 Renhao Xue
Renhao Xue Hao Meng2,3
Hao Meng2,3 Zhitao Hu
Zhitao Hu Huisheng Liu
Huisheng Liu- 1Shanghai Key Laboratory of Maternal Fetal Medicine, Clinical and Translational Research Center of Shanghai First Maternity & Infant Hospital, School of Life Sciences and Technology, Tongji University, Shanghai, China
- 2Guangzhou Laboratory, Guangzhou, China
- 3Bioland Laboratory, Guangzhou Regenerative Medicine and Health Guangdong Laboratory, Guangzhou, China
- 4Clem Jones Centre for Ageing Dementia Research (CJCADR), Queensland Brain Institute, The University of Queensland, Brisbane, QLD, Australia
Exocytosis is a Ca2+-regulated process that requires the participation of Ca2+ sensors. In the 1980s, two classes of Ca2+-binding proteins were proposed as putative Ca2+ sensors: EF-hand protein calmodulin, and the C2 domain protein synaptotagmin. In the next few decades, numerous studies determined that in the final stage of membrane fusion triggered by a micromolar boost in the level of Ca2+, the low affinity Ca2+-binding protein synaptotagmin, especially synaptotagmin 1 and 2, acts as the primary Ca2+ sensor, whereas calmodulin is unlikely to be functional due to its high Ca2+ affinity. However, in the meantime emerging evidence has revealed that calmodulin is involved in the earlier exocytotic steps prior to fusion, such as vesicle trafficking, docking and priming by acting as a high affinity Ca2+ sensor activated at submicromolar level of Ca2+. Calmodulin directly interacts with multiple regulatory proteins involved in the regulation of exocytosis, including VAMP, myosin V, Munc13, synapsin, GAP43 and Rab3, and switches on key kinases, such as type II Ca2+/calmodulin-dependent protein kinase, to phosphorylate a series of exocytosis regulators, including syntaxin, synapsin, RIM and Ca2+ channels. Moreover, calmodulin interacts with synaptotagmin through either direct binding or indirect phosphorylation. In summary, calmodulin and synaptotagmin are Ca2+ sensors that play complementary roles throughout the process of exocytosis. In this review, we discuss the complementary roles that calmodulin and synaptotagmin play as Ca2+ sensors during exocytosis.
Introduction
Ca2+ is an important signaling molecule that mediates a variety of cellular functions including exocytosis, gene transcription, differentiation, apoptosis, etc. Hence, Ca2+-binding proteins are required to serve as sensors to transpose Ca2+ signals in these Ca2+-dependent processes. A typical Ca2+-regulated event is exocytosis, which is a key functional cellular action, as it is responsible for a wide range processes such as the secretion of hormones from endocrine cells, the release of neurotransmitters from presynaptic neurons, the acrosome reaction during fertilization, and the delivery of plasma membrane-bound receptors (Jahn and Sudhof, 1994; Rothman, 1994; Sudhof, 1995, 2012; Wassarman, 1999). During exocytosis, secretory vesicles are directed to release sites at the plasma membrane and become a ready-to-release state through trafficking, docking and priming processes. The well-prepared vesicles subsequently discharge their contents via membrane fusion (Lin and Scheller, 2000; Burgoyne and Morgan, 2003). The fusion process is triggered by Ca2+ influx and requires the participation of Ca2+ sensors. In addition to triggering the final fusion, Ca2+ signal is also involved in the steps prior to fusion, indicating that Ca2+-binding proteins potentially contribute to earlier stages of vesicle exocytosis (Regehr, 2012).
Two classes of Ca2+-binding proteins emerged as Ca2+ sensors for exocytosis: EF-hand proteins and C2 domain proteins. A typical EF-hand protein is calmodulin (CaM), which binds Ca2+ via four EF-hands. CaM is a highly conserved protein ubiquitously and abundantly expressed in eukaryotic cells (Stevens, 1983; Chin and Means, 2000). The C2 domain proteins bind Ca2+ via the C2 domains that consist of eight β-strands connected by seven loops, two of which normally coordinate calcium ions. The C2 domains have been found in a variety of proteins, such as phospholipase C (PLC), protein kinase C (PKC), and synaptotagmin (Syt).
Since the 1980s, tremendous studies have started seeking Ca2+ sensor for exocytosis. At the beginning, researchers focused on both EF-hand and C2 domain proteins because of their Ca2+ binding properties. Later studies have found that it is the C2 domain protein Syt, but not the EF-hand protein, acts as the primary Ca2+ sensor for membrane fusion. This notion was confirmed by studies in many species, including mouse (Geppert et al., 1994; Nishiki and Augustine, 2004), Drosophila (Lee et al., 2013), zebra fish (Wen et al., 2010), as well as the recent studies in the nematode Caenorhabditis elegans (Li et al., 2018, 2021). Meanwhile, EF-hand protein CaM, has been found to play an active role in exocytotic steps prior to fusion via interaction with multiple functional proteins in a Ca2+-dependent manner (Junge et al., 2004; Zikich et al., 2008). Interestingly, CaM and Syt also has crosstalk during exocytosis. In this review, we will summarize the functions of the two types Ca2+-binding proteins, CaM and Syt, in membrane fusion and pre-fusion steps to provide a full map describing the role of Ca2+ sensors throughout the process of exocytosis.
Molecular Mechanisms Underlying Dynamic Steps of Exocytosis
As described above, exocytosis comprises dynamic steps, including vesicle trafficking, docking, priming, and fusion (Sudhof, 2004; Becherer and Rettig, 2006; Figure 1). Over the past few decades, molecular mechanisms underlying each specific step have been revealed. During trafficking, vesicles wrapped in a lipid bilayer membrane and loaded with secretory cargos, such as hormones or neurotransmitters, are transported from the inner cytosol to the subplasmalemmal region along the microtubule and F-actin tracks by motor proteins (e.g., dynamin and myosin) (Loubery and Coudrier, 2008; Trifaro et al., 2008). Upon arrival, secretory vesicles get tethered to the release sites, and in a following process termed docking, are brought into close contact with the plasma membrane, allowing the vesicle protein VAMP (vesicle-associated membrane protein, also named as synaptobrevin) to interact with the plasma membrane protein syntaxin and the cytoplasmic protein SNAP-25 (synaptosomal-associated protein of 25 kD). Together, VAMP, syntaxin and SNAP-25 can form a protein complex known as the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex (Sollner, 2003; Verhage and Sorensen, 2008). After docking, vesicles are primed (or mature) in an ATP (adenosine 5’-triphosphate)-dependent manner to a readily releasable state (Chen et al., 2001). Munc13 is known to act as an essential vesicle priming factor (Augustin et al., 1999) that promotes the assembly of SNARE complex by interacting with syntaxin (Betz et al., 1997) and switching it to an active configuration (Ma et al., 2011). Finally, when cells are excited, the boost of [Ca2+]i (intracellular free Ca2+ concentration) triggers the fusion of the vesicle and plasma membrane, and subsequent release of secretory cargos. This membrane fusion is driven by the SNARE complex as a core release machinery (Jahn et al., 2003): the H3 helix of syntaxin interacts with the VAMP coiled-coil domain and two SNAP-25 helices to form coiled-coil bundles. The complex then twists itself to proceed down the energy gradient and bring the lipid bilayers of the vesicle membrane and the plasma membrane sufficiently close to each other to overcome the hydration barrier, and initiate fusion (Burgoyne and Morgan, 2003).
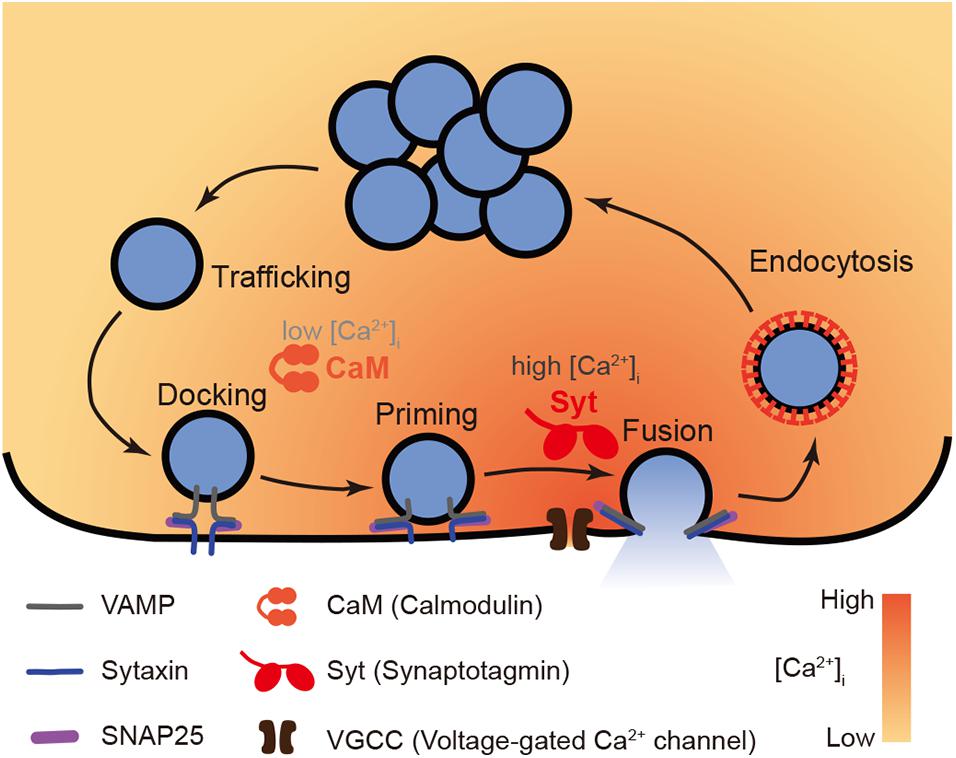
Figure 1. A model of functions of Ca2+ sensor CaM vs. Syt in exocytosis. The whole process of Ca2+ dependent exocytosis includes vesicle trafficking, docking, priming, and fusion. The final membrane fusion is trigger by Ca2+ influx through VGCC that induces a boost of [Ca2+]i up to micromolar level. The low affinity Ca2+ sensor Syt (specifically Syt1 or Syt2) is suitable to convey such high Ca2+ signals to vesicle fusion events. Meanwhile, in early steps prior to fusion, the high affinity Ca2+ sensor CaM plays a role to facilitate vesicle trafficking, docking, and priming in submicromolar [Ca2+]i.
Clearly, membrane fusion, the final step, is triggered by Ca2+ signals. As neither v-SNARE (vesicle SNARE: VAMP) nor t-SNAREs (target-SNAREs: syntaxin and SNAP-25) demonstrate direct interactions with Ca2+; the final process that releases secretory cargos requires a Ca2+ sensor (or Ca2+ sensors) to couple the Ca2+ signals to the SNARE-driven membrane fusion. Meanwhile, pre-fusion steps are also mediated by a [Ca2+]i slightly above resting level, although not synchronized with the boost of [Ca2+]i upon activation. Hence, Ca2+-binding proteins might also play roles in secretory vesicle lifespan before the final release.
The Ca2+ Sensor for Membrane Fusion: Syt, Rather Than CaM
Given the critical roles of Ca2+ in triggering the final fusion, it must require Ca2+ sensor proteins to initiate this step. In this scenario, we will highlight the process of how Syt was identified as the functional Ca2+ sensor for fusion, and why CaM was ruled out in this step.
Why Is CaM Ruled Out as the Ca2+ Sensor for Membrane Fusion?
CaM is a common Ca2+-binding protein that consists of 148 amino acids (16.7 KDa) and coordinates four calcium ions via four EF-hands. Two of these motifs form a globular domain at the N-terminal while the other two constitute an almost symmetrical structure at the C-terminal. A flexible helical linker connects these two globular domains. In response to Ca2+ coordinating with the Ca2+-binding loops in the EF-hand motifs, a hydrophobic group in each globular domain is exposed, switching CaM to an “open” conformation that is functionally activated (Stevens, 1983; Chin and Means, 2000). CaM is probably the most popular cellular Ca2+ binding partner, with diverse functions that vary from buffering intracellular Ca2+ to transduction of Ca2+ signals. Interestingly, it also interacts with the v-SNARE protein (Quetglas et al., 2000), suggesting a putative role in vesicle exocytosis.
In the 1980s, CaM was shown to be involved in glucose-stimulated insulin release from pancreatic beta cells using a pharmacological inhibitor: trifluoperazine (Krausz et al., 1980; Steinberg et al., 1984). Thereafter, CaM antagonists, including trifluoperazine, W-7 or ophiobolin, were also found to impair catecholamine secretion from bovine chromaffin cells (Clapham and Neher, 1984; Matsumura et al., 1999) and PC12 cells (Quetglas et al., 2002; Ando et al., 2013), inhibit rat parotid amylase exocytosis (Tojyo et al., 1989), affect the mouse acrosome reaction (Bendahmane et al., 2001), diminish neurotransmitter release from various types of nerve terminals (Cazalis et al., 1987; Ando et al., 2013), block the expansion and release of trichocyst matrix during exocytosis in Paramecium (Garofalo et al., 1983) and suppress exocytosis in mast cells (Douglas and Nemeth, 1982; Gigl et al., 1987). The involvement of CaM in exocytosis was established in various experimental systems, but two key questions were raised: whether CaM acts as the Ca2+ sensor for membrane fusion, and whether CaM actively mediates pre-fusion steps.
The above findings revealed that CaM is a functional mediator of exocytosis. Together with its Ca2+-binding properties, CaM was originally speculated to act as a Ca2+ sensor for membrane fusion (Brown et al., 1985). However, a clear conclusion cannot be reached simply based on these observations using pharmacological approaches. There are three major arguments against this notion. First, the primary Ca2+ sensor for fusion should have an essential effect on vesicle release, with a severe secretion defect being expected if the sensor is blocked. However, inhibition of CaM unlikely induces a strong reduction of release. Second, insufficiency of solid evidence to support the idea that the Ca2+-binding activity of CaM is crucial for fusion. Lastly but most importantly, intrinsic Ca2+-binding properties make CaM unsuitable to convey Ca2+ signals upon [Ca2+]i boost. Moreover, Syt, another type of Ca2+-binding protein, has been found to meet almost all criteria as a primary Ca2+ sensor for membrane fusion.
Firstly, the primary Ca2+ sensor for membrane fusion should be an essential factor of vesicle release, that it is unlikely for CaM. Indeed, in some research systems for exocytosis, contradictory results have been reported. For example, a CaM antagonist was found to have no effect on Ca2+-dependent amylase release from the rat parotid gland (Spearman and Butcher, 1983). In digitonin-permeabilized adrenal chromaffin cells, neither the CaM inhibitor (Wilson and Kirshner, 1983) nor the restoration of reconstituted CaM protein (Sarafian et al., 1987) led to any significant changes in Ca2+-evoked secretory activity. Another experiment also revealed dual effects of CaM in transmitter release at neuromuscular junctions (Branisteanu et al., 1989). In mast cells, CaM-mediated disassembly of cortical F-actin was not required for secretion (Sullivan et al., 2000). In human parathyroid adenoma, a CaM inhibitor was found to increase, rather than decrease, parathyroid hormone release (Lu et al., 2011). These findings demonstrated non-identical effect of CaM in different cell types, questioning an essential role for CaM in secretion. It seems like CaM only plays an auxiliary role instead of acting as the primary cellular Ca2+ sensing apparatus in the final fusion step.
Secondly, limited direct evidence support that CaM is directly associated with the membrane fusion events in a Ca2+-dependent manner. Although the majority of pharmacological studies supported an active role of CaM in exocytosis, mechanism studies were required to elucidate whether it transposes Ca2+ signals during fusion. Since the 1990s, with the development of techniques, much more detailed information could be collected from experiments to further elucidate mechanisms underlying observations. Kibble and Burgoyne (1996) measured the rate, instead of quantity of catecholamine release from adrenal chromaffin cells and found that CaM increases the initial release rate, while Watkins and Cooperstein (1997) found using electron microscopy that CaM modulates the interaction between the secretory vesicles and plasma membrane in the parotid gland. These studies began to explore the mechanisms underlying CaM-mediated exocytosis but were not sufficient to draw a final conclusion. It was also reported that CaM functions as a calcium sensitizing factor for cortical granule exocytosis during fertilization of sea urchin eggs, as the Ca2+ sensitivity of cortical granule release was largely diminished if CaM was masked by antibodies (Steinhardt and Alderton, 1982). However, a following study denied this conclusion as they claimed that brain extracts failed to restore Ca2+ sensitivity in the same system after a brief heat-shock, ruling out the involvement of CaM, given CaM considered to be relatively heat-stable (Sasaki, 1992). Hence, the protein factor that confers Ca2+ sensitivity to exocytosis in this system remains an open question. Observation of Paramecium using electron microscopy revealed that CaM is essential for the assembly of links that connect the plasma and trichocyst membranes, instead of playing a Ca2+-dependent role in stimulus-exocytosis coupling (Kerboeuf et al., 1993). These findings reduced the probability of CaM acting as the membrane fusion Ca2+ sensor. However, it should be noted that in some unique cellular systems, it is still possible that CaM is the major Ca2+ sensor for fusion. For example, in the fusion of vacuoles (Peters and Mayer, 1998) and sperm-specific membranous organelle (Shang et al., 2013).
Finally, Burgoyne and Clague (2003) pointed out that CaM is a relatively high affinity Ca2+ sensor that is fully activated at submicromolar [Ca2+]i (∼0.5 μM). This Ca2+ concentration is much lower than the boost of [Ca2+]i in most activated cells (e.g., neurons), which is above the micromolar level. If CaM serves as the Ca2+ sensor, membrane fusion would theoretically occur even in the absence of the [Ca2+]i boost. Therefore, the real Ca2+ sensor that triggers exocytotic membrane fusion should have a lower affinity to Ca2+ than CaM.
How Is Syt Identified as the Ca2+ Sensor for Membrane Fusion?
Although considerable attention was driven to CaM, a significant body of research elucidated that the other class of putative Ca2+ sensor, Syt, is indeed responsible for the transit of Ca2+ signals to membrane fusion. Syt is a family of C2 domain-containing proteins with 17 isoforms (Craxton, 2004). Most of Syt isoforms consist of two C2 domains. The best studied isoform is Syt1 (synaptotagmin I), which is a membrane protein that binds with Ca2+ through its tandem C2 domains (C2A and C2B) with a much lower affinity than that of CaM (Kd > 10 μM) (Shao et al., 1998; Fernandez-Chacon et al., 2001). Hence, this Ca2+ sensor can only be switched on by robust Ca2+ signals such as the Ca2+ influx triggered by action potentials at presynaptic nerve terminals. Syt also binds with both t-SNARE proteins [syntaxin (Bennett et al., 1992) and SNAP25 (Schiavo et al., 1997; Zhou et al., 2015)], and these interactions are Ca2+ dependent (Chapman et al., 1995; Gerona et al., 2000). This intrinsic property makes Syt1 a perfect candidate of Ca2+ sensor for membrane fusion in the final step of exocytosis.
Unlike CaM, considerable evidence supports the role of Syt as major Ca2+ sensors for membrane fusion. In the 1990s, various studies revealed that inactivation of Syt by mutation, inhibitory peptide or knockout, largely impaired neurotransmitter release in the nematode C. elegans (Nonet et al., 1993), Drosophila (Littleton et al., 1993), squid Loligo pealei (Bommert et al., 1993), and mouse (Geppert et al., 1994), suggesting an essential role of Syt conservative through the evolution. Early in this century, advanced electrophysiological technique together with molecular biological approaches allowed researchers to modify proteins via various mutations and measure the release rate of synaptic vesicles driven by modified proteins. It was found that changes in the Ca2+ coordination activity of Syt via Ca2+ ligand mutants in the C2 domains lead to dramatic changes in the rate of release and its cooperativity with the extracellular Ca2+ concentration (Fernandez-Chacon et al., 2001; Mackler et al., 2002; Robinson et al., 2002; Stevens and Sullivan, 2003; Nishiki and Augustine, 2004). These findings provided direct evidence that the Ca2+-binding activity of Syt involves membrane fusion. Despite this, doubts remained regarding whether Syt simply synchronize vesicle release via interactions with voltage-gated Ca2+ channels (VGCC) and therefore does not indeed convey Ca2+ signals to release events (Neher and Penner, 1994). To answer this question, studies using flash photolysis of caged Ca2+ as Ca2+ source to bypass Ca2+ channels ruled out this possibility (Sun et al., 2007; Burgalossi et al., 2010). These studies confirmed Syt as the major Ca2+ sensor for fusion. Syt1 and Syt2 (synaptotagmin 2) are the two Syt isoforms that demonstrate the lowest Ca2+ affinity (Sugita et al., 2002; Pinheiro et al., 2016). These isoforms are also functional in neurons which exhibit an enormous and sharp [Ca2+]i peak when excited. Syt1 is active in hippocampal excitatory neurons (Geppert et al., 1994) while Syt2 is functional in cortical inhibitory neurons (Chen et al., 2017). In the case of endocrinal cells, Syt1 and Syt7 (synaptotagmin 7) are overlapping Ca2+ sensors that both trigger large dense core vesicle (LDCV) fusion in chromaffin cells (Voets et al., 2001; Schonn et al., 2008). Syt7 is responsible for insulin vesicle fusion in pancreatic beta cells (Gustavsson et al., 2008). These findings suggested a general role of Syt as a Ca2+ sensor in a variety of cell types. Moreover, when Syt1 was chemico-genetically engineered to sense Sr2+, a non-physiological metal, Sr2+-dependent exocytosis was observed in cultured neurons, confirming that Syt1 acts as a functional metal sensor for release (Evans et al., 2015). Together, these lines of evidence support the idea that Syt functions as the primary Ca2+ sensor, driving the Ca2+-evoked fusion events of synaptic vesicles (Figure 1).
CaM Is Unlikely the Ca2+ Sensor for Membrane Fusion Even at Low [Ca2+]i
As described above, CaM is not a Ca2+ sensor for final fusion due to its high Ca2+ affinity. Nonetheless, it should be noted that the final release event does not always coincident with a [Ca2+]i boost above the micromolar level, but sometimes also occurs at submicromolar [Ca2+]i. For example, in neurons, asynchronous release of synaptic vesicles does not synchronize with the peak of Ca2+ signal, but accompanies a post-peak [Ca2+]i which is only a little above the resting [Ca2+]i. Moreover, secretion of neurotransmitters can even occur spontaneously at a [Ca2+]i close to basal level. Hence, it is still questionable whether CaM senses Ca2+ for fusion at low [Ca2+]i during asynchronous or spontaneous exocytosis.
An electrophysiological study that measured the release rate and [Ca2+]i simultaneously suggested an allosteric model for the release Ca2+ sensor (Lou et al., 2005). According to this model, Ca2+ sensors that mediate release at distinct [Ca2+]i are structurally distinct. Hence, it is likely that different Ca2+ sensors with different Ca2+ affinities mediate different types of release. Using the same method, it was found that removal of Syt2, the Syt with low Ca2+ affinity, affects the release rate only at high [Ca2+]i, but not at submicromolar [Ca2+]i, in the calyx of Held (Sun et al., 2007). Consistently, knockout of Syt1 or Syt2, the two synaptotagmins with low Ca2+ affinity, only eliminated synchronous neurotransmitter release, without reducing (actually, even increasing) asynchronous or spontaneous release (Geppert et al., 1994; Liu et al., 2009). This led to the conclusion that Syt1 and Syt2 were Ca2+ sensors only for synchronous vesicle fusion in neurons.
As a high affinity Ca2+ sensor, CaM is plausibly suitable for triggering asynchronous or spontaneous synaptic vesicle release. However, the fact that knockdown of CaM failed to reduce asynchronous neurotransmitter release in Syt1 knockout cortical neurons (Pang et al., 2010b), directly contradicted this idea. Evidence subsequently emerged to indicate that Syt7, a high Ca2+ affinity Syt isoform (Sugita et al., 2002) might act as a Ca2+ sensor for asynchronous release (Bacaj et al., 2013) and double C2-like domain-containing protein (Doc2), another C2 domain protein with relatively high Ca2+ affinity, senses Ca2+ signal in both asynchronous (Yao et al., 2011; Xue et al., 2015) and spontaneous release (Groffen et al., 2010). Hence, CaM is unlikely to act as a Ca2+ sensor that directly triggers membrane fusion. Nevertheless, it is still possible that it plays a secondary role by mediating key proteins, such as Syt.
CaM Plays Ca2+-Dependent Roles in Pre-Fusion Steps
The early processes of vesicle exocytosis, namely trafficking, docking and priming are crucial preparations for the final release of the contents of vesicles, but are not associated with the peak of the [Ca2+]i boost upon cell excitation. Hence, low affinity Ca2+ sensors, such as Syt1 and Syt2, are not activated, whereas high affinity Ca2+ sensors, i.e., CaM, might be functionally involved in these pre-fusion steps due to a [Ca2+]i below the micromolar level (Burgoyne and Clague, 2003).
Through these early steps, secretory vesicles are delivered to the release sites and conveyed into a mature state ready for release. These mature vesicles constitute a readily releasable pool (RRP) (Rizzoli and Betz, 2005). The maintenance of the RRP before release and the subsequent replenishment of this pool after release are crucial determinants of the extent of final release. Using a pharmacological inhibitor, it was found that CaM is involved in the delivery of vesicle into the RRP in the calyx of Held (Sakaba and Neher, 2001) and hippocampal neurons (Liu H. et al., 2014). CaM also mediates short-term synaptic plasticity at least partly through the recovery of RRP (Junge et al., 2004; Lipstein et al., 2013). It has also been reported that CaM supports the recovery from short-term depression at retinal cone ribbon synapses, but the mechanisms underlying this acceleration and its functional implications for vision are unknown (Van Hook et al., 2014). In summary, CaM clearly plays a role in exocytotic actions prior to final vesicular fusion (Figure 1).
How does CaM complete its role? Once activated by Ca2+ binding, CaM may mediate the function of multiple key regulatory exocytotic proteins through direct interactions or modulation by phosphorylation via activation of some important kinases (Creutz et al., 1983; Watkins and White, 1985).
CaM Direct Interactions With Key Regulatory Proteins in Pre-fusion Steps
CaM may contribute to the early steps of exocytosis via direct binding with components of the SNARE complex. As described above, the SNARE complex, consisting of v-SNARE VAMP, and t-SNARE SNAP25 and syntaxin, serves as the core machinery of membrane fusion. It has been reported that CaM binds to VAMP2, the v-SNARE, but not to syntaxin or SNAP25, the t-SNAREs, and that this direct binding mediates the lipid interaction activity of VAMP2 (Quetglas et al., 2000). If this binding is diminished, the formation of the SNARE complex is impaired and hence the hormone secretion from chromaffin cells is inhibited (Quetglas et al., 2002). Another study also revealed that Ca2+/calmodulin mediates SNARE assembly by transferring VAMP from cis-membrane to trans-membrane (de Haro et al., 2004). These findings suggested that CaM is an active mediator of secretory vesicle docking and priming via interaction with of v-SNARE.
The interaction partners of CaM are not limited to the SNARE proteins. Another important binding partner of CaM is myosin V (Espindola et al., 1992). Myosin-V is a key molecular motor that drives vesicle trafficking along F-actin (Reck-Peterson et al., 2000). In the presence of CaM and Ca2+, syntaxin-VAMP-myosin V form a complex (Ohyama et al., 2001). At the switch point where vesicles are unloaded from cytoskeleton tracks and tethered to the plasma membrane, submicromolar [Ca2+]i releases CaM from myosin V, the syntaxin-1A interaction site in myosin V is exposed. Then syntaxin-1A binds with myosin V and consequently vesicle docking is completed (Watanabe et al., 2005). This CaM-myosin interaction underscores the role of CaM during trafficking and docking.
Munc13, which plays an essential role in vesicle priming (Augustin et al., 1999), also exhibits robust binding property with CaM. The two major Munc13 isoforms, Munc13-1 and ubMunc13-2, bind with CaM in the region between the first C2 domain and the C1 domain. Active mutants at this region which eliminate CaM binding activity also impair short-term plasticity, at least partly by inhibiting the replenishment of the RRP (Junge et al., 2004). These results reveal a key role of CaM-Munc13 binding in Munc13-induced vesicle priming. A similar functional interaction has also been observed in chromaffin cells (Zikich et al., 2008). UNC-13, the homolog of Munc13 in C. elegans, also binds with CaM to accelerate the release (Hu et al., 2013). These findings reveal a crucial mechanism underlying CaM-mediated vesicle priming.
CaM also interacts with synapsin, GAP43, Rab3, and Syt. Synapsin is a functional protein that is involved in synaptic vesicle trafficking, docking, and release in presynaptic nerve terminals and modulates synaptic plasticity (Greengard et al., 1993; Cesca et al., 2010). CaM directly binds to synapsin 1 (Hayes et al., 1991) and synapsin 2 (Nicol et al., 1997), the two major isoforms of synapsin family. GAP43 is a regulator of exocytosis that directly interacts with the core complex of exocytosis and Syt (Haruta et al., 1997). CaM binding with GAP43 (Baudier et al., 1991) may impair the priming step by inhibiting phosphorylation of GAP43 (Misonou et al., 1998). Rab3 is a small GTPase that is involved in exocytosis as an inhibitory regulator (Johannes et al., 1994; Geppert et al., 1997). CaM binds with Rab3 (Park et al., 1997) and such interaction was found to abolish the Rab3-induced inhibition of vesicle release from catecholamine and insulin secreting cells (Coppola et al., 1999), but not in PC12 cell (Schluter et al., 2002). CaM also promotes GTP binding to Rab3A (Park et al., 1997), forming an active GTP-bound Rab3A-Ca2+/CaM complex (Park et al., 2002), and the Rab3A-CaM interaction involved in insulin secretion from pancreatic beta cells (Kajio et al., 2001), and acrosome exocytosis (Yunes et al., 2002). In summary, CaM directly interacts with a variety of functional proteins, usually in a Ca2+-dependent manner, to regulate early steps of exocytosis (Figure 2).
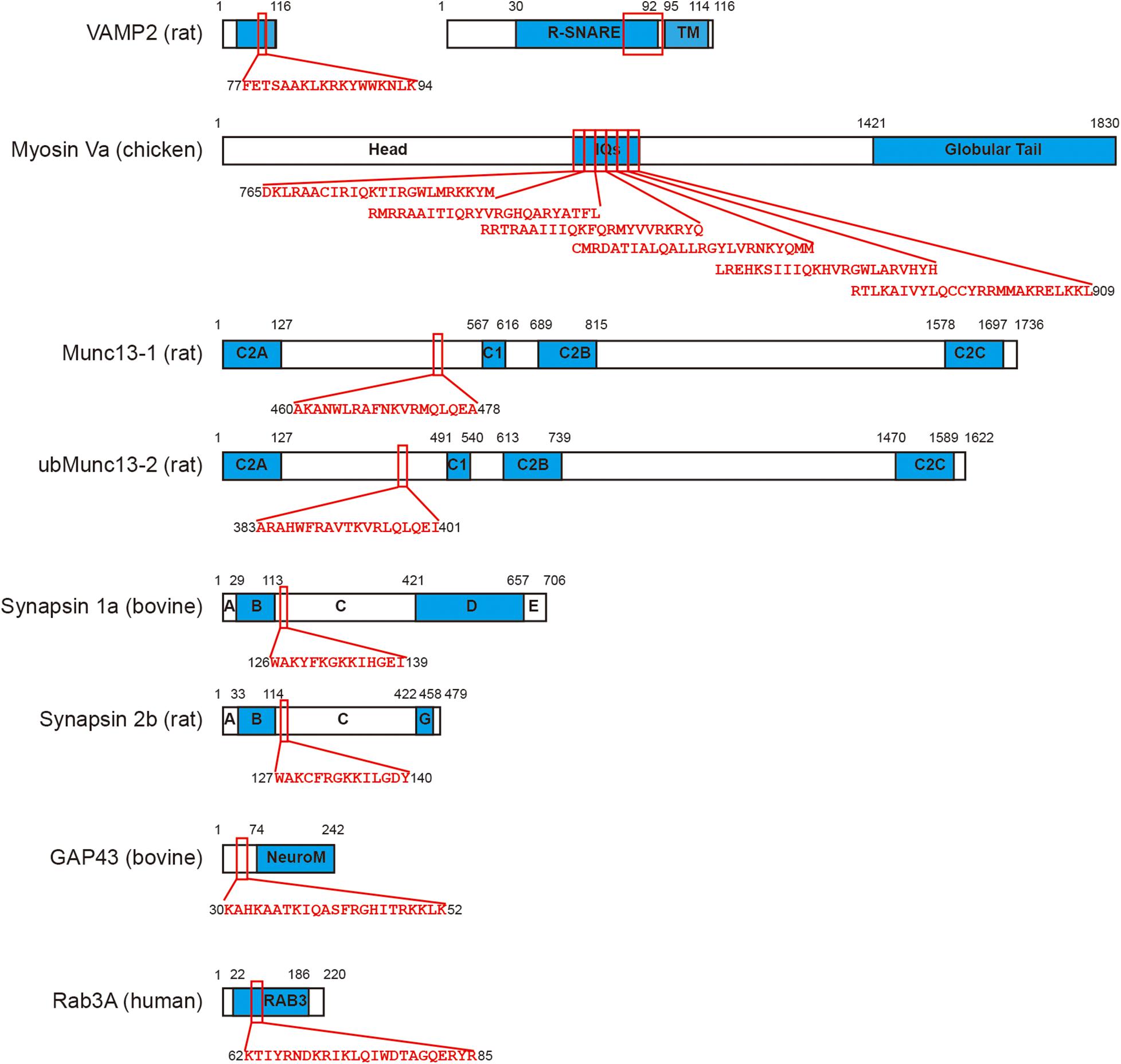
Figure 2. CaM directly binds with a series of functional proteins in exocytosis. VAMP2 (Quetglas et al., 2000), myosin Va (Espindola et al., 1992), Munc13-1, ubMunc13-2 (Junge et al., 2004), synapsin 1a, synapsin 2b (Nicol et al., 1997), GAP43 (Baudier et al., 1991), and Rab3A (Park et al., 1997) are all binding partners of CaM. The CaM binding motifs in each protein are labeled using red squares and the corresponding sequence of amino acid is shown. Please note myosin Va consists of six tandem IQ motifs that interact with CaM. Since the size of VAMP2 is relatively small, a 5-times amplification is displayed on its right.
CaM Induces Phosphorylation of Regulatory Proteins in Pre-fusion Steps by Activating Key Kinases
Some of the key functional proteins in exocytosis are substrates of kinases that need to be activated by CaM (Cooperstein and Watkins, 1995). A typical kinase related to CaM is type II Ca2+/CaM dependent protein kinase (CaMKII). CaMKII (Colbran, 1992) is important for exocytosis (Ammala et al., 1993), including GABA release at inhibitory synapses (Sitges et al., 1995), insulin release from pancreatic beta cells (Ashcroft et al., 1994), catecholamine release from PC12 cells (Schweitzer et al., 1995), calcium-evoked dendritic exocytosis (Maletic-Savatic et al., 1998), postsynaptic secretion of BDNF (brain-derived neurotrophic fact) and neurotrophin-3 from hippocampal neurons (Kolarow et al., 2007), acetylcholine and GABA release at the C. elegans neuromuscular junctions (Liu et al., 2007), and neurotransmitter release in cultured neurons (Pang et al., 2010a). CaMKII also contributes to plasma membrane resealing in response to cell membrane injury, by facilitating membrane fusion through vesicle exocytosis (Steinhardt et al., 1994). Activated CaMKII phosphorylates multiple proteins to switch on their functions in regulation of exocytosis.
Interestingly, the most important substrate of CaMKII is itself. Twelve CaMKII molecules usually form a functional dodecamer. Upon interaction with CaM in the presence of Ca2+, the CaMKII dodecamers undergo autophosphorylation and become activated (Miller and Kennedy, 1986; Meyer et al., 1992; Hanson et al., 1994). Such autophosphorylation has been found to mediate the plasticity of synaptic vesicle release (Giese et al., 1998). CaMKII directly binds to syntaxin 1A at plasma membrane in a Ca2+-and ATP-dependent manner, with autophosphorylation of CaMKII largely facilitating this interaction (Ohyama et al., 2002; Nomura et al., 2003; Watanabe et al., 2013). These CaMKII-syntaxin-1A interactions have been found to be important for regulated exocytosis (Ohyama et al., 2002).
Some SNARE proteins are also substrates of CaMKII. Although there is no evidence to indicate that syntaxin-1A is directly phosphorylated by CaMKII, syntaxin-3, another isoform of syntaxin, was identified as a substrate of CaMKII (Risinger and Bennett, 1999). In ribbon synapses of the retina, CaMKII-induced phosphorylation of syntaxin-3B mediated the exocytosis of synaptic vesicles by modulating the assembly of the SNARE complex (Liu X. et al., 2014). Furthermore, CaMKII can also phosphorylate the v-SNARE protein VAMP (Nielander et al., 1995).
Besides the autophosphorylation and phosphorylation of SNARE proteins, CaMKII also phosphorylates a variety of regulatory exocytotic proteins, such as synapsin (Baines and Bennett, 1985), Rab3-interacting molecule (RIM) (Sun et al., 2003), Ca2+ channels (Sun et al., 2003), and even Syt (Popoli, 1993; Hilfiker et al., 1999). For example, RIM is an important mediator of synaptic vesicle release (Wang et al., 1997; Schoch et al., 2002; Deng et al., 2011). Its serine sites are phosphorylated by Ca2+/CaM, which promotes the interaction of 14-3-3 with the N-terminal of RIM (Sun et al., 2003). CaMKII also phosphorylates the voltage-gated Ca2+ channels, Cav2.1 and Cav2.2, and subsequently modulates their interaction with SNARE proteins (Yokoyama et al., 2005).
Moreover, Ca2+/CaM dependent kinases are not limited to CaMKII. There are still other kinases that are functionally involved in exocytosis in a Ca2+-and CaM-dependent manner, such as myosin light chain kinase (MLCK). This kinase was found to play a role in thyroid hormone secretion via phosphorylation of the myosin light chain (Tawata et al., 1984). It also mediates the release of secretory granules from the pituitary gland (Nelson et al., 1987), and exocytosis of amylase from the parotid gland (Hashioka and Kato, 1990). Another study using bovine chromaffin cells established the role of MLCK in the priming of secretory vesicles (Matsumura et al., 1999). MLCK also enhances the size of the RRP without affecting the release probability in post-tetanic potentiated calyx of Held synapses (Lee et al., 2008). This is consistent with the idea that CaM is mainly involved in pre-fusion steps of exocytosis.
Concurrent Effects of Direct Interaction and Phosphorylation Modification
As described above, CaM can either directly interact with or indirectly phosphorylate (via CaM-dependent kinases) multiple regulatory proteins that are important for exocytosis. Notably, such direct interaction and indirect phosphorylation are not always isolated events. A good example is synapsin I. In order to play its role in exocytosis, synapsin I must be phosphorylated and it also serves as a substrate of CaMKII (Baines and Bennett, 1985). Interestingly, this phosphorylation is dependent on Ca2+-dependent binding of CaM with the head domain of synapsin 1 (Hayes et al., 1991; Goold and Baines, 1994). Upon CaM-synapsin I binding, CaMKII is activated and phosphorylates the synapsin I. This in turn mediates the interactions between synapsin I and cytoskeletal proteins or small synaptic vesicles (Petrucci et al., 1991), and mobilizes synaptic vesicles (Chi et al., 2003), thereby playing a role in exocytosis, such as facilitating synapses if dynamin-dependent vesicle recycling is impaired (Lou et al., 2012), mediating insulin vesicle release (Yamamoto et al., 2003), or modulating fusion pore kinetics of dense core vesicles in PC12 cells (Yang et al., 2021). In this situation, binding with CaM does not directly activate the protein but induces its phosphorylation via a CaM-dependent kinase and subsequent functionalization. Clearly, these two mechanisms are highly correlative. Besides synapsin, there are other proteins that are both binding partners of CaM and substrates of CaM-dependent kinase, such as VAMP and Syt. These proteins can be modulated by CaM through both mechanisms. However, it is still unclear whether these two mechanisms act synergistically or independently for each individual protein.
It should be noted that, in addition to direct binding and indirect phosphorylation, CaM is also involved in other cellular processes under physiological conditions. For instance, CaM enhances ribbon replenishment and shapes filtering of synaptic transmission by functioning as an endogenous buffer of intracellular Ca2+ (Van Hook et al., 2014). It has also been reported that CaM plays a dominant role in inhibiting vesicular release and modulating short-term synaptic plasticity (Timofeeva and Volynski, 2015).
In summary, CaM is an active mediator of exocytosis that is mainly involved in vesicle trafficking, docking and priming steps prior to membrane fusion. The major mechanism underlying this function is modulation of key regulatory proteins through Ca2+-dependent direct binding and phosphorylation via Ca2+/CaM activated-kinases. In general, a submicromolar [Ca2+]i level is required for CaM to act. Hence, CaM is considered as a high affinity Ca2+ sensor for the pre-fusion steps, while Syt is a low affinity Ca2+ sensor for the final fusion (Figure 1).
Interactions Between CaM and Syt
Finally, it is worth paying some attention to the direct interplay between the two major classes of Ca2+-binding proteins. CaM, the EF-hand protein, can interact with Syt, the C2 domain protein. Hence, to some extent, the function of the Ca2+ sensor in membrane fusion might also be modulated by CaM. Perin (1996) demonstrated that multiple isoforms of Syt bind with CaM via a neuroxin-binding region close to the C-terminal. However, the physiological function of this interaction remained unknown until recent report that CaM-Syt7 binding is essential for the Syt7-mediated refilling of the RRP during synaptic depression (Liu H. et al., 2014) and the replenishment of insulin vesicle pool in pancreatic beta cells (Dolai et al., 2016). These findings reinforced an important role of CaM in vesicle trafficking, docking and priming, but it still remains unclear whether the EF-hands in CaM or the C2 domains in Syt7 are the major active Ca2+ sensing modules in the CaM-Syt7 complex.
In addition to direct binding, it has been reported that Syt can be phosphorylated by CaMKII, which is activated by Ca2+-bound CaM (Popoli, 1993; Hilfiker et al., 1999; Celano et al., 2003). This phosphorylation facilitates its Ca2+-dependent interaction with t-SNAREs (Verona et al., 2000). In light of these observations, CaM is highly likely to be involved in the modulation of Syt’s function during exocytosis via either direct binding or indirect phosphorylation. It is still unknown whether the indirect phosphorylation is dependent on the direct binding, as for synapsin. Future studies are required to address this question.
CaM and Syt can also inhibit each other. It has been reported that Ca2+/CaM suppresses the expression of Syt2 in cortical neurons via an unknown mechanism (Pang et al., 2010b). In an in vitro liposome fusion assay, CaM was also found to inhibit, rather than facilitate, membrane fusion in the absence of Syt, with these inhibitory effects being abolished in the presence of Syt (Di Giovanni et al., 2010). These studies demonstrate that the crosstalk between CaM and Syt is complicated. Further studies are therefore required to fully elucidate the mechanism and function of this remarkable protein-protein interaction.
Conclusion and Perspective
In conclusion, during exocytosis, Syt, but not CaM, senses Ca2+ signals and consequently triggers the final fusion events. Although not the Ca2+ sensor for fusion, CaM might act as a putative high affinity Ca2+ sensor that is switched on at low [Ca2+]i, which is close to resting [Ca2+]i, thereby working in temporal disassociation from the [Ca2+]i boost or spatially distant to the Ca2+ micro/nano domain. Hence, it mainly mediates early exocytotic steps prior to membrane fusion, such as vesicle trafficking, docking and priming, via direct interactions with and/or indirect phosphorylation of key regulatory exocytotic proteins.
To further understand the roles of Ca2+-binding proteins CaM during exocytosis, several key questions need to be addressed in future studies. Does CaM play a secondary role in final membrane fusion? What is the impact of the CaM-Syt interaction in exocytosis? Are any interaction partners of CaM or substrates of CaM-dependent kinases still to be identified? Addressing these questions will enhance our understanding of the entire process of exocytosis and further illuminate how exocytosis is precisely regulated by calcium ions and a plethora of proteins.
Author Contributions
RX, HM, JY, JX, ZH, and HL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by grants from the National Key Research and Development Program of China (2020YFA0908200), the Bioland Laboratory program (1102101207 and 1101304101-8), the National Health and Medical Research Council Project Grant (GNT1122351 to ZH), the National Natural Science Foundation of China (32071229), the Shanghai Science and Technology Innovation Action Plan (20ZR1444100), and the Shanghai Pujiang Program (20PJ1412600).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Rowan Tweedale for critically reading the manuscript.
References
Ammala, C., Eliasson, L., Bokvist, K., Larsson, O., Ashcroft, F. M., and Rorsman, P. (1993). Exocytosis elicited by action-potentials and voltage-clamp calcium currents in individual mouse pancreatic b-cells. J. Physiol. Lond. 472, 665–688. doi: 10.1113/jphysiol.1993.sp019966
Ando, K., Kudo, Y., Aoyagi, K., Ishikawa, R., Igarashi, M., and Takahashi, M. (2013). Calmodulin-dependent regulation of neurotransmitter release differs in subsets of neuronal cells. Brain Res. 1535, 1–13. doi: 10.1016/j.brainres.2013.08.018
Ashcroft, F. M., Proks, P., Smith, P. A., Ammala, C., Bokvist, K., and Rorsman, P. (1994). Stimulus-secretion coupling in pancreatic beta-cells. J. Cell. Biochem. 55, 54–65. doi: 10.1002/jcb.240550007
Augustin, I., Rosenmund, C., Sudhof, T. C., and Brose, N. (1999). Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461. doi: 10.1038/22768
Bacaj, T., Wu, D., Yang, X., Morishita, W., Zhou, P., Xu, W., et al. (2013). Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80:947959. doi: 10.1016/j.neuron.2013.10.026
Baines, A. J., and Bennett, V. (1985). Synapsin-i is a spectrin-binding protein immunologically related to erythrocyte protein-4.1. Nature 315, 410–413. doi: 10.1038/315410a0
Baudier, J., Deloulme, J. C., Vandorsselaer, A., Black, D., and Matthes, H. W. D. (1991). Purification and characterization of a brain-specific protein-kinase-c substrate, neurogranin (p17) - identification of a consensus amino-acid-sequence between neurogranin and neuromodulin (gap43) that corresponds to the protein-kinase-c phosphorylation site and the calmodulin-binding domain. J. Biol. Chem. 266, 229–237.
Becherer, U., and Rettig, J. (2006). Vesicle pools, docking, priming, and release. Cell Tissue Res. 326, 393–407. doi: 10.1007/s00441-006-0243-z
Bendahmane, M., Lynch, C., and Tulsiani, D. R. P. (2001). Calmodulin signals capacitation and triggers the agonist-induced acrosome reaction in mouse spermatozoa. Arch. Biochem. Biophys. 390, 1–8. doi: 10.1006/abbi.2001.2364
Bennett, M. K., Calakos, N., and Scheller, R. H. (1992). Syntaxin - a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257, 255–259. doi: 10.1126/science.1321498
Betz, A., Okamoto, M., Benseler, F., and Brose, N. (1997). Direct interaction of the rat unc-13 homologue Munc13-1 with the N terminus of syntaxin. J. Biol. Chem. 272, 2520–2526. doi: 10.1074/jbc.272.4.2520
Bommert, K., Charlton, M. P., Debello, W. M., Chin, G. J., Betz, H., and Augustine, G. J. (1993). Inhibition of neurotransmitter release by c2-domain peptides implicates synaptotagmin in exocytosis. Nature 363, 163–165. doi: 10.1038/363163a0
Branisteanu, D. D., Branisteanu, D. D., and Haulica, I. (1989). Calmodulin and the transmitter release at the neuromuscular junction. Contradictory results obtained with trifluoperazine. Physiologie (Bucarest) 26, 261–266.
Brown, B. L., Walker, S. W., and Tomlinson, S. (1985). Calcium calmodulin and hormone-secretion. Clin. Endocrinol. 23, 201–218. doi: 10.1111/j.1365-2265.1985.tb00216.x
Burgalossi, A., Jung, S. Y., Meyer, G., Jockusch, W. J., Jahn, O., Taschenberger, H., et al. (2010). SNARE protein recycling by alpha snap and beta snap supports synaptic vesicle priming. Neuron 68, 473–487. doi: 10.1016/j.neuron.2010.09.019
Burgoyne, R. D., and Clague, M. J. (2003). Calcium and calmodulin in membrane fusion. BBA Mol. Cell Res. 1641, 137–143. doi: 10.1016/s0167-4889(03)00089-2
Burgoyne, R. D., and Morgan, A. (2003). Secretory granule exocytosis. Physiol. Rev. 83, 581–632. doi: 10.1152/physrev.00031.2002
Cazalis, M., Dayanithi, G., and Nordmann, J. J. (1987). Requirements for hormone-release from permeabilized nerve-endings isolated from the rat neurohypophysis. J. Physiol. Lond. 390, 71–91. doi: 10.1113/jphysiol.1987.sp016687
Celano, E., Tiraboschi, E., Consogno, E., D’Urso, G., Mbakop, M. P., Gennarelli, M., et al. (2003). Selective regulation of presynaptic calcium/calmodulin-dependent protein kinase II by psychotropic drugs. Biol. Psychiatry 53, 442–449. doi: 10.1016/s0006-3223(02)01491-9
Cesca, F., Baldelli, P., Valtorta, F., and Benfenati, F. (2010). The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 91, 313–348. doi: 10.1016/j.pneurobio.2010.04.006
Chapman, E. R., Hanson, P. I., An, S., and Jahn, R. (1995). CA2+ regulates the interaction between synaptotagmin and syntaxin-1. J. Biol. Chem. 270, 23667–23671. doi: 10.1074/jbc.270.40.23667
Chen, C., Arai, I., Satterfield, R., Young, S. M. Jr., and Jonas, P. (2017). Synaptotagmin 2 is the fast Ca2+ sensor at a central inhibitory synapse. Cell Rep. 18, 723–736. doi: 10.1016/j.celrep.2016.12.067
Chen, Y. A., Scales, S. J., Duvvuri, V., Murthy, M., Patel, S. M., Schulman, H., et al. (2001). Calcium regulation of exocytosis in PC12 cells. J. Biol. Chem. 276, 26680–26687. doi: 10.1074/jbc.M103522200
Chi, P., Greengard, P., and Ryan, T. A. (2003). Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38, 69–78. doi: 10.1016/s0896-6273(03)00151-x
Chin, D., and Means, A. R. (2000). Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10, 322–328. doi: 10.1016/s0962-8924(00)01800-6
Clapham, D. E., and Neher, E. (1984). Trifluoperazine reduces inward ionic currents and secretion by separate mechanisms in bovine chromaffin cells. J. Physiol. Lond. 353, 541–564. doi: 10.1113/jphysiol.1984.sp015350
Colbran, R. J. (1992). Regulation and role of brain calcium calmodulin-dependent protein kinase-II. Neurochem. Int. 21, 469–497. doi: 10.1016/0197-0186(92)90080-b
Cooperstein, S. J., and Watkins, D. T. (1995). Ca++-calmodulin-dependent phosphorylation and dephosphorylation of rat parotid secretion granules. Biochem. Biophys. Res. Commun. 215, 75–81. doi: 10.1006/bbrc.1995.2435
Coppola, T., Perret-Menoud, V., Luthi, S., Farnsworth, C. C., Glomset, J. A., and Regazzi, R. (1999). Disruption of Rab3-calmodulin interaction, but not other effector interactions, prevents Rab3 inhibition of exocytosis. EMBO J. 18, 5885–5891. doi: 10.1093/emboj/18.21.5885
Craxton, M. (2004). Synaptotagmin gene content of the sequenced genomes. BMC Genomics 5:43. doi: 10.1186/1471-2164-5-43
Creutz, C. E., Dowling, L. G., Sando, J. J., Villarpalasi, C., Whipple, J. H., and Zaks, W. J. (1983). Characterization of the chromobindins - soluble-proteins that bind to the chromaffin granule membrane in the presence of Ca-2+. J. Biol. Chem. 258, 4664–4674.
de Haro, L., Ferracci, G., Opi, S., Iborra, C., Quetglas, S., Miquelis, R., et al. (2004). Ca2+/calmodulin transfers the membrane-proximal lipid-binding domain of the v-SNARE synaptobrevin from cis to trans bilayers. Proc. Natl. Acad. Sci. U.S.A. 101, 1578–1583. doi: 10.1073/pnas.0303274101
Deng, L. B., Kaeser, P. S., Xu, W., and Sudhof, T. C. (2011). RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69, 317–331. doi: 10.1016/j.neuron.2011.01.005
Di Giovanni, J., Iborra, C., Maulet, Y., Leveque, C., El Far, O., and Seagar, M. (2010). Calcium-dependent regulation of SNARE-mediated membrane fusion by calmodulin. J. Biol. Chem. 285, 23665–23675. doi: 10.1074/jbc.M109.096073
Dolai, S., Xie, L., Zhu, D., Liang, T., Qin, T., Xie, H., et al. (2016). Synaptotagmin-7 functions to replenish insulin granules for exocytosis in human islet beta-cells. Diabetes 65, 1962–1976. doi: 10.2337/db15-1436
Douglas, W. W., and Nemeth, E. F. (1982). On the calcium receptor activating exocytosis - inhibitory effects of calmodulin-interacting drugs on rat mast-cells. J. Physiol. Lond. 323, 229–244. doi: 10.1113/jphysiol.1982.sp014070
Espindola, F. S., Espreafico, E. M., Coelho, M. V., Martins, A. R., Costa, F. R. C., Mooseker, M. S., et al. (1992). Biochemical and immunological characterization of p190-calmodulin complex from vertebrate brain - a novel calmodulin-binding myosin. J. Cell Biol. 118, 359–368. doi: 10.1083/jcb.118.2.359
Evans, C. S., Ruhl, D. A., and Chapman, E. R. (2015). An engineered metal sensor tunes the kinetics of synaptic transmission. J. Neurosci. 35, 11769–11779. doi: 10.1523/JNEUROSCI.1694-15.2015
Fernandez-Chacon, R., Konigstorfer, A., Gerber, S. H., Garcia, J., Matos, M. F., Stevens, C. F., et al. (2001). Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49. doi: 10.1038/35065004
Garofalo, R. S., Gilligan, D. M., and Satir, B. H. (1983). Calmodulin antagonists inhibit secretion in paramecium. J. Cell Biol. 96, 1072–1081. doi: 10.1083/jcb.96.4.1072
Geppert, M., Goda, Y., Hammer, R. E., Li, C., Rosahl, T. W., Stevens, C. F., et al. (1994). Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727. doi: 10.1016/0092-8674(94)90556-8
Geppert, M., Goda, Y., Stevens, C. F., and Sudhof, T. C. (1997). The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature 387, 810–814. doi: 10.1038/42954
Gerona, R. R. L., Larsen, E. C., Kowalchyk, J. A., and Martin, T. F. J. (2000). The C terminus of SNAP25 is essential for Ca2+-dependent binding of synaptotagmin to SNARE complexes. J. Biol. Chem. 275, 6328–6336. doi: 10.1074/jbc.275.9.6328
Giese, K. P., Fedorov, N. B., Filipkowski, R. K., and Silva, A. J. (1998). Autophosphorylation at Thr(286) of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873. doi: 10.1126/science.279.5352.870
Gigl, G., Hartweg, D., Sanchezdelgado, E., Metz, G., and Gietzen, K. (1987). Calmodulin antagonism - a pharmacological approach for the inhibition of mediator release from mast-cells. Cell Calcium 8, 327–344. doi: 10.1016/0143-4160(87)90008-x
Goold, R., and Baines, A. J. (1994). Evidence that 2 nonoverlapping high-affinity calmodulin-binding sites are present in the head region of synapsin-I. Eur. J. Biochem. 224, 229–240. doi: 10.1111/j.1432-1033.1994.tb20016.x
Greengard, P., Valtorta, F., Czernik, A. J., and Benfenati, F. (1993). Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259, 780–785. doi: 10.1126/science.8430330
Groffen, A. J., Martens, S., Arazola, R. D., Cornelisse, L. N., Lozovaya, N., de Jong, A. P. H., et al. (2010). Doc2b Is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327, 1614–1618. doi: 10.1126/science.1183765
Gustavsson, N., Lao, Y., Maximov, A., Chuang, J.-C., Kostromina, E., Repa, J. J., et al. (2008). Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc. Natl. Acad. Sci. U.S.A. 105, 3992–3997. doi: 10.1073/pnas.0711700105
Hanson, P. I., Meyer, T., Stryer, L., and Schulman, H. (1994). Dual role of calmodulin in autophosphorylation of multifunctional cam kinase may underlie decoding of calcium signals. Neuron 12, 943–956. doi: 10.1016/0896-6273(94)90306-9
Haruta, T., Takami, N., Ohmura, M., Misumi, Y., and Ikehara, Y. (1997). Ca2+-dependent interaction of the growth-associated protein GAP-43 with the synaptic core complex. Biochem. J. 325, 455–463. doi: 10.1042/bj3250455
Hashioka, T., and Kato, M. (1990). Effects of inhibitors of intracellular messenger systems on amylase release from rat parotid gland. Aichi Gakuin Dent. Sci. 3, 59–66.
Hayes, N. V. L., Bennett, A. F., and Baines, A. J. (1991). Selective Ca2+-dependent interaction of calmodulin with the head domain of synapsin-1. Biochem. J. 275, 93–97. doi: 10.1042/bj2750093
Hilfiker, S., Pieribone, V. A., Nordstedt, C., Greengard, P., and Czernik, A. J. (1999). Regulation of synaptotagmin I phosphorylation by multiple protein kinases. J. Neurochem. 73, 921–932. doi: 10.1046/j.1471-4159.1999.0730921.x
Hu, Z., Tong, X.-J., and Kaplan, J. M. (2013). UNC-13L, UNC-13S, and Tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. Elife 2:e00967. doi: 10.7554/eLife.00967
Jahn, R., and Sudhof, T. C. (1994). Synaptic vesicles and exocytosis. Annu. Rev. Neurosci. 17, 219–246. doi: 10.1146/annurev.neuro.17.1.219
Jahn, R., Lang, T., and Sudhof, T. C. (2003). Membrane fusion. Cell 112, 519–533. doi: 10.1016/s0092-8674(03)00112-0
Johannes, L., Lledo, P. M., Roa, M., Vincent, J. D., Henry, J. P., and Darchen, F. (1994). The gtpase Rab3A negatively controls calcium-dependent exocytosis in neuroendocrine cells. EMBO J. 13, 2029–2037. doi: 10.1002/j.1460-2075.1994.tb06476.x
Junge, H. J., Rhee, J. S., Jahn, O., Varoqueaux, F., Spiess, J., Waxham, M. N., et al. (2004). Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 118, 389–401. doi: 10.1016/j.cell.2004.06.029
Kajio, H., Olszewski, S., Rosner, P. J., Donelan, M. J., Geoghegan, K. F., and Rhodes, C. J. (2001). A low-affinity Ca2+-dependent association of calmodulin with the Rab3A effector domain inversely correlates with insulin exocytosis. Diabetes 50, 2029–2039. doi: 10.2337/diabetes.50.9.2029
Kerboeuf, D., Leberre, A., Dedieu, J. C., and Cohen, J. (1993). Calmodulin is essential for assembling links necessary for exocytotic membrane-fusion in paramecium. EMBO J. 12, 3385–3390. doi: 10.1002/j.1460-2075.1993.tb06012.x
Kibble, A. V., and Burgoyne, R. D. (1996). Calmodulin increases the initial rate of exocytosis in adrenal chromaffin cells. Pflugers Arch. Eur. J. Physiol. 431, 464–466.
Kolarow, R., Brigadski, T., and Lessmann, V. (2007). Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium-calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J. Neurosci. 27, 10350–10364. doi: 10.1523/jneurosci.0692-07.2007
Krausz, Y., Wollheim, C. B., Siegel, E., and Sharp, G. W. G. (1980). Possible role for calmodulin in insulin release - studies with trifluoperazine in rat pancreatic-islets. J. Clin. Invest. 66, 603–607. doi: 10.1172/jci109893
Lee, J. S., Kim, M. H., Ho, W. K., and Lee, S. H. (2008). Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of held synapse. J. Neurosci. 28, 7945–7953. doi: 10.1523/jneurosci.2165-08.2008
Lee, J., Guan, Z., Akbergenova, Y., and Littleton, J. T. (2013). Genetic analysis of synaptotagmin C2 domain specificity in regulating spontaneous and evoked neurotransmitter release. J. Neurosci. 33, 187–200. doi: 10.1523/JNEUROSCI.3214-12.2013
Li, L., Liu, H., Krout, M., Richmond, J. E., Wang, Y., Bai, J., et al. (2021). A novel dual Ca2+ sensor system regulates Ca2+-dependent neurotransmitter release. J. Cell Biol. 220:e202008121. doi: 10.1083/jcb.202008121
Li, L., Liu, H., Wang, W., Chandra, M., Collins, B. M., and Hu, Z. (2018). SNT-1 functions as the ca(2+) sensor for tonic and evoked neurotransmitter release in caenorhabditis elegans. J. Neurosci. 38, 5313–5324. doi: 10.1523/JNEUROSCI.3097-17.2018
Lin, R. C., and Scheller, R. H. (2000). Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 16, 19–49. doi: 10.1146/annurev.cellbio.16.1.19
Lipstein, N., Sakaba, T., Cooper, B. H., Lin, K.-H., Strenzke, N., Ashery, U., et al. (2013). Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca2+-Calmodulin-Munc13-1 signaling. Neuron 79, 82–96. doi: 10.1016/j.neuron.2013.05.011
Littleton, J. T., Stern, M., Schulze, K., Perin, M., and Bellen, H. J. (1993). Mutational analysis of drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell 74, 1125–1134. doi: 10.1016/0092-8674(93)90733-7
Liu, H., Bai, H., Hui, E., Yang, L., Evans, C. S., Wang, Z., et al. (2014). Synaptotagmin 7 functions as a Ca2+-sensor for synaptic vesicle replenishment. Elife 3:e01524. doi: 10.7554/eLife.01524
Liu, H., Dean, C., Arthur, C. P., Dong, M., and Chapman, E. R. (2009). Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J. Neurosci. 29, 7395–7403. doi: 10.1523/jneurosci.1341-09.2009
Liu, Q., Chen, B., Ge, Q., and Wang, Z. W. (2007). Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J. Neurosci. 27, 10404–10413. doi: 10.1523/jneurosci.5634-06.2007
Liu, X., Heidelberger, R., and Janz, R. (2014). Phosphorylation of syntaxin 3B by CaMKII regulates the formation of t-SNARE complexes. Mol. Cell. Neurosci. 60, 53–62. doi: 10.1016/j.mcn.2014.03.002
Lou, X. L., Scheuss, V., and Schneggenburger, R. (2005). Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature 435, 497–501. doi: 10.1038/nature03568
Lou, X., Fan, F., Messa, M., Raimondi, A., Wu, Y., Looger, L. L., et al. (2012). Reduced release probability prevents vesicle depletion and transmission failure at dynamin mutant synapses. Proc. Natl. Acad. Sci. U.S.A. 109, E515–E523. doi: 10.1073/pnas.1121626109
Loubery, S., and Coudrier, E. (2008). Myosins in the secretory pathway: tethers or transporters? Cell. Mol. Life Sci. 65, 2790–2800. doi: 10.1007/s00018-008-8350-5
Lu, M., Berglund, E., Larsson, C., Hoog, A., Farnebo, L.-O., and Branstrom, R. (2011). Calmodulin and calmodulin-dependent protein kinase II inhibit hormone secretion in human parathyroid adenoma. J. Endocrinol. 208, 31–39. doi: 10.1677/joe-10-0123
Ma, C., Li, W., Xu, Y., and Rizo, J. (2011). Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol. 18, 542–U206. doi: 10.1038/nsmb.2047
Mackler, J. M., Drummond, J. A., Loewen, C. A., Robinson, I. M., and Reist, N. E. (2002). The C2BCa2+-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418, 340–344. doi: 10.1038/nature00846
Maletic-Savatic, M., Koothan, T., and Malinow, R. (1998). Calcium-evoked dendritic exocytosis in cultured hippocampal neurons. Part II: mediation by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 18, 6814–6821. doi: 10.1523/JNEUROSCI.18-17-06814.1998
Matsumura, C., Kuwashima, H., and Kimura, T. (1999). Myosin light chain kinase inhibitors and calmodulin antagonists inhibit Ca2+- and ATP-dependent catecholamine secretion from bovine adrenal chromaffin cells. J. Auton. Pharmacol. 19, 115–121. doi: 10.1046/j.1365-2680.1999.00125.x
Meyer, T., Hanson, P. I., Stryer, L., and Schulman, H. (1992). Calmodulin trapping by calcium-calmodulin dependent protein-kinase. Science 256, 1199–1202. doi: 10.1126/science.256.5060.1199
Miller, S. G., and Kennedy, M. B. (1986). Regulation of brain type-II Ca-2+ calmodulin-dependent protein-kinase by autophosphorylation - a Ca-2+-triggered molecular switch. Cell 44, 861–870. doi: 10.1016/0092-8674(86)90008-5
Misonou, H., Ohara-Imaizumi, M., Murakami, T., Kawasaki, M., Ikeda, K., Wakai, T., et al. (1998). Protein kinase C controls the priming step of regulated exocytosis in adrenal chromaffin cells. Cell. Mol. Neurobiol. 18, 379–390. doi: 10.1023/a:1022593330685
Neher, E., and Penner, R. (1994). Neurobiology – mice sans synaptotagmin. Nature 372, 316–317. doi: 10.1038/372316a0
Nelson, T. Y., Lorenson, M. Y., Jacobs, L. S., and Boyd, A. E. (1987). Distribution of calmodulin and calmodulin-binding proteins in bovine pituitary - association of myosin light chain kinase with pituitary secretory granule membranes. Mol. Cell. Biochem. 74, 83–94.
Nicol, S., Rahman, D., and Baines, A. J. (1997). Ca2+-dependent interaction with calmodulin is conserved in the synapsin family: identification of a high-affinity site. Biochemistry 36, 11487–11495. doi: 10.1021/bi970709r
Nielander, H. B., Onofri, F., Valtorta, F., Schiavo, G., Montecucco, C., Greengard, P., et al. (1995). Phosphorylation of vamp synaptobrevin in synaptic vesicles by endogenous protein-kinases. J. Neurochem. 65, 1712–1720. doi: 10.1046/j.1471-4159.1995.65041712.x
Nishiki, T., and Augustine, G. J. (2004). Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J. Neurosci. 24, 8542–8550. doi: 10.1523/jneurosci.2545-04.2004
Nomura, K., Ohyama, A., Komiya, Y., and Igarashi, M. (2003). Minimal residues in linker domain of syntaxin 1A required for binding affinity to Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. Res. 72, 198–202. doi: 10.1002/jnr.10563
Nonet, M. L., Grundahl, K., Meyer, B. J., and Rand, J. B. (1993). Synaptic function is impaired but not eliminated in c-elegans mutants lacking synaptotagmin. Cell 73, 1291–1305. doi: 10.1016/0092-8674(93)90357-v
Ohyama, A., Hosaka, K., Komiya, Y., Akagawa, K., Yamauchi, E., Taniguchi, H., et al. (2002). Regulation of exocytosis through Ca2+/ATP-dependent binding of autophosphorylated Ca2+/calmodulin-activated protein kinase II to syntaxin 1A. J. Neurosci. 22, 3342–3351. doi: 10.1523/JNEUROSCI.22-09-03342.2002
Ohyama, A., Komiya, Y., and Igarashi, M. (2001). Globular tail of myosin-V is bound to VAMP/synaptobrevin. Biochem. Biophys. Res. Commun. 280, 988–991. doi: 10.1006/bbrc.2001.4236
Pang, Z. P., Cao, P., Xu, W., and Sudhof, T. C. (2010a). Calmodulin controls synaptic strength via presynaptic activation of calmodulin kinase II. J. Neurosci. 30, 4132–4142. doi: 10.1523/jneurosci.3129-09.2010
Pang, Z. P., Xu, W., Cao, P., and Suedhof, T. C. (2010b). Calmodulin suppresses synaptotagmin-2 transcription in cortical neurons. J. Biol. Chem. 285, 33930–33939. doi: 10.1074/jbc.M110.150151
Park, J. B., Farnsworth, C. C., and Glomset, J. A. (1997). Ca2+/calmodulin causes Rab3A to dissociate from synaptic membranes. J. Biol. Chem. 272, 20857–20865. doi: 10.1074/jbc.272.33.20857
Park, J. B., Kim, J. S., Lee, J. Y., Kim, J., Seo, J. Y., and Kim, A. R. (2002). GTP binds to Rab3A in a complex with Ca2+/calmodulin. Biochem. J. 362, 651–657.
Perin, M. S. (1996). Mirror image motifs mediate the interaction of the COOH terminus of multiple synaptotagmins with the neurexins and calmodulin. Biochemistry 35, 13808–13816. doi: 10.1021/bi960853x
Peters, C., and Mayer, A. (1998). Ca2+/calmodulin signals the completion docking and triggers a late step of vacuole fusion. Nature 396, 575–580. doi: 10.1038/25133
Petrucci, T. C., Macioce, P., and Paggi, P. (1991). Axonal-transport kinetics and posttranslational modification of synapsin-i in mouse retinal ganglion-cells. J. Neurosci. 11, 2938–2946.
Pinheiro, P. S., Houy, S., and Sorensen, J. B. (2016). C2-domain containing calcium sensors in neuroendocrine secretion. J. Neurochem. 139, 943–958. doi: 10.1111/jnc.13865
Popoli, M. (1993). Synaptotagmin is endogenously phosphorylated by Ca-2+ calmodulin protein kinase-II in synaptic vesicles. FEBS Lett. 317, 85–88. doi: 10.1016/0014-5793(93)81496-m
Quetglas, S., Iborra, C., Sasakawa, N., De Haro, L., Kumakura, K., Sato, K., et al. (2002). Calmodulin and lipid binding to synaptobrevin regulates calcium-dependent exocytosis. EMBO J. 21, 3970–3979. doi: 10.1093/emboj/cdf404
Quetglas, S., Leveque, C., Miquelis, R., Sato, K., and Seagar, M. (2000). Ca2+-dependent regulation of synaptic SNARE complex assembly via a calmodulin- and phospholipid-binding domain of synaptobrevin. Proc. Natl. Acad. Sci. U.S.A. 97, 9695–9700. doi: 10.1073/pnas.97.17.9695
Reck-Peterson, S. L., Provance, D. W., Mooseker, M. S., and Mercer, J. A. (2000). Class V myosins. BBA Mol. Cell Res. 1496, 36–51. doi: 10.1016/s0167-4889(00)00007-0
Regehr, W. G. (2012). Short-Term presynaptic plasticity. Cold Spring Harb. Perspect. Biol. 4:a005702. doi: 10.1101/cshperspect.a005702
Risinger, C., and Bennett, M. K. (1999). Differential phosphorylation of syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) isoforms. J. Neurochem. 72, 614–624. doi: 10.1046/j.1471-4159.1999.0720614.x
Rizzoli, S. O., and Betz, W. J. (2005). Synaptic vesicle pools. Nat. Rev. Neurosci. 6, 57–69. doi: 10.1038/nrn1583
Robinson, I. M., Ranjan, R., and Schwarz, T. L. (2002). Synaptotagmins I and IV promote transmitter release independently of Ca2+ binding in the C(2)A domain. Nature 418, 336–340. doi: 10.1038/nature00915
Rothman, J. E. (1994). Mechanisms of intracellular protein transport. Nature 372, 55–63. doi: 10.1038/372055a0
Sakaba, T., and Neher, E. (2001). Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 32, 1119–1131. doi: 10.1016/s0896-6273(01)00543-8
Sarafian, T., Aunis, D., and Bader, M. F. (1987). Loss of proteins from digitonin-permeabilized adrenal chromaffin cells essential for exocytosis. J. Biol. Chem. 262, 16671–16676.
Sasaki, H. (1992). A protein factor extracted from murine brains confers physiological Ca-2+ sensitivity to exocytosis in sea-urchin eggs. FEBS Lett. 304, 207–210. doi: 10.1016/0014-5793(92)80620-v
Schiavo, G., Stenbeck, G., Rothman, J. E., and Sollner, T. H. (1997). Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc. Natl. Acad. Sci. U.S.A. 94, 997–1001. doi: 10.1073/pnas.94.3.997
Schluter, O. M., Khvotchev, M., Jahn, R., and Sudhof, T. C. (2002). Localization versus function of Rab3 proteins - Evidence for a common regulatory role in controlling fusion. J. Biol. Chem. 277, 40919–40929. doi: 10.1074/jbc.M203704200
Schoch, S., Castillo, P. E., Jo, T., Mukherjee, K., Geppert, M., Wang, Y., et al. (2002). RIM1 alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415, 321–326. doi: 10.1038/415321a
Schonn, J. S., Maximov, A., Lao, Y., Suedhof, T. C., and Sorensen, J. B. (2008). Synaptotagmin-1 and-7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc. Natl. Acad. Sci. U.S.A. 105, 3998–4003. doi: 10.1073/pnas.0712373105
Schweitzer, E. S., Sanderson, M. J., and Wasterlain, C. G. (1995). Inhibition of regulated catecholamine secretion from PC12 cells by the Ca2+/calmodulin kinase-II inhibitor kn-62. J. Cell Sci. 108, 2619–2628.
Shang, Y., Chen, L., Liu, Z., Wang, X., Ma, X., and Miao, L. (2013). Cytosolic Ca2+ as a multifunctional modulator is required for spermiogenesis in Ascaris suum. Protein Cell 4, 456–466. doi: 10.1007/s13238-013-3019-8
Shao, X. G., Fernandez, I., Sudhof, T. C., and Rizo, J. (1998). Solution structures of the Ca2+-free and Ca2+-bound C(2)A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry 37, 16106–16115. doi: 10.1021/bi981789h
Sitges, M., Dunkley, P. R., and Chin, L. M. (1995). A role for calcium/calmodulin kinase(s) in the regulation of gaba exocytosis. Neurochem. Res. 20, 245–252. doi: 10.1007/bf00969539
Sollner, T. H. (2003). Regulated exocytosis and SNARE function (Review). Mol. Membr. Biol. 20, 209–220. doi: 10.1080/0968768031000104953
Spearman, T. N., and Butcher, F. R. (1983). The effect of calmodulin antagonists on amylase release from the rat parotid-gland invitro. Pflugers Arch. Eur. J. Physiol. 397, 220–224. doi: 10.1007/bf00584361
Steinberg, J. P., Leitner, J. W., Draznin, B., and Sussman, K. E. (1984). Calmodulin and cyclic-amp - possible different sites of action of these 2 regulatory agents in exocytotic hormone-release. Diabetes 33, 339–345. doi: 10.2337/diabetes.33.4.339
Steinhardt, R. A., and Alderton, J. M. (1982). Calmodulin confers calcium sensitivity on secretory exocytosis. Nature 295, 154–155. doi: 10.1038/295154a0
Steinhardt, R. A., Bi, G. Q., and Alderton, J. M. (1994). Cell-membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 263, 390–393. doi: 10.1126/science.7904084
Stevens, C. F., and Sullivan, J. M. (2003). The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron 39, 299–308. doi: 10.1016/s0896-6273(03)00432-x
Stevens, F. C. (1983). Calmodulin: an introduction. Can. J. Biochem. Cell Biol. 61, 906–910. doi: 10.1139/o83-115
Sudhof, T. C. (1995). The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375, 645–653. doi: 10.1038/375645a0
Sudhof, T. C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. doi: 10.1146/annurev.neuro.26.041002.131412
Sudhof, T. C. (2012). Calcium control of neurotransmitter release. Cold Spring Harb. Perspect. Biol. 4:a011353. doi: 10.1101/cshperspect.a011353
Sugita, S., Shin, O. H., Han, W. P., Lao, Y., and Sudhof, T. C. (2002). Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 21, 270–280. doi: 10.1093/emboj/21.3.270
Sullivan, R., Burnham, M., Torok, K., and Koffer, A. (2000). Calmodulin regulates the disassembly of cortical F-actin in mast cells but is not required for secretion. Cell Calcium 28, 33–46. doi: 10.1054/ceca.2000.0127
Sun, J., Pang, Z. P., Qin, D., Fahim, A. T., Adachi, R., and Sudhof, T. C. (2007). A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450, 676–682. doi: 10.1038/nature06308
Sun, L., Bittner, M. A., and Holz, R. W. (2003). Rim, a component of the presynaptic active zone and modulator of exocytosis, binds 14-3-3 through its N terminus. J. Biol. Chem. 278, 38301–38309. doi: 10.1074/jbc.M212801200
Tawata, M., Totsuka, Y., Nielsen, T., and Field, J. B. (1984). Cellular-distribution of thyroid myosin. Endocrinology 114, 962–968. doi: 10.1210/endo-114-3-962
Timofeeva, Y., and Volynski, K. E. (2015). Calmodulin as a major calcium buffer shaping vesicular release and short-term synaptic plasticity: facilitation through buffer dislocation. Front. Cell. Neurosci. 9:239. doi: 10.3389/fncel.2015.00239
Tojyo, Y., Matsumoto, Y., Okumura, K., and Kanazawa, M. (1989). The role of calmodulin in rat parotid amylase secretion - effects of calmodulin antagonists on secretion and acinar cell structure. Jpn. J. Pharmacol. 50, 149–157. doi: 10.1254/jjp.50.149
Trifaro, J. M., Gasman, S., and Gutierrez, L. M. (2008). Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol. 192, 165–172. doi: 10.1111/j.1748-1716.2007.01808.x
Van Hook, M. J., Parmelee, C. M., Chen, M., Cork, K. M., Curto, C., and Thoreson, W. B. (2014). Calmodulin enhances ribbon replenishment and shapes filtering of synaptic transmission by cone photoreceptors. J. Gen. Physiol. 144, 357–378. doi: 10.1085/jgp.201411229
Verhage, M., and Sorensen, J. B. (2008). Vesicle docking in regulated exocytosis. Traffic 9, 1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x
Verona, M., Zanotti, S., Schafer, T., Racagni, G., and Popoli, M. (2000). Changes of synaptotagmin interaction with t-SNARE proteins in vitro after calcium/calmodulin-dependent phosphorylation. J. Neurochem. 74, 209–221. doi: 10.1046/j.1471-4159.2000.0740209.x
Voets, T., Moser, T., Lund, P. E., Chow, R. H., Geppert, M., Sudhof, T. C., et al. (2001). Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc. Natl. Acad. Sci. U.S.A. 98, 11680–11685. doi: 10.1073/pnas.201398798
Wang, Y., Okamoto, M., Schmitz, F., Hofmann, K., and Sudhof, T. C. (1997). Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 388, 593–598. doi: 10.1038/41580
Wassarman, P. M. (1999). Mammalian fertilization: molecular aspects of gamete adhesion, exocytosis, and fusion. Cell 96, 175–183. doi: 10.1016/s0092-8674(00)80558-9
Watanabe, M., Nomura, K., Ohyama, A., Ishikawa, R., Komiya, Y., Hosaka, K., et al. (2005). Myosin-Va regulates exocytosis through the submicromolar Ca2+-dependent binding of syntaxin-1A. Mol. Biol. Cell 16, 4519–4530. doi: 10.1091/mbc.E05-03-0252
Watanabe, Y., Katayama, N., Takeuchi, K., Togano, T., Itoh, R., Sato, M., et al. (2013). Point mutation in syntaxin-1A causes abnormal vesicle recycling, behaviors, and short term plasticity. J. Biol. Chem. 288, 34906–34919. doi: 10.1074/jbc.M113.504050
Watkins, D. T., and Cooperstein, S. J. (1997). Effects of calcium and calmodulin on the binding of rat parotid secretion granules to the plasma membrane. J. Dent. Res. 76, 744–753. doi: 10.1177/00220345970760030601
Watkins, D., and White, B. A. (1985). Identification and characterization of calmodulin-binding proteins in islet secretion granules. J. Biol. Chem. 260, 5161–5165.
Wen, H., Linhoff, M. W., McGinley, M. J., Li, G. L., Corson, G. M., Mandel, G., et al. (2010). Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc. Natl. Acad. Sci. U.S.A. 107, 13906–13911. doi: 10.1073/pnas.1008598107
Wilson, S. P., and Kirshner, N. (1983). Calcium-evoked secretion from digitonin-permeabilized adrenal-medullary chromaffin cells. J. Biol. Chem. 258, 4994–5000.
Xue, R., Gaffaney, J. D., and Chapman, E. R. (2015). Structural elements that underlie Doc2 beta function during asynchronous synaptic transmission. Proc. Natl. Acad. Sci. U.S.A. 112, E4316–E4325. doi: 10.1073/pnas.1502288112
Yamamoto, H., Matsumoto, K., Araki, E., and Miyamoto, E. (2003). New aspects of neurotransmitter release and exocytosis: involvement of Ca2+/calmodulin-dependent phosphorylation of synapsin I in insulin exocytosis. J. Pharmacol. Sci. 93, 30–34. doi: 10.1254/jphs.93.30
Yang, H. J., Chen, P. C., Huang, C. T., Cheng, T. L., Hsu, S. P., Chen, C. Y., et al. (2021). The phosphoprotein Synapsin Ia regulates the kinetics of dense-core vesicle release. J. Neurosci. 41, 2828–2841. doi: 10.1523/jneurosci.2593-19.2021
Yao, J., Gaffaney, J. D., Kwon, S. E., and Chapman, E. R. (2011). Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell 147, 666–677. doi: 10.1016/j.cell.2011.09.046
Yokoyama, C. T., Myers, S. J., Fu, J., Mockus, S. M., Scheuer, T., and Catterall, W. A. (2005). Mechanism of SNARE protein binding and regulation of Ca(v)2 channels by phosphorylation of the synaptic protein interaction site. Mol.Cell. Neurosci. 28, 1–17. doi: 10.1016/j.mcn.2004.08.019
Yunes, R., Tomes, C., Michaut, M., De Blas, G., Rodriguez, F., Regazzi, R., et al. (2002). Rab3A and calmodulin regulate acrosomal exocytosis by mechanisms that do not require a direct interaction. FEBS Lett. 525, 126–130. doi: 10.1016/s0014-5793(02)03102-2
Zhou, Q. J., Lai, Y., Bacaj, T., Zhao, M. L., Lyubimov, A. Y., Uervirojnangkoorn, M., et al. (2015). Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525, 62–67. doi: 10.1038/nature14975
Keywords: calmodulin, exocytosis, Ca2+ sensor, synaptotagmin, vesicles
Citation: Xue R, Meng H, Yin J, Xia J, Hu Z and Liu H (2021) The Role of Calmodulin vs. Synaptotagmin in Exocytosis. Front. Mol. Neurosci. 14:691363. doi: 10.3389/fnmol.2021.691363
Received: 06 April 2021; Accepted: 19 July 2021;
Published: 05 August 2021.
Edited by:
Han Wang, Soochow University, ChinaReviewed by:
Peng Cao, National Institute of Biological Sciences (NIBS), ChinaIgor Medina, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2021 Xue, Meng, Yin, Xia, Hu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renhao Xue, eHVlcmVuaGFvQHRvbmdqaS5lZHUuY24=; Huisheng Liu, TGl1X2h1aXNoZW5nQGdybWgtZ2RsLmNu