinant AAV delivers cargo DNA to the nucleus, but in contrast to the native AAV, does not integrate the transgene in the genome. The transgene persists as an episome in the nucleus of the transduced cell (Naso et al., 2017). Expression of Grn in neurons via adeno-associated viral delivery was able to correct neuropathological and behavioral abnormalities in heterozygous and homozygous Grn mutant mice. Interestingly, the vector carrying the Grn gene was only introduced after the onset of pathology in aged mice (10–12 months of age; Arrant et al., 2018). However, in a different study, T cell-mediated hippocampal degeneration was reported as a consequence of adeno-associated viral delivery of the Grn gene (Amado et al., 2019). The group used a different AAV9 strain, an alternative (intraventricular) route of transgene delivery and analyzed the animals at a later time point (Amado et al., 2019). In order to facilitate the translation of AAV-based gene therapy to GRN haploinsufficient FTD patients, Hinderer et al., supported by biotech company Passage Bio, have evaluated several GRN-expressing AAV vectors in rhesus macaques by direct intra-cisterna magna delivery (Hinderer et al., 2020). Despite a transduction efficiency of less than 1 percent of neurons after a single administration, a prolonged increase of GRN levels was seen, up to 10-fold (and even 40-fold for AAV1) compared to normal human GRN CSF levels. Neuropathological examination of the animals showed no signs of hippocampal degeneration but revealed mild axonal degeneration of sensory neurons in the posterior columns of the spinal cord, a side effect previously reported in other AAV studies using different transgenes (Hinderer et al., 2020). These results have supported the development of an AAV1-GRN treatment, called PBFT02, which will be studied in a Phase 1 study. Passage Bio plans to conduct the phase 1 trial in 2021 to assess the safety, tolerability, and efficacy of PBFT02 (ClinicalTrials.gov Identifier: NCT04747431). In July 2020, Prevail Therapeutics started the first GRN-AAV Phase 1/2 clinical trial in 15 people with FTD due to a GRN mutation. The study evaluates a different GRN-AAV9 vector, administered in combination with immunosuppressive drugs (PR006, ClinicalTrials.gov Identifier: NCT04408625).
As an alternative to gene therapy, direct intracerebroventricular delivery of recombinant GRN could allow a more controlled and dosed delivery of GRN. Whereas this delivery method has been safely tested before for other recombinant proteins (Van Damme et al., 2020), it still remains an invasive and expensive method of drug delivery.
To allow intravenous protein therapy, a recombinant human GRN protein has been developed with improved blood–brain-barrier (BBB) penetrance. The protein named PTV:PGRN (Protein Transport Vehicle) was engineered to bind to the human transferrin receptor, facilitating transport across the BBB. Preliminary reports state that intravenously administered PTV:PGRN can correct inflammatory and lysosomal lipid alterations in Grn null mice (Logan et al., 2020). Figure 4 illustrates the methods for exogenous GRN expression.
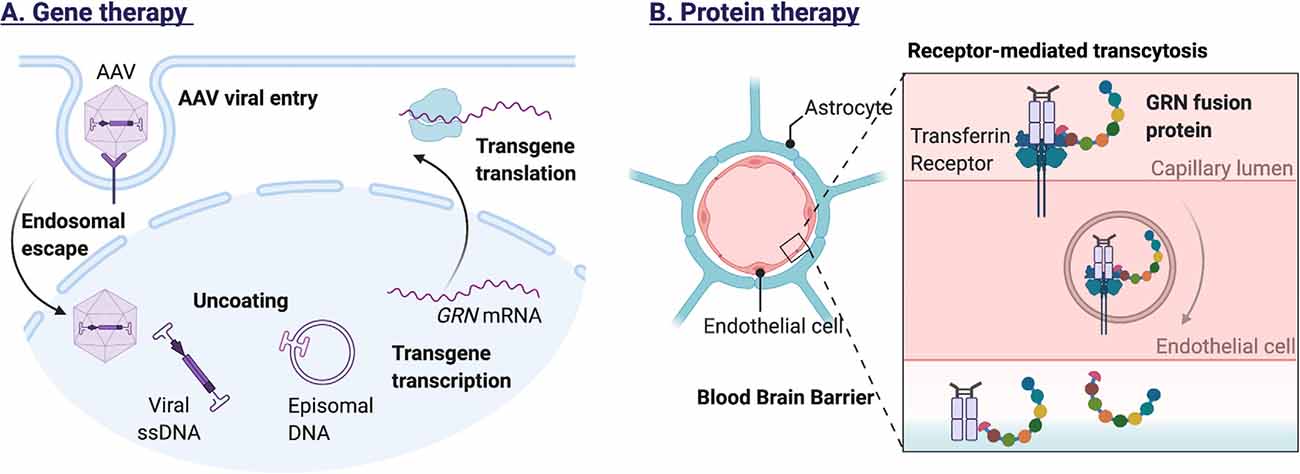
Figure 4. Methods for exogenous GRN expression. (A) Adeno-associated viral vectors, modified to carry the GRN gene, can infect a wide range of cells in the central nervous system. Recognition by cell surface receptors of the host cell triggers endocytosis and endosomal escape of the AAV particle. After nuclear import, the viral DNA is released, allowing transgene expression. Because the viral DNA persists as a nonintegrating stable episome, transgene expression can be prolonged, especially in non-dividing cells (e.g., neurons). (B) To allow intravenous protein therapy, a recombinant human GRN protein has been developed with improved blood–brain-barrier (BBB) penetrance. The GRN fusion protein binds the human transferrin receptor, facilitating receptor-mediated transcytosis across the blood-brain barrier.
Research conducted by Capell et al. (2014) provided insights into the translational regulation of GRN expression. The group identified different GRN transcripts with short and long 5′ untranslated regions (UTR). Expression of the long 5′UTR, containing an upstream open reading frame, stalled the ribosomal translation of GRN and affected mRNA stability. Similar repression mechanisms by 5′UTRs have been demonstrated for the mRNAs of BACE1 and ADAM10. Selective repression of GRN translation could contribute to the differential expression of for example neurons and microglia, although to our knowledge, this has not been investigated.
Several microRNAs have been identified in the past years that can affect GRN mRNA stability and translation. The 3′UTR of progranulin contains a predicted miR-29b binding site and the knockdown of miR-29b was shown to increase the secretion of GRN in NIH3T3 cells (Jiao et al., 2010). GRN was identified as the strongest target for mIR-107 in an RNA immunoprecipitation technique coupled to microarray. Interestingly the expression of mIR-107 itself was glucose-sensitive and mIR-107 is downregulated in Alzheimer’s disease (Wang et al., 2010). A third miRNA affecting GRN expression was discovered when sequencing the GRN gene of more than three hundred FTLD patients with no known GRN mutation. A single-nucleotide polymorphism (SNP), rs5848, in the 3’UTR of the GRN gene was overrepresented in FTLD cases and was predicted in silico to increase the binding of miRNA-659, resulting in decreased GRN expression (Rademakers et al., 2008). However, in a different FTLD cohort, the SNP rs5848 risk allele was not reported (Simon-Sanchez et al., 2009). The recent finding that the risk allele is specifically associated with pathological subtype type A of FTLD-TDP and not with other subtypes (type B or C) could explain these differences (Pottier et al., 2019). Interestingly, the same SNP rs5848 has been described as a risk factor for Alzheimer’s disease and as a disease modifier for ALS as well (van Blitterswijk et al., 2014). Presence of the rs5848 risk allele was associated with decreased survival after onset in carriers of a C9orf72 repeat expansion.
Targeting miRNAs could be a potential therapeutic approach to recover GRN expression. To achieve miRNA inhibition, engineered oligonucleotides can be developed to mask the miRNA target sequence in the 3′ UTR of the target gene, preventing the miRNA/mRNA interaction. Alternatively, oligonucleotides can be developed that interfere directly with mature miRNAs and consequently compete with target genes, preventing target gene mRNA destruction (Atri et al., 2019).
Shortly after the discovery of mutations in the GRN gene, TDP-43 was identified as the key constituent of the tau-negative neuronal inclusions (Neumann et al., 2006). TDP-43 is an RNA-binding protein, involved in pre-mRNA splicing, mRNA transport, stability, and translation (Tziortzouda et al., 2021). As an RNA-binding protein, TDP-43 was suspected to regulate GRN levels. The relationship between TDP-43 and GRN however, is not straightforward. While studies have demonstrated that TDP-43 binds to the 3′ untranslated region of GRN mRNA, promoting mRNA instability (Colombrita et al., 2012; Fukushima et al., 2019), GRN protein levels were not decreased. The observed difference in transcript and protein levels could be explained by the concurrent effect of TDP-43 on SORT1 mRNA (Gumina et al., 2019). Depletion of TDP-43 in human cells was found to increase the production of a splice variant of sortilin, that is secreted and not bound to the plasma membrane (Prudencio et al., 2012), impeding the normal uptake and delivery of GRN to the lysosome (Hu et al., 2010). The identification of additional RNA-binding proteins that regulate the stability of the GRN transcript, would provide novel therapeutic targets to modulate GRN gene expression. Figure 5 illustrates the mechanisms influencing GRN translation.
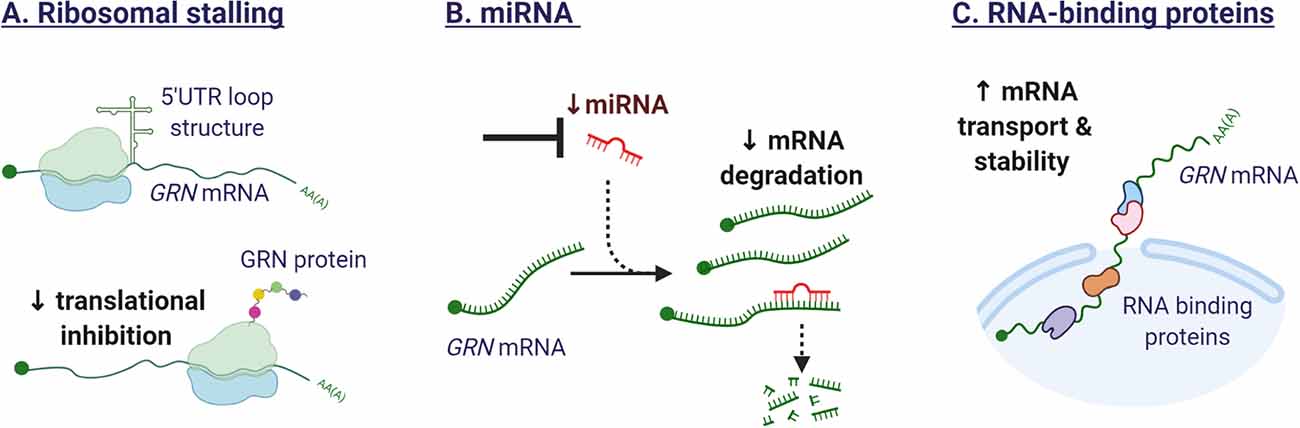
Figure 5. Mechanisms affecting GRN translation. (A) The secondary structure of the 5′ untranslated regions (UTR) in the GRN transcript can affect ribosomal function. The formation of a loop structure can hinder efficient translation. (B) Several miRNA have been identified that target GRN mRNA and repress GRN protein synthesis. Synthetic oligonucleotides could sequester endogenous miRNAs or mask miRNA target sequences in the GRN mRNA, leading to enhanced gene expression. (C) RNA-binding proteins are potential therapeutic targets as they affect the splicing, mRNA transport, stability, and translation of GRN mRNA.
An N-terminal signaling sequence directs the nascent GRN protein chain to the endoplasmatic reticulum, where the protein undergoes extensive post-translational modifications. Proper protein maturation is essential for the biological activity of GRN and includes steps of signal sequence cleavage, the addition of N-linked glycans, and protein folding by chaperones in the lumen. Full–length GRN consists of 593 amino acid residues with a predicted molecular weight of 68.5 kDa, but an observed mass of approximately 75–80 kDa due to heavy glycosylation (Bateman and Bennett, 1998). Fucosylated oligosaccharides are the major N-linked glycans found in GRN (Songsrirote et al., 2010). The regulation and functional consequences of the different glycosylation sites are currently poorly understood.
Full-length GRN consists of seven and a half cysteine-rich granulin modules. Each granulin module folds into a prototypic structure of four β-hairpins, joined together by six disulfide bridges (He and Bateman, 2003). The proper folding and transport of GRN through the ER-Golgi pathway is assured by several ER Ca2+-binding chaperones including calreticulin, Binding immunoglobulin protein (BiP), GRP94, and members of the protein disulfide isomerase family (Almeida et al., 2011). Modulating the ER chaperone network has been proposed as a therapeutic strategy to increase the level of extracellular GRN.
The presence of an amino-terminal secretion signal directs mature GRN through the trans-golgi network into secretory vesicles, in which it undergoes regulated exocytosis (Petoukhov et al., 2013). GRN can be secreted as a soluble protein or associated with exosomes (Benussi et al., 2016). Alternatively, by binding sortilin or through an interaction with prosaposin, GRN can be diverted from the secretory pathway to the endolysosomal system (Hu et al., 2010; Zhou X. et al., 2015). The interaction between GRN and prosaposin and sortilin will be discussed further in the next section.
In neurons, GRN was found to be co-transported with BDNF and recruited to synapses for secretion in an activity-dependent matter (Petoukhov et al., 2013). Alkalizing drugs and vacuolar ATPase inhibitors (like chloroquine or bafilomycin) can increase GRN levels within the secretory pathway, with increased levels in the ER and the Golgi network as well as extracellular (Capell et al., 2011). Chloroquine is commonly used as an anti-malarial drug and to treat autoimmune diseases. In an exploratory clinical study, three FTD patients known to be GRN deficient, received chloroquine treatment for a year, with a reported rescue of GRN CSF levels and a reported improvement in cognitive abilities (Körtvelyessy et al., 2017). However, to our knowledge, there are currently no clinical trials further investigating chloroquine as a treatment option for FTD.
While mutations in GRN can be predicted based on a reduced blood GRN levels, this does not explain the large variability in plasma GRN levels seen in healthy controls (Finch et al., 2009; Meeter et al., 2016). Genetic studies have therefore sought to reveal additional regulators of GRN expression. A repurposed genome-wide association study (GWAS) of late-onset Alzheimer’s Dementia, revealed two SNPs on chromosome 1 linked to lower plasma GRN levels. The most significant SNP, rs646776, was associated with a modest reduction (16 percent) in plasma GRN levels and increased expression of the sortilin gene (SORT1) (Carrasquillo et al., 2010). Sortilin, a lysosomal sorting and trafficking receptor, was simultaneously identified in a screen for GRN binding partners using monkey kidney cells (unable to bind GRN) overexpressing a cDNA library (Hu et al., 2010). Knocking out SORT1 severely reduced the binding of GRN to the cell surface with a concomitant rise in extracellular levels of GRN (Hu et al., 2010).
Initially, sortilin was thought to be the long sought-after GRN receptor, mediating the neurotrophic effect. Sortilin was known to interact with the low-affinity neurotrophin receptor p75NTR and mediate proneurotrophin-induced apoptosis (Nykjaer et al., 2004). In addition, the extracellular part of sortilin binds granulin E (Zheng et al., 2011), the most C-terminal part of GRN and a granulin known to exert neurotrophic properties on cortical and motor neurons (Van Damme et al., 2008). However, despite the deletion of the sortilin binding site, granulin E was capable of promoting the survival of mouse motor and cortical neurons (De Muynck et al., 2013). Likewise, GRN could still stimulate neurite outgrowth in hippocampal cultures of Sort1 null mice, suggesting GRN acted independently of sortilin (Gass et al., 2012).
Nevertheless, the identification of sortilin as a receptor and regulator of GRN levels, did instigate the therapeutic targeting of the GRN-sortilin axis. As sortilin targets GRN for destruction in the lysosome (Hu et al., 2010), preventing GRN uptake and destruction could protect neurons and improve disease. Small molecule MPEP, known to decrease sortilin levels, has been shown to increase extracellular GRN levels but not intracellular GRN in iPSC-derived neurons harboring the GRN S116X mutation (Lee et al., 2014). A different tactic is the development of a sortilin antibody, a therapeutic approach currently pursued by Alector in partnership with Abbvie. AL001, a monoclonal human recombinant anti-human sortilin antibody, has progressed to Phase 2 and Phase 3 studies (ClinicalTrials.gov Identifier: NCT03987295 and NCT04374136). The completed Phase 1 study (NCT03636204) demonstrated that AL001 treatment could normalize GRN CSF levels in symptomatic and asymptomatic GRN mutation carriers (Haynes et al., 2020). Eight weeks after the last dose, a modest reduction of plasma neurofilament light chain (NfL) was observed, hinting at a possible beneficial effect on neuronal integrity. NfL, a neuron-derived cytoskeletal protein, is a promising biomarker of neurodegeneration and has been shown to increase over time in symptomatic GRN mutation carriers (van der Ende et al., 2019).
At the cellular level, the impact of redirecting GRN via inhibition of sortilin is incompletely understood. A number of studies have recently accentuated the role of full-length GRN and granulin peptides in the lysosome (Holler et al., 2017; Lee et al., 2017; Zhou et al., 2017a). Inhibiting the endocytosis of GRN could therefore further deplete GRN and the granulins in the lysosome, possibly affecting lysosomal homeostasis. Then again, blocking sortilin could increase the availability of GRN to alternative receptors (e.g., the EphA2 receptor) (Neill et al., 2016). This duality underscores that continued mechanistic research is needed, in parallel with the development of therapies controlling GRN levels.
Linking whole-genome sequencing data with plasma GRN levels in a large Mexican-American cohort, led to the confirmation of prosaposin (PSAP) as a regulator of GRN (Nicholson et al., 2016). GRN and prosaposin were known to be structurally and functionally very similar and the interaction between GRN and prosaposin had been identified through a proteomic screen, one year prior (Zhou X. et al., 2015). Prosaposin can, like progranulin, be cleaved into cysteine-rich subunits called saposins and homozygous loss-of-function mutations lead to dysfunction in lysosomal sphingolipid metabolism (sphingolipidosis) (Zhou X. et al., 2015). Prosaposin binds the mannose-6-phosphate receptor (M6PR) or low-density lipoprotein receptor-related protein 1 (LRP1) to gain access to lysosomes. By forming heterodimers, GRN and prosaposin were shown to use each other’s transport receptors to travel to the lysosome (Zhou X. et al., 2015; Nicholson et al., 2016). The finding provided a new perspective on the role of sortilin as a GRN signaling receptor. Could GRN have stimulated neurite outgrowth and survival independent of sortilin (Gass et al., 2012; De Muynck et al., 2013), because there is a functional interchangeability of the trafficking receptors in the presence of prosaposins? The functional redundancy could indicate a fail-safe mechanism, underscoring the importance of the pathway.
An international collaboration identified variants on chromosome 7p21, associated with increased TMEM106B expression, as susceptibility loci for FTLD-TDP (Van Deerlin et al., 2010). Interestingly, the association of the risk locus was stronger in GRN mutation carriers (Van Deerlin et al., 2010). Later it was shown that the risk locus correlated with decreased plasma GRN levels (Finch et al., 2011) and elevated TMEM106B protein levels in patient FTLD brain samples (Chen-Plotkin et al., 2012). The clinical importance of variants in TMEM106B for GRN mutation carriers was recently demonstrated by Pottier et al. (2018). Carriers of the protective haplotype, associated with lower levels of TMEM106B, are estimated to have 50% less chance of developing disease symptoms (Pottier et al., 2018). In ALS patients, the protective allele was associated with preserved cognition (Vass et al., 2011). The role of the lysosomal transmembrane protein TMEM106B as an aggravating risk factor has been explored and a reciprocal regulation between GRN and TMEM106B levels has become evident. GRN deficiency leads to increased TMEM106B levels in aged mice (Zhou et al., 2017b). Overexpression of TMEM106B in a cell model caused abnormalities in late endosome-lysosome morphology and led to the sequestration of GRN (Chen-Plotkin et al., 2012).
Complete loss of Tmem106b however, was not beneficial in Grn knockout mice models (Feng et al., 2020; Werner et al., 2020; Zhou et al., 2020). Double knockout mice demonstrated worsened neuropathology with increased TDP-43 accumulation, gliosis, and lysosomal dysfunction. Additionally, the mice developed motor deficits, a phenotype not present in single knockout models (Feng et al., 2020; Werner et al., 2020; Zhou et al., 2020). In knockout models that retained a low residual expression of Tmem106b, a phenotypic delay was seen with motor deficits emerging 2 months later (Zhou et al., 2020). Residual Tmem106b expression could explain the partial rescue phenotype observed by Klein et al. (2017). In that study, removing Tmem106b reversed neuronal death and a hyperactive behavioral phenotype associated with complete loss of Grn. Both Tmem106b deletion (Klein et al., 2017) as well as overexpression impaired lysosome acidification (Chen-Plotkin et al., 2012). While the possibility to manipulate TMEM106B levels remains an interesting new therapeutic avenue, these functional studies indicate the importance of accurately balancing TMEM106B levels. One potential additional drawback to manipulating TMEM106B is the identification of de novo mutations as a rare cause for hypomyelinating leukodystrophy (Simons et al., 2017). Further study is needed to fully gauge the consequences of the TMEM106B variants in GRN-FTLD and to determine how GRN and TMEM106B interact.
Extracellular GRN levels are regulated through endocytosis and proteolytic degradation. Individual six kDA granulins can be excised from the full-length GRN protein by extracellular proteases, for example, metalloproteinases MMP-9, MMP-12, MMP-14, and ADAMTS-7 (De Muynck and Van Damme, 2011). Both full-length GRN and the released granulin peptides are biologically active but the cellular effects can differ from granulin to granulin and can often antagonize the actions of full-length GRN (Bateman and Bennett, 2009). The granulins were in fact identified before the discovery of the intact precursor protein, in extracts of granulocytes, hence the origin of the name granulins (Bateman et al., 1990). They were later shown to possess pro-inflammatory properties and to negatively impact wound healing in contrast to their common precursor (Zhu et al., 2002). Granulins regulate cell growth but with opposing effects as demonstrated on breast cancer cell lines for GRN A and GRN F (Tolkatchev et al., 2008; Bateman and Bennett, 2009). Granulin E, the most C-terminal part of full-length GRN exerts neurotrophic properties on cortical and motor neurons (Van Damme et al., 2008). The precursor protein is protected from cleavage by association with the high-density lipoprotein (HDL)/Apolipoprotein A-I complex (Okura et al., 2010), or by binding to the secretory leukocyte protease inhibitor (SLPI; Zhu et al., 2002). Figure 6 illustrates the intracellular and extracellular processing of the GRN protein.
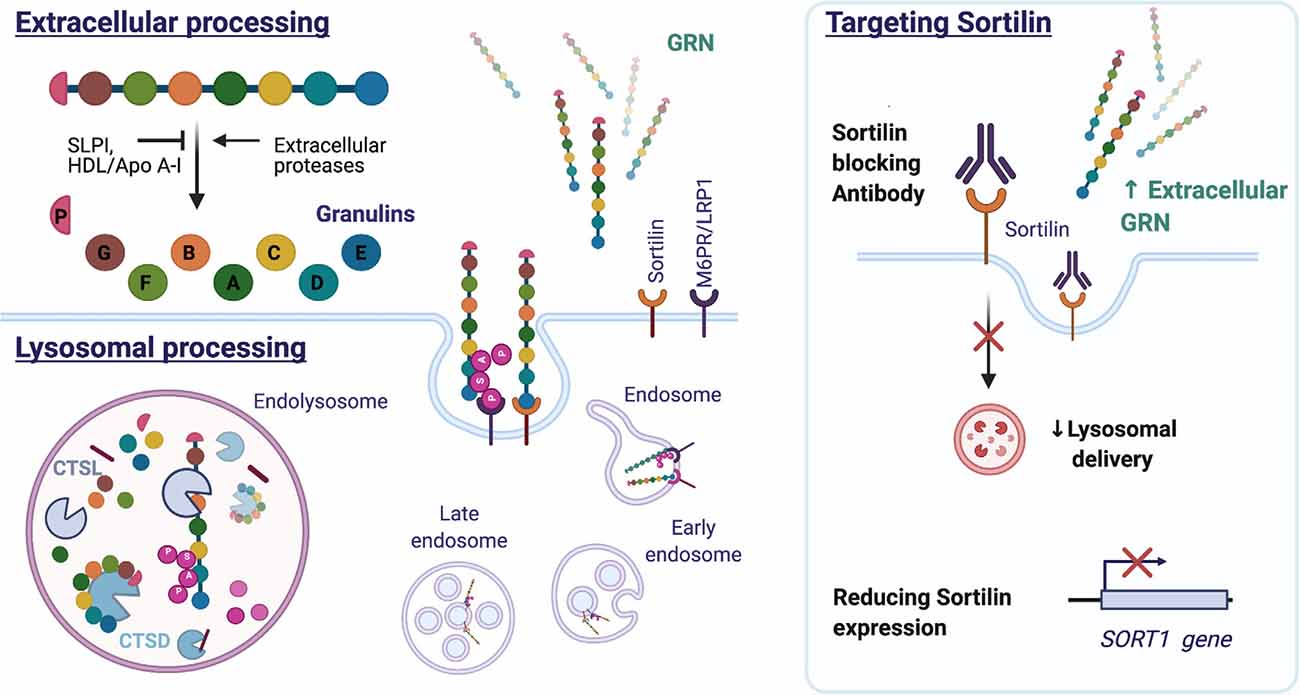
Figure 6. Structure and processing of mammalian GRN. GRN consists of seven and a half granulin domains. The linker sequences can be cleaved by extracellular and intracellular proteases, releasing individual granulins or a combination of GRN modules. Both SLPI and HDL/Apo A-I can bind GRN and inhibit GRN processing. Extracellular GRN is rapidly endocytosed and targeted to the lysosome directly by binding sortilin or indirectly by forming a heterodimer with prosaposin, which utilizes the M6PR or LRP1 receptors. In the lysosome, GRN is cleaved into granulins by Cathepsin L (CTSL) and possibly other hydrolases. GRN contributes to lysosomal homeostasis by increasing the stability of lysosomal enzymes [e.g., Cathepsin D (CTSD) and consequently affects the processing of prosaposin (PSAP)]. Reducing sortilin (SORT1) gene expression or preventing GRN-sortilin interaction with blocking antibodies can increase extracellular GRN levels and prevent lysosomal degradation of GRN (HDL: high-density lipoprotein, Apo A-I: apolipoprotein A-I, M6PR: mannose 6-phosphate receptor, LRP1: lipoprotein receptor-related protein 1).
Extracellular GRN is rapidly endocytosed and delivered to the lysosome by SORT1 (Hu et al., 2010) or in association with prosaposin (PSAP) (Zhou X. et al., 2015). While the lysosome was previously considered a degradation endpoint for GRN, research attention is currently focused on probing the lysosomal function of the protein. Cathepsin L, a lysosomal cysteine protease, was found responsible for the lysosomal processing of GRN (Holler et al., 2017; Lee et al., 2017; Zhou et al., 2017a). Excised granulins were shown to be more stable and to persist in the lysosome long after endocytosis and the disappearance of full-length GRN (Holler et al., 2017). The function of granulins in the lysosome remains to be further elucidated (reviewed in Zhou et al., 2021).
Apart from being a substrate of lysosomal enzymes, full-length GRN functions as a chaperone to lysosomal hydrolases like cathepsin D and β-glucocerebrosidase (Jian et al., 2016; Beel et al., 2017). Interestingly, mutations in GBA1, the gene encoding β-glucocerebrosidase, are common genetic risk factors for the development of Parkinson disease, and homozygous mutations are associated with Gaucher disease, a juvenile onset lysosomal storage disease (Paushter et al., 2018).
Selective inhibition of GRN proteases could be of great therapeutic value, but requires a better understanding of GRN protein catabolism.
Growth factor signaling cascades are known to be important for neuronal survival and function, but GRN turns out to be far from a conventional growth factor. While GRN activates classical growth factor signaling cascades, including the mitogen-activated protein (MAP) kinase and phosphoinositide 3 (PI3) kinase pathways, it does not bind a classical growth factor receptor nor does the mode of growth factor signaling resemble that of other growth factors like PDGF or EGF (Bateman and Bennett, 2009). In IGFI-R-deficient fibroblasts, GRN was able to elicit a prolonged growth factor signal that carried the fibroblast through both S phase (DNA synthesis phase) and M phase (cell division phase) (Zanocco-Marani et al., 1999). GRN is therefore simultaneously both a competence and a progression factor, which is a unique feature as classical growth factors normally require the assistance from other growth factors to complete both S- and M-phase. In neurons, the MAP kinase pathway and PI3K pathways are involved in GRN-dependent neuroprotection (Xu et al., 2011; Almeida et al., 2012). Apart from evoking classical growth factor signaling cascades, GRN additionally promotes tyrosine phosphorylation of focal adhesion kinase (FAK) (He and Bateman, 2003). FAK is a critical mediator of integrin signaling and could therefore potentially explain GRN’s influence on cell migration, cell cycle progression, and prevention of anoikis.
In 2011, using a yeast-two-hybrid screen, GRN was reported as a new ligand and antagonist of tumor necrosis factor (TNF) receptors (Tang et al., 2011). The finding was not replicated, possibly due to technical differences and the conformation of the recombinant GRN protein used in the study (Chen et al., 2013). Later studies confirmed GRN as a ligand of TNFR, implicating GRN in many inflammatory diseases (e.g., contact dermatitis, osteoarthritis, colitis, etc.). Wang et al. (2015) provide an overview of the confirmation experiments and initial controversies regarding the GRN-TNFR interaction. Atsttrin, a more stable GRN-like engineered protein, was shown to suppresses TNF-α-mediated inflammation in arthritis models (Wei et al., 2017).
Additionally, GRN has been shown to interact with the toll-like receptor 9 (Park et al., 2011), a key regulator of the immune response to foreign DNA. GRN also bound the extracellular domain of Notch receptors and affected Notch target gene expression (Altmann et al., 2016). The Notch signaling system has distinct roles in innate immunity and cell development. DLK1, a modulator of Notch signaling, was found to interact with GRN in a yeast-two-hybrid screen (Baladron et al., 2002).
In 2016, EphA2 was identified as a functional high-affinity GRN receptor, using a phospho-RTK array and a human urinary bladder carcinoma cell line. Silencing the receptor or blocking the ligand interaction, affected capillary lumen formation by human umbilical vein endothelial cells (HUVEC) as well as GRN transcriptional autoregulation (Neill et al., 2016). Analysis of downstream signaling events [activation of MAPK and protein kinase B (Akt) pathways], suggested GRN antagonizes Ephrin/Eph signaling (Neill et al., 2016). Considering the known involvement of Ephrin signaling in axon guidance and synaptogenesis, the finding poses questions for future study. The same research group identified drebrin, an F-actin–binding protein, and perlecan, an ECM proteoglycan, as GRN binding partners (Gonzalez et al., 2003; Xu et al., 2015).
The interaction of GRN with a multitude of extracellular, transmembrane, intracellular, and nuclear proteins is remarkable (De Muynck and Van Damme, 2011; Cui et al., 2019; Zhou et al., 2021) However, the multitude of GRN binding partners is the result of researching GRN biology in diverse histological contexts (peripheral inflammation, vasculogenesis, carcinogenesis and so forth). Regarding GRN biology in the CNS, it seems likely that concerted actions of different binding partners contribute to the neurotrophic effect.
In recent years, significant progress has been made towards the development of therapies modulating GRN levels. While raising GRN has always seemed straightforward, the multitude of mechanisms at play in regulating GRN levels pose extra challenges for the development of therapeutic agents. With a common goal to raise GRN levels, every therapeutic intervention differs in the extent of action depending on the biological strategy used (e.g., affected by viral vector tropism or depending on SORT1 expression) and by mode of administration (e.g., systemic or CNS restricted delivery). The resulting pattern of (ectopic) GRN expression will therefore influence the biological effect and possible side-effects. Close monitoring of GRN protein levels will be needed to ensure expression within a physiological range, as elevated GRN levels have been associated with cancer, metabolic disease, and chronic inflammatory states.
The current development and utilization of GRN therapy in FTD patients with GRN mutations provides a unique starting point for further research. The recent extensive documentation of fluid, neuroimaging and cognitive biomarkers for FTD will undoubtedly prove to be crucial in monitoring treatment response and guiding therapeutic development (Benussi et al., 2021; Panman et al., 2021). Additionally, specific biomarkers for the biological effects of GRN are needed to identify candidate diseases for GRN therapy. The pleiotropy of the GRN protein ensures that the applicability of GRN as a therapeutic target reaches far beyond the ALS-FTD spectrum.
JT, CV, and PVD wrote the manuscript. All authors made substantial contributions to the discussion of the content, reviewed and edited the article. All authors contributed to the article and approved the submitted version.
The authors were supported by grants from KU Leuven (C1-C14-17-107), Opening the Future Fund (KU Leuven), the Fund for Scientific Research Flanders (FWO-Flanders), the ALS Liga Belgium, the KU Leuven funds “Een Hart voor ALS”, “Laeversfonds voor ALS Onderzoek” and the “Valéry Perrier Race against ALS Fund”, the Alzheimer Research Foundation (SAO-FRA 2017/023 and SAO-FRA 2019/0014), The Alzheimer Association (AARG-NTF-19-616888), the Flemish Government initiated Flanders Impulse Program on Networks for Dementia Research (VIND 135043), Flanders Innovation and Enterpreneurship (IWT grants Project MinE and iPSCAF), the Belgian National Lottery, the Latran Foundation, the European Union’s Horizon 2020 research and innovation programme (755094), and the European Union’s ERA-Netfor Research Programmes on Rare Diseases (INTEGRALS). PVD holds a senior clinical investigatorship of FWO-Vlaanderen and was supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
All figures presented in this manuscript were made using BioRender.com.
Abella, V., Pino, J., Scotece, M., Conde, J., Lago, F., Gonzalez-Gay, M. A., et al. (2017). Progranulin as a biomarker and potential therapeutic agent. Drug Discov. Today 22, 1557–1564. doi: 10.1016/j.drudis.2017.06.006
Ahmed, Z., Sheng, H., Xu, Y. F., Lin, W. L., Innes, A. E., Gass, J., et al. (2010). Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 177, 311–324. doi: 10.2353/ajpath.2010.090915
Almeida, S., Zhang, Z., Coppola, G., Mao, W., Futai, K., Karydas, A., et al. (2012). Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2, 789–798. doi: 10.1016/j.celrep.2012.09.007
Almeida, S., Zhou, L., and Gao, F. B. (2011). Progranulin, a glycoprotein deficient in frontotemporal dementia, is a novel substrate of several protein disulfide isomerase family proteins. PLoS One 6:e26454. doi: 10.1371/journal.pone.0026454
Altmann, C., Vasic, V., Hardt, S., Heidler, J., Haussler, A., Wittig, I., et al. (2016). Progranulin promotes peripheral nerve regeneration and reinnervation: role of notch signaling. Mol. Neurodegener. 11:69. doi: 10.1186/s13024-016-0132-1
Amado, D. A., Rieders, J. M., Diatta, F., Hernandez-Con, P., Singer, A., Mak, J. T., et al. (2019). AAV-mediated progranulin delivery to a mouse model of progranulin deficiency causes T cell-mediated toxicity. Mol. Ther. 27, 465–478. doi: 10.1016/j.ymthe.2018.11.013
Arechavaleta-Velasco, F., Perez-Juarez, C. E., Gerton, G. L., and Diaz-Cueto, L. (2017). Progranulin and its biological effects in cancer. Med. Oncol. 34:194. doi: 10.1007/s12032-017-1054-7
Arrant, A. E., Filiano, A. J., Patel, A. R., Hoffmann, M. Q., Boyle, N. R., Kashyap, S. N., et al. (2019). Reduction of microglial progranulin does not exacerbate pathology or behavioral deficits in neuronal progranulin-insufficient mice. Neurobiol. Dis. 124, 152–162. doi: 10.1016/j.nbd.2018.11.011
Arrant, A. E., Filiano, A. J., Unger, D. E., Young, A. H., and Roberson, E. D. (2017). Restoring neuronal progranulin reverses deficits in a mouse model of frontotemporal dementia. Brain 140, 1447–1465. doi: 10.1093/brain/awx060
Arrant, A. E., Onyilo, V. C., Unger, D. E., and Roberson, E. D. (2018). Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis. J. Neurosci. 38, 2341–2358. doi: 10.1523/JNEUROSCI.3081-17.2018
Atri, C., Guerfali, F. Z., and Laouini, D. (2019). “MicroRNAs in diagnosis and therapeutics,” in AGO-Driven Non-Coding RNAs, ed B. Mallick (Amsterdam, Netherlands: Academic Press an imprint of Elsevier), 137–177.
Baba, T., Hoff, H. B., Nemoto, H., Lee, H., Orth, J., Arai, Y., et al. (1993). Acrogranin, an acrosomal cysteine-rich glycoprotein, is the precursor of the growth-modulating peptides, granulins and epithelins and is expressed in somatic as well as male germ cells. Mol. Reprod. Dev. 34, 233–243. doi: 10.1002/mrd.1080340302
Baizabal-Carvallo, J. F., and Jankovic, J. (2016). Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 12, 175–185. doi: 10.1038/nrneurol.2016.14
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi: 10.1038/nature05016
Baladron, V., Ruiz-Hidalgo, M. J., Bonvini, E., Gubina, E., Notario, V., and Laborda, J. (2002). The EGF-like homeotic protein dlk affects cell growth and interacts with growth-modulating molecules in the yeast two-hybrid system. Biochem. Biophys. Res. Commun. 291, 193–204. doi: 10.1006/bbrc.2002.6431
Bandey, I., Chiou, S. H., Huang, A. P., Tsai, J. C., and Tu, P. H. (2015). Progranulin promotes Temozolomide resistance of glioblastoma by orchestrating DNA repair and tumor stemness. Oncogene 34, 1853–1864. doi: 10.1038/onc.2014.92
Banzhaf-Strathmann, J., Claus, R., Mucke, O., Rentzsch, K., van der Zee, J., Engelborghs, S., et al. (2013). Promoter DNA methylation regulates progranulin expression and is altered in FTLD. Acta Neuropathol. Commun. 1:16. doi: 10.1186/2051-5960-1-16
Bateman, A., Belcourt, D., Bennett, H., Lazure, C., and Solomon, S. (1990). Granulins, a novel class of peptide from leukocytes. Biochem. Biophys. Res. Commun. 173, 1161–1168. doi: 10.1016/s0006-291x(05)80908-8
Bateman, A., and Bennett, H. P. (1998). Granulins: the structure and function of an emerging family of growth factors. J. Endocrinol. 158, 145–151. doi: 10.1677/joe.0.1580145
Bateman, A., and Bennett, H. P. (2009). The granulin gene family: from cancer to dementia. Bioessays 31, 1245–1254. doi: 10.1002/bies.200900086
Beel, S., Herdewyn, S., Fazal, R., De Decker, M., Moisse, M., Robberecht, W., et al. (2018). Progranulin reduces insoluble TDP-43 levels, slows down axonal degeneration and prolongs survival in mutant TDP-43 mice. Mol. Neurodegener. 13:55. doi: 10.1186/s13024-018-0288-y
Beel, S., Moisse, M., Damme, M., De Muynck, L., Robberecht, W., Van Den Bosch, L., et al. (2017). Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum. Mol. Genet. 26, 2850–2863. doi: 10.1093/hmg/ddx162
Belcastro, V., Siciliano, V., Gregoretti, F., Mithbaokar, P., Dharmalingam, G., Berlingieri, S., et al. (2011). Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Res. 39, 8677–8688. doi: 10.1093/nar/gkr593
Benussi, L., Ciani, M., Tonoli, E., Morbin, M., Palamara, L., Albani, D., et al. (2016). Loss of exosomes in progranulin-associated frontotemporal dementia. Neurobiol. Aging 40, 41–49. doi: 10.1016/j.neurobiolaging.2016.01.001
Benussi, L., Ghidoni, R., Pegoiani, E., Moretti, D. V., Zanetti, O., and Binetti, G. (2009). Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol. Dis. 33, 379–385. doi: 10.1016/j.nbd.2008.11.008
Benussi, A., Premi, E., Gazzina, S., Brattini, C., Bonomi, E., Alberici, A., et al. (2021). Progression of behavioral disturbances and neuropsychiatric symptoms in patients with genetic frontotemporal dementia. JAMA Netw. Open 4:e2030194. doi: 10.1001/jamanetworkopen.2020.30194
Boxer, A., Moebius, H., Harris, B., Boeve, B., Borroni, B., and Swieten, J. (2019). Phase 2A randomized, double-blind, placebo-controlled trial of HDACI, FRM-0334, in asymptomatic carriers of, or patients with FTLD due to progranulin gene mutations: Alzheimer’s Association International Conference. Alzheimers Dement. (N Y) 15, P1231–P1232. doi: 10.1016/j.jalz.2019.06.4746
Brouwers, N., Nuytemans, K., van der Zee, J., Gijselinck, I., Engelborghs, S., Theuns, J., et al. (2007). Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch. Neurol. 64, 1436–1446. doi: 10.1001/archneur.64.10.1436
Capell, A., Fellerer, K., and Haass, C. (2014). Progranulin transcripts with short and long 5′ untranslated regions (UTRs) are differentially expressed via posttranscriptional and translational repression. J. Biol. Chem. 289, 25879–25889. doi: 10.1074/jbc.M114.560128
Capell, A., Liebscher, S., Fellerer, K., Brouwers, N., Willem, M., Lammich, S., et al. (2011). Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J. Neurosci. 31, 1885–1894. doi: 10.1523/JNEUROSCI.5757-10.2011
Carrasquillo, M. M., Nicholson, A. M., Finch, N., Gibbs, J. R., Baker, M., Rutherford, N. J., et al. (2010). Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am. J. Hum. Genet. 87, 890–897. doi: 10.1016/j.ajhg.2010.11.002
Cenik, B., Sephton, C. F., Dewey, C. M., Xian, X., Wei, S., Yu, K., et al. (2011). Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J. Biol. Chem. 286, 16101–16108. doi: 10.1074/jbc.M110.193433
Chen-Plotkin, A. S., Unger, T. L., Gallagher, M. D., Bill, E., Kwong, L. K., Volpicelli-Daley, L., et al. (2012). TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J. Neurosci. 32, 11213–11227. doi: 10.1523/JNEUROSCI.0521-12.2012
Chen, X., Chang, J., Deng, Q., Xu, J., Nguyen, T. A., Martens, L. H., et al. (2013). Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J. Neurosci. 33, 9202–9213. doi: 10.1016/j.jchemneu.2021.102000
Chitramuthu, B. P., Baranowski, D. C., Kay, D. G., Bateman, A., and Bennett, H. P. (2010). Progranulin modulates zebrafish motoneuron development in vivo and rescues truncation defects associated with knockdown of Survival motor neuron 1. Mol. Neurodegener. 5:41. doi: 10.1186/1750-1326-5-41
Chitramuthu, B. P., Kay, D. G., Bateman, A., and Bennett, H. P. (2017). Neurotrophic effects of progranulin in vivo in reversing motor neuron defects caused by over or under expression of TDP-43 or FUS. PLoS One 12:e0174784. doi: 10.1093/nar/gkab582
Colombrita, C., Onesto, E., Megiorni, F., Pizzuti, A., Baralle, F. E., Buratti, E., et al. (2012). TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 287, 15635–15647. doi: 10.1074/jbc.M111.333450
Cruts, M., Gijselinck, I., van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. doi: 10.1038/nature05017
Cui, Y., Hettinghouse, A., and Liu, C. J. (2019). Progranulin: a conductor of receptors orchestra, a chaperone of lysosomal enzymes and a therapeutic target for multiple diseases. Cytokine Growth Factor Rev. 45, 53–64. doi: 10.1016/j.cytogfr.2019.01.002
Daniel, R., He, Z., Carmichael, K. P., Halper, J., and Bateman, A. (2000). Cellular localization of gene expression for progranulin. J. Histochem. Cytochem. 48, 999–1009. doi: 10.1177/002215540004800713
De Muynck, L., Herdewyn, S., Beel, S., Scheveneels, W., Van Den Bosch, L., Robberecht, W., et al. (2013). The neurotrophic properties of progranulin depend on the granulin E domain but do not require sortilin binding. Neurobiol. Aging 34, 2541–2547. doi: 10.1016/j.neurobiolaging.2013.04.022
De Muynck, L., and Van Damme, P. (2011). Cellular effects of progranulin in health and disease. J. Mol. Neurosci. 45, 549–560. doi: 10.1007/s12031-011-9553-z
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Egashira, Y., Suzuki, Y., Azuma, Y., Takagi, T., Mishiro, K., Sugitani, S., et al. (2013). The growth factor progranulin attenuates neuronal injury induced by cerebral ischemia-reperfusion through the suppression of neutrophil recruitment. J. Neuroinflammation 10:105. doi: 10.1186/1742-2094-10-105
Feng, T., Mai, S., Roscoe, J. M., Sheng, R. R., Ullah, M., Zhang, J., et al. (2020). Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep. 21:e50219. doi: 10.15252/embr.202050219
Ferrari, R., Hernandez, D. G., Nalls, M. A., Rohrer, J. D., Ramasamy, A., Kwok, J. B., et al. (2014). Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 13, 686–699. doi: 10.1016/S1474-4422(14)70065-1
Finch, N., Baker, M., Crook, R., Swanson, K., Kuntz, K., Surtees, R., et al. (2009). Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132, 583–591. doi: 10.1093/brain/awn352
Finch, N., Carrasquillo, M. M., Baker, M., Rutherford, N. J., Coppola, G., Dejesus-Hernandez, M., et al. (2011). TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76, 467–474. doi: 10.1212/WNL.0b013e31820a0e3b
Frew, J., Baradaran-Heravi, A., Balgi, A. D., Wu, X., Yan, T. D., Arns, S., et al. (2020). Premature termination codon readthrough upregulates progranulin expression and improves lysosomal function in preclinical models of GRN deficiency. Mol. Neurodegener. 15:21. doi: 10.1186/s13024-020-00369-5
Fukushima, M., Hosoda, N., Chifu, K., and Hoshino, S. I. (2019). TDP-43 accelerates deadenylation of target mRNAs by recruiting Caf1 deadenylase. FEBS Lett. 593, 277–287. doi: 10.1002/1873-3468.13310
Gao, X., Joselin, A. P., Wang, L., Kar, A., Ray, P., Bateman, A., et al. (2010). Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3β. Protein Cell 1, 552–562. doi: 10.1007/s13238-010-0067-1
Gass, J., Lee, W. C., Cook, C., Finch, N., Stetler, C., Jansen-West, K., et al. (2012). Progranulin regulates neuronal outgrowth independent of sortilin. Mol. Neurodegener. 7:33. doi: 10.1186/1750-1326-7-33
Ghidoni, R., Benussi, L., Glionna, M., Franzoni, M., and Binetti, G. (2008). Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71, 1235–1239. doi: 10.1212/01.wnl.0000325058.10218.fc
Gijselinck, I., van der Zee, J., Engelborghs, S., Goossens, D., Peeters, K., Mattheijssens, A., et al. (2008). Progranullin locus deletion in frontotemporal dementia. Hum. Mutat. 29, 53–58. doi: 10.1002/humu.20651
Gonzalez, E. M., Mongiat, M., Slater, S. J., Baffa, R., and Iozzo, R. V. (2003). A novel interaction between perlecan protein core and progranulin: potential effects on tumor growth. J. Biol. Chem. 278, 38113–38116. doi: 10.1074/jbc.C300310200
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Gotzl, J. K., Mori, K., Damme, M., Fellerer, K., Tahirovic, S., Kleinberger, G., et al. (2014). Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 127, 845–860. doi: 10.1007/s00401-014-1262-6
Guha, R., Yue, B., Dong, J., Banerjee, A., and Serrero, G. (2021). Anti-progranulin/GP88 antibody AG01 inhibits triple negative breast cancer cell proliferation and migration. Breast Cancer Res. Treat. 186, 637–653. doi: 10.1007/s10549-021-06120-y
Gumina, V., Onesto, E., Colombrita, C., Maraschi, A., Silani, V., and Ratti, A. (2019). Inter-species differences in regulation of the progranulin-sortilin axis in TDP-43 cell models of neurodegeneration. Int. J. Mol. Sci. 20:5866. doi: 10.3390/ijms20235866
Haynes, B. A., Rhinn, H., Yeh, F., Long, H., Ward, M., Mackenzie, H., et al. (2020). AL001 restores CSF PGRN levels and normalizes disease-associated biomarkers in individuals with frontotemporal dementia due to heterozygous mutations in the progranulin gene. Alzheimers Dement. 16:e046114. doi: 10.1002/alz.046114
He, Z., and Bateman, A. (1999). Progranulin gene expression regulates epithelial cell growth and promotes tumor growth in vivo. Cancer Res. 59, 3222–3229.
He, Z., and Bateman, A. (2003). Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J. Mol. Med. (Berl.) 81, 600–612. doi: 10.1007/s00109-003-0474-3
He, Z., Ong, C. H., Halper, J., and Bateman, A. (2003). Progranulin is a mediator of the wound response. Nat. Med. 9, 225–229. doi: 10.1038/nm816
Herdewyn, S., De Muynck, L., Van Den Bosch, L., Robberecht, W., and Van Damme, P. (2013). Progranulin does not affect motor neuron degeneration in mutant SOD1 mice and rats. Neurobiol. Aging 34, 2302–2303. doi: 10.1016/j.neurobiolaging.2013.03.027
Hinderer, C., Miller, R., Dyer, C., Johansson, J., Bell, P., Buza, E., et al. (2020). Adeno-associated virus serotype 1-based gene therapy for FTD caused by GRN mutations. Ann. Clin. Transl. Neurol. 7, 1843–1853. doi: 10.1002/acn3.51165
Holler, C. J., Taylor, G., Deng, Q., and Kukar, T. (2017). Intracellular proteolysis of progranulin generates stable, lysosomal granulins that are haploinsufficient in patients with frontotemporal dementia caused by GRN mutations. eNeuro 4:ENEURO.0100-17.2017. doi: 10.1523/ENEURO.0100-17.2017
Holler, C. J., Taylor, G., McEachin, Z. T., Deng, Q., Watkins, W. J., Hudson, K., et al. (2016). Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia. Mol. Neurodegener. 11:46. doi: 10.1186/s13024-016-0114-3
Hu, F., Padukkavidana, T., Vægter, C. B., Brady, O. A., Zheng, Y., Mackenzie, I. R., et al. (2010). Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667. doi: 10.1016/j.neuron.2010.09.034
Huin, V., Barbier, M., Bottani, A., Lobrinus, J. A., Clot, F., Lamari, F., et al. (2020). Homozygous GRN mutations: new phenotypes and new insights into pathological and molecular mechanisms. Brain 143, 303–319. doi: 10.1093/brain/awz377
Jian, J., Tian, Q. Y., Hettinghouse, A., Zhao, S., Liu, H., Wei, J., et al. (2016). Progranulin recruits HSP70 to β-glucocerebrosidase and is therapeutic against gaucher disease. EBioMedicine 13, 212–224. doi: 10.1016/j.ebiom.2016.10.010
Jiao, J., Herl, L. D., Farese, R. V., and Gao, F. B. (2010). MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS One 5:e10551. doi: 10.1371/journal.pone.0010551
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., et al. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U S A 108, 4441–4446. doi: 10.1073/pnas.1100650108
Khan, O., and La Thangue, N. B. (2008). Drug insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat. Clin. Pract. Oncol. 5, 714–726. doi: 10.1038/ncponc1238
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Klein, Z. A., Takahashi, H., Ma, M., Stagi, M., Zhou, M., Lam, T. T., et al. (2017). Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 95, 281.e6–296.e6. doi: 10.1016/j.neuron.2017.06.026
Kleinberger, G., Capell, A., Haass, C., and Van Broeckhoven, C. (2013). Mechanisms of granulin deficiency: lessons from cellular and animal models. Mol. Neurobiol. 47, 337–360. doi: 10.1007/s12035-012-8380-8
Kleinberger, G., Wils, H., Ponsaerts, P., Joris, G., Timmermans, J. P., Van Broeckhoven, C., et al. (2010). Increased caspase activation and decreased TDP-43 solubility in progranulin knockout cortical cultures. J. Neurochem. 115, 735–747. doi: 10.1111/j.1471-4159.2010.06961.x
Körtvelyessy, P., Huchtemann, T., Heinze, H. J., and Bittner, D. M. (2017). Progranulin and its related microRNAs after status epilepticus: possible mechanisms of neuroprotection. Int. J. Mol. Sci. 18:490. doi: 10.3390/ijms18030490
Krabbe, G., Minami, S. S., Etchegaray, J. I., Taneja, P., Djukic, B., Davalos, D., et al. (2017). Microglial NFκB-TNFα hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc. Natl. Acad. Sci. U S A 114, 5029–5034. doi: 10.1073/pnas.1700477114
Laird, A. S., Van Hoecke, A., De Muynck, L., Timmers, M., Van den Bosch, L., Van Damme, P., et al. (2010). Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One 5:e13368. doi: 10.1371/journal.pone.0013368
Le Ber, I., Camuzat, A., Hannequin, D., Pasquier, F., Guedj, E., Rovelet-Lecrux, A., et al. (2008). Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 131, 732–746. doi: 10.1093/brain/awn012
Lee, C. W., Stankowski, J. N., Chew, J., Cook, C. N., Lam, Y. W., Almeida, S., et al. (2017). The lysosomal protein cathepsin L is a progranulin protease. Mol. Neurodegener. 12:55. doi: 10.1186/s13024-017-0196-6
Lee, W. C., Almeida, S., Prudencio, M., Caulfield, T. R., Zhang, Y. J., Tay, W. M., et al. (2014). Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum. Mol. Genet. 23, 1467–1478. doi: 10.1093/hmg/ddt534
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Logan, T., DeVos, S., Simon, M. J., Davis, S., Wang, J., Low, R., et al. (2020). A brain penetrant progranulin biotherapeutic rescues lysosomal and inflammatory phenotypes in the brain of GRN knockout mice. Alzheimers Dement. 16:e040602. doi: 10.1002/alz.040602
Lui, H., Zhang, J., Makinson, S. R., Cahill, M. K., Kelley, K. W., Huang, H. Y., et al. (2016). Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 165, 921–935. doi: 10.1016/j.cell.2016.04.001
Mackenzie, I. R. (2007). The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 114, 49–54. doi: 10.1007/s00401-007-0223-8
Mackenzie, I. R., Baker, M., Pickering-Brown, S., Hsiung, G. Y., Lindholm, C., Dwosh, E., et al. (2006). The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129, 3081–3090. doi: 10.1093/brain/awl271
Martens, L. H., Zhang, J., Barmada, S. J., Zhou, P., Kamiya, S., Sun, B., et al. (2012). Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest. 122, 3955–3959. doi: 10.1172/JCI63113
Matsubara, T., Mita, A., Minami, K., Hosooka, T., Kitazawa, S., Takahashi, K., et al. (2012). PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 15, 38–50. doi: 10.1016/j.cmet.2011.12.002
Matsumura, N., Mandai, M., Miyanishi, M., Fukuhara, K., Baba, T., Higuchi, T., et al. (2006). Oncogenic property of acrogranin in human uterine leiomyosarcoma: direct evidence of genetic contribution in in vivo tumorigenesis. Clin. Cancer Res. 12, 1402–1411. doi: 10.1158/1078-0432.CCR-05-2003
Meeter, L. H., Patzke, H., Loewen, G., Dopper, E. G., Pijnenburg, Y. A., van Minkelen, R., et al. (2016). Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement. Geriatr. Cogn. Dis. Extra 6, 330–340. doi: 10.1159/000447738
Minami, S. S., Min, S. W., Krabbe, G., Wang, C., Zhou, Y., Asgarov, R., et al. (2014). Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 20, 1157–1164. doi: 10.1038/nm.3672
Moore, K. M., Nicholas, J., Grossman, M., McMillan, C. T., Irwin, D. J., Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 19, 145–156. doi: 10.1016/S1474-4422(19)30394-1
Moreno-Yruela, C., Fass, D. M., Cheng, C., Herz, J., Olsen, C. A., and Haggarty, S. J. (2019). Kinetic tuning of HDAC inhibitors affords potent inducers of progranulin expression. ACS Chem. Neurosci. 10, 3769–3777. doi: 10.1021/acschemneuro.9b00281
Mukherjee, O., Pastor, P., Cairns, N. J., Chakraverty, S., Kauwe, J. S., Shears, S., et al. (2006). HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann. Neurol. 60, 314–322. doi: 10.1002/ana.20963
Naso, M. F., Tomkowicz, B., Perry, W. L., and Strohl, W. R. (2017). Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 31, 317–334. doi: 10.1007/s40259-017-0234-5
Neill, T., Buraschi, S., Goyal, A., Sharpe, C., Natkanski, E., Schaefer, L., et al. (2016). EphA2 is a functional receptor for the growth factor progranulin. J. Cell Biol. 215, 687–703. doi: 10.1083/jcb.201603079
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nguyen, A. D., Nguyen, T. A., Zhang, J., Devireddy, S., Zhou, P., Karydas, A. M., et al. (2018). Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay. Proc. Natl. Acad. Sci. U S A 115, E2849–E2858. doi: 10.1073/pnas.1722344115
Nicholson, A. M., Finch, N. A., Almeida, M., Perkerson, R. B., van Blitterswijk, M., Wojtas, A., et al. (2016). Prosaposin is a regulator of progranulin levels and oligomerization. Nat. Commun. 7:11992. doi: 10.1038/ncomms11992
Nykjaer, A., Lee, R., Teng, K. K., Jansen, P., Madsen, P., Nielsen, M. S., et al. (2004). Sortilin is essential for proNGF-induced neuronal cell death. Nature 427, 843–848. doi: 10.1038/nature02319
Okura, H., Yamashita, S., Ohama, T., Saga, A., Yamamoto-Kakuta, A., Hamada, Y., et al. (2010). HDL/apolipoprotein A-I binds to macrophage-derived progranulin and suppresses its conversion into proinflammatory granulins. J. Atheroscler. Thromb. 17, 568–577. doi: 10.5551/jat.3921
Olney, N. T., Spina, S., and Miller, B. L. (2017). Frontotemporal dementia. Neurol. Clin. 35, 339–374. doi: 10.1016/j.ncl.2017.01.008
Palfree, R. G., Bennett, H. P., and Bateman, A. (2015). The evolution of the secreted regulatory protein progranulin. PLoS One 10:e0133749. doi: 10.1371/journal.pone.0133749
Palmer, D. N., Barry, L. A., Tyynelä, J., and Cooper, J. D. (2013). NCL disease mechanisms. Biochim. Biophys. Acta 1832, 1882–1893. doi: 10.1016/j.bbadis.2013.05.014
Panman, J. L., Venkatraghavan, V., van der Ende, E. L., Steketee, R. M. E., Jiskoot, L. C., Poos, J. M., et al. (2021). Modelling the cascade of biomarker changes in GRN-related frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 92, 494–501. doi: 10.1136/jnnp-2020-323541
Park, B., Buti, L., Lee, S., Matsuwaki, T., Spooner, E., Brinkmann, M. M., et al. (2011). Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity 34, 505–513. doi: 10.1016/j.immuni.2011.01.018
Paushter, D. H., Du, H., Feng, T., and Hu, F. (2018). The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 136, 1–17. doi: 10.1007/s00401-018-1861-8
Petkau, T. L., Kosior, N., de Asis, K., Connolly, C., and Leavitt, B. R. (2017). Selective depletion of microglial progranulin in mice is not sufficient to cause neuronal ceroid lipofuscinosis or neuroinflammation. J. Neuroinflammation 14:225. doi: 10.1186/s12974-017-1000-9
Petkau, T. L., Neal, S. J., Orban, P. C., MacDonald, J. L., Hill, A. M., Lu, G., et al. (2010). Progranulin expression in the developing and adult murine brain. J. Comp. Neurol. 518, 3931–3947. doi: 10.1002/cne.22430
Petoukhov, E., Fernando, S., Mills, F., Shivji, F., Hunter, D., Krieger, C., et al. (2013). Activity-dependent secretion of progranulin from synapses. J. Cell Sci. 126, 5412–5421. doi: 10.1242/jcs.132076
Philips, T., De Muynck, L., Thu, H. N. T., Weynants, B., Vanacker, P., Dhondt, J., et al. (2010). Microglial upregulation of progranulin as a marker of motor neuron degeneration. J. Neuropathol. Exp. Neurol. 69, 1191–1200. doi: 10.1097/NEN.0b013e3181fc9aea
Plowman, G. D., Green, J. M., Neubauer, M. G., Buckley, S. D., McDonald, V. L., Todaro, G. J., et al. (1992). The epithelin precursor encodes two proteins with opposing activities on epithelial cell growth. J. Biol. Chem. 267, 13073–13078. doi: 10.1016/s0021-9258(18)42382-4
Pottier, C., Ravenscroft, T. A., Sanchez-Contreras, M., and Rademakers, R. (2016). Genetics of FTLD: overview and what else we can expect from genetic studies. J. Neurochem. 138, 32–53. doi: 10.1111/jnc.13622
Pottier, C., Ren, Y., Perkerson, R. B., Baker, M., Jenkins, G. D., van Blitterswijk, M., et al. (2019). Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. 137, 879–899. doi: 10.1007/s00401-019-01962-9
Pottier, C., Zhou, X., Perkerson, R. B., Baker, M., Jenkins, G. D., Serie, D. J., et al. (2018). Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol. 17, 548–558. doi: 10.1016/S1474-4422(18)30126-1
Prudencio, M., Jansen-West, K. R., Lee, W. C., Gendron, T. F., Zhang, Y. J., Xu, Y. F., et al. (2012). Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc. Natl. Acad. Sci. U S A 109, 21510–21515. doi: 10.1073/pnas.1211577110
Rabinovici, G. D., and Miller, B. L. (2010). Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24, 375–398. doi: 10.2165/11533100-000000000-00000
Rademakers, R., Eriksen, J. L., Baker, M., Robinson, T., Ahmed, Z., Lincoln, S. J., et al. (2008). Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum. Mol. Genet. 17, 3631–3642. doi: 10.1093/hmg/ddn257
Rascovsky, K., and Grossman, M. (2013). Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int. Rev. Psychiatry 25, 145–158. doi: 10.3109/09540261.2013.763341
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Sha, S. J., Miller, Z. A., Min, S. W., Zhou, Y., Brown, J., Mitic, L. L., et al. (2017). An 8-week, open-label, dose-finding study of nimodipine for the treatment of progranulin insufficiency from. Alzheimers Dement (N Y) 3, 507–512. doi: 10.1016/j.trci.2017.08.002
Shankaran, S. S., Capell, A., Hruscha, A. T., Fellerer, K., Neumann, M., Schmid, B., et al. (2008). Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J. Biol. Chem. 283, 1744–1753. doi: 10.1074/jbc.M705115200
She, A., Kurtser, I., Reis, S. A., Hennig, K., Lai, J., Lang, A., et al. (2017). Selectivity and kinetic requirements of HDAC inhibitors as progranulin enhancers for treating frontotemporal dementia. Cell Chem. Biol. 24, 892–906.e5. doi: 10.1016/j.chembiol.2017.06.010
Simon-Sanchez, J., Seelaar, H., Bochdanovits, Z., Deeg, D. J., van Swieten, J. C., and Heutink, P. (2009). Variation at GRN 3’-UTR rs5848 is not associated with a risk of frontotemporal lobar degeneration in Dutch population. PLoS One 4:e7494. doi: 10.1371/journal.pone.0007494
Simons, C., Dyment, D., Bent, S. J., Crawford, J., D’Hooghe, M., Kohlschütter, A., et al. (2017). A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain 140, 3105–3111. doi: 10.1093/brain/awx314
Smith, K. R., Damiano, J., Franceschetti, S., Carpenter, S., Canafoglia, L., Morbin, M., et al. (2012). Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 90, 1102–1107. doi: 10.1016/j.ajhg.2012.04.021
Songsrirote, K., Li, Z., Ashford, D., Bateman, A., and Thomas-Oates, J. (2010). Development and application of mass spectrometric methods for the analysis of progranulin N-glycosylation. J. Proteomics 73, 1479–1490. doi: 10.1016/j.jprot.2010.02.013
Suárez-Calvet, M., Capell, A., Araque Caballero, M., Morenas-Rodríguez, E., Fellerer, K., Franzmeier, N., et al. (2018). CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM2, neurodegeneration and cognitive decline. EMBO Mol. Med. 10:e9712. doi: 10.15252/emmm.201809712
Tang, W., Lu, Y., Tian, Q. Y., Zhang, Y., Guo, F. J., Liu, G. Y., et al. (2011). The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484. doi: 10.1126/science.1199214
Tanimoto, R., Lu, K. G., Xu, S. Q., Buraschi, S., Belfiore, A., Iozzo, R. V., et al. (2016). Mechanisms of progranulin action and regulation in genitourinary cancers. Front. Endocrinol. (Lausanne) 7:100. doi: 10.3389/fendo.2016.00100
Thurner, L., Preuss, K. D., Fadle, N., Regitz, E., Klemm, P., Zaks, M., et al. (2013). Progranulin antibodies in autoimmune diseases. J. Autoimmun. 42, 29–38. doi: 10.1016/j.jaut.2012.10.003
Tolkatchev, D., Malik, S., Vinogradova, A., Wang, P., Chen, Z., Xu, P., et al. (2008). Structure dissection of human progranulin identifies well-folded granulin/epithelin modules with unique functional activities. Protein Sci. 17, 711–724. doi: 10.1110/ps.073295308
Tziortzouda, P., Van Den Bosch, L., and Hirth, F. (2021). Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat. Rev. Neurosci. 22, 197–208. doi: 10.1038/s41583-021-00431-1
van Blitterswijk, M., Mullen, B., Wojtas, A., Heckman, M. G., Diehl, N. N., Baker, M. C., et al. (2014). Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol. Neurodegener. 9:38. doi: 10.1186/1750-1326-9-38
Van Damme, P., Tilkin, P., Mercer, K. J., Terryn, J., D’Hondt, A., Herne, N., et al. (2020). Intracerebroventricular delivery of vascular endothelial growth factor in patients with amyotrophic lateral sclerosis, a phase I study. Brain Commun. 2:fcaa160. doi: 10.1093/braincomms/fcaa160
Van Damme, P., Van Hoecke, A., Lambrechts, D., Vanacker, P., Bogaert, E., van Swieten, J., et al. (2008). Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 181, 37–41. doi: 10.1083/jcb.200712039
Van Deerlin, V. M., Sleiman, P. M., Martinez-Lage, M., Chen-Plotkin, A., Wang, L. S., Graff-Radford, N. R., et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239. doi: 10.1038/ng.536
van der Ende, E. L., Meeter, L. H., Poos, J. M., Panman, J. L., Jiskoot, L. C., Dopper, E. G. P., et al. (2019). Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 18, 1103–1111. doi: 10.1016/S1474-4422(19)30354-0
Van Kampen, J. M., Baranowski, D., and Kay, D. G. (2014). Progranulin gene delivery protects dopaminergic neurons in a mouse model of Parkinson’s disease. PLoS One 9:e97032. doi: 10.1371/journal.pone.0097032
Vandenberghe, R. (2016). Classification of the primary progressive aphasias: principles and review of progress since 2011. Alzheimers Res. Ther. 8:16. doi: 10.1186/s13195-016-0185-y
Vass, R., Ashbridge, E., Geser, F., Hu, W. T., Grossman, M., Clay-Falcone, D., et al. (2011). Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 121, 373–380. doi: 10.1007/s00401-010-0782-y
Wang, B. C., Liu, H., Talwar, A., and Jian, J. (2015). New discovery rarely runs smooth: an update on progranulin/TNFR interactions. Protein Cell 6, 792–803. doi: 10.1007/s13238-015-0213-x
Wang, W. X., Wilfred, B. R., Madathil, S. K., Tang, G., Hu, Y., Dimayuga, J., et al. (2010). miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 177, 334–345. doi: 10.2353/ajpath.2010.091202
Ward, M. E., Chen, R., Huang, H. Y., Ludwig, C., Telpoukhovskaia, M., Taubes, A., et al. (2017). Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med. 9:eaah5642. doi: 10.1126/scitranslmed.aah5642
Wei, J. L., Fu, W., Ding, Y. J., Hettinghouse, A., Lendhey, M., Schwarzkopf, R., et al. (2017). Progranulin derivative Atsttrin protects against early osteoarthritis in mouse and rat models. Arthritis Res. Ther. 19:280. doi: 10.1186/s13075-017-1485-8
Werner, G., Damme, M., Schludi, M., Gnorich, J., Wind, K., Fellerer, K., et al. (2020). Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep. 21:e50241. doi: 10.15252/embr.202050241
Wils, H., Kleinberger, G., Pereson, S., Janssens, J., Capell, A., Van Dam, D., et al. (2012). Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J. Pathol. 228, 67–76. doi: 10.1002/path.4043
Xu, J., Xilouri, M., Bruban, J., Shioi, J., Shao, Z., Papazoglou, I., et al. (2011). Extracellular progranulin protects cortical neurons from toxic insults by activating survival signaling. Neurobiol. Aging 32, e2325–e2326. doi: 10.1016/j.neurobiolaging.2011.06.017
Xu, S. Q., Buraschi, S., Morcavallo, A., Genua, M., Shirao, T., Peiper, S. C., et al. (2015). A novel role for drebrin in regulating progranulin bioactivity in bladder cancer. Oncotarget 6, 10825–10839. doi: 10.18632/oncotarget.3424
Yin, F., Banerjee, R., Thomas, B., Zhou, P., Qian, L., Jia, T., et al. (2010). Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med. 207, 117–128. doi: 10.1084/jem.20091568
Youn, B. S., Bang, S. I., Kloting, N., Park, J. W., Lee, N., Oh, J. E., et al. (2009). Serum progranulin concentrations may be associated with macrophage infiltration into omental adipose tissue. Diabetes 58, 627–636. doi: 10.2337/db08-1147
Yu, C. E., Bird, T. D., Bekris, L. M., Montine, T. J., Leverenz, J. B., Steinbart, E., et al. (2010). The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch. Neurol. 67, 161–170. doi: 10.1001/archneurol.2009.328
Zanocco-Marani, T., Bateman, A., Romano, G., Valentinis, B., He, Z. H., and Baserga, R. (1999). Biological activities and signaling pathways of the granulin/epithelin precursor. Cancer Res. 59, 5331–5340.
Zheng, Y., Brady, O. A., Meng, P. S., Mao, Y., and Hu, F. (2011). C-terminus of progranulin interacts with the beta-propeller region of sortilin to regulate progranulin trafficking. PLoS One 6:e21023. doi: 10.1371/journal.pone.0021023
Zhou, B., Li, H., Liu, J., Xu, L., Guo, Q., Sun, H., et al. (2015). Progranulin induces adipose insulin resistance and autophagic imbalance via TNFR1 in mice. J. Mol. Endocrinol. 55, 231–243. doi: 10.1530/JME-15-0075
Zhou, X., Sun, L., Bastos de Oliveira, F., Qi, X., Brown, W. J., Smolka, M. B., et al. (2015). Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J. Cell Biol. 210, 991–1002. doi: 10.1083/jcb.201502029
Zhou, J., Gao, G., Crabb, J. W., and Serrero, G. (1993). Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J. Biol. Chem. 268, 10863–10869. doi: 10.1016/s0021-9258(18)82064-6
Zhou, X., Brooks, M., Jiang, P., Koga, S., Zuberi, A. R., Baker, M. C., et al. (2020). Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep. 21:e50197. doi: 10.15252/embr.202050197
Zhou, X., Kukar, T., and Rademakers, R. (2021). Lysosomal dysfunction and other pathomechanisms in FTLD: evidence from progranulin genetics and biology. Adv. Exp. Med. Biol. 1281, 219–242. doi: 10.1007/978-3-030-51140-1_14
Zhou, X., Paushter, D. H., Feng, T., Sun, L., Reinheckel, T., and Hu, F. (2017a). Lysosomal processing of progranulin. Mol. Neurodegener. 12:62. doi: 10.1186/s13024-017-0205-9
Zhou, X., Sun, L., Brady, O. A., Murphy, K. A., and Hu, F. (2017b). Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol. Commun. 5:9. doi: 10.1186/s40478-017-0412-1
Keywords: progranulin (GRN), frontotemporal dementia (FTD), frontotemporal lobar degeneration (FTLD), amyotrophic lateral sclerosis (ALS), neurodegeneration, gene therapy, therapeutic target
Citation: Terryn J, Verfaillie CM and Van Damme P (2021) Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front. Mol. Neurosci. 14:713031. doi: 10.3389/fnmol.2021.713031
Received: 21 May 2021; Accepted: 07 July 2021;
Published: 23 July 2021.
Edited by:
Sheng Chen, Shanghai Jiao Tong University, ChinaReviewed by:
Xiaolai Zhou, Sun Yat-sen University, ChinaCopyright © 2021 Terryn, Verfaillie and Van Damme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip Van Damme, cGhpbGlwLnZhbmRhbW1lQGt1bGV1dmVuLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.