 Christopher A. Piggott
Christopher A. Piggott Yishi Jin
Yishi Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci. , 08 July 2021
Sec. Neuroplasticity and Development
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.709390
This article is part of the Research Topic Regulation of Synaptic Structure and Function View all 20 articles
Contacts between the endoplasmic reticulum (ER) and plasma membrane (PM) contain specialized tethering proteins that bind both ER and PM membranes. In excitable cells, ER–PM contacts play an important role in calcium signaling and transferring lipids. Junctophilins are a conserved family of ER–PM tethering proteins. They are predominantly expressed in muscles and neurons and known to simultaneously bind both ER- and PM-localized ion channels. Since their discovery two decades ago, functional studies using junctophilin-deficient animals have provided a deep understanding of their roles in muscles and neurons, including excitation-contraction coupling, store-operated calcium entry (SOCE), and afterhyperpolarization (AHP). In this review, we highlight key findings from mouse, fly, and worm that support evolutionary conservation of junctophilins.
Close contact sites between membrane compartments are observed universally in all cell types (Prinz et al., 2020). Junctional Membrane Complexes (JMCs) refer to stable contacts between the endoplasmic reticulum (ER) and plasma membrane (PM) found in excitable cells, particularly muscles and neurons. JMCs are composed of unique proteins that couple PM electrical excitation to ER calcium release. This review focuses on the junctophilin protein family, originally identified from rabbit muscle JMCs. Extensive work has now established that junctophilins are the primary component responsible for the generation of JMCs in skeletal and cardiac muscles. Junctophilins directly bind PM- and ER-localized calcium channels, and enable efficient trans-membrane signaling. Here, we will begin with a historical overview on the discovery of junctophilins, then cover functional studies of mammalian junctophilins in muscles and neurons, and end with emerging evidence supporting evolutionary conservation of junctophilins.
Muscles have specialized structures known as transverse tubules (t-tubules) that are tubular invaginations of the PM. T-tubules extend the PM deep into the muscle cell where they make JMCs with the sarcoplasmic reticulum (SR, muscle equivalent of ER). In skeletal muscle, t-tubules are sandwiched between two SR compartments. Under electron microscopy, the t-tubule and adjacent SR appear as three compartments in a row and hence are called “triads.” In cardiac muscle, t-tubules are adjacent to only one SR compartment at a time and hence these are called “diads.”
Triads and diads are critical for excitation-contraction coupling in muscle contraction. In skeletal muscle, PM depolarization causes the voltage sensing L-type calcium channel (LTCC) to undergo a conformational change. Through direct physical coupling, the LTCCs trigger the opening of calcium-activated calcium channels called ryanodine receptors (RyRs) on the SR surface. RyRs release calcium from SR stores and binding of calcium to actin-myosin filaments enables muscle contraction. In skeletal muscle, LTCCs and RyRs are physically coupled and localize to triads. In cardiac muscle, LTCCs and RyRs localize to diads, but LTCCs are not physically coupled to RyRs. Instead, entry of extracellular calcium through LTCCs activates and opens RyRs, which is called calcium-induced calcium release.
In the late 1990s, the molecular basis of triad and diad formation was unclear. Knocking out the LTCC and RyRs in mouse skeletal muscle reduced the number of triad junctions, but the structure of formed triads was morphologically normal (Franzini-Armstrong et al., 1991; Ikemoto et al., 1997). The SR transmembrane proteins triadin and junctin localize to triads, but hydropathy and topology analysis placed the bulk of both proteins in the SR lumen, making it unlikely that the small cytoplasmic portions could directly interact with the PM (Knudson et al., 1993; Jones et al., 1995). These observations suggested that other, yet undiscovered, molecules were the primary structural component of triad junctions.
To identify proteins involved in triad formation, Takeshima et al. (1998) generated monoclonal antibodies against isolated SR vesicles enriched for junctional membranes from rabbit skeletal muscle. Using these antibodies for immunostaining rabbit skeletal muscle cryosections, they identified an antibody that labeled transverse rows corresponding to the location of triad junctions. Further screening of a protein-expression library using this antibody revealed a novel protein, named junctophilin 1 (JPH1) (Takeshima et al., 2000). Through cross-hybridization with mouse cDNA libraries, two other junctophilins were identified which were named junctophilin 2 (JPH2) and junctophilin 3 (JPH3). A fourth junctophilin, junctophilin 4 (JPH4), was later identified by sequence homology (Nishi et al., 2003). Importantly, electron microscopy studies with immunogold labeling confirmed that JPH1 localizes to JMCs in rabbit skeletal muscle (Takeshima et al., 2000).
mRNA and protein analyses in mouse and human tissue samples revealed that the four mammalian junctophilins have different expression patterns in excitable tissues. Skeletal muscle expresses both JPH1 and JPH2 at similar levels, while JPH2 is the primary isotype in heart and smooth muscle (Nishi et al., 2000; Takeshima et al., 2000; Ito et al., 2001; Minamisawa et al., 2004; Pritchard et al., 2019; Saeki et al., 2019). JPH3 and JPH4 are broadly expressed in neurons of the brain and nervous system (Takeshima et al., 2000; Nishi et al., 2003, 2000). In addition, JPH3 is expressed in pancreatic beta cells (Li et al., 2016) and JPH4 is expressed in T-cells (Woo et al., 2016), both of which are excitable cell types.
Junctophilins are conserved from C. elegans to humans (Figure 1A). Their domain structure supports their role as an ER–PM contact site protein (Garbino et al., 2009). All junctophilins have eight N-terminal MORN (Membrane Occupation and Recognition Nexus) motifs, which are 14 amino acid motifs with the consensus sequence YxGxWxxGKRHGYG (Figure 1A) (Takeshima et al., 2000; Garbino et al., 2009). Expression studies in amphibian embryos using full-length and truncated rabbit JPH1 showed that MORN motifs are required for targeting JPH1 to the PM (Takeshima et al., 2000). Lipid binding assays showed that recombinant versions of JPH1 and JPH2 that lack the transmembrane domain can bind directly to phospholipids, particularly types enriched in the PM, suggesting that junctophilins can bind to the PM through their MORN motifs (Kakizawa et al., 2008; Bennett et al., 2013).
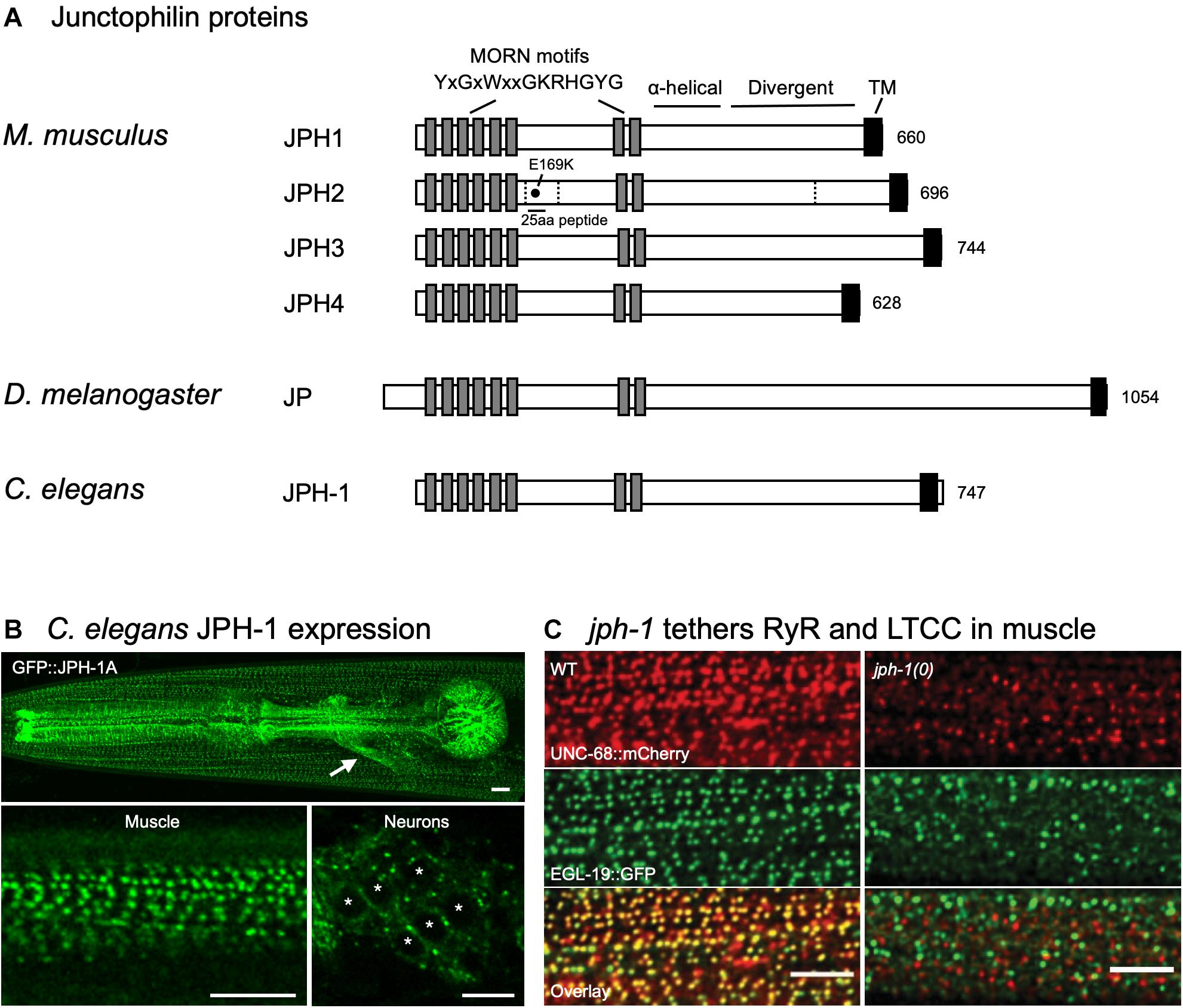
Figure 1. Domain structures of junctophilin proteins and C. elegans junctophilin localization and function. (A) Domain structures of the four Mus musculus (mouse) junctophilins and sole members in Drosophila melanogaster (fly) and Caenorhabditis elegans (worm). Dashed lines indicate JPH2 cleavage sites. E169K mutation blocks binding to RyR and a 25 aa peptide flanking E169 prevents spontaneous RyR channel opening. Gene accession numbers are: M. musculus JPH1 (NP_065629.1), M. musculus JPH2 (NP_001192005.1), M. musculus JPH3 (NP_065630.1), M. musculus JPH4 (NP_796023.2), D. melanogaster Jp (NP_523525.2), and C. elegans JPH-1 (NP_492193.2). (B) Expression of GFP-tagged JPH-1 shows localization to JMCs in C. elegans muscles and neurons. Top: Head of a C. elegans animal showing GFP::JPH-1 in the pharyngeal muscle and body wall muscle. The white arrow indicates GFP::JPH-1 expression in a bundle of neuronal processes known as the nerve ring. Bottom left: In body wall muscle, JPH-1 localizes to rows of puncta, each one a JMC. Bottom right: In neurons, JPH-1 labels JMCs that form at the periphery of the soma. White asterisks mark neuronal nuclei. (C) jph-1 is required for the co-localization of UNC-68/RyR and EGL-19/LTCC in body wall muscle. jph-1(0) is a complete knockout. Scale bars, 5 μm. Panels (B,C) reproduced from Piggott et al. (2021).
Following the MORN motifs is a predicted α-helical domain of approximately 70 amino acids that would provide a flexible linker 10.5 nm long, enough to span the 10–12 nm JMCs (Garbino et al., 2009). The divergent region (Figure 1A) has high conservation when comparing the same isotype in different species – for example, >80% sequence identity when comparing human, mouse, and rat isotypes – but low conservation between JPH isotypes, with <20% sequence identity across isotypes in human, mouse, and rat (Garbino et al., 2009). The divergent region might be important for the isotype-specific functions of junctophilin, such as tissue-specific binding sites (Garbino et al., 2009).
A hydrophobic transmembrane domain resides at the C-terminus of junctophilin and anchors junctophilin in the ER/SR membrane (Takeshima et al., 2000). Collectively, these domains allow junctophilins to simultaneously bind both PM and ER membranes and localize to ER–PM JMCs.
Junctophilins localize to ER–PM membrane contact sites by simultaneously binding both the ER and PM, thereby functioning as membrane tethers. Expression of JPH1 in amphibian embryos generated ER–PM contacts as well as unnatural ER stacks visible by electron microscopy, demonstrating that junctophilins are capable of tethering membranes (Takeshima et al., 2000). JPH2 knockout is embryonic lethal due to cardiac failure (Takeshima et al., 2000). Embryonic cardiomyocytes from JPH2 knockout mice had fewer 12 nm JMCs, suggesting that JPH2 is required for ER–PM coupling (Takeshima et al., 2000). Using an inducible heart-specific JPH2 knockdown to bypass embryonic lethality, van Oort et al. (2011) showed that JPH2 knockdown reduces ER–PM contact site number. Conversely, heart-specific overexpression of JPH2 in mouse increased ER–PM contact site area and also generated convoluted membrane structures visible by electron microscopy (Guo et al., 2014), reminiscent of the ER stacks generated by JPH1 overexpression in amphibian embryos (Takeshima et al., 2000). Taken together, these early studies provide strong evidence that junctophilin acts as an ER–PM tether.
Over the past two decades, extensive studies using knockdown and knockout mice and relevant cell lines have supported critical roles of junctophilins in various types of muscle. Below, we will review functional studies of each junctophilin (summarized in Table 1).
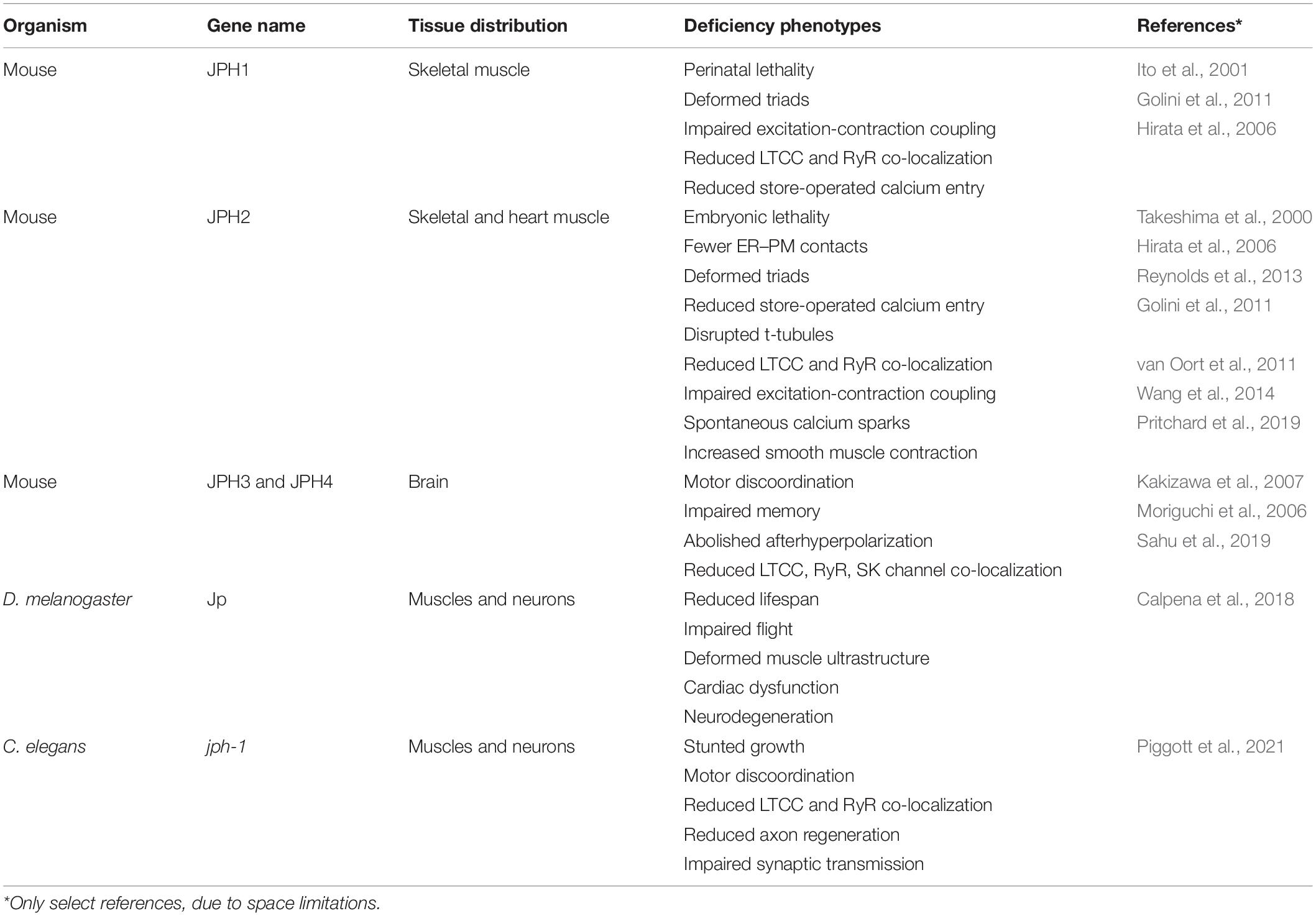
Table 1. Functions of junctophilins from invertebrates to mammals.
JPH1 was first identified as a protein that localizes to triad junctions in rabbit skeletal muscle (Takeshima et al., 2000). Skeletal muscle from newborn JPH1 knockout mice has abnormal SR morphology and fewer triads, consistent with a role for JPH1 in triad formation (Ito et al., 2001). JPH1 knockout mice also have weaker muscle contraction and die within a day of birth due to suckling defects, suggesting that JPH1-mediated triad assembly is critical for muscle function (Ito et al., 2001). In adult mice, dual knockdown of JPH1 and JPH2 disrupts existing triads (Hirata et al., 2006). Therefore, JPH1 and JPH2 are required for both triad development and stabilization. Junctophilin’s ability to directly bind membranes likely contributes to this role. However, interactions with JMC-localized proteins such as the LTCC and RyRs, which will be discussed later in this article, may contribute to both membrane tethering and targeting JPH1 and JPH2 to triads.
Extensive studies have shown that JPH2 is also involved in the development and stabilization of t-tubules. The arrival of JPH2 at the rat cardiomyocyte PM coincides with the start of membrane invagination at P10 (Ziman et al., 2010). Constitutive heart-specific knockdown of JPH2 impedes t-tubule development (Chen et al., 2013; Reynolds et al., 2013). Knockdown of JPH2 in rat myocytes disrupts the organization of existing t-tubules (Wei et al., 2010). In an inducible mouse heart failure model, where JPH2 undergoes proteolytic cleavage, t-tubules are disrupted, suggesting that intact JPH2 is required for t-tubule maintenance (Wu et al., 2014). JPH2 overexpression has the opposite effect on t-tubule organization. Heart-specific JPH2 overexpression in mouse accelerates t-tubule development (Reynolds et al., 2013). In trans-aortic banded mice, which normally exhibit t-tubule disruption, JPH2 overexpression protects t-tubule organization (Guo et al., 2014). Given their ability to simultaneously bind t-tubule and SR membranes, it is conceivable that JPH2 generates and maintains t-tubule organization by tethering t-tubules to the SR.
These findings highlight junctophilin’s role as a structural protein that shapes the muscle ultrastructure. However, as we will discuss next, junctophilins also play a key role in positioning and regulating ion channels to facilitate excitation-contraction coupling.
The co-localization of ER- and PM-localized calcium channels at JMCs is essential for effective excitation-contraction coupling. In skeletal muscle, depolarization of the PM causes LTCCs to trigger the opening of physically linked RyRs and release calcium from SR stores. In heart muscle, PM depolarization opens LTCCs and the resulting calcium influx triggers the opening of nearby RyRs. In both cases, coupling of PM-localized LTCCs and SR-localized RyRs is crucial for effective conversion of PM depolarization to SR calcium release which drives muscle contraction.
Pioneering studies showed that embryonic cardiomyocytes isolated from JPH2 knockout mice exhibited random, unsynchronized calcium transients (Takeshima et al., 2000). Calcium transients occurred even when extracellular calcium was removed, indicating that JPH2 is required for coupling SR calcium release to extracellular calcium entry (Takeshima et al., 2000). In cardiomyocytes and intact hearts from JPH2 knockdown mice, electrical stimulation induced calcium transients that were smaller and irregular compared to wild-type (Chen et al., 2013; Reynolds et al., 2013). This occurred with no change to LTCC and RyR protein levels, suggesting that LTCCs and and/or RyRs were mis-localized or mis-regulated when JPH2 was depleted.
Multiple studies have demonstrated that junctophilins interact with both RyRs and the LTCC. Super-resolution microscopy of rat cardiomyocytes showed that roughly 80% of RyRs and JPH2 co-localize (Jayasinghe et al., 2012). Co-immunoprecipitation studies in rabbit skeletal muscle, mouse heart muscle, and transiently transfected HEK293 cells demonstrated binding between JPH1 or JPH2 and RyRs (Phimister et al., 2007; Golini et al., 2011; van Oort et al., 2011; Beavers et al., 2013). Similarly, in rabbit skeletal muscle binding was seen between JPH1 or JPH2 and the LTCC (Golini et al., 2011). Moreover, cardiomyocytes isolated from heart-specific JPH2 knockdown mice had reduced LTCC and RyR co-localization (van Oort et al., 2011). These cardiomyocytes had normal depolarization-stimulated calcium influx through LTCCs but smaller cytosolic transients. These findings suggest that JPH2 is required for co-localizing LTCCs and RyRs so that extracellular calcium entry through LTCCs can efficiently stimulate RyR-mediated SR calcium release.
Another elegant study addressed the role of direct protein interactions in localizing LTCCs (Nakada et al., 2018). In GLT myotubes, expression of LTCCs with a point mutation that blocked binding to JPH1 and JPH2 caused LTCCs to become diffuse and no longer co-localize with RyR. Fewer myotubes produced calcium transients in response to electric field stimulation, and those that produced transients had smaller amplitudes. Moreover, mis-localizing LTCCs by introducing a truncated JPH1 lacking the transmembrane domain that itself was mis-localized resulted in reduced calcium amplitude and reduced contraction strength in mouse skeletal muscle. Therefore, JPH2 likely mediates excitation-contraction coupling by binding to and localizing LTCCs near RyRs.
Altogether, this substantial body of work, performed in multiple systems, shows that JPH1 and JPH2 directly bind the LTCC and RyRs to facilitate their co-localization and enable efficient excitation-contraction coupling.
A growing body of evidence suggests that junctophilins not only couple ER- and PM-localized calcium channels but also play a role in channel gating. Cardiomyocytes have spontaneous local increases in cytosolic calcium called “calcium sparks” that are caused by opening of RyRs (Cheng et al., 1993). Cardiomyocytes from inducible JPH2 knockdown mouse hearts have larger and more frequent spontaneous calcium sparks with no change in RyR expression level, suggesting that JPH2 is required for keeping RyRs closed (van Oort et al., 2011; Wang et al., 2014). Cardiomyocytes from mice expressing JPH2 with the E169K mutation, a residue required for RyR binding, also have more frequent spontaneous calcium sparks, suggesting that JPH2 directly binds and gates RyRs (Figure 1A) (Beavers et al., 2013). Supporting this, addition of a 25aa peptide flanking E169 to permeabilized cardiomyocytes from JPH2 knockdown mice abolishes spontaneous calcium release (Beavers et al., 2013).
Further evidence that JPH2-binding controls RyR gating comes from in vitro single-channel recordings using microsomes extracted from mouse hearts and reconstituted in planar lipid bilayers. RyRs from inducible JPH2 knockdown mouse hearts have higher open probability than RyRs from wild-type mice, suggesting that JPH2 prevents RyR opening (Wang et al., 2014). The 25aa JPH2-derived peptide flanking E169 reduces RyR opening probability to wild-type levels (Beavers et al., 2013). Conversely, JPH2 knockdown reduced spontaneous calcium spark frequency in HL-1 immortalized cardiomyocytes (Landstrom et al., 2011). This response, which was the opposite of results obtained from cardiomyocytes from JPH2 knockdown mice, may have been different due to the immortalized nature and altered calcium handling of HL-1 cells.
JPH1 and JPH2 dual knockdown in C2C12 myotubes in vitro has been shown to impair calcium influx through LTCC (Nakada et al., 2018). These findings were seen with unchanged membrane expression of LTCC and suggest that junctophilins may gate LTCC, though could also be explained by attenuation of a retrograde signal from RyR (Nakai et al., 1996).
When ER calcium stores are depleted, they can be replenished through a process called store-operated calcium entry (SOCE). ER calcium sensor STIM1 senses the drop in calcium concentration and relocalizes to ER–PM contacts, where it activates PM-localized calcium channel Orai1 to allow the entry of extracellular calcium. Sarco/ER calcium transport ATPase (SERCA) pumps then take up the calcium into the ER (Serwach and Gruszczynska-biegala, 2020).
In vitro and in vivo studies in skeletal muscle have demonstrated roles for junctophilins in SOCE. Knockdown of both JPH1 and JPH2 in cultured myotubes reduced pharmacologically-induced SOCE, measured by the quenching of intracellular Fura-2 by the entry of extracellular Mn2+ through SOCE channels (Hirata et al., 2006; Li et al., 2010). Adenovirus-mediated knockdown of both JPH1 and JPH2 in mouse skeletal muscle also reduced SOCE, indicated by the reduction of calcium in t-tubules (Hirata et al., 2006). This was associated with a slower recovery of voltage-induced calcium release after SR calcium depletion, consistent with impaired SOCE (Hirata et al., 2006). Skeletal muscle from these mice was found to have deformed triads (Hirata et al., 2006). As STIM1 and Orai1 interact at ER–PM contacts, an appealing hypothesis is that JPH1 and JPH2 are required for SOCE in skeletal muscle because STIM1-Orai1 interactions occur at JMCs generated by junctophilins.
Like cardiac and skeletal muscle, smooth muscle contraction is primarily regulated by an increase in cytosolic calcium concentration (Allen and Walsh, 1994). However, the regulation of cytosolic calcium in smooth muscle is a complex process that can be modulated by various signaling pathways (Kuo and Ehrlich, 2015). Similar to cardiac muscle, membrane depolarization triggers calcium-induced calcium release involving the LTCC and RyRs (Sanders, 1985). Alternatively, extracellular ligands bind to PM-localized receptors which generate 1,4,5-trisphosphate (IP3). IP3 diffuses across the cytosol and activates SR-localized calcium channels known as IP3 receptors, which release SR calcium (Sanders, 1985). Both calcium-induced and IP3-induced calcium release would appear to benefit from close coupling of the ER and PM. Indeed, LTCCs and RyRs co-localize at JMCs in smooth muscle (Moore et al., 2004). However, LTCCs and RyRs in smooth muscle exhibit “loose” coupling, wherein LTCC opening does not necessarily trigger RyR activation, and RyRs can open spontaneously to generate calcium sparks (Collier et al., 2000). Furthermore, unlike cardiac and skeletal muscle, smooth muscle lacks t-tubules to enhance ER–PM contact area. Therefore, JMCs, and by extension, junctophilins, may not be as critical for excitation-contraction coupling in smooth muscle as in cardiac or skeletal muscle.
Studies on junctophilins in smooth muscle have focused on coupling between RyRs and large conductance calcium-activated potassium channels, commonly called BK channels (short for “big potassium”). Calcium sparks from spontaneous opening of RyRs activate nearby BK channels and generate an outward potassium current (Nelson et al., 1995). This hyperpolarizes the PM, suppressing depolarization-dependent calcium influx and relaxing the muscle (Bolton and Imaizumi, 1996). JPH2 is the most abundant junctophilin isotype in vascular smooth muscle (Pritchard et al., 2019; Saeki et al., 2019). JPH2 localization overlaps with RyR and BK channels in vascular smooth muscle, where it directly binds BK, suggesting JPH2 couples RyRs and BK channels (Pritchard et al., 2019; Saeki et al., 2019). Calcium sparks, visualized using TIRF microscopy and the fluorescent calcium indicator Fluo-4 AM, occur near JPH2 clusters (Saeki et al., 2019). The outward current activated by spontaneous calcium sparks is reduced when JPH2 is knocked down, consistent with a role for JPH2 in coupling RyRs and BK channels (Pritchard et al., 2019; Saeki et al., 2019). Further supporting this, JPH2 knockdown causes increased vascular smooth muscle contraction (Pritchard et al., 2019; Saeki et al., 2019). Therefore, JPH2 plays an important role in maintaining vascular smooth muscle resting tone by coupling RyRs to BK channels. As JPH2 expression was detected in mouse stomach and lung (Takeshima et al., 2000), which contain smooth muscle, further study will be required to determine if this function of JPH2 is conserved across smooth muscle in different tissues.
Excessive muscle contraction and heart stress can cause prolonged elevation of cytosolic calcium (Gissel, 2000; Stary and Hogan, 2000; Dhalla et al., 2008). Such increases in calcium have been shown to disrupt excitation-contraction coupling in muscle fibers without altering LTCC or RyR levels (Lamb et al., 1995). Evoking high calcium levels was found to cause proteolysis of JPH1 and JPH2 in mouse skeletal muscle, raising the possibility that disrupted excitation-contraction coupling was due to junctophilin degradation (Murphy et al., 2013). In support of this, exposure of rat muscle fiber to high calcium degraded JPH1 and reduced the muscle’s contractile force (Murphy et al., 2013).
Junctophilin cleavage was found to be dependent on the calcium-activated protease calpain, as administering a calpain inhibitor to an inducible mouse model of heart failure blocked JPH2 cleavage and the associated t-tubule disruption and abnormal calcium handling (Wu et al., 2014). Subsequent studies revealed three putative calpain cleavage sites in JPH2 (Figure 1A) (Guo et al., 2015). Moreover, an N-terminal cleavage product of JPH2 (JPH2-NT) produced by heart stress is directed to the nucleus where it directly binds DNA (Guo et al., 2018). Microarray analysis of cultured cardiomyocytes overexpressing JPH2-NT demonstrated that JPH2-NT could alter transcription, likely by competing with transcription factors for DNA binding. Guo et al. further showed that JPH2-NT overexpression protected mouse hearts against stress, and in the converse experiment, preventing nuclear accumulation (by deleting the NLS in endogenous JPH2) exacerbated symptoms of heart stress.
Disrupted excitation-contraction coupling is considered to be a common step in the progression of heart failure (Ibrahim et al., 2011). It can be argued that reducing excitation-contraction coupling by cleaving JPH2 provides a temporary solution to excessive cardiac muscle contraction, which can lead to hypertrophy and eventually heart failure (Hunter and Chien, 1999). The cleavage product JPH2-NT has an additional protective role in inducing transcriptional reprogramming and attenuating the progression of heart failure (Guo et al., 2018). Terminating heart failure-inducing signals in a mouse model results in normalization of JPH2 levels, improvements in excitation-contraction coupling, and reversal of heart failure (Wu et al., 2014). Therefore, while JPH2 cleavage may be harmful under conditions of prolonged heart stress, it may be a beneficial response to short-term stress. Additional studies in other models will be required to test this model.
JPH3 and JPH4 are broadly expressed in neurons of the brain and nervous system (Takeshima et al., 2000; Nishi et al., 2003, 2000). A growing body of evidence suggests that JPH3 and JPH4 have overlapping roles mediating learning and motor control through the regulation of intracellular calcium signaling in neurons. JPH3 or JPH4 knockout mice show slight impairment of motor coordination, which progresses with aging in the case of JPH3 knockout (Nishi et al., 2002; Kakizawa et al., 2007; Seixas et al., 2012). In contrast, JPH3 and JPH4 double knockout (JPH DKO) mice die 3-4 weeks after birth, though this can be prevented by switching their food from dry pellets to a wet paste, suggesting lethality is due to defects in the circuitry controlling saliva secretion (Moriguchi et al., 2006). JPH DKO causes severe defects in motor coordination, learning, and memory, suggesting that JPH3 and JPH4 have important overlapping roles in the brain (Moriguchi et al., 2006; Kakizawa et al., 2007).
The molecular basis for the neurological defects in JPH DKO may lie in the production of afterhyperpolarization (AHP) currents. The depolarization and repolarization phases of an action potential are followed by AHP, where the neuron’s membrane potential falls below the normal resting potential, and is one factor determining action potential frequency (Andrade et al., 2012). Slice recordings in hippocampal CA1 neurons obtained from JPH DKO mice showed that AHP is absent (Moriguchi et al., 2006). Inhibitors of NMDA receptor cation channels, RyR channels, and small conductance calcium-activated potassium (SK) channels abolished AHP in wild-type neurons but had no additional effect on currents in JPH DKO neurons, leading the authors to propose that neuronal junctophilins are required for the production of AHP currents by coupling NMDA receptors, RyRs, and SK channels (Moriguchi et al., 2006). Slice recordings in cerebellar Purkinje cells obtained from JPH DKO mice or treated with channel inhibitors showed similar effects on AHP (Kakizawa et al., 2007). Moreover, addition of the SK channel enhancer EBIO restored AHP in Purkinje cells from JPH DKO animals, suggesting that SK channels are functional in JPH DKO but require junctophilin-mediated coupling to RyR for activation (Kakizawa et al., 2007).
Unlike in muscle, neuronal junctophilin knockout caused no disruption to ER–PM membrane contact sites detectable by electron microscopy (Nishi et al., 2002; Moriguchi et al., 2006; Kakizawa et al., 2007). Therefore, it appears that junctophilin’s main role in neurons is not to tether membranes but facilitate channel localization. Indeed, in cultured hippocampal CA1 neurons, studies using super-resolution microscopy showed that JPH3 and JPH4 are required to maintain the co-assembly of LTCCs, RyRs, and SK channels, and that disruption of this co-assembly leads to impaired AHP and more frequent action potentials (Sahu et al., 2019). These results suggest a model where junctophilins couple PM-localized cation channels (e.g., NMDA receptor, CaV1.3), ER-localized RyRs, and PM-localized SK channels for intracellular communication to link membrane depolarization to AHP current generation and ultimately control action potential frequency.
It is currently unclear how altered AHP might manifest as motor coordination and learning defects. Recordings from Purkinje cell and hippocampal CA1 neurons have shown that JPH DKO causes defects in long term potentiaton and long term depression (Moriguchi et al., 2006; Kakizawa et al., 2007). Future work will be required to determine the mechanism causing behavioral defects in JPH DKO animals.
Shortly after the discovery of junctophilins, a CAG/CTG repeat expansion in an alternatively spliced exon of JPH3 was found to cause Huntington disease-like 2 (HDL2), a disease clinically indistinguishable from Huntington’s disease (Holmes et al., 2001). Based on studies in cultured cells and transgenic mice, three disease mechanisms have been proposed: (1) sequestration of JPH3 mRNA carrying the expanded repeat and subsequent loss of function (Seixas et al., 2012), (2) toxic gain of function by JPH3 mRNA carrying the expanded repeat (Rudnicki et al., 2007), and (3) toxic gain of function peptides translated from either the sense or antisense strand carrying the expanded repeat (Wilburn et al., 2011). Additional studies will be required to determine if HDL2 pathogenesis involves the loss of endogenous JPH3 function, is caused by CUG repeats which merely happen to be located at the JPH3 locus, or a combination of both.
Recent studies from the invertebrates C. elegans and D. melanogaster have demonstrated that the functions and mechanisms of junctophilins are highly conserved. Invertebrates have only one junctophilin, facilitating the study of junctophilin without concerns of redundancy (Garbino et al., 2009) (Figure 1A).
In D. melanogaster, knockdown or overexpression of the sole junctophilin caused structural defects in skeletal and cardiac muscle structure which were accompanied by functional deficits (Calpena et al., 2018). However, the localization of fly junctophilin is not yet reported; therefore, its relationship to ER–PM calcium channels remains unaddressed. Interestingly, junctophilin overexpression appears to be protective against neuronal degeneration caused by expression of human huntingtin exon 1 carrying expanded polyglutamine repeats (Calpena et al., 2018). Genetic interaction experiments suggest that this role of junctophilin may involve the Notch signaling pathway.
In C. elegans, expression of GFP-tagged JPH-1 under the control of the jph-1 promoter showed that jph-1 is expressed in all muscles, and likely most neurons (Figure 1B) (Piggott et al., 2021). In muscles, JPH-1 localizes to longitudinal rows of puncta that match the pattern of repeating sarcomere units. These JPH-1 puncta co-localize with both the ER-localized UNC-68/RyR and PM-localized LTCC subunit EGL-19, implying that JPH-1 localizes to ER–PM contact sites. In neurons, JPH-1 co-localizes with the ER–PM contact site protein Extended-SYnaptoTagmin 2 (ESYT-2). Together, these data demonstrate that C. elegans junctophilin is expressed in excitable tissues and localizes to ER–PM contact sites, a feature that is conserved from C. elegans to mammals.
jph-1 knockout animals are viable, allowing the study of junctophilin function in whole animals (Piggott et al., 2021). jph-1 knockout animals show stunted growth and slow and uncoordinated movement. The stunted growth is likely due to reduced nutrient intake caused by weak contraction of the pharyngeal muscle, the organ that draws in and crushes bacteria for eating, as expression of JPH-1 in the pharyngeal muscle restored both muscle contraction and animal growth. The impaired movement is likely due to defective body wall muscle contraction, as expression of JPH-1 in body wall muscle rescued movement. Consistent with a role of jph-1 in coupling ER- and PM-localized calcium channels, jph-1 knockout abolishes co-localization between LTCC subunit EGL-19 and UNC-68/RyR in muscles (Figure 1C). Interestingly, precise subcellular localization of JPH-1 in both muscle and neurons depends on unc-68/RyR. It was reported that in rat cardiomyocytes, RyR localization to muscle triads precedes JPH2 arrival, suggesting that the targeting of junctophilins by RyRs may be conserved (Ziman et al., 2010). Mammalian JPH1 and JPH2 directly bind to RyR (Phimister et al., 2007; van Oort et al., 2011). Thus, it is possible that junctophilin targeting may involve directly binding to RyR already localized at MCSs.
Studies from C. elegans have also opened new directions for junctophilin research, particularly in neurons. jph-1 knockout animals were found to have reduced axon regrowth after injury (Piggott et al., 2021). Regrowth was rescued by expression of JPH-1 in pharyngeal muscle, raising the intriguing possibility that gut nutrients may impact neuronal injury response. In cholinergic neurons, JPH-1 surrounds synaptic release sites labeled by the vesicular acetylcholine transporter UNC-17. Pharmacological assays showed that jph-1 is required for synaptic transmission at the neuromuscular junction, a previously undescribed role for junctophilin. The ER–PM contact site protein ESYT-2 was also found to be required for synaptic transmission, echoing findings from D. melanogaster (Kikuma et al., 2017). Unexpectedly, mutating both jph-1 and esyt-2 restored wild-type synaptic transmission, in a display of mutual suppression (Piggott et al., 2021). While the underlying mechanism remains to be addressed, jph-1 and esyt-2 appear to have antagonistic roles in neuromuscular synaptic transmission. These observations hint at a delicate balance of different classes of tethering molecules at ER–PM junctions.
Gene duplication events provide the opportunity for isotypes to evolve specialized roles (Ohno, 1970). As vertebrates have four junctophilins and invertebrates have one, the ancestral function of junctophilin is likely closer to invertebrate junctophilins. Studies in C. elegans have shown that junctophilins likely have a conserved role in coupling ER- and PM-localized calcium channels, suggesting this is an ancestral role. Although a role in ER–PM tethering has yet to be experimentally demonstrated in fly or C. elegans, the high conservation of the overall domain structure and MORN motifs across species suggests that ER–PM tethering may also be an ancestral role.
Since the discovery of junctophilins two decades ago, great progress has been made toward revealing their cell biology and functional importance. Compared to membrane contact sites, junctophilins are unique in that each isotype displays critical roles in a cell-type specific manner. JPH1 and JPH2 are expressed in muscle and contribute to muscle structure by generating JMCs, triads/diads, and t-tubules. JPH1 and JPH2 bind to LTCCs and RyRs to facilitate their co-localization and enable efficient excitation-contraction coupling. In addition to these well-established roles, there is evidence that JPH1 and JPH2 regulate SOCE in skeletal muscle, JPH2 controls gating of RyRs and acts as a transcriptional regulator in heart muscle, and JPH2 couples BK and RyR channels in smooth muscle. JPH3 and JPH4 are required in neurons for the generation of AHP currents and have roles in motor coordination and learning.
Emerging findings have begun to show additional roles for JPH3 and JPH4 in other cell types. JPH3 is required for glucose-stimulated insulin release in pancreatic beta cells (Li et al., 2016). JPH4 is required for SOCE in T-cells and dorsal root ganglia, showing that a role in SOCE is conserved in at least three out of four junctophilin isotypes (Woo et al., 2016; Hogea et al., 2021). JPH4 was found to facilitate SOCE by recruiting STIM1 and Orai1 to ER–PM contacts, providing a possible mechanism for how JPH1 and JPH2 might regulate SOCE in skeletal muscle (Woo et al., 2016; Hogea et al., 2021). In vitro studies demonstrated that JPH3 and JPH4 interact with Cav2.1 P/Q-type calcium channels and Cav2.2 N-type calcium channels and modify their inactivation rates (Perni and Beam, 2021). This study also found that JPH3 and JPH4 differentially interact with RyR isotypes, which may explain why the brain expresses two different junctophilin isotypes.
The roles of junctophilin in neurons are among the biggest open questions that remain to be addressed. More specifically: (1) Does impaired AHP, whichis observed in neurons obtained from JP3 and JPH4 knockout mice, cause the motor coordination and learning defects found in JPH3 and JPH4 knockout animals? And if so, what is the mechanism? (2) While JPH3 and JPH4 are broadly expressed in the nervous system, they have only been studied in cerebellar Purkinje cells, hippocampal CA1 neurons, and certain sensory neurons. Given that different neurons have different ion channel expression patterns, what are the functions of JPH3 and JPH4 in unexamined neurons? (3) Studies in C. elegans demonstrated that junctophilin and extended-synaptotagmin have antagonistic roles in synaptic transmission. The mechanism behind their roles in synaptic transmission and genetic interaction remain to be investigated. (4) What is the disease mechanism responsible for HDL2 in patients with JPH3 trinucleotide repeats? Addressing these questions will not only further our understanding of junctophilins, but also how calcium signaling in neurons can regulate neuronal function.
CP wrote the original draft, along with inputs and edits from YJ. Both authors contributed to the article and approved the submitted version.
This work in our lab was supported by grants from NIH (NS R37 035546 and NS R01 093588).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank our lab members for their valuable input throughout our work on characterizing C. elegans JPH-1, which stimulated our ideas for this review.
Allen, B. G., and Walsh, M. P. (1994). The biochemical basis of the regulation of smooth-muscle contraction. Trends Biochem. Sci. 19, 362–368. doi: 10.1016/0968-x0004(94)90112-0
Andrade, R., Foehring, R. C., and Tzingounis, A. V. (2012). The calcium-activated slow AHP: cutting through the Gordian Knot. Front. Cell. Neurosci. 6:47. doi: 10.3389/fncel.2012.00047
Beavers, D. L., Wang, W., Ather, S., Voigt, N., Garbino, A., Dixit, S. S., et al. (2013). Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J. Am. Coll. Cardiol. 62, 2010–2019. doi: 10.1016/j.jacc.2013.06.052
Bennett, H. J., Davenport, J. B., Collins, R. F., Trafford, A. W., Pinali, C., and Kitmitto, A. (2013). Human junctophilin-2 undergoes a structural rearrangement upon binding PtdIns(3,4,5)P3 and the S101R mutation identified in hypertrophic cardiomyopathy obviates this response. Biochem. J. 456, 205–217. doi: 10.1042/BJ20130591
Bolton, T. B., and Imaizumi, Y. (1996). Spontaneous transient outward currents in smooth muscle cells. Cell Calcium 20, 141–152. doi: 10.1016/S0143-4160(96)90103-7
Calpena, E., Del Amo, V. L., Chakraborty, M., Llamusi, B., Artero, R., Espinós, C., et al. (2018). The Drosophila junctophilin gene is functionally equivalent to its four mammalian counterparts and is a modifier of a Huntingtin poly-Q expansion and the Notch pathway. DMM Dis. Model. Mech. 11:dmm029082. doi: 10.1242/dmm.029082
Chen, B., Guo, A., Zhang, C., Chen, R., Zhu, Y., Hong, J., et al. (2013). Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc. Res. 100, 54–62. doi: 10.1093/cvr/cvt180
Cheng, H., Lederer, W., and Cannell, M. (1993). Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science (80-) 262, 740–744. doi: 10.1126/science.8235594
Collier, M. L., Ji, G., Wang, Y. X., and Kotlikoff, M. I. (2000). Calcium-induced calcium release in smooth muscle: loose coupling between the action potential and calcium release. J. Gen. Physiol. 115, 653–662. doi: 10.1085/jgp.115.5.653
Dhalla, N. S., Saini-Chohan, H. K., and Duhamel, T. A. (2008). Strategies for the regulation of intracellular calcium in ischemic heart disease. Future Cardiol. 4, 339–345. doi: 10.2217/14796678.4.4.339
Franzini-Armstrong, C., Pincon-Raymond, M., and Rieger, F. (1991). Muscle fibers from dysgenic mouse in vivo lack a surface component of peripheral couplings. Dev. Biol. 146, 364–376. doi: 10.1016/0012-1606(91)90238-X
Garbino, A., van Oort, R. J., Dixit, S. S., Landstrom, A. P., Ackerman, M. J., and Wehrens, X. H. T. (2009). Molecular evolution of the junctophilin gene family. Physiol. Genomics 37, 175–186. doi: 10.1152/physiolgenomics.00017.2009
Gissel, H. (2000). Ca2+ accumulation and cell damage in skeletal muscle during low frequency stimulation. Eur. J. Appl. Physiol. 83, 175–180. doi: 10.1007/s004210000276
Golini, L., Chouabe, C., Berthier, C., Cusimano, V., Fornaro, M., Bonvallet, R., et al. (2011). Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J. Biol. Chem. 286, 43717–43725. doi: 10.1074/jbc.M111.292755
Guo, A., Hall, D., Zhang, C., Peng, T., Miller, J. D., Kutschke, W., et al. (2015). Molecular determinants of calpain-dependent cleavage of junctophilin-2 protein in cardiomyocytes. J. Biol. Chem. 290, 17946–17955. doi: 10.1074/jbc.M115.652396
Guo, A., Wang Yihui, Chen, B., Wang Yunhao, Yuan, J., Zhang, L., et al. (2018). E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science (80-) 362, 1–14. doi: 10.1126/science.aan3303
Guo, A., Zhang, X., Iyer, V. R., Chen, B., Zhang, C., Kutschke, W. J., et al. (2014). Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc. Natl. Acad. Sci. U.S.A. 111, 12240–12245. doi: 10.1073/pnas.1412729111
Hirata, Y., Brotto, M., Weisleder, N., Chu, Y., Lin, P., Zhao, X., et al. (2006). Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys. J. 90, 4418–4427. doi: 10.1529/biophysj.105.076570
Hogea, A., Shah, S., Jones, F., Carver, C. M., Hao, H., Liang, C., et al. (2021). Junctophilin-4 facilitates inflammatory signalling at plasma membrane-endoplasmic reticulum junctions in sensory neurons. J. Physiol. 599, 2103–2123. doi: 10.1113/JP281331
Holmes, S. E., O’hearn, E., Rosenblatt, A., Callahan, C., Hwang, H. S., Ingersoll-Ashworth, R. G., et al. (2001). A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat. Genet. 29, 377–378. doi: 10.1038/ng760
Hunter, J. J., and Chien, K. R. (1999). Signaling pathways for cardiac hypertrophy and failure. N. Engl. J. Med. 344, 1276–1283. doi: 10.1056/NEJM199910213411706
Ibrahim, M., Gorelik, J., Yacoub, M. H., and Terracciano, C. M. (2011). The structure and function of cardiac t-tubules in health and disease. Proc. R. Soc. B 278, 2714–2723. doi: 10.1098/rspb.2011.0624
Ikemoto, T., Komazaki, S., Takeshima, H., Nishi, M., Noda, T., Iino, M., et al. (1997). Functional and morphological features of skeletal muscle from mutant mice lacking both type 1 and type 3 ryanodine receptors. J. Physiol. 501, 305–312. doi: 10.1111/j.1469-7793.1997.305bn.x
Ito, K., Komazaki, S., Sasamoto, K., Yoshida, M., Nishi, M., Kitamura, K., et al. (2001). Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J. Cell Biol. 154, 1059–1067. doi: 10.1083/jcb.200105040
Jayasinghe, I. D., Baddeley, D., Kong, C. H. T., Wehrens, X. H. T., Cannell, M. B., and Soeller, C. (2012). Nanoscale organization of junctophilin-2 and ryanodine receptors within peripheral couplings of rat ventricular cardiomyocytes. Biophys. J. 102, L19–L21. doi: 10.1016/j.bpj.2012.01.034
Jones, L. R., Zhang, L., Sanborn, K., Jorgensen, A. O., and Kelley, J. (1995). Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J. Biol. Chem 270, 30787–30796. doi: 10.1074/jbc.270.51.30787
Kakizawa, S., Kishimoto, Y., Hashimoto, K., Miyazaki, T., Furutani, K., Shimizu, H., et al. (2007). Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J. 26, 1924–1933. doi: 10.1038/sj.emboj.7601639
Kakizawa, S., Moriguchi, S., Ikeda, A., Iino, M., and Takeshima, H. (2008). Functional crosstalk between cell-surface and intracellular channels mediated by junctophilins essential for neuronal functions. Cerebellum 7, 385–391. doi: 10.1007/s12311-008-0040-1
Kikuma, K., Li, X., Kim, D., Sutter, D., and Dickman, D. K. (2017). Extended synaptotagmin localizes to presynaptic ER and promotes neurotransmission and synaptic growth in drosophila. Genetics 207, 993–1006. doi: 10.1534/genetics.117.300261
Knudson, C. M., Stang, K. K., Moomaw, C. R., Slaughter, C. A., and Campbell, K. P. (1993). Primary structure and topological analysis of a skeletal muscle-specific junctional sarcoplasmic reticulum glycoprotein (triadin). J. Biol. Chem. 268, 12646–12654. doi: 10.1016/s0021-9258(18)31437-6
Kuo, I. Y., and Ehrlich, B. E. (2015). Signaling in muscle contraction. Cold Spring Harb Perspect. Biol. 7, 1–14. doi: 10.1101/cshperspect.a006023
Lamb, G. D., Junankar, P. R., and Stephenson, D. G. (1995). Raised intracellular [Ca2+] abolishes excitation-contraction coupling in skeletal muscle fibres of rat and toad. J. Physiol. 489, 349–362. doi: 10.1113/jphysiol.1995.sp021056
Landstrom, A. P., Kellen, C. A., Dixit, S. S., van Oort, R. J., Garbino, A., Weisleder, N., et al. (2011). Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ. Hear Fail. 4, 214–223. doi: 10.1161/CIRCHEARTFAILURE.110.958694
Li, H., Ding, X., Lopez, J. R., Takeshima, H., Ma, J., Allen, P. D., et al. (2010). Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J. Biol. Chem. 285, 39171–39179. doi: 10.1074/jbc.M110.149690
Li, L., Pan, Z. F., Huang, X., Wu, B. W., Li, T., Kang, M. X., et al. (2016). Junctophilin 3 expresses in pancreatic beta cells and is required for glucose-stimulated insulin secretion. Cell Death. Dis. 7, 1–10. doi: 10.1038/cddis.2016.179
Minamisawa, S., Oshikawa, J., Takeshima, H., Hoshijima, M., Wang, Y., Chien, K. R., et al. (2004). Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem. Biophys. Res. Commun. 325, 852–856. doi: 10.1016/j.bbrc.2004.10.107
Moore, Voigt, T., Kobayashi, Y. M., Isenberg, G., Fay, F. S., Gallitelli, M. F., et al. (2004). Organization of Ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys. J. 87, 1836–1847. doi: 10.1529/biophysj.104.044123
Moriguchi, S., Nishi, M., Komazaki, S., Sakagami, H., Miyazaki, T., Masumiya, H., et al. (2006). Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc. Natl. Acad. Sci. U.S.A. 103, 10811–10816. doi: 10.1073/pnas.0509863103
Murphy, R. M., Dutka, T. L., Horvath, D., Bell, J. R., Delbridge, L. M., and Lamb, G. D. (2013). Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J. Physiol. 591, 719–729. doi: 10.1113/jphysiol.2012.243279
Nakada, T., Kashihara, T., Komatsu, M., Kojima, K., Takeshita, T., and Yamada, M. (2018). Physical interaction of junctophilin and the CaV1.1 C terminus is crucial for skeletal muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 115, 4507–4512. doi: 10.1073/pnas.1716649115
Nakai, J., Dirksent, R. T., Nguyen, H. T., Pessahll, I. N., Beamt, K. G., and Allen, P. D. (1996). Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380, 72–75. doi: 10.1038/380072a0
Nelson, M. T., Cheng, H., Rubart, M., Santana, L. F., Bonev, A. D., Knot, H. J., et al. (1995). Relaxation of arterial smooth muscle by calcium sparks. Science (80-) 270, 633–637. doi: 10.1126/science.270.5236.633
Nishi, M., Mizushima, A., Nakagawara, Ki, and Takeshima, H. (2000). Characterization of human junctophilin subtype genes. Biochem. Biophys. Res. Commun. 273, 920–927. doi: 10.1006/bbrc.2000.3011
Nishi, M., Sakagami, H., Komazaki, S., Kondo, H., and Takeshima, H. (2003). Coexpression of junctophilin type 3 and type 4 in brain. Mol. Brain Res. 118, 102–110. doi: 10.1016/S0169-328X(03)00341-3
Nishi, M., Takeshima, H., Hashimoto, K., Kano, M., Hashimoto, K., Kano, M., et al. (2002). Motor discoordination in mutant mice lacking junctophilin type 3. Biochem. Biophys. Res. Commun. 292, 318–324. doi: 10.1006/bbrc.2002.6649
Perni, S., and Beam, K. G. (2021). Neuronal junctophilins recruit specific Cav and RyR isoforms to ER-PM junctions and functionally alter Cav2.1 and Cav2.2. Elife 10, 1–31. doi: 10.7554/eLife.64249
Phimister, A. J., Lango, J., Eun, H. L., Ernst-Russell, M. A., Takeshima, H., Jianjie, M., et al. (2007). Conformation-dependent stability of junctophilin 1 (JP1) and Ryanodine Receptor Type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J. Biol. Chem. 282, 8667–8677. doi: 10.1074/jbc.M609936200
Piggott, C. A., Wu, Z., Nurrish, S., Xu, S., Kaplan, J. M., Chisholm, A. D., et al. (2021). Caenorhabditis elegans Junctophilin has tissue-specific functions and regulates neurotransmission with extended-synaptotagmin. Genetics 19:iyab063. doi: 10.1093/genetics/iyab063
Prinz, W. A., Toulmay, A., and Balla, T. (2020). The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 21, 7–24. doi: 10.1038/s41580-019-0180-9
Pritchard, H. A. T., Griffin, C. S., Yamasaki, E., Thakore, P., Lane, C., Greenstein, A. S., et al. (2019). Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc. Natl. Acad. Sci. U.S.A. 116, 21874–21881. doi: 10.1073/pnas.1911304116
Reynolds, J. O., Chiang, D. Y., Wang, W., Beavers, D. L., Dixit, S. S., Skapura, D. G., et al. (2013). Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc. Res. 100, 44–53. doi: 10.1093/cvr/cvt133
Rudnicki, D. D., Holmes, S. E., Lin, M. W., Thornton, C. A., Ross, C. A., and Margolis, R. L. (2007). Huntington’s disease-like 2 is associated with CUG repeat-containing RNA foci. Ann. Neurol. 61, 272–282. doi: 10.1002/ana.21081
Saeki, T., Suzuki, Y., Yamamura, H., Takeshima, H., and Imaizumi, Y. (2019). A junctophilin-caveolin interaction enables efficient coupling between ryanodine receptors and BKCa channels in the Ca2+ microdomain of vascular smooth muscle. J. Biol. Chem. 294, 13093–13105. doi: 10.1074/jbc.RA119.008342
Sahu, G., Wazen, R. M., Colarusso, P., Chen, S. R. W., Zamponi, G. W., and Turner, R. W. (2019). Junctophilin Proteins Tether a Cav1-RyR2-KCa3.1 Tripartite Complex to Regulate Neuronal Excitability. Cell Rep. 28, 2427.e–2442.e. doi: 10.1016/j.celrep.2019.07.075
Sanders, K. M. (1985). Invited review: mechanisms of calcium handling in smooth muscles. J. Appl. Physiol. 91, 1438–1449. doi: 10.1152/jappl.2001.91.3.1438
Seixas, A. I., Holmes, S. E., Takeshima, H., Pavlovich, A., Sachs, N., Pruitt, J. L., et al. (2012). Loss of junctophilin-3 contributes to huntington disease-like 2 pathogenesis. Ann. Neurol. 71, 245–257. doi: 10.1002/ana.22598
Serwach, K., and Gruszczynska-biegala, J. (2020). Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurosci. 13:617422. doi: 10.3389/fnmol.2020.617422
Stary, C. M., and Hogan, M. C. (2000). Impairment of Ca2+ release in single Xenopus muscle fibers fatigued at varied extracellular PO2. J. Appl. Physiol. 88, 1743–1748. doi: 10.1152/jappl.2000.88.5.1743
Takeshima, H., Komazaki, S., Nishi, M., Iino, M., and Kangawa, K. (2000). Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22. doi: 10.1016/s1097-2765(00)00003-4
Takeshima, H., Shimuta, M., Komazaki, S., Ohmi, K., Nishi, M., Uno, M., et al. (1998). Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem. J. 331, 317–322. doi: 10.1042/bj3310317
van Oort, R. J., Garbino, A., Wang, W., Dixit, S. S., Landstrom, A. P., Gaur, N., et al. (2011). Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123, 979–988. doi: 10.1161/CIRCULATIONAHA.110.006437
Wang, W., Landstrom, A. P., Wang, Q., Munro, M. L., Beavers, D., Ackerman, M. J., et al. (2014). Reduced junctional Na+/Ca2+-exchanger activity contributes to sarcoplasmic reticulum Ca2+ leak in junctophilin-2-deficient mice. Am. J. Physiol. Hear Circ. Physiol. 307, H1317–H1326. doi: 10.1152/ajpheart.00413.2014
Wei, S., Guo, A., Chen, B., Kutschke, W., Xie, Y. P., Zimmerman, K., et al. (2010). T-tubule remodeling during transition from hypertrophy to heart failure. Circ. Res. 107, 520–531. doi: 10.1161/CIRCRESAHA.109.212324
Wilburn, B., Rudnicki, D. D., Zhao, J., Weitz, T. M., Cheng, Y., Gu, X., et al. (2011). An Antisense CAG Repeat Transcript at JPH3 Locus Mediates Expanded Polyglutamine Protein Toxicity in Huntington’s Disease-like 2 Mice. Neuron 70, 427–440. doi: 10.1016/j.neuron.2011.03.021
Woo, J. S., Srikanth, S., Nishi, M., Ping, P., Takeshima, H., and Gwack, Y. (2016). Junctophilin-4, a component of the endoplasmic reticulum-plasma membrane junctions, regulates Ca2+ dynamics in T cells. Proc. Natl. Acad. Sci. U.S.A. 113, 2662–2767. doi: 10.1073/pnas.1524229113
Wu, C. Y. C., Chen, B., Jiang, Y. P., Jia, Z., Martin, D. W., Liu, S., et al. (2014). Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J. Am. Heart Assoc. 3, 1–18. doi: 10.1161/JAHA.113.000527
Keywords: membrane contact site proteins, ER–PM tethers, calcium channels, RyR channels, jph-1, synaptic transmission, muscle excitation
Citation: Piggott CA and Jin Y (2021) Junctophilins: Key Membrane Tethers in Muscles and Neurons. Front. Mol. Neurosci. 14:709390. doi: 10.3389/fnmol.2021.709390
Received: 13 May 2021; Accepted: 15 June 2021;
Published: 08 July 2021.
Edited by:
Zhiyong Shao, Fudan University, ChinaReviewed by:
Anbing Shi, Huazhong University of Science and Technology, ChinaCopyright © 2021 Piggott and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher A. Piggott, Y3BpZ2dvdHRAdWNzZC5lZHU=; Yishi Jin, eWlqaW5AdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.